Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The miR-21 PTEN AZIN2-sv/miR-214...
The MALATA1/miR-92a network
The MEG3/miR-125b/miR-26a network
The TUG1/miR-124 network
The KCNQ1OT1/miR-152 network
The miR-590/miR-199a network
The miR-155 network
The circHIPK3 network
Other lncRNA/miRNA networks
Other miRNA networks
Perspectives and challenges
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(8):3194-3208. doi:10.7150/ijbs.69671 This issue Cite
Review
The Networks of Noncoding RNAs and Their Direct Molecular Targets in Myocardial Infarction
Shiqi Wang, MMed1,2, Yang Wang, MMed1,2, Hongxin Cheng, BS1,2, Qing Zhang, MMed1,2, Chenying Fu, PhD3,4 ![]() , Chengqi He, MD1,2, Quan Wei, MD1,2
, Chengqi He, MD1,2, Quan Wei, MD1,2 ![]()
1. Rehabilitation Medicine Center and Institute of Rehabilitation Medicine, West China Hospital, Sichuan University, Chengdu, Sichuan, PR China.
2. Key Laboratory of Rehabilitation Medicine in Sichuan Province, Chengdu, Sichuan, PR China.
3. National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, Sichuan, PR China.
4. Aging and Geriatric Mechanism Laboratory, West China Hospital, Sichuan University, Chengdu, Sichuan, PR China.
Received 2021-12-2; Accepted 2022-4-14; Published 2022-5-1
Abstract

Noncoding RNAs are closely related to the development of myocardial infarction (MI), and their specific roles in MI are still being carefully studied. Researchers can select the literature they are interested in according to their own wishes in traditional reviews, which results in a certain amount of selection bias. A data-driven approach was used to organize this review to understand the ncRNAs in MI in the past five years. Here, we reveal important networks of interactions between noncoding RNAs and their direct targets. Our review gives an unbiased description of the role of noncoding RNAs in MI. Key information, such as carrier selection, treatment time window, treatment dose and possible side effects of ncRNA therapy, needs to be further determined. In short, the interactions between coding and noncoding genes play indispensable roles in the occurrence and development of MI and still deserve great attention from researchers in this field. The rational application of ncRNAs is expected to become a target for the treatment of MI.
Keywords: ncRNAs, miRNAs, lncRNAs, circRNAs, myocardial infarction, data-driven
Introduction
Noncoding RNAs (ncRNAs), which account for approximately 99% of the human genome, can be transcribed but do not encode proteins and have been proven to be related to a variety of human diseases [1]. Myocardial infarction (MI) is the most severe type of coronary artery disease and one of the leading causes of death in human beings today [2]. In recent years, many studies have been looking for effective strategies for targeted treatment of MI. An increasing number of studies have suggested that ncRNAs, especially microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), are closely related to the development of MI, and their specific roles in MI are still being carefully studied. From this perspective, it is essential and desirable to understand the relationship between ncRNAs and their direct targets, as well as their regulatory effects on the pathophysiology after MI.
The main approach of ncRNA research is to screen one or more ncRNAs from clinical genomics, then search for the corresponding molecular targets and explore their relationships with disease phenotypes. In recent years, many traditional reviews of ncRNAs and MI have been published. However, researchers can select the literature they are interested in according to their own wishes in traditional reviews, which results in a certain amount of selection bias [3, 4]. Therefore, a data-driven approach was used to organize this review to understand the ncRNAs in MI in the past five years and to discuss the characteristics of ncRNAs and their molecular targets objectively [5].
Previous researchers explored the networks of ncRNAs and their molecular targets in breast cancer in a similar approach [6]. This paper referred to their screening methods and retrieved all the literatures related to ncRNAs (especially miRNAs) and MI in PubMed in the past five years (Table S1). Then, the literatures were screened according to the characteristics of the basic research in MI (Figure S1). The details of the molecules in the networks are shown in Table 1. Finally, the networks of ncRNAs and their direct molecular targets were constructed, and a network is defined as an independent cluster of at least four molecules. Boxes and white ellipses represent ncRNAs and coding genes, respectively, lines with flat red arrows indicate negative interactions or inhibitions, while black arrows indicate positive interactions or activations, and solid lines represent direct effects, while dotted lines represent indirect effects.
List of ncRNA-target and the type of interaction present in networks.
| ncRNA | Name | Direct target | Direct effect | PMID | Function* |
|---|---|---|---|---|---|
| miRNA | let-7i | FASLG | neg | 28350318 | anti-apoptosis |
| miRNA | let-7i | CCND2 | neg | 30679264 | Inhibition of cell proliferation |
| miRNA | let-7i | E2F2 | neg | 30679264 | |
| miRNA | miR-124 | HIC5 | neg | 29864957 | Pro-apoptosis |
| miRNA | miR-124 | DHCR24 | neg | 31100313 | |
| miRNA | miR-125b | BAK1 | neg | 29122578 | Anti-apoptosis |
| miRNA | miR-125b | KLF13 | neg | 29122578 | |
| miRNA | miR-125b | p53 | neg | 29921652 | |
| miRNA | miR-125b | p53 | neg | 30613290 | |
| miRNA | miR-125b | BAK1 | neg | 30613290 | |
| miRNA | miR-142 | APC | neg | 32327611 | Anti-apoptosis Activation of cardiac fibroblasts |
| miRNA | miR-142 | TP53INP1 | neg | 31085811 | |
| miRNA | miR-143 | IGF-IR | neg | 32368046 | Pro-angiogenesis |
| miRNA | miR-143 | ERG1 | neg | 31900593 | Inhibition ventricular arrhythmias |
| miRNA | miR-143 | SPRY3 | neg | 30878395 | Pro-fibrosis |
| miRNA | miR-144 | mTOR | neg | 30084039 | Pro-fibrosis |
| miRNA | miR-144 | PTEN | neg | 31737623 | Pro-autophagy |
| miRNA | miR-152 | DNMT1 | neg | 29358670 | Promotion of cell proliferation |
| miRNA | miR-152 | p27 | neg | 29358670 | |
| miRNA | miR-152 | ATG12 | neg | 28350318 | |
| miRNA | miR-155 | SOS1 | neg | 28129114 | Anti-fibrosis |
| miRNA | miR-155 | SOCS1 | neg | 28129114 | |
| miRNA | miR-155 | QKI | neg | 31191799 | Pro-apoptosis |
| miRNA | miR-155 | RAC1 | neg | 32112145 | Anti-angiogenesis |
| miRNA | miR-155 | PAK2 | neg | 32112145 | |
| miRNA | miR-155 | SIRT1 | neg | 32112145 | |
| miRNA | miR-155 | AMPKα2 | neg | 32112145 | |
| miRNA | miR-181a | STAT1 | neg | 28597963 | Anti-inflammation, Anti-apoptosis |
| miRNA | miR-181a | c-Fos | neg | 28597963 | |
| miRNA | miR-181a | ADAMTS1 | neg | 32304626 | Inhibition of cardiomyocytes hypertrophy |
| miRNA | miR-199a | HOMER1 | neg | 28077443 | Promotion of cell proliferation |
| miRNA | miR-199a | HOPX | neg | 30125571 | |
| miRNA | miR-21 | PTEN | neg | 26978580 | Anti-apoptosis |
| miRNA | miR-21 | KBTBD7 | neg | 29991775 | Anti-inflammation |
| miRNA | miR-21 | PDCD4 | neg | 31149048 | Anti-apoptosis |
| miRNA | miR-21 | PDCD4 | neg | 30684569 | |
| miRNA | miR-21 | PTEN | neg | 30684569 | |
| miRNA | miR-21 | STRN | neg | 32297634 | Disorder of nitrogen metabolism |
| miRNA | miR-21 | SPRY1 | neg | 33273841 | Pro-fibrosis |
| miRNA | miR-210 | CXCR4 | neg | 29710553 | Reduction of hypoxia-induced damage |
| miRNA | miR-210 | AIFM3 | neg | 32513270 | Anti-apoptosis |
| miRNA | miR-210 | EFNA3 | neg | 28249798 | Pro-angiogenesis |
| miRNA | miR-214 | DIO3 | neg | 27014189 | Protection of cardiac systolic function |
| miRNA | miR-214 | PTEN | neg | 29584819 | Promotion of cell proliferation |
| miRNA | miR-214 | PTEN | neg | 30545799 | |
| miRNA | miR-214 | GSK3β | neg | 30947518 | Promotion of cardiac regeneration |
| miRNA | miR-221 | PTEN | neg | 32586406 | Pro-angiogenesis Anti-apoptosis |
| miRNA | miR-221 | PTEN | neg | 32432109 | |
| miRNA | miR-221 | FOXO3 | neg | 32629000 | Anti-fibrosis |
| miRNA | miR-26a | ATM | neg | 31990056 | Anti-apoptosis, Anti-fibrosis |
| miRNA | miR-26a | USP15 | neg | 31512556 | Pro-autophagy |
| miRNA | miR-26a | BAK1 | neg | 32464547 | Anti-apoptosis |
| miRNA | miR-590 | CLIC5 | neg | 28077443 | Promotion of cell proliferation |
| miRNA | miR-590 | HOMER1 | neg | 28077443 | |
| miRNA | miR-590 | ZEB1 | neg | 31675172 | Anti-fibrosis |
| miRNA | miR-92a | KLF2 | neg | 32939962 | Anti-angiogenesis |
| miRNA | miR-92a | ATG4A | neg | 30571345 | Anti-autophagy |
| miRNA | miR-92a | ABCA8B | neg | 30571345 | Lipometabolic disturbance |
| miRNA | miR-92a | CD36 | neg | 30571345 | |
| lncRNA | AZIN2-sv | miR-214 | neg | 29584819 | Inhibition of cell proliferation |
| lncRNA | AZIN2-sv | PTEN | pos | 29584819 | |
| lncRNA | AZIN2-sv | miR-214 | neg | 30545799 | |
| lncRNA | AZIN2-sv | TLN1 | neg | 30545799 | Anti-angiogenesis |
| lncRNA | GAS5 | SEMA3A | neg | 30099044 | Anti-apoptosis |
| lncRNA | GAS5 | miR-142 | neg | 31085811 | Pro-apoptosis |
| lncRNA | H19 | YB-1 | neg | 31588235 | Pro-fibrosis |
| lncRNA | H19 | miR-22 | neg | 31755219 | Reduction of cardiomyocytes damage |
| lncRNA | H19 | miR-675 | pos | 31119268 | Anti-apoptosis, Pro-angiogenesis |
| lncRNA | KCNQ1OT1 | RUNX3 | neg | 31625414 | Pro-inflammation |
| lncRNA | KCNQ1OT1 | DNMT1 | pos | 31625414 | Pro-apoptosis |
| lncRNA | KCNQ1OT1 | miR-466k | neg | 32422306 | Cardiomyocytes damage |
| lncRNA | KCNQ1OT1 | miR-466i | neg | 32422306 | |
| lncRNA | MALAT1 | miR-320 | neg | 29990866 | Pro-apoptosis |
| lncRNA | MALAT1 | miR-558 | neg | 30536615 | Anti-apoptosis |
| lncRNA | MALAT1 | miR-497 | neg | 32393784 | |
| lncRNA | MALAT1 | miR-145 | neg | 30146700 | Pro-fibrosis |
| lncRNA | MALAT1 | miR-92a | neg | 32939962 | Pro-angiogenesis |
| lncRNA | MEG3 | FUS | pos | 30287867 | Pro-apoptosis |
| lncRNA | MEG3 | p53 | pos | 31631486 | |
| lncRNA | TUG1 | miR-124 | neg | 29864957 | Anti-apoptosis |
| lncRNA | TUG1 | miR-9a | neg | 31787746 | Pro-apoptosis |
| circRNA | circHIPK3 | miR-17 | neg | 31595165 | Increase of Ca2+ in cardiomyocytes |
| circRNA | circHIPK3 | N1ICD | pos | 32736292 | Promotion of cell proliferation |
| circRNA | circHIPK3 | miR-133a | neg | 32736292 | Pro-angiogenesis |
| circRNA | circHIPK3 | miR-29a | neg | 32733638 |
* Function refers to the pathophysiological changes after MI caused by upregulation of the ncRNA expression.
In this review, the networks and their components and interactions are discussed, a deeper understanding is gained of the effects of ncRNAs on MI and related mechanisms, and the prospects of the clinical application of ncRNA therapy and its challenges are discussed. It is worth noting that some studies not included in the networks will be referenced for accurate descriptions and discussions to comprehensively understand the overall situation of each ncRNA in the networks.
The miR-21 PTEN AZIN2-sv/miR-214 network
PTEN is an essential molecular hub that mediates apoptosis, proliferation, migration, fibrosis, and angiogenesis after MI [7]. Figure 1 shows that PTEN plays a crucial role in this network, connecting miR-21 to the cluster composed of AZIN2-SV and miR-214. MiR-21 plays a pivotal role in cardiomyocyte apoptosis, myocardial fibrosis, and inflammation after MI. MiR-21 enhances the inhibition of cardiomyocyte apoptosis through the PTEN/Akt-P-P38-caspase-3 signaling pathway [8]. MiR-21 can also inhibit PDCD4 expression and reduce cell apoptosis [9, 10], and the overexpression of miR-21 can significantly inhibit the release of MI-induced inflammatory cytokines. Bejerano T et al. found that when nanoparticles of miR-21 mimics were delivered to cardiac macrophages, the level of the anti-inflammatory gene SOCS1 was significantly increased, suggesting that the increase in the expression level of miR-21 in cardiac macrophages after MI could lead to a change in phenotype from pro-inflammatory to anti-inflammatory [11]. Moreover, miR-21 targets KBTBD7 and inhibits the activation of the P38 and NF-κB signaling pathways directly, thereby reducing inflammation after MI [12]. However, the upregulation of miR-21 does not bring full benefits because it targets SPRY1 to inhibit the activation of the ERK signal, thus promoting myocardial fibrosis [13]. The authors found that miR-21 knockout mice developed more pronounced cardiac fibrosis, as did in vitro studies based on primary cardiac fibroblasts. Under hypoxic conditions, the expression of miR-21 is significantly upregulated, and it targets and downregulates the level of STRN and regulates the expression of NOS3, resulting in the disorder of nitrogen metabolism [14]. Currently, a specific anti-miR-21 has been demonstrated to effectively improve cardiac remodeling in a porcine MI model [15]. However, inhibition of miR-21 inactivates the profibrotic pathway, which is not conducive to the repair of infarction scars and may even prevent the body from inhibiting myocardial cell apoptosis [16]. Therefore, although the regulation of miR-21 expression is a potential method, its application in clinical practice still has limitations.
The miR-21 PTEN AZIN2-sv/miR-214 network. The orange box represents lncRNA, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, while black arrows indicate positive interactions or activations, and the solid lines represent direct effects.
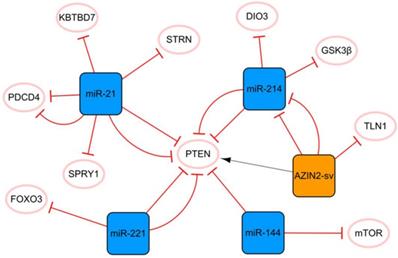
AZIN2-sv and miR-214 form a cluster with a clear targeting relationship. AZIN2-sv can inhibit angiogenesis by promoting ubiquitin-dependent degradation of TLN1 after MI [17]. AZIN2-sv acts as a miR-214 sponge to release PTEN, blocks the activation of the PI3K/Akt signaling pathway, and inhibits cardiomyocyte proliferation [18]. In addition, we know that lncRNAs in the cytoplasm regulate signaling by interacting with specific proteins. Li X et al. identified relevant proteins interacting with AZIN2-sv in cardiomyocytes by an RNA pull-down assay and found that AZIN2-sv could increase the stability of PTEN through direct binding [18]. Previous studies have shown that GSK3β inhibition has strong angiogenic and anti-apoptosis effects and plays an important role in endothelial angiogenesis [19]. The upregulation of miR-214 can inhibit the expression of Gsk3β and promote the activity of β-catenin, thus promoting cardiac regeneration and functional recovery after MI [20]. It is worth mentioning that triiodothyronine (T3) is an essential regulator of cardiac contractility and metabolism. DIO3 can convert T3 into diiodothyronine (3,3′-T2). The upregulation of miR-214 leads to a decrease in DIO3, which limits the reduction in cardiac T3 signaling and protects cardiac systolic function [21]. It is not difficult to see that miR-214 plays a protective role in MI, and most of the current evidence shows that miR-214 has a positive effect on the cellular behavior of cardiomyocytes and endothelial cells. However, it has also been found that overexpression of miR-214 in cardiomyocytes may induce dilated cardiomyopathy. This again suggests that the dosage and timing of ncRNA therapy are critical.
In this network, two miRNA axes are also strung through PTEN, namely, miR-221 and miR-144. Circulating miR-221 has been shown to be increased in patients with AMI, and many studies have confirmed that upregulated miR-221 from different sources can improve angiogenesis and cell proliferation and inhibit apoptosis through the PTEN/Akt pathway [22, 23]. In addition, the upregulation of miR-221 negatively regulates FOXO3, thereby inhibiting ATG7 transcription and reducing the occurrence of fibrosis after MI [24]. The level of serum miR-144 was significantly correlated with MI and was found to be highly expressed in the infarcted area of the porcine MI model. Compared with the function of miR-221 in alleviating myocardial fibrosis, there is some contradiction in the phenotype of miR-144 in myocardial fibrosis. It was reported that miR-144 enhanced myocardial fibrosis after MI by targeting PTEN [25]. However, Li J et al. reported that the increase in miR-144 decreased the size of infarction and collagen scarring, and miR-144 could regulate fibrosis signaling at least in part by downregulating TGF-β signaling [26]. For such different results, we think that the main reason may be related to the different animal models used in the two studies (Yuan X et al. established the MI model in pigs, while Li J et al. established in mice). There are great differences in structure, heart rate and oxygen consumption between mouse hearts and human hearts, while pig hearts are closer to human heart, especially in terms of their coronary distribution characteristics, and both have less collateral circulation [27]. Therefore, more rigorous preclinical studies are needed to treat miR-144 as a therapeutic target for MI. In addition, cardiomyocytes are long-lived postdivision cells with little ability to differentiate and regenerate. Therefore, autophagy plays a vital role in maintaining cardiac function and vitality. MTOR is a crucial negative regulator of the autophagy pathway, and miR-144 can inhibit mTOR from enhancing autophagy signaling, which may also be related to cardiac protection [26].
The MALATA1/miR-92a network
This subnetwork is primarily constructed by connecting MALAT1 and miR-92a, and there is a clear targeting relationship between them (Figure 2). Upregulation of MALAT1 can inhibit the expression of miR-92a in the left ventricular myocardium after MI and indirectly promote the expression of KLF2, thus promoting neovascularization [28].
The MALATA1/miR-92a network. The orange box represents lncRNA, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, and the solid lines represent direct effects.
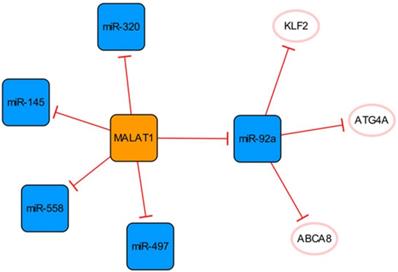
The expression of MALAT1 is increased in patients with MI and has been confirmed to activate the hypoxia pathway [29, 30]. MALAT1 can be used as a sponge to regulate the expression of miR-145 and promote myocardial fibrosis mediated by the TGF-β1 pathway [29]. MALAT1 can inhibit apoptosis by targeting miR-497 [31]. However, MALAT1, as the competing endogenous RNA (ceRNA) of miR-320, regulates the expression of PTEN in mouse cardiomyocytes. Through the miR-320/PTEN axis, overexpression of MALAT1 can promote cardiomyocyte apoptosis [32]. Although Wu Q et al. identified the binding of MALAT1 to miR-497, they did not know how much MALAT1 needed to be delivered to the infarcted heart to fully bind and inhibit miR-497. Meanwhile, their findings were at the cellular level, and their studies used neonatal rat cardiomyocytes, which did not fully reflect the final results of adult cardiomyocytes [31]. They also admitted that the cardioprotective effect and potential mechanism of MALAT1 need to be verified in infarcted hearts, while Hu H et al. explored a lncRNA MALAT1 gene knockdown in post-MI mice [31, 32]. Furthermore, miR-558 is also a direct target of MALAT1, which can protect cardiomyocytes from isoproterenol-induced apoptosis by enhancing UlK1-mediated protective autophagy through sponging miR-558 [33]. The function of the MALAT1 was further demonstrated in knockout mice, while the mice disease model was not established by ligation of coronary arteries in the study, but by injection of isoproterenol. Therefore, based on the specific methods of various studies and research results, we consider that it is difficult to determine the specific effect of MALTA1 on cardiomyocytes apoptosis. In addition, the upregulation of miR-497 eliminate the role of MALAT1 in promoting endothelial cell (EC) tube formation, suggesting that the high expression of MALAT1 can promote angiogenesis by inhibiting the expression of miR-497 [31].
It is known that an appropriate autophagy level can maintain cell homeostasis and promote cell survival. ECs enhance their ability to use their organelles as fuel substrates by increasing autophagic flux [34]. Meanwhile, the arrangement of ECs under physiological blood flow also depends on autophagy [35]. MiR-92a targets and inhibits the expression of ATG4A and limits endothelial autophagy. In addition, miR-92a targets ABCA8B and CD36, interfering with cholesterol transport, reducing the level of high-density lipoprotein, and limiting myocardial fatty acid uptake [34]. In conclusion, high expression of miR-92a will aggravate injury after MI, and targeted inhibition of miR-92a expression can be used as a new therapeutic option, which has clinical application value in alleviating tissue damage in patients with MI.
The MEG3/miR-125b/miR-26a network
This network is closely associated with apoptosis (Figure 3). MEG3 transcribed by imprinted genes is a lncRNA that is significantly related to apoptosis [36]. Under hypoxic conditions, MEG3 can be directly upregulated by p53 and participate in the regulation of apoptosis through direct binding to the RNA binding protein FUS [36]. Moreover, MEG3 knockout can inhibit the expression of p53 and reduce the apoptosis induced by endoplasmic reticulum stress after MI [37]. Connected by p53 is another protagonist in this subnetwork, miR-125b, which is also significantly related to apoptosis. MiR-125b has antiapoptotic effects on ischemic cardiomyocytes in vivo and in vitro by inhibiting p53 and BAK1 [38]. MiR-125b acts as a protective miRNA by inhibiting pro-apoptotic KLF13 in cardiomyocytes [39]. It is well known that the appropriate level of autophagy has a robust protective effect. Blocking autophagy can enlarge the area of MI, but excessive autophagy can promote apoptosis and necrosis [40]. Regarding the specific anti-apoptosis mechanism of miR-125b, further studies have found that it interferes with p53/Bnip3 signal transduction, inhibits autophagic flux, and reduces cell death [41]. In this subnetwork, miR-26a is connected to miR-125b through the proapoptotic gene BAK1, and miR-26 plays an antiapoptotic role by targeting BAK1 [42]. Overexpression of miR-26a activates autophagy by targeting USP15, which reduces cardiomyocyte death induced by ischemic stress [43]. In addition, ATM is a target for the treatment of MI, and miR-26a can minimize the development of MI by inhibiting the expression of ATM in MI [44]. The ncRNA in this network has a consistent conclusion on the regulation of apoptosis after MI, with no obvious contradictory conclusion; therefore, this network may be the preferred network for the target selection of anti-apoptosis strategies of ncRNA therapy.
The MEG3/miR-125b/miR-26a network. The orange box represents lncRNA, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, while black arrows indicate positive interactions or activations, and the solid lines represent direct effects.
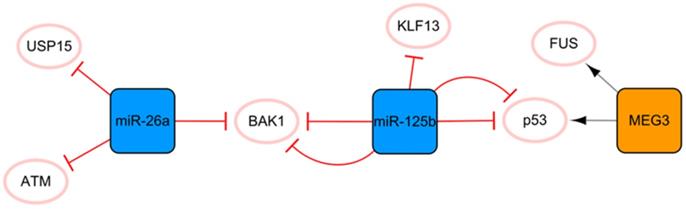
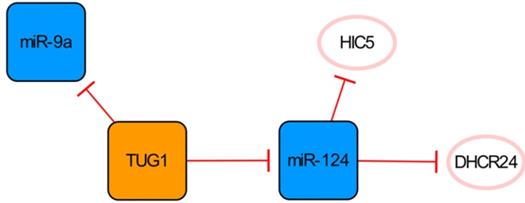
The TUG1/miR-124 network
LncRNA TUG1 and its two targets constitute a network significantly related to apoptosis (Figure 4). Downregulation of the lncRNA TUG1 gene significantly improves cardiac function in mice with MI. MiR-9a exerts an anti-cardiomyocyte apoptosis effect by inhibiting KLF5. As a competitive endogenous RNA of miR-9a, overexpressed TUG1 further aggravates myocardial cell apoptosis by downregulating the expression of miR-9a [45], while conversely, Jiang N et al. have found that TUG1 seems to have anti-apoptotic ability because the overexpression of HIC5 can enhance the viability, migration and invasion of cells induced by hypoxia injury and then reduce the apoptosis of cells [46]. MiR-124, one of the direct targets of TUG1, can inhibit the expression of HIC5 and aggravate the damage to cardiomyocytes caused by hypoxia. Therefore, overexpression of TUG1 can downregulate miR-124 and upregulate the expression of HIC5 indirectly, thereby increasing the expression of SP1 and SURVIVIN and reducing cardiomyocyte damage and apoptosis [46]. As also shown in this network, DHCR24 is another target of miR-124, and the overexpression of miR-124 can significantly increase cardiomyocyte apoptosis by targeting the inhibition of DHCR24 [47]. The regulatory functions of miR-9a and miR-124 in this network on apoptosis are completely opposite. Interestingly, they are all direct targets of TUG1, which leads to conflicting results. However, it is worth mentioning that Yang D et al. observed the pro-apoptotic effect of TUG1 on the animal level in their study, which might be more convincing [45].
The TUG1/miR-124 network. The orange box represents lncRNA, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, and the solid lines represent direct effects.
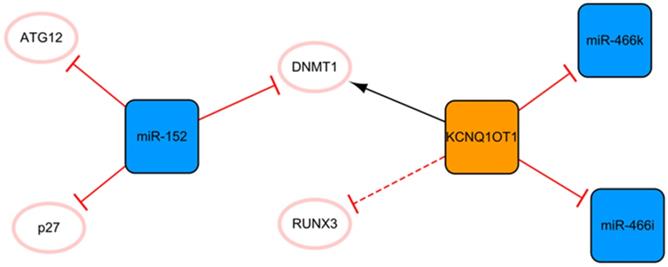
The KCNQ1OT1/miR-152 network
The network shown in Figure 5 demonstrates the long noncoding KCNQ1OT1 and miR-152. These ncRNAs are connected through DNMT1. KCNQ1OT1 can promote RUNX3 methylation by recruiting DNMT1, which leads to the downregulation of RUNX3 expression and regulation of cardiac microvascular endothelial cell activity and the inflammatory response in mice after MI [48]. After KCNQ1OT1 gene knockout, the proliferation of cardiac microvascular ECs after AMI is promoted, while apoptosis is inhibited, accompanied by a decrease in the level of inflammatory cytokines. These trends can also be realized by RUNX3 overexpression via the Notch pathway [48]. Overexpression of KCNQ1OT1 can also target miR-466k and miR-466i to play sponge roles and trigger cardiomyocyte injury in the process of MI [49]. At present, the role of KCNQ1OT1 after MI is not known, so the inhibition of KCNQ1OT1 as the target may be a potential direction of ncRNA therapy. There are relatively fewer studies on miR-152 in MI, and only two articles studying it were retrieved using our search methods; however, miR-152 is expected to become a new target for MI therapy in the future. MiR-152 inhibits the expression of DNMT1 and the cell cycle inhibitory protein p27, leading to cardiomyocyte proliferation [50]. In addition, a luciferase reporter assay confirmed that miR-152 targets ATG12, suggesting that it might have a potential antiapoptotic effect mediated by regulating autophagy [51]. Of course, this also needs to be proven by subsequent experiments.
The KCNQ1OT1/miR-152 network. The orange box represents lncRNA, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, while black arrows indicate positive interactions or activations. The solid lines represent direct effects, while dotted lines represent indirect effects.
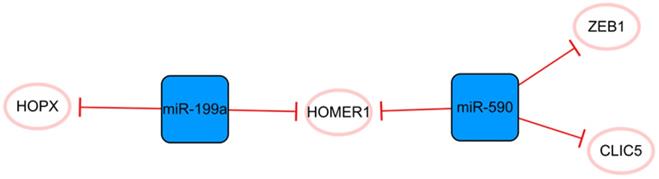
The miR-590/miR-199a network
This network consists only of miRNAs strung by HOMER1 (Figure 6). Some studies have demonstrated that miR-199a and miR-590 can promote the proliferation of cardiomyocytes, which may be due to their cumulative effects on multiple cellular mRNA targets [52]. The common target HOMER1 is involved in the regulation of calcium signaling in cardiomyocytes and HOPX, and the direct target of miR-199a is a key negative regulator of cardiomyocyte proliferation [53-55]. CLIC5 is also a direct target of miR-590, but its role in MI is not completely clear [52]. It is only known that CLIC5 plays a direct role in regulating the production of mitochondrial reactive oxygen species and is associated with the actin-based cytoskeleton [56, 57]. In brief, miR-199a and miR-590 inhibit the expression of these functional molecules, produce cumulative effects and stimulate the proliferation of cardiomyocytes [52, 55]. Lesizza P et al. further found that viral vector-based expression of miR-199a and miR-590 in mouse hearts could induce cardiac regeneration after MI [58]. In addition, miR-590 has been found to significantly inhibit the proliferation and migration of human cardiac fibroblasts and reduce the mRNA and protein expression levels of α-SMA, Col1A1, and Col3A [59]. The specific mechanism is that miR-590 directly targets the 3'UTR of ZEB1, thus inhibiting the translation of ZEB1 [59]. Interference with the expression of ZEB1 decreases cell proliferation, migration activity, and the expression of the abovementioned related proteins. Overall, this network is closely associated with cell proliferation, and the targeting relationship between each molecule is relatively clear. The overexpression of miRNAs in this network can promote the proliferation of cardiomyocytes and effectively inhibit the proliferation and transformation of fibroblasts.
The miR-590/miR-199a network. The blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, and the solid lines represent direct effects.
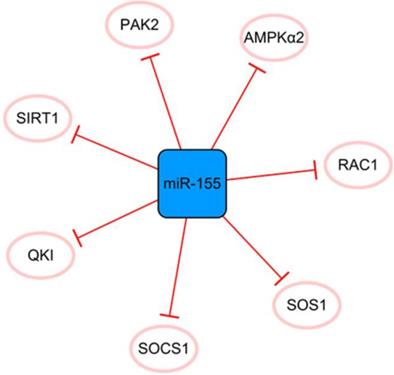
The miR-155 network
MiR-155 has many direct targets that constitute a separate network (Figure 7). MiR-155 inhibits the proliferation of fibroblasts and alleviates fibrosis after MI to some extent [60]. Fibroblast proliferation may not bring complete harm to the infarcted heart. The extracellular matrix secreted by fibroblasts after activation can effectively promote the healing of the heart after infarction and prevent the occurrence of ventricular rupture; therefore, it is crucial to effectively control the expression of miR-155 [61]. However, the disadvantages of miR-155 overexpression far outweigh its contribution to ameliorating fibrosis after MI. It was reported that miR-155 could promote MI-induced apoptosis by targeting QKI [60]. Furthermore, the level of SOCS1 in cardiac fibroblasts is decreased by miR-155 mimics and promotes inflammation [62]. It inhibits angiogenesis and exacerbates cardiac dysfunction by downregulating its target genes, including RAC1, PAK2, SIRT1, and AMPKα2 [63]. It can be seen that if we want to achieve the goal of treating MI with miR-155 as a regulatory target, it is necessary to regulate the specific expression of miR-155 in different target cells and define its specific treatment time window. In general, miR-155 is like a double-edged sword, and reasonable application is expected to become a target for the treatment of MI.
The miR-155 network. The blue box represents miRNA, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, and the solid lines represent direct effects.
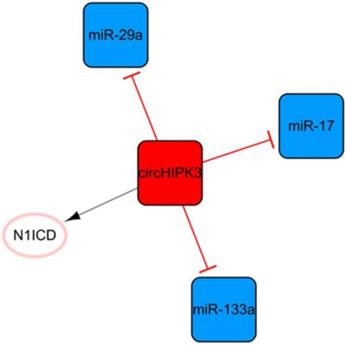
The circHIPK3 network
Circular RNA (circRNA) is another kind of ncRNA that is mainly produced by the back-splicing of exons and covalent bonds [64]. CircHIPK3, the only circRNA in this ncRNA network, is transcribed from the second exon of HIPK3 and plays pivotal roles in cell growth and angiogenesis (Figure 8) [65, 66]. CircHIPK3-rich exosomes can be released during hypoxia in cardiomyocytes. CircHIPK3 can increase the expression of VEGFA by inhibiting the expression of miR-29a, thus promoting the cell cycle progression and proliferation of cardiac ECs and changing the proliferation, migration, and tube formation of cardiac ECs [67]. Meanwhile, circHIPK3 can act as a sponge of miR-133a as well, indirectly promoting the expression of connective tissue growth factors, activating ECs, and improving angiogenesis after MI [68]. CircHIPK3 enhances the stability of N1ICD by acetylation and promotes the proliferation of cardiomyocytes [68]. In addition, the upregulation of circHIPK3 expression after epinephrine administration can temporarily improve cardiac function by increasing the peak concentration of Ca2+ in cardiomyocytes via the circHIPK3/miR-17/ADCY6 axis [69]. However, downregulation of circHIPK3 can alleviate fibrosis after MI in mice and maintain cardiac function [69]. This means that short-term upregulation of circHIPK3 can temporarily improve cardiac function; while long-term high expression may aggravate cardiac fibrosis and decrease cardiac function.
The circHIPK3 network. The red box represents circRNA, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, while black arrows indicate positive interactions or activations, and the solid lines represent direct effects.
Other lncRNA/miRNA networks
In addition to the networks mentioned above, there are also some networks composed of lncRNAs and miRNAs that deserve our attention (Figure 9). The regulatory role of lncRNA GAS5 in cancer cells has been widely reported [70], and it forms a network with miR-142 (Figure 9A). RNA immunoprecipitation revealed that GAS5 is the molecular sponge of miR-142. TP53INP1 is the target gene of miR-142. GAS5 silencing promotes the activation of the PI3K/AKT and MEK/ERK signaling pathways in a miR-142-dependent manner and weakens the cardiomyocyte apoptosis induced by hypoxia [71]. In short, the high expression of GAS5 may aggravate cardiomyocyte apoptosis, and many other studies have confirmed this point of view [72, 73]. However, another study showed that GAS5 could negatively regulate the expression of SEMA3A protein and reduce cardiomyocyte apoptosis [74], and we consider that different sources of cardiomyocytes and modeling methods may lead to two opposing outcomes. In addition, it can be found in this network that APC, a negative regulator of the WNT signaling pathway, is another target of miR-142. It was found that miR-142 could directly target and inhibit the expression of APC and promote the activation of the WNT signaling pathway and cardiac fibroblasts [75].
Other lncRNA/miRNA networks. (A) The GAS5/miR-142 network. (B) The H19/miR-22 network. The orange boxes represent lncRNAs, the blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, while black arrows indicate positive interactions or activations, and the solid lines represent direct effects.
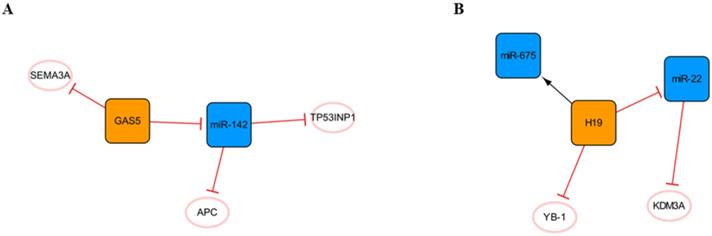
The H19 network is shown in Figure 9B. H19, known as a fetal gene, is downregulated after birth[76]. H19 polymorphism has been shown to be significantly associated with the risk of coronary artery disease, suggesting that H19 plays an important role in cardiac pathogenesis [77]. Choong OK et al. found that H19 was gradually upregulated within a week after MI, suggesting that H19 might play a role in the early stage of cardiac remodeling. Subsequent studies on the function of H19 in the early postinfarction period illustrated that the early ectopic expression of H19 in the mouse heart led to severe cardiac dilation and fibrosis. This may be related to the direct interaction of H19 with YB-1 under hypoxia, and the inhibition of YB-1 can promote the expression of COL1A1 and lead to myocardial fibrosis [78]. Another study found that miR-22 is the direct target of H19 and that miR-22 targets the inhibition of KDM3A directly. H19 regulates the expression of KDM3A in a miR-22-dependent manner to reduce myocardial injury [79]. In addition, H19 in exosomes derived from bone marrow mesenchymal stem cells can be upregulated by the clinically common lipid-lowering drug atorvastatin calcium. The upregulation of H19 can regulate the expression of miR-675, activate VEGF and the mediators of intercellular adhesion molecule-1, promote angiogenesis, and reduce myocardial cell apoptosis [80]. Briefly, the high expression of H19 in the early stage after MI can alleviate a series of injuries caused by acute ischemia to a certain extent, promote angiogenesis, reduce myocardial apoptosis, and achieve protective effects. However, every coin has two sides, and high expression of H19 can also promote the occurrence of myocardial fibrosis. Therefore, inhibiting the interaction between H19 and YB-1 while upregulating the high expression of H19 in the early stage of MI may be a therapeutic strategy that is worthy of further study.
Other miRNA networks
Other networks, which consist only of miRNAs and their coding genes, have also attracted the attention of researchers (Figure 10). The main cause of MI is ischemic hypoxia, and miR-210 is considered a ncRNA closely related to hypoxia [81]. There is increasing evidence that miR-210 can be used as an independent indicator of MI severity [82]. It has been found that miR-210 is upregulated in hypoxic cardiomyocytes, which can alleviate hypoxia-induced damage by targeting CXCR4 inhibition [83]. Hypoxia can also induce the upregulation of miR-210 expression in exosomes derived from mesenchymal stem cells. Overexpression of miR-210 inhibits AIFM3, activates PI3K/AKT and p53 signal transduction, reduces apoptosis, and plays a role in cell protection [84]. In addition, miR-210 has been found to promote angiogenesis in mice with MI, which may be related to the targeted inhibition of EFNA3 as a protein regulating angiogenesis [85]. In short, miRNA-210 has great potential in the treatment of cardiovascular diseases, and it is necessary to further study the cardiovascular protective mechanism of miR-210 and its future applications.
Other miRNA networks. (A) The miR-210 network. (B) The leti-7i network. (C) The miR-143 network. (D) The miR-181a network. The blue boxes represent miRNAs, and the white ellipses represent the protein coding genes. The lines with flat red arrows indicate negative interactions or inhibitions, and the solid lines represent direct effects.
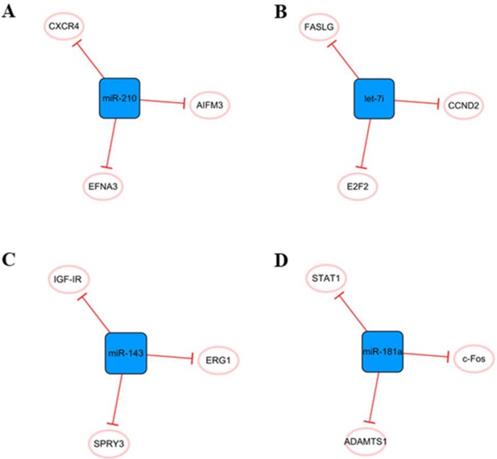
Elevated miR-143 level was detected in human MI samples [86]. When the expression of miR-143 is upregulated, it can directly target SPRY3, activate the P38, ERK, and JNK pathways, and promote the proliferation, migration, and transformation of human cardiac fibroblasts and the excessive accumulation of extracellular matrix, thus aggravating the occurrence of myocardial fibrosis [86]. Overexpression of miRNA-143 can inhibit IGF-IR, which has a negative effect on angiogenesis [87]. Moreover, downregulation of miR-143 can regulate EGR1, improve the dysfunction of INa and IK1 currents in ventricular myocytes, and effectively inhibit ventricular arrhythmias [88]. Therefore, downregulation of miR-143 expression after MI seems to be a feasible therapeutic strategy.
The let-7 family is one of the earliest discovered miRNAs and plays an important role in the regulation of cardiovascular disease [89]. Let-7i, a member of the let-7 family, is abundant in cardiomyocyte-derived exosomes under hypoxia and has a potential anti-apoptosis effect by targeting FASLG expression [51]. In addition, let-7i is involved in the regulation of the cardiomyocyte cell cycle and further affects proliferation, and E2F2 and CCND2 are two direct targets of let-7i. After the inhibition of let-7i, the expression of E2F2 and CCND2 is significantly upregulated, which can promote the proliferation of cardiomyocytes[90]. In fact, the let-7 family is involved in many cellular processes, including proliferation, inflammation and apoptosis [91], but their roles in MI have not been well studied. Therefore, it is necessary to further study their roles in cardiovascular diseases.
We believe that miR-181a may be an effective therapeutic target to reduce inflammation and improve cardiac remodeling after MI. The aldosterone-mineralocorticoid receptor pathway is activated during MI [92]. Garg An et al. found that overexpression of miR-181a could significantly inhibit aldosterone-mineralocorticoid-induced cardiomyocyte hypertrophy. The mechanism might be related to the direct targeting of miR-181a to inhibit the expression of ADAMTS1 and then regulate the level of Ngala [93]. Moreover, MI was accompanied by severe inflammation, and the expression of miR-181a was found to be increased in dendritic cells. Overexpression of miR-181a can reduce the maturation of dendritic cells and the production of inflammatory cytokines, prevent the occurrence of severe inflammation, and inhibit the apoptosis of cardiomyocytes under hypoxia by inhibiting STAT1 and c-Fos [94].
Perspectives and challenges
NcRNAs have been well studied in cellular processes and cell types associated with the cardiovascular system [95]. After MI, the internal environment of cell survival changes, and the expression of some ncRNAs changes. The expression of different ncRNAs affects the prognosis of MI [96]. They provide not only potential biomarkers for the diagnosis of MI but also new possibilities and opportunities for the selection of therapeutic targets [96]. Applying ncRNA therapy in the clinic is a very attractive prospect. However, before translating the therapy into the clinic, a large number of preclinical studies are needed to ensure its efficacy and safety in patients with MI. In fact, some ncRNAs in these networks have been subjected to some preclinical studies. MiR-199a can promote the proliferation of endogenous cardiomyocytes to realize heart repair in pigs [97], and the use of miR-144 inhibitors can also reduce the occurrence of fibrosis after MI in pigs [25].
However, there are still some challenges in the clinical application of ncRNA therapy. First, ncRNAs have opposite effects on cells in different tissues. NcRNAs exist widely in different cells, and some are thought to be therapeutic targets for MI, which may aggravate the occurrence and development of other diseases. H19, mentioned above, is a typical example, which reduces cardiomyocyte apoptosis and promotes angiogenesis in the early stage after MI, but overexpression of H19 promotes the invasion, migration and epithelial-mesenchymal transformation of ovarian cancer cells. Second, ncRNAs can have different effects on different cells in the same tissue [98]. The cellular composition of heart tissue is complex. In addition to cardiomyocytes, there are many cellular components, such as macrophages, cardiac fibroblasts, and endothelial cells. NcRNAs play different roles in different cells, which make targeted treatment of MI difficult [99]. The above mentioned miR-21 plays a key role in anti-cardiomyocyte apoptosis and inflammation after infarction, but its overexpression may promote the proliferation and differentiation of cardiac fibroblasts and aggravate fibrosis after MI. All these conditions increase the clinical complexity and difficulty of the application of ncRNA therapy.
In addition, high-throughput sequencing found that there are a large number of changes in ncRNA levels in cells or tissues under pathological conditions, and repairing the expression levels of single or several kinds of ncRNAs may not necessarily reach the ideal level of treatment. More importantly, some of the effects of ncRNAs have opposite conclusions in the heart, such as the regulation of cardiomyocyte apoptosis by TUG1 and MALAT1 [31, 32, 45, 46]. This may be related to the selection of cells and targeted regulatory molecules in vitro, but large animal experiments are also urgently needed to further determine their specific roles in vivo.
Controlling the expression of ncRNAs is also very important for the clinical application of ncRNA therapy. At present, the ncRNA delivery carriers used in some studies are liposomes, which may lead to the overexpression of ncRNAs in the cardiovascular system and produce a nonspecific interferon response [4]. Therefore, a new vector, such as adenovirus, can be chosen to avoid ncRNA overexpression in vivo. To regulate the specific expression profiles of ncRNAs in different tissues, focusing on the use of different biomedical technologies like nanoparticle-hydrogel systems is also a future research direction [100]. Meanwhile, the treatment dose, treatment time window and possible side effects of ncRNA therapy need to be further determined.
Conclusions
Our review reveals important networks of interactions between ncRNAs and their direct targets and provides an unbiased description of the roles of ncRNAs in MI over the last five years. First, our review manually searched and screened all the literature on noncoding RNAs related to MI in the last five years available on PubMed. Second, only the original studies of ncRNA-targeting relationships or interactions confirmed by laboratory methods in vitro and/or in vivo could be included. Finally, the relationships between the noncoding RNAs and their direct molecular targets in the networks were discovered and described.
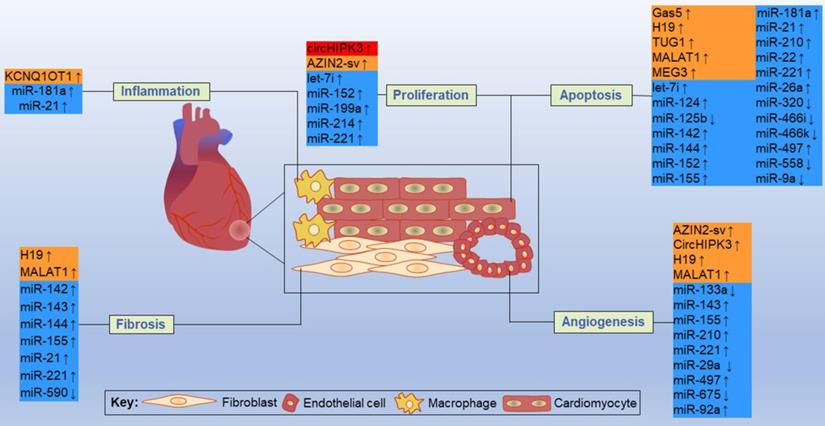
By acting on direct targets, ncRNAs in these networks play various functions in major resident cell types of the heart, such as cardiomyocytes, ECs, fibroblasts and macrophages, and play crucial roles in cell proliferation, apoptosis, fibrosis, inflammation and other processes (Figure 11). Inhibition or overexpression of certain ncRNAs or their targets may help alleviate cardiac tissue injury, promote angiogenesis, and prevent cardiac remodeling after MI. However, some ncRNAs may have opposite effects on different targets; therefore, how to rationally utilize these ncRNAs and their targets has become an urgent problem to be solved. Key information, such as carrier selection, treatment time window, treatment dose and possible side effects of ncRNA therapy, needs to be further determined. In short, the interactions between coding and noncoding genes play indispensable roles in the occurrence and development of MI and still deserve great attention from researchers in this field. The rational application of ncRNAs is expected to become a target for the treatment of MI.
Some representative ncRNAs in the network involved in various functions of the heart after MI are reviewed. The up and down arrows indicate changes in ncRNAs after MI. The red box represents circRNA, the orange boxes represent lncRNAs, and the blue boxes represent miRNAs.
Abbreviations
ceRNA: competing endogenous RNA; circRNAs: circular RNAs; ECs: endothelial cells; lncRNAs: long noncoding RNAs; MI: myocardial infarction; miRNAs: microRNAs; ncRNAs: noncoding RNAs.
Supplementary Material
Supplementary figure and table.
Acknowledgements
Funding
This work was supported by National Key R&D Program of China (Grant No. 2020YFC2008500 and 2020YFC2008502) and National Natural Science Foundation of China (Grant No. 8157223 and 82172534) and the Project of Department of Science and Technology of Sichuan Province (Grant Nos 2019YJ0119, 2020YFS0277) and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (Grant No. ZYJC21038). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions
Quan Wei and Chenying Fu contributed to the study conception and design. Literature search and data analysis were performed by Shiqi Wang, Yang Wang, and Hongxin Cheng. The first draft of the manuscript was written by Shiqi Wang, Yang Wang, Hongxin Cheng, Qing Zhang, Chenying Fu, and Quan Wei. Chenying Fu and Chengqi He revised manuscript. All authors listed have made a substantial contribution to the work. All authors read and approved the final manuscript.
Availability of data and materials
All data analyzed during this study are included in this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M. et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. 2018;39:2704-16
2. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP. et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56-e528
3. Sun T, Dong YH, Du W, Shi CY, Wang K, Tariq MA. et al. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int J Mol Sci. 2017 18
4. Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nature reviews Cardiology. 2015;12:135-42
5. Corrà F, Agnoletto C, Minotti L, Baldassari F, Volinia S. The Network of Non-coding RNAs in Cancer Drug Resistance. Front Oncol. 2018;8:327
6. Crudele F, Bianchi N, Reali E, Galasso M, Agnoletto C, Volinia S. The network of non-coding RNAs and their molecular targets in breast cancer. Mol Cancer. 2020;19:61
7. Ghafouri-Fard S, Abak A, Shoorei H, Mohaqiq M, Majidpoor J, Sayad A. et al. Regulatory role of microRNAs on PTEN signaling. Biomed Pharmacother. 2021;133:110986
8. Huang W, Tian SS, Hang PZ, Sun C, Guo J, Du ZM. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol Ther Nucleic Acids. 2016;5:e296
9. Song Y, Zhang C, Zhang J, Jiao Z, Dong N, Wang G. et al. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics. 2019;9:2346-60
10. Xing Y, Li L. Gastrodin protects rat cardiomyocytes H9c2 from hypoxia-induced injury by up-regulation of microRNA-21. Int J Biochem Cell Biol. 2019;109:8-16
11. Bejerano T, Etzion S, Elyagon S, Etzion Y, Cohen S. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018;18:5885-91
12. Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu Z. et al. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018;9:769
13. Li D, Mao C, Zhou E, You J, Gao E, Han Z. et al. MicroRNA-21 Mediates a Positive Feedback on Angiotensin II-Induced Myofibroblast Transformation. J Inflamm Res. 2020;13:1007-20
14. Kang JY, Kim H, Mun D, Yun N, Joung B. Therapeutic potential of miR-21 regulation by human peripheral blood derived-small extracellular vesicles in myocardial infarction. Clin Sci (Lond). 2020;134:985-99
15. Hinkel R, Ramanujam D, Kaczmarek V, Howe A, Klett K, Beck C. et al. AntimiR-21 Prevents Myocardial Dysfunction in a Pig Model of Ischemia/Reperfusion Injury. Journal of the American College of Cardiology. 2020;75:1788-800
16. Aimo A, Egea OI, Emdin M, Bayes-Genis A. Overlapping Effects of miR-21 Inhibition and Drugs for Idiopathic Pulmonary Fibrosis: Rationale for Repurposing Nintedanib as a Novel Treatment for Ischemia/Reperfusion Injury. Journal of cardiovascular pharmacology. 2021;77:332-3
17. Li X, Sun Y, Huang S, Chen Y, Chen X, Li M. et al. Inhibition of AZIN2-sv induces neovascularization and improves prognosis after myocardial infarction by blocking ubiquitin-dependent talin1 degradation and activating the Akt pathway. EBioMedicine. 2019;39:69-82
18. Li X, He X, Wang H, Li M, Huang S, Chen G. et al. Loss of AZIN2 splice variant facilitates endogenous cardiac regeneration. Cardiovasc Res. 2018;114:1642-55
19. Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J Mol Cell Cardiol. 2006;40:138-47
20. Huang S, Li X, Zheng H, Si X, Li B, Wei G. et al. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration After Myocardial Infarction in Adult Mice. Circulation. 2019;139:2857-76
21. Janssen R, Zuidwijk MJ, Muller A, van Mil A, Dirkx E, Oudejans CB. et al. MicroRNA 214 Is a Potential Regulator of Thyroid Hormone Levels in the Mouse Heart Following Myocardial Infarction, by Targeting the Thyroid-Hormone-Inactivating Enzyme Deiodinase Type III. Front Endocrinol (Lausanne). 2016;7:22
22. Hao C, Lu Z, Zhao Y, Chen Z, Shen C, Ma G. et al. Overexpression of GATA4 enhances the antiapoptotic effect of exosomes secreted from cardiac colony-forming unit fibroblasts via miRNA221-mediated targeting of the PTEN/PI3K/AKT signaling pathway. Stem Cell Res Ther. 2020;11:251
23. Sun L, Zhu W, Zhao P, Zhang J, Lu Y, Zhu Y. et al. Down-Regulated Exosomal MicroRNA-221 - 3p Derived From Senescent Mesenchymal Stem Cells Impairs Heart Repair. Front Cell Dev Biol. 2020;8:263
24. Li F, Long TY, Bi SS, Sheikh SA, Zhang CL. circPAN3 exerts a profibrotic role via sponging miR-221 through FoxO3/ATG7-activated autophagy in a rat model of myocardial infarction. Life Sci. 2020;257:118015
25. Yuan X, Pan J, Wen L, Gong B, Li J, Gao H. et al. MiR-144-3p Enhances Cardiac Fibrosis After Myocardial Infarction by Targeting PTEN. Frontiers in cell and developmental biology. 2019;7:249
26. Li J, Cai SX, He Q, Zhang H, Friedberg D, Wang F. et al. Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res Cardiol. 2018;113:36
27. Kumar M, Kasala ER, Bodduluru LN, Dahiya V, Sharma D, Kumar V. et al. Animal models of myocardial infarction: Mainstay in clinical translation. Regulatory toxicology and pharmacology: RTP. 2016;76:221-30
28. Shyu KG, Wang BW, Fang WJ, Pan CM, Lin CM. Hyperbaric oxygen-induced long non-coding RNA MALAT1 exosomes suppress MicroRNA-92a expression in a rat model of acute myocardial infarction. J Cell Mol Med. 2020;24:12945-54
29. Huang S, Zhang L, Song J, Wang Z, Huang X, Guo Z. et al. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J Cell Physiol. 2019;234:2997-3006
30. Choudhry H, Mole DR. Hypoxic regulation of the noncoding genome and NEAT1. Brief Funct Genomics. 2016;15:174-85
31. Wu Q, Wang J, Tan WLW, Jiang Y, Wang S, Li Q. et al. Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 2020;11:354
32. Hu H, Wu J, Li D, Zhou J, Yu H, Ma L. Knockdown of lncRNA MALAT1 attenuates acute myocardial infarction through miR-320-Pten axis. Biomed Pharmacother. 2018;106:738-46
33. Guo X, Wu X, Han Y, Tian E, Cheng J. LncRNA MALAT1 protects cardiomyocytes from isoproterenol-induced apoptosis through sponging miR-558 to enhance ULK1-mediated protective autophagy. J Cell Physiol. 2019;234:10842-54
34. Rogg EM, Abplanalp WT, Bischof C, John D, Schulz MH, Krishnan J. et al. Analysis of Cell Type-Specific Effects of MicroRNA-92a Provides Novel Insights Into Target Regulation and Mechanism of Action. Circulation. 2018;138:2545-58
35. Vion AC, Kheloufi M, Hammoutene A, Poisson J, Lasselin J, Devue C. et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A. 2017;114:E8675-e84
36. Wu H, Zhao ZA, Liu J, Hao K, Yu Y, Han X. et al. Long noncoding RNA Meg3 regulates cardiomyocyte apoptosis in myocardial infarction. Gene Ther. 2018;25:511-23
37. Li X, Zhao J, Geng J, Chen F, Wei Z, Liu C. et al. Long non-coding RNA MEG3 knockdown attenuates endoplasmic reticulum stress-mediated apoptosis by targeting p53 following myocardial infarction. J Cell Mol Med. 2019;23:8369-80
38. Zhu LP, Tian T, Wang JY, He JN, Chen T, Pan M. et al. Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics. 2018;8:6163-77
39. Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y. et al. A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol. 2018;114:72-82
40. Zhu JH, Gusdon AM, Cimen H, Van Houten B, Koc E, Chu CT. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 2012;3:e312
41. Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y. et al. Transplanted Mesenchymal Stem Cells Reduce Autophagic Flux in Infarcted Hearts via the Exosomal Transfer of miR-125b. Circ Res. 2018;123:564-78
42. Su X, Lv L, Li Y, Fang R, Yang R, Li C. et al. lncRNA MIRF Promotes Cardiac Apoptosis through the miR-26a-Bak1 Axis. Mol Ther Nucleic Acids. 2020;20:841-50
43. Liang H, Su X, Wu Q, Shan H, Lv L, Yu T. et al. LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy. 2020;16:1077-91
44. Chiang MH, Liang CJ, Lin LC, Yang YF, Huang CC, Chen YH. et al. miR-26a attenuates cardiac apoptosis and fibrosis by targeting ataxia-telangiectasia mutated in myocardial infarction. J Cell Physiol. 2020;235:6085-102
45. Yang D, Yu J, Liu HB, Yan XQ, Hu J, Yu Y. et al. The long non-coding RNA TUG1-miR-9a-5p axis contributes to ischemic injuries by promoting cardiomyocyte apoptosis via targeting KLF5. Cell Death Dis. 2019;10:908
46. Jiang N, Xia J, Jiang B, Xu Y, Li Y. TUG1 alleviates hypoxia injury by targeting miR-124 in H9c2 cells. Biomed Pharmacother. 2018;103:1669-77
47. Han F, Chen Q, Su J, Zheng A, Chen K, Sun S. et al. MicroRNA-124 regulates cardiomyocyte apoptosis and myocardial infarction through targeting Dhcr24. J Mol Cell Cardiol. 2019;132:178-88
48. Wang Y, Yang X, Jiang A, Wang W, Li J, Wen J. Methylation-dependent transcriptional repression of RUNX3 by KCNQ1OT1 regulates mouse cardiac microvascular endothelial cell viability and inflammatory response following myocardial infarction. FASEB J. 2019;33:13145-60
49. Liao B, Dong S, Xu Z, Gao F, Zhang S, Liang R. LncRNA Kcnq1ot1 renders cardiomyocytes apoptosis in acute myocardial infarction model by up-regulating Tead1. Life Sci. 2020;256:117811
50. Wang X, Ha T, Liu L, Hu Y, Kao R, Kalbfleisch J. et al. TLR3 Mediates Repair and Regeneration of Damaged Neonatal Heart through Glycolysis Dependent YAP1 Regulated miR-152 Expression. Cell Death Differ. 2018;25:966-82
51. Zhang J, Ma J, Long K, Qiu W, Wang Y, Hu Z. et al. Overexpression of Exosomal Cardioprotective miRNAs Mitigates Hypoxia-Induced H9c2 Cells Apoptosis. International journal of molecular sciences. 2017 18
52. Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S. et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376-81
53. Pouliquin P, Dulhunty AF. Homer and the ryanodine receptor. Eur Biophys J. 2009;39:91-102
54. Trivedi CM, Zhu W, Wang Q, Jia C, Kee HJ, Li L. et al. Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation. Dev Cell. 2010;19:450-9
55. Chen G, Li H, Li X, Li B, Zhong L, Huang S. et al. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J Mol Cell Cardiol. 2018;122:152-64
56. Ponnalagu D, Gururaja Rao S, Farber J, Xin W, Hussain AT, Shah K. et al. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion. 2016;27:6-14
57. Li F, Yin J, Yue T, Liu L, Zhang H. The CLIC5 (chloride intracellular channel 5) involved in C2C12 myoblasts proliferation and differentiation. Cell Biol Int. 2010;34:379-84
58. Lesizza P, Prosdocimo G, Martinelli V, Sinagra G, Zacchigna S, Giacca M. Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function After Myocardial Infarction. Circ Res. 2017;120:1298-304
59. Yuan X, Pan J, Wen L, Gong B, Li J, Gao H. et al. MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J Cell Mol Med. 2020;24:227-37
60. Guo J, Liu HB, Sun C, Yan XQ, Hu J, Yu J. et al. MicroRNA-155 Promotes Myocardial Infarction-Induced Apoptosis by Targeting RNA-Binding Protein QKI. Oxid Med Cell Longev. 2019;2019:4579806
61. Chen W, Bian W, Zhou Y, Zhang J. Cardiac Fibroblasts and Myocardial Regeneration. Frontiers in bioengineering and biotechnology. 2021;9:599928
62. Wang C, Zhang C, Liu L, A X, Chen B, Li Y. et al. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Molecular therapy: the journal of the American Society of Gene Therapy. 2017;25:192-204
63. Liu S, Chen J, Shi J, Zhou W, Wang L, Fang W. et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res Cardiol. 2020;115:22
64. Kai D, Yannian L, Yitian C, Dinghao G, Xin Z, Wu J. Circular RNA HIPK3 promotes gallbladder cancer cell growth by sponging microRNA-124. Biochem Biophys Res Commun. 2018;503:863-9
65. Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B. et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215
66. Shan K, Liu C, Liu BH, Chen X, Dong R, Liu X. et al. Circular Noncoding RNA HIPK3 Mediates Retinal Vascular Dysfunction in Diabetes Mellitus. Circulation. 2017;136:1629-42
67. Wang Y, Zhao R, Shen C, Liu W, Yuan J, Li C. et al. Exosomal CircHIPK3 Released from Hypoxia-Induced Cardiomyocytes Regulates Cardiac Angiogenesis after Myocardial Infarction. Oxid Med Cell Longev. 2020;2020:8418407
68. Si X, Zheng H, Wei G, Li M, Li W, Wang H. et al. circRNA Hipk3 Induces Cardiac Regeneration after Myocardial Infarction in Mice by Binding to Notch1 and miR-133a. Mol Ther Nucleic Acids. 2020;21:636-55
69. Deng Y, Wang J, Xie G, Zeng X, Li H. Circ-HIPK3 Strengthens the Effects of Adrenaline in Heart Failure by MiR-17-3p - ADCY6 Axis. Int J Biol Sci. 2019;15:2484-96
70. Li J, Yang C, Li Y, Chen A, Li L, You Z. LncRNA GAS5 suppresses ovarian cancer by inducing inflammasome formation. Biosci Rep. 2018 38
71. Du J, Yang ST, Liu J, Zhang KX, Leng JY. Silence of LncRNA GAS5 Protects Cardiomyocytes H9c2 against Hypoxic Injury via Sponging miR-142-5p. Mol Cells. 2019;42:397-405
72. Xie MY, Hou LJ. LncRNA GAS5 aggravates myocardial infarction by sponging miR-26b. Int J Cardiol. 2021;331:210
73. Zhang JC, Xia L, Jiang Y, Wu DQ, Liu SC, Zhou XN. et al. Effect of lncRNA GAS5 on rats with acute myocardial infarction through regulating miR-21. Eur Rev Med Pharmacol Sci. 2019;23:8573-9
74. Hao S, Liu X, Sui X, Pei Y, Liang Z, Zhou N. Long non-coding RNA GAS5 reduces cardiomyocyte apoptosis induced by MI through sema3a. Int J Biol Macromol. 2018;120:371-7
75. Cai L, Chao G, Li W, Zhu J, Li F, Qi B. et al. Activated CD4(+) T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast. Aging (Albany NY). 2020;12:7380-96
76. Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153-5
77. Gómez J, Lorca R, Reguero JR, Martín M, Morís C, Alonso B. et al. Genetic variation at the long noncoding RNA H19 gene is associated with the risk of hypertrophic cardiomyopathy. Epigenomics. 2018;10:865-73
78. Choong OK, Chen CY, Zhang J, Lin JH, Lin PJ, Ruan SC. et al. Hypoxia-induced H19/YB-1 cascade modulates cardiac remodeling after infarction. Theranostics. 2019;9:6550-67
79. Zhang BF, Jiang H, Chen J, Hu Q, Yang S, Liu XP. et al. LncRNA H19 ameliorates myocardial infarction-induced myocardial injury and maladaptive cardiac remodelling by regulating KDM3A. J Cell Mol Med. 2020;24:1099-115
80. Huang P, Wang L, Li Q, Tian X, Xu J, Xu J. et al. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovascular research. 2020;116:353-67
81. Ivan M, Huang X. miR-210: fine-tuning the hypoxic response. Adv Exp Med Biol. 2014;772:205-27
82. Guan Y, Song X, Sun W, Wang Y, Liu B. Effect of Hypoxia-Induced MicroRNA-210 Expression on Cardiovascular Disease and the Underlying Mechanism. Oxidative medicine and cellular longevity. 2019;2019:4727283
83. Feng M, Li Z, Wang D, Wang F, Wang C, Wang C. et al. MicroRNA-210 aggravates hypoxia-induced injury in cardiomyocyte H9c2 cells by targeting CXCR4. Biomed Pharmacother. 2018;102:981-7
84. Cheng H, Chang S, Xu R, Chen L, Song X, Wu J. et al. Hypoxia-challenged MSC-derived exosomes deliver miR-210 to attenuate post-infarction cardiac apoptosis. Stem Cell Res Ther. 2020;11:224
85. Wang N, Chen C, Yang D, Liao Q, Luo H, Wang X. et al. Mesenchymal stem cells-derived extracellular vesicles, via miR-210, improve infarcted cardiac function by promotion of angiogenesis. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2085-92
86. Li C, Li J, Xue K, Zhang J, Wang C, Zhang Q. et al. MicroRNA-143-3p promotes human cardiac fibrosis via targeting sprouty3 after myocardial infarction. Journal of molecular and cellular cardiology. 2019;129:281-92
87. Geng T, Song ZY, Xing JX, Wang BX, Dai SP, Xu ZS. Exosome Derived from Coronary Serum of Patients with Myocardial Infarction Promotes Angiogenesis Through the miRNA-143/IGF-IR Pathway. Int J Nanomedicine. 2020;15:2647-58
88. Li J, Xu C, Liu Y, Li Y, Du S, Zhang R. et al. Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Res Cardiol. 2020;115:9
89. Bao MH, Feng X, Zhang YW, Lou XY, Cheng Y, Zhou HH. Let-7 in cardiovascular diseases, heart development and cardiovascular differentiation from stem cells. Int J Mol Sci. 2013;14:23086-102
90. Hu Y, Jin G, Li B, Chen Y, Zhong L, Chen G. et al. Suppression of miRNA let-7i-5p promotes cardiomyocyte proliferation and repairs heart function post injury by targetting CCND2 and E2F2. Clinical science (London, England: 1979). 2019;133:425-41
91. Bernstein DL, Jiang X, Rom S. let-7 microRNAs: Their Role in Cerebral and Cardiovascular Diseases, Inflammation, Cancer, and Their Regulation. Biomedicines. 2021 9
92. Buonafine M, Bonnard B, Jaisser F. Mineralocorticoid Receptor and Cardiovascular Disease. Am J Hypertens. 2018;31:1165-74
93. Garg A, Foinquinos A, Jung M, Janssen-Peters H, Biss S, Bauersachs J. et al. MiRNA-181a is a novel regulator of aldosterone-mineralocorticoid receptor-mediated cardiac remodelling. Eur J Heart Fail. 2020;22:1366-77
94. Zhu J, Yao K, Guo J, Shi H, Ma L, Wang Q. et al. miR-181a and miR-150 regulate dendritic cell immune inflammatory responses and cardiomyocyte apoptosis via targeting JAK1-STAT1/c-Fos pathway. J Cell Mol Med. 2017;21:2884-95
95. Blanco-Domínguez R, Sánchez-Díaz R, de la Fuente H, Jiménez-Borreguero LJ, Matesanz-Marín A, Relaño M. et al. A Novel Circulating MicroRNA for the Detection of Acute Myocarditis. The New England journal of medicine. 2021;384:2014-27
96. Zhang L, Ding H, Zhang Y, Wang Y, Zhu W, Li P. Circulating MicroRNAs: Biogenesis and Clinical Significance in Acute Myocardial Infarction. Frontiers in physiology. 2020;11:1088
97. Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I. et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019;569:418-22
98. Xu H, Ding Y, Yang X. Overexpression of Long Noncoding RNA H19 Downregulates miR-140-5p and Activates PI3K/AKT Signaling Pathway to Promote Invasion, Migration and Epithelial-Mesenchymal Transition of Ovarian Cancer Cells. BioMed research international. 2021;2021:6619730
99. Park M, Shen YT, Gaussin V, Heyndrickx GR, Bartunek J, Resuello RR. et al. Apoptosis predominates in nonmyocytes in heart failure. American journal of physiology Heart and circulatory physiology. 2009;297:H785-91
100. Bheri S, Davis ME. Nanoparticle-Hydrogel System for Post-myocardial Infarction Delivery of MicroRNA. ACS nano. 2019;13:9702-6
Author contact
![]() Corresponding authors: Chenying Fu (fcying_2004com) and Quan Wei (weiquanedu.cn). West China Hospital, Sichuan University, Chengdu, Sichuan Province, China.
Corresponding authors: Chenying Fu (fcying_2004com) and Quan Wei (weiquanedu.cn). West China Hospital, Sichuan University, Chengdu, Sichuan Province, China.