Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Mechanisms of NAFLD/NASH
3. Mouse models of NAFLD
4. Use of mouse models to study...
5. Use of mouse models to study...
6. Concluding remarks and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(15):5681-5697. doi:10.7150/ijbs.65044 This issue Cite
Review
Mouse models of nonalcoholic fatty liver disease (NAFLD): pathomechanisms and pharmacotherapies
Tingyu Fang1, Hua Wang2, Xiaoyue Pan3, Peter J. Little4, Suowen Xu1 ![]() , Jianping Weng1
, Jianping Weng1 ![]()
1. Department of Endocrinology, Institute of Endocrine and Metabolic Diseases, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, Clinical Research Hospital of Chinese Academy of Sciences (Hefei), University of Science and Technology of China, Hefei 230001, China.
2. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, 230022, Anhui, China.
3. Department of Foundations of Medicine, New York University Long Island School of Medicine, Mineola, New York, NY 11501, USA.
4. School of Pharmacy, Pharmacy Australia Centre of Excellence, The University of Queensland, Woolloongabba, Queensland, 4102 Australia.
Received 2021-7-16; Accepted 2022-6-29; Published 2022-9-6
Abstract

The prevalence of non-alcoholic fatty liver disease (NAFLD) increases year by year, and as a consequence, NAFLD has become one of the most prevalent liver diseases worldwide. Unfortunately, no pharmacotherapies for NAFLD have been approved by the United States Food and Drug Administration despite promising pre-clinical benefits; this situation highlights the urgent need to explore new therapeutic targets for NAFLD and for the discovery of effective therapeutic drugs. The mouse is one of the most commonly used models to study human disease and develop novel pharmacotherapies due to its small size, low-cost and ease in genetic engineering. Different mouse models are used to simulate various stages of NAFLD induced by dietary and/or genetic intervention. In this review, we summarize the newly described patho-mechanisms of NAFLD and review the preclinical mouse models of NAFLD (based on the method of induction) and appraises the use of these models in anti-NAFLD drug discovery. This article will provide a useful resource for researchers to select the appropriate model for research based on the research question being addressed.
Keywords: Drug discovery, mouse model, NAFLD, NASH, pharmacotherapy
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is one of the most prevalent liver diseases worldwide. In the coming decades, NAFLD will become the major cause of end-stage liver disease. NAFLD affects both adults and children [1]. Between 2016 and 2030, the growth rate of total NAFLD cases is between 0-and 30% [2]. A meta-analysis showed that the overall prevalence of NAFLD in Asia was 29.62% [3]. The annual incidence of primary liver cancer in the Asian NAFLD population is 1.8‰ [3].
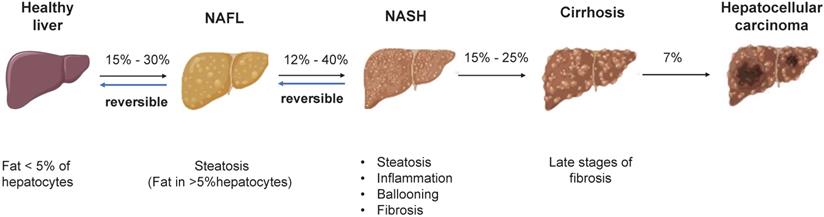
NAFLD is an umbrella term that covers a wide spectrum of liver diseases ranging from nonalcoholic fatty liver (NAFL), liver fibrosis, cirrhosis to hepatocellular carcinoma (HCC) (Figure 1) [4]. NAFLD begins with the abnormal accumulation of triglycerides in the liver, which triggers lipotoxicity, ER stress and inflammatory response and progresses to cirrhosis even liver cancer. Nonalcoholic steatohepatitis (NASH) is the advanced form of NAFLD characterized by the presence of hepatic steatosis, inflammation, ballooning and/or fibrosis [5]. Despite the deepened understanding of the pathogenesis and therapeutic targets of NAFLD/NASH [6, 7], no drug with this indication has been approved by the US-FDA [8]. By using mouse models, the study of human diseases and the identification of new therapeutic targets have been significantly facilitated. Therefore, a summary of the existing preclinical models can not only expedite the research of new targets but also contribute to the refinement of animal models. The purpose of this review was to summarize the pathogenesis, preclinical animal models, susceptibility genes and the latest research advances of NAFLD, providing useful information to identify new therapeutic targets and develop new pharmacotherapies.
The pathogenesis of NAFLD. There is reversibility between normal liver and NAFL, NAFL and NASH. When progressed to NASH, steatosis, inflammation, ballooning degeneration with fibrosis and eventually cirrhosis will occur. The progression of NASH to HCC is an irreversible process and therefore treatment and intervention for NAFLD is suggested to target the reversible stage of disease. Abbreviations: NAFL: Non-alcoholic fatty liver; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; HCC: hepatocellular carcinoma.
2. Mechanisms of NAFLD/NASH
2.1 Role of hepatic steatosis in NAFLD
The lack of approved pharmacotherapies for NAFLD/NASH indicate the need of suitable preclinical animal models which recapitulate human NAFLD pathologies. The liver is an important metabolic organ where de novo fatty acid (FA) synthesis occurs. Fatty liver tends to form when excessive FA synthesis causes dysregulated lipid metabolism. Liver fatty mainly derived from hepatic de novo lipogenesis (DNL) in human patients [9]. DNL related genes include ATP citrate lyase (Acly), acetyl-CoA carboxylase 1 (Acc1), fatty acid synthase (Fasn), and so on. Many therapeutic drugs targeting these DNL related genes are currently in different stages of clinical trials [9]. Recently, Huh et al. [10] found that TANK binding kinase 1 (TBK1) affected the mitochondrial localization of acyl-CoA synthetase long-chain family member 1 (ACSL1) and the oxidation of FA in hepatocytes by binding to ACSL1 during FA oxidation. Besides, the silencing of methylation-controlled J protein (MCJ) in the liver reduces NAFLD pathogenesis by increasing mitochondrial FA oxidation [11]. Molecules related to lipid metabolism not only represent therapeutic targets but also serve as potential new biomarkers for NAFLD. For example, the serum level of thrombospondin-1 (TSP1) in patients with fatty liver was increased before treatment, but the serum TSP1 level was decreased after hepatic fat-lowering therapy [12]. In addition, microRNAs can regulate hepatic lipid metabolism by regulating the transcriptional activity of key nuclear receptors, such as Liver X receptor alpha (LXRα) and Farnesoid X receptor (FXR) [13]. For example, miR-552-3p regulated LXRα and FXR expression by binding to the AGGTCA conserved motif [14]. MicroRNA-regulated expression of metabolic receptors offers a promising avenue to search for potential targets for treating fatty liver disease. By feeding mice with a high-fat diet (HFD) to establish a NAFLD model, Zhao et al. [15] verified that the inhibition of miR-122 up-regulated Sirt1 expression and the activation of the AMP-activated protein kinase (AMPK) pathway to inhibit adipogenesis. In addition to enzymes, serum proteins, non-coding RNA, and ion channels also affect lipid metabolism. For example, cyclin and CBS domain divalent metal cation transport mediator 4 (CNNM4) is a Mg2+ transporter. Knockdown of CNNM4 invoked intracellular Mg2+ accumulation, reduced endoplasmic reticulum (ER) stress, and promoted hepatic lipid clearance [16].
2.2 Autophagy in NAFLD
In hepatocytes, large lipid droplets (LDs) were divided into small LDs by cytoplasmic lipolysis, followed by lipophagy-dependent degradation [17].
A recent study has demonstrated that liver-specific knockdown of acyl-coenzyme A (CoA) oxidase 1 (Acox1) protected mice from starvation- or HFD-induced hepatic steatosis by inducing autophagic degradation of LD [18]. Acox1 deficiency in the liver reduced the total cytoplasmic acetyl-CoA level, which led to decreased lysosomal localization of mammalian target of rapamycin (mTOR) [19]. Acox1 attenuates mice NAFLD via different mechanisms illustrating that Acox1 is a potential target for NAFLD.
Dynamin-related GTPases for division (Drp1) phosphorylation activated autophagy. When Drp1 was abrogated in the liver, insufficient splitting of mitochondria caused mitophagy and p62 (autophagy ligase protein) upregulation to recruit two subunits of the E3 ligase complex, Keap1 and Rbx1, to promote ubiquitination of mitochondrial proteins [20]. This is similar to diet-induced NAFLD in which mitochondria size expands and accumulates mitochondrial autophagic intermediates.
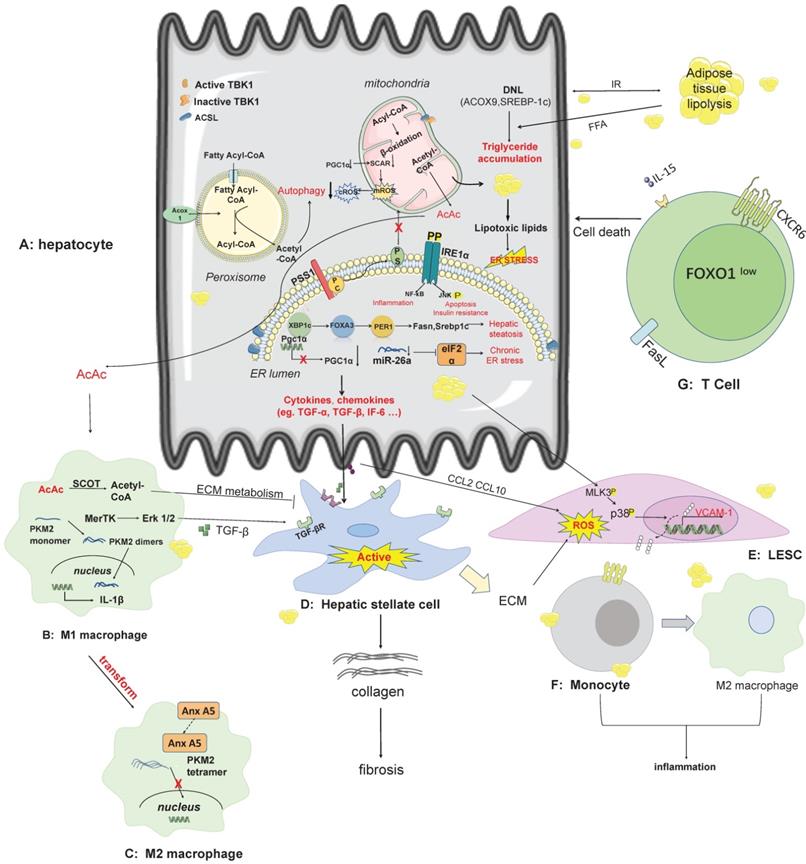
The pathogenesis and therapeutic targets of NAFLD/NASH. A: In hepatocytes, lipid synthesis, mitochondrial energy homeostasis, endoplasmic reticulum stress, novel mechanisms of peroxisomal involvement in the pathogenesis of NAFLD. The lipotoxicity arising from free fatty acid produced in hepatocytes elevates the secretion of cytokines and chemokines that affect other non-parenchymal cells, including kupffer cells, HSC, LESC. B-C: AcAc secreted by hepatocyte mitochondria can enter macrophages across cells, and metabolites of ECM can inhibit HSC activation when catalyzed by SCOT. Some pathways such as the MerTK-ERK1/2 pathway, produce TGF-β, which stimulates the activation of HSC. Anx A5 is a protein that influences the phenotypic switch from M1 to M2 in hepatic macrophages, which secrete cytokines with pro-inflammatory effects. D: HSC is stimulated by secreted proteins from other cell types and switches from a resting state to an activated state, producing collagen which in turn causes fibrosis. E-F: LSEC secretes VCAM-1 in a lipotoxic environment, and monocytes have receptors for VCAM-1 on their surface. Adhesion of LSEC to monocytes can contribute to the conversion of monocytes to M2 macrophages. G: CD8 T-cell production of auto-aggression involves IL-15-driven transcriptional programming. Self-attack by CD8 T cells in the liver is involved in liver injury via cell death pathways. Abbreviations: DNL: de novo lipogenesis; ECM: extra cellular matrix; IRE1a: nucleus signaling 1; LSEC: Liver sinusoids endothelial cell; PGC1a: PPAR-gamma co-activator-1 alpha; PSS1: phosphatidylserine synthase1; SCOT: succinyl-CoA:3-ketoacid CoA transferase; TBK1: TANK-binding kinase 1; VCAM1: vascular cell adhesion molecule-1.
2.3 ER stress in NAFLD
Hyperlipidemia, inflammation, virus infection, and drugs can disrupt metabolic homeostasis in the ER of hepatocytes [21]. ER controls liver protein and lipid homeostasis through unfolded protein response (UPR) [22]. UPR was originally perceived as a normal physiological mechanism to tackle aberrant protein turnover, but it subsequently transformed into chronic stress and impaired hepatocyte function to stimulate inflammation and cell death in NAFLD.
In a recent study, Liu et al. [23] have identified a novel role of the Xbp1-Foxa3- Period1/Srebp1c signaling axis in NAFLD. Mechanistic investigations showed that Foxa3 directly regulated Period1 transcription in mice, which in turn promoted the expression of lipogenic genes and led to NAFLD development [23].
In addition to Foxa3, some microRNAs and circular RNAs (circRNAs) serve as the bridge molecule between ER stress and NAFLD. For example, overexpression of miR-26a mitigated palmitate-triggered ER stress and lipid accumulation in human hepatoma cells and murine primary hepatocytes [24]. Targeted delivery of the newly-identified circRNA-SCAR inhibited the generation of mitochondrial reactive oxygen species (ROS) and the proinflammatory phenotype of NASH [25]. These findings provide further evidence supporting that mitochondrial ROS mediated the proinflammatory phenotype in hepatocytes after exposure to lipotoxic stimuli.
2.4 Ferroptosis in NAFLD
Deregulated iron metabolism also accelerates the progression of NAFL to NASH. A clinical study has shown that hepatic iron loading affects hepatic fat content in dialysis patients [26]. Researchers also found that ferroptosis in an ethionine-supplemented diet (CDE) model was involved in the initial necrotic cell death and induced steatohepatitis [27]. In contrast, ferroptosis inhibitors can slow the progression of NAFLD [28, 29]. Sorafenib, antifibrotic drug, trigger ferroptosis via HIF-1αonly in hepatic stellate cells (HSCs) but not hepatocytes [30]. Ferroptosis is still a less studied pathogenic mechanism, and further research is warranted to discover novel ferroptosis-related targets in NAFLD.
2.5 Inflammasome activation in NAFLD
In methionine- and choline-deficient diet (MCD) and HFD induced model of NAFLD, mRNA level of NLRP3 was higher than control group [31]. A recent study has demonstrated that palmitic acid (PA) promoted the activation of NLRP3 inflammasome in hepatocytes, and NLRP3 inflammasome activation was inhibited by adiponectin through the AMPK/JNK/ERK1/2-NFκB/ROS signaling pathway [32]. NLRP3 inflammasome amplified inflammation and triggered pyroptosis. Hepatocyte pyroptosis and the release of inflammatory components are new mechanisms for the development of liver injury and liver fibrosis [33].
2.6 Role of immune regulators in NAFLD
Emerging evidence has shown that immune regulators play essential roles in NAFLD. For example, mitochondrial antiviral signaling protein (MAVS) is a part of innate immunity and antiviral response [34]. In NASH, mitochondrial damage extends to MAVS, resulting in a decrease in the induction of type I interferons [35]. MAVS is the downstream adaptor protein of RIG-I/ Melanoma differentiation-related gene 5 (MDA5, also known as Ifih1). After binding, it can induce the production of type 1 interferon and activate NF-κB, IRF-3 and other pathways. MDA5, as the target of apoptosis signal-related kinase 1 (ASK1), is an important inhibitor of NASH induced by an HFD/ high-fat and high-cholesterol diet (HFHC) [36]. Knolle et al. [37] reported the mechanism of T-cell response to NASH-associated liver injury in a CD-HFD mouse model. Forkhead box O1 (FOXO1) was downregulated and C-X-C motif chemokine ligand 6 was upregulated after IL-15 induction in T cells, leaving the CXCR6+CD8 T cell susceptible to metabolic stimuli. ATP released triggered rapid Fas ligand (FasL) upregulation and self-attack in CD8 T cells, causing liver injury. Meanwhile, Pfister et al. [38] used the same animal model to discover the involvement of CD8 T cells in the induction of HCC due to NAFLD. Patients with advanced HCC found that immunotherapy was ineffective in improving HCC due to NAFLD. This condition is probably due to unconventional activation of T cells and auto-attack, resulting in compromised immune system function.
2.7 Extracellular vesicles (EVs) in the development of NAFLD
It is well established that almost all cell types in the liver can secrete exosomes, such as hepatocytes, macrophages, neutrophils and so on [39]. Other metabolic organs, such as adipose tissues, can also secrete exosomes. The cargos transported by exosomes include lipids, proteins and nucleic acid [40]. The exchange of EVs between neutrophils and liver cells can improve or promote NASH dependent on the transported cargo. The increment of miR-223 in liver cells is due to the preferential uptake of miR-223-rich EVs derived from neutrophils. miR-223 improve NASH in the mouse model by upregulating the expression of low-density lipoprotein receptor (LDLR) [41]. Overexpression of CXCL1 and aldo-keto-reductase 1B7 (Akr1b7) can also induce NASH [42, 43]. Noteworthy, deletion of Akr1b7 (from adipocyte-secreted exosome which generated by ER stress) protected mouse liver from NASH induced by feeding with HFD and MCD. In vivo, CXCL1 overexpression in HFD-fed mice increased the mtDNA content in EV. Lysophosphatidylcholine (LPC), cholesterol and other lipid mediators induced hepatocytes to secrete exosomes which contain integrins and microRNAs. EVs from hepatocytes undergoing lipotoxic stress are rich in integrin β1 (ITGβ1) and microRNA 192-5p [44, 45]. The adhesion of monocytes to liver sinusoidal endothelial cells (LSECs) mainly depends on ITGβ1, while miR-192-5p plays a role in the activation of proinflammatory macrophages and NAFLD pathogenesis by regulating Rictor/Akt/FoxO1 signaling.
2.8 Gut microbiota in NAFLD
The intestinal mucosa of healthy people is intact. This helps maintain the integrity of the intestinal flora composition and resists bacterial invasion [46]. In recent years, emerging evidence has shown that the gut microbiota regulates the pathogenesis of NAFLD. Mice after a short-term of HFD feeding (1 week), experience gut vascular barrier (GVB) damage and bacterial translocation into the liver [47]. In turn, transplanting fecal bacteria from these HFD-fed mice to specific pathogen-free recipients also caused GVB damage [47]. The composition of the gut microbiota changes with the pathogenesis of NAFLD [48]. Gut microbiota and its metabolites promote the formation of an immunosuppressive environment [34].
2.9 Fibrogenesis and NAFLD
Fibrosis is the late stage of NAFLD, with the activation of HSCs being the core cellular mechanism. After activation, HSC express high levels of extracellular matrix proteins leading to the formation of fibrosis. Farnesoid X receptor (FXR) is a bile acid receptor. It regulates bile acid secretion, which can down-regulate the expression of crucial lipogenesis-associated genes in the liver and ultimately reduce hepatic lipid content [49]. A previous study has shown that SUMOylation inhibitors can increase the inhibitory effect of prophylactic obeticholic acid (OCA, an FXR agonist) on HSC activation and fibrosis [50]. Acetyl-CoA carboxylase (ACC) is an important target for regulating DNL. Inhibition of ACC reduces the lipotoxicity of liver cells and inhibits the activation of HSCs [51].
3. Mouse models of NAFLD
“Multiple-hit” hypothesis is a well-accepted one for NASH development [52]. To enhance our understanding of NAFLD's pathomechanisms, the use of animal models is very helpful. Animal models are also helpful for exploring new therapeutic modalities. An ideal animal model should be able to reproduce the complex pathomechanisms of human disease pathology with high fidelity. This section summarizes the most commonly used animal models in the study of NAFLD, highlighting the major advantages and disadvantages of each model (Table 1).
Comparison of mouse models of NAFLD/NASH
| Animal model | Phenotype | Fibrosis | Advantage | Disadvantage | References | |||
|---|---|---|---|---|---|---|---|---|
| IR | Obesity | Steatosis | Inflammation | |||||
| MCD | No | Weight loss | +++ | +++ | Yes | Short period; Easy to operate; High Reproducibility | No NAFLD-related metabolic syndrome | [65, 159] |
| CDAA | No | Weight loss | +++ | ++ | Yes | Gain of weight; unclear increase in hepatic; peripheral insulin sensitivity | Long-period; high costs | [160] |
| High-fat diet (HFD) | Yes | Yes | +++ | + | Yes | Low costs; easy to operate | Requires large sample size; difficult comparison between groups and protocols. | [161] |
| High-fructose diet | Yes | No | +++ | ++ | No | Develop Metabolic syndrome. | It does not develop into advanced fibrosis or hepatocellular carcinoma; A high-fructose diet alone does not produce a Nash phenotype in the liver | [162-164] |
| High-cholesterol diet (HCD) | Yes | Yes | +++ | ++ | Yes | Phenotype very similar to the clinical features of NASH in patients with metabolic syndrome | Not common in humans; Cholesterol in the diet may not be physiological | [165, 166] |
| WD+CCl4 | No | No | +++ | +++ | Yes | Short-term; Progression to advanced liver disease; Simulating the histological, immunological and transcriptional characteristics of human NASH | IR was not observed in some models | [95] |
| HFD/HCD+CCl4 | Yes | No | Yes | ++ | Yes | |||
| STAM | Yes | No | +++ | ++ | + | The whole process of NAFLD can be simulated with short time | Not include full range of human disease features | [100] |
| ob/ob | Yes | Yes (extremely) | +++ | + | No | A number of features similar to human NAFLD appear | Rarely develop NASH without diet challenge | [167, 168] |
| db/db | Yes | Yes | +++ | + | No | Same as above | Difficult to progress to advanced NASH | [169] |
| foz/foz | Yes | Yes | ++ | + | Yes | Phenotype very similar to the clinical features of NASH in patients with metabolic syndrome | Phenotype may be strain-dependent | [170] |
Abbreviations: CDAA: Choline-deficient L-amino-defined diet; MCD: methionine- and choline-deficient diet; WD: Western diet;
3.1 Dietary model
3.1.1 High-fat diet (HFD)
There are different types of HFD in terms of nutrient composition, and this difference may also be caused by different sources (animal or vegetable) and amounts of fat. Conventional HFD usually contains 60 kcal% or 45 kcal% fat and mice fed with HFD will develop obesity, insulin resistance (IR) and hepatic inflammation after 16 weeks of feeding [53]. Mice fed HFD for 1 week can develop diet-induced ecological dysbiosis, leading to bacterial translocation to the liver [47]. Therefore, HFD is an appropriate choice to study the effect of gut microbiota on the development of NAFLD. The HFD-fed mouse model can be used to verify the mechanism of action of some drugs such as water extract of shepherds purse (WESP) [54] and ursolic acid in alleviating NAFLD development [55]. In mice, different strains and genders show different sensitivities to HFD. Therefore, variables in selecting this model include dietary fat content, duration and strains. The model also displays similar features to human NAFLD/NASH development and is widely used in metabolic research, but the pathological outcome was less severe than human NASH, particularly the fibrogenesis process [56].
3.1.2 High-fat high-fructose diet (HFFD)
The second frequently-used model is HFFD model by adding 10% fructose to the HFD. However, the extent of steatosis, inflammation and fibrosis were ameliorated in fructokinase knockout mice [57]. Fats can come from animal or plant sources [58, 59]. Excessive fat and fructose intake cause IR, inflammation and ER stress through different mechanisms, such as free fatty acid (FFA)-derived lipotoxicity, and the expression of proinflammatory cytokines and chemokines [60-62]. Accordingly, the HFFD model is a good choice when studying the role of ER stress and lipid steatosis in NAFLD. A recent study has compared the differential effect of types of diets on NAFLD in mice with that in human patients, and conclude that HFFD model best recapitulates the human phenotype of NAFLD [63].
3.1.3 Methionine- and choline-deficient diet (MCD)
MCD is rich in sucrose, which provides the right amount of fat but lacks methionine and choline. Methionine deficiency can lead to the blockade of protein synthesis in the body [64]. Choline is a strong organic base, a constituent of lecithin and a precursor of acetylcholine. Recently, the MCD model has been used to study the role of gut microbiota and immune response in NAFLD. The activation of macrophages involved the interaction of hypoxia-inducible factors 1 alpha (HIF-1α) and autophagy, which promoted the proinflammatory overactivation of in a preclinical model induced by MCD [65]. Additionally, in kupffer cells residing in the liver, the stimulator of IFN genes (such as the stimulator of interferon genes (STING) induced inflammation through the IRF and NF-κB pathway [66]. Symbiotic microbiota was found to have hepatoprotective effects in MCD-induced steatohepatitis. Unfortunately, the composition of the intestinal flora of MCD diet-fed mice is not similar to that of humans [67]. In addition, MCD model is a commonly used model for studying ferroptosis. MCD-fed mice have iron accumulation in the liver and serum. Ferroptosis inhibitors can reduce liver damage and liver fibrosis caused by MCD feeding [28], while ferroptosis inducers increased lipoxygenase and apoptosis-inducing factors and affect lipid oxidation and other types of cell death [29].
3.1.4 Choline-deficient L-amino-defined diet (CDAA)
Dietary choline deficiency, L-amino acid defined diet (CDAA) is another popular model for pharmacological and genetic research of NAFLD [68, 69]. Like MCD, CDAA is a choline-deficient diet in which sulfhydryl-containing amino acids are less restricted and ample energy is supplied by the HFD. The methionine level in the semisynthetic CDAA diet was normal or only moderately reduced, while the protein content in the formula was replaced by an L-amino acid mixture [70]. After 12 weeks of feeding with CDAA, steatohepatitis developed with fibrosis and an approximately three-fold increment in liver collagen level was observed. After 21 weeks of feeding, the fibrosis progresses to a moderate stage. In addition, CDAA model showed liver cancer-associated fibrosis, thereby accelerating the study of disease progression from NASH to HCC [71].
3.1.5 Choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD)
Compared with the above models, choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) is another widely-used model in NAFLD/NASH studies. This model simulates human NAFLD pathology much more closely [72]. The most commonly used formula for CDAHFD is a high-fat, choline-deficient diet, including 0.1% methionine and 45% fat [73]. After 6 weeks of feeding, the CDAHFD model exhibited hepatic steatosis, liver damage, and inflammation [73]. CDAHFD feeding for 6 weeks triggers liver fibrosis evidenced by picosirus red staining [74]. Furthermore, C57BL/6J mice fed with CDAHFD can develop steatohepatitis without affecting body weight [74]. Interesting features of this model also include i) CDAHFD can induce steatohepatitis within one week, together with mitochondrial dysfunction and severe oxidative stress, but without fibrosis. ii) The content of methionine in the formula determines whether or not the mouse can become obese [75, 76]. This is a unique feature that other models do not have, and it simulates some of the characteristics of human NAFLD. The CDAHFD model has significant applications in studying the role of novel therapeutic targets (such as TSP-1, transient receptor potential canonical (TRPC) [77, 78], traditional Chinese medicine (such as nobiletin, astaxanthin) [79, 80] or western medicine (such as metformin and PPAR agonist) in NAFLD/NASH development [74, 81].
3.1.6 High-fat high-cholesterol diet (HFHC)
Dietary cholesterol is an important factor associated with the progression of steatohepatitis and liver inflammation in mouse models and humans [82]. Wang et al. found that hepatocyte cholesterol activated TAZ (a transcriptional regulator that promotes fibrosis) leading NASH and liver fibrosis. Mice fed with 30% fat, 1.25% cholesterol, and 0.5% cholate have a ballooning-like expansion of hepatocytes, an essential characteristic of human NASH [83].
The high-fat and high-cholesterol diet (HFHC) model was widely used in NAFLD/NASH research [84]. This model can be used for drug evaluation and probing molecular mechanisms of NAFLD. For example, cordycepin, a potentially new natural AMPK activator for treating NASH, relieves hepatic steatosis, inflammation, liver injury and fibrosis in the NASH mouse model established by feeding mice with HFHC diet [85]. In addition, cholesterol diets induced modification in intestinal microflora and metabolites to promote NAFLD development [86]. Thus, HFHC feeding is also used to study the influence of intestinal microorganisms on NAFLD [86, 87].
3.1.7 High-fat, fructose, and cholesterol model (HFFC)
The high-fat, high-fructose, and high-cholesterol diet (HFFC) model is frequently used in establishing animal models of metabolic syndrome. HFFC diet usually contains 40% fat, 20% fructose, and 2% cholesterol, which was previously known as the Amylin Liver NASH (AMLN) diet [88]. After 34-36 weeks, liver TG but not serum TG has increased in mice [89]. Liver injury was observed evidenced by elevated levels of serum AST/ALT [89]. Compared to the control group, expression of fibrosis-related genes, such as collagen 1α1 (Col1a1), α-SMA (Acta2), and lysyl oxidase-like 2 (Loxl2) were elevated and prominent collagen accumulation was detected by picosirius red staining [90]. Recently, Song et al. found 25-hydroxylanosterol (25-HL) has preventive and curative effects on NASH mice fed with HFFC diet [91]. Mechanistically, 25-HL binds to insulin-induced gene, promoting SCAR-SREBP retention in ER, thereby reducing the level of cholesterol and TG. Another unique feature of this model is that HFFC diet-treated Ldlr-/- mice simultaneously induced the development NASH and atherosclerosis [91]. Therefore, this model has tremendous utility in developing new therapeutic targets as well as new drugs which ameliorate NASH and atherosclerosis in light of the fact that most NAFLD patients die from extrahepatic complications, such as atherosclerosis.
3.1.8 High-fat diet plus carbon tetrachloride (CCl4) induction
Carbon tetrachloride (CCl4) is a hepatotoxic chemical which causes liver injury, liver fibrosis and cirrhosis in experimental animals. Repeated administration of CCl4 to HFD-fed obese mice successfully induced chronic oxidative stress, triggered inflammation and led to liver fibrosis [92]. Notably, under feeding with Western diet (WD) supplemented with 5% fructose (WDF), CCl4 reduced the induction time and aggravated liver fibrosis in FATZO mice [93]. In comparison to CDAA- treated mice, additional treatment with a single dose of CCl4 resulted in not merely hepatic lipid deposition but also peri-hepatocellular fibrosis [94]. HFFC diet combined with a low-dose of CCl4 injection intraperitoneally to establish a murine NASH model with extensive fibrosis and rapid HCC progression [95]. The advantages of this model are i) generalization from simple steatosis to inflammation, fibrosis and hepatic cancer. ii) simplicity and reproducibility of the model facilitate the study of disease pathogenesis and the tests of new treatments. Similar model has been established in rats by HFFC diet and CCl4 treatment [96]. This is a preclinical model of moderate and advanced NASH that mimics human disease and exhibits almost all the characteristics of advanced human NASH after 10 weeks and cirrhotic NASH after 24 weeks. However, very few evidence exists as to whether NASH induced by this model can be regressed by diet switch.
3.1.9 STAM model
Like HFFC plus CCl4 model, STAM model also induced NASH and even HCC by combining diet and chemical treatment. Two-day-old nascent C57BL/6J male mice were injected low-dose streptozotocin (STZ) (200 μg per mouse) [97]. Mice were fed a high-fat/high-calorie diet after 4 weeks [98]. Those mice developed liver steatosis at 7 weeks, reached cirrhosis at 12 weeks, and developed HCC within 20 weeks [98]. Steatohepatitis, inflammation, ballooning and fibrosis were observed in those mice [99]. Pathological analysis revealed that treated mice have mild steatosis, more severe inflammation and 62.5% of mice have severe ballooning [98]. However, the limitation of STAM model lies in the administration of STZ, which does not mimic human NAFLD conditions [100].
3.2 Genetic models
3.2.1 CXCL1 plus HFD mouse model
Up-regulation of chemokine production by liver neutrophils is characteristic of human NASH. HFD feeding alone is difficult to trigger the development of NASH in mice. However, overexpression of the chemokine CXCL2 in the liver can induce NASH in mice fed a HFD [42]. When CXCL1 is overexpressed in the livers of HFD-fed mice, infiltrating and activated neutrophils produce a high level of reactive oxygen species that contribute to liver injury under chronic inflammatory conditions in NASH. CXCL1 overexpression led to p38α activation that induces the cleavage of caspase-3, C/EBP Homologous Protein (CHOP) expression, and BCL2 phosphorylation, thereby exacerbating hepatocyte death [101]. However, mice fed a HFD only showed weak p38α activation, which upregulated genes involved in fatty acid β-oxidation that may act to compensate for hepatocyte lipid accumulation [101]. The new NASH model established by hepatic overexpression of CXCL1plus HFD feeding also mimics human NASH pathology. High level of CXCL1 compensated the effects caused by other factors to a certain extent. Together, based on the HFD model, the liver-specific overexpression of the key factors in driving NASH progression can be a new means to establish the NASH model.
3.2.2 microRNA deficiency-induced mouse model
MicroRNA has been increasingly regarded as a critical regulator of metabolism and inflammation in the liver. For example, microRNA 122 (miR-122) is a liver -enriched miRNA, accounting for 70% of the total liver microRNA [102]. Long-term inhibition of miR-122a in the liver reproduced human liver pathology, but the short-term decrease of miR-122a expression is beneficial [103]. In another study using the NASH mouse model and serum samples from NASH patients, the level of miR-223 is up-regulated [104, 105]. MiR-223-KO mice developed NASH after feeding a HFD or MCD for 3 months [106]. Genetic deletion of miR-223 induces a full spectrum of NAFLD in long-term HFD-fed mice. In terms of mechanism, miR-223 prevents the progression of steatosis to NASH by inhibiting the expression of Cxcl10 and Taz in the liver [106].
3.2.3 ob/ob mouse model
Obesity is a significant risk factor for NAFLD. The most prevalent genetic models of obesity are leptin (ob/ob) and leptin receptor (db/db) deficient mice [107]. Ob/ob mice have spontaneous mutations in the OB gene (encoding leptin), resulting in leptin deficiency. When fed a normal chow diet, ob/ob mice are hyperphagic, inactive, and extremely obese and have many features such as IR, hyperinsulinemia, and spontaneous liver steatosis similar to human NAFLD [62, 108, 109]. An experiment conducted to explore different NASH mouse models suggests that ob/ob mice fed fast-food diet or HFD showed metabolic, histological and transcriptome dysfunction similar to human NASH [110].
3.2.4 db/db mouse model
Db/db mice deficient in the DB gene (encoding leptin receptor), are leptin resistant with or without hyperleptinemia [111]. In terms of fat accumulation and adipose inflammation, db/db fed with chow diet is comparable to C57BL/6J mice fed with HFD. Db/db mice can show the characteristics of human type 2 diabetes [112]. NAFLD pathology in db/db mice can be accelerated by feeding mice with a western-type diet [113].
3.2.5 Foz/foz mouse model
Foz/foz mice carry a mutation in Alström syndrome protein 1 (Alms1) which leads to primary ciliary dysfunction. Patients with Alström syndrome are more likely to develop childhood type 2 diabetes mellitus (T2DM) than patients with other syndromes, suggesting that ALMS1 may be of significance in the cell function and/or peripheral insulin signaling pathway [114]. A study has shown increased obesity, glucose intolerance and accelerated NASH pathology in foz/foz mice after 6 weeks of HFD feeding [115]. Foz/foz mice fed with HFD developed NASH with fibrosis after 12 weeks, which was alleviated when the diet was switched to a normal diet. As with other genetically NAFLD mouse models, the foz/foz model requires challenge with HFD to induce the occurrence of late-stage of NAFLD phenotype.
Because the pathogenesis of fatty liver is diverse and heterogenous, the mouse models available have similar pathological features but not necessarily the same processes as we see in the clinic. The ARRIVE guideline which was established by the editors of major biomedical journals provides some principles for selecting the right type of animal models in scientific research [94].
4. Use of mouse models to study the mechanisms of NAFLD
The establishment of these preclinical animal models can also be used to investigate the impact of disease-relevant targets on the development of NAFLD and the search for potential therapeutic agents (Table 2).
Established genetic targets of NAFLD/NASH: evidence from animal models and clinical trials
| Gene | Animal model | Clinical trial number* and status |
|---|---|---|
| PNPLA3 | HFD[171-174] | NCT04640324 (Completed, 2020) |
| NCT01966627 (Completed, 2017) | ||
| NCT02116192 (Completed, 2018) | ||
| TM6SF2 | HFD [175] | NCT04640324 (Completed, 2020) |
| High-fructose diet model [176] | NCT04501042 (Recruiting, 2020) | |
| GCKR 2 | HFD [177] | NCT01966627 (Completed, 2017) |
| HSD17B13 | Western Diet model [130] | NCT04565717 (Recruiting, 2020) |
| High-fat diet model [130] | ||
| TAZ | High-fat, high-glucose [178] | / |
| High-fat diet model [179] | ||
| CDAHFD[180] | ||
| Genetically deficient mouse models[181] |
4.1. PNPLA3
Patatin-like phospholipase domain 3 (PNPLA3) is located on chromosome 22 which encodes protein adiponutrin. Point mutation of PNPLA3 (I148M) promoted steatosis by inhibiting lipid droplets (LDs) degradation via cofactor CGI-58 [116]. A study by Yang et al. [117] revealed a stronger binding affinity of PNPLA3 with adipose triglyceride lipase (ATGL) (compared to CGI-58), which reduced lipid degradation. However, the PNPLA3I148M mutant has a stronger binding affinity to CGI-58 than ATGL and PNPLA2 by suppressing its ubiquitination and protein degradation [116-118]. Studies have shown that PNPLA3I148M mutant, but not wild-type PNPLA3, shows fatty liver phenotype and other metabolic characteristics similar to human patients [119]. Based on this, Lindén et al. [120] constructed a mouse model of human PNPLA3I148M knock-in mutation. The injection of liver-targeted GalNAC-conjugated antisense oligonucleotides (ASO) that mediates Pnpla3 silencing improved all the characteristics of NAFLD in this mouse model, including liver fibrosis [120]. In addition to PNPLA3148M, the PNPLA3 rs738409 variant is associated with the early stage of NAFLD diagnosis [121].
4.2. TM6SF2
Genetic mutation of transmembrane 6 superfamily member 2 (TM6SF2) plays a role in liver diseases. Emopamil binding protein (EBP) is a cholesterol biosynthetic enzyme. TM6SF2 was confirmed to be homologous to EBP and shared the same domain structure [122]. In addition, ATP-binding cassette sub-family G member 5 (ABCG5) promoted reverse cholesterol transport in the liver of TM6SF2 knockout mice [123]. TM6SF2 also affects the levels of VLDL and ApoB. Kozlitina et al. [124] simulated the effects of TM6SF2-167 Lys mutation on hepatic triglyceride content and blood lipids in high sucrose diets model. Tm6sf2 increased triglyceride (TG) content in the liver of mice and decreased the secretion of VLDL. Studies performed in TM6SF2 depleted or TM6SF2 knockout mice suggest that TM6SF2 reduced ApoB levels and led to lipid accumulation in the liver [125].
4.3. HSD17B13
Hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) was initially named as SCDR9 and was cloned from a human liver cDNA library for the first time in 2007 [126]. Most of the HSD17B family members are involved in regulating the biological activity of sex hormones, FA metabolism, cholesterol biosynthesis and the production of bile acid (BA) [127]. Human genetic research has shown that the splice variant of HSD17B13 (rs72613567: TA) prevented NAFLD development [128]. In addition, a meta-analysis has demonstrated that SNP rs72613567 reduced the severity of NAFLD in humans [129]. The above studies have indicated that mutating or inactivating HSD17B13 in humans can resist NAFLD. Ma et al. [130] used a murine model to study the in vivo function of HSD17B13. The authors compared Hsd17b13 knockout (KO) mice and wild-type (WT) littermate controls fed with a regular diet, HFD, WD, or NIAAA alcohol exposure models (a mouse model of chronic and binge ethanol feeding). Compared with WT mice, KO mice showed higher body weight and liver mass; furthermore, KO mice on an obese diet had larger lipid droplet size. Additionally, HSD17B13 KO mice fed a soy-free, natural-ingredient diet showed steatosis and inflammation [131].
4.4. TAZ
Higher transcriptional activity of TAZ (also known as WW-domain containing transcriptional regulator 1, WWTR1) was observed in human and mouse NASH hepatocytes compared with normal or simple steatotic hepatocytes. Back in 2016, Wang et al. [132] found that TAZ depletion prevented or reversed liver inflammation, hepatocyte death and fibrosis, but not steatosis in mice fed a high-fat and high- fructose diet. After feeding mice with NASH diet for 9 weeks, targeted delivery of siTAZ to the liver can reduce the expression of inflammation and fibrosis associated marker genes, Col1a1, Col3a1 and α-SMA [133]. These findings indicate that TAZ can be a therapeutic target for NASH.
In addition, hepatocyte-specific TAZ deletion downregulates p62 expression in NASH models and plays a role in inflammation and liver injury [134]. Of translational relevance, rosemary acid (RA), a natural product isolated from medicinal plants was reported to slow down the pathogenesis of NAFLD by downregulating TAZ and upregulating PPARγ and PGC-1α in HFD-induced NAFLD model [135].
5. Use of mouse models to study the pharmacological actions of drug candidates in NAFLD clinical trials
5.1 Selonsertib (GS-4997)
The activity of ASK1 is increased in mouse and human NAFLD samples and is finely tuned by extracellular and intracellular signals. Selonsertib (SEL) is an inhibitor of ASK1. SEL inhibited NASH in established NASH models, such as MCD model and CDAHFD model. SEL (30 mg/kg, q.d.) reduced collagen area and inflammation but did not affect steatosis in MCD and CDAHFD models [136]. The improvement of inflammation and fibrosis by SEL was also verified in CCl4 plus MCD mouse model [136].
STELLAR-4 is a phase III (NCT03053063), randomized, double-blind, placebo-controlled study to evaluate the safety and effectiveness of SEL in patients with compensated liver cirrhosis (F4) caused by NASH. In short, although SEL improved steatosis and fibrosis in diet-induced mouse models, it did not improve liver fibrosis in patients with advanced NAFLD.
5.2 Saroglitazar
Saroglitazar (SAR) is a dual PPARα/γ agonist. In CDAHFD-induced NASH mouse model, SAR reduced liver steatosis and inflammation and reduced the levels of biomarkers of liver injury and inflammation [137]. SAR outperforms pioglitazone in HFFC model despite no significant influence on liver and body weight was observed [138]. The authors observed that SAR (3 mg/kg, oral administration) treatment for 12 weeks could reverse NASH development. SAR (3 mg/kg, oral) also mitigate the expression of pro-inflammatory genes (Tnfα, Mcp-1), fibrotic genes (α-SMA, Col1a1 and Ctgf), and collagen area in CDAHFD induced NASH model [137]. Notably, the decrement of fibrosis-related expression wasn't observed in fenofibrate (100 mg/kg) or pioglitazone (30 mg/kg).
In March 2020, the Drug Control Center of India approved SAR's new drug application, which is the world's first drug for the treatment of non-cirrhotic NASH. The drug met the primary and secondary endpoints in the phase II clinical trial (NCT03863574). However, the sample size of this trial is small, including only 16 participants.
5.3 Obeticholic acid (OCA)
Farnesol X receptor (FXR) is highly expressed in the liver and plays a role in enterohepatic circulation and BA synthesis. OCA is a FXR agonist. In a NASH model, OCA had higher efficacy and longer half-life than the dual FXR/TGR5 agonist INT-767 [139]. Diet-induced dysbiosis occurred after one week of HFD feeding [47]. In one study, OCA treatment prevented the gut vascular barrier (GVB) integrity. Besides, melanocortin 4 receptor-deficient (MC4R-KO) mice can develop NASH when fed a HFD. This model developed obesity and IR and was used to validate the anti-fibrotic effects of OCA [140]. OCA also alleviated inflammation and the progression of fibrosis in HFD-fed Ldlr-/- Leiden mice by reducing collagen deposition and limiting de novo lipogenesis [141]. Steatosis and fibrosis occurred in FATZO mice when fed a WD supplemented with fructose (WDF) [142]. FATZO mice were used to evaluate the effect of OCA on NASH progression. In addition, in another model with ob/ob mice fed an AMLN diet for 15 weeks, the effect of OCA and elafibranor (ELA) in combination on liver histological changes was evaluated. The results showed that OCA reduced liver weight in mice compared to ELA, reflecting an improvement in steatosis by OCA.
In one clinical trial (NCT01265498), patients were randomly assigned to receive OCA treatment (n=141), or placebo treatment (n=142). OCA improved the histopathological characteristics of NASH [143]. After receiving OCA treatment, some patients have increased harmful cholesterol in their bodies. While, the FXR target reduced circulating cholesterol by inducing anti-cholesterol transport proteins such as scavenger receptor B1 (SCARB1/SR-B1) and the ATP binding cassette G8 transporter in mice model [144, 145]. These differences were also confirmed in human hepatocyte chimeric mice [146].
5.4 Liraglutide
Glucagon-like peptide 1 (GLP-1) is an insulin-stimulating hormone produced in L cells and secreted after food ingestion [147]. Liraglutide is an analog of GLP-1, which can improve the function of pancreatic β-cells and reduce the body weight of obese patients [148].
Interestingly, liraglutide has been proven to be effective in mouse models of NASH. For example, Moreira et al. [149] evaluated the effects of liraglutide on obesity and NAFLD in two obese mouse models (the ob/ob mice and the HFD mice). Liraglutide improved NAFLD pathologies in mice with induced diabetes by modulating inflammatory signaling pathways [150]. In addition, liraglutide prevented the accumulation of ceramide/sphingomyelin in MCD-fed mouse liver [151]. Similarly, in db/db mice, ob/ob mice and HFD mouse models, liraglutide has appreciable therapeutic effects on NAFLD. The underlying mechanism is related to the alteration of the composition and diversity of intestinal microbes [149, 152]. The therapeutic effects of liraglutide exhibited in the diet-induced obesity (DIO)-NASH and ob/ob-NASH mouse models are translated into observed clinical benefits in NASH clinical trials [153].
5.5 Cenicriviroc
Cenicriviroc (CVC) is a C-C chemokine receptor type 2 (CCR2) and type 5 (CCR5) dual antagonist that inhibits liver fibrosis [154]. In high-fat, high-fructose models, CVC (0.1% wt/wt, a target dose of 100 mg/kg/day, 8 weeks) administration with diet inhibited liver inflammation and fibrogenesis, facilitating the development of NASH and fibrosis [155]. According to the phase IIb clinical trial (CENTAUR study, NCT02217475), fibrosis was improved after 1 year of CVC treatment compared to placebo treatment [156]. Thus, CVC has shown anti-fibrosis ability both in preclinical models and clinical trials.
The full list of drugs in clinical stage of development is summarized in Table 3.
Emerging pharmacotherapies for NAFLD/NAS
| Target category | Drug name | Action | Animal model | Clinical trial number and status | Phase | Current Primary Outcome |
|---|---|---|---|---|---|---|
| Nuclear receptor | Pioglitazone | PPAR-γ agonist | HFD model[182] CDAA diet model[183] | NCT03950505 (Recruiting, 2020) | 4 | In liver fibroscan, liver stiffness (kPa) as a marker of fibrosis and CAP (dB/m) as a marker of steatosis will be estimated. |
| NCT03646292 (Recruiting, 2020) | 3 | More sensitive than the biopsy-based steatosis grade assessment in confirming liver fat changes. | ||||
| Elafibranor | PPARα/δ agonist | 3D Spheroids[184] Ob/Ob mice model[185] | NCT02704403 (Terminated, 2020) | 3 | Composite long-term outcome composed of all-cause mortality, cirrhosis, and liver-related clinical outcomes. | |
| Saroglitazar | PPARα/γ agonist | High fat western diet and ad lib sugar water (WDSW)[138] High-Fat Emulsion/LPS Model[186] HFHC model[187] | NCT03061721 (Completed, 2021) | 2 | Percentage change from baseline in serum ALT levels at Week 16. | |
| NCT03863574 (Completed, 2020) | 3 | NAS Score (NAFLD Activity Score) | ||||
| Obeticholic acid | FXR agonist | HFD model[47] HFHC model[50] | NCT02548351 (Active, not recruiting, 2021) | 3 | To evaluate the effect of OCA on liver histology in non-cirrhotic NASH subjects with stage 2 or 3 fibrosis. | |
| NCT03439254 (Active, not recruiting, 2021) | 3 | Percentage of subjects with improvement in fibrosis by at least 1 stage with no worsening of NASH, using NASH Clinical Research Network (CRN) scoring system. | ||||
| Incretin | Liraglutide | GLP 1 analogues | HFCC-CDX model[188] Ob/Ob mice model[149] HFD model[150] | NCT01237119 (Completed, 2016) | 2,3 | Liver histological improvement. |
| Semaglutide | GLP 1 analogues | Diet-induced obesity (DIO) mice[189] (40% fat, 40% carbohydrate and 2% cholesterol) | NCT03919929 (Recruiting, 2020) | 2,3 | Change in hepatic fat fraction. | |
| NCT04822181 (Recruiting, 2021) | 3 | Resolution of steatohepatitis and no worsening of liver fibrosis; Improvement in liver fibrosis and no worsening of steatohepatitis; Time to first liver-related clinical event (composite endpoint). | ||||
| NCT02970942 (Completed, 2020) | 2 | Percentage of Participants with Non- Alcoholic Steatohepatitis (NASH) Resolution Without Worsening of Fibrosis After 72 Weeks. | ||||
| Sitagliptin | DPP4 inhibitor | MCD model[190] HFD model[191] CDAA model[192] | NCT02263677 (Withdrawn, 2016) | 4 | Change in liver steatosis. | |
| Anti-inflammatory and antioxidant effects | Alpha-lipoic acid | Nutraceutical | / | NCT04475276 (Not yet recruiting,2020) | 4 | Change in fatty liver grading in NAFLD assessed by abdominal ultrasound. |
| Immunomodulator | Selonsertib[175] | ASK1 inhibitor | / | NCT03053063 (Terminated, 2020) | 3 | Percentage of Participants Who Achieve a ≥ 1-Stage Improvement in Fibrosis According to the NASH Clinical Research Network (CRN) Classification Without Worsening of NASH. |
| Inflammation | Cenicriviroc | CCR2/5 antagonist | HFHF model[155] | NCT03028740 (Terminated, 2021) | 3 | Superiority of CVC compared to placebo on liver histology at Month 12 relative to the Screening biopsy; Superiority of CVC compared to placebo on the composite endpoint of histopathologic progression to cirrhosis, liver-related clinical outcomes, and all-cause mortality. |
6. Concluding remarks and future perspectives
The pathogenesis of NAFLD involves rather a complex interaction among different mechanisms, based on which the “multiple-hit” theory is the predominant theory. Mouse models are widely used because they are inexpensive, and convenient for genetic engineering. However, they also face the shortcomings due to the inability to fully replicate the characteristics of human NAFLD/NASH. As discussed, each mouse model has its own advantages or disadvantages, and continued efforts to establish new mouse models that can better recapitulate the process and mechanism of human NAFLD are urgently needed.
At the same time, the development of new technologies, such as 3-D organoid derived from human induced pluripotent stem cells (hiPSC), combined with animal models, will greatly accelerate the translation from basic science to clinical discoveries since hepatocytes are terminally differentiated. For example, Wang et al. [157] established a human NAFLD-like model based on a liver organoid chip system derived from hiPSC. This system can phenocopy the pathological features of NAFLD in liver organoids by prolonged exposure to FFA in perfused 3D-cultures. Likewise, Jarai et al. [158] co-cultured primary human hepatocytes with HSCs, Kupffer cells and liver sinusoid endothelial cells to produce a 3D human liver microtissue (3D-hLMT) system with NASH-like features. Due to the negative impact of this disease on human health and economic burden, NAFLD/NASH will become a high-priority area for future research worldwide.
Abbreviations
ABCG5: ATP-binding cassette sub-family G member 5; ASK1: apoptosis signal-related kinase 1; ATGL: adipose triglyceride lipase; CDAA: choline-deficient L-amino-defined diet; CDHFD: choline-deficient HFD; CVC: cenicriviroc; DCGI: Drug Control Center of India; DMN: dimethylnitrosamine; EBP: emopamil binding protein; ELA: elafibranor; FXR: farnesol X receptor; GVB: gut vascular barrier; GWAS: genome-wide association study; HFD: high-fat diet; HFE: high-fat emulsion; HFFD: high-fat and high-fructose diet; HFHC: high-fat and high-cholesterol diet; hiPSC: human induced pluripotent stem cells; HLF: human lung fibroblasts; HSC: hepatic stellate cell; HTGC: hepatic triglyceride content; IRS2: insulin receptor substrate 2; JNK: c-Jun N-terminal kinase; LDs: lipid droplets; LPS: lipopolysaccharides; LSEC: liver sinusoidal endothelial cells; MCD: methionine- and choline-deficient diet; MC4R-KO: melanocortin 4 receptor-deficient; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; OCA: obeticholic acid; PNPLA3: patatin-like phospholipase domain 3; PPARs: peroxisome proliferator-activated receptors; RA: rosemary acid; SAR: saroglitazar; SEL: selonsertib; TLR4: toll-like receptor 4; WD: western diet; WDF: WD supplemented with fructose; 3D-hLMT: three-dimensional (3D) human liver microtissue.
Acknowledgements
The authors are grateful to Prof. Chengxue Helena Qin from Monash Institute of Pharmaceutical Sciences, Monash University, for critical reading and constructive comments on this manuscript.
Funding
This study was supported by grants from National Natural Science Foundation of China (Grant Nos. 81941022, 81530025, 82070464) and Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB38010100). This work was also supported by the Program for Innovative Research Team of The First Affiliated Hospital of USTC (Grant No. CXGG02), Anhui Provincial Key Research and Development Program (Grant No. 202104j07020051), Anhui Province Science Fund for Distinguished Young Scholars (Grant No. 2208085J08) and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant No. 2017BT01S131).
Author contributions
TF conducted literature survey and wrote the paper under the supervision of JW and SX. HW, XP, and PJL provide insightful discussions and edits on this manuscript. JW and SX conceptualized and edit the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11-20
2. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J. et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 2018;69:896-904
3. Li J, Zou B, Yeo YH, Feng Y, Xie X, Lee DH. et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2019;4:389-98
4. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908-22
5. Kleiner DE, Makhlouf HR. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin Liver Dis. 2016;20:293-312
6. Kanda T, Goto T, Hirotsu Y, Masuzaki R, Moriyama M, Omata M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020 21
7. Abdelmalek MF. Nonalcoholic fatty liver disease: another leap forward. Nat Rev Gastro Hepat. 2021;18:85-6
8. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. 2018;53:362-76
9. Softic S, Cohen DE, Kahn CR. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig Dis Sci. 2016;61:1282-93
10. Huh JY, Reilly SM, Abu-Odeh M, Murphy AN, Mahata SK, Zhang JY. et al. TANK-Binding Kinase 1 Regulates the Localization of Acyl-CoA Synthetase ACSL1 to Control Hepatic Fatty Acid Oxidation. Cell Metab. 2020 32
11. Barbier-Torres L, Fortner KA, Iruzubieta P, Delgado TC, Giddings E, Chen Y. et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat Commun. 2020;11:3360
12. Gwag T, Reddy Mooli RG, Li D, Lee S, Lee EY, Wang S. Macrophage-derived thrombospondin 1 promotes obesity-associated non-alcoholic fatty liver disease. JHEP Rep. 2021;3:100193
13. Fan L, Lai RT, Ma NN, Dong YX, Li Y, Wu Q. et al. miR-552-3p modulates transcriptional activities of FXR and LXR to ameliorate hepatic glycolipid metabolism disorder. Journal of Hepatology. 2021;74:8-19
14. Fan L, Lai R, Ma N, Dong Y, Li Y, Wu Q. et al. miR-552-3p modulates transcriptional activities of FXR and LXR to ameliorate hepatic glycolipid metabolism disorder. J Hepatol. 2021;74:8-19
15. Long JK, Dai W, Zheng YW, Zhao SP. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol Med. 2019;25:26
16. Simon J, Goikoetxea-Usandizaga N, Serrano-Macia M, Fernandez-Ramos D, Saenz de Urturi D, Gruskos JJ. et al. Magnesium accumulation upon cyclin M4 silencing activates microsomal triglyceride transfer protein improving NASH. J Hepatol. 2021;75:34-45
17. Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA. et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019;218:3320-35
18. He A, Chen X, Tan M, Chen Y, Lu D, Zhang X. et al. Acetyl-CoA Derived from Hepatic Peroxisomal beta-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol Cell. 2020;79:30-42 e4
19. He AY, Chen XW, Tan M, Chen YL, Lu DL, Zhang XY. et al. Acetyl-CoA Derived from Hepatic Peroxisomal beta-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Molecular Cell. 2020;79:30 -+
20. Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A. et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018;28:588-604 e5
21. Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69:927-47
22. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081-6
23. Liu C, Zhou B, Meng M, Zhao W, Wang D, Yuan Y. et al. FOXA3 induction under endoplasmic reticulum stress contributes to non-alcoholic fatty liver disease. J Hepatol. 2021;75:150-62
24. Xu H, Tian Y, Tang D, Zou S, Liu G, Song J. et al. An Endoplasmic Reticulum Stress-MicroRNA-26a Feedback Circuit in NAFLD. Hepatology (Baltimore, Md). 2021;73:1327-45
25. Zhao Q, Liu J, Deng H, Ma R, Liao JY, Liang H. et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell. 2020;183:76-93 e22
26. Rostoker G, Loridon C, Griuncelli M, Rabate C, Lepeytre F, Urena-Torres P. et al. Liver Iron Load Influences Hepatic Fat Fraction in End-Stage Renal Disease Patients on Dialysis: A Proof of Concept Study. EBioMedicine. 2019;39:461-71
27. Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M. et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10:449
28. Li X, Wang TX, Huang X, Li Y, Sun T, Zang S. et al. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020;40:1378-94
29. Qi J, Kim JW, Zhou Z, Lim CW, Kim B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation-Mediated Cell Death in Mice. Am J Pathol. 2020;190:68-81
30. Yuan S, Wei C, Liu G, Zhang L, Li J, Li L. et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1alpha/SLC7A11 pathway. Cell Prolif. 2022;55:e13158
31. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (Baltimore, Md). 2011;54:133-44
32. Dong Z, Zhuang Q, Ye X, Ning M, Wu S, Lu L. et al. Adiponectin Inhibits NLRP3 Inflammasome Activation in Nonalcoholic Steatohepatitis via AMPK-JNK/ErK1/2-NFkappaB/ROS Signaling Pathways. Front Med (Lausanne). 2020;7:546445
33. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA. et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74:156-67
34. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669-82
35. Csak T, Dolganiuc A, Kodys K, Nath B, Petrasek J, Bala S. et al. Mitochondrial Antiviral Signaling Protein Defect Links Impaired Antiviral Response and Liver Injury in Steatohepatitis in Mice. Hepatology (Baltimore, Md). 2011;53:1917-31
36. Zhang X, Yang H, Zeng S, Tian S, Hu S, Yang L. et al. Melanoma-Differentiation-Associated gene 5 protects against nonalcoholic steatohepatitis in mice. Hepatology (Baltimore, Md). 2021
37. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M. et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021
38. Pfister D, Nunez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M. et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021
39. Royo F, Gil-Carton D, Gonzalez E, Mleczko J, Palomo L, Perez-Cormenzana M. et al. Differences in the metabolite composition and mechanical properties of extracellular vesicles secreted by hepatic cellular models. J Extracell Vesicles. 2019;8:1575678
40. Sung S, Kim J, Jung Y. Liver-Derived Exosomes and Their Implications in Liver Pathobiology. Int J Mol Sci. 2018 19
41. He Y, Rodrigues RM, Wang X, Seo W, Ma J, Hwang S. et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J Clin Invest. 2021 131
42. Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J. et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology (Baltimore, Md). 2020;72:412-29
43. Gu H, Yang K, Shen Z, Jia K, Liu P, Pan M. et al. ER stress-induced adipocytes secrete-aldo-keto reductase 1B7-containing exosomes that cause nonalcoholic steatohepatitis in mice. Free Radic Biol Med. 2021;163:220-33
44. Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A. et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol. 2019;71:1193-205
45. Liu XL, Pan Q, Cao HX, Xin FZ, Zhao ZH, Yang RX. et al. Lipotoxic Hepatocyte-Derived Exosomal MicroRNA 192-5p Activates Macrophages Through Rictor/Akt/Forkhead Box Transcription Factor O1 Signaling in Nonalcoholic Fatty Liver Disease. Hepatology (Baltimore, Md). 2020;72:454-69
46. Hendrikx T, Schnabl B. Antimicrobial proteins: intestinal guards to protect against liver disease. J Gastroenterol. 2019;54:209-17
47. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R. et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol. 2019;71:1216-28
48. Behary J, Amorim N, Jiang XT, Raposo A, Gong L, McGovern E. et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. 2021;12:187
49. Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE. et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671-84 e4
50. Zhou J, Cui S, He Q, Guo Y, Pan X, Zhang P. et al. SUMOylation inhibitors synergize with FXR agonists in combating liver fibrosis. Nat Commun. 2020;11:240
51. Bates J, Vijayakumar A, Ghoshal S, Marchand B, Yi S, Kornyeyev D. et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J Hepatol. 2020;73:896-905
52. Peng C, Stewart AG, Woodman OL, Ritchie RH, Qin CX. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front Pharmacol. 2020;11:603926
53. Cao L, Xu E, Zheng R, Zhangchen Z, Zhong R, Huang F. et al. Traditional Chinese medicine Lingguizhugan decoction ameliorate HFD-induced hepatic-lipid deposition in mice by inhibiting STING-mediated inflammation in macrophages. Chin Med. 2022;17:7
54. Lu Y, Wu Y, Chen X, Yang X, Xiao H. Water extract of shepherd's purse prevents high-fructose induced-liver injury by regulating glucolipid metabolism and gut microbiota. Food Chem. 2021;342:128536
55. Mukonowenzou NC, Dangarembizi R, Chivandi E, Nkomozepi P, Erlwanger KH. Administration of ursolic acid to new-born pups prevents dietary fructose-induced non-alcoholic fatty liver disease in Sprague Dawley rats. J Dev Orig Health Dis. 2021;12:101-12
56. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22:1707-18
57. Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C. et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology (Baltimore, Md). 2013;58:1632-43
58. Pummoung S, Werawatganon D, Chayanupatkul M, Klaikeaw N, Siriviriyakul P. Genistein Modulated Lipid Metabolism, Hepatic PPARgamma, and Adiponectin Expression in Bilateral Ovariectomized Rats with Nonalcoholic Steatohepatitis (NASH). Antioxidants (Basel). 2020 10
59. Khlifi R, Dhaouefi Z, Toumia IB, Lahmar A, Sioud F, Bouhajeb R. et al. Erica multiflora extract rich in quercetin-3-O-glucoside and kaempferol-3-O-glucoside alleviates high fat and fructose diet-induced fatty liver disease by modulating metabolic and inflammatory pathways in Wistar rats. J Nutr Biochem. 2020;86:108490
60. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y. et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752-61
61. Chen Q, Wang T, Li J, Wang S, Qiu F, Yu H. et al. Effects of Natural Products on Fructose-Induced Nonalcoholic Fatty Liver Disease (NAFLD). Nutrients. 2017 9
62. Diehl AM. Lessons from animal models of NASH. Hepatol Res. 2005;33:138-44
63. Im YR, Hunter H, de Gracia Hahn D, Duret A, Cheah Q, Dong J. et al. A Systematic Review of Animal Models of NAFLD Finds High-Fat, High-Fructose Diets Most Closely Resemble Human NAFLD. Hepatology (Baltimore, Md). 2021;74:1884-901
64. Ramadori P, Weiskirchen R, Trebicka J, Streetz K. Mouse models of metabolic liver injury. Lab Anim. 2015;49:47-58
65. Wang X, de Carvalho Ribeiro M, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A. et al. Macrophage-Specific Hypoxia-Inducible Factor-1alpha Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology (Baltimore, Md). 2019;69:545-63
66. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. 2019;129:546-55
67. Schneider KM, Mohs A, Kilic K, Candels LS, Elfers C, Bennek E. et al. Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis. Int J Mol Sci. 2019 20
68. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A. et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages(). J Hepatol. 2020;73:757-70
69. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simao AL, Gaspar MM, Majdi A. et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2020
70. Nakae D, Mizumoto Y, Andoh N, Tamura K, Horiguchi K, Endoh T. et al. Comparative changes in the liver of female Fischer-344 rats after short-term feeding of a semipurified or a semisynthetic L-amino acid-defined choline-deficient diet. Toxicol Pathol. 1995;23:583-90
71. Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N. et al. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology (Baltimore, Md). 2019;69:2241-57
72. Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C. et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int J Exp Pathol. 2013;94:93-103
73. Ulmasov B, Noritake H, Carmichael P, Oshima K, Griggs DW, Neuschwander-Tetri BA. An Inhibitor of Arginine-Glycine-Aspartate-Binding Integrins Reverses Fibrosis in a Mouse Model of Nonalcoholic Steatohepatitis. Hepatol Commun. 2019;3:246-61
74. Okishio S, Yamaguchi K, Ishiba H, Tochiki N, Yano K, Takahashi A. et al. PPARalpha agonist and metformin co-treatment ameliorates NASH in mice induced by a choline-deficient, amino acid-defined diet with 45% fat. Sci Rep. 2020;10:19578
75. Sugasawa T, Ono S, Yonamine M, Fujita SI, Matsumoto Y, Aoki K. et al. One Week of CDAHFD Induces Steatohepatitis and Mitochondrial Dysfunction with Oxidative Stress in Liver. Int J Mol Sci. 2021 22
76. Suzuki-Kemuriyama N, Abe A, Nakane S, Uno K, Ogawa S, Watanabe A. et al. Nonobese mice with nonalcoholic steatohepatitis fed on a choline-deficient, l-amino acid-defined, high-fat diet exhibit alterations in signaling pathways. FEBS Open Bio. 2021;11:2950-65
77. Min-DeBartolo J, Schlerman F, Akare S, Wang J, McMahon J, Zhan YT. et al. Thrombospondin-I is a critical modulator in non-alcoholic steatohepatitis (NASH). PloS one. 2019 14
78. Nishiyama K, Toyama C, Kato Y, Tanaka T, Nishimura A, Nagata R. et al. Deletion of TRPC3 or TRPC6 Fails to Attenuate the Formation of Inflammation and Fibrosis in Non-alcoholic Steatohepatitis. Biological & pharmaceutical bulletin. 2021;44:431-6
79. Li S, Li X, Chen F, Liu M, Ning L, Yan Y. et al. Nobiletin mitigates hepatocytes death, liver inflammation, and fibrosis in a murine model of NASH through modulating hepatic oxidative stress and mitochondrial dysfunction. J Nutr Biochem. 2021;100:108888
80. Yang M, Kimchi ET, Staveley-O'Carroll KF, Li G. Astaxanthin Prevents Diet-Induced NASH Progression by Shaping Intrahepatic Immunity. Int J Mol Sci. 2021 22
81. Liu L, Liu C, Zhao M, Zhang Q, Lu Y, Liu P. et al. The pharmacodynamic and differential gene expression analysis of PPAR alpha/delta agonist GFT505 in CDAHFD-induced NASH model. PloS one. 2020;15:e0243911
82. Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta. 2015;1852:1765-78
83. Omagari K, Asakawa E, Sasao M, Narita S, Hisano M, Fukuda A. et al. Age-Related Alterations of Nonalcoholic Steatohepatitis in Sprague-Dawley Rats Fed a High-Fat and High-Cholesterol Diet. J Nutr Sci Vitaminol (Tokyo). 2019;65:349-56
84. Kumar S, Verma AK, Rani R, Sharma A, Wang J, Shah SA. et al. Hepatic Deficiency of Augmenter of Liver Regeneration Predisposes to Nonalcoholic Steatohepatitis and Fibrosis. Hepatology (Baltimore, Md). 2020;72:1586-604
85. Lan T, Yu Y, Zhang J, Li H, Weng Q, Jiang S. et al. Cordycepin Ameliorates Nonalcoholic Steatohepatitis via Activation of AMP-Activated Protein Kinase Signaling Pathway. Hepatology (Baltimore, Md). 2021
86. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX. et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2021;70:761-74
87. Liang H, Jiang F, Cheng R, Luo Y, Wang J, Luo Z. et al. A high-fat diet and high-fat and high-cholesterol diet may affect glucose and lipid metabolism differentially through gut microbiota in mice. Exp Anim. 2021;70:73-83
88. Perakakis N, Joshi A, Peradze N, Stefanakis K, Li G, Feigh M. et al. The Selective Peroxisome Proliferator-Activated Receptor Gamma Modulator CHS-131 Improves Liver Histopathology and Metabolism in a Mouse Model of Obesity and Nonalcoholic Steatohepatitis. Hepatol Commun. 2020;4:1302-15
89. Kannt A, Wohlfart P, Madsen AN, Veidal SS, Feigh M, Schmoll D. Activation of thyroid hormone receptor-beta improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br J Pharmacol. 2021;178:2412-23
90. Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao CY. et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology. 2020;159:1487-503 e17
91. Jiang SY, Yang X, Yang Z, Li JW, Xu MQ, Qu YX. et al. Discovery of an insulin-induced gene binding compound that ameliorates nonalcoholic steatohepatitis by inhibiting sterol regulatory element-binding protein-mediated lipogenesis. Hepatology (Baltimore, Md). 2022
92. Kubota N, Kado S, Kano M, Masuoka N, Nagata Y, Kobayashi T. et al. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin Exp Pharmacol Physiol. 2013;40:422-30
93. Zhang G, Wang X, Chung TY, Ye W, Hodge L, Zhang L. et al. Carbon tetrachloride (CCl4) accelerated development of non-alcoholic fatty liver disease (NAFLD)/steatohepatitis (NASH) in MS-NASH mice fed western diet supplemented with fructose (WDF). BMC Gastroenterol. 2020;20:339
94. Sasaki R, Takami T, Fujisawa K, Matsumoto T, Ishikawa T, Yamamoto N. et al. Trans-portal hepatic infusion of cultured bone marrow-derived mesenchymal stem cells in a steatohepatitis murine model. J Clin Biochem Nutr. 2020;67:274-82
95. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S. et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol. 2018;69:385-95
96. Maeso-Diaz R, Boyer-Diaz Z, Lozano JJ, Ortega-Ribera M, Peralta C, Bosch J. et al. New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease. Cells. 2019 8
97. Lee S, Woo DC, Kang J, Ra M, Kim KH, Lee SR. et al. The Role of the Histone Methyltransferase EZH2 in Liver Inflammation and Fibrosis in STAM NASH Mice. Biology (Basel). 2020 9
98. Oniciu DC, Hashiguchi T, Shibazaki Y, Bisgaier CL. Gemcabene downregulates inflammatory, lipid-altering and cell-signaling genes in the STAM model of NASH. PloS one. 2018;13:e0194568
99. Segal-Salto M, Barashi N, Katav A, Edelshtein V, Aharon A, Hashmueli S. et al. A blocking monoclonal antibody to CCL24 alleviates liver fibrosis and inflammation in experimental models of liver damage. JHEP Rep. 2020;2:100064
100. Marquez-Quiroga LV, Arellanes-Robledo J, Vasquez-Garzon VR, Villa-Trevino S, Muriel P. Models of nonalcoholic steatohepatitis potentiated by chemical inducers leading to hepatocellular carcinoma. Biochemical pharmacology. 2022;195:114845
101. Hwang S, Wang X, Rodrigues RM, Ma J, He Y, Seo W. et al. Protective and Detrimental Roles of p38alpha Mitogen-Activated Protein Kinase in Different Stages of Nonalcoholic Fatty Liver Disease. Hepatology (Baltimore, Md). 2020;72:873-91
102. Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA. et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106-13
103. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R. et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884-97
104. Katsura A, Morishita A, Iwama H, Tani J, Sakamoto T, Tatsuta M. et al. MicroRNA profiles following metformin treatment in a mouse model of non-alcoholic steatohepatitis. Int J Mol Med. 2015;35:877-84
105. Pirola CJ, Fernandez Gianotti T, Castano GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M. et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64:800-12
106. He Y, Hwang S, Cai Y, Kim SJ, Xu M, Yang D. et al. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology (Baltimore, Md). 2019;70:1150-67
107. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG. et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197-208
108. Lindstrom P. The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal. 2007;7:666-85
109. Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y. et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16:44-54
110. Abe N, Kato S, Tsuchida T, Sugimoto K, Saito R, Verschuren L. et al. Longitudinal characterization of diet-induced genetic murine models of non-alcoholic steatohepatitis with metabolic, histological, and transcriptomic hallmarks of human patients. Biol Open. 2019 8
111. Martin SS, Qasim A, Reilly MP. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008;52:1201-10
112. Burke SJ, Batdorf HM, Burk DH, Noland RC, Eder AE, Boulos MS. et al. db/db Mice Exhibit Features of Human Type 2 Diabetes That Are Not Present in Weight-Matched C57BL/6J Mice Fed a Western Diet. J Diabetes Res. 2017;2017:8503754
113. Kang I, Park M, Yang SJ, Lee M. Lipoprotein Lipase Inhibitor, Nordihydroguaiaretic Acid, Aggravates Metabolic Phenotypes and Alters HDL Particle Size in the Western Diet-Fed db/db Mice. Int J Mol Sci. 2019 20
114. Girard D, Petrovsky N. Alstrom syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol. 2011;7:77-88
115. Legry V, Van Rooyen DM, Lambert B, Sempoux C, Poekes L, Espanol-Suner R. et al. Endoplasmic reticulum stress does not contribute to steatohepatitis in obese and insulin-resistant high-fat-diet-fed foz/foz mice. Clin Sci (Lond). 2014;127:507-18
116. Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology (Baltimore, Md). 2019;69:2427-41
117. Yang A, Mottillo EP, Mladenovic-Lucas L, Zhou L, Granneman JG. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat Metab. 2019;1:560-9
118. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A. 2019;116:9521-6
119. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T. et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130-44
120. Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC. et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab. 2019;22:49-61
121. Walker RW, Belbin GM, Sorokin EP, Van Vleck T, Wojcik GL, Moscati A. et al. A common variant in PNPLA3 is associated with age at diagnosis of NAFLD in patients from a multi-ethnic biobank. J Hepatol. 2020;72:1070-81
122. Sanchez-Pulido L, Ponting CP. TM6SF2 and MAC30, new enzyme homologs in sterol metabolism and common metabolic disease. Front Genet. 2014;5:439
123. Fan Y, Lu H, Guo Y, Zhu T, Garcia-Barrio MT, Jiang Z. et al. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology. 2016;150:1208-18
124. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A. et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352-6
125. Li BT, Sun M, Li YF, Wang JQ, Zhou ZM, Song BL. et al. Disruption of the ERLIN-TM6SF2-APOB complex destabilizes APOB and contributes to non-alcoholic fatty liver disease. PLoS Genet. 2020;16:e1008955
126. Liu S, Huang C, Li D, Ren W, Zhang H, Qi M. et al. Molecular cloning and expression analysis of a new gene for short-chain dehydrogenase/reductase 9. Acta Biochim Pol. 2007;54:213-8
127. Saloniemi T, Jokela H, Strauss L, Pakarinen P, Poutanen M. The diversity of sex steroid action: novel functions of hydroxysteroid (17beta) dehydrogenases as revealed by genetically modified mouse models. J Endocrinol. 2012;212:27-40
128. Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P. et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med. 2018;378:1096-106
129. Wang P, Wu CX, Li Y, Shen N. HSD17B13 rs72613567 protects against liver diseases and histological progression of nonalcoholic fatty liver disease: a systematic review and meta-analysis. Eur Rev Med Pharmacol Sci. 2020;24:8997-9007
130. Ma Y, Brown PM, Lin DD, Ma J, Feng D, Belyaeva OV. et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Deficiency Does Not Protect Mice From Obesogenic Diet Injury. Hepatology (Baltimore, Md). 2021;73:1701-16
131. Adam M, Heikela H, Sobolewski C, Portius D, Maki-Jouppila J, Mehmood A. et al. Hydroxysteroid (17beta) dehydrogenase 13 deficiency triggers hepatic steatosis and inflammation in mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2018;32:3434-47
132. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B. et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016;24:848-62
133. Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE. et al. A Therapeutic Silencing RNA Targeting Hepatocyte TAZ Prevents and Reverses Fibrosis in Nonalcoholic Steatohepatitis in Mice. Hepatol Commun. 2019;3:1221-34
134. Yang X, Sheng S, Du X, Su W, Tian J, Zhao X. Hepatocyte-specific TAZ deletion downregulates p62/ Sqstm1 expression in nonalcoholic steatohepatitis. Biochem Biophys Res Commun. 2021;535:60-5
135. Luo C, Sun H, Peng J, Gao C, Bao L, Ji R. et al. Rosmarinic acid exerts an antagonistic effect on nonalcoholic fatty liver disease by regulating the YAP1/TAZ-PPARgamma/PGC-1alpha signaling pathway. Phytother Res. 2021;35:1010-22
136. Puengel T, De Vos S, Hundertmark J, Kohlhepp M, Guldiken N, Pujuguet P. et al. The Medium-Chain Fatty Acid Receptor GPR84 Mediates Myeloid Cell Infiltration Promoting Steatohepatitis and Fibrosis. J Clin Med. 2020 9
137. Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R. et al. Dual PPARalpha/gamma agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018;38:1084-94
138. Kumar DP, Caffrey R, Marioneaux J, Santhekadur PK, Bhat M, Alonso C. et al. The PPAR alpha/gamma Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci Rep. 2020;10:9330
139. Anfuso B, Tiribelli C, Adorini L, Rosso N. Obeticholic acid and INT-767 modulate collagen deposition in a NASH in vitro model. Sci Rep. 2020;10:1699
140. Goto T, Itoh M, Suganami T, Kanai S, Shirakawa I, Sakai T. et al. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci Rep. 2018;8:8157
141. Morrison MC, Verschuren L, Salic K, Verheij J, Menke A, Wielinga PY. et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr-/-.Leiden Mice. Hepatol Commun. 2018;2:1513-32
142. Sun G, Jackson CV, Zimmerman K, Zhang LK, Finnearty CM, Sandusky GE. et al. The FATZO mouse, a next generation model of type 2 diabetes, develops NAFLD and NASH when fed a Western diet supplemented with fructose. BMC Gastroenterol. 2019;19:41
143. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF. et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956-65
144. Lambert G, Amar MJ, Guo G, Brewer HB Jr, Gonzalez FJ, Sinal CJ. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. The Journal of biological chemistry. 2003;278:2563-70
145. Li G, Thomas AM, Williams JA, Kong B, Liu J, Inaba Y. et al. Farnesoid X receptor induces murine scavenger receptor Class B type I via intron binding. PloS one. 2012;7:e35895
146. Papazyan R, Liu X, Liu J, Dong B, Plummer EM, Lewis RD 2nd. et al. FXR activation by obeticholic acid or nonsteroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. Journal of lipid research. 2018;59:982-93
147. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696-705
148. Vilsboll T, Garber AJ. Non-glycaemic effects mediated via GLP-1 receptor agonists and the potential for exploiting these for therapeutic benefit: focus on liraglutide. Diabetes Obes Metab. 2012;14(Suppl 2):41-9
149. Moreira GV, Azevedo FF, Ribeiro LM, Santos A, Guadagnini D, Gama P. et al. Liraglutide modulates gut microbiota and reduces NAFLD in obese mice. J Nutr Biochem. 2018;62:143-54
150. Luo Y, Yang P, Li Z, Luo Y, Shen J, Li R. et al. Liraglutide Improves Non-Alcoholic Fatty Liver Disease In Diabetic Mice By Modulating Inflammatory Signaling Pathways. Drug Des Devel Ther. 2019;13:4065-74
151. Somm E, Montandon SA, Loizides-Mangold U, Gaia N, Lazarevic V, De Vito C. et al. The GLP-1R agonist liraglutide limits hepatic lipotoxicity and inflammatory response in mice fed a methionine-choline deficient diet. Transl Res. 2021;227:75-88
152. Liu Q, Cai BY, Zhu LX, Xin X, Wang X, An ZM. et al. Liraglutide modulates gut microbiome and attenuates nonalcoholic fatty liver in db/db mice. Life Sci. 2020;261:118457
153. Tolbol KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP. et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J Gastroenterol. 2018;24:179-94
154. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z. et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology (Baltimore, Md). 2020;72:892-905
155. Wang Z, Park H, Bae EJ. Efficacy of evogliptin and cenicriviroc against nonalcoholic steatohepatitis in mice: a comparative study. Korean J Physiol Pharmacol. 2019;23:459-66
156. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J. et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (Baltimore, Md). 2018;67:1754-67
157. Wang Y, Wang H, Deng P, Tao T, Liu H, Wu S. et al. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater Sci Eng. 2020;6:5734-43
158. Mukherjee S, Zhelnin L, Sanfiz A, Pan J, Li Z, Yarde M. et al. Development and validation of an in vitro 3D model of NASH with severe fibrotic phenotype. Am J Transl Res. 2019;11:1531-40
159. Larter CZ, Yeh MM, Williams J, Bell-Anderson KS, Farrell GC. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J Hepatol. 2008;49:407-16
160. Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C. et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502-9
161. Baumgardner JN, Shankar K, Hennings L, Badger TM, Ronis MJ. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a high-polyunsaturated fat diet. Am J Physiol Gastrointest Liver Physiol. 2008;294:G27-38
162. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology (Baltimore, Md). 2009;50:1094-104
163. Bergheim I, Weber S, Vos M, Kramer S, Volynets V, Kaserouni S. et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol. 2008;48:983-92
164. Wada T, Kenmochi H, Miyashita Y, Sasaki M, Ojima M, Sasahara M. et al. Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology. 2010;151:2040-9
165. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300-8
166. Valenzuela R, Videla LA. Impact of the Co-Administration of N-3 Fatty Acids and Olive Oil Components in Preclinical Nonalcoholic Fatty Liver Disease Models: A Mechanistic View. Nutrients. 2020 12
167. Maity B, Basak M, Das K, Mahata T, Sengar AS, Verma SK. et al. RGS7-ATF3-Tip60 complex promotes hepatic steatosis and fibrosis by directly inducing TNFalpha. Antioxid Redox Signal. 2022
168. Polyzos SA, Kountouras J, Mantzoros CS. Leptin in nonalcoholic fatty liver disease: a narrative review. Metabolism: clinical and experimental. 2015;64:60-78
169. Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127-8
170. Arsov T, Larter CZ, Nolan CJ, Petrovsky N, Goodnow CC, Teoh NC. et al. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem Biophys Res Commun. 2006;342:1152-9
171. Kanda T, Goto T, Hirotsu Y, Masuzaki R, Moriyama M, Omata M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. Int J Mol Sci. 2020 21
172. Velazquez AM, Bentanachs R, Sala-Vila A, Lazaro I, Rodriguez-Morato J, Sanchez RM. et al. ChREBP-driven DNL and PNPLA3 Expression Induced by Liquid Fructose are Essential in the Production of Fatty Liver and Hypertriglyceridemia in a High-Fat Diet-Fed Rat Model. Mol Nutr Food Res. 2022;66:e2101115
173. Banini BA, Kumar DP, Cazanave S, Seneshaw M, Mirshahi F, Santhekadur PK. et al. Identification of a Metabolic, Transcriptomic, and Molecular Signature of Patatin-Like Phospholipase Domain Containing 3-Mediated Acceleration of Steatohepatitis. Hepatology (Baltimore, Md). 2021;73:1290-306
174. Tardelli M, Bruschi FV, Fuchs CD, Claudel T, Auer N, Kunczer V. et al. Absence of Adiponutrin (PNPLA3) and Monoacylglycerol Lipase Synergistically Increases Weight Gain and Aggravates Steatohepatitis in Mice. Int J Mol Sci. 2021 22
175. Newberry EP, Hall Z, Xie Y, Molitor EA, Bayguinov PO, Strout GW. et al. Liver-Specific Deletion of Mouse Tm6sf2 Promotes Steatosis, Fibrosis, and Hepatocellular Cancer. Hepatology (Baltimore, Md). 2021;74:1203-19
176. Kong L, Lu Y, Zhang S, Nan Y, Qiao L. Role of nutrition, gene polymorphism, and gut microbiota in non-alcoholic fatty liver disease. Discov Med. 2017;24:95-106
177. Shen H, Pollin TI, Damcott CM, McLenithan JC, Mitchell BD, Shuldiner AR. Glucokinase regulatory protein gene polymorphism affects postprandial lipemic response in a dietary intervention study. Hum Genet. 2009;126:567-74
178. Wu X, Zhang Y, Xing Y, Zhao B, Zhou C, Wen Y. et al. High-fat and high-glucose microenvironment decreases Runx2 and TAZ expression and inhibits bone regeneration in the mouse. J Orthop Surg Res. 2019;14:55
179. Wang L, Wang S, Shi Y, Li R, Gunther S, Ong YT. et al. YAP and TAZ protect against white adipocyte cell death during obesity. Nat Commun. 2020;11:5455
180. Hyun J, Al Abo M, Dutta RK, Oh SH, Xiang K, Zhou X. et al. Dysregulation of the ESRP2-NF2-YAP/TAZ axis promotes hepatobiliary carcinogenesis in non-alcoholic fatty liver disease. J Hepatol. 2021;75:623-33
181. Wang X, Zeldin S, Shi H, Zhu C, Saito Y, Corey KE. et al. TAZ-induced Cybb contributes to liver tumor formation in non-alcoholic steatohepatitis. J Hepatol. 2022;76:910-20
182. Hsiao PJ, Chiou HC, Jiang HJ, Lee MY, Hsieh TJ, Kuo KK. Pioglitazone Enhances Cytosolic Lipolysis, beta-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci Rep. 2017;7:9030
183. Tsuchiya S, Amano Y, Isono O, Imai M, Shimizu F, Asada M. et al. Pharmacological evaluation of pioglitazone and candesartan cilexetil in a novel mouse model of non-alcoholic steatohepatitis, modified choline-deficient, amino acid-defined diet fed low-density lipoprotein receptor knockout mice. Hepatol Res. 2017;47:584-92
184. Sumida Y, Okanoue T, Nakajima A, Japan Study Group of N. Phase 3 drug pipelines in the treatment of non-alcoholic steatohepatitis. Hepatol Res. 2019;49:1256-62
185. Roth JD, Veidal SS, Fensholdt LKD, Rigbolt KTG, Papazyan R, Nielsen JC. et al. Combined obeticholic acid and elafibranor treatment promotes additive liver histological improvements in a diet-induced ob/ob mouse model of biopsy-confirmed NASH. Sci Rep. 2019;9:9046
186. Hassan NF, Nada SA, Hassan A, El-Ansary MR, Al-Shorbagy MY, Abdelsalam RM. Saroglitazar Deactivates the Hepatic LPS/TLR4 Signaling Pathway and Ameliorates Adipocyte Dysfunction in Rats with High-Fat Emulsion/LPS Model-Induced Non-alcoholic Steatohepatitis. Inflammation. 2019;42:1056-70
187. Jain MR, Giri SR, Trivedi C, Bhoi B, Rath A, Vanage G. et al. Saroglitazar, a novel PPARalpha/gamma agonist with predominant PPARalpha activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol Res Perspect. 2015;3:e00136
188. Duparc T, Briand F, Trenteseaux C, Merian J, Combes G, Najib S. et al. Liraglutide improves hepatic steatosis and metabolic dysfunctions in a 3-week dietary mouse model of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2019;317:G508-G17
189. Liu C, Li C, Cai X, Zou Y, Mo J, Chen B. et al. Discovery of a novel GLP-1/GIP dual receptor agonist CY-5 as long-acting hypoglycemic, anti-obesity agent. Bioorg Chem. 2021;106:104492
190. Samano-Hernandez L, Fierro R, Marchal A, Gueant JL, Gonzalez-Marquez H, Gueant-Rodriguez RM. Beneficial and deleterious effects of sitagliptin on a methionine/choline-deficient diet-induced steatohepatitis in rats. Biochimie. 2021;181:240-8
191. Kawaguchi T, Nakano D, Koga H, Torimura T. Effects of a DPP4 Inhibitor on Progression of NASH-related HCC and the p62/ Keap1/Nrf2-Pentose Phosphate Pathway in a Mouse Model. Liver Cancer. 2019;8:359-72
192. Shimozato N, Namisaki T, Kaji K, Kitade M, Okura Y, Sato S. et al. Combined effect of a farnesoid X receptor agonist and dipeptidyl peptidase-4 inhibitor on hepatic fibrosis. Hepatol Res. 2019;49:1147-61
Author contact
![]() Corresponding authors: Jianping Weng, wengjpedu.cn (ORCID ID: 0000-0002-7889-1697); Suowen Xu, sxu1984edu.cn (ORCID ID: 0000-0002-5488-5217)
Corresponding authors: Jianping Weng, wengjpedu.cn (ORCID ID: 0000-0002-7889-1697); Suowen Xu, sxu1984edu.cn (ORCID ID: 0000-0002-5488-5217)