Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(3):865-879. doi:10.7150/ijbs.79355 This issue Cite
Research Paper
G protein inhibitory α subunit 2 is a molecular oncotarget of human glioma
Yin Wang1,2#, Fang Liu3#, Jiang Wu4#, Mei-qing Zhang1,2#, Jin-long Chai1,2, Cong Cao1,2,5 ![]()
1. Institute of Neuroscience, Soochow University, Institute for Excellence in Clinical Medicine of Kunshan First People's Hospital and Soochow University, Suzhou, China.
2. Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, Suzhou, China.
3. Department of Neurosurgery, The affiliated Changzhou No.2 People's Hospital of Nanjing Medical University, Changzhou, China.
4. Department of Neurosurgery, the First Affiliated Hospital of Soochow University, Suzhou, China.
5. The Affiliated Eye Hospital, Nanjing Medical University, Nanjing, China.
#Co-first authors.
Received 2022-9-28; Accepted 2022-12-27; Published 2023-1-9
Abstract

Identification of novel therapeutic oncotargets for human glioma is extremely important. Here we tested expression, potential functions and underlying mechanisms of G protein inhibitory α subunit 2 (Gαi2) in glioma. Bioinformatics analyses revealed that Gαi2 expression is significantly elevated in human glioma, correlating with poor patients' survival, higher tumor grade and wild-type IDH status. Moreover, increased Gαi2 expression was also in local glioma tissues and different glioma cells. In primary and immortalized (A172) glioma cells, Gαi2 shRNA or knockout (KO, by Cas9-sgRNA) potently suppressed viability, proliferation, and mobility, and induced apoptosis. Ectopic Gαi2 overexpression, using a lentiviral construct, further augmented malignant behaviors in glioma cells. p65 phosphorylation, NFκB activity and expression of NFκB pathway genes were decreased in Gαi2-depleted primary glioma cells, but increased following Gαi2 overexpression. There was an increased binding between Gαi2 promoter and Sp1 (specificity protein 1) transcription factor in glioma tissues and different glioma cells. In primary glioma cells Gαi2 expression was significantly reduced following Sp1 silencing, KO or inhibition. In vivo studies revealed that Gαi2 shRNA-expressing AAV intratumoral injection hindered growth of subcutaneous glioma xenografts in nude mice. Moreover, Gαi2 KO inhibited intracranial glioma xenograft in nude mice. Gαi2 depletion, NFκB inhibition and apoptosis induction were observed in subcutaneous and intracranial glioma xenografts with Gαi2 depletion. Together, overexpressed Gαi2 is important for glioma cell growth possibly by promoting NFκB cascade activation.
Introduction
Current traditional treatments have very limited effects on improving survival of glioblastoma (GBM) and other high-grade glioma (HGG) [1-3]. Multiple signaling cascades are dysregulated in glioma, including EGFR, the type III mutations, VEGFR and CDK, which are essential for the uncontrolled glioma growth and malignant progression [4-8]. Targeted therapies for specific molecules on the signaling pathways have become the research hotspot for glioma [4-6, 9].
G protein inhibitory α subunits (Gαi proteins) have three members, Gαi1, Gαi2 and Gαi3 [10]. Gαi proteins bind to GPCR and inhibit adenylate cyclase (AC), thereby depleting cyclic AMP (cAMP) [10]. Such process can be blocked by pertussis toxin [11, 12]. Pertussis toxin was shown to potently suppress HGG cell migration and proliferation [13]. In addition, co-administration of pertussis toxin and temozolomide led to robust anti-glioma effect [14]. Moreover, Gαi proteins activation increased malignant growth of glioma cells [12]. These sporadic studies revealed a potential role of Gαi proteins in glioma growth and progression.
Interestingly, our group has shown that Gαi1 and Gαi3 are vital proteins mediating signaling transduction for receptor tyrosine kinases (RTKs), including VEGFR2 [15], EGFR [16] and FGFR [17] as well as KGFR [18] and TrkB [19]. Gαi1 or Gαi3 are recruited to ligand-activated RTKs, mediating downstream signaling cascades (Akt-mTORC1/Erk-MAPK) activation [15-19]. We found that Gαi1/3-RTKs association was important for Akt-mTORC1 activation, and more importantly, glioma growth. Contrarily, Gαi1/3 silencing, Cas9-sgRNA knockout (KO) or mutation hindered Akt-mTOR activation and suppressed malignant behaviors of glioma cells [17, 20, 21]. Moreover, Gαi1 and Gαi3 upregulation in human glioma correlates with patients' clinical parameters [17, 20, 21].
Intriguingly, depletion of Gαi2 was unable to prevent downstream signaling activation by RTKs [15-19]. Yet a potential function of Gαi2 in carcinogenesis and tumor progression has been reported. Gαi2 is elevated in colitis-associated cancer (CAC), correlating with decreased relapse-free survival [22]. Conversely, conditional knockdown of Gαi2 in CD11c+ cells reduced CAC carcinogenesis [22]. Yin et al., reported that Gαi2 is important for epithelial ovarian cancer cell growth [23]. Conversely, microRNA-222-3p silenced Gαi2 to arrest epithelial ovarian cancer cell growth [23]. Zhang et al., proposed a pivotal role of Gαi2 in the development of nonalcoholic steatohepatitis [24]. Gαi2 expression was upregulated in liver tissues of NASH patients [24]. Importantly, hepatocytes specific Gαi2-deficient mice were resistant to the development of steatohepatitis [24]. Here we will show that overexpressed Gαi2 is important for glioma cell growth possibly by promoting activation of NFκB (nuclear factor kappa B) cascade.
Materials and methods
Reagents
Polybrene, BAY-11-7082, mithramycin A, antibiotics, serum, puromycin and cell culturing medium were from Sigma-Aldrich (St. Louis, MO). Antibodies and fluorescence probes were reported early [17, 20, 21, 25].
Bioinformatics studies
The RNA-seq data, including 166 GBM (glioblastoma multiforme), 523 LGG (low grade glioma) tissues and 1157 normal tissues, along with the clinical data, were provided from UCSC XENA (https://xenabrowser.net/datapages/). Normalized gene expression was measured as transcripts per million reads plus the log2-based transformation. The overall survival of GBM and LGG patients was assessed through Kaplan-Meier analysis using the “Survival” along with “SurvMiner” R packages. The accuracy evaluation of the prognostic of Gαi2 was carried out by ROC curves using the R packages “Survival ROC” and “time ROC”. TCGA LGGGBM cohorts were thereafter analyzed and Gαi2-associated differentially expressed gene (DEGs) were retrieved. KEGG analyses were carried out to explore the enrichment pathways. Chinese glioma functional genomic data were retrieved from the Chinese Glioma Genome Atlas (CGGA) [26]. The RNA sequencing of Diffuse Gliomas was through the Illumina Hiseq 2000. Clinical data were also retrieved from the CGGA data portal. RNA sequencing data and Clinical data were analyzed using R. software. The "Survival" package, “SurvMiner” package and "ggpubr" package were used.
Human tissues and cells
Human tissues were reported previously [17, 20, 21, 25] and were tested as reported [17, 21]. The primary human glioma cells (“P1-P5”, derived from five different patients), the primary human astrocytes (“Astrocytes1/2”), glioma cell lines (A172/U87MG/U251MG/SHG-44) were reported early [17, 21, 27, 28].
shRNA or gene overexpression
Verified Gαi2 shRNA sequence, Sp1 shRNA sequence, Gαi2 cDNA or Sp1 cDNA [NM_138473.3] was packaged into a GV369 construct (no GFP) (from Genechem). The constructs were each transfected to HEK-293T cells together with the lentivirus envelope constructs (Genechem). Thereafter, the viral particles were filtered, enriched and transfected (at MOI = 15) to the indicated glioma cells or astrocytes. Cells were maintained under polybrene-containing complete medium, and stable cells formed after puromycin treatment for 96h. Overexpression or silencing of targeted genes was verified. Alternatively, Gαi2 shRNA/shC (“the scramble control non-sense shRNA” [20, 25]) was packed into the described adeno-associated virus (AAV) construct [20, 25], and shRNA AAV generated.
Cas9-sgRNA (single guide RNA)-induced gene knockout
Cells were transfected with the pLV-hUbC-dCas9-VP64 lentiviral construct (GeneChem), and dCas9-expressing glioma cells were established after selection [29]. Next, the verified sgRNA-CRISPR/dCas-9-Gαi2 lentiviral construct (Genechem) or the verified sgRNA-CRISPR/dCas-9-Sp1 lentiviral construct (Genechem) was transduced to dCas9-expressing cells, and stable cells formed following puromycin-mediated selection (for 96h). Gαi2/Sp1 KO was verified at mRNA and protein levels. The control cells were with the lenti-CRISPR/dCas-9 empty vector (“Cas9C”) [20, 25].
Cellular functions and gene/protein detection
CCK-8, colony formation, “Transwell” cell migration and “Matrigel Transwell” invasion assays, were reported early [21, 30-34]. The caspase activity assay, Histone DNA ELISA, nuclear TUNEL fluorescence staining and JC-1 monomer staining of mitochondrial depolarization were reported early [21, 30-35]. mRNA detection by qRT-PCR and protein detection by Western blotting were reported previously [21, 30-34]. Figure S1 listed the uncropped blotting images.
NFκB activity
Briefly, the nuclear proteins were extracted through high-speed centrifugation. The TransAM™ ELISA kit (Active Motif) was utilized to examine the NFκB (p65) DNA-binding activity. In brief, 0.25 μg of nuclear extracts were tested for the binding of p65 to the specific DNA sequence. Following colorimetric reaction, the optical density (OD) value was tested through ELISA at 450 nm.
Chromatin immunoprecipitation (ChIP)
The detailed protocols of ChIP assay were reported in our previous study [25, 30]. Briefly, the cell and human tissue lysates were homogenized [36] and fragmented DNA was achieved. Lysates were immunoprecipitated (IP) with the anti-Sp1 antibody. Sp1-bound DNA with the Gαi2 promoter site [37] was tested by the quantitative PCR method.
Xenograft studies
The nude mice were reported previously [17, 21]. P1 cells or A172 cells were subcutaneously injected into the right flanks of nude mice and glioma xenografts were formed. The mice were intratumorally injected with the Gαi2 shRNA-containing AAV or control shRNA AAV (1×109 PFU). Alternatively, P1 glioma cells were intracranially injected to the brains of the nude mice as described [38] and intracranial P1 glioma xenografts were formed. The protocols of this study were approved by Soochow University's IACUC and Ethics Committee.
Statistical analyses
Statistical methods were reported in our previous studies [20, 25]. The data in this study were normally distributed and were shown as mean ± standard deviation (SD). All in vitro experiments were repeated five times, and each time similar results obtained.
Results
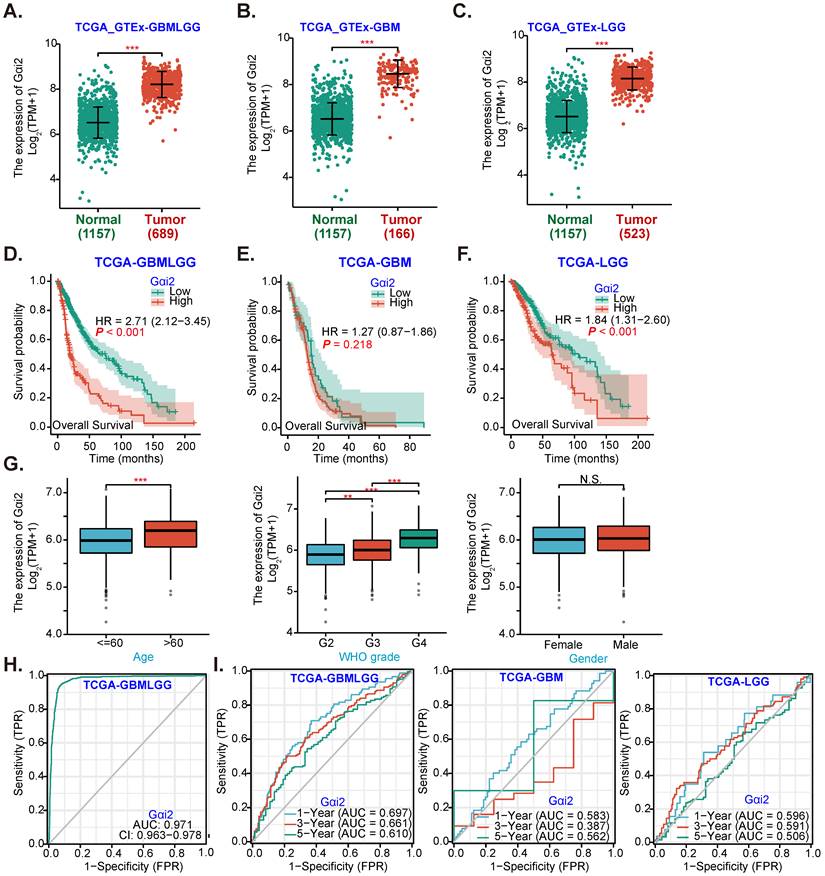
Gαi2 overexpression in human glioma
Gαi2 expression data were retrieved from TCGA and Genotype-Tissue Expression (GTEx) project through UCSC XENA. A total of 689 glioma tissues (“Tumor”), including 166 glioblastoma (GBM) tissues and 523 low grade glioma (LGG) tissues, as well as 1157 normal brain tissues (“Normal”) were retrieved. Gαi2 transcript number in 689 glioma tissues (GBMLGG, “Tumor”) was higher than it in the normal brain tissues (P < 0.001, Figure 1A). Further analyses revealed that Gαi2 transcripts were significantly elevated in both GBM tissues (Figure 1B) and LGG tissues (Figure 1C).
The Kaplan-Meier survival and univariate Cox analysis from TCGA revealed that high Gαi2 expression in glioma tissues (GBMLGG) was correlated with poor patients' overall survival (P < 0.001, Figure 1D). Compared to Gαi2-low GBM patients, Gαi2-high GBM patients tend to have poor overall survival (Figure 1E), although no significant different was detected (P = 0. 218, Figure 1E). Importantly, Gαi2-high LGG patients' overall survival is significantly lower (Figure 1F). Moreover, Gαi2 overexpression in GBMLGG tissues is correlated with patients' age and higher tumor grade (Figure 1G), but was not correlated with patients' gender (Figure 1G).
Alignment Diagram (Nomogram) prediction map based on Gαi2 expression showed that Gαi2 overexpression had a significant value in predicting poor survival probability of patients with GBMLGG (Figure 1H), with area under the survival curve (AUC) at: 0.971 (Figure 1H). Moreover, Gαi2 overexpression could predict poor 1/3/5-year survival probability of patients with GBMLGG, GBM or LGG (Figure 1I). Bioinformatics analyses show that Gαi2 is significantly elevated in human glioma, correlating with patients' poor survival and higher grade of the tumors.
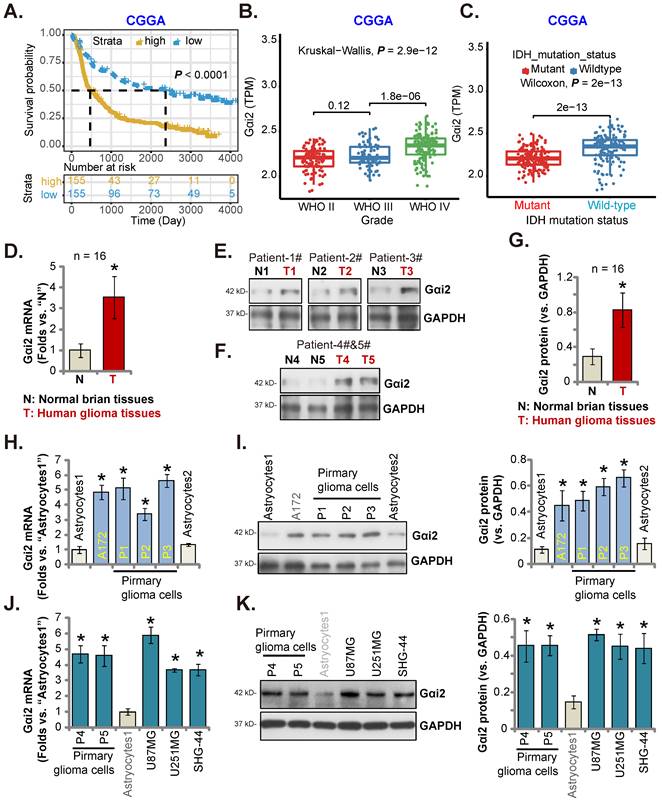
Gαi2 is overexpressed in local glioma tissues and cells
CGGA database revealed that high-Gαi2 expression glioma patients have lower survival (P < 0.001, Figure 2A). Moreover, in HGG tissues Gαi2 expression was significantly higher than that in low grade (grade II) glioma tissues (Figure 2B). Importantly, in glioma tissues Gαi2 overexpression correlated with wild-type (WT) IDH status (P < 0.001, Figure 2C), and low Gαi2 expression was detected in IDH mutant glioma tissues (Figure 2C).
Gαi2 overexpression in human glioma. TCGA cohorts plus GTEx project revealed Gαi2 transcripts in 689 glioma tissues (“Tumor”), including 166 glioblastoma (GBM) tissues and 523 low grade glioma (LGG) tissues as well as in 1157 normal brain tissues (“Normal”) (A-C). Kaplan Meier Survival analyses of TCGA cohorts based on Gαi2 expression in glioma (GBMLGG) patients (D), GBM patients (E) and LGG patients (F). The subgroup analyzing Gαi2 expression and glioma patients' clinical characteristics in TCGA GBMLGG cohorts were shown (G). Nomogram for high glioma Gαi2 expression in predicting overall survival probability of GBMLGG patients (H), and in predicating 1-/3-/5-year overall survival probability of GBMLGG/GBM/LGG patients was shown (I). “TPM” stands for transcripts per million. “AUC” stands for area under curve. ***P < 0.001; **P < 0.001; *P < 0.05; “N. S.” means P > 0.05.
In local human glioma tissues Gαi2 expression was tested. Sixteen (n = 16) HGG tissues (labeled as “T”) and matched adjacent normal brain tissues (labeled as “N”) [17, 20, 21] were tested. Figure 2D revealed that Gαi2 mRNA levels in glioma tissues were increased significantly. In five representative HGG patients (Patient-1#-5#), Gαi2 protein expression was elevated in the glioma tissues (Figure 2E and F). Combining all 16 pairs of Gαi2 expression data showed that Gαi2 protein was significantly upregulated in glioma tissues (Figure 2G).
We next studied whether Gαi2 was upregulated in different human glioma cells, including primary human glioma cells (“P1-P5”, derived from five patients [21]) and A172 cells. Gαi2 mRNA levels in the tested glioma cells were significantly higher than those in the primary human astrocytes (“Astrocytes1/2”) (Figure 2H). Gαi2 protein upregulation was also in primary and immortalized glioma cells (Figure 2I). Gαi2 mRNA (Figure 2J) and protein (Figure 2K) expression was also significantly elevated in other primary glioma cells-derived from other patients (“P4”/“P5”) and in immortalized cell lines, including U87MG, U251MG and SHG-44. These results clearly show that Gαi2 is overexpressed in local glioma tissues/cells.
Gαi2 is overexpressed in local glioma tissues and cells. Kaplan Meier Survival analyses from Chinese Glioma Genome Atlas (CGGA) database based on Gαi2 expression in glioma patients were shown (A). CGGA cohorts showed that Gαi2 mRNA overexpression in glioma patients correlated with high tumor grade (B) and WT IDH status (C). Gαi2 expression in glioma tissues (“T”) and adjacent normal brain tissues (“N”) of 16 local HGG patients was shown (D-G). Gαi2 mRNA and protein expression in astrocytes (“Astrocytes1/2”), the immortalized (A172, U87MG, U251MG and SHG-44) and primary (“P1”, “P2”, “P3”, “P4” and “P5”) glioma cells was shown (H-K). *P < 0.05 versus “N” tissues/“Astrocytes1”.
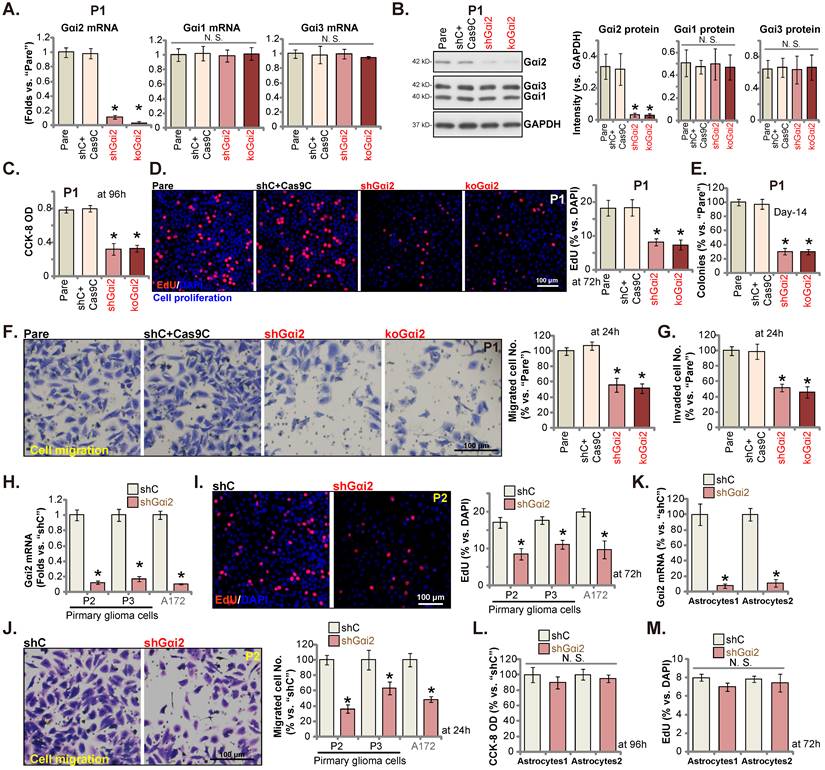
Gαi2 depletion leads to robust anti-glioma cell activity
We next tested Gαi2's potential functions. P1 primary human glioma cells, as reported previously [17, 20, 21, 25], were infected with Gαi2 shRNA-expressing lentiviral particles. Stable P1 glioma cells were thereafter formed after selection, namely“shGαi2” cells. Alternatively, the lentiviral particles with the CRISPR/dCas9-Gαi2-KO construct were added to P1 glioma cells with dCas9, and stable cells formed and named “koGαi2” cells. The control P1 glioma cells were with the scramble control shRNA (non-sense) plus the CRISPR/dCas9 empty construct (“shC+Cas9C”). As shown, Gαi2 mRNA and protein (Figure 3A and B) levels were significantly decreased in shGαi2 and koGαi2 P1 glioma cells, and Gαi1/3 expression unchanged (Figure 3A and B). CCK-8 viability was decreased significantly in shGαi2 and koGαi2 P1 glioma cells (Figure 3C). Gαi2 shRNA or KO largely inhibited P1 glioma cell proliferation and significantly decreased the EdU-stained nuclei (Figure 3D). In addition, genetic depletion of Gαi2 prevented P1 glioma cell colony formation (Figure 3E), further supporting the anti-proliferative activity.
Next, the in vitro cell migration and the in vitro cell invasion of P1 glioma cells were tested separately by “Transwell” (Figure 3F) and “Matrigel Transwell” (Figure 3G) assays. Following Gαi2 depletion, the mobility of P1 glioma cells was largely inhibited (Figure 3F and G). Notably, the “shC+Cas9C” treatment failed to significantly change Gαi1/2/3 expression (Figure 3A and B) as well as P1 glioma cell functions (Figure 3C-G).
Gαi2 depletion provokes apoptosis in glioma cells. Puromycin-selected stable P1 glioma cells, with the lentiviral Gαi2 shRNA (“shGαi2”), the CRISPR/dCas9-Gαi2-KO construct (“koGαi2”) or the scramble non-sense control shRNA (“shC”) plus the CRISPR/dCas9 empty construct (“Cas9C”), were formed. After culturing for designated time periods, the caspase-3/-9 activities (A and B), apoptosis-associated proteins (C) and Histone DNA contents (D) were measured, with JC-1 green monomers measured (E); Cell apoptosis was examined by nuclear TUNEL staining (F). Puromycin-selected stable P2/ P3 primary glioma cells, A172 glioma cells (G and H), or primary human astrocytes (“Astryocyte1” and “Astryocyte2”, I and J), with shGαi2 or shC, were formed and cultured, the caspase-3 activity (G and I) and cell apoptosis (H and J) were similarly tested. “Pare” are parental control cells. * P < 0.05 vs. “Pare” cells /“shC” treatment. “N.S.” means P > 0.05. Scale bar = 100 μm.
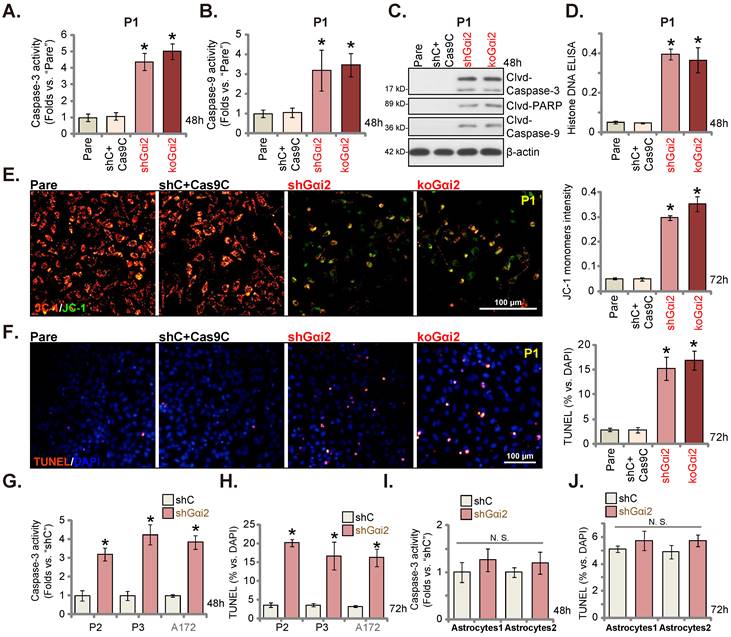
Other primary human glioma cells, “P2” and “P3” (see our previous studies [17, 20, 21, 25]), as well as the immortalized A172 glioma cells were again infected Gαi2 shRNA-expressing lentiviral particles, and stable cells (labeled as “shGαi2”) were established after selection, showing depleted Gαi2 mRNA (Figure 3H). Gαi2 shRNA significantly inhibited cell proliferation and decreased EdU incorporation in these primary and immortalized glioma cells (Figure 3I). Moreover, in vitro cell migration was significantly slowed following Gαi2 silencing (Figure 3J). These results clearly supported that Gαi2 depletion led to robust anti-glioma cell activity.
Gαi2 shRNA lentiviral particles were also added to the primary human astrocytes (“Astryocyte1” and “Astryocyte2”) [17, 20, 21], and stable “shGαi2” astrocytes were formed after puromycin selection. Robust Gαi2 mRNA silencing was detected in shGαi2 astrocytes (Figure 3K). Gαi2 shRNA, however, did not alter CCK-8 viability (Figure 3L) and proliferation/EdU incorporation (Figure 3M) in the primary astrocytes.
Gαi2 depletion provokes apoptosis in glioma cells
Gαi2 depletion led to robust anti-glioma cell activity, causing viability reduction, proliferation inhibition, G1-S arrest and mobility reduction. Its role on cell apoptosis was tested as well. In both shGαi2 and koGαi2 P1 glioma cells (see Figure 3), the activity of caspase-3/-9 was augmented significantly (Figure 4A and B). Caspase-3, caspase-9 and poly (ADP-ribose) polymerase-1 (PARP) cleavages were induced in Gαi2-silenced/KO P1 glioma cells (Figure 4C), and levels of histone-bound DNA were augmented (Figure 4D). Gαi2 shRNA/KO induced mitochondrial depolarization in P1 cells, causing the transition of JC-1 red fluorescence to green fluorescence (monomers) (Figure 4E). Significantly, Gαi2 depletion provoked apoptosis in P1 cells and the TUNEL stained nuclei were increased in shGαi2 and koGαi2 cells (Figure 4F). The shC+Cas9C control treatment, as expected, failed to induce caspase activation (Figure 4A-D), mitochondrial depolarization (Figure 4E) and apoptosis (Figure 4F) in P1 glioma cells.
Gαi2 depletion leads to robust anti-glioma cell activity. Puromycin-selected stable P1 glioma cells, with the lentiviral Gαi2 shRNA (“shGαi2”), the CRISPR/dCas9-Gαi2-KO construct (“koGαi2”) or the scramble non-sense control shRNA (“shC”) plus the CRISPR/dCas9 empty construct (“Cas9C”), were formed, and expression of listed mRNAs and proteins was tested (A and B). After culturing for designated time periods, cell viability (C), nuclear EdU incorporation (D) and colony formation (E), as well as cell in vitro cell migration (F) and invasion (G) were tested, and results quantified. Puromycin-selected stable P2/P3 primary glioma cells, A172 glioma cells (H-J), or primary human astrocytes (“Astryocyte1” and “Astryocyte2”, K-M), with shGαi2 or shC, were formed, Gαi2 mRNA expression was shown (H and K). After culturing for designated time periods, cellular functions including EdU incorporation (I and M), in vitro cell migration (J) and cell viability (L) were tested. “Pare” are parental control cells. * P < 0.05 vs. “Pare” cells /“shC” treatment. “N.S.” means P > 0.05. Scale bar = 100 μm.
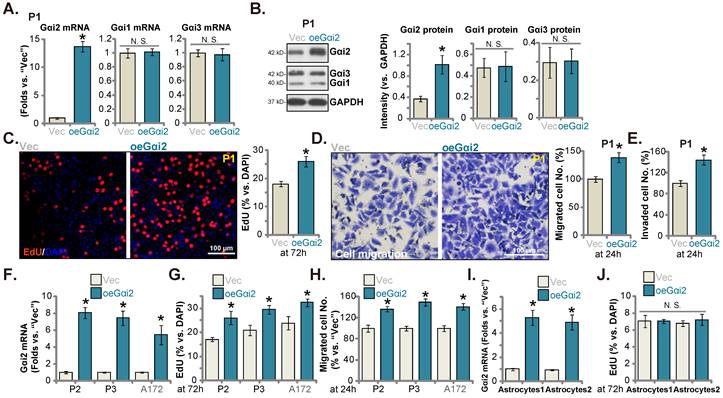
Ectopic Gαi2 overexpression promotes glioma cell growth. Puromycin-selected stable P1 glioma cells, with the lentiviral Gαi2-expressing construct (“oeGαi2”) or the vector (“Vec”), were formed, and listed mRNAs and proteins were shown (A and B). After culturing for indicated time periods, cellular functions, including nuclear EdU incorporation (C), in vitro cell migration (D) and invasion (E) were tested. Puromycin-selected stable P2/P3 primary glioma cells, A172 glioma cells (F-H), or primary human astrocytes (“Astryocyte1” and “Astryocyte2”, I and J), with oeGαi2 or Vec, were formed and Gαi2 mRNA expression was tested (F and I); After culturing, EdU incorporation (G and J) and in vitro cell migration (H) were tested similarly, with results quantified. * P < 0.05 vs. “Vec” cells. “N.S.” means P > 0.05. Scale bar = 100 μm.
Similar results were obtained in P2/P3 primary human glioma cells and A172 cells. As shown shRNA-induced silencing of Gαi2 (“shGαi2”, see Figure 3) increased caspase-3 activity (Figure 4G) and the TUNEL nuclei number (Figure 4H). Whereas in the “Astryocyte1” and “Astryocyte2”, Gαi2 shRNA (“shGαi2”, see Figure 3) failed to boost the caspase-3 activity (Figure 4I) and nuclear TUNEL staining ratio (Figure 4J).
Ectopic Gαi2 overexpression promotes glioma cell growth
Ectopic Gαi2 overexpression could possibly further enhance glioma cell progression. Therefore, the lentiviral particles packaging the Gαi2-expressing construct were transfected to P1 cells. Stable cells were again formed after selection: “oeGαi2”. Gαi2 mRNA level was significantly augmented in oeGαi2 cells and was over 13-fold higher than that of vector control P1 glioma cells (“Vec”) (Figure 5A). Figure 5B confirmed Gαi2 protein upregulation in oeGαi2 P1 glioma cells. Gαi1/3 expression was not changed (Figure 5A-B). oeGαi2 promoted P1 glioma cell proliferation and increased nuclear EdU incorporation (Figure 5C). Cell in vitro migration and invasion (Figure 5D-E) were accelerated following ectopic Gαi2 overexpression. The Gαi2-expressing construct lentiviral particles were also added to P2/P3 primary cells and A172 cells, resulting in robust Gαi2 mRNA elevation (“oeGαi2”) (Figure 5F). In the tested primary and immortalized glioma cells, ectopic Gαi2 overexpression augmented cell proliferation (EdU incorporation, Figure 5G) and accelerated in vitro cell migration (Figure 5H). Thus, Gαi2 overexpression resulted in pro-glioma cell activity. Whereas in the “Astryocyte1” and “Astryocyte2”, ectopic Gαi2 overexpression, by the same lentiviral Gαi2-expressing construct, led to Gαi2 mRNA upregulation (“oeGαi2”, Figure 5I). It however failed to increase proliferation (EdU incorporation, Figure 5J) in the astrocytes.
Gαi2 is important for NFκB activation in glioma cells
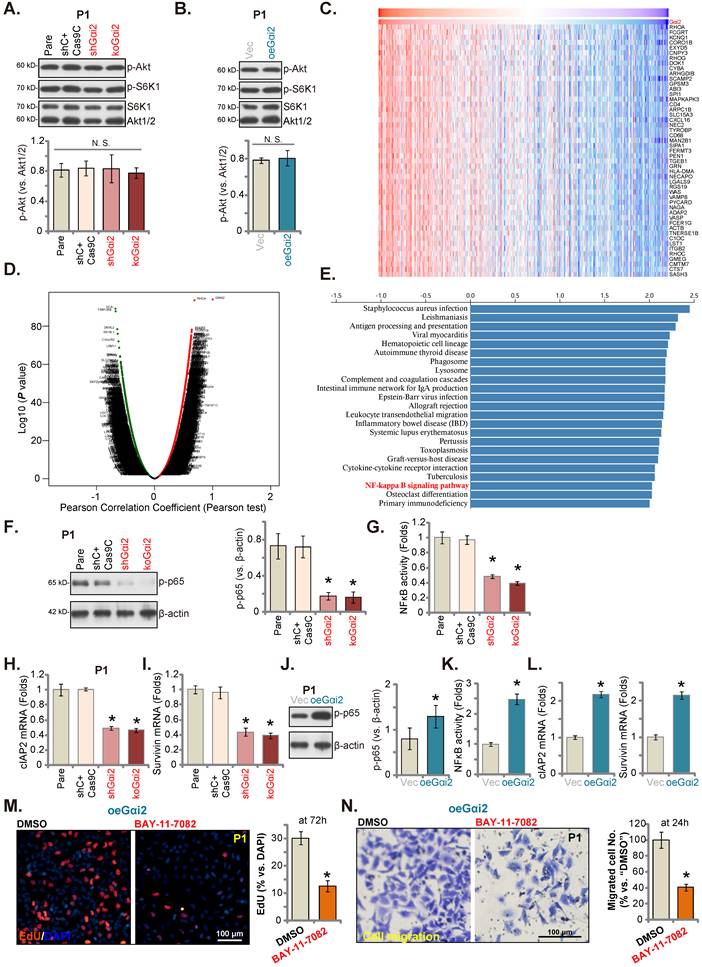
Gαi1/3 association with multiple RTKs (and non-RTK receptors) mediates Akt-mTOR cascade activation [15, 16, 18, 19, 21, 31, 39]. In P1 glioma cells, Gαi2 shRNA or KO however failed to significantly inhibit phosphorylation of Akt and S6K1 (Figure 6A). Moreover, Akt-S6K1 phosphorylation was unchanged following ectopic Gαi2 overexpression (Figure 6B). TCGA LGGGBM cohorts were thereafter analyzed and Gαi2-associated differentially expressed gene (DEGs) were retrieved (Figure 6C). The volcano map of Gαi2-assocaited DEGs was presented in Figure 6D (|LogFC|>1, Adjust P-value < 0.05). KEGG enrichment pathway analyses found that Gαi2-associated DEGs were enriched in multiple signaling cascades (Figure 6E). One key cascade is NFκB, important for glioma tumorigenesis and progression [40-42].
Gαi2 is important for NFκB activation in glioma cells. Expression of listed mRNAs/proteins in the stable P1 glioma cells with designated Gαi2 genetic modifications was tested (A, B, F, H-J and L); The p65 DNA-binding activity was tested as well (G and K). Differentially expressed gene (DEGs) based on Gαi2 expression in TCGA LGGGBM cohorts were shown (C) and the volcano map of DEGs was presented (D); KEGG analyses of Gαi2-associated DEGs and the corresponding enriched pathways were listed (E). The oeGαi2 P1 glioma cells were treated with BAY-11-7082 (15 μM) and cultured for designated hours, cell proliferation (nuclear EdU staining, M) and in vitro cell migration (N) were measured. *P < 0.05 versus “Pare” cells/“Vec” cells or “DMSO” treatment. Scale bar = 100 μm.
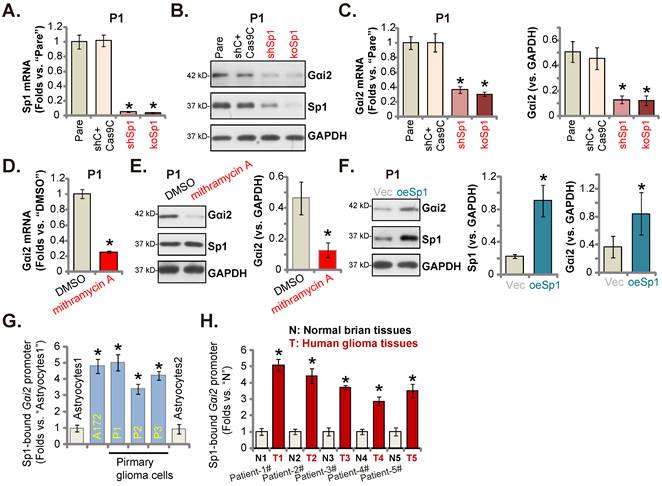
Sp1 and Gαi2 promoter binding increases in glioma tissues and cells. Puromycin-selected stable P1 glioma cells, with the lentiviral Sp1 shRNA (“shSp1”), the CRISPR/dCas9-Sp1-KO construct (“koSp1”) or “shC plus Cas9C”, the lentiviral Sp1-expressing construct (“oeSp1”) or “Vec” were formed, and listed mRNAs and proteins were tested (A-C and F). P1 primary glioma cells were treated with mithramycin A (200 nM) or DMSO (0.1%) for 24h, and listed mRNAs and proteins were shown (D and E). Chromosome IP (ChIP) showed the relative amount of Gαi2 promoter DNA binding to the transcription factor Sp1 in the listed cells (G) and human tissues (H). *P < 0.05 versus “Pare”/“Vec”/“DMSO”/“Astrocytes”/“N” tissues.
We thus tested NFκB activation in glioma cells. As shown, in both shGαi2 and koGαi2 P1 glioma cells (see Figure 3 and 4), phosphorylated p65 levels were robustly decreased (Figure 6F), the NFκB (p65) DNA-binding activity was also reduced in Gαi2-depleted P1 glioma cells (Figure 6G). Moreover, NFκB-dependent oncogenic genes, including cIAP2 and survivin [43-47], were downregulated in shGαi2 and koGαi2 P1 glioma cells (Figure 6H and I). Conversely, phosphorylated p65 (Figure 6J), NFκB (p65) DNA-binding activity (Figure 6K) and cIAP2-survivin mRNA levels (Figure 6L) were significantly augmented in oeGαi2 P1 glioma cells. Thus, Gαi2 is important for NFκB activation in glioma cells. BAY-11-7082, a well-known NFκB blocker [48, 49], substantially suppressed proliferation (Figure 6M) and migration (Figure 6N) of oeGαi2 P1 glioma cells. Thus, Gαi2-driven glioma cell progression is mediated, at least in part, through promoting NFκB cascade activation.
Sp1 and Gαi2 promoter binding increases in glioma tissues and cells
Since Gαi2 mRNA/protein levels were both elevated in glioma, it could be due to the transcriptional mechanism. Recent studies have implied that Sp1 (specificity protein 1) could be an important transcription factor of Gαi2 [37, 50]. We first tested whether Sp1 was important for Gαi2 expression in glioma cells. To this purpose, lentiviral particles with Sp1 shRNA were added to P1 cells, and stable cells (“shSp1”) formed. Alternatively, the lentiviral particles with the CRISPR/dCas9-Sp1-KO construct was added to the dCas9-expressing P1 cells, and stable Sp1 KO cells (“koSp1”) formed after selection. Sp1 mRNA (Figure 7A) and protein (Figure 7B) expression was robustly decreased in shSp1 and koSp1 P1 glioma cells. Importantly, Gαi2 mRNA/protein (Figure 7B and C) levels were reduced in Sp1-depleted P1 glioma cells. Mithramycin A, a compound that can prevent Sp1 binding to GC boxes in DNA [50], also decreased Gαi2 mRNA/protein levels in P1 cells (Figure 7D and E).
Next, the lentiviral particles packaging Sp1-overexpressing construct were added to P1 glioma cells, and stable cells established (“oeSp1”). Sp1 protein levels were remarkably upregulated in oeSp1 P1 glioma cells (Figure 7F). Following Sp1 overexpression, Gαi2 protein expression was increased as well (Figure 7F). Remarkably, Sp1 ChIP results revealed that Sp1-Gαi2 promoter binding [51] in various glioma cells (“P1-P3” primary cells and A172 cells) was substantially higher than it in Astrocytes1/2 (Figure 7G). Moreover, in human glioma tissues of five representative GBM patients, Sp1 binding to the Gαi2 promoter was robustly increased (Figure 7H). Therefore, Sp1 and Gαi2 promoter binding increasing could be an important mechanism of Gαi2 upregulation in human glioma tissues and cells.
Gαi2 silencing inhibits subcutaneous glioma xenograft growth in nude mice
P1 glioma cells (five million cells per mouse) were subcutaneously (s.c.) injected to nude mice. Twenty days after cell injection, the subcutaneous P1 glioma xenografts were formed and each xenograft was close to 100 mm3 (“Day-0”). AAV with Gαi2 shRNA (“AAV-sh-Gαi2”) were intratumorally injected to P1 glioma xenografts daily (for ten days), and control mice intratumorally injected with AAV-shC. Every six days tumor volumes were recorded. As shown, AAV-sh-Gαi2 injection remarkably hindered subcutaneous P1 glioma xenograft growth (Figure 8A) and reduced the estimated daily tumor growth [33, 52]. Intratumoral AAV-sh-Gαi2 injection slowed P1 glioma xenograft growth (Figure 8B). All P1 xenografts were carefully isolated at Day-42 and were tested. AAV-sh-Gαi2 xenografts were much smaller and lighter than AAV-shC xenografts (Figure 8C). No significant difference was observed in the mice body weights (Figure 8D). Thus, Gαi2 silencing inhibited subcutaneous P1 glioma xenograft growth in nude mice.
Gαi2 silencing inhibits subcutaneous glioma xenograft growth in nude mice. The subcutaneous P1 glioma xenograft-bearing nude mice were daily intratumorally injected with Gαi2 shRNA-expressing AAV (“AAV-sh-Gαi2”) or AAV-shC. The volumes of the xenografts (A) and animal body weights (D) were recorded. The estimated daily growth was calculated and was expressed at mm3 per day (B). At Day-42, all P1 glioma xenografts were isolated and measured (C). Listed mRNAs and proteins in the described P1 glioma xenograft tissues were tested (E, F, H and I). The representative IHC images of Gαi2 in the described P1 glioma xenograft slides were presented (G). Nuclear TUNEL fluorescence staining in the described P1 xenograft slides were presented (J). The subcutaneous A172 glioma xenograft-bearing nude mice were subject to daily intratumoral injection of AAV-sh-Gαi2 or AAV-shC. At Day-35, all A172 glioma xenografts were isolated, and were measured (K and L). In the xenograft tissues listed mRNAs and proteins were examined, and results quantified (M and N). *P < 0.05 versus “AAV-shC” group. “N.S.” means P > 0.05. Scale bar = 100 μm.
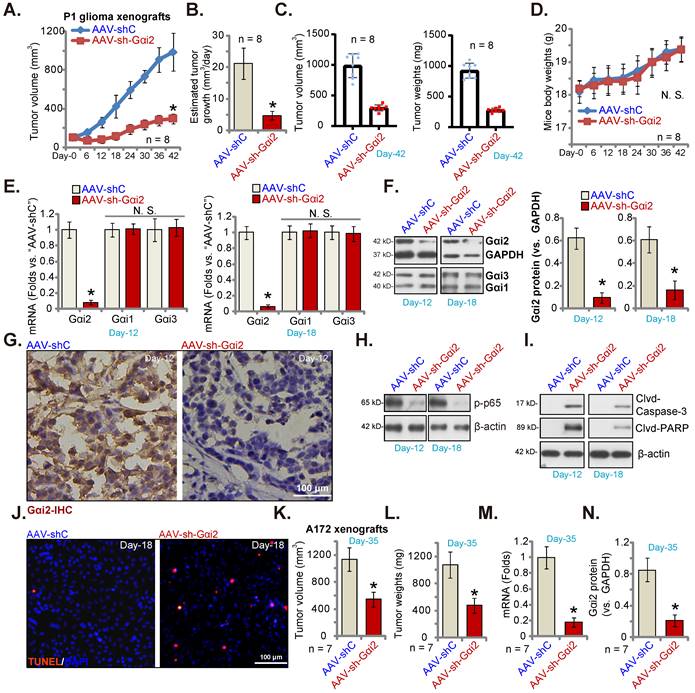
On “Day-12” and “Day-18” of the animal experiment, we separated one P1 glioma xenograft from each group. Gαi2 mRNA/protein (Figure 8E and F) levels were robustly decreased in AAV-sh-Gαi2-injected xenograft tissues. Contrarily, Gαi1 and Gαi3 expression was not changed (Figure 8E and F). IHC studies confirmed Gαi2 protein silencing in AAV-sh-Gαi2 xenograft slides (at “Day-12”, Figure 8G). Levels of phosphorylated p65, the indicator of NFκB activation, was significantly decreased in P1 glioma xenografts with AAV-sh-Gαi2 injection (Figure 8H), whereas caspase-3 and PARP cleavages were augmented (Figure 8I). Tissue immunofluorescence staining showed that TUNEL-stained nuclei were significantly increased in AAV-sh-Gαi2-injected P1 glioma xenografts (at “Day-18”, Figure 8J), supporting apoptosis activation.
Next, A172 glioma cells (six million cells in each mouse) were s.c. injected to the nude mice, and A172 glioma xenografts formed after two weeks (100 mm3 at “Day-0”). The A172 xenograft-bearing nude mice were then subject to the same AAV-sh-Gαi2 injection or AAV-shC injection (daily for 10 days). After five weeks (“Day-35”), all A172 xenografts were isolated. As demonstrated, A172 xenografts with AAV-sh-Gαi2 injection were significantly smaller (Figure 8K) and lighter (Figure 8L) than AAV-shC-injected xenografts. Analyzing A172 xenograft tissues confirmed Gαi2 mRNA (Figure 8M) and protein (Figure 8N) silencing in the AAV-sh-Gαi2 A172 xenografts.
Gαi2 knockout hinders intracranial glioma xenograft growth in nude mice
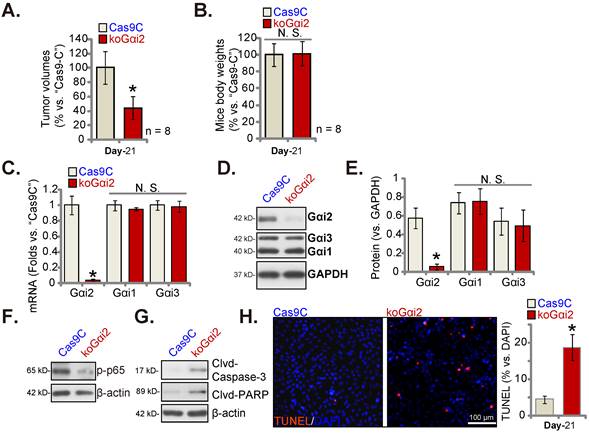
Lastly, using the described protocol [17] CRISPR/dCas9-Gαi2-KO construct (“koGαi2”)-expressing P1 glioma cells or Cas9C control cells were injected intracranially into the brains of the nude mice. Five days later, the intracranial glioma xenograft was established [17, 20, 25]. After 21 days (“Day-21”) the first Cas9C group mouse showed apparent symptoms. All mice were sacrificed and intracranial xenografts were isolated [17]. The koGαi2 intracranial glioma xenografts were smaller than Cas9C intracranial xenografts (Figure 9A). Mice body weights were indifferent (Figure 9B).
Gαi2 mRNA/protein expression was significantly decreased in koGαi2 intracranial P1 glioma xenograft tissues (Figure 9C-E). Gαi1/3 expression was again unchanged (Figure 9C-E). In koGαi2 xenografts, phosphorylated p65 was decreased, indicating NFκB inactivation (Figure 9F). Whereas caspase-3-PARP cleavages were significantly increased (Figure 9G). Moreover, TUNEL-stained nuclei were robustly increased in koGαi2 intracranial P1 glioma xenografts (Figure 9H), supporting apoptosis activation. These results showed that Gαi2 knockout hindered intracranial P1 glioma xenograft growth in nude mice.
Gαi2 knockout hinders intracranial glioma xenograft growth in nude mice. P1 primary human glioma cells, at half million cells of each mouse, with the CRISPR/dCas9-Gαi2-KO construct (“koGαi2”) or control (“Cas9C”), were intracranially injected to nude mice's brains; After 21 days (“Day-21”), animals were decapitated and intracranial glioma xenografts were isolated, the tumor volumes (A) and mice body weights (B) were shown. The listed mRNAs and proteins were measured (C-G); Nuclear TUNEL fluorescence staining in the described intracranial P1 glioma xenograft slides were presented (H). *P < 0.05 versus “Cas9C” group. “N.S.” means P > 0.05. Scale bar = 100 μm.
Discussion
GBM and other HGG are most aggressive and lethal malignant tumors that originate in the brain [53, 54]. Currently, there is a lack of effective treatments [5, 6, 55, 56]. Compared with traditional treatment methods, molecular targeted therapies [57, 58] could have better selectivity and specificity against glioma [5, 6, 55, 56]. We showed that Gαi2 could be an important therapeutic oncotarget of glioma. Bioinformatics analyses revealed that Gαi2 transcripts are significantly elevated in human glioma, and its overexpression correlates with poor patients' survival, higher tumor grade and WT-IDH status. Moreover, Gαi2 upregulation is also detected in local glioma tissues and various human glioma cells.
In primary and immortalized (A172) glioma cells, Gαi2 shRNA or KO substantially suppressed viability, cell proliferation and mobility. Silence of Gαi2 by targeted shRNA however failed to inhibit viability and proliferation in non-cancerous human astrocytes. In addition, Gαi2 shRNA or KO provoked caspase activation, mitochondrial depolarization and apoptosis in the primary and A172 glioma cells. Whereas Gαi2 silencing failed to provoke caspase-apoptosis activation in human astrocytes. Contrarily, ectopic Gαi2 overexpression, using the lentiviral construct, further promoted malignant behaviors of primary and immortalized glioma cells, enhancing cell proliferation, migration and invasion. Gαi2 overexpression was however not effective in human astrocytes. Importantly, daily intratumoral Gαi2 shRNA AAV injection largely hindered subcutaneous P1 xenograft growth in nude mice. Moreover, the growth of intracranial P1 xenografts was largely inhibited after Gαi2 KO. Therefore, overexpressed Gαi2 is important for glioma cell growth.
Activation of NFκB cascade is important for carcinogenesis and progression of human glioma [40-42]. Xu et al., have shown that cullin-7 (CUL7), a DOC domain-containing cullin family protein, promoted gliomagenesis by promoting MST1 protein degradation and activating NF-κB pathway [42]. Conversely, CUL7 silencing inhibited NF-κB activation and prevented growth of glioma cells [42]. Chai et al., reported that overexpressed YTHDF2 promoted glioma cell growth by activating NF-κB activation [41]. YTHDF2 dictated degradation of UBX domain protein 1 (UBXN1) mRNA through methyltransferase-like 3 (METTL3)-dependent m6A modification, which in turn activated NF-κB cascade [41]. Chang et al., also reported that METTL3 promoted the malignant progression of IDH-WT glioma possibly by enhancing NF-κB activation [40]. Ji et al., reported that elevated TRIM22 (tripartite motif 22) promoted GBM cell proliferation by activating NF-κB signaling [59].
Early studies have implied that Gαi2 could be important for NF-κB cascade activation. Conditional disruption of Gαi2 in CD11c+ DCs and MDSCs prevented NF-κB and STAT3 activation [22]. Gαi2-depletion-induced NF-κB inactivation was possibly due to blocking IL-6 signaling [22]. Under hepatic ischemia-reperfusion injury, increased Gαi2 expression promoted NF-κB pathway activation through phosphorylating mixed-lineage protein kinase 3 (MLK3) [60]. In the present study, we found that Gαi2 was important for NFκB activation in glioma cells. Indeed, p65 phosphorylation, NFκB (p65) DNA-binding activity and expression of NFκB-dependent genes (cIAP2 and survivin) were significantly decreased in Gαi2-depleted primary glioma cells, but were increased following Gαi2 overexpression. BAY-11-7082, the NFκB inhibitor, largely suppressed proliferation and migration of Gαi2-overexpressed P1 glioma cells. Importantly, decreased p65 phosphorylation was observed in subcutaneous and intracranial glioma xenografts with Gαi2 depletion. Therefore, promoting NFκB cascade activation should be one important mechanism of Gαi2-driven glioma cell growth.
Studies have proposed that Sp1 is an important transcription factor for the malignant progression of glioma. Yu et al., found that Sp1 enhanced NLR family pyrin domain containing 6 (NLRP6) transcription to promote immune evasion, malignant behaviors and radio-resistance in glioma cells. Contrarily, Sp1 silencing suppressed in vitro glioma cell growth and tumorigenesis in vivo [61]. Li et al., reported that Sp1 upregulated the LncRNA LBX2-AS1 to promote proliferation and EMT in glioma cells [62]. Tan et al., discovered that miR-150-3p silenced Sp1 to hinder glioma cell growth [63]. Our results supported that Sp1-dependent Gαi2 transcription was increased in glioma tissues and cells, which might be one primary mechanism of Gαi2 upregulation in glioma. In glioma cells Gαi2 expression was downregulated after Sp1 silencing, KO or inhibition. It was however increased following Sp1 overexpression. Therefore, the increase of Sp1-dependent transcription should be one key mechanism of Gαi2 overexpression in human glioma.
Here, TCGA LGGGBM cohorts were analyzed and Gαi2-asscoateid DEGs in glioma tissues were retrieved, including a significant number of genes with unknown functions in human glioma. Moreover, KEGG analyses showed that Gαi2-asscoateid DEGs were enriched in NFκB and other signaling cascades. Further studies will be needed to explore expression and potential functions of these Gαi2-asscoateid DEGs in glioma, and to test these enriched pathways in the progression of glioma. Their connection with Gαi2 should also be analyzed.
Conclusion
Together, overexpressed Gαi2 is important for glioma cell growth possibly by promoting NFκB cascade activation. Gαi2 is possibly a novel and promising therapeutic oncotarget of glioma.
Supplementary Material
Supplementary figure.
Acknowledgements
Funding
This work was generously supported by Key Research and Development Program of Jiangsu Province (No. BE2019652), National Natural Science Foundation of China (81922025, 81802511, 82171461, 81771457, 82171294) and Development Program of Changzhou City (CE20205024). Changzhou international cooperation program (CZ20200039). A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Ethical approval and consent to participate
This study was approved by the Ethics Committee of Soochow University.
Author contributions
All authors conceived the idea and designed the work, contributed to acquisition of data.
Availability of data and material
All data generated during this study are included in this published article. Data will be made available upon request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ellis HP, Greenslade M, Powell B, Spiteri I, Sottoriva A, Kurian KM. Current Challenges in Glioblastoma: Intratumour Heterogeneity, Residual Disease, and Models to Predict Disease Recurrence. Front Oncol. 2015;5:251
2. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055-60
3. Little SE, Popov S, Jury A, Bax DA, Doey L, Al-Sarraj S. et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012;72:1614-20
4. Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011;12:495-508
5. Wen PY, Reardon DA. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nat Rev Neurol. 2016;12:69-70
6. Reardon DA, Wen PY. Glioma in 2014: unravelling tumour heterogeneity-implications for therapy. Nat Rev Clin Oncol. 2015;12:69-70
7. Zhou Q, van den Berg NS, Rosenthal EL, Iv M, Zhang M, Vega Leonel JCM. et al. EGFR-targeted intraoperative fluorescence imaging detects high-grade glioma with panitumumab-IRDye800 in a phase 1 clinical trial. Theranostics. 2021;11:7130-43
8. Xiao J, Jin X, Zhang C, Zou H, Chang Z, Han N. et al. Systematic analysis of enhancer regulatory circuit perturbation driven by copy number variations in malignant glioma. Theranostics. 2021;11:3060-73
9. Kwiatkowska A, Symons M. Signaling Determinants of Glioma Cell Invasion. Adv Exp Med Biol. 2020;1202:129-49
10. Fan H, Li P, Zingarelli B, Borg K, Halushka PV, Birnbaumer L. et al. Heterotrimeric Galpha(i) proteins are regulated by lipopolysaccharide and are anti-inflammatory in endotoxemia and polymicrobial sepsis. Biochim Biophys Acta. 2011;1813:466-72
11. Chen YJ, Wang CJ, Yang KD, Chang PR, Huang HC, Huang YT. et al. Pertussis toxin-sensitive Galphai protein and ERK-dependent pathways mediate ultrasound promotion of osteogenic transcription in human osteoblasts. FEBS Lett. 2003;554:154-8
12. Luo Y, Kokkonen GC, Wang X, Neve KA, Roth GS. D2 dopamine receptors stimulate mitogenesis through pertussis toxin-sensitive G proteins and Ras-involved ERK and SAP/JNK pathways in rat C6-D2L glioma cells. J Neurochem. 1998;71:980-90
13. Gilder AS, Wang L, Natali L, Karimi-Mostowfi N, Brifault C, Gonias SL. Pertussis Toxin Is a Robust and Selective Inhibitor of High Grade Glioma Cell Migration and Invasion. PLoS One. 2016;11:e0168418
14. Magana-Maldonado R, Manoutcharian K, Hernandez-Pedro NY, Rangel-Lopez E, Perez-De la Cruz V, Rodriguez-Balderas C. et al. Concomitant treatment with pertussis toxin plus temozolomide increases the survival of rats bearing intracerebral RG2 glioma. J Cancer Res Clin Oncol. 2014;140:291-301
15. Sun J, Huang W, Yang SF, Zhang XP, Yu Q, Zhang ZQ. et al. Galphai1 and Galphai3mediate VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis. Theranostics. 2018;8:4695-709
16. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS. et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2:ra17
17. Liu YY, Chen MB, Cheng L, Zhang ZQ, Yu ZQ, Jiang Q. et al. microRNA-200a downregulation in human glioma leads to Galphai1 over-expression, Akt activation, and cell proliferation. Oncogene. 2018;37:2890-902
18. Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q. et al. Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol. 2015;135:181-91
19. Marshall J, Zhou XZ, Chen G, Yang SQ, Li Y, Wang Y. et al. Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc Natl Acad Sci U S A. 2018;115:E3549-E58
20. Liu F, Chen G, Zhou L-N, Wang Y, Zhang Z-q, Qin X. et al. YME1L overexpression exerts pro-tumorigenic activity in glioma by promoting Gαi1 expression and Akt activation. Protein & Cell. 2022 10.1093/procel/pwac011
21. Wang Y, Liu YY, Chen MB, Cheng KW, Qi LN, Zhang ZQ. et al. Neuronal-driven glioma growth requires Galphai1 and Galphai3. Theranostics. 2021;11:8535-49
22. Li ZW, Sun B, Gong T, Guo S, Zhang J, Wang J. et al. GNAI1 and GNAI3 Reduce Colitis-Associated Tumorigenesis in Mice by Blocking IL6 Signaling and Down-regulating Expression of GNAI2. Gastroenterology. 2019;156:2297-312
23. Fu X, Li Y, Alvero A, Li J, Wu Q, Xiao Q. et al. MicroRNA-222-3p/GNAI2/AKT axis inhibits epithelial ovarian cancer cell growth and associates with good overall survival. Oncotarget. 2016;7:80633-54
24. Zhang Z, Ji Z, He J, Lu Y, Tian W, Zheng C. et al. Guanine Nucleotide-Binding Protein G(i) Subunit Alpha 2 Exacerbates NASH Progression by Regulating Peroxiredoxin 1-Related Inflammation and Lipophagy. Hepatology. 2021;74:3110-26
25. Guo YZ, Chen G, Huang M, Wang Y, Liu YY, Jiang Q. et al. TIMM44 is a potential therapeutic target of human glioma. Theranostics. 2022;12:7586-602
26. Zhao Z, Zhang KN, Wang Q, Li G, Zeng F, Zhang Y. et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genomics Proteomics Bioinformatics. 2021;19:1-12
27. Shao NY, Wang DX, Wang Y, Li Y, Zhang ZQ, Jiang Q. et al. MicroRNA-29a-3p Downregulation Causes Gab1 Upregulation to Promote Glioma Cell Proliferation. Cell Physiol Biochem. 2018;48:450-60
28. Cai S, Li Y, Bai JY, Zhang ZQ, Wang Y, Qiao YB. et al. Galphai3 nuclear translocation causes irradiation resistance in human glioma cells. Oncotarget. 2017;8:35061-8
29. Yang J, Xia A, Zhang H, Liu Q, You H, Ding D. et al. Up-Regulating ERIC by CRISPR-dCas9-VPR Inhibits Cell Proliferation and Invasion and Promotes Apoptosis in Human Bladder Cancer. Frontiers In Molecular Biosciences. 2021;8:654718
30. Yao J, Wu XY, Yu Q, Yang SF, Yuan J, Zhang ZQ. et al. The requirement of phosphoenolpyruvate carboxykinase 1 for angiogenesis in vitro and in vivo. Sci Adv. 2022;8:eabn6928
31. Bian ZJ, Shan HJ, Zhu YR, Shi C, Chen MB, Huang YM. et al. Identification of Galphai3 as a promising target for osteosarcoma treatment. Int J Biol Sci. 2022;18:1508-20
32. Shan HJ, Zhu LQ, Yao C, Zhang ZQ, Liu YY, Jiang Q. et al. MAFG-driven osteosarcoma cell progression is inhibited by a novel miRNA miR-4660. Mol Ther Nucleic Acids. 2021;24:385-402
33. Lv Y, Wang Y, Song Y, Wang SS, Cheng KW, Zhang ZQ. et al. LncRNA PINK1-AS promotes G alpha i1-driven gastric cancer tumorigenesis by sponging microRNA-200a. Oncogene. 2021;40:3826-44
34. Gao YY, Ling ZY, Zhu YR, Shi C, Wang Y, Zhang XY. et al. The histone acetyltransferase HBO1 functions as a novel oncogenic gene in osteosarcoma. Theranostics. 2021;11:4599-615
35. Zhang J, Yin DP, Zhang Y, Zhang JN, Yang Y, Zhang ZQ. et al. Identification of Galphai3 as a novel molecular therapeutic target of cervical cancer. Int J Biol Sci. 2022;18:5667-80
36. He L, Fan X, Li Y, Chen M, Cui B, Chen G. et al. Overexpression of zinc finger protein 384 (ZNF 384), a poor prognostic predictor, promotes cell growth by upregulating the expression of Cyclin D1 in Hepatocellular carcinoma. Cell Death Dis. 2019;10:444
37. Menzaghi C, Paroni G, De Bonis C, Soccio T, Marucci A, Bacci S. et al. The -318 C>G single-nucleotide polymorphism in GNAI2 gene promoter region impairs transcriptional activity through specific binding of Sp1 transcription factor and is associated with high blood pressure in Caucasians from Italy. J Am Soc Nephrol. 2006;17:S115-9
38. Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS. et al. Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J Clin Invest. 2012;122:253-66
39. Xu G, Qi L-n, Zhang M-q, Li X-y, Chai J-l, Zhang Z-q. et al. Gαi1/3 mediation of Akt-mTOR activation is important for RSPO3-induced angiogenesis. Protein & Cell. 2022 10.1093/procel/pwac035
40. Chang YZ, Chai RC, Pang B, Chang X, An SY, Zhang KN. et al. METTL3 enhances the stability of MALAT1 with the assistance of HuR via m6A modification and activates NF-kappaB to promote the malignant progression of IDH-wildtype glioma. Cancer Lett. 2021;511:36-46
41. Chai RC, Chang YZ, Chang X, Pang B, An SY, Zhang KN. et al. YTHDF2 facilitates UBXN1 mRNA decay by recognizing METTL3-mediated m(6)A modification to activate NF-kappaB and promote the malignant progression of glioma. J Hematol Oncol. 2021;14:109
42. Xu J, Zhang Z, Qian M, Wang S, Qiu W, Chen Z. et al. Cullin-7 (CUL7) is overexpressed in glioma cells and promotes tumorigenesis via NF-kappaB activation. J Exp Clin Cancer Res. 2020;39:59
43. Cui X, Shen D, Kong C, Zhang Z, Zeng Y, Lin X. et al. NF-kappaB suppresses apoptosis and promotes bladder cancer cell proliferation by upregulating survivin expression in vitro and in vivo. Sci Rep. 2017;7:40723
44. Tracey L, Perez-Rosado A, Artiga MJ, Camacho FI, Rodriguez A, Martinez N. et al. Expression of the NF-kappaB targets BCL2 and BIRC5/Survivin characterizes small B-cell and aggressive B-cell lymphomas, respectively. J Pathol. 2005;206:123-34
45. Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M. et al. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int J Cancer. 2005;115:967-74
46. Jiang XJ, Chen ZW, Zhao JF, Liao CX, Cai QH, Lin J. cIAP2 via NF-kappaB signalling affects cell proliferation and invasion in hepatocellular carcinoma. Life Sci. 2021;266:118867
47. Kingeter LM, Schaefer BC. Malt1 and cIAP2-Malt1 as effectors of NF-kappaB activation: kissing cousins or distant relatives? Cell Signal. 2010;22:9-22
48. Irrera N, Vaccaro M, Bitto A, Pallio G, Pizzino G, Lentini M. et al. BAY 11-7082 inhibits the NF-kappaB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin Sci (Lond). 2017;131:487-98
49. Chen L, Ruan Y, Wang X, Min L, Shen Z, Sun Y. et al. BAY 11-7082, a nuclear factor-kappaB inhibitor, induces apoptosis and S phase arrest in gastric cancer cells. J Gastroenterol. 2014;49:864-74
50. Arinze IJ, Kawai Y. Sp family of transcription factors is involved in valproic acid-induced expression of Galphai2. J Biol Chem. 2003;278:17785-91
51. Guo S, Zhang Y, Zhou T, Wang D, Weng Y, Wang L. et al. Role of GATA binding protein 4 (GATA4) in the regulation of tooth development via GNAI3. Sci Rep. 2017;7:1534
52. Chen MB, Liu YY, Xing ZY, Zhang ZQ, Jiang Q, Lu PH. et al. Itraconazole-Induced Inhibition on Human Esophageal Cancer Cell Growth Requires AMPK Activation. Mol Cancer Ther. 2018;17:1229-39
53. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
54. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
55. Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol. 2019;15:405-17
56. Watts C, Price SJ, Santarius T. Current concepts in the surgical management of glioma patients. Clin Oncol (R Coll Radiol). 2014;26:385-94
57. Chen S, Zhang Z, Zhang B, Huang Q, Liu Y, Qiu Y. et al. CircCDK14 Promotes Tumor Progression and Resists Ferroptosis in Glioma by Regulating PDGFRA. Int J Biol Sci. 2022;18:841-57
58. Li H, Wang D, Yi B, Cai H, Wang Y, Lou X. et al. SUMOylation of IGF2BP2 promotes vasculogenic mimicry of glioma via regulating OIP5-AS1/miR-495-3p axis. Int J Biol Sci. 2021;17:2912-30
59. Ji J, Ding K, Luo T, Zhang X, Chen A, Zhang D. et al. TRIM22 activates NF-kappaB signaling in glioblastoma by accelerating the degradation of IkappaBalpha. Cell Death Differ. 2021;28:367-81
60. Sun Q, He Q, Xu J, Liu Q, Lu Y, Zhang Z. et al. Guanine nucleotide-binding protein G(i)alpha2 aggravates hepatic ischemia-reperfusion injury in mice by regulating MLK3 signaling. FASEB J. 2019;33:7049-60
61. Yu Y, Cao F, Xiong Y, Zhou H. SP1 transcriptionally activates NLRP6 inflammasome and induces immune evasion and radioresistance in glioma cells. Int Immunopharmacol. 2021;98:107858
62. Li W, Soufiany I, Lyu X, Lu C, Wei Y, Shi Z. et al. SP1-upregulated LBX2-AS1 promotes the progression of glioma by targeting the miR-491-5p/LIF axis. J Cancer. 2021;12:6989-7002
63. Tan Z, Jia J, Jiang Y. MiR-150-3p targets SP1 and suppresses the growth of glioma cells. Biosci Rep. 2018 38
Author contact
![]() Corresponding author: Prof. Cong Cao, Ph.D, M.D. E-mail: caocongedu.cn.
Corresponding author: Prof. Cong Cao, Ph.D, M.D. E-mail: caocongedu.cn.