Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(8):3167-3177. doi:10.7150/ijbs.69155 This issue Cite
Research Paper
Gamma synuclein promotes cancer metastasis through the MKK3/6-p38MAPK cascade
Jieya Liu1*, Ting Shao1,6*, Jin Zhang1, Qianyi Liu1, Hui Hua2, Hongying Zhang1, Jiao Wang3, Ting Luo4, Yuenian Eric Shi5, Yangfu Jiang1 ![]()
1. Laboratory of Oncogene, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, China
2. Laboratory of Stem Cell Biology, West China Hospital, Sichuan University. Chengdu, China
3. School of Basic Medicine, Chengdu University of Traditional Chinese Medicine. Chengdu, China
4. Cancer center, West China Hospital, Sichuan University. Chengdu, China
5. Chimigen Bio, Chengdu, China
6. Current address: BeiGene (Shanghai) Co., Ltd, Shanghai, China
*These authors contributed equally to this work.
Received 2021-11-16; Accepted 2022-4-17; Published 2022-5-1
Abstract

Gamma synuclein (SNCG) is a neuronal protein that is also aberrantly overexpressed in various types of human cancer. SNCG overexpression promotes cancer invasion and metastasis. However, the mechanisms that drive cancer metastasis upon SNCG expression remain elusive. Elucidation of the mechanisms underlying the promotion of cancer metastasis by SNCG may help discover therapeutic avenues for SNCG-overexpressed cancer. Here, we show that SNCG promotes transforming growth factor-β (TGF-β)-induced p38 mitogen-activated protein kinase (MAPK) phosphorylation. Mechanistically, SNCG promotes p38MAPK phosphorylation by interacting with the MAPK kinase 3/6 (MKK3/6) and prevents their degradation. SNCG knockdown leads to a decrease in TGF-β-induced phosphorylation of MKK3/6; and abrogates the induction of matrix metalloproteinase (MMP)-9 expression by TGF-β and its target gene Twist1. Furthermore, p38MAPK inhibition abrogates the promotion of MMP-9 expression and cancer cell invasion by SNCG. Both p38MAPK and MMP inhibitors can suppress the promotion of cancer cell invasion by SNCG. Finally, overexpression of SNCG in liver cancer cells promotes lung metastasis, which can be suppressed by the p38MAPK inhibitor. Together, our data uncover a previously unknown role of SNCG in promoting TGF-β-MKK3/6-p38MAPK signaling. This study highlights the critical role of p38MAPK in the promotion of cancer metastasis by SNCG, and indicates that p38MAPK inhibitor may serve as a potential therapeutic for SNCG-overexpressed cancer.
Introduction
Regional lymph node spread and distant metastasis are inherent features of malignant tumors. While early-stage cancer may be effectively treated by multiple procedures such as surgical resection, radiotherapy and chemotherapy, it is still hard to treat metastatic cancer. Hence, the prognosis of patients with metastatic cancer remains to be very poor. The dissemination of cancer cells and development of organ-specific metastasis involve multiple steps, including tumor cells detachment from the primary site, migration, invasion, intravasation, survival in the circulation, extravasation, colonization at distant sites and metastatic outgrowth [1]. Oncogenes, growth factors, inflammatory factors and cancer microenvironment may jointly promote cancer metastasis by enhancing cancer cell motility, invasion, colonization and angiogenesis [1-5]. A better understanding the mechanisms of cancer metastasis may help uncover novel biomarkers and therapeutic targets [6].
Transforming growth factor-β (TGF-β) is a well-known growth factor that may promote cancer metastasis, stemness and immune evasion, whilst it may inhibit cell growth at the early stage of tumorigenesis [7, 8]. TGF-β can induce the expression of many genes by activating mothers against decapentaplegic homolog (SMADs). Whereas the TGF-β responsive p21Cip1 and p15Ink4b may inhibit cell proliferation, Twist1, snail, ZEB1 can induce epithelial-mesenchymal transition (EMT), a process that is tightly involved in cancer metastasis [7]. On the other hand, the non-SMAD pathways, such as extracellular signal-activated kinase (Erk), Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38MAPK), can be activated by TGF-β receptor signaling [9]. p38MAPK is a growth factors-, cytokines- and sresses-activated kinase that has complex roles in tumorigenesis [10, 11]. Among the 4 members of p38MAPK family, p38α is universially expressed in most cell types, whereas p38β, p38γ and p38δ expression are restricted in specific cell types. Whilst p38MAPK activation may inhibit the proliferation of some types of cells, it is often subverted by cancer cells to promote tumor growth, metastasis and drug resistance.
Previously, we reported that gamma synuclein (SNCG), one of three members of the synuclein family (α-synuclein, β-synuclein and SNCG) that are preferentially expressed in the nervous system, is a TGF-β responsive gene that can be induced by SMAD-mediated Twist1 expression [12]. Of note, overexpression of SNCG is detected in numerous types of malignant tumors, including breast, ovary, liver, and cervical carcinoma [13-16]. Previous studies have demonstrated that SNCG is more frequently upregulated in late stage of cancers, and its expression correlates with cancer metastasis and poor prognosis [13, 14, 16-19]. Overexpression of SNCG in breast cancer cells promotes metastasis in a murine model of breast carcinoma [20], while SNCG knockdown suppresses perineural invasion and liver metastasis in mouse models of pancreatic carcinoma [21]. Mechanistically, SNCG may promote breast and ovarian cancer cells migration by activating Erk and phosphoinositide 3-kinase (PI3K)/Akt [22-24]. Moreover, secreted SNCG can enhance the motility of colorectal carcinoma cells by activating β1 integrin-focal adhesion kinase signaling [25].
While SNCG is induced by TGF-β in SMAD-dependent manner, it is unclear whether SNCG feeds back to regulate the non-SMAD pathways in TGF-β signaling, and how SNCG may mediate the pro-metastasis effect of TGF-β. In the current study, we investigate the effects of SNCG on TGF-β-induced activation of MAPK pathways. Herein, we report that SNCG promotes TGF-β-induced phosphorylation of p38MAPK by stabilizing MAPK kinase 3/6 (MKK3/6). The upregulation of p38MAPK by SNCG leads to increased MMP-9 expression, which promotes cancer cell invasion. Overexpression of SNCG in HCC cells promotes lung metastasis in mice. Treatment with p38MAPK inhibitor suppresses the promotion of HCC metastasis by SNCG.
Materials and Methods
Reagents
TGF-β was purchased from PeproTech (Rocky Hill, NJ, USA). The p38MAPK inhibitor SB203580 and MMP inhibitor BB-94 were from MedChemExpress (NJ, USA). The proteasome inhibitor MG132 was from Merk-Millipore (Darmstadt, Germany). Cycloheximide was from Beyotime Biotechnology (Shanghai, China). The antibodies used were as follows: anti-Twist1, anti-FLAG, anti-phosphorylated MKK3 (S189) and anti-phosphorylated MKK6 (S207) (Abcam, Cambridge, UK); anti-SNCG and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-Erk1/2, anti-phosphorylated Erk1/2 (T202/Y204) and anti-phosphorylated p38MAPK (T180/Y182) (Cell Signaling Technology, Beverly, MA, USA); anti-p38α and anti-vimentin (Proteintech, Rosemont, IL, USA); anti-MMP-9, anti-SMAD3, anti-MKK3/6, anti-fibronection, anti-N-cadherin (Abways Technology, Shanghai, China). The pcDNA3-Twist1 and PCI-SNCG plasmids were prepared as described previously [12, 20].
Preparation of FLAG-tagged SNCG plasmids
The C-terminal truncated SNCG expression plasmid (PCI-SNCG∆106-127) was constructed by mutagenesis of Gln 106 site (CAA) into the stop codon (UAA) using Mut Express II Fast Mutagenesis Kit V2 Kit (Vazyme, Nanjing, China). The primers for site-directed mutagenesis were 5′-TCTGCCCCCtAACAGGAGGGTGAGGCATCCAA-3′ (forward) and 5′-TCCTGTTaGGGGGCAGATGGCCTCAAGTCCTC-3′ (reverse). Both the full-length SNCG and truncated SNCG constructs were then cloned into pcDNA3.1-3xFlag plasmid ClonExpress II using One Step Cloning Kit (Vazyme, Nanjing, China), generating expression plasmids for FLAG-tagged SNCG (SNCG-FLAG) and truncated SNCG (SNCG∆106-127-FLAG). The forward PCR primer for constructing SNCG-FLAG and SNCG∆106-127-FLAG were 5′-AACGGGCCCTCTAGACTCGAGATGGATGTCTTCAAGAAGGGCTT-3′. The reverse primers for SNCG-FLAG and SNCG∆106-127-FLAG were 5′-CTTGGTACCGAGCTCGGATCCAAGTCTCCCCCACTCTGGGC-3′ and 5′-AACGGGCCCTCTAGACTCGAGATGGATGTCTTCAAGAAGGGCTT-3′, respectively.
Cell culture
Liver cancer cell line HepG2 and cervical cancer cell line HeLa were obtained from Cell Lines Bank, Chinese Academy of Science (Shanghai, China). The cells were cultured in DMEM supplemented with 10% new born calf serum (Thermo Fisher Scientific, Waltham, MA, USA) as described [26].
RNA interference
All siRNAs were custom-synthesized products of Ribobio Co. Ltd. (Guangzhou, China). The siR-Ribo negative control (siControl) was used for all siRNA experiments. The target sequences for SNCG and p38α knockdown are listed in Table S1. siRNA transfection was conducted as described [12].
Western blot analysis and immunoprecipitation
Protein extracts were prepared by lysis of cells in ice-cold radioimmunoprecipitation assay (RIPA) buffer. About 30 µg of total proteins were resolved by SDS-PAGE, followed by western blotting as described [12]. For immunoprecipitation, protein extracts were prepared by lysis buffer for immunoprecipitation containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 2 mM sodium phosphate, 25 mM β-glycerophosphate, 1 mM EDTA and proteinases inhibitors (Beyotime). One miligram of total proteins were incubated with primary antibodies or normal immunoglobulin G at 4°C overnight, and then incubated with 30 μl protein G agarose beads for 2 h at 4°C. The agarose-bound proteins were detected by western blotting.
Quantitative reverse transcription (RT)-PCR analysis
The forward and reverse primers used in this study are shown in Table S2. Quantitative RT-PCRs were performed as described [26]. The relative levels of target genes expression were normalized with the endogenous reference gene GAPDH.
Cell migration and invasion assay
Cell migration and invasion were determined by wound-healing and transwell assays as described previously [6, 12].
Mouse model of lung metastasis
All animal care and experiments were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of West China Hospital of Sichuan University (approvement number: 20211268A). All animal experimentation strictly adhered to the ARRIVE guidelines and the protocol approved by IACUC of West China Hospital. Five-week-old athymic nude male mice provided by Vital River Laboratories (Beijing, China) were used in this study. To generate lung metastasis models, the empty vector- or SNCG expression plasmid-transfected HepG2 cells (2 × 106/mL; 0.15 mL per mice) were injected into the tail vein. Three days later, both the control and SNCG-overexpression groups were randomly divided into groups (6-7 mice/group) that were treated with intraperitoneal injection of vehicle or SB203580 (1 mg/kg). The body weight was measured every 3 days. After 6 weeks, the mice were sacrificed, and all lungs were harvested. The lung tissues were fixed with 4% paraformaldehyde, embedded in paraffin and cut into 5 μm sections. The lung sections were then subjected to hematoxylin-eosin staining. The metastatic foci in lung sections were checked under a microscopy. The area of metastatic foci was measured by ImageJ.
Immunohistochemical staining
The lung tissues from murine model of metastasis were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into 5 µm sections. The expression of SNCG, MKK3/6 and phosphorylated p38MAPK were detected by immunohistochemical staining as described previously [10]. The antibodies for SNCG (1 mg/ml), MKK3/6 (1.3 mg/ml) and phosphorylated p38MAPK (33 μg/ml) were used at 1:50, 1:200 and 1:800 dilutions, respectively. Images of the stained sections were captured under a microscope at a magnification of 400×.
Statistical analysis
One-way analysis of variance with post hoc tests or Student's t-test (two-tailed) was used in statistical analysis. Each group size in all analysis is at least n = 3 (exact size is indicated in the figure legends). Data are presented as the mean ± standard deviation (SD) for in vitro studies or mean ± SEM for animal study. Differences are considered statistically significant if p < 0.05.
Results
SNCG knockdown reduces TGF-β-induced phosphorylation of p38 MAPK
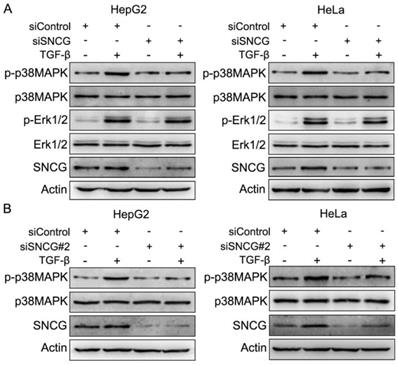
TGF-β may initiate SMAD and non-SMAD pathways including MAPK pathways. To determine whether SNCG regulates MAPK pathways in TGF-β signaling, we detected the effects of SNCG knockdown on TGF-β-induced p38MAPK, JNK and Erk1/2 phosphorylation. While TGF-β induced SNCG expression and p38MAPK phosphorylation, SNCG knockdown by two different sets of siRNA consistently abrogated the induction of p38MAPK phosphorylation by TGF-β in HepG2 and HeLa cells (Fig. 1). However, SNCG knockdown did not suppress the induction of Erk1/2 phosphorylation by TGF-β in both HepG2 and HeLa cells (Fig. 1). In addition, SNCG knockdown had no effect on the induction of JNK phosphorylation by TGF-β (Fig. S1). Together, these data demonstrate that SNCG is not only a TGF-β-responsive protein, but also a positive regulator of TGF-β-induced p38MAPK phosphorylation.
SNCG knockdown suppresses TGF-β-induced p38MAPK phopshorylation. (A) HepG2 and HeLa cells were transfected with siControl or siSNCG. Twenty-four hours later, the cells were treated with 5 ng/ml TGF-β for another 48h, followed by western blot analysis of indicated proteins. (B) HepG2 and HeLa cells were transfected with siControl or siSNCG#2. Twenty-four hours later, the cells were treated with 5 ng/ml TGF-β for another 48h, followed by western blot analysis of indicated proteins.
SNCG stabilizes MKK3/6 to promote p38MAPK phosphorylation
While TGF-β-induced activation of TAK1-TRAF6-TAB1 is responsible for p38MAPK, Erk and JNK pnosphorylation, MKK3/6, MKK1 and MKK4 mediate the induction of p38MAPK, Erk and JNK phosphorylation by TGF-β-TAK1 signaling, respectively. Since SNCG knockdown predominantly suppresses TGF-β-induced p38MAPK phosphorylation, we then detected whether SNCG could regulate MKK3/6. Indeed, SNCG knockdown led to a decrease in the levels of total MKK3/6 and abrogated the induction of MKK3/6 phosphorylation by TGF-β in both HepG2 and HeLa cells (Fig. 2A). However, SNCG knockdown did not affect the levels of MKK3/6 transcripts, suggesting that SNCG may affect MKK3/6 at protein level (Fig. 2B).
SNCG stabilizes MKK3/6 and promotes TGF-β-induced p38MAPK phopshorylation. (A) HepG2 and HeLa cells were transfected with siControl or siSNCG. Twenty-four hours later, the cells were treated with 5 ng/ml TGF-β for another 48h, followed by western blot analysis of indicated proteins. (B) HepG2 cells were transfected with 50 nM of siControl or siSNCG for 24h, followed by qRT-PCR analysis of SNCG, MKK3 and MKK6 transcription. The levels of transcripts in siControl-transfected cells were set as 1. Values represent mean ± SD. (n = 3). ***, p <0.001, compared with siControl-transfected cells. (C) HepG2 lysates were subjected to immunoprecipitation with anti-SNCG, anti-MKK3/6 or normal IgG, followed by western blot analysis of MEK3/6 and SNCG. (D) HepG2 cells were transfected with siControl or siSNCG. 24h later, the cells were treated with protein translation inhibitor cycloheximide (CHX, 100 μg/mL) for indicated periods, followed by western blot analysis of MKK3/6 and SNCG. The relative levels of MKK3/6 were plotted. For both siControl- and siSNCG-transfected cells, the levels of MKK3/6 at 0 h were set as 100%. (E) HepG2 cells were transfected with the empty vector or SNCG expression plasmid, and treated with CHX for indicated periods, followed by western blot analysis of MKK3/MKK6 and SNCG. The relative levels of MKK3/6 were plotted. For both vector- and SNCG-transfected cells, the levels of MKK3/6 at 0 h were set as 100%.
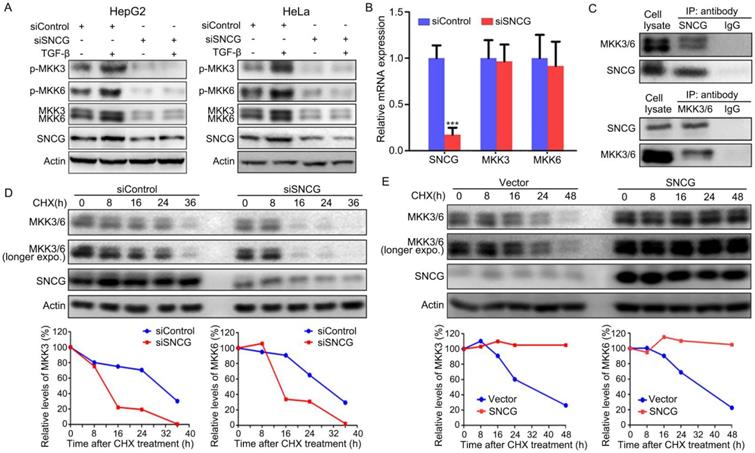
Next, we detected whether SNCG physically interacted with MKK3/6. To this end, HepG2 cell lysates were subjected to immunoprecipitation with anti-SNCG antibody, followed by western blot analysis of SNCG and MKK3/6. Immunoprecipitation of SNCG co-precipitated both MKK3 and MKK6, and vice versa (Fig. 2C). Meanwhile, SNCG did not interact with TAK1, TRAF6 and p38MAPK (Fig. S2). To determine whether SNCG affects the stability of MKK3/6 proteins, HepG2 cells were transfected with control siRNA or SNCG siRNA, followed by treatment with cycloheximide to inhibit protein synthesis and chasing the levels of MKK3/6 for different periods. After treating with cycloheximide, the levels of MKK3/6 proteins dropped more rapidly in SNCG-knockdown cells than that in non-knockdown cells (Fig. 2D). In contrast, overexpression of SNCG stabilized MKK3/6 proteins (Fig. 2E). Moreover, heat shock resulted in an increase in SNCG expression and modest decrease in the levels of MKK3 in HepG2 cells, while it did not affect MKK6 (Fig. S3). SNCG knockdown slightly accelerated the decrease in MKK3 levels following heat shock (Fig. S3).
SNCG regulates MKK3/6 ubiquitination and proteasomal degradation
To determine whether SNCG regulates proteasomal degradation of MKK3/6, HepG2 and HeLa cells were transfected with or without siSNCG, and treated with or without the proteasome inhibitor MG132, followed by western blot analysis of MKK3/6 levels. Treatment with MG132 abrogated the down-regulation of MKK3/6 expression by SNCG knockdown in both HepG2 and HeLa cells (Fig. 3A). Knockdown of SNCG by another set of siRNA consistently inhibited MKK3/6 expression, which was also reversed by MG132 (Fig. S4). Collectively, these data suggest that SNCG may inhibit proteasomal degradation of MKK3/6.
SNCG silencing promotes MKK3/6 ubiquitination and proteasomal degradation. (A) HepG2 and HeLa cells were transfected with siControl or siSNCG. 24h later, the cells were treated with or without proteasome inhibitor MG132 (5 μM) for another 48 h, followed by western blot analysis of MKK3/6 and SNCG. (B) HepG2 cells were transfected with siControl or siSNCG. Cell lysates were subject to immunoprecipitation with the MKK3/6 antibody, followed by western blot analysis of MKK3/6, SNCG and ubiuqitinated MKK3/6 (poly-Ub). HC, heavy chain. The indicated proteins in cell lysate were also shown (input). (C) The FLAG-tagged full-length SNCG (SNCG-FLAG) and C-terminal-truncated SNCG (SNCG△106-127-FLAG) were overexpressed in HepG2 cells, and validated by western blot analysis. The MKK3/6 immunoprecipitates were subject to western blot analysis of MKK3/6 and FLAG-tagged SNCG. LC, light chain. (D) HepG2 cells were transfected with empty vector, SNCG-FLAG and SNCG△106-127-FLAG, followed by treatment with CHX for indicated periods. The levels of MKK3/6 and FLAG-tagged SNCG were detected by western blotting.
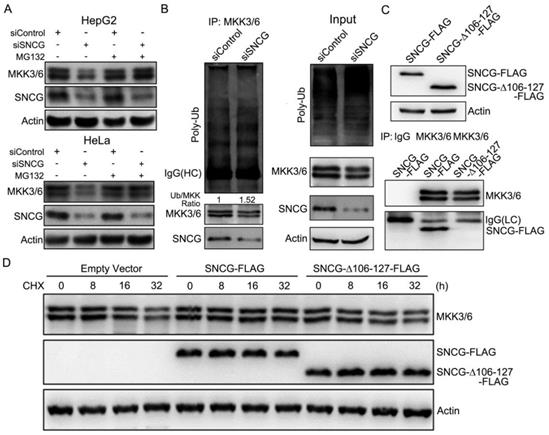
Furthermore, immunoprecipitation of MKK3/6 protein demonstrated that the relative levels of MKK3/6 ubiquitination were increased by SNCG knockdown (Fig. 3B). SNCG usually interacts with its targets via the C-terminal region of SNCG [30]. We then prepared a FLAG-tagged construct for the expression of C-terminal-truncated SNCG (SNCG△106-127-FLAG) (Fig. 3C). Immunoprecipitation of MKK3/6 co-precipitated FLAG-tagged SNCG but not SNCG△106-127, indicating that the C-terminal region of SNCG is required for interaction with MKK3/6 (Fig. 3C). While overexpression of the full-length SNCG stabilized MKK3/6, overexpression of the C-terminal-truncated SNCG did not affect MKK3/6 stability (Fig. 3D), indicating that the C-terminal region of SNCG is critical for stabilizing MKK3/6.
SNCG promotes cell migration and invasion through p38MAPK
To determine whether p38MAPK contributes to TGF-β-induced cell migration, HepG2 cells were treated with or without TGF-β and the p38MAPK inhibitor SB203580, followed by wound healing assays. Treatment with TGF-β promoted HepG2 cells migration (Fig. S5). SB203580 not only inhibited HepG2 cells migration, but also abrogated the promotion of cell migration by TGF-β (Fig. S5). In addition, HepG2 cells were transfected with the empty vector or SNCG-expressing plasmid, and treated with or without SB203580, followed by wound-healing assays. Overexpression of SNCG promoted HepG2 cells migration (Fig. 4A). SB203580 abrogated the promotion of cell migration by SNCG (Fig. 4A). Moreover, p38α knockdown abrogated the promotion of cell migration by SNCG in both HepG2 and HeLa cells (Figs. 4B and C).
SNCG promotes cell migration and invasion through p38MAPK. (A) HepG2 cells were transfected with 1 μg/ml of the empty vector or SNCG expression plasmid, and then treated with or without 50 μM of SB203580 for another 24h, followed by wound healing assays. The relative wound closure rate was plotted. Values represent mean ± SD (n = 3). *, p < 0.05. **, p <0.01. ***, p <0.001. In parallel, the efficiency of SNCG overexpresion was detected by western blot analysis. (B-C) HepG2 or HeLa cells were transfected with the empty vector or SNCG plasmid, and then transfected with siControl or siP38α, followed by wound healing assays. Values represent mean ± SD (n = 3). *, p < 0.05. **, p <0.01. ***, p <0.001. In parallel, the efficiency of SNCG overexpresion and p38α knockdown was detected by western blot analysis. (D-E) HepG2 or HeLa cells were transfected with the empty vector or SNCG plasmid, and then transfected with siControl or siP38α, followed by cell invasion assays. The relative cell invasion rate was plotted. Values represent mean ± SD (n = 3). The invasion rate in vehicle-treated and vector-transfected cells was set as 1. *, p < 0.05. **, p <0.01. ***, p <0.001, compared with vehicle-treated and vector-transfected cells. In parallel, the efficiency of SNCG overexpresion and p38α knockdown was detected by western blot analysis.
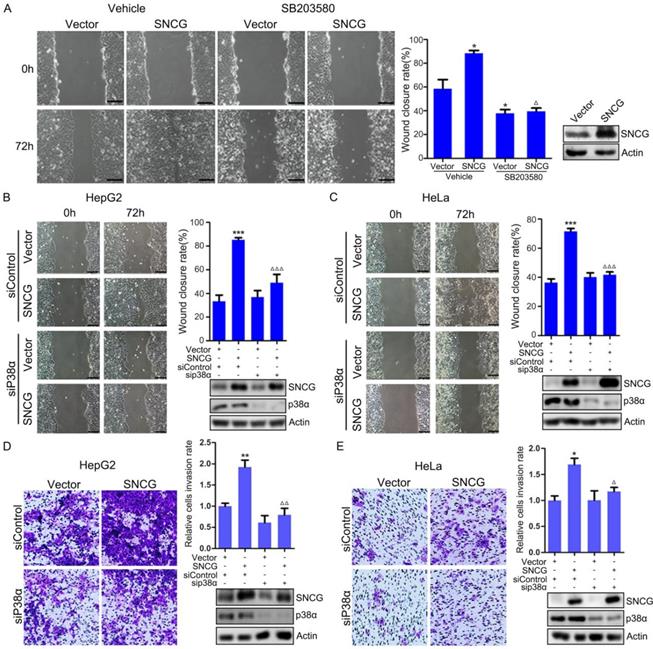
Next, we determined whether SNCG could promote cell invasion through p38MAPK. To this end, HepG2 cells were transfected with or without SNCG expression plasmid and p38α siRNA, followed by detecting cell invasion in matrigeal-coated transwell chambers. Overexpression of SNCG promoted HepG2 cells invasion (Fig. 4D). p38α knockdown abrogated the promotion of HepG2 cells invasion by SNCG (Fig. 4D). Similar effects were detected in HeLa cells (Fig. 4E). Collectively, these data indicate that p38MAPK mediates the promotion of cancer cells migration and invasion by SNCG.
p38MAPK mediates the promotion of MMP-9 expression by SNCG
Previous studies indicate that p38MAPK may promote cancer cell invasion through up-regulating MMP-9 [27]. We then detected whether SNCG up-regulated MMP-9 through p38MAPK. Indeed, overexpression of SNCG in HepG2 and HeLa cells induced p38MAPK phosphorylation and MMP-9 expression (Fig. 5A). Treatment with p38MAPK inhibitor abrogated the induction of MMP-9 by SNCG (Fig. 5A). Consistently, p38α knockdown abrogated the induction of MMP-9 by SNCG in both HepG2 and HeLa cells (Fig. 5B). In addition, SNCG knockdown suppressed the induction of MMP-9 by TGF-β in both HepG2 and HeLa cells, while the closely related MMP-2 was not affected by SNCG silencing (Fig. 5C). Knockdown of SNCG by another set of siRNA consistently abrogated the induction of MMP-9 by TGF-β (Fig. S6). Since Twist1 mediates the induction of SNCG expression by TGF-β, we also detected the role of SNCG in the induction of MMP-9 expression by Twist1. Overexpression of Twist1 in HepG2 and HeLa cells up-regulated both SNCG and MMP-9 expression. SNCG knockdown abrogated the induction of MMP-9 by Twist1 (Fig. 5D).
SNCG promotes MMP-9 expression through p38MAPK. (A) HepG2 and HeLa cells were transfected with 1 μg/ml of the empty vector or SNCG expression plasmid, and then treated with or without 50 μM of SB203580 for another 48 h, followed by western blot analysis of indicated proteins. (B) HepG2 and HeLa cells were transfected with 1 μg/ml of the empty vector or SNCG expression plasmid, and then transfected with siControl or siP38α, followed by western blot analysis of indicated proteins. (C) HepG2 and HeLa cells were transfected with 50 nM of siControl or siSNCG. 24h later, the cells were treated with or without 5 ng/ml of TGF-β for another 48h, followed by western blot analysis of indicated proteins. (D) HepG2 and HeLa cells were transfected with 1 μg/ml of the empty vector or Twist1 expression plasmid, and then transfected with siControl or siSNCG, followed by western blot analysis of indicated proteins.

Since EMT is involved in TGF-β-induced cell migration and invasion, we then evaluated whether SNCG might promote EMT. While TGF-β induced the expression of EMT markers including fibronectin, N-cadherin and vimentin, SNCG knockdown had no effects on TGF-β-induced expression of these key EMT markers, suggesting that SNCG was not involved in TGF-β-induced EMT (Fig. S7).
Inhibition of p38MAPK or MMP-9 abrogates the promotion of cancer cell invasion by SNCG
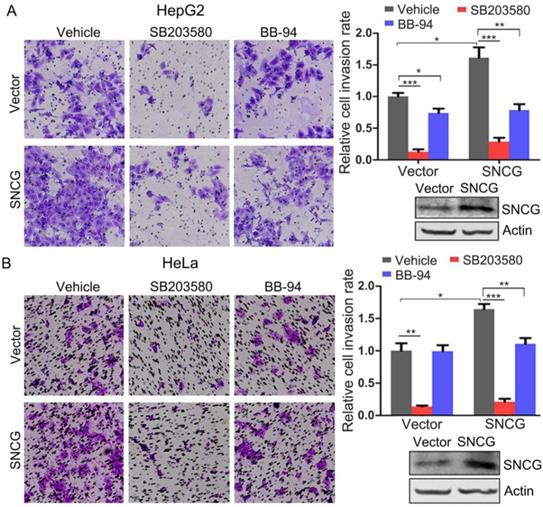
The above-described data demonstrate that p38MAPK and MMP-9 mediate the promotion of cancer cells invasion by SNCG, suggesting that pharmacological inhibition of p38MAPK or MMP-9 may be potential approach for antagonizing the promotion of cancer cell invasion by SNCG. Indeed, treatment with a well-validated p38MAPK inhibitor SB203580 not only inhibited HepG2 and HeLa cells invasion, but also suppressed the promotion of cells invasion by SNCG. Similarly, treatment of HepG2 cells with the MMP inhibitor BB-94 also inhibited cells invasion, albeit to a lesser extent. BB-94 abrogated the promotion of HepG2 and HeLa cells invasion by SNCG (Fig. 6). Together, these data suggest that p38MAPK inhibitor may be promising agent to counteract SNCG-stimulated cancer cells invasion.
Inhibition of p38MAPK or MMP abrogates the promotion of cell invasion by SNCG. (A) HepG2 cells were transfected with 1 μg/ml of the empty vector or SNCG expression plasmid, and then treated with or without 50 μM of SB203580 and 10 μM of BB-94 for another 24h, followed by cell invasion assays. The relative cell invasion rate was plotted. The invasion rate in vehicle-treated and vector-transfected cells was set as 1. (B) HeLa cells were transfected with the empty vector or SNCG expression plasmid, and then treated with or without 50 μM of SB203580 and 10 μM of BB-94 for another 24h, followed by cell invasion assays. The relative cell invasion rate was plotted. Values represent mean ± SD (n = 3). *, p < 0.05;**, p <0.01; ***, p <0.001. In parallel, the efficiency of SNCG overexpresion was detected by western blotting.
p38MAPK inhibition abrogates the promotion of HCC metastasis by SNCG
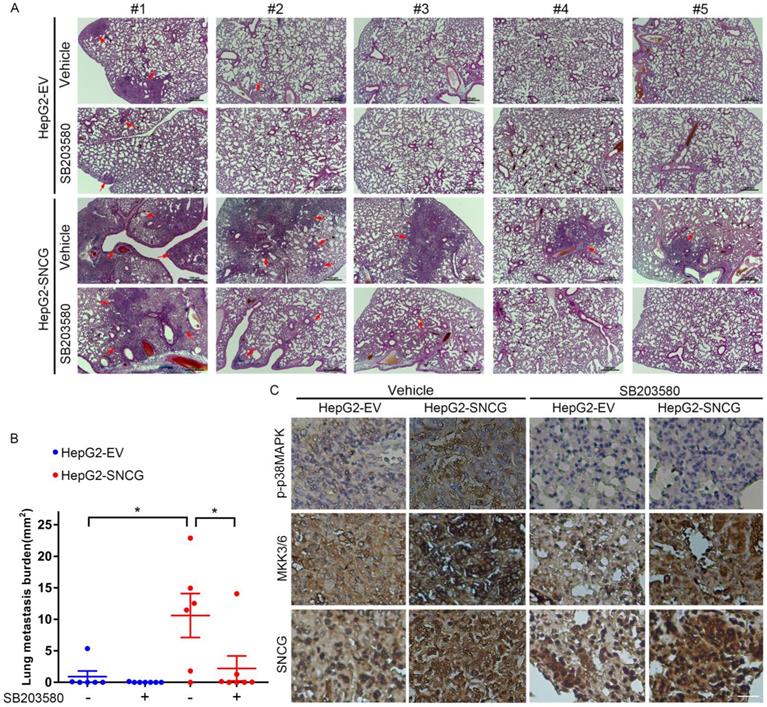
To determine the effect of SNCG on HCC metastasis and the metastasis-suppressing effect of p38MAPK inhibitor, we proceeded to test the in vivo effect of SNCG overexpression and SB203580 in an experimental metastasis model. Forty days after tail vein injection of HepG2 cells stably transfected with empty vector (HepG2-EV) or SNCG expression plasmid (HepG2-SNCG) into nude mice, the lung metastasis of HepG2 cells was evaluated. Although there were no obvious nodules in the lung surface of all mice, microscopic analysis of hematoxylin-eosin-stained lung sections demonstrated that there were diffuse- or micro-metastasis in two of six mice injected with HepG2-EV cells, one of seven mice injected with HepG2-EV cells and treated with SB203580, and all mice injected with HepG2-SNCG cells (Figs. 7A and B). Overexpression of SNCG induced larger or more metastatic lesions in the lungs (Figs. 7A and B). Treatment with SB203580 suppressed the promotion of pulmonary metastasis by SNCG (Figs. 7A and B). Immunohistochemical analysis demonstrated that MKK3/6 expression and p38MAPK phosphorylation were increased in HepG2-SNCG micro-metastases compared with HepG2-EV (Fig. 3C). SB203580 inhibited p38MAPK phosphorylation in both HepG2-EV and HepG2-SNCG micro-metastases (Fig. 7C). Together, these data demonstrate that SNCG can promote the metastasis of HCC, and pharmacological inhibition of p38MAPK is an approach to suppressing the pro-metastasis effect of SNCG.
Pharmacological inhibition of p38MAPK abrogates the promotion of lung metastasis by SNCG. (A) HepG2 cells stably transfected with empty vector (EV) or SNCG expression plasmid were injected into the mice tail veins, and then treated with or without SB203580 daily. The mice were sacrificed 42 days after injection. The sections from paraffin-embedded lung tissues were subjected to HE staining. The red arrows-indicated metastatic foci in representative lung sections were shown. Bars, 100 μm. (B) The area of metastic foci in lung sections was measured. The lung metastasis burden in each group was plotted. Bars, SEM. *, p < 0.05. (C) The phosphorylation of p38MAPK and expression of MKK3/6 and SNCG in lung micrometastases were detected by immunohistochemistry. Bar, 25 μm.
Discussion
SNCG is an oncogene that can be induced by both IGF and TGF-β, which are critically involved in tumor progression [12, 28, 29]. It is well-defined that TGF-β is a contextual tumor suppressor or promoter, depending on the tumor types and stages. Whereas TGF-β may inhibit tumor cell growth at the early stage, it usually promotes cancer metastasis at late stages. As a TGF-β- and IGF-responsive gene, SNCG is more frequently overexpressed in advanced cancers and involved in the stabilization or activation of some oncogenes such as IGF-IR and Akt, which have critical roles in tumorigenesis and drug resistance [2, 28, 30, 31]. Preclinical studies have demonstrated that SNCG can promote cancer metastasis [14, 20, 21, 24]. Hence, SNCG may be one of the mediators of TGF-β- or IGF-induced cancer metastasis.
TGF-β signaling is mediated by multiple pathways, including SMAD and non-SMAD pathways. While SMAD proteins directly mediate the genomic response to TGF-β, the non-canonical TGF-β signaling is mediated by SMAD-independent protein kinases such as MAPKs and JNK [32]. Previous studies have demonstrated that there is cross-talk between the SMAD and non-SMAD pathways. For example, TGF-β-induced activation of MAPKs and JNK may phosphorylate SMADs to promote the transcription of oncogenes [32]. On the other hand, SMADs can mediate TGF-β-induced activation of MAPKs. Expression of dominant-negative SMAD3 and SMAD4 attenuates TGF-β-induced activation of p38MAPK and ERK1/2, respectively [33]. While SNCG is up-regulated by TGF-β-SMAD-Twist1 axis [12], the current study demonstrates that it also stimulates p38MAPK signaling, a non-SMAD pathway in TGF-β signaling. Thus, SNCG may be a novel link between the SMAD and non-SMAD pathways in TGF-β signaling.
p38MAPK activation is directly catalysed by upstream MAPK kinases including MKK3 and MKK6. TGF-β-activated protein kinase 1 (TAK1) and TAK1-binding protein 1 work together to mediate TGF-β-induced MKK3/6-p38MAPK signaling [34]. Our current study demonstrates that SNCG can prevent proteasomal degradation of MKK3/6 proteins. Whilst little is known about the regulation of MKK3 stability, previous study has demonstrated that the stability of MKK6 protein is regulated by FBXO31-mediated ubiquitination at Lys82 residue [35]. Lys48-linked ubiquitination of MKK6 leads to proteasomal degradation [35]. It remains to know whether other ubiquitin ligases may be involved in regulating MKK3/6 stability. SNCG appears to be a novel regulator of MKK3/6 stability, while it warrants further study to decipher the mechanism.
Previous studies have demonstrated that p38MAPK promotes the migration and invasion of variety types of human tumor cells [36, 37]. Mechanistically, p38MAPK may activate MAPK-activated protein kinase 2, which in turn phosphorylates HSP27 and then promotes cytoskeletal remodelling, leading to increased cell motility and invasiveness [38, 39]. In addition, p38MAPK may promote cancer metastasis by activating NF-kB and stabilizing FOXC1 [40, 41]. Moreover, MMPs have important roles in cell invasion and cancer metastasis [42]. TGF-β also upregulates MMPs to promote cell invasion through extracellular matrix remodeling. One mechanism underlying the up-regulation of MMP-9 by TGF-β is dependent on p38MAPK [43]. The up-regulation of MMP-9 by p38MAPK may involve transcription factors including AP1 and Sp1 [44, 45]. While TGF-β induces both MMP-2 and MMP-9 expression, SNCG knockdown only abrogates the induction of MMP-9 but not MMP-2. The promotion of MMP-9 expression by SNCG is dependent on p38MAPK. P38MAPK-mediated induction of MMP-9 expression may contribute, in part, to the promotion of cancer cell invasion by SNCG.
The prevention and treatment of cancer metastasis remain to be a great challenge. Pharmacological inhibition of p38MAPK may suppress the metastasis of breast cancer and melanoma [46, 47]. The oral p38MAPK inhibitor ralimetinib (LY2228820) has undergone clinical trials and demonstrated acceptable tolerability [48]. A phase 1b/2 clinical trial indicates that the addition of ralimetinib to gemcitabine-carboplatin regimen may moderately improve the progression-free survival in patients with recurrent platinum-sensitive ovarian carcinoma [49]. Except for ralimetinib, there are other p38MAPK inhibitors under development, such as BIRB796 and its derivatives [50]. Given that inhibition of p38MAPK may suppress the metastasis of SNCG-overexpressing cancer cells, targeting p38MAPK may be an approach for treating SNCG-positive cancers.
In summary, our findings indicate that SNCG is a regulator of the MKK3/6-p38MAPK pathway in TGF-β signaling. SNCG interacts with MKK3/6 and enhances their stability, thereby promoting TGF-β-induced p38MAPK activation, cell migration and invasion. Our finding that p38MAPK deficiency restricts SNCG-induced cell migration and invasion provides insights into mechanisms underlying the promotion of tumor progression by SNCG. Targeting p38MAPK may be considered for treating SNCG-overexpressed cancer.
Abbreviations
EMT: epithelial-mesenchymal transition; Erk: extracellular signal-activated kinase; JNK: Jun N-terminal kinase; MAPK: mitogen-activated protein kinase; MKK3/6: MAPK kinase 3/6; MMP: matrix metalloproteinase; PI3K: phosphoinositide 3-kinase; SMAD: mothers against decapentaplegic homolog; SNCG: gamma synuclein; TAK1: TGF-β-activated protein kinase 1; TGF-β: transforming growth factor-β.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81872388).
Author Contributions
TS, JL and YJ designed the experiments. JL, TS, JZ, QL, HH, HZ, JW, and TL performed experiments. JL, YS and YJ interpreted the data, wrote and revised the manuscript. YJ received funding and supervised this study. All authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guo S, Deng CX. Effect of stromal cells in tumor microenvironment on metastasis initiation. Int J Biol Sci. 2018;14:2083-93
2. Hua H, Kong Q, Yin J, Zhang J, Jiang Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy. J Hematol Oncol. 2020;13:64
3. Chen Q, Lei JH, Bao J, Wang H, Hao W, Li L. et al. BRCA1 deficiency impairs mitophagy and promotes inflammasome activation and mammary tumor metastasis. Adv Sci (Weinh). 2020;7:1903616
4. Li X, Wang J. Mechanical tumor microenvironment and transduction: cytoskeleton mediates cancer cell invasion and metastasis. Int J Biol Sci. 2020;16:2014-28
5. Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15:34
6. Wang X, Wang C, Guan J, Chen B, Xu L, Chen C. Progress of breast cancer basic research in China. Int J Biol Sci. 2021;17:2069-79
7. Yang S, Zhang H, Yang H, Zhang J, Wang J, Luo T. et al. SEPHS1 promotes SMAD2/3/4 expression and hepatocellular carcinoma cells invasion. Exp Hematol Oncol. 2021;10:17
8. Wang X, Thiery JP. Harnessing carcinoma cell plasticity mediated by TGF-β signaling. Cancers (Basel). 2021;13:3397
9. Aashaq S, Batool A, Mir SA, Beigh MA, Andrabi KI, Shah ZA. TGF-β signaling: A recap of SMAD-independent and SMAD-dependent pathways. J Cell Physiol. 2022;237:59-85
10. Yang S, Yang S, Zhang H, Hua H, Kong Q, Wang J. et al. Targeting Na+ /K+ -ATPase by berbamine and ouabain synergizes with sorafenib to inhibit hepatocellular carcinoma. Br J Pharmacol. 2021;178:4389-407
11. Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 pathway: From biology to cancer therapy. Int J Mol Sci. 2020;21:1913
12. Shao T, Song P, Hua H, Zhang H, Sun X, Kong Q. et al. Gamma synuclein is a novel Twist1 target that promotes TGF-beta-induced cancer cell migration and invasion. Cell Death Dis. 2018;9:625
13. Bruening W, Giasson BI, Klein-Szanto AJ, Lee VM, Trojanowski JQ, Godwin AK. Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer. 2000;88:2154-63
14. Hu H, Sun L, Guo C, Liu Q, Zhou Z, Peng L. et al. Tumor cell-microenvironment interaction models coupled with clinical validation reveal CCL2 and SNCG as two predictors of colorectal cancer hepatic metastasis. Clin Cancer Res. 2009;15:5485-93
15. Hua H, Xu L, Wang J, Jing J, Luo T, Jiang Y. Up-regulation of gamma-synuclein contributes to cancer cell survival under endoplasmic reticulum stress. J Pathol. 2009;217:507-15
16. Liu H, Liu W, Wu Y, Zhou Y, Xue R, Luo C. et al. Loss of epigenetic control of synuclein-gamma gene as a molecular indicator of metastasis in a wide range of human cancers. Cancer Res. 2005;65:7635-43
17. Wu K, Quan Z, Weng Z, Li F, Zhang Y, Yao X. et al. Expression of neuronal protein synuclein gamma gene as a novel marker for breast cancer prognosis. Breast Cancer Res Treat. 2007;101:259-67
18. Winder AD, Maniar KP, Wei JJ, Liu D, Scholtens DM, Lurain JR. et al. Synuclein-γ in uterine serous carcinoma impacts survival: An NRG Oncology/Gynecologic Oncology Group study. Cancer. 2017;123:1144-55
19. Takemura Y, Ojima H, Oshima G, Shinoda M, Hasegawa Y, Kitago M. et al. Gamma-synuclein is a novel prognostic marker that promotes tumor cell migration in biliary tract carcinoma. Cancer Med. 2021;10:5599-613
20. Jia T, Liu YE, Liu J, Shi YE. Stimulation of breast cancer invasion and metastasis by synuclein gamma. Cancer Res. 1999;59:742-7
21. Hibi T, Mori T, Fukuma M, Yamazaki K, Hashiguchi A, Yamada T. et al. Synuclein-gamma is closely involved in perineural invasion and distant metastasis in mouse models and is a novel prognostic factor in pancreatic cancer. Clin Cancer Res. 2009;15:2864-71
22. Zhuang Q, Liu C, Qu L, Shou C. Synuclein-γ promotes migration of MCF7 breast cancer cells by activating extracellular-signal regulated kinase pathway and breaking cell-cell junctions. Mol Med Rep. 2015;12:3795-800
23. Pan ZZ, Bruening W, Godwin AK. Involvement of RHO GTPases and ERK in synuclein-gamma enhanced cancer cell motility. Int J Oncol. 2006;29:1201-5
24. Zhang J, Liu XH, Li C, Wu XX, Chen YL, Li WW. et al. SNCG promotes the progression and metastasis of high-grade serous ovarian cancer via targeting the PI3K/AKT signaling pathway. J Exp Clin Cancer Res. 2020;39:79
25. Liu C, Qu L, Zhao C, Shou C. Extracellular gamma-synuclein promotes tumor cell motility by activating β1 integrin-focal adhesion kinase signaling pathway and increasing matrix metalloproteinase-24, -2 protein secretion. J Exp. Clin Cancer Res. 2018;37:117
26. Zhang H, Yang S, Wang J, Jiang Y. Blockade of AMPK-mediated cAMP-PKA-CREB/ATF1 signaling synergizes with aspirin to inhibit hepatocellular carcinoma. Cancers (Basel). 2021;13:1738
27. Radziwon-Balicka A, Santos-Martinez MJ, Corbalan JJ, O'Sullivan S, Treumann A, Gilmer JF. et al. Mechanisms of platelet-stimulated colon cancer invasion: role of clusterin and thrombospondin 1 in regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis. 2014;35:324-32
28. Li M, Yin Y, Hua H, Sun X, Luo T, Wang J. et al. The reciprocal regulation of gamma-synuclein and IGF-I receptor expression creates a circuit that modulates IGF-I signaling. J Biol Chem. 2010;285:30480-8
29. Leivonen SK, Kähäri VM. Transforming growth factor-beta signaling in cancer invasion and metastasis. Int J Cancer. 2007;121:2119-24 (2007)
30. Liang W, Miao S, Zhang B, He S, Shou C, Manivel P. et al. Synuclein gamma protects Akt and mTOR and renders tumor resistance to Hsp90 disruption. Oncogene. 2015;34:2398-405
31. Hua H, Zhang H, Chen J, Wang J, Liu J, Jiang Y. Targeting Akt in cancer for precision therapy. J Hematol Oncol. 2021;14:128
32. Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413-20
33. Simeone DM, Zhang L, Graziano K, Nicke B, Pham T, Schaefer C. et al. SMAD4 mediates activation of mitogen-activated protein kinases by TGF-beta in pancreatic acinar cells. Am J Physiol Cell Physiol. 2001;281:C311-319
34. Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol. 2007;292:F1471-8
35. Liu J, Han L, Li B, Yang J, Huen MS, Pan X. et al. F-box only protein 31 (FBXO31) negatively regulates p38 mitogen-activated protein kinase (MAPK) signaling by mediating lysine 48-linked ubiquitination and degradation of mitogen-activated protein kinase kinase 6 (MKK6). J Biol Chem. 2014;289:21508-18
36. Hsieh YH, Wu TT, Huang CY, Hsieh YS, Hwang JM, Liu JY. p38 mitogen-activated protein kinase pathway is involved in protein kinase Calpha-regulated invasion in human hepatocellular carcinoma cells. Cancer Res. 2007;67:4320-7
37. Junttila MR, Ala-Aho R, Jokilehto T, Peltonen J, Kallajoki M, Grenman R. et al. p38alpha and p38delta mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene. 2007;26:5267-79
38. Pavan S, Musiani D, Torchiaro E, Migliardi G, Gai M, Di Cunto F. et al. HSP27 is required for invasion and metastasis triggered by hepatocyte growth factor. Int J Cancer. 2014;134:1289-99
39. Cheung S, Jain P, So J, Park SI, Lee YS. p38 MAPK inhibition mitigates hypoxia-induced AR signaling in castration-resistant prostate cancer. Cancers (Basel). 2021;13:831
40. Wang Z, Ao X, Shen Z, Ao L, Wu X, Pu C. et al. TNF-α augments CXCL10/CXCR3 axis activity to induce Epithelial-Mesenchymal Transition in colon cancer cell. Int J Biol Sci. 2021;17:2683-702
41. Zhang Y, Liao Y, Chen C, Sun W, Sun X, Liu Y. et al. p38-regulated FOXC1 stability is required for colorectal cancer metastasis. J Pathol. 2020;250:217-30
42. Larsson P, Syed Khaja AS, Semenas J, Wang T, Sarwar M, Dizeyi N. et al. The functional interlink between AR and MMP9/VEGF signaling axis is mediated through PIP5K1α/pAKT in prostate cancer. Int J Cancer. 2020;146:1686-99
43. Takahashi Y, Maki T, Liang AC, Itoh K, Lok J, Osumi N. et al. p38 MAP kinase mediates transforming-growth factor-beta1-induced upregulation of matrix metalloproteinase-9 but not -2 in human brain pericytes. Brain Res. 2014;1593:1-8
44. Park SL, Won SY, Song JH, Kambe T, Nagao M, Kim WJ. et al. EPO gene expression promotes proliferation, migration and invasion via the p38MAPK/AP-1/MMP-9 pathway by p21WAF1 expression in vascular smooth muscle cells. Cell Signal. 2015;27:470-8
45. Xu K, Shu HK. EGFR activation results in enhanced cyclooxygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res. 2007;67:6121-9
46. Zonneville J, Colligan S, Grant S, Miller A, Wallace P, Abrams SI. et al. Blockade of p38 kinase impedes the mobilization of protumorigenic myeloid populations to impact breast cancer metastasis. Int J Cancer. 2020;147:2279-92
47. Tang YM, Cao QY, Guo XY, Dong SH, Duan JA, Wu QN. et al. Inhibition of p38 and ERK1/2 pathways by Sparstolonin B suppresses inflammation-induced melanoma metastasis. Biomed Pharmacother. 2018;98:382-9
48. Patnaik A, Haluska P, Tolcher AW, Erlichman C, Papadopoulos KP, Lensing JL. et al. A first-in-human phase I study of the oral p38 MAPK inhibitor, ralimetinib (LY2228820 dimesylate), in patients with advanced cancer. Clin Cancer Res. 2016;22:1095-102 (2016)
49. Vergote I, Heitz F, Buderath P, Powell M, Sehouli J, Lee CM. et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol Oncol. 2020;156:23-31
50. Haller V, Nahidino P, Forster M, Laufer SA. An updated patent review of p38 MAP kinase inhibitors (2014-2019). Expert Opin Ther Pat. 2020;30:453-66
Author contact
![]() Corresponding author: Yangfu Jiang, Laboratory of Oncogene, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University. Chengdu 610041, China. E-mail: jyangfuedu.cn
Corresponding author: Yangfu Jiang, Laboratory of Oncogene, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University. Chengdu 610041, China. E-mail: jyangfuedu.cn