Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
The mechanism of MMR gene...
MMR gene polymorphism and cancer
The function of MMR in the...
Prospect
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(7):2821-2832. doi:10.7150/ijbs.71714 This issue Cite
Review
The role of DNA mismatch repair in immunotherapy of human cancer
Yuchen He1,2*, Luyuan Zhang3*, Ruoyu Zhou2*, Yumin Wang4,5 ![]() , Hao Chen1
, Hao Chen1 ![]()
1. Department of General Surgery, The First Affiliated Hospital of USTC; Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui, China.
2. Xiangya School of Medicine, Central South University, Changsha, Hunan, China.
3. Department of Neurosurgery, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China.
4. Department of Otolaryngology Head and Neck Surgery, Xiangya Hospital, Central South University, Changsha, Hunan, China.
5. National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, Hunan, China.
*These authors contributed equally to this work.
Received 2022-2-4; Accepted 2022-3-22; Published 2022-4-4
Abstract

DNA mismatch repair (MMR) is an important pathway which helps to maintain genomic stability. Mutations in DNA MMR genes are found to promote cancer initiation and foster tumor progression. Deficiency or inactivation of MMR results in microsatellite instability (MSI) which triggers neoantigen generation and impairs tumor growth. Immunotherapies targeting MMR can increase the burden of neoantigens in tumor cells. While MSI has been regarded as an important predictor of sensitivity and drug resistance for immunotherapy-based strategies. Different approaches targeting genomic instability have been demonstrated to be promising in malignancies derived from different tissues. Underlying MMR deficiency-associated immunogenicity is important for improving the therapeutic efficacy of immunotherapies. In this review we provide an overview of the MMR systems, their role in tumorigenesis, drug resistance, prognostic significance and potential targets for therapeutic treatment in human cancers, especially in hematological malignancies.
Keywords: MMR, Development, Immunotherapy, DNA repair, Cancer
Background
Cancer ranks first in the cause of morbidity in the world. The global cancer burden is expected to be 28.4 million cases in 2040, a 47% rise from 2020, with a larger increase in transitioning (64% to 95%) versus transitioned (32% to 56%) countries due to demographic changes [1]. The rising prominence of cancer makes it a leading cause of death which partly reflects marked declines in mortality rates of stroke and coronary heart disease [2]. Overall incidence was from 2‐fold to 3‐fold higher in transitioned versus transitioning countries for both sexes [3, 4]. Cancer incidence and mortality are rapidly growing worldwide, with a predicted 22 million new cancer cases and 13 million cancer-related deaths occurring annually by 2030 [5].
The occurrence and development of cancer is a complex process of multi-stage, multi-factor, long-term exposure and multi-channel accumulation, caused by genetic and environmental factors [6,7]. The hallmarks of cancer have provided a framework for a deeper molecular understanding of cancer which include sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprogramming of energy metabolism, tumor-promoting inflammation, avoiding immune destruction [8]. Genome instability, which generates the genetic diversity that expedites their acquisition, participates in the regulation of these hallmarks and inflammation.
As its name indicates, DNA mismatch repair (MMR) is responsible for correcting the mismatched nucleotides caused by polymerase misincorporation errors, recombination between heteroallelic parental DNAs, and chemical or physical damages [9]. To achieve the repairment, MMR goes through an excision-resynthesis process that requires the cooperation of MMR protein complex, DNA replicative polymerase and DNA ligase [10]. In addition to its roles in editing replication errors, the MMR system is also implicated in the DNA damage response (DDR) that triggers cell cycle arrest and apoptosis, avoiding tumorigenesis caused by unrepairable damages [11]. Mutations of human MMR genes were linked to common human cancers [12-14]. According to the data from The Cancer Genome Atlas (TCGA), most mutations found in human tumors are single base substitutions [15]. This high mutational burden renders tumors immunogenic and sensitive to programmed cell death-1 (PD-1) immune checkpoint inhibitors [16]. Besides, patients with MMR-deficient tumors experience highly variable responses, and roughly half are refractory to treatment [17]. These discoveries solidify a role for MMR in human tumorigenesis and provide support for the hypothesis that mutators might be driving the large numbers of mutations found in cancer. This article intends to review the function and mutation of MMR genes and their roles in cancer immunotherapy.
The mechanism of MMR gene causing tumor
The MMR gene is a group of genetic susceptibility genes isolated from hereditary non-polyposis colorectal cancer (HNPPC) [18]. Fisher isolated and cloned the first human MMR gene hMSH2 in 1993. At present, totally nine genes were found involved in the function of mismatch repair: mutS homologs (MSH2, MSH3, MSH4, MSH5, MSH6), mutL homologues (MLH1, MLH3), and postmeiotic segregation increased (PMS1, PMS2) [19]. In the process of biological evolution, MMR genes remain conservative, mainly functioning to correct the base mismatch that are generated during DNA recombination and replication. Properly functioned MMR genes ensure the integrity and stability of genetic material, induce apoptosis of DNA damaged cells and eliminate the formation of mutant cancerous cells [20]. Any mutations of this gene family will lead to defects in the mismatch repair function, cause genetic instability, and eventually lead to tumorigenesis [21] (Figure 1).
Functions of DNA Mismatch repair in tumorigenesis of human cancer.
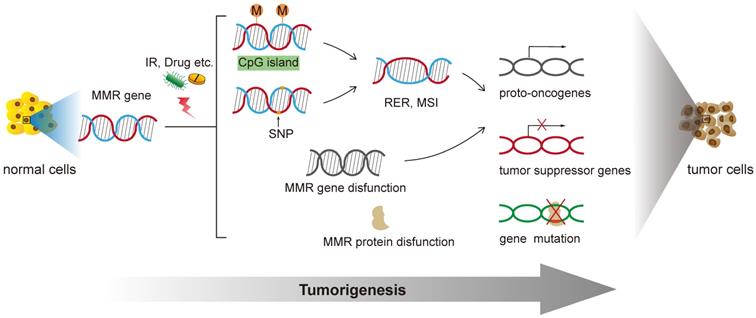
MMR gene promoter methylation
Some carcinogens, such as microorganisms and alkylating agents, can inhibit the MMR gene transcription process by methylating the CpG island in the promoter region and preventing the action of transcription factors [22]. Analysis results from 32 patients showed that the methylation rate of hM-SH2 promoter region in esophageal cancer tissue was 34.4%, and no methylation was found in normal esophageal tissue [23]. In liver cancer specimens, the methylation rate of CpG island in hMSH2 promoter is up to 68.4%, making it a common early genetic change in the occurrence and development of liver cancer [24]. The methylation rate of the hMLH1 promoter CpG island was 72.9% in gastric cancer tissues compared with 20% in non-gastric cancer tissues [25]. This significant difference indicates that methylation is highly related to the occurrence and development of gastric cancer [26]. Methylation of hMLH1 gene could be detected in 89% of patients with endometrial cancer [27], 18.0% in elderly colon cancer samples by Polymerase chain reaction (PCR) and 51.2% through the heterozygous deletion of microsatellite markers [28]. These studies have shown that the promoter methylation of DNA mismatch repair genes exists in most tumors, especially hMSH2 and hMLH1.
Correlation between MMR genes and clinical characteristics in human cancer
| Gene name | Gene Position | Tumor type | Related characteristics |
|---|---|---|---|
| MSH2 | 2p21-p16.3 | Colorectal Cancer; NSCLC; Thyroid Cancer; Breast Cancer; Liver Cancer; et al. | Overall survival; Drug resistance |
| MSH3 | 5q14.1 | Colorectal Cancer; Prostate Cancer; Breast Cancer; Thyroid Cancer; Prostate Cancer; Esophageal Cancer; Liver Cancer; et al. | Overall survival; Cancer risk; Chemotherapy resistance (Cisplatin, PARPi et al.) |
| MSH4 | 1p31.1 | Head and neck Cancer; Thyroid Cancer; et al. | Anti PDL1 treatment; Overall survival |
| MSH5 | 6p21.33 | Lung Cancer, non-Hodgkin's lymphoma | Cancer risk |
| MSH6 | 2p16.3 | Colorectal Cancer; Prostate Cancer; Breast Cancer; Thyroid Cancer; Prostate Cancer; Esophageal Cancer; Pancreatic Cancer; Liver Cancer; et al. | Overall survival; Cancer risk; Chemotherapy resistance; Radiotherapy resistance; Immunotherapy; |
| MLH1 | 3p22.2 | Colon Cancer; Gallbladder Cancer; Lung Cancer; Breast Cancer; Pancreatic Cancer; et al. | Metastasis; Overall survival; Chemotherapy resistance (TKI drug, PARPi et al.); Radiotherapy resistance; Immunotherapy |
| MLH3 | 14q24.3 | Endometrial Cancer. Colorectal Cancer; Liver Cancer. | Overall survival; Cancer risk; |
| PMS2 | 7p22.1 | Colorectal Cancer; Prostate; Breast Cancer; Thyroid Cancer; prostate Cancer; Esophageal Cancer; Pancreatic Cancer; Liver Cancer; et al. | Metastasis; Overall survival; Chemotherapy resistance; Radiotherapy resistance; Immunotherapy |
| PMS1 | 2q32.2 | Colon Cancer; Breast Cancer; et al. | DNA repair; Cancer risk |
MMR gene polymorphism and microsatellite instability
A single nucleotide polymorphism (SNP) refers to an orthologous nucleotide position that is variable across the genomes. Generally they are base-pair differences among chromosome sequences which are caused by mutations that convert, transverse, insert or delete single bases [29]. More than 90% essential manifestation of human genetic information is caused by genetic polymorphism [30]. SNPs in MMR genes are permanent changes that can impair mismatch repair function. When this happens, MMR system cannot repair mismatched bases and insertion/deletion loops. As a results, a large number of replication errors (RER) and MSI are generated during DNA replication [31]. The non-repetitive single-base DNA sequences are mainly affected by base-base mismatches. In addition, the insertion or deletion of short repeat sequences such as insertion-deletion loops is another reason for the occurrence of MSI [32].
MSI is a common phenomenon observed across different solid tumor types. Examples of common cancers that have MSI-H frequency >10% include colorectal cancer, endometrial cancer, and gastric cancer. Cancers with MSI-H frequency between 2% and 10% include ovarian cancer, cervical cancer, and thyroid cancer [21]. The polymorphisms of hMLH1 at -93G-A were detected in 165 patients with non-small cell lung cancer and 193 healthy controls [33]. Results showed that the homozygous variant A/A genotype was associated with a significantly increased risk for lung cancer. The patients with a homozygous variant A/A genotype had a trend toward poorer prognoses compared with other patients [33]. PCR-SSCP was applied to detect the MSI gene status of small intestinal adenocarcinoma tissues and adjacent tissues. Among them, MSI occurred in 19 cases (32.76%) of cancer tissues, and 9 cases had hMLH1 and hMSH2 gene mutations. The total rate was 47.37%, but no mutations were found in those who did not have MSI. Therefore, hMLH1 and hMSH2 gene mutations have clinical significance for the early diagnosis of small intestinal adenocarcinoma.
Change the mutation frequency of oncogenes and/or tumor suppressor genes
Besides oncogenes and tumor suppressor genes, MMR genes are the third type of genes that are closely related to tumorigenesis. Studies have shown that MMR dysfunction leads to the loss of repairment of the mutated coding region, directly or indirectly leading to the activation of proto-oncogenes and the inactivation of tumor suppressor genes [9]. As a result, cells begin to proliferate and differentiate indefinitely, eventually causing tumorigenesis. It's reported that people who positively express hMSH2 genes have a lower expression level of p53 compared with those who negatively express hMSH2 genes [34]. Speculating that the hMSH2 gene could be used as a protective factor, it might function via p53-dependent pathways.
Defects in MMR protein function
Of the 9 human MMR genes, six are involved in the mismatch repair process, namely hMSH2, hMSH3, hMSH6, hMLHl, hPMSl and hPMS2. MSH2 couples with either MSH6 or MSH3 (forming MutSα and MutSβ complexes, respectively), and MLH1 couples with PMS2 or MLH3 (forming MutLα, MutLβ or MutLγ complexes, respectively). hMutL forms a temporary complex with hMutS and bound to the DNA strand, responsible for the recognition of mismatches and insertion-deletion loops [35, 36]. Then recruitment of the MLH1/PMS2 complex will degrade the mutated stretch and initiates resynthesis. The newly formed DNA strand replaces the excised mismatched DNA strand to complete the repair process.
The reduction or deletion of MMR leads to defects in DNA repair, which affects the normal progress of DNA replication and increases the risk of developing tumors. Mismatch repair gene SNPs were found in most individuals with MMR gene expression defects [37]. In addition, the expression of hMSH2 was positively correlated with the malignant level of liver cancer [38]. MMR gene mutations may also form truncated proteins, stopping the transcription and translation.
Other mechanisms
Related mechanisms also include activity changes of cytokines caused by the mismatch repair gene SNPs such as IFN-beta [39]. These cytokines control the regulation of cell cycle and inflammation. Once lost, malignant proliferation of tumor cells would get started. However, the detailed mechanism still needs to be researched in future.
MMR gene polymorphism and cancer
The structure of hMSH2 and its relationship with cancer
The hMSH2 gene is in the chromosomal region 2p21, which covers a 73Kb segment with 16 exons. hMSH2 gene has molecular weight corresponding to 104,7 KDa, encoding a nuclear localization protein containing 934aa [40]. MMR is a powerful, evolutionary conserved, mutation avoidance mechanism. The identity between hMSH2 and the corresponding region of the yeast protein is 85%. hMSH2 protein can specifically bind the mismatch of G-T and A-C in the DNA double-strand and can bind to (CA) n and 14-base insertion-deletion protruding loops. In humans, DNA mismatches are recognized by one of two heterodimers, both of which contain MSH2. MutSα (MSH2-MSH6) preferentially recognizes and repairs base-base mismatches as well as small insertion and deletion loops, whereas MutSβ (MSH2-MSH3) recognizes and repairs small insertion and deletion loops [41]. The intron mutation rate of hMSH2 gene is higher than that of exon, so the existing reports are mostly concentrated in the intron region.
Mutations in the MSH2 gene are linked with the Lynch syndrome (LS), also known as HNPPC, hematological malignancy, gastrointestinal, urinary tract and ovary cancers [42-44]. Besides, MSH2 mutations are found in patients with endometrial carcinoma and adenocarcinoma of the colon. Besides, MSH2 mutation carriers have an increased risk of breast cancer (BC) with or without a LS family history [45]. A homozygous G to A transition mutation in the invariant G of the intron 10 splice acceptor of the MSH2 gene is associated with leukemia and multiple cafe´-au-lait spots, a feature of neurofibromatosis type 1 [46]. Three sites of hMSH2 gene: rs2303428, rs4952887 and rs2059520 in liver cancer samples. Results showed that in the development of liver cancer, polymorphism of rs2303428 site of hMSH2 gene was related to HBsAg positive and hepatic tumor family history. There is no interaction between rs4952887 and rs2059520.
Mechanically, researchers have found that MSH2 mutations altered the regulating pathways of nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 is an important regulator in modulating DNA mismatch repair (MMR) gene in acute myeloid leukemia (AML). Studies have found that patients with Nrf2 overexpression had a higher frequency of gene mutation and drug resistance. Mechanism behind is that Nrf2 overexpression inhibited MSH2 protein expression in a ROS-independent manner via JNK/c-Jun signaling, which caused DNA MMR deficiency and induced gene instability-dependent drug resistance in AML [47].
The structure of hMLH1 and its relationship with cancers
The hMLH1 gene is a mismatch repair gene discovered after hMSH2 in 1994 [48]. It is located on chromosome 3p21 and is homologous to the MutL in bacteria. Its genome is 58 kb in length and contains 19 exons. The total length of cDNA of hMLH1 is 2484bp. The 2268bp development reading frame of hMLH1 encodes protein containing 756 amino acids, and 41% homology with hMLH1 of yeast [49]. hMLH1 is a potential functional SNP. The expression of hMLH1 protein is affected by SNP and is closely related to the occurrence and development of tumors. At present, research on hMLH1 and human cancers are relatively extensive.
A homozygous germ-line MLH1 mutation was found to cause a mutator phenotype characterized by leukemia and/or lymphoma associated with neurofibromatosis type 1 [50]. In addition to mutations, hypermethylation of the MLH1 gene promoter and subsequent mismatch repair deficiency were involved in the pathogenesis of hematological malignancies such as acute T-cell leukemia/lymphoma (ATL) [51, 52]. Moreover, a study of 453 cases showed that after adjusted for age and gender, individuals with AG and GG genotypes in hMLH1 gene rs1800734 had higher risk than that of AA genotype in developing liver cancer. The frequency of allele A in the case group was higher than that in the control group. There are different major binding features for MMR genes. MutS alpha complex prefers to bind mismatch while MutSbeta prefers to bind the loop structure (Figure 2).
Molecular mechanism of DNA mismatch repair with represent MMR genes.
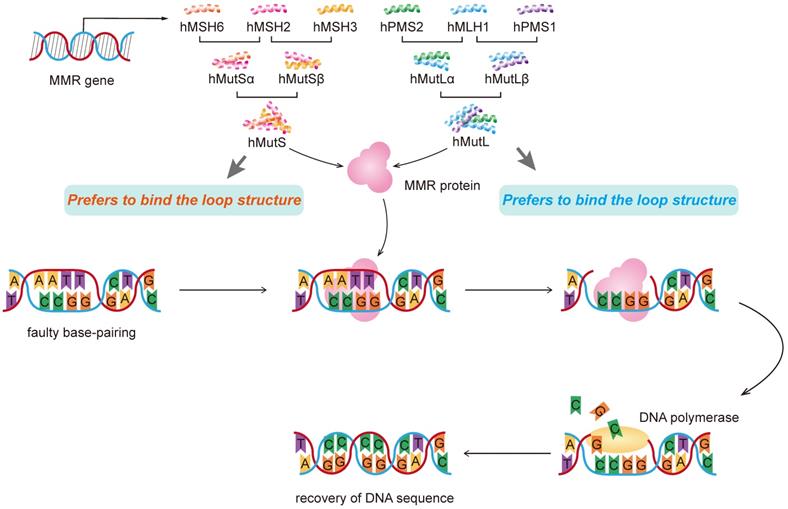
In addition, there may be a gene-environment interaction between the hMLH1 gene polymorphism and HBV infection and family history of tumors but not with drinking. In the occurrence and development of liver cancer, the polymorphism of hMLH1 locus rs1800734 gene is related with tumor family history and HBsAg positive [53]. The expressions of hMSH2 and hMLH1 are positively correlated with the degree of differentiation of liver cancer tissues. The higher the degree of differentiation, the higher the expression level. Reduced expression of hMLH1 gene was found in high-grade hepatocellular carcinoma [54].
The structure of hMSH6 and its relationship with cancer
In 1995, it was discovered that one of the members of the MMR family is hMSH6, which combined with hMSH2 protein to constitute the mismatch repair hMutSα protein complex [55]. hMSH6 protein is responsible for G/T mismatch, naming as G/T mismatch binding protein (GTBP). hMSH6 gene is 23806 bp in length and located on chromosome 2 p15-16, containing 10 exons and an untranslated region of 83 bp. The full-length cDNA is 4.2 kb, and the protein is 160kD. Studies had found that it mainly affected the stability of the human genome, and most of the mutations appear as small base mutations [56].
MSH6 mutations are related to several diseases. For example, MMR-deficiency (MMR-D) syndrome is characterized by childhood brain tumors, hematological and/or gastrointestinal malignancies, and signs of neurofibromatosis type 1 (NF1). Using an RNA-based mutation detection assay, researchers found a homozygous complex MSH6 splicing alteration in the index patients of a family with children suspected to MMR-D syndrome [57]. A germline de novo 2p16.3 deletion of MSH6 was found in a boy with neurodevelopmental delay and a diffuse large B-cell lymphoma (DLBCL) [58]. A case-control study involving 250 patients showed that the CT genotype of hMSH6 (rs1042821) reduced the risk of liver cancer. Most studies on hMSH6 gene are accompanied with hMSH2 gene, mainly because hMSH2 protein and hMSH6 protein combine to form heterodimer hMutSα protein to achieve the mismatch repair function. The polymorphism of hMSH6 gene alone is not enough to cause an increase in cancer susceptibility.
In addition to influencing tumorigenesis, MSH6 mutations are also involved in drug resistance. Relapse-specific heterozygous deletions in MSH6 results in a hypermutator phenotype associated with generation of secondary mutations that contributes to the development of thiopurine resistance in pediatric B-lymphoblastic leukemia [59].
The function of other MMR genes and the relationship with cancer
Other five genes, hMSH3, hMSH4, hMSH5, hPMS2 and hMLH3, achieve their function via working with other proteins: hMSH3 and hMSH2 form a complex hMmtSβ, which mainly recognizes 2-4 base mismatches; hMSH4 and hMSH5 mainly participate in meiotic recombination [60]; hPMS1, hPMS2 and hMLH1 form complexes hMutLβ and hMutLα, respectively; hMLH3 and hMLH1 form complexes [61].
The function of MMR in the immunotherapy of human cancer
Development of immunotherapy in cancer
Historically, conventional chemotherapies and radiotherapies have been identified to inhibit the proliferation or cause the death of cancer cells based on their uncontrolled relentless proliferation capacity. However, these therapies are cytotoxic with non-specific targets, such as DNA itself or enzymes required for DNA synthesis and repair. These targets are not restricted to malignant cells but rather are common to most cell types, limiting the application [62]. Because of this, people turn to human immune system, hoping to find some tumor-specific antigens as therapeutic targets. The concept that the immune system can specifically recognize and control tumor growth can be traced back to 1893 when William Coley used live bacteria as an immune stimulant to treat cancer [63]. Tumor-specific antigens that are generated by somatic mutation can influence immune system response to carcinoma cells and contribute to tumor shrinkage. In the past decade there has been an explosion of new approaches and technologies to explore the human immune system with unprecedented precision. Tremendous progress has been made in the understanding of how immune system recognize cancer cells and how cancer evades the immune surveillance. These findings in turn offers new ways to stop cancer immune evasion and facilitate cleaning cancer cells [64].
Mammals have two major sub-immune systems. The innate immune system provides an immediate, but non-specific response, targeting broad groups of situations and stimuli. While the adaptive immune system provides an antigen-specific response and requires the recognition of specific "non-self" antigens during a process called antigen presentation [65]. CD8+ cytotoxic T cells are important immune cells of adaptive immune system. They can recognize and target cancer cells that present tumor-specific antigens and drive the effect of tumor shrinkage. These antigens include but not limited to cancer testis antigens and somatic neoantigens [66]. To activate tumor-specific immune responses, two fundamental requirements must be simultaneously fulfilled. First, cancer cells must express antigens that can be recognized by a circulating naive T cell clone. Second, malignant cells must deliver adjuvant-like danger signals to antigen-presenting cells (APC) in the form of exogenous microbe-associated molecular patterns (MAMPs) or as endogenous damage-associated molecular patterns (DAMPs) [67]. The immune system encompasses inhibitory mechanisms to prevent excessive reactions and limit immune responses. These inhibitory mechanisms are necessary for balancing immunity in normal homeostasis. PD-1 is a member of the CD28/CTLA-4 family. This is a family of co-stimulatory receptors that expressed on the surface of natural killer cells, dendritic cells (DCs), activated monocytes, B cells and T cells. PD-1 contributes to the immune tolerance of self-antigens by conveying an inhibitory signal to T cells and suppressing immune response [68]. However, in the presence of a growing malignancy, the balance is disrupted and skewed towards excessive inhibition of immune reactivity due to tumor-induced immune suppression and enhanced immunologic tolerance [69]. Immunotherapies against inhibiting receptors, such as PD-1/PD-L1 antibodies, can block the combination of tumor PD-L1 and T cell PD-1, eliminating this immunosuppressive effect [70].
Current immunotherapies in cancer
Immune checkpoint inhibitors are rapidly developing immunotherapy methods in recent years, which boost T-cell activity against patient-specific neoantigens, restoring the suppressed immune function of the body and killing tumor cells [71]. During immune process, CD4+ regulatory T cells (Treg) accumulate, accompanied with CD8+ T cell activation. They regulate the duration and intensity of immune reaction by suppressing the function of effector CD8+ T cells [72]. This mechanism is known as immune checkpoint, which helps to avoid excessive immune response. Antibody-based checkpoint blockade immunotherapy mainly acts by boosting the immune system to target tumor cells through releasing CD8+ T cells from immunosuppressive activity of Treg. Monoclonal antibodies targeting co-inhibitory immune checkpoints (e.g., PD-1 and CTLA-4) have demonstrated clinical activity in several malignances, including melanoma, non-small cell lung cancer, renal cell carcinoma, bladder cancer, head and neck squamous cell carcinoma, MSI-high colorectal carcinoma, Merkel cell carcinoma, and Hodgkin lymphoma [73]. Approved treatments now include anti-PD-1 (nivolumab and pembrolizumab), anti-CTLA-4 (ipilimumab), and combination anti-PD-1/CTLA-4 regimens (nivolumab-ipilimumab) [74].
Cancer vaccines are another method to regulate immune system activity. Different from checkpoint inhibitors, cancer vaccines boost the immune system's ability to recognize and kill cancer cells by injecting cancer-specific elements into patients to [75]. After injection, DCs take up and process the introduced antigens and display the antigen on the cell surface through major histocompatibility complex (MHC) class I or II molecules. Then DCs present these antigens to resting T cells and active them. Activated T cells start to proliferate and differentiate into CD8+ cytotoxic T cells and target antigens displayed on the tumor cell surface [76]. One of the most widely adopted cancer vaccination is the design of MHC class I restricted peptide epitopes which are derived from shared tumor-associated antigens. Such vaccinations have been applied as experimental treatments for metastatic melanoma, clear cell renal cell cancer, melanoma/breast cancer and other tumor types. However, the attempt to use vaccines against chronic myelogenous leukemia (CML) by targeting the BCR-ABL fusion oncoprotein failed to prove a clear clinical benefit [77, 78]. Reasons for the failure could be low antigen expression/presentation on these tumors that are not enough for T cells to initiate an appropriate immune response. Another reason could be that the tumor is rapidly able to adapt to immunologic selection via an immunoediting mechanism [79]. Based on this, researchers started to use multiple, carefully selected shared neoantigens to see if it could increase the chance of inducing meaningful T-cell reactivity. Theoretically, this strategy could also lower the chances of tumor clones escaping from immune system elimination [80]. Besides using tumor-associated peptides, other alternative antigen sources include antitumor dendritic cells, whole tumor cell mRNA extracts and tumor cell extracellular vesicles such as exosomes [81].
MMR and cancer immunotherapy
The mutation rate during replication is rather low, approximately once for every 104 and 105 nucleotides. Still, each time a cell divides, about 100,000 polymerase errors occur. Although DNA polymerases are able to correct part of these errors, some errors always escape proofreading, which need the help of MMR system [82]. MMR gene mutation is common among different type of cancers. Due to these defects, cancer cells start to produce and secret mutated non-self-proteins which are termed as neoantigens. These neoantigens are recognized by the immune system and stimulate the activation of immune cells. These self-produced neoantigens can trigger a more robust and long-lasting immune response, suppressing tumorigenesis more effectively than externally injected neoantigen vaccines [83]. Besides, MMR deficient tumors are immunogenic and sensitive to programmed cell death-1 (PD-1) immune checkpoint inhibitors [84, 85]. An upregulation of genes involved in the immune response, such as proinflammatory cytokines and cytotoxic mediators is observed in MMR deficiency tumors, resulting in an increased secretion of soluble mediators in the tumor microenvironment with the subsequent activation of the PD-1 pathway [86]. Studies on several types of cancers (colorectal, gastric, ovarian, upper urinary tract urothelial cancer, biliary tract cancers) have identified that MMR deficiency has a more favorable prognosis with a lower tendency to lymph node spread and better overall survival [87-89]. Based on these characters, MMR has emerged as an important predictor of sensitivity for immunotherapy-based strategies [90, 91] (Figure 3).
Mechanism of MMR deficiency tumor cell with immune checkpoint drug like PD1 antibody.
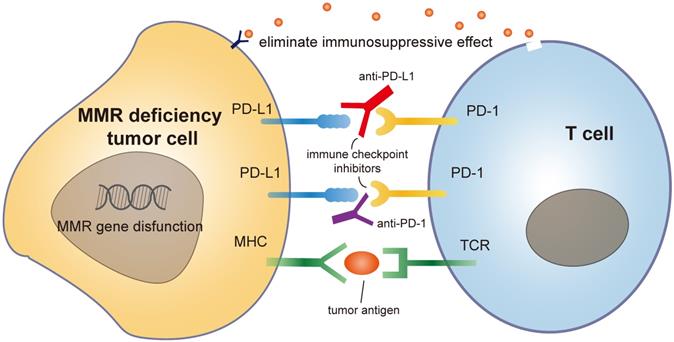
Moreover, MMR genes augment tumor immunity via affecting certain kinds of chemotherapies. Cancer chemotherapy was previously seemed as immune suppressive. Nowadays, chemotherapies are found to promote tumor immunity by disrupting strategies that tumors use to evade immune recognition [92]. Different drugs can influence the immune response to cancer through a wide variety of mechanisms such as inducing immunogenic cell death, changing antigen-presentation, activating tumor cell targes and depleting immunosuppressive cells [93]. It is known that MSI status of MMR gene may predict cancer response/resistance to certain chemotherapies. MMR deficient tumors are commonly resistant to methylating agents, platinum compounds and fluoropyrimidines [94, 95]. A possible explanation is that under unrepairable DNA damage, DNA damage response proteins (i.e., ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related protein (ATR)) are recruited by MMR proteins and induce cell cycle arrest, DNA repair, or apoptosis through DNA damage checkpoint proteins activation. While MMR deficiency might alter this mechanism and fail to remove these transferred cells. Accumulation of these DNA damage and transferred cells in turn confer resistance to many chemotherapies [96]. Identifying the immunological changes associated with chemotherapy and MMR mutations is important in combining chemotherapy with checkpoint blockade and translating promising preclinical data into successful treatments for cancer patients.
MMR gene mutations are common in hematological malignancies and correlated with genome instability as well as tumorigenesis [97]. Besides, chemotherapy-induced MMR mutations, such as thiopurine treatment, facilitate drug resistance in hematological malignant cells [98, 99]. At present, immunotherapeutic methods targeting MMR are implicated in solid tumors. For example, two PD1-blocking antibodies, pembrolizumab and nivolumab, have shown efficacy in patients with metastatic colorectal cancers (CRCs) and have been granted accelerated FDA approval [100, 101]. However, studies on hematologic malignancies are limited. Priyanjali et al. found 6 out of 536 plant derived biomolecules that may have anticancer properties against the tumors driven by deregulated MMR-pathways in blood-related cancers [102]. Barthelemy et al. found that somatic deletions of 4 genes (FRAP1, HERC1, PRKCZ, PIK3C2B) recapitulated the MSH2 protein deficiency by enhancing MSH2-degradation, leading to significant reduction in MMR expression and increased resistance to thiopurines in human leukemia cells [41]. Nuclear factor erythroid 2-related factor 2 (Nrf2, also called NFE2L2) overexpression protected acute myeloid leukemia (AML) cells from apoptosis induced by cytarabine via inhibiting MSH2 expression in a ROS-independent manner [103]. Nikki et al. showed that MSH6 haploinsufficiency at relapse contributed to the development of thiopurine resistance in pediatric B-lymphoblastic leukemia [59]. Other strategies such as upregulation of cGAS/STING, neoantigen-based vaccinations and immune checkpoint inhibitors could be effective ways to conquer MMR deficiency related tumors [39, 104].
Prospect
There are at least six DNA repair pathways involved in repairing specific types of DNA damage: 1) the DNA MMR pathway repairs base-base mismatches and insertion/deletion mis-pairs; 2) the base excision repair (BER) pathway corrects single-strand breaks and homologous recombination (HR); 3) the non-homologous end joining (NHEJ) pathway repairs double-strand breaks; 4) the nucleotide excision repair (NER) pathway repairs DNA adducts; 5) the Fanconi anemia (FA) pathway fixes inter-strand crosslinks; 6) the O-6-methylguanine DNA methyltransferase (MGMT) pathway repairs O-6-methylguanine adducts [105]. Among these pathways, mutational inactivation of MMR is the most typical and high-frequent character in tumor cells which allows cancer cells to accumulate thousands of mutations.
Nowadays, a plethora of drugs or drug combinations, including chemotherapy, targeted therapies, immunotherapies, immune checkpoint inhibitors, and CAR-T cells (chimeric antigen receptor-T cells) are applying to cancer treatment. However, malignant hematopoietic cells consistently develop cellular strategies to adapt to and survive from currently available therapies. This is regarded as a general hallmark and major drawback of leukemia and lymphoma, and significantly accounts for relapse and failure of treatments [106]. Therefore, figuring out the mechanisms of resistance to therapies and finding new therapeutic targets are required in hematological malignancies treatment.
Epidemiological studies have shown that the occurrence of tumors is different among individuals, and the main determinant of this susceptibility is genetic differences [107]. Somatic mutations in genomes happen when cells are exposed to mutational factors, including exogenous chemicals and physical agents as well as endogenous reactive metabolites such as reactive oxygen and nitrogen species (ROS and RNS) [108]. Other sources of DNA damages are errors that occur during normal DNA metabolism or aberrant DNA processing reactions. Base substitutions, small insertions and deletions (indels), genome rearrangements and chromosome copy-number changes may happen during DNA replication, recombination, and repair [109]. Thus, high-fidelity DNA replication is crucial to preserve the genomic integrity of eukaryotic cells and organismal health [110]. Normal somatic cells possess DNA damage repair system to help them correct the mutations. While in most cancer cells, mutational inactivation of DNA repair genes was found which resulted in a profound repair defect and progressive accumulation of mutations throughout the genome [111].
The genomes of MMR deficient cancers are characterized by sequence alterations in microsatellites and thousands of mutations. These mutations are predicted to produce lots of mutation-associated neoantigens that might be recognized by the immune system and promote tumor destruction [112]. Therefore, cancers with MMR deficiency were sensitive to PD-1 immune checkpoint blockade [113]. Therapies with immune checkpoint inhibitors, such as anti-PD-1 antibodies, showed a potent and durable anti-tumor response regardless of the cancers' tissue of origin [114]. Besides, the genetic diversity of MMR deficient cancers also influences the extension of anti-PD-1 immunotherapy response [115]. In addition to immune checkpoint blockade, blocking the Nedd8-mediated degradation pathway with MLN4924 is another method to induce immunogenic cell death in MMR deficient cancer cells. Because of the proteome instability, an abundance of misfolded protein aggregates in MMR deficient tumors [116]. To compensate, tumor cells utilize a Nedd8-mediated degradation pathway to facilitate clearance of misfolded proteins. Blocking this Nedd8 clearance pathway causes accumulation of misfolded protein, ultimately inducing immunogenic cell death [117]. Abnormal activation of the cGAS-STING pathway due to the loss of MutLα-specific regulation of exonuclease 1 (Exo1) during DNA repair also facilizes the clearance of MMR deficient tumor cells [118].
Hematological malignancies are characterized by genetic defect in the form of chromosomal translocation or breakpoint/fusion, exposing cells to genomic instability [119]. Loss of MMR function exacerbates genomic instabilities and results in the production of neoantigens [120]. To escape form immunological surveillance, cancer cells highly express PD-1 antigen to suppress T cell function, which may contribute to refractory and relapsed acute myeloid leukemia [121]. Therefore, applying checkpoint inhibitors is a promising strategy to combat cancer and improve survival rate through inducing genetic instability in cancer cells [119]. Therapeutic cancer vaccines such as monoclonal proteasome-targeting antibodies, agonists for co-stimulatory molecules, adoptive transfer of genetically modified T cells like chimeric antigen receptor CAR-T cells [122], as well as agents that suppress negative regulatory pathways of T cells are all under active clinical investigation to provide longer survival time and fewer adverse reactions [123-125]. Despite higher requirements and more challenges putting forward, we believe more new drugs will be coming out, bringing new treatment options to patients suffering from carcinoma all over the world.
Abbreviations
MMR: mismatch repair; MSI: microsatellite instability; ROS: reactive oxygen species; RNS: reactive nitrogen species; BER: base excision repair; HR: homologous recombination; NHEJ: non-homologous end joining; NER: nucleotide excision repair; FA: Fanconi anemia; MGMT: O-6-methylguanine DNA methyltransferase; PD: programmed cell death; HNPPC: hereditary non-polyposis colorectal cancer; hMSH: Human mutS homolog protein; hMSL: Human mutL homolog protein; hPMS: Human PMS1 homolog protein; PMS: postmeiotic segregation increased; PCR: Polymerase chain reaction; SNP: single nucleotide polymorphism; RER: replication errors; LS: Lynch syndrome; AML: acute myeloid leukemia; Nrf2: nuclear factor erythroid 2-related factor 2; ATL: acute T-cell leukemia/lymphoma; GTBP: G/T mismatch binding protein; NF1: neurofibromatosis type 1; DLBCL: diffuse large B-cell lymphoma; APC: antigen-presenting cells; MAMPs: microbe-associated molecular patterns; DAMPs: damage-associated molecular patterns; DC: dendritic cell; MHC: major histocompatibility complex; CML: chronic myelogenous leukemia; ATM: ataxia telangiectasia mutated; ATR: ataxia telangiectasia and Rad3-related protein; CAR-T: chimeric antigen receptor-T cells; Exo1: exonuclease 1.
Acknowledgements
We would like to thank Dr. Meihan Duan for the discussion of manuscript.
Availability of data and materials
All relevant data are provided in the manuscript.
Funding
This Research was funded by Anhui Provincial Natural Science Foundation (No. 2108085MH285), the Fundamental Research Funds for the Central Universities (No. WK9110000154), Hunan Postdoctoral Program for Innovative Talent (2021RC2017), Natural Science Foundation of Hunan Province (2021JJ41027) and China Postdoctoral Science Foundation funded project (2021M693567, 2021TQ0374). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
YW, YH and HC contributed to all of the literature review in this work. LZ and RZ participated in the process of finalizing the final manuscript. All authors have read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209-49
2. Mathers CD. History of global burden of disease assessment at the World Health Organization. Arch Public Health. 2020;78:77
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68:394-424
4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2021;71:209-49
5. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-E86
6. Pilleron S, Sarfati D, Janssen-Heijnen M, Vignat J, Ferlay J, Bray F. et al. Global cancer incidence in older adults, 2012 and 2035: A population-based study. Int J Cancer. 2019;144:49-58
7. Fidler MM, Bray F, Soerjomataram I. The global cancer burden and human development: A review. Scand J Public Health. 2018;46:27-36
8. Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
9. Gupta D, Heinen CD. The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair (Amst). 2019;78:60-9
10. Fishel R. Mismatch repair. J Biol Chem. 2015;290:26395-403
11. Li Z, Pearlman AH, Hsieh P. DNA mismatch repair and the DNA damage response. DNA repair. 2016;38:94-101
12. Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of lynch syndrome: 1895-2015. Nat Rev Cancer. 2015;15:181-94
13. Tamura K, Kaneda M, Futagawa M, Takeshita M, Kim S, Nakama M. et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011
14. Ijsselsteijn R, Jansen JG, de Wind N. DNA mismatch repair-dependent DNA damage responses and cancer. DNA Repair (Amst). 2020;93:102923
15. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214-8
16. Lee V, Murphy A, Le DT, Diaz LA Jr. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist. 2016;21:1200-11
17. Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science (New York, NY). 2019;364:485-91
18. Müller A, Fishel R. Mismatch repair and the hereditary non-polyposis colorectal cancer syndrome (HNPCC). Cancer Invest. 2002;20:102-9
19. Marti TM, Kunz C, Fleck O. DNA mismatch repair and mutation avoidance pathways. J Cell Physiol. 2002;191:28-41
20. Liu D, Keijzers G, Rasmussen LJ. DNA mismatch repair and its many roles in eukaryotic cells. Mutation Research/Reviews in Mutation Research. 2017;773:174-87
21. Baretti M, Le DT. DNA mismatch repair in cancer. Pharmacol Ther. 2018;189:45-62
22. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa J-PJ. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci. 1999;96:8681-6
23. Zhang H, Zhang S, Cui J, Zhang A, Shen L, Yu H. Expression and promoter methylation status of mismatch repair gene hMLH1 and hMSH2 in epithelial ovarian cancer. Australian and New Zealand Journal of Obstetrics and Gynaecology. 2008;48:505-9
24. Herath NI, Leggett BA, MacDonald GA. Review of genetic and epigenetic alterations in hepatocarcinogenesis. Journal of gastroenterology and hepatology. 2006;21:15-21
25. Ebrahimi V, Soleimanian A, Ebrahimi T, Azargun R, Yazdani P, Eyvazi S. et al. Epigenetic modifications in gastric cancer: Focus on DNA methylation. Gene. 2020;742:144577
26. Ye P, Shi Y, Li A. Association between hMLH1 promoter methylation and risk of gastric cancer: a meta-analysis. Frontiers in physiology. 2018;9:368
27. Buchanan DD, Tan YY, Walsh MD, Clendenning M, Metcalf AM, Ferguson K. et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. Journal of Clinical Oncology. 2014;32:90
28. Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA. et al. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837-45
29. Leaché AD, Oaks JR. The utility of single nucleotide polymorphism (SNP) data in phylogenetics. Annual Review of Ecology, Evolution, and Systematics. 2017;48:69-84
30. Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177:26-31
31. Caja F, Vodickova L, Kral J, Vymetalkova V, Naccarati A, Vodicka P. DNA mismatch repair gene variants in sporadic solid cancers. Int J Mol Sci. 2020;21:5561
32. Belfield EJ, Ding ZJ, Jamieson FJ, Visscher AM, Zheng SJ, Mithani A. et al. DNA mismatch repair preferentially protects genes from mutation. Genome Res. 2018;28:66-74
33. Shih CM, Chen CY, Lee IH, Kao WT, Wang YC. A polymorphism in the hMLH1 gene (-93G->A) associated with lung cancer susceptibility and prognosis. Int J Mol Med. 2010;25:165-70
34. Spagnoletti I, Pizzi C, Galietta A, Di Maio M, Mastranzo P, Daniele S. et al. Loss of hMSH2 expression in primary breast cancer with p53 alterations. Oncol Rep. 2004;11:845-51
35. Jiricny J. The multifaceted mismatch-repair system. Nature reviews Molecular cell biology. 2006;7:335-46
36. Jiricny J. MutLα: at the cutting edge of mismatch repair. Cell. 2006;126:239-41
37. Pardini B, Corrado A, Paolicchi E, Cugliari G, Berndt SI, Bezieau S. et al. DNA repair and cancer in colon and rectum: Novel players in genetic susceptibility. Int J Cancer. 2020;146:363-72
38. Helal TE, Khamis NS, El-Sharkawy TM, Nada OH, Radwan NA. Immunohistochemical expression of mismatch repair genes (hMSH2 and hMLH1) in hepatocellular carcinoma in Egypt. Apmis. 2010;118:934-40
39. Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T. et al. DNA sensing in mismatch repair-deficient tumor cells is essential for anti-tumor immunity. Cancer Cell. 2021;39:96-108.e6
40. da Silva Calixto P, Lopes OS, dos Santos Maia M, Herrero SST, Longui CA, Melo CGF. et al. Single-nucleotide polymorphisms of the MSH2 and MLH1 genes, potential molecular markers for susceptibility to the development of basal cell carcinoma in the brazilian population. Pathology & Oncology Research. 2018;24:489-96
41. Diouf B, Cheng Q, Krynetskaia NF, Yang W, Cheok M, Pei D. et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nature medicine. 2011;17:1298-303
42. Hirano K, Yamashita K, Yamashita N, Nakatsumi Y, Esumi H, Kawashima A. et al. Non-Hodgkin's lymphoma in a patient with probable hereditary nonpolyposis colon cancer. Diseases of the colon & rectum. 2002;45:273-9
43. Joost P, Therkildsen C, Dominguez-Valentin M, Jönsson M, Nilbert M. Urinary tract cancer in Lynch syndrome; increased risk in carriers of MSH2 mutations. Urology. 2015;86:1212-7
44. Provenzale D, Gupta S, Ahnen DJ, Bray T, Cannon JA, Cooper G. et al. Genetic/familial high-risk assessment: colorectal version 1.2016, NCCN clinical practice guidelines in oncology. Journal of the National Comprehensive Cancer Network. 2016;14:1010-30
45. Wu B, Peng Y, Eggert J, Alexov E. Novel genetic markers for early detection of elevated breast cancer risk in women. Int J Mol Sci. 2019 20
46. Whiteside D, McLeod R, Graham G, Steckley JL, Booth K, Somerville MJ. et al. A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple café-au-lait spots. Cancer Res. 2002;62:359-62
47. Liu P, Ma D, Wang P, Pan C, Fang Q, Wang J. Nrf2 overexpression increases risk of high tumor mutation burden in acute myeloid leukemia by inhibiting MSH2. Cell Death Dis. 2021;12:20
48. Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK. et al. Mutation in the DNA mismatch repair gene homologue hMLH 1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258-61
49. Quehenberger. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. Journal of Medical Genetics. 2005;42:491-6
50. Ricciardone MD, Özçelik T, Cevher B, Özdağ H, Tuncer M, Gürgey A. et al. Human MLH1 deficiency predisposes to hematological maligancy and neurofibromatosis type 1. Cancer Res. 1999;59:290
51. Matsushita M, Takeuchi S, Yang Y, Yoshino N, Tsukasaki K, Taguchi H. et al. Methylation of the MLH1 gene in hematological malignancies. Oncol Rep. 2005;14:191-4
52. Daino K, Ishikawa A, Suga T, Amasaki Y, Kodama Y, Shang Y. et al. Mutational landscape of T-cell lymphoma in mice lacking the DNA mismatch repair gene Mlh1: no synergism with ionizing radiation. Carcinogenesis. 2019;40:216-24
53. Matsukura S, Soejima H, Nakagawachi T, Yakushiji H, Ogawa A, Fukuhara M. et al. CpG methylation of MGMT and hMLH1 promoter in hepatocellular carcinoma associated with hepatitis viral infection. British Journal of Cancer. 2003;88:521-9
54. Wani Y, Notohara K, Tsukayama C, Okada S. Reduced expression of hMLH1 and hMSH2 gene products in high-grade hepatocellular carcinoma. Acta Medica Okayama. 2001;55:65
55. Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT. et al. hMSH2 forms specific mispair-binding complexes with hMSH3andhMSH6. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13629-34
56. Gras E, Catasus L, Argüelles R, Moreno-Bueno G, Palacios J, Gamallo C. et al. Microsatellite instability, MLH-1 promoter hypermethylation, and frameshift mutations at coding mononucleotide repeat microsatellites in ovarian tumors. Cancer. 2015;92:2829-36
57. Etzler J, Peyrl A, Zatkova A, Schildhaus HU, Ficek A, Merkelbach-Bruse S. et al. RNA-based mutation analysis identifies an unusual MSH6 splicing defect and circumvents PMS2 pseudogene interference. Hum Mutat. 2008;29:299-305
58. van Engelen N, van Dijk F, Waanders E, Buijs A, Vermeulen MA, Loeffen JLC, et al. Constitutional 2p16.3 deletion including MSH6 and FBXO11 in a boy with developmental delay and diffuse large B-cell lymphoma. Fam Cancer. 2021
59. Evensen NA, Madhusoodhan PP, Meyer J, Saliba J, Chowdhury A, Araten DJ. et al. MSH6 haploinsufficiency at relapse contributes to the development of thiopurine resistance in pediatric B-lymphoblastic leukemia. Haematologica. 2018;103:830-9
60. Snowden T, Acharya S, Butz C, Berardini M, Fishel R. hMSH4-hMSH5 recognizes Holliday Junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol Cell. 2004;15:437-51
61. Salem ME, Bodor JN, Puccini A, Xiu J, Goldberg RM, Grothey A. et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int J Cancer. 2020;147:2948-56
62. Schirrmacher V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int J Oncol. 2019;54:407-19
63. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51-81
64. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335-7
65. Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol. 2017;17:21-9
66. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269-81
67. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nature Reviews Clinical Oncology. 2020;17:725-41
68. Muenst S, Schaerli AR, Gao F, Däster S, Trella E, Droeser RA. et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Research and Treatment. 2014;146:15-24
69. Palomba ML. Active immunotherapy: current state of the art in vaccine approaches for NHL. Current oncology reports. 2012;14:433-40
70. Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. 2018;4:418-28
71. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Experimental & Molecular Medicine. 2018;50:1-11
72. Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Annals of the New York Academy of Sciences. 2018;1417:104-15
73. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707-23
74. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9-16
75. Desrichard A, Snyder A, Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res. 2016;22:807
76. Kastenmüller W, Kastenmüller K, Kurts C, Seder RA. Dendritic cell-targeted vaccines-hope or hype? Nat Rev Immunol. 2014;14:705-11
77. Jain N, Reuben JM, Kantarjian H, Li C, Gao H, Lee BN. et al. Synthetic tumor-specific breakpoint peptide vaccine in patients with chronic myeloid leukemia and minimal residual disease: a phase 2 trial. Cancer. 2009;115:3924-34
78. Rojas JM, Knight K, Wang L, Clark RE. Clinical evaluation of BCR-ABL peptide immunisation in chronic myeloid leukaemia: results of the EPIC study. Leukemia. 2007;21:2287-95
79. Pan C, Liu H, Robins E, Song W, Liu D, Li Z. et al. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol. 2020;13:29 -
80. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C. et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nature medicine. 2012;18:1254-61
81. Harari A, Graciotti M, Bassani-Sternberg M, Kandalaft LE. Antitumour dendritic cell vaccination in a priming and boosting approach. Nature Reviews Drug Discovery. 2020;19:635-52
82. Bebenek K, Kunkel TA. Functions of DNA polymerases. Advances in Protein Chemistry: Academic Press. 2004 p. 137-65
83. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342-50
84. Schwitalle Y, Linnebacher M, Ripberger E, Gebert J, von Knebel Doeberitz M. Immunogenic peptides generated by frameshift mutations in DNA mismatch repair-deficient cancer cells. Cancer Immunity Archive. 2004 4
85. Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science (New York, NY). 2019;364:485
86. Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science (New York, NY). 2019;364:485-91
87. Cloyd JM, Chun YS, Ikoma N, Vauthey JN, Aloia TA, Cuddy A. et al. Clinical and genetic implications of DNA mismatch repair deficiency in biliary tract cancers associated with Lynch syndrome. Journal of gastrointestinal cancer. 2018;49:93-6
88. Kato M, Takano M, Miyamoto M, Sasaki N, Goto T, Tsuda H. et al. DNA mismatch repair-related protein loss as a prognostic factor in endometrial cancers. Journal of gynecologic oncology. 2015;26:40
89. Wensink GE, Elferink MAG, May AM, Mol L, Hamers PAH, Bakker SD. et al. Survival of patients with deficient mismatch repair metastatic colorectal cancer in the pre-immunotherapy era. Br J Cancer. 2021;124:399-406
90. Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12:1-14
91. Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12:54
92. Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunology Research. 2015;3:436
93. Cook AM, Lesterhuis WJ, Nowak AK, Lake RA. Chemotherapy and immunotherapy: mapping the road ahead. Curr Opin Immunol. 2016;39:23-9
94. Smyth EC, Wotherspoon A, Peckitt C, Gonzalez D, Hulkki-Wilson S, Eltahir Z. et al. Mismatch repair deficiency, microsatellite instability, and survival: an exploratory analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial. JAMA oncology. 2017;3:1197-203
95. Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nature reviews Clinical oncology. 2010;7:197
96. Viale G, Trapani D, Curigliano G. Mismatch repair deficiency as a predictive biomarker for immunotherapy efficacy. Biomed Res Int. 2017;2017:4719194
97. Gaymes TJ, Mohamedali AM, Patterson M, Matto N, Smith A, Kulasekararaj A. et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica. 2013;98:1397-406
98. Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y. et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood. 2020;135:41-55
99. Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K. et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469
100. Lizardo DY, Kuang C, Hao S, Yu J, Huang Y, Zhang L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim Biophys Acta Rev Cancer. 2020;1874:188447
101. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH. et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16:361-75
102. Bhattacharya P, Patel TN. A study of deregulated MMR pathways and anticancer potential of curcuma derivatives using computational approach. Sci Rep. 2021;11:10110
103. Liu P, Ma D, Wang P, Pan C, Fang Q, Wang J. Nrf2 overexpression increases risk of high tumor mutation burden in acute myeloid leukemia by inhibiting MSH2. Cell Death Dis. 2021;12:20
104. Westdorp H, Kolders S, Hoogerbrugge N, de Vries IJM, Jongmans MCJ, Schreibelt G. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Lett. 2017;403:159-64
105. Ruiz-Bañobre J, Goel A. DNA mismatch repair deficiency and immune checkpoint inhibitors in gastrointestinal cancers. Gastroenterology. 2019;156:890-903
106. Auberger P, Tamburini-Bonnefoy J, Puissant A. Drug resistance in hematological malignancies. International journal of molecular sciences. 2020;21:6091
107. Sud A, Kinnersley B, Houlston RS. Genome-wide association studies of cancer: current insights and future perspectives. Nature Reviews Cancer. 2017;17:692-704
108. Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85-98
109. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y. et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94-101
110. Ganai RA, Johansson E. DNA replication—a matter of fidelity. Mol Cell. 2016;62:745-55
111. Peltomäki P. DNA mismatch repair and cancer. Mutation Research/Reviews in Mutation Research. 2001;488:77-85
112. Supek F, Lehner B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature. 2015;521:81-4
113. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450-61
114. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-13
115. Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science (New York, NY). 2019;364:485-91
116. Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F. et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116-20
117. McGrail DJ, Garnett J, Yin J, Dai H, Shih DJH, Lam TNA. et al. Proteome instability is a therapeutic vulnerability in mismatch repair-deficient cancer. Cancer Cell. 2020;37:371-86.e12
118. Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L. et al. MLH1 deficiency-triggered DNA hyperexcision by exonuclease 1 activates the cGAS-STING pathway. Cancer Cell. 2021;39:109-21.e5
119. Alhmoud JF, Mustafa AG, Malki MI. Targeting DNA repair pathways in hematological malignancies. Int J Mol Sci. 2020;21:7365
120. Roerden M, Nelde A, Walz JS. Neoantigens in hematological malignancies-ultimate targets for immunotherapy? Frontiers in immunology. 2019;10:3004 -
121. Mao G, Yuan F, Absher K, Jennings CD, Howard DS, Jordan CT. et al. Preferential loss of mismatch repair function in refractory and relapsed acute myeloid leukemia: potential contribution to AML progression. Cell Res. 2008;18:281-9
122. Zhao J, Wu M, Li Z, Su S, Wen Y, Zhang L. et al. Chimeric antigen receptor therapy in hematological malignancies: antigenic targets and their clinical research progress. Ann Hematol. 2020;99:1681-99
123. Vasekar M, Rizvi S, Liu X, Vrana KE, Zheng H. Novel immunotherapies for hematological malignancies. Curr Mol Pharmacol. 2016;9:264-71
124. Han Y, Liu Z, Liu J, Yan W, Xia Y, Yue S. et al. Antibody-based immunotherapeutic strategies for the treatment of hematological malignancies. Biomed Res Int. 2020;2020:4956946
125. Miller Z, Lee W, Kim KB. The immunoproteasome as a therapeutic target for hematological malignancies. Curr Cancer Drug Targets. 2014;14:537-48
Author contact
![]() Corresponding authors: Hao Chen, E-mail: ch2446edu.cn & Yumin Wang, E-mail: wangyvmincom.
Corresponding authors: Hao Chen, E-mail: ch2446edu.cn & Yumin Wang, E-mail: wangyvmincom.