Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(3):1065-1078. doi:10.7150/ijbs.68591 This issue Cite
Review
Calcium and calcium-related proteins in endometrial cancer: opportunities for pharmacological intervention
Ting Huang, Jingyi Zhou ![]() , Jianliu Wang
, Jianliu Wang ![]()
Department of Obstetrics and Gynecology, Peking University People's Hospital, Beijing 100044, P.R. China.
Received 2021-10-31; Accepted 2021-12-12; Published 2022-1-1
Abstract

Intracellular calcium ions are key second messengers and play an important role in malignant transformation and cancer progression. Estrogen can evoke intracellular calcium increases through membrane-initiated effects and activate subsequent kinase cascades within minutes in normal and cancerous epithelial cells. Ca2+-related proteins are expressed in normal epithelial cells or endometrial cancer cells, some of which are upregulated by estrogen. Both estrogen-induced transient calcium increases and long-term changes in protein expression levels may be involved in regulating cancer initiation, progression and metastasis. Calcium channel blockers are reported to regulate both the rapid estrogen-induced intracellular Ca2+ increase and cell proliferation, apoptosis and migration, thus having the potential for pharmacological modulators to be repurposed for the treatment of endometrial cancer.
Keywords: Endometrial cancer, Calcium, Estrogen, Calcium channel blocker
Introduction
Endometrial cancer (EC) is one of the most common gynecological malignancies worldwide and is a global threat to women's health and well-being. Approximately 420000 patients were newly diagnosed with EC in 2020, accounting for 4.5% of all cases in women [1]. Calcium ions are ubiquitous intracellular messengers that regulate a myriad of cellular processes and can affect cell survival. Our previous studies have identified serum calcium ion level as a potential predictor for lymph node metastasis and positive peritoneal cytology in endometrial cancer, suggesting that calcium ions participate in the progression of EC [2, 3].
Calcium homeostasis is a crucial determinant of cellular function and survival. Calcium ions in the cytosol are dynamically regulated by the plasma membrane, endoplasmic reticulum and mitochondria. A sustained calcium increase from extracellular calcium entry or release from calcium stores can affect cancer cell growth, migration, distant metastasis, and survival [4-6]. The cytosolic calcium concentration is tightly regulated by ion channels, pumps and exchangers [7]. Our previous work suggested that inhibitors of L-type voltage-gated calcium ion channels and transient receptor potential vanilloid 4 (TRPV4) could partly block calcium influx in EC cells [8, 9]. In this review, we summarize the latest studies on calcium signaling, differentially expressed calcium-related proteins and their functions in cancer progression as well as the potential for pharmacological application.
Relationship between estrogen and calcium signal
Clinically, prolonged estrogen stimulation without progesterone antagonism increases the risk of endometrial cancer and estrogen-dependent cases represent 75-90% of all endometrial cancers [10]. Our previous work suggested that endometrioid carcinoma had higher serum calcium levels than non-endometrioid carcinoma, the subtype that usually suffers from estrogen deprivation [11]. Estrogen is an important hormone that rapidly induces calcium mobilization and regulates calcium-related protein expression in the endometrium [12-14]. On the one hand, estrogen stimulus drives calcium influx, and rapidly augmented calcium triggers a series of reactions, which are a main part of non-genomic effects. Any dysfunction of this process may contribute to cancerous transformation and progression. Often, the rapid Ca2+ surge causes photophosphorylation of kinase cascades and regulates transcription factors, thus affecting cellular processes and function. On the other hand, long-term estrogen stimulus causes some calcium-related protein changes in normal endometrial epithelial cells (EECs) and cancer cells [14, 15]. Sustained high Ca2+ exerts its influence on both epithelial cells and the microenvironment [16, 17]. However, it remains uncertain whether estrogen-induced short-term effects and long-term regulation of calcium function together in the occurrence and progression of endometrial cancer.
Estrogen-induced rapid intracellular calcium increases in endometrial cells
Intracellular calcium is measured by the accumulation of 45Ca2+ and calcium fluorescent probes, such as fluo-2, fluo-3, fluo-4 and fluo-8. Estrogen-induced rapid increases in intracellular calcium have been detected in endometrial epithelial cells isolated from the uteri of rats and RL95-2 cells (a poorly polarized human endometrial cancer cell line) [12, 13]. Moreover, Zhang LL et al demonstrated that both E2 and its membrane-impermeable conjugate, estrogen and bovine serum albumin (E2-BSA), can elicit calcium influx in Ishikawa cells, a moderately polarized endometrial cancer cell line [18]. It appears that estrogens may act directly at the cell membrane and exert effects on the activity of ligand-gated ion channels. Transient increases in intracellular free calcium are reported to precede or trigger the cell cycle and growth, which is exactly the end-point of estrogen effects on the endometrium [19]. Figure 1 illustrates estrogen-induced intracellular calcium increases in endometrial cancer.
Estrogen-induced rapid calcium changes in endometrial cells. Estrogen can rapidly trigger an increase in intracellular calcium in endometrial cells through membrane-initiated signaling. Intracellular calcium mobilization is mediated by different signals: ① GPER1- Gα-CACNA1D; ② GPER1- Gα-PLC-IP3-IP3R-Ca2+ store; and ③ mER-PLC-IP3-IP3R-Ca2+ store. The degree of increase in cytosolic free Ca2+ and duration of maintenance vary in different cells. Increased intracellular calcium activates the MAPK pathway and regulates the expressions of Bcl-2, CREB, and so on. (Created with BioRender.com)
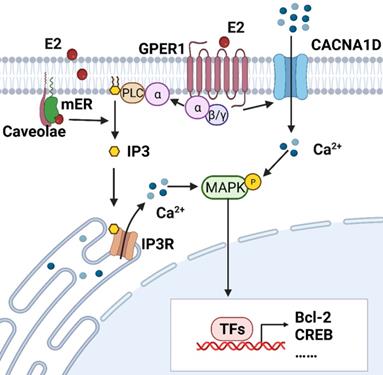
The addition of 17β-estradiol at a final concentration of 1 nM increased 45Ca2+ uptake, reaching a peak at about 230% of baseline within 30 minutes. 45Ca2+ uptake was gradually restored to baseline in rat EECs [13]. About 15% of RL95-2 cells were responsive to 17β-estradiol stimulus and showed transient intracellular calcium rises within 10 minutes with a maximal value about 130% of baselines obtained at a concentration of 10 nM [12]. Intracellular calcium peaked at about 300 seconds and lasted for 600 seconds after adding E2 or E2-BSA to Ishikawa cells. Interestingly, calcium waves showed two peaks under E2-BSA stimulation [18]. The different amplitudes and rates may be partly due to the concentration of estrogen.
Source of estrogen-induced intracellular calcium increase
The estrogen-evoked Ca2+ increase may occur as a result of Ca2+ entry from the extracellular milieus or Ca2+ release from intracellular sources. Morley demonstrated that the estrogen-triggered calcium surge was not affected by incubating the cells in Ca2+-free solution or pretreating cells with calcium channel blockers (CCBs) but was abolished by incubating cells with inhibitors of inositol phospholipid hydrolysis in chicken granulosa cells [20]. Teresa et al. demonstrated that E2 could induce a rise in intracellular calcium in the presence or absence of extracellular calcium [21]. These results imply that estrogen induces Ca2+ mobilization mainly from intracellular stores.
However, some studies identified that an estrogen-induced calcium rise had a critical dependence on external calcium. Estrogen was also reported to increase the activity of plasma membrane calcium pumps in distal tubule kidney cells [22]. Additionally, E2-induced rapid Ca2+ influx in hippocampal neurons and endometrial cancer cells could be significantly inhibited by nifedipine, a calcium channel blocker [18, 23]. Wu TW et al found that the initial influx of Ca2+ through the L-type calcium channel is necessary for E2 activation of downstream signals [23]. Moreover, 17β-estradiol increased intracellular Ca2+ in a biphasic manner through extracellular calcium entry and endoplasmic reticulum release in rat osteoblasts [24].
In normal EECs, E2 induced 45Ca2+ uptake from the medium [13]. In RL95-2 cells, 17β-estradiol could not induce calcium transients when the RL95-2 cells were bathed in external Ca2+-free medium, implying a dependence on calcium entry. However, the calcium surge was significantly increased by depletion of intracellular calcium stores and decreased after treatment with an inhibitor of protein kinase C (PKC), which suggested that calcium release from intracellular stores via the PKC-sensitive pathway contributed to E2-induced intracellular calcium increases [12]. Similarly, E2-BSA elicited a dual peak in Ishikawa cells, the first coming from external calcium influx and the second releasing from intracellular calcium stores [18]. In summary, the two sources may also coexist in endometrial cells.
Receptors that mediate estrogen-induced calcium increase in endometrial cells
There is no consensus regarding which receptors should be responsible for the rapid Ca2+ rise. As a membrane-initiated effect, membrane estrogen receptor (mER) is widely investigated. Endometrial cells show abundant binding to estrogen on the cell surface [25]. Several variants of ERα and ERβ as well as estrogen receptor G protein-coupled estrogen receptor (GPER1, also known as GRP30) have been reported to be associated with non-genomic estrogen signaling [26-28].
Classical mERs are identical to nuclear estrogen receptor (nER) in not only spectra, weight and affinity to estrogen, but also in their protein epitopes [29, 30]. ERα and ERβ were detected in endothelial cell caveolae [31, 32]. Without the transmembrane domain, classical ERs may interact with the plasma membrane by covalent binding to membrane proteins such as caveolin-1 [33]. ERαs translocation to the membrane is dependent on direct binding to caveolin-1. The complex formed by ERα, Src homology and collagen homology (Shc), and insulin-like growth factor receptor-1 (IGF-1R) increases the mERα levels within caveolae rafts of the plasma membrane once stimulated by estrogen [34]. In Ishikawa cells, rapid estrogen-induced calcium mobilization could be partly inhibited by the ER antagonist, ICI182780 [18]. E2/ERα activates phospholipase C (PLC)-dependent inositol 1,4,5-trisphosphate (IP3) production mediated by Gαi/o proteins, thus causing Ca2+ store release. E2-induced calcium mobilization was completely blocked by U73122, a PLC inhibitor in ERα-overexpressing COS7 cells [35, 36]. In addition, the pattern of testosterone action offers another possibility of regulating calcium flux in prostate cells. The N-terminal region of androgen receptor (AR) has specific sites for transient receptor potential melastatin 8 (TRPM8) and the accumulation of the TRPM8-AR complex in lipid rafts mediates testosterone-induced cell migration [37]. A previous study identified several calcium binding sites in the ERα ligand binding domain [38]. Moreover, calmodulin regulates the calcium-dependent activation of ERα by directly binding to ERα at several sites [39].
As a novel estrogen receptor, GPER1 is reported to participate in estrogen-triggered non-genomic effects in ovarian cancer [40], ER-negative breast cancer [41] and thyroid cancer cells [42]. GPER1 can regulate intracellular free calcium by (1) activating membrane ion channels, (2) regulating Ca2+-calmodulin interactions or (3) triggering Ca2+ store release [43, 44]. In endometrial cancer cells, the GPER agonist G1 facilitated the expression of CACNA1D, while E2-BSA-activated CACNA1D was blocked after silencing the GPER1 gene [8]. Likewise, E2 promoted CACNA1D expression in a time-dependent and dose-dependent manner and triggered Ca2+ influx through GPER1 in breast cancer cells [45]. GPER1 regulates the activity of L-type VGCCs through coupling with Gαs and Gαi/o and triggers subsequent Ca2+ entry [43]. GPER1 can also be directly regulated by the Ca2+-calmodulin complex because of the existence of four distinct calmodulin-binding domains in GPER1, as feedback to the E2-induced calcium increase [46]. As a seven-transmembrane receptor, GPER1 stimulates the production of PLCβ and IP3 through coupling with Gβγ. The latter binds to its receptor IP3R1 situated on the endoplasmic reticulum and evokes Ca2+ release [47].
Estrogen might directly bind to certain calcium channels. Estrogen, as a lipophilic hormone, affects membrane fluidity, induces membrane fusion and modifies ion channel activity [48]. As early as 1986, Kenji et al reported that calcium channels of the estrogen groups in the rat uterus were at a high affinity state, without changing the numbers of calcium channels [49]. E2 was reported to directly activate the Maxi-K+ channel [50]. In hippocampal neurons, estrogen could directly interact with the L-type calcium channel alpha 1C subunit (CACNA1C) at the dihydropyridine site in an estrogen receptor-independent way [51].
Activated kinase cascades by estrogen-induced calcium signaling
Mitogen-activated protein kinase (MAPK) cascades mainly consist of four pathways: Extracellular-signal-regulated kinase (ERK) 1/2, ERK5, p38 MAPK and c-Jun N-terminal kinase (JNK) [52]. E2-induced activation of MAPK cascades can be observed among four endometrial cancer cell lines [12, 18, 53]. Accumulating evidences show that E2 can rapidly activate MAPK and in an ER-dependent manner in mammalian cells [54-56]. However, the molecular mechanisms underlying E2-triggered MAPK activation and its biological effects remain to be explained.
In MCF-7 breast cancer cells, MAPK activation is preceded by a rapid increase in cytosolic calcium from Ca2+ stores in response to estrogen stimulus [21]. In Ishikawa cells, either E2 or E2-BSA could induce ERK1/2 phosphorylation [18]. Notably, c-Src activation triggered by estrogen could lead to parallel activation of ERK1/2 and Akt signals [57]. The same hippocampal neurons were successively stained with a calcium probe and immunocytochemistry for pERK. The results confirmed that the E2-triggered calcium increase was coincident with pERK [23].
Activated MAPKs can translocate into the nucleus and regulate gene transcription, thus playing critical roles in cell proliferation, the cell cycle and apoptosis. Rapid activation of MAPK induced nuclear factor kappa B (NF-κB) activation, CD1 transcription and subsequent cell cycle progression in Swiss 3T3 cells [58]. Treatment with a MAPK inhibitor significantly suppressed E2-facilitated proliferation in lactotrophs and breast cancer cells, indicating an important role of the MAPK pathways in E2-dependent progression [30, 59]. Ca2+/Src/ERK signaling is required for the E2-induced activation of B-cell lymphoma-2 (Bcl-2), an apoptosis regulatory protein [23]. Interestingly, E2 results in increased mitochondrial sequestration of Ca2+ to attenuate cytosolic Ca2+ and a subsequent increase in Bcl-2 expression, aiming to promote mitochondrial tolerance and cell survival in response to glutamate [60]. nERs also interacted with MAPK cascades. In MCF-7 cells, ERK2 and ERK5 interacted with different regions of nERα. Upon E2 exposure, activated ERK2 and ERK5 localize with nERα and modulate estrogen-dependent gene transcription and cell proliferation programs [61, 62]. In addition to directly binding to ERα, MAPK cascades can indirectly regulate ERα transcriptional activity by targeting several cofactors [63]. P38 MAPK and ERK1/2 are involved in hormone-induced activation of c-fos in rat intestinal cells [64]. Transient calcium markedly upregulated the expression of semaphorin 3A through the MAPK/activator protein (AP)-1 axis in keratinocytes [65].
There are other E2-induced kinases that are closely associated with the intracellular Ca2+ rise. In myometrial cells, the G1-induced intracellular calcium increase occurred prior to myosin light-chain kinase (MLCK) phosphorylation. Thereafter, MLCK became desensitized to Ca2+/calmodulin and began dephosphorylation [66]. These results described the dynamic change in estrogen-induced calcium and its effects on cell movement. Cytoskeletal rearrangements induced by E2 and tamoxifen could be blocked by a Src inhibitor, implying the important role of Src kinase in estrogen-induced rapid effects in endometrial cancer cells [53].
Alterations in calcium channels/pumps/exchangers in endometrial cells
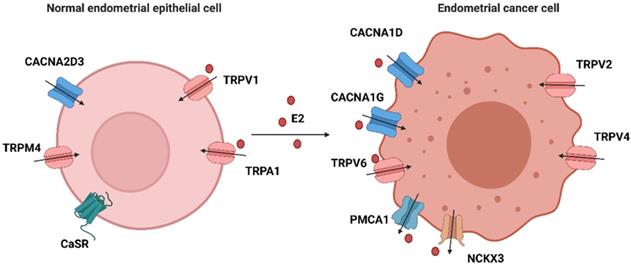
Cytosolic Ca2+ signaling is coordinately controlled by both intracellular and extracellular stores. In most mammalian cells, external stimuli bind to ligand-engaged G protein-coupled receptors (GPCRs), causing subsequent synthesis of IP3 and activation of the IP3 receptor at the endoplasmic reticulum membrane, resulting in the release of calcium from the endoplasmic reticulum [67, 68]. Extracellular calcium ions can enter the cytosol through multiple voltage-gated calcium ion channels (VGCC) and transient receptor potential (TRP) family channels. Two main ATP-dependent systems extrude Ca2+ from the cytosol: plasma membrane Ca2+ ATPases (PMCAs) and sarcoendoplasmic reticular Ca2+ ATPases (SERCAs), the former expelling Ca2+ to the extracellular space and the latter accumulating Ca2+ within the endoplasmic reticulum [69]. Together, these calcium channels/pumps/exchangers struggle to maintain dynamic homeostasis. Any dysfunction of these calcium-related proteins may result in a disruption of calcium balance. Therefore, we summarize the expression of calcium-related genes in tissue or cell line(s) of endometrium, their effects on biological behaviors and associations with E2 in Table 1 and Figure 2.
Altered Ca2+ channels/pumps and their functions in endometrial cells
| Calcium channel/pumps | Differential expression in EC tissues comparing to normal | Effects on biological behavior | E2 effects on Ca2+ channels/pumps | ref | ||
|---|---|---|---|---|---|---|
| E2 stimulation | Cell line(s) | |||||
| VGCC | CACNA1D | ↑ | promotes proliferation, migration and apoptosis and estrogen-induced Ca2+ influx in ISK cells | ↑ | ISK | [8] |
| CACNA1G | ND | promotes proliferation, migration and apoptosis | ↑ | ISK | [72] | |
| CACNA2D3 | ↓ | suppresses cell proliferation and migration, and induce apoptosis and Ca2+ influx in ISK and RL95-2 cells | ND | ND | [73] | |
| TRP | TRPV1 | ND | reduces viability of Ishikawa and Hec50co cells | ↑ | Rat EEC | [14] |
| TRPV2 | ↑ in non-endometrioid tissues | promotes migration and chemo-sensitivity in ISK cells | ND | ND | [5] | |
| TRPV4 | ↑ | promotes migration in ISK and Hec-1A cells | ND | ND | [9] | |
| TRPV6 | ND | ND | ↑ | ISK | [15] | |
| TRPM4 | ↓ | suppresses proliferation and migration in AN3CA cells | ND | ND | [83] | |
| TRPA1 | ND | ND | ↑ | Rat EEC | [14] | |
| Pump(s) | PMCA1 | ND | ND | ↑ | ISK | [15] |
| NCKX3 | ND | ND | ↑ | ISK | [84] | |
↑ increased levels in cancer sample; ↓ decreased levels in cancer samples; ND, not determined; ISK, Ishikawa; EEC, endometrial epithelial cell; EC: endometrial cancer; VGCC: Voltage-gated calcium ion channels; CACNA1C: Calcium channel alpha1C; CACNA1D: Calcium channel alpha1D; CACNA1G: Calcium channel alpha1G; CACNA2D3: Calcium channel alpha 2 delta subunit 3; TRP: Transient receptor potential; TRPV1: TRP vanilloid 1; TRPV2: TRP vanilloid 2; TRPV4: TRP vanilloid 4; TRPV6: TRP vanilloid 6; TRPM4: TRP melastatin 4; TRPA1: TRP ankyrin 1; PMCAs: Plasma membrane Ca2+ ATPases; NCKX3: potassium-dependent sodium/calcium exchanger 3.
The expression of calcium-related proteins in EEC and EC cells. Calcium-related proteins in endometrial cells are divided into four categories: ① that are regulated by E2 in normal epithelial cells: TRPV1 and TRPA1; ② that are regulated by E2 in endometrial cancer cells: CACNA1D, CACNA1G, TRPV6, PMCA1 and NCKX3; ③ that are highly expressed in normal epithelial cells: CACNA2D3, TRPM4 and CaSR; and ④ that are highly expressed in cancer cells: TRPV2 and TRPV4. (Created with BioRender.com)
Voltage-gated calcium channels
Although VCGGs are ubiquitously expressed in excitable cells, they are also detected in many kinds of malignant cells [70]. VGCCs are subdivided into L-type, T-type, P/Q-type, R-type and N-type. CACNA1D is an auxiliary member of the alpha-1 subunit family of the VGCC complex and is involved in androgen-stimulated Ca2+ influx and androgen receptor transactivation in prostate cancer [71]. 17β-estradiol was added to the medium after pretreatment with nifedipine, a blocker targeting CACNA1D and other L-type calcium channels. The mRNA expression of CACNA1D returned to normal at 30 minutes and protein expression started to rise after 60 minutes in Hec-1A cells, suggesting that estrogen regulates the expression of CACNA1D in a rapid manner [72]. Next, the effect of CACNA1D on the estrogen-induced intracellular calcium increase was investigated. After knocking down CACNA1D, the intercellular free calcium concentration was significantly reduced in Ishikawa cells compared to the negative control group. Compared to the benign endometrial tissues, atypical hyperplasia and carcinoma tissues have a higher expression of CACNA1D. Moreover, genetic knockdown of CACNA1D inhibited the estrogen-induced growth and migration of Ishikawa cells [8]. Calcium channel alpha1G (CACNA1G), a subunit of T-type VGCCs, is also regulated rapidly by estrogen in Hec-1A cells [72].
In contrast, another VGCC, calcium channel alpha 2 delta subunit 3 (CACNA2D3), suppressed cell proliferation and migration, and induced cell apoptosis and Ca2+ influx in EC by acting as the downstream of progesterone. The expression of CACNA2D3 was downregulated in EC tissues and cells compared with noncancer tissues or endometrial epithelial cells [73].
TRP ion channels
TRP ion channels consist of a superfamily of several cation channels (TRPC, TRPV, TRPM, TRPA, TRPP, and TRPML) and can be activated by various stimuli [74, 75]. TRP vanilloid 1 (TRPV1) is involved in the reduction in cell viability and the activation of the apoptotic pathway induced by its agonist cannabinoids (CB) in endometrial cancer cells. After exposure to CB, a rapid increase in intercellular calcium levels was detected and a TRPV1 antagonist was able to reverse these effects [76]. In rat endometrial cells, about 11% of cells were responsive to capsaicin, the TRPV1 activator, and showed a rapid calcium influx. E2 and ERα/ERβ agonists both upregulated TRPV1 mRNA [14]. Notably, E2, not E2-BSA, prevented capsaicin from activating TRPV1 channels through ERβ signaling in neurons [77]. Therefore, the E2/ERβ complex might regulate TRPV1 activity and modulate rapid calcium entry in some endometrial cells.
TRP vanilloid 2 (TRPV2) has an increased expression in type II endometrial cancer and correlates with worse progression-free survival. Ishikawa cells with TRPV2 overexpression showed a high migratory ability and sensitivity to cisplatin [78].
TRPV4 expression was higher in the EC group than in the normal epithelium group. Furthermore, TRPV4 could regulate migration and metastasis both in vitro and in vivo through cytoskeleton regulation and the Rho protein pathway [9]. In Hec-1A and Hec-1B cells, E2 rapidly induced cytoskeletal remodeling, which was mediated by ERα signaling [53]. However, there is no direct evidence indicating that E2 could activate TRPV4 and drive calcium current. In addition, the expression of transient receptor potential vanilloid 6 (TRPV6) was upregulated by E2 in both primary epithelial cells and Ishikawa cells and the increases were completely reversed with an ER antagonist [15].
TRP melastatin 4 (TRPM4) expression has been reported in several cancers and is involved in malignant transformation and immunity modulation [79-81]. Bioinformatics analysis based on The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) gene expression data of EC tissue and normal endometrial tissue identified TRPM4 as a protective prognostic gene [82]. In detail, lower expression of TRPM4 was associated with a higher clinical stage, a more advanced grade, positive lymph node metastasis, myometrial invasion, worse recurrence free survival and overall survival [83]. Another membrane of the TRP family, transient receptor potential ankyrin 1 (TRPA1) was also positively regulated by E2 in rat endometrial cells. More studies on its mechanism are needed.
Ca2+ pumps and exchangers
PMCA1 and potassium-dependent sodium/calcium exchanger 3 (NCKX3) are also crucial components of intracellular calcium homeostasis mainly by extruding calcium out of the cytosol. PMCA1 and NCKX3 were richly expressed in the endometrium especially in endometrial and glandular epithelial cells, while their expression was significantly increased at the proliferative phase compared to other phases. PMCA1 and NCKX3 were also detected in Ishikawa cells and their mRNA levels were markedly increased following E2 treatment [15, 84]. The E2-induced increase in PMCA1 mRNA levels was completely reversed after pretreatment with ER antagonist [15]. All these results indicated that estrogen and its receptors might participate in the regulation of PMCA1 and NCKX3 levels both in endometrial epithelial cells and cancer cells and thus maintain calcium homeostasis.
Other calcium-related genes are involved in the carcinogenesis or progression of EC. Calcium sensing receptor (CaSR) is a membrane of G-protein-coupled receptors. It can activate PLC and respond to intracellular Ca2+ fluctuations. CaSR might serve as a tumor suppressor because overexpression induced apoptosis but inhibited invasion of Ishikawa cells [85].
Ca2+ signal and endometrial cancer initiation and progression
Calcium signaling could not only regulate the biological behaviors of cancer cells [86, 87], but also involve the carcinogenic process [88]. The oncogenic transformation of epithelial cells is a multistage process during which normal cells shift toward a cancerous state characterized by unlimited proliferation. As we can see in this review, estrogen regulates calcium signaling through rapid calcium influx and alteration of calcium-related protein expression in both normal epithelial and cancerous cells. Therefore, we will discuss the role of Ca2+ signals in carcinogenesis and cancer progression (e.g., proliferation, metastasis, cancer cell death and drug resistance).
Cancer initiation
Both extracellular and intracellular calcium ions have been demonstrated to play important roles in cancerous transformation. The interaction between oncogenic K-Ras and calmodulin is crucial for tumorigenicity through the suppression of the Wnt-Ca2+ signaling pathway [89]. In addition, intracellular and extracellular calcium ions at high concentrations could enhance ERα transcriptional activity in breast cancer in different ways [90]. First, intracellular calcium at physiological concentrations (μM) confers calmodulin (CaM) an active conformation to interact with ERα and enhances receptor-mediated transcription [91]. Second, intracellular calcium at hypercalcemic concentrations (mM), may directly bind to ERα [38]. Finally, high extracellular Ca2+ concentrations (>10 mM) found in metastatic bone lesions increase transcriptional activity of ERα by binding to CaSR at the cell membrane [92]. Depleting extracellular calcium in the growth medium by chelation or using calcium-depleted medium inhibited the neoplastic transformation of mouse JB6 epidermal cells. Such a transformation could also be inhibited by nifedipine, an L-type CCB [93]. Furthermore, multiple drugs triggering calcium fluxes have been reported to reactivate epigenetically mediated suppression of tumor suppressor genes in colon cancer cells [94]. Based on these evidences, the association between CCBs usage (mainly nifedipine, amlodipine, verapamil and diltiazem) and the risk of neoplasia has been widely discussed since the 1990s [95, 96]. However, there is no definitive evidence involving the association between cancer and CCBs use.
Cell proliferation
Calcium ions are closely related to cell proliferation. As early as the 1970s, calcium was thought to be a short regulator of cell growth [97]. Interestingly, normal cells require higher external free Ca2+ concentrations to induce proliferation than preneoplastic and neoplastic cells [98, 99]. External stimuli such as hormones, chemokines and neurotransmitters invoke intracellular calcium increases. Such increases in free calcium ions mainly bind to calmodulin (CaM) and form a Ca2+-CaM complex, which subsequently activates calmodulin-dependent protein kinases (CaMK) and leads to the transcriptional activation of NF-κB, nuclear factor of activated T-cells (NFAT) and cAMP response element-binding protein (CREB) [100-102]. In addition, CaMs and CaMK were reported to interact with cyclin-dependent protein kinases (CDKs) and regulate cell cycle events, thus affecting cell survival [103, 104].
Calcium influx via CACNA1D in Ishikawa cells was considered to induce the phosphorylation of ERK1/2 and activation of CREB [8]. CACNA1G has been proposed as a key regulator of cell cycle progression and survival [105]. Mibefradil is a T-type VGCC inhibitor and has been reported to reduce the viability of Hec-1A cells and stimulate proapoptotic factors [72]. The downregulation of TRPM4 resulted in an increase in the proportion of AN3CA cells in G2/M phase [83].
Metastasis
Calcium signaling has been demonstrated to be crucial for regulating processes that occur during metastasis, including cancer cell migration and invasion. Adding calcium to the culture medium increased the migration of Ishikawa and AN3CA cells, which was reversed by a calcium chelating agent [9]. Coordinated rearrangements of the cytoskeleton and cell-matrix adhesion are required for cell migration. Sustained or transient increases in intracellular calcium ions activate Ca2+-dependent effectors, which can regulate focal adhesion proteins including integrins, paxillin, vinculin, talin, focal adhesion kinase (FAK) and Src family kinases [106]. Mechanosensitive TRPV4 channel could interact directly with cytoskeletons and subsequently induce rapid morphological changes [107]. Silencing of TRPV4 or pharmacological inhibition with its antagonist modulates the RhoA/ROCK1/LIMK/cofilin pathway and further regulates the actin cytoskeleton and paxillin in Ishikawa cells and ultimately decreases the metastatic ability of this cell line [9].
Degradation of extracellular matrix (ECM) is essential for cancer invasion and distant metastasis. Ca2+ influx via TRPV2 can upregulate the expression of some invasive enzymes, such as matrix metalloproteinases and cathepsin B, which can degrade ECM and provide conditions for cancer invasion [108]. In addition, Ca2+ signaling may regulate epithelial-mesenchymal transition (EMT) induction [109]. TRPM4 silencing promotes AN3CA cell progression via the induction of several EMT markers, including E-cadherin, vimentin and N-cadherin [83].
Cancer cell death
Either intracellular Ca2+ overload or perturbation of Ca2+ compartmentalization can cause toxicity to the cells and lead to cell death in the form of apoptosis, autophagy and necrosis [110]. Therefore, the regulation of activity and expression of certain calcium channels or pumps may be exploited for cancer treatment. It is generally believed that severe calcium dysregulation promotes necrotic death, whereas a moderate Ca2+ increase facilitates cell death through autography or apoptosis [111, 112]. Massive Ca2+ influx results in the activation of hydrolysis enzymes, the subsequent loss of membrane integrity and finally cell death through necrosis [113].
Aberrant Ca2+ concentrations always activate endoplasmic reticulum stress (ERS). Stress signals are directly or indirectly relayed from endoplasmic reticulum to the mitochondria and trigger cell apoptosis. On the one hand, Ca2+ released form the endoplasmic reticulum activates numerous pathways and subsequently evokes the release of caspase cofactors from mitochondria and promotes cell death [114]. On the other hand, the endoplasmic reticulum may communicate with mitochondria by direct contacts at mitochondria-associated endoplasmic reticulum membranes (MAMs) [112]. Ca2+ handling proteins at the MAM regions control Ca2+ transfer and affect cell apoptosis [112].
In addition, Ca2+ is a regulator of autophagy. Pharmacological application of the L-type VGCC inhibitor, nifedipine, promotes autophagy through mammalian target of rapamycin (mTOR) and the Beclin1 pathway in Hec-1A cells. Interestingly, adding the autophagy inhibitor 3-MA decreased the protein expression of CACNA1D and augmented nifedipine-induced cell apoptosis, suggesting that autophagy might serve as a protective mechanism for cell survival [115]. In summary, CACNA1D inhibitors are considered as potential drug candidates in EC treatment. Cannabidiol increased the expression of cleaved poly ADP-ribose polymerase (c-PARP) and C/EBP homologous protein (CHOP) through TRPV1 activation in Ishikawa cells. c-PARP is an enzyme involved in DNA repair and CHOP plays a key role in ERS-mediated apoptosis.
Drug resistance
Medications for endometrial cancer mainly include chemotherapeutic drugs and hormone therapy. The platinum-based chemotherapy regimen is the most commonly used in endometrial carcinoma, but the effective rate is not satisfactory [116]. Overexpression of TRPV2 increased the cisplatin cytotoxic effect in Ishikawa cells. The TRPV2 activator, cannabidiol, also enhanced the cell-killing effect of cisplatin in TRPV2-transfected cells [78]. The detailed mechanism is still unknown. Usually, the mechanisms of chemotherapy resistance involving calcium-dependent pathways include drug efflux, evasion of cell death, increased DNA damage tolerance and dysregulation of certain critical genes [117]. Cannabidiol increased the drug retention of several chemotherapeutic agents and synergized with them to induce the apoptosis of glioblastoma cells via TRPV2-dependent Ca2+ influx [118]. In addition, cannabidiol induced the differentiation of glioma stem-like cells, activated autophagy and overcame carmustine resistance in a TRPV2-dependent manner [119]. Cannabidiol was also found to decrease the phosphorylation of nitric oxide synthase 3 (NOS3), increase the production of reactive oxygen species and thus reverse oxaliplatin resistance [120].
Since both rapid and slow regulation of calcium by estrogens exist in endometrial cells, it is interesting to investigate whether calcium ions link these two modes of estrogen. Interestingly, activation of protein kinase A plays an important role in regulating transient receptor potential (TRP) channel functions [121]. In addition, a two-pulse regimen of estrogen treatments has been developed to study the association between rapid and slow estrogen actions in human neuroblastoma cells. E2-BSA given in the first pulse for 20 minutes was followed by 17β-estradiol in the second pulse for 2 hours. The results showed that E2-BSA could enhance the transcription of estrogen response element (ERE) initiated by the later administration of 17β-estradiol. This transcriptional activity was blocked by Ca2+ chelator, suggesting that calcium plays an important role in coupling the rapid and slow estrogen actions [122]. From this perspective, we consider the possibility of inhibiting estrogen actions by using CCBs.
Modulators of calcium-related proteins
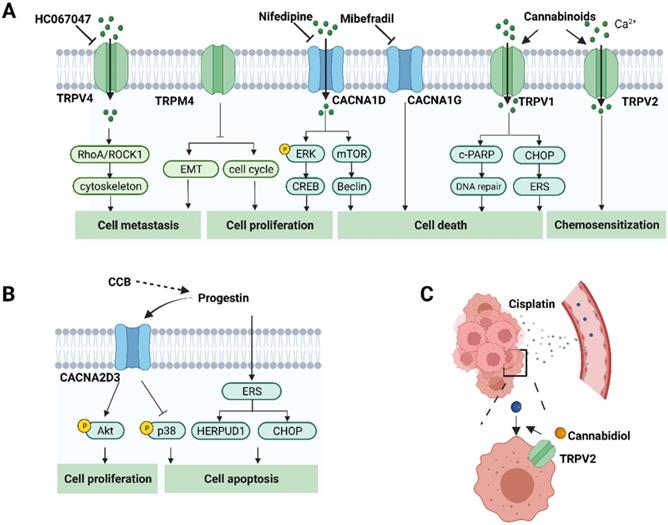
Given the involvement of Ca2+ signaling in carcinogenesis and progression, specific pharmacological agents modulating Ca2+ channels, pumps and exchangers are regarded as druggable. The application of inhibitors or activators depends on whether the resultant alteration to Ca2+ promotes cell survival or death [123]. Strategies targeting Ca2+ signaling in endometrial cancer are illustrated in Figure 3. Pharmacological modulation of calcium channels, pumps or exchangers can affect cell functions and suppress tumor progression by disrupting calcium homeostasis in cancer cells [124, 125]. As described above, a variety of calcium permeable ion channels are involved in uterine carcinogenesis and progression. The modulators of calcium channels that are altered in endometrial cancer cells are expected to be potential therapeutic drugs and are outlined in Table 2.
The role of calcium-related proteins in EC progression and potential strategies for EC treatment. A. Calcium channels affect cancer cell proliferation, cell death, metastasis and response to chemotherapeutic agents through different pathways in EC. Therefore, inhibitors or activators, have the potential for cancer treatment. It also shows the corresponding modulators of these calcium channels. B. Progestin also promotes cell apoptosis by regulating CACNA2D3 and ERS. Calcium channel blockers might generate synergistic anti-tumor effects with progestin in EC. C. Calcium channel modulators might enhance the delivery to the tumor site via modulating relaxation of vasculatures. TRPV2 activation by cannabidiol augments the effects of cisplatin in EC cells. (Created with BioRender.com)
Studies on drugs targeting EC-related Ca2+ channels/ exchangers/ pumps in cancer cells
| Target | Drug | Activator/Inhibitor | Effects on EC | Effects on other cancer cells | ||
|---|---|---|---|---|---|---|
| Studies in EC cells | ref | Studies in non-EC cells | ref | |||
| L-type VGCC | Nifedipine | inhibitor | Nifedipine reduced the proliferation, invasion, apoptosis and promoted autophagy in Hec-1A cells | [72,115] | Nifedipine suppressed colon cancer progression | [147] |
| T-type VGCC | Mibefradil | inhibitor | Mibefradil reduced the proliferation, invasion, apoptosis in Hec-1A cells | [72] | Inhibited proliferation and induced apoptosis in leukemia cells and glioblastoma cells. | [126,128] |
| NNC 55-0396 | inhibitor | ND | NNC 55-0396 suppressed tumor growth in glioblastoma | [129] | ||
| TRPV1/2 | Cannabinoids | inhibitor | Cannabinoids reduced cell viability, activated apoptosis in type I cells (Ishikawa, MFE-280, HEC-1a and PCEM002 cell lines) and autophagy in mixed type EC cells (PCEM004a and PCEM004b cell lines), inhibited migration ability of ISK, PCEM004a and PCEM004b cells and improved chemotherapeutic drugs cytotoxic effects in ISK cells. | [76, 78] | Cannabinoids inhibited cell growth, migration and invasion of several cancer types, including brain, breast and prostate cancers | [132-133] |
| TRPV4 | GSK1016790A | activator | GSK1016790A increased motility of ISK cells | [9] | GSK1016790A reduced the proliferation of tumor endothelial cells | [134] |
| HC067047 | inhibitor | HC067047 led to decreased motility of Hec-1A cells and peritoneal spreading sites in vivo. | [9] | HC067047 suppressed glioma migration and invasion | [137] | |
| SOR-C13 | inhibitor | ND | SOR-C13 reduced ovarian tumor growth in a mouse model. | [138] | ||
| TRPA1 | HC-030031 | inhibitor | ND | HC-030031 alleviated pain in cancer patients. | [139] | |
| PMCA | [Pt(O,O′-acac)(γ-acac)(DMS)] | inhibitor | ND | [Pt(O,O′-acac)(γ-acac)(DMS)] triggers rapid apoptosis in MCF-7 cells. | [141] | |
ND, not determined; EC: endometrial cancer; VGCC: Voltage-gated calcium ion channels; TRPV1: TRP vanilloid 1; TRPV2: TRP vanilloid 2; TRPV4: TRP vanilloid 4; TRPA1: TRP ankyrin 1; PMCAs: Plasma membrane Ca2+ ATPases.
VGCC inhibitors
Clinically, VGCC inhibitors are used in the treatment of hypertension and other cardiovascular diseases by blocking calcium influx. These drugs are mainly divided into two categories: dihydropyridines (DHPs), such as nifedipine and amlodipine, and non-DHPs, such as diltiazem and verapamil. As increasing evidences suggest the important roles of VGCCs in many cancers, numerous investigators have attempted to repurpose FDA-approved VGCC inhibitors for cancer treatment [126]. As early as the 1990s, nifedipine, verapamil and diltiazem were found to inhibit the growth of human astrocytoma U-373 MG cells and human neuroblastoma SK-N-MC cells [127]. Mibefradil, a T- and L-type Ca2+ channel blocker, was approved by FDA for hypertension in 1997 and then withdrawn due to its interaction with other drugs. Surprisingly, mibefradil showed a promising potential to reduce tumor size and improve the survival rate in glioma animal models and pancreatic cancer xenografts [126, 128]. Therefore, mibefradil was repurposed for high-grade glioma cancer and pancreatic cancer treatment. NNC-55-0396, a derivative of mibefradil, was developed to overcome the side effects of its patient and inhibit tumor-induced angiogenesis in vitro and in vivo, thus appearing to be a promising drug [129].
In our previous studies, nifedipine was involved in the estrogen-induced calcium mobilization and phosphorylation of ERK in Ishikawa cells [18, 130]. In addition to the rapid response, nifedipine and mibefradil affect the proliferation, migration and apoptosis of endometrial carcinoma Hec-1A cells [72]. Nifedipine also regulated autophagy through the mTOR and Beclin1 pathways in Hec-1A cells [115]. These results suggest that these CCBs may serve as drug candidates in targeted therapy of endometrial cancer.
TRP channel regulators
CBs have been widely studied for their potential anticancer effects since the 1970s [131]. In addition to the two Gi/o-coupled CB receptors, CB1 and CB2, CBs could also pharmacologically target TRPV1, TRPV2, TRPA1 and TRPM8 [132]. CBs may exert their antitumor effects in a CB1/2-independent manner, as shown in the human pancreatic cancer cell line MIA PaCa-2 [133]. Several kinds of CBs, including endocannabinoid, anandamide and cannabidiol, have been reported to induce cancer cell death in Ishikawa and Hec50co cells through TRPV1-mediated apoptosis [76]. Besides, high TRPV2 expression or its activation by cannabidiol was able to enhance chemotherapeutic drug effects in Ishikawa cells [78].
As TRPV4 was reported to normalize the tumor vasculature in vivo, its newly developed agonist, GSK1016790A, may help improve therapy efficacy by augmenting the delivery of cytotoxic agents to the tumor mass [134]. Interestingly, TRPV4 exerts an impact on cell migration by regulating the actin cytoskeleton in gastric cancer, ovarian cancer, glioma cancer cells and endometrial cancer [9, 135-137]. Pharmacological inhibition with HC067047 or knockdown of TRPV4 inhibits endometrial cancer metastasis, as shown in glioma cells, thus having the potential to be repurposed for EC therapy [9, 137]. SOR-C13, a TRPV6 calcium channel inhibitor, significantly reduces ovarian tumor growth in vivo and thus enters a phase I human clinical trial in patients with advanced ovarian cancers overexpressing TRPV6 [138]. HC-030031 is a potent blocker of TRPA1 and can relieve ongoing pain in a breast cancer pain model [139].
Ca2+-ATPase inhibitors
PMCAs play an important role in pumping Ca2+ out of the cell, therefore, they can be targeted by certain inhibitors to generate toxic Ca2+ concentrations for cell death [140]. [Pt(O,O′-acac)(γ-acac)(DMS)], a selective PMCA inhibitor, was shown to alter intracellular calcium homeostasis and trigger rapid apoptosis in MCF-7 cells [141].
Potential applications of calcium channel modulators for cancer treatment
Currently, the potential application of CCBs is focused on combination with existing treatments, such as chemotherapy or immunotherapy. Targeting Ca2+ signaling of stromal cells in the TME, such as immune cells and tumor endothelial cells, is an emerging strategy and can augment the effects of immunotherapy and chemotherapy [134, 142]. The synergistic administration of DHPs (lercanidipine and amlodipine) and chemotherapeutic drugs (doxorubicin, vincristine and topotecan) has been reported to induce cell apoptosis and autophagy in gastric cancer cells, neuroblastoma cells and multidrug-resistant leukemia cells [143-145]. Verapamil has been well-known to reverse multidrug resistance by directly binding to P-glycoprotein (P-gp) and thus decreasing its expression [146]. Another reason for the synergistic efficacy might be that blocking Ca2+ signaling in vascular endothelial cells increases the delivery of chemotherapeutic agents to the tumor site. The combination of an activator of TRPV4 with cisplatin could increase the delivery of cytotoxic agents to the tumor site and significantly suppress tumor growth [134]. Programmed death 1 (PD-1) and its ligand (PD-L1) are important targets of immunotherapy. Nifedipine and amlodipine could enhance the effects of immunotherapy by depleting PD-L1 expression, and the former even inhibited the expression of PD-1 in T lymphocytes. By mimicking the role of PD-1/PD-L1 inhibitors in tumors, CCBs cooperate with anti-PD-1 therapy in breast cancer, colorectal cancer and colon cancer [142, 147]. Chemotherapy and immunotherapy are also important adjuvant therapeutic methods for endometrial cancer. Appropriate antihypertensive drugs in patients with hypertension might benefit cancer treatment.
Progestin is a viable option for fertility-sparing treatment of patients with early EC and palliative treatment of women with advanced EC. Progestin is reported to rapidly activate intracellular calcium increases in multiple cancer cells, such as triple negative breast cancer [148], oral squamous cancer [149] and endometrial cancer [73]. Ishikawa cells treated with medroxyprogesterone acetate (MPA) showed increased activation of the ERS pathway. Meanwhile, the ERS-related molecules, CHOP and HERPUD1, were significantly upregulated [150]. Alterations in intracellular calcium concentrations often induce ERS and activate downstream pathways. Severe and prolonged ERS leads to cell death. The VGCC inhibitors, verapamil and mibefradil were reported to facilitate cell death via ERS activation in myeloma cells and C2C12 myoblasts [151, 152]. Furthermore, progesterone inhibited cell growth and promoted apoptosis via CACNA2D3 in Ishikawa cells [73]. In addition, four progesterone derivatives were reported to have binding sites on P-gp, which were distinct and nonexclusive with the modulating sites of verapamil. Progesterone, in combination with verapamil, exhibited synergetic activities to induce P-gp ATPase activity and further reverse MDR in a highly resistant tumor cell line [153]. These results provide a foundation for future application of coadministration of progesterone and calcium channel blockers.
The application of CCBs in endometrial cancer treatment depends on a variety of factors. The tissue distribution of calcium channels/pumps/exchangers and possible side reactions might be an important consideration. Drugs targeting certain factors with widespread expression are likely to be associated with generalized toxicity, as they will damage normal cells. After all, VGCC inhibitors have a long history in the clinical treatment of hypertension by blocking VGCCs on vascular endothelial cells. In the majority of studies, repurposing VGCC inhibitors for cancer treatment alone is usually at a much higher dose than is traditionally used to treat hypertension. Therefore, new drug delivery and formulation methods should be exploited. Nanoscale therapeutic delivery systems wrapping CCBs are expected as potential future medicines by increasing accumulation at the tumor site.
Conclusion
Dysregulated Ca2+ homeostasis plays an important role in the occurrence and progression of endometrial cancer. E2 rapidly induces an increase in intracellular calcium and upregulates some of the calcium channels/pumps/exchangers afterwards. Membrane estrogen receptors and downstream kinase cascades participate in the rapid response and affect cell function mainly by activating gene transcription. The role of the Ca2+ signaling in tumor onset and progression goes beyond the cancer cell itself and may also involve the regulation of the TME. Certain calcium channel modulators are involved in both rapid E2-induced intracellular calcium increases and Ca2+-related biological behavior changes in endometrial cancer. It has the potential for CCBs to be repurposed alone or in combination with existing toxic agents for endometrial cancer therapy. Furthermore, structure-based rational transformation of CCBs, aiming to target specific cancer cells and reduce their side effects, will likely provide promising leads for EC treatment in the future.
Abbreviations
EC: Endometrial cancer; TRPV4: Transient receptor potential vanilloid 4; EEC: Endometrial epithelial cells; E2-BSA: Estrogen and bovine serum albumin; CCBs: Calcium channel blockers; PKC: Protein kinase C; mER: Membrane estrogen receptor; nER: nuclear estrogen receptor; GPER1: G protein-coupled estrogen receptor; Shc: Src homology and collagen homology; IGF-1R: Insulin-like growth factor receptor-1; PLC: Phospholipase C; IP3: Inositol 1,4,5-trisphosphate; MAPK: Mitogen-activated protein kinase; ERK: Extracellular-signal-regulated kinase; AP: Activator protein; Bcl-2: B-cell lymphoma-2; JNK: c-Jun N-terminal kinase; NF-κB: Nuclear factor kappa B; MLCK: Myosin light-chain kinase; CaM: Calmodulin; CaMK: Calmodulin-dependent protein kinases; NFAT: nuclear factor of activated T-cells; CREB: cAMP response element-binding protein; CDKs: cyclin-dependent protein kinases; FAK: focal adhesion kinase; ECM: Extracellular matrix; EMT: Epithelial-mesenchymal transition; ERS: Endoplasmic reticulum stress; MAM: mitochondria-associated endoplasmic reticulum membranes; mTOR: Mammalian target of rapamycin; c-PARP: poly ADP-ribose polymerase; CHOP: C/EBP homologous protein; NOS3: Nitric oxide synthase 3; TME: Tumor microenvironment; CAFs: Cancer-associated fibroblast cells; GPCR: G protein-coupled receptors; AR: Androgen receptor; VGCC: Voltage-gated calcium ion channels; CACNA1C: Calcium channel alpha1C; CACNA1D: Calcium channel alpha1D; CACNA1G: Calcium channel alpha1G; CACNA2D3: Calcium channel alpha 2 delta subunit 3; TRP: Transient receptor potential; TRPM8: Transient receptor potential melastatin 8; TRPV1: TRP vanilloid 1; TRPV2: TRP vanilloid 2; TRPV4: TRP vanilloid 4; TRPV6: TRP vanilloid 6; TRPM4: TRP melastatin 4; TRPA1: TRP ankyrin 1; ER: Endoplasmic reticulum; PMCAs: Plasma membrane Ca2+ ATPases; SERCAs: Sarcoendoplasmic reticular Ca2+ ATPases; NCKX3: potassium-dependent sodium/calcium exchanger 3; CaSR: Calcium sensing receptor; CB: Cannabinoid; TCGA: the Cancer Genome Atlas; GEO: Gene Expression Omnibus; DHPs: Dihydropyridines; PD-1: Programmed death 1; PD-L1: Programmed death ligand 1.
Acknowledgements
Funding
This work was supported by the National Key Technology R&D Program of China (no. 2019YFC1005200 and 2019YFC1005201), National Natural Science Foundation of China (no. 81874108, 81672571, and 82072861) and the Beijing Municipal Natural Science Foundation (no. 7202213).
Author Contributions
T.H. and JY.Z. planned and wrote the manuscript. JL.W. revised the manuscript. All authors read and approved the final revision of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Li XC, Dong YY, Cheng Y. et al. Increased Serum Calcium Level Promotes the Risk of Lymph Node Metastasis in Endometrial Cancer. Cancer Manag Res. 2020;12:5023-30
3. Li X, Cheng Y, Dong Y. et al. An elevated preoperative serum calcium level is a significant predictor for positive peritoneal cytology in endometrial carcinoma. Chin J Cancer Res. 2019;31:965-73
4. Jing Z, Sui X, Yao J. et al. SKF-96365 activates cytoprotective autophagy to delay apoptosis in colorectal cancer cells through inhibition of the calcium/CaMKIIγ/AKT-mediated pathway. Cancer Lett. 2016;372:226-38
5. Wei JY, Li WM, Zhou LL. et al. Melatonin induces apoptosis of colorectal cancer cells through HDAC4 nuclear import mediated by CaMKII inactivation. J Pineal Res. 2015;58:429-38
6. Zhang Z, Liu X, Feng B. et al. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene. 2016;35:6043
7. Wang W, Yu S, Huang S. et al. A Complex Role for Calcium Signaling in Colorectal Cancer Development and Progression. Mol Cancer Res. 2019;17:2145-53
8. Hao J, Bao X, Jin B. et al. Ca2+ channel subunit α 1D promotes proliferation and migration of endometrial cancer cells mediated by 17β-estradiol via the G protein-coupled estrogen receptor. FASEB J. 2015;29:2883-93
9. Li X, Cheng Y, Wang Z. et al. Calcium and TRPV4 promote metastasis by regulating cytoskeleton through the RhoA/ROCK1 pathway in endometrial cancer. Cell Death Dis. 2020;11:1009
10. Kriseman M, Monsivais D, Agno J. et al. Uterine double-conditional inactivation of Smad2 and Smad3 in mice causes endometrial dysregulation, infertility, and uterine cancer. Proc Natl Acad Sci U S A. 2019;116:3873-82
11. Lin Y, Zhou J, Cao L. et al. Serum calcium is a novel parameter to assess metabolic syndrome in endometrial carcinoma. J Gynecol Oncol. 2019;30:e12
12. Perret S, Dockery P, Harvey BJ. 17beta-oestradiol stimulates capacitative Ca2+ entry in human endometrial cells. Mol Cell Endocrinol. 2001;176:77-84
13. Pietras RJ, Szego CM. Endometrial cell calcium and oestrogen action. Nature. 1975;253:357-9
14. Pohóczky K, Kun J, Szalontai B. et al. Estrogen-dependent up-regulation of TRPA1 and TRPV1 receptor proteins in the rat endometrium. J Mol Endocrinol. 2016;56:135-49
15. Yang H, Choi KC, Hyun SH. et al. Coexpression and estrogen-mediated regulation of TRPV6 and PMCA1 in the human endometrium during the menstrual cycle. Mol Reprod Dev. 2011;78:274-82
16. Clapham DE. Calcium signaling. Cell. 2007;131:1047-58
17. Jaffe LF. A calcium-based theory of carcinogenesis. Adv Cancer Res. 2005;94:231-63
18. Zhang L, Li X, Zhao L. et al. Nongenomic effect of estrogen on the MAPK signaling pathway and calcium influx in endometrial carcinoma cells. J Cell Biochem. 2009;106:553-62
19. Whitaker M, Patel R. Calcium and cell cycle control. Development. 1990;108:525-42
20. Morley P, Whitfield JF, Vanderhyden BC. et al. A new, nongenomic estrogen action: the rapid release of intracellular calcium. Endocrinology. 1992;131:1305-12
21. Improta-Brears T, Whorton AR, Codazzi F. et al. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci U S A. 1999;96:4686-91
22. Dick IM, Liu J, Glendenning P. et al. Estrogen and androgen regulation of plasma membrane calcium pump activity in immortalized distal tubule kidney cells. Mol Cell Endocrinol. 2003;212:11-8
23. Wu TW, Wang JM, Chen S. et al. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59-72
24. Lieberherr M, Grosse B, Kachkache M. et al. Cell signaling and estrogens in female rat osteoblasts: a possible involvement of unconventional nonnuclear receptors. J Bone Miner Res. 1993;8:1365-76
25. Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. 1977;265:69-72
26. Barton M, Filardo EJ, Lolait SJ. et al. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J Steroid Biochem Mol Biol. 2018;176:4-15
27. Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995;9:404-10
28. Rainville J, Pollard K, Vasudevan N. Membrane-initiated non-genomic signaling by estrogens in the hypothalamus: cross-talk with glucocorticoids with implications for behavior. Front Endocrinol (Lausanne). 2015;6:18
29. Saczko J, Michel O, Chwiłkowska A. et al. Estrogen Receptors in Cell Membranes: Regulation and Signaling. Adv Anat Embryol Cell Biol. 2017;227:93-105
30. Soltysik K, Czekaj P. Membrane estrogen receptors - is it an alternative way of estrogen action. J Physiol Pharmacol. 2013;64:129-42
31. Chambliss KL, Yuhanna IS, Anderson RG. et al. ERbeta has nongenomic action in caveolae. Mol Endocrinol. 2002;16:938-46
32. Razandi M, Oh P, Pedram A. et al. ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol. 2002;16:100-15
33. Chambliss KL, Yuhanna IS, Mineo C. et al. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res. 2000;87:E44-52
34. Evinger AJ 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361-3
35. Revankar CM, Cimino DF, Sklar LA. et al. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625-30
36. Simoncini T, Hafezi-Moghadam A, Brazil DP. et al. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538-41
37. Grolez GP, Gordiendko DV, Clarisse M. et al. TRPM8-androgen receptor association within lipid rafts promotes prostate cancer cell migration. Cell Death Dis. 2019;10:652
38. Divekar SD, Storchan GB, Sperle K. et al. The role of calcium in the activation of estrogen receptor-alpha. Cancer Res. 2011;71:1658-68
39. Zhang Y, Li Z, Sacks DB. et al. Structural basis for Ca2+-induced activation and dimerization of estrogen receptor α by calmodulin. J Biol Chem. 2012;287:9336-44
40. Albanito L, Madeo A, Lappano R. et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67:1859-66
41. Lappano R, Rosano C, De Marco P. et al. Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol Cell Endocrinol. 2010;320:162-70
42. Vivacqua A, Bonofiglio D, Albanito L. et al. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol. 2006;70:1414-23
43. Tran QK. Reciprocality Between Estrogen Biology and Calcium Signaling in the Cardiovascular System. Front Endocrinol (Lausanne). 2020;11:568203
44. Tran QK, Firkins R, Giles J. et al. Estrogen Enhances Linkage in the Vascular Endothelial Calmodulin Network via a Feedforward Mechanism at the G Protein-coupled Estrogen Receptor 1. J Biol Chem. 2016;291:10805-23
45. Ji Y, Han Z, Shao L. et al. Ultrasound-targeted microbubble destruction of calcium channel subunit α 1D siRNA inhibits breast cancer via G protein-coupled receptor 30. Oncol Rep. 2016;36:1886-92
46. Tran QK, Vermeer M. Biosensor-based approach identifies four distinct calmodulin-binding domains in the G protein-coupled estrogen receptor 1. PLoS One. 2014;9:e89669
47. Zeng W, Mak DO, Li Q. et al. A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gbetagamma. Curr Biol. 2003;13:872-6
48. Schmidt BM, Gerdes D, Feuring M. et al. Rapid, nongenomic steroid actions: A new age. Front Neuroendocrinol. 2000;21:57-94
49. Ishii K, Kano T, Ando J. Calcium channel, Ca++ mobilization, and mechanical reactivity of estrogen- and progesterone-treated rat uterus. Jpn J Pharmacol. 1986;41:47-54
50. Simoncini T, Genazzani AR. Non-genomic actions of sex steroid hormones. Eur J Endocrinol. 2003;148:281-92
51. Sarkar SN, Huang RQ, Logan SM. et al. Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A. 2008;105:15148-53
52. Guo YJ, Pan WW, Liu SB. et al. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19:1997-2007
53. Acconcia F, Barnes CJ, Kumar R. Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cancer cells. Endocrinology. 2006;147:1203-12
54. Endoh H, Sasaki H, Maruyama K. et al. Rapid activation of MAP kinase by estrogen in the bone cell line. Biochem Biophys Res Commun. 1997;235:99-102
55. Migliaccio A, Di Domenico M, Castoria G. et al. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292-300
56. Singer CA, Figueroa-Masot XA, Batchelor RH. et al. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455-63
57. Haynes MP, Li L, Sinha D. et al. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. 2003;278:2118-23
58. Sée V, Rajala NK, Spiller DG. et al. Calcium-dependent regulation of the cell cycle via a novel MAPK-NF-kappaB pathway in Swiss 3T3 cells. J Cell Biol. 2004;166:661-72
59. Yamakawa K, Arita J. Cross-talk between the estrogen receptor-, protein kinase A-, and mitogen-activated protein kinase-mediated signaling pathways in the regulation of lactotroph proliferation in primary culture. J Steroid Biochem Mol Biol. 2004;88:123-30
60. Nilsen J, Diaz Brinton R. Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc Natl Acad Sci U S A. 2003;100:2842-7
61. Madak-Erdogan Z, Lupien M, Stossi F. et al. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol. 2011;31:226-36
62. Madak-Erdogan Z, Ventrella R, Petry L. et al. Novel roles for ERK5 and cofilin as critical mediators linking ERα-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol Cancer Res. 2014;12:714-27
63. Arnal JF, Lenfant F, Metivier R. et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol Rev. 2017;97:1045-87
64. Pardo VG, Boland R, de Boland AR. 1alpha,25(OH)(2)-Vitamin D(3) stimulates intestinal cell p38 MAPK activity and increases c-Fos expression. Int J Biochem Cell Biol. 2006;38:1181-90
65. Kamata Y, Tominaga M, Umehara Y. et al. Calcium-Inducible MAPK/AP-1 Signaling Drives Semaphorin 3A Expression in Normal Human Epidermal Keratinocytes. J Invest Dermatol. 2020;140:1346-54.e5
66. Word RA, Tang DC, Kamm KE. Activation properties of myosin light chain kinase during contraction/relaxation cycles of tonic and phasic smooth muscles. J Biol Chem. 1994;269:21596-602
67. Jain R, Watson U, Vasudevan L. et al. ERK Activation Pathways Downstream of GPCRs. Int Rev Cell Mol Biol. 2018;338:79-109
68. Prole DL, Taylor CW. Structure and Function of IP3 Receptors. Cold Spring Harb Perspect Biol. 2019 11
69. Marchi S, Giorgi C, Galluzzi L. et al. Ca2+ Fluxes and Cancer. Mol Cell. 2020;78:1055-69
70. Phan NN, Wang CY, Chen CF. et al. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol Lett. 2017;14:2059-74
71. Chen R, Zeng X, Zhang R. et al. Cav1.3 channel α1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol Oncol. 2014;32:524-36
72. Bao XX, Wang JL, Wei LH. [Impact of calcium channel antagonists for estrogen action on the endometrial carcinoma HEC-1A cells]. Zhonghua Fu Chan Ke Za Zhi. 2012;47:212-7
73. Kong X, Li M, Shao K. et al. Progesterone induces cell apoptosis via the CACNA2D3/Ca2+/p38 MAPK pathway in endometrial cancer. Oncol Rep. 2020;43:121-32
74. Canales J, Morales D, Blanco C. et al. A TR(i)P to Cell Migration: New Roles of TRP Channels in Mechanotransduction and Cancer. Front Physiol. 2019;10:757
75. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619-47
76. Fonseca BM, Correia-da-Silva G, Teixeira NA. Cannabinoid-induced cell death in endometrial cancer cells: involvement of TRPV1 receptors in apoptosis. J Physiol Biochem. 2018;74:261-72
77. Xu S, Cheng Y, Keast JR. et al. 17beta-estradiol activates estrogen receptor beta-signalling and inhibits transient receptor potential vanilloid receptor 1 activation by capsaicin in adult rat nociceptor neurons. Endocrinology. 2008;149:5540-8
78. Marinelli O, Morelli MB, Annibali D. et al. The Effects of Cannabidiol and Prognostic Role of TRPV2 in Human Endometrial Cancer. Int J Mol Sci. 2020 21
79. Ashida S, Nakagawa H, Katagiri T. et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res. 2004;64:5963-72
80. Guinamard R, Demion M, Launay P. Physiological roles of the TRPM4 channel extracted from background currents. Physiology (Bethesda). 2010;25:155-64
81. Loo SK, Ch'ng ES, Md Salleh MS. et al. TRPM4 expression is associated with activated B cell subtype and poor survival in diffuse large B cell lymphoma. Histopathology. 2017;71:98-111
82. Liu L, Lin J, He H. Identification of Potential Crucial Genes Associated With the Pathogenesis and Prognosis of Endometrial Cancer. Front Genet. 2019;10:373
83. Li XC, Cheng Y, Yang X. et al. Decreased expression of TRPM4 is associated with unfavorable prognosis and aggressive progression of endometrial carcinoma. Am J Transl Res. 2020;12:3926-39
84. Yang H, Kim TH, Lee HH. et al. Distinct expression of the calcium exchangers, NCKX3 and NCX1, and their regulation by steroid in the human endometrium during the menstrual cycle. Reprod Sci. 2011;18:577-85
85. Xin X, Zeng X, Feng D. et al. The suppressive role of calcium sensing receptor in endometrial cancer. Sci Rep. 2018;8:1076
86. Lee H, Kim JW, Kim DK. et al. Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells. Int J Mol Sci. 2020 21
87. Li P, Zhong D, Gong PY. Synergistic effect of paclitaxel and verapamil to overcome multi-drug resistance in breast cancer cells. Biochem Biophys Res Commun. 2019;516:183-8
88. Izquierdo-Torres E, Hernández-Oliveras A, Fuentes-García G. et al. Calcium signaling and epigenetics: A key point to understand carcinogenesis. Cell Calcium. 2020;91:102285
89. Wang MT, Holderfield M, Galeas J. et al. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell. 2015;163:1237-51
90. Leclercq G. Calcium-induced activation of estrogen receptor alpha-New insight. Steroids. 2012;77:924-7
91. Bouhoute A, Leclercq G. Modulation of estradiol and DNA binding to estrogen receptor upon association with calmodulin. Biochem Biophys Res Commun. 1995;208:748-55
92. Journé F, Dumon JC, Kheddoumi N. et al. Extracellular calcium downregulates estrogen receptor alpha and increases its transcriptional activity through calcium-sensing receptor in breast cancer cells. Bone. 2004;35:479-88
93. Smith BM, Gindhart TD, Colburn NH. Extracellular calcium requirement for promotion of transformation in JB6 cells. Cancer Res. 1986;46:701-6
94. Raynal NJ, Lee JT, Wang Y. et al. Targeting Calcium Signaling Induces Epigenetic Reactivation of Tumor Suppressor Genes in Cancer. Cancer Res. 2016;76:1494-505
95. Pahor M, Guralnik JM, Ferrucci L. et al. Calcium-channel blockade and incidence of cancer in aged populations. Lancet. 1996;348:493-7
96. Rosenberg L, Rao RS, Palmer JR. et al. Calcium channel blockers and the risk of cancer. JAMA. 1998;279:1000-4
97. McKeehan WL, Ham RG. Calcium and magnesium ions and the regulation of multiplication in normal and transformed cells. Nature. 1978;275:756-8
98. Boynton AL, Whitfield JF. Different calcium requirements for proliferation of conditionally and unconditionally tumorigenic mouse cells. Proc Natl Acad Sci U S A. 1976;73:1651-4
99. Boynton AL, Whitfield JF, Isaacs RJ. et al. Different extracellular calcium requirements for proliferation of nonneoplastic, preneoplastic, and neoplastic mouse cells. Cancer Res. 1977;37:2657-61
100. Humeau J, Bravo-San Pedro JM, Vitale I. et al. Calcium signaling and cell cycle: Progression or death. Cell Calcium. 2018;70:3-15
101. Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611-4
102. Ma H, Groth RD, Cohen SM. et al. γCaMKII shuttles Ca²⁺/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell. 2014;159:281-94
103. Kahl CR, Means AR. Calcineurin regulates cyclin D1 accumulation in growth-stimulated fibroblasts. Mol Biol Cell. 2004;15:1833-42
104. Keith C, DiPaola M, Maxfield FR. et al. Microinjection of Ca++-calmodulin causes a localized depolymerization of microtubules. J Cell Biol. 1983;97:1918-24
105. Kuga T, Kobayashi S, Hirakawa Y. et al. Cell cycle-dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res. 1996;79:14-9
106. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609-18
107. Goswami C, Kuhn J, Heppenstall PA. et al. Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells. PLoS One. 2010;5:e11654
108. Monet M, Lehen'kyi V, Gackiere F. et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010;70:1225-35
109. Davis FM, Azimi I, Faville RA. et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33:2307-16
110. Zhivotovsky B, Orrenius S. Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium. 2011;50:211-21
111. Kania E, Pająk B, Orzechowski A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed Res Int. 2015;2015:352794
112. Pinton P, Giorgi C, Siviero R. et al. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407-18
113. Pressman BC. Biological applications of ionophores. Annu Rev Biochem. 1976;45:501-30
114. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013-30
115. Bao XX, Xie BS, Li Q. et al. Nifedipine induced autophagy through Beclin1 and mTOR pathway in endometrial carcinoma cells. Chin Med J (Engl). 2012;125:3120-6
116. Moxley KM, McMeekin DS. Endometrial carcinoma: a review of chemotherapy, drug resistance, and the search for new agents. Oncologist. 2010;15:1026-33
117. Santoni G, Morelli MB, Marinelli O. et al. Calcium Signaling and the Regulation of Chemosensitivity in Cancer Cells: Role of the Transient Receptor Potential Channels. Adv Exp Med Biol. 2020;1131:505-17
118. Nabissi M, Morelli MB, Santoni M. et al. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis. 2013;34:48-57
119. Nabissi M, Morelli MB, Amantini C. et al. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int J Cancer. 2015;137:1855-69
120. Jeong S, Kim BG, Kim DY. et al. Cannabidiol Overcomes Oxaliplatin Resistance by Enhancing NOS3- and SOD2-Induced Autophagy in Human Colorectal Cancer Cells. Cancers (Basel). 2019 11
121. Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726-41
122. Vasudevan N, Kow LM, Pfaff DW. Early membrane estrogenic effects required for full expression of slower genomic actions in a nerve cell line. Proc Natl Acad Sci U S A. 2001;98:12267-71
123. Roberts-Thomson SJ, Chalmers SB, Monteith GR. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb Perspect Biol. 2019 11
124. Takahashi N, Chen HY, Harris IS. et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell. 2018;33:985-1003.e7
125. Peters AA, Jamaludin S, Yapa K. et al. Oncosis and apoptosis induction by activation of an overexpressed ion channel in breast cancer cells. Oncogene. 2017;36:6490-500
126. Kale VP, Amin SG, Pandey MK. Targeting ion channels for cancer therapy by repurposing the approved drugs. Biochim Biophys Acta. 2015;1848:2747-55
127. Lee YS, Sayeed MM, Wurster RD. Inhibition of cell growth and intracellular Ca2+ mobilization in human brain tumor cells by Ca2+ channel antagonists. Mol Chem Neuropathol. 1994;22:81-95
128. Ignacio Garrido-Laguna ACT, Maria Cristina Villarroel NVR, Belen Rubio-Viqueira LGaMH. Garrido-Laguna I, Tan A.C, Villarroel M.C, Rajeshkumar N, Rubio-Viqueira B, Gray L. Activity of the T-type calcium channel antagonist Mibefradil in pancreatic cancer xenografts. Clin Cancer Res. 2008;14 [B49-B49]. Clin Cancer Res. 2008; 14 [B49-B49]
129. Kim KH, Kim D, Park JY. et al. NNC 55-0396, a T-type Ca2+ channel inhibitor, inhibits angiogenesis via suppression of hypoxia-inducible factor-1α signal transduction. J Mol Med (Berl). 2015;93:499-509
130. Zhang LL, Wang JL. [Nongenomic effects of estrogen on extracellular signal-regulated kinases through initiating transient calcium flux in endometrial cancer]. Beijing Da Xue Xue Bao Yi Xue Ban. 2015;47:489-93
131. Dariš B, Tancer Verboten M, Knez Ž. et al. Cannabinoids in cancer treatment: Therapeutic potential and legislation. Bosn J Basic Med Sci. 2019;19:14-23
132. De Petrocellis L, Ligresti A, Moriello AS. et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479-94
133. Fogli S, Nieri P, Chicca A. et al. Cannabinoid derivatives induce cell death in pancreatic MIA PaCa-2 cells via a receptor-independent mechanism. FEBS Lett. 2006;580:1733-9
134. Adapala RK, Thoppil RJ, Ghosh K. et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene. 2016;35:314-22
135. Wang H, Zhang B, Wang X. et al. TRPV4 Overexpression Promotes Metastasis Through Epithelial-Mesenchymal Transition in Gastric Cancer and Correlates with Poor Prognosis. Onco Targets Ther. 2020;13:8383-94
136. Wang K, Feng X, Zheng L. et al. TRPV4 is a Prognostic Biomarker that Correlates with the Immunosuppressive Microenvironment and Chemoresistance of Anti-Cancer Drugs. Front Mol Biosci. 2021;8:690500
137. Ou-Yang Q, Li B, Xu M. et al. TRPV4 promotes the migration and invasion of glioma cells via AKT/Rac1 signaling. Biochem Biophys Res Commun. 2018;503:876-81
138. Xue H, Wang Y, MacCormack T.J, Lutes T, Rice C, Davey M, Dugourd D, Ilenchuk T.T, Stewart J.M. (2018). Inhibition of Transient Receptor Potential Vanilloid 6 channel, elevated in human ovarian cancers, reduces tumour growth in a xenograft model. J Cancer 9, 3196-3207.
139. de Almeida AS, Pereira GC, Brum E. et al. Role of TRPA1 expressed in bone tissue and the antinociceptive effect of the TRPA1 antagonist repeated administration in a breast cancer pain model. Life Sci. 2021;276:119469
140. Curry MC, Roberts-Thomson SJ, Monteith GR. Plasma membrane calcium ATPases and cancer. Biofactors. 2011;37:132-8
141. Muscella A, Calabriso N, Vetrugno C. et al. The platinum (II) complex [Pt(O,O'-acac)(γ-acac)(DMS)] alters the intracellular calcium homeostasis in MCF-7 breast cancer cells. Biochem Pharmacol. 2011;81:91-103
142. Li C, Yao H, Wang H. et al. Repurposing screen identifies Amlodipine as an inducer of PD-L1 degradation and antitumor immunity. Oncogene. 2021;40:1128-46
143. Panneerpandian P, Rao DB, Ganesan K. Calcium channel blockers lercanidipine and amlodipine inhibit YY1/ERK/TGF-β mediated transcription and sensitize the gastric cancer cells to doxorubicin. Toxicol In vitro. 2021;74:105152
144. Taghizadehghalehjoughi A, Sezen S, Hacimuftuoglu A. et al. Vincristine combination with Ca+2 channel blocker increase antitumor effects. Mol Biol Rep. 2019;46:2523-8
145. Li X, Lu WL, Liang GW. et al. Effect of stealthy liposomal topotecan plus amlodipine on the multidrug-resistant leukaemia cells in vitro and xenograft in mice. Eur J Clin Invest. 2006;36:409-18
146. Wang L, Sun Y. Efflux mechanism and pathway of verapamil pumping by human P-glycoprotein. Arch Biochem Biophys. 2020;696:108675
147. Wu L, Lin W, Liao Q. et al. Calcium Channel Blocker Nifedipine Suppresses Colorectal Cancer Progression and Immune Escape by Preventing NFAT2 Nuclear Translocation. Cell Rep. 2020;33:108582
148. Cantonero C, Salido GM, Rosado JA. et al. PGRMC1 Inhibits Progesterone-Evoked Proliferation and Ca2+ Entry Via STIM2 in MDA-MB-231 Cells. Int J Mol Sci. 2020 21
149. Palmerini CA, Mazzoni M, Radicioni G. et al. Antagonistic Effect of a Salivary Proline-Rich Peptide on the Cytosolic Ca2+ Mobilization Induced by Progesterone in Oral Squamous Cancer Cells. PLoS One. 2016;11:e0147925
150. Cao W, Gao W, Zheng P. et al. Medroxyprogesterone acetate causes the alterations of endoplasmic reticulum related mRNAs and lncRNAs in endometrial cancer cells. BMC Med Genomics. 2019;12:163
151. Meister S, Frey B, Lang VR. et al. Calcium channel blocker verapamil enhances endoplasmic reticulum stress and cell death induced by proteasome inhibition in myeloma cells. Neoplasia. 2010;12:550-61
152. Chen M, Li S, Hao M. et al. T-type calcium channel blockade induces apoptosis in C2C12 myotubes and skeletal muscle via endoplasmic reticulum stress activation. FEBS Open Bio. 2020;10:2122-36
153. Orlowski S, Mir LM, Belehradek J Jr. et al. Effects of steroids and verapamil on P-glycoprotein ATPase activity: progesterone, desoxycorticosterone, corticosterone and verapamil are mutually non-exclusive modulators. Biochem J. 1996;317( Pt 2):515-22
Author contact
![]() Corresponding authors: Jianliu Wang, MD, Department of Gynecology and Obstetrics, Peking University People's Hospital, Beijing, China; Tel: 86-10-88324278; E-mail address: wangjianliuedu.cn; Jingyi Zhou, PhD, Department of Gynecology and Obstetrics, Peking University People's Hospital, Beijing, China; Phone: +86 15210829207; E-mail address: sy_zhoujingyipku.edu.cn.
Corresponding authors: Jianliu Wang, MD, Department of Gynecology and Obstetrics, Peking University People's Hospital, Beijing, China; Tel: 86-10-88324278; E-mail address: wangjianliuedu.cn; Jingyi Zhou, PhD, Department of Gynecology and Obstetrics, Peking University People's Hospital, Beijing, China; Phone: +86 15210829207; E-mail address: sy_zhoujingyipku.edu.cn.