Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Ubiquitination
SUMOylation
Phosphorylation
Acetylation
Other PTMs
Conclusions and Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(14):4047-4059. doi:10.7150/ijbs.64628 This issue Cite
Review
Post-Translational Modifications of PCNA in Control of DNA Synthesis and DNA Damage Tolerance-the Implications in Carcinogenesis
Siyi Zhang1#, Tingting Zhou1#, Zhuo Wang1, Fei Yi1, Chunlu Li1, Wendong Guo1, Hongde Xu1, Hongyan Cui1, Xiang Dong1, Jingwei Liu1, Xiaoyu Song1 ![]() , Liu Cao2
, Liu Cao2 ![]()
1. Institute of Health Sciences, China Medical University, Shenyang, Liaoning Province, 110122, PR China.
2. College of Basic Medical Science, Key Laboratory of Cell Biology of Ministry of Public Health, Key Laboratory of Medical Cell Biology of Ministry of Education, Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, China Medical University, Shenyang, Liaoning Province, 110122, PR China.
#These authors contributed equally to this work.
Received 2021-7-6; Accepted 2021-9-19; Published 2021-9-23
Abstract

The faithful DNA replication is a critical event for cell survival and inheritance. However, exogenous or endogenous sources of damage challenge the accurate synthesis of DNA, which causes DNA lesions. The DNA lesions are obstacles for replication fork progression. However, the prolonged replication fork stalling leads to replication fork collapse, which may cause DNA double-strand breaks (DSB). In order to maintain genomic stability, eukaryotic cells evolve translesion synthesis (TLS) and template switching (TS) to resolve the replication stalling. Proliferating cell nuclear antigen (PCNA) trimer acts as a slide clamp and encircles DNA to orchestrate DNA synthesis and DNA damage tolerance (DDT). The post-translational modifications (PTMs) of PCNA regulate these functions to ensure the appropriate initiation and termination of replication and DDT. The aberrant regulation of PCNA PTMs will result in DSB, which causes mutagenesis and poor response to chemotherapy. Here, we review the roles of the PCNA PTMs in DNA duplication and DDT. We propose that clarifying the regulation of PCNA PTMs may provide insights into understanding the development of cancers.
Keywords: replication, DDT, ubiquitination, phosphorylation, acetylation, SUMOylation, NEDDylation, ISGylation
Introduction
Accurate DNA replication is essential for appropriate cell division. Human cells suffer from various “stress” at the replication fork, where the stress derives from replication obstacles and sources of damage. The replication obstacles contain re-replication, misregulated origin licensing, nucleotide depletion, and oncogene activation. The damage sources include reactive oxygen species (ROS), ultraviolet (UV), ionizing radiation (IR), and DNA damaging chemotherapy drugs. During replication, DNA synthesis stalls when replication machinery encounters lesions. Persistent replication stalling will cause DNA double-strand break (DSB). Because the failure of post-replication DNA damage repair leads to cell death or genomic instability, organisms evolve DNA damage tolerance (DDT) mechanisms to bypass DNA lesions. DDT pathways mainly consist of the error-prone translesion synthesis (TLS) and error-free template switching (TS) pathway.
Proliferating cell nuclear antigen (PCNA) plays a central role in DNA synthesis and DDT. PCNA serves as a moving platform recruiting various factors for replication or DDT. A conserved motif named PCNA-interacting protein box (PIP box) has been found in PCNA-binding partners. The core element is a consensus sequence Q1-x2-x2-h4-x5-x6-a7-a8, where “h” represents hydrophobic residues (L, M, I, or V) and “a” being aromatic amino acids (F, Y, or H) [1].
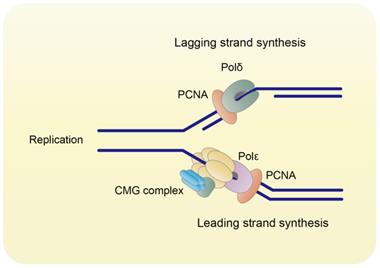
During DNA replication, three monomers of PCNA form a ring-shaped homo-trimer (Figure 1), which encircles DNA and recruits B-family DNA polymerases (Polδ and Polε) to carry out the high fidelity DNA synthesis (Figure 2) [2]. In eukaryotes, Polδ is dominant over Polε in association with PCNA [3]. Polδ is responsible for the lagging strand synthesis, while Polε acts on the leading strand for replication [4, 5].
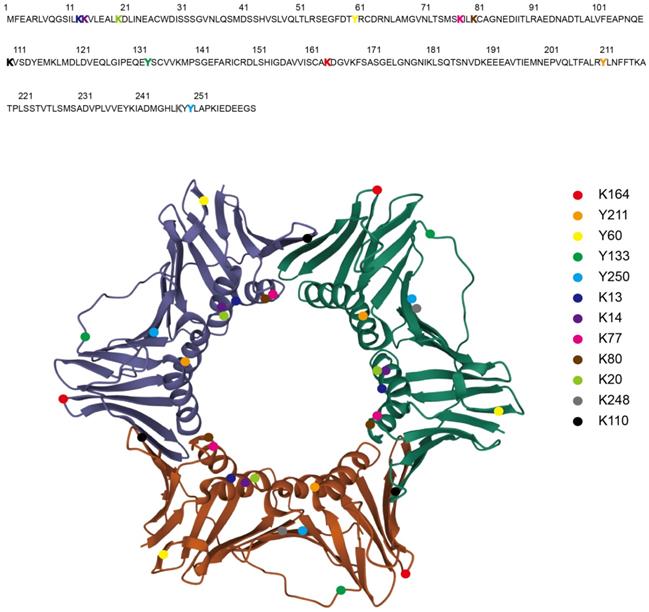
The Structure of PCNA Homo-trimer and the PTMs Sites. The crystal structure of Homo sapiens PCNA homo-trimer is shown as each monomer with different color (Protein Data Bank: 1VYM) [158]. The PTMs sites are highlighted in the amino acid sequence as colored characters (upper) and in the homo-trimer as colored balls (lower). Ubiquitination (K164), SUMOylation (K164), phosphorylation (Y211, Y60, Y133, Y250), acetylation (K13, K14, K77, K80, K20), methylation (K248, K110).
PCNA orchestrates DNA replication. PCNA binds to Polδ and Polε to orchestrate DNA lagging strand and leading strand synthesis, respectively.
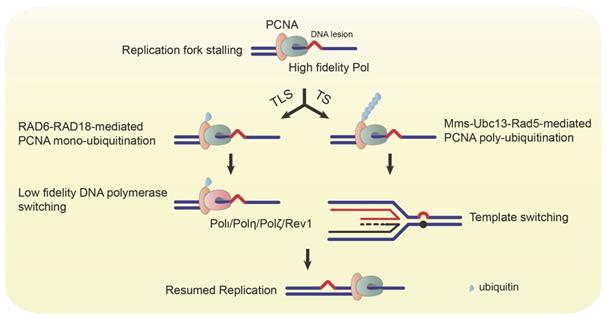
The replication fork stalls when the replication machinery confronts DNA lesions, and PCNA is mono-ubiquitinated at lysine residue (K) 164 by RAD6-RAD18 complex [6]. The mono-ubiquitinated PCNA switches its association from high fidelity polymerases to low fidelity ones, such as Polι, Polη, REV1, and Polζ [7-9]. These polymerases help the replication fork bypass the damaged bases to continue DNA synthesis (Figure 3) [10-14]. However, every coin has two sides. The TLS pathway ensures uninterrupted replication and protects cells from DSB, while it also increases the frequency of mutations due to the low accuracy of TLS polymerases. In addition to the RAD6-RAD18 complex, the K63 poly-ubiquitination of PCNA at K164 requires the Mms2-Ubc13-Rad5 complex. The poly-ubiquitinated PCNA triggers the error-free TS (Figure 3) [15]. Thus, dysregulation of replication and the TLS/TS pathway will result in genomic instability and tumorigenesis.
Ubiquitinated PCNA mediates TLS and TS. When the replication fork encounters a DNA lesion, the replication machinery stalls, and PCNA is mono-ubiquitinated by the RAD6-RAD18 complex. The mono-ubiquitinated PCNA switches recruitment of low fidelity DNA polymerases (Polι, Polη, Polζ, and Rev1) to bypass the lesion. The mono-ubiquitinated PCNA can also be poly-ubiquitinated by the Mms-Ubc13-Rad5 complex. The poly-ubiquitinated PCNA mediates the template switching to resume replication.
Post-translational modifications (PTMs) possess various functions such as modulating protein activity, localization, stability, and interactions [16]. This review discusses the recent studies about the PTMs in regulating PCNA functions and the implications in tumor development.
Ubiquitination
Ubiquitin is an 8.5 kDa small protein with 76 amino acids. Protein ubiquitination, discovered in the early 1980s, is sequentially catalyzed by the ubiquitin system containing an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin ligase (E3). Initially, studies reported that ubiquitination mainly guides the tagged protein for 26S proteasome degradation.
As the investigation develops in-depth, proteins are either modified with a single ubiquitin (mono-ubiquitination) or polymeric ubiquitin chains (poly-ubiquitination). Mono-ubiquitination serves as a signal for DNA damage response, such as mono-ubiquitinated H2AX/H2A/H2B and PCNA [17]. Additionally, it acts as a critical step for subsequent assembly of poly-ubiquitination [18]. Based on the different lysine (K) sites of the ubiquitin conjugation, poly-ubiquitination is diverse in various types, including K6, K11, K27, K29, K33, K48, and K63 poly-ubiquitination [19, 20]. Different ubiquitin linkages result in a diversity of cellular signals. The most well-studied types of poly-ubiquitination are the K48- and K63-ubiquitin chains. The K48 poly-ubiquitination labels substrates for proteasome degradation [21], while the K63 poly-ubiquitination participates in DNA damage response [22]. Protein ubiquitination is a reversible process that can be influenced by the ubiquitin-cleavage activity of deubiquitinating enzymes (DUBs).
In response to stalled replication forks, PCNA is mono-ubiquitinated by a sequential process mediated by an E1, RAD6 (E2), and RAD18 (E3) [6]. The human homolog of RAD6 is HHR6A and HHR6B [23]. The human RAD18, an approximately 54kDa protein consisting of 484 amino acids, is corresponded to yeast RAD18 [24]. PCNA is mono-ubiquitinated specifically at K164 during TLS [6, 12]. When PCNA is mono-ubiquitinated, it switches its interaction from high fidelity Polδ or Polε to low fidelity TLS polymerases (Polη, Polι, Polκ, Polλ, Polζ, or REV1). Then, these TLS polymerases proceed to bypass the lesions [10-14, 25].
High expressions of RAD6 and RAD18 are positively related to the development and therapy resistance of various tumors [26-36]. Sanders et al. identified the small molecular inhibitors of RAD6. Among them, SMI#9 shows a potent inhibition effect on the proliferation and migration of breast cancer cells [37]. It has been demonstrated that SMI#9 attenuates chemotherapy agent-induced PCNA mono-ubiquitination and enhances chemosensitivity of triple-negative breast cancer (TNBC) or ovarian cancer [35, 36, 38]. Inhibition of RAD18 has also been found to suppress gastric cancer progression and sensitize gastric cancer to chemotherapy via reduction of PCNA mono-ubiquitination [31]. However, the small molecular inhibitors of RAD18 have not been designed, which needs further studies to find effective ones for tumor treatment.
The K63-linked poly-ubiquitination of PCNA at its K164 site is involved in error-free lesion bypass via TS [15]. In addition to the RAD6-RAD18 complex, PCNA poly-ubiquitination requires RING-finger E3 ligase Rad5, which recruits Ubc13 and Mms2 as an E2 complex [39]. The human homolog of Rad5 contains the helicase-like transcription factor (HLTF) and the SNF2 histone linker PHD RING helicase (SHPRH) to mediate PCNA poly-ubiquitination [40]. The mechanism of poly-ubiquitinated PCNA regulating lesion bypass has not been fully understood. One possibility is that poly-ubiquitinated PCNA may induce replication fork reversion to promote a recombination-like process using the undamaged DNA duplex as a template.
DNA binding is critical for PCNA mono-ubiquitination, as mono-ubiquitination of free PCNA is inhibited by the overabundance of free replication protein A (RPA) through its interaction with RAD18 under native conditions [41]. RPA, the main ssDNA-binding protein, is required for DNA replication, recombination, DNA damage repair, cell cycle checkpoints, and DNA damage checkpoints [42]. RPA undergoes a conformational change during replication stress when it binds to ssDNA and forms filaments at TLS sites. RAD6-RAD18 complex is recruited to the RPA filaments and then promotes the mono-ubiquitination of PCNA on DNA [41, 43].
The chromatin microenvironment modulated by various factors may be crucial for the regulation of PCNA ubiquitination. The chromatin structure modulated by histone modifications and nucleosome remodeling is essential for the control of replication, transcription, and DNA damage repair [44-46]. The cells with the mutations of histone H3/H4, which participated in packaging DNA into chromatin, showed a reduced level of PCNA mono- and di-ubiquitination after treating with methyl methanesulfonate (MMS) or UV. Accordingly, the H3/H4 mutants exhibited decreased MMS-induced mutagenesis compared to wild-type ones [47].
WRN protein is one member of the RecQ helicase family. The mutation of the WRN gene causes Werner Syndrome (WS) with premature aging and predisposition to cancer [48, 49]. In response to DNA damage agents, WRN forms discrete nuclear foci only during the S phase [50, 51], implying the essential roles of WRN in replication-post DNA damage repair. WRN interacts with PCNA in the absence of DNA damage. ATM-mediated phosphorylation of WRN causes its degradation in response to DNA damage. This degradation results in the release of PCNA from WRN that probably promotes PCNA ubiquitination. The WS and WRN knockdown cells show high-level ubiquitination of PCNA even without DNA damage [51]. These results indicate that WRN may inhibit aging and carcinogenesis via PCNA ubiquitination regulation. A small molecular inhibitor NSC19630 against WRN effectively inhibits tumor cell growth and induces apoptosis via impairing the response to DNA damage and replication stress [52]. Novel small molecular inhibitors targeting WRN have also been screened out, prohibiting the proliferation of cancer cells [53]. However, the underlying mechanisms require further investigation.
The human PTIP protein, the homolog of the Xenopus Swift with six BRCT domains participating in DNA damage repair, is an adaptor for ATM/ATR kinase [54-56]. The investigation from PTIP/Swift deleted mouse and yeast models reveals that PTIP/Swift plays a significant role in DNA damage repair during replication [57, 58]. Depleting PTIP/Swift in Xenopus and human cells compromised the mono- and di-ubiquitination of PCNA in response to UV [59], raising the potential role of PTIP/Swift in DDT.
RNF8 is an E3 ligase and a key DNA damage response factor [60]. It was identified as a novel ligase for mono- and poly-ubiquitination of PCNA using UbcH5c and Ubc13/Uev1a as an E2 ubiquitin-conjugating enzyme, respectively [60]. Similar to the RAD6-RAD18 complex, RNF8-UbcH5c-mediated mono-ubiquitination of PCNA serves as a substrate for poly-ubiquitination [60]. In medulloblastoma cells, knockdown of RNF8 inhibited IR-induced PCNA ubiquitination while enhanced cell cycle arrest and apoptosis [61]. However, the deletion of RNF8 barely affected PCNA ubiquitination in DT40 chicken cells after exposure to UV [62]. The effects of RNF8 on PCNA ubiquitination call for further research in different species of cells.
The insulin-like growth factor (IGF) signal pathway primarily functions in cell proliferation, survival, apoptosis, differentiation, metabolism, and migration [63]. It also participates in PCNA ubiquitination. After exposure to UVB, the skin of individuals over the age of 65 shows an increased level of PCNA mono-ubiquitination [64]. The geriatric skin contains a decreased expression of IGF-1 and reduced activation of IGF-1 receptor (IGF-1R) [65]. Both deprivation of IGF-1 and inhibition of IGF-1R result in elevated UVB-induced PCNA mono-ubiquitination in human skin and keratinocytes [64]. Exposure to UV is the primary cause of non-melanoma skin cancer, which possesses mutagenic photoproducts in DNA. These findings suggest that the IGF signaling pathway may prevent UV-induced carcinogenesis by regulating PCNA ubiquitination.
Phosphorylation kinases are emerging as targets attracting the most attention for cancer therapy development [66, 67]. By screening the library of kinase inhibitors, the inhibitor targeting pro-survival kinase Akt has been shown to impair the mono-ubiquitination of PCNA in cells after UV exposure. Persistent inhibition of Akt resulted in the blockage of TLS Polη recruitment to DNA damage sites and further perturbation of DNA replication processivity, which led to synthetic lethality in homologous recombination (HR)-deficient cells [68]. However, this study did not unravel whether Akt could phosphorylate PCNA to regulate its ubiquitination. Nevertheless, it points to the field of modification crosstalk targeting PCNA for TLS regulation.
The deubiquitination enzyme USP1 reversely regulates PCNA ubiquitination in the S phase, which is critical to prevent unwanted mutagenesis. After the cells are exposed to UV, USP1 is downregulated by autocleavage and accompanied by upregulation of PCNA ubiquitination [69]. USP1-induced deubiquitination of PCNA is positively regulated by ELG1, the component of the RFC complex. ELG1 directs USP1-UAF1 complex for PCNA [70]. USP1 is upregulated in BRCA1 deficient tumors. The USP1-mediated restriction of PCNA ubiquitination protects replication fork in BRCA1-deficient cells because persistent ubiquitination of PCNA recruits Polκ and REV1, which contributes to replication fork instability. Thus, the USP1 inhibitor may be beneficial for a subset of PARP inhibitor-resistant BRCA1-deficient tumors [71].
Kashiwaba et al. have identified another DUB USP7, which is responsible for the reduction of H2O2-induced PCNA mono-ubiquitination. Suppression of USP7 increases H2O2-induced mutagenesis in cells [72]. It has also been found that USP7 removes K63 poly-ubiquitination of PCNA [73]. Surprisingly, USP7 can also facilitate UV-induced mono-ubiquitination of PCNA via deubiquitinating and stabilizing Polη [74, 75]. Hence, further studies need to clarify whether USP7 affects PCNA ubiquitination in cells under different stresses. Moreover, ubiquitinated PCNA can also be reverted by Ubp2, Ubp10, Ubp12, and Ubp15 in the S phase [76-78].
Thus, timely regulation of ubiquitination and deubiquitination of PCNA is essential to limit DDT and ensure appropriate DNA replication.
SUMOylation
The SUMOylation is a reversible and dynamic PTM that conjugates small ubiquitin-related modifier (SUMO) to the lysine of target proteins. The nascent SUMO is proteolytically cleaved by SUMO-specific proteases (SUPs) to expose the C-terminal glycine-glycine (GG) motif. The SUMO E1 enzyme activates the mature SUMO. Then, it is transferred by the SUMO E2 and E3 enzyme to the substrate. SUMOylation is involved in several biological processes, including protein-protein interactions, protein-DNA binding, subcellular localization, transcriptional regulation, and DNA repair [79]. The aberrant regulation of SUMOylation is frequently related to cancer, cardiac disease, and neurodegenerative disease [80-82].
The human SUMO molecules contain four paralogs (SUMO1-4). PCNA can be conjugated with both SUMO1 and SUMO2 [83-85]. PCNA is SUMOylated at K164 and K127. Distinctively from K164 being both ubiquitinated and SUMOylated, K127 is only SUMOylated but cannot be ubiquitinated [6]. By co-expressing ectopic PCNA with SUMO1, SUMO2, and SUMO3 in human cells, it was found that SUMO1 is predominantly conjugated to PCNA, while SUMO2 or SUMO3 is weakly attached to PCNA even with UV treatment. Furthermore, the in vitro PCNA SUMOylation assay revealed that the E1 SUMO-activating enzyme (SAE1/SAE2), the E2 SUMO-conjugating enzyme (Ubc9), and the E3 SUMO-ligases (Pias1, Pias2, Pias3, and Pias4) participate in SUMO1 conjugating to PCNA [84].
PCNA SUMOylation occurs during the S phase or in the presence of DNA damage. In yeast, the SUMOylated PCNA cooperates with the helicase Srs2 to inhibit HR repair during the S phase [86]. The recruitment of Srs2 by SUMOylated PCNA at replication sites disrupts Rad51 nucleoprotein filaments and dissociates Polδ/Polη from the repair synthesis machinery [87]. This mechanism controls the spontaneous HR [88], which contributes to maintaining genomic stability.
The replication factor C (RFC) is a conserved chaperone-like complex, which loads PCNA onto chromatin. PCNA SUMOylation by SUMO1 is facilitated by RFC. The association of PCNA to RFC is the prerequisite for SUMOylation, while DNA binding is not necessary [84]. The human SUMO1-PCNA has also been found to suppress inappropriate HR by recruiting the helicase PARI (Figure 4A) [83]. Furthermore, it has been demonstrated that the expression of SUMO1-PCNA fusion protein in human cells inhibits HR in response to DSB and reduces MMS-induced γH2A foci and fragmented double-stranded DNA. The mutation at K164 for PCNA SUMOylation in Rad18-/- cells eliminates the ubiquitination at this site and increases the MMS-induced γH2A foci [84]. However, rather than generating DSB directly, MMS-induced DSB derives from replication fork stalling [89]. Thus, these observations indicate that the SUMOylation of PCNA by SUMO1 may be critical in preventing the replication fork collapse to DSB to maintain genome stability.
The Functions of SUMOylated PCNA. (A) The prolonged stalling of the replication fork induces the SAE1/2-Ubc9-mediated SUMOylation of PCNA by SUMO1. RFC facilitates the SUMOylation of PCNA. The SUMOylated PCNA recruits PARI to inhibit HR. (B) The TRC promotes the RECQ5-induced SUMOylation of PCNA by SUMO2. SUMO2-PCNA removes RNAPⅡ by FACT-mediated removal of transcriptionally repressive histone and CAF1-mediated deposition of transcriptionally active histone.
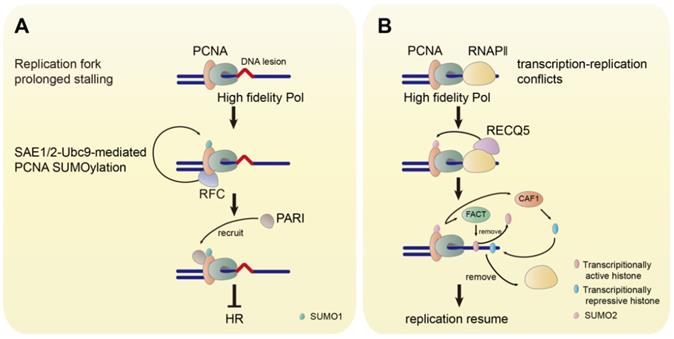
DSB can also occur in unperturbed growing cells induced by transcription-replication conflicts (TRCs). The TRCs cause replication fork stalling leading to replication fork collapse, which results in DSB formation and genomic instability [90, 91]. The E3 ligase TRIM28 catalyzes the SUMO2 conjugation to PCNA, which is triggered by transcription to resolve the TRCs [92]. The SUMOylation of PCNA by SUMO2 is induced by the RNA polymerase Ⅱ (RNAPII)-bound helicase RECQ5, which suppresses transcription-related DSB and functions as a tumor suppressor [93-97]. SUMO2-PCNA temporarily dissociates RNAPⅡ in the collision path by enriching the histone chaperone FACT to remove parental histones ahead of the replication fork and CAF1 to deposit repressive histone marks at the replication site, that ensures the progression of the replication fork avoiding DSB risks (Figure 4B) [97].
The SUMOylation can also serve as a signal for ubiquitination. The SUMO-targeting ubiquitin ligases (STUbLs) harboring SUMO-interaction motifs (SIMs) mediate the ubiquitination of SUMO moieties or SUMOylated proteins [98, 99]. The mutant of the K164/K127 of PCNA or SIM of RAD18 reduces PCNA ubiquitination, suggesting that the SUMOylation of PCNA can direct RAD18 to target PCNA [100]. This observation implies the crosstalk between SUMOylation and ubiquitination of PCNA.
Phosphorylation
Phosphorylation, discovered in the mid-1950s, is a principal way to regulate most cellular functions, including protein stability, subcellular localization, and protein-protein interaction. It reversibly confers phosphate group to substrate proteins at a serine, threonine, or tyrosine residue [101].
The chromatin binding is essential for PCNA to exert DNA replication and DDT functions. PCNA is phosphorylated when it binds to DNA synthesis sites [102, 103]. In 1995, Zophonias O. Jonsson et al. found that the Y211 site of PCNA is critical for chromatin loading, as the Y211A mutation of PCNA completely abolishes its trimer formation, which then cannot be loaded onto DNA by RFC [104]. Subsequently, it was found that the Y211 site of PCNA can be phosphorylated by the epidermal growth factor (EGF) receptor (EGFR) to stabilize the chromatin-bound PCNA. The increased PCNA Y211 phosphorylation is frequently correlated to prostate cancer and breast cancer [105, 106]. Blocking PCNA Y211 phosphorylation by the Y211F PCNA peptide, the proliferation of prostate cancer and TKI-resistant triple-negative breast cancer (TNBC) cells is significantly restrained [106, 107]. The treatment of cells with EGFR inhibitors such as AG1478 or lapatinib suppresses Y211 phosphorylation of PCNA and downregulates chromatin-bound PCNA [108].
In the absence of EGFR, the phosphorylation of PCNA at the Y211 site is also catalyzed by the non-receptor tyrosine kinase c-Abl when the upstream Ron receptor is activated by its ligand hepatocyte growth factor-like protein (HGFL). Imatinib, a pharmacological inhibitor of c-Abl, suppresses the HGFL-induced proliferation of breast cancer cells [88]. Thus, the Y211 site of PCNA may be an effective target to develop cancer therapy.
PCNA can also be phosphorylated at Y60, Y133, and Y250 sites by the nuclear IGF-1R (nIGF-1R). The phosphorylation of PCNA leads to the recruitment of E2 conjugating enzyme (UBC13) and E3 ligases (RAD18, SHPRH, and HLTF) for PCNA mono- and poly-ubiquitination. In IGF-1R negative cells, DNA damage induces G1 arrest and S phase replication fork stalling, while the nIGF-1R activation can rescue the stalled replication fork [109]. IGF-1R and PCNA are colocalized in many cancer types. The colocalization increases in the tumor tissues compared to the adjacent normal tissues. This colocalization is frequently lost in carcinomas with poor chemotherapy response and a majority of metastatic lesions. However, the stronger colocalization of IGF-1R and PCNA is correlated to the higher overall survival in cancer patients [110].
These findings suggest that the phosphorylation of PCNA is crucial for its loading onto chromatin, which serves as a prerequisite for the succeeding functions of PCNA. Moreover, the phosphorylation may also have crosstalk with the ubiquitination of PCNA.
Acetylation
The acetylation was first identified on histones in 1964, followed by the discovery of acetylation of non-histone proteins such as high-mobility group (HMG) proteins, tubulin, and p53 [111-115]. Lysine acetylation is dynamically controlled by two groups of enzymes. The enzymes are lysine acetyltransferases (HATs) and lysine deacetylases (HDACs) [116]. The non-histone protein acetylation is implicated in diverse functions, including gene transcription, cell cycle regulation, DNA damage repair, protein stability, protein folding, protein aggregation, cytoskeleton organization, RNA processing, RNA stability, and autophagy [117].
Hoyun Lee and colleagues found that mammalian PCNA is not regulated by phosphorylation but by acetylation. They found that PCNA may be acetylated and deacetylated by p300 and HDAC1, respectively [118]. The acetylated PCNA shows a high affinity to Polβ and Polδ, which is important for DNA replication. The acetylation of PCNA at K14 is induced after UV damage and promotes PCNA for degradation to inhibit DNA replication [119]. Ennio Prosperi and colleagues further clarified that CBP and p300 are required for the acetylation of PCNA. CBP- and p300-mediated acetylation of PCNA at K13, K14, K77, and K80 promotes the removal of chromatin-bound PCNA and its degradation after DNA synthesis [120]. Thus, the acetylation of PCNA avoids the excessive retention of PCNA on chromatin, preventing genomic instability.
In response to MMS-induced DNA damage, the sliding surface of PCNA is acetylated at K20 by Eco1, which stimulates HR and suppresses the DNA damage sensitivity of the DDT mutant yeast cells [121]. Depleting HAT3 in Leishmania donovani caused the loss of PCNA ubiquitination in response to UV [122]. This observation indicates that the HAT3-mediated PCNA acetylation may be a prerequisite for PCNA ubiquitination. This study links the acetylation to the ubiquitination of PCNA in eukaryotes.
Other PTMs
Methylation
The understanding of protein methylation is expanding these years since the first report of flagellar protein methylation in 1959. The methyl group is transferred by methyltransferase from S-adenosylmethionine (SAM) to lysine, arginine, aspartic acid, or glutamic acid. The methylation is reversible as the methyl group can be removed from substrates by demethylases.
PCNA was found to be methylated at K248 by histone H4K20 methyltransferase SETD8, and this methylation allows the stabilization of PCNA expression [123, 124]. SETD8-mediated PCNA methylation increased the interaction between PCNA and flap endonuclease FEN1. This effect is essential for DNA replication and repair. The methylation-inactive mutation of PCNA causes impaired DNA replication and induced DNA damage [123]. Depleting SETD8 results in decreased cell proliferation and increased DNA damage during the S phase [125]. SETD8 is overexpressed in various types of cancers. The expression of SETD8 and PCNA are correlated in cancer samples, implying the crucial role of SETD8-mediated methylation of PCNA in tumor development [123]. Various SETD8 inhibitors have been discovered, such as Nahuoic acid A, UNC0379, SPS8I1-3, MS453 [126-129]. Among these inhibitors, UNC0379 shows an anti-growth effect on neuroblastoma and ovarian cancer [130, 131]. The functional effect of other inhibitors on tumor development and whether they target the regulatory sites of PCNA by SETD8 requires further identification.
The methyltransferase enhancer of zeste homolog 2 (EZH2) is responsible for the tri-methylation of histone H3 at K27 [132-134]. EZH2 is positively related to cancer cell proliferation and localizes at the replication fork in response to DNA damage [135-138]. EZH2 directly binds to PCNA via PIP-box and methylates PCNA at K110. The EZH2-mediated methylation of PCNA stabilizes the PCNA trimer to enhance its interaction with Polδ and DNA replication [139]. Inhibitors targeting EZH2 such as 3-Deazaneplanocin A (DZNep), GSK126, EI1, and UNC1999 exhibit a proliferation suppression effect on various tumor cells or cancer models, including Acute Myeloblastic Leukemia (AML), lymphoma, gastric cancer, breast cancer, and MLL-rearranged leukemia [140-145]. Additionally, the inhibitor CPI-1205 being in clinical trials has shown a potent anti-tumor effect [146].
NEDDylation
The neuronal precursor cell-expressed developmentally down-regulated protein 8 (NEDD8) is a small ubiquitin-like molecule. NEDD8 is reversibly conjugated to a lysine residue of target proteins [147]. Similar to the ubiquitin system, NEDDylation is also a process triggered by the NEDD8-activating enzyme (E1), NEDD8-conjugating enzyme (E2), and NEDD8 ligase (E3). NEDDylation participates in DNA damage response, such as the decision between non-homologous end joining (NHEJ) and HR [148-150]. PCNA can be NEDDylated by RAD18. The deNEDDylation of PCNA is catalyzed by the deNEDDylase NEDP1. In response to H2O2-induced oxidative stress, NEDP1 dissociates with PCNA causing elevated RAD18-dependent NEDDylation of PCNA. The NEDDylation antagonizes the ubiquitination of PCNA. In addition, the NEDDylation of PCNA disrupts the interaction between PCNA and Polη. The impairment of PCNA NEDDylation reduces the cell sensitivity to oxidative stress and vice versa [151]. This crosstalk between NEDDylation and ubiquitination of PCNA may be a mechanism to confine the error-prone TLS to maintain genomic stability.
ISGylation
ISGylation resembles the ubiquitination process. The interferon-stimulated gene 15 (ISG15) ubiquitin-like modifier is conjugated sequentially by an E1 (ubiquitin-activating enzyme E1-like protein, Ube1L), an E2 (ubiquitin-carrier protein H8, UbcH8), and an E3 (HECT domain and RCC1-like domain-containing protein 5, HERC5, or estrogen-responsive finger protein, EFP) [152-156]. The primary function of ISGylation is to impede the infection of intracellular pathogens. ISGylated proteins are also involved in physiological processes such as TLS, autophagy regulation, protein synthesis, cytoskeletal dynamics, and secretion. Moreover, the dysregulation of ISGylation is closely related to cancer proliferation, migration, and sensitivity to chemotherapy [157]. Upon UV irradiation, the ISGylation of PCNA is induced by EFP, which binds to mono-ubiquitinated PCNA. The ISGylated PCNA recruits USP10 to deubiquitinate PCNA, which results in Polη disassociation from PCNA. This dissociation leads to the termination of TLS. Ultimately, PCNA is deISGylated by UBP43 for reloading of replicative DNA polymerases to resume normal replication. By ISGylation-defective mutation in PCNA or silencing ISG15/EFP, the compromise of PCNA ISGylation causes persistent retention of PCNA and Polη to nuclear foci. The prolonged recruitment of PCNA and Polη on chromatin increases mutation frequency [77]. This investigation indicates that ISGylation of PCNA is essential for controlling the appropriate TLS and preventing mutagenesis.
Conclusions and Perspectives
PCNA is the master of DNA replication and DDT during the S phase. The faithful replication and timely lesion bypass if the replication fork encounters lesions are indispensable events for cell survival and proliferation. PCNA trimer serves as a coordinator recruiting DNA polymerases and slides on the DNA to facilitate DNA synthesis. When replication fork stalls induced by endogenous or exogenous sources of damage, PCNA is ubiquitinated and transfers to bind DDT DNA polymerases or mediate template switching. This process ensures the replication processivity. The ubiquitination of PCNA is removed by DUBs to restrict unwanted mutagenesis [69, 71, 75]. Thus, the appropriate regulation of PCNA in the control of DNA replication and DDT is critical for normal cellular function and preventing mutagenesis.
PTMs dynamically regulate the functions of target proteins to fine-tune biological processes in response to environmental changes. In this review, we discuss the regulation and functions of PCNA PTMs. PCNA can be modified by ubiquitination, SUMOylation, phosphorylation, acetylation, methylation, NEDDylation, and ISGylation (Table 1), and the modification sites are presented in Figure 1. Chromatin loading is a requisite for PCNA to exert replication and repair functions. PCNA phosphorylation catalyzed by EGFR or c-Abl is critical for PCNA loading onto chromatin [88, 105]. The affinity to Polδ, responsible for DNA replication, is regulated by PCNA acetylation [118]. The acetylation of PCNA also promotes its degradation to suppress DNA synthesis in response to DNA damage [119], which may provide time for DNA damage response and repair.
PTMs and the regulators of PCNA
| PTM | enzyme | site | regulator | Function |
|---|---|---|---|---|
| Ubiquitination | Rad6-Rad18; Rad5-Ubc13-Mms2 | K164 | free RPA | inhibition of free PCNA mono-ubiquitination (ref. [41]) |
| DNA-bound RPA | promotion of chromatin-bound PCNA mono-ubiquitination (ref. [41, 43]) | |||
| H3/H4 | promotion of PCNA ubiquitination (ref. [47]) | |||
| WRN | inhibition of PCNA ubiquitination (ref. [51]) | |||
| PTIP | promotion of PCNA ubiquitination (ref. [59]) | |||
| IGF-1/IGF-1R | inhibition of PCNA ubiquitination (ref. [64]) | |||
| Akt | promotion of PCNA ubiquitination (ref. [68]); promotion of chromatin recruitment of Polη (ref. [68]) | |||
| RNF8-UbcH5c/Ubc13/Uev1a | K164 | promotion of PCNA ubiquitination (ref. [60]) | ||
| SUMOylation | SAE1/SAE2, Ubc9, Pias1-4 | K164 | RFC | promotion of SUMO1-PCNA (ref. [84]) |
| TRIM28 | K164 | RECQ5 | promotion of SUMO2-PCNA (ref.[92, 97]) | |
| Phosphorylation | EGFR | Y211 | stabilization of the chromatin-bound PCNA (ref. [105, 88]) | |
| c-Abl | ||||
| nIGF-1R | Y60, Y133, Y250 | rescue stalled replication fork (ref. [109]); promotion of PCNA ubiquitination (ref. [109]) | ||
| Acetylation | CBP, p300 | K13, 14, 77, 80 | chromatin-bound PCNA removal (ref. [120]); PCNA degradation (ref. [120]) | |
| Eco1 | K20 | HR stimulation (ref. [121]) | ||
| HAT3 | promotion of PCNA ubiquitination (ref. [122]) | |||
| Methylation | SETD8 | K248 | stabilization of PCNA expression (ref. [123]); promotion of interaction between PCNA and FEN1 (ref. [123]) | |
| EZH2 | K110 | stabilization of PCNA trimer (ref. [139]); promotion of interaction between PCNA and Polδ (ref. [139]) | ||
| NEDDylation | RAD18 | disruption of interaction between PCNA and Polη (ref. [151]); inhibition of PCNA ubiquitination (ref. [151]) | ||
| ISGylation | EFP | inhibition of PCNA ubiquitination (ref. [77]); Polη dissociation from PCNA (ref. [77]) |
When the replication fork stalls, the ubiquitination allows PCNA to recruit TLS polymerases or promote TS to tolerate the DNA lesions. The DNA binding of PCNA and the histones are essential for the ubiquitination of PCNA [41, 47]. Several DNA damage response factors (WRN, PTIP, and RNF8) and kinases (IGF-1R and Akt) related to cell growth signal function in regulating PCNA ubiquitination [51, 59-61, 64, 68].
The SUMOylation of PCNA with SUMO1 is regulated by RFC [84]. The SUMO1-PCNA negatively regulates HR to suppress genome rearrangements and DSB [83, 86]. The SUMO2 is conjugated to PCNA by TRIM28 E3 ligase and induced by RECQ5 [92, 97]. The SUMO2-PCNA ensures replication progression by resolving the collision between transcription and replication machinery [97].
SETD8 and EZH2, which are positively correlated to tumor development, can catalyze PCNA methylation. The methylation causes PCNA stabilization and increases its interaction with FEN1 and Polδ for DNA replication and DDT [123, 139]. The NEDDylation of PCNA is dynamically regulated by the E3 ligase RAD18 and the deNEDDylase NEDP1. The NEDDylation of PCNA impairs the association between PCNA and Polη [151], which may be essential to control TLS.
The modification crosstalk also takes place between the SUMOylation and the ubiquitination of PCNA. The SUMOylation serves as a signal for PCNA ubiquitination, as it activates RAD18 to target SUMO-PCNA [100]. The nuclear IGF-1R-mediated phosphorylation of PCNA promotes its ubiquitination by recruiting UBC13, RAD18, SHPRH, and HLTF. The colocalization of PCNA and IGF-1R is deficient in cancer tissues from patients with poor chemotherapy response, suggesting that the crosstalk between phosphorylation and ubiquitination of PCNA may predict the high overall survival of cancer patients [110]. PCNA ubiquitination is deficient in acetyltransferase HAT3-null Leishmania donovani [122]. This result indicates that acetylation may promote the ubiquitination of PCNA. In contrast, NEDDylation suppresses the ubiquitination of PCNA in response to oxidative stress [151], which suggests a negative regulation of the NEDDylation for ubiquitination of PCNA. In addition, ISGylation antagonizes the ubiquitination of PCNA directly. The ISGylated PCNA recruits USP10 to reduce the ubiquitination of PCNA and leads to the termination of TLS subsequently [77].
The PTMs of PCNA and the related regulators described above have several implications in carcinogenesis. These modifications can be aberrantly utilized by cancer cells to exert unlimited growth and resistance to chemotherapy (Table 2). Consequently, elucidating the regulation and mechanism of PCNA PTMs will provide insights to develop cancer therapies. As summarized in Table 3, the Y211F PCNA peptide targeting the phosphorylation site Y211 of PCNA inhibits the proliferation of prostate cancer and TNBC cells effectively [106, 107]. A variety of small molecular inhibitors against the enzymes or regulators of PCNA PTMs, including SMI#9 (RAD6, ubiquitination), AG1478 (phosphorylation, EGFR), and Lapatinib (phosphorylation, EGFR), have been identified that they suppress cell proliferation or enhance chemosensitivity via inhibition of these PCNA PTMs [35-38]. Although WRN, c-Abl, SETD8, and EZH2 show regulation of PCNA PTMs (Table 1) and potent anti-growth effects on various tumors (Table 2), the anti-tumor mechanisms directly correlated to the regulation of PCNA PTMs need further investigation. Moreover, novel inhibitors or specific blocking peptides suppressing tumor development via inhibition of PCNA PTMs require identification.
The implications of PCNA PTMs in carcinogenesis
| PTM | Site in PCNA | Implication in Carcinogenesis | Reference |
|---|---|---|---|
| Ubiquitination | K164 | RAD6, RAD18→development and therapy resistance of tumors. | [26-36] |
| H3/H4→MMS-induced mutagenesis. | [47] | ||
| WRN mutation→predisposition to cancer | [49] | ||
| RNF8→cell cycle arrest and apoptosis of medulloblastoma cells | [61] | ||
| IGF signal→cell proliferation, survival, apoptosis, differentiation, metabolism, migration | [63] | ||
| SUMOylation | K164, K127 | SUMO1-PCNA→prevention of the replication fork collapse to DSB | [83, 84, 89] |
| SUMO2-PCNA→avoidance of the TRCs-induced DSB | [90-92, 97] | ||
| Phosphorylation | Y211 | Increased phosphorylation→prostate and breast cancer | [105, 106] |
| Y60, Y133, Y250 | IGF-1R and PCNA colocalization→many cancer types, lost in tumors with poor chemotherapy response, high overall survival | [109, 110] | |
| Acetylation | K13, K14, K77, K80 | Removal of chromatin-bound PCNA→avoidance of excessive retention of PCNA and genomic instability | [120] |
| Methylation | K248 | SETD8→cell proliferation, high expression in many types of cancers | [125, 123] |
| K110 | EZH2→cancer cell proliferation | [135-138] | |
| NEDDylation | Cell sensitivity to oxidative stress, maintain genomic stability | [151] | |
| ISGylation | Control of appropriate TLS and prevention of mutagenesis | [77, 157] |
Inhibitors or blocking peptides relating to PTMs and regulators for cancer therapy
| Inhibitor | Affected PTM | Target | Cancer Type/Cancer Cell Type | Reference |
|---|---|---|---|---|
| SMI#9 | Ubiquitination | RAD6 | breast cancer, ovarian cancer | [35-38] |
| NSC19630 | Ubiquitination | WRN | HeLa cells | [52] |
| Y211F PCNA peptide | Phosphorylation | Y211 site in PCNA | prostate cancer, breast cancer | [106, 107] |
| AG1478, Lapatinib | Phosphorylation | EGFR | MDA-MB-468 cells | [108] |
| Imatinib | Phosphorylation | c-Abl | breast cancer | [88] |
| UNC0379 | Methylation | SETD8 | neuroblastoma, ovarian cancer | [130, 131] |
| DZNep, GSK126, EI1, UNC1999, CPI-1205 | Methylation | EZH2 | AML, lymphoma, gastric cancer, breast cancer, MLL-rearranged leukemia | [140-146] |
According to these investigations, the regulation outline of PCNA PTMs gradually becomes clear. The modulations and the crosstalk in these PTMs of PCNA orchestrate its functions in DNA replication and DDT. However, the dysregulation of PCNA PTMs will lead to mutagenesis and ultimately result in carcinogenesis. Thus, clarifying the mechanisms of PCNA PTMs is critical for understanding tumor development, which may provide effective targets for cancer therapy.
Abbreviations
PTMs: post-translational modifications; PCNA: proliferating cell nuclear antigen; TLS: translesion synthesis; TS: template switching; ROS: reactive oxygen species; UV: ultraviolet; IR: ionizing radiation; DSB: DNA double-strand break; DDT: DNA damage tolerance; PIP: PCNA-interacting protein box; K: lysine; DUBs: deubiquitination enzymes; HLTF: helicase like transcription factor; SHPRH: SNF2 histone linker PHD RING helicase; RPA: replication protein A; MMS: methyl methanesulfonate; WS: Werner Syndrome; IGF: insulin-like growth factor; IGF-1 receptor: IGF-1R; HR: homologous recombination; SUMO: small ubiquitin-related modifier; SUPs: SUMO-specific proteases; RFC: replication factor C; STUbLs: SUMO-targeting ubiquitin ligases; SIMs: SUMO-interaction motifs; TRCs: transcription-replication conflicts; RNAPⅡ: RNA polymerase Ⅱ; EGF: epidermal growth factor; EGFR: EGF receptor; TNBC: triple-negative breast cancer; HGFL: hepatocyte growth factor-like protein; nIGF-1R: nuclear IGF-1R; HMG: high-mobility group; HATs: lysine acetyltransferases; HDACs: lysine deacetylases; SAM: S-adenosylmethionine; EZH2: zeste homolog 2; DZNep: 3-Deazaneplanocin A; AML: Myeloblastic Leukemia; NEDD8: neuronal precursor cell-expressed developmentally down-regulated protein 8; NHEJ: non-homologous end joining; ISG15: interferon stimulated gene 15.
Acknowledgements
We receive fundings from National Key R&D Program of China (2016YFC1302400), National Natural Science Foundation of China (82030091, 81770001, and Z18-4-021), and the Ministry of Education Innovation Team Development Plan (IRT_17R107&IRT13101) for L.C.; National Natural Science Foundation of China (81502438) for T.Z., (81502414) for F.Y.; Foundation of Liaoning Province of China (LFWK201725) for X.S., (QN2019039) for T.Z., (QN2019038) for F.Y., (LQNK201747) for Z.W.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Warbrick E. PCNA binding through a conserved motif. Bioessays. 1998;20:195-9
2. Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665-79
3. Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J. et al. Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat Struct Mol Biol. 2014;21:664-70
4. Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004;18:2764-73
5. Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127-30
6. Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135-41
7. Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491-500
8. Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886-96
9. Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B. et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821-4
10. Zhang Y, Yuan F, Wu X, Taylor JS, Wang Z. Response of human DNA polymerase iota to DNA lesions. Nucleic Acids Res. 2001;29:928-35
11. Maga G, Villani G, Ramadan K, Shevelev I, Tanguy Le Gac N, Blanco L. et al. Human DNA polymerase lambda functionally and physically interacts with proliferating cell nuclear antigen in normal and translesion DNA synthesis. J Biol Chem. 2002;277:48434-40
12. Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci U S A. 2005;102:18361-6
13. Pence MG, Blans P, Zink CN, Hollis T, Fishbein JC, Perrino FW. Lesion bypass of N2-ethylguanine by human DNA polymerase iota. J Biol Chem. 2009;284:1732-40
14. Tonzi P, Yin Y, Lee CWT, Rothenberg E, Huang TT. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. Elife. 2018 7
15. Giannattasio M, Zwicky K, Follonier C, Foiani M, Lopes M, Branzei D. Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Biol. 2014;21:884-92
16. The UniProt C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158-D69
17. Sigismund S, Polo S, Di Fiore PP. Signaling through monoubiquitination. Curr Top Microbiol Immunol. 2004;286:149-85
18. Windheim M, Peggie M, Cohen P. Two different classes of E2 ubiquitin-conjugating enzymes are required for the mono-ubiquitination of proteins and elongation by polyubiquitin chains with a specific topology. Biochem J. 2008;409:723-9
19. Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18:520-8
20. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. 'Protein Modifications: Beyond the Usual Suspects' review series. EMBO Rep. 2008;9:536-42
21. Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK. et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576-83
22. Liu P, Gan W, Su S, Hauenstein AV, Fu TM, Brasher B. et al. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci Signal. 2018 11
23. Koken MH, Reynolds P, Jaspers-Dekker I, Prakash L, Prakash S, Bootsma D. et al. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc Natl Acad Sci U S A. 1991;88:8865-9
24. Xin H, Lin W, Sumanasekera W, Zhang Y, Wu X, Wang Z. The human RAD18 gene product interacts with HHR6A and HHR6B. Nucleic Acids Res. 2000;28:2847-54
25. Northam MR, Garg P, Baitin DM, Burgers PM, Shcherbakova PV. A novel function of DNA polymerase zeta regulated by PCNA. EMBO J. 2006;25:4316-25
26. Wong RP, Aguissa-Toure AH, Wani AA, Khosravi S, Martinka M, Martinka M. et al. Elevated expression of Rad18 regulates melanoma cell proliferation. Pigment Cell Melanoma Res. 2012;25:213-8
27. Xie C, Wang H, Cheng H, Li J, Wang Z, Yue W. RAD18 mediates resistance to ionizing radiation in human glioma cells. Biochem Biophys Res Commun. 2014;445:263-8
28. Zou S, Yang J, Guo J, Su Y, He C, Wu J. et al. RAD18 promotes the migration and invasion of esophageal squamous cell cancer via the JNK-MMPs pathway. Cancer Lett. 2018;417:65-74
29. Wu B, Wang H, Zhang L, Sun C, Li H, Jiang C. et al. High expression of RAD18 in glioma induces radiotherapy resistance via down-regulating P53 expression. Biomed Pharmacother. 2019;112:108555
30. Xie C, Lu D, Xu M, Qu Z, Zhang W, Wang H. Knockdown of RAD18 inhibits glioblastoma development. J Cell Physiol. 2019;234:21100-12
31. Baatar S, Bai T, Yokobori T, Gombodorj N, Nakazawa N, Ubukata Y. et al. High RAD18 Expression is Associated with Disease Progression and Poor Prognosis in Patients with Gastric Cancer. Ann Surg Oncol. 2020;27:4360-8
32. Li P, He C, Gao A, Yan X, Xia X, Zhou J. et al. RAD18 promotes colorectal cancer metastasis by activating the epithelialmesenchymal transition pathway. Oncol Rep. 2020;44:213-23
33. Lyakhovich A, Shekhar MP. RAD6B overexpression confers chemoresistance: RAD6 expression during cell cycle and its redistribution to chromatin during DNA damage-induced response. Oncogene. 2004;23:3097-106
34. Somasagara RR, Tripathi K, Spencer SM, Clark DW, Barnett R, Bachaboina L. et al. Rad6 upregulation promotes stem cell-like characteristics and platinum resistance in ovarian cancer. Biochem Biophys Res Commun. 2016;469:449-55
35. Somasagara RR, Spencer SM, Tripathi K, Clark DW, Mani C, Madeira da Silva L. et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene. 2017;36:6680-90
36. Haynes B, Gajan A, Nangia-Makker P, Shekhar MP. RAD6B is a major mediator of triple negative breast cancer cisplatin resistance: Regulation of translesion synthesis/Fanconi anemia crosstalk and BRCA1 independence. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165561
37. Sanders MA, Brahemi G, Nangia-Makker P, Balan V, Morelli M, Kothayer H. et al. Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification, and functional characterization. Mol Cancer Ther. 2013;12:373-83
38. Saadat N, Liu F, Haynes B, Nangia-Makker P, Bao X, Li J. et al. Nano-delivery of RAD6/Translesion Synthesis Inhibitor SMI#9 for Triple-negative Breast Cancer Therapy. Mol Cancer Ther. 2018;17:2586-97
39. Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000;19:3388-97
40. Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X. et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A. 2008;105:12411-6
41. Hedglin M, Aitha M, Pedley A, Benkovic SJ. Replication protein A dynamically regulates monoubiquitination of proliferating cell nuclear antigen. J Biol Chem. 2019;294:5157-68
42. Zou Y, Liu Y, Wu X, Shell SM. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J Cell Physiol. 2006;208:267-73
43. Li M, Sengupta B, Benkovic SJ, Lee TH, Hedglin M. PCNA Monoubiquitination Is Regulated by Diffusion of Rad6/Rad18 Complexes along RPA Filaments. Biochemistry. 2020;59:4694-702
44. House NC, Koch MR, Freudenreich CH. Chromatin modifications and DNA repair: beyond double-strand breaks. Front Genet. 2014;5:296
45. Poli J, Gasser SM, Papamichos-Chronakis M. The INO80 remodeller in transcription, replication and repair. Philos Trans R Soc Lond B Biol Sci. 2017 372
46. Sinha M, Watanabe S, Johnson A, Moazed D, Peterson CL. Recombinational repair within heterochromatin requires ATP-dependent chromatin remodeling. Cell. 2009;138:1109-21
47. Hayashi M, Keyamura K, Yoshida A, Ariyoshi M, Akanuma G, Hishida T. A Conserved Histone H3-H4 Interface Regulates DNA Damage Tolerance and Homologous Recombination during the Recovery from Replication Stress. Mol Cell Biol. 2021 41
48. Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM. et al. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17:100-3
49. Yu CE, Oshima J, Wijsman EM, Nakura J, Miki T, Piussan C. et al. Mutations in the consensus helicase domains of the Werner syndrome gene. Werner's Syndrome Collaborative Group. Am J Hum Genet. 1997;60:330-41
50. Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001;6:421-30
51. Kobayashi J, Okui M, Asaithamby A, Burma S, Chen BP, Tanimoto K. et al. WRN participates in translesion synthesis pathway through interaction with NBS1. Mech Ageing Dev. 2010;131:436-44
52. Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci U S A. 2011;108:1525-30
53. Sommers JA, Kulikowicz T, Croteau DL, Dexheimer T, Dorjsuren D, Jadhav A. et al. A high-throughput screen to identify novel small molecule inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PLoS One. 2019;14:e0210525
54. Shimizu K, Bourillot PY, Nielsen SJ, Zorn AM, Gurdon JB. Swift is a novel BRCT domain coactivator of Smad2 in transforming growth factor beta signaling. Mol Cell Biol. 2001;21:3901-12
55. Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68-76
56. Jowsey PA, Doherty AJ, Rouse J. Human PTIP facilitates ATM-mediated activation of p53 and promotes cellular resistance to ionizing radiation. J Biol Chem. 2004;279:55562-9
57. Cho EA, Prindle MJ, Dressler GR. BRCT domain-containing protein PTIP is essential for progression through mitosis. Mol Cell Biol. 2003;23:1666-73
58. Hanway D, Chin JK, Xia G, Oshiro G, Winzeler EA, Romesberg FE. Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proc Natl Acad Sci U S A. 2002;99:10605-10
59. Gohler T, Munoz IM, Rouse J, Blow JJ. PTIP/Swift is required for efficient PCNA ubiquitination in response to DNA damage. DNA Repair (Amst). 2008;7:775-87
60. Zhang S, Chea J, Meng X, Zhou Y, Lee EY, Lee MY. PCNA is ubiquitinated by RNF8. Cell Cycle. 2008;7:3399-404
61. Li F, Liu B, Zhou X, Xu Q. Silencing of E3 Ubiquitin Ligase RNF8 Enhances Ionizing Radiation Sensitivity of Medulloblastoma Cells by Promoting the Deubiquitination of PCNA. Oncol Res. 2018;26:1365-73
62. Mohiuddin Kobayashi S, Keka IS Guilbaud G, Sale J Narita T. et al. The role of HERC2 and RNF8 ubiquitin E3 ligases in the promotion of translesion DNA synthesis in the chicken DT40 cell line. DNA Repair (Amst). 2016;40:67-76
63. LeRoith D, Roberts CT Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127-37
64. Hutcherson RJ, Gabbard RD, Castellanos AJ, Travers JB, Kemp MG. Age and insulin-like growth factor-1 impact PCNA mono-ubiquitination in UVB-irradiated human skin. J Biol Chem. 2021: 100570.
65. Lewis DA, Travers JB, Somani AK, Spandau DF. The IGF-1/IGF-1R signaling axis in the skin: a new role for the dermis in aging-associated skin cancer. Oncogene. 2010;29:1475-85
66. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422-39
67. Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res. 2019;144:19-50
68. Villafanez F, Garcia IA, Carbajosa S, Pansa MF, Mansilla S, Llorens MC. et al. AKT inhibition impairs PCNA ubiquitylation and triggers synthetic lethality in homologous recombination-deficient cells submitted to replication stress. Oncogene. 2019;38:4310-24
69. Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W. et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339-47
70. Lee KY, Yang K, Cohn MA, Sikdar N, D'Andrea AD, Myung K. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J Biol Chem. 2010;285:10362-9
71. Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA. et al. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell. 2018;72:925-41 e4
72. Kashiwaba S, Kanao R, Masuda Y, Kusumoto-Matsuo R, Hanaoka F, Masutani C. USP7 Is a Suppressor of PCNA Ubiquitination and Oxidative-Stress-Induced Mutagenesis in Human Cells. Cell Rep. 2015;13:2072-80
73. Masuda Y, Kanao R, Kawai H, Kukimoto I, Masutani C. Preferential digestion of PCNA-ubiquitin and p53-ubiquitin linkages by USP7 to remove polyubiquitin chains from substrates. J Biol Chem. 2019;294:4177-87
74. Durando M, Tateishi S, Vaziri C. A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 2013;41:3079-93
75. Qian J, Pentz K, Zhu Q, Wang Q, He J, Srivastava AK. et al. USP7 modulates UV-induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene. 2015;34:4791-6
76. Gallego-Sanchez A, Andres S, Conde F, San-Segundo PA, Bueno A. Reversal of PCNA ubiquitylation by Ubp10 in Saccharomyces cerevisiae. PLoS Genet. 2012;8:e1002826
77. Park JM, Yang SW, Yu KR, Ka SH, Lee SW, Seol JH. et al. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol Cell. 2014;54:626-38
78. Alvarez V, Vinas L, Gallego-Sanchez A, Andres S, Sacristan MP, Bueno A. Orderly progression through S-phase requires dynamic ubiquitylation and deubiquitylation of PCNA. Sci Rep. 2016;6:25513
79. Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. 2012;13:755-66
80. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17:184-97
81. Da Silva-Ferrada E, Ribeiro-Rodrigues TM, Rodriguez MS, Girao H. Proteostasis and SUMO in the heart. Int J Biochem Cell Biol. 2016;79:443-50
82. Mun MJ, Kim JH, Choi JY, Kim MS, Jang WC, Lee JJ. et al. Polymorphisms of small ubiquitin-related modifier genes are associated with risk of Alzheimer's disease in Korean: A case-control study. J Neurol Sci. 2016;364:122-7
83. Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ. et al. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell. 2012;45:75-86
84. Gali H, Juhasz S, Morocz M, Hajdu I, Fatyol K, Szukacsov V. et al. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012;40:6049-59
85. Hendriks IA, D'Souza RC, Chang JG, Mann M, Vertegaal AC. System-wide identification of wild-type SUMO-2 conjugation sites. Nat Commun. 2015;6:7289
86. Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005;436:428-33
87. Burkovics P, Sebesta M, Sisakova A, Plault N, Szukacsov V, Robert T. et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 2013;32:742-55
88. Zhao H, Chen MS, Lo YH, Waltz SE, Wang J, Ho PC. et al. The Ron receptor tyrosine kinase activates c-Abl to promote cell proliferation through tyrosine phosphorylation of PCNA in breast cancer. Oncogene. 2014;33:1429-37
89. Nikolova T, Ensminger M, Lobrich M, Kaina B. Homologous recombination protects mammalian cells from replication-associated DNA double-strand breaks arising in response to methyl methanesulfonate. DNA Repair (Amst). 2010;9:1050-63
90. French S. Consequences of replication fork movement through transcription units in vivo. Science. 1992;258:1362-5
91. Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533-43
92. Li M, Xu X, Chang CW, Liu Y. TRIM28 functions as the SUMO E3 ligase for PCNA in prevention of transcription induced DNA breaks. Proc Natl Acad Sci U S A. 2020;117:23588-96
93. Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L. et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007;21:3073-84
94. Aygun O, Svejstrup J, Liu Y. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc Natl Acad Sci U S A. 2008;105:8580-4
95. Kanagaraj R, Huehn D, MacKellar A, Menigatti M, Zheng L, Urban V. et al. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 2010;38:8131-40
96. Li M, Xu X, Liu Y. The SET2-RPB1 interaction domain of human RECQ5 is important for transcription-associated genome stability. Mol Cell Biol. 2011;31:2090-9
97. Li M, Xu X, Chang CW, Zheng L, Shen B, Liu Y. SUMO2 conjugation of PCNA facilitates chromatin remodeling to resolve transcription-replication conflicts. Nat Commun. 2018;9:2706
98. Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci U S A. 2004;101:14373-8
99. Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I. Specification of SUMO1- and SUMO2-interacting motifs. J Biol Chem. 2006;281:16117-27
100. Parker JL, Ulrich HD. A SUMO-interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO-modified PCNA. Nucleic Acids Res. 2012;40:11380-8
101. Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127-30
102. Prosperi E, Stivala LA, Sala E, Scovassi AI, Bianchi L. Proliferating cell nuclear antigen complex formation induced by ultraviolet irradiation in human quiescent fibroblasts as detected by immunostaining and flow cytometry. Exp Cell Res. 1993;205:320-5
103. Prosperi E, Scovassi AI, Stivala LA, Bianchi L. Proliferating cell nuclear antigen bound to DNA synthesis sites: phosphorylation and association with cyclin D1 and cyclin A. Exp Cell Res. 1994;215:257-62
104. Jonsson ZO, Podust VN, Podust LM, Hubscher U. Tyrosine 114 is essential for the trimeric structure and the functional activities of human proliferating cell nuclear antigen. EMBO J. 1995;14:5745-51
105. Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC. et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359-68
106. Zhao H, Lo YH, Ma L, Waltz SE, Gray JK, Hung MC. et al. Targeting tyrosine phosphorylation of PCNA inhibits prostate cancer growth. Mol Cancer Ther. 2011;10:29-36
107. Yu YL, Chou RH, Liang JH, Chang WJ, Su KJ, Tseng YJ. et al. Targeting the EGFR/PCNA signaling suppresses tumor growth of triple-negative breast cancer cells with cell-penetrating PCNA peptides. PLoS One. 2013;8:e61362
108. Lo YH, Ho PC, Wang SC. Epidermal growth factor receptor protects proliferating cell nuclear antigen from cullin 4A protein-mediated proteolysis. J Biol Chem. 2012;287:27148-57
109. Waraky A, Lin Y, Warsito D, Haglund F, Aleem E, Larsson O. Nuclear insulin-like growth factor 1 receptor phosphorylates proliferating cell nuclear antigen and rescues stalled replication forks after DNA damage. J Biol Chem. 2017;292:18227-39
110. Yang C, Zhang Y, Chen Y, Ragaller F, Liu M, Corvigno S. et al. Nuclear IGF1R interact with PCNA to preserve DNA replication after DNA-damage in a variety of human cancers. PLoS One. 2020;15:e0236291
111. Phillips DM. The presence of acetyl groups of histones. Biochem J. 1963;87:258-63
112. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786-94
113. Sterner R, Vidali G, Allfrey VG. Studies of acetylation and deacetylation in high mobility group proteins. Identification of the sites of acetylation in HMG-1. J Biol Chem. 1979;254:11577-83
114. L'Hernault SW, Rosenbaum JL. Chlamydomonas alpha-tubulin is posttranslationally modified in the flagella during flagellar assembly. J Cell Biol. 1983;97:258-63
115. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595-606
116. Gil J, Ramirez-Torres A, Encarnacion-Guevara S. Lysine acetylation and cancer: A proteomics perspective. J Proteomics. 2017;150:297-309
117. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20:156-74
118. Naryzhny SN, Lee H. The post-translational modifications of proliferating cell nuclear antigen: acetylation, not phosphorylation, plays an important role in the regulation of its function. J Biol Chem. 2004;279:20194-9
119. Yu Y, Cai JP, Tu B, Wu L, Zhao Y, Liu X. et al. Proliferating cell nuclear antigen is protected from degradation by forming a complex with MutT Homolog2. J Biol Chem. 2009;284:19310-20
120. Cazzalini O, Sommatis S, Tillhon M, Dutto I, Bachi A, Rapp A. et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014;42:8433-48
121. Billon P, Li J, Lambert JP, Chen Y, Tremblay V, Brunzelle JS. et al. Acetylation of PCNA Sliding Surface by Eco1 Promotes Genome Stability through Homologous Recombination. Mol Cell. 2017;65:78-90
122. Kumar D, Saha S. HAT3-mediated acetylation of PCNA precedes PCNA monoubiquitination following exposure to UV radiation in Leishmania donovani. Nucleic Acids Res. 2015;43:5423-41
123. Takawa M, Cho HS, Hayami S, Toyokawa G, Kogure M, Yamane Y. et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012;72:3217-27
124. Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005;19:1455-65
125. Jorgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON. et al. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol. 2007;179:1337-45
126. Williams DE, Dalisay DS, Li F, Amphlett J, Maneerat W, Chavez MA. et al. Nahuoic acid A produced by a Streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org Lett. 2013;15:414-7
127. Ma A, Yu W, Li F, Bleich RM, Herold JM, Butler KV. et al. Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J Med Chem. 2014;57:6822-33
128. Blum G, Ibanez G, Rao X, Shum D, Radu C, Djaballah H. et al. Small-molecule inhibitors of SETD8 with cellular activity. ACS Chem Biol. 2014;9:2471-8
129. Butler KV, Ma A, Yu W, Li F, Tempel W, Babault N. et al. Structure-Based Design of a Covalent Inhibitor of the SET Domain-Containing Protein 8 (SETD8) Lysine Methyltransferase. J Med Chem. 2016;59:9881-9
130. Veschi V, Liu Z, Voss TC, Ozbun L, Gryder B, Yan C. et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell. 2017;31:50-63
131. Wada M, Kukita A, Sone K, Hamamoto R, Kaneko S, Komatsu M. et al. Epigenetic Modifier SETD8 as a Therapeutic Target for High-Grade Serous Ovarian Cancer. Biomolecules. 2020 10
132. Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185-96
133. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039-43
134. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893-905
135. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-9
136. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323-35
137. Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE. et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107:18475-80
138. Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R. et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371-8
139. A P, Xu X, Wang C, Yang J, Wang S, Dai J. et al. EZH2 promotes DNA replication by stabilizing interaction of POLdelta and PCNA via methylation-mediated PCNA trimerization. Epigenetics Chromatin. 2018;11:44
140. Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A. et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733-43
141. Lue JK, Prabhu SA, Liu Y, Gonzalez Y, Verma A, Mundi PS. et al. Precision Targeting with EZH2 and HDAC Inhibitors in Epigenetically Dysregulated Lymphomas. Clin Cancer Res. 2019;25:5271-83
142. Yang Y, Zhu F, Wang Q, Ding Y, Ying R, Zeng L. Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells. Onco Targets Ther. 2018;11:8455-63
143. Puppe J, Opdam M, Schouten PC, Jozwiak K, Lips E, Severson T. et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin Cancer Res. 2019;25:4351-62
144. Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R. et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360-5
145. Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG. et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346-57
146. Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M. et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J Med Chem. 2016;59:9928-41
147. Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272:28557-62
148. Ma T, Chen Y, Zhang F, Yang CY, Wang S, Yu X. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol Cell. 2013;49:897-907
149. Brown JS, Lukashchuk N, Sczaniecka-Clift M, Britton S, le Sage C, Calsou P. et al. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015;11:704-14
150. Jimeno S, Fernandez-Avila MJ, Cruz-Garcia A, Cepeda-Garcia C, Gomez-Cabello D, Huertas P. Neddylation inhibits CtIP-mediated resection and regulates DNA double strand break repair pathway choice. Nucleic Acids Res. 2015;43:987-99
151. Guan J, Yu S, Zheng X. NEDDylation antagonizes ubiquitination of proliferating cell nuclear antigen and regulates the recruitment of polymerase eta in response to oxidative DNA damage. Protein Cell. 2018;9:365-79
152. Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20:362-71
153. Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol. 2004;24:9592-600
154. Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA. et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc Natl Acad Sci U S A. 2004;101:7578-82
155. Wong JJ, Pung YF, Sze NS, Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci U S A. 2006;103:10735-40
156. Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem. 2006;281:3989-94
157. Villarroya-Beltri C, Guerra S, Sanchez-Madrid F. ISGylation - a key to lock the cell gates for preventing the spread of threats. J Cell Sci. 2017;130:2961-9
158. Kontopidis G, Wu SY, Zheleva DI, Taylor P, McInnes C, Lane DP. et al. Structural and biochemical studies of human proliferating cell nuclear antigen complexes provide a rationale for cyclin association and inhibitor design. Proc Natl Acad Sci U S A. 2005;102:1871-6
Author contact
![]() Corresponding authors: Liu Cao, E-mail: lcaoedu.cn; Xiaoyu Song, E-mail: xysongedu.cn; College of Basic Medical Science, Key Laboratory of Cell Biology of Ministry of Public Health, Key Laboratory of Medical Cell Biology of Ministry of Education, Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, China Medical University, Shenyang, Liaoning Province, 110122, PR China. Tel: +86 24 31939636, Fax: +86 24 31939636.
Corresponding authors: Liu Cao, E-mail: lcaoedu.cn; Xiaoyu Song, E-mail: xysongedu.cn; College of Basic Medical Science, Key Laboratory of Cell Biology of Ministry of Public Health, Key Laboratory of Medical Cell Biology of Ministry of Education, Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, China Medical University, Shenyang, Liaoning Province, 110122, PR China. Tel: +86 24 31939636, Fax: +86 24 31939636.