Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(13):5019-5037. doi:10.7150/ijbs.72367 This issue Cite
Research Paper
The interactions between integrin α5β1 of liver cancer cells and fibronectin of fibroblasts promote tumor growth and angiogenesis
Zheng Peng1, Meng Hao1, Haibo Tong1, Hongmei Yang1, Bin Huang1, Zhigang Zhang2, Kathy Qian Luo1,3 ![]()
1. Faculty of Health Sciences, University of Macau, Taipa, Macao SAR, China.
2. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
3. Ministry of Education-Frontiers Science Center for Precision Oncology, University of Macau, Taipa, Macao SAR, China.
Received 2022-2-24; Accepted 2022-6-10; Published 2022-8-1
Abstract

Hepatocellular carcinoma (HCC) progression is closely related to pathological fibrosis, which involves heterotypic intercellular interactions (HIIs) between liver cancer cells and fibroblasts. Here, we studied them in a direct coculture model, and identified fibronectin from fibroblasts and integrin-α5β1 from liver cancer cells as the primary responsible molecules utilizing CRISPR/Cas9 gene-editing technology. Coculture led to the formation of 3D multilayer microstructures, and obvious fibronectin remodeling was caused by upregulated integrin-α5β1, which greatly promoted cell growth in 3D microstructures. Integrin-α5 was more sensitive and specific than integrin-β1 in this process. Subsequent mechanistic exploration revealed the activation of integrin-Src-FAK, AKT and ERK signaling pathways. Importantly, the growth-promoting effect of HIIs was verified in a xenograft tumor model, in which more blood vessels were observed in bigger tumors derived from the coculture group than that derived from monocultured groups. Hence, we conducted triculture by introducing human umbilical vein endothelial cells, which aligned to and differentiated along multilayer microstructures in an integrin-α5β1 dependent manner. Furthermore, fibronectin, integrin-α5, and integrin-β1 were upregulated in 52 HCC tumors, and fibronectin was related to microvascular invasion. Our findings identify fibronectin, integrin-α5, and integrin-β1 as tumor microenvironment-related targets and provide a basis for combination targeted therapeutic strategies for future HCC treatment.
Keywords: Hepatocellular carcinoma, tumor growth, fibronectin, integrin-α5, integrin-β1, angiogenesis
Introduction
HCC comprises 75%-85% of primary liver cancer cases and is the leading cause of cancer-related mortality [1-3]. The development of HCC is believed to be closely related to pathological fibrosis [4, 5], which involves extracellular matrix (ECM) remodeling and heterotypic intercellular interactions (HIIs) between liver cancer cells and surrounding fibroblasts [6, 7]. Currently, many studies have focused on this field in the hope of finding key tumor microenvironment (TME)-related targets for combination targeted therapy [6-13], given the unsatisfactory outcomes of current therapies.
As a major product of fibroblasts and a versatile ECM component in the TME, fibronectin (Fn) carries out many pivotal physiological and pathological functions, since it provides a provisional scaffold for the assembly of other ECM proteins [12, 14-20]. Integrin-α5β1, as the primary fibronectin receptor and a widely distributed dimeric transmembrane protein [21], can serve as a cellular “hand” that exerts tensional force upon naturally coiled fibronectin to expose cryptic binding domains inside fibronectin [21-28], which could greatly promote fibrillogenesis [14, 29]. Importantly, vascular endothelial growth factor-A (VEGFA) can bind to these exposed domains [30-35], and exert a more potent proangiogenic effect than free VEGFA [20, 36, 37]. Taken together, the above findings suggest the neglected roles of HIIs between fibronectin from fibroblasts and integrins from cancer cells in the development of pathological fibrosis and angiogenesis in HCC.
To investigate the HIIs between liver cancer cells and fibroblasts, we established a direct coculture system. After finding that HIIs caused 3D multilayer microstructure formation, we performed CRISPR/Cas9 gene-editing to determine primary responsible targets and mechanistically analyzed how these targets exert their effects. In vivo experiments were performed to confirm our findings. Then, focusing on the biological functions of HIIs, we introduced human umbilical vein endothelial cells (HUVECs) and further explored the roles of these targets. Finally, clinical significance was explored by immunohistochemistry staining (IHC-staining) and KMplot database analysis.
Material and methods
Cell lines and cell culture
MEFs and MEF-FN-/- cells were derived from mouse kidneys and immortalized by lentiviral transduction of SV40 (Simian vacuolating virus 40) large T antigen, and generously provided by Professor Reinhard Fässler and his assistant Dr. Ralph Böttcher (Max-Planck Institute for Biochemistry, Martinsried, Germany). The human hepatocellular carcinoma cell line HepG2 and the HUVEC cell line CRL-1730™ were purchased from ATCC (American Type Culture Collection; Rockville, MD, USA). Huh7 cells were kindly provided by Prof. Chuxia Deng from University of Macau. HepG2-α5-/-, HepG2-β1-/-, HUVEC-α5-/-, and HUVEC-β1-/- were single cell-derived gene-knockout clones generated by the CRISPR/Cas9 system. All monocultured and cocultured cells were cultured in DMEM (Dulbecco's modified Eagle's medium) containing 10% fetal bovine serum (FBS) and 100 U/ml penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and maintained in a humidified incubator with 5% CO2 at 37 °C.
Reagents
Mowiol® 4-88 powder was ordered from Sigma-Aldrich (St. Louis, MO, USA). Sunitinib, apatinib, dasatinib, dactolisib, trametinib, and Y15 were purchased from Selleck Chemicals (Houston, TX, USA). Recombinant human VEGFA (rhVEGFA) was ordered from R&D Systems (Minneapolis, MN, USA).
Coculture and triculture systems
After trypsinization and cell counting, liver cancer cells and MEF cells were mixed at a ratio of 1:1 for the coculture assay. For triculture, liver cancer cells, MEF cells, and HUVCs were mixed at a ratio of 5:3:2 before being seeded in cell culture dishes.
Cell density assay
Cells were plated at 2.5 × 105 cells/dish (60 mm in diameter) and cultured for 1 to 10 days in DMEM supplemented with 10% FBS. The culture medium was changed every day, and cell numbers were counted. Cell density was calculated by dividing the total cell number by the total culture surface area. Each value represents the mean ± S.D. of triplicate experiments.
Protein extraction and Western blot
After washing cells with PBS, cellular proteins were extracted using RIPA lysis buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). After sonication and centrifugation, the supernatant was collected and quantified using Protein Assay Dye Reagent concentrate (Bio-Rad). After denaturation (95 °C, 5 min), equal amounts of protein (30 μg) from different samples were separated by SDS-PAGE and then electrotransferred (Mini-PROTEAN system, Bio-Rad) to a nitrocellulose membrane (Bio-Rad). After blocking with 5% Blotting-Grade Blocker (Bio-Rad) for 1 hr, the membrane was probed with a specific primary antibody (4°C, overnight) and then incubated with the HRP-conjugated secondary antibody (room temperature, 1 hr). Finally, after incubation with chemiluminescent substrate (Clarity Western ECL Substrate, Bio-Rad), the membrane was exposed using a ChemiDoc™ Touch Imaging System (Bio-Rad). The primary antibodies used in this study are listed in Table S2.
Fluorescence live-cell microscopy
Fluorescence live-cell imaging was performed using a Zeiss Axio Observer microscope equipped with a Zeiss Axiocam 512 monocamera and a set of objectives and filters, which could capture the right size pictures and separate signals from different channels, and captured images were then analyzed and exported by ZEN imaging software (Carl Zeiss Vision GmbH, München, Germany).
Two-photon fluorescence microscopy
Two-photon fluorescence imaging was performed using a Nikon A1RMP confocal and multiphoton microscope equipped with lasers (820-1300 nm), NDD detectors, and a set of objectives and then analyzed and exported by NIS-Elements AR 5.2 software (Nikon Instrument Inc., Melville, NY).
Immunofluorescence staining (IF-staining)
Cells were washed in PBS before fixation with 4% paraformaldehyde for 15 min and then permeabilized with 0.2% Triton-X 100 for 10 min at room temperature (RT). After blocking with 3% BSA for 1 hr, cells were incubated with primary antibody at the recommended dilution, followed by Alexa-Fluor 594- or 488-conjugated secondary antibodies (Invitrogen, Thermo Fisher Scientific) at a 1:100 dilution for 1 hr. Nuclei were labeled with Hoechst-33342 (Thermo Fisher Scientific) before cells were mounted onto a microscope slide using Mowiol® 4-88. Finally, IF-staining images were captured with a Zeiss Axio Observer microscope. The primary antibodies used in this study are listed in Table S2.
Enzyme-linked immunosorbent assay (ELISA)
The protein levels of mouse VEGFA in the cell culture supernatants were quantified using an ELISA kit from R&D Systems (Minneapolis) according to the manufacturers' instructions. Briefly, after capture antibody coating overnight and subsequent blocking for 1 hr, the prepared 96-well microplates were added to 100 μl of sample or standards and incubated for 2 hr at RT. After 3 washes, the detection antibodies were added to each well and incubated for 2 hr at RT. After 3 washes, streptavidin-HRP was added to each well and incubated for 20 min at RT. After 3 washes, substrate solution was added and incubated for 20 min at RT. Finally, the stop solution was added to each well. After gentle mixing, the optical density value of each well was determined by a plate reader immediately at 450 nm and 540 nm.
Wound healing plus IF-staining
Liver cancer cells and MEF-clover cells were seeded separately (3000 cells per well) in a 2 well silicone culture-insert (iBidi, Martin Reid, Gräfelfing, Germany) mounted on a glass slide to create a 500 μm cell-free gap between these two cell types. The culture-insert was then removed on day 3 after they reached confluence, and the glass slide was placed in a culture dish to allow these two cell types to grow toward each other. Finally, the glass slide was removed from the dish on day 5 and subjected to IF-staining procedures.
Species-specific primers
First, the human and mouse mRNA sequences of genes of interest were obtained from the NIH website. The alignment of human and mouse mRNA sequences was conducted by using SnapGene software (Version 3.1; GSL Biotech, snapgene.com), with differentiated parts marked out. At least 3 independent species-specific primers were designed on the basis of differentiated mRNA sequences if possible. The specificity of the primers was first examined using the “primer designing tool” to check whether there were products in the other specie's genome. Then, these primer sequences were sent to and synthesized by Beijing Genomics Institute (BGI, China) in Guangzhou. Finally, the validation of these primers was performed by checking the melt curves after RT-qPCR and running electrophoresis in 1.5% agarose gels with the q-PCR products. The primers used in this study are listed in Table S3.
Real-time quantitative polymerase chain reaction (RT-qPCR)
First, total RNA was extracted using Trizol® (Thermo Fisher Scientific) and isopropanol (Sigma-Aldrich) precipitation and then reverse transcribed into cDNA by an iScript™ cDNA Synthesis Kit (Bio-Rad). Finally, RT-qPCR was conducted in triplicate using cDNA, primers, and iTaq™ Universal SYBR® Green Supermix (Bio-Rad) on a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). Fold changes in mRNA levels of genes of interest were compared using the ΔΔCt method with the ΔΔCt of GAPDH as the internal control.
Human hepatocellular carcinoma xenograft tumor model in nude mice
All animal experiments were conducted in accordance with the guidelines approved by the Animal Research Ethics Committee of the University of Macau. Five-week-old nude mice were injected subcutaneously with 2 million cells in the right flank. Body weight and xenograft volume were monitored every 3 days beginning on day 5. Xenograft volume (mm3) was calculated by using the following formula: π/6 × length × width2. All mice were sacrificed on day 29, and tumor xenografts were dissected using a surgical blade and weighed before taking images by a digital camera and an Olympus MVX10 macro-zoom fluorescence microscope (Olympus, Hamburg, Germany). Finally, xenografts were fixed in 4% paraformaldehyde and embedded in paraffin blocks before tissue sectioning.
Immunohistochemistry staining (IHC-staining)
Tissue microarray (TMA) slides were generously provided by Prof. Zhang Zhigang (Shanghai Cancer Institute, China). After deparaffinization and hydration in Milli-Q water, antigens were retrieved by boiling TMA slides in citrate buffer for 20 min. Then, endogenous peroxidase activity and nonspecific binding activity were blocked before incubation with the primary antibody at the recommended dilution (4 °C, overnight) in a wet box. Finally, immunoreactivity was visualized using the Mouse and Rabbit specific HRP/DAB (ABC) Detection IHC Kit (Abcam, Cambridge, MA, USA) in accordance with the manufacturer's instructions. Nuclei were counterstained with hematoxylin (Sigma-Aldrich). Images were taken by a NanoZoomer S60 slide scanner (Hamamatsu Photonics, Shizuoka, Japan) at a magnification of 20×. The expression levels of corresponding targets were determined by quantitative analysis. IHC scores were graded according to the percentage of stained cells as previously described [38]. Briefly, the scoring was assigned as follows: the percentage scores: 0, <5% of positively stained cells; 1, 5-50%; 2, 51-100%; the intensity scores: 0, absent or faint; 1, weak; 2, moderate; 3, strong.
Tube formation assay (TFA)
A total of 2.5 × 105 HUVEC-i670 cells were resuspended in 50 μl medium containing 0.5% FBS and plated onto Matrigel-coated µ-slides (iBidi). After incubation for 6 hr, images were taken on a Zeiss Axio Observer microscope.
Image quantitative analysis
For quantitative analysis of the percentage of area occupied by MEF-clover cells, images were analyzed by ImageJ 1.53c software (National Institute of Health, Bethesda, MD, USA) to obtain the percentage of area of interest. For quantitative analysis of the colocalized area of two types of fluorescent cells, images were analyzed by AutoQuant X3 software (Media Cybernetics Inc., Rockville, MD, USA) to obtain the percentage of colocalized area.
Quantitative analysis of angiogenesis characteristics
Far red channel live-cell fluorescence images were analyzed by AngioTool software (National Cancer Institute, Bethesda, MD, USA). From the analysis, the number of junctions, number of tubules, total tubule length, and average tubule length are presented.
Lentivirus production and transfection
Core plasmids were transfected into Lenti-X 293T cells by polyethylenimine (PEI, Polysciences, Warrington, PA, USA) to produce lentivirus, together with envelope plasmid (dR8.2) and virus packaging plasmid (VSV-G). Then, the produced viral particles were concentrated and collected at 48 hr and 72 hr and added to target cells for transfection. The primary core plasmids used in this study are listed in Table S4.
Fluorescence activated cell sorting (FACS)
The fluorescent protein-transfected cell suspension was kept at 1-10 million/ml in PBS after trypsinization. Cells were filtered immediately before sorting by a BD FACSAria™ III digital flow cytometer (San Diego, CA, USA). During the sorting process, the top 10% of cells were collected for fluorescence intensity. Then, this process was repeated after the collected cells reached enough numbers for the next round of sorting. On the whole, 2-4 rounds of sorting yielded a fluorescent cell line.
Generation of single cell-derived gene-knockout clones
After trypsinization, transfected cells were counted and diluted to a concentration of 200 cells/ml, and serial dilutions were performed in a flat bottom 96-well plate. Every well was observed under a microscope, and wells containing a single cell were marked. Allow single cells to expand to microcolonies. Split cells when they reach confluence. Continue expanding them until they proliferate to a sufficient number. Finally, the gene-knockout effect was validated using Western blotting.
Statistical analysis
All experiments were conducted at least 3 times. All the data are presented as the mean ± SD (standard deviation). The difference between groups was analyzed using the relevant test through GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA). The following probability values were deemed statistically significant: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Results
Coculture of liver cancer cells and MEFs led to the formation of 3D multilayer microstructures
Inspired by our previous study [39], to determine the HIIs between liver cancer cells and fibroblasts, we established a coculture system using liver cancer cells and mouse embryo fibroblasts (MEFs). To distinguish these two cell types, we separately introduced plasmids to express tdTomato (tdT) and clover and obtained HepG2-tdT, Huh7-tdT, and MEF-clover cell lines. Then, we observed the HIIs between two cell types by comparing coculture and monoculture systems using live-cell fluorescence images of the same spot over 10 sequential days. A ratio of 1:1 between the two cell types was set as the best combination, and coculture without fluorescent protein expression was performed to exclude possible confounding factors (Fig. S1A-D, and Fig. S3A).
From the chronological images in Fig. 1A, we observed that after the introduction of HIIs by coculture, 3D multilayer microstructures began to form on day 4 and became obvious after day 5. The overall zoomed-out images corresponding to the enlarged parts on day 1 and day 10 clearly reveal the dramatic effects induced by HIIs. However, monocultured cells formed only small aggregates (Fig. 1C and D). Moreover, from the cell density comparison shown in Fig. 1B, we observed that coculture slightly increased cell density on day 4 and had a more significant effect after day 7. On day 10, the cocultured cell density was increased by more than 20% compared with monocultured cells, probably because 3D microstructures improved growth environment.
Coculture of HepG2 cells and MEFs led to the formation of 3D multilayer microstructures. (A) Representative live-cell fluorescence images demonstrating the formation of 3D multilayer microstructures from day 1 to day 10. HepG2-tdT and MEF-clover cells were cocultured at a ratio of 1:1. Gridded glass coverslips were employed to take images at the same spots. Zoomed-out views of the images on day 1 and day 10 are shown below. (B) A cell density curve of monoculture and coculture systems from day 1 to day 10. HepG2-tdT and MEF-clover cells were monocultured or cocultured in 2.5 × 105 cells/60-mm culture dishes, and the cell density was assessed every day. Two-way ANOVA and Tukey's multiple comparison test were performed. (C and D) Representative images of monocultured HepG2-tdT (C) and MEF-clover cells (D) at the same spots. (E) Representative 3D XYZ reconstructed images of cocultured cells on day 1 and day 10. Images were generated from Z-stack images taken on a two-photon microscope. (F) A height curve of 3D multilayer microstructures formed during coculture. Height data were extracted from the 3D volume-rendering images. One-way ANOVA and Tukey's multiple comparison test were performed, using the height on day 1 as the reference point. (G) XY-plane images at different heights of a 3D multilayer microstructure formed in coculture on day 10. Scale bars, 100 µm. All data are the mean ± SD from 3 independent experiments. * P<0.05, ** P<0.01, **** P<0.0001, and ns, not significant.
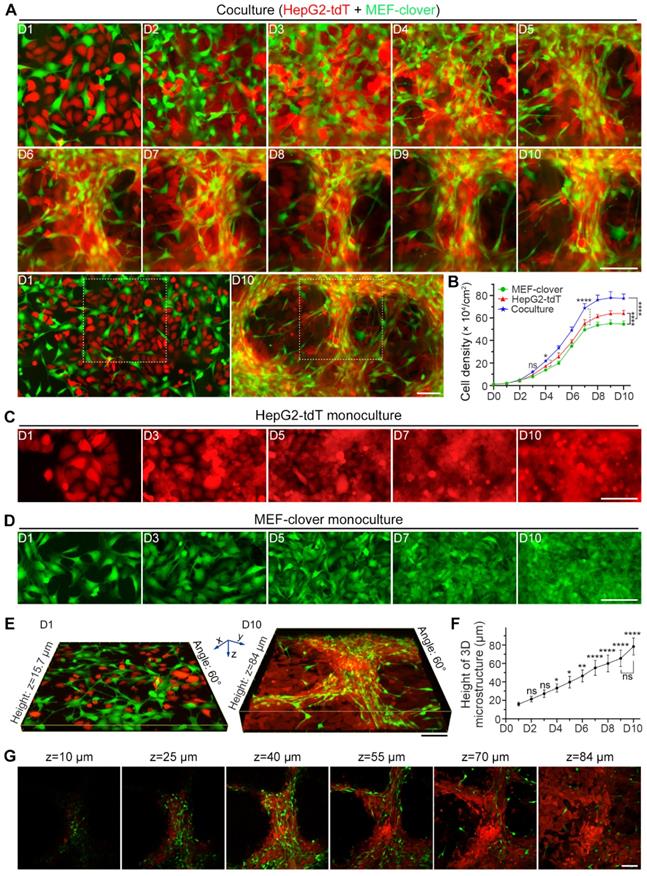
To characterize microstructures, we reconstructed 3D volume-rendering images and found that in contrast to their tendency to spread in monoculture, the cocultured MEFs tended to gather, and HepG2 cells displayed higher cell density surrounding MEFs, which caused two cell types to converge. Interestingly, unlike the wide distribution of HepG2 cells, MEFs were mainly distributed along 3D multilayer microstructures after day 5, suggesting their active role in microstructure formation. Moreover, the morphology of most fibroblasts shifted from a flat spindle-like shape to an elongated shape (Fig. 1A, Fig. S2A and E).
Next, we generated a height curve using the height extracted from two-photon images (Fig. 1E and F), and observed that compared with the height on day 1 (15.9 μm), the height had doubled around day 4 (33.3 μm) and peaked on day 10 (78.4 μm), which reflected the effects of HIIs as shown in Fig. 1A. However, the height fluctuated around 35.9 μm and 20.9 μm on day 10 for monocultured HepG2-tdT and MEF-clover cells, respectively (Fig. S2B-D).
To determine the topological relationship between two cell types inside 3D microstructures, XY-plane images at different depths on day 10 were examined and are shown in Fig. 1G. Combined with results in Fig. S2A, we observed that the relationship between the two cell types shifted from their random distribution to their interweaving in 3D microstructures, with MEFs mainly distributed in upper layers, while HepG2 cells mainly distributed in bottom layers.
Similar results were obtained in the coculture of Huh7 cells and MEFs (Fig. S3A-D). Taken together, we concluded that HIIs promoted the aggregation of liver cancer cells and fibroblasts during coculture, and caused the formation of 3D multilayer microstructures.
Within 3D microstructures, integrin-α5 and integrin-β1 were upregulated in liver cancer cells, while fibronectin and collagen I assembly were increased in fibroblasts
Integrins and ECM proteins are probably involved in microstructure formation, since they are the primary players in the formation of focal adhesions which bring different cells together to form microstructures. Then, we performed Western blot (WB) assays to determine the expression of major integrins and ECM proteins.
From WB results in Fig. 2A, we saw integrin-α5 displayed a higher increase compared with integrin-β1 or integrin-αV during coculture, in which integrin-α5 increased by 11.6-fold, while integrin-β1 and integrin-αV increased by 2.8- and 2.1-fold on day 10, respectively; fibronectin showed a higher increase than collagen I on day 10 (7.4-fold vs. 4.2-fold). Moreover, integrin-α5 upregulation occurred earlier than other integrins and quickly reached its maximum level of 12.2-fold on day 5. Similarly, fibronectin upregulation occurred earlier than collagen I upregulation. These results indicated that integrin-α5 and fibronectin are probably the primary mediators.
Integrins and ECM proteins were enriched in 3D multilayer microstructures with more activated fibroblasts. (A) WB results showing the levels of related proteins in cocultured cells and monocultured cells from day 1 to day 10. (B) Representative IF-staining images of related proteins in cocultured cells and monocultured cells. (C and D) Representative IF-staining images of α-SMA in monocultured MEF-clover cells and coculture cells on day 5 (C), and subsequent quantification of the proportion of α-SMA-positive MEFs under two conditions (D). The results were quantified based on the same area in the 20 × observation field (2120 × 1416 pixels, 0.31 µm × 0.31 µm per pixel) from 3 independent experiments, and data are shown as the mean ± SD. Unpaired t test was performed. **** P<0.0001. (E) WB results of the levels of α-SMA in monocultured MEF-clover cells and cocultured cells (HepG2 and MEF-clover cells) on day 10. Nuclei were visualized using Hoechst-33342 staining. Scale bars, 100 µm.
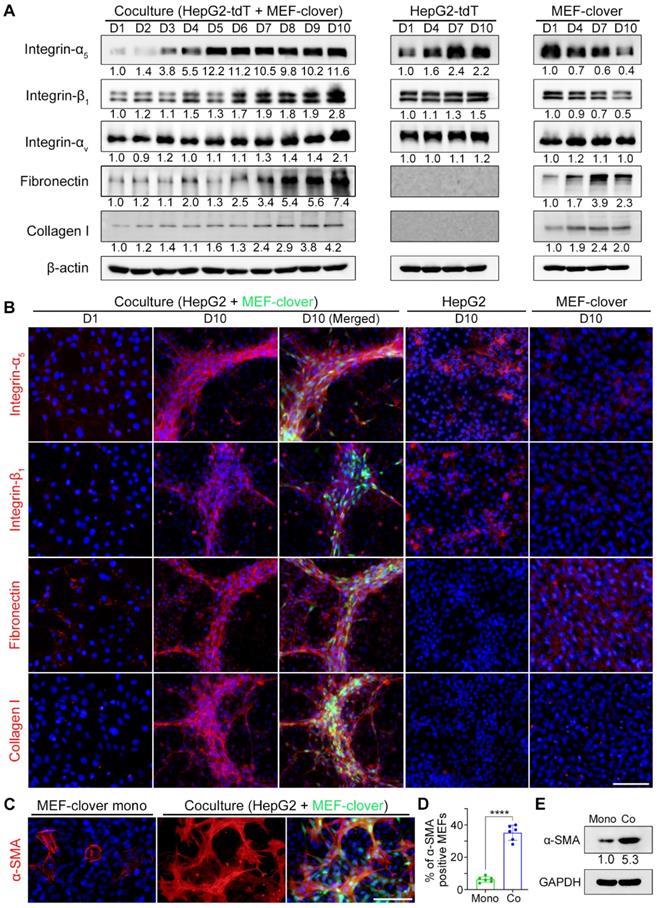
Based on WB results in Fig. 2A, we knew fibronectin and collagen I were mainly from fibroblasts, as supported by q-PCR results (Fig. S4A) using species-specific primers that only recognize either human- or mouse-derived corresponding mRNAs. For integrin-α5 and integrin-β1, although both types of cells displayed a basal protein level, mRNA levels of both genes from MEFs in coculture displayed downward trends as in monoculture. However, mRNA levels of both genes in cocultured HepG2 cells showed upward trends, with ITGA5 and ITGB1 mRNA levels on day 10 increasing by 469.4% and 42.9% compared with day 1, respectively. Moreover, both genes' mRNA levels in cocultured HepG2 cells displayed significant increases compared with monocultured HepG2 cells on day 10, as ITGA5 displayed a higher increase than ITGB1 (129.4% vs. 8.8%) (Fig. S4A). These results illustrated that upregulated integrin-α5 and integrin-β1 were mainly from cocultured HepG2 cells, and integrin-α5 was more sensitive to HIIs than integrin-β1.
Since our interest is in microstructure formation, the structural and distributional changes of these proteins during microstructure formation at different timepoints were investigated by immunofluorescence staining (IF-staining). Monocultured HepG2 and MEF-clover cells on day 10 were adopted as controls (Fig. S4B and Fig. 2B). Little bleed-through was observed using Alexa-Fluor 594 labeled secondary antibodies and suitable filter sets (Fig. S5A). From IF results, we observed that integrin-α5 and integrin-β1 underwent dramatic distributional changes during coculture, shifting from a scattered pattern to a clustered pattern with enriched signals distributed in 3D microstructures, while in monoculture, the weak signals were irregularly distributed with small aggregates. In addition to similar distributional changes, fibronectin and collagen I underwent dramatic structural changes, shifting from small thin fibers to long thick bundles, while no such changes were observed in monocultures (Fig. 2B and Fig. S5B). Similar results were also obtained in the IF staining and WB results of Huh7 and MEFs coculture (Fig. S6 and Fig. S7). These changes clearly illustrated the existence of HIIs and its effects on fibronectin and collagen I remodeling. The results were repeated using cells not expressing fluorescent proteins (Fig. S5B and Fig. S7B).
To highlight fibroblasts, we merged the MEF-clover cells with target proteins. Interestingly, we observed good colocalization (Fig. 2B and Fig. S6), suggesting the active role of fibroblasts in microstructure formation. Additionally, the distribution pattern of fibroblasts in coculture was also confirmed by IF-staining using cells not expressing fluorescent proteins through their elliptical, speckled nuclei (Fig. S5B-D).
Furthermore, results from wound healing plus IF-staining experiments showed that where the two cell types converged, integrin-α5, integrin-β1, and fibronectin signals were obviously stronger than those of either cell type's territory (Fig. S8A-C). These results further verified that HIIs are the driving force for these proteins' upregulation.
Since MEFs underwent significant morphological changes during coculture, we also tested related markers and found that fibroblast growth factor receptor-1 (FGFR1) and α-smooth muscle actin (α-SMA) were upregulated in cocultured MEFs compared with monocultured MEFs. Importantly, from the quantified results of α-SMA, we found that the percentage of α-SMA-positive MEFs was significantly increased from 6% in monoculture to 35% in coculture, which was supported by a 5.3-fold protein level increase (Fig. 2C-E and Fig. S8D).
Collectively, above results suggest that fibronectin from fibroblasts and integrin-α5 and integrin-β1 from liver cancer cells are probably the main players in 3D microstructure formation.
Fibronectin, integrin-α5 and integrin-β1 are essential in promoting 3D microstructure formation
To verify our hypothesis and determine the essential roles of these targets in 3D microstructure formation, we used fibronectin-knockout MEFs (MEF-FN-/-), integrin-α5-knockout HepG2 (HepG2-α5-/-) and integrin-β1-knockout HepG2 (HepG2-β1-/-) cells, and labeled them with fluorescent proteins (Fig. S9A-C). Gene-knockout was confirmed by WB analysis (Fig. 3A).
The formation of 3D multilayer microstructures required fibronectin, integrin-α5, and integrin-β1. (A) WB analysis of the levels of fibronectin, collagen I, integrin-α5, and integrin-β1 in the following samples: MEF-clover cells and MEF-FN-/--clover cells; HepG2-tdT cells, HepG2-α5-/--tdT cells, and HepG2-β1-/--tdT cells; cocultured HepG2-tdT and MEF-clover cells (coculture-ctrl), cocultured HepG2-tdT and MEF-FN-/--clover cells (coculture-FN-/-), cocultured HepG2-α5-/--tdT and MEF-clover cells (coculture-α5-/-), and cocultured HepG2-β1-/--tdT cells and MEF-clover cells (coculture-β1-/-). (B to E) Representative live-cell fluorescence images of coculture-ctrl (B), coculture-FN-/- (C), coculture-α5-/- (D), and coculture-β1-/- (E) groups at the same spots from day 1 to day 10. (F) The percentages of XY-plane growth space occupied by fibroblasts per 10 × observation field (4248 × 2832 pixels, 0.31 µm × 0.31 µm per pixel) were measured using ImageJ in the following four coculture groups: coculture-ctrl, coculture-FN-/-, coculture-α5-/-, and coculture-β1-/-. (G) The percentages of colocalized area between two cell types per 10 × observation field were measured in the four coculture groups with AutoQuant X3 software. (H) Zoomed-out view (10 × observation field) of the image from day 10 in panel e showing the obviously uneven distribution of fibroblasts in coculture-β1-/- group. Scale bars, 100 µm. All data are the mean ± SD from 3 independent experiments. Two-way ANOVA and Tukey's multiple comparison test were performed. * P<0.05, **** P<0.0001, and ns, not significant.
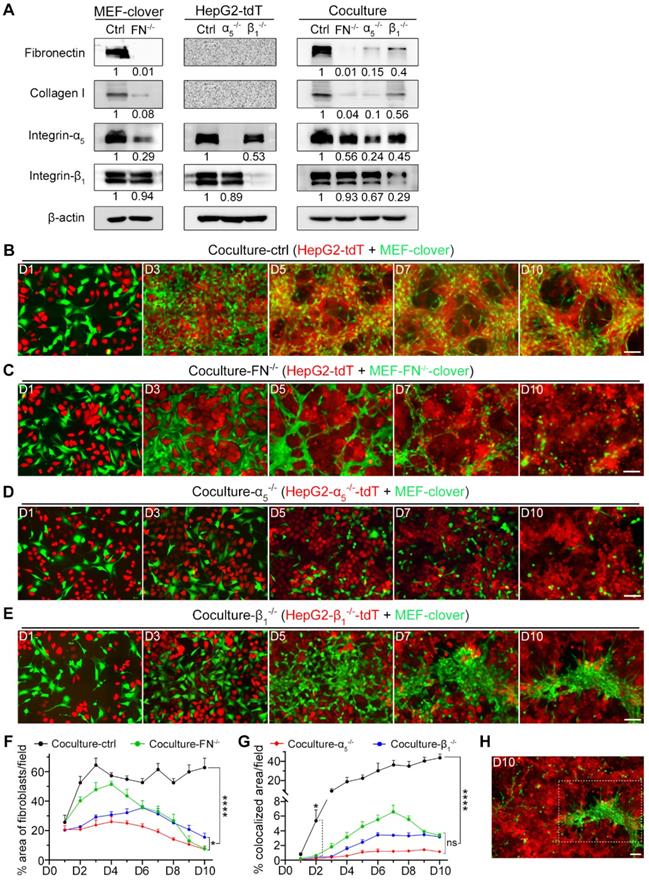
Next, we cocultured different combinations. Unlike coculture-ctrl group in Fig. 3B, all three gene-knockout groups showed obvious interruption of HIIs after day 5, as MEFs and HepG2 cells grew separately without obvious microstructure formation. Fibroblasts gradually died after day 5, probably due to the loss of HIIs stimulation and HepG2 cells as an anchor (Fig. 3C-E). Furthermore, these observations were supported by 2D histograms, as the convergence tendency between two types of pixels was only shown in coculture-ctrl group between day 5 to day 10 (Fig. S9D).
Since fibronectin and collagen I were mainly from fibroblasts, the percentage area occupied by fibroblasts in different groups was quantified. To exclude selection bias as shown in Fig. 3E and Fig. 3H on day 10, three 10 × images from each group were analyzed. Fig. 3F shows that although there is fluctuation, this parameter in coculture-ctrl group remained above 50% beginning on day 2, while for the other groups, this parameter continuously decreased after day 5. On day 10, coculture-FN-/- and coculture-α5-/- groups showed a larger decrease than coculture-ctrl group (7.71% and 7.32% vs. 62.82%), while coculture-β1-/- group showed a secondary decrease (15.47% vs. 62.82%), which indicated that there were fewer dead fibroblasts in coculture-β1-/- group than in the other two gene-knockout groups.
Moreover, as our focus was on HIIs, which would naturally result in the overlap of the two cell types during microstructure formation, the percentage of colocalized areas was then marked and calculated by AutoQuant X3 (Fig. S10A). Fig. 3G shows that all three gene-knockout coculture groups differed significantly from coculture-ctrl group beginning on day 2 in this parameter, which clearly reflected HIIs interruption after gene-knockout.
Next, we tried to explain the observations by WB analysis (Fig. 3A). From results with MEF-FN-/--clover cells, we discovered that fibronectin-knockout not only greatly reduced fibronectin from 1 to 0.01 but also dramatically reduced collagen I from 1 to 0.08. Furthermore, fibronectin-knockout greatly reduced integrin-α5 from 1 to 0.29 but had little impact on integrin-β1. Similar results were observed in coculture-FN-/- group. From the WB analysis of coculture-α5-/- and coculture-β1-/- groups, we found that compared with integrin-β1, integrin-α5 in HepG2 cells had a greater influence on fibronectin expression (0.15 vs. 0.4) and collagen I expression (0.10 vs. 0.56). These results illustrated that integrin-α5 had a stronger influence on fibronectin and collagen I expression than integrin-β1, which explained why coculture-α5-/- group exhibited stronger interference with microstructure formation than coculture-β1-/- group. Although integrin-α5β1 is the only known dimer containing integrin-α5, integrin-β1 also possesses a complex relationship with other α-class integrins [40], and integrin-β1-knockout would inevitably influence other integrins' expression. Indeed, integrin-β1-knockout was reported to upregulate other β-class integrins, such as integrin-β3 [41-45], which would compensate for its gene-knockout effects. In contrast, integrin-α5 is only related to integrin-β1, which makes integrin-α5 a more specific target than integrin-β1 in terms of regulating integrin-α5β1's expression.
We also performed IF-staining to observe the distributions of these four proteins in the three gene-knockout coculture groups and found that with the disappearance of microstructures in fibronectin-knockout or integrin-α5-knockout coculture groups, IF signals of these proteins were obviously reduced, but in integrin-β1-knockout coculture group, the other three proteins weakly clustered around the aggregated fibroblasts (Fig. S10B).
Similar results were obtained in Huh7 and MEFs coculture, after fibronectin-knockout in MEFs or integrin-α5-knockout in Huh7 cells (Fig. S11A-D). Collectively, above results indicate that fibronectin from fibroblasts, and integrin-α5β1 from liver cancer cells are the primary mediators of HIIs during coculture. Additionally, HIIs not only stimulated cancer cell proliferation but also supported fibroblast survival.
HIIs activated integrin-α5β1 downstream signaling
Next, we examined the downstream molecules of integrin-α5β1 signaling by IF-staining and found that the levels of paxillin, p-Src, p-FAK, and AKT surrounding microstructures formed during the coculture were obviously higher than the adjacent region or monoculture controls (Fig. 4A, Fig. S6, and Fig. S7A). After fibronectin-knockout in MEFs or integrin-α5-knockout in HepG2 cells, the above increased signals disappeared with microstructures. However, in the integrin-β1-knockout group, weak signals surrounding aggregated MEFs were still observed (Fig. 4A), probably because HIIs was preserved to some extent. Additionally, WB analysis in Fig. S12A showed that downstream signaling molecules, including p-paxillin, p-ERK, and p-AKT, were also affected to a certain extent after gene-knockout in coculture.
Integrin downstream signaling pathways were activated by fibronectin-integrin-α5β1-mediated HIIs during coculture. (A) Representative IF-staining images on day 10. Nuclei were visualized using Hoechst-33342 staining. Scale bar, 100 µm. (B) WB analysis showed that the treatment of coculture-ctrl cells with a FAK inhibitor (Y15, 5 µM) or a Src inhibitor (dasatinib, 0.1 µM) for 10 days reduced p-paxillin, p-Src, p-AKT, and p-ERK1/2 levels. (C and D) WB results showed that the treatment of coculture-ctrl cells with an MEK inhibitor (trametinib, 0.1 µM) (C) or a PI3K inhibitor (dactolisib, 0.1 µM) (D) for 10 days reduced p-ERK1/2 or p-AKT levels. (E) Diagram depicting heterotypic intercellular interactions (HIIs) between HepG2 cells and MEFs mediated by fibronectin-integrin-α5β1 and the proposed signaling pathways involved during coculture.
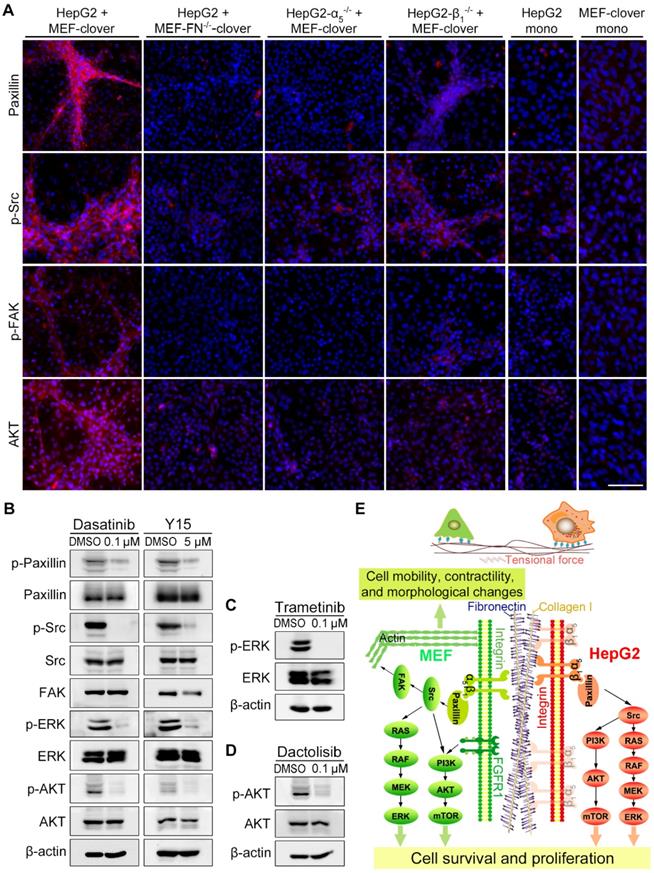
To assess the functional involvement of downstream molecules in microstructure formation, we added following inhibitors to coculture system: the FAK inhibitor Y15, the Src inhibitor dasatinib, the MEK inhibitor trametinib, and the PI3K inhibitor dactolisib. WB results showed that the inhibitors obviously blocked the phosphorylation of corresponding targets and their downstream molecules (Fig. 4B-D), resulting in the blockade of microstructure formation, which was similar to the results of gene-knockout cocultures (Fig. S12B vs. Fig. 3C-E). Interestingly, there are indeed some reports on the synergistic application of above inhibitors and other anticancer drugs for the treatment of HCC [46-49], which supported the original intention of our study, i.e., to find key TME-related targets for the combination targeted therapy. Collectively, we confirmed that the integrin-FAK-Src, MEK-ERK, and PI3K-AKT pathways were involved in microstructure formation.
Additionally, the effect of stretch forces on HepG2 cells was observed by IF-staining using a human-specific integrin-α5β1 antibody, which clearly illustrated morphological changes in certain HepG2 cells in coculture (from a typical round shape to a long, unidirectionally patterned shape) (Fig. S12C). The colocalization of fibronectin with integrin-α5/integrin-β1 was also verified (Fig. S12D and E). In Fig. 4E, we summarized the HIIs mechanism and signaling pathways during coculture.
HIIs-primed liver cancer cells displayed higher tumorigenic ability
To determine the situation in vivo, we established a xenograft tumor model in nude mice as shown in Fig. 5A. The xenograft volume of each mouse was monitored every 3 days until day 29. The xenografts (Fig. 5B-E) show that although MEFs have no tumorigenic ability, they helped HepG2 cells form larger tumors, with the tumor volume and tumor weight in coculture-ctrl group increasing by 6.0-fold and 5.2-fold, respectively, compared with monocultured HepG2 group. These data indicated that HepG2 cells primed with HIIs exhibited greater tumor formation and tumor growth ability, which were significantly inhibited in gene-knockout groups, with tumor volume and tumor weight in all three groups decreasing by more than 75% and 79%, respectively. Furthermore, fibronectin-knockout and integrin-α5-knockout had a better inhibitory effect than integrin-β1-knockout (Fig. 5B-E). A more significant difference was found using Huh7 and MEFs, in which monocultured Huh7 cell failed to grow into xenografts tumors, while cocultured Huh7 cells grew into tumors with a mean volume of 0.18 cm3 or a mean weight of 0.15 g (Fig. S13A-C).
HIIs-primed HepG2 cells exhibited greater tumor-formation ability than the controls. (A) Schematic of the procedure used to establish the HCC xenograft nude mouse model. Cells in each group were cultured for 4 days before subcutaneous injections. For each mouse, 2 × 106 cells in 100 µl were injected. (B) Representative bright-field images of xenografts in different groups on day 29. Scale bar, 1 cm. (C) Representative fluorescent images of xenografts in different groups on day 29. Scale bar, 5 mm. (D) Quantification of the xenograft tumor volume in different groups. The tumor volume data were obtained every 3 days from day 5 to day 29 (tumor volume was assigned to 0.1 cm3 on day 1). Two-way ANOVA and Tukey's multiple comparison test were performed. (E) Quantification of the xenograft tumor weight in different groups. The tumor weight data were obtained on day 29 after sacrifice and dissection. One-way ANOVA and Tukey's multiple comparisons test were performed. (F) Representative H&E-stained images showing the abundance of blood vessels inside the tumor xenografts in different groups. Scale bars, 100 µm. All data were obtained from 6 mice in each group and are represented as the mean ± SD. * P<0.05, *** P<0.001, **** P<0.0001.
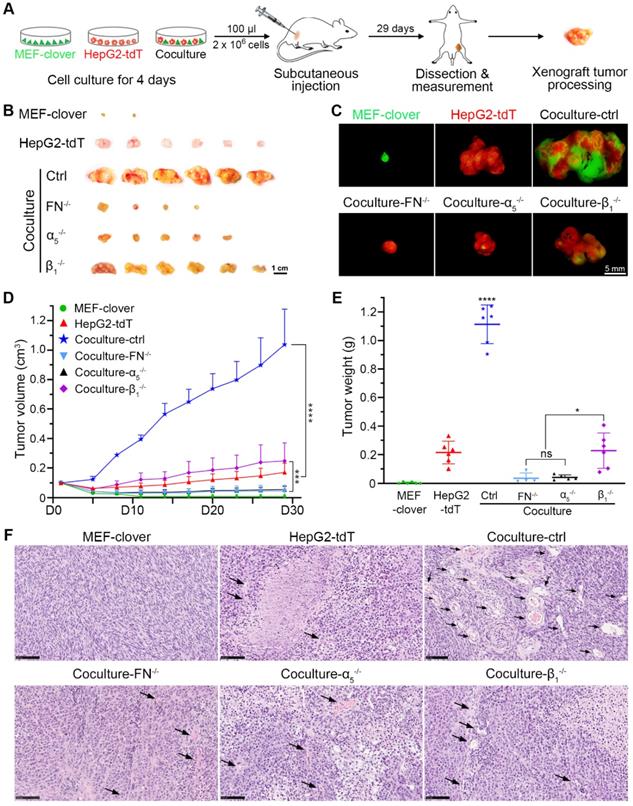
Based on above results, we further hypothesized that the vascular density in coculture-ctrl group was higher than that of the other groups. Next, hematoxylin and eosin (H&E) staining was performed using paraffin-embedded sections, and phase images were captured and used to compare vascular abundance between different groups. As shown in Fig. 5F, coculture-ctrl groups had 6-fold more vessel-like structures containing red blood cells per 20 × observation field than monocultured groups. Moreover, this parameter was greatly decreased in all three gene-knockout groups. Collectively, these results suggested the potential involvement of HIIs in pathological angiogenesis.
Fibronectin and collagen I remodeling induced by HIIs promoted HUVEC alignment and elongation
Based on above in vivo experiments, we introduced HUVECs into our coculture system to form a triculture system. To visualize HUVECs, we first labeled them with a near-infrared fluorescent protein (iRFP), iRFP-670 (i670), and installed a suitable filter to exclude bleed-through. We then tricultured three cell types and found that the 5:3:2 ratio produced the best outcome on day 10, as HUVECs had the best differentiation (Fig. S14A).
Chronological fluorescent images taken at the same spots showed that compared with monocultured HUVECs in Fig. 6B, HUVECs in triculture underwent dramatic morphological changes; not only was the cell body elongated, but long and thin branches extended (Fig. 6A). Interestingly, HUVECs aligned with 3D microstructures and colocalized with MEFs, as certain extended branches perfectly colocalized with elongated MEFs (Fig. S14B), suggesting the role of fibroblasts in HUVEC migration and differentiation. Additionally, 3D volume-rendering images obtained by two-photon microscopy are shown in Fig. 6C, which provides us with a more comprehensive view. Similar results were obtained in the triculture of Huh7 cells (Fig. S15A and B).
HUVECs aligned to and elongated along microstructures in triculture. (A) Representative images showing tricultured cells at the same spots from day 1 to day 10. Corresponding HUVEC-i670 morphology is listed below. Zoomed-in image in the right-upper corner on day 10 showing the small tube. Scale bar, 100 µm. (B) Representative images of monocultured HUVEC-i670 cells at the same spots from day 1 to day 10. Scale bar, 100 µm. (C) Representative two-photon images of tricultured cells on day 1 and day 10. Scale bar, 100 µm. (D and E) Representative zoomed-out and zoomed-in images of HUVEC-i670 in triculture on day 10 (D) or HUVEC-i670 after incubation on Matrigel for 6 hr (E). Scale bars, 200 µm. (F) Angiogenic parameters of HUVEC-i670 cells in triculture on day 10 (left) and tube formation assay (TFA) at 6 hr (right). Values were quantified over 5 × observation fields with AngioTool software. Unpaired t test was performed. (G and H) Representative images of the following triculture groups from day 1 to day 10: triculture-FN-/- (G) and triculture-α5-/- (H). Scale bar, 100 µm. (I) Representative images showing HUVEC-i670 morphology in different triculture groups (for triculture-β1-/- group, please refer to Fig. S9E). Scale bar, 100 µm. (J) Quantification of the average tube length of HUVECs in different triculture groups on day 10. Values were quantified over 10 × observation fields. One-way ANOVA was performed. All data are the mean ± SD from 3 independent experiments. ** P<0.01, *** P<0.001, **** P<0.0001, and ns, not significant.
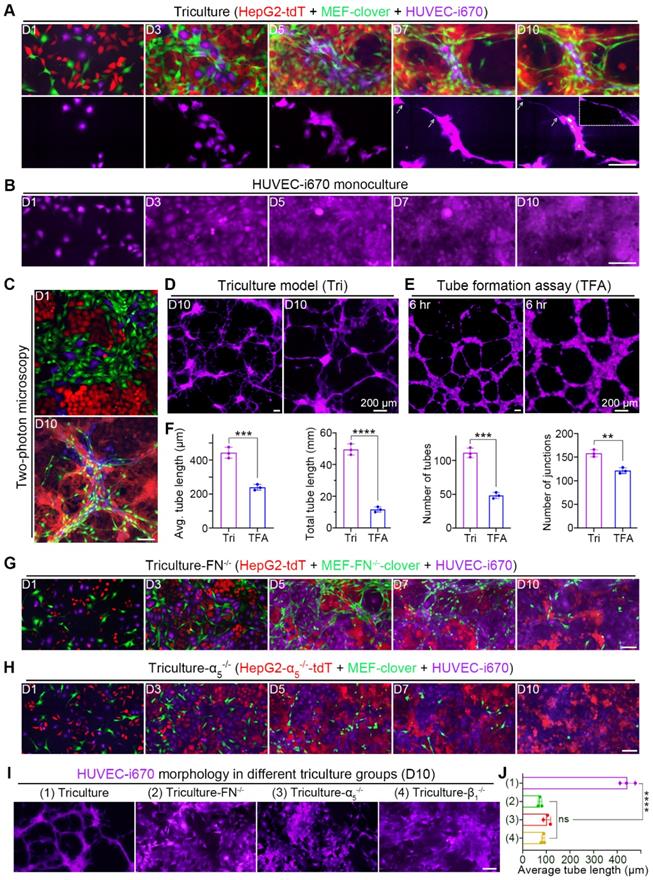
Since no obvious differentiation was observed in monocultured HUVECs (Fig. 6B), we compared tricultured HUVECs with HUVECs in a classical tube-formation assay (TFA) (Fig. 6D-E). After quantification using AngioTool software, features of angiogenesis (the length, number and junction of the tubes from HUVECs) were compared. From the results, we observed that HUVEC differentiation in triculture was better than that in TFA, with more and longer branches (Fig. 6F).
Furthermore, the coculture results in Fig. S14C and D suggested that HUVEC differentiation required HIIs between HepG2 cells and MEFs. To verify this assumption, we further performed triculture using gene-knockout cell types to interrupt HIIs. As expected, the patterned alignment and elongation of HUVECs were interrupted with the disappearance of microstructures in these gene-knockout triculture groups, leaving HUVECs to develop into colonies (Fig. 6G and H, Fig. S14E). To compare the morphology of HUVECs in these groups with that of HUVECs in normal triculture group, far-red channel images of all groups were collectively shown in Fig. 6I and then analyzed using AngioTool software. The average tube length of HUVECs in these groups was used to illustrate the effects of disrupting HIIs between HepG2 cells and MEFs on HUVEC differentiation (Fig. 6J). From these results, we observed that HUVEC elongation was significantly reduced after gene-knockout, with the average tube length decreasing from 442.3 μm to below 100 μm in all gene-knockout triculture groups. These results confirmed that the alignment and elongation of HUVECs required HIIs between liver cancer cells and MEFs.
HUVEC alignment and elongation in triculture were dependent on integrin-α5 and integrin-β1 and facilitated by ECM-bound VEGFA secreted by MEFs
Based on above results, we further explored how HIIs provide the “route” and guide HUVEC. A VEGFA concentration gradient was reported to be involved [50-52]. Next, we performed q-PCR using species-specific primers to quantify VEGFA mRNA changes during coculture, and found that the mRNA level was increased by 69.4% in cocultured MEFs compared with monocultured MEFs on day 10 (Fig. S16A). To confirm these results, we measured mouse VEGFA (mVEGFA) levels in conditioned medium (CM) by a mouse-specific ELISA kit. The levels of cumulative mVEGFA in the medium of coculture and triculture on day 3 were increased by more than 2-fold compared with those of monocultured MEFs (Fig. S16B). Given that ECM-bound VEGFA is reported more potent than free VEGFA [52-54], evidence of ECM-bound VEGFA would lay a solid basis for the above unknown questions of how HIIs provide the “route” and guide HUVECs. Additionally, as the primary VEGFA mediator, VEGFR2 activation could promote endothelial cell differentiation and proliferation [55-57].
Based on above results and analysis, we then performed IF-staining of related targets in triculture cells on day 10. From Fig. 7A, Fig. S16C, and Fig. S16D, we observed that HUVECs colocalized with fibronectin, collagen I, and matrix-bound mVEGFA during triculture. The obviously elevated p-VEGFR2 IF signals were supported by WB analysis showing an obvious upregulation of p-VEGFR2 level in tricultured HUVECs after day 5, with the level on day 10 comparable to that of monocultured HUVECs treated with 25 ng/ml VEGFA for 5 min (Fig. 7B). Furthermore, we applied clinically approved small-molecule drugs targeting VEGFR2 to triculture system. The administration of either sunitinib or apatinib at a concentration of 10 nM for 10 days almost completely abolished VEGFR2 phosphorylation (Fig. 7B) and greatly inhibited HUVEC differentiation, with the average tube length decreasing significantly from 442.3 μm to 65.7 μm or 92.3 μm, respectively (Fig. S16E and F). These results indicated the potential application of our triculture system in screening antiangiogenic agents for HCC treatment.
HUVEC migration to 3D multilayer microstructures enriched with ECM-bound VEGFA required integrin-α5β1. (A) Representative IF-staining images. (B) WB results showing the levels of p-VEGFR2 in monocultured HUVEC-i670 on day 10, the positive control (monocultured HUVEC-i670 with 25 ng/ml rhVEGFA treatment for 5 min), tricultured cells from day 1 to day 10, and the tricultured cells on day 10 administrated with sunitinib (10 nM) or apatinib (10 nM) for 10 days. Drugs were added to the medium beginning on day 2, and the medium was changed daily. (C) Representative IF-staining images. (D) WB analysis showed the levels of integrin-α5 and integrin-β1 in HUVEC-i670, HUVEC-α5-/--i670, and HUVEC-β1-/--i670 cells. (E) Representative fluorescence live-cell images at the same spots. The far-red channel in the right-lower corner shows HUVEC-α5-/--i670 morphology in triculture on day 10. (F) Representative fluorescent images of the following types of HUVECs in triculture with HepG2-tdT and MEF-clover cells on day 10: HUVECs-i670, HUVECs-α5-/--i670, and HUVECs-β1-/--i670 (please refer to Fig. S11C). (G) Quantification of the average tube length of the above types of tricultured HUVECs on day 10. Values were quantified over a 10 × observation field from 3 independent experiments. Data are mean ± SD. One-way ANOVA and Tukey's multiple comparisons test were performed. **** P<0.0001, and ns, not significant. Scale bars, 100 µm.
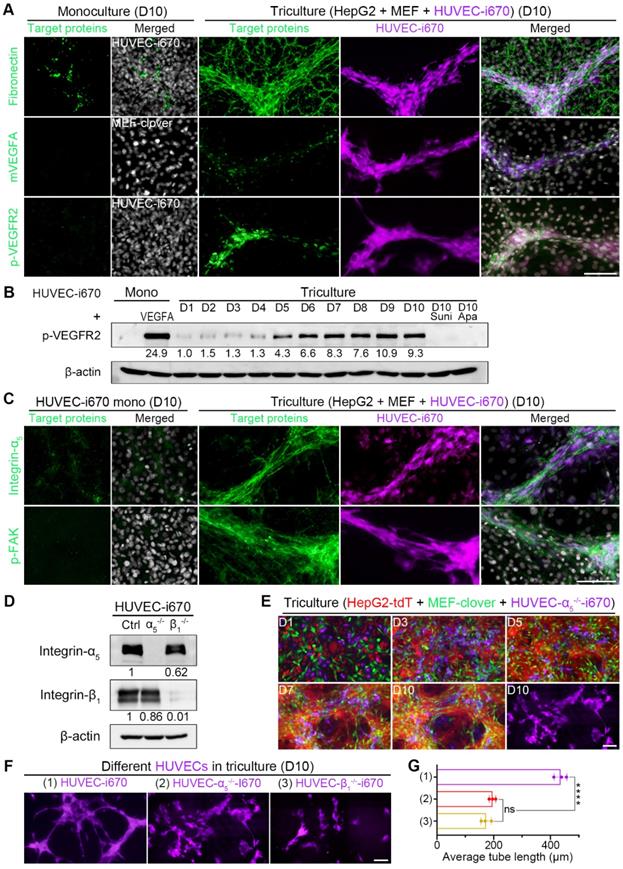
Since integrin-α5β1 can mediate cell attachment and migration on fibronectin [58-61], we next explored whether HUVEC migration and differentiation utilized integrin-α5β1. From IF-staining results, we found that the enhanced integrin-α5, integrin-β1, and p-FAK signals colocalized with HUVEC distribution in 3D microstructures (Fig. 7C, Fig. S16C and D), suggesting the involvement of integrin-α5 and integrin-β1 in HUVEC migration, which activated integrin-mediated downstream signaling. To verify this hypothesis, we generated integrin-α5-knockout HUVECs (HUVEC-α5-/-) or integrin-β1-knockout HUVECs (HUVEC-β1-/-) (Fig. 7D, Fig. S17A and B). The migration of these two cell types to 3D microstructures was interrupted to a certain extent when tricultured with HepG2-tdT and MEF-clover cells (Fig. 7E and Fig. S17C). More importantly, when we compared the far-red channel images on day 10 from these two groups with those of the normal triculture group (Fig. 7F), we observed that HUVEC differentiation was also affected. After analysis with AngioTool software, we found that the average tube length decreased by more than 55% in the two groups (Fig. 7G), which illustrated that HUVEC migration and elongation required integrin-α5 and integrin-β1 in HUVECs and further highlighted the role of 3D microstructures in HUVEC differentiation. Apart from the apparent reason that their migration was affected after gene-knockout, another reason might be that the cross-activation between VEGFR2 and integrins was also affected [62-64].
Clinical relevance of fibronectin, collagen I, integrin-α5, and integrin-β1 and their downstream signaling molecules in HCC
To assess the clinical significance of fibronectin, collagen I, integrin-α5, integrin-β1 and their downstream signaling molecules in HCC, we performed IHC-staining in 52 HCC tumors and paired adjacent nontumorous tissues. The main characteristics of these 52 patients with HCC were summarized in Table S1.
As shown in representative images and quantified results, fibronectin expression was significantly higher in HCC tumors than in adjacent tissues (Fig. 8A). To investigate the clinical significance of fibronectin in HCC, we classified these 52 patients into 2 groups: fibronectin high (IHC score≥3) and low (IHC score≤2), and analyzed the association between fibronectin expression status and certain clinicopathological characteristics. The results indicated that fibronectin high expression was closely correlated with microvascular invasion (MVI), tumor size, and TNM stages (Table 1). We also observed several MVI cases in our IHC result analysis (Fig. 8B), and more importantly, higher mRNA levels of FN1 genes were found corresponding to a shorter survival time in HCC cases with MVI in the KMplot dataset (Fig. 8C). However, no significant difference was found in collagen I (Fig. S17D). The results suggested that fibronectin might be a better marker to indicate MVI or HCC metastasis than collagen I.
Correlation analysis of fibronectin expression with clinicopathological characteristics
| Variables | IHC scores of fibronectin (n) | P value | |
|---|---|---|---|
| High (≥3) | Low (≤2) | ||
| Microvascular invasion | |||
| No | 5 | 28 | P<0.01 |
| Yes | 11 | 8 | |
| Tumor size | |||
| ≤5 cm | 3 | 23 | P<0.01 |
| >5 cm | 13 | 13 | |
| TNM stage | |||
| I | 10 | 23 | P<0.05 |
| II | 4 | 1 | |
| III | 2 | 12 | |
Pearson's chi squared test was used.
Clinical significance of fibronectin, integrin-α5, integrin-β1 and their downstream signaling molecules in HCC tumors. (A) Representative IHC-staining images of fibronectin expression in adjacent tissues and HCC tumors from HCC patients (left) and violin plots of IHC scores from these 52 patients (right). (B) Representative images of microvascular invasion (MVI) events found by IHC-staining for fibronectin with high (≥3) and low (≤2) IHC scores. (C) Correlation of FN1 mRNA levels with the survival rate of HCC patients with MVI events in the KMplot dataset. (D to G) Representative IHC-staining images of integrin-α5 expression (D), integrin-β1 expression (E), paxillin expression (F), and Src expression (G) in adjacent tissues and HCC tumors from HCC patients (left), and violin plots of IHC scores from these 52 patients (right). Scale bars, 20 µm. Data are shown as medians and quartiles. Unpaired t test was performed. HR, hazard ratio. ** P<0.01, *** P<0.001, **** P<0.0001, and ns, not significant. (H to K) Correlation of ITGA5 (H), ITGB1 (I), PXN (J), and SRC (K) mRNA levels with the overall survival rate of HCC patients in the KMplot dataset.
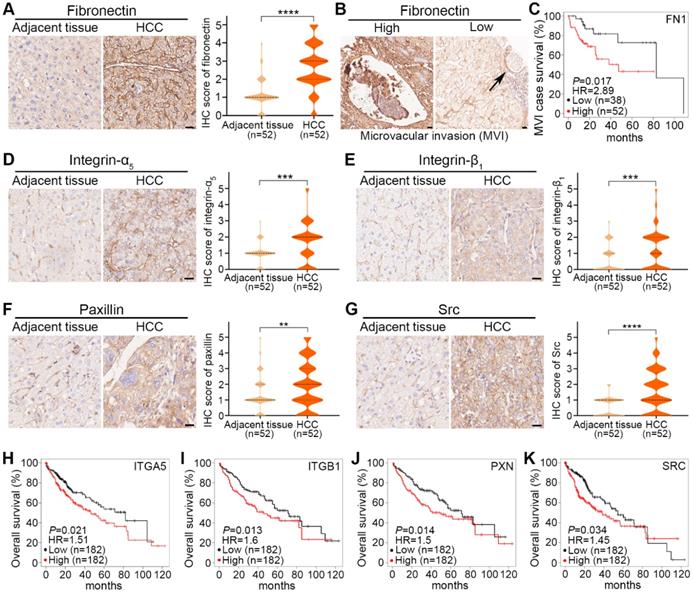
For integrin-α5, integrin-β1, paxillin, or Src, similar increased expression in HCC tumors compared with adjacent tissues is shown in representative images and quantified results (Fig. 8D-G). Additionally, a shorter survival time was correlated with higher mRNA levels of ITGA5, ITGB1, PXN, or SRC in the KMplot dataset obtained from HCC patients (Fig. 8H-K). No significant difference in FAK was found (Fig. S17E).
Discussion
The development of nonsurgical therapies for HCC is relatively slow compared with those for other cancers, largely due to the presence of cirrhosis or other chronic liver diseases, which provides a constant niche suitable for the maintenance of cancer cell stemness and the subsequent recurrence [8, 65]. Moreover, the localization of secondary tumors also seems to be orchestrated by the TME [11]. Hence, understanding the HIIs between liver cancer cells and major stromal cells in the TME, such as fibroblasts and endothelial cells, is of great importance for the development of combination targeted therapies.
The best way to study HIIs between cancer cells and stromal cells is through coculture. However, due to the difficulties in separating different cell types after coculture, many studies avoided direct coculture [66-69]. Here, we tried to solve this issue by coculturing liver cancer cells from humans with fibroblasts from mice, and then exploited the difference between them to detect the protein or mRNA level changes of targets in either cell type after coculture. Moreover, unlike previous studies, we extended coculture time to 10 days to fully examine HIIs and discovered microstructure formation. Additionally, we established a direct triculture system for HCC and discovered the effects of microstructures on angiogenesis induction. Furthermore, we found that in addition to serving as a receptor, integrin-α5β1 could mediate HIIs and remodel fibronectin and collagen I, which became the foundation for tumorous structure formation. Although the mechanism of ECM remodeling and the many roles of remodeled ECM have been well studied [70-73], the functions of the remodeled ECM in microstructure formation and angiogenesis have not been reported. Finally, we first reported that HUVECs could migrate and differentiate along microstructures containing ECM-bound VEGFA, which is dependent on integrin-α5β1 in HUVECs.
Nevertheless, there are also some shortcomings in this study. First, as mentioned above, it is very difficult to separate fibroblasts from liver cancer cells after coculture, and commercially available species-specific antibodies are few, which makes performing related experiments inconvenient. Second, it takes 10 days for our direct coculture and triculture systems to be established, which is rather time-consuming compared with others models, but we believe that our systems are closer to the natural process of tumor formation in animal models. Moreover, the generation of purified gene-knockout cell lines in this study was rather time-consuming.
Conclusion
In summary, this study explored HIIs in HCC and revealed that HIIs mediated by fibronectin and integrin-α5β1 could promote 3D growth of cancer cells by forming 3D microstructures. In this process, fibronectin and collagen I were remodeled by upregulated integrins in cancer cells, and VEGFA was enriched in remodeled ECM and induced endothelial cell migration and differentiation. Furthermore, the expression level of fibronectin, integrin-α5 and integrin-β1 were higher in 52 HCC tumors than in paired adjacent tissues. These findings lay a solid basis for the future development of combination targeted therapies for HCC.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We sincerely thank Professor Reinhard Fässler and Dr. Ralph Böttcher for providing us with the MEF and MEF-FN-/- cell lines. We thank Prof. Chuxia Deng for providing us with the Huh7 cell line. We thank Dr. Hao Jia, Renfei Wu, and Koukou Li from KQL's group for their suggestions and comments. We thank Dr. Kai Miao from Prof. Chuxia Deng' group and Dr. Zhiming Zhang from Prof. Tzu-Ming Liu' group for their help in lentiviral production and two-photon microscopy. We thank Prof. Wakam Chang and Sandy from Prof. Simon Ming Yuen Lee's group for providing integrin-β1 antibody and rhVEGFA.
Funding
This work was supported by the University of Macau Multi-year Research Grant (MYRG2017-00019-FHS).
Author contributions
KQL conceptualized the idea. ZP and KQL designed the research studies. ZP conducted the experiments, and MH, HT, HY, and BH helped with the experiments. KQL provided resource, and ZZ helped resource. ZP and KQL analyzed the data. ZP and KQL wrote the manuscript.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68:394-424
2. Bruix J, Sherman M. Management of hepatocellular carcinoma: An update. Hepatology. 2011;53:1020-2
3. Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nature Reviews Gastroenterology & Hepatology. 2010;7:448-58
4. Affo S, Yu L-X, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annual Review of Pathology: Mechanisms of Disease. 2017;12:153-86
5. Tahmasebi Birgani M, Carloni V. Tumor microenvironment, a paradigm in hepatocellular carcinoma progression and therapy. International Journal of Molecular Sciences. 2017;18:405
6. Massalha H, Bahar Halpern K, Abu-Gazala S, Jana T, Massasa EE, Moor AE. et al. A single cell atlas of the human liver tumor microenvironment. Molecular Systems Biology. 2020;16:e9682
7. Lacina L, Plzak J, Kodet O, Szabo P, Chovanec M, Dvorankova B. et al. Cancer microenvironment: What can we learn from the stem cell niche. International Journal of Molecular Sciences. 2015;16:24094-110
8. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nature Reviews Cancer. 2006;6:674-87
9. Zhang J, Liu J. Tumor stroma as targets for cancer therapy. Pharmacology & Therapeutics. 2013;137:200-15
10. Pietras K, Östman A. Hallmarks of cancer: interactions with the tumor stroma. Experimental Cell Research. 2010;316:1324-31
11. Borovski T, De Sousa E Melo F, Vermeulen L, Medema JP. Cancer Stem Cell Niche: The Place to Be. Cancer Research. 2011;71:634-9
12. Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. Journal of Cell Biology. 2012;196:395-406
13. Schwager SC, Taufalele PV, Reinhart-King CA. Cell-Cell Mechanical Communication in Cancer. Cellular and Molecular Bioengineering. 2019;12:1-14
14. Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biology. 2005;24:389-99
15. Saunders JT, Schwarzbauer JE. Fibronectin matrix as a scaffold for procollagen proteinase binding and collagen processing. Molecular Biology of the Cell. 2019;30:2218-26
16. Ingham KC, Brew SA, Erickson HP. Localization of a cryptic binding site for tenascin on fibronectin. Journal of Biological Chemistry. 2004;279:28132-5
17. Mezzenga R, Mitsi M. The Molecular Dance of Fibronectin: Conformational Flexibility Leads to Functional Versatility. Biomacromolecules. 2019;20:55-72
18. Schwarzbauer JE, DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harbor Perspectives in Biology. 2011;3:a005041
19. Zollinger AJ, Smith ML. Fibronectin, the extracellular glue. Matrix Biology. 2017;60-61:27-37
20. Zhu J, Clark RA. Fibronectin at select sites binds multiple growth factors and enhances their activity: expansion of the collaborative ECM-GF paradigm. Journal of Investigative Dermatology. 2014;134:895-901
21. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature Reviews Cancer. 2010;10:9
22. Pankov R, Cukierman E, Katz BZ, Matsumoto K, Lin DC, Lin S. et al. Integrin dynamics and matrix assembly: tensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis. Journal of Cell Biology. 2000;148:1075-90
23. Clark K, Pankov R, Travis MA, Askari JA, Mould AP, Craig SE. et al. A specific α5β1-integrin conformation promotes directional integrin translocation and fibronectin matrix formation. Journal of Cell Science. 2005;118:291-300
24. Kubow KE, Vukmirovic R, Zhe L, Klotzsch E, Smith ML, Gourdon D. et al. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nature Communications. 2015;6:8026
25. Smith ML, Gourdon D, Little WC, Kubow KE, Eguiluz RA, Luna-Morris S. et al. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biology. 2007;5:e268
26. Ohashi T, Erickson HP. Revisiting the mystery of fibronectin multimers: the fibronectin matrix is composed of fibronectin dimers cross-linked by non-covalent bonds. Matrix Biology. 2009;28:170-5
27. Bharadwaj M, Strohmeyer N, Colo GP, Helenius J, Beerenwinkel N, Schiller HB. et al. αV-class integrins exert dual roles on α5β1 integrins to strengthen adhesion to fibronectin. Nature Communications. 2017;8:14348
28. Strohmeyer N, Bharadwaj M, Costell M, Fässler R, Müller DJ. Fibronectin-bound α5β1 integrins sense load and signal to reinforce adhesion in less than a second. Nature Materials. 2017;16:1262
29. Tomasini-Johansson BR, Annis DS, Mosher DF. The N-terminal 70-kDa fragment of fibronectin binds to cell surface fibronectin assembly sites in the absence of intact fibronectin. Matrix Biology. 2006;25:282-93
30. Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nature Reviews Clinical Oncology. 2011;8:292-301
31. Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z. et al. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circulation Research. 2006;99:853-60
32. Mitsi M, Hong Z, Costello CE, Nugent MA. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry. 2006;45:10319-28
33. Sewell-Loftin MK, Bayer SVH, Crist E, Hughes T, Joison SM, Longmore GD. et al. Cancer-associated fibroblasts support vascular growth through mechanical force. Scientific Reports. 2017;7:1-12
34. Ambesi A, McKeown-Longo PJ. Conformational remodeling of the fibronectin matrix selectively regulates VEGF signaling. Journal of Cell Science. 2014;127:3805-16
35. Usuelli M, Meyer T, Mezzenga R, Mitsi M. VEGF and VEGFR2 bind to similar pH-sensitive sites on fibronectin, exposed by heparin-mediated conformational changes. Journal of Biological Chemistry. 2021 296
36. Buczek-Thomas JA, Rich CB, Nugent MA. Hypoxia Induced Heparan Sulfate Primes the Extracellular Matrix for Endothelial Cell Recruitment by Facilitating VEGF-Fibronectin Interactions. International Journal of Molecular Sciences. 2019;20:5065
37. Wang K, Andresen Eguiluz RC, Wu F, Seo BR, Fischbach C, Gourdon D. Stiffening and unfolding of early deposited-fibronectin increase proangiogenic factor secretion by breast cancer-associated stromal cells. Biomaterials. 2015;54:63-71
38. Li K, Wu R, Zhou M, Tong H, Luo KQ. Desmosomal proteins of DSC2 and PKP1 promote cancer cells survival and metastasis by increasing cluster formation in circulatory system. Science Advances. 2021;7:eabg7265
39. Chiew GGY, Fu A, Low KP, Luo KQ. Physical supports from liver cancer cells are essential for differentiation and remodeling of endothelial cells in a HepG2-HUVEC co-culture model. Scientific Reports. 2015;5:10801
40. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673-87
41. Jeanes AI, Wang P, Moreno-Layseca P, Paul N, Cheung J, Tsang R. et al. Specific β-containing integrins exert differential control on proliferation and two-dimensional collective cell migration in mammary epithelial cells. Journal of Biological Chemistry. 2012;287:24103-12
42. Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Molecular Biology of the Cell. 2013;24:3449-59
43. Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Devedec SE. et al. β1 integrin inhibition elicits a prometastatic switch through the tgfβ-mir-200-zeb network in e-cadherin-positive triple-negative breast cancer. Science Signaling. 2014;7:ra15-ra
44. Notni J, Steiger K, Hoffmann F, Reich D, Kapp TG, Rechenmacher F. et al. Complementary, Selective PET Imaging of Integrin Subtypes α5β1 and αvβ3 Using 68Ga-Aquibeprin and 68Ga-Avebetrin. Journal of Nuclear Medicine. 2016;57:460-6
45. Bui T, Rennhack J, Mok S, Ling C, Perez M, Roccamo J. et al. Functional Redundancy between β1 and β3 Integrin in Activating the IR/Akt/mTORC1 Signaling Axis to Promote ErbB2-Driven Breast Cancer. Cell Reports. 2019;29:589-602.e6
46. Cao W, Liu X, Zhang Y, Li A, Xie Y, Zhou S. et al. BEZ235 Increases the Sensitivity of Hepatocellular Carcinoma to Sorafenib by Inhibiting PI3K/AKT/mTOR and Inducing Autophagy. BioMed Research International. 2021. 2021
47. Yu C-C, Huang S-Y, Chang S-F, Liao K-F, Chiu S-C. The Synergistic Anti-Cancer Effects of NVP-BEZ235 and Regorafenib in Hepatocellular Carcinoma. Molecules. 2020;25:2454
48. Kim R, Tan E, Wang E, Mahipal A, Chen DT, Cao B. et al. A phase I trial of trametinib in combination with sorafenib in patients with advanced hepatocellular cancer. The Oncologist. 2020;25:e1893
49. Thomas HE, Mercer CA, Carnevalli LS, Park J, Andersen JB, Conner EA. et al. mTOR Inhibitors Synergize on Regression, Reversal of Gene Expression, and Autophagy in Hepatocellular Carcinoma. Science Translational Medicine. 2012;4:139ra84-ra84
50. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nature Reviews Cancer. 2002;2:795-803
51. Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. Journal of Cell Communication and Signaling. 2016;10:347-54
52. Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. Journal of Cell Biology. 2010;188:595-609
53. Sawicka KM, Seeliger M, Musaev T, Macri LK, Clark RA. Fibronectin interaction and enhancement of growth factors: importance for wound healing. Advances in Wound Care. 2015;4:469-78
54. Sack KD, Teran M, Nugent MA. Extracellular matrix stiffness controls VEGF signaling and processing in endothelial cells. Journal of Cellular Physiology. 2016;231:2026-39
55. Morse MA, Sun W, Kim R, He AR, Abada PB, Mynderse M. et al. The Role of Angiogenesis in Hepatocellular Carcinoma. Clinical Cancer Research. 2019;25:912-20
56. Zhang Z, Neiva KG, Lingen MW, Ellis LM, Nör JE. VEGF-dependent tumor angiogenesis requires inverse and reciprocal regulation of VEGFR1 and VEGFR2. Cell Death & Differentiation. 2010;17:499-512
57. Ou J, Yu Z, Qiu M, Dai Y, Dong Q, Shen J. et al. Knockdown of VEGFR2 inhibits proliferation and induces apoptosis in hemangioma-derived endothelial cells. European Journal of Histochemistry: EJH. 2014 58
58. Huck L, Pontier S, Zuo D, Muller W. β1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proceedings of the National Academy of Sciences. 2010;107:15559-64
59. Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration - the actin connection. Journal of Cell Science. 2009;122:199-206
60. Michaelis UR. Mechanisms of endothelial cell migration. Cellular and Molecular Life Sciences. 2014;71:4131-48
61. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circulation Research. 2007;100:782-94
62. Somanath PR, Ciocea A, Byzova TV. Integrin and Growth Factor Receptor Alliance in Angiogenesis. Cell Biochemistry and Biophysics. 2009;53:53-64
63. Mahabeleshwar GH, Feng W, Reddy K, Plow EF, Byzova TV. Mechanisms of integrin-vascular endothelial growth factor receptor cross-activation in angiogenesis. Circulation Research. 2007;101:570-80
64. De S, Razorenova O, McCabe NP, O'Toole T, Qin J, Byzova TV. VEGF-integrin interplay controls tumor growth and vascularization. Proceedings of the National Academy of Sciences. 2005;102:7589-94
65. Zhong C, Tao B, Tang F, Yang X, Peng T, You J. et al. Remodeling cancer stemness by collagen/fibronectin via the AKT and CDC42 signaling pathway crosstalk in glioma. Theranostics. 2021;11:1991
66. Medeiros M, Ribeiro AO, Lupi LA, Romualdo GR, Pinhal D, de Almeida Chuffa LG. et al. Mimicking the tumor microenvironment: Fibroblasts reduce miR-29b expression and increase the motility of ovarian cancer cells in a co-culture model. Biochemical and Biophysical Research Communications. 2019;516:96-101
67. Dean T, Li NT, Cadavid JL, Ailles L, McGuigan AP. A TRACER culture invasion assay to probe the impact of cancer associated fibroblasts on head and neck squamous cell carcinoma cell invasiveness. Biomaterials Science. 2020;8:3078-94
68. Liu T, Lin B, Qin J. Carcinoma-associated fibroblasts promoted tumor spheroid invasion on a microfluidic 3D co-culture device. Lab on a Chip. 2010;10:1671-7
69. Koh B, Jeon H, Kim D, Kang D, Kim KR. Effect of fibroblast co-culture on the proliferation, viability and drug response of colon cancer cells. Oncology Letters. 2019;17:2409-17
70. Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Current Opinion in Cell Biology. 2006;18:463-71
71. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139:891-906
72. Mohan V, Das A, Sagi I. Emerging roles of ECM remodeling processes in cancer. Seminars in Cancer Biology: Elsevier. 2020 p. 192-200
73. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L. et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. Journal of Cell Biology. 2017;216:3799-816
Author contact
![]() Corresponding author: Kathy Qian Luo, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR, China. Tel: 853-8822-4233; Fax: 853-8822-2314; Email address: kluoedu.mo.
Corresponding author: Kathy Qian Luo, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR, China. Tel: 853-8822-4233; Fax: 853-8822-2314; Email address: kluoedu.mo.