Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(3):760-771. doi:10.7150/ijbs.77495 This issue Cite
Research Paper
The ADAM9/WISP-1 axis cooperates with osteoblasts to stimulate primary prostate tumor growth and metastasis
An-Chen Chang1, Liang-Wei Lin2, Yen-Chen Chen1, Po-Chun Chen1,3,4, Shan-Chi Liu5, Huai-Ching Tai6,7, Hsi-Chin Wu8,9,10, Shian-Ying Sung11,12,13,14,15,16, Tien-Huang Lin17,18 ![]() , Chih-Hsin Tang2,8,19,20
, Chih-Hsin Tang2,8,19,20 ![]()
1. Translational Medicine Center, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan.
2. Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan.
3. Department of Life Science, National Taiwan Normal University, Taipei, Taiwan.
4. Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan.
5. Department of Medical Education and Research, China Medical University Beigang Hospital, Yunlin, Taiwan.
6. School of Medicine, Fu-Jen Catholic University, New Taipei City, Taiwan.
7. Department of Urology, Fu-Jen Catholic University Hospital, New Taipei City, Taiwan.
8. School of Medicine, China Medical University, Taichung, Taiwan.
9. Department of Urology, China Medical University Hospital, Taichung, Taiwan.
10. Department of Urology, China Medical University Beigang Hospital, Beigang, Yunlin, Taiwan.
11. Graduate Institute of Clinical Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan.
12. International Ph.D. Program for Translational Science, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan.
13. The Ph.D. Program for Translational Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan.
14. TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, Taiwan.
15. Office of Human Research, Taipei Medical University, Taipei, Taiwan.
16. TMU-Research Center of Urology and Kidney, Taipei Medical University, Taipei, Taiwan.
17. Department of Urology, Buddhist Tzu Chi General Hospital Taichung Branch, Taichung, Taiwan.
18. School of Post-Baccalaureate Chinese Medicine, Tzu Chi University, Hualien, Taiwan.
19. Chinese Medicine Research Center, China Medical University, Taichung, Taiwan.
20. Department of Biotechnology, Asia University, Taichung, Taiwan.
Received 2022-7-26; Accepted 2022-12-14; Published 2023-1-1
Abstract

Background: Metastatic prostate cancer (PCa) predicts a poor prognosis and lower likelihood of survival. Osteoblasts (OBs) are known to be responsible for the synthesis and mineralization of bone, although it is unclear as to whether PCa in the prostate gland cooperates with OBs in bone to promote PCa malignant transformation. We aimed to elucidate how primary PCa cells cooperate with distal OBs and contribute to the vicious cycle that leads to metastatic PCa.
Methods: N-cadherin, E-cadherin, and Twist protein expression were measured by Western blot. Twist translocation into the nucleus was detected by the immunofluorescence (IF) assay. Enzyme-linked immunosorbent assay (ELISA) detected protein levels in human serum samples. Levels of candidate protein expression were examined by the human cytokine array. Prostate tumor growth and metastasis were analyzed by orthotopic and metastatic prostate cancer models, respectively. Immunohistochemistry (IHC) staining was used to observe ADAM metallopeptidase domain 9 (ADAM9) and WNT1 inducible signaling pathway protein 1 (WISP-1) expression in tissue.
Results: Our in vitro and in vivo analyses have now discovered that primary PCa expressing ADAM9 protein enables the transformation of OBs into PCa-associated osteoblasts (PCa-OBs), inducing WISP-1 secretion from PCa-OBs in the bone microenvironment. The upregulation of WISP-1 in bone provided feedback to primary PCa and promoted PCa cell aggressiveness via epithelial-mesenchymal transition (EMT) activity. Elevated levels of WISP-1 expression were detected in the serum of patients with PCa. ADAM9 levels were overexpressed in tumor tissue from PCa patients; ADAM9 blockade interrupted OB-induced release of WISP-1 and also suppressed primary tumor growth and distal metastasis in orthotopic PCa mouse models.
Conclusion: Our study suggests that the ADAM9/WISP-1 axis assists with metastatic PCa progression. Thus, targeting the ADAM9/WISP-1 axis may help to prevent the malignant phenotypes of PCa cells.
Keywords: Prostate cancer, Osteoblast, WISP-1, ADAM9, Metastasis
Introduction
Prostate cancer (PCa) commonly affects the urinary tract of men, whose susceptibility for this cancer increases with age [1]. Around 77% of newly diagnosed PCa cases present with localized disease; 11% and 5% of cases present with disease that has metastasized to the regional lymph nodes and distant metastasis, respectively [2]. Metastatic PCa has a low 5-year survival rate of 30%, compared with nearly 100% for local or regional disease [3]. A clear understanding of the mechanism of malignant progression in PCa would enable researchers to develop early prevention and intervention strategies.
Secretion of androgen by testicular Leydig cells plays a pivotal role in prostate epithelial cell growth and differentiation [4] and PCa development [5]. The requirement for androgen in PCa progression has led to the development of androgen deprivation therapy (ADT) by orchiectomy or luteinizing hormone-releasing hormone (LHRH) agonists and LHRH antagonists [6]. However, ADT is not ideal, because patients typically develop castration-resistant prostate cancer that progresses to metastatic disease, with a poor survival time of only 16-18 months [7, 8]. An alternative approach is needed to prevent and manage PCa progression. The growth factor-rich environment in bone increases tumor growth [9]. The finding that PCa-derived extracellular vesicles support the formation of a premetastatic niche in bone suggests that the primary PCa cooperates with bone to regulate future metastasis in distal bone [10]. However, it remains unclear as to whether bone cell-derived factors initiate the malignant transformation of primary PCa cells and stimulate the development of metastases.
Osteoblasts (OBs) play a critical role in the repair and regeneration of the bone matrix [11]. Growing evidence suggests that OBs release a range of different molecules including bone morphogenic proteins (BMPs), alkaline phosphatase (ALP), osteopontin (OPN) that stimulates osteoblastogenesis [12, 13], and also vascular endothelial growth factor A (VEGF-A) and hypoxia-inducible factor 1-alpha (HIF-1α), which both regulate bone marrow angiogenesis [14-16]. OBs also produce WISP-1 to drive new sites of bone formation during embryonic development [17]. WISP-1 reportedly functions as an oncogene by mediating the transformation of normal kidney fibroblasts into tumors in vivo [18]. Overexpression of WISP-1 promotes the motility and invasiveness of cancer cells [19, 20], triggers EMT activity [21], and enhances experimental tumorigenesis and distal metastasis [20, 22]. Moreover, WISP-1 expression correlates positively with clinical staging, lymphatic metastasis and poor clinical outcomes in human cancers [23, 24], although it is not yet known whether OB-derived WISP-1 facilitates the malignant transformation of primary PCa tumors.
In this study, our data indicate that primary PCa cells may distally influence OBs in bone, transforming OBs into PCa-OBs, with subsequent increases in WISP-1 expression in the bone microenvironment. Positive feedback from PCa-OBs expressing WISP-1 elevates the invasiveness of primary PCa cells via EMT functioning. WISP-1 enhances EMT activity in PCa cells by suppressing the epithelial marker E-cadherin and inducing the mesenchymal markers N-cadherin and Twist. Cytokine array analysis revealed that soluble ADAM9 (ADAM9-S) is a main mediator secreted from primary PCa cells, capable of regulating PCa-OB formation and WISP-1 expression. Higher ADAM9 levels were detected in clinical PCa tumor tissue compared with normal tissue specimens, while ADAM9 blockade suppressed PCa tumor growth and reduced the formation of distant metastasis in orthotopic PCa mouse models. Our results not only elucidate the mechanism underlying cooperation between primary PCa tumor cells and distal OBs, but also indicate that targeting the ADAM9/WISP-1 axis may interrupt the progression to metastatic disease.
Materials and Methods
Cell culture
The human osteoblastic-like cell line MG-63 was grown in DMEM medium. The human prostate epithelial cell line (PZ-HPV-7) was grown in keratinocyte serum-free medium supplemented with bovine pituitary extract. All PCa cell lines (LNCaP, PC-3 and DU145) were grown in RPMI-1640 medium. All cell types were cultivated at 37°C in a 5% CO2 atmosphere.
Collection of conditioned medium (CM)
MG-63 cells, human prostate epithelial cells (PZ-HPV-7), and PCa cell lines (LNCaP, PC-3 and DU145) were seeded (at a rate of 2x106/10 mL each) in a 10 cm dish with fetal bovine serum (FBS) for 24 h, then the medium was replaced with serum-free medium. After 48 h, CMs were collected as osteoblast CM (OBCM), PZ-HPV-7 CM, LNCaP CM, PC-3 CM, and DU145 CM.
In another series of experiments, MG-63 cells (2x106) were seeded in a 10 cm dish for 24 h, then incubated with serum-free medium for another 24 h. The cells were treated with 30% PZ-HPV-7 (PZ) CM, PC-3 CM, or DU145 CM for 24 h, then incubated in serum-free medium for 48 h. CMs were collected as PZ-OBCM, PC-3-OBCM, and DU145-OBCM. Long-term OBCM (OBCM-L) was also collected without PCa CM pretreatment (Figure 1A).
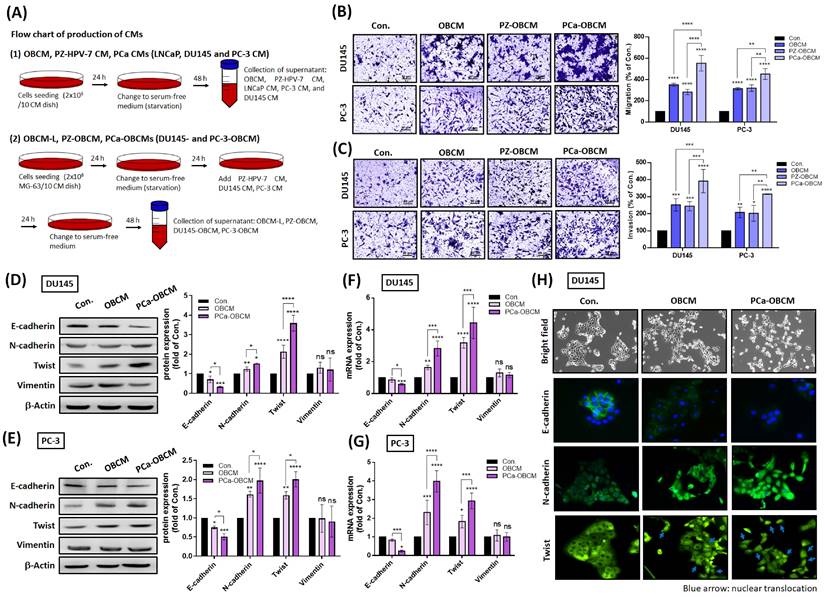
PCa cells corporate with osteoblasts to facilitate invasiveness. (A) A schematic diagram showing the collection of different CMs from OBs, PCa cells, and PCa-OBs. (B&C) Treatment of PC-3 and DU145 cells with OBCM, PZ-OBCM or PCa-OBCM for 24 h. The Transwell assay examined cell migration and invasion activities. (D-G) PCa cells were incubated with OBCM or PCa-OBCM for 24 h, then examined for EMT markers using Western blot and qRT-PCR assays. ImageJ software quantified levels of EMT protein marker expression. (H) The cell scattering phenotype was imaged by bright-field microscopy. Levels of E-cadherin, N-cadherin and Twist translocation into the nucleus were examined by the immunofluorescence (IF) assay. The nuclear location of DU145 cells was determined by DAPI staining. Blue arrow: nuclear translocation of Twist. All data are expressed as the mean ± the S.D. of three samples. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Analysis of ADAM9 expression in PCa patients
Correlations between ADAM9 expression and promoter methylation levels for normal and tumor tissues were analyzed using The Cancer Genome Atlas (TCGA), Oncomine and UALCAN tools.
In vitro cell migration and invasion assay
Cell migratory and invasive activity was evaluated through Transwell inserts in 24-well dishes (Costar, NY, USA), as according to our previous research [25]. Migratory and invasive cells were imaged under ×200 magnification using an Eclipse Ti2 microscope (Nikon, Tokyo, Japan).
Quantitative real-time polymerase chain reaction (qRT-PCR) assay
A Magic RT Master Mix cDNA synthesis kit (Bio-Genesis Technologies, lnc., Hsinchu, Taiwan) was used to convert cDNA from RNA fragment, according to the manufacturer's recommendations. Sequence-specific primers (N-cadherin, E-cadherin, Twist, vimentin, and GAPDH) and Taqman one-step PCR Master Mix (Applied Biosystems, CA, USA) were used to perform qRT-PCR analysis, as according to our previous study [26].
Human cytokine array
A human cytokine array (R&D Systems, MN, USA) containing a total of 32 different cytokine antibodies spotted in duplicate on two membranes was used to analyze protein expression in CM, according to the manufacturer's recommendations. Levels of 32 proteins were detected through enhanced chemiluminescence and quantified by UN-SCAN-IT gel 6.1 software.
Analysis of ADAM9 protein levels in CMs by ELISA assay
The ADAM9 ELISA kit (PeproTech, NJ, USA) assayed ADAM9 protein concentrations in CM, according to the manufacturer's instructions.
Orthotopic and metastatic prostate cancer models
Six‐week‐old male nude mice, bought from BioLASCO Taiwan Co., Ltd. (Taipei, Taiwan), were injected in the anterior prostate with 5 × 105 PC-3 cells suspended in 50 μL Matrigel via a 22-gauge needle. Tumor development and distal metastasis was monitored using the IVIS Spectrum scanner (Xenogen, AZ, USA). After 2 weeks, the mice in the orthotopic model (n=7) were humanely sacrificed in a CO2 chamber. The tumors and tibias were collected before immunostaining. Mice in the metastatic model (n=9) were humanely sacrificed after 5 weeks and distant organs, including the liver and lungs, were harvested. Hematoxylin and eosin (H&E) staining was used to investigate distal metastasis in liver and lung specimens.
Ethics statement
All procedures in the animal studies were approved by the Institutional Animal Care and Use Committee and performed according to the Guidelines of Animal Experimentation issued by Shin Kong Wu Ho-Su Memorial Hospital (Approval No: 109MOST0016). Serum samples were collected from 11 patients undergoing surgical resection in China Medical University Hospital, under approval granted by the Institutional Review Board of China Medical University Hospital (Approval No: CMUH109-REC2-142). Written informed consent was obtained from each study participant before enrollment.
Statistical analysis
A statistical comparison between two samples was performed using the Student's t-test. One-way ANOVA followed by Bonferroni's post hoc comparison tests compared the means of more than two groups. The results are reported using means ± standard deviations. Statistical significance was indicated if P < 0.05.
Results
PCa cells collaborate with OBs to promote cell invasiveness via EMT induction
In order to determine whether primary PCa cells cooperate with OBs to promote the malignant transformation of PCa, samples of PCa CM, OBCM, PZ-OBCM and PCa-OBCM were collected for Transwell assay analyses of cancer cell motility and invasion (Fig. 1A). Our stimulation of PCa cell lines DU145 and PC-3 with PCa-OBCM revealed that PCa-OBCM significantly enhanced PCa cell migration and invasion compared with OBCM and PZ-OBCM treatment (Fig. 1B&C). As EMT is critical for tumor initiation, progression and metastasis [27], we examined whether EMT is involved in PCa-OB-induced mediation of PCa cell invasiveness. As shown in Fig. 1D-H, PCa-OBCM enhanced EMT function by upregulating N-cadherin and Twist expression, and suppressing E-cadherin expression; levels of vimentin, Snail, ZEB1, and ZEB2 expression were unchanged (Fig. 1D-G; Supplementary Fig. 1A&B). Moreover, nuclear translocation of the Twist transcription factor was observed after PCa-OBCM treatment (Fig. 1H). We also found that collection of OBCM under a prolonged period of time (OBCM-L; the same amount of collection time as for PCa-OBCM) did not further increase N-cadherin or Twist expression, or inhibit E-cadherin expression when compared with OBCM-induced values, suggesting that OBCM-induced EMT activation is limited in PCa cells (Supplementary Fig. 1C). These findings suggest that prolonged exposure to PCa-OBCM stimulates the EMT phenomenon and thus increases the invasiveness and aggressiveness of PCa cells.
Primary PCa promotes WISP-1 expression in distal OBs according to in vitro and in vivo analyses
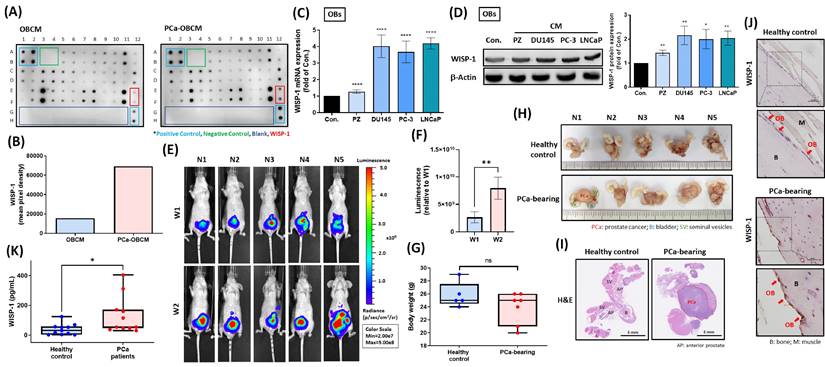
A cytokine array that examined levels of candidate protein secretion from PCa-OBs showed higher WISP-1 expression in PCa-OBCM compared with OBCM (Fig. 2A&B). Treatment of OBs with different PCa CMs (DU145, PC-3 and LNCaP) promoted WISP-1 mRNA and protein expression (Fig. 2C&D). Orthotopic PCa tumors were established for 2 weeks in nude mice to verify WISP-1 expression in distal OBs. IVIS imaging demonstrated that at 2 weeks, tumors were larger in size compared with tumors at 1 week (Fig. 2E&F); body weight was not affected by tumor growth (Fig. 2G). Orthotopic PCa tumors and healthy prostate glands were then removed and stained with H&E via whole-mount imaging to confirm tumor development (Fig. 2H&I). Histopathological examination of WISP-1 expression revealed higher levels in distal OBs located on the tibia bone surface in mice orthotopically implanted with PCa cell lines compared with mice that were not (Fig. 2J). Similarly, analyses of healthy human serum and PCa serum found higher WISP-1 expression in the PCa serum (Fig. 2K). Our investigations indicate that primary PCa tumors may influence the transformation of distal OBs into PCa-OBs capable of promoting WISP-1 secretion in the bone microenvironment.
Primary PCa tumors promote WISP-1 expression in distal OBs. (A) Levels of candidate protein expression were examined by the cytokine array. (B) WISP-1 expression levels were quantified by UN-SCAN-IT gel 6.1. (C&D) Treatment of OBs with PZ CM or different PCa CMs (DU145, PC-3, LNCaP) for 24 h. WISP-1 mRNA and protein expression was measured using qRT-PCR and Western blot assays, respectively. ImageJ software quantified levels of WISP-1 protein expression. (E&F) Luminescence imaging of tumor-bearing mice was performed weekly using the IVIS spectrum system, and the luminescent intensity of photons emitted from each tumor was quantified. (G) Body weights for healthy controls and tumor-bearing mice were recorded manually at the end of week 2. (H) Images of excised tumors obtained by bright-field microscopy. (I) H&E-stained images from whole-mount tumor tissue. (J) IHC staining revealed WISP-1 expression in OBs from tibia specimens. Red arrows: OBs. (K) WISP-1 levels in healthy human serum and PCa serum were detected by ELISA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
PCa-OB-derived WISP-1 confers aggressiveness in PCa cancer cells
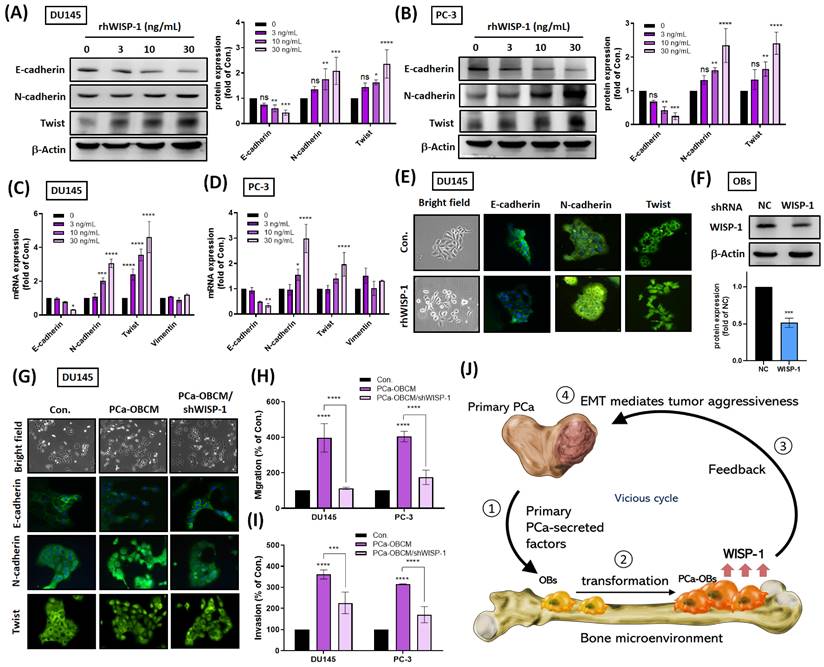
To examine WISP-1 protumor functions, recombinant human WISP-1 (rhWISP-1) protein was used to measure EMT function in PCa cells. We observed that rhWISP-1 treatment elevated N-cadherin and Twist expression and reduced E-cadherin expression in PCa cell lines DU145 and PC-3 (Fig. 3A-E). Immunofluorescent staining revealed that rhWISP-1 promoted the translocation of Twist into the nucleus (Fig. 3E). Incubating PCa cells with PCa-OBCM promoted EMT activity, cell migration and invasion, while knockdown WISP-1 inhibited PCa-OBCM-mediated invasiveness in OBs (Fig. 3F-I). It appears that PCa-OBs regulate cancer cell invasiveness via WISP-1 upregulation and that WISP-1 blockade in OBs reduces this phenomenon. The schematic diagram in Figure 3J summarizes the results from Figures 1-3, showing how primary PCa promotes the transformation of distal OBs into PCa-OBs and is accompanied by increasing levels of WISP-1 expression in the bone microenvironment, subsequently triggering PCa cell migratory and invasive activities via EMT activation.
PCa-OB-derived WISP-1 increases PCa cell invasiveness. (A-D) After incubation of PCa cells with rhWISP-1 (0, 3, 10, or 30 ng/mL) for 24 h, levels of E-cadherin, N-cadherin and Twist protein were quantified by Western blot and ImageJ software, respectively; mRNA expression was analyzed by qRT-PCR. (E&G) DU145 cells were stimulated with rhWISP-1 (30 ng/mL), PCa-OBCM, or PCa-OBCM/shWISP-1 for 24 h, before imaging the cell scattering phenotypes by microscope. Levels of E-cadherin, N-cadherin and Twist translocation into the nucleus were examined by the IF assay. (F) The knockdown efficiency of WISP-1 shRNA on OBs was quantified by Western blot and ImageJ software, respectively. (H&I) The Transwell assay examined levels of PCa cell migration and invasion after PCa-OBCM or PCa-OBCM/shWISP-1 treatment for 24 h. (J) Modeling shows how primary PCa cooperates with distal PCa-OBs to constitute a vicious cycle with increasing PCa aggressiveness via EMT activation. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
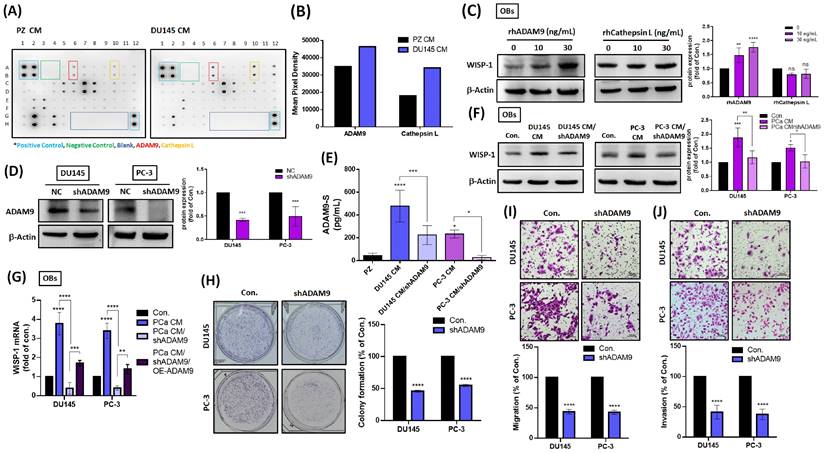
ADAM9 supports the formation of a vicious cycle between primary PCa cells and OBs
Next, we sought to determine what factors are released by primary PCa cells that initiate crosstalk between primary PCa cells and distal OBs. Cytokine array data revealed that the soluble forms of ADAM9 (ADAM9-S) and cathepsin L (cathepsin L-S) levels were elevated in DU145 CM compared with PZ CM (Fig. 4A&B). Treatment of OBs with different concentrations of rhADAM9 dose-dependently increased WISP-1 protein expression, whereas rhCathepsin L had no significant effect (Fig. 4C). To investigate whether the amount of ADAM9-S secreted from PCa cells affects WISP-1 expression in OBs, subsequent experiments quantified shRNA-mediated depletion of ADAM9 in PCa cells. Firstly, total ADAM9 protein and ADAM9-S levels were detected by Western blot and ELISA assays, respectively, to confirm the efficiency of ADAM9 depletion in PCa cells (Fig. 4D&E). CM from PCa cells with or without ADAM9 depletion was collected and incubated with OBs to measure WISP-1 expression. The resulting data showed that ADAM9 depletion suppressed PCa-induced increases in WISP-1 protein and mRNA expression (Fig. 4F&G), while ADAM9 overexpression (OE-ADAM9) rescued WISP-1 mRNA expression in OBs (Fig. 4G). ADAM9 depletion also interrupted colony formation, cell motility and invasiveness of PCa cells (Fig. 4H-J).
ADAM9 is responsible for the initiation of a vicious cycle between primary PCa and OBs. (A) Cytokine arrays were used to analyze levels of candidate proteins. (B) Levels of ADAM9 expression were quantified by UN-SCAN-IT gel 6.1. (C) PCa cells were stimulated with rhADAM9 (0, 10, or 30 ng/mL) and rhCathepsin L (0, 10, or 30 ng/mL) for 24 h, then WISP-1 protein levels were determined using the Western blot assay. (D) The knockdown efficiency of ADAM9 shRNA upon both DU145 and PC-3 cells was determined by Western blot. (E) DU145 and PC-3 cells were transfected with or without ADAM9 shRNA (1 µg/µL) for 24 h, then CM was collected and quantified for ADAM9-S levels by ELISA. (F) PCa CMs and PCa CM/shADAM9 were added to OBs for 24 h, then examined for WISP-1 protein by Western blot. (G) OBs were treated with PCa CMs, PCa CM/shADAM9, or PCa CM/shADAM9/OE-ADAM9 for 24 h. WISP-1 mRNA expression was analyzed by qRT-PCR assays. (H-J) PCa cells were transfected with ADAM9 shRNA (1 µg/µL) to observe colony formation, migratory and invasive activity. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
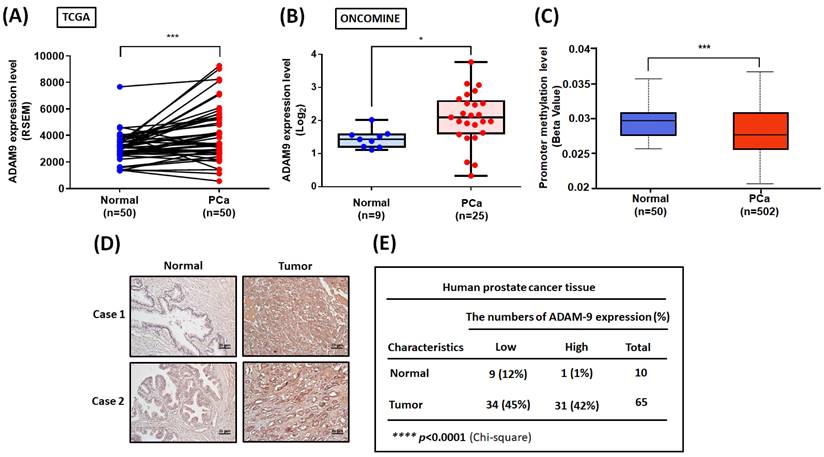
To further examine the clinical importance of ADAM9, we analyzed ADAM9 levels in PCa tissue samples obtained from the TCGA and ONCOMINE databases. We found high levels of ADAM9 mRNA expression in PCa tissue compared with healthy tissue samples (Fig. 5A&B). Interestingly, the ADAM9 promoter region exhibited low levels of methylation, indicating high transcriptional activity of the ADAM9 gene in PCa (Fig. 5C). A positive correlation was observed between ADAM9 and WISP-1 mRNA levels in PCa tissues (Pearson correlation: 0.2; P=9.236e-6) (Supplementary Fig. 2A). Histopathological examination of ADAM9 protein levels also revealed higher levels in PCa tissue sections compared with healthy control samples (Fig. 5D&E). This evidence indicates that ADAM9 is overexpressed in PCa and may facilitate PCa aggressiveness.
ADAM9 expression in human PCa tissue. (A) Levels of ADAM9 expression in paired PCa and adjacent normal tissue samples (N=50) were determined using a paired t-test. (B) Tissue samples from the ONCOMINE database were measured for levels of ADAM9 expression in normal (N=9) and PCa tissue (N=25). (C) Methylation in the ADAM9 promoter region was analyzed using the UALCAN web server. (D) Representative images of IHC staining for ADAM9 in tissue samples from healthy individuals and PCa patients. (E) Tumor tissues were categorized as exhibiting high or low levels of ADAM9 protein expression and significant between-group differences were analyzed by the chi-square test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
ADAM9 blockade inhibits PCa growth and metastasis in the orthotopic model
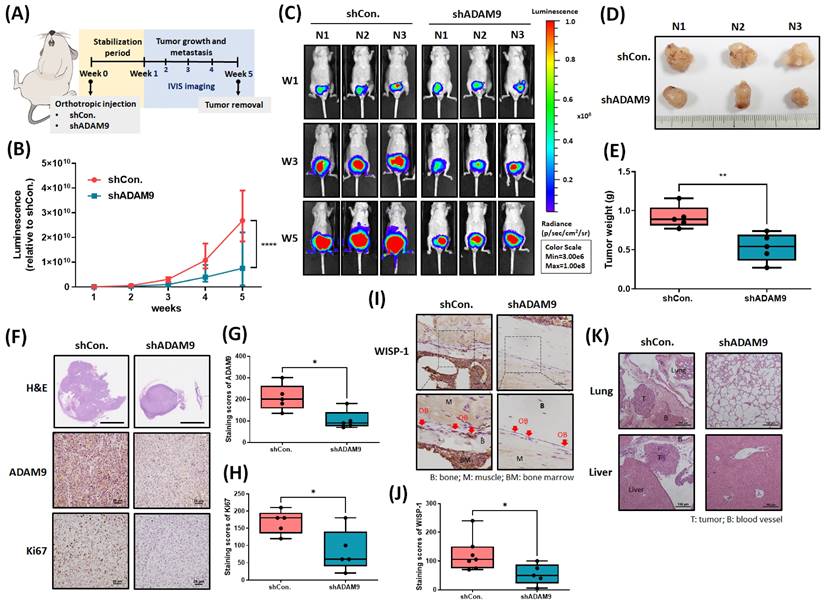
Next, we addressed whether ADAM9 blockade enhances the therapeutic efficacy of PCa in tumor xenograft mouse models. First, we implanted PC-3 cells stably expressing pLenti CMV V5-Luc into nude mouse anterior prostates, then sacrificed the mice 5 weeks later (Fig. 6A). ADAM9 blockade significantly suppressed tumor growth, according to IVIS imaging (Fig. 6B&C) and manual tumor weight measurements (Fig. 6D&E). IHC staining demonstrated that ADAM9 blockade significantly suppressed levels of ADAM9 expression and of the proliferation marker protein Ki67 (Fig. 6F-H). Importantly, ADAM9 blockade also enhanced the reduction of WISP-1 expression in PCa-OBs, indicating that ADAM9 depletion in PCa interrupted the cooperation between primary PCa and PCa-OBs (Fig. 6I&J) and prevented distant metastases in the lung and liver (Fig. 6K). These results support the targeting of ADAM9 for reducing the development of metastatic PCa.
Targeting ADAM9 prevents PCa growth and metastasis in vivo. (A) The flowchart shows how to establish an in vivo model of orthotopic PCa mice. (B&C) Luminescence imaging of tumor-bearing mice was performed weekly using the IVIS spectrum system, and the luminescent intensity of photons emitted from each tumor was quantified. (D) Bright-field imaging of excised tumor tissue. (E) Tumor weights were recorded manually. (F-H) Tumor sections were stained with H&E via whole-mount imaging. IHC staining exhibited ADAM9 and Ki67 expression in tumor tissues. (I&J) IHC staining revealed WISP-1 expression in OBs of tibia specimens. Red arrows: OBs. (K) H&E staining was used to examine liver and lung metastases. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
The present study determined that primary PCa cells cooperate with distal PCa-OBs to constitute a vicious cycle that leads to metastatic PCa. shRNA-mediated depletion of ADAM9 in primary PCa cells suppressed the formation of PCa-OBs in bone, as well as PCa tumor growth and distal metastasis in an orthotopic PCa mouse model. ADAM9 blockade prevented this vicious cycle, suggesting that this may be a beneficial therapeutic strategy in response to metastatic PCa progression.
Tumor-derived factors and extracellular vesicles predominantly support the creation of the tumor microenvironment (TME) and promote tumor development [28]. Numerous nonmalignant cells, including fibroblasts, mesenchymal stem cells, adipocytes and immune cells, are recruited into the TME and are educated to work for the benefit of cancer cells, leading to immunosuppression, malignant transformation and distal metastasis [29-31]. Our results indicate that PCa tumors not only influence surrounding nonmalignant cells, but also act in concert with distant OBs located on the bone surface, encouraging metastatic PCa progression. After analyzing 37 candidates from a human cytokine array, we found that PCa-released ADAM9-S is a key regulator that contributes to the transformation of distal OBs into PCa-OBs with WISP-1 overexpression. ADAM9 can present as either a membrane-anchored or soluble form and is responsible for proteolysis of functional proteins, extracellular matrix degradation and cell-cell interactions [32, 33]. For example, ADAM9 cleaves the latency-associated peptide to active transforming growth factor β1 (TGF-β1), which subsequently promotes T helper 17 (Th17) cell differentiation [34]. TGF-β1 is known to play critical roles in matrix production and osteoblast differentiation [13, 35] and is an important upstream mediator that increases WISP-1 expression [36]. Whether TGF-β1 is involved in the ADAM9-induced mediation of WISP-1 levels in PCa-OBs is worth further study. Marked upregulation of ADAM9 in cancer cells and tissues is associated with several different cancers, such as lung, prostate, breast, and brain cancers [37]. In triple-negative breast cancer (TNBC), higher ADAM9 expression correlates significantly with poorer outcomes, while inhibition of ADAM9 expression lowers TNBC cell aggressiveness by suppressing AKT/NF-κB signaling [38]. In androgen-independent PCa, ADAM9 can form a complex with N-α-acetyltransferase 10 protein to maintain ADAM9 protein stability and thus promote tumor cell invasiveness and in vivo tumor progression [39]. ADAM9 also serves as a significant and independent prognostic marker associated with prostate-specific antigen relapse-free survival in PCa patients treated with ADT [40]. In clinical PCa specimens, higher ADAM9 protein expression has been found in malignant prostatic hyperplasia (Gleason scores 6 and 7) compared with benign prostatic tissues [41]. ADAM9 expression is even elevated in PCa tissue with low metastatic potential (M1 stage) compared with nonmetastatic (M0) tissue samples [42]. Notably, enhanced expression levels of ADAM9-S have been detected in human PCa serum [42], suggesting that ADAM9 may be a predictive biomarker of cancer progression in PCa clinical and serum specimens. This evidence supports the important impact of ADAM9 upon tumor progression.
IMGC936 (an anti-ADAM9 antibody drug conjugate developed by MacroGenics) is undergoing Phase 1 investigations involving patients with TNBC, nonsquamous non-small cell lung cancer, colorectal cancer, gastroesophageal cancer, or pancreatic cancer (NCT04622774). Identifying novel ADAM9 inhibitors from cell-based assays may encourage the design of more selective compounds that help to block ADAM9-induced promotion of cancer [43]. According to our study results revealing protumor functions of ADAM9 in PCa, IMGC936 and other novel pharmacological agents that inhibit ADAM9 should be explored in preclinical investigations and clinical trials with PCa patients.
The initiation of metastasis requires tumor cells to exhibit an invasive ability, which is enabled by the development of the EMT process in cancer cells [44, 45]. In this study, we provide evidence in support of cooperation between primary PCa and PCa-OBs caused as a result of EMT activation in PCa cells. PCa-OBs expressing WISP-1 increased the levels of N-cadherin and Twist expression, with a concomitant loss of E-cadherin, increasing the aggressiveness and metastatic ability of PCa cells via EMT activity. Similarly, WISP-1 exhibits a powerful impact on activating EMT via the miR-153-3p/Snail axis in oral squamous cell carcinoma [21], or through stimulating AKT and MEK/ERK signaling pathways in melanoma [20]. Targeting WISP-1 expression may therefore prevent the development of EMT-related tumor aggressiveness. An intriguing observation from our study is that WISP-1 upregulated both total Twist protein and nuclear translocation expression. However, levels of Twist nuclear translocation are not necessarily dependent upon increases in total Twist protein. Whereas some research has shown that Hsp90β elevates levels of Twist expression by increasing Twist protein stability that ultimately influence Twist translocation from the cytoplasm to the nucleus in hepatocellular carcinoma [46], other research has reported that antiapoptotic Bcl-2 switches on Twist nuclear localization signals and results in the transcriptional activation of EMT under hypoxic conditions [47]. Studies have shown that EMT can be induced by ADT in metastatic PCa, so EMT is capable of conferring an invasive potential in tumour epithelial cells [48, 49]. In other research, CD44+ cancer stem-like cells can switch to EMT cells via TGFβ1-CD44 signaling and drive PCa metastasis [50]. Our findings are consistent with these reports, showing that EMT functions have a marked, comprehensive influence upon metastatic PCa progression. Notably, an ongoing neoadjuvant Phase 2 study including patients with PCa is comparing the effects of ADT with or without chemotherapeutic agents against SRC or MEK (degarelix, enzalutamide, trametinib, or dasatinib), by evaluating the inhibition of mesenchymal markers N-cadherin and vimentin (NCT01990196). Numerous clinical trials are investigating EMT therapeutic approaches in combination with known cancer-targeted therapies, immunotherapies, and/or novel compounds [51].
Since PCa-derived factors are favorable for OB differentiation and mineralization in the bone microenvironment, PCa bone metastases are mainly characterized as osteoblastic (forming new bone), according to radiographic and/or pathologic appearances of the lesions [52]. Similarly, we found that primary PCa cells influence the transformation of OBs into PCa-OBs, with subsequent increases in the release of WISP-1 from PCa-OBs in the bone microenvironment. However, we did not evaluate whether PCa-OBs expressing WISP-1 regulate cancer-related osteoblastogenesis. This aspect deserves further investigation. Transgenic mice overexpressing human WISP-1 in preosteoblasts reportedly exhibit enhanced bone mineral density compared with their wild-type controls, which suggests that WISP-1 positively regulates osteoblastogenesis in vivo [53]. In addition, it appears that PCa bone metastases require both osteoclastic and osteoblastic activity [54]. Confirmation is needed as to whether primary PCa influences distal OBs and osteoclasts in bone to advance its own malignant characteristics.
Conclusion
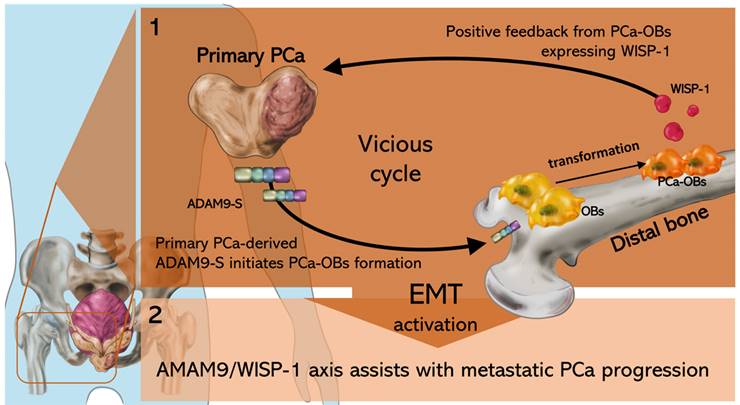
In conclusion, our study demonstrated that the ADAM9/WISP-1 axis triggers primary prostate tumor growth and distant metastasis by cooperating with OBs in bone. Interfering with ADAM9 expression in primary PCa interrupts the formation of the vicious cycle between primary PCa cells and OBs, impairing PCa-OB formation, tumor growth and metastatic spread (Fig. 7).
The schematic diagram shows how the ADAM9/WISP-1 system creates a vicious cycle between primary PCa and OBs that stimulates metastatic PCa progression.
Abbreviations
ADAM9: ADAM metallopeptidase domain 9
ADT: androgen deprivation therapy
ALP: alkaline phosphatase
BMP: bone morphogenic protein
CM: conditioned medium
ELISA: enzyme-linked immunosorbent assay
EMT: epithelial-mesenchymal transition
H&E: hematoxylin and eosin
HIF-1α: hypoxia-inducible factor 1-alpha
IHC: immunohistochemistry
LHRH: luteinizing hormone-releasing hormone
OBs: osteoblasts
OPN: osteopontin
PCa: prostate cancer
PSA: prostate-specific antigen
qRT-PCR: quantitative real-time polymerase chain reaction
TCGA: The Cancer Genome Atlas
TME: tumor microenvironment
TNBC: triple-negative breast cancer
VEGF-A: vascular endothelial growth factor A
WISP-1: WNT1 inducible signaling pathway protein 1
Supplementary Material
Supplementary materials and methods, figures.
Acknowledgements
We thank the Taiwan Animal Consortium for their help with IVIS imaging. We gratefully acknowledge the data from the TCGA research network (https://www.cancer.gov/tcga). We would like to thank Iona J. MacDonald (China Medical University) for her editing of this manuscript.
Funding
This work was supported by the Ministry of Science and Technology, Taiwan (MOST-109-2320-B-341-002-, MOST-110-2314-B-341-001-MY2), Shin Kong Wu Ho-Su Memorial Hospital (2022SKHBND002), China Medical University (CMU111-NSTC-03, CMU111-NSTC-04, CMU111-ASIA-04), and China Medical University Beigang Hospital (CMUBHR 108-012, 1-CMUBHR109-007).
Author contributions
Conceptualization, ACC and CHT; Data curation, LWL and SCL; Formal analysis, THL and HCW; Investigation, ACC, LWL and YCC; Methodology, HCT, PCC, SYS and LWL; Project administration, ACC and PCC; Resource management, SCL, SYS and HCT; Software, ACC, LWL and YCC; Supervision, HCW and CHT; Writing (original draft), ACC; Review, THL and CHT. All authors read the manuscript and approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Force USPST, Grossman DC, Curry SJ, Owens DK, Bibbins-Domingo K, Caughey AB. et al. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1901-13
2. Siegel DA, O'Neil ME, Richards TB, Dowling NF, Weir HK. Prostate Cancer Incidence and Survival, by Stage and Race/Ethnicity - United States, 2001-2017. MMWR Morb Mortal Wkly Rep. 2020;69:1473-80
3. Taitt HE. Global Trends and Prostate Cancer: A Review of Incidence, Detection, and Mortality as Influenced by Race, Ethnicity, and Geographic Location. Am J Mens Health. 2018;12:1807-23
4. Waltregny D, Leav I, Signoretti S, Soung P, Lin D, Merk F. et al. Androgen-driven prostate epithelial cell proliferation and differentiation in vivo involve the regulation of p27. Mol Endocrinol. 2001;15:765-82
5. Dai C, Heemers H, Sharifi N. Androgen Signaling in Prostate Cancer. Cold Spring Harb Perspect Med. 2017;7:a030452
6. Crawford ED, Heidenreich A, Lawrentschuk N, Tombal B, Pompeo ACL, Mendoza-Valdes A. et al. Androgen-targeted therapy in men with prostate cancer: evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019;22:24-38
7. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501-11
8. Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4:365-80
9. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-64
10. Furesi G, Rauner M, Hofbauer LC. Emerging Players in Prostate Cancer-Bone Niche Communication. Trends Cancer. 2021;7:112-21
11. Florencio-Silva R, Sasso GR, Sasso-Cerri E, Simoes MJ, Cerri PS. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed Res Int. 2015;2015:421746
12. Morinobu M, Ishijima M, Rittling SR, Tsuji K, Yamamoto H, Nifuji A. et al. Osteopontin expression in osteoblasts and osteocytes during bone formation under mechanical stress in the calvarial suture in vivo. J Bone Miner Res. 2003;18:1706-15
13. Chen G, Deng C, Li YP. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8:272-88
14. Wan C, Gilbert SR, Wang Y, Cao X, Shen X, Ramaswamy G. et al. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc Natl Acad Sci U S A. 2008;105:686-91
15. Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018;6:16
16. Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR. et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117:1616-26
17. French DM, Kaul RJ, D'Souza AL, Crowley CW, Bao M, Frantz GD. et al. WISP-1 is an osteoblastic regulator expressed during skeletal development and fracture repair. Am J Pathol. 2004;165:855-67
18. Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ. WISP-1 is a Wnt-1- and beta-catenin-responsive oncogene. Genes Dev. 2000;14:585-95
19. Chuang JY, Chang AC, Chiang IP, Tsai MH, Tang CH. Apoptosis signal-regulating kinase 1 is involved in WISP-1-promoted cell motility in human oral squamous cell carcinoma cells. PLoS One. 2013;8:e78022
20. Deng W, Fernandez A, McLaughlin SL, Klinke DJ 2nd. WNT1-inducible signaling pathway protein 1 (WISP1/CCN4) stimulates melanoma invasion and metastasis by promoting the epithelial-mesenchymal transition. J Biol Chem. 2019;294:5261-80
21. Chang AC, Lien MY, Tsai MH, Hua CH, Tang CH. WISP-1 Promotes Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma Cells Via the miR-153-3p/Snail Axis. Cancers (Basel). 2019;11:1903
22. Liu Y, Song Y, Ye M, Hu X, Wang ZP, Zhu X. The emerging role of WISP proteins in tumorigenesis and cancer therapy. J Transl Med. 2019;17:28
23. Wang QY, Feng YJ, Ji R. High expression of WISP1 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2020;24:10445-51
24. Xie D, Nakachi K, Wang H, Elashoff R, Koeffler HP. Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers associated with more advanced features. Cancer Res. 2001;61:8917-23
25. Chou KY, Chang AC, Ho CY, Tsai TF, Chen HE, Chen PC. et al. Thrombospondin-4 promotes bladder cancer cell migration and invasion via MMP2 production. J Cell Mol Med. 2021;25:6046-6055
26. Chang AC, Chen PC, Lin YF, Su CM, Liu JF, Lin TH. et al. Osteoblast-secreted WISP-1 promotes adherence of prostate cancer cells to bone via the VCAM-1/integrin alpha4beta1 system. Cancer Lett. 2018;426:47-56
27. Son H, Moon A. Epithelial-mesenchymal Transition and Cell Invasion. Toxicol Res. 2010;26:245-52
28. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79:4557-66
29. Denton AE, Roberts EW, Fearon DT. Stromal Cells in the Tumor Microenvironment. Adv Exp Med Biol. 2018;1060:99-114
30. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15:669-82
31. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84
32. Duffy MJ, Mullooly M, O'Donovan N, Sukor S, Crown J, Pierce A. et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin Proteomics. 2011;8:9
33. Nath D, Slocombe PM, Webster A, Stephens PE, Docherty AJ, Murphy G. Meltrin gamma(ADAM-9) mediates cellular adhesion through alpha(6)beta(1 )integrin, leading to a marked induction of fibroblast cell motility. J Cell Sci. 2000;113( Pt 12):2319-28
34. Umeda M, Yoshida N, Hisada R, Burbano C, Orite SYK, Kono M. et al. ADAM9 enhances Th17 cell differentiation and autoimmunity by activating TGF-beta1. Proc Natl Acad Sci U S A. 2021;118:e2023230118
35. Zhang Z, Zhang X, Zhao D, Liu B, Wang B, Yu W. et al. TGFbeta1 promotes the osteoinduction of human osteoblasts via the PI3K/AKT/mTOR/S6K1 signalling pathway. Mol Med Rep. 2019;19:3505-18
36. Jia H, Janjanam J, Wu SC, Wang R, Pano G, Celestine M. et al. The tumor cell-secreted matricellular protein WISP1 drives pro-metastatic collagen linearization. EMBO J. 2019;38:e101302
37. Chou CW, Huang YK, Kuo TT, Liu JP, Sher YP. An Overview of ADAM9: Structure, Activation, and Regulation in Human Diseases. Int J Mol Sci. 2020 21
38. Zhou R, Cho WCS, Ma V, Cheuk W, So YK, Wong SCC. et al. ADAM9 Mediates Triple-Negative Breast Cancer Progression via AKT/NF-kappaB Pathway. Front Med (Lausanne). 2020;7:214
39. Lin YW, Wen YC, Chu CY, Tung MC, Yang YC, Hua KT. et al. Stabilization of ADAM9 by N-alpha-acetyltransferase 10 protein contributes to promoting progression of androgen-independent prostate cancer. Cell Death Dis. 2020;11:591
40. Fritzsche FR, Jung M, Tolle A, Wild P, Hartmann A, Wassermann K. et al. ADAM9 expression is a significant and independent prognostic marker of PSA relapse in prostate cancer. Eur Urol. 2008;54:1097-106
41. Sung SY, Kubo H, Shigemura K, Arnold RS, Logani S, Wang R. et al. Oxidative stress induces ADAM9 protein expression in human prostate cancer cells. Cancer Res. 2006;66:9519-26
42. Le TT, Hsieh CL, Lin IH, Chu CY, Do AD, Chen SH. et al. The ADAM9/UBN2/AKR1C3 axis promotes resistance to androgen-deprivation in prostate cancer. Am J Cancer Res. 2022;12:176-97
43. Maretzky T, Swendeman S, Mogollon E, Weskamp G, Sahin U, Reiss K. et al. Characterization of the catalytic properties of the membrane-anchored metalloproteinase ADAM9 in cell-based assays. Biochem J. 2017;474:1467-79
44. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49:361-74
45. Khan MI, Adhami VM, Lall RK, Sechi M, Joshi DC, Haidar OM. et al. YB-1 expression promotes epithelial-to-mesenchymal transition in prostate cancer that is inhibited by a small molecule fisetin. Oncotarget. 2014;5:2462-74
46. Meng J, Chen S, Lei YY, Han JX, Zhong WL, Wang XR. et al. Hsp90beta promotes aggressive vasculogenic mimicry via epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene. 2019;38:228-43
47. Sun T, Sun BC, Zhao XL, Zhao N, Dong XY, Che N. et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma. Hepatology. 2011;54:1690-706
48. Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S. et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72:527-36
49. Izumi K, Fang LY, Mizokami A, Namiki M, Li L, Lin WJ. et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol Med. 2013;5:1383-401
50. Shang Z, Cai Q, Zhang M, Zhu S, Ma Y, Sun L. et al. A switch from CD44(+) cell to EMT cell drives the metastasis of prostate cancer. Oncotarget. 2015;6:1202-16
51. Santamaria PG, Moreno-Bueno G, Portillo F, Cano A. EMT: Present and future in clinical oncology. Mol Oncol. 2017;11:718-38
52. Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21-8
53. Ono M, Inkson CA, Kilts TM, Young MF. WISP-1/CCN4 regulates osteogenesis by enhancing BMP-2 activity. J Bone Miner Res. 2011;26:193-208
54. Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem. 2004;91:718-29
Author contact
![]() Corresponding authors: Tien-Huang Lin, MD., Department of Urology, Buddhist Tzu Chi General Hospital Taichung Branch, Taichung, Taiwan; No. 88, Section 1, Fengxing Road, Tanzi Dist., Taichung, Taiwan. Tel: (886) 4-36060666; E-mail: thlincom. Chih-Hsin Tang, Ph.D., Department of Pharmacology, School of Medicine, China Medical University, Taichung, Taiwan; No. 91, Hsueh-Shih Road, Taichung, Taiwan. Tel: (886) 4-22052121 Ext. 7726; Fax: (886) 4-22333641; E-mail: chtangcmu.edu.tw
Corresponding authors: Tien-Huang Lin, MD., Department of Urology, Buddhist Tzu Chi General Hospital Taichung Branch, Taichung, Taiwan; No. 88, Section 1, Fengxing Road, Tanzi Dist., Taichung, Taiwan. Tel: (886) 4-36060666; E-mail: thlincom. Chih-Hsin Tang, Ph.D., Department of Pharmacology, School of Medicine, China Medical University, Taichung, Taiwan; No. 91, Hsueh-Shih Road, Taichung, Taiwan. Tel: (886) 4-22052121 Ext. 7726; Fax: (886) 4-22333641; E-mail: chtangcmu.edu.tw