Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. The effect and regulation of...
2. The role and regulation of...
3. Malfunctions of...
4. m6A in the tumor...
5. m6A regulator as a...
Conclusion and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(11):4432-4451. doi:10.7150/ijbs.73093 This issue Cite
Review
Novel insights into m6A modification of coding and non-coding RNAs in tumor biology: From molecular mechanisms to therapeutic significance
Jinlin Jia1*, Suwen Wu2*, Zimo Jia3, Chang Wang1, Chenxi Ju1, Jinxiu Sheng1, Fucheng He1, Mingxia Zhou4 ![]() , Jing He5
, Jing He5 ![]()
1. Department of Medical Laboratory, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China
2. Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200011, China
3. Department of Biochemistry and Molecular Biology, Hebei Medical University, Shijiazhuang 050017, China
4. Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China
5. Department of Breast Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China
*These authors contributed equally to this work.
Received 2022-3-23; Accepted 2022-6-12; Published 2022-7-4
Abstract

Accumulating evidence has revealed that m6A modification, the predominant RNA modification in eukaryotes, adds a novel layer of regulation to the gene expression. Dynamic and reversible m6A modification implements sophisticated and crucial functions in RNA metabolism, including generation, splicing, stability, and translation in messenger RNAs (mRNAs) and non-coding RNAs (ncRNAs). Furthermore, m6A modification plays a determining role in producing various m6A-labeling RNA outcomes, thereby affecting several functional processes, including tumorigenesis and progression. Herein, we highlighted current advances in m6A modification and the regulatory mechanisms underlying mRNAs and ncRNAs in distinct cancer stages. Meanwhile, we also focused on the therapeutic significance of m6A regulators in clinical cancer treatment.
Keywords: m6A, non-coding RNA, tumor microenvironment, therapeutic target
Introduction
Extensively investigations have revealed that m6A RNA modification is a reversible and dynamic complex biological process, among which a series of proteins are recruited to the bound regions to execute their functions [1]. m6A modification typically accounts for 0.1%-0.4% of total cellular RNA adenosines. Previous studies have validated that m6A modification sites are mainly located close to stop codons, or the 5'- and 3'-untranslated regions (UTRs) [2, 3]. The m6A modification is frequently observed in the long internal exon in the vast genome of mRNA, which indicates that it is intrinsically connected with mRNA functions [1, 4-6]. Additionally, m6A modification sites also exist in the unique motifs of diverse ncRNAs, including miRNAs, long non-coding RNAs, and circRNAs [7, 8]. The m6A modification complements are associated with dysregulation of RNAs, including mRNA and ncRNAs, responsible for cancer's hallmarks, such as sustaining proliferative signaling, resisting cell death, evading growth suppressors, inducing angiogenesis, avoiding immune destruction, etc. [9-14]. Therefore, we can conclude that there are intricate potential interaction networks between m6A and mRNA or ncRNA, by which m6A and mRNA or ncRNA co-modulate target mRNAs via counteraction, orchestration or both.
Although still in inception, outstanding efforts have been devoted to scrutinizing the complicated crosstalk between m6A modification and RNAs. In this review, we summarized the latest proceedings concerned with the interaction between m6A modification and RNAs by describing the biological effect and underlying regulatory mechanisms of m6A modified RNAs and the influences of m6A-RNAs on human malignancies. Finally, the prospect of m6A in cancer diagnosis and therapy was heavily discussed.
1. The effect and regulation of m6A in mRNA
1.1 m6A writers
The biological function of m6A modification is dedicatedly governed by m6A methyltransferase (also called m6A writers), m6A demethylase (also called m6A erasers), and m6A binding protein (also called m6A readers) [15]. The first widespread dynamic mRNA modification, in conjunction with other RNA chemical modifications, has coined the epitranscriptomics field and m6A has become the spotlight in this emerging territory. Nowadays, compelling evidence has validated a series of m6A molecular compositions.
M6A methyltransferase-like3 (METTL3) and methyltransferase-like 14 (METTL4) form the multicomponent m6A methyltransferase complex (MTC), which executes the m6A modification function [16]. The former binds to the methyl donor S-adenosyl methionine (SAM) and catalyzes methyl group transfer depending on its highly active methyltransferase domain. Meanwhile the latter is essential for m6A deposition by forming a heterodimer with METTL3 and substrate recognition, contributing to the methylase activity [17]. At the same time, auxiliary protein participates in the MTC assembly to ensure the stability of the METTL3-METTL14 complex. Wilms tumor 1-associated protein (WTAP) was the first identified auxiliary protein to reciprocate with the METTL3-METTL14 heterodimer [4]. Moreover, MTC consists of key regulatory factors, including via-like m6A methyltransferase associated (VIRMA, also known as KIAA1429), zinc finger CCCH domain-containing protein 13 (ZC3H13), and RNA-binding motif protein 15 (RAB15/15B), which play critical roles in stabilizing core complex [18]. However, the precise mechanisms by which these adaptor proteins control MTC formation remain to be elucidated.
Distinctive from what has been observed within the m6A chemical mark catalyzed by MTC, recent research has pinpointed that methyltransferases like 5 (METTL5), methyltransferases like 16 (METTL16), and zinc finger CCHC-type containing 4 (ZCCHC4) execute m6A methyltransferase function in an MTC-independent manner [19-21]. For example, the methyltransferase activity of ZCCHC4 is independently mediated by an integrated 28S RNA-binding surface, comprising of three domains [19]. Except for the typical ZCCHC4 methyltransferase domains, there are two specific structures involved in the RNA-binding site: The N-terminal specific zinc finger domain and the C-terminal CCHC domain [19]. We can deduce that this ubiquitous m6A chemical remark harbors multiple complicated mechanisms, in contrast to except for what has been observed in the METTL family.
1.2 m6A erasers
The m6A modification has been shown to be a reversible and dynamic biological process ever since the functional identification of the fat mass and obesity-associated protein (FTO), the first reported human m6A erasers to be described [22]. ALKB homolog 5 (ALKBH5) has been identified as the second m6A eraser, which is also capable of demethylating m6A modification [23]. The demethylation characters embedded in FTO and ALKBH5 can remove the m6A remark labeled in mRNAs and ncRNAs. They promote demethylation reactions and ebb m6A modification level in cellular mRNA, which is essential for dynamic balance in the m6A regulation network. In contrast to being exclusively localized in cell nuclei or nucleoplasm, FTO is partly distributed in a spotty form throughout the nucleoplasm and partially colocalized within the nuclear splicing speckles [22]. These splicing speckles such as RNA polymerase II phosphorylated at Ser2 (Pol II-S2P) functions in the spliceosome assembly [22]. Moreover, FTO is partially located in nuclei and could be recruited to the assembly spliceosome center by communicating with its nuclear speckles partner, finally facilitating mRNA processing [22]. Similar to FTO, ALKBH5 has been identified as a factor in mRNA export, connecting RNA metabolism to nuclear speckle. Interestingly, ALKBH5 has been certified as a key determinant in spermatogenesis and the p53 signal pathway network [23].
Maintaining the stability of m6A modification levels in endogenous RNAs is indispensable for diverse cellular activities under physiological situations. The equilibrium between m6A modification deposition and removal serves a functional purpose in the dynamic interactive network. As a result, mutation or deregulation of the m6A composer is intrinsically linked to human cancers. Overpowering research has validated the oncogenic and tumor-suppressing effects of these writers and erasers in cancer. In addition, the non-homeostasis caused by writers alone, erasers alone, or both could result in the aberrant m6A modification in the critical and functional RNA transcripts which have a significant role in tumorigenesis and progression.
1.3 m6A readers
Besides writers and erasers, the m6A regulation system also contains one type of unique RNA-binding protein, called m6A readers. There have been distinct complex regulatory mechanisms modulating gene expression during transcription, post-transcription, and following gene expression procedures such as translation. The m6A modification can occur in infant RNA transcripts in the nucleus during transcription. The m6A methylation installed in RNA also impacts the subsequent gene expression mediated by diverse mechanisms, such as altering RNA local structure or recruiting selective m6A readers to the bound m6A remarked RNA [16].
The mRNA alternative splicing (AS) event occurs in nucleus speckles, where m6A writers and erasers to accumulate [4, 24]. Most recent studies have revealed that m6A writers and erasers directly or indirectly interact with nearby splicing speckles that function as critical mRNA splicing factors in AS or pre-mRNA processing [4, 24]. This cellular location pattern demonstrates that m6A modification is closely linked to mRNA splicing. RNA transcripts distributed in the nucleus and nucleoplasm possess fundamentally distinct fates. Regarding the nucleus, nascent transcripts tend to produce mature signals devoid of introns. In the cytoplasm, mRNA can be stored in specific functional carriages, including messenger ribonucleoprotein (mRNP) foci, actively translating, stabilizing, or degrading. Active protein complexes involving gene expression vary from nuclei such as mRNA AS and export to cytoplasm such as translation and decay. Based on this, there has been one distinct m6A reader mechanism notion: m6A readers could be categorized as either nuclear readers, such as heterogeneous nuclear ribonucleoproteins A2/B1 (HNRNPA2B1) and YTH domain-containing protein 1 (YTHDC1) or cytoplasmic readers such as YTH domain family proteins (YTHDF1-3), which bind specific to a m6A pocket within the nucleus or cytoplasm, respectively [25].
Nuclear m6A readers
The RNA binding protein YTHDC1 is located in YT bodies, which are spatially in proximity with nuclear speckles recruiting AS factors, implying that YTHDC1 participates in mRNA splicing [26]. SR family proteins have been demonstrated to function as a crucial splicing factor by activating exon inclusion or exon skipping [27]. The splicing mechanism between SRSF3 and SRSF10 is distinct from each other. SRSF3 facilitates exon inclusion in trans-acting splicing. Interestingly, the C-terminal RS domain of SRSF3 and SRSF10 competitively binds with the N-terminal YTHDC1 competitively [28]. In the nucleus, YTHDC1 recruits SRSF3 to form a protein-protein interaction complex and brings SRSF3 to the mRNA-binding element by binding to the m6A-bearing mRNA, changing the splicing pattern [28].
Meanwhile, the interaction between YTHDC1 and SRSF10 impedes exon skipping of SRSF10 in the mRNA-binding element [28]. In addition, research has reported that YTHDC1 shortens the residence time of m6A modification mRNA in the nucleus, thereby reducing the subcellular abundance of target transcripts in the nucleus and conversely promoting the accumulation of transcripts in the cytoplasm [29]. YTHDC1 regulates the distribution of cellular transcripts and facilities m6A-mRNA transport from the nucleus to the cytoplasm by interacting with SRSF3 [29]. Indeed, nucleus reader proteins binding m6A possess other protein-protein interaction domains, which communicate with the bound downstream responding elements to specify the key activities.
Besides engaging in AS, YTHDC1 has been validated to fine-tune transcription and gene expression by different mechanisms. YTHDC1 binding with m6A-modified transcripts recruits H3K9me2 demethylase, which diminishes transcriptional repression in the chromatin domains of m6A-mRNA [30]. Moreover, it tunes chromatin accessibility and transcription by facilitating chromosome-associated regulatory RNAs (carRNAs) degradation, mediated by nuclear exosome targeting-mediated degradation [31]. Also, YTHDC1 governs transcription and gene expression by interacting with enhancer RNAs (eRNAs), or ncRNAs [32]. YTHDC1 increases enhancer activity by perceiving m6A modified eRNA and recruits transcriptional activators BRD4, which is implicated in phase-separated transcriptional condensates [33]. ncRNAs will be discussed in the following section. Herein, we can conclude that the complex and dynamic roles of YTHDC1 in the nucleus may be associated with RNA types such as typical mRNAs, car RNAs, and lncRNAs.
Cytoplasm reader
This working mechanism gets along with cytoplasm reader proteins well. According to translational activities, mRNA present in the cytoplasm is divided into three function pools: non-ribosome mRNA-protein particles (the lowest translational viability, mRNA decay sites), translatable mRNA pool mRNPs related to translation effectors (middle translational viability), and actively translating polysome (high translation viability) [34]. The C-terminal domain of YTHDF2 has been confirmed to recognize the core motif of G(m6A)C in the bound mRNA. This interaction transfers target mRNA from translatable pool to processing bodies or other mRNA degrading compartments mediated by the N-terminal domain [35]. However, YTHDF1 preferentially delivers m6A-bearing mRNAs into the translation pool and increases the translation initiation efficiency of m6A remarked mRNA loading with ribosomes by interacting with ribosomes and eIF3 in an RNA-independent manner [36]. Simultaneously, YTHDF1 collaborates with trans-acting regulators such as insulin-like growth factor 2 mRNA-binding protein1 (IGF2BP1), YBX1, G3BP1 and poly(A) binding protein cytoplasmic 2 (PCBP2) in an RNA-dependent pattern. Translation and decay are two adverse gene expression outputs. The translation-promotion influence of YTHDF1 and the decay-promotion effect of YTHDF2 on mRNA fate are two opposite mechanisms. Indeed, YTHDF2 and YTHDF1 share abundant target transcripts besides their specific target transcripts [36]. The latter combines target m6A sites located in these common mRNAs earlier than the former. YTHDF2 and YTHDF1 corroborate to modulate the translation efficiency of dynamic transcripts [36]. Recent research has suggested that YTHDF3 facilitates protein synthesis by combining with YTHDF1 and promotes mRNA decay by interacting with YTHDF2 [37]. We can conclude from all these studies that three YTHDF proteins contribute to cytoplasmic m6A mRNA metabolism in the cytoplasm in an integrated and cooperative mechanism.
Growing evidence has demonstrated that emerging m6A readers such as IGF2BPs (IGF2BP1/2/3) and HNRNPA2B1 recognize m6A sites and modulate mRNA life cycle [25, 38]. As is well established, IGF2BP elevates the stability of m6A modified mRNA and promotes translation by its K homology domains (KH domain)- containing m6A binding site [38]. METTL3 has been identified as an m6A reader as studies have confirmed that it promotes mRNA translation in an m6A-independent manner [39]. Furthermore, it interacts with eIF3 subunit h (eIF3h) to form the METTL3-eIF3h loop, enhancing protein synthesis by ribosome cycling in a manner analogous to eIF4G-PABPC1 (poly(A)-binding protein) mediated mRNA looping [40]. Notably, mRNA metabolism events in the nuclear and cytoplasm do not occur independently. Diverse mechanisms have allowed frequent information communications and crosstalk between the nucleus and cytoplasm. We speculate that m6A modification plays a key role in coordinating RNA fundamental biological processes between the nucleus and cytoplasm.
In addition to participating in mRNA metabolism, m6A alters local secondary RNA structure. It regulates the accessibility of RNA binding motifs (RBM), suggesting that m6A affects the RNA-protein interaction to produce a widespread and far-ranging influence on biological processes [41, 42]. This working mechanism is also termed the m6A switch. For example, m6A modification in mRNA increases the spatial affinity of its surrounding protein binding elements to the RBM of HNRNP C, which is best known to be associated with pre-mRNA processing in AS [43]. The accessibility of mRNA to bind Arg-Gly-Gly repeats included in the low-complexity domain of HNRNPG is enhanced once upon the target mRNA is exposed to m6A, changing the AS pattern of the m6A modified mRNA [43]. Rather than directly binding m6A modification, HNRNPG recognizes the m6A methyl group that has remodeled its surrounding RNA sequence by m6A switch regulation, thus affecting the mRNA maturation [44].
Furthermore, the final fate of the m6A labeled mRNA depends on the location of m6A modification in mRNA transcripts and the type of m6A readers recognizing the m6A site (Figure 1). The identification of functional and novel m6A regulators as well as the deep deciphering of working mechanisms will require more advanced and mature high-throughput approaches for profiling RNA-protein interactions.
Functions of m6A modification in mRNA. Writer proteins deposit m6A remarks in newborn mRNAs transcript from DNA.The m6A signal deposited in mRNA can adjust the local flanking sequences by the m6A flag installed in the mRNA and recruit YTHDC1, which manipulates mRNA alternative splicing and nuclear export. The mRNA labeling m6A can recruit IGF2BPs to stabilize the mRNA in the nucleus. After penetrating the cytoplasm, the m6A-labeled mRNA produces sophisticated and far-reaching biological effects. IGF2BP1/2/3 proteins recognize the m6A remark embedded in mRNA, influencing the mRNA stability. On the contrary, m6A deposited in mRNA can recruit YTHDF2/3, promoting target mRNA degradation. In addition, the presence of an m6A modified mRNA regulates target mRNA translation and, consequently protein synthesis. The binding of eIF3 and m6A pockets launches m7G-independent translation. The interplay between YTHDF2 and m6A sites existing in mRNA enhances translation elongation. YTHDF1/3 or YTHDC2 perceive m6A pockets, and promotes target mRNA translation initiation.
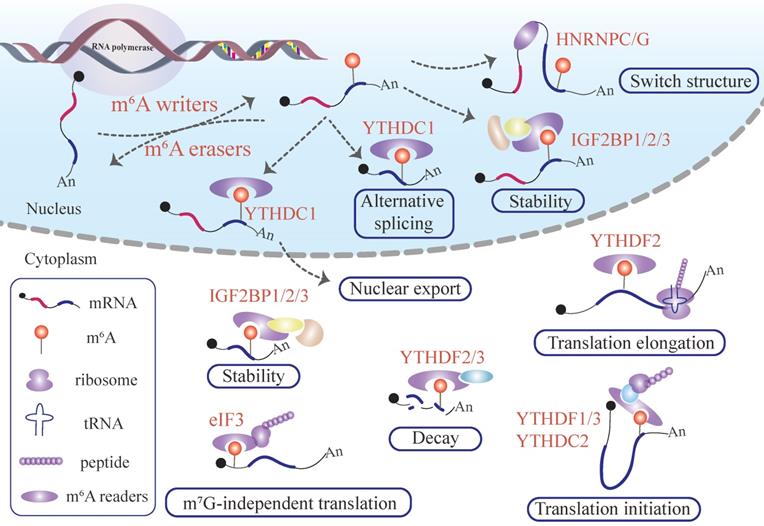
1.4 Emerging m6A -specific regulatory mechanisms
To date, most studies have mainly focused on the delicate and complex function of m6A proteins. However, the internal regulatory mechanisms of these writers, erasers and readers remain largely unknown. Exemplifying the upstream regulatory mechanism of these proteins will enable a better comprehension of the far-ranging biological effects of m6A and provide guidelines for m6A-associated diseases. A recent study has found the methyltransferase activity of METLL3 decreases following SUMOylation modification [45]. However, other characteristics, including stability, localization, and interplay with METTL14 and WTAP do not undergo substantial alteration [45].
Further research has revealed that extensive SUMOylation occurring in the 177/211/212/215 lysine sites of METTL3 spatially affects its interaction with substrate mRNAs, which alternatively reduces methyltransferase activity [45]. Subsequently, the authors have again validated that the SUMOylation accepted by the 571-lysine site of YTHDF2 enhances its binding affinity with m6A-bearing mRNA, which contributes to the decay-promoting effect of YTHDF2. In addition, this SUMOylation could be activated by microenvironmental hypoxia [46]. Herein, we theorize that m6A regulators undergo further post-translational modifications (ubiquitination, phosphorylation, acetylation, and so on). These special modifications may play a direct role in the function of m6A modified RNAs under specific stress conditions or pathological conditions.
Surprisingly, site-specific m6A modifications mechanisms and their predilection to occur in the CDS and 3'UTR of mRNA have been recently uncovered. Research has found that H3K36me3 histone modification directly recognizes METTL14 (serves as an m6A reader), thereby co-transcriptionally governing m6A deposition [47]. However, it is notable that almost all were actively transcribing genes with substantial H3K36me3 modifications, whereas m6A modification varies widely across mRNAs. How do they achieve their selectivity? The lack of a structural domain for METTL14 to recognize H3K36me3, indicates that a novel recognition mechanism achieves required to bind H3K36me3 to METTL14, and the structural biological basis of this interaction must to be deeply explored.
2. The role and regulation of m6A in ncRNAs
2.1 m6A in miRNA
miRNA accounts for most ncRNA and exert diverse functions in gene expression. miRNA biogenesis originates from the primary transcripts of miRNA transcribed from DNA (pri-miRNA) processing under the guideline of the microprocessor complex [48]. The microprocessor complex subunit DGCR8 while ribonuclease type III DROSHA mediate primary mi-RNA processing [49]. Specifically, DGCR8 recognizes the dsRNA-ssRNA junction of the pri-miRNA hairpin by its heme-bounding and RNA binding domains. It then recruits DROSHA, which releases hairpin-shaped pre-miRNA by cleaving 11 bp away from the junction [50]. Furthermore, METTL3 endows pre-miRNAs with m6A modification and recruits DGCR8, facilitating pre-miRNA processing and mature miRNA output by triggering a global co-transcriptional procedure microprocessor machinery governing pri-miRNA processing [51] (Figure 2).
The roles of m6A modification in non-coding RNAs. METTL3 and hNRNPA2B1 promote pri-miRNA processing and mature miRNA output in the m6A tone. The m6A modification existing in long noncoding RNA influences its binding affinity with target miRNA, influencing the target mRNA of boundary miRNA (also called competitive endogenous RNA working mechanisms). YTHDC1 zooms lncRNA XIST -mediated gene silence. The m6A modification acts as a controlling element to regulate AS lncRNA functions. The m6A label in lncRNA also change the secondary structure via the m6A switch. The m6A signal existing in circRNA could be recognized as “self” circRNA, distinguishing itself from foreign circRNA and escaping RNA immunity. CircRNA launches cap-independent translation mediated by the YTHDF3/eIF4G2/eIF3A complex in an m6A dependent fashion.
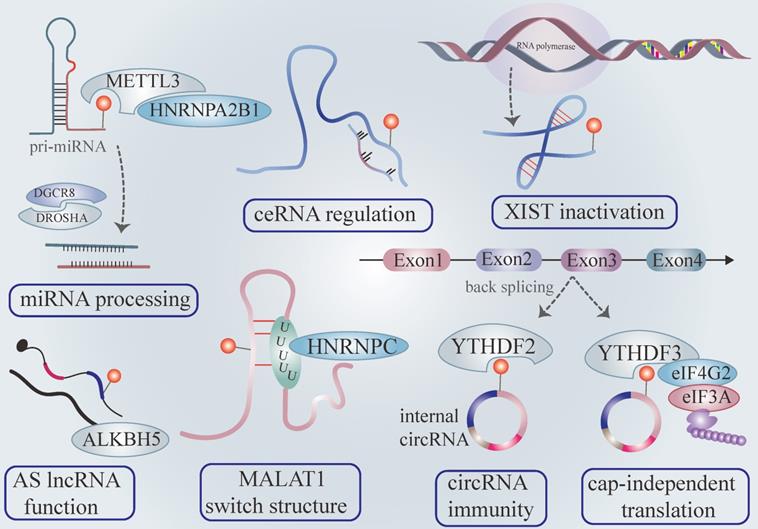
Consistent with the above findings, HNRNPA2B1 selectively recognizes the m6A motif of pri-miRNAs marked by METTL3 and interacts with DGCR8, enhancing pri-miRNA output via microprocessor machinery [25]. The depletion of hNRNPA2B1 or METTL3 attenuates pri-miRNA processing [25] (Figure 2). Recent research has validated that METTL14 plays a manipulative role in pri-miRNA processing [52]. It is widely accepted that miRNA directly binds 3'UTR of mRNA to exert biological function. Interestingly, the m6A peaks are strongly enriched in 3'UTR, implying a region-specific regulatory mechanism. Previous studies have revealed that m6A influences the binding affinity of miRNA with the 3'UTR mRNA [8]. Furthermore, the miRNA or miRNA enzyme Dicer adversely regulates overall m6A level and individual m6A abundance of mRNA via interacting with the mRNA-METTL3 complex in an m6A-dependent pattern [53]. The accumulation of mutation miRNA in mRNA binding sites conceives the novel m6A modification on the previously unmethylated sequence. Indeed, the augmentation of m6A production is indispensable for pluripotent cell programming [53].
2.2 m6A in lncRNA
Long non-coding RNA has been found to modulate gene expression by interacting with proteins, cross-talking with other RNA molecules, or remodeling chromatin despite lacking an open reading framework. The m6A mapping researchers have identified the chemical m6A modification present in the lncRNA sequence and confirmed that m6A manipulates the local structure and function [8, 41, 54]. LncRNA X-inactive specific transcript (XIST) is well-known to mediate gene silence of the X chromosome at the transcriptional level by recruiting slicing proteins. RBM15/15B serves as a scaffold in anchoring the WTAP-METTL3 methylation complex to XIST, which catalyzes m6A modification in both adjacent and distant binding sites, depending on its three-dimension structure [55]. The m6A mark of XIST can recruit YTHDC1, whose binding with XIST is indispensable for X-linked gene silence. Furthermore, METTL3 knockdown decreases m6A abundance and triggers X-linked gene silence [55]. It is reasonable to assume that m6A modification is required for the proper XIST function (Figure 2). However, the molecular mechanism by which YTHDC1 binds to XIST to mediate a series of repressive chromosome programs remains unclear.
LncRNA binds to miRNA by RNA-RNA interaction, which relieves the suppression of miRNA on target mRNA, thereby alternatively enhancing mRNA functions [56]. This mechanism is also the termed competitive endogenous mechanism (ceRNA). The m6A modification could fine-tune the ceRNA working mechanism (Figure 2). METTL3 endows linc1281 with an m6A signal and this m6A modification contributes to the binding of linc1281 with miR-let7, which indirectly increases target mRNA Lin28 abundance [57]. As previously described, METTL3 can enhance the output of mature miRNA. It's important to distinguish the enhancement of binding affinity of linc1281 with miR-let7 is caused by METTL3 or m6A modification. The hairpin-stem structure of metastasized associated lung adenocarcinoma transcript (MALAT1) prefers to be m6A modified in 2577 residues, which destabilizes the opposing U5-tract structure within lncRNA MALAT1 [41, 58]. This enhances the accessibility of the U5-tract with HNRNP C, which intensifies the interactions between MALAT1 and HNRNP C via m6A-switch mechanism [41] (Figure 2). ALKBH5 up-regulates FOXM1 expression by removing its m6A modification and FOXM1-AS facilitates this regulation relationship [59]. GATA3-AS, the lncRNA anti-sense to GATA3, contributes to the interaction between KIAA1429 and GATA3 nascent transcript [60]. Here, we noted that interference with anti-sense lncRNA, transcribed from the antisense strand of the target mRNA, seems to function as a cis-acting element by regulating the preferential interaction between m6A proteins and pre-mRNA (Figure 2). However, how these anti-sense lncRNAs interact with m6A proteins remains elusive. Notably, over 70% of gene transcriptomes have been confirmed to produce anti-sense transcripts [61]. Identifying the existence of other anti-sense lncRNA containing m6A modification will aid in understanding the distinct m6A level in certain genes.
2.3 m6A in circRNA
CircRNA is produced from a series of lariats back splicing events rather than the canonical linear splicing [62]. Recently, overpowering evidence has shown that m6A modification abundance across different types of circRNA is determined by its exon feature [63]. CircRNA consisting of long single exons contains more enriched m6A peaks than circRNA composed of multi-exons [63]. The m6A-circRNA is created by mRNA exon lacking m6A, demonstrating that m6A-circRNA shares similar writers and readers (YTH proteins) with mRNA [16, 63]. However, m6A-circRNA originated from a methylated exon that can recognize YTHDF2 inversely promotes parental mRNA degradation rather than circRNA [63]. It appears that this regulatory mechanism guarantees a stable m6A-circRNA output. Research results have indicated that some circRNAs are intrinsically connected with polysomes, suggesting endogenous circRNA can be translated and carries coding potential [64, 65]. Extensive m6A peaks in circRNA stimulate cap-independent translation by recruiting YTHDF3, eIF4G2, and eIF3A [66] (Figure 2). Interestingly, the authors discovered that this circRNA translation could significantly increase under heat shock conditions, where circRNA translation driven by m6A plays a key role in stress response [66]. The exact function of protein or peptide generated by cap-independent m6A-circRNA translation needs to be further exemplified.
RNA immune system defines the internal and foreign RNA transcripts by checking the 5'm7G cap structure and RNA pattern recognition receptor I (RIG-I) necessary for internal immunity senses 5'-triphosphate in the ends [67]. Large endogenous circRNA can escape this screening due to lacking ends. Indeed, the circRNA immune system can distinguish internal circRNA from foreign ones. Since internal circRNA was generated, it is marked by m6A [68]. YTHDF2 binds to the m6A site to prevent removal of the m6A mark, thereby allowing the circRNA recognized to be as "self" (Figure 2).
In contrast, foreign circRNA is discriminated by RIG I and drives innate immunity owing to the lack of m6A modification [68]. Interestingly, foreign circRNA can adjust itself to escape the RIG I screening by m6A modification, which makes foreign circRNA changed to be "self" [68]. RNA chemical modifications besides m6A likely modulate circRNA immunity and further studies are warranted to elucidate their role.
3. Malfunctions of m6A of mRNA in cancer
It is widely accepted that m6A plays a key role in various biological processes such as neurogenesis development, directional differentiation of hemopoietic stem and progenitor cell, spermatogenesis, stress response, and RNA metabolism as previously described [23, 69, 70]. Extensive efforts have been devoted to identifying the complex influence of m6A modification and its underlying regulatory mechanisms in multiple cancers (Figure 3). In addition, the deregulation of m6A proteins impacts multiple steps of tumorigenesis including initiation, metastasis, relapse, drug resistance, etc. [71, 72].
Malfunctions of m6A regulators in various human cancers. Red regulators suggest an oncogenic character. In contrast, blue regulators suggest a tumor-suppressive character and the roles of green regulators are hard to define (controversial portrayal reported in the specific cancer type).
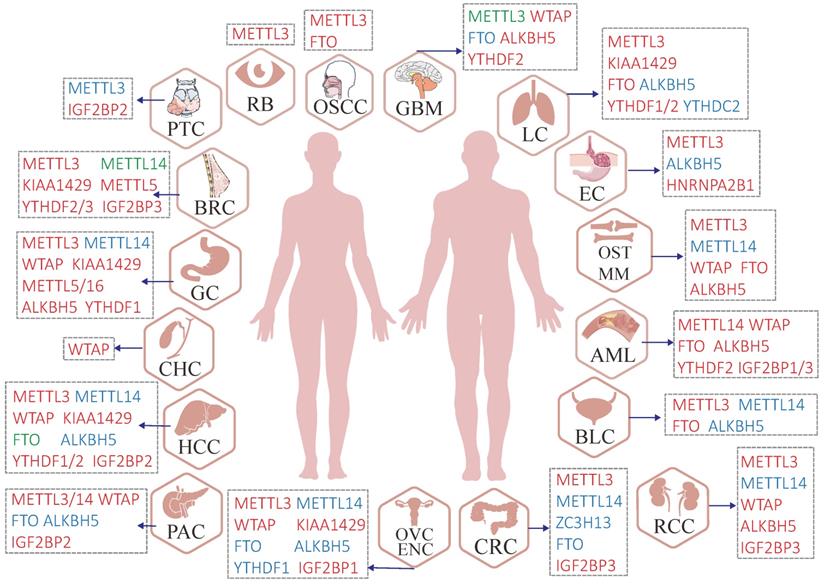
3.1 Malfunction of m6A writers in cancer
Oncogene
METTL3 has been reported to serve as an oncogene in most cancers, including HCC, CRC, AML, PAC, PRC, BRC, OVC, NSCLC, ESCC, etc. For instance, METTL3 facilitates NSCLC growth by increasing YAP translation through recruiting YTHDF1/3 and eIF3a. In addition, METTL3 enhances MALAT1 stability in an m6A manner, which promotes YAP expression by removing the suppression of miR-1914-3p [102]. Accumulating evidence has demonstrated the oncogenic effect of METTL14 in AML, PAC, and NSCLC. METTL14 upregulation mediated by oncogenic protein SPI1 catalyzes m6A reaction on target mRNA MYB and MYC, attenuating HSPCs myeloid differentiation and enhancing self-renewal of leukemia stem/initiation cells (LSC/LICs), ultimately accelerating AML development [115].
WTAP has been identified to exhibit an oncogenic role in most tumors (Table 1). WTAP facilitates GC progression by binding with the 3'UTR m6A site of HK2 mRNA, which elongates HK2 transcript half-life to drive cell proliferation and glycolytic capacity [139]. KIAA1429 is an oncogene in NSCLC, where KIAA1429 regulates m6A modification of death-associated protein kinase 3 (DAPK3) by YTHDF2/3-mediated degradation and enhances cell growth [148]. Progressively, it has been reported that 18S RNA m6A methyltransferase METTL5 is highly expressed in BRC tissues and cells and promotes BRC development [151]. Nevertheless, the underlying mechanisms remain an enigma. Emerging studies have documented that METTL16 is up-regulated in GC tissues and cells and related with a poor prognosis [153].
Roles of m6A writers in human cancer
| Cancer | Role | Expression | Target genes | Source of experimental evidence | Ref | |
|---|---|---|---|---|---|---|
| METTL3 | BLC | Oncogene | Up | PTEN, AFF4/NF-KB/MYC, SETD7/ KLF4, ITGA6 | BLC tissues and cells, mouse | [9, 73-75] |
| BRC | Oncogene | Up by HBXIP | miR-let-7, P21, miR-221-3p, KRT7 | BRC tissues and cells | [76-79] | |
| CC | Oncogene | Up | HK2/YTHDF1, RAB2B, RAGE | CC tissues and cells, mouse | [10, 80, 81] | |
| CRC | Oncogene | Up | GLUT1/mTORC, miR-1246/SPRED2 /MAPK, IGFBP2, YPEL5, CCNE1 | CRC tissues and cells, mouse | [82-86] | |
| ENC | Oncogene | Up | Akt | ENC tissues and cells, mouse | [87] | |
| ESCC | Oncogene | Up | Notch, APC | ESCC tissues and cells, mouse | [88, 89] | |
| GBM | Tumor suppressor/Oncogene | Down/Up | SRSF, ADAR1 | GBM tissues and cells, mouse | [90-92] | |
| GC | Oncogene | Up by H3K27ac | HDGF/IGFBP2/GLUT4/ENO2, ZMYM1/Hur/CtBP/LSD1, MYC, miR-17-92/PTEN/TMEM127 | GC tissues and cells, mouse | [93-96] | |
| HCC | Oncogene | Up | FOXO3, SOCS2, Snail, CRNNB1 | HCC tissues and cells, mouse | [97-99] | |
| LC | Oncogene | Up | JUNB, miR-143-3p/VASH1 | LC cells | [100, 101] | |
| NSCLC | Oncogene | Up | MALAT1/miR-1914-3p/YAP, ABHD11-AS1, ATG5/7 | NSCLC tissues and cells, mouse | [102-104] | |
| OSCC | Oncogene | Up | c-MYC/YTHDF1, BMI1/IGF2BP1 | OSCC tissues and cells, mouse | [105, 106] | |
| OST | Oncogene | Up | ATAD2 | OST cell | [107] | |
| OVC | Oncogene | Up | miR-126-5p/PTEN/PI3K/Akt/mTor, miR-1246/CCNG2 | OVC tissues and cells, mouse | [108, 109] | |
| PRC | Oncogene | Up | Akt, ITGB1, USP4 | PRC cells, mouse | [110, 111] | |
| PTC | Tumor suppressor | Down | c-Rel/IL-8 | PTC tissues and cells, mouse | [112] | |
| RB | Oncogene | Up | PI3K/Akt/mTOR | RB tissues and cells, mouse | [113] | |
| RCC | Oncogene | Up | ABCD1 | RCC tissues and cells, mouse | [114] | |
| METTL14 | AML | Oncogene | Up by SPI 1 | MYB/MYC | AML cells, mouse | [115] |
| BLC | Tumor suppressor | Down | Notch1 | BLC tissues and cells, | [116] | |
| BRC | Oncogene/Tumor suppressor | Up/down | CXCR4/CTP1B1, | BRC tissues and cells, mouse | [11, 117, 118] | |
| CC | Tumor suppressor | Down | Akt | CC cells, mouse | [87] | |
| CRC | Tumor suppressor | Down | miR-375, lncRNA XIST, SOX4 | CRC tissues and cells, mouse | [119-121] | |
| GC | Tumor suppressor | Down | PI3K/AKT/mTOR | GC tissues and cells, mouse | [122, 123] | |
| HCC | Tumor suppressor | Down | DGCR8/miR-126, EGFR/PI3K/AKT | HCC tissues and cells, mouse | [52, 124, 125] | |
| NSCLC | Oncogene | Up | Twist | NSCLC tissues and cells | [126] | |
| OST | Tumor suppressor | Down | cas3 | OST tissues and cells | [127] | |
| PAC | Oncogene | Up | PERP, mTOR | PAC tissues and cells, mouse | [128, 129] | |
| RCC | Tumor suppressor | Down | BPTF, lncRNA NEAT1 | RCC tissues and cells, mouse | [130, 131] | |
| WTAP | AML | Oncogene | Up | Hsp90 | AML cells and mouse | [132, 133] |
| CHC | Oncogene | Up | MMP7/MMP28 | CHC tissues and cells, mouse | [134] | |
| ENC | Oncogene | Up | CAV-1/NF-κB | ENC tissues and cells | [135] | |
| GBM | Oncogene | Up | EGFR | GBM tissues and cells, mouse | [136, 137] | |
| GC | Oncogene | Up | HK2 | GC tissues and cells, mouse | [138, 139] | |
| HCC | Oncogene | Up | ETS1/p21/27 | HCC tissues and cells, mouse | [140, 141] | |
| OST | Oncogene | Up | HMBOX1/ PI3K/AKT | OST tissues and cells, mouse | [142] | |
| PAC | Oncogene | Up | Fak | PAC tissues and cells, mouse | [143] | |
| RCC | Oncogene | Up | CDK2 | RCC tissues and cells, mouse | [144] | |
| KIAA1429 | BRC | Oncogene | Up | CDK1 | BRC tissues and cells,mouse | [145] |
| GC | Oncogene | Up | c-Jun | GC tissues and cells, mouse | [146] | |
| GCT | Oncogene | Up | DNA repair | GCT tissues and cells, mouse | [147] | |
| HCC | Oncogene | Up | GATA3/HuR | HCC tissues and cells, mouse | [60] | |
| NSCLC | Oncogene | Up | DAPK3 | NSCLC tissues and cells,mouse | [148] | |
| PRC | Oncogene | Up | Lnc CCAT1 and CCAT2 | PRC tissues and cells | [149] | |
| ZC3H13 | CRC | Tumor suppressor | Down | Ras-ERK | CRC tissues and cells | [150] |
| METTL5 | BRC | Oncogene | Up | BRC tissues and cells | [151] | |
| GC | Tumor suppressor | Down | GC tissues | [152] | ||
| METTL16 | GC | Oncogene | Up | CDK1 | GC tissues and celle,mouse | [153] |
Tumor suppressor
As shown in Table 1, METTL14 and ZC3H13 have been reported to function as tumor suppressors in most cancer. Of note, METTL3 remodels neutrophil infiltration in the tumor microenvironment by regulating the m6A/c-Rel/IL-8 network, thereby inhibiting PTC progression [112]. METTL14 is rarely expressed in BLC tissues and tumor-initiating cells (TIC) and knocking out METTL14 promotes cell survival, self-renewal, migration, and invasion by regulating Notch1 mRNA stability, which plays an important role in TIC-driven BLC tumorigenesis [116]. METTL14 has been verified to be weakly enriched in GC tissues and a low METTL14 level predicts a poor prognosis [122]. METTL14 slumps CRC progression by triggering m6A modification on oncogenic lncRNA XIST, and the m6A label in XIST is subsequently recognized by YTHDF2, which finally mediates XIST degradation [120]. Likewise, ZC3H13 has been identified to suppress CRC tumorigenesis by regulating the Ras-Erk pathway [150]. However, whether this tumor-inhibiting effect of ZC3H13 is dependent on its m6A modification or not remains unclear. Another study has revealed that METTL5 is downregulated in GC tissues and inhibits cancer development [152].
The dual effects of m6A writers on cancer
Notably, the exact role of METTL3 in GBM is intricate and difficult to define. In GBM, overexpressing METTL3 reduces CD44 expression and sphere-formation rate of glioblastoma stem cells (GSCs), indicating that it suppresses GSC growth and self-renewal, whose presence confers cell resistance to radiotherapy and chemotherapy [90]. On the contrary, another study has reported that knocking down METTL3 promoted the SRSF-mediated nonsense-mediated mRNA decay effect, adversely altering BCL-X and NCOR2 splicing patterns, thereby leading to GSC apoptosis and differentiation [91]. These discrepancies and controversies regarding METTL3 in GBM may depend on variable m6A reader protein and research contexts such as the origin of tumor sample, compensatory genetic mutation, and epigenetic influence of GSC cells in distinctive growth backgrounds. Similarly, the role of METTL14 in BRC is complex and hard to determine. METTL14 exhibits both oncogenic and tumor-suppressor effects in BRC according to different study data [11, 117, 118]. Systematic and comprehensive BRC research is needed to clarify the actual influence of METTL14 in BRC.
There are currently a very limited number of investigations attempting to identify the influence of MTC adaptor proteins excluding WTAP and m6A methyltransferase in human cancers. A cross-sectional and comprehensive study is required to decipher the magic code of m6A modification in caner.
3.2 Malfunction of m6A erasers in cancer
Oncogene
FTO has been confirmed as an oncogene in cancers including AML, MM, OSCC, and NSCLC (Table 2). For example, FTO reduces the m6A level of E2F1 and increases E2F1 expression in NSCLC, which facilitates cell proliferation and metastasis by activating neural epidermal growth factor-like 2 (NELL2) transcription, and thus promoting tumorigenesis [162]. ALKBH5 has been reported as an oncogene in AML, GC, OST, ENC, etc. (Table 2). Homeobox protein Hox-A10 (HOXA10) interacts with the TAAA region of ALKBH5 promoter as TF and expedites ALKBH5 expression, which adversely drives HOXA10 expression by mediating its stability [177]. This ALKBH5-HOXA10 regulating loop promotes JAK2 demethylation, insulating the binding affinity of the m6A site in the JAK2 transcript with YTHDF2, which enhances JAK2 stability and upregulates OVC cell resistance to cisplatin through activating STAT3 phosphorylation reaction [177].
Roles of m6A erasers in human cancer
| Cancer | Role | Expression | Target genes | Source of experimental evidence | Ref | |
|---|---|---|---|---|---|---|
| FTO | AML | Oncogene | Up | ASB2/RARA | AML cells and mouse | [154] |
| BLC | Oncogene | Up | MALAT/miR-384/MAL2 | BLC tissues and cells, mouse | [155] | |
| CRC | Tumor suppressor | Down | MTA1 | CRC tissues and cells, mouse | [156, 157] | |
| GBM | Tumor suppressor | Down | FOXO3a | GBM tissues and cells, mouse | [158] | |
| HCC | Tumor suppressor/ Oncogene | Down/Up | PKM2 | HCC tissues and cells.mouse | [159, 160] | |
| MM | Oncogene | Up | PD-1, CXCR4, SOX10 | MM tissues and cells, mouse | [161] | |
| NSCLC | Oncogene | Up | E2F1/NELL2 | NSCLC tissues and cells, mouse | [162] | |
| OSCC | Oncogene | Up | eIF4G1 | OSCC tissues and cells, mouse | [163] | |
| OVC | Tumor suppressor | Down | cAMP | OVC tissues and cells, mouse | ||
| PAC | Tumor suppressor | Down | PJA2, Wnt | PAC tissues and cells, | [164] | |
| ALKBH5 | AML | Oncogene | Up | TACC3 | AML cells | [165] |
| BLC | Tumor suppressor | Down | CK2α | BLC tissues and cells, mouse | [166] | |
| EC | Tumor suppressor | Down | pri-miR-194-2/ RAI1 | EC tissues and cells, mouse | [167] | |
| ENC | Oncogene | Up | IGF1R | ENC tissues and cells | [168] | |
| GBM | Oncogene | Up | HR | GBM cells | [169] | |
| GC | Oncogene | Up | NEAT1 | GC tissues and cells, | [170] | |
| HCC | Tumor suppressor | Down | LYPD1 | HCC tissues and cells, mouse | [171] | |
| NSCLC | Tumor suppressor | Down | miR-107/LATS2/YAP | NSCLC tissues and cells, mouse | [172] | |
| OST | Oncogene | Up | PVT1, pre-miR-181b-1/YAP | OST tissues and cells, mouse | [173, 174] | |
| OVC | Oncogene | Up | miR-7/BCL-2, NF-κB, HOXA10 / JAK2 | OVC tissues and cells, mouse | [175-177] | |
| PAC | Tumor suppressor | Down | PER1, WIF-1/ Wnt signaling, KCNK15-AS1 | PAC tissues and cells, mouse | [178-180] | |
| RCC | Oncogene | Up | AURKB | RCC tissues and cells, mouse | [181] |
Tumor suppressor
To date, FTO has been confirmed to inhibit PAC, CRC, OVC, and GBM progression (Table 2). It prevents PAC initialization by restricting Wnt signal-mediated cell proliferation and metastasis through downregulating Praja ring finger ubiquitin ligase 2 (PJA2) mRNA decay [164]. In CRC, FTO level is negatively associated with recurrence and prognosis [156]. ALKBH5 acts as a tumor suppressor in PAC, BLC, EC, NSCLC, and HCC (Table 2). In PAC, ALKBH5 stymies tumor outgrowth and metastasis by stimulating PER1 transcription in an m6A-YTHDF2-mediated mRNA degradation style, which causes ATM phosphorylation and G2/M arrest by ATM/CHK2/p53/CDC25 signal axis [178]. The p53 signal adversely intensifies ALKBH5 transcription, forming a feedback loop consisting of PER1-hampered p53 expression and p53-magnified ALKBH5 expression in regulating PAC cells malignancies [178].
The dual effect of m6A writers on cancer
The role of FTO in HCC is hard to define. One research team used long-term diethyl nitrosamine (DEN) to induce HCC in the hepatic FTO-deficient mice [159]. Subsequently, they found that depletion of FTO amplifies HCC burden and that FTO exerts a protective function in HCC initiation by regulating m6A modification of CUL4A [159]. In contrast, another study revealed that FTO is upregulated in HCC tissues and cells while silencing FTO prohibits cell growth and induces G0/G1 cycle arrest by remodeling PKM2 translation in HCC [160].
3.3 Malfunction of m6A readers in cancer
Oncogene
As presented in Table 3, most reader proteins seem to promote cancer progression. For instance, YTHDF1 is associated with HCC maintenance, where HIF-1α swells YTHDF1 transcription by mediating HIF-1α-binding site 1 (HBS1) and HBS3 site of HIF-1α interacting with the YTHDF1 promoter region under hypoxic conditions, which zooms autophagy-related tumor growth and metastasis [183]. Compared with normal neural stem cell (NSC), GSC exhibits preferential YTHDF2 expression and dependency while YTHDF2 strikingly increases the stability of oncogene MYC and VEGFA transcripts by regulating m6A modification. This activates the IGFBP3 responding element to ameliorate GSC growth [189]. Notably, in GSC, YTHDF2 maintains the target mRNA MYC and VEGFA stability, which is inconsistent with the findings of this study: the role of YTHDF2 in mRNA destabilization. There might be additional unknown regulatory factors working differently for various types of cells. We can inter that at least in certain cancer stem cells, YTHDF2 might indirectly stabilize some transcripts by destabilizing mRNAs encoding suppressors of other mRNAs. Research has also revealed that novel independent-cap translation mediated by m6A binding with 5'UTR of YTHDF3 lead to the strong enrichment of YTHDF3 in brain metastases in BRC [192]. YTHDC2 is consistently deposited in radioresistant NPC cells and physically interacts with insulin-like growth factor 1 receptor mRNA (IGF1R), which subsequently activates downstream IGF1R-AKT/S6 signaling response elements urged by translation initiation of IGF1R in an m6A dependent tone [194].
Roles of m6A readers in human cancer
| Cancer | Role | Expression | Target genes | Source of experimental evidence | Ref | |
|---|---|---|---|---|---|---|
| YTHDF1 | GC | Oncogene | Up | FZD7 | GC tissues and cells and mouse | [182] |
| HCC | Oncogene | Up | ATG2A, ATG14, FZD5 | HCC tissues and cells and mouse | [183, 184] | |
| NSCLC | Oncogene | Up | CDK2, CDK4 | NSCLC tissues and cells and mouse | [185] | |
| OVC | Oncogene | Up | EIF3C | OVC tissues and cells and mouse | [186] | |
| YTHDF2 | AML | Oncogene | Up | Tnfrsf2 | AML cells, mouse | [187] |
| BRC | Oncogene | Up | MYC | BRC cells, | [188] | |
| GBM | Oncogene | Up | MYC, LXRα/HIVEP2 | GBM tissues and cells, mouse | [189, 190] | |
| HCC | Oncogene | Up | OCT4 | HCC tissues and cells, mouse | [12] | |
| LUAD | Oncogene | Up | AXIN1/Wnt/β-catenin | LUAD tissues and cells, mouse | [191] | |
| PRC | Oncogene | Up | Akt | PRC tissues and cells, mouse | [110] | |
| YTHDF3 | BRC | Oncogene | Up | ST6GALNAC5/GJA1/EGFR | BRC tissues and cells, mouse | [192] |
| YTHDC2 | LC | Tumor suppressor | Down | SLC7A11 | LC tissues and cells, mouse | [193] |
| NPC | Oncogene | Up | IGF1R/AKT/S6 | NPC cells | [194] | |
| HNRNPA2B1 | EC | Oncogene | Up | ACLY, ACC1 | EC tissues and cells | [195] |
| IGF2BP1 | ENC | Oncogene | Up | PEG10 | ENC tissues and cells, mouse | [196] |
| AML | Oncogene | Up | HOXB4, MYB, ALDH1A1 | AML cells, mouse | [197] | |
| OVC | Oncogene | Up | SRC/MAPK | OVC tissues and cells, mouse | [198] | |
| IGF2BP2 | HCC | Oncogene | Up | FEN1 | HCC tissues and cells, mouse | [199] |
| PAC | Oncogene | Up | PI3K/Akt | HCC tissues and cells, mouse | [200] | |
| PTC | Oncogene | Up | ERBB2 | PTC cells, | [201] | |
| IGF2BP3 | AML | Oncogene | Up | MYC, CDK6 | AML cells, mouse | [202] |
| BRC | Oncogene | Up | miR-3614/ TRIM25 | BRC tissues and cells, mouse | [203] | |
| CRC | Oncogene | Up | CCND1, VEGF | CRC tissues and cells, mouse | [204] | |
| RCC | Oncogene | Up | DMDRMR/CDK4 | CRC tissues and cells, mouse | [205] | |
HNRNPA2B1 depletion significantly prohibits cell proliferation by repressing cellular lipid increase mediated by fatty acid synthetic enzymes ACLY and ACC1 [195]. IGF2BP1 is highly enriched in high-grade serous ovarian cancer. It advances SRC activation through a protein-ligand- independent RNA form, which can expedite EMT by elevating ERK2 expression based on RNA-binding in OVC cells [198]. In HCC, IGF2BP2 directly perceives the m6A pocket embedded in FEN1, which prolongs FEN1 mRNA stability and facilitates HCC progression [199]. IGF2BP3 enhances DNA methylation-deregulated and RNA m6A reader-cooperating lncRNA (DMDRMR) stability, which reinforces the G1-S transition in the cell cycle [205]. In addition, the interaction between DMDRMR and IGF2BP3 increases the stability of target genes such as extracellular matrix genes (COL6A1, LAMA5, and FN1) and CDK4 by m6A modification, thus promoting CRC development [205].
Tumor suppressor
YTHDC2 is marginally expressed in LUAD and associated with poor clinically prognosis [193]. It executes a tumor-suppressive function in LUAD via enlarging solute carrier 7A11 (SLC7A11) degeneration. YTHDC2 blocks cystine uptake and perturbs the following antioxidant project, which curbs the sensitivity of the PDX mouse models to antioxidant administration [193].
4. m6A in the tumor microenvironment
Recently, the tumor environment (TME) characterized by hypoxia, aberrant metabolism, immune escape, and chronic inflammation has been reported to affect anti-tumor response. Accumulating evidence has demonstrated that metabolic dysfunction promotes the shift of immune cells from an anti-tumor state to a tumor-active state, facilitating cancer cells escaping from the monitoring system in TME. The intrinsic m6A modification has been confirmed to remodel TME by regulating the integral crosstalk and communications between various TME cellular components such as T cell, natural killer cell (NK), dendritic cell (DC), tumor-associated with macrophages (TAM), tumor-associated with fibroblast (TAF), and so on, affecting the anti-tumor immune response. This indicates m6A can be a novel immunotherapeutic target [206].
Tumor-infiltrating T cell plays a pivotal role in the anti-tumor response. CD8+T cell subpopulations within the TME could be sorted into five discrete subpopulations: effector-like T cell (Teff), progenitor T cell, exhausted T cell (Tex), cycling exhausted T cell, and transitory T cell without enough activation according to their distinct signature gene profiles [207]. C1q+ TAM subpopulation-specific METTL14 deficiency promotes tumor-infiltrating CD8+T cell dysfunction by reducing cytokine subunit Ebi3 mRNA stability, promoting CD8+T cell exhaustion by increasing Tex cell and increasing Teff cell count, dampening CD8+T cell to eliminate tumors [207] (Figure 4A). We can conclude that m6A plays a key role in deciding T cell fate within the TME.
The interplay between m6A modification and the tumor microenvironment (TME). Several remarkable examples were reviewed below: (A) C1q+ TAM subpopulation-specific METTL14 deficiency induced tumor-infiltrating CD8+ T cell dysfunction by decreasing cytokine subunit Ebi3 mRNA stability, facilitating CD8+T cell exhaustion by increasing Tex cell and increasing Teff cell, and impeding CD8+T cells to eliminate tumors. (B) TGF-β inhibits METTL3 expression in tumor-infiltrating NK cells. By targeting SHP-2, METTL3 elevates the response of NK cells to IL-15 via the AKT-mTOR and MAPK-ERK signaling pathways, thus promoting the immunosurveillance of NK cells. (C) METTL3-deficiency in DC modulates co-stimulatory molecules (CD40 and CD80) and the translational efficiency of TLR signaling adaptor Tirap, which blocks T cell activation and responses by inhibiting cytokine IL-12 production mediated by TLR4/NF-κB signaling. (D) Hypoxia-induced GBM cell-specific ALKBH5 dramatically amplifies IL-8 production by eliminating m6A modification in lncRNA NEAT1, which triggers transcriptional suppressing factor SPFQ relocation from IL-8 promoter sequence to paraspeckles assembly mediated by NEAT1 stability.
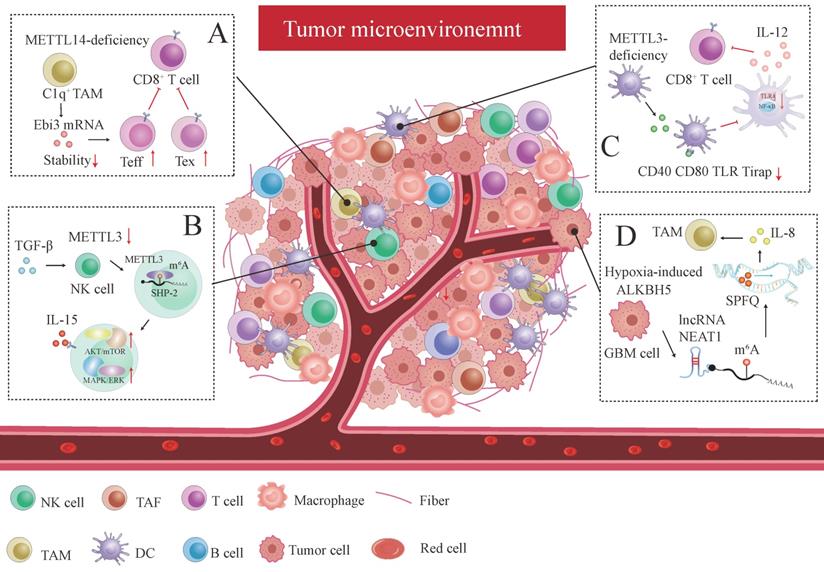
METTL3 is downregulated in tumor-infiltrating NK cells within the TME caused by TGF-β, which activates m6A-mediated fragile effector function and limits the terminal differentiation of peripheral NK cells [208]. METTL3 surges NK cells homeostasis and immunosurveillance by targeting SHP-2, which increases NK cell response to IL-15 via the AKT-mTOR and MAPK-ERK signaling pathways [208] (Figure 4B). Knocking out ALKBH5 in mouse model treated with GVAX/anti-PD-1 immunotherapy prohibited the nuclear export of metabolite content lactate, which latterly reduced the amounts of Treg cell and myeloid-derived suppressor cell (MDSC) and promotes DC infiltration by regulating mRNA MCT4/Slc16a3 expression in an m6A dependent fashion [209]. YTHDF1 depletion enhances antitumor response and immunosurveillance by increasing the number of CD8+ cytotoxic T cells and infiltrating NK cells [210]. DC-specific YTHDF1 enhances the neoantigen-specific immunity of CD8+T cells and inhibits the cross-presentation of engulfed tumor neoantigens by perceiving m6A pocket embedded in lysosomal proteases transcript, and enlarging translational output of lysosomal cathepsins [210].
Similarly, another research has uncovered that specific METTL3 absence in DC inhibits functional maturation and activation of DC by regulating co-stimulatory molecules CD40, CD80 and the translational efficiency of the TLR signaling adaptor Tirap, which curtails T cell activation and responses by suppressing cytokine IL-12 production mediated by TLR4/NF-κB signaling [211] (Figure 4C). Both studies have demonstrated that immune activation and tolerance induced by aberrant T cell response are closely associated with DC dysregulation. Adoptive infusion of DC vaccines could be particularly effective in cancer immunotherapy.
m6A affects immune checkpoint blockade (ICB) therapy and the efficacy of immunotherapy. Research has also revealed that the ALKBH5 expression pattern is consistent with the hypoxic phenotype signature-associated gene pattern in GBM cells [212]. GBM cell-specific ALKBH5 induced by hypoxia stimulus significantly amplifies IL-8 production by clearing m6A modification in lncRNA NEAT1, provoking transcriptional suppressing factor SPFQ relocation from the IL-8 promoter sequence to paraspeckles assembly mediated by NEAT1 stability [212] (Figure 4D). The indirect IL-8-promoting effect mediated by hypoxic ALKBH5 expedites TAM recruitment within the TME and stymies the immunosuppressive microenvironment in anti-tumor response [212].
5. m6A regulator as a therapeutic target in cancer
Developed small molecules targeting m6A writer and erasers, especially erasers in therapeutic applications have been designed and put into clinical practice considering the predominant role of m6A in cancer initiation and progression (Figure 5). Research groups have developed a bioavailable inhibitor of METTL3, namely STM2457, with highly potent and selective first-in-grade catalytic activity [213]. Administration of STM2457 in AML re-transplantation experiments fazes tumor growth and facilitates survival by suppressing crucial stem cell flux and reversing AML phenotypes by inducing m6A-associated cellular effects including suppressing known leukemogenic mRNAs expression [213]. According to a recent investigation, the small-molecule inhibitors FB23 and FB23-2 targeting FTO significantly suppress the proliferation of AML cell flux and primary LSCs in Xeon-transplanted mice by directly reducing m6A demethylase activity of FTO in target mRNA including MYC, CEBPA, RARA and ASB2 [214]. Two newly identified effective FTO inhibitors (CS1 and CS2) significantly repress the growth rate of LSC/LIC cells in vivo and in vitro by conquering the demethylation bag of FTO and hampering the handcuff of FTO in m6A-modified RNAs such as MYC [215]. Notably, AML patients with high FTO expression phenotypes are highly responsive and sensitivite to C1 and C2 anti-leukemic treatments.
Therapeutic implications of m6A regulators in cancer. Given the attractive prospect of targeting FTO and METTL3 in cancer treatment, numerous inhibitors of m6A modifiers have been developed.
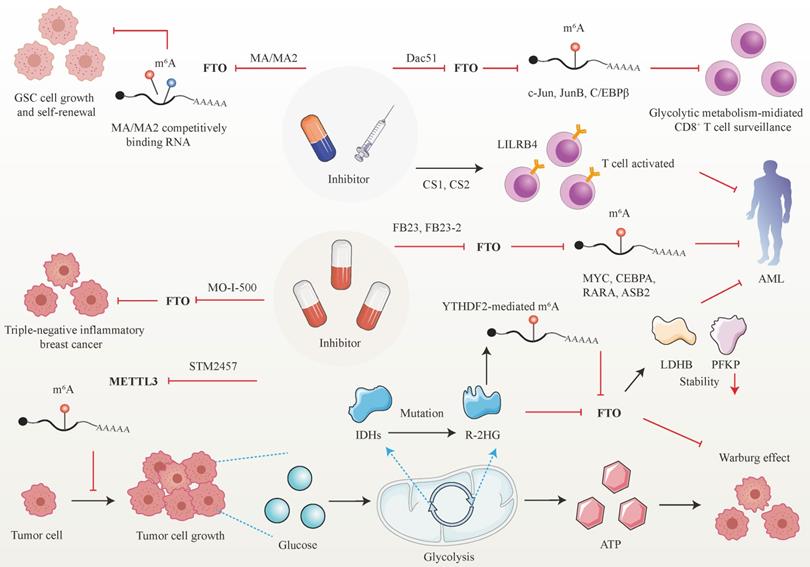
Interestingly, applying small-molecule compounds CS1/CS2 in AML cells catalyzes the cytotoxicity of the activated T cells to AML cells by regulating LILRB4, which serves as key intrinsic immune checkpoint genes to mediate immune evasion [215]. Interestingly, C1 and C2 exhibit anti-tumor activity that is at least tenfold more potent than that of FB23 and FB23-2 in AML cells with IC50 values below nanomolar levels [215]. Another small-molecule FTO inhibitor, Dac51, has also been confirmed to serve as an essential reprogramming RNA epitranscriptome factor to prevent tumor cells from evading CD8+T cells surveillance mediated by glycolytic metabolism, which is induced by transcription factors c-Jun, JunB, and C/EBPβ mediated by FTO in an m6A dependent tone [216].
Besides the above small-molecule inhibitors targeting m6A regulators, others targeting FTO have exhibited anti-tumor response in cancer therapy. For instance, R-2-hydroxyglutarate (R-2HG), a mutation of critical enzymes Isocitrate dehydrogenases (IDHs) in the aerobic glycolysis of cancer cells, has been reported to effectively blunt cancer metabolism by silencing the FTO-induced flux of transcription response associated with the Warburg effect [217]. In R-2HG-sensitive leukemia cells, R-2HG suppresses FTO activity and decreases the stability of target mRNA phosphofructokinase platelet (PFKP) and lactate dehydrogenase B (LDHB) by YTHDF2. This minimizes aerobic glycolysis compared with healthy CD34+ hematopoietic progenitor cells, finally hampering AML metabolism and tumor growth [217]. Consistent with the above findings, R-2HG also executes an anti-leukemic function by governing the FTO/m6A/MYC/CEBPA signal network. Moreover, R-2HG inhibits GBM tumor outgrowth and ameliorates tumor survival in vitro and in vivo by hindering FTO activity [218]. The Saikosaponin D inhibitor targeting FTO has been indicated to decrease AML cell proliferation and tumor outgrowth, as well as resistance to tyrosine kinase inhibitors in vitro and in vivo by enhancing the overall m6A level of the downstream target genes [219]. Pharmacological FTO inhibitor MO-I-500 suppresses cell proliferation and survival in triple-negative inflammatory breast cancer [220, 221]. Meclofenamic acid (MA/MA2), a highly selective inhibitor of FTO, has been proved to faze GSC growth and self-renewal in vitro by competitively binding m6A-containing mRNA [90, 222].
Conclusion and future perspectives
The latest proceedings in m6A epitranscriptome and technological advances in m6A associated methodology have conclusively validated that the dynamic and reversible m6A regulation network is indispensable for RNA metabolism including alternative splicing, nuclear export, stability, and translation. m6A emerges as a novel potential mechanisms of tumor-associated gene regulation. Remarkably, the dysregulation of m6A regulators in human cancer has been extensively documented. According to current reports, m6A writers, erasers, and readers are aberrantly expressed in most human cancer tissues and cells. These m6A regulatory factors manipulate malignancies such as unrestricted proliferation, active tissue metastasis and invasion, dysfunctional energy metabolism, skilled immune escape, and emerging resistance to chemotherapy and radiotherapy by governing oncogene or tumor-suppressor expression pattern in m6A dependent manner. However, how m6A writers and erasers distinctively perceive the onco-transcripts or tumor-suppressor transcripts and belatedly reshape most tumor-associated gene expression patterns remains an enigma. Fortunately, emerging studies have attempted to establish an explanation: the distinct cellular distribution of oncogenic or tumor-associated transcript and uneven dynamic m6A deposition, the cellular location of m6A readers and their variable amplification, cellular context and genetic background, and other additional unknown factors, these cross-talking elements orchestrate with each other to produce the m6A balance and exert diverse function. Most m6A proteins appear to facilitate tumor progression irrespective of the type of cancer. Intrinsically, extensive efforts have been made to elucidate the m6A-mRNA interaction. However, little is known about m6A modified ncRNA. Determining the interplay between m6A and ncRNA metabolism including ncRNA production, maturation, cellular location, and the cross-talks between m6A and ncRNA function in cancer cells will offer a profound understanding of the far-reaching influence of m6A and undervalued ncRNA. Fortunately, current report has highlighted the presence of m6A modification in promoter-related RNA, enhancer RNA, and other chromosome-associated RNA [31].
The tissue-specific m6A profiles of certain transcript and transcript loci could be promising biomarkers for the diagnosis and prognosis of cancer. More importantly, considering the critical and complex functions of m6A in cancer such as stemness, immune escape, energy metabolism, therapy resistance, and so on, endowing therapeutic target with m6A regulator goes one step further in clinical application. Current studies regarding m6A in cancer therapy seems extensively focused on AML and relevant studies performed on the other solid tumors are limited and remain largely unexploited. Moreover, the small molecules targeting m6A regulators including MO-I-500, MA, FB23-2, R-2HG, and CS1/CS2 are restricted to FTO. Therefore, potent and specific small molecule targeting m6A writers such as STM2457 based on small molecule inhibitor compound library screening should be of utmost relevance, considering that m6A writers mediate large m6A modified mRNAs in the whole genome. Furthermore, discoveries in cancer therapy targeting m6A readers controlling mRNA fate should not be ignored in the m6A research wave. Primary cell-derived from the target tissues, organoids, and animal models should be developed to test novel inhibitors' anti-tumor response, cellular toxicity, and pharmacokinetics. In the end, it is foreseeable that clinical trials of perfect small-molecule inhibitors targeting m6A regulators will be initiated and the pharmacological efficiency of mono/combination therapy in cancer patients with dysregulation of m6A profiles will be explored.
Abbreviations
ncRNA: noncoding RNA; UTR: untranslated region; METTL3: methyltransferase-like3; METTL14: methyltransferase-like 14; MTC: methyltransferase complex; WTAP: Wilms tumor 1-associated protein; KIAA1429: via-like m6A methyltransferase associated; ZC3H13: zinc finger CCCH domain-containing protein 13; RAB15/15B: RNA-binding motif protein 15; METTL5: methyltransferase like 5; METTL16: methyltransferases like16; ZCCHC4: zinc finger CCHC-type containing 4; FTO: fat mass and obesity-associated protein; ALKBH5: ALKB homolog 5; AS: alternative splicing; mRNP: messenger ribonucleoprotein; HNRNPA2B1: heterogeneous nuclear ribonucleoproteins A2/B1; YTHDC1: YTH domain-containing protein 1; YTHDF1-3: YTH domain family proteins; IGF2BP1: insulin-like growth factor 2 mRNA-binding protein1; RBM: RNA binding motifs; XIST: X-inactive specific transcript; MALAT1: metastasized associated lung adenocarcinoma transcript 1; AML: acute myeloid leukemia; BLC: bladder cancer; BRC: breast cancer; CC: cervical cancer; CHC: cholangiocarcinoma; CRC: colorectal cancer; ENC: endometrial cancer; ESCC: esophageal squamous cell carcinoma; GBM: glioblastoma; GC: gastric cancer; GCT: germ cell tumors; HCC: hepatocellular carcinoma; LC: lung cancer; NSCLC: non-small cell lung cancer; OSCC: oral squamous cell carcinoma; OST: osteosarcoma; OVC: ovarian cancer; PAC: pancreatic cancer; PRC: prostatic cancer; PTC: papillary thyroid carcinoma; RB: retinoblastoma; RCC: renal cell carcinoma; LSC/LICs: leukemia stem/initiation cells; GSCs: glioblastoma stem cells; NPC: nasopharyngeal carcinoma; TME: tumor environment; NK cell: natural killer cell; DC: dendritic cell; TAM: tumor-associated with macrophages; TAF: tumor-associated with fibroblast; R-2HG: R-2-hydroxyglutarate; MA/MA2: meclofenamic acid.
Acknowledgements
We thank Home for Researchers (www.home-for-researchers.com) for their assistance in editing this manuscript.
Funding
The current study is supported by the Natural Science Foundation of Henan Province (222300420564) and Key Medical Science & Technology Program of Henan Province (SBGJ202101008).
Author Contributions
J.H., M.Z. and F.H. organized the whole topic. J.J. and S.W. drafted the manuscript. Z.J., C.W., C.J., J.S. and H.L. revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat Rev Genet. 2014;15:293-306
2. Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5' terminus of HeLa cell messenger RNA. Cell. 1975;4:379-86
3. Rottman F, Shatkin AJ, Perry RP. Sequences containing methylated nucleotides at the 5' termini of messenger RNAs: possible implications for processing. Cell. 1974;3:197-9
4. Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177-89
5. Fustin J-M, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M. et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793-806
6. Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990;18:5735-41
7. Coker H, Wei G, Brockdorff N. m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim Biophys Acta Gene Regul Mech. 2019;1862:310-8
8. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149:1635-46
9. Han J, Wang J-Z, Yang X, Yu H, Zhou R, Lu H-C. et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18:110
10. Wang Q, Guo X, Li L, Gao Z, Su X, Ji M. et al. N-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell death & disease. 2020;11:911
11. Sun T, Wu Z, Wang X, Wang Y, Hu X, Qin W. et al. LNC942 promoting METTL14-mediated mA methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39:5358-72
12. Zhang C, Huang S, Zhuang H, Ruan S, Zhou Z, Huang K. et al. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene. 2020;39:4507-18
13. Xu J, Wan Z, Tang M, Lin Z, Jiang S, Ji L. et al. N-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating β-catenin signaling. Mol Cancer. 2020;19:163
14. Shulman Z, Stern-Ginossar N. The RNA modification N-methyladenosine as a novel regulator of the immune system. Nat Immunol. 2020;21:501-12
15. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187-200
16. Huang H, Weng H, Chen J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer cell. 2020;37:270-88
17. Śledź P, Jinek M. Structural insights into the molecular mechanism of the m(6)A writer complex. Elife. 2016 5
18. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z. et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575-8
19. Ren W, Lu J, Huang M, Gao L, Li D, Wang GG. et al. Structure and regulation of ZCCHC4 in mA-methylation of 28S rRNA. Nat Commun. 2019;10:5042
20. Ruszkowska A, Ruszkowski M, Dauter Z, Brown JA. Structural insights into the RNA methyltransferase domain of METTL16. Sci Rep. 2018;8:5311
21. Oerum S, Meynier V, Catala M, Tisné C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021;49:7239-55
22. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885-7
23. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18-29
24. Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3'-end processing. Nucleic Acids Res. 2017;45:11356-70
25. Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299-308
26. Nayler O, Hartmann AM, Stamm S. The ER repeat protein YT521-B localizes to a novel subnuclear compartment. J Cell Biol. 2000;150:949-62
27. Bradley T, Cook ME, Blanchette M. SR proteins control a complex network of RNA-processing events. RNA. 2015;21:75-92
28. Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F. et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. 2016;61:507-19
29. Roundtree IA, Luo G-Z, Zhang Z, Wang X, Zhou T, Cui Y. et al. YTHDC1 mediates nuclear export of N-methyladenosine methylated mRNAs. Elife. 2017 6
30. Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M. et al. N-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat Genet. 2020;52:870-7
31. Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM. et al. -methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science. 2020;367:580-6
32. Widagdo J, Anggono V, Wong JJL. The multifaceted effects of YTHDC1-mediated nuclear mA recognition. Trends Genet. 2022;38:325-32
33. Lee J-H, Wang R, Xiong F, Krakowiak J, Liao Z, Nguyen PT. et al. Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol Cell. 2021 81
34. Shenton D, Smirnova JB, Selley JN, Carroll K, Hubbard SJ, Pavitt GD. et al. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem. 2006;281:29011-21
35. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
36. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388-99
37. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ. et al. YTHDF3 facilitates translation and decay of N-methyladenosine-modified RNA. Cell Res. 2017;27:315-28
38. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. et al. Recognition of RNA N-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285-95
39. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335-45
40. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J. et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556-60
41. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560-4
42. Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR. et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. 2018;9:420
43. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051-63
44. Yang Y, Hsu PJ, Chen Y-S, Yang Y-G. Dynamic transcriptomic mA decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616-24
45. Du Y, Hou G, Zhang H, Dou J, He J, Guo Y. et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46:5195-208
46. Hou G, Zhao X, Li L, Yang Q, Liu X, Huang C. et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Res. 2021;49:2859-77
47. Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M. et al. Histone H3 trimethylation at lysine 36 guides mA RNA modification co-transcriptionally. Nature. 2019;567:414-9
48. Denli AM, Tops BBJ, Plasterk RHA, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231-5
49. Kwon SC, Nguyen TA, Choi Y-G, Jo MH, Hohng S, Kim VN. et al. Structure of Human DROSHA. Cell. 2016;164:81-90
50. Nguyen TA, Jo MH, Choi Y-G, Park J, Kwon SC, Hohng S. et al. Functional Anatomy of the Human Microprocessor. Cell. 2015;161:1374-87
51. Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482-5
52. Ma J-Z, Yang F, Zhou C-C, Liu F, Yuan J-H, Wang F. et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529-43
53. Chen T, Hao Y-J, Zhang Y, Li M-M, Wang M, Han W. et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289-301
54. Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Science. 2016;352:1408-12
55. Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369-73
56. Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M. et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358-69
57. Yang D, Qiao J, Wang G, Lan Y, Li G, Guo X. et al. N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. Nucleic Acids Res. 2018;46:3906-20
58. Kierzek E, Kierzek R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003;31:4472-80
59. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z. et al. mA Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer cell. 2017 31
60. Lan T, Li H, Zhang D, Xu L, Liu H, Hao X. et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18:186
61. Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M. et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564-6
62. Chen L-L. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205-11
63. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N. et al. Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep. 2017;20:2262-76
64. Tang C, Xie Y, Yu T, Liu N, Wang Z, Woolsey RJ. et al. mA-dependent biogenesis of circular RNAs in male germ cells. Cell Res. 2020;30:211-28
65. Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE. et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365-79
66. Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y. et al. Extensive translation of circular RNAs driven by N-methyladenosine. Cell Res. 2017;27:626-41
67. Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE. et al. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol Cell. 2017 67
68. Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L. et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell. 2019 76
69. Yoon K-J, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D. et al. Temporal Control of Mammalian Cortical Neurogenesis by mA Methylation. Cell. 2017 171
70. Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D. et al. mA modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549:273-6
71. Haruehanroengra P, Zheng YY, Zhou Y, Huang Y, Sheng J. RNA modifications and cancer. RNA biology. 2020;17:1560-75
72. Zhang M, Song J, Yuan W, Zhang W, Sun Z. Roles of RNA Methylation on Tumor Immunity and Clinical Implications. Frontiers in immunology. 2021;12:641507
73. Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M. et al. The mA methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38:3667-80
74. Xie H, Li J, Ying Y, Yan H, Jin K, Ma X. et al. METTL3/YTHDF2 m A axis promotes tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer. J Cell Mol Med. 2020;24:4092-104
75. Jin H, Ying X, Que B, Wang X, Chao Y, Zhang H. et al. N-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195-207
76. Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z. et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer letters. 2018;415:11-9
77. Cheng L, Zhang X, Huang Y-Z, Zhu Y-L, Xu L-Y, Li Z. et al. Metformin exhibits antiproliferation activity in breast cancer via miR-483-3p/METTL3/mA/p21 pathway. Oncogenesis. 2021;10:7
78. Pan X, Hong X, Li S, Meng P, Xiao F. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6A-dependent manner. Exp Mol Med. 2021 53
79. Chen F, Chen Z, Guan T, Zhou Y, Ge L, Zhang H. et al. -Methyladenosine Regulates mRNA Stability and Translation Efficiency of KRT7 to Promote Breast Cancer Lung Metastasis. Cancer research. 2021;81:2847-60
80. Hu Y, Li Y, Huang Y, Jin Z, Wang C, Wang H. et al. METTL3 regulates the malignancy of cervical cancer via post-transcriptional regulation of RAB2B. Eur J Pharmacol. 2020;879:173134
81. Li R, Song Y, Chen X, Chu M, Wang Z-W, Zhu X. METTL3 increases cisplatin chemosensitivity of cervical cancer cells via downregulation of the activity of RAGE. Mol Ther Oncolytics. 2021;22:245-55
82. Chen H, Gao S, Liu W, Wong C-C, Wu J, Wu J. et al. RNA N-Methyladenosine Methyltransferase METTL3 Facilitates Colorectal Cancer by Activating the mA-GLUT1-mTORC1 Axis and Is a Therapeutic Target. Gastroenterology. 2021 160
83. Peng W, Li J, Chen R, Gu Q, Yang P, Qian W. et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38:393
84. Li T, Hu P-S, Zuo Z, Lin J-F, Li X, Wu Q-N. et al. METTL3 facilitates tumor progression via an mA-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112
85. Zhou D, Tang W, Xu Y, Xu Y, Xu B, Fu S. et al. METTL3/YTHDF2 m6A axis accelerates colorectal carcinogenesis through epigenetically suppressing YPEL5. Mol Oncol. 2021;15:2172-84
86. Zhu W, Si Y, Xu J, Lin Y, Wang J-Z, Cao M. et al. Methyltransferase like 3 promotes colorectal cancer proliferation by stabilizing CCNE1 mRNA in an m6A-dependent manner. J Cell Mol Med. 2020;24:3521-33
87. Liu J, Eckert MA, Harada BT, Liu S-M, Lu Z, Yu K. et al. mA mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074-83
88. Han H, Yang C, Zhang S, Cheng M, Guo S, Zhu Y. et al. METTL3-mediated mA mRNA modification promotes esophageal cancer initiation and progression via Notch signaling pathway. Molecular therapy Nucleic acids. 2021;26:333-46
89. Wang W, Shao F, Yang X, Wang J, Zhu R, Yang Y. et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N-methyladenosine-dependent YTHDF binding. Nat Commun. 2021;12:3803
90. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G. et al. mA RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622-34
91. Li F, Yi Y, Miao Y, Long W, Long T, Chen S. et al. N-Methyladenosine Modulates Nonsense-Mediated mRNA Decay in Human Glioblastoma. Cancer research. 2019;79:5785-98
92. Tassinari V, Cesarini V, Tomaselli S, Ianniello Z, Silvestris DA, Ginistrelli LC. et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021;22:51
93. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J. et al. METTL3-mediated mA modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193-205
94. Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z. et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142
95. Yang D-D, Chen Z-H, Yu K, Lu J-H, Wu Q-N, Wang Y. et al. METTL3 Promotes the Progression of Gastric Cancer via Targeting the MYC Pathway. Frontiers in oncology. 2020;10:115
96. Sun Y, Li S, Yu W, Zhao Z, Gao J, Chen C. et al. N-methyladenosine-dependent pri-miR-17-92 maturation suppresses PTEN/TMEM127 and promotes sensitivity to everolimus in gastric cancer. Cell death & disease. 2020;11:836
97. Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X. et al. RNA m A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020;39:e103181
98. Chen M, Wei L, Law C-T, Tsang FH-C, Shen J, Cheng CL-H. et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254-70
99. Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W. et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10:5671-86
100. Wanna-Udom S, Terashima M, Lyu H, Ishimura A, Takino T, Sakari M. et al. The m6A methyltransferase METTL3 contributes to Transforming Growth Factor-beta-induced epithelial-mesenchymal transition of lung cancer cells through the regulation of JUNB. Biochem Biophys Res Commun. 2020;524:150-5
101. Wang H, Deng Q, Lv Z, Ling Y, Hou X, Chen Z. et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol Cancer. 2019;18:181
102. Jin D, Guo J, Wu Y, Du J, Yang L, Wang X. et al. mA mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. Journal of hematology & oncology. 2019;12:135
103. Xue L, Li J, Lin Y, Liu D, Yang Q, Jian J. et al. m A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J Cell Physiol. 2021;236:2649-58
104. Liu S, Li Q, Li G, Zhang Q, Zhuo L, Han X. et al. The mechanism of mA methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell death & disease. 2020;11:969
105. Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y. et al. METTL3 Facilitates Oral Squamous Cell Carcinoma Tumorigenesis by Enhancing c-Myc Stability via YTHDF1-Mediated mA Modification. Molecular therapy Nucleic acids. 2020 20
106. Liu L, Wu Y, Li Q, Liang J, He Q, Zhao L. et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 mA Methylation in Oral Squamous Cell Carcinoma. Mol Ther. 2020;28:2177-90
107. Zhou L, Yang C, Zhang N, Zhang X, Zhao T, Yu J. Silencing METTL3 inhibits the proliferation and invasion of osteosarcoma by regulating ATAD2. Biomed Pharmacother. 2020;125:109964
108. Bi X, Lv X, Liu D, Guo H, Yao G, Wang L. et al. METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther. 2021;28:335-49
109. Bi X, Lv X, Liu D, Guo H, Yao G, Wang L. et al. METTL3 promotes the initiation and metastasis of ovarian cancer by inhibiting CCNG2 expression via promoting the maturation of pri-microRNA-1246. Cell Death Discov. 2021;7:237
110. Li J, Xie H, Ying Y, Chen H, Yan H, He L. et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Mol Cancer. 2020;19:152
111. Chen Y, Pan C, Wang X, Xu D, Ma Y, Hu J. et al. Silencing of METTL3 effectively hinders invasion and metastasis of prostate cancer cells. Theranostics. 2021;11:7640-57
112. He J, Zhou M, Yin J, Wan J, Chu J, Jia J. et al. METTL3 restrains papillary thyroid cancer progression via mA/c-Rel/IL-8-mediated neutrophil infiltration. Mol Ther. 2021;29:1821-37
113. Zhang H, Zhang P, Long C, Ma X, Huang H, Kuang X. et al. mA methyltransferase METTL3 promotes retinoblastoma progression via PI3K/AKT/mTOR pathway. J Cell Mol Med. 2020
114. Shi Y, Dou Y, Zhang J, Qi J, Xin Z, Zhang M. et al. The RNA N6-Methyladenosine Methyltransferase METTL3 Promotes the Progression of Kidney Cancer via N6-Methyladenosine-Dependent Translational Enhancement of ABCD1. Frontiers in cell and developmental biology. 2021;9:737498
115. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L. et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA mA Modification. Cell Stem Cell. 2018 22
116. Gu C, Wang Z, Zhou N, Li G, Kou Y, Luo Y. et al. Mettl14 inhibits bladder TIC self-renewal and bladder tumorigenesis through N-methyladenosine of Notch1. Mol Cancer. 2019;18:168
117. Gong P-J, Shao Y-C, Yang Y, Song W-J, He X, Zeng Y-F. et al. Analysis of N6-Methyladenosine Methyltransferase Reveals METTL14 and ZC3H13 as Tumor Suppressor Genes in Breast Cancer. Frontiers in oncology. 2020;10:578963
118. Dong X-F, Wang Y, Huang B-F, Hu G-N, Shao J-K, Wang Q. et al. Downregulated METTL14 Expression Correlates with Breast Cancer Tumor Grade and Molecular Classification. Biomed Res Int. 2020;2020:8823270
119. Chen X, Xu M, Xu X, Zeng K, Liu X, Sun L. et al. METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol Ther. 2020;28:599-612
120. Yang X, Zhang S, He C, Xue P, Zhang L, He Z. et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19:46
121. Chen X, Xu M, Xu X, Zeng K, Liu X, Pan B. et al. METTL14-mediated N6-methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Mol Cancer. 2020;19:106
122. Shimura T, Kandimalla R, Okugawa Y, Ohi M, Toiyama Y, He C. et al. Novel evidence for mA methylation regulators as prognostic biomarkers and FTO as a potential therapeutic target in gastric cancer. Br J Cancer. 2021
123. Liu X, Xiao M, Zhang L, Li L, Zhu G, Shen E. et al. The m6A methyltransferase METTL14 inhibits the proliferation, migration, and invasion of gastric cancer by regulating the PI3K/AKT/mTOR signaling pathway. J Clin Lab Anal. 2021;35:e23655
124. Zhou T, Li S, Xiang D, Liu J, Sun W, Cui X. et al. m6A RNA methylation-mediated HNF3γ reduction renders hepatocellular carcinoma dedifferentiation and sorafenib resistance. Signal Transduct Target Ther. 2020;5:296
125. Shi Y, Zhuang Y, Zhang J, Chen M, Wu S. METTL14 Inhibits Hepatocellular Carcinoma Metastasis Through Regulating EGFR/PI3K/AKT Signaling Pathway in an m6A-Dependent Manner. Cancer Manag Res. 2020;12:13173-84
126. Yang F, Yuan W-Q, Li J, Luo Y-Q. Knockdown of METTL14 suppresses the malignant progression of non-small cell lung cancer by reducing Twist expression. Oncol Lett. 2021;22:847
127. Liu Z, Liu N, Huang Z, Wang W. Overexpression Promotes Osteosarcoma Cell Apoptosis and Slows Tumor Progression via Activation. Cancer Manag Res. 2020;12:12759-67
128. Wang M, Liu J, Zhao Y, He R, Xu X, Guo X. et al. Upregulation of METTL14 mediates the elevation of PERP mRNA N adenosine methylation promoting the growth and metastasis of pancreatic cancer. Mol Cancer. 2020;19:130
129. Kong F, Liu X, Zhou Y, Hou X, He J, Li Q. et al. Downregulation of METTL14 increases apoptosis and autophagy induced by cisplatin in pancreatic cancer cells. Int J Biochem Cell Biol. 2020;122:105731
130. Zhang C, Chen L, Liu Y, Huang J, Liu A, Xu Y. et al. Downregulated METTL14 accumulates BPTF that reinforces super-enhancers and distal lung metastasis via glycolytic reprogramming in renal cell carcinoma. Theranostics. 2021;11:3676-93
131. Liu T, Wang H, Fu Z, Wang Z, Wang J, Gan X. et al. METTL14 suppresses growth and metastasis of renal cell carcinoma by decreasing long non-coding RNA NEAT1. Cancer Sci. 2021
132. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Proia D. et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28:1171-4
133. Kuai Y, Gong X, Ding L, Li F, Lei L, Gong Y. et al. Wilms' tumor 1-associating protein plays an aggressive role in diffuse large B-cell lymphoma and forms a complex with BCL6 via Hsp90. Cell Commun Signal. 2018;16:50
134. Jo H-J, Shim H-E, Han M-E, Kim H-J, Kim K-S, Baek S. et al. WTAP regulates migration and invasion of cholangiocarcinoma cells. J Gastroenterol. 2013;48:1271-82
135. Li Q, Wang C, Dong W, Su Y, Ma Z. WTAP facilitates progression of endometrial cancer via CAV-1/NF-κB axis. Cell Biol Int. 2021;45:1269-77
136. Xi Z, Xue Y, Zheng J, Liu X, Ma J, Liu Y. WTAP Expression Predicts Poor Prognosis in Malignant Glioma Patients. J Mol Neurosci. 2016;60:131-6
137. Jin D-I, Lee SW, Han M-E, Kim H-J, Seo S-A, Hur G-Y. et al. Expression and roles of Wilms' tumor 1-associating protein in glioblastoma. Cancer Sci. 2012;103:2102-9
138. Li H, Su Q, Li B, Lan L, Wang C, Li W. et al. High expression of WTAP leads to poor prognosis of gastric cancer by influencing tumour-associated T lymphocyte infiltration. J Cell Mol Med. 2020;24:4452-65
139. Yu H, Zhao K, Zeng H, Li Z, Chen K, Zhang Z. et al. N-methyladenosine (mA) methyltransferase WTAP accelerates the Warburg effect of gastric cancer through regulating HK2 stability. Biomed Pharmacother. 2021;133:111075
140. Chen Y, Peng C, Chen J, Chen D, Yang B, He B. et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18:127
141. Zhuo Z-J, Hua R-X, Chen Z, Zhu J, Wang M, Yang Z. et al. Gene Variants Confer Hepatoblastoma Susceptibility: A Seven-Center Case-Control Study. Mol Ther Oncolytics. 2020;18:118-25
142. Chen S, Li Y, Zhi S, Ding Z, Wang W, Peng Y. et al. WTAP promotes osteosarcoma tumorigenesis by repressing HMBOX1 expression in an mA-dependent manner. Cell death & disease. 2020;11:659
143. Li B-Q, Liang Z-Y, Seery S, Liu Q-F, You L, Zhang T-P. et al. WT1 associated protein promotes metastasis and chemo-resistance to gemcitabine by stabilizing Fak mRNA in pancreatic cancer. Cancer letters. 2019;451:48-57
144. Tang J, Wang F, Cheng G, Si S, Sun X, Han J. et al. Wilms' tumor 1-associating protein promotes renal cell carcinoma proliferation by regulating CDK2 mRNA stability. J Exp Clin Cancer Res. 2018;37:40
145. Qian J-Y, Gao J, Sun X, Cao M-D, Shi L, Xia T-S. et al. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. 2019;38:6123-41
146. Miao R, Dai C-C, Mei L, Xu J, Sun S-W, Xing Y-L. et al. KIAA1429 regulates cell proliferation by targeting c-Jun messenger RNA directly in gastric cancer. J Cell Physiol. 2020;235:7420-32
147. Miranda-Gonçalves V, Lobo J, Guimarães-Teixeira C, Barros-Silva D, Guimarães R, Cantante M. et al. The component of the mA writer complex VIRMA is implicated in aggressive tumor phenotype, DNA damage response and cisplatin resistance in germ cell tumors. J Exp Clin Cancer Res. 2021;40:268
148. Xu Y, Chen Y, Yao Y, Xie H, Lu G, Du C. et al. VIRMA contributes to non-small cell lung cancer progression via N-methyladenosine-dependent DAPK3 post-transcriptional modification. Cancer letters. 2021;522:142-54
149. Barros-Silva D, Lobo J, Guimarães-Teixeira C, Carneiro I, Oliveira J, Martens-Uzunova ES. et al. VIRMA-Dependent N6-Methyladenosine Modifications Regulate the Expression of Long Non-Coding RNAs CCAT1 and CCAT2 in Prostate Cancer. Cancers (Basel). 2020 12
150. Zhu D, Zhou J, Zhao J, Jiang G, Zhang X, Zhang Y. et al. ZC3H13 suppresses colorectal cancer proliferation and invasion via inactivating Ras-ERK signaling. J Cell Physiol. 2019;234:8899-907
151. Rong B, Zhang Q, Wan J, Xing S, Dai R, Li Y. et al. Ribosome 18S mA Methyltransferase METTL5 Promotes Translation Initiation and Breast Cancer Cell Growth. Cell Rep. 2020;33:108544
152. Wang Z, Liu J, Yang Y, Xing C, Jing J, Yuan Y. Expression and prognostic potential of ribosome 18S RNA mA methyltransferase METTL5 in gastric cancer. Cancer Cell Int. 2021;21:569
153. Wang X-K, Zhang Y-W, Wang C-M, Li B, Zhang T-Z, Zhou W-J. et al. METTL16 promotes cell proliferation by up-regulating cyclin D1 expression in gastric cancer. J Cell Mol Med. 2021;25:6602-17
154. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C. et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N-Methyladenosine RNA Demethylase. Cancer cell. 2017;31:127-41
155. Tao L, Mu X, Chen H, Jin D, Zhang R, Zhao Y. et al. FTO modifies the m6A level of MALAT and promotes bladder cancer progression. Clin Transl Med. 2021;11:e310
156. Ruan D-Y, Li T, Wang Y-N, Meng Q, Li Y, Yu K. et al. FTO downregulation mediated by hypoxia facilitates colorectal cancer metastasis. Oncogene. 2021;40:5168-81
157. Relier S, Ripoll J, Guillorit H, Amalric A, Achour C, Boissière F. et al. FTO-mediated cytoplasmic mA demethylation adjusts stem-like properties in colorectal cancer cell. Nat Commun. 2021;12:1716
158. Tao B, Huang X, Shi J, Liu J, Li S, Xu C. et al. FTO interacts with FOXO3a to enhance its transcriptional activity and inhibits aggression in gliomas. Signal Transduct Target Ther. 2020;5:130
159. Mittenbühler MJ, Saedler K, Nolte H, Kern L, Zhou J, Qian S-B. et al. Hepatic FTO is dispensable for the regulation of metabolism but counteracts HCC development in vivo. Mol Metab. 2020;42:101085
160. Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am J Transl Res. 2019;11:6084-92
161. Yang S, Wei J, Cui Y-H, Park G, Shah P, Deng Y. et al. mA mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782
162. Wang Y, Li M, Zhang L, Chen Y, Zhang S. m6A demethylase FTO induces NELL2 expression by inhibiting E2F1 m6A modification leading to metastasis of non-small cell lung cancer. Mol Ther Oncolytics. 2021;21:367-76
163. Wang F, Liao Y, Zhang M, Zhu Y, Wang W, Cai H. et al. N6-methyladenosine demethyltransferase FTO-mediated autophagy in malignant development of oral squamous cell carcinoma. Oncogene. 2021;40:3885-98
164. Zeng J, Zhang H, Tan Y, Wang Z, Li Y, Yang X. m6A demethylase FTO suppresses pancreatic cancer tumorigenesis by demethylating and inhibiting Wnt signaling. Molecular therapy Nucleic acids. 2021;25:277-92
165. Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L. et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell. 2020 27
166. Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J. et al. ALKBH5 Inhibited Cell Proliferation and Sensitized Bladder Cancer Cells to Cisplatin by m6A-CK2α-Mediated Glycolysis. Molecular therapy Nucleic acids. 2021;23:27-41
167. Chen P, Li S, Zhang K, Zhao R, Cui J, Zhou W. et al. N-methyladenosine demethylase ALKBH5 suppresses malignancy of esophageal cancer by regulating microRNA biogenesis and RAI1 expression. Oncogene. 2021;40:5600-12
168. Pu X, Gu Z, Gu Z. ALKBH5 regulates IGF1R expression to promote the Proliferation and Tumorigenicity of Endometrial Cancer. J Cancer. 2020;11:5612-22
169. Kowalski-Chauvel A, Lacore MG, Arnauduc F, Delmas C, Toulas C, Cohen-Jonathan-Moyal E. et al. The m6A RNA Demethylase ALKBH5 Promotes Radioresistance and Invasion Capability of Glioma Stem Cells. Cancers (Basel). 2020 13
170. Zhang J, Guo S, Piao H-Y, Wang Y, Wu Y, Meng X-Y. et al. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J Physiol Biochem. 2019;75:379-89
171. Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R. et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via mA-guided epigenetic inhibition of LYPD1. Mol Cancer. 2020;19:123
172. Jin D, Guo J, Wu Y, Yang L, Wang X, Du J. et al. mA demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol Cancer. 2020;19:40
173. Chen S, Zhou L, Wang Y. ALKBH5-mediated mA demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020;20:34
174. Yuan Y, Yan G, He M, Lei H, Li L, Wang Y. et al. ALKBH5 suppresses tumor progression via an mA-dependent epigenetic silencing of pre-miR-181b-1/YAP signaling axis in osteosarcoma. Cell death & disease. 2021;12:60
175. Zhu H, Gan X, Jiang X, Diao S, Wu H, Hu J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J Exp Clin Cancer Res. 2019;38:163
176. Jiang Y, Wan Y, Gong M, Zhou S, Qiu J, Cheng W. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-κB pathway. J Cell Mol Med. 2020;24:6137-48
177. Nie S, Zhang L, Liu J, Wan Y, Jiang Y, Yang J. et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J Exp Clin Cancer Res. 2021;40:284
178. Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang Y. et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 2020;19:91
179. Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi Y. et al. mA demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol Cancer. 2020;19:3
180. He Y, Yue H, Cheng Y, Ding Z, Xu Z, Lv C. et al. ALKBH5-mediated mA demethylation of KCNK15-AS1 inhibits pancreatic cancer progression via regulating KCNK15 and PTEN/AKT signaling. Cell death & disease. 2021;12:1121
181. Zhang X, Wang F, Wang Z, Yang X, Yu H, Si S. et al. ALKBH5 promotes the proliferation of renal cell carcinoma by regulating AURKB expression in an mA-dependent manner. Ann Transl Med. 2020;8:646
182. Pi J, Wang W, Ji M, Wang X, Wei X, Jin J. et al. YTHDF1 Promotes Gastric Carcinogenesis by Controlling Translation of FZD7. Cancer research. 2021;81:2651-65
183. Li Q, Ni Y, Zhang L, Jiang R, Xu J, Yang H. et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6:76
184. Liu X, Qin J, Gao T, Li C, He B, Pan B. et al. YTHDF1 Facilitates the Progression of Hepatocellular Carcinoma by Promoting FZD5 mRNA Translation in an m6A-Dependent Manner. Molecular therapy Nucleic acids. 2020;22:750-65
185. Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L. et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019;10:4892
186. Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y. et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020;48:3816-31
187. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I. et al. Targeting the RNA mA Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell. 2019 25
188. Einstein JM, Perelis M, Chaim IA, Meena JK, Nussbacher JK, Tankka AT. et al. Inhibition of YTHDF2 triggers proteotoxic cell death in MYC-driven breast cancer. Mol Cell. 2021 81
189. Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q. et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov. 2021;11:480-99
190. Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi H. et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun. 2021;12:177
191. Li Y, Sheng H, Ma F, Wu Q, Huang J, Chen Q. et al. RNA mA reader YTHDF2 facilitates lung adenocarcinoma cell proliferation and metastasis by targeting the AXIN1/Wnt/β-catenin signaling. Cell death & disease. 2021;12:479
192. Chang G, Shi L, Ye Y, Shi H, Zeng L, Tiwary S. et al. YTHDF3 Induces the Translation of mA-Enriched Gene Transcripts to Promote Breast Cancer Brain Metastasis. Cancer cell. 2020 38
193. Ma L, Chen T, Zhang X, Miao Y, Tian X, Yu K. et al. The mA reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biol. 2021;38:101801
194. He J-J, Li Z, Rong Z-X, Gao J, Mu Y, Guan Y-D. et al. mA Reader YTHDC2 Promotes Radiotherapy Resistance of Nasopharyngeal Carcinoma via Activating IGF1R/AKT/S6 Signaling Axis. Frontiers in oncology. 2020;10:1166
195. Guo H, Wang B, Xu K, Nie L, Fu Y, Wang Z. et al. mA Reader HNRNPA2B1 Promotes Esophageal Cancer Progression via Up-Regulation of ACLY and ACC1. Frontiers in oncology. 2020;10:553045
196. Zhang L, Wan Y, Zhang Z, Jiang Y, Gu Z, Ma X. et al. IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics. 2021;11:1100-14
197. Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V. et al. RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia. 2020;34:1354-63
198. Bley N, Schott A, Müller S, Misiak D, Lederer M, Fuchs T. et al. IGF2BP1 is a targetable SRC/MAPK-dependent driver of invasive growth in ovarian cancer. RNA biology. 2021;18:391-403
199. Pu J, Wang J, Qin Z, Wang A, Zhang Y, Wu X. et al. IGF2BP2 Promotes Liver Cancer Growth Through an m6A-FEN1-Dependent Mechanism. Frontiers in oncology. 2020;10:578816
200. Xu X, Yu Y, Zong K, Lv P, Gu Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J Exp Clin Cancer Res. 2019;38:497
201. Sa R, Liang R, Qiu X, He Z, Liu Z, Chen L. IGF2BP2-dependent activation of ERBB2 signaling contributes to acquired resistance to tyrosine kinase inhibitor in differentiation therapy of radioiodine-refractory papillary thyroid cancer. Cancer letters. 2021;527:10-23
202. Palanichamy JK, Tran TM, Howard JM, Contreras JR, Fernando TR, Sterne-Weiler T. et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J Clin Invest. 2016;126:1495-511
203. Wang Z, Tong D, Han C, Zhao Z, Wang X, Jiang T. et al. Blockade of miR-3614 maturation by IGF2BP3 increases TRIM25 expression and promotes breast cancer cell proliferation. EBioMedicine. 2019;41:357-69
204. Yang Z, Wang T, Wu D, Min Z, Tan J, Yu B. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J Exp Clin Cancer Res. 2020;39:203
205. Gu Y, Niu S, Wang Y, Duan L, Pan Y, Tong Z. et al. -Mediated Regulation of mA-Modified by mA Reader IGF2BP3 Drives ccRCC Progression. Cancer research. 2021;81:923-34
206. Gu Y, Wu X, Zhang J, Fang Y, Pan Y, Shu Y. et al. The evolving landscape of N-methyladenosine modification in the tumor microenvironment. Mol Ther. 2021;29:1703-15
207. Dong L, Chen C, Zhang Y, Guo P, Wang Z, Li J. et al. The loss of RNA N-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8 T cell dysfunction and tumor growth. Cancer cell. 2021 39
208. Song H, Song J, Cheng M, Zheng M, Wang T, Tian S. et al. METTL3-mediated mA RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun. 2021;12:5522
209. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H. et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159-70
210. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R. et al. Anti-tumour immunity controlled through mRNA mA methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270-4
211. Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L. et al. Mettl3-mediated mRNA mA methylation promotes dendritic cell activation. Nat Commun. 2019;10:1898
212. Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X. et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer research. 2021;81:5876-88
213. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597-601
214. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H. et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer cell. 2019 35
215. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M. et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer cell. 2020 38
216. Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z. et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021 33
217. Qing Y, Dong L, Gao L, Li C, Li Y, Han L. et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/mA/PFKP/LDHB axis. Mol Cell. 2021 81
218. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y. et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/mA/MYC/CEBPA Signaling. Cell. 2018 172
219. Sun K, Du Y, Hou Y, Zhao M, Li J, Du Y. et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/mA signaling. Theranostics. 2021;11:5831-46
220. Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS One. 2016;11:e0159072
221. Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME. et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5:658-65
222. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373-84
Author contact
![]() Corresponding authors: Jing He, Department of Breast Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. E-mail: jinghe16edu.cn; Mingxia Zhou, Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. E-mail: mingxiazhou0906com.
Corresponding authors: Jing He, Department of Breast Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. E-mail: jinghe16edu.cn; Mingxia Zhou, Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. E-mail: mingxiazhou0906com.