Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(10):4171-4186. doi:10.7150/ijbs.69332 This issue Cite
Research Paper
CCDC65, a Gene Knockout that leads to Early Death of Mice, acts as a potentially Novel Tumor Suppressor in Lung Adenocarcinoma
Ziyan Zhang1#, Ping Xu2,3#, Zhe Hu1#, Zhaojian Fu4#, Tongyuan Deng1#, Xiaojie Deng1, Lanzhu Peng1, Yingying Xie1, Lingzhi Long5, Dayong Zheng1, Peng Shen6, Mengmin Zhang1, Bin Gong1, Zhibo Zhu1, Junhao Lin1, Rui Chen7, Zhen Liu1,9 ![]() , Huilin Yang2,8
, Huilin Yang2,8 ![]() , Rong Li2,6
, Rong Li2,6 ![]() , Weiyi Fang1
, Weiyi Fang1 ![]()
1. Cancer Center, Integrated Hospital of Traditional Chinese Medicine, Southern Medical University, 510315 Guangzhou, China.
2. Cancer Research Institute, Southern Medical University, Guangzhou, Guangdong, 510515, China.
3. Respiratory Department, Peking University Shenzhen Hospital, Shenzhen, 518034, China.
4. Department of Oncology, Dali Bai Autonomous Prefecture People's Hospital, Dali, Yunnan, 671000, China.
5. Department of Pulmonary and Critical Care Medicine, The Third Xiangya Hospital of Central South University, Changsha, 410000, China.
6. Department of Oncology, Nanfang Hospital, Southern Medical University, 510515 Guangzhou, China.
7. Department of Hepatobiliary Surgery, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, 710061, China.
8. School of Pharmacy, Guangdong Medical University, Dongguan, Guangdong 523808, China.
9. Affiliated Cancer Hospital & Institute of Guangzhou Medical University, Guangzhou Municipal and Guangdong Provincial Key Laboratory of Protein Modification and Degradation, State Key Laboratory of Respiratory Disease, School of Basic Medical Sciences, Guangzhou Medical University Guangzhou 510095, Guangdong, China.
#These authors contributed equally to this work.
Received 2021-11-22; Accepted 2022-6-6; Published 2022-6-27
Abstract

CCDC65 is a member of the coiled-coil domain-containing protein family and was only reported in gastric cancer by our group. We first observed that it is downregulated in lung adenocarcinoma based on the TCGA database. Reduced CCDC65 protein was shown as an unfavorable factor promoting the clinical progression in lung adenocarcinoma. Subsequently, CCDC65-/- mice were found possibly dead of hydrocephalus. Compared with the CCDC65+/+ mice, the downregulation of CCDC65 in CCDC65+/- mice significantly increased the formation ability of lung cancer induced by urethane. In the subsequent investigation, we observed that CCDC65 functions as a tumor suppressor repressing cell proliferation in vitro and in vivo. Molecular mechanism showed that CCDC65 recruited E3 ubiquitin ligase FBXW7 to induce the ubiquitination degradation of c-Myc, an oncogenic transcription factor in tumors, and reduced c-Myc binding to ENO1 promoter, which suppressed the transcription of ENO1. In addition, CCDC65 also recruited FBXW7 to degrade ENO1 protein by ubiquitinated modulation. The downregulated ENO1 further reduced the phosphorylation activation of AKT1, which thus inactivated the cell cycle signal. Our data demonstrated that CCDC65 is a potential tumor suppressor by recruiting FBWX7 to suppress c-Myc/ENO1-induced cell cycle signal in lung adenocarcinoma.
Keywords: CCDC65, c-Myc, ENO1, Knockdown mice, Ubiquitin degradation, Cell cycle
Introduction
Lung cancer has a high incidence and the highest mortality among tumors. In recent years, adenocarcinoma has become the most common type among all types of lung cancer [1]. Currently, the treatment of lung adenocarcinoma (LUAD) has gradually become “individualized”. Through the detection of specific gene mutations, patients can be treated with precision therapy using personalized treatments, such as the use of EGFR-TKI drugs in EGFR mutation NSCLC. Targeting the EGFR [2, 3] has been proven to have a longer median duration of progression-free survival compared to traditional chemotherapy. Thus, exploring novel important biomarkers of LUAD could significantly improve the prevention, diagnosis, and treatment of LUAD.
The coiled-coil domain (CCD)-containing (CCDC) family plays a key role in multiple important biological functions such as regulating T cell activity [4], motile cilia function [5], erythroid iron levels, heme trafficking [6], and DNA sensors [7]. Meanwhile, CCDC also modulates the development of malignant tumor proliferation and metastasis [8-11].
In our previous research, we cloned and revised the coding sequence of NESG1 (also known as CFAP45 and CCDC19) and subsequently confirmed that it acts as a potential tumor suppressor in NPC and non-small cell lung cancer [12-14]. Yeast Two-Hybrid experiments showed that CCDC65 was one of the candidates interacting proteins. Although the further experiment confirmed that CCDC65 did not interact with NESG1. Interestingly, CCDC65 was found significantly downregulated in nasopharyngeal carcinoma and lung cancer.
CCDC65, known as CFAP250, DRC2, FAP250, and NYD-SP28, is located on the long arm of chromosome 12. It is firstly identified in the sperm tail [15]. Subsequently, the specific functions of CCDC65 revealed that it was an essential component of the nexin-dynein regulatory complex participating in the formation of motile cilia. Mutations in CCDC65 caused primary ciliary dyskinesia (PCD), a disease characterized by impaired ciliary function leading to chronic sinopulmonary disease [16, 17]. In tumor areas, only our research group recently reported that CCDC65 played the role of a tumor suppressor in gastric cancer [18]. However, the role of CCDC65 in LUAD remains unclear.
Here, we detected the expression of CCDC65 in LUAD, evaluated the tumor suppressor role of CCDC65 in human LUAD cells, CRISPR/Cas9 mouse model and nude mice.
Materials and methods
Cell culture and reagents
Human lung adenocarcinoma cancer cell lines used were: A549, H1299, H1975, SPC-A1 and H460. Human normal bronchial epithelial cell line used was 16HBE. Lung adenocarcinoma cancer cell lines were cultured in RPMI 1640 Medium (Vivacell, Shanghai, China) containing 10% fetal bovine serum (FBS) (Biowest, Nuaillé, France), HEK293T and 16HBE were cultured in DMEM (Vivacell, Shanghai, China) containing 10% FBS (Biowest, Nuaillé, France) at 37 °C in a 5% CO2 atmosphere. Culture dishes were purchased from Jet Biofil (Guangzhou, China).
Western blotting and antibodies
Western blotting was performed according to standard methods. Antibodies of CCDC65 (24376-1-AP), ENO1 (11204-1-AP), c-Myc (10828-1-AP), CCND1 (60186-1-Ig), p-AKT (Ser473) (66444-1-Ig), DYKDDDDK Tag (80010-1-RR), mTOR(66888-1-Ig), p-mTOR(Ser2448) (677778-1-Ig), GSK3β(22104-1-AP), p-GSK3β(Ser9) (67558-1-Ig), were purchased from Proteintech (Rosemont, IL, USA). Anti-P-Akt1 (Ser473) (#9018), Mouse IgG (#3420), normal Rabbit IgG (#2729), Anti-p27(#3686), Anti-p21(#2947) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-AKT1 (48884) was purchased from Signalway Antibody (College Park, MD, USA). Anti-flag (F1804) was purchased from Sigma (St. Louis, MO, USA); Anti-β-actin (AP0060) and anti-GAPDH (AP0063) were purchased from Bioworld (Bloomington, MN, USA). Antibodies were diluted with SuperKine™ Enhanced Antibody Dilution Buffer (Abbkine, Wuhan, China). Western blotting was performed by Mini-PROTEAN Tetra- and Mini Trans-Blot (Bio-Rad, Hercules, CA, USA). The images were captured with Chemiluminescence Imaging System (Minichemi, Beijing, China).
Reverse transcription and quantitative real-time polymerase chain reaction (RT-qPCR)
Total RNA from cells and animal tissues were extracted using the Total RNA Isolation Kit (Foregene, Chengdu, China). Using Reverse Transcription Reagent Kit (Vazyme, Nanjing, China), mRNA was transcribed into cDNA, which acted as a template to amplify target genes in subsequent qPCR tests. Quantitative real-time PCR was detected by CFX96 Real-Time System (Bio-Rad). Primers are shown in supplementary materials (Supplementary Table S1).
Tissue samples and immunohistochemical staining
Seven human lung adenocarcinoma and corresponding para-carcinoma tissue specimens as well as twenty cases of un-paired para-carcinoma tissues were obtained from TCM-Integrated Hospital of Southern Medical University. The use of clinical materials was approved by the Ethics Committees of TCM-Integrated Hospital of Southern Medical University. Tissue microarray (HLugA180Su08) was purchased from Outdo Biotech (Shanghai, China). Streptavidin/Peroxidase kit (ZSGB BIO, Beijing, China) and DAB kit (ZSGB BIO) were used for immunohistochemical staining. Immunohistochemical staining was performed according to standard methods. Scoring rule: multiply the score of staining intensity by the score of the proportion of positive cells (0-12 points). Proportion score: Positive cells less than 25% (0 points), 25-50% (1 points), 50-75% (2 points), 75% above (3 points). Strength score: - (0 points), + (1 points), ++ (2 points), +++ (3 points). Total score: 0-6 scores (low expression), 7-12 scores (high expression). Staining scores were performed with the assistance of two senior doctors in the Department of Pathology.
Genotyping
Mice were genotyped by DNA sequence and standard PCR. Primers were shown in supplementary materials (Supplementary Table S1). Agarose gel was prepared with 2% agarose and TAE (Leagene, Beijing, China). The PCR product was mixed with DNA loading buffer and Gelred (Biosharp, Anhui, China). After electrophoresis, the images were captured with GelDoc XR+ (Bio-Rad).
Lung adenocarcinoma model mouse and HE staining
C57BL/6J CCDC65+/- mice were purchased from Nanjing BioMedical Research Institute of Nanjing University (Nanjing, China). CCDC65 engineered mice were constructed by CRISPR/Cas9 technology, gRNA was designed and transcribed in vitro. The fertilized eggs of mice were simultaneously injected with Cas9 and all 4 gRNA within half a day. After embryo transfer, F0 generation mice were born about 19 days later. C57BL/6J wild-type mice were purchased from Animal Research Center of Southern Medical University (Guangzhou, China). Mice were housed and bred in a specific pathogen-free (SPF) environment. Mice were divided into 2 groups: CCDC65+/+ and CCDC65+/-, 15 mice per group (6-8 weeks old, 8 males and 7 females). The lung adenocarcinoma mouse model was achieved by intraperitoneal injection of urethane (1 g/kg in 0.9% NaCl solution, Sigma) once a week for 20 weeks [19]. Mice were sacrificed and lungs were harvested 20 weeks after the end of urethane treatment. The tissues were dehydrated, paraffin-embedded, and prepared into 3-μm paraffin sections. Slides were dewaxed using xylene and gradient alcohol solutions and then treated with hematoxylin for 5 min and eosin for 1 min. After dehydration, the slices were sealed.
Transfection
When cells reached 40% confluence, transfection was performed using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the operating instructions. The RNA of cells were harvested 24-48 hours after transfection and protein of cells were harvested 48-72 hours after transfection.
Lentivirus infection
The full-length sequences of CCDC65 and FLAG-Tag were inserted into the lentivirus (Genechem, Shanghai, China) which contains green fluorescent protein (GFP). To build stably over-expressing CCDC65 cell lines, A549, H1975, and SPC-A1 were infected with GFP empty vector or full-length CCDC65 GFP vector.
Cell Counting Kit 8 assay
Cells were seeded into 94-well plates at 3000 cells/well, and each condition was performed in quadruplicate. Cell Counting Kit (CCK8) (Vazyme) was added every 24 hours for 4 days and after being treated with CCK8 for 2 hours, the optical density (OD) value of each well was measured at 450 nm.
Colony formation assay
Cells were seeded into 6-well culture plates at 200 cells/well. Cells were incubated for two weeks at 37 °C in a 5% CO2 atmosphere and the medium was changed every 4 days. After incubation, colonies were washed twice with PBS and fixed with methanol, and finally stained with Giemsa.
5-Ethynyl-2'-deoxyuridine staining
Cells were seeded into 96-well plates at 5000 cells/well and experiments were performed in triplicate for each condition in each group. At 12 hours after seeding, the culture medium was changed to FBS-free or low FBS medium for 24 hours to synchronize the cell cycle. 5-Ethynyl-2'-deoxyuridine (EdU) staining was performed using the Cell-Light EdU Apollo567 in vitro Kit (Ribobio, Guangdong, China) and 4',6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Beyotime Biotechnology, Shanghai, China) according to manufacturer's instructions.
Cell-cycle analysis
A total of 107 cells were harvested 48 hours after transfection and washed twice with PBS. The cells were fixed with 70% ethanol. Then, cells were treated with 10 mg/mL propidium iodide (PI) and 0.5 mg/mL RNase A for 15 min at 37 °C. After washing three times with PBS, A FACS caliber flow cytometer (BD Biosciences) was used to measure the DNA content of treated cells.
Tumor xenograft experiments
Four-week-old nude mice were purchased from SPF (Beijing) Biotechnology Co., Ltd. (Beijing, China). The mice were housed in the SPF environment. Cells were infected with lentiviral vectors encoding CCDC65 or control vectors. A total of 5×106 cells were placed into 100 μL PBS. Controls cells were subcutaneously injected into the left back and CCDC65 infected cells were subcutaneously injected into the right back. After 28 days, the mice were sacrificed and tumor nodules were weighed, dehydrated, and paraffin-embedded.
Co-immunoprecipitation assay
Cells were treated with immunoprecipitation (IP) lysis buffer (Thermo Scientific, Waltham, Mass, USA). Cell lysates were exposed to specific antibodies or Normal IgG (final concentration of 20 μg/mL) at 4 °C overnight and stirred using a rotator. The protein-antibody compound followed incubation with protein A/G magnetic beads (Bimake, Shanghai, China) for 30 min at room temperature. The precipitate was washed with washing buffer (50 mM Tris, 150 mM NaCl, 0.2% TritonX-100, 0.2% Tween, pH 7.5) five times. The bound proteins were resolved in SDS loading buffer and boiled for 10 minutes and subjected to immunoblotting.
Immunofluorescence staining
Cells were harvested 48 hours after transfection, seeded into laser confocal petri dishes for 12 hours, and then washed twice with PBS. The cells were then fixed with paraformaldehyde and 2 mg/mL glycine for 15 min and permeabilized with 0.2% Triton X-100 for 5 min. After blocking with 5% BSA for 30 min and incubating with specific primary antibodies at 4°C overnight, the cells were washed three times with PBST and stained with Alexa Fluor secondary antibodies (Earthox, Millbrae, CA, USA) and DAPI (Beyotime, Biotechnology).
Cycloheximide chase assay and ubiquitination assay
The cycloheximide (CHX) chase assay and ubiquitination assay were performed as described previously [20, 21]. Cells were treated with CHX (50 μg/mL) (Selleck, Shanghai, China) for Gradient time. Proteins were extracted using RIPA lysis buffer and mixed with SDS loading buffer and boiled for 10 minutes, and then were subjected to immunoblotting. For the ubiquitination assay, cells were treated with MG132 (20 μM) (Selleck) for 4 hours before the proteins were harvested. The proteins were extracted using IP lysis buffer and immunoprecipitation was performed with anti-c-Myc or anti-ENO1. Harvested proteins were resolved in SDS loading buffer and boiled for 10 minutes, then subjected to immunoblotting.
High-throughput sequencing and bioinformatics analysis
High-throughput mRNA sequencing and subsequent bioinformatics analysis between CCDC65+/+ and CCDC65+/- mice lung tissues were performed by the Oebiotech Biomedical Technology Co., Ltd (Shanghai, China). The sequencing and analysis in A549, SPC-A1 and 5-8F were performed by Genergy Inc (Shanghai, China). Detailed transcriptome mice sequencing information was available in the SRA database (PRJNA695129).
Chromatin immunoprecipitation assay
The experiments were performed with Pierce Magnetic ChIP Kit and according to the manufacturer's manual (Thermo Scientific, Waltham, Mass, USA). Briefly, cells were cross-linked by the final concentration of 1% formaldehyde. Cross-linked cells were treated with membrane extraction buffer and MNase to remove membrane proteins and expose the nucleic acid. Four micrograms of c-Myc antibody and 2 μg normal rabbit IgG antibody were added to the supernatant containing the digested chromatin, as the experimental group and control group respectively, and IP reactions were incubated overnight at 4 °C with mixing. After incubation, ChIP grade protein A/G magnetic beads were added to each IP reaction and further incubated for 2 hours at 4 °C with mixing. Cleaned the beads and eluted the nucleic acid-proteins complex among the beads. Proteinase K was used to remove the proteins in the nucleic acid-proteins complex. DNA fragments were purified after being washed in the DNA clean-up column. JASPAR database was used to predict promoter binding sequences [22].
Statistical analysis
Statistical analyses were performed with the IBM SPSS Statistics 25 (IBM, Armonk, NYC, USA). Comparisons between two groups were performed using two independent samples t-test. Comparisons between multiple groups were performed using one-way ANOVA. Survival plots were performed using the log-rank test. Cox regression was used to screen out single and multiple risk/protective factors. Spearman correlation coefficient was used for grade data analysis. All statistical tests were two-sided and the asterisk indicated statistical significance. *P<0.05, **P<0.01, ***P<0.001.
Results
CCDC65 was downregulated in human LUAD and correlated with the clinical characteristics of LUAD patients
Analysis of The Cancer Genome Atlas (TCGA) lung cancer database showed that CCDC65 was downregulated in primary lung tumors (Fig. 1a, b). Due to CCDC65 being a component of cilia, which is always affected by smoking, we divided datasets from TCGA into three groups based on the smoking status (Fig. 1c). The results showed that CCDC65 was expressed at lower levels in primary lung tumor samples compared to normal lung tissues, regardless of smoking history. We then constructed survival plots using the KM plotter [23] (Fig. 1d, e). The results showed that the survival probability of patients with high CCDC65 expression was higher than patients with low CCDC65 expression. Subsequently, we detected the mRNA expression of CCDC65 in a normal bronchial epithelial cell line and four LUAD cell lines (Fig. 1f). Furthermore, the protein expression of CCDC65 in seven human LUAD and corresponding para-carcinoma tissue were also compared (Fig. 1g). Immunohistochemistry staining was performed in tissue microarray and clinical tissue samples (Fig. 1h, 1i). Statistical analysis was used in immunohistochemical staining scores to compare the expression of CCDC65 among different sample types. The results showed that CCDC65 was significantly downregulated in LUAD both in cell lines and human tissues.
Higher expression of CCDC65 inhibited lung adenocarcinoma and was associated with a better prognosis. (a) The heatmap of CCDC65 mRNA expression was derived based on samples from the TCGA lung cancer database (n =1299). (b) Comparison of the expression of CCDC65 mRNA in primary tumors and normal tissues. (c) Comparison of the expression of CCDC65 mRNA in primary tumors and solid normal tissues stratified according to smoking history. The data were analyzed using UCSC Xena (http://xena.ucsc.edu/). Kaplan-Meier survival curves relative to CCDC65 expression were generated from lung cancer (Kaplan-Meier plotter, http://kmplot.com/analysis). The Affymetrix IDs validated were (d) 1552321_a_at CCDC65 and (e) 1552320_a_at CCDC65. (f) qRT-PCR analysis of CCDC65 expression in lung adenocarcinoma cell lines and immortalized human bronchial epithelium cell line. (g) Expression of CCDC65 protein in 7 paired lung adenocarcinoma and adjacent nontumor tissues were detected by western blotting. P: para-carcinoma tissues C: cancer tissues. (h) The CCDC65 expression in lung cancer and para-carcinoma tissues (20 cases of para-carcinoma tissues from clinical samples and 5 cases of para-carcinoma tissues from tissue microarray, 97 cases of lung cancer from tissue microarray). (i) Tissues microarray immunohistochemical analysis of CCDC65 protein expression. (j) The survival curve was determined by Kaplan-Meier analysis based on the tissue microarray immunohistochemical score.
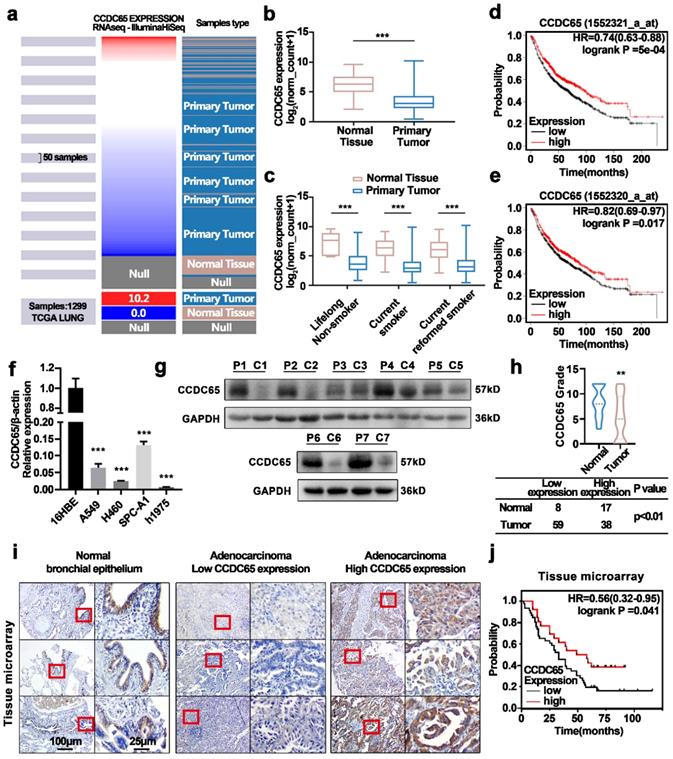
Based on the detailed clinical data associated with the tissue microarray samples IHC scores, One Way ANOVA analysis revealed that the expression of CCDC65 was associated with N stage (P=0.041) in LUAD patients (Table 1). The univariate Cox regression analysis showed that lymph node metastasis (P=0.002 HR=3.22 95%CI=1.517-6.835), T stage (P=0.028 HR=1.509 95%CI=1.044-2.180), N stage (P<0.001 HR=1.437 95%CI=1.310-2.303), and the expression of CCDC65 (P=0.047 HR=0.551 95%CI=0.306-0.993) were associated with the patients' survival. Multivariate Cox regression analysis showed that the expression of CCDC65 was an independently protective prognostic factor (P=0.049 HR=0.513 95%CI=0.264-0.998) (Table 2). Finally, we drew survival plots based on the information from the tissue microarray (Fig. 1j). The results were in line with the database and indicated that the survival probability of patients with high CCDC65 expression was better than patients with low CCDC65 expression.
Correlation between the clinicopathologic characteristics and expression of CCDC65 protein in lung adenocarcinoma
| Characteristics | The expression of CCDC65 | Chi square value | P value | |
|---|---|---|---|---|
| Low expression | High expression | |||
| Age | 0.000 | 0.994 | ||
| ≥50 | 47 | 27 | ||
| <50 | 7 | 4 | ||
| Sex | 0.097 | 0.755 | ||
| Male | 33 | 20 | ||
| Female | 21 | 11 | ||
| T stage* | 0.155 | 0.694 | ||
| 1-2 | 38 | 20 | ||
| 3-4 | 14 | 9 | ||
| N stage* | 4.157 | 0.041 | ||
| 0-1 | 28 | 24 | ||
| 2-3 | 16 | 4 | ||
| Clinical stage* | 2.360 | 0.124 | ||
| 1-2 | 24 | 20 | ||
| 3-4 | 21 | 8 | ||
*Some patients are not included in the statistics due to the missing data.
Summary of univariate and multivariate Cox regression analysis of overall survival duration
| Characteristics | Univariate Cox | Multivariate Cox | ||||
|---|---|---|---|---|---|---|
| Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | |
| Sex | 0.92 | 0.534-1.584 | 0.763 | |||
| Age (y) | 0.908 | 0.361-2.283 | 0.837 | |||
| Pathologic stage | 0.987 | 0.586-1.664 | 0.961 | |||
| Lymph metastasis | 3.22 | 1.517-6.835 | 0.002 | 1.78 | 0.439-73214 | 0.419 |
| T stage | 1.509 | 1.044-2.180 | 0.028 | 1.252 | 0.817-1.917 | 0.302 |
| N stage | 1.437 | 1.310-2.303 | <0.001 | 1.336 | 0.822-2.170 | 0.242 |
| Expression of CCDC65 | 0.551 | 0.306-0.993 | 0.047 | 0.513 | 0.264-0.998 | 0.049 |
The CCDC65-/- mice experienced early mortality probably due to hydrocephalus
CCDC65 engineered mice were generated by CRISPR/Cas9 technology and the strategies were presented in supplementary materials (Supplementary Fig. S1a). CCDC65+/- mice were allowed to copulate (female: male=2:1). DNA was extracted from the mouse tail at the age of 1 week for genotyping (Supplementary Fig. S1b). We detected the mRNA and protein expression of CCDC65 in the lung tissue of different groups of mice, and the results identified the knockout of CCDC65 (Supplementary Fig. S1c-e).
Mice were then divided into three groups: CCDC65+/+, CCDC65+/-, and CCDC65-/-. And the sex ratio and genotype ratio (Supplementary Fig. S2a) were determined six weeks after the mice were born. Results showed that the birth rate of CCDC65-/- mice was lower than the expected rate from Mendelian law. The CCDC65-/- mice died at three weeks of age (Supplementary Fig. S2b) and developed slowly compared with the CCDC65+/+ mice or CCDC65+/- mice (Supplementary Fig. S2c, S2d). The mice were dissected to explore the reason. Except for the possible cardiac transposition heart (red arrow), the lung, liver, kidney, stomach, and intestines of CCDC65-/- mice showed no significant differences compared with CCDC65+/+ mice (Supplementary Fig. S2e). Remarkably, the morphological structure of brain tissue in the CCDC65-/- group was significantly different from that in the CCDC65+/+ and CCDC65+/- groups. The craniocerebral diameter ratio of mice in the CCDC65-/- group was significantly lower than that in CCDC65+/+ and CCDC65+/- mice (Supplementary Table S2), and the brain tissue of mice in the CCDC65-/- group was sunken, while the brain tissue of mice in CCDC65+/+ group and CCDC65+/- the group was full. Similar changes were observed from mice raised in different cages (Supplementary Fig. S2f, g, h). Paraffin-embedded brain tissue and HE staining revealed that the lateral ventricle of CCDC65-/- mice were significantly larger than that in the CCDC65+/+ mice or CCDC65+/- mice (Supplementary Fig. S2i). Based on the known functions of CCDC65 in cilia[16, 17], we supposed that CCDC65-/- died of hydrocephalus. However, this was not the main point we focused on, therefore, in-depth research was not performed.
The mice in the CCDC65-/- group did not live more than three weeks. However, the mRNA levels of CCDC65 in the CCDC65+/- group decreased significantly compared with the CCDC65+/+ group, even at different ages (Supplementary Fig. S1c). And the protein expression also decreased (Supplementary Fig. S1d, S1e). Consequently, CCDC65+/+ and CCDC65+/- mice were used to reveal the function of CCDC65 in tumorigenesis.
The downregulation of CCDC65 promoted lung tumorigenesis in vivo
To investigate the role of CCDC65 in LUAD, we used a mouse model of urethane-induced LUAD to confirm our hypothesis [19, 24, 25].
Mice aged 6-8 weeks in the CCDC65+/+ and CCDC65+/- groups were injected intraperitoneally once weekly with 1g/kg urethane for 20 weeks to induce a LUAD model (Fig. 2a). Twenty weeks after the end of the urethane injection, the mice lung tissues were harvested (Fig. 2b).
CCDC65 inhibited urethane-induced lung carcinogenesis. (a) The mice were genotyped at the age of two weeks and then CCDC65+/+ and CCDC65+/- groups were treated with urethane (1 g/kg in 0.9% NaCl solution) intraperitoneally for 20 weeks. (b) Lungs were collected 6 months after the last urethane treatment. (c) The number of tumor nodules. (d) The HE staining of the harvested lung samples. (e) The average area of tumor nodules.
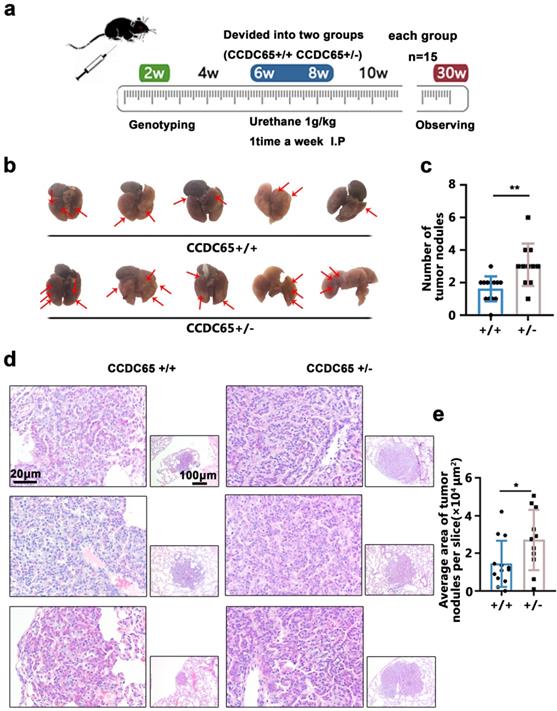
The results showed that the average number of tumor nodules in the urethane-induced LUAD CCDC65+/- group (3.09±1.30) was greater than that in the CCDC65+/+ group (1.62±0.77) (Fig. 2c). Subsequently, these lungs were excised, paraffin-embedded and used for HE staining (Fig. 2d), as well as the average area of the LUAD/lung adenoma on the sections were calculated. The results showed that the average area of LUAD/lung adenoma on the sections from CCDC65+/- mice (2.71±1.60 ×104 μm2) was larger than that from CCDC65+/+ mice tissue sections (1.44±1.23 ×104 μm2) (Fig. 2e).
Overexpression of CCDC65 inhibited the cell cycle and attenuated proliferation of LUAD cells both in vitro and in vivo
The expression of CCDC65 in H1975 and A549 cells were lower compared with other LUAD cell lines. The two cell lines were selected for subsequent experiments. The lentivirus were used to engineer steady CCDC65 upregulated cells. Two siCCDC65 sequences were used to engineer CCDC65 knockdown cells. The efficiency of overexpression or knockdown was presented in supplementary materials (Supplementary Fig. S3a-c).
The CCK8 assay was used to detect changes in cell proliferation in CCDC65 over-expressing cells. The results showed that, compared with the control group, CCDC65 over-expressing cells produced a lower absorption peak at 450 nm. Conversely, when compared with the control group, CCDC65-knockdown cells exhibited a higher absorption peak at 450nm (Fig. 3a). Colony formation showed that, compared with the control group, CCDC65 over-expressing cells produced less cell colony numbers (Fig. 3b).
CCDC65 inhibited H1975 and A549 cell proliferation and cell cycle transition, in vitro and in vivo. (a) The establishment of CCDC65 over-expressing and knocking down cell lines. The effects of CCDC65 on the cell proliferation of H1975 and A549 cells were examined by (a) the CCK8 assay, (b) the Colony formation assay. (c) Enriched pathways based on the differential gene expression of cell lines (A549, SPC-A1 and 5-8F) whit CCDC65 or empty vector by using GSEA analysis. The effects of CCDC65 on the G1 to S transition of H1975 and A549 cells were examined by (d) EdU incorporation assay and (e) cell cycle flow analysis, Mean±SD (n=3). *P<0.05; **P<0.01.; ***P<0.005. #To prevent the phenomenon that the proportion of S phase cells were too high for us to do statistical analysis, we reduced incubation time in the knockdown experiment. (f and g) ENO1, c-Myc, CCND1 and CCDC65 were detected following transfection with CCDC65 lentivirus and siRNA. β-actin or GAPDH served as a loading control. (h) Excised tumors 28 days after implantation and measured the corresponding tumor volume. (i) Representative Ki67 and PCNA IHC staining of excised tumor tissues of A549-CON and A549-CCDC65 were shown.
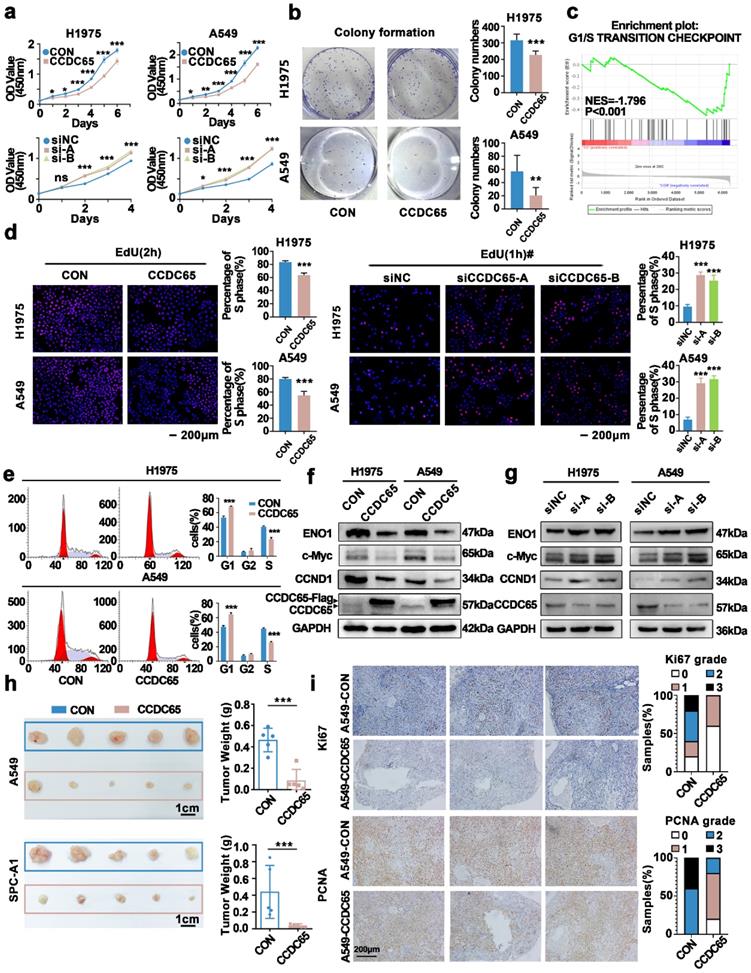
Two lung adenocarcinoma cell lines A549, SPC-A1 and one nasopharyngeal carcinoma 5-8F were transfected with CCDC65 and empty vector and were analyzed by high-throughput sequencing. Gene Set Enrichment Analysis(GSEA) showed that CCDC65 was associated with multiple signaling pathways (Supplementary Table S3). The G1/S transition checkpoint pathway was screened out due to its relation to cell proliferation and most DEGs (differential expressed genes from high-throughput sequencing) were involved in it (Fig. 3c).
To further confirm the influence of CCDC65 on the G1/S transition, we evaluated the cell cycle by EdU assay. The results showed that the G1 to S cell cycle transition of cells were significantly inhibited in CCDC65 over-expressing cells, while the G1 to S cell cycle transition of cells were enhanced when CCDC65 was silenced (Fig. 3d). Next, Flow cytometry showed the higher G1 phase proportion and lower S phase proportion in CCDC65 over-expressing cells compared with control cells (Fig. 3e).
ENO1, which was reported to be regulated by CCDC65[18], and the cell cycle-related proteins CCND1 and c-Myc were evaluated by western blotting. The data showed that ENO1, CCND1 and c-Myc were downregulated in CCDC65 over-expressing cells (Fig. 3f). To further confirm the effect of CCDC65, cell lines were transfected with siCCDC65, and the results showed that ENO1, CCND1 and c-Myc were upregulated in CCDC65 silenced cells (Fig. 3g).
Next, we performed an in vivo tumor formation experiment. Stably over-expressing CCDC65 cells and corresponding control cells were subcutaneously injected into the eight-week-old nude mice. Since the SPC-A1 cell line was used in the subsequent experiments, we selected SPC-A1 as one of the cell lines in the animal experiments. After implantation for 28 days, mice were sacrificed and the tumor nodules were harvested. The tumor nodules obtained were smaller in the CCDC65 group compared with that of the CON group (Fig. 3h). Ki67, PCNA immunohistochemical staining suggested that the proliferation of A549-CCDC65 cells were weaker than the A549-CON cells (Fig. 3i).
CCDC65 interacted with c-Myc
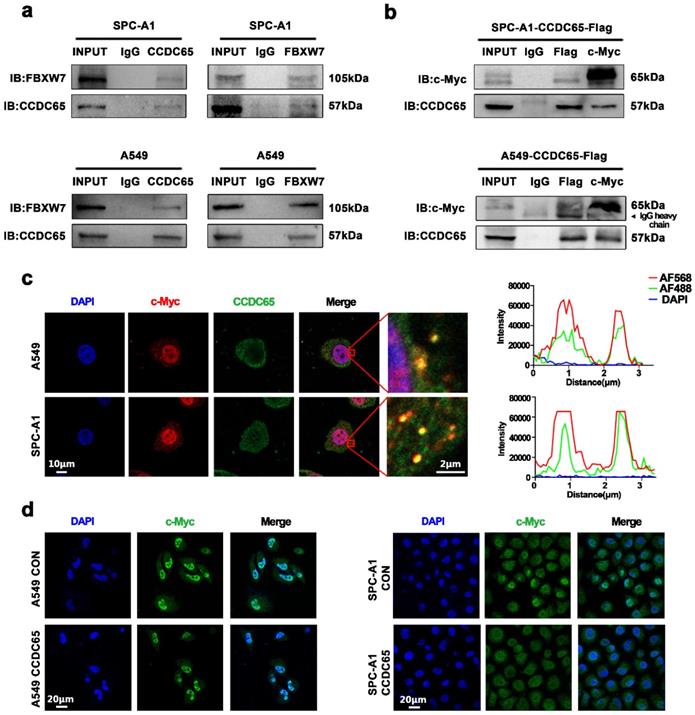
In previous studies, we have confirmed that CCDC65 mediated the ubiquitination degradation of ENO1 by recruiting FBXW7. FBXW7, which acts as a critical tumor suppressor of human cancers, controls proteasome-mediated degradation of oncoproteins. However, c-Myc is one of those oncoproteins [26-28] and was inhibited by the overexpression of CCDC65 (Fig. 3f). Therefore, we attempted to explore the relationship between CCDC65, c-Myc and FBXW7.
Since the expression of CCDC65 in H1975 was significantly lower than that in other LUAD cells, SPC-A1 with relatively high expression of CCDC65 was selected for mechanism research. Firstly, Co-immunoprecipitation experiment was performed to verify the interaction between CCDC65 and FBXW7 in LUAD cells and the result confirmed their interaction relation. (Fig. 4a). And results also showed that CCDC65 interacted with c-Myc in both A549 and SPC-A1 cells (Fig. 4b). As a transcription factor, c-Myc was recognized as expressed in both cytoplasm and nucleus. Based on the IHC staining results shown before, CCDC65 was mainly expressed in the cytoplasm (Fig. 1i). Confocal microscopy experiments indicated that CCDC65 and c-Myc co-localized in the cytoplasm (Fig. 4c). In addition, immunohistochemical staining showed that the overexpression of the CCDC65 reduced the nuclear transportation of c-Myc in both A549 and SPC-A1 (Fig. 4d).
CCDC65 interacted with c-Myc. (a) Co-IP and western blotting assays indicated the interaction between CCDC65 and FBXW7. (b) Co-IP and western blotting assays indicated the interaction between CCDC65 and c-Myc. (c) The representative images of immunofluorescent staining of CCDC65 (green) and c-Myc (red). (d) Immunofluorescent staining showed CCDC65 reduced the expression and nuclear transportation of c-Myc.
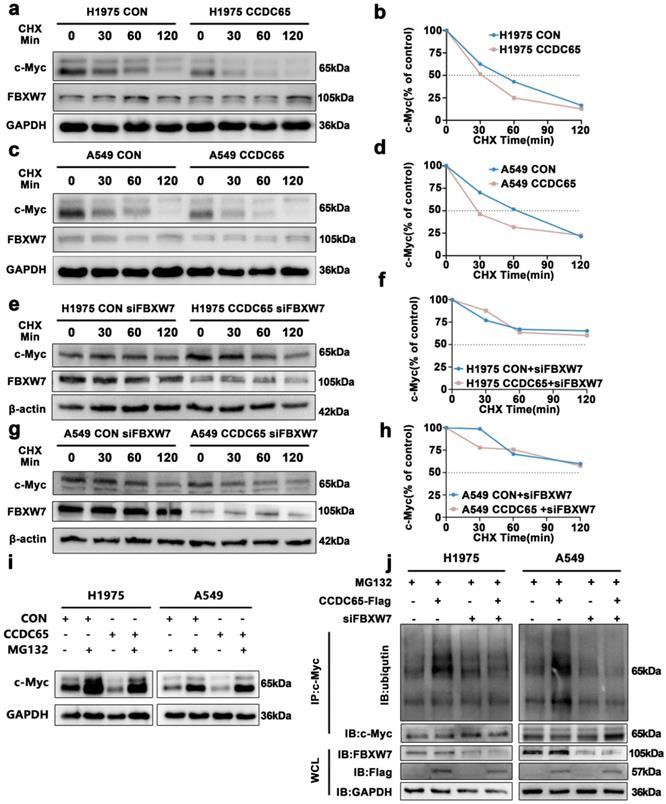
CCDC65 mediated the ubiquitination degradation of c-Myc through the recruitment of FBXW7
To further explore the influence of CCDC65 on c-Myc, ubiquitination research was performed. The CHX chase assay revealed that the c-Myc half-life in CCDC65 over-expressing cells had significantly shorter than that in control cells (Fig. 5a-d, Supplementary Fig. S3d, 3e). And the change of c-Myc half-life induced by CCDC65 was counteracted after being treated with siFBXW7 (Fig. 5e-h, Supplementary Fig. S3f, 3g). After treated with MG132 for 4 hours, the downregulation of c-Myc induced by CCDC65 was reversed (Fig. 5i, Supplementary Fig. S3h). Then the results of immunoprecipitation and western blotting determined that, depending on FBXW7, the overexpression of CCDC65 promoted c-Myc ubiquitination (Fig. 5j). These results suggested that CCDC65 mediated the ubiquitination degradation of c-Myc through the recruitment of FBXW7.
CCDC65 mediated the ubiquitination degradation of c-Myc by recruiting FBXW7. (a, c) CHX chase analysis of c-Myc protein half-life in CCDC65 over-expressing and control group in H1975 and A549 cells. CHX (50 µg/ml). (e, g) CHX chase analysis detected the effects of FBXW7 knockdown on protein stability of c-Myc. (b,d,f,h) The half-life curves of c-Myc protein in H1975 and A549 cells. (i) The effects of DMSO or MG132 (20 µM) treatment on the stability of the c-Myc protein in the control and CCDC65 overexpression groups. (j) Co-IP and western blotting assays were used to detect the effect of CCDC65 overexpression and treated with siFBXW7 on the ubiquitination level of c-Myc.
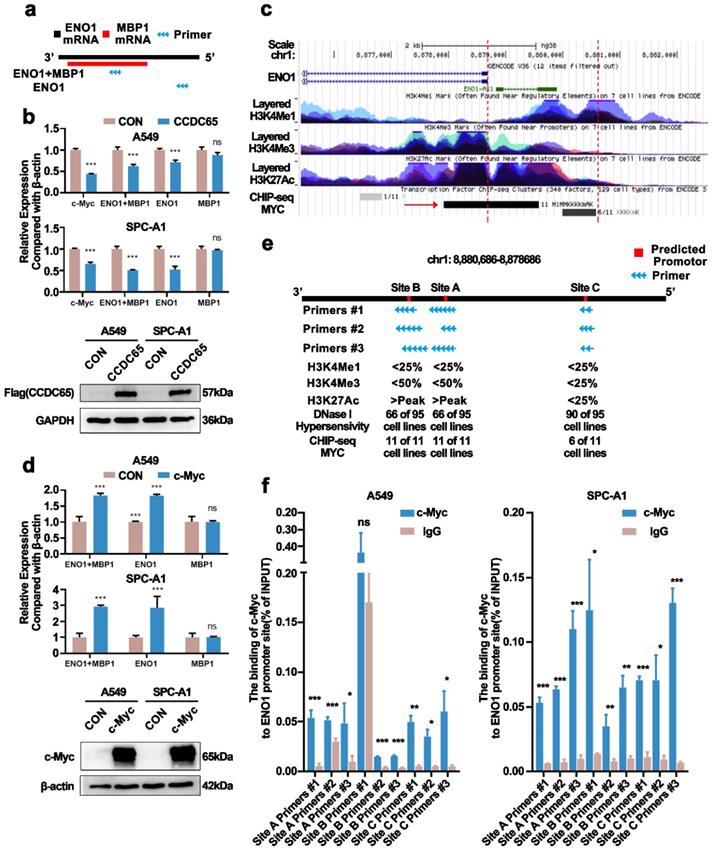
c-Myc bound to the ENO1 transcriptional regulatory region and promoted its transcription
In previous studies, we have confirmed that CCDC65 only downregulated the protein levels of ENO1 in gastric cancer [18]. Myc promoter-binding protein-1 (MBP-1), a shorter protein variant of ENO1, was reported as a negative regulator of c-Myc expression [29]. To eliminate the distractions MBP-1 probably brought about, we constructed ENO1 specific primer sequence (Fig. 6a). Nonetheless, we found that the mRNA levels of ENO1 and c-Myc were downregulated after the overexpression of CCDC65 in LUAD, which was different from CCDC65 in gastric cancer (Fig. 6b). Therefore, we speculated that CCDC65 inhibited ENO1 expression through different mechanisms. Strikingly, based on the transcription factor ChIP-Seq clusters (A549 cell line, ENCSR000DYC) from ENCODE (MYC) [30, 31] and UCSC Genome Browser, we found that there was a potential MYC binding region in the upstream of ENO1 coding region at 2000 bp (Fig. 6c). More details of MYC ChIP-seq clusters were presented in supplementary materials (Supplementary Table S4). Thus we transfected c-Myc into cells and found that the ENO1, instead of MBP-1, mRNA levels were upregulated (Fig. 6d). Combined with ChIP-seq and the result that c-Myc promoted the expression of ENO1 mRNA, we hypothesized c-Myc as a transcription factor binding to the ENO1 transcriptional regulatory region and regulating the expression of ENO1. Using the JASPAR database [32], we predicted the sequences that MYC might bind in 2000 bp upstream and 100 bp downstream of ENO1. The top 5 sequences were screened out (Supplementary Table S5). Nevertheless, among these 5 sequences, two sets of sequences were only a few bp apart, three sets of primers can be designed. In addition, we obtained the H3K4Me1, H3K4Me3, H3K27Ac marks on 7 cells (without lung adenocarcinoma cell lines) and DNase I Hypersensitivity clusters in 125 cell types (containing A549 cell line) from ENCODE (Supplementary Table S6) for predicting promotor region. The data showed that several cell lines had a high H3K27Ac mark on Site A and Site B and most of the cell lines were hypersensitive to being cut by the DNase enzyme on all sites (Fig. 6e). Chromatin immunoprecipitation assay and qPCR assay were performed and the results showed that both three sites were binding with c-Myc (Fig. 6f). Therefore, CCDC65 could inhibit ENO1 transcription by downregulating c-Myc. In addition, we tested whether CCDC65 mediated the ubiquitin degradation of ENO1. The results revealed that CCDC65 promoted the ubiquitin degradation of ENO1, which was in keeping with the previous studies (Supplementary Fig. S4).
c-Myc bound to the ENO1 transcriptional regulatory region and promoted its transcription. (a) Diagram of the ENO1 specific primer sequence and ENO1/MBP1 common fragment primer sequence. (b) The overexpression of CCDC65 inhibited the transcription of c-Myc and ENO1. The expression of MBP-1 was calculated by (ENO1+MBP1)/ENO1. “ns” means no statistical significance. (c) The diagram of ENO1 in Genome Browser (http://genome.ucsc.edu/). The area between the red dotted lines means the upstream 2000bp of ENO1. (d) The overexpression of c-Myc promoted the transcription of ENO1. The expression of MBP-1 was calculated by (ENO1+MBP1)/ENO1. “ns” means no statistical significance. (e) The diagram of upstream 2000bp of ENO1. Red areas: Predicted promotor. Blue arrow: Primers designed according to the predicted promoter. (f) Ch-IP-qPCR detected the binding between c-Myc and the predicted promoter of ENO1.
CCDC65 played the role of tumor suppressor by regulating the c-Myc/ENO1/AKT1 pathway
Several studies revealed that ENO1 promotes cancer phenotype by activating AKT signaling pathway [18, 33, 34]. We observed the influence of CCDC65 on AKT1 phosphorylation and its downstream (Supplementary Fig. S5a). And the high-throughput mRNA sequencing between CCDC65+/+ and CCDC65+/- mice normal lung tissues were conducted. Strikingly, KEGG pathway enrichment analysis suggested the AKT signaling pathway was affected by CCDC65 (Supplementary Fig. S5b, Supplementary Table S7).
We proposed that CCDC65 played the role of tumor suppressor by regulating the c-Myc/ENO1/AKT1 pathway. Rescue experiments were performed to test this hypothesis. C-Myc was introduced into stably over-expressing CCDC65 cells, CCK8 and EdU assays (Supplementary Fig. S6a,6b) showed that c-Myc reversed the cell cycle arrest and the change of cell cycle-related protein induced by CCDC65 (Supplementary Fig. S6c). When ENO1 was transfected into stably over-expressing CCDC65 cells, CCK8 and EdU assays(Supplementary Fig. S7a, S7b) were used to test changes in cell proliferation and cell cycle. The results showed that the effects on cell proliferation and cell cycle inhibited by CCDC65 were reversed after ENO1 overexpression. In addition, the western blotting showed that the expression levels of c-Myc and CCND1 inhibited by CCDC65 were also reversed (Supplementary Fig. S7c). Then, cells were treated with MK2206 (an AKT phosphorylation inhibitor) [35, 36] and siCCDC65. CCK8 (Supplementary Fig. S8a) and EdU assays (Supplementary Fig. S8b) results showed that the increase in cell proliferation induced by siCCDC65 was reversed after exposure to MK2206. CCND1, c-Myc, AKT1 and p-AKT1 were subsequently detected in treated cells, and the results showed that the up-regulation of CCND1 and c-Myc caused by siCCDC65 was also reversed (Supplementary Fig. S8c). To further clarify the relationship between CCDC65, ENO1, and c-Myc, the mRNA levels of which from the GDC TCGA LUAD database were analyzed. The results showed that, both in tumor tissues and normal tissues, CCDC65 was negatively correlated with ENO1 or c-Myc expression (Supplementary Fig.S9a). Meanwhile, ENO1 was positively correlated with c-Myc expression in tumors (Supplementary Fig.S9b). In addition, IHC was performed on the lung adenocarcinoma tissues from CCDC65 engineered mice (Supplementary Fig. S9c). After statistical analysis (Supplementary Fig. S9d) and protein correlation analysis based on the IHC score (Supplementary Fig. S9e-g), the results showed that ENO1 was strongly positively correlated with c-Myc, while ENO1 and c-Myc were negatively correlated with CCDC65. Because a large number of tissue slices had been used for HE staining in tumor area statistics, only a small amount of point-to-point tumor staining could be caught. Although the latter result was not statistically significant, the trends of those protein expression correlations could still be seen.
Discussion
In the present study, we found that CCDC65, a member of the CCDC family, expressed in lower levels in LUAD tissues than in normal lung tissue based on the TCGA database. We also found the downregulation of CCDC65 mRNA expression in LUAD cell lines compared with normal bronchial epithelial cell lines. Consistent with the in vitro results from cell lines and the TCGA database, we found CCDC65 protein expression decreased in LUAD tissues compared with normal lung tissues. In addition, the expression of CCDC65 protein was negatively correlated with lymph node metastasis in LUAD patients. Furthermore, univariate Cox regression analysis demonstrated the downregulation of CCDC65 was an independent poor prognostic factor in LUAD patients. These data preliminarily suggested that CCDC65 exerted a tumor suppressive role in LUAD pathogenesis.
To clarify the role of CCDC65 in adenocarcinoma, we first engineered a CCDC65-/- mouse model using CRISPR/Cas9 gene-editing technology. Serendipitously found that CCDC65-/- mice had a larger head size, while their bodies were smaller and all CCDC65-/- mice died within three weeks after birth. According to the sagittal plane of the whole brain and the known function of CCDC65 [16, 17], we supposed the death of CCDC65-/- mice was possibly relevant to hydrocephalus. But, this was not the point that we focused on, and in-depth research was not performed. Although CCDC65-/- mice could not survive more than three weeks, there were neither phenotypic differences nor DFS differences between CCDC65+/- mice and CCDC65+/+ mice. However, the mRNA and protein levels of CCDC65 in lung tissues of CCDC65+/- mice were half as much as that in CCDC65+/+ mice. Hence, CCDC65+/- and CCDC65+/+ mice were used as urethane-induced LUAD models. We found that urethane induced LUAD nodules were more numerous and larger in CCDC65+/- mice than in CCDC65+/+ mice.
To further validate the tumor-suppressive activity of CCDC65, we established a CCDC65 over-expressing and knock-down human LUAD cell line model and used high-throughput sequencing for further research. The findings indicated that CCDC65 significantly inhibited the proliferation of LUAD cells in vivo and in vitro. GSEA showed G1/S transition checkpoint was the potential research direction. C-Myc and CCND1 were two key G1-S transition proteins involved in the regulation of the cell cycle [37-40]. C-Myc was considered as one of the major driving factors of human tumorigenesis, which was essential for cell proliferation [41, 42]. The mutation of c-Myc causes the death of mice and the knockdown of which significantly inhibits cell proliferation [43]. As a transcription factor, c-Myc drives tumorigenesis through transcriptional regulation and CCND1 is one of the target genes regulated by c-Myc [44]. ENO1 was reported to be inhibited by CCDC65 in gastric cancer and participated in the regulation of the cell cycle [18]. Western blot showed the expression of c-Myc, CCND1 and ENO1 was downregulated by the overexpression of CCDC65. These data further supported that CCDC65 acted as a tumor suppressor via suppression of cell cycle G1-S transition signaling in LUAD.
In previous studies, we have confirmed that CCDC65 mediated the ubiquitination degradation of ENO1 by recruiting FBXW7 in gastric cancer [18]. FBXW7, as a critical tumor suppressor of human cancers, controls proteasome-mediated degradation of oncoproteins. However, c-Myc is one of those oncoproteins [26-28] and was downregulated by the overexpression of CCDC65. Then we attempted to analyze the relationship between CCDC65 and c-Myc. The data showed that CCDC65 interacted with c-Myc and further mediated the ubiquitination degradation of c-Myc. And this degradation process depended on the FBXW7.
ENO1 is a multifunctional protein which implicated in critical biological progression pathways in cancers. As a glycolytic enzyme, ENO1 contributes to the Warburg effect and provides ATP to promote cancer development and progression [45]. In addition, ENO1 promotes the plasminogen activation system and upregulates integrin to enhance the migration and invasion of cancers [46-48]; ENO1 regulates gastric cancer cells stemness by stimulating glycolysis [49]. Our previous studies found that ENO1 promoted tumor proliferation, migration, and invasion in non-small cell lung cancer [50]. What conflicted with previous reports were that CCDC65 downregulated the mRNA levels of ENO1. Thus, we further explore how CCDC65 inhibited the transcription of ENO1. C-Myc was noticed because of its predicted binding to the upstream of ENO1 coding region by ChIP-seq and as a transcription factor which was downregulated by CCDC65. Moreover, ENO1 mRNA levels were upregulated after the introduction of c-Myc. Hence, we hypothesized that c-Myc was involved in ENO1 transcriptional regulation. Three potential ENO1 promoter binding sites were predicted by the JASPAR database and evaluated by the levels of H3K4me1, H3K4me3, H3K27ac, DNase I Hypersensitivity. Histone modification markers are often used as markers of transcriptional regulatory sites, such as H3K4me1 and H3K27Ac for enhancers or promoters, H3K4me3 for repressed regions [15, 51]. DNase I hypersensitive sites are generic markers of regulatory DNA [52]. Experimental results showed that c-Myc was separately bound to all three potential ENO1 promoter binding sites, which meant CCDC65 inhibited ENO1 mRNA levels by mediating c-Myc degradation. The current study reported a shorter protein variant of ENO1, which was named Myc promoter-binding protein-1 (MBP-1). MBP-1 is a negative regulator of c-Myc expression [53, 54]. Our study proposed the transcriptional regulation of ENO1 by c-Myc, which complemented the precise regulation between ENO1 and c-Myc. Previous research reported that CCDC65 promoted ENO1 ubiquitination in gastric cancer [18]. We also tested it in lung cancer and experiment results confirmed the interaction between CCDC65 and ENO1 and the overexpression of CCDC65 promoted the ubiquitination of ENO1.
CCDC65 downregulated ENO1 mRNA levels by decreasing the stability of c-Myc protein and downregulated ENO1 protein levels by mediating its ubiquitination degradation. Previous reports revealed that ENO1 could promote the phosphorylation of AKT1 [33, 50]. AKT1, as a member of the AKT family, plays a key role in multiple tumor signaling pathways, the phosphorylation of which promotes cell proliferation, cell cycle and cell steaminess [55-57]. Our results also showed that CCDC65 decreased the phosphorylation of AKT1, but did not modulate total protein levels. Based on the different genes between CCDC65+/+ and CCDC65+/- mice lung tissues, KEGG pathway enrichment analysis also supported that CCDC65 affected the AKT signaling pathway. The rescue experiments suggested that CCDC65 inhibited the proliferation of LUAD by inhibiting ENO1 and AKT1 phosphorylation.
In summary, this study showed that CCDC65 was downregulated in LUAD, which promoted the pathogenesis of LUAD based on evidence derived from in vivo and in vitro experiments. Molecular mechanism showed that CCDC65 recruited E3 ubiquitin ligase FBXW7 to induce the ubiquitination degradation of c-Myc, an oncogenic transcription factor in tumors, and reduced the binding between c-Myc and ENO1 promoter and thus suppressed the expression of ENO1 in mRNA levels. In addition, CCDC65 also recruited FBXW7 to degrade ENO1 protein through ubiquitinated modulation. The downregulated ENO1 further reduced the phosphorylation activation of AKT1, which thus inactivated the cell cycle signal. Our data demonstrated that CCDC65 was a potential tumor suppressor by recruiting FBWX7 to suppress c-Myc/ENO1-induced cell cycle signal in lung adenocarcinoma.
Abbreviations
CCD: Coiled-coil domain; CCDC65: Coiled-coil domain 65; ENO1: Alpha-enolase; PCD: Primary ciliary dyskinesia; SPF: specific pathogen-free; WT: Wild-type; HET: Heterozygous; KO: Knockout; CHX: Cycloheximide; NSCLC: Non-Small Cell Lung Cancer; LUAD: Lung adenocarcinoma; HR: Hazard ratio; CON: Control group; RT-qPCR: Reverse transcriptase-quantitative real-time PCR; IF: Immunofluorescence; Co-IP: Co-immunoprecipitation; Ch-IP: Chromatin-immunoprecipitation.
Supplementary Material
Supplementary figures.
Supplementary tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81772872), the Nature Science Foundation of Guangdong Province (Grant No. 2022A1515012624, 2020A1515010412). Shenzhen Science and technology innovation Commission Foundation (Grant No. JCYJ20190809103203711, JCYJ20210324105411031). We thank Dr. Ziqing Wu and his pathology team for their kind help in the knowledge of pathology.
Ethics Declarations
The present study was approved by the Ethics Committees of the TCM-Integrated Hospital of Southern Medical University. The tissue microarray was approved by the Ethics Committees of Shanghai Outdo Biotech Company (No. YB M-05-02). The procedures for care and use of animals were approved by the Ethics Committee of the TCM-Integrated Hospital of Southern Medical University and all applicable institutional and governmental regulations concerning the ethical use of animals were followed.
Author Contributions
Ziyan Zhang, Ping Xu, Tongyuan Deng, Zhe Hu, Zhaojian Fu contributed equally to this work.
ZZY, XP and DTY designed and performed main experiments, analyzed data and wrote the manuscript; HZ, FZJ and LLZ performed some of the experiments; DXJ designed the experiment; XYY provided the immunofluorescence technical support; PLZ, ZMM and GB assisted in the study. LJH and CR analyzed the data. ZDY, YHL, LR and SP reviewed the data. ZZB provided financial and technical support. FWY and LZ conceived of the study, secured research funding, supervised all aspects of the work. All authors read and approved the final paper.
Data Availability
CCDC65+/+ and CCDC65+/- mice lung tissue transcriptome sequencing data are available from the SRA database (PRJNA695129). The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
2. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS. et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med. 2017;376:629-40
3. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y. et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med. 2020;382:41-50
4. Kennedy JM, Fodil N, Torre S, Bongfen SE, Olivier JF, Leung V. et al. CCDC88B is a novel regulator of maturation and effector functions of T cells during pathological inflammation. The Journal of experimental medicine. 2014;211:2519-35
5. Becker-Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan-Brown J. et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet. 2011;43:79-84
6. Sobh A, Loguinov A, Zhou J, Jenkitkasemwong S, Zeidan R, El Ahmadie N. et al. Genetic screens reveal CCDC115 as a modulator of erythroid iron and heme trafficking. American journal of hematology. 2020;95:1085-98
7. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J. et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583:133-8
8. Hu X, Zhao Y, Wei L, Zhu B, Song D, Wang J. et al. CCDC178 promotes hepatocellular carcinoma metastasis through modulation of anoikis. Oncogene. 2017;36:4047-59
9. Radulovich N, Leung L, Ibrahimov E, Navab R, Sakashita S, Zhu CQ. et al. Coiled-coil domain containing 68 (CCDC68) demonstrates a tumor-suppressive role in pancreatic ductal adenocarcinoma. Oncogene. 2015;34:4238-47
10. Wang H, Zhang CZ, Lu SX, Zhang MF, Liu LL, Luo RZ. et al. A Coiled-Coil Domain Containing 50 Splice Variant Is Modulated by Serine/Arginine-Rich Splicing Factor 3 and Promotes Hepatocellular Carcinoma in Mice by the Ras Signaling Pathway. Hepatology. 2019;69:179-95
11. Wang J, Wu X, Dai W, Li J, Xiang L, Tang W. et al. The CCDC43-ADRM1 axis regulated by YY1, promotes proliferation and metastasis of gastric cancer. Cancer letters. 2020;482:90-101
12. Cheng C, Li W, Peng X, Liu X, Zhang Z, Liu Z. et al. miR-1254 induced by NESG1 inactivates HDGF/DDX5-stimulated nuclear translocation of beta-catenin and suppresses NPC metastasis. Molecular therapy Methods & clinical development. 2021;20:615-24
13. Liang Z, Liu Z, Cheng C, Wang H, Deng X, Liu J. et al. VPS33B interacts with NESG1 to modulate EGFR/PI3K/AKT/c-Myc/P53/miR-133a-3p signaling and induce 5-fluorouracil sensitivity in nasopharyngeal carcinoma. Cell death & disease. 2019;10:305
14. Liu Z, Liu J, Li Y, Wang H, Liang Z, Deng X. et al. VPS33B suppresses lung adenocarcinoma metastasis and chemoresistance to cisplatin. Genes & diseases. 2021;8:307-19
15. Zheng Y, Zhang J, Wang L, Zhou Z, Xu M, Li J. et al. Cloning and characterization of a novel sperm tail protein, NYD-SP28. Int J Mol Med. 2006;18:1119-25
16. Austin-Tse C, Halbritter J, Zariwala MA, Gilberti RM, Gee HY, Hellman N. et al. Zebrafish Ciliopathy Screen Plus Human Mutational Analysis Identifies C21orf59 and CCDC65 Defects as Causing Primary Ciliary Dyskinesia. Am J Hum Genet. 2013;93:672-86
17. Horani A, Brody SL, Ferkol TW, Shoseyov D, Wasserman MG, Ta-shma A. et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PloS one. 2013;8:e72299
18. Deng T, Shen P, Li A, Zhang Z, Yang H, Deng X. et al. CCDC65 as a new potential tumor suppressor induced by metformin inhibits activation of AKT1 via ubiquitination of ENO1 in gastric cancer. Theranostics. 2021;11:8112-28
19. Huang L, Carney J, Cardona DM, Counter CM. Decreased tumorigenesis in mice with a Kras point mutation at C118. Nature communications. 2014;5:5410
20. Hou R, Li Y, Luo X, Zhang W, Yang H, Zhang Y. et al. ENKUR expression induced by chemically synthesized cinobufotalin suppresses malignant activities of hepatocellular carcinoma by modulating beta-catenin/c-Jun/MYH9/USP7/c-Myc axis. Int J Biol Sci. 2022;18:2553-67
21. Zhan P, Wang Y, Zhao S, Liu C, Wang Y, Wen M. et al. FBXW7 negatively regulates ENO1 expression and function in colorectal cancer. Laboratory investigation; a journal of technical methods and pathology. 2015;95:995-1004
22. Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87-D92
23. Lanczky A, Gyorffy B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res. 2021;23:e27633
24. Wang Q, Gao G, Zhang T, Yao K, Chen H, Park MH. et al. TRAF1 Is Critical for Regulating the BRAF/MEK/ERK Pathway in Non-Small Cell Lung Carcinogenesis. Cancer research. 2018;78:3982-94
25. Feng K, Ma C, Liu Y, Yang X, Yang Z, Chen Y. et al. Encapsulation of LXR ligand by D-Nap-GFFY hydrogel enhances anti-tumorigenic actions of LXR and removes LXR-induced lipogenesis. Theranostics. 2021;11:2634-54
26. King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P. et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552-66
27. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H. et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116-25
28. Li Z, Liu J, Chen T, Sun R, Liu Z, Qiu B. et al. HMGA1-TRIP13 axis promotes stemness and epithelial mesenchymal transition of perihilar cholangiocarcinoma in a positive feedback loop dependent on c-Myc. J Exp Clin Cancer Res. 2021;40:86
29. Feo S, Arcuri D, Piddini E, Passantino R, Giallongo A. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 2000;473:47-52
30. Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC. et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016;44:D726-32
31. Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC. et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798-812
32. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic acids research. 2022;50:D165-D73
33. Dai J, Zhou Q, Chen J, Rexius-Hall ML, Rehman J, Zhou G. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nature communications. 2018;9:3850
34. Li HJ, Ke FY, Lin CC, Lu MY, Kuo YH, Wang YP. et al. ENO1 Promotes Lung Cancer Metastasis via HGFR and WNT Signaling-Driven Epithelial-to-Mesenchymal Transition. Cancer research. 2021;81:4094-109
35. Li J, Xie H, Ying Y, Chen H, Yan H, He L. et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Molecular cancer. 2020;19:152
36. Tang YQ, Li ZW, Feng YF, Yang HQ, Hou CL, Geng C. et al. MK2206 attenuates atherosclerosis by inhibiting lipid accumulation, cell migration, proliferation, and inflammation. Acta pharmacologica Sinica. 2022;43:897-907
37. Farria AT, Plummer JB, Salinger AP, Shen J, Lin K, Lu Y. et al. Transcriptional Activation of MYC-Induced Genes by GCN5 Promotes B-cell Lymphomagenesis. Cancer research. 2020;80:5543-53
38. Tang Z, Chen W, Xu Y, Lin X, Liu X, Li Y. et al. miR-4721, Induced by EBV-miR-BART22, Targets GSK3beta to Enhance the Tumorigenic Capacity of NPC through the WNT/beta-catenin Pathway. Mol Ther Nucleic Acids. 2020;22:557-71
39. Kurnit KC, Draisey A, Kazen RC, Chung C, Phan LH, Harvey JB. et al. Loss of CD73 shifts transforming growth factor-beta1 (TGF-beta1) from tumor suppressor to promoter in endometrial cancer. Cancer letters. 2021;505:75-86
40. Liu JH, Yang HL, Deng ST, Hu Z, Chen WF, Yan WW. et al. The small molecule chemical compound cinobufotalin attenuates resistance to DDP by inducing ENKUR expression to suppress MYH9-mediated c-Myc deubiquitination in lung adenocarcinoma. Acta pharmacologica Sinica. 2022
41. Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L. et al. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell. 2015;28:129-40
42. Huang J, Tian F, Song Y, Cao M, Yan S, Lan X. et al. A feedback circuit comprising EHD1 and 14-3-3zeta sustains beta-catenin/c-Myc-mediated aerobic glycolysis and proliferation in non-small cell lung cancer. Cancer letters. 2021;520:12-25
43. Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671-82
44. Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J. 1999;18:5310-20
45. Qiao G, Wu A, Chen X, Tian Y, Lin X. Enolase 1, a Moonlighting Protein, as a Potential Target for Cancer Treatment. Int J Biol Sci. 2021;17:3981-92
46. Ceruti P, Principe M, Capello M, Cappello P, Novelli F. Three are better than one: plasminogen receptors as cancer theranostic targets. Experimental hematology & oncology. 2013;2:12
47. Jiang K, Dong C, Yin Z, Li R, Mao J, Wang C. et al. Exosome-derived ENO1 regulates integrin alpha6beta4 expression and promotes hepatocellular carcinoma growth and metastasis. Cell death & disease. 2020;11:972
48. Principe M, Borgoni S, Cascione M, Chattaragada MS, Ferri-Borgogno S, Capello M. et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. Journal of hematology & oncology. 2017;10:16
49. Yang T, Shu X, Zhang HW, Sun LX, Yu L, Liu J. et al. Enolase 1 regulates stem cell-like properties in gastric cancer cells by stimulating glycolysis. Cell death & disease. 2020;11:870
50. Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong SW. et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. Journal of hematology & oncology. 2015;8:22
51. Hou C, Zhao X, Tian GG, Wu J. Stella Regulates the Development of Female Germline Stem Cells by Modulating Chromatin Structure and DNA Methylation. Int J Biol Sci. 2022;18:3006-18
52. Meuleman W, Muratov A, Rynes E, Halow J, Lee K, Bates D. et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature. 2020;584:244-51
53. Ray R, Miller DM. Cloning and characterization of a human c-myc promoter-binding protein. Mol Cell Biol. 1991;11:2154-61
54. Ray RB, Steele R, Seftor E, Hendrix M. Human breast carcinoma cells transfected with the gene encoding a c-myc promoter-binding protein (MBP-1) inhibits tumors in nude mice. Cancer research. 1995;55:3747-51
55. Duggal S, Jailkhani N, Midha MK, Agrawal N, Rao KVS, Kumar A. Defining the Akt1 interactome and its role in regulating the cell cycle. Scientific reports. 2018;8:1303
56. Toulany M, Rodemann HP. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Seminars in cancer biology. 2015;35:180-90
57. Li J, Wang J, Xie D, Pei Q, Wan X, Xing HR. et al. Characteristics of the PI3K/AKT and MAPK/ERK pathways involved in the maintenance of self-renewal in lung cancer stem-like cells. Int J Biol Sci. 2021;17:1191-202
Author contact
![]() Corresponding authors: Prof. Zhen Liu, E-mail: narcisuss_janecom; Prof. Huilin Yang, E-mail: 112699355com; Prof. Rong Li, E-mail: nflirongcom; Prof. Weiyi Fang, E-mail: fangweiyi1975com.
Corresponding authors: Prof. Zhen Liu, E-mail: narcisuss_janecom; Prof. Huilin Yang, E-mail: 112699355com; Prof. Rong Li, E-mail: nflirongcom; Prof. Weiyi Fang, E-mail: fangweiyi1975com.