Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Statistical analysis
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(2):571-592. doi:10.7150/ijbs.70211 This issue Cite
Research Paper
Sesn2 Serves as a Regulator between Mitochondrial Unfolded Protein Response and Mitophagy in Intervertebral Disc Degeneration
Wen-Ning Xu1,2*, Chun Liu1*, Huo-Liang Zheng2*, Hai-Xia Xu1, Run-Ze Yang3, Sheng-Dan Jiang2 ![]() , Li-Xin Zhu1
, Li-Xin Zhu1 ![]()
1. Department of Spinal Surgery, Orthopedic Medical Center, Zhujiang Hospital, Southern Medical University, Guangzhou 510280, China.
2. Department of Clinic of Spine Center, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200082, China.
3. Department of Orthopedics, Orthopedic Research Institute, West China Hospital, Sichuan University.
*These authors contributed equally to this work.
Received 2021-12-18; Accepted 2022-10-30; Published 2023-1-1
Abstract

Mitochondrial unfold protein response (UPRmt) can induce mitophagy to protect cell from unfold protein. However, how UPRmt induces mitophagy to protect cell is not yet clear. Herein, Sesn2 was considered to be a key molecule that communicated UPRmt and mitophagy in the intervertebral disc. Silencing of Sesn2 was able to reverse the protective effects of Nicotinamide riboside (NR) on nucleus pulposus (NP) cells and inhibit mitophagy induced by UPRmt. UPRmt upregulated Sesn2 through Eif2ak4/eIF2α/Atf4, and further induced mitophagy. Sesn2 promoted the translocation of cytosolic Parkin and Sqstm1 to the defective mitochondria respectively, thereby enhancing mitophagy. The translocation of cytosolic Sqstm1 to the defective mitochondria was dependent on Parkin. The two functional domains of Sesn2 were necessary for the interaction of Sesn2 with Parkin and Sqstm1. The cytosolic interaction of Sesn2 between Parkin and Sqstm1 was independent on Pink1 (named as PINK1 in human) but the mitochondrial translocation was dependent on Pink1. Sesn2-/- mice showed a more severe degeneration and NR did not completely alleviate the intervertebral disc degeneration (IVDD) of Sesn2-/- mice. In conclusion, UPRmt could attenuate IVDD by upregulation of Sesn2-induced mitophagy. This study will help to further reveal the mechanism of Sesn2 regulating mitophagy, and open up new ideas for the prevention and treatment of IVDD.
Keywords: Intervertebral disc degeneration, Mitochondrial unfold protein response, Sesn2, Mitophagy.
Introduction
Intervertebral disc degeneration (IVDD) is a pathogenic process mediated by apoptotic cells [1]. Apoptosis of nucleus pulposus (NP) and annulus cells plays an important role in IVDD. The apoptosis of intervertebral disc cells destroys the homeostasis and metabolism of the intervertebral disc, including the synthesis of collagen and the production of extracellular matrix, and ultimately the intervertebral disc tissue cannot maintain the biological and mechanical integrity [2-4]. Apoptosis and autophagy are common in degenerative intervertebral discs. It is caused by many reasons, such as inflammatory nutrient depletion, biological and abiotic stress, and viral infection [5]. Therefore, the study of the mechanism of programmed intervertebral disc cell death will be a meaningful work.
Mitochondrial unfolded protein response (UPRmt) refers to the response that when the mitochondrial dysfunction occurs, the mitochondrial matrix will accumulate a large number of unfolded, misfolded, and ineffective protein-induced changes in gene transcription and cellular activity [6-8]. After oxidative stress-induced mitochondrial dysfunction, the mitochondrial matrix accumulates a large number of unfolded, misfolded, and ineffective proteins, leading to nuclear encoding of targeted mitochondrial chaperone proteins, such as heat shock protein family A member 9 (Hspa9, named as HSPA9 in human), heat shock protein 70 (Hspa1a, named as HSP70 in human), and proteases including caseinolytic mitochondrial matrix peptidase proteolytic subunit (Clpp, named as CLPP in human), YME1-like 1 ATPase (YME1-like 1 ATPase, Yme1l1, named as YME1L1 in human), Mitochondrial Lon protease-Like protein (Lonp1, named as LONP1 in human) and other proteins are up-regulated, which promotes the repair of mitochondrial function [9, 10], and delays aging [11].
Data from the past 5 years indicate that Atf4 is related to the integrated stress response (ISR) [12, 13]. Atf4 requires phosphorylation of eukaryotic translation initiation factor 2 α subunit (eIF2α), which is catalyzed by four kinases that respond to different cellular stresses. eIF2α kinase is activated by Eif2ak4 (named as GCN2 in human) and Eif2ak3 in the amino acid consumption and endoplasmic reticulum dysfunction, which in turn causes eIF2α to be phosphorylated in the molecular structure, and the number of phosphorylated eIF2α increases [14, 15]. The increase in phosphorylation of eIF2α leads to a decrease in overall protein synthesis and preferential translation of open reading frame mRNA in the 5'untranslated region, such as Atf4 mRNA [14]. A number of studies have shown that different forms of mitochondrial stress induce the expression of Atf4 and genes involved in UPRmt, such as mitochondrial protein homeostasis and metabolic remodeling genes [16]. Atf4 and Eif2ak3(named as PERK in human) have been shown to mediate the transcriptional induction of mitochondrial protein suppressor genes and the expression of genes involved in metabolic remodeling [17].
Previous studies have reported that Eif2ak3/Eif2ak4-eIF2α-Atf4 can induce Sesn2 (named as SESN2 in human) expression [18]. For example, nefinavir therapy induces Sesn2 expression [19] Atf4 ectopic expression induces transcriptional upregulation of Sesn2 expression, whereas Atf3 and Chop ectopic expression does not, suggesting that Atf4 regulates Sesn2 expression [20]. In some epithelial cancer cell lines, inhibitors of mitochondrial respiratory chain complex III (Myxothiazol) and I (Piericidin A) induce transcription of the P53-responsive Sesn2 gene, which plays an important role in stress response and homeostasis regulation [20, 21]. UPRmt and mitophagy eliminate severely damaged mitochondria and can be activated at the same time as a response to mitochondrial dysfunction. The events engineered by UPRmt and mitophagy are fairly clear (transcriptional adaptation promotes restoration of mitochondrial activity and degradation of severely damaged mitochondria) [22]. Yang et al have reported that mitochondrial ROS and endoplasmic reticulum stress (ERS) induced by denervation or rotenone can cause an increased expression of Sesn2 to mediate unfold protein response (UPR) in ER and mitophagy, which protect against denervated muscle atrophy [23]. However, how they coordinate to restore the mitochondrial network remains to be determined.
The selective removal or degradation of damaged mitochondria by autophagy is called mitophagy. Damaged mitochondria require timely and precise excision to maintain normal mitochondrial function [24, 25]. Mitophagy can play a defensive role by selectively removing damaged and dysfunctional mitochondria from cells, maintaining mitochondrial mass and thus maintaining mitochondrial physiological functions. In conclusion, mitophagy is essential for cells to clear abnormal mitochondria under stress conditions. Increased or accumulated mitochondrial damage is associated with the development and pathogenesis of many diseases [26]. Mitophagy is a double-edged sword. Excessive mitophagy aggravates advanced intervertebral disc degeneration [27, 28], while moderate mitophagy delays early and mid-term intervertebral disc degeneration [29-31]. Therefore, it is of great significance to study the role of UPRmt and mitophagy in IVDD and its potential connection.
In summary, we propose a research hypothesis: under pathological conditions, UPRmt activates mitophagy and antioxidant activity through Sesn2 to protect the survival of Nucleus Pulposus (NP) cells. The upregulation of UPRmt in the intervertebral disc cells may attenuates IVDD.
Materials and Methods
Patients and samples
4 Pfirrmann grade Ⅰ tissue samples were obtained from congenital scoliosis patient who had undergone HR (Hemivertebra Resection). 11 Pfirrmann grade Ⅱ tissue samples were obtained from patients who had undergone PELD (Percutaneous Endoscopic Lumbar Discectomy). 11 Pfirrmann grade Ⅲ tissue samples were obtained from patients who had undergone PELD or TLIF (Transforaminal Lumbar Interbody Fusion). 8 Pfirrmann grade Ⅳ tissue samples were obtained from patients who had undergone TLIF. IVDD samples were collected from 30 patients (14 males and 16 females; age 56.9 ± 10.8 years) with degenerative disc disease. The control samples were collected from 4 patients (3 males and 1 female; age 8.5 ± 3.0 years). Above patients signed the informed consent. This study was approved by the Human Ethics Committee of Xinhua Hospital and all protocols were performed according to Declaration of Helsinki.
Animals
Sesn2 knockout (Sesn2-/-) mice was generated by CRISPR/Cas9 system (Fig 1). To obtain Sesn2-/- mice, single-guide RNAs (sgRNAs) were designed targeting exon3-exon8 by using MEGAshortscriptTM Kit (Thermo Fisher Scientific, Waltham, MA, USA). The sequence of sgRNAs were listed in Table 1. In order to microinject Cas9 protein and sgRNA into fertilized eggs, male C57BL6/J mice were individually caged for 1 week before mating. Female C57BL6/J mice were given 5 IU of serum gonadotropin and 5 IU of human chorionic gonadotropin (HCG) to pregnant horses for superovulation. After C57BL6/J male and female mice were injected with HCG, the fertilized eggs of male and female mice were taken for mating. sgRNA (40ng/μl) and Cas9 protein (40ng/μl) were injected into the cytoplasm of the fertilized egg at the single-cell stage using continuous flow injection microinjection. The surviving two-cell embryo was implanted into the fallopian tube of a pseudo-pregnant female. The creation mice (F0) were analyzed in the born pups. F0 mice were mated with C57BL6/J wild-type mice, and F1 mice were bred to obtain F2 homozygous mutants. In order to maintain a stable inbred background, female mice will be backcrossed to their parental inbred strains every 10 generations.
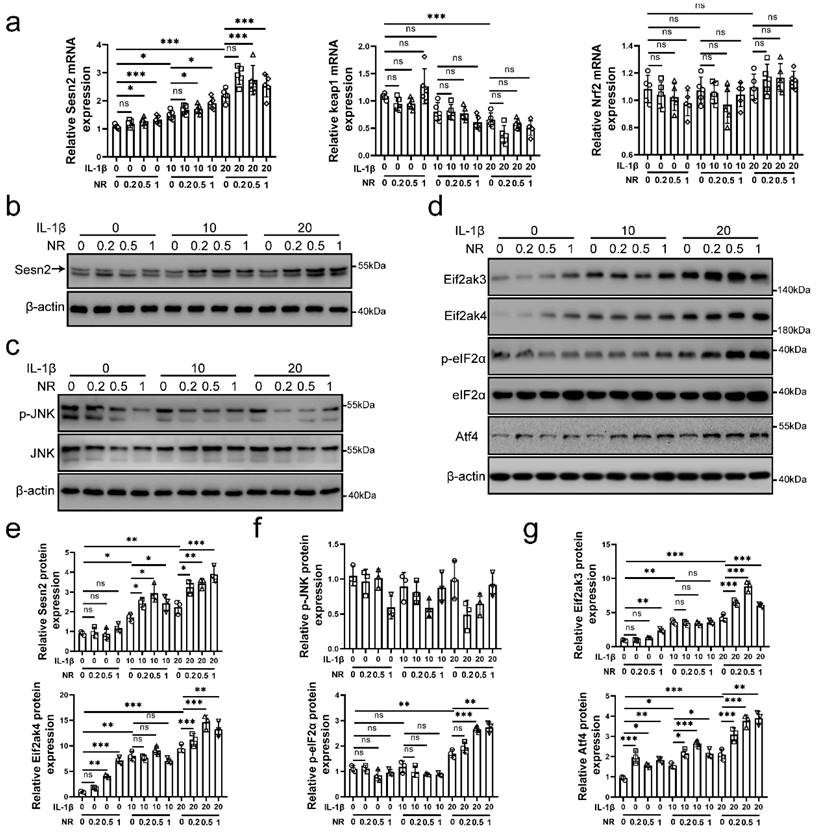
Sesn2 and eIF2α/Atf4 signal pathway was activated by IL-1β and NR. NP cells were divided into 3 groups and treated with 0 ng/ml, 10 ng/ml and 20 ng/ml IL-1β respectively. Each group was subdivided into 4 groups and treated with 0, 0.2, 0.5- and 1-mM NR respectively after IL-1β treatments. The experimental grouping settings were indicated. (a) The mRNA expression of Sesn2, Keap1 and Nrf2 in NP cells were detected by qRT-PCR (n = 5). (b) Sesn2 and β-actin protein expression in NP cells were detected by Western blot (n = 3). (c) p-JNK, JNK and β-actin in NP cells were detected by Western blot (n = 3). (d) Eif2ak3, Eif2ak4, p-eIF2α, eIF2α, Atf4 and β-actin in NP cells (n = 3). (e) The quantitative analysis of Sesn2 protein expression were showed. (f) The quantitative analysis of p-JNK protein expression were showed. (g) The quantitative analysis of Eif2ak3, Eif2ak4, p-eIF2α and Atf4 protein expression were showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
The sequences of sgRNA
| sgRNA | SgRNA sequence (5'-3') | PAM |
|---|---|---|
| SgRNA1 | AAGAACTAAGAATGGTCGCT | GGG |
| SgRNA2 | AGCTAGAGTACTTTGTTTGC | TGG |
| SgRNA3 | AAGAATGGTCGCTGGGACTG | GGG |
| SgRNA4 | AGTACTTTGTTTGCTGGAGT | AGG |
Cell culture
NP cells were separated from Sprague-Dawley rat disc tissues. The separation methods were performed according to our previous study [32]. Rat tissues were gained from male rats (6 weeks old and 200-250 g in weight). Disc was sliced as fragments. Fragments of rat dis tissues were digested with 0.1% collagenase for 4h. And then the tissues were removed as explants and placed in complete medium (DMEM/F12 and 10% fetal bovine serum (FBS) supplemented with antibiotics) at 37 °C in a 5% CO2 environment. Tissues were removed form plates after 1 week. Finally, primary passage cells were harvested by 0.25% trypsin-EDTA (1 mM) solution and replanted in appropriate culture plates once confluent.
Plasmids
The Flag-Sestrin2, Flag-Sestrin2-NTD (N-terminal domain, 1-220), Flag-Sestrin2-linker-CTD+L (C-terminal domain + linker, 220-480) and HA-Sestrin2-linker-CTD+L (C-terminal domain + linker, 220-480) mutants were constructed by cloning the cDNA of PGMLV-CMV-MCS-PGK-Puro.The Myc-Sqstm1 and Myc-Parkin were constructed by cloning the cDNA of PGMLV-CMV-MCS-PGK-Puro. Small interfering RNAs to Atf4, Sesn2 and Pink1 (si-Atf4, si-Sesn2 and si-Parkin) were designed and purchased from Genomeditech (Shanghai, China). Si-Pink1 and Si-Sqstm1 were purchased from GenePharma (Shanghai, China). The sequences of si-Atf4, si-Sesn2 and si-Parkin were listed in Table 2. Si-Pink1 and Si-Sqstm1 were listed in Table 3. Briefly, cDNA, si-RNAs and Lipofectamine 3000 (Thermo Fisher, USA) were added to Opti-MEM medium (Thermo Fisher, USA) respectively. And then they were mixed as mixtures and subsequently incubated for 20 min. Finally, the mixtures were added to plates.
The sequences of siRNA
| siRNA | Sense siRNA sequence (5'-3') | Antiense siRNA sequence (5'-3') |
|---|---|---|
| Si-Sesn2-1 | GCGAGAUCAACAAAUUACU tt | AGUAAUUUGUUGAUCUCGC tt |
| Si-Sesn2-2 | CCACGUUUGGAUAUGAAGA tt | UCUUCAUAUCCAAACGUGG tt |
| Si-Sesn2-3 | GAAAGGAACCUCAAGAUCU tt | AGAUCUUGAGGUUCCUUUC tt |
| Si-Atf4-1 | GCCUGACUCUGCUGCUUAU tt | AUAAGCAGCAGAGUCAGGC tt |
| Si-Atf4-2 | GCCUAGGUCUCUUAGAUGA tt | UCAUCUAAGAGACCUAGGC tt |
| Si-Atf4-3 | GAGUUAGUGUGACAGCUAA tt | UUAGCUGUCACACUAACUC tt |
| Si-Parkin-1 | CCAAAUUGCUCAGGAAUAA tt | UUCUGA GUCACUCUUGCUG tt |
| Si-Parkin-2 | CCAACUCCCUGAUUAAAGA tt | UCUUUAAUCAGGGAGUUGG tt |
The sequences of siRNA
| siRNA | siRNA sequence (5'-3') |
|---|---|
| Si-Pink1-1 | CGCAAATGTGCTTCATCTA |
| Si-Pink1-2 | CCTATGAAATCTTCGGGCT |
| Si-Sqstm1-1 | GCAUCUACAUUAAAGAGAA |
| Si-Sqstm1-2 | GUGGGAACU CGCUAUAAGU |
| Si-Sqstm1-3 | CCAACGUGAUUUGUGAUGG |
Lentivirus transfection
NP cells were grown until 80% confluence in six-well plates. After polybrene was added to the medium, 1 μM mRFP-GFP-LC-3 and Sesn2 lentivirus were added into plates and incubated for 48 h. NP cells transfected with mRFP-GFP-LC-3 lentivirus were used to determine the flux of autophagy in follow-up experiments. The green dot represents the initial autophagosome, and the red dot represents the mature autophagosome. NP cells transfected with Sesn2 lentivirus were used to overexpress the expression of Sesn2.
Quantitative Real-time PCR
Total RNA was obtained from NP cells and disc tissues by TRIzol reagent (TaKaRa, Inc., Dalian, China). Reverse transcription was performed to gain the first strand cDNA by using the PrimeScript RT Master Mix cDNA Synthesis Kit (TaKaRa, Inc., Dalian, China). The real-time PCR was operated on ABI 7500 system Applied Biosystems, Foster City, CA, USA) using SYBR1 Premix (TaKaRa, Inc., Dalian, China) according to manufacturer's protocol. The relative expression of target genes was quantified the 2-ΔΔCt methods after normalization to GAPDH. The sequence of specific primers was listed as Table 4.
The sequence of primers
| Gene | Forward (5'-3') | Reverse (5'-3') |
|---|---|---|
| Hspd1 | CACCACCACTGCCACTGTTCTG | CAACAGCCAACATCACACCTCTCC |
| Clpp | ACTGTGCACACGGACAGTAG | CTGGATCTTTGGCCGGACTT |
| Lonp1 | GGAGAAGACCATTGCGGCTA | AGATGTCGCGGTAGTGTTCC |
| Cox5a | TTGATGCCTGGGAGTTGCGTAAAG | ACAACCTCCAAGATGCGAACAGC |
| Sdhc | GCAAGAACCAAGACCGTCACTCC | TCCGACACTTGATGTGGGACCTAG |
| Ndufb5 | CCGTTGCCTACCCTACTTCC | GGAGCGACAGTCTTCGGAAA |
| SESN2 | CCGCTACATGACCTGACTCC | CTGCACATCACACACAAGCC |
| Sesn2 | ACACCCCGACTACCTTAGCA | AACTCGGTCATGTGGGAACC |
| ATF4 | TCAAACCTCATGGGTTCTCC | GTGTCATCCAACGTGGTCAG |
| GCN2 | GCAGCAGCATGTGTGTGAAA | CTTGCAAAAGGGATCCGCAG |
| Keap1 | AGCGCTACGATGTGGAAACA | GTCCAGGAACGTGTGACCAT |
| Nrf2 | GACCTAAAGCACAGCCAACACAT | CTCAATCGGCTTGAATGTTTGTC |
Mitochondrial protein extraction
Mitochondrial extraction was performed by Cell Mitochondrial Isolation Kit (Beyotime, Cat. No. C3601, China). NP cells were washed with PBS, digested with Trypsin-EDTA solution, centrifugated at room temperature for 5-10 minutes. After cells were collected, Cells were added with 1-2.5ml of mitochondrial separation reagent and place in an ice bath for 10-15 minutes. And then the suspension was transferred to an appropriately sized glass homogenizer for about 10-30 homogenates. The cell homogenate was centrifuged at 600g at 4℃ for 10 min. The supernatant was transferred carefully to another centrifuge tube and centrifuged at 11,000g for 4 min. The supernatant was removed carefully and the precipitate was the isolated mitochondria. The separated mitochondrial samples were lysed by RIPA containing PMSF and were used for Western blot.
Co-Immunoprecipitation
NP cells were added with appropriate amount of cell lysis buffer (containing protease inhibitor), lysed on ice for 30 minutes, and centrifuged at 4°C at maximum speed for 30 minutes and then collected the supernatant. A small amount of lysate was used to perform Western blot analysis and the remaining lysate were added 1μg of the corresponding antibody, and then incubated with slow shaking at 4°C overnight. 10μl of protein A agarose beads were washed 3 times with an appropriate amount of lysis buffer, and centrifuged at 3,000 rpm for 3 minutes each time. The cell lysate that had been incubated with the antibody overnight were added with the pretreated 10μl protein A agarose beads and incubated with slow shaking at 4°C for 2-4 h to couple the antibody to the protein A agarose beads. After the immunoprecipitation reaction, the lysate was centrifuged at 3,000 rpm at 4°C for 3 minutes. The supernatant was carefully aspirated and the agarose beads were washed with 1ml of lysis buffer 3-4 times. Finally, 15µl of 2×SDS loading buffer was added and boiled for 5 minutes. The samples were used to Western blot analysis.
Western blot analysis
30 µg protein was separated by SDS-polyacrylamide gel electrophoresis and electroblotted onto polyvinylidene difluoride membranes. Membranes containing target proteins were blocked using 5% non-fat milk for 2 hours and incubated with primary antibodies overnight at 4 ℃. The information primary antibodies were listed in Table 5. Membranes were washed with TBST three times and then incubated with corresponding horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 hours. The membranes of target protein were developed with ECL plus reagent (Millipore) on the ChemiDocTM XRS + System (Bio- Rad, USA).
Antibody information
| Antibodies human (mouse or rat) | Source | Item No |
|---|---|---|
| Anti-SESN2 (Sesn2) | Proteintech | Cat No. 10795-1-AP |
| Anti-Bcl-2 | Proteintech | Cat No. 26593-1-AP |
| Anti-Bax | Proteintech | Cat No. 50599-2-Ig |
| Anti-Sqstm1 | Proteintech | Cat. No. 18420-1-AP |
| Anti-Tom20 | Proteintech | Cat. No. 11802-1-AP |
| Anti-eIF2α | Cell Signaling Technology | Cat. No. #5324 |
| Anti-p-eIF2α | Cell Signaling Technology | Cat. No. #3398 |
| Anti-ATF4 (Atf4) | Cell Signaling Technology | Cat. No. #11815 |
| Anti-GCN2 (Eif2ak3) | Cell Signaling Technology | Cat. No. #3302 |
| Anti-p-JNK | Cell Signaling Technology | Cat. No. #4668 |
| Anti-JNK | Cell Signaling Technology | Cat. No. #9252 |
| Anti-Cleaved-Caspase 3 | Cell Signaling Technology | Cat. No. #9661 |
| Anti-Cleaved-Caspase 9 | Cell Signaling Technology | Cat. No. #9507 |
| Anti-Ubiquitin | Cell Signaling Technology | Cat. No. #3936 |
| Anti-Parkin | Abcam | Cat. No. ab77924 |
| Anti-Nqo1 | Abcam | Cat. No. ab80588 |
| Anti-Hmox1 | Abcam | Cat. No. ab13248 |
| Anti-Tubulin | Abcam | Cat. No. ab6160 |
| Anti-LC3 | Novus | Cat. No. NB100-2220 |
| Mouse Anti-IgG | Cell Signaling Technology | Cat No. #5946 |
| Mouse Anti-Sesn2 | Proteintech | Cat No. 66297-1-Ig |
| Anti-β-actin | Beyotime Bitotechnology | Cat. No. AF0003 |
| Mouse Anti-Flag | Cell Signaling Technology | Cat. No. #8146 |
| Rabbit Anti-Flag | Cell Signaling Technology | Cat. No. #14793 |
| Anti-Myc | abcam | Cat. No. ab32 |
| Anti-HA | Sigma | Cat. No. H6533 |
Flow cytometry assays
NP cell apoptosis was detected by an Annexin V-FITC apoptosis detection kit (BD, Cat. No. 556547, USA) according to manufacturer's protocol. After various treatments, NP cells were obtained and then washed by ice TBS for twice. Subsequently, 300 μl of 1 × binding buffer were added to resuspend the cells. Next, 5 μl of Annexin V-FITC and PI were added. The cells were vortexed gently, incubated for 30 min in the dark and then analyzed by flow cytometry.
TUNEL assays for apoptosis
NP cells were seeded on coverslips in six-well plates. After various treatments, cells were washed by PBS. Cells were fixed with 4% paraformaldehyde for 30 minutes and then washed once with PBS. Cells were permeabilized by PBS containing 0.3% Triton X-100 at room temperature for 5 minutes. Cells were washed by PBS for twice. Adding 50 μL of TUNEL detection solution to the samples and incubating at 37 °C for 60 minutes in the dark were performed. And then samples were added with DAPI and incubated for 5 minutes. Finally, images were taken by fluorescence microscopy (Olympus BX51).
ROS assay
Reactive oxygen species detection kit (Beyotime, China) was used to detect ROS production according to the manufacturer's protocol. After various treatments, NP cells were washed with PBS, incubated with DCFH-DA-containing serum-free medium for 20 min at 37°C, and then DCFH-DA was removed and washed 3 times with serum-free medium. The DCF fluorescence distribution of cells was detected by fluorescence microscopy analysis (Olympus Fluoview, Japan). Positive cells glow green.
JC-1 assay for mitochondrial membrane potential
The mitochondrial membrane potential (mtDY) was detected by JC-1 kit (C2006; Beyotime, China) according to the manufacturer's instructions. The NP cells were obtained and re-suspended in 500ul JC-1 staining fluid, and then incubated in the dark at 37 ℃ for 20 min. Cells were re-suspended in 500ul ice-cold staining buffer and detected by fluorescence microplate after washing with ice-cold staining buffer twice and centrifugation. In normal cells, mitochondria have a high mtDY and JC-1 formed orange-red fluorescent J-aggregates, while in cells with depolarized or damaged mitochondria, the sensor dye appeared as green fluorescent monomers.
Surgical procedure
Disc degenerative model were performed in Sesn2-/- mice as previously described [27]. All the experiments were abided by International Guiding Principles for Biomedical Research Involving Animals and approved by Ethics Committee of Xinhua Hospital Affiliated with Shanghai Jiao Tong University School of Medicine. Eighteen 8-week healthy adult mice were randomly divided into three groups: Sesn2 WT group + control Lentivirus, Sesn2-/- + control Lentivirus and Sesn2-/- + Lenti-Sesn2. The disc tissues of mice were obtained in 4 months old. Twenty-four 8-week healthy adult WT and Sesn2-/- mice were randomly divided into four groups: the sham operation group (skin incision), the sham operation + NR (skin incision + NR), the disc degenerative (surgical procedure) group and the disc degenerative + NR (surgical procedure + NR) group. Mice were fed with pellets containing vehicle or NR (400 mg/kg/day) for 2 months according to previous study [33]. The pellets were prepared by mixing powdered chow diet [34] with water or with NR dissolved in water. Pellets were dried under a laminar flow hood for 48 hours. The surgical procedure was performed according to previous study [27]. In brief, animals were anesthetized intraperitoneally using 0.1ml 1% pentobarbital (50 mg/kg). After 5 min, the spinous processes, sacrospinal muscles, interspinous ligaments, supraspinous ligaments, and posterolateral 1/2 of bilateral zygapophysial joints of the lumbar spine were removed through a dorsal medial approach. Skin incision served as a control. 2 months after surgery, the disc tissues of rats and mice were obtained in 4 months old.
Lentivirus-Sesn2 transfection in vivo
Lentivirus Sesn2 transfection was performed according to our previous study [27]. Surgical procedure after one weeks, the L4-5 discs were confirmed by a trial radiograph. Needles were used to puncture the L4-5 discs through the annulus fibrosus. The needles were fixed into the nucleus pulposus and 2 ul Lentivirus was then injected into the nucleus pulposus each two weeks until the rats were sacrificed. Finally, the needles were pulled out gently and slowly.
Safranin O-fast green staining
The disc specimens were decalcified with EDTA and fixed in 4% formaldehyde. The disc specimens were dehydrated in gradient solutions of ethyl alcohol and then embedded in paraffin. The specimens were cut into 5-μm sections continuously and then were stained with safranin O-fast green (S-O). The morphology of disc was evaluated by a blinded and experienced histological researcher.
Statistical analysis
The data are presented as the mean ± SD (standard deviation). Statistical analyses were performed on GraphPad prism 8. Multiple comparisons of data among the groups were analyzed by one-way ANOVA and Tukey's post hoc test. Independent-samples t-tests were used to analyze the differences between two groups. Differences were considered statistically significant when the P value was <0.05. ***p < 0.001, **p < 0.01, *p < 0.05. All experiments were performed at least three.
Results
Sesn2 might act as a bridge between mitochondrial unfolded protein response and mitophagy
Interleukin (IL)-1β is thought to lead to the senescence and apoptosis of NP cells, resulting in IVDD [35, 36]. As a NAD+-boosting compound, Nicotinamide riboside (NR) was reported to improving the level of UPRmt, which alleviates aging in mice [11, 37, 38]. Therefore, we performed 0.2, 0.5 and 1mM NR to treat NP cells for 6h after IL-1β treatments (0, 10 and 20 ng/ml) [39]. Sestrin 2 (Sesn2) has been reported to regulate the mitophagy [40-43]. Sesn2 senses oxidative stress from mitochondria, attenuates senescence and improves the life span in mammals [44, 45]. To investigate whether Sesn2 is involved in UPRmt, we detected the expression of Sesn2 in NP cells. As shown in Fig. 1a, b and e, IL-1β increased Sesn2 mRNA and protein expression. NR further enhanced Sesn2 mRNA and protein expression. These data showed that Sesn2 might be involved in UPRmt. However, the correlation between UPRmt and Sesn2 are still not clear.
In recent years, some studies have reported that Sesn2 promotes p62-dependent autophagy to degrade Keap1, thereby upregulating Nrf2 signaling and reducing ROS accumulation [46-48]. However, the results in this study showed that Keap1 and Nrf2 mRNA expression was not changed after NR treatments (Fig. 1a). These results confirmed that UPRmt did not induce mitophagy by Sesn2/keap1/Nrf2 signal pathway. JNK signal has been reported to regulate Sesn2 directly and UPRmt also regulate JNK [49, 50]. Western blot results disavowal above conjecture (Fig. 1c and f). JNK was down-regulated after IL-1β treatments and NR treatments. Eif2ak3/Eif2ak4-induced eIF2α-Atf4 signal pathway was regulated by UPRmt. UPRmt promotes the UPRmt markers including Clpp, Lonp1, Hspd1 (named as HSP60 in human) and so on via Eif2ak4-eIF2α-Atf4 signal pathway [10, 16, 17]. Our results confirmed this point that IL-1β increased Eif2ak3, Eif2ak4, p-eIF2α and Atf4 protein expression in NP cells and UPRmt inducer NR further enhanced their expression (Fig. 1d and g). Above results demonstrated that UPRmt, Eif2ak4-eIF2α-Atf4 signal pathway and Sesn2 might have some correlation.
SESN2 expression was decreased in human tissues of IVDD
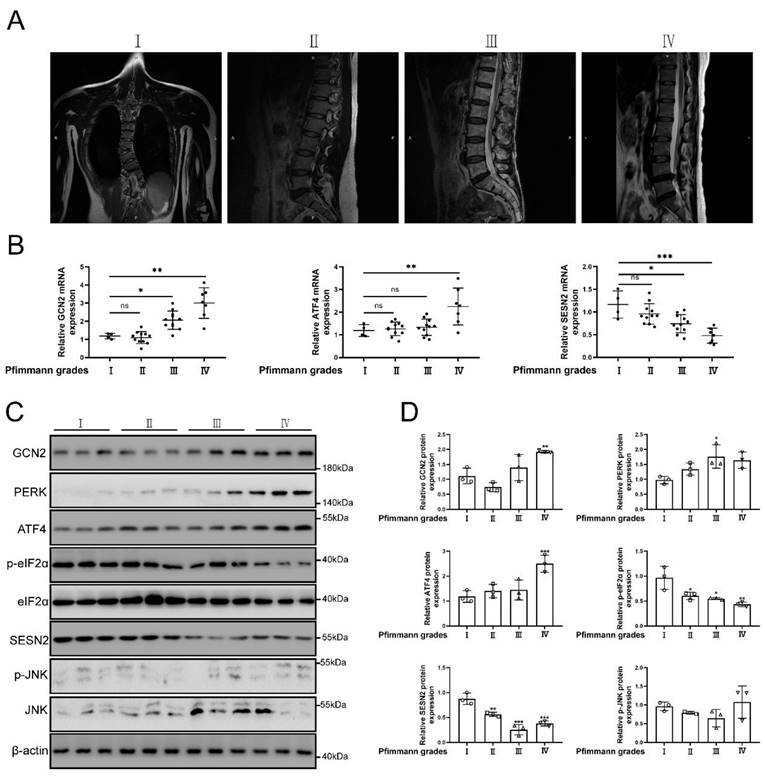
Grades of MRI imaging for IVDD referenced to the Pfirrmann scoring system [51]. The source of normal and degenerative intervertebral disc tissues in human was according to previous study [52]. The classification of intervertebral disc tissue according to the grades of Pfirrmann scoring system. The representative MRI of patients were shown in Fig. 2a. As the degree of degeneration increased, SESN2 and p-eIF2α (named as Sesn2 in rat and mouse) expression was decreased in human tissues of degenerative intervertebral discs (Fig. 2b, c and d). The expression of GCN2, PERK and ATF4 (named as Atf4 in rat and mouse) increased in human tissues of discs. The expression of JNK did not change significantly in human tissues of discs (Fig. 2b, c and d). This was an interesting finding. Eif2ak3/Eif2ak4-eIF2α-Atf4 signal pathway was also reported to regulate Sesn2 signal pathway [18, 20, 21, 53]. Based on these studies, the hypothesis that UPRmt activated Sesn2 through Eif2ak3/Eif2ak4-eIF2α-Atf4 signal pathway needed to be verified according to follow-up experiments.
The protein expression GCN2, ATF4 and SESN2 were confirmed. (a) The representing graphics of each Pfirrman grade was shown. (b) The mRNA expression of GCN2, ATF4 and SESN2 in the tissue of human IVDD were detected by qRT-PCR (n = 5). (c) The protein expression of GCN2, PERK, ATF4, p-eIF2α, eIF2α, SESN2, p-JNK, JNK and β-actin in the tissue of human IVDD were confirmed by Western blot (n = 3). (d) The quantitative analysis of GCN2, PERK, ATF4, p-eIF2α, SESN2 and p-JNK protein expression were showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
Silencing of Sesn2 reduced the protective effect of Nicotinamide riboside on NP cells
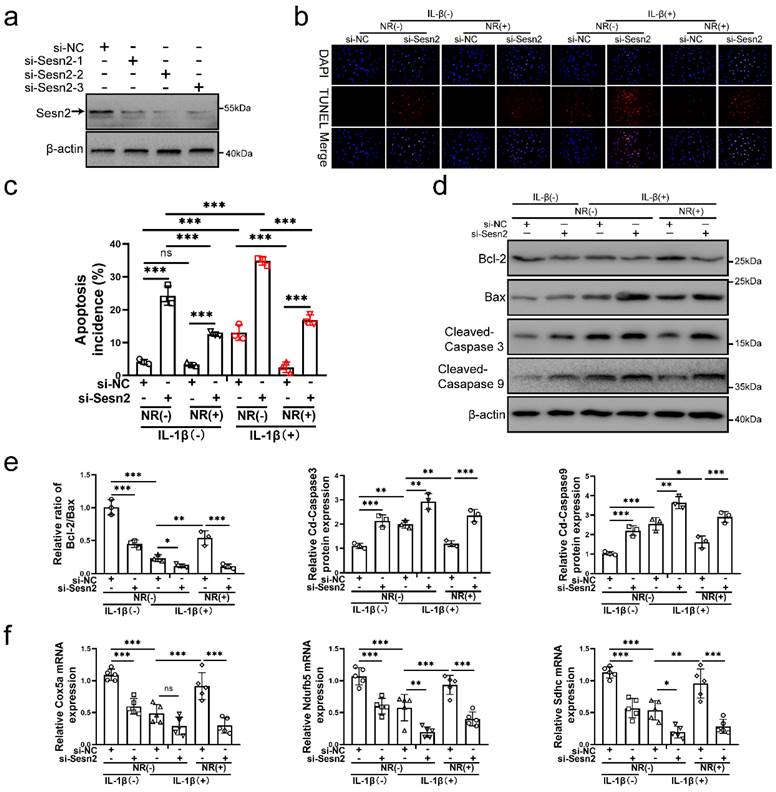
To confirm whether UPRmt protects NP cells via upregulating Sesn2, Sesn2 was silenced in NP cells before IL-1β and NR treatment. Sesn2 was silenced by si-Sesn2-1, si-Sesn2-2 and si-Sesn2-3 in NP cells (Fig. 3a). After Sesn2 was silenced, NR could not protect NP cells from IL-1β (Fig. 3b and c). Reduction of apoptosis by NR were partially destroyed in NP cells after Sesn2 knockdown. Regardless of whether it was treated by IL-1β, knocking down the expression of Sesn2 significantly increased the TUNEL positive rate of NP cells (Fig. 3b). Flow cytometry showed that Sesn2 knockdown increased the apoptosis rate (late + early) from 4.19 ± 0.37% to 24.30 ± 1.93% in the IL-1β (-) + NR (-) groups. IL-1β increased the rate of apoptosis (late + early) from 4.19 ± 0.37% to 13.08 ± 1.96%. NR reduced apoptosis (late + early) induced by IL-1β from 13.08 ± 1.96% to 2.51 ± 0.50%. However, Sesn2 knockdown increased the apoptosis rate (late + early) from 2.51 ± 0.50% to 16.88 ± 2.83% (Fig. 3c and Fig. S1a). The protein expression of Bcl-2, Bax and Cleaved-caspase3, Cleaved-caspase 9 bore out these results (Fig. 3d and e). To confirm the role of Sesn2 in mitochondria, some methods were used to determine mitochondrial function. Sesn2 knockdown could prevent repair of mitochondrial function induced by NR (Fig. 3f). Cox5a, Ndufb5 and Sdhc protein expression represents changes in mitochondrial function. The promotion of Cox5a, Ndufb5 and Sdhc protein expression caused by NR was reduced in NP cells after Sesn2 knockdown. The results of ROS Assay showed that Sesn2 knockdown significantly increased ROS production induced by IL-1β (Fig. S2a). JC-1 results also confirmed that Sesn2 knockdown could further increase the damage of mitochondrial membrane potential (Fig. S2b). These results showed Sesn2 served as an important role in the protection of UPRmt on NP cells.
Sesn2 knockdown reduced the protective effect of NR in NP cells. (a) Western blot to Sesn2 and β-actin in NP cells (n = 3). NP cells were transfected with si-NC, si-Sesn2-1, si-Sesn2-2 and si-Sesn2-3 respectively. (b-f) NP cells were transfected with si-NC or si-Sesn2, treated with 0 ng/ml and 20 ng/ml IL-1β for 48h and then treated with 0- and 1-mM NR for 6h. The experimental grouping settings were indicated. (b) TUNEL assays to NP cell apoptosis (n = 3). (c) The bar-chart of flow cytometry to NP cell apoptosis (n = 3). (d) Western blot to Bcl-2, Bax, Cleaved-caspase 3, Cleaved-caspase 9 and β-actin in NP cells (n = 3). (e) The quantitative analysis of Bcl-2/Bax, Cleaved-Caspase 3 and Cleaved-Caspase 9 protein expression were showed. (f) Mitochondrial functional mNRA expression including Cox5a, Ndufb5 and Sdhc were detected by qRT-PCR (n = 5). Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
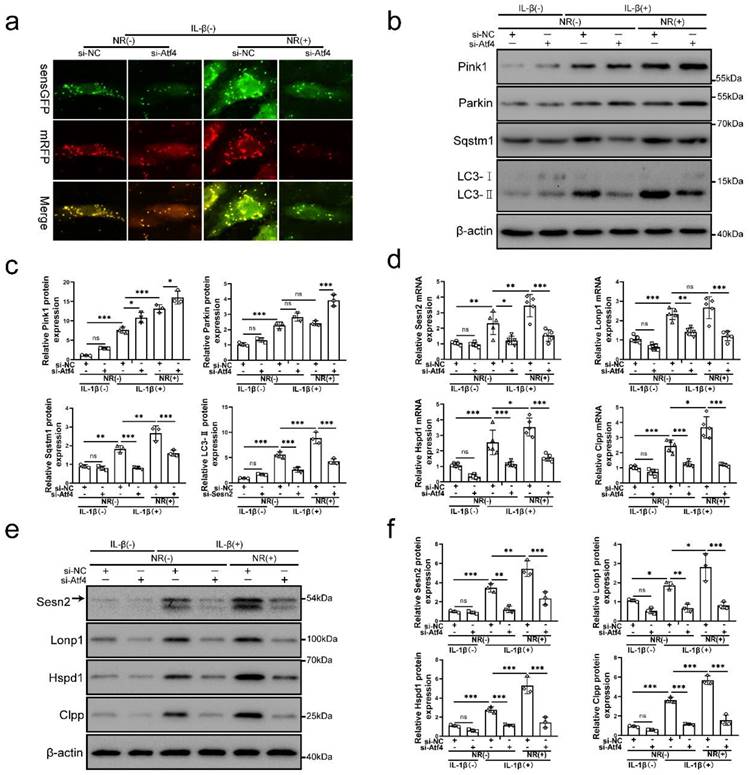
Silencing of Sesn2 prevented mitophagy induced by mitochondrial unfolded protein response
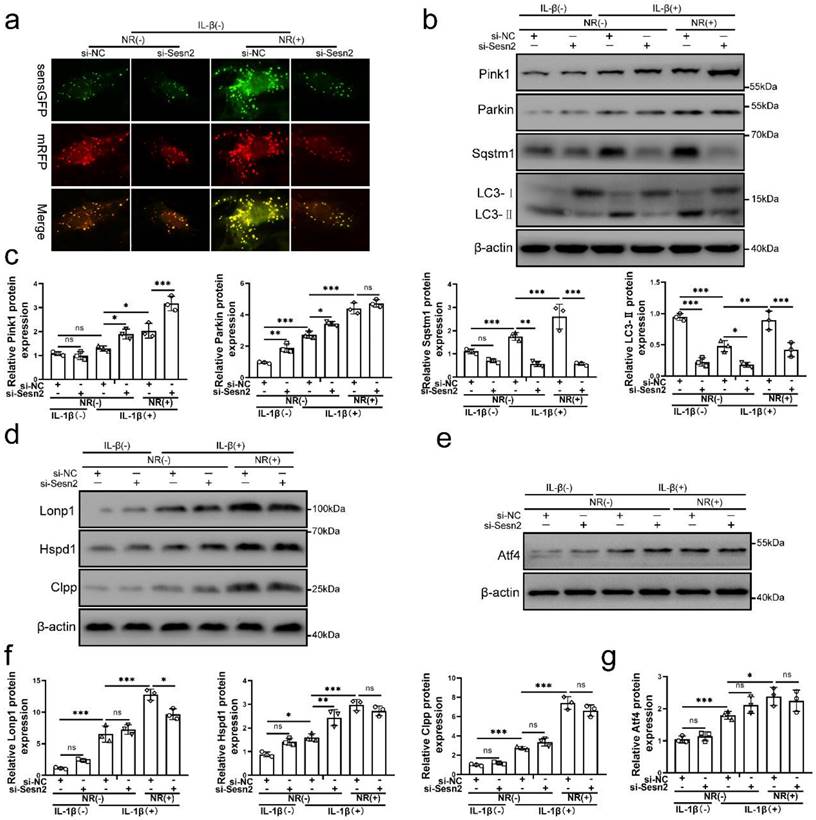
Sesn2 knockdown was able to inhibit the protective effect of UPRmt on NP cells. Importantly, NR-induced mitophagy was restraint by si-Sesn2 (Fig. 4a, b and c). However, the reduction in LC3-Ⅱ and Sqstm1 caused by si-Sesn2 was not accompanied by the low expression of Pink1 and Parkin. Silencing of Sesn2 caused an increased expression of Pink1 and Parkin, which intrigued us (Fig. 4b and c). When Sesn2 was silenced, the protein level of UPRmt such as Lonp1, Hspd1 and Clpp was not changed in NP cells (Fig. 4d and f). Silencing of Sesn2 did not change the protein expression of Atf4 in NP cells (Fig. 4e and g). These results suggested that UPRmt induced mitophagy to protect NP cells via Sesn2. According to our hypothesis, UPRmt might independently regulate Sesn2 and UPRmt marker molecules through Atf4. Sesn2 did not regulate UPRmt marker molecules. Sesn2-regulated mitophagy was one of the many downstream of UPRmt. Although the mechanism is not yet clear, UPRmt could target mitophagy via Sesn2 pathway.
Silencing of Sesn2 inhibited mitophagy induced by UPRmt in NP cells. (a) NP cells stably expressing the stubRFP-sensGFP-LC3 fusion protein were established and observed by the fluorescence microscope (n = 3). sensGFP is sensitive to the pH changes owing to the fusion of autophagosomes and lysosomes, whereas mRFP is stable. When autophagy was induced, autophagosomes and lysosomes were fused, sensGFP was quenched and mRFP was increased. The experimental grouping settings were indicated. (b-g) NP cells were transfected with si-NC or si-Sesn2, treated with 0 ng/ml and 20 ng/ml IL-1β for 48h and then treated with 0- and 1-mM NR for 6h. The experimental grouping settings were indicated. (b) Pink1, Parkin, Sqstm1, LC3 and β-actin were confirmed by Western blot (n = 3). (c) The quantitative analysis of Pink1, Parkin, Sqstm1 and LC3-Ⅱ protein expression were showed. (d) The protein expression of UPRmt markers including Lonp1, Hspd1, Clpp and β-actin were confirmed by Western blot (n = 3). (e) Atf4 and β-actin were confirmed by Western blot (n = 3). (f) The quantitative analysis of Lonp1, Hspd1 and Clpp protein expression were showed. (g) The quantitative analysis of Atf4 protein expression were showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
UPRmt upregulated Sesn2-mediated mitophagy through Eif2ak3/Eif2ak4-eIF2α-Atf4 signal pathway
To verify the role of Atf4 in UPRmt, Atf4 was silenced in NP cells before IL-1β and NR treatments. Atf4 was silenced by si-Atf4-1 and si-Atf4-2 in NP cells (Fig. 5a). NR could not protect NP cells from IL-1β after Atf4 knockdown (Fig. 5b and c). Reduction of apoptosis by NR were destroyed completely in NP cells after Atf4 knockdown. Regardless of whether it was treated by IL-1β, knocking down the expression of Atf4 also significantly increased the TUNEL positive rate of NP cells (Fig. 5b). Atf4 knockdown increased the apoptosis rate (late + early) from 3.83 ± 0.51% to 27.46 ± 1.19% in the IL-1β (-) + NR (-) groups. IL-1β increased the rate of apoptosis (late + early) from 3.83 ± 0.51% to 12.11 ± 2.90%. NR reduced apoptosis (late + early) induced by IL-1β from 12.11 ± 2.90% to 3.74 ± 0.38%. However, Atf4 knockdown increased the apoptosis rate (late + early) from 3.74 ± 0.38% to 33.09 ± 1.75% (Fig. 5c and Fig. S3a). The protein expression of Bcl-2, Bax and Cleaved-caspase3, Cleaved-caspase 9 bore out these results (Fig. 5d and e). These results revealed that Atf4 also reduced the protective effect of UPRmt on NP cells. Knockdown of Atf4 damaged NP cells more severely than Sesn2, which meant in addition to regulating Sesn2, Atf4 might protect NP cells through UPRmt markers or other ways. NR could protect partially NP cells after Sesn2 knockdown while NR could not protect completely NP cells after Atf4 knockdown.
Atf4 knockdown decreased the protective effect of NR in NP cells. (a) Western blot of Atf4 and β-actin in NP cells (n = 3). NP cells were transfected with si-NC, si-Atf4-1 and si-Atf4-2 respectively. (b-f) NP cells were transfected with si-NC or si-Atf4, treated with 0 ng/ml and 20 ng/ml IL-1β for 48h and then treated with 0- and 1-mM NR for 6h. The experimental grouping settings were indicated. (b) TUNEL assays to NP cell apoptosis (n = 3). (c) The bar-chart of flow cytometry to NP cell apoptosis (n = 3). (d) Western blot to Bcl-2, Bax, Cleaved-caspase 3, Cleaved-caspase 9 and β-actin in NP cells (n = 3). (e) The quantitative analysis of Bcl-2/Bax, Cleaved-Caspase 3 and Cleaved-Caspase 9 protein expression were showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
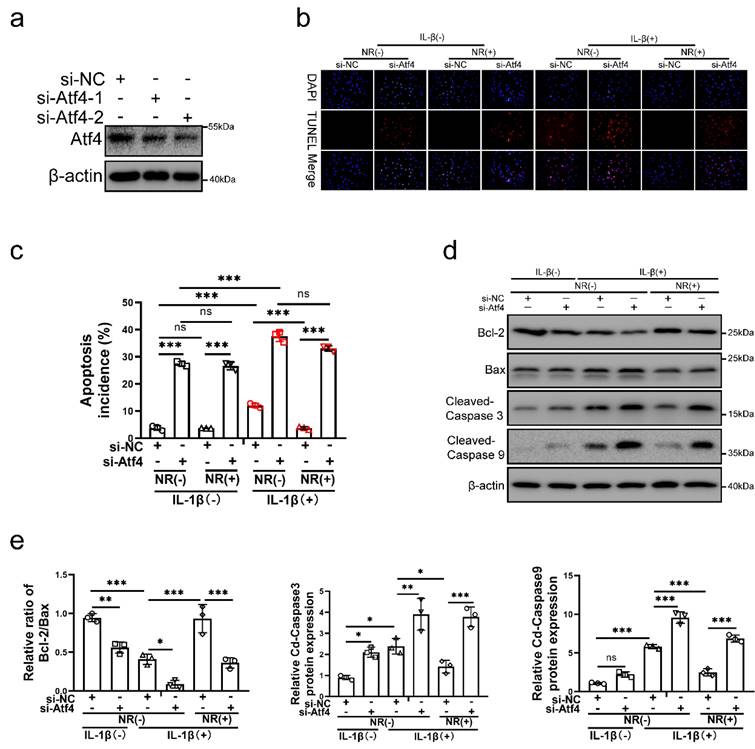
Atf4 was the common upstream of mitochondrial unfolded protein response marker molecules and Sesn2
To further investigate the underlying mechanism of Atf4 in this process, the level of mitophagy, Sesn2 and UPRmt markers were confirmed in NP cells after Atf4 knockdown. Atf4 knockdown inhibited mitophagy induced by IL-1β and NR (Fig. 6a, b and c). Atf4 knockdown reduced Sqstm1 and LC3-Ⅱ in NP cells after IL-1β and NR treatments. However, Pink1 and Parkin expression was increased in NP cells after Atf4 knockdown (Fig. 6b and c). This phenomenon was consistent with the findings that increased expression of Pink1 and Parkin were observed after Sesn2 knockdown (Fig. 5b and c). On the contrary, silencing of Atf4 could reduce the mRNA level of UPRmt markers and Sesn2 (Fig. 6d). Silencing of Atf4 also caused a reduction of UPRmt markers and Sesn2 protein expression (Fig. 6e and f). In summary, these results demonstrated that UPRmt upregulated Sesn2 expression through Eif2ak3/Eif2ak4-eIF2α-Atf4 signal pathway. Atf4 was the common upstream of UPRmt marker molecules and Sesn2. Atf4 not only protect NP cells through Sesn2-induced autophagy, it might be protected by UPRmt markers.
Atf4 served as an upstream of Sesn2, mitophagy and UPRmt markers. (a) NP cells stably expressing the stubRFP-sensGFP-LC3 fusion protein were established and observed by the fluorescence microscope (n = 3). (b-g) NP cells were transfected with si-NC or si-Atf4, treated with 0 ng/ml and 20 ng/ml IL-1β for 48h and then treated with 0- and 1-mM NR for 6h. The experimental grouping settings were indicated. (b) Pink1, Parkin, Sqstm1, LC3 and β-actin were confirmed by Western blot (n = 3). (c) The quantitative analysis of Pink1, Parkin, Sqstm1 and LC3-Ⅱ protein expression were showed. (d) The mRNA expression of Sesn2, UPRmt markers including Lonp1, Hspd1, Clpp were detected by qRT-PCR. (e) The protein expression of Sesn2, UPRmt markers including Lonp1, Hspd1, Clpp and β-actin were confirmed by Western blot (n = 3). (f) The quantitative analysis of Sesn2, Lonp1, Hspd1 and Clpp protein expression were showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
NR cannot attenuate the IVDD of Sesn2 KO mice through mitophagy
For determining the role of Sesn2 between UPRmt and mitophagy in vivo, we built Sesn2-/- mice and established IVDD model of Sesn2-/- mice (Fig. 7a). Sesn2 was not expressed in Sesn2-/- mice (Fig. S4a and b). Sesn2 knockout could not decrease UPRmt genes Clpp, Hspa1a and Lonp1 expression (Fig. 7b and c). Interestingly, Sesn2-/- mice with IVDD and treated with NR demonstrated an increased expression of UPRmt markers (Fig. 7b and c). These results proved in vivo that Sesn2 could not regulate UPRmt markers.
NR could not attenuate IVDD of Sesn2 KO mice. (a) Schematic diagram of animal experimental schedule. (b) UPRmt markers including Clpp, Lonp1, Hspa1a and β-actin protein expression were confirmed by Western blot. (c) The quantitative analysis of Clpp, Lonp1 and Hspa1a protein expression were showed. (d) Safranin O-fast green staining showed the structure of the intervertebral disc of mice (n = 6). (e) Western blot to Sesn2, Bcl-2, Bax, Nqo1, Homx1 and β-actin in the tissue samples of mouse discs (n = 3). (f) The quantitative analysis of Sesn2, Bcl-2/Bax, Nqo1 and Homx1 protein expression were showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
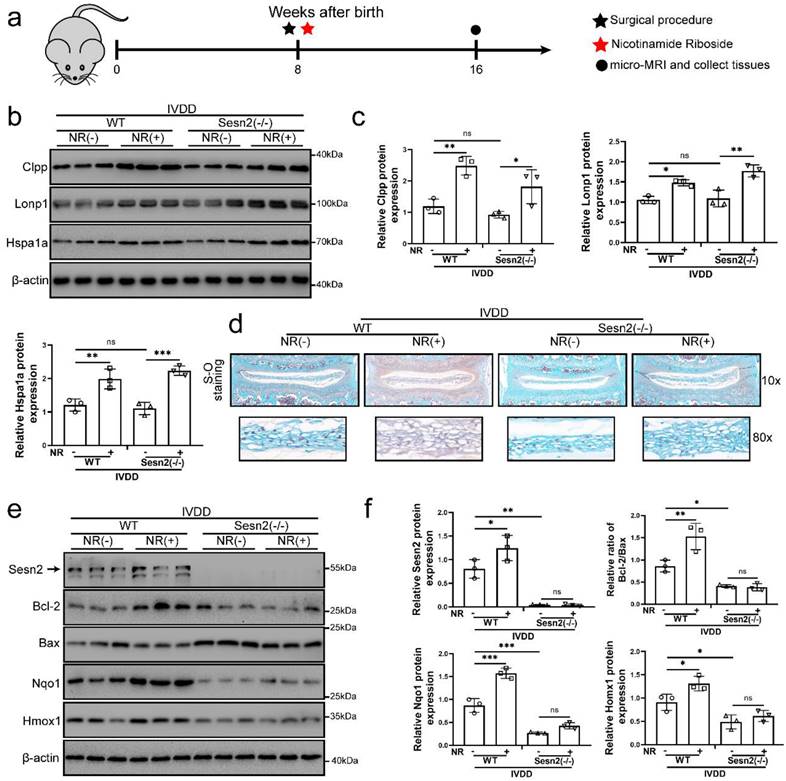
Normal nucleus pulposus of intervertebral disc is rich in proteoglycan. As a result, normal nucleus pulposus is stained red by safranin O. When the nucleus pulposus degenerates or is injured, proteoglycans in the nucleus pulposus are reduced, resulting in light or no staining of safranin O. In figure 7d, the proteoglycan in the nucleus pulposus of intervertebral disc was significantly decreased and the height of the nucleus pulposus was significantly reduced after surgery procedure. Intervertebral discs form of lumbar in WT mice fed with NR exhibited a more well-developed NP and a higher disc height than WT mice without NR treatments after surgery procedure (Fig. 7d). NR also promoted the levels of proteoglycan. Intervertebral discs from Sesn2-/- mice after surgery procedure suffered worse histological disrupted. Knockout of Sesn2 further reduced the level of proteoglycan and the height of the nucleus pulposus. Importantly, NR was able to attenuate the degeneration of intervertebral discs in WT mice but not Sesn2-/- mice (Fig. 7d). Meanwhile, NR could not protect intervertebral disc cell from apoptosis and could not enhance antioxidant genes expression too (Fig. 7e and f). There was a remarkable reduction in Nqo-1 and Homx-1 as well as increase in apoptotic markers in Sesn2-/- mice and Sesn2-/- + NR. Furthermore, NR could not promote mitophagy in the intervertebral disc of Sesn2-/- mice (Fig. S4c and d). These results confirmed that Sesn2 could not regulate UPRmt in the intervertebral disc but served as a downstream of UPRmt. Sesn2 was a bridge between UPRmt and mitophagy.
Sqstm1 and LC3-Ⅱ expression were decreased in Sesn2-/- mice while Parkin and Pink1 were increased in Sesn2-/- mice (Fig. S4e and f). To further confirm the function of Sesn2 in the association of UPRmt and mitophagy, Sesn2 was replied to express in the intervertebral disc of Sesn2-/- mice via injection of Lent-Sesn2. Recovery of mitophagy and reduction of apoptosis were observed in the intervertebral discs of Sesn2-/- mice after Sesn2 overexpression (Fig. S4e and f). Apoptotic genes Cleaved-caspase 3 and Cleaved-caspase 9 was increased in Sesn2-/- mice (Fig. S4e and f). Sesn2 overexpression increased Parkin, Sqstm1 and LC3-Ⅱ expression in Sesn2-/- (Fig. S4e and f). These results showed that mitophagy was decreased in the intervertebral disc of Sesn2-/- mice.
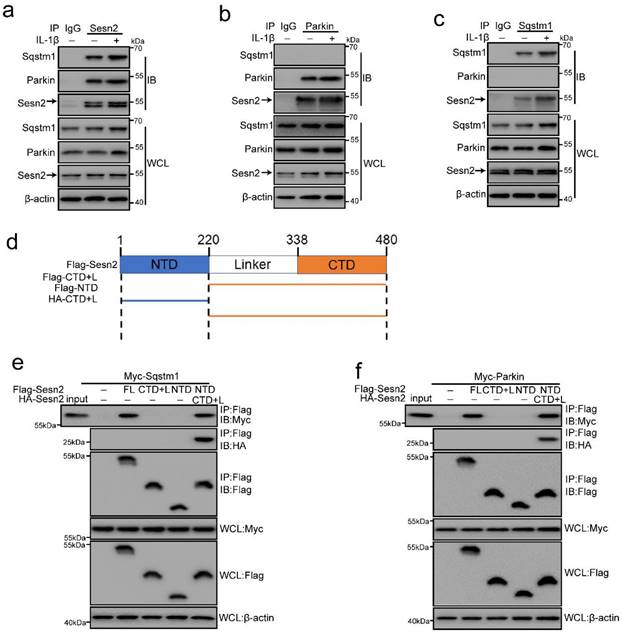
Sesn2 interacted with Sqstm1 and Parkin respectively in NP cells
It is reported in the literature that Sesn2 can regulate autophagy and mitophagy through Sqstm1 and Parkin [41, 42, 47]. Sesn2 can degrade Keap1 through Sqstm1-dependent autophagy, thereby increasing the level of Nrf2 in the cell [47]. Sesn2 can also promote the translocation of Parkin to mitochondria and induce mitophagy [41]. However, the interaction mechanism between Sesn2, Sqstm1 and Parkin has not yet been elucidated. Herein, Sesn2 was found to pull down Sqstm1 and Parkin (Fig. 8a). IL-1β enhanced the interaction Sesn2 with Sqstm1 and Parkin. This indicates that there might be an interaction between the three. However, it was interesting that Sqstm1 could pull down Sesn2, but not Parkin (Fig. 8b). Parkin was also able to pull down Sesn2, but not Sqstm1 (Fig. 8c). It showed that Sesn2, Sqstm1 and Parkin were not interacted together, but Sesn2 combined with Sqstm1 and Parkin respectively.
Sesn2 served as a Parkin and Sqstm1 interacting protein. (a) The interaction between Sesn2 and Parkin, Sqstm1 in NP cells treated with 0 or 20 ng/ml IL-1β for 48h was determined by Co-IP assays. Whole cell lysate (WCL) was used to perform IP of control serum (IgG) or anti-Sesn2 antibody, and then immunoblotting (IB) with indicator antibody to confirm the presence of a specific protein in the lysate. (b) The interaction between Parkin and Sesn2, Sqstm1 in NP cells treated with 0 or 20 ng/ml IL-1β for 48h was determined by Co-IP assays. WCL was used to perform IP of control serum (IgG) or anti-Parkin antibody, and then immunoblotting (IB) with indicator antibody to confirm the presence of a specific protein in the lysate. (c) The interaction between Sqstm1 and Sesn2, Parkin in NP cells treated with 0 or 20 ng/ml IL-1β for 48h was determined by Co-IP assays. WCL was used to perform IP of control serum (IgG) or anti-Sqstm1 antibody, and then immunoblotting (IB) with indicator antibody to confirm the presence of a specific protein in the lysate. (d) Sesn2 is a 55kda monomer, full alpha-helix, globular protein, containing different N-terminal [NTD, residues 66-220] and C-terminal [CTD, residues 339-480] domains connected by a partially disordered, partially helical linker region [linker, remaining 221-338]. (e) Immunoprecipitation of Sesn2 N- and C- terminal fragments. Transient transfection of full-length Flag-Sesn2 (FL), Flag-Sesn2-NTD (N-terminal domain,1-220), Flag-Sesn2-CTD + L (C-terminal domain plus Linker, 220-480) or Flag-Sesn2-NTD+HA-Sesn2-CTD +L and Myc-Sqstm1 to NP cells were treated with 20 ng/ mL IL-1β to prepare immunoprecipitation cell lysate. Immunoprecipitation and WCL were analyzed by Western blot for indicator protein detection results as shown (n = 3). (f) Immunoprecipitation of Sesn2 N- and C- terminal fragments. Transient transfection of full-length Flag-Sesn2 (FL), Flag-Sesn2-NTD (N-terminal domain,1-220), flag-Sesn2-CTD + L (C-terminal domain plus Linker, 220-480) or Flag-Sesn2-NTD+HA-Sesn2-CTD +L and Myc-Parkin to NP cells were treated with 20 ng/ mL IL-1β to prepare immunoprecipitation cell lysate. Immunoprecipitation and WCL were analyzed by western blotting for indicator protein detection results as shown (n = 3).
Sesn2 is a 55kda monomer, full alpha-helix, globular protein, containing different N-terminal [NTD, residues 66-220] and C-terminal [CTD, residues 339-480] domains connected by a partially disordered, partially helical linker region [linker, remaining 221-338] (Fig. 8d) [54]. In order to test the importance of intramolecular contact between the two domains of Sesn2, we expressed the two region of the labeled N- and C-terminus as separate polypeptides separately or together, and performed co-immunoprecipitation analysis. Although neither domain alone could bind Sqstm1 and Parkin, when the two separated domains are expressed together, they had strong binding to each other (Fig. 8e and f). Similarly, although the two regions of Sesn2 alone could not bind to Sqstm1 and Parkin, the two co-expressed regions could bind to Sqstm1 and Parkin (Fig. 8e and f). Therefore, the stable interaction between the N-terminal and C-terminal domains of Sesn2 was necessary for the interaction with Sqstm1 and Parkin.
Sesn2 promoted the translocation of cytosolic Parkin and Sqstm1 to the defective mitochondria to induce mitophagy
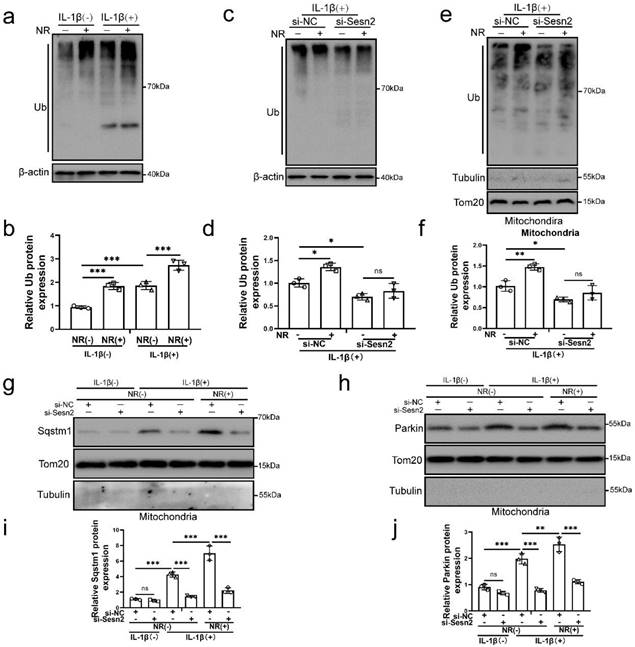
Mitochondrial ubiquitination plays an important role in mitophagy. Parkin is an E3 ubiquitin ligase. When mitochondrial fusion is blocked, mitochondrial subgroups are damaged and lose membrane potential, Parkin will translocation to damaged mitochondrial subgroups and mediate mitochondrial ubiquitination, thereby mediating mitophagy [55-57]. The results of our study found that NR and IL-1β increased the ubiquitination level of NP cells (Fig. 9a and b). After Sesn2 was knockdown, the ubiquitinated level in NP cells was weakened (Fig. 9c and d). Importantly, knockdown of Sesn2 reduced the mitochondrial ubiquitination of NP cells induced by NR and IL-1β (Fig. 9e and f). In order to further determine the relationship between Sesn2, Sqstm1 and Parkin in mitophagy, mitochondrial proteins were extracted from NP cells. Silencing of Sesn2 reduced the level of Sqstm1 and Parkin on the mitochondrial (Fig. 9g, h, i and j). These results demonstrated that Sesn2 interacted the cytosolic Parkin and Sqstm1 and translocated them to the defective mitochondria to induce mitochondrial ubiquitination, thereby increasing mitophagy.
Sesn2 improved the ubiquitination of mitochondria by promoting the transfer of Sqtms1 and Parkin to mitochondria. (a) The level of ubiquitination in NP cells was determined by Western blot (n = 3). The NP cells were divided into 2 groups, which were treated with 0 ng/ mL and 20 ng/ mL IL-1β for 48h, respectively, and then each group was further divided into 2 groups, which were treated with 0- and 1-mm NR for 6h, respectively. (b) The quantitative analysis of Ub protein expression was showed. (c) The level of ubiquitination in NP cells was determined (n = 3). NP cells were divided into two groups, transfected with si-NC and si-Sesn2, and all groups were received 20 ng/ml IL-1β treatment for 48h. And then each group was divided into two groups, treated with 0- and 1-mM NR for 6h. (d) The quantitative analysis of Ub protein expression was showed. (e) The ubiquitination level of mitochondria in NP cells was detected (n = 3). NP cells were divided into two groups, transfected with si-NC and si-Sesn2, and all groups were received 20 ng/ml IL-1β treatment for 48h. And then each group was divided into two groups, treated with 0- and 1-mM NR for 6h. (f) The quantitative analysis of Ub protein expression was showed. (g-j) NP cells were transfected with si-NC or si-Sesn2, treated with 0 ng/ml and 20 ng/ml IL-1β for 48h and then treated with 0- and 1-mM NR for 6h. The experimental grouping settings were indicated. (g) The protein expression of Sqstm1 was determined by Western blot (n = 3). (h) The protein expression of Parkin was determined by Western blot (n = 3). (i) The quantitative analysis of Sqstm1 protein expression was showed. (j) The quantitative analysis of Parkin protein expression was showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
The interaction of Sesn2 with Parkin and Sqstm1 was independent of each other
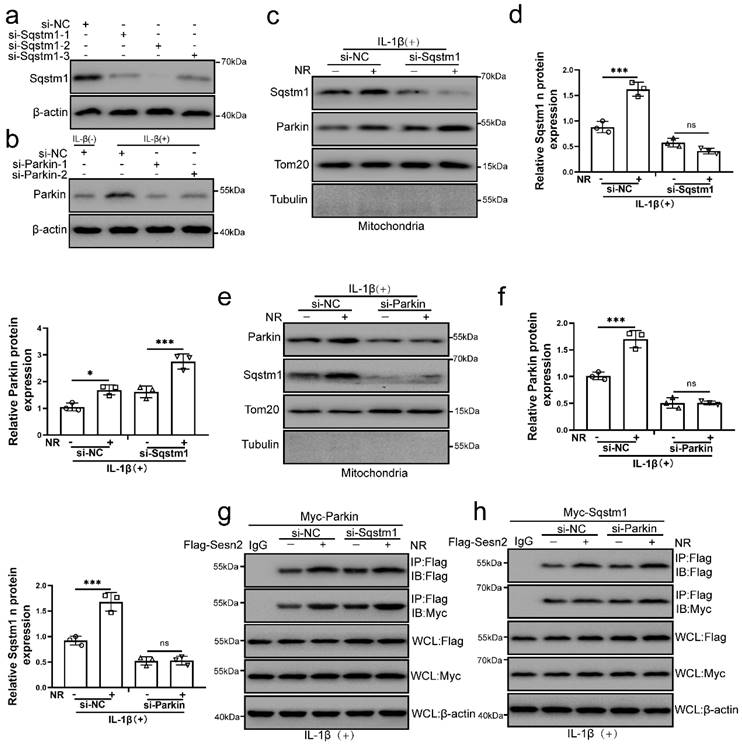
The main role of Sqstm1 in mitophagy is to promote mitochondrial aggregation by positioning in the mitochondrial membrane, and further amplify the efficiency and effect of mitophagy [58, 59]. Parkin is a key molecule that mediates mitochondrial autophagy [57]. It was found that NR and IL-1β promoted the localization of Sqstm1 and Parkin on the mitochondria of NP cells. To investigate whether Sqstm1 and Parkin play a role in the interaction process of Sesn2, Sqstm1 and Parkin was silenced by si-Sqstm1 and si-Parkin respectively (Fig. 10a). We subsequently found that Sqstm1 knockdown would not affect the translocation of Parkin to mitochondria, while Parkin knockdown would affect the translocation of Sqstm1 to mitochondria (Fig. 10b, c, d and e). These results indicated that Parkin was an essential molecule for the translocation of Sqstm1 to mitochondria. Interestingly, Sqstm1 knockdown did not affect the interaction of Sesn2 and Parkin, meanwhile Parkin knockdown did not affect the interaction of Sesn2 and Sqstm1 too (Fig. 10f and g). The interaction of Sesn2 with Parkin and Sqstm1 was independent of each other.
The interaction of Sesn2 with Parkin and Sqstm1 was independent of each other. (a) NP cells were transfected with si-NC, si-Sqstm-1, si-Sqstm-2 and si-Sqstm-3 respectively (n = 3). (b) NP cells were transfected with si-NC, si-Parkin-1 and si-Parkin-2 respectively (n = 3). NP cells were treated with 0 or 20 ng/ mL IL-1β for 48h and then 0- and 1-mm NR for 6h. The experimental grouping settings were indicated. (c) The protein level of Sqstm1 and Parkin in the mitochondria of NP cells were detected by Western blot (n = 3). NP cells were divided into two groups, transfected with si-NC and si-Sqstm1, and all groups were received 20 ng/ml IL-1β treatment for 48h. And each group was divided into two groups, treated with 0- and 1-mM NR for 6h. (d) The quantitative analysis of Sqstm1 and Parkin protein expression was showed. (e) The protein level of Sqstm1 and Parkin on the mitochondria of NP cells were detected (n = 3). NP cells were divided into two groups, transfected with si-NC and si-Parkin, and all groups were received 20 ng/ml IL-1β treatment for 48h. And then each group was divided into two groups, treated with 0- and 1-mM NR for 6h. (f) The quantitative analysis of Sqstm1 and Parkin protein expression was showed. (g) The level of Flag and Myc in WCL, IgG-IP and Flag-IP were determined respectively (n = 3). Firstly, Flag-Sesn2 and Myc-Parkin were transfected into NP cells and then transfected with si-NC and si-Pink1. All groups were received 20 ng/ml IL-1β treatment for 48h and then treated with 0 and 1mM NR for 6h. The experimental grouping settings were indicated. (h) The level of Flag and Myc in WCL, IG-IP and Flag-IP were determined respectively (n = 3). Firstly, Flag-Sesn2 and Myc-Sqstm1 were transfected into NP cells and then transfected with si-NC and si-Pink1. All groups were received 20 ng/ml IL-1β treatment for 48h and then treated with 0 and 1mM NR for 6h. The experimental grouping settings were indicated. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
The interaction of Sesn2 with Parkin and Sqstm1 were independent of Pink1
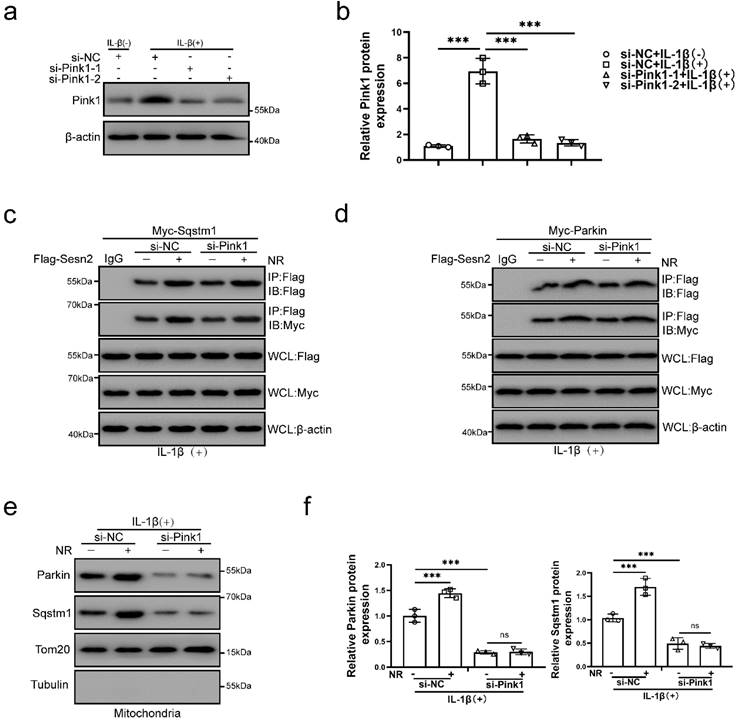
Pink1 is a key molecule that induces Parkin to localize to damage mitochondria [60]. To investigate the role of Pink1 in the interaction process, Pink1 was silenced by si-Pink1 in NP cells (Fig. 11a and b). However, we found that Pink1 knockdown did not affect the interaction of Sesn2 and Parkin (Fig. 11c and d). Silencing of Pink1 could reduce the location of Parkin and Sqstm1 on mitochondria (Fig. 11e and f). Based on the above results, we believed that Sesn2 promoted the translocation of Parkin and Sqstm1 to mitochondria, while Pink1 was the key molecule that attracted Parkin and Sqstm1 to transfer to mitochondria, but Pink1 did not affect the binding between Sesn2 with Parkin and Sqstm1. These results indicated that the interaction of Sesn2 with Parkin and Sqstm1 were independent of Pink1.
The interaction of Sesn2 with Parkin and Sqstm1 were independent of Pink1. (a) NP cells were transfected with si-NC, si-Pink1-1 and si-Pink1-2 respectively (n = 3). NP cells were treated with 0 or 20 ng/ml IL-1β treatment for 48h. The experimental grouping settings were indicated. (b) The quantitative analysis of Pink1 protein expression was showed. (c) The level of Flag and Myc in WCL, IG-IP and Flag-IP were detected respectively (n = 3). Firstly, Flag-Sesn2 and Myc-Sqstm1 were transfected into NP cells and then transfected with si-NC and si-Pink1. All groups were received 20 ng/ml IL-1β treatment for 48h and then treated with 0 and 1mM NR for 6h. The experimental grouping settings were indicated. (d) The level of Flag and Myc in WCL, IgG-IP and Flag-IP were detected respectively (n = 3). Firstly, Flag-Sesn2 and Myc-Parkin were transfected into NP cells and then transfected with si-NC and si-Pink1. All groups were received 20 ng/ml IL-1β treatment for 48h and then treated with 0 and 1mM NR for 6h. The experimental grouping settings were indicated. (e) The protein level of Sqstm1 and Parkin on the mitochondria of NP cells were detected (n = 3). NP cells were divided into two groups, transfected with si-NC and si-Pink1, and all groups were received 20 ng/ml IL-1β treatment for 48h. And then each group was divided into two groups, treated with 0- and 1-mM NR for 6h. (f) The quantitative analysis of Sqstm1 and Parkin protein expression was showed. Statistical significance was analyzed by one-way ANOVA followed by a post hoc Tukey's test. All data were presented as mean ± SEM (error bars). *p < 0.05; **p < 0.01; ***p<0.001.
Discussion
Both UPRmt and mitophagy play an important role in mitochondrial quality control [6]. During the mitochondrial dysfunction, UPRmt has the protective effect on cellular and organismal physiology, as well as protects the mitochondria by mitochondrial stress responses such as mitophagy [61]. UPRmt and mitophagy, eliminating severe impaired mitochondrial, can be activated simultaneously because both have a response in similar forms of mitochondrial dysfunction [62-64]. However, how they coordinate the recovery of the mitochondrial network remains to be determined.
The mechanism by which UPRmt induces mitophagy is not yet clear. UPRmt promotes the repair of mitochondrial function, improves the homeostasis of mitochondria, and removes unfolded, misfolded and ineffectively folded proteins within the mitochondria [65]. What has caught our attention is that since the UPRmt can protect the mitochondria, why does the UPRmt inducer promote the activation of mitophagy? After mitochondrial dysfunction, UPRmt induces molecular chaperone proteins, proteolytic enzymes, and mitophagy to repair mitochondrial functions and remove excessively damaged mitochondria [10]. Multiple studies have shown that different forms of mitochondrial stress induce Atf4 and other protein molecules to participate in the expression of UPRmt genes such as mitochondrial protein homeostasis and metabolic remodeling genes [66, 67]. Atf4 and Eif2ak3 have been shown to mediate transcriptional induction of mitochondrial protein suppressor genes and the expression of genes involved in metabolic remodeling. However, it is not clear how transcription factor regulates mitochondrial dysfunction to achieve a specific mitochondrial stress response. Previous literatures have reported that Eif2ak3/Eif2ak4-eIF2α-Atf4 can induce the expression of Sesn2 [18, 20, 21]. The ectopic expression of Atf4 caused the transcriptional upregulation of Sesn2, while the ectopic expression of Atf3 and Chop did not cause the transcriptional upregulation of Sesn2, indicating that Atf4 regulates the expression of Sesn2[19]. The role of Atf4 in upregulation of Sesn2 gene induced by mitochondrial dysfunction were confirmed [20]. Sesn2 is considered to be a key molecule that regulates mitophagy. Down-regulation of Sesn2 prevents Parkin from accumulating in damaged mitochondria [20]. Sesn2 can be used as a positive regulator of Parkin-mediated mitophagy [42]. In summary, we speculated that the UPRmt mediated mitophagy through Eif2ak3/Eif2ak4-eIF2α-Atf4-Sesn2 signal pathway.
More and more studies have shown that IL-1β participates in the inflammatory response in the process of IVDD [68, 69]. IL-1β is closely related to mechanical load, chronic inflammation, and participates in the vicious circle of IVDD [70]. IL-1β, one of the commonly used inflammatory factors, is widely used to simulate NP cell in vitro [36, 71]. Therefore, we used IL-1β to treat NP cells. To prove our hypothesis, we used the UPRmt inducer NR to further activate the UPRmt of NP cells. We found that IL-1β could induce the high expression of Sesn2, and NR could further promote the expression of Sesn2. At the same time, the expression trend of Eif2ak3/Eif2ak4-eIF2α-Atf4 signaling pathway is consistent with Sesn2. However, surprisingly, the expression of GCN2 and ATF4 in human IVDD tissues was inconsistent with the expression of SESN2. As the degree of IVDD increased, the expression of SESN2 gradually decreased while the expression of GCN and ATF4 gradually increased. It is reported in the literature that the eIF2α-Atf4 signaling pathway has the effect of protecting intervertebral disc cells in the early stage, and it can aggravate the damage of intervertebral disc cells in the late stage of degeneration [72]. In order to further confirm our hypothesis, Sesn2 was silenced in NP cells and found that NR did not inhibit IL-1β-induced apoptosis after Sesn2 knockdown. But Sesn2 knockdown did not completely prevent the effect of NR. This suggested that UPRmt does not completely protect NP cells through Sesn2. At the same time, the ability of NR to promote mitophagy was reduced. Silencing of Sesn2 did not change the level of UPRmt and Atf4 protein expression. These results indicated that Sesn2 might be a bridge between UPRmt and mitophagy. Knockdown of Atf4 has the same effect. However, Atf4 knockdown could completely destroy the effect of NR. Atf4 knockdown also reduced the protein level of UPRmt marker such as Lonp1, Hspd1 and Clpp. Moreover, Atf4 knockdown also inhibit the protein expression of Sesn2 in NP cells. More importantly, in Sens2-/- mice, disc degeneration was more severe than in WT mice, and the expression level of mitophagy genes was reduced. After NR feeding, the degeneration of the disc in Sens2-/- mice was not alleviated. NR could promote the level of UPRmt in the disc degeneration of Sesn2-/- mice, but could not increase the level of mitophagy in the disc degeneration of Sesn2-/- mice. It showed that UPRmt induced mitophagy through Sesn2 to alleviate the degeneration of the intervertebral disc in vivo. It is reported in the literature that the Eif2ak3/Eif2ak4-eIF2α-Atf4 signaling pathway conducts the mediating signal pathway of the UPRmt [6]. Mitochondrial dysfunction mediates UPRmt through Eif2ak3/Eif2ak4-eIF2α-Atf4. The expression of UPRmt genes restores mitochondrial function [6]. This indicated that Sesn2 might be one of the signaling pathways mediated by the Eif2ak3/Eif2ak4-eIF2α-Atf4 signaling pathway. Atf4 was a key molecule of UPRmt and Mitophagy coordinating effect, which communicates with mitophagy by Sesn2.
How does Sesn2 regulate mitophagy in NP cells? It is reported in the literature that Sesn2 can bind to Sqstm1-dependent autophagy to degrade Keap1, indicating that Sesn2 can bind to Sqstm1. Sesn2 can also accelerate the translocation of Parkin to mitochondria to promote mitophagy. We found that Sesn2 could interact with Sqstm1 and Parkin respectively. Although no single domain of Sesn2 could bind Parkin, when the two separated domains of Sesn2 were expressed together and they had strong binding to each other and to Parkin. Similarly, although the two separated domains of Sesn2 alone could not bind to Sqstm1, the two co-expressed domains were able to interact with Sqstm1. Therefore, the N-terminal and C-terminal domains of Sesn2 interacted stably, which were required for the interaction with Sqstm1 and Parkin. Sesn2 promoted the translocation of Sqstm1 and Parkin from the cytoplasm to the mitochondria. Parkin promoted the translocation of Sqstm1 to mitochondria, but Parkin did not affect the interaction of Sesn2 and Sqstm1. Pink1 also did not affect the interaction of Sesn2, Sqstm1 and Parkin in the cytoplasm. However, the translocation of Parkin and Sqstm1 to mitochondria needed to be mediated by Pink1.
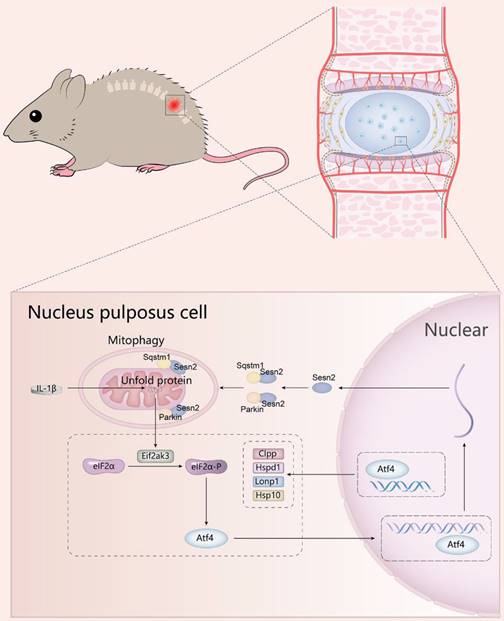
The translocation of Parkin to uncoupled mitochondria to induce mitophagy requires the activation of Pink1[73]. Pink1 is expressed and imported into all mitochondria, and then rapidly degraded by proteolysis and maintained at a very low level [74]. When a group of mitochondria is damaged, Pink1 proteolysis is inhibited, and Pink1 is allowed to accumulate in the damaged mitochondria, and Parkin is then recruited to the mitochondria. Combined with our discovery, Pink1 was the key to regulating the translocation of Parkin to mitochondria. Pink1 accumulated on impaired mitochondria, attracting Parkin aggregation and Sesn2 could accelerate the aggregation of Parkin into the mitochondrial. Subsequently the aggregation of Parkin promoted Sqstm1 located on mitochondria and Sesn2 was also able to accelerate this process. The interaction of Sesn2, Parkin and Sqstm1 was independent of each other. Pink1 did not regulate the interaction of them (Fig. 12).
A schematic diagram for the underlying mechanism of UPRmt in regulating IVDD. Mitochondrial unfold proteins stimulate UPRmt marker genes and Sesn2 through Eif2ak3/Eif2ak4-eIF2α-Atf4 signal pathway. The whole process is called the mitochondrial unfolded protein response. Sesn2 acts as a bridge between UPRmt and mitophagy. Sesn2 promoted the translocation of cytosolic Parkin and Sqstm1 to the defective mitochondria respectively, thereby enhancing mitophagy.
Why the protein expression of Pink1 and Parkin increased after Sesn2 knockdown? It is reported in the literature that Pink1 cannot successfully recruit Parkin into mitochondria, causing the expression of Pink1 and Parkin to gradually accumulate in the process of IVDD [75]. Pink1 accumulates in continuously damaged mitochondria, while Parkin cannot accumulate in damaged mitochondria. Negative feedback regulation further promotes the expression of Parkin and Pink1. On the other hand, the further high expression mediates excessive mitophagy. Our previous research findings confirmed this phenomenon. In the late stage of IVDD, other factors induced excessive mitophagy may aggravate the IVDD [27]. These two points explain why the expression of Pink1 and Parkin in human IVDD tissue samples increased with the increase of the degree of degeneration.
This study has the following points worthy of further improvement. Firstly, the role of UPRmt maker genes including Clpp, Hspd1, Lonp1 and so on in the protection of IVDD need to be confirmed. Secondly, how Pink1 regulates the adsorption of Sqstm1 and Parkin on mitochondria needs to be further clarified in NP cells.
Conclusion
UPRmt induced mitophagy to reduce apoptosis and improve the metabolism of NP cells via Sesn2 signaling pathway, and then alleviates disc degeneration. Disturbances in the regulatory mechanisms of UPRmt and mitophagy might be a risk factor in IVDD. UPRmt agonist NR has the potential to become a drug for the treatment of IVDD.
Abbreviations
UPRmt: Mitochondrial unfold protein response; IVDD: Intervertebral disc degeneration; NP: Nucleus pulposus; NR: Nicotinamide riboside; YME1L1: YME1-like 1 ATPase; Lonp1: Mitochondrial Lon protease-Like protein 1; CLPP: Caseinolytic mitochondrial matrix peptidase proteolytic subunit; HSPA9: Heat shock protein family A member 9; HSP70: Heat shock protein 70; ISR: Integrated stress response; eIF2α: Eukaryotic translation initiation factor 2 α subunit; PELD: Percutaneous Endoscopic Lumbar Discectomy; TLIF: Transforaminal Lumbar Interbody Fusion; HCG: human chorionic gonadotropin; CTD: C-terminal domain; NTD: N-terminal domain; S-O: safranin O-fast green; MRI: magnetic resonance imaging.
Supplementary Material
Supplementary figures.
Acknowledgements
Thanks for to all the colleagues in Room 402, Science and Education Building, Xinhua Hospital, Shanghai Jiaotong University School of Medicine. The study was supported by the National Natural Science Foundation of China (81672206, 8227090465 to SD Jiang) and 81974329 to LX Zhu) and Postdoctoral Research Foundation of China (2021M701630 to WN Xu).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Feng C, Yang M, Lan M, Liu C, Zhang Y, Huang B. et al. ROS: Crucial Intermediators in the Pathogenesis of Intervertebral Disc Degeneration. Oxidative medicine and cellular longevity. 2017;2017:5601593
2. Xi Y, Ma J, Chen Y. PTEN promotes intervertebral disc degeneration by regulating nucleus pulposus cell behaviors. Cell biology international. 2020;44:583-92
3. Lu L, Hu J, Wu Q, An Y, Cui W, Wang J. et al. Berberine prevents human nucleus pulposus cells from IL-1β-induced extracellular matrix degradation and apoptosis by inhibiting the NF-κB pathway. International journal of molecular medicine. 2019;43:1679-86
4. Liu Z, Zhang Z, Zhang A, Zhang F, Du W, Zhang Y. et al. Osteogenic protein-1 alleviates high glucose microenvironment-caused degenerative changes in nucleus pulposus cells. Bioscience reports. 2019 39
5. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell death and differentiation. 2015;22:367-76
6. Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nature reviews Molecular cell biology. 2018;19:109-20
7. Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response-synchronizing genomes. Current opinion in cell biology. 2015;33:74-81
8. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. The EMBO journal. 2002;21:4411-9
9. Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y. et al. The Mitochondrial Unfolded Protein Response Is Mediated Cell-Non-autonomously by Retromer-Dependent Wnt Signaling. Cell. 2018;174:870-83.e17
10. Melber A, Haynes CM. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell research. 2018;28:281-95
11. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P. et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science (New York, NY). 2016;352:1436-43
12. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M. et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11:619-33
13. Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. The Journal of biological chemistry. 2010;285:33165-74
14. Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S. et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Molecular and cellular biology. 2002;22:6681-8
15. Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D. et al. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15920-5
16. Bozi LHM, Campos JC, Gross ER, Ferreira JCB. Mitochondrial Unfolded Protein Response (UPR(mt)) Activation in Cardiac Diseases: Opportunities and Challenges. Journal of the American College of Cardiology. 2019;74:1011-2
17. Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK. et al. The Unfolded Protein Response Mediator PERK Governs Myeloid Cell-Driven Immunosuppression in Tumors through Inhibition of STING Signaling. Immunity. 2020;52:668-82.e7
18. Jin HR, Du CH, Wang CZ, Yuan CS, Du W. Ginseng metabolite Protopanaxadiol induces Sestrin2 expression and AMPK activation through GCN2 and PERK. Cell death & disease. 2019;10:311
19. Brüning A, Rahmeh M, Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Molecular oncology. 2013;7:1012-8
20. Garaeva AA, Kovaleva IE, Chumakov PM, Evstafieva AG. Mitochondrial dysfunction induces SESN2 gene expression through Activating Transcription Factor 4. Cell cycle (Georgetown, Tex). 2016;15:64-71
21. Kim HJ, Joe Y, Kim SK, Park SU, Park J, Chen Y. et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2α-ATF4 pathway. Free radical biology & medicine. 2017;110:81-91
22. Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233-40
23. Yang X, Xue P, Yuan M, Xu X, Wang C, Li W. et al. SESN2 protects against denervated muscle atrophy through unfolded protein response and mitophagy. Cell death & disease. 2021;12:805
24. Campos JC, Bozi LH, Bechara LR, Lima VM, Ferreira JC. Mitochondrial Quality Control in Cardiac Diseases. Frontiers in physiology. 2016;7:479
25. Springer MZ, Macleod KF. In Brief: Mitophagy: mechanisms and role in human disease. The Journal of pathology. 2016;240:253-5
26. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241-52
27. Xu WN, Zheng HL, Yang RZ, Liu T, Yu W, Zheng XF. et al. Mitochondrial NDUFA4L2 attenuates the apoptosis of nucleus pulposus cells induced by oxidative stress via the inhibition of mitophagy. Experimental & molecular medicine. 2019;51:1-16
28. Xu WN, Yang RZ, Zheng HL, Yu W, Zheng XF, Li B. et al. PGC-1α acts as an mediator of Sirtuin2 to protect annulus fibrosus from apoptosis induced by oxidative stress through restraining mitophagy. International journal of biological macromolecules. 2019;136:1007-17
29. Song Y, Lu S, Geng W, Feng X, Luo R, Li G. et al. Mitochondrial quality control in intervertebral disc degeneration. Experimental & molecular medicine. 2021;53:1124-33
30. Hu S, Zhang C, Qian T, Bai Y, Chen L, Chen J. et al. Promoting Nrf2/Sirt3-Dependent Mitophagy Suppresses Apoptosis in Nucleus Pulposus Cells and Protects against Intervertebral Disc Degeneration. Oxidative medicine and cellular longevity. 2021;2021:6694964
31. Lin J, Zhuge J, Zheng X, Wu Y, Zhang Z, Xu T. et al. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free radical biology & medicine. 2020;150:109-19
32. Xu WN, Zheng HL, Yang RZ, Jiang LS, Jiang SD. HIF-1alpha Regulates Glucocorticoid-Induced Osteoporosis Through PDK1/AKT/mTOR Signaling Pathway. Frontiers in endocrinology. 2019;10:922
33. Li Y, Jin K, van Pelt GW, van Dam H, Yu X, Mesker WE. et al. c-Myb Enhances Breast Cancer Invasion and Metastasis through the Wnt/beta-Catenin/Axin2 Pathway. Cancer research. 2016;76:3364-75
34. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P. et al. NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science (New York, NY). 2016;352:1436-43
35. Cheng X, Zhang L, Zhang K, Zhang G, Hu Y, Sun X. et al. Circular RNA VMA21 protects against intervertebral disc degeneration through targeting miR-200c and X linked inhibitor-of-apoptosis protein. Annals of the rheumatic diseases. 2018;77:770-9
36. Chen F, Jiang G, Liu H, Li Z, Pei Y, Wang H. et al. Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone research. 2020;8:10
37. Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D. et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017;552:187-93
38. Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H. et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell metabolism. 2016;24:566-81
39. Luo X, Huan L, Lin F, Kong F, Sun X, Li F. et al. Ulinastatin Ameliorates IL-1β-Induced Cell Dysfunction in Human Nucleus Pulposus Cells via Nrf2/NF-κB Pathway. Oxidative medicine and cellular longevity. 2021;2021:5558687
40. Yao RQ, Ren C, Xia ZF, Yao YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2020:1-17
41. Kumar A, Shaha C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Scientific reports. 2018;8:615
42. Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J. et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12:1272-91
43. Wu D, Zhang H, Wu Q, Li F, Wang Y, Liu S. et al. Sestrin 2 protects against LPS-induced acute lung injury by inducing mitophagy in alveolar macrophages. Life sciences. 2021;267:118941
44. Budanov AV, Lee JH, Karin M. Stressin' Sestrins take an aging fight. EMBO molecular medicine. 2010;2:388-400
45. Piochi LF, Machado IF, Palmeira CM, Rolo AP. Sestrin2 and mitochondrial quality control: Potential impact in myogenic differentiation. Ageing research reviews. 2021;67:101309
46. Ding S, Ma N, Liu H, Tang M, Mei J. Sesn2 attenuates the damage of endothelial progenitor cells induced by angiotensin II through regulating the Keap1/Nrf2 signal pathway. Aging. 2020;12:25505-27
47. Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN. et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell metabolism. 2013;17:73-84
48. Lee DH, Park JS, Lee YS, Han J, Lee DK, Kwon SW. et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy. 2020;16:1949-73
49. Zhang XY, Wu XQ, Deng R, Sun T, Feng GK, Zhu XF. Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cellular signalling. 2013;25:150-8
50. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nature reviews Drug discovery. 2013;12:465-83
51. Pfirrmann CW, Metzdorf A, Zanetti M, Hodler J, Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine. 2001;26:1873-8
52. Stirling A, Worthington T, Rafiq M, Lambert PA, Elliott TS. Association between sciatica and Propionibacterium acnes. Lancet (London, England). 2001;357:2024-5
53. Ye J, Palm W, Peng M, King B, Lindsten T, Li MO. et al. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes & development. 2015;29:2331-6
54. Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T. et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science (New York, NY). 2016;351:53-8
55. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795-803
56. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nature reviews Molecular cell biology. 2018;19:93-108
57. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nature cell biology. 2018;20:1013-22
58. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090-106
59. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cell biology. 2010;12:119-31
60. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J. et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS biology. 2010;8:e1000298
61. Eckl EM, Ziegemann O, Krumwiede L, Fessler E, Jae LT. Sensing, signaling and surviving mitochondrial stress. Cellular and molecular life sciences: CMLS. 2021;78:5925-51
62. Gitschlag BL, Kirby CS, Samuels DC, Gangula RD, Mallal SA, Patel MR. Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C. elegans. Cell metabolism. 2016;24:91-103
63. Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature. 2016;533:416-9
64. Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nature chemical biology. 2017;13:136-46
65. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Molecular cell. 2016;61:654-66
66. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell. 2000;6:1099-108
67. Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. The Journal of biological chemistry. 2008;283:7064-73
68. Jia J, Nie L, Liu Y. Butyrate alleviates inflammatory response and NF-κB activation in human degenerated intervertebral disc tissues. International immunopharmacology. 2020;78:106004
69. Jimbo K, Park JS, Yokosuka K, Sato K, Nagata K. Positive feedback loop of interleukin-1beta upregulating production of inflammatory mediators in human intervertebral disc cells in vitro. Journal of neurosurgery Spine. 2005;2:589-95
70. Walter BA, Korecki CL, Purmessur D, Roughley PJ, Michalek AJ, Iatridis JC. Complex loading affects intervertebral disc mechanics and biology. Osteoarthritis and cartilage. 2011;19:1011-8
71. Dong W, Liu J, Lv Y, Wang F, Liu T, Sun S. et al. miR-640 aggravates intervertebral disc degeneration via NF-κB and WNT signalling pathway. Cell proliferation. 2019;52:e12664
72. Chang H, Cai F, Zhang Y, Xue M, Liu L, Yang A. et al. Early-stage autophagy protects nucleus pulposus cells from glucose deprivation-induced degeneration via the p-eIF2α/ATF4 pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;89:529-35
73. Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB. et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. The Journal of biological chemistry. 2010;285:27879-90
74. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA. et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. The Journal of cell biology. 2010;189:211-21
75. Zhang Z, Xu T, Chen J, Shao Z, Wang K, Yan Y. et al. Parkin-mediated mitophagy as a potential therapeutic target for intervertebral disc degeneration. Cell death & disease. 2018;9:980
Author contact
![]() Corresponding authors: Sheng-Dan Jiang (jiangshengdancom.cn) and Li-Xin Zhu (zhulixin1966com); Tel: 13917924984; Postal Address: 510280.
Corresponding authors: Sheng-Dan Jiang (jiangshengdancom.cn) and Li-Xin Zhu (zhulixin1966com); Tel: 13917924984; Postal Address: 510280.