Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(4):1508-1520. doi:10.7150/ijbs.68861 This issue Cite
Research Paper
Identification of Gαi3 as a promising target for osteosarcoma treatment
Zheng-jun Bian1,2#, Hua-jian Shan1#, Yun-Rong Zhu3#, Ce Shi4#, Min-bin Chen5, Yu-min Huang6 ![]() #, Xiao-dong Wang7
#, Xiao-dong Wang7 ![]() , Xiao-zhong Zhou1
, Xiao-zhong Zhou1 ![]() , Cong Cao1,8,9
, Cong Cao1,8,9 ![]()
1. Department of Orthopedics, Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, Suzhou, China.
2. Department of Orthopedics, The Affiliated Hospital of Yangzhou University, Yangzhou University, Yangzhou, China.
3. Department of Orthopedics, Affiliated Jiangyin Hospital of Xuzhou Medical University, Jiangyin, China.
4. Department of Orthopedics, The Affiliated Suqian Hospital of Xuzhou Medical University, Suqian, China.
5. Department of Radiotherapy and Oncology, Kunshan First People's Hospital Affiliated to Jiangsu University, Kunshan, China.
6. Department of Orthopedics, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China.
7. Department of Orthopedics, Children's Hospital of Soochow University, Suzhou, China.
8. Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, Suzhou, China.
9. North District, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Suzhou, China.
# Co-first authors.
Received 2021-11-11; Accepted 2021-12-31; Published 2022-1-24
Abstract

Sustained activation of multiple receptor tyrosine kinases (RTKs) simultaneously is vital for tumorigenesis and progression of osteosarcoma (OS). Gαi proteins recruitment to various RTKs mediates downstream oncogenic signaling activation. The expression, functions and underlying mechanisms of Gαi3 in human OS were examined. Expression of Gαi3 is robustly elevated in human OS tissues and is correlated with a poor overall survival. In patient-derived primary OS cells and immortalized lines (MG63 and U2OS), Gαi3 depletion, by shRNA and CRISPR/Cas9 strategies, robustly suppressed cell viability, proliferation and migration, while provoking G1-S arrest and apoptosis activation. Conversely, Gαi3 overexpressing ectopically can accelerate proliferation and migration of OS cells. In OS cells, Gαi3 immunoprecipitated with VEGFR2, FGFR, PGDFR and EGFR, mediating downstream cascade transduction. Akt-mTOR activation in primary OS cells was potently inhibited by Gαi3 shRNA, knockout or dominant negative mutation, but augmented after Gαi3 overexpression. In vivo studies showed that Gαi3 shRNA AAV (adeno-associated viruses) intratumoral injection largely inhibited the growth of subcutaneous xenografts of primary OS cells. Moreover, the growth of the Gαi3-knockout primary OS xenografts was much slower than that of OS xenografts with empty vector. In Gαi3-depleted OS xenografts tissues, Gαi3 downregulation and Akt-mTOR inactivation were detected. Taken together, overexpressed Gαi3 mediates RTK-Akt signaling to drive OS progression.
Keywords: Osteosarcoma, Gαi3, multiple receptor tyrosine kinase, Akt-mTOR, Signaling
Introduction
For the advanced osteosarcoma (OS) patients with metastatic, recurrent or therapy-resistant OS, the prognosis is poor [1]. Further exploring the novel targeted therapeutics for OS is thus extremely important [2-7]. Due to various gene mutations, overexpression and/or over-activation of multiple receptor tyrosine kinases (RTKs) and its downstream oncogenic cascades are essential for initiation, progression and therapy-resistance of OS [4, 8, 9]. Several RTKs, including VEGFR, PDGFR, RET, EGFR and IGFR, as well as KIT and FGFR, are key drivers for the cancerous behaviors of OS [4, 8, 9]. Concurrent activation of multiple RTKs shall provoke sustained activation of oncogenic cascades, causing persistent cancer growth [8, 9]. Therefore, targeting one or few RTKs, using genetic methods and/or pharmacological inhibitors, could only achieve minimal anti-OS efficiency [8, 9]. The novel stragies targeting multiple RTKs simultaneously should achieve better anti-OS outcome [8, 9].
The inhibitory guanine nucleotide regulatory proteins, Gαi proteins, consists of three subunits, including Gαi1, Gαi2 and Gαi3 [10]. It is known that GPCRs (G protein-coupled receptors) binding to Gαi proteins and the β and γ complexes will hinder adenylate cyclase, causing cyclic AMP (cAMP) depletion [10]. Such actions would be reversed by pertussis toxin [11]. Few studies have explored the expression, function and potential signaling mechanism of Gαi proteins in OS. Pine et al., have shown that Gαi proteins are important for the agonist-induced cAMP production in osteosarcoma cells that were derived from rat [12]. Wang et al., showed that pertussis toxin can inhibit bradykinin-induced Ca2+ mobilization in MG63 OS cells [13].
Our group has identified an essential role of Gαi proteins in transducing signals for multiple RTKs [14-20]. We found that EGFR-induced Akt-mTOR activation was abolished after Gαi1/3 double knockout (DKO) or silencing [20]. With VEGF stimulation, Gαi1/3 associated with VEFGR2, promoting VEGFR2 endocytosis and downstream signaling activation [16]. Similarly, Gαi1/3 mediated BDNF (brain-derived neurotrophic factor)-stimulated Akt-mTOR activation [17]. Therefore, by mediating signaling transduction of multiple RTKs, Gαi proteins could be important oncogenic genes and therapeutic targets for human cancer [14, 18, 21]. Indeed, we have previously shown that Gαi proteins are upregulated in glioma and gastric cancers, required for cancer growth [14, 18, 21]. The current study explored Gαi3 expression and potential functions in human OS.
Methods
Ethics
The protocols of the present study were reviewed and approved by the Ethics Committee of Soochow University and were in according to the principle of Helsinki declaration.
Reagents
LY294002 was from Sigma Aldrich (St Louis, M.O.). The antibodies were described in our previous studies [18]. All the primers, sequences, constructs and virus were synthesized by Shanghai Genechem Co. (Shanghai, China), or mentioned otherwise.
Cells and tissues
The immortalized OS lines, MG63 and U2OS, as well as the established hFOB1.19 osteoblastic cells were cultivated as described [22-24]. Patient-derived primary OS patients, namely pOS-1 and pOS-2, were described previously [22, 23]. Patient-derived human osteoblasts (pOsteoblasts) were differentiated and cultured using the previously-descried protocols [25, 26]. The human tissues, including the OS tumor tissue specimens and the adjacent normal bone tissue specimens [22, 23], were obtained from the written-informed consent OS patients who were all administrated at the Affiliated Hospitals of Soochow University.
Gene detection
Protein detection by Western blotting, RNA assays by qRT-PCR and co-immunoprecipitation (co-IP) examining protein-protein interaction were extensively described early [17, 20]. When necessary, lysates from the same set of the experiment were detected in the parallel gels to test different proteins. The primers were described early [16].
Gαi3 shRNA
The lentivirus-encoded Gαi3 shRNAs and in vitro cell infection were described early [16]. Stable cells were selected by puromycin-containing complete medium (with FBS) for additional 96h. Gαi3 silencing (with over 95-98% knockdown efficiency) in the stable cells was always verified. The control cells were infected with non-sense scramble control shRNA lentivirus (“sh-C”) [16]. For the in vivo studies, the Gαi3 shRNA sequence or shC sequence was sub-cloned into an established adenoviral vector, adeno-associated virus 9/AAV9 construct (Genechem). Through Lipofectamine 3000 the construct was thereafter transfected to HEK-293T cells, and the shRNA-expressing AAV virus was generated and was injected to xenograft tumors.
CRISPR/Cas9-induced Gαi3 knockout (KO)
OS cells were transfected with Cas9-expressing construct (Genechem) by Lipofectamine 3000 (Invitrogen, Shanghai, China) to establish stable cells. Next, the lenti-CRISPR/Cas-9 Gαi3 KO construct [14, 16], was transduced to Cas9-expressing OS cells, with stable cells established by using puromycin-containing medium for additional 96h. Gαi3 KO screening was carried out and thereafter the Gαi3 KO cells were eventually established. The control OS cells were with a lenti-CRISPR/Cas-9 empty vector with non-sense small guide RNA (“Cas9-C”).
Gαi3 overexpression and dominant negative mutation, stable cells establishment and verification were described in our previous studies [14, 16].
Constitutively-active mutant Akt1
OS cells were infected with the constitutively-active Akt1 (caAkt1, S473D)-expressing adenovirus (provided by Dr. Li [27, 28]) for 48h. Puromycin was thereafter added for 96h to establish the stable OS cells, where expression of the caAkt1 was confirmed by Western blotting.
Akt1/2 shRNA
The commercial available Akt1/2 shRNA lentiviral particles (sc-43609-V, Santa Cruz Biotech) were added and transfected to cultured OS cells. After 48h, cells were cultured and selected in puromycin-containing medium for another 96h. Akt1/2 silencing was always examined.
Cellular functional studies
The cell viability detection by cell counting kit-8 (CCK-8), the EdU nuclear staining assaying of cell proliferation, propidium iodide (PI)-FACS, “Transwell” assays were carried out using the previously described protocols [18, 21, 25, 29-33].
Apoptosis detection
Apoptosis-related assays, including the TUNEL nuclear staining, 7-AAD and Annexin V double staining, caspase-3/-9 activities measurement, and ELISA testing the cellular ssDNA (single strand DNA) contents were described early [21, 25, 31-33].
Xenograft studies
The primary pOS1 cells (five million cells in every mouse) were subcutaneously (s.c.) injected to the nude mice (18.5-19.5g, half female and half male, please refer to our previous studies [22, 23]). Tumor-bearing mice were then subject to the designated treatments. Tumor volumes [(length × width2)/2] and animal body weights were weekly recorded. Soochow University's Ethics Committee and Institutional Animal Care and Use Committee (IACUC) reviewed and approved the protocols of animal studies.
Statistical analyses
Statistical analyses were described previously [22, 23]. The numerical data in the bar graphs indicated the mean and standard deviation (S.D.). P-values < 0.05 were statistically significant.
Results
In human OS Gαi3 is upregulated
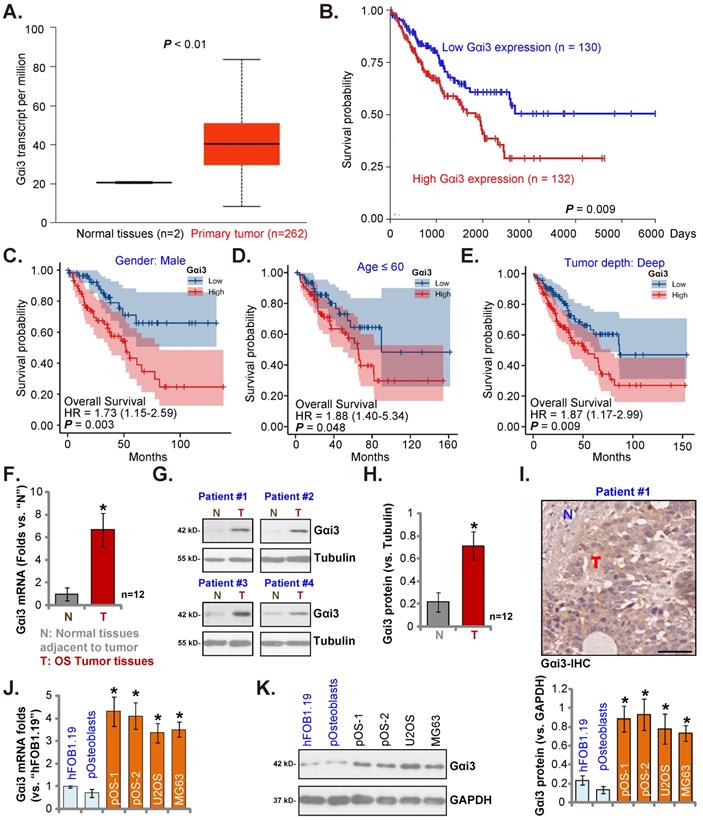
The Cancer Genome Atlas (TCGA) database (available on the public domain https://portal.gdc.cancer.gov) was first consulted to analyze Gαi3 transcripts in human sarcoma tissues. Total 264 samples (HTSeq-FPKM) were collected, including the two normal specimens and the 262 sarcoma specimens. As shown, Gαi3 transcripts in the retrieved sarcoma tissue specimens were higher than those in the retrieved normal tissue specimens (Figure 1A). The Kaplan-Meier survival, Figure 1B, verified that high Gαi3 expression in sarcoma patients was correlated with a poor prognosis (HR = 1.73, P = 0.008). Subgroup analysis by different clinical features demonstrated that Gαi3-high expression was associated with a poor prognosis in male sarcoma patients (P = 0.003, Figure 1C), age≤60 (P = 0.048, Figure 1D), Tumor depth: deep (P = 0.009, Figure 1E).
Next, we tested Gαi3 expression in local OS tissues. As previously described [22, 23], OS tissue specimens and matched adjacent bone normal tissue specimens were retrieved from 10 different OS patients. Figure 1F showed that Gαi3 mRNA in the OS tissue specimens (“T”) was more than six-fold higher than that in the adjacent normal bone tissues (“N”). Gαi3 protein expression was tested as well. In the OS tissues of four representative OS patients (Patient #1/#2/#3/#4), protein expression of Gαi3 was significantly elevated (Figure 1G). When combining all blotting results of the ten sets tissues, Gαi3 protein was found to be significantly upregulated in the OS tissues (P < 0.001 versus “N” tissues, Figure 1H). The immunohistochemistry (IHC) staining assay results, in Figure 1I, verified Gαi3 protein elevation in OS tumor tissues (Figure 1I), While low expression of Gαi3 was detected in the adjacent normal tissue specimens (Figure 1I).
The expression of Gαi3 in different human OS cells was examined. Patient-derived primary OS cells (pOS-1/2, from two different OS patients), as well as the immortalized cells (MG63 and U2OS lines), were cultured. The expression of Gαi3 mRNA in different primary and immortalized OS cells was dramatically higher than that in hFOB1.19 human osteoblastic cells and patient-derived human osteoblasts (“pOsteoblasts”) (Figure 1J). In addition, Gαi3 protein upregulation was also shown in different primary and established OS cells (Figure 1K). Taken together, in OS tissues and cells Gαi3 is upregulated.
Gαi3 silencing exerts anti-tumorigenic activity in cultured OS cells
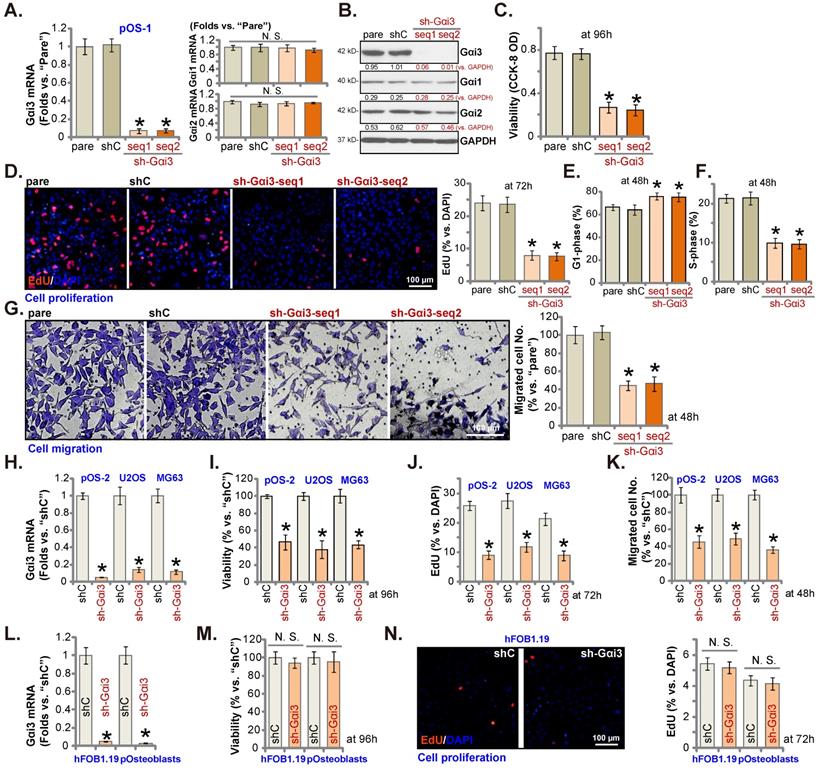
pOS-1 primary cells were separately transfected with two different Gαi3 lentiviral shRNAs, sh-Gαi3-seq1/sh-Gαi3-seq2 [22, 23]. After puromycin-induced selection, Gαi3-silenced stable OS cells were established. Gαi3 mRNA expression decreased over 90-95% in sh-Gαi3-expressing stable pOS-1 cells (Figure 2A). Expression of Gαi1 and Gαi2 mRNA was unaffected (Figure 2A). Gαi3 protein silencing was detected in the stable pOS-1 cells with Gαi3 shRNAs. Gαi1 and Gαi2 protein expression was again unchanged (Figure 2B).
CCK-8 assays showed that Gαi3 shRNA potently decreased pOS-1 cell viability (CCK-8 OD, Figure 2C). Moreover, Gαi3 shRNA potently inhibited pOS-1 cell proliferation, as the ratio of EdU positively stained nuclei was robustly decreased in sh-Gαi3-expressing pOS-1 cells (Figure 2D). The PI-FACS assays were employed to test cell cycle progression. The ratio of G1-phase cells was significantly increased in pOS-1 cells expressing Gαi3 shRNA (Figure 2E), where S-phase pOS-1 cell percentage was decreased (Figure 2F). These results implied that the designated Gαi3 shRNAs induced G1-S arrest in primary OS cells. The “Transwell” results demonstrated that Gαi3 shRNA dramatically inhibited pOS-1 cell in vitro migration (Figure 2G). The scramble control shRNA, or shC, did not significantly alter Gαi1/2/3 expression (Figure 2A-B) and affect OS cellular behaviors (Figure 2C-G).
In human OS Gαi3 is upregulated. TCGA cohorts show Gαi3 mRNA transcripts in 262 sarcoma cases (“Primary Tumor”) and two normal tissue cases (A). The Kaplan Meier Survival curve of Gαi3-low (n = 130, blue) and Gαi3-high (n = 132, red) sarcoma patients was presented (B). Subgroup analyses, based on the different clinical features of the OS patients, were performed as well (C-E). The expression of Gαi3 (both mRNA and protein) in twelve (n = 12) pairs of OS tumor tissue specimens (“T”) and adjacent normal bone tissue specimens (“N”) was tested (F-H). The representative human tissue Gαi3 IHC were presented as well (I). The expression of Gαi3 mRNA and protein in the mentioned cells was measured (J-K). *P < 0.05 versus “N” tissues (F and H) or hFOB1.19 cells (J and K). Scale bar = 100 µm (I).
Next, OS cells that were derived from another OS patient, pOS2 [22, 23], and the immortalized lines (MG63 and U2OS), were stably transduced with the lentiviral Gαi3 shRNA (sh-Gαi3-seq1, “sh-Gαi3”). As shown, the designated shRNA resulted in robust Gαi3 mRNA downregulation in both primary and immortalized OS cells (Figure 2H). Gαi3 shRNA largely suppressed CCK-8 viability (Figure 2I), proliferation (the ratio of EdU positively stained nuclei reduction, Figure 2J) and migration (Figure 2K) in pOS2 primary cells and established lines.
The potential effect of Gαi3 shRNA on the Gαi3-low human osteoblasts (see Figure 1J-K) was tested as well. hFOB1.19 osteoblastic cell line or patient-derived primary osteoblasts (pOsteoblasts) were stably transduced with the lentiviral sh-Gαi3-seq1, causing dramatic Gαi3 mRNA downregulation (“sh-Gαi3”, Figure 2L). Interestingly, in hFOB1.19 cells and pOsteoblasts, Gαi3 shRNA failed to significantly inhibit CCK-8 viability (Figure 2M) and cell proliferation (by measuring EdU positively stained nuclei ratio, Figure 2N), supporting a cancer cell-specific effect by Gαi3 shRNA.
Gαi3 silencing provokes apoptosis activation in OS cells
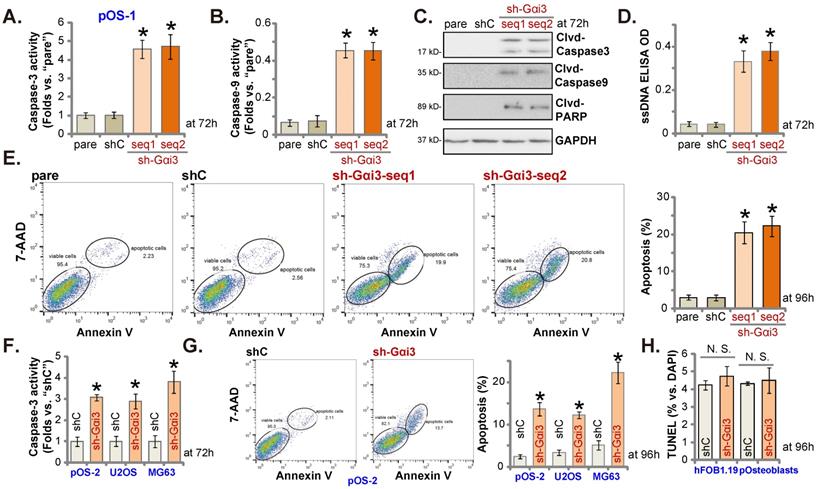
Since Gαi3 silencing induced viability reduction, growth arrest, proliferation inhibition, we therefore tested its potential function on apoptosis in OS cells. Results showed that the relative activities of caspase-3 and caspase-9 (Figure 3A and B) were augmented in sh-Gαi3-expressing stable pOS-1 cells. Figure 3C showed that Gαi3 shRNA induced cleavages of caspase-3, PARP and caspase-9 in pOS-1 primary cells. Confirming increased DNA breaks in pOS-1 cells with Gαi3 shRNA, we showed that ssDNA contents were dramatically increased (tested by the ELISA assays, Figure 3D). Moreover, FACS assay results in Figure 3E showed that Gαi3 silencing increased the pOS-1 cell number with Annexin V-7-AAD double positive staining, confirming apoptosis activation. As expected, shC did not provoke caspase-apoptosis activation in the primary pOS-1 cells (Figure 3A-E).
In pOS-2 cells and immortalized lines (MG63 and U2OS), Gαi3 shRNA (sh-Gαi3-seq1, “sh-Gαi3”, see Figure 2) similarly augmented the relative caspase-3 activity (Figure 3F). Apoptosis induction was observed as well in the sh-Gαi3-seq1-expressing pOS-2 cells and immortalized cell lines, evidenced by the significantly increased Annexin V-7-AAD double staining (Figure 3G). Conversely, in the osteoblastic cell line hFOB1.19 and pOsteoblasts, shRNA-induced silencing Gαi3 (“sh-Gαi3”, see Figure 2) failed to significantly induce apoptosis (by quantifying TUNEL-positively stained nuclei, Figure 3H).
Gαi3 silencing exerts anti-tumorigenic activity in cultured OS cells. Patient-derived primary human OS cells (“pOS-1/2”), the immortalized OS lines (MG63 and U2OS), the immortalized hFOB1.19 cells or patient-derived primary osteoblasts (“pOsteoblasts”) were stably transduced with the designated Gαi3 lentivirus shRNA (sh-Gαi3-seq1/sh-Gαi3-seq2, with two different sequences) or the non-sense control shRNA (“shC”), the expression of indicated mRNAs and proteins were examined (A, B, H and L). After culturing for the designated hours, CCK-8 viability (C, I and M), and cell proliferation (testing the ratio of EdU positively stained nuclei , D, J and N), as well as cell cycle progression (PI-FACS assays, results quantified in E and F) and cell migration (“Transwell” assays, G and K) were tested by the listed assays. “pare” indicated the parental control OS cells. *P < 0.05 versus “shC” group. “N. S.” indicated no statistical difference (P > 0.05, A, M and N). Each single experiment was repeated for five times. Scale bar = 100 µm (D, G and N).
Gαi3 silencing provokes apoptosis activation in OS cells. Patient-derived primary human OS cells (“pOS-1/2”), the immortalized OS lines (MG63 and U2OS), the immortalized hFOB1.19 cells or patient-derived primary osteoblasts (“pOsteoblasts”) were stably transduced with the designated Gαi3 lentivirus shRNA (sh-Gαi3-seq1/sh-Gαi3-seq2, two different sequences) or the non-sense control shRNA (“shC”). After culturing for the designated hours, the caspase-PARP activation was tested (A-C, F), with DNA breaks tested by ssDNA ELISA assays (D); Cell apoptosis was examined by Annexin V-7-AAD double staining FACS (E and G) and nuclear TUNEL staining (results quantified in H) assays. All blotting data in this Figure were repeated five times. “pare” indicated the parental control OS cells. *P < 0.05 versus “shC” group. “N. S.” indicated no statistical difference (P > 0.05, H). Each single experiment was repeated for five times.
Gαi3 knockout potently inhibits OS cell progression in vitro
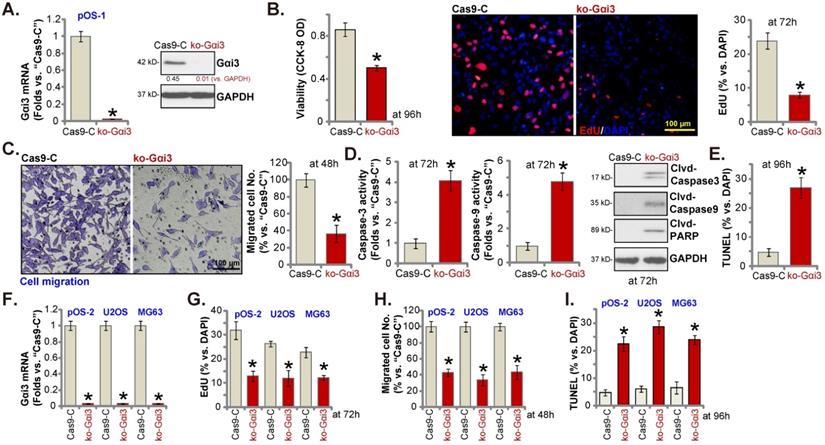
Next, a previously-described CRISPR/Cas9-Gαi3-KO-puro construct [15, 16] was transduced to the Cas9-expressing pOS-1 primary cells. Thereafter, single stable pOS-1 cells with the Gαi3 KO construct, or the ko-Gαi3 cells, were established after Gαi3 KO screening, and Gαi3 mRNA and protein (Figure 4A) depletion was detected. Gαi3 KO largely decreased CCK-8 viability (Figure 4B) and inhibited cell proliferation (the ratio of EdU positively stained nuclei decreasing, Figure 4B) and migration (Figure 4C) in pOS-1 cells. On the contrast, caspase-3 and caspase-9 activities (Figure 4D) were augmented in ko-Gαi3 pOS-1 cells, where caspase-3, PARP and caspase-9 cleavages were induced (Figure 4D). In addition, apoptosis induction was detected in the ko-Gαi3 pOS-1 cells, as the ratio of TUNEL-positively stained nuclei was increased significantly (see the quantified results in Figure 4E).
The Gαi3-KO construct was employed to knockout Gαi3 in primary pOS-2 cells and immortalized lines (MG63 and U2OS), and stable cells established (“ko-Gαi3”) after screening (Figure 4F). As shown, Gαi3 KO inhibited cell proliferation (the ratio of EdU positively stained nuclei reduction, Figure 4G) and in vitro migration (Figure 4H) in pOS-2 and established OS cells. Increased TUNEL-positive nuclei ratio confirmed apoptosis activation in ko-Gαi3 pOS-2 cells and immortalized lines (Figure 4I). Together, Gαi3 KO, by the CRISPR/Cas9 strategy, resulted in profound anti-OS cell activity.
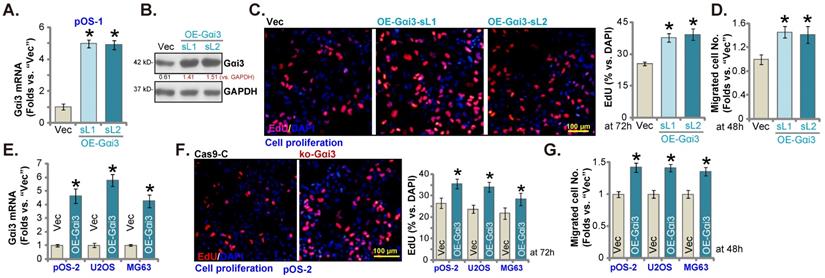
Further promoting OS cell growth by Gαi3 ectopic overexpression
Next, a Gαi3-expressing lentiviral construct (see our previous studies [14, 16]) was transduced to pOS-1 cells. Following selection Gαi3-overexpressed stable pOS-1 cells were thereafter established: namely “OE-Gαi3-sL1” and “OE-Gαi3-sL2” (two selections). Gαi3 mRNA and protein expression levels (Figure 5A and B) were robustly elevated in the OE-Gαi3 pOS-1 cells. Gαi3 overexpression accelerated pOS-1 cell proliferation (the ratio of EdU positively stained nuclei increasing, Figure 5C) and in vitro migration (“Transwell” assays, results quantified in Figure 5D). In pOS-2 cells and the immortalized lines (MG63 and U2OS), ectopic overexpression of Gαi3 using the same construct (“OE-Gαi3”, Figure 5E) enhanced cell proliferation (the ratio of EdU positively stained nuclei increasing, Figure 5F) and in vitro migration (see the quantified results in Figure 5G). Therefore, these results again supported the key cancer-promoting function of Gαi3 in OS cells.
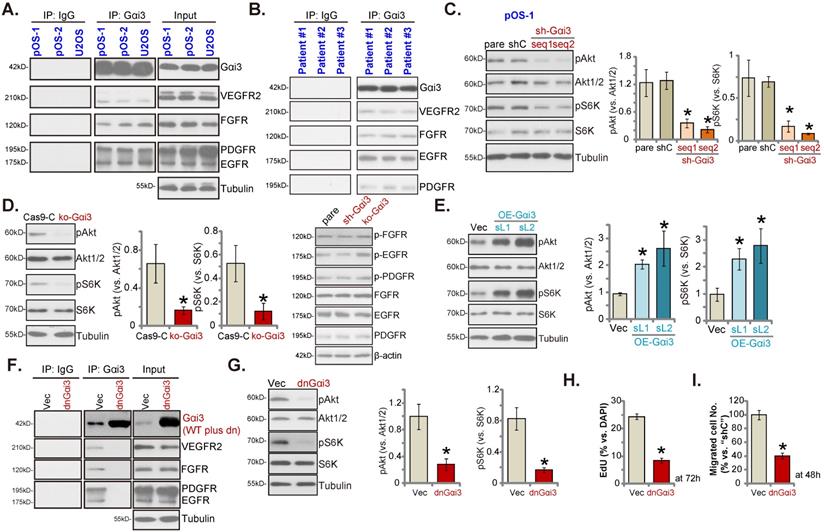
Gαi3 immunoprecipitates with RTKs and is key to Akt-mTOR activation in OS cells
Our previous studies have shown that Gαi proteins associated with several oncogenic RTKs (EGFR, VEGFR2, TrkB, FGFR and KGFR), mediating downstream signaling activation [14, 16-21]. Co-IP (co-immunoprecipitation) assays, Figure 6A, demonstrated that Gαi3 immunoprecipitated with VEGFR2, FGFR, PGDFR and EGFR in primary OS cells (“pOS-1/2”) and immortalized U2OS cells. Moreover, the association between Gαi3 and multiple RTKs (VEGFR2, FGFR, PGDFR and EGFR) was detected in the human OS tissues from three representative patients (Figure 6B). When testing downstream Akt-mTOR signaling, we showed that levels of phosphorylated-Akt (Ser-473) and phosphorylated-S6K (Thr-389) were dramatically decreased in pOS-1 cells bearing Gαi3 shRNAs (Figure 6C). Moreover, CRISPR/Cas9-induced Gαi3 KO (see Figure 4) largely inhibited Akt-S6K phosphorylations in pOS-1 cells (Figure 6D). Notably, RTKs (FGFR, PGDFR and EGFR) expression and phosphorylation were unaffected by Gαi3 shRNA (sh-Gαi3-seq1) or Gαi3 KO (Figure 6D). Conversely, ectopic overexpression of Gαi3 (see Figure 5) significantly increased Akt-S6K activation in pOS-1 cells (Figure 6E).
Gαi3 knockout potently inhibits OS cell progression in vitro. Patient-derived OS cells (“pOS-1/-2”) or immortalized OS lines (MG63 and U2OS), bearing the CRISPR/Cas9-Gαi3-KO-puro construct (“ko-Gαi3”) or the corresponding vector (“Cas9-C”), were established, expression of listed mRNAs and proteins were examined (A and F); After culturing for the designated hours, CCK-8 viability (B), proliferation (testing the ratio of EdU positively stained nuclei , B and G), cell migration (“Transwell” assays, C and H), caspase-PARP activation was tested (D), with cell apoptosis measured through quantifying the TUNEL-positively stained nuclei ratio (E and I). All blotting data in this Figure were repeated five times. *P < 0.05 versus “Cas9-C” group. Each single experiment was repeated for five times. Scale bar = 100 µm (B and C).
Further promoting OS cell growth by Gαi3 ectopic overexpression. Patient-derived primary OS cells (“pOS-1/-2”) or immortalized OS lines (MG63 and U2OS), bearing the lentiviral construct encoding wild-type Gαi3 (“OE-Gαi3”) or the corresponding vector (“Vec”), were established and cultivated, expression of listed mRNAs and proteins were measured (A, B and E); After culturing for the designated hours, cell proliferation (testing the ratio of EdU positively stained nuclei , C and F) and cell migration (“Transwell” assays, D and G) were measured. All blotting data in this Figure were repeated five times. *P < 0.05 versus “Vec” group. Each single experiment was repeated for five times. Scale bar = 100 µm (C and F).
Gαi3 immunoprecipitates with RTKs and is key to Akt-mTOR activation in OS cells. The association between Gαi3 and the designated RTKs (VEGFR2, FGFR, PGDFR and EGFR) in patient-derived primary human OS cells (“pOS-1/2”) and U2OS line (A, cultured in FBS-containing medium for 5 min) as well as in OS tissues of the representative patients (B) was examined by the co-immunoprecipitation (Co-IP) assays. The pOS-1 primary cells stably expressing the Gαi3 shRNA (sh-Gαi3-seq1/sh-Gαi3-seq2), the CRISPR/Cas9-Gαi3-KO-puro construct (“ko-Gαi3”), the Gαi3-expressing lentiviral construct (“OE-Gαi3”), or their corresponding controls (“shC”, “Cas9-C” or “Vec”) were established, and expression of listed proteins tested (C-E). The pOS-1 primary cells, stably expressing the lentiviral dominant negative Gαi3 construct (dnGαi3) or the empty vector (“Vec”), were established, the association between Gαi3 and RTKs (VEGFR2, FGFR, PGDFR and EGFR) as well as their expression were examined (F and G). After culturing for the designated hours, cell proliferation and migration were separately examined by EdU staining (H) and “Transwell” (I) assays. All blotting data in this Figure were repeated five times. “pare” indicated the parental control OS cells. *P < 0.05 versus “shC”/“Cas9-C”/“Vec” group. Each single experiment was repeated for five times.
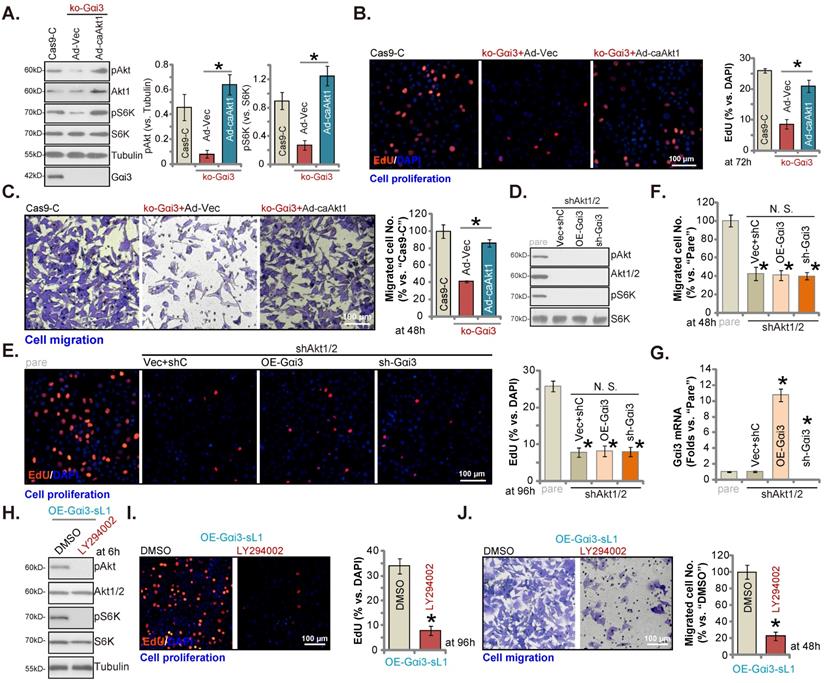
Akt-mTOR inhibition contributes to Gαi3 depletion-induced anti-OS cell activity. The pOS-1 cells bearing the CRISPR/Cas9-Gαi3-KO-puro construct (“ko-Gαi3”) were further infected with the constitutively-active Akt1 adenovirus (“Ad-caAkt1”) or the adenovirus with the empty vector (“Ad-Vec”), control cells were with the CRISPR/Cas9 empty vector (“Cas9-C”), listed proteins were shown (A). Cells were cultured for designated hours, cell proliferation (B, EdU assays) and migration (C, “Transwell” assays) were tested. pOS-1 cells stably bearing the lentiviral Akt1/2 shRNA (“shAkt1/2”) were further transduced with a wild-type Gαi3 (“OE-Gαi3”) lentiviral construct, the lentiviral sh-Gαi3-seq1 (“sh-Gαi3”) or their control construct (“Vec+shC”), stable cells were established. Gαi3 mRNA and listed proteins were shown (D and G). After culturing for the designated hours, cell proliferation (E, by measuring EdU positively stained nuclei ratio) and migration (F) were measured. pOS-1 cells, bearing the lentiviral construct encoding wild-type Gαi3 (“OE-Gαi3-sL1”) were treated with LY294002 (150 nM) or the vehicle control (0.1% DMSO), and cultured for designated time periods, listed proteins were shown (H), with cell proliferation (I) and migration (J) examined as well. All blotting data in this Figure were repeated five times. “pare” indicated the parental control OS cells. *P < 0.05 (A-C). *P < 0.05 versus “pare” cells (E-G). *P < 0.05 versus “DMSO” (I and J). “N. S.” indicated no statistical difference (P > 0.05, E and F). Each single experiment was repeated for five times. Scale bar = 100 µm (B, C, E, I and J).
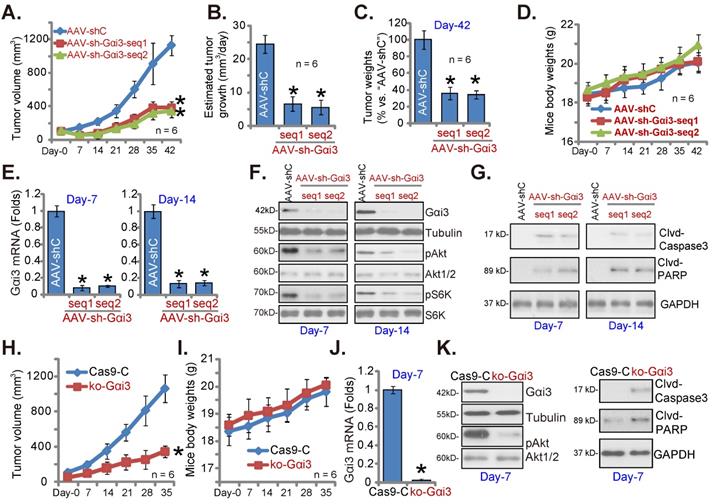
Gαi3 depletion inhibits OS cell growth in vivo. The pOS-1 xenografts-bearing nude mice were intratumorally injected (daily) with the AAV-packed Gαi3 shRNAs (AAV-sh-Gαi3-seq1 or AAV-sh-Gαi3-seq2, two different sequences) or AAV-packed control shRNA (AAV-shC) for 12 days. The mice images was shown (A); The tumor volumes (A) and the mice body weights (D) were recorded weekly (“Day-0” to “Day-42”, total 42 days). The estimated daily tumor growth was calculated and presented (B). At Day-42, all pOS-1 xenograft tumors were isolated and weighted (C). Expression of listed genes and proteins in indicated tumor tissues lysates was shown (E-G) assays. The ko-Gαi3 pOS-1 cells and Ca9-C control cells were s.c. injected to the nude mice. After 20 days, tumor volumes recordings were started (“Day-0”). Weekly tumor volumes (H) and the mice body weights (I) were presented. Expression of listed genes and proteins in the described tumor lysates was tested (J and K). All blotting data in this Figure were repeated five times. *P < 0.05 versus “AAV-shC”/“Ca9-C” group.
To block Gαi3-RTKs association, the lentiviral dominant negative (dn) Gαi3 mutant construct was stably transduced into pOS-1 cells. The dnGαi3 mutant will replace the conserved Gly (G) residue with the Thr (T) residue in the G3 box, thereby preventing Gαi1/3 interaction with the associated proteins [19, 20]. Results show that dnGαi3 disrupted the association between Gαi3 and multiple RTKs (VEGFR2, FGFR, PGDFR and EGFR) in pOS-1 cells (Figure 6F). Expression of RTKs was however unchanged (Figure 6F, “Input”). Importantly, dnGαi3 largely inhibited Akt-S6K phosphorylations in pOS-1 primary cells (Figure 6G). The dnGαi3 largely suppressed pOS-1 cell in vitro proliferation and migration, examined through the nuclear EdU staining (see the quantified results in Figure 6H) and “Transwell” (Figure 6I) assays, respectively.
Akt-mTOR inhibition contributes to Gαi3 depletion-induced anti-OS cell activity
To support that Akt-mTOR inhibition was the main mechanism of Gαi3 depletion-caused anti-OS cell activity, we expressed the constitutively active Akt1 (caAkt1) [34] adenovirus (“Ad-caAkt1”) that could rescue Akt and S6K phosphorylation in koGαi3 pOS-1 cells (Figure 7A). Significantly, Ad-caAkt1 restored proliferation (by quantifying EdU-positively stained nuclei, Figure 7B) and in vitro migration (“Transwell” assays, Figure 7C) of koGαi3 pOS-1 cells. Furthermore, shRNA-induced silencing of Akt1/2 (Figure 7D) blocked Akt-S6K phosphorylations (Figure 7D) and mimicked Gαi3 depletion-induced actions, suppressing pOS-1 cell proliferation (Figure 7E) and in vitro migration (Figure 7F). Significantly, re-introducing the Gαi3 shRNA lentivirus or the Gαi3-expresing construct (Figure 7G) was unable to further influence cell proliferation (Figure 7E) and migration (see quantified results in Figure 7F) in Akt1/2-silenced pOS-1 cells. Moreover, in the Gαi3-overexpressed pOS-1 cells (OE-Gαi3-sL1), treatment with LY294002, a PI3K-Akt inhibitor [35], blocked Akt-S6K phosphorylation (Figure 7H) and inhibited cell proliferation (Figure 7I) and in vitro cell migration (Figure 7J). Thus, Akt-mTOR inhibition should be responsible for Gαi3 depletion-induced anti-OS cell activity.
Gαi3 depletion inhibits OS cell growth in vivo
At last, pOS-1 cells were subcutaneously (s.c.) injected to the nude mice. Within 20 days of cell inoculation, the subcutaneous pOS-1 xenograft tumors were established and each tumor volume was close to 100 mm3 (“Day-0”). The xenograft-bearing mice were thereafter assigned into three different groups randomly, with six mice in every group (n = 6). Afterwards, the mice were intratumorally injected daily with the AAV-packed Gαi3 shRNAs (AAV-sh-Gαi3-seq1 or AAV-sh-Gαi3-seq2, two different sequences in the AAV9 construct) or AAV-packed control shRNA (AAV-shC), for 12 consecutive days. Figure 8A, recording tumor growth, demonstrated that the growth of pOS-1 xenograft tumors was robustly mitigated after AAV-sh-Gαi3 injection. The volumes of AAV-sh-Gαi3-injected pOS-1 xenografts were dramatically lower than those of AAV-shC-injected pOS-1 xenografts (Figure 8A). The estimated daily tumor growth (see previous studies [22, 23]) results demonstrated that subcutaneous pOS-1 xenograft growth was dramatically suppressed with AAV-sh-Gαi3 injection in the nude mice (Figure 8B). At Day-42, all the animals were anaesthetized and decapitated, and pOS-1 xenografts carefully isolated and weighted. The pOS-1 tumors with AAV-sh-Gαi3 injection were significantly lighter than pOS-1 tumors with the control shRNA virus injection (Figure 8C). Among the three mice groups there was no significant difference in the mice body weights (Figure 8D).
At experimental Day-7 and Day-14, 3h after virus injection, one tumor of each group was carefully isolated, and total six tumors were obtained. Gαi3 mRNA was dramatically decreased in AAV-sh-Gαi3-injected pOS-1 xenograft tissues (Figure 8E), where Gαi3 protein downregulation as well as p-Akt and p-S6K inhibition were detected (Figure 8F). Supporting apoptosis activation, we showed that cleaved-caspsae-3/cleaved-PARP levels were augmented in Gαi3-silenced pOS-1 xenograft tissues (Figure 8G). Thus, intratumoral injection of AAV-packed Gαi3 shRNA suppressed Akt-mTOR activation and provoked apoptosis in pOS-1 xenografts.
In addition the ko-Gαi3 pOS-1 cells and the Ca9-C control cells (see Figure 4) were injected to the nude mice, forming subcutaneous xenografts. After 20 days, tumor recordings were started (“Day-0”). As shown ko-Gαi3 pOS-1 xenograft growth was slower than the Ca9-C xenografts (Figure 8H), while animal body weights were indifferent (Figure 8I). At experimental Day-7, we carefully isolated one tumor xenograft per group. Gαi3 mRNA and protein (Figure 8J-K) expression was completely depleted in ko-Gαi3 pOS-1 xenograft tissues, where p-Akt was decreased (Figure 8K). The cleaved-caspsae-3 and the cleaved-PARP levels were increased in Gαi3-KO xenograft tissues (Figure 8K), supporting apoptosis induction in vivo.
Discussion
The GPCR superfamily is composed of the immense structural and functional different proteins, participating in various biological processes and signals in the bone [36]. Due to gene mutations, depletion or overexpression, GPCR components are dysregulated in human OS [36]. Their roles in OS progression have been established [36]. For example, Iyer et al., found that A3 adenosine receptor (A3AR) depletion activated protein kinase A (PKA)-Akt-nuclear factor (NF)-κB signaling to promote OS cell growth [37]. High GPR56 (G protein-coupled receptor 56) expression is an unfavorable prognostic factor, promoting invasion and proliferation of OS cells [38]. Liu et al. demonstrated that GPR110 (G protein-coupled receptor 110) silencing inhibited OS cell growth [39].
Importantly, studies have reported that Gαi-coupled GPCRs, including Apelin receptors [40], CXCR4 [41-43], melatonin receptors [44], are important contributor for OS progression. Due to various gene mutations (or overexpression), concurrent and sustained activation of multiple different RTKs in OS will provoke sustained activation of oncogenic signaling, leading to persistent OS growth and progression [4, 8, 9]. Interestingly, we have previously shown that Gαi proteins are essential for signalings by several important oncogenic RTKs (including EGFR [20], VEGFR2 [16], KGFR [19], FGFR [18] and TrkB [17]) as well as the non-RTK receptor (IL-4R [15]).
After showing the essential role of Gαi 1/3 in activation of oncogenic signalings by RTKs, we previously explored Gαi 1/3 in different human cancers. Gαi 1 and Gαi 3 expression is elevated in glioma, correlating with tumor stage [14, 18]. Gαi1 can form a complex with multiple RTKs (including FGFR, PDGFR and EGFR), transducing downstream Akt-mTOR activation in glioma tissues and cells [18]. Conversely, Gαi1 silencing or mutation inhibited glioma cell growth [18]. In the mouse brain, the orthotopic growth of patient-derived glioma xenografts was arrested after Gαi 1/3 depletion, whereas forced overexpression of Gαi 1/3 enhanced growth [14]. We also showed that Gαi 1 upregulation in human gastric cancer was correlated with poor overall survival [21]. Gαi 1 silencing or knockout inhibited Akt-mTOR activation and gastric cancer cell growth [21]. These previous studies supported that Gαi 1/3 could be important oncogenic genes and promising therapeutic targets of human cancer.
Gαi3 should be a vital gene for OS progression. TCGA database shows that transcripts of Gαi3 are significantly upregulated in sarcoma tissues, and high-Gαi3 expression in sarcoma correlating with the poor overall survival. Gαi3 elevation was observed in local OS tissues as well as in different immortalized and primary OS cells, while low expression was observed in cancer-surrounding normal bone tissues and in immortalized and primary osteoblasts. Functional studies showed that in different OS cells, Gαi3 depletion, by shRNA or CRISPR/Cas9 strategies, robustly suppressed cell survival, proliferation and cell migration, and provoking G1-S arrest and apoptosis. Contrarily, ectopic Gαi3 overexpression can further accelerate OS cell growth. In vivo, Gαi3 shRNA AAV intratumoral injection potently suppressed the growth of the patient-derived OS xenografts in nude mice. Moreover, the growth of primary OS xenografts of the Gαi3 KO cells was largely suppressed.
We have previously discovered that Gαi1/3 association with multiple RTKs was required for downstream signaling activation. For instance, Gαi1/3 are key proteins in mediating VEGF-induced VEGFR2 signaling [16]. Following VEGF stimulation, Gαi1/3 were in the VEGFR2 endocytosis complex, required for VEGFR2 endocytosis and subsequent activation of downstream signalings [16]. Similarly, Gαi1/3 proteins are indispensable signaling molecule for EGF- and KGF-induced Akt-mTORC1 signaling activation [19, 20]. In addition, brain-derived neurotrophic factor (BDNF)-induced signaling and anti-depressive actions required Gαi1/3 [17]. Gαi 1/3 silencing inhibited BDNF-induced TrkB endocytosis and activation of the downstream signaling [17].
The present study implied that Gαi3-driven OS cell growth was primarily through mediating Akt-mTOR cascade activation. In OS cell and tissues Gαi3 associated with RTKs (VEGFR2, FGFR, PGDFR and EGFR), essential for downstream Akt-mTOR activation. In OS cells Akt-S6K activation was robustly suppressed by Gαi3 shRNA/KO, but augmented after Gαi3 overexpression. In vivo, Akt-S6K phosphorylations were decreased in Gαi3-depleted OS xenografts tissues. Importantly, disrupting Gαi3-RTKs association, through dnGαi3, largely inhibited Akt-S6K activation and OS cell proliferation and migration. Restoring Akt-S6K activation, by caAkt1, rescued proliferation and migration of Gαi3-KO OS cells. Conversely, mimicking Gαi3 depletion-induced actions, Akt1/2 silencing inhibited OS cell proliferation and migration. Significantly, exogenously altering Gαi3 expression failed to affect proliferation and migration in Akt1/2-silenced cells. Therefore, Gαi3-driven OS cell growth was possibly due to mediating RTKs-Akt signaling.
Conclusion
Over three-fifths of bone sarcoma are OS [45, 46]. The standard chemotherapy of OS in clinic is the combination of methotrexate, doxorubicin, and cisplatin [45, 47, 48], showing limited success in metastatic and other OS patients with advanced diseases [45, 47, 48]. Further exploring key pathologic mechanisms and the driving signaling molecule for advanced OS is therefore important [45, 47, 48]. The results of this study showed that overexpressed Gαi3 mediated RTKs signaling to drive OS progression, serving as a novel and promising treatment molecular target for patients with OS.
Acknowledgements
This work was generously supported by grants from the National Natural Science Foundation of China (81922025, 81974388, 81873995, 81974334, 81571282 and 81771457), Development Fund of the Affiliated Hospital of Xuzhou Medical University (XYFM2021043), and Suqian Sci&Tech Program (K202016), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethics Statement
This study was approved by Ethics Committee of Soochow University.
Data Availability Statement
All data are available upon request.
Author Contributions
All the listed authors conceived, designed, and supervised the study. All listed authors collected samples, performed the experiments and analyzed the data. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Anderson ME. Update on Survival in Osteosarcoma. Orthop Clin North Am. 2016;47:283-92
2. Zhao J, Dean DC, Hornicek FJ, Yu X, Duan Z. Emerging next-generation sequencing-based discoveries for targeted osteosarcoma therapy. Cancer Lett. 2020;474:158-67
3. Bishop MW, Janeway KA, Gorlick R. Future directions in the treatment of osteosarcoma. Curr Opin Pediatr. 2016;28:26-33
4. Denduluri SK, Wang Z, Yan Z, Wang J, Wei Q, Mohammed MK. et al. Molecular pathogenesis and therapeutic strategies of human osteosarcoma. J Biomed Res. 2015 30
5. Liang X, Wang X, He Y, Wu Y, Zhong L, Liu W. et al. Acetylation dependent functions of Rab22a-NeoF1 Fusion Protein in Osteosarcoma. Theranostics. 2020;10:7747-57
6. Hu XK, Rao SS, Tan YJ, Yin H, Luo MJ, Wang ZX. et al. Fructose-coated Angstrom silver inhibits osteosarcoma growth and metastasis via promoting ROS-dependent apoptosis through the alteration of glucose metabolism by inhibiting PDK. Theranostics. 2020;10:7710-29
7. Wang S, Zhong L, Li Y, Xiao D, Zhang R, Liao D. et al. Up-regulation of PCOLCE by TWIST1 promotes metastasis in Osteosarcoma. Theranostics. 2019;9:4342-53
8. Tian Z, Niu X, Yao W. Receptor Tyrosine Kinases in Osteosarcoma Treatment: Which Is the Key Target? Front Oncol. 2020;10:1642
9. Rettew AN, Getty PJ, Greenfield EM. Receptor tyrosine kinases in osteosarcoma: not just the usual suspects. Adv Exp Med Biol. 2014;804:47-66
10. Fan H, Li P, Zingarelli B, Borg K, Halushka PV, Birnbaumer L. et al. Heterotrimeric Galpha(i) proteins are regulated by lipopolysaccharide and are anti-inflammatory in endotoxemia and polymicrobial sepsis. Biochim Biophys Acta. 2011;1813:466-72
11. Broxmeyer HE, Youn BS, Kim C, Hangoc G, Cooper S, Mantel C. Chemokine regulation of hematopoiesis and the involvement of pertussis toxin-sensitive G alpha i proteins. Ann N Y Acad Sci. 2001;938:117-27 discussion 27-8
12. Pines M, Santora A, Gierschik P, Menczel J, Spiegel A. The inhibitory guanine nucleotide regulatory protein modulates agonist-stimulated cAMP production in rat osteosarcoma cells. Bone Miner. 1986;1:15-26
13. Wang JW, Su W, Law YP, Lu CH, Chen YC, Wang JL. et al. Mechanism of bradykinin-induced Ca(2+) mobilization in MG63 human osteosarcoma cells. Horm Res. 2001;55:265-70
14. Wang Y, Liu YY, Chen MB, Cheng KW, Qi LN, Zhang ZQ. et al. Neuronal-driven glioma growth requires Galphai1 and Galphai3. Theranostics. 2021;11:8535-49
15. Bai JY, Li Y, Xue GH, Li KR, Zheng YF, Zhang ZQ. et al. Requirement of Galphai1 and Galphai3 in interleukin-4-induced signaling, macrophage M2 polarization and allergic asthma response. Theranostics. 2021;11:4894-909
16. Sun J, Huang W, Yang SF, Zhang XP, Yu Q, Zhang ZQ. et al. Galphai1 and Galphai3mediate VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis. Theranostics. 2018;8:4695-709
17. Marshall J, Zhou XZ, Chen G, Yang SQ, Li Y, Wang Y. et al. Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc Natl Acad Sci U S A. 2018;115:E3549-E58
18. Liu YY, Chen MB, Cheng L, Zhang ZQ, Yu ZQ, Jiang Q. et al. microRNA-200a downregulation in human glioma leads to Galphai1 over-expression, Akt activation, and cell proliferation. Oncogene. 2018;37:2890-902
19. Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q. et al. Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol. 2015;135:181-91
20. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS. et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2:ra17
21. Lv Y, Wang Y, Song Y, Wang SS, Cheng KW, Zhang ZQ. et al. LncRNA PINK1-AS promotes G alpha i1-driven gastric cancer tumorigenesis by sponging microRNA-200a. Oncogene. 2021;40:3826-44
22. Shan HJ, Zhu LQ, Yao C, Zhang ZQ, Liu YY, Jiang Q. et al. MAFG-driven osteosarcoma cell progression is inhibited by a novel miRNA miR-4660. Mol Ther Nucleic Acids. 2021;24:385-402
23. Gao YY, Ling ZY, Zhu YR, Shi C, Wang Y, Zhang XY. et al. The histone acetyltransferase HBO1 functions as a novel oncogenic gene in osteosarcoma. Theranostics. 2021;11:4599-615
24. Guo S, Chen C, Ji F, Mao L, Xie Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem Biophys Res Commun. 2017;487:660-5
25. Zhu CY, Yao C, Zhu LQ, She C, Zhou XZ. Dexamethasone-induced cytotoxicity in human osteoblasts is associated with circular RNA HIPK3 downregulation. Biochem Biophys Res Commun. 2019;516:645-52
26. Fan JB, Liu W, Zhu XH, Cui SY, Cui ZM, Zhao JN. microRNA-7 inhibition protects human osteoblasts from dexamethasone via activation of epidermal growth factor receptor signaling. Mol Cell Biochem. 2019;460:113-21
27. Zhang D, Xia H, Zhang W, Fang B. The anti-ovarian cancer activity by WYE-132, a mTORC1/2 dual inhibitor. Tumour Biol. 2016;37:1327-36
28. Yang H, Zhao J, Zhao M, Zhao L, Zhou LN, Duan Y. et al. GDC-0349 inhibits non-small cell lung cancer cell growth. Cell Death Dis. 2020;11:951
29. Lv Y, Si M, Chen N, Li Y, Ma X, Yang H. et al. TBX2 over-expression promotes nasopharyngeal cancer cell proliferation and invasion. Oncotarget. 2017;8:52699-707
30. Shao NY, Wang DX, Wang Y, Li Y, Zhang ZQ, Jiang Q. et al. MicroRNA-29a-3p Downregulation Causes Gab1 Upregulation to Promote Glioma Cell Proliferation. Cell Physiol Biochem. 2018;48:450-60
31. Zhang X, Shan H, Zhang P, C S, XZ Z. LncRNA EPIC1 protects human osteoblasts from dexamethasone-induced cell death. Biochemical and biophysical research communications. 2018;503:2255-62
32. Chen MB, Liu YY, Xing ZY, Zhang ZQ, Jiang Q, Lu PH. et al. Itraconazole-Induced Inhibition on Human Esophageal Cancer Cell Growth Requires AMPK Activation. Mol Cancer Ther. 2018;17:1229-39
33. Wang SS, Lv Y, Xu XC, Zuo Y, Song Y, Wu GP. et al. Triptonide inhibits human nasopharyngeal carcinoma cell growth via disrupting Lnc-RNA THOR-IGF2BP1 signaling. Cancer Lett. 2019;443:13-24
34. Zuo Y, Wang Y, Hu H, Cui W. Atorvastatin Protects Myocardium Against Ischemia-Reperfusion Injury Through Inhibiting miR-199a-5p. Cell Physiol Biochem. 2016;39:1021-30
35. Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1996;15:5256-67
36. Luo J, Sun P, Siwko S, Liu M, Xiao J. The role of GPCRs in bone diseases and dysfunctions. Bone Res. 2019;7:19
37. Iyer SV, Ranjan A, Elias HK, Parrales A, Sasaki H, Roy BC. et al. Genome-wide RNAi screening identifies TMIGD3 isoform1 as a suppressor of NF-kappaB and osteosarcoma progression. Nat Commun. 2016;7:13561
38. Chen Z, Gao P, Li Z. Expression of G Protein-coupled Receptor 56 Is an Unfavorable Prognostic Factor in Osteosarcoma Patients. Tohoku J Exp Med. 2016;239:203-11
39. Liu Z, Zhang G, Zhao C, Li J. Clinical Significance of G Protein-Coupled Receptor 110 (GPR110) as a Novel Prognostic Biomarker in Osteosarcoma. Med Sci Monit. 2018;24:5216-24
40. Cui L, Zhang JY, Ren ZP, Zhao HJ, Li GS. APLNR promotes the progression of osteosarcoma by stimulating cell proliferation and invasion. Anticancer Drugs. 2019;30:940-7
41. Guan G, Zhang Y, Lu Y, Liu L, Shi D, Wen Y. et al. The HIF-1alpha/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells. Cancer Lett. 2015;357:254-64
42. Laverdiere C, Hoang BH, Yang R, Sowers R, Qin J, Meyers PA. et al. Messenger RNA expression levels of CXCR4 correlate with metastatic behavior and outcome in patients with osteosarcoma. Clin Cancer Res. 2005;11:2561-7
43. Zhang P, Dong L, Yan K, Long H, Yang TT, Dong MQ. et al. CXCR4-mediated osteosarcoma growth and pulmonary metastasis is promoted by mesenchymal stem cells through VEGF. Oncol Rep. 2013;30:1753-61
44. Lu KH, Lin RC, Yang JS, Yang WE, Reiter RJ, Yang SF. Molecular and Cellular Mechanisms of Melatonin in Osteosarcoma. Cells. 2019 8
45. Isakoff M, Bielack S, Meltzer P, R G. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:3029-35
46. Lin Y, Jewell B, Gingold J, L L, R Z, LL W. et al. Osteosarcoma: Molecular Pathogenesis and iPSC Modeling. Trends in molecular medicine. 2017;23:737-55
47. Luetke A, Meyers P, Lewis I, H J. Osteosarcoma treatment - where do we stand? A state of the art review. Cancer treatment reviews. 2014;40:523-32
48. Zhang Y, Yang J, Zhao N, C W, S K, Y Z. et al. Progress in the chemotherapeutic treatment of osteosarcoma. Oncology letters. 2018;16:6228-37
Author contact
![]() Corresponding authors: Dr. Yu-min Huang, Department of Orthopedics The First Affiliated Hospital of Nanjing Medical University, Nanjing, 210029, China. Email: xiaoan19860820com; Prof. Xiao-dong Wang, Department of Orthopedics, Children's Hospital of Soochow University, Suzhou, China, 215100. E-mail: xiaodongwangszcom. Prof. Xiao-zhong Zhou, Department of Orthopedics, The Second Affiliated Hospital of Soochow University, San-xiang Rd, Suzhou, China, 215003. Email: zhouxzedu.cn. Prof. Cong Cao, Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, 199 Ren-ai Road, Suzhou, Jiangsu 215123, China. Tel/Fax: +86-0512-65883658. E-mail: caocongedu.cn.
Corresponding authors: Dr. Yu-min Huang, Department of Orthopedics The First Affiliated Hospital of Nanjing Medical University, Nanjing, 210029, China. Email: xiaoan19860820com; Prof. Xiao-dong Wang, Department of Orthopedics, Children's Hospital of Soochow University, Suzhou, China, 215100. E-mail: xiaodongwangszcom. Prof. Xiao-zhong Zhou, Department of Orthopedics, The Second Affiliated Hospital of Soochow University, San-xiang Rd, Suzhou, China, 215003. Email: zhouxzedu.cn. Prof. Cong Cao, Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, 199 Ren-ai Road, Suzhou, Jiangsu 215123, China. Tel/Fax: +86-0512-65883658. E-mail: caocongedu.cn.