Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The IL-6/IL-6R system and signal...
The role of IL-6 in wound healing
IL-6-induced cellular processes...
The role of IL-6 in fibrosis in...
Conclusions and the future
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(14):5405-5414. doi:10.7150/ijbs.75876 This issue Cite
Review
The Role of IL-6 in Fibrotic Diseases: Molecular and Cellular Mechanisms
Yanxia Li, Jing Zhao, Yuan Yin, Ke Li, Chenchen Zhang, Yajuan Zheng ![]()
Department of Ophthalmology, The Second Hospital of Jilin University, Jilin University, Changchun, China
Received 2022-6-6; Accepted 2022-8-23; Published 2022-8-29
Abstract

Fibrosis is a detrimental outcome of most chronic inflammatory disorders and is defined by the buildup of excess extracellular matrix (ECM) components, which eventually leads to organ failure and death. Interleukin 6 (IL-6) is promptly produced by immune cells in response to tissue injuries and has a wide range of effects on cellular processes such as acute responses, hematopoiesis, and immune reactions. Furthermore, high levels of IL-6 have been found in a variety of chronic inflammatory disorders characterized by fibrosis, and this factor plays a significant role in fibrosis in various organs via Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) activation. Here, we review what is known about the role of IL-6 in fibrosis and why targeting IL-6 for fibrotic disease treatment makes sense.
Keywords: Interleukin 6, JAK, STAT3, fibrosis
Introduction
Fibrosis or scarring, which is defined as the excessive accumulation of fibrous connective tissue, is a common pathological condition resulting from a dysregulated tissue repair response, most notably during chronic inflammatory disorders [1]. Myofibroblasts, the most important cell type initiating the wound healing response when tissues are injured, are characterized by the expression of alpha-smooth muscle actin (α-SMA); these cells have high contractility, secrete inflammatory factors, and deposit extracellular matrix (ECM) components, such as fibronectin and collagen [2]. Fibrosis affects nearly all tissues and organs in the body. Although fibrogenesis is a major cause of adverse prognosis in most tissue repair processes or chronic inflammatory disorders, few therapeutic options are available to prevent or reverse this process [3]. As a result, more efficient antifibrotic therapies to reduce mortality and morbidity and developing a greater understanding of the molecular mechanisms related to fibrosis deserve further attention.
Interleukin-6 (IL-6) is a multifunctional cytokine that is involved in a variety of biological processes, including inflammation, immunological response, and hematopoiesis [4]. Most importantly, IL-6 is crucial for the inflammatory phase and the switch to a reparative environment during the resolution of wound healing [5]. However, the consequences of repair will eventually lead to fibrosis if the switch to the proliferative phase is uncontrolled. IL-6 is a key regulator of inflammation and repair, and its role in scar formation has been demonstrated in previous studies.
In this review, we summarized recent research investigating the role of IL-6 in fibrosis and discuss how to block IL-6 signaling to reduce fibrosis.
The IL-6/IL-6R system and signal transduction
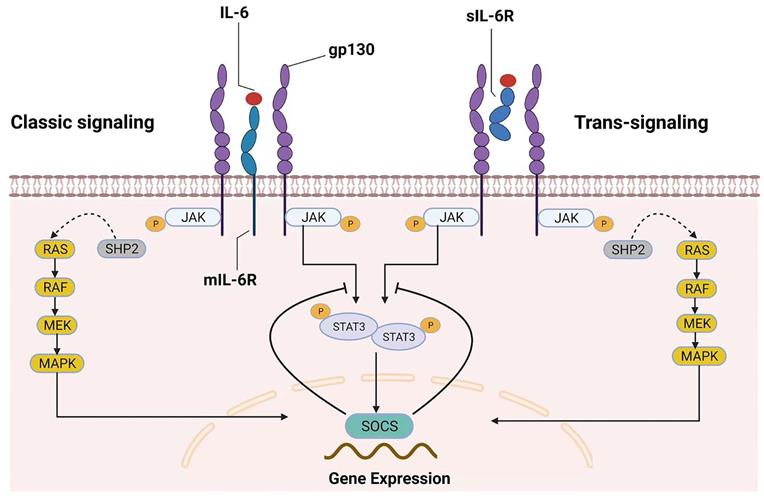
Because some excellent reviews on this topic have examined IL-6 signaling in great detail, the general mechanisms of IL-6 signaling are only briefly introduced here. Many cell types, including monocytes, macrophages, fibroblasts, keratinocytes, astrocytes, and endothelial cells, can secrete IL-6, which is a four-helical cytokine of 184 amino acids [6]. The IL-6 receptor (IL-6R) and glycoprotein 130 (gp130) are responsible for the biological effects of IL-6[7]. Although gp130 is expressed universally, IL-6R is expressed primarily by hepatocytes, leukocytes, and megakaryocytes [6]. There are two types of IL-6R, membrane-bound IL-6R (mIL-6R) and soluble IL-6R (sIL-6R), which mediate the classic signaling pathway and trans-signaling pathway, respectively [7-9]. IL-6 binding to mIL-6R induces gp130 homodimerization, which leads to the formation of a high-affinity functional receptor complex of IL-6-mIL-6R-gp130 in the classic signaling pathway. sIL-6R can also bind to IL-6, and the sIL-6R-IL-6-gp130 complex can result in intracellular signaling similar to that of mIL-6R. Importantly, the presence of sIL-6R can stimulate downstream signals in cells that do not express mIL-6R; this unique receptor signaling mechanism has been called the IL-6 trans-signaling pathway [10].
Generally, the pathway that involves tyrosine kinases in the Janus kinase (JAK) family and transcription factors in the signal transducer and activator of transcription (STAT) family (the JAK/STAT pathway) and the mitogen-activated protein kinase (MAPK) signaling cascades are activated once the IL-6 receptor complex is engaged [11]. In the JAK/STAT pathway, STAT proteins are recruited and phosphorylated by JAKs in response to IL-6 stimulation. Subsequently, activated STAT proteins form dimers and translocate to the nucleus to trigger the transcription of target genes [7], such as those encoding regulators of cellular proliferation (such as cyclin D1, cyclin B1 and c-myc) and survival (such as survivin, BCL-2 and BCL-xL), as well as angiogenic factors (such as VEGF and HIF1α) and cytokines (such as IL-10, IL-11 and IL-17) [10, 12]. Interestingly, suppressor of cytokine signaling (SOCS), one of the target genes of the JAK/STAT pathway, can inhibit JAK activity and thus negatively regulate this pathway, suggesting that this signaling pathway has an autoregulatory mechanism [7, 13]. On the other hand, SHP-2 is recruited to the phosphorylated Tyr759 residue of gp130 and is then phosphorylated by JAKs [7, 11, 14]. Subsequently, SHP-2 interacts with the growth-factor-receptor-bound protein 2/Son of Sevenless (Grb2-SOS) complex [7, 11, 15], which is a GDT/GTP exchanger for Ras. Finally, the Raf-Ras-MAPK cascade is activated after IL-6 stimulation. IL-6 binding to mIL-6R/sIL-6R activates the downstream signaling pathway, as shown in Figure 1.
Two different modes of IL‑6 signaling. IL-6 can bind to both mIL‑6R (classic signaling) and sIL-6R (trans-signaling). After homodimerization of the signal-transducing receptor subunit gp130 on the plasma membrane, the two signaling pathways converge, triggering the intracellular JAK/STAT and MAPK signaling cascades. SOCS is the product of JAK/STAT3 signaling pathway activation and is also a protein associated with the negative-feedback reaction that leads to the inhibition of this signaling pathway.
The role of IL-6 in wound healing
An overly active wound healing response frequently results in tissue fibrosis, and wound repair has been divided into three primary stages [16]. First, the innate immune system is activated and inflammatory cytokines are released in the early inflammatory phase; the proliferative phase is initiated by an influx of fibroblasts and the transdifferentiation of fibroblasts to myofibroblasts, resulting in wound contraction and ECM deposition; with wound closure, type III collagen degrades and type I collagen synthesis increases, the tissue gains strength and flexibility, and the remodeling phase begins and can last for months to years [5, 16]. To engage in wound repair, IL-6 is expressed during two periods in wound sites. It is rapidly upregulated following injury, and robust levels peak at approximately 12 hours, but by 24 hours, IL-6 levels return to near baseline levels [17, 18]. After approximately 48 hours, the second phase of IL-6 expression in the wound commences, and cytokine levels progressively reach a stable state at 3-7 days postinjury, after which the concentration returns to normal levels in the remodeling phase [17, 18].
Inflammation can be collectively activated by the clotting response, necrotic debris, and invading bacteria after injury. Neutrophils, monocytes, and other innate immune cells initially accumulate at the wound site to clear cell debris and infectious organisms and secrete proinflammatory cytokines and growth factors to aid in the tissue healing response. The wound inflammatory response, on the other hand, can become dysregulated or chronic, leading to pathological fibrosis or scarring, which can disrupt normal tissue architecture and function [19]. As a significant modulator of inflammatory and reparative processes, IL-6 plays a critical role in wound healing. Compared to wild-type mice, transgenic IL-6-deficient mice exhibited impaired wound healing as well as a reduction in leukocyte infiltration, angiogenesis, re-epithelialization, and collagen accumulation at damage sites [20, 21]. Furthermore, in wild-type mice, treatment with a neutralizing anti-IL-6 monoclonal antibody significantly delayed wound closure [21]. Specifically, IL-6 is expressed in neutrophils, macrophages and fibroblasts [22] and participates in wound healing by regulating the activity of these cells. sIL-6R can be shed from the surface of neutrophils into the wound [23]. IL-6/sIL-6R complexes further trigger the recruitment of macrophages into wound tissues by increasing the expression of certain chemokines, such as monocyte chemoattractant protein 1 (MCP-1) [24, 25]. IL-6 then promotes the polarization of proinflammatory M1 macrophages to reparative M2 macrophages, as well as their proliferation [6, 26]. Importantly, a switch in macrophages from the M1 to the M2 phenotype is a significant step in the transition of wound healing from the inflammatory phase to the proliferative phase. Furthermore, IL-6 can drive fibroblast migration to injury sites and regulate fibroblast differentiation to myofibroblasts through the JAK/ERK pathway or via paracrine production of transforming growth factor β (TGF-β) at wound sites [5].
IL-6-induced cellular processes associated with fibrosis
The roles of various cellular mechanisms in the pathogenesis of fibrosis, as well as their relationship with IL-6, are discussed below.
The process by which fibroblasts transform into myofibroblasts is known as fibroblast-to-mesenchymal transition (FMT), which is characterized by the expression of α-SMA and excessive deposition of ECM. Accumulating evidence suggests that FMT could be induced by IL-6 in fibroblasts derived from different tissues. In cultured cardiac fibroblasts exposed to exogenous recombinant hypoxia-induced mitogenic factor, the increased production of IL-6 induced cell proliferation, migration, and myofibroblast differentiation through the MAPK and Ca2+/calmodulin-dependent protein kinase II (CaMKII)-STAT3 pathways [27]. Hepatic stellate cells (HSCs) are mesenchymal cells that retain features of fibroblasts, which are the major cellular source of myofibroblasts and the major driver of liver fibrosis [28]. HSCs treated with IL-6 upregulate the expression of α-SMA and collagen and induce the phenotypic transition of quiescent HSCs toward myofibroblast-like cells [29]. Wheeler [30] also suggested that IL-6 trans-signaling might be involved in regulating fibrosis genes in lung mesenchymal cells, causing increased invasion and fibrotic differentiation. These findings indicate that IL-6 might drive fibrosis in various human fibrotic diseases.
Epithelial-mesenchymal transition (EMT) is another biological process induced by IL-6 in fibrotic diseases, in which epithelial cells lose their epithelial phenotypes and gain mesenchymal phenotypes with increased migration and invasion capacities [31]. EMT plays a crucial role in embryonic development and wound healing and contributes pathologically to fibrosis and cancer progression. Exogenous addition of IL-6 to ovarian cancer cells activated STAT3 and mesenchymal cell marker expression and enhanced cell motility [32]. Furthermore, neutralizing IL-6 signaling was sufficient to reverse the EMT characteristics of human proximal tubular epithelial cells induced by PM2.5 [33]. In peritoneal fibrosis, IL-6 also stimulates EMT [34].
Apoptosis is a type of programmed cell death that maintains the homeostasis of many adult tissues by regulating cell numbers. Generally, myofibroblasts are cleared from wound sites through apoptosis as repair is completed under normal conditions. Thus, myofibroblast apoptosis is a critical process in the resolution of fibrosis. However, myofibroblasts from pathological fibrotic tissues are usually resistant to apoptosis, leading to persistent accumulation of ECM and contraction of the tissue. IL-6, which is a growth or survival factor, has been shown to play a critical role in apoptosis resistance by inducing antiapoptotic proteins, such as survivin, Bcl-xL and Mcl-1 [35]. In non-small cell lung cancer (NSCLC), the addition of IL-6 to NSCLC cells could improve their resistance to apoptosis [35]. IL-6 can also have different effects on apoptosis in different types of cells, tissues and organs. Moodley et al. [36] demonstrated that in fibroblasts without lung disease, IL-6 could enhance FasL-induced apoptosis via STAT3. Treatment of idiopathic pulmonary fibrosis (IPF) fibroblasts with IL-6, on the other hand, conferred resistance to FasL-induced apoptosis. It was thought that the contrasting effects of IL-6 were linked to differential activation of STAT3 and ERK.
Autophagy is a process that maintains cell survival by delivering dysfunctional cellular cytoplasmic components to lysosomes for degradation [37]. Recently, evidence has indicated that autophagy dysregulation is related to diverse types of pathologic conditions, including fibrotic processes. Autophagy defects in human microvascular endothelial cells (HMVECs) induce EMT [38]. Furthermore, the IL-6 level was significantly higher due to autophagy dysregulation. In vitro experiments also showed that endothelial-specific autophagy related 5 (atg5)-knockout mice developed kidney and heart fibrosis with an increase in serum IL-6 levels [38]. In Takayasu's arteritis, IL-6 significantly promotes autophagy and fibrosis via the JAK1 pathway [39].
The role of IL-6 in fibrosis in various tissues
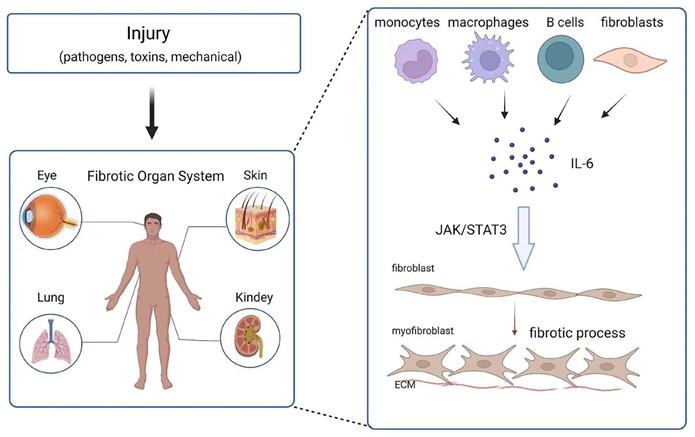
Fibrosis can affect any system and organ in the body. The role of IL-6 in fibrosis in different tissues and organs has been reported (Figure 2). In the following section, the contribution of IL-6 to the development of fibrosis in various tissues is detailed.
Effect and mechanism of IL-6 on organ fibrosis. Chronic inflammation is caused by injury (pathogens, toxins, mechanical injury), and monocytes, macrophages, B cells and fibroblasts release IL-6 at the wound site. IL-6 promotes the transformation of fibroblasts into myofibroblasts through the JAK/STAT3 signaling pathway.
IL-6 and lung fibrosis
Pulmonary fibrosis occurs in a wide range of clinical contexts, is a leading cause of morbidity and mortality, and is a major unmet medical need [40]. The morphological characteristics of pulmonary fibrosis are diffuse inflammatory infiltration and alveolar septal thickening with collagen accumulation and myofibroblast proliferation [41]. IPF is a chronic and progressive interstitial pneumonia of unknown cause [42] and is a representative disease of pulmonary fibrosis. IL-6 was shown to be upregulated in IPF patients [43] and animal models of pulmonary fibrosis [44]; furthermore, JAK2 and STAT3 were increased and activated in the hyperplastic alveolar cells of IPF patients [42], which suggested that IL-6/JAK/STAT3 plays a role in lung fibrosis. In animal studies, Saito et al. [41] found that IL-6-deficient mice had relatively attenuated fibrosis following bleomycin administration, which is a well-characterized animal model of pulmonary fibrosis, in comparison to the wild-type controls. Consistently, IL-6 overexpression could increase the fibrotic response to bleomycin, such as collagen production, lung elasticity and fibrotic scores [45]. In terms of cellular mechanisms, alveolar epithelial cell damage is a key stage in the pathogenesis of lung fibrosis, as it can lead to dysregulated activation and establish a profibrotic microenvironment [46, 47]. Type II alveolar epithelial cell injury can be reduced by inhibiting the STAT3 pathway [47], indicating that IL-6 may participate in pulmonary fibrosis through alveolar epithelial cells. Lung fibroblasts are another important cell type involved in the pathogenesis of pulmonary fibrosis. Pechkovsky showed that collagen I secretion in IPF lung fibroblasts was regulated by STAT3 and enhanced collagen I expression, which might be responsible for their fibrogenic phenotype [48]. These results suggest that IL-6/JAK/STAT3 may be a potential therapeutic target for treating pulmonary fibrosis.
The global outbreak of coronavirus disease 2019 (COVID-19) has seriously endangered healthcare systems worldwide. Fibrotic manifestations resulting from lung injury [49, 50] are commonly associated with severe injury, and some of the molecular markers, such as TGF-β and IL-6, are increased in the peripheral blood of severe COVID-19 patients [51]. These findings suggest that pulmonary fibrosis plays a role in the progression of COVID-19. Indeed, it is possible that COVID-19-induced lung injury in patients with severe disease might trigger an abnormal wound healing response that leads to the deposition of fibrotic tissue and eventually develops into pulmonary fibrosis [52]. Several studies suggest that an elevated level of IL-6 correlates with a severe response to COVID-19 [53, 54]; that is, IL-6 may be a predictor for COVID-19 patients who develop pulmonary fibrosis. In fact, tocilizumab, a humanized anti-IL-6R antibody that binds to both mIL-6R and sIL-6R for total IL-6 signal transduction suppression, has been used to treat critically ill patients with COVID-19 [55]. Pulmonary computed tomography imaging in 23 patients with severe COVID-19 who were treated with tocilizumab showed improvements without adverse events according to a retrospective study from China [56]. Another observational study reported that patients with severe COVID-19 who received an intravenous tocilizumab dose of 8 mg/kg (maximum 800 mg) had survival rates comparable to those in the nonsevere group [57]. These findings point to the therapeutic potential of tocilizumab in preventing long-term fibrotic repercussions by reducing lung damage, which warrants further investigation.
IL-6 and renal fibrosis
Because of the high morbidity and mortality associated with chronic kidney disease (CKD), CKD is a serious public health concern [58]. Renal fibrosis, which represents the common final pathway of CKD, is characterized by fibroblast activation and ECM deposition and results in renal parenchyma injuries and renal function loss [59]. Recently, a large body of experimental evidence suggested that IL-6 plays a role in renal fibrosis, although this finding remains controversial. IL-6 was significantly increased in an animal model of unilateral ureteral obstruction (UUO)-induced kidney fibrosis; nevertheless, IL-6-knockout mice exhibited comparable expression levels of ECM proteins in the kidneys following obstructive damage compared to wild-type animals [60]. Contrary to these results, IL-6-deficient mice had significantly attenuated angiotensin II-induced collagen staining and total collagen levels [61]. In another study, Chen et al. [59] used Fc-gp130, which binds to sIL-6R, to block IL-6 trans-signaling in the UUO mouse model of renal fibrosis and found significantly reduced renal fibrosis and anti-inflammatory effects, as evidenced by decreased ECM protein synthesis and immune cell infiltration. A plausible explanation for the contradictory findings is that the classic signaling pathway is anti-inflammatory and the trans-signaling pathway is proinflammatory, and the effects of the two pathways may counteract each other in IL-6-knockout mice following obstructive injury; although renal fibrosis might be reduced due to the absence of proinflammatory trans-signaling, injured renal tissue also lacks the anti-inflammatory effects of classic IL-6 signaling, and fibrosis can probably be exacerbated by an increase in other inflammatory processes [59].
In end-stage renal disease, peritoneal dialysis, which is a common therapeutic method, has drawn increasing attention [62]. However, long-term use of the patient's peritoneal membrane as a dialyzer filter is unphysiological and results in chronic inflammation, which eventually develops into peritoneal fibrosis [63]. In patients with peritoneal dialysis, it was reported that elevated circulating levels of IL-6 indicated a poor outcome [64]. In addition, IL-6 has been shown to promote EMT in human peritoneal mesothelial cells, which are involved in fibrotic processes. For example, Xiao et al. [34] found that IL-6 overexpression may cause morphological changes in fibroblast-like cells among human peritoneal mesothelial cells through the JAK2/STAT3 signaling pathway. Another study illustrated the mechanism of IL-6-induced EMT from an epigenetic standpoint. The authors found that exposing human peritoneal mesothelial cells to IL-6 increased the expression of histone deacetylase 6, which not only regulated IL-6 downstream of JAK2/STAT3 signaling but also activated TGF-β/Smad3 signaling, resulting in a change in the phenotype and increased cell migration [65].
IL-6 and dermal fibrosis
Hypertrophic scars and keloids represent an aberrant response of the skin to the wound healing process [66]. One main histological characteristic of hypertrophic scars is the hypercellularity caused by persistent inflammation during early wound healing and after wound closure [67]. In patients with hypertrophic scars, Tyr705 STAT3 phosphorylation is elevated [68]. Furthermore, IL-6/sIL-6R administration to fibroblasts derived from hypertrophic scars and the nonburned area of the same patient both resulted in the production of ECM and the upregulation of the cellular proliferation markers cyclin D1, Bcl-Xl and c-Myc [68]. However, hypertrophic scar fibroblasts have enhanced sensitivity to IL-6 trans-signaling. These results implicated the IL-6 trans-signaling-STAT3 pathway in hypertrophic scar pathogenesis. Unlike hypertrophic scars, keloids can emerge several years later and grow in a tumor-like manner beyond the borders of the original lesion [66, 69]. In fibroblasts isolated from patients with keloids, increased gene expression and protein production of IL-6 was identified [70, 71]. Furthermore, inhibiting IL-6 with the corresponding antibodies in keloid fibroblast culture resulted in a dose-dependent decrease in the expression of collagen type I and fibronectin [72]. In brief, the IL-6 signaling pathway has been implicated in both hypertrophic scars and keloid pathogenesis.
Systemic sclerosis (scleroderma, SSc) is an autoimmune connective tissue disease characterized by the production of autoantibodies, alterations in the vasculature and increased deposition of ECM in the skin and internal organs. Numerous studies have shown a high level of IL-6 in the serum of patients with SSc [73], and IL-6 expression is highly upregulated in SSc skin fibroblasts [74]. In a hypochlorous acid-induced mouse model of systemic sclerosis, the mRNA expression of IL-6 was upregulated in skin and lung tissues [75]. Trans-signaling of IL-6 through the JAK2/STAT3 and ERK pathways has been shown to have a significant profibrotic effect in vitro [76]. As a result, IL-6 could be a therapeutic target for patients with SSc. A phase 2 trial of tocilizumab by Khanna et al. [77] showed preliminary evidence of the efficacy of this strategy in SSc. Recently, a phase 3 trial further assessed the safety and efficacy of tocilizumab in treating skin fibrosis and systemic sclerosis-associated interstitial lung disease (SSc-ILD). Although the primary skin fibrosis endpoint was not met, the secondary endpoint of the percentage of predicted forced vital capacity indicated that tocilizumab might preserve lung function in people with early SSc-ILD. Overall, these findings suggest that tocilizumab has a positive risk/benefit profile in SSc, warranting further investigation.
IL-6 and ocular fibrosis
IL-6 has been implicated in numerous fibrotic eye diseases, including glaucoma filtration surgery (GFS), posterior capsular opacification (PCO) and proliferative vitreoretinopathy (PVR). We summarize several recent developments in the role and mechanism of IL-6 in fibrotic eye diseases as follows.
Glaucoma is a chronic progressive optic neuropathy that is sensitive to elevated intraocular pressure (IOP) and can lead to permanent loss of peripheral or central vision [78]. Currently, the only available evidence-based treatment is to reduce IOP with topical drugs, lasers and surgery [79]. GFS, which involves draining the aqueous humor into the filtration bleb in the conjunctiva, is widely used to reduce IOP. However, fibrosis and scarring in the subconjunctival space leading to the recurrence of elevated IOP are the major causes of surgical failure [80]. RNA sequencing showed that the IL-6 gene was significantly upregulated in fibrotic fibroblasts isolated from patients with previous glaucoma surgery [81]. Furthermore, compared to eyes with surgical failure, eyes with filtration surgical success, which is defined as an IOP less than 21 mm Hg without antiglaucoma medication, have significantly lower levels of IL-6, suggesting that following GFS, increased levels of IL-6 in the aqueous humor through the filtering passage into the sub-Tenon's space may enhance postoperative inflammation and contribute to fibrosis [82]. Watanabe-Kitamura et al. [83] found that the expression levels of genes encoding IL-6 at the surgical site were elevated at 3 h following GFS, whereas the levels of fibrotic genes such as TGF-β and α-SMA were elevated at 3 days. In addition, TGF-β-induced α-SMA expression was inhibited by IL-6 trans-signaling, suggesting that IL-6 can delay the phase transition from inflammation to proliferation. The findings in the present study suggest that IL-6 is important for scar wound repair in GFS. However, further research is needed to understand the mechanism of IL-6 in GFS fibrosis.
PCO, which is the most common complication of cataract surgery, is a consequence of surgical injury initiating a wound healing response that ultimately results in a reduction in visual quality [84]. PCO occurs when residual lens epithelial cells undergo fibrotic changes, such as hyperproliferation, migration, ECM deposition and epithelial cell transdifferentiation to a myofibroblast phenotype [84]. PCO is more severe and frequent in young patients than in elderly patients. Human capsular bags of young and elderly individuals were cultured in serum-free Eagle's minimum essential medium, and a significant increase in IL-6 was observed in young counterparts [85]. TGF-β2, which is an important factor in the development of PCO, can be upregulated in the presence of IL-6 via a JAK/STAT3 signaling-dependent mechanism [86]. Moreover, inhibiting JAK/STAT3 signaling effectively prevented PCO development in vitro and in vivo.
PVR, which is a scarring process characterized by the formation of fibrous membranes on the epiretinal surface, is a severe complication of ocular trauma, retinal detachment and inflammatory vitreoretinopathies. It was found that the concentration levels of IL-6 are significantly upregulated in the vitreous of PVR patients [87]. Consistently, Symeonidis et al. [88] demonstrated that IL-6 levels were increased in the subretinal fluid and were positively correlated with PVR grade. PVR membranes are composed of a variety of cell types, including fibroblasts, macrophages, and retinal pigment epithelial (RPE) cells. Of these cell types, RPE cells account for the largest proportion and are considered to play a crucial role in the pathogenesis of PVR by transitioning from the epithelial to mesenchymal phenotype. It was reported that treating RPE cells with IL-6 promoted cell proliferation, induced morphological changes from epithelial to fibroblast-like cells, and upregulated mesenchymal markers by activating the JAK1/STAT3 signaling pathway in vitro [89]. In addition, IL-6 knockout in a PVR mouse model markedly prevented PVR progression [89]. The present evidence suggests that blocking IL-6 may be a promising strategy for the prevention and treatment of PVR.
Conclusions and the future
In this review, we gathered evidence about the role of IL-6 in fibrosis and recent research on IL-6 in different fibrotic tissues and organs. According to previous research results, we can conclude that IL-6, which is a pleiotropic cytokine that plays an important role in inflammation, immunity and hematopoiesis, is a critical cytokine that drives the development of fibrosis. Given its possible role in the pathophysiology of fibrosis, IL-6 has been suggested to be a useful fibrotic biomarker. Previous studies on fibrosis have mostly focused on TGF-β and its downstream signaling pathway; however, the safety and efficacy of drugs targeting TGF-β for the treatment of fibrosis are not yet clear. Although nintedanib and pirfenidone have been approved by the Food and Drug Administration (FDA) for fibrotic disease treatment, these drugs only delay disease progression [90]. Therefore, it is very important to find new antifibrotic targets, and inhibitors of IL-6 and its signaling pathway may be new agents for resisting fibrosis. Understanding the signaling pathways and role of IL-6 in fibrosis provides an opportunity to develop therapies to attenuate the development of fibrotic diseases and facilitate the resolution of fibrotic injury.
The FDA has approved agents that target IL-6 and downstream signaling pathways, such as IL-6, IL-6R, and JAKs, for the treatment of inflammatory diseases and myeloproliferative neoplasms [10]. Novel inhibitors of the IL-6/JAK/STAT3 pathway are also being developed, including STAT3-selective medicines, and early phase clinical trials are now underway. Many of these drugs are also being investigated as potential treatments for other pathological conditions. In addition, immune-mediated adverse effects are one concern of IL-6 treatments. As we discussed above, proinflammatory IL-6 trans-signaling plays an important role in the development of fibrosis. Therefore, in future drug research and development, to reduce side effects, targeting the trans-signaling pathway of IL-6 to alleviate fibrosis should be a focus of attention.
In conclusion, inhibiting the IL-6/JAK/STAT3 signaling axis, which has previously been shown to be effective in the treatment of fibrosis, holds considerable promise for inhibiting fibrotic development. In an attempt to optimize the general application and efficacy of medicines targeting the IL-6/JAK/STAT3 signaling pathway and their use in precision medicine for patients with fibrosis, predictive biomarkers and rational combination therapy based on the immunological and genetic profiles of fibrosis are needed.
Abbreviations
ECM: extracellular matrix; IL-6: interleukin 6; JAK: Janus kinase; STAT3: signal transducer and activator of transcription 3; α-SMA: alpha-smooth muscle actin; IL-6R: IL-6 receptor; gp130: glycoprotein 130; mIL-6R: membrane-bound IL-6R; sIL-6R: soluble IL-6R; MAPK: mitogen-activated protein kinase; SOCS: suppressor of cytokine signaling; Grb2-SOS: growth-factor-receptor-bound protein 2/Son of Sevenless; MCP-1: monocyte chemoattractant protein 1; TGF-β: transforming growth factor β; FMT: fibroblast-to-mesenchymal transition; CaMKII: Ca2+/calmodulin-dependent protein kinase I; EMT: epithelial-mesenchymal transition; NSCLC: non-small cell lung cancer; IPF: idiopathic pulmonary fibrosis; HMVECs: human microvascular endothelial cells; atg5: autophagy-related 5; COVID-19: coronavirus disease 2019; CKD: chronic kidney disease; SSc: systemic sclerosis; SSc-ILD: systemic sclerosis-associated interstitial lung disease; GFS: filtration surgery; PCO: posterior capsular opacification; PVR: proliferative vitreoretinopathy; IOP: intraocular pressure; RPE: retinal pigment epithelial; FDA: Food and Drug Administration.
Acknowledgements
The work was supported by the Science and Technology Development Program of Jilin Province (20200201431JC) and the Health Special Project of Jilin Provincial Finance Department (2020SCZT030).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parola M, Pinzani M. Pathophysiology of Organ and Tissue Fibrosis. Mol Aspects Med. 2019;65:1
2. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587:555-66
3. Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov. 2020;19:57-75
4. Tanaka T, Narazaki M, Kishimoto T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb Perspect Biol. 2018;10:a028456
5. Johnson BZ, Stevenson AW, Prêle CM, Fear MW, Wood FM. The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines. 2020;8:101
6. Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. 2018;17:395-412
7. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). 2012;122:143-59
8. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16:448-57
9. Rose-John S, Neurath MF. IL-6 trans-signaling: the heat is on. Immunity. 2004;20:2-4
10. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234-48
11. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1-20
12. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798-809
13. Starr R, Hilton DJ. SOCS: suppressors of cytokine signalling. Int J Biochem Cell Biol. 1998;30:1081-5
14. Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D. et al. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J. 1998;335( Pt 3):557-65
15. Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T. et al. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity. 1996;5:449-60
16. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314-21
17. Gallucci RM, Lee EG, Tomasek JJ. IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J Invest Dermatol. 2006;126:561-8
18. Mateo RB, Reichner JS, Albina JE. Interleukin-6 activity in wounds. Am J Physiol. 1994;266:R1840-4
19. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. 2017;356:1026-30
20. Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, Sugawara T. et al. Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J. 2000;14:2525-31
21. Lin ZQ, Kondo T, Ishida Y, Takayasu T, Mukaida N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol. 2003;73:713-21
22. Sato Y, Ohshima T. The expression of mRNA of proinflammatory cytokines during skin wound healing in mice: a preliminary study for forensic wound age estimation (II). Int J Legal Med. 2000;113:140-5
23. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3
24. Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N. et al. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705-14
25. Marin V, Montero-Julian FA, Grès S, Boulay V, Bongrand P, Farnarier C. et al. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol. 2001;167:3435-42
26. Braune J, Weyer U, Hobusch C, Mauer J, Brüning JC, Bechmann I. et al. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J Immunol. 2017;198:2927-34
27. Kumar S, Wang G, Zheng N, Cheng W, Ouyang K, Lin H. et al. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension. 2019;73:1058-70
28. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27-42
29. Kagan P, Sultan M, Tachlytski I, Safran M, Ben-Ari Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS One. 2017;12:e0176173
30. Wheeler DS, Misumi K, Walker NM, Vittal R, Combs MP, Aoki Y. et al. Interleukin 6 trans-signaling is a critical driver of lung allograft fibrosis. Am J Transplant. 2021;21:2360-71
31. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M. et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21:341-52
32. Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, Findlay J. et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer. 2009;100:134-44
33. Lin CH, Wan C, Liu WS, Wang HH. PM2.5 Induces Early Epithelial Mesenchymal Transition in Human Proximal Tubular Epithelial Cells through Activation of IL-6/STAT3 Pathway. Int J Mol Sci. 2021;22:12734
34. Xiao J, Gong Y, Chen Y, Yu D, Wang X, Zhang X. et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am J Physiol Renal Physiol. 2017;313:F310-F8
35. Dalwadi H, Krysan K, Heuze-Vourc'h N, Dohadwala M, Elashoff D, Sharma S. et al. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin Cancer Res. 2005;11:7674-82
36. Moodley YP, Misso NL, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Laurent GJ. et al. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol. 2003;29:490-8
37. Li W, He P, Huang Y, Li YF, Lu J, Li M. et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics. 2021;11:222-56
38. Takagaki Y, Lee SM, Dongqing Z, Kitada M, Kanasaki K, Koya D. Endothelial autophagy deficiency induces IL6 - dependent endothelial mesenchymal transition and organ fibrosis. Autophagy. 2020;16:1905-14
39. Chen R, Sun Y, Cui X, Ji Z, Kong X, Wu S. et al. Autophagy promotes aortic adventitial fibrosis via the IL-6/Jak1 signaling pathway in Takayasu's arteritis. J Autoimmun. 2019;99:39-47
40. Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756-62
41. Saito F, Tasaka S, Inoue K, Miyamoto K, Nakano Y, Ogawa Y. et al. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am J Respir Cell Mol Biol. 2008;38:566-71
42. Milara J, Hernandez G, Ballester B, Morell A, Roger I, Montero P. et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir Res. 2018;19:24
43. Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PloS One. 2010;5:e9224
44. Pedroza M, Schneider DJ, Karmouty-Quintana H, Coote J, Shaw S, Corrigan R. et al. Interleukin-6 contributes to inflammation and remodeling in a model of adenosine mediated lung injury. PloS One. 2011;6:e22667
45. Ayaub EA, Dubey A, Imani J, Botelho F, Kolb MRJ, Richards CD. et al. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Sci Rep. 2017;7:13281
46. Horowitz JC, Thannickal VJ. Epithelial-mesenchymal interactions in pulmonary fibrosis. Semin Respir Crit Care Med. 2006;27:600-12
47. Pedroza M, Le TT, Lewis K, Karmouty-Quintana H, To S, George AT. et al. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016;30:129-40
48. Pechkovsky DV, Prêle CM, Wong J, Hogaboam CM, McAnulty RJ, Laurent GJ. et al. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am J Pathol. 2012;180:1398-412
49. Ojo AS, Balogun SA, Williams OT, Ojo OS. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm Med. 2020;2020:6175964
50. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med. 2020;8:807-15
51. Zhang C, Wu Z, Li JW, Tan K, Yang W, Zhao H. et al. Discharge may not be the end of treatment: Pay attention to pulmonary fibrosis caused by severe COVID-19. J Med Virol. 2021;93:1378-86
52. Montero P, Milara J, Roger I, Cortijo J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. Int J Mol Sci. 2021;22:6211
53. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y. et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated With Drastically Elevated Interleukin 6 Level in Critically Ill Patients With Coronavirus Disease 2019. Clin Infect Dis. 2020;71:1937-42
54. Gao Y, Li T, Han M, Li X, Wu D, Xu Y. et al. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J Med Virol. 2020;92:791-6
55. Copaescu A, Smibert O, Gibson A, Phillips EJ, Trubiano JA. The role of IL-6 and other mediators in the cytokine storm associated with SARS-CoV-2 infection. J Allergy Clin Immunol. 2020;146:518-34.e1
56. Xu X, Han M, Li T, Sun W, Wang D, Fu B. et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A. 2020;117:10970-5
57. Price CC, Altice FL, Shyr Y, Koff A, Pischel L, Goshua G. et al. Tocilizumab Treatment for Cytokine Release Syndrome in Hospitalized Patients With Coronavirus Disease 2019: Survival and Clinical Outcomes. Chest. 2020;158:1397-408
58. Mehta RL, Cerdá J, Burdmann EA, Tonelli M, García-García G, Jha V. et al. International Society of Nephrology's 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385:2616-43
59. Chen W, Yuan H, Cao W, Wang T, Chen W, Yu H. et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. 2019;9:3980-91
60. Yang J, Chen J, Yan J, Zhang L, Chen G, He L. et al. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PloS One. 2012;7:e52415
61. Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C. et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59:136-44
62. Yeates K, Zhu N, Vonesh E, Trpeski L, Blake P, Fenton S. Hemodialysis and peritoneal dialysis are associated with similar outcomes for end-stage renal disease treatment in Canada. Nephrol Dial Transplant. 2012;27:3568-75
63. Balzer MS. Molecular pathways in peritoneal fibrosis. Cell Signal. 2020;75:109778
64. Pecoits-Filho R, Bárány P, Lindholm B, Heimbürger O, Stenvinkel P. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol Dial Transplant. 2002;17:1684-8
65. Shi Y, Tao M, Ni J, Tang L, Liu F, Chen H. et al. Requirement of Histone Deacetylase 6 for Interleukin-6 Induced Epithelial-Mesenchymal Transition, Proliferation, and Migration of Peritoneal Mesothelial Cells. Front Pharmacol. 2021;12:722638
66. Berman B, Maderal A, Raphael B. Keloids and Hypertrophic Scars: Pathophysiology, Classification, and Treatment. Dermatol Surg. 2017;43(Suppl 1):S3-S18
67. Ogawa R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int J Mol Sci. 2017;18:606
68. Ray S, Ju X, Sun H, Finnerty CC, Herndon DN, Brasier AR. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J Invest Dermatol. 2013;133:1212-20
69. Al-Attar A, Mess S, Thomassen JM, Kauffman CL, Davison SP. Keloid pathogenesis and treatment. Plast Reconstr Surg. 2006;117:286-300
70. Xue H, McCauley RL, Zhang W. Elevated interleukin-6 expression in keloid fibroblasts. J Surg Res. 2000;89:74-7
71. Tosa M, Ghazizadeh M, Shimizu H, Hirai T, Hyakusoku H, Kawanami O. Global gene expression analysis of keloid fibroblasts in response to electron beam irradiation reveals the involvement of interleukin-6 pathway. J Invest Dermatol. 2005;124:704-13
72. Ghazizadeh M, Tosa M, Shimizu H, Hyakusoku H, Kawanami O. Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J Invest Dermatol. 2007;127:98-105
73. Hasegawa M, Fujimoto M, Matsushita T, Hamaguchi Y, Takehara K, Sato S. Serum chemokine and cytokine levels as indicators of disease activity in patients with systemic sclerosis. Clin Rheumatol. 2011;30:231-7
74. Feghali CA, Bost KL, Boulware DW, Levy LS. Mechanisms of pathogenesis in scleroderma. I. Overproduction of interleukin 6 by fibroblasts cultured from affected skin sites of patients with scleroderma. J Rheumatol. 1992;19:1207-11
75. Meng M, Tan J, Chen W, Du Q, Xie B, Wang N. et al. The Fibrosis and Immunological Features of Hypochlorous Acid Induced Mouse Model of Systemic Sclerosis. Front Immunol. 2019;10:1861
76. Khan K, Xu S, Nihtyanova S, Derrett-Smith E, Abraham D, Denton CP. et al. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann Rheum Dis. 2012;71:1235-42
77. Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME. et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387:2630-40
78. Stein JD, Khawaja AP, Weizer JS. Glaucoma in Adults-Screening, Diagnosis, and Management: A Review. JAMA. 2021;325:164-74
79. Schuster AK, Erb C, Hoffmann EM, Dietlein T, Pfeiffer N. The Diagnosis and Treatment of Glaucoma. Dtsch Arztebl Int. 2020;117:225-34
80. Wolters JEJ, van Mechelen RJS, Al Majidi R, Pinchuk L, Webers CAB, Beckers HJM. et al. History, presence, and future of mitomycin C in glaucoma filtration surgery. Curr Opin Ophthalmol. 2021;32:148-59
81. Yu-Wai-Man C, Owen N, Lees J, Tagalakis AD, Hart SL, Webster AR. et al. Genome-wide RNA-Sequencing analysis identifies a distinct fibrosis gene signature in the conjunctiva after glaucoma surgery. Sci Rep. 2017;7:5644
82. Cvenkel B, Kopitar AN, Ihan A. Inflammatory molecules in aqueous humour and on ocular surface and glaucoma surgery outcome. Mediators Inflamm. 2010;2010:939602
83. Watanabe-Kitamura F, Ogawa A, Fujimoto T, Iraha S, Inoue-Mochita M, Watanabe T. et al. Potential roles of the IL-6 family in conjunctival fibrosis. Exp Eye Res. 2021;210:108708
84. Wormstone IM, Wormstone YM, Smith AJO, Eldred JA. Posterior capsule opacification: What's in the bag? Prog Retin Eye Res. 2021;82:100905
85. Dawes LJ, Duncan G, Wormstone IM. Age-related differences in signaling efficiency of human lens cells underpin differential wound healing response rates following cataract surgery. Invest Ophthalmol Vis Sci. 2013;54:333-42
86. Ma B, Yang L, Jing R, Liu J, Quan Y, Hui Q. et al. Effects of Interleukin-6 on posterior capsular opacification. Exp Eye Res. 2018;172:94-103
87. Ni Y, Qin Y, Huang Z, Liu F, Zhang S, Zhang Z. Distinct Serum and Vitreous Inflammation-Related Factor Profiles in Patients with Proliferative Vitreoretinopathy. Adv Ther. 2020;37:2550-9
88. Symeonidis C, Papakonstantinou E, Androudi S, Georgalas I, Rotsos T, Karakiulakis G. et al. Comparison of interleukin-6 and matrix metalloproteinase expression in the subretinal fluid and the vitreous during proliferative vitreoretinopathy: correlations with extent, duration of RRD and PVR grade. Cytokine. 2014;67:71-6
89. Chen X, Yang W, Deng X, Ye S, Xiao W. Interleukin-6 promotes proliferative vitreoretinopathy by inducing epithelial-mesenchymal transition via the JAK1/STAT3 signaling pathway. Mol Vis. 2020;26:517-29
90. Suryadevara V, Ramchandran R, Kamp DW, Natarajan V. Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. Int J Mol Sci. 2020;21:4257
Author contact
![]() Corresponding author: Yajuan Zheng, Department of Ophthalmology, The Second Hospital of Jilin University, Jilin University, 218 Ziqiang Street, Changchun, Jilin 130041, China. E-mail: yjzhengedu.cn
Corresponding author: Yajuan Zheng, Department of Ophthalmology, The Second Hospital of Jilin University, Jilin University, 218 Ziqiang Street, Changchun, Jilin 130041, China. E-mail: yjzhengedu.cn