Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Gut microbiota and GI cancer...
Gut microbiota and therapies for...
Future directions
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(10):4101-4117. doi:10.7150/ijbs.69331 This issue Cite
Review
Gut microbiome in gastrointestinal cancer: a friend or foe?
Yang Liu1, Yoshifumi Baba2,3, Takatsugu Ishimoto2,4, Xi Gu1, Jun Zhang2,4, Daichi Nomoto2, Kazuo Okadome2, Hideo Baba2,5, Peng Qiu6 ![]()
1. Department of Oncology, Shengjing Hospital of China Medical University, Shenyang, 110004, Liaoning province, China.
2. Department of Gastroenterological Surgery, Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan.
3. Department of Next-Generation Surgical Therapy Development, Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan.
4. Gastrointestinal Cancer Biology, International Research Center for Medical Sciences, Kumamoto University, Kumamoto, Japan.
5. Center for Metabolic Regulation of Healthy Aging, Kumamoto University, Kumamoto, Japan.
6. Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, 110004, Liaoning Province, China.
Received 2021-11-22; Accepted 2022-6-4; Published 2022-6-21
Abstract

The impact of the gut microbiome on host health is becoming increasingly recognized. To date, there is growing evidence that the complex characteristics of the microbial community play key roles as potential biomarkers and predictors of responses in cancer therapy. Many studies have shown that altered commensal bacteria lead to cancer susceptibility and progression in diverse pathways. In this review, we critically assess the data for gut microbiota related to gastrointestinal cancer, including esophageal, gastric, pancreatic, colorectal cancer, hepatocellular carcinoma and cholangiocarcinoma. Importantly, the underlying mechanisms of gut microbiota involved in cancer occurrence, prevention and treatment are elucidated. The purpose of this review is to provide novel insights for applying this understanding to the development of new therapeutic strategies in gastrointestinal cancer by targeting the microbial community.
Keywords: gut microbiota, GI cancer, carcinogenesis, chemotherapy
Introduction
The human symbiotic microbial community consists of >100 trillion microorganisms, including bacteria, viruses, fungi and protozoa, which mainly live on the surface of human epithelia, including the skin and digestive and respiratory tracts [1]. The gut microbiota is the main components of the human microbial community, which has the largest number of bacteria and the highest diversity compared to the microbiome in other parts of the body. The gastrointestinal (GI) tract is an extension of our natural environment, providing a suitable living environment and rich nutrition for the microbiota. The gut microbiota produces short-chain fatty acids (SCFAs) by metabolizing dietary fiber, synthesizing vitamins B and K [2], metabolizing a variety of compounds such as sterols and exogenous substances, and regulating immune function, which have beneficial effects on the human body [3]. Microbes in the GI tract can play a role in the maintenance of host physiological and immune functions and contribute to the pathogenesis of a variety of chronic diseases by interfering with the immune system [4]. Numerous risk factors, including pathogens, are thought to be associated with the development of human cancer [5-7]. A mechanistic link between commensal gut microbiota and cancer has emerged in recent years, including convincing evidence of the gut microbiome in cancer onset, development and regulation of therapeutic response [8-10]. In this review article, we have reviewed the effect of the gut microbiome on GI carcinogenesis and discuss the role and implication of gut microbiota in the treatment of GI cancer. Modulation of the gut microbiome may be used as an adjunct strategy for anticancer therapy. Thus, the genetic background of the patients and lifestyle factors such as diet and exercise also affect the diversity of the intestinal microbiome and ultimately influence cancer occurrence and treatment.
Gut microbiota and GI cancer carcinogenesis
With the rapid development of next-generation high-throughput sequencing (NGS), the role of microbial communities in the host ecosystem has been comprehensively characterized [11, 12]. Microbes in the GI tract provide protection and maintain a balance in the host by regulating a large number of basic biological processes [13] (Fig. 1). Microbiome disturbance, known as dysbiosis, is associated with a variety of pathological conditions, such as neurological and behavioral disorders, diabetes, obesity, rheumatic and inflammatory diseases, metabolic syndrome, liver cirrhosis and even various cancers [14, 15]. Dysbiosis leads to microecological changes and activates inflammatory factors in the GI mucous membranes, such as the activation of oxidative stress, the release of nitric oxide (NO), the production and secretion of pro-inflammatory cytokines and cyclo-oxygenase 2 (COX-2). Harmful microbial metabolites can affect the normal state of extra-intestinal organs in many ways, with adverse impacts on the gut-brain axis and gut-liver axis [16, 17]. With regard to carcinogenesis, it is believed that dysbiosis should be considered as a continuous deviation of host-microbiota from a health-related and homeostatic state for promoting and/or sustaining various cancer phenotypes [18]. Well-balanced gut microbiota plays a crucial role in a healthy life, while dysbiosis can have inflammatory consequences that aggravate the development of cancer [19]. Numerous preclinical studies have revealed the role of gut microbiota in the occurrence and progression of cancer through different mechanisms.
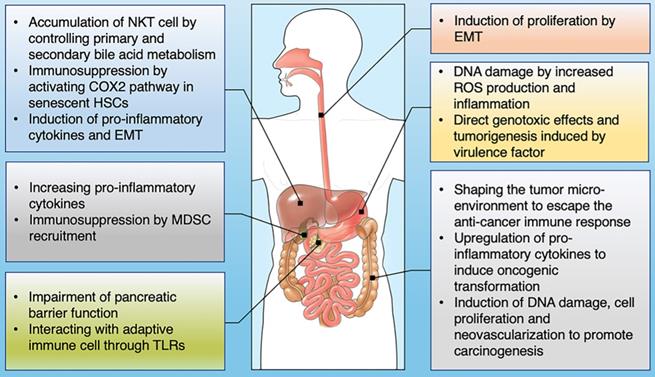
The influence of gut microbiome on the development of GI cancer. Shown are the main mechanisms through which the gut microbiota is proposed to affect the tumorigenesis across GI cancer types, including esophagus, stomach, colon, liver, pancreas and bile duct.
Esophageal squamous cell carcinoma (ESCC)
The esophageal mucosa has a large surface area that is located between the oropharynx and stomach with a large number and diverse microbiota. Microbes can easily enter the esophagus by swallowing and reflux. Blackett et al. found a significant increase in the abundance of Campylobacter in patients with gastroesophageal reflux disease (GERD) and Barrett's esophagus [20]. Campylobacter is considered to induce inflammation of the esophageal mucosa, followed by epithelial metaplasia, eventually leading to malignant transformation [21]. Elliott et al. found that microbial diversity in esophageal adenocarcinoma (EAC) decreases while the relative abundance of Lactobacillus fermentum increases, and some species of Lactobacillus are enriched in tumors in about 50% of EAC patients [22]. Zaidi et al. found that Escherichia coli (E. coli) is abundant in EAC. The expression of certain Toll-like receptors (TLR1, 3, 6, 7 and 9) in tumor tissue from a rat model of EAC was also significantly up-regulated [23]. The pathogenesis of Helicobacter pylori (H. pylori) in EAC is still controversial. Etiological studies have shown that H. pylori might reduce the incidence of EAC by inhibiting gastric acid secretion to reduce reflux esophagitis, and alter the number of T cells [24]. In contrast, H. pylori has been demonstrated to induce the occurrence of GERD [25]. Several studies have found that Tannerella forsythia is associated with a higher risk of EAC, symbiotic Neisseria and Streptococcus pneumoniae are associated with a lower risk of EAC and enrichment of Porphyromonas gingivalis (P. gingivalis) is an important risk factor for ESCC [26-29]. P. gingivalis triggers the nuclear factor (NF)-κB signaling pathway to induce proliferation and metastasis of ESCC cells [30], and it induces epithelial-mesenchymal transformation (EMT) through transforming-growth-factor (TGF)-dependent Smad/YAP/TAZ signaling pathway [31] (Fig. 2). Fusobacterium nucleatum (F. nucleatum) is associated with the stage of ESCC and poor prognosis and could be used as a biomarker for the outcome of ESCC. It has also been revealed by KEGG enrichment analysis that F. nucleatum activates the chemokine CCL20 to promote tumor invasiveness [32].
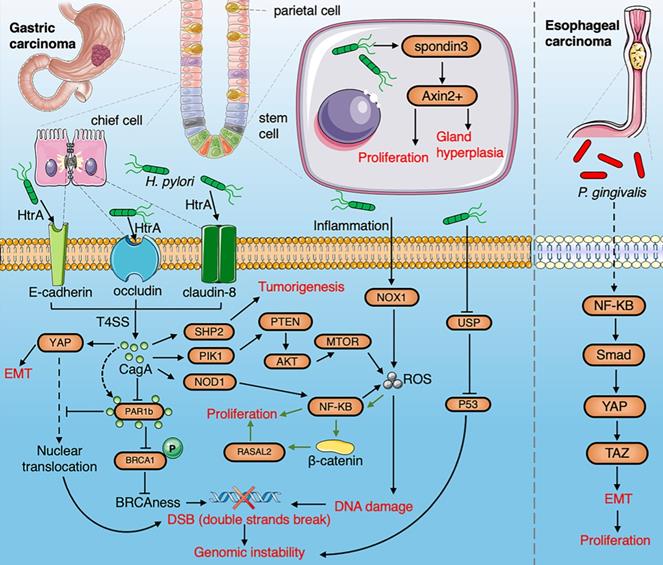
A summarized figure demonstrating the linkage between the gut microbiome and gastric cancer and esophageal cancer. Section 1 Gastric cancer: HtrA protease secreted by H.pylori produce CagA protein by cleaving occludin, claudin-8 and E-cadherin. The cagA protein promotes EMT by activating the YAP pathway and inducing tumorigenesis through activation of NF-κB, PTEN, and SHP2 pathways. The production of ROS induced by H.pylori via the NF-κB pathway and inflammation contributes to DNA damage. H. pylori infection induces gastric stem cells to proliferate and stimulate gland hyperplasia through R-spondin 3 and Axin2. The interaction of CagA and PAR1b induces genomic instability by inhibiting the nuclear translocation of BRCA1 and YAP. The hypermethylation of USF1 by H. pylori degrades the p53 proteasome to promote gene instability. H.pylori-induced inflammation upregulates NOX1/ROS signaling pathway to promote the stemness of gastric cancer. H. pylori promotes the proliferation of cancer cell infection by activating the RASAL2/β-catenin signaling pathway. Section 2 ESCC: P. gingivalis trigger activation of NF-κB to induce EMT through the Smad/YAP/TAZ signaling pathway.
Gastric cancer
H. pylori infection is one of the major risk factors for the development of gastric adenocarcinoma [33]. Among the patients infected with H. pylori, the diversity of gastric microbiota is lower than that of healthy people and successful elimination of H. pylori from the GI tract using antibiotics reduces the risk of gastric cancer by 75% [34]. Colonization of the human gastric mucosa by H. pylori affects the plasticity and homeostasis of gastric epithelial cells and promotes the carcinogenesis of epithelial cells [35]. HtrA protease secreted by H. pylori destroys the protective layer of epithelial cells by cleaving three proteins: closed protein (occludin), tight junction protein-8 (claudin-8) and epithelial cadherin (E-cadherin), and secreted CagA protein reprograms host cells and induces carcinogenesis [36]. The stem cell driving signal R-spondin controls the renewal of two types of gastric stem cells after H.pylori infection via the Wnt pathway [37]. Intragastric colonization of the gut microbiota has been shown to promote H. pylori-associated gastric cancer [38, 39]. The microbial community in patients with gastric cancer increases nitrosation, which is consistent with increased genotoxicity potential [40]. Recent studies have also highlighted that miRNA-mediated regulation and epigenetic modifications, through DNA methylation, are key events in H. pylori-induced tumorigenesis, resulting in a stronger carcinogenic activity of the CagA protein [41, 42]. The CagA protein of H. pylori promotes genetic instability, EMT and carcinogenesis by activating novel CagA‐dependent pathways including YAP [43]. CagA-positive H.pylori inhibits the partitioning-defective 1b (PAR1b). The interaction of CagA and PAR1b inhibits the nuclear translocation of BRCA1 and YAP, which in turn induces genomic instability [44]. H.pylori can lead to the hypermethylation of the upstream promoter region of transcription factor (USF1), degrades the p53 proteasome, and promotes the accumulation of gene mutations, thereby accelerating the occurrence of gastric cancer [45]. H.pylori-induced inflammation upregulates NF-κB through the nicotinamide adenine dinucleotide phosphate oxidase 1 (NOX1)/ROS signaling pathway, thereby promoting the uncontrolled proliferation of gastric epithelial stem cells [46]. H. pylori infection activates ras protein activator like 2 (RASAL2) by β-catenin to promote the proliferation of cancer cell [47]. H. pylori also induces methylation of CpG islands [48]. Gastric colonization by other intestinal bacteria such as Peptostreptococcus stomatis, Streptococcus anginosus, Parvimonas micra (P. micra), Slackia exigua and Dialister pneumosintes affects the risk of developing gastric cancer [49]. Intragastric colonization by the gut microbiota has been shown to promote the occurrence of H. pylori-associated gastric cancer [50]. Following the eradication of H. pylori, other gastric microbes can play a potential role in the development and persistence of gastric precancerous lesions by functional pathways (Fig. 2), which might be used as therapeutic targets for the prevention of gastric cancer [51].
Hepatocellular carcinoma (HCC)
The gut-liver axis composed of the portal system and the biliary system is the anatomical basis for the reciprocal interaction of gut microbiota and liver [52]. The dysbiosis and gut leakage induced by various chronic pathogenic factors such as viruses, alcohol and metabolic abnormalities make the liver exposed to the gut microbiota and the metabolites. The liver regulates the host metabolism, immune response and affects intestinal function through bile secretion and enterohepatic circulation [53]. Gut microbiota (mainly pathogenic bacteria) related molecular patterns and metabolites, such as deoxycholic acid (DCA) and lipopolysaccharide (LPS), can promote liver inflammation, fibrosis and genotoxicity, activate antiapoptotic signaling pathways and trigger immune responses which contribute to HCC development[54] (Fig. 3). Therefore, the gut microbiota plays important role in promoting the development of HCC, increasing the abundance of LPS-producing bacteria that activate the NF-κB signaling pathway and producing a variety of pro-inflammatory cytokines (TNF-α, IL-6 and IL-1), leading to liver inflammation and oxidative damage [55]. Ren et al. have found that 13 genera including Gemmiger and Parabacteroides are significantly enriched in patients with early HCC compared with liver cirrhosis patients. Butyrate is a kind of SCFAs produced by the intestinal microbiome, which plays a very important role in maintaining the integrity of intestinal epithelial cells, inhibiting intestinal inflammation and tumorigenesis [56, 57]. The decreasing of butyrate-producing bacteria leads to the destruction of the intestinal mucosa and tumorigenesis of HCC [58]. Activation of TLR4 induced by LPS can enhance the invasiveness of HCC and induce EMT [59]. It has been shown that primary bile acid enhances the expression of chemokine CXCL16, which increases the accumulation of natural killer (NK) T cells, resulting in inhibition of HCC [60]. Lipoteichoic acid (LTA) in gut microbiota collaboratively with DCA can upregulate the expression of senescence-associated secretory phenotype (SASP) and COX-2 through TLR2 in hepatic stellate cells, while COX-2-mediated prostaglandin E2 (PGE2) inhibits anti-tumor immunity through prostaglandin EP4 receptors [61]. Clostridium metabolizes bile acid into DCA, thus increasing the serum level of DCA in HCC [62]. Yoshimoto et al. have proved that inhibiting DCA production or modulating gut microbiota efficiently prevented the tumorigenesis of HCC [63]. Diet fat, cholesterol, fiber or carbohydrate could modulate gut microbiome composition by a number of metabolic pathways to contribute to the development of non-alcoholic steatohepatitis (NASH) and non-alcoholic fatty liver disease (NAFLD)-HCC [64]. Dysbiosis caused by a high-fat diet can affect the expression of specific miRNA in HCC [65]. Dietary cholesterol induces an increase in taurocholic acid and a decrease in indole propionic acid which drive the occurrence of NAFLD-HCC [66]. It was found that the intake of soluble cellulose can lead dysbiosis and induce cholestatic liver cancer [67]. Dietery fructose was found to promote the liver lipid synthesis and re-shape the microbial composition with an increased abundance of Bacteroides in NASH which increases the risk of NAFLD-HCC [68, 69]. Liu et al. investigated the differences in gut microbes between hepatitis B virus (HBV)-related HCC (B-HCC) and non-HBV non-hepatitis-C-virus-associated HCC (NBNC-HCC). They found a greater increase in pro-inflammatory bacteria (Escherichia shigella, Enterococcus) in the feces of NBNC-HCC patients, and decreased levels of Faecalibacterium, Ruminiclostridium and Ruminococcus, which led to a decrease in the potential of anti-inflammatory SCFAs.
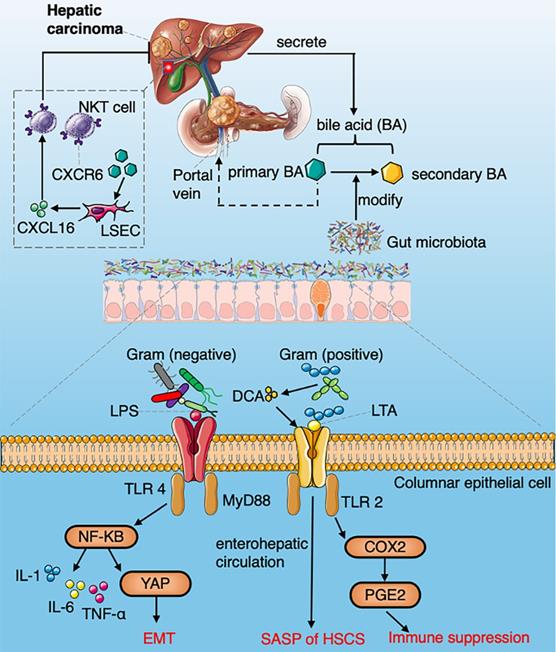
A summarized figure demonstrating the mechanisms of gut microbiome regulating HCC development. Section 1 Commensal bacteria regulate the metabolism of primary and secondary bile acids to control NKT accumulation via CXCL16. Section 2 LPS arising from gram-negative bacteria activate the NF-κB signaling pathway and produce a variety of pro-inflammatory cytokines. LTA in gram-positive bacteria collaboratively with DCA upregulate SASP and COX-2 through TLR2 in HSCs, COX2 mediated PGE2 attenuate anti-tumor immunity through PTGER4.
Compared with NBNC-HCC patients, B-HCC patients exhibit markedly contrasting results in terms of bacterial composition and biological pathways, which suggest that modifying specific microbes could provide therapeutic benefits for B-HCC and NBNC-HCC [70]. E. coli impaired and penetrated the gut vascular barrier, and colonized the liver to recruit immune cells, thus facilitating the formation of the premetastatic niche and promoting liver metastasis [71]. The enrichment of F. nucleatum reduced the diversity of gut microbiota in mice leading to dysbiosis of intestinal microbiota. F. nucleatum significantly increased the serum level of pro-inflammatory cytokines and reduced the cytotoxicity of immune cells to promote liver metastasis in mice [72]. Current data from preclinical and clinical studies pointing to the targeting gut microbiota and gut-liver axis can be used to monitor and prevent the progression of HCC.
Colorectal cancer (CRC)
CRC is one of the most common forms of malignancy with high morbidity and mortality [73]. Although the main gut microbiota involved in CRC has not been fully determined, the importance of F. nucleatum, E. coli and Bacteroides fragilis (B. fragilis) in CRC has been proved, which could promote tumor progression by inducing DNA damage and other signaling pathways [74] (Fig. 4). The Fap2 protein can combine with the Gal-GalNAc enriched on the surface of CRC cells to facilitate F. nucleatum colonization in the host cell [75]. F. nucleatum binds to E-cadherin of intestinal epithelial cells through FadA, activates the β-catenin signaling pathway, induces NF-κB pathway activation, upregulates proinflammatory cytokines (TNF-α, IL-6, IL-8 and IL-1β), and induces Fap2 to bind to TIGIT receptors on NK cells and other tumor infiltrating lymphocytes (TILs) thus promoting cancer progression and immune escape [76, 77]. It has the capacity to activate the NF-κB pathway via TLR4 and MyD88 to promote the proliferation and invasion of CRC [78]. Bullman et al. have demonstrated that F. nucleatum is enriched in primary lesions of CRC, and can be found in liver metastasis [79]. B. fragilis and E. coli strain psk+ penetrate the mucous layer on the surface of the colon, which usually performs important barrier functions [80]. The toxins released by E. coli and B. fragilis damage double-stranded DNA and promote cellular inflammation and oncogenic mutation [81, 82]. EspF, a targeted mitochondrial effector protein secreted by enteropathogenic E. coli, removes DNA mismatch repair proteins from the host, thereby reducing DNA repair activity and increasing mutation rates in patients with CRC. E. coli also increases the level of reactive oxygen species (ROS) and promotes the accumulation of spontaneous mutations [83]. Peptostreptococcus anaerobius (P. anaerobius) is enriched in CRC and promotes carcinogenesis by increasing the level of ROS via TLR2 and/or TLR4 signaling pathways in mice [84]. Based on an analysis of 526 metagenomic samples from a multinational cohort, seven bacteria (B. fragilis, F. nucleatum, Porphyromonas asaccharolytica, P. micra, Prevotella intermedia, Alistipes finegoldii, and Thermanaerovibrio acidaminovorans enriched in CRC were identified among different populations. The potential bacterial diagnostic markers were robust among these populations. The bacteria enriched in CRC were associated with LPS-induced inflammation and energy biosynthesis pathways [85]. Yachida et al. have shown that the abundance of Atopobium parvulum and Actinomyces odontolyticus increases in early CRC, and enrichment of these two bacteria promotes the occurrence of CRC, which suggests that it could be used as a biomarker for early detection of CRC [86]. Enterotoxigenic B. fragilis (ETBF) has been found to induce IL-17 and trigger the NF-κB pathway to enhance the production of C-X-C chemokine. Myeloid-derived Suppressor Cells (MDSCs) are recruited by ETBF infection which inhibits the activity of cytotoxic CD8 + T cells and ETBF induces the expression of matrix metalloproteinase 9 (MMP9) and vascular endothelial growth factor A (VEGFA) thus triggering CXCL1 and CXCL2 via IL-17 [87, 88] (Fig. 4). Oral administration of CRC-associated Streptococcus gallolyticus in mice with dextran-sodium-sulfate-induced CRC results in increased tumor burden, selective recruitment of CD11b + myeloid cells and increased expression of cytokines (including IL-6 and IL-8) [89]. Gut microbiota is also involved in regulating the expression of plasma membrane transporter SLC5A8, cell-surface G-protein-coupled receptor GPR109A and GPR43 in the colon, enhancing the memory potential of CD8+ cells to manipulate the immune response and tumor cell proliferation and apoptosis [90-92]. Therefore, the strategy of promoting transformation from a “cold tumor” to a “hot tumor” (characterized by infiltration of CD8+ T cells) by modulating the gut microbiota will be a novel, timely and interesting therapeutic approach.
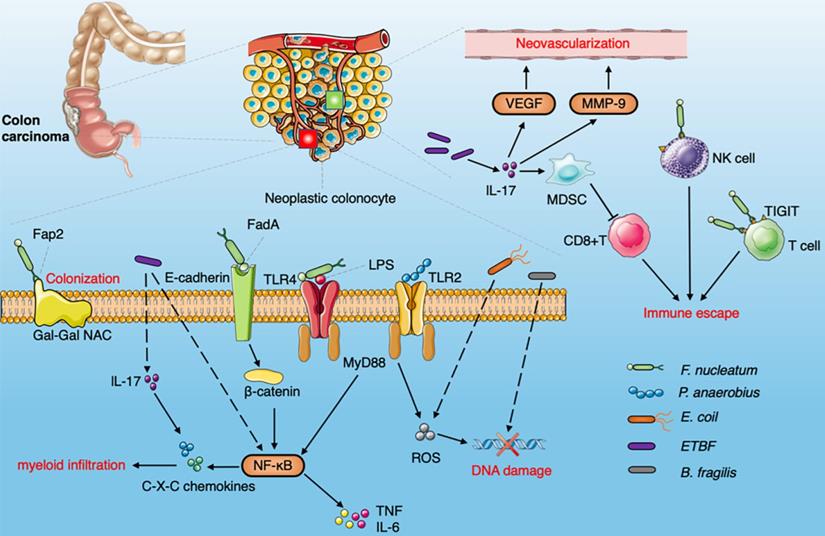
Diagram summarizing the oncogenic interaction between the gut microbiome and CRC. The Fap2 protein combined with Gal-GalNAc is enriched on the surface of CRC cells to promote the colonization of F.nucleatum. F. nucleatum binds to E-cadherin on intestinal epithelial cells through FadA , activates the β-catenin signaling pathway, induces NF-κB pathway activation and upregulates pro-inflammatory cytokines, produces Fap2 to bind to TIGIT receptors on NK cells and other TILs. F.nucleatum activate NF-κB pathway via TLR4 and MYD88 to promote cell proliferation and invasion. Peptostreptococcus anaerobius increase the level of ROS via TLR2 and/or TLR4 signaling pathways. ETBF induce IL-17 and NF-κB pathway to enhance the production of C-X-C chemokine. MDSCs recruited by ETBF via IL-17 to inhibit the activity of cytotoxic CD8 + T cells. ETBF enhances the expression of MMP9 and VEGFA to induce neovascularization through IL-17. E. coli can also increase the level of ROS and promote the accumulation of spontaneous mutations. B. fragilis induces DNA damage.
Cholangiocarcinoma
The occurrence of cholangiocarcinoma is related to gut dysbiosis [93]. Chng et al. found that the colonized flora in cholangiocarcinoma tissue was significantly different from paracancerous tissue and normal liver tissue, and Pseudomonadaceae is enriched in tumor lesions. The abundance of Bifidobacteriaceae, Enterobacteriaceae and Enterococcaceae is associated with the development of Opisthorchis viverrini (O. viverrini) cholangiocarcinoma [94]. Di Carlo et al showed that the patients with cholangiocarcinoma had a poor prognosis when infected with Klebsiella pneumonia (K. pneumonia) [95]. Jia et al. showed that Actinomyces, Lactobacillus, Peptostreptococcaceae and Alloscardovia were significantly enriched in intrahepatic cholangiocarcinoma. The enrichment of glycoursodeoxycholic acid and tauroursodeoxycholic acid (TUDCA) plasma-stool ratios (PSRs) can be used as biomarkers to distinguish intrahepatic cholangiocarcinoma and HCC. The increase of Ruminococcaceae was related to the concentration of plasma IL-4, IL-6 and Chenodeoxycholic acid (CDCA) in intrahepatic cholangiocarcinoma [96]. Avilés-Jiménez et al. reported that Nesterenkonia was decreased in extrahepatic cholangiocarcinoma, while the enrichment of Actinomyces, Novosphingobium, Methylophilaceae, Fusobacterium, Prevotella and H. pylori increased compared with normal biliary tract [97]. H. pylori has been proved to promote EMT of bile duct epithelial cells, which is related to the occurrence of cholangiocarcinoma [97]. Gram-negative bacteria and LPS induce CXCR2+ polymorphonuclear-MDSC accumulation through TLR4-dependent CXCL1 production to control hepatocytes to form an immunosuppressive microenvironment, thereby promoting cholangiocarcinogenesis [98].
Pancreatic ductal adenocarcinoma (PDAC)
Researchers have gradually gained an in-depth understanding of PDAC, and there is evidence that the occurrence, development and treatment of PDAC are related to the gut microbiota. Fan et al. have shown that enrichment of P. gingivalis and Actinobacillus actinomycetemcomitans is associated with a high risk of PDAC [99]. Patients with high antibody levels to P. gingivalis had a more than a two-fold increased risk of developing PDAC compared with patients with normal antibody levels [100]. Thirteen phyla of bacteria were found in PDAC tissues, mainly including Proteobacteria (45%), Bacteroidetes (31%) and Firmicutes (22%) [101]. At present, there are several mechanisms for the bacteria to enter the pancreas from the digestive tract, such as the portal circulation, mesenteric lymph nodes and the lower digestive tract. Farrell and colleagues have demonstrated that Neisseria elongata and Streptococcus mitis were significantly decreased in PDAC patients compared with healthy people [102]. In a mouse model, bacteria were involved in the accelerated development of PDAC, which might be mediated by bacterial metabolites. Another possible mechanism is that bacteria may transfer from the intestinal or oral cavity to the pancreas, accompanied by impaired pancreatic barrier function, and become colonized in the pancreas to reset immune tolerance and promote the progression of PDAC. In a small number of patients with long-term survival of PDAC, the gut microbiota in the tumor were significantly more diverse than that in the tumors of short-term survival patients, while Pseudoxanthomonas, Saccharopolyspora, Streptomyces and Bacillus Clausii (B. Clausii) were significantly enriched in tumors of long-term survival patients. The number of mature CD8+ T cells and granular B cells in long-term survival patients was significantly more than that in short-term survival patients, which was positively correlated with the amounts of Pseudoxanthomonas, Saccharopolyspora, Streptomyces in the gut. The bacteria in these tumors may promote an anti-tumor immune response via recruiting and activating of CD8+ T cells, and fecal microbiota transplant (FMT) from long-term survivors promotes an immune response and suppresses tumors in mouse models by changing the tumor microbiome [103]. Preclinical models have shown that intestinal flora could induce transcriptome changes, interact with adaptive immune cells, and induce immunosuppression of PDAC cells through TLR2 and TLR5 pathways in tumor-associated macrophages (TAMs), which plays an important role in the occurrence and development of PDAC [101, 104, 105].
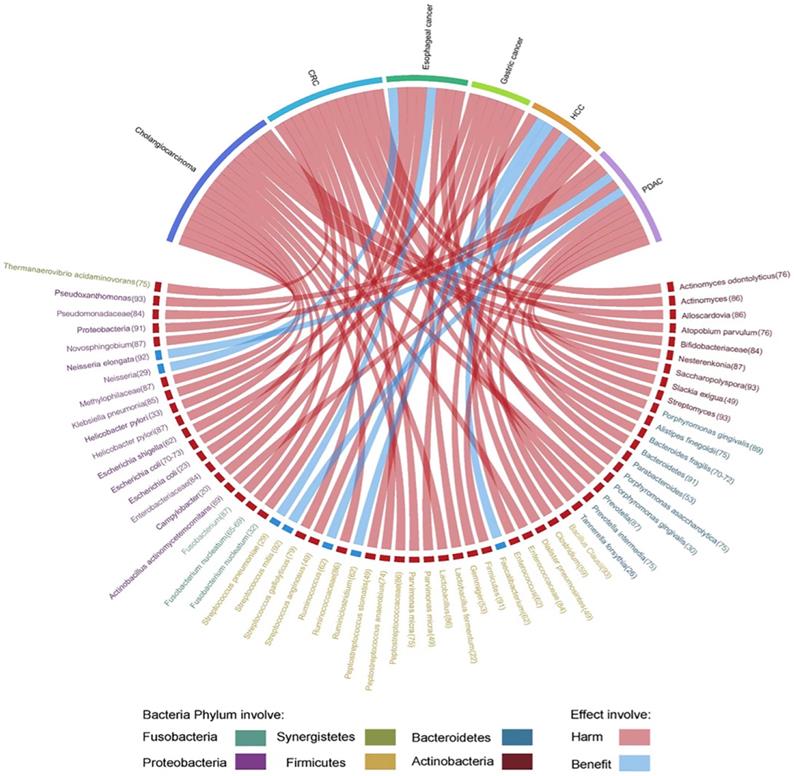
Herein, we highlight the effect of gut microbiota across the continuum of GI cancer and discuss the correlation of bacteria with tumorigenesis by circos plots (Fig. 5), however, subtle complexities exist, and many questions remain concerning the potential for exploiting the gut microbiota that regulate key mechanisms in carcinogenesis.
Evidence of the gut microbiota enriched in GI cancer playing a crucial role in carcinogenesis. Circos plots illustrating the correlation of bacteria with tumorigenesis in GI cancer. The red ribbons represent the harmful effects of bacteria on GI cancer development. The blue ribbons represent the beneficial effects of bacteria on GI cancer development. The causality of the microbiota in GI cancer has not yet been fully elucidated. Different taxa are divided into six groups and colored by their phylum. Numbers indicate the references that highlight these relationships.
Gut microbiota and therapies for GI cancer
Gut microbiota in chemotherapy for GI cancer
The gut microbiota regulates host responses to chemotherapeutic drugs by promoting drug efficacy, influencing anticancer effects and toxicity (Table 1) [106]. The interaction between gut microbiota and chemotherapeutic drugs can be manipulated by the TIMER mechanistic framework through translocation, immunomodulation, metabolism, enzyme degradation, and ecological variation [107]. Yamamura et al. have found that the DNA of intratumoral F. nucleatum is associated with the response to neoadjuvant chemotherapy in ESCC patients [108]. Liu et al. have found that F. nucleatum is an intracellular bacteria that survives in ESCC cells and confers chemoresistance via autophagy [109]. F. nucleatum targets the TLR4/MyD88 signaling pathway and reduces expression of miRNA-4802 and miRNA-18a, activating autophagy and upregulating expression of autophagy-related genes, which induces chemoresistance in CRC. It has been found that F. nucleatum infection reduces chemosensitivity to 5-FU by regulating baculoviral IAP repeat containing 3 (BIRC3) via the TLR4/NF-κB signaling pathway in adjuvant chemotherapy of CRC patients [110, 111]. Gemcitabine can be metabolized into inactive 2',2'-difluorodeoxyuridine by Mycoplasm hyorhinis through CDDL gene in a mouse CRC model. Furthermore, it has been shown that most of the bacteria in PDAC belong to the Gammaproteobacteria, which have the CDDL gene that metabolizes gemcitabine, and this effect can be antagonized by ciprofloxacin [112]. A variety of commensal microbiota can affect the efficacy of conventional chemotherapy by modulating the tumor microenvironment. The absence of Lactobacillus decreases the cytotoxicity of oxaliplatin by reducing the production of ROS [113]. Cyclophosphamide (CTX) induces its anticancer effect by interfering with various immune signaling cascades. A preclinical study has found that CTX-induced immune activation requires the participation of some bacterial species such as Enterococcus hirae (E.hirae). CTX induces the bacteria to translocate to lymph nodes and the spleen to stimulate the host immune response. Subsequent studies have also found that E. hirae and Barnesiella intestinihominis are necessary for the anti-tumor effect of CTX [114, 115]. Irinotecan is converted into its active form (SN-38) through cleavage of the side chains of carboxylesterase in plasma, intestinal mucosa, liver and tumor cells. Liver UDP-glucuronosyltransferase (UGT)A1 and UGT1A9 and extrahepatic UGT1A7 play a major role in the detoxification of SN-38. The inactive form of bile secretion in the intestinal cavity, SN-38G, is transformed into active metabolite SN-38, by β-glucuronidase produced by gut microbiota. Intestinal mucosal injury and diarrhea are directly caused by irinotecan [116]. Butyrate could promote the efficacy of oxaliplatin by modulating the antitumor cytotoxic CD8+ T cell responses via activating the IL-12 signaling pathway [117]. A prospective study suggested that the gut microbiota may be used as potential biomarkers for predicting the response to neoadjuvant chemoradiotherapy of patients with locally advanced rectal cancer (LARC) [118]. Further baseline analysis of samples before neoadjuvant radiotherapy and chemotherapy found that some bacteria related to SCFA metabolism were significantly enriched in the LARC neoadjuvant chemotherapy response group, while Clostridium was significantly enriched in the non-response group. In addition, the beneficial microflora of the response group was more enriched than that of the non-response group, while the pathogenic bacteria of the non-response group were more enriched than the response group [119].
Gut microbiota involved in therapeutic responses and side-effects of GI cancer
| Treatment | Gut Microbiota | Cancer type | Drug | Response/effect | Ref. |
|---|---|---|---|---|---|
| Response effect in chemotherapy | F. nucleatum | CRC | 5-Fu Oxaliplatin | poor | [109,110,111] |
| ESCC | CDDP Docetaxel | ||||
| Gammaprotect-obacteria | CRC | Gemcitabine | poor | [112] | |
| Clostridium | LARC | poor | [119] | ||
| Enterococcus hirae | CRC | CTX | favorable | [115] | |
| Barnesiella intestinihominis | CRC | CTX | favorable | [115] | |
| Response effect In ICIs | Prevotella | CRC | anti-PD-1/PD-L1 | favorable | [143] |
| Ruminococcaceae | CRC | anti-PD-1/PD-L1 | favorable | [143] | |
| Lachnospiraceae | CRC | anti-PD-1/PD-L1 | favorable | [143] | |
| Eubacterium | CRC | anti-PD-1/PD-L1 | favorable | [143] | |
| Lactobacillus | CRC | anti-PD-1/PD-L1 | favorable | [143] | |
| Streptococcus | CRC | anti-PD-1/PD-L1 | favorable | [143] | |
| Bifidobacterium pseudolongum | CRC | anti-CTLA-4/anti-PD-L1 | favorable | [149] | |
| Therapeutic side-effect | Lactobacillus | oxaliplatin | favorable | [113] | |
| Roseburia | radiotherapy | poor | [123] | ||
| Clostridium IV | radiotherapy | poor | [123] | ||
| Faecalibacterium | radiotherapy | poor | [123] | ||
| Lactobacillus rhamnosus | radiotherapy | favorable | [125] | ||
| Lachnospiraceae | radiotherapy | favorable | [126] | ||
| Enterococcaceae | radiotherapy | favorable | [126] |
Gut microbiota in radiotherapy for GI cancer
The mechanism of intestinal microflora regulating radiotherapy response is still unclear. Radiotherapy can promote tumor cytotoxicity and the systemic immune response regulated by the immune system [120]. Because the gut microbiota participates in the regulation of immunogenic cytotoxicity in traditional anticancer therapy strategies, it may also play a similar role in radiotherapy-mediated immune responses (Table 1). The gut microbiota of patients and experimental mice receiving radiotherapy is destroyed, which could cause diarrhea and colitis, partly mediated by IL-1β [121]. Radiotherapy can also lead to intestinal cell apoptosis and destruction of intestinal barrier function, these changes regulate intestinal immune response, leading to intestinal inflammation [122]. The low diversity of the gut microbiota is related to late radiation-induced bowel disease, and the high enrichment of Roseburia, Clostridium IV, and Faecalibacterium was significantly related to radiation enteropathy, which suggests that the microbiota influence susceptibility to GI adverse effects after radiotherapy [123]. The intake of probiotics can inhibit radiation-induced cell injury [124]. Lactobacillus rhamnosus can induce mesenchymal stem cell pre-migration through the TLR2 pathway, repair the radiation-damaged intestinal mucosa, and protect the normal intestinal recess, but it has no protective effect on transplanted tumor tissue [125]. Long-term surviving mice exposed to high doses of radiation have unique microbial characteristics, in which increased abundance of Lachnospiraceae and Enterococcaceae plays a radiation protective role in reducing hematopoietic and GI tissue damage after radiation. Further studies have shown that SCFAs (especially propionate) and specific tryptophan metabolites produced by the microbiota mediate the radioprotective effect of the microflora. The microbiome and its metabolites promote hematopoiesis and intestinal injury repair, thus, helping the host resist radiation-induced injury and death [126]. FMT can improve the survival rate of irradiated mice and alleviate the radiation-induced damage [127]. 3-propionic acid derived from FMT is a key metabolite to decrease radiation-induced intestinal toxicity [128]. Commensal bacteria can regulate host responses to ionizing radiation and repair the damage induced by radiation. A better understanding of the non-targeted mechanism of radiotherapy and the regulation of bacteria is of inestimable value for improving the effectiveness of radiotherapy, reducing the adverse outcomes of radiotherapy in cancer, and optimizing the treatment of patients exposed to radiotherapy. These findings provide a potential therapeutic target for alleviation of radiation-induced injury and reduction of the adverse effects of radiotherapy in cancer.
Gut microbiota in ICIs for GI cancer
The influence of the gut microbiota goes beyond the GI tract itself and affects human immune cell dynamics [129, 130]. In addition to the direct regulation of bacteria, bacterial metabolites can be translocated from the intestinal cavity to the lamina propria of the intestinal mucosa, affecting the expression of host immune-related genes [131]. LPS and peptidoglycan are important components of the outer membrane of gram-negative bacteria. They induced intestinal immunoregulation by activating host TLRs, which are mainly expressed by intestinal epithelial cells and dendritic cells (DCs). TLRs are actively involved in mediating T cell responses to tumor cells [132-134]. Studies have revealed that the gut microbiota plays an important role in modulating the therapeutic response to ICIs [135-137]. B. fragilis contributes to the maturation of regulatory cells (Tregs) and secretion of anti-inflammatory cytokine IL-10 by activating TLR2-dependent plasmacytoid dendritic cells [138, 139]. Bifidobacterium stimulates the production of anti-CD47 antibodies by activating the STING signaling pathway, which might alter the tumor microenvironment to achieve the effect of immunotherapy [140]. It also can alleviate Checkpoint inhibitors (ICIs)-associated colitis by modulating gut microbiota composition and inhibiting Tregs cell by IL-10 [141]. Combined with anti-programmed death ligand 1 (PD-L1) therapy, oral Bifidobacterium can enhance the function of DCs, promote the initiation and accumulation of CD8 + T cells, and interact with the tumor microenvironment [142]. Patients with advanced GI cancer who received anti-PD-1/PD-L1 therapy were recruited in a comprehensive analysis study. The ratio of Prevotella/Bacteroides in the feces increased, which was associated with prolongation of progression-free survival, and the relative abundance of Prevotella, Ruminococcaceae, and Lachnospiraceae in the responders were higher than that in the non-responders [143]. The differential functional pathways of the gut microbiota, including nucleoside biosynthesis, lipid biosynthesis, glucose metabolism, and abundance of SCFAs, were related to the different clinical responses to ICI therapy. The propionate increases the differentiation and function of Tregs in the gut[144]. Tregs remodel a proinflammatory state to an anti-inflammatory system by producing anti-inflammatory cytokines in TME. The propionate causes Foxp3 + Tregs to produce IL-10, and through the GPR43 signaling pathway, inhibiting histone deacetylase (HDAC) activity to prevent colitis. The butyrate promotes the production of Foxp3+ Tregs in peripheral tissues by inhibiting HDAC. Clostridium-produced butyrate acetylates the Foxp3 gene promoter histone H3 and accelerates Foxp3+ Tregs differentiation [145]. SCFA producing bacteria (Eubacterium, Lactobacillus and Streptococcus) were positively associated with favorable outcomes of ICI therapy [143]. Kenya et al. isolated 11 bacterial strains from the feces of healthy donors. A combination of commensal bacteria can enhance the immune response against infection and cancer by increasing the level of CD8+T cells in a mouse model of CRC [146]. Cytotoxic T lymphocyte-associated antigen (CTLA)-4 blockade has been successfully used in tumor immunotherapy. The efficacy is influenced by the composition of the microbial community (B. fragilis, Bacteroides thetaiotaomicron and Burkholderiales), which highlights the important role of bacteria in the efficacy of CTLA-4 blockade [147]. Gut microbiota controls the synthesis of bile acid metabolites, thereby regulating the amount of intestinal RORγ+ Tregs and regulating intestinal homeostasis[148]. Another study identified two novel secondary bile acids (ω-MCA and isoDCA) that are potent in inducing Tregs differentiation in vitro. The isoDCA interacts with farnesoid X receptor (FXR) in dendritic cells. The interaction of FXR enhances the Tregs-inducing effect to inhibit the occurrence of CRC [56]. The metabolite Inosine derived from Bifidobacterium pseudolongum promotes Th1 cell differentiation and enhances the therapeutic effect of ICIs, and this process is mediated by T-cell specific adenosine A2A receptor signaling in a mouse model of CRC [149].
Current studies have confirmed that the gut microbiota has the potential to regulate the response to ICI therapy, which implies that targeting the gut microbiota is crucial for ICI therapy (Table 1).
Modulation of gut microbiota for GI cancer
Targeted bacterial modulations such as FMT, probiotics, prebiotics and indirect metabolites modulation by dietary and engineered bacteria have shown the potential to optimize cancer treatment and are expected to be used in personalized medicine, while fungi, yeast, viruses, and archaea are gradually emerging in the impact of GI cancer treatment.
FMT involves the transfer of functional microbiota from healthy donor feces into a patient's GI tract, rebalancing the intestinal microflora, repairing the intestinal mucosal barrier, regulating the body's immunity and inflammatory response, and providing a potential strategy for enhanced cancer therapy [103, 150]. FMT can improve the efficacy of tumor immunotherapy [19, 151] and relieve the adverse side effects of cancer treatment. One case report showed that two cancer patients had significantly abrogated colitis caused by ICIs after FMT. Preliminary data analysis has shown that the application of healthy gut microbes can eliminate ICI-associated colitis, restore the gut microenvironment and increase the density of Tregs in the colonic mucosa. FMT has been shown to have a positive therapeutic impact on immune-related adverse events [152]. There are many controversial views about FMT in cancer [153, 154], therefore, more research and clinical data are needed to demonstrate the safety and effectiveness of FMT. In order to ensure that the benefits outweigh the risks, future research should detect harmful bacteria such as multidrug-resistant bacteria in the donated feces. Although at the present we cannot be certain about what the composition of a healthy intestinal microflora should be, it is possible to identify the beneficial gut microbiota and study them in detail. This will allow us to determine the specific bacterial composition in the fecal donors, which is helpful for more detailed and in-depth studies of beneficial therapeutic responses. More active cooperation among basic, translational and clinical research and epidemiological analysis will bring many new opportunities and challenges to the potential exploration of FMT in the field of cancer treatment [155]. In addition, due to the short application time of this technology and the lack of long-term safety data, it is important to closely track and carefully record the wellbeing of patients after FMT. Therefore, we also need high-quality clinical data from properly designed trials to further study the practicability and effectiveness of FMT.
Probiotics are involved in modulating the gut microbiota, enhancing the integrity of the intestinal barrier, and inhibiting the growth of pathogenic bacteria [156]. A prospective intervention study in patients with CRC has shown that the use of probiotics containing Lactobacillus acidophilus and Bifidobacterium lactobacillus have a potential role to be a promising supplementary for the prevention and treatment of CRC. Probiotics increase the number of butyrate-producing bacteria (such as Faecalibacterium and Clostridiales spp) and reduce the number of CRC-related genera (including Clostridium and Streptococcus) [157]. Some Researchers have designed a probiotic therapy that may improve the safety of tumor immunotherapy, including immunotherapy targeting PD-L1 and CTLA-4. The probiotic drugs are released by bacteria and attack the tumor, which promotes the immune response and eventually leads to tumor regression [158]. Although probiotics are generally safe, their administration in immunocompromised cancer patients may have a potential risk of opportunistic infections and conferment of antibiotic resistance. In areas such as cancer and immune diseases [159], there is no doubt that the controversy about probiotics will continue, and individualized probiotics may be the treatment strategy of the future.
Prebiotics are indigestible food ingredients that can promote the growth of probiotics. The chemopreventive properties of prebiotics are due to the production of SCFAs, which enhance host immunity [160]. Recent studies have shown that two prebiotics, mucin and inulin, can produce different bacterial populations, suggesting that the different activity modes mediated by the two prebiotics stimulate the emergence of anti-tumor immunity in mice [161, 162]. Prebiotic spores (spore-dex) may be prepared by host-guest reaction between commercial Clostridium butyricum (C. butyricum) and chemically modified probiotic dextran. Recent research has shown that spore-dex is specifically enriched in the tumor lesions after oral administration. The dextran is fermented by C. butyricum to produce anticancer SCFAs in CRC lesions [163].
Gut microbiota-derived metabolites can alter cellular metabolism and gene regulation, thereby positively impacting the efficiency of tumor therapy, particularly SCFAs can be used as predictive biomarkers for tumor immunotherapy [1]. Industrialization, westernization, and food refining have led to dysbiosis which impairs SCFAs production. One study showed that volunteers were fed a high-protein, low-carbohydrate, and low-fiber diet resulting in low levels of butyrate produced bacteria (Roseburia and Eubacterium rectale), a lower proportion of butyrate in fecal SCFAs, and reducing of the intestinal free phenolic acid [164]. Fermentation of dietary fiber from fruits, vegetables and grains is recommended during therapy which might be supposed to affect the outcome of cancer treatment [165].
By modifying the E. coli MG1655 expressing nitric oxide synthase to bind carbon nitride (C3N4) on its surface, the photoelectron produced by carbon nitride can be transferred to E. coli under light irradiation, so that E.coli can better enrich the tumor lesions. Photo-controlled bacterial metabolite therapy (PMT) can inhibit 80% of tumor growth in CRC [166]. Niobium carbide (Nb2C)/Au nanocomposites and phototherapy enable “chemical” and “physical” regulation of bacteria, and synergistic microbiota regulation can alter the abundance and diversity of the intratumoral microbiota and disrupt metabolic pathways in the TME. The combination of bacterial manipulation and anti-tumor necrosis factor-α (TNF-α) drugs can synergistically alleviate bacterial-induced inflammation, meanwhile disrupt the metabolism of intratumoral flora to reverse drug resistance, and significantly enhance the response of cancer cells to phototherapy [167].
Intratumoral fungi in pancreatic cancer, especially Malassezia, may be highly correlated with the pathogenesis of PDA. Anti-Malassezia treatment reduces pancreatic cancer incidence by 40% in mice [168]. Fungus stimulates PDAC cells to secrete IL-33, recruits and activates type 2 innate lymphocytes (ILC2), and promotes tumor progression. IL-33 or antifungal drugs may be new targets for pancreatic cancer therapeutic targets [169]. A Fungi-based acetaldehyde generator was prepared by modifying alcohol dehydrogenase-loaded metal-organic framework nanocarriers onto the surface of Saccharomyces cerevisiae (S. cerevisiae), which can target the TAMs by mannose, induce tumor cell apoptosis and promote TAMs polarization to anti-tumor phenotype, and further enhance macrophage-mediated immunotherapy by combining anti-CD47 antibody, significantly inhibit the growth of CRC cell [170]. After intratumoral injection, yeast-derived nanoparticles can be effectively recognized by dendritic cells, and can effectively migrate to the lymphatic drainage area, so as to reverse the immunosuppressive microenvironment and inhibit tumor growth. The combination of yeast-derived nanoparticles and PD-L1 antibody has shown a superior antitumor effect in CRC. It can not only effectively eliminate in situ tumor, but also inhibit the growth of distal spreading cancer cells [171].
Oncolytic viruses have been proven to be an ideal therapeutic approach to enhance the response of ICIs [172]. The mechanisms of oncolytic viruses include carcinolysis, TME modulation, TILs recruitment, angiogenesis and initiation of immune response. Oncolytic viruses are able to enhance immunogenic cell death, which further leads to the recruitment of innate immune cells to form tumor-specific T cells [173]. Bacteriophages have the ability to modulate immunity and kill specific bacteria, and the use of phages may become a precise treatment plan that can target “cancer-promoting bacteria” [174]. Bacteriophages loaded with silver nanoparticles selectively eliminate F. nucleatum, providing a favorable microenvironment for tumor immunotherapy [175].
There are growing evidence that archaea are involved in human cancers [176]. Archaea produces a variety of metabolites by utilizing unique metabolic pathways, and cancer-related metabolites (such as polyamines and SCFAs) of archaea are prevalent and diverse in oral and gut, which are mainly related to the TACK superphylum and Euryarchaeota, especially methanogenic archaea [177]. However, there are very few studies to elucidate how archaea regulate cancer treatment up to now.
Future directions
The ecological imbalance within the individual appears to be a precursor to tumorigenesis, and maintaining an optimal composition of the microbial community is potentially the key to preventing tumorigenic events. Scientists are currently working to enhance treatment response and eliminate treatment-related toxicity by modulating the gut microbiota. Microbes can be modified through the management of FMT, probiotics, diet and lifestyle changes, and targeted regulation using customized antibiotic therapy or bacteriophages. In the future, it is likely that we will be able to combine pharmacogenomic analysis with customized microbiota or their specific metabolites to develop more precise and personalized cancer treatments. Moreover, we need to focus on the regulation of gut microbes, which requires a better understanding of the carcinogenic mechanisms. At present, the carcinogenic mechanisms of the microbiome have only been confirmed as causal in some GI cancers, and more plausible mechanisms need to be confirmed by more experimental and experiential evidence, including systematic reviews, high-quality randomized controlled trials and high-powered cohort studies. Future preclinical studies should use appropriate animal models to elucidate the role of microbes in the occurrence, progression and therapeutic response of GI cancer. These studies should be followed up with translational studies to meet the challenge of individualized treatment. With the increasing depth and breadth of research in this field, the microbiota will become an important part of cancer prevention and treatment. More large-scale and international cohort studies need to be carried out in the future, not only cross-sectional studies but also prospective longitudinal cohort studies. Currently, however, it is critical to carry out observational research and campaign for interventional research, combined with multi-omics approaches to carry out deep phenotyping of the microbiome, to make the transition from correlation to causal research.
Conclusions
Numerous studies have provided evidence to support the concept that manipulation of the gut microbiome can be used as an adjunct strategy to improve the outcome of cancer treatment, although many of these studies are still in the early stages. The effect of anticancer drugs depends, to a large extent, on the gut microbiota, which can affect the response of intestinal and extraintestinal organs to anticancer chemotherapy. The composition of the gut microbiota can also be used as a potential biomarker for tumorigenesis and a target for cancer management. It can shed light on new directions for cancer prevention and the development of personalized treatment strategies. The interaction among host, drug and gut microbiota is complex; therefore, a personalized approach will be necessary for precision treatment. Additionally, the prevalence of excess body weight resulting in dysbiosis increase the risk of various cancers, thus, a balanced low-fat, low sugar, high fiber diet consisting of unprocessed foods and supplementation of safe probiotics and prebiotics have the potential for prevention of cancer, improving the efficacy of cancer treatment, and reducing adverse effects of anticancer drugs.
Abbreviations
B-HCC: hepatitis B virus-related HCC; BIRC3: baculoviral IAP repeat containing 3; B.fragilis: Bacteroides fragilis; B. Clausii: Bacillus Clausii; COX-2: cyclo-oxygenase 2; CTX: Cyclophosphamide; CTLA: cytotoxic T lymphocyte-associated antigen; C. butyricum: Clostridium butyricum; DCs: dendritic cells; DCA: deoxycholic acid; EAC: esophageal adenocarcinoma; E.coli: Escherichia coli; E.hirae: Enterococcus hirae; ESCC: esophageal squamous cell carcinoma; ETBF: Enterotoxigenic B. fragilis; EMT: epithelial-mesenchymal transformation; E-cadherin: epithelial-cadherin; FMT: fecal microbiota transplant; F.nucleatum: Fusobacterium nucleatum; FXR: farnesoid X receptor; H. pylori: Helicobacter pylori; GERD: gastroesophageal reflux disease; GI: Gastrointestinal; ICIs: immune checkpoint inhibitors; LPS: lipopolysaccharide; LARC: locally advanced rectal cancer; LTA: lipoteichoic acid; MDSCs: myeloid-derived suppressor cells; NGS: next-generation sequencing; NBNC-HCC: non-HBV non-hepatitis-C-virus- associated HCC; ROS: reactive oxygen species; SCFAs: short-chain fatty acids; SASP: senescence-associated secretory phenotype; S. cerevisiae: Saccharomyces cerevisiae; TLR: toll-like receptors; TGF: transforming-growth-factor; Tregs: regulatory cells; TNF-α: tumor necrosis factor-α; TAMs: tumor-associated macrophages; P. gingivalis: Porphyromonas gingivalis; PDAC: pancreatic ductal adenocarcinoma; PD-L1: programmed death ligand 1; UGT: UDP-glucuronosyltransferase; PAR1b: partitioning-defective 1b; P. micra: Parvimonas micra; NOX1: nicotinamide adenine dinucleotide phosphate oxidase 1; RASAL2: ras protein activator like 2; TUDCA: tauroursodeoxycholic acid; PSRs: plasma-stool ratios; CDCA: chenodeoxycholic acid; ILC2: type 2 innate lymphocytes; MMP9: matrix metalloproteinase 9; VEGFA: vascular endothelial growth factor A; CRC: colorectal cancer; HCC: hepatocellular carcinoma; NASH: non-alcoholic steatohepatitis; NAFLD: non-alcoholic fatty liver disease; NF: nuclear factor; O. viverrini: Opisthorchis viverrini; K. pneumonia: Klebsiella pneumonia; HDAC: histone deacetylase; PMT: photo-controlled bacterial metabolite therapy; Nb2C: niobium carbide; TILs: tumor infiltrating lymphocytes.
Acknowledgements
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Funding
This work was supported by the National Natural Science foundation of China (Grant No. 82103468 to YL), Natural Science foundation of Liaoning Province of China (Grant No.2020-MS-178 to XG) and 345 Talent Project of Shengjing Hospital of China Medical University (M0961 to YL).
Author contributions
Conception and design: YL, YB, TI, HB, XG, JZ. Drafting of the manuscript: YL.DN.KO. Drawing of figures: YL. PQ. Conceiving and critical revision of the manuscript for important intellectual content: YB, TI, HB, BD. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Malczewski AB, Navarro S, Coward JI. et al. Microbiome-derived metabolome as a potential predictor of response to cancer immunotherapy. J Immunother Cancer. 2020 8
2. Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207-14
3. Ramakrishna BS. Role of the gut microbiota in human nutrition and metabolism. J Gastroenterol Hepatol. 2013;28(Suppl 4):9-17
4. Allen-Vercoe E, Coburn B. A Microbiota-Derived Metabolite Augments Cancer Immunotherapy Responses in Mice. Cancer Cell. 2020;38:452-3
5. Ocvirk S, O'Keefe SJD. Dietary fat, bile acid metabolism and colorectal cancer. Semin Cancer Biol. 2020
6. Simon TG, Kim MN, Luo X. et al. Physical activity compared to adiposity and risk of liver-related mortality: Results from two prospective, nationwide cohorts. J Hepatol. 2020;72:1062-9
7. Sung H, Siegel RL, Torre LA. et al. Global patterns in excess body weight and the associated cancer burden. CA Cancer J Clin. 2019;69:88-112
8. Wang F, Meng W, Wang B. et al. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345:196-202
9. Di Domenico EG, Cavallo I, Pontone M. et al. Biofilm Producing Salmonella Typhi: Chronic Colonization and Development of Gallbladder Cancer. Int J Mol Sci. 2017 18
10. Tsilimigras MC, Fodor A, Jobin C. Carcinogenesis and therapeutics: the microbiota perspective. Nat Microbiol. 2017;2:17008
11. Bhatt AP, Redinbo MR, Bultman SJ. The role of the microbiome in cancer development and therapy. CA Cancer J Clin. 2017;67:326-44
12. Church DL, Cerutti L, Gürtler A. et al. Performance and Application of 16S rRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin Microbiol Rev. 2020 33
13. Lagier J-C, Dubourg G, Million M. et al. Culturing the human microbiota and culturomics. Nature Reviews Microbiology. 2018;16:540-50
14. Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260-70
15. Schulz MD, Atay C, Heringer J. et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature. 2014;514:508-12
16. Rothhammer V, Borucki DM, Tjon EC. et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557:724-8
17. Schwabe RF, Greten TF. Gut microbiome in HCC - Mechanisms, diagnosis and therapy. J Hepatol. 2020;72:230-8
18. Scott AJ, Alexander JL, Merrifield CA. et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut. 2019;68:1624-32
19. Helmink BA, Khan MAW, Hermann A. et al. The microbiome, cancer, and cancer therapy. Nat Med. 2019;25:377-88
20. Blackett KL, Siddhi SS, Cleary S. et al. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett's and oesophageal carcinoma: association or causality? Aliment Pharmacol Ther. 2013;37:1084-92
21. Kaakoush NO, Castaño-Rodríguez N, Man SM. et al. Is Campylobacter to esophageal adenocarcinoma as Helicobacter is to gastric adenocarcinoma? Trends Microbiol. 2015;23:455-62
22. Elliott DRF, Walker AW, O'Donovan M. et al. A non-endoscopic device to sample the oesophageal microbiota: a case-control study. Lancet Gastroenterol Hepatol. 2017;2:32-42
23. Zaidi AH, Kelly LA, Kreft RE. et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer. 2016;16:52
24. Wang Z, Shaheen NJ, Whiteman DC. et al. Helicobacter pylori Infection Is Associated With Reduced Risk of Barrett's Esophagus: An Analysis of the Barrett's and Esophageal Adenocarcinoma Consortium. Am J Gastroenterol. 2018;113:1148-55
25. Howden CW. H. pylori and Barrett's Esophagus: Implications for Populations and Practice. Am J Gastroenterol. 2018;113:1119-20
26. Chen X, Winckler B, Lu M. et al. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. PLoS One. 2015;10:e0143603
27. Gao S, Li S, Ma Z. et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect Agent Cancer. 2016;11:3
28. Peters BA, Wu J, Pei Z. et al. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res. 2017;77:6777-87
29. Deshpande NP, Riordan SM, Castano-Rodriguez N. et al. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome. 2018;6:227
30. Meng F, Li R, Ma L. et al. Porphyromonas gingivalis promotes the motility of esophageal squamous cell carcinoma by activating NF-κB signaling pathway. Microbes Infect. 2019;21:296-304
31. Sun T, Wu Z, Wang X. et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39:5358-72
32. Yamamura K, Baba Y, Nakagawa S. et al. Human Microbiome Fusobacterium Nucleatum in Esophageal Cancer Tissue Is Associated with Prognosis. Clin Cancer Res. 2016;22:5574-81
33. Guo Y, Zhang Y, Gerhard M. et al. Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut. 2020;69:1598-607
34. Kumar S, Metz DC, Ellenberg S. et al. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter pylori Infection: A Large Cohort Study. Gastroenterology. 2020;158:527-36.e7
35. Yao X, Smolka AJ. Gastric Parietal Cell Physiology and Helicobacter pylori-Induced Disease. Gastroenterology. 2019;156:2158-73
36. Tegtmeyer N, Wessler S, Necchi V. et al. Helicobacter pylori Employs a Unique Basolateral Type IV Secretion Mechanism for CagA Delivery. Cell Host Microbe. 2017;22:552-60.e5
37. Sigal M, Logan CY, Kapalczynska M. et al. Stromal R-spondin orchestrates gastric epithelial stem cells and gland homeostasis. Nature. 2017;548:451-5
38. Smyth EC, Nilsson M, Grabsch HI. et al. Gastric cancer. Lancet. 2020;396:635-48
39. Kim YH, Shin SW. Helicobacter pylori and Prevention of Gastric Cancer. N Engl J Med. 2018;378:2244
40. Ailloud F, Estibariz I, Suerbaum S. Evolved to vary: genome and epigenome variation in the human pathogen Helicobacter pylori. FEMS Microbiol Rev. 2021 45
41. Saju P, Murata-Kamiya N, Hayashi T. et al. Host SHP1 phosphatase antagonizes Helicobacter pylori CagA and can be downregulated by Epstein-Barr virus. Nat Microbiol. 2016;1:16026
42. Hayashi T, Senda M, Suzuki N. et al. Differential Mechanisms for SHP2 Binding and Activation Are Exploited by Geographically Distinct Helicobacter pylori CagA Oncoproteins. Cell Rep. 2017;20:2876-90
43. Li N, Feng Y, Hu Y. et al. Helicobacter pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway. J Exp Clin Cancer Res. 2018;37:280
44. Imai S, Ooki T, Murata-Kamiya N. et al. Helicobacter pylori CagA elicits BRCAness to induce genome instability that may underlie bacterial gastric carcinogenesis. Cell Host Microbe. 2021;29:941-58 e10
45. Costa L, Corre S, Michel V. et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut. 2020;69:1582-91
46. Echizen K, Horiuchi K, Aoki Y. et al. NF-kappaB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene. 2019;38:4250-63
47. Cao L, Zhu S, Lu H. et al. Helicobacter pylori-induced RASAL2 Through Activation of Nuclear Factor-kappaB Promotes Gastric Tumorigenesis via beta-catenin Signaling Axis. Gastroenterology. 2022
48. Choi J, Kim SG, Kim BG. et al. Helicobacter pylori Eradication Modulates Aberrant CpG Island Hypermethylation in Gastric Carcinogenesis. Korean J Gastroenterol. 2016;68:253-9
49. Coker OO, Dai Z, Nie Y. et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67:1024-32
50. Lertpiriyapong K, Whary MT, Muthupalani S. et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63:54-63
51. Sung JJY, Coker OO, Chu E. et al. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut. 2020;69:1572-80
52. Ohtani N, Hara E. Gut-liver axis-mediated mechanism of liver cancer: A special focus on the role of gut microbiota. Cancer Sci. 2021;112:4433-43
53. Ozeke LG, Mann IR, Murphy KR. et al. Explaining the apparent impenetrable barrier to ultra-relativistic electrons in the outer Van Allen belt. Nat Commun. 2018;9:1844
54. Yang X, Lu D, Zhuo J. et al. The Gut-liver Axis in Immune Remodeling: New insight into Liver Diseases. Int J Biol Sci. 2020;16:2357-66
55. Darnaud M, Faivre J, Moniaux N. Targeting gut flora to prevent progression of hepatocellular carcinoma. J Hepatol. 2013;58:385-7
56. Campbell C, McKenney PT, Konstantinovsky D. et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature. 2020;581:475-9
57. Schulthess J, Pandey S, Capitani M. et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity. 2019;50:432-45 e7
58. Ren Z, Li A, Jiang J. et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014-23
59. Jing YY, Han ZP, Sun K. et al. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 2012;10:98
60. Ma C, Han M, Heinrich B. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018 360
61. Loo TM, Kamachi F, Watanabe Y. et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017;7:522-38
62. Buffie CG, Bucci V, Stein RR. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205-8
63. Yoshimoto S, Loo TM, Atarashi K. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97-101
64. Pan Y, Zhang X. Diet and gut microbiome in fatty liver and its associated liver cancer. J Gastroenterol Hepatol. 2022;37:7-14
65. Blasco-Baque V, Coupe B, Fabre A. et al. Associations between hepatic miRNA expression, liver triacylglycerols and gut microbiota during metabolic adaptation to high-fat diet in mice. Diabetologia. 2017;60:690-700
66. Zhang X, Coker OO, Chu ES. et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2021;70:761-74
67. Singh V, Yeoh BS, Chassaing B. et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell. 2018;175:679-94 e22
68. Zhao S, Jang C, Liu J. et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. 2020;579:586-91
69. Boursier J, Mueller O, Barret M. et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764-75
70. Liu Q, Li F, Zhuang Y. et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019;11:1
71. Bertocchi A, Carloni S, Ravenda PS. et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell. 2021;39:708-24 e11
72. Yin H, Miao Z, Wang L. et al. Fusobacterium nucleatum promotes liver metastasis in colorectal cancer by regulating the hepatic immune niche and altering gut microbiota. Aging (Albany NY). 2022;14:1941-58
73. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16:713-32
74. Tilg H, Adolph TE, Gerner RR. et al. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell. 2018;33:954-64
75. Abed J, Emgard JE, Zamir G. et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe. 2016;20:215-25
76. Rubinstein MR, Wang X, Liu W. et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195-206
77. Gur C, Ibrahim Y, Isaacson B. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344-55
78. Yang Y, Weng W, Peng J. et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology. 2017;152:851-66.e24
79. Bullman S, Pedamallu CS, Sicinska E. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. 2017;358:1443-8
80. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A. et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature. 2020;580:269-73
81. Allen-Vercoe E, Jobin C. Fusobacterium and Enterobacteriaceae: important players for CRC? Immunol Lett. 2014;162:54-61
82. Dejea CM, Fathi P, Craig JM. et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359:592-7
83. Maddocks OD, Scanlon KM, Donnenberg MS. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. mBio. 2013;4:e00152-13
84. Tsoi H, Chu ESH, Zhang X. et al. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology. 2017;152:1419-33.e5
85. Dai Z, Coker OO, Nakatsu G. et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome. 2018;6:70
86. Yachida S, Mizutani S, Shiroma H. et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25:968-76
87. Thiele Orberg E, Fan H, Tam AJ. et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017;10:421-33
88. Chung L, Thiele Orberg E, Geis AL. et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe. 2018;23:203-14 e5
89. Zhang Y, Weng Y, Gan H. et al. Streptococcus gallolyticus conspires myeloid cells to promote tumorigenesis of inflammatory bowel disease. Biochem Biophys Res Commun. 2018;506:907-11
90. Ganapathy V, Thangaraju M, Prasad PD. et al. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol. 2013;13:869-74
91. Bachem A, Makhlouf C, Binger KJ. et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8(+) T Cells. Immunity. 2019;51:285-97.e5
92. Chen D, Jin D, Huang S. et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020;469:456-67
93. Rao B, Ren T, Wang X. et al. Dysbiosis in the Human Microbiome of Cholangiocarcinoma. Front Physiol. 2021;12:715536
94. Chng KR, Chan SH, Ng AHQ. et al. Tissue Microbiome Profiling Identifies an Enrichment of Specific Enteric Bacteria in Opisthorchis viverrini Associated Cholangiocarcinoma. EBioMedicine. 2016;8:195-202
95. Di Carlo P, Serra N, D'Arpa F. et al. The microbiota of the bilio-pancreatic system: a cohort, STROBE-compliant study. Infect Drug Resist. 2019;12:1513-27
96. Jia X, Lu S, Zeng Z. et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology. 2020;71:893-906
97. Aviles-Jimenez F, Guitron A, Segura-Lopez F. et al. Microbiota studies in the bile duct strongly suggest a role for Helicobacter pylori in extrahepatic cholangiocarcinoma. Clin Microbiol Infect. 2016;22:178 e11- e22
98. Zhang Q, Ma C, Duan Y. et al. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov. 2021;11:1248-67
99. Fan X, Alekseyenko AV, Wu J. et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018;67:120-7
100. Michaud DS, Izard J, Wilhelm-Benartzi CS. et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut. 2013;62:1764-70
101. Pushalkar S, Hundeyin M, Daley D. et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018;8:403-16
102. Farrell JJ, Zhang L, Zhou H. et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut. 2012;61:582-8
103. Riquelme E, Zhang Y, Zhang L. et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019;178:795-806 e12
104. Sethi V, Kurtom S, Tarique M. et al. Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology. 2018;155:33-7 e6
105. Thomas RM, Gharaibeh RZ, Gauthier J. et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis. 2018;39:1068-78
106. Matson V, Chervin CS, Gajewski TF. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology. 2021;160:600-13
107. Alexander JL, Wilson ID, Teare J. et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14:356-65
108. Yamamura K, Izumi D, Kandimalla R. et al. Intratumoral Fusobacterium Nucleatum Levels Predict Therapeutic Response to Neoadjuvant Chemotherapy in Esophageal Squamous Cell Carcinoma. Clin Cancer Res. 2019;25:6170-9
109. Liu Y, Baba Y, Ishimoto T. et al. Fusobacterium nucleatum confers chemoresistance by modulating autophagy in oesophageal squamous cell carcinoma. Br J Cancer. 2021;124:963-74
110. Yu T, Guo F, Yu Y. et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170:548-63 e16
111. Zhang S, Yang Y, Weng W. et al. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J Exp Clin Cancer Res. 2019;38:14
112. Geller LT, Barzily-Rokni M, Danino T. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156-60
113. Viaud S, Daillere R, Boneca IG. et al. Gut microbiome and anticancer immune response: really hot Sh*t!. Cell Death Differ. 2015;22:199-214
114. Viaud S, Saccheri F, Mignot G. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971-6
115. Daillère R, Vétizou M, Waldschmitt N. et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity. 2016;45:931-43
116. Wallace BD, Wang H, Lane KT. et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831-5
117. He Y, Fu L, Li Y. et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8(+) T cell immunity. Cell Metab. 2021;33:988-1000 e7
118. Yi Y, Shen L, Shi W. et al. Gut Microbiome Components Predict Response to Neoadjuvant Chemoradiotherapy in Patients with Locally Advanced Rectal Cancer: A Prospective, Longitudinal Study. Clin Cancer Res. 2021;27:1329-40
119. Yi Y, Shen L, Shi W. et al. Gut microbiome components predict response to neoadjuvant chemoradiotherapy in locally advanced rectal cancer patients: a prospective, longitudinal study. Clin Cancer Res. 2020
120. Uribe-Herranz M, Rafail S, Beghi S. et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest. 2020;130:466-79
121. Gerassy-Vainberg S, Blatt A, Danin-Poleg Y. et al. Radiation induces proinflammatory dysbiosis: transmission of inflammatory susceptibility by host cytokine induction. Gut. 2018;67:97-107
122. Ferreira MR, Muls A, Dearnaley DP. et al. Microbiota and radiation-induced bowel toxicity: lessons from inflammatory bowel disease for the radiation oncologist. Lancet Oncol. 2014;15:e139-47
123. Reis Ferreira M, Andreyev HJN, Mohammed K. et al. Microbiota- and Radiotherapy-Induced Gastrointestinal Side-Effects (MARS) Study: A Large Pilot Study of the Microbiome in Acute and Late-Radiation Enteropathy. Clin Cancer Res. 2019;25:6487-500
124. Fuccio L, Guido A. Probiotics supplementation for the prevention of gastrointestinal radiation-induced side effects: the time is now. Am J Gastroenterol. 2013;108:277
125. Riehl TE, Alvarado D, Ee X. et al. Lactobacillus rhamnosus GG protects the intestinal epithelium from radiation injury through release of lipoteichoic acid, macrophage activation and the migration of mesenchymal stem cells. Gut. 2019;68:1003-13
126. Guo H, Chou WC, Lai Y. et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science. 2020 370
127. Cui M, Xiao H, Li Y. et al. Faecal microbiota transplantation protects against radiation-induced toxicity. EMBO Mol Med. 2017;9:448-61
128. Xiao HW, Cui M, Li Y. et al. Gut microbiota-derived indole 3-propionic acid protects against radiation toxicity via retaining acyl-CoA-binding protein. Microbiome. 2020;8:69
129. Schluter J, Peled JU, Taylor BP. et al. The gut microbiota is associated with immune cell dynamics in humans. Nature. 2020;588:303-7
130. Andrews MC, Duong CPM, Gopalakrishnan V. et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med. 2021
131. Yatsunenko T, Rey FE, Manary MJ. et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222-7
132. Bryant CE, Spring DR, Gangloff M. et al. The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol. 2010;8:8-14
133. Reynolds JM, Pappu BP, Peng J. et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32:692-702
134. Rojas ER, Billings G, Odermatt PD. et al. The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature. 2018;559:617-21
135. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321-30
136. Clavel T, Gomes-Neto JC, Lagkouvardos I. et al. Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes. Immunol Rev. 2017;279:8-22
137. Schmidt TSB, Raes J, Bork P. The Human Gut Microbiome: From Association to Modulation. Cell. 2018;172:1198-215
138. Kayama H, Takeda K. Polysaccharide A of Bacteroides fragilis: actions on dendritic cells and T cells. Mol Cell. 2014;54:206-7
139. Geis AL, Fan H, Wu X. et al. Regulatory T-cell Response to Enterotoxigenic Bacteroides fragilis Colonization Triggers IL17-Dependent Colon Carcinogenesis. Cancer Discov. 2015;5:1098-109
140. Meng LD, Shi GD, Ge WL. et al. Linc01232 promotes the metastasis of pancreatic cancer by suppressing the ubiquitin-mediated degradation of HNRNPA2B1 and activating the A-Raf-induced MAPK/ERK signaling pathway. Cancer Lett. 2020;494:107-20
141. Sun S, Luo L, Liang W. et al. Bifidobacterium alters the gut microbiota and modulates the functional metabolism of T regulatory cells in the context of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2020;117:27509-15
142. Sivan A, Corrales L, Hubert N. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084-9
143. Peng Z, Cheng S, Kou Y. et al. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol Res. 2020;8:1251-61
144. Park J, Kim M, Kang SG. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8:80-93
145. Kayama H, Okumura R, Takeda K. Interaction Between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annu Rev Immunol. 2020;38:23-48
146. Tanoue T, Morita S, Plichta DR. et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565:600-5
147. Vétizou M, Pitt JM, Daillère R. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079-84
148. Song X, Sun X, Oh SF. et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature. 2020;577:410-5
149. Mager LF, Burkhard R, Pett N. et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369:1481-9
150. Wargo JA. Modulating gut microbes. Science. 2020;369:1302-3
151. Elinav E, Garrett WS, Trinchieri G. et al. The cancer microbiome. Nat Rev Cancer. 2019;19:371-6
152. Wang Y, Wiesnoski DH, Helmink BA. et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24:1804-8
153. DeFilipp Z, Bloom PP, Torres Soto M. et al. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N Engl J Med. 2019;381:2043-50
154. Blaser MJ. Fecal Microbiota Transplantation for Dysbiosis - Predictable Risks. N Engl J Med. 2019;381:2064-6
155. Li SS, Zhu A, Benes V. et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science. 2016;352:586-9
156. Britton RA, Hoffmann DE, Khoruts A. Probiotics and the Microbiome: How Can We Help Patients Make Sense of Probiotics? Gastroenterology. 2020
157. Hibberd AA, Lyra A, Ouwehand AC. et al. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017;4:e000145
158. Gurbatri CR, Lia I, Vincent R. et al. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci Transl Med. 2020 12
159. Yelin I, Flett KB, Merakou C. et al. Genomic and epidemiological evidence of bacterial transmission from probiotic capsule to blood in ICU patients. Nat Med. 2019;25:1728-32
160. Dalile B, Van Oudenhove L, Vervliet B. et al. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol. 2019;16:461-78
161. Li Y, Elmen L, Segota I. et al. Prebiotic-Induced Anti-tumor Immunity Attenuates Tumor Growth. Cell Rep. 2020;30:1753-66 e6
162. Han K, Nam J, Xu J. et al. Generation of systemic antitumour immunity via the in situ modulation of the gut microbiome by an orally administered inulin gel. Nat Biomed Eng. 2021
163. Zheng DW, Li RQ, An JX. et al. Prebiotics-Encapsulated Probiotic Spores Regulate Gut Microbiota and Suppress Colon Cancer. Adv Mater. 2020;32:e2004529
164. Russell WR, Gratz SW, Duncan SH. et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93:1062-72
165. Tuli HS, Mittal S, Aggarwal D. et al. Path of Silibinin from diet to medicine: A dietary polyphenolic flavonoid having potential anti-cancer therapeutic significance. Semin Cancer Biol. 2021;73:196-218
166. Zheng DW, Chen Y, Li ZH. et al. Optically-controlled bacterial metabolite for cancer therapy. Nat Commun. 2018;9:1680
167. Kong F, Fang C, Zhang Y. et al. Abundance and Metabolism Disruptions of Intratumoral Microbiota by Chemical and Physical Actions Unfreeze Tumor Treatment Resistance. Adv Sci (Weinh). 2022;9:e2105523
168. Aykut B, Pushalkar S, Chen R. et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264-7
169. Alam A, Levanduski E, Denz P. et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell. 2022;40:153-67 e11
170. Zhang Y, Zheng DW, Li CX. et al. Temulence Therapy to Orthotopic Colorectal Tumor via Oral Administration of Fungi-Based Acetaldehyde Generator. Small Methods. 2022;6:e2100951
171. Xu J, Ma Q, Zhang Y. et al. Yeast-derived nanoparticles remodel the immunosuppressive microenvironment in tumor and tumor-draining lymph nodes to suppress tumor growth. Nat Commun. 2022;13:110
172. Moehler M, Delic M, Goepfert K. et al. Immunotherapy in gastrointestinal cancer: Recent results, current studies and future perspectives. Eur J Cancer. 2016;59:160-70
173. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18:498-513
174. Sroussi M, Fluckiger A, Zitvogel L. [A bacteriophage from the intestinal microbiota induces an anti-tumoral response by a cross-reactivity mechanism]. Med Sci (Paris). 2021;37:970-2
175. Dong X, Pan P, Zheng DW. et al. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor-immune microenvironment against colorectal cancer. Sci Adv. 2020;6:eaba1590
176. Coker OO, Wu WKK, Wong SH. et al. Altered Gut Archaea Composition and Interaction With Bacteria Are Associated With Colorectal Cancer. Gastroenterology. 2020;159:1459-70 e5
177. Cai M, Kandalai S, Tang X. et al. Contributions of Human-Associated Archaeal Metabolites to Tumor Microenvironment and Carcinogenesis. Microbiol Spectr. 2022: e0236721.
Author contact
![]() Corresponding author: Peng Qiu, Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, 110004, Liaoning Province, China. E-mail: storm0513com. ORCID ID number: 0000-0001-7085-080X.
Corresponding author: Peng Qiu, Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, 110004, Liaoning Province, China. E-mail: storm0513com. ORCID ID number: 0000-0001-7085-080X.