Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Mitochondrial function in cancer...
Mitochondrial function in cancer...
Mitochondria in the tumor...
Targeting mitochondria for...
Conclusion and perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(3):897-915. doi:10.7150/ijbs.81609 This issue Cite
Review
An Overview: The Diversified Role of Mitochondria in Cancer Metabolism
Yu'e Liu1 ![]() #, Yihong Sun1#, Yadong Guo2#, Xiaoyun Shi3#, Xi Chen4, Wenfeng Feng1, Lei-Lei Wu5, Jin Zhang6, Shibo Yu7, Yi Wang8, Yufeng Shi1,9
#, Yihong Sun1#, Yadong Guo2#, Xiaoyun Shi3#, Xi Chen4, Wenfeng Feng1, Lei-Lei Wu5, Jin Zhang6, Shibo Yu7, Yi Wang8, Yufeng Shi1,9 ![]()
1. Tongji University Cancer Center, Shanghai Tenth People's Hospital of Tongji University, School of Medicine, Tongji University, Shanghai 200092, China.
2. Department of Urology, Shanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai, China.
3. Institute of Artificial Intelligence, Hefei Comprehensive National Science Center, Hefei, China.
4. Xi Chen, Children's Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, Houston, TX, 77030, USA.
5. Department of Thoracic Surgery, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, 200433, Shanghai, China.
6. Department of Pharmacology and Toxicology, University of Mississippi Medical Center, 39216, Jackson, Mississippi, USA.
7. Department of Pathology and Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
8. Department of Critical Care Medicine, Sichuan Academy of Medical Science and Sichuan Provincial People's Hospital, University of Electronic Science and Technology of China, Chengdu, China.
9. Clinical Center for Brain and Spinal Cord Research, Tongji University, Shanghai 200092, China.
#These authors contributed equally to this work.
Received 2022-12-6; Accepted 2023-1-4; Published 2023-1-16
Abstract

Mitochondria are intracellular organelles involved in energy production, cell metabolism and cell signaling. They are essential not only in the process of ATP synthesis, lipid metabolism and nucleic acid metabolism, but also in tumor development and metastasis. Mutations in mtDNA are commonly found in cancer cells to promote the rewiring of bioenergetics and biosynthesis, various metabolites especially oncometabolites in mitochondria regulate tumor metabolism and progression. And mutation of enzymes in the TCA cycle leads to the unusual accumulation of certain metabolites and oncometabolites. Mitochondria have been demonstrated as the target for cancer treatment. Cancer cells rely on two main energy resources: oxidative phosphorylation (OXPHOS) and glycolysis. By manipulating OXPHOS genes or adjusting the metabolites production in mitochondria, tumor growth can be restrained. For example, enhanced complex I activity increases NAD+/NADH to prevent metastasis and progression of cancers. In this review, we discussed mitochondrial function in cancer cell metabolism and specially explored the unique role of mitochondria in cancer stem cells and the tumor microenvironment. Targeting the OXPHOS pathway and mitochondria-related metabolism emerging as a potential therapeutic strategy for various cancers.
Keywords: mitochondria, cancer, tumor metastasis, tumor metabolism
Introduction
Mitochondria, known as the “powerhouse of the cell”, are one of the most crucial organelles inside the cell. They evolved from bacteria and participated in the eukaryotic cell via the process named endosymbiosis, thus improving cell energy production and generating adenosine triphosphate (ATP) via the respiratory chain. Mitochondria have their translation system and genetic material mitochondrial DNA (mtDNA) without introns which are similar to bacterial DNA. These maternally inherited mtDNAs present in multiple copies which are easily mutated due to replication errors, oxidative stress or inefficient DNA repair. Usually, cancer cells harbor somatic mutations in mtDNA and mutations of mtDNA showed advantages of tumor growth. Now the somatic mutations in mtDNA can be tracked by single-cell RNA or assay for transposase accessible chromatin (ATAC) sequencing [1].
Mitochondria are the metabolic hub of tumor cell proliferation, survival and metastasis due to their bioenergetic and biosynthetic function [2]. The oxidative phosphorylation (OXPHOS) metabolic pathway generates ATP by transporting electrons through a series of complexes in inner membrane, known as the Electron Transport Chain (ETC) [3] (Figure 1). Proteins and complexes in the OXPHOS pathway have been identified as cancer targets, thus a myriad of inhibitors was developed in recent years [4, 5]. Moreover, mitochondria are dynamic organelles, they are undergoing fusion and fission to adjust morphology to enhance cell survival. Mitochondrial trafficking is crucial during cell division, proliferation and apoptosis. To maintain the consistent dynamic condition and to keep cell homeostasis, mitochondria can execute mitophagy to degrade the damaged mitochondria [6, 7].
The OXPHOS pathway. Mitochondria produce ATP via OXPHOS carried out by ETC and synthase. In humans, the OXPHOS is comprised of five complexed embedded in the inner mitochondria membrane (IMM), the ETC (complex I-IV) are responsible for the transfer of electrons from NADH and FADH2 to oxygen which is the final electron acceptor. This electron transportation generates the electrochemical gradient across the IMM. The proton gradient drives the translocation of protons from intermembrane space back into the matrix via the ATP synthase (Complex V (CV)) that catalyzes the conversion of ADP to ATP.
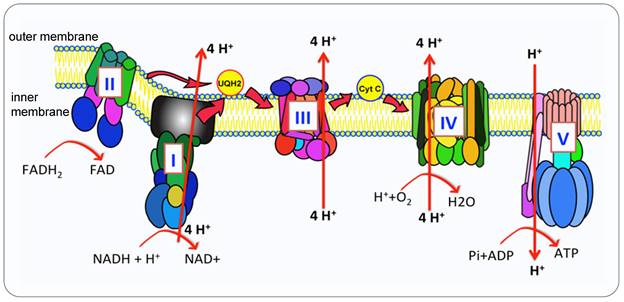
This review covers several fundamental functions of mitochondria, intending to introduce non-experts to understand the unique role of mitochondria and exploring the possible therapeutic target related to mitochondria.
Mitochondrial function in cancer cell metabolism
Mitochondria are the major cellular source of NADH and house parts of the pyrimidine and lipid biosynthetic pathways, it plays various roles in cancer cell metabolism including but not limited to ATP metabolism, lipid metabolism and nucleic acid metabolism. Many different metabolites or intermediates in the tricarboxylic acid (TCA) cycle are critical in cancer cell proliferation, metastasis and apoptosis.
Mitochondrial function in ATP metabolism
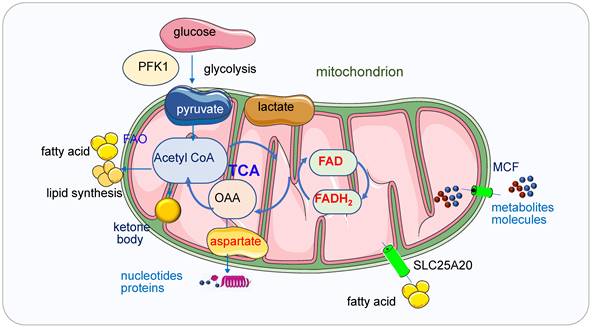
ATP is the central energy source in the cell, it can be generated via two pathways: glycolysis in cytostome and OXPHOS in mitochondria [8]. ATP production and consumption play vital roles in biosynthesis, metabolism regulation and cellular maintenance [9]. Altered energy metabolism representing one of the “hallmarks of cancer” is a biochemical fingerprint of cancer cells. Glycolysis regulated by phosphorfuctokinase1 (PFK1) produces pyruvate and NADH from glucose and then the pyruvate enters the mitochondria under aerobic conditions and fuels the TCA cycle which produces produce metabolites [10] (Figure 2). These metabolites or enzymes such as isocitrate dehydrogenase (IDH), fumarate hydratase (FH), succinate dehydrogenase (SDH) and α-ketoglutarate dehydrogenase (α-KGDHC) are often mutated or deregulated in human cancers. Both glycolysis and OXPHOS pathway have been demonstrated as the targets in cancer treatment.
Mitochondria in ATP, lipid, and nucleic acid metabolism. Glycolysis regulated by PFK1 produces pyruvate and NADH from glucose and then the pyruvate enters the mitochondria under aerobic conditions and fuels the TCA cycle which produces produce metabolites. Mitochondria generate ATP through OXPHOS mainly by using pyruvate derived from glycolysis. In the absence of glucose, ATP will be produced via the degradation of fatty acids and proteins. Ketone bodies will be produced from FAO when the glucose is insufficient. Ketone bodies are generated in the mitochondrial matrix of liver cells and are subsequently exported via the blood to other organs to cover the energy demands. When the glucose transporters or ETC complexes deficiency happens, the glucose metabolism and the oxidation of pyruvate in mitochondria are bypassed, and cellular energy supply will be shifted from glucose to ketone bodies. Fatty acids are transferred from the cytosol to mitochondria by SLC25A20. In the TCA cycle, oxaloacetate generated from malic acid is transaminated to aspartate under the catalysis of transaminase and aspartate is the intermediate for synthesizing both purine and pyrimidine bases. Small metabolites or molecules are transported to the matrix of mitochondria by MCFs, which transport ADP into the mitochondrial matrix for ATP synthesis and ATP out to reach high cytosolic ATP concentrations for energy-requiring reactions.
Due to the heterogeneity, tumors exhibit different metabolic phenotypes. In gliomas, there are both OXPHOS-dependent cells and glycolytic-dependent cells, the mechanism lies in the lactate dehydrogenase (LDH), the former expressed both LDH-A and -B isoforms whereas the latter only expressed LDH-B and LDH-B would be expected to be essential for the use of extracellular lactate to fuel cell activities [11]. Even proliferating tumor cells prefer a shift towards increasing in glycolytic metabolism from OXPHOS in the presence of O2 (known as the Warburg effect), slow-cycling tumor cells may prefer mitochondrial respiration as a primary source of energy [12, 13]. Mitochondria play essential roles in these slow-cycling tumor cells and thus targeting OXPHOS will be a promising therapy to these cancer cells.
Compared with glycolysis, OXPHOS produces ATP in a higher yield but the lower rate (30 or 32 mol ATP versus 2 mol per mole of glucose) [14]. NADH and FADH2 generated from glycolysis and TCA cycle entered the matrix in mitochondria, the first four complexes embedded in the inner mitochondrial membrane transfer electrons from complex I or II to complex IV, the oxidization process of NADH and FADH coupling with the phosphorylation of ADP generate ATP, supplying energy for all the cell activities, this process is called OXPHOS [15] (Figure 1). Many diverse types of cancer cells depend on OXPHOS instead of glycolysis to obtain their ATP to promote tumorigenic potential [4, 16]. Cancer cells upregulate OXPHOS and TCA cycle to obtain more ATP than their surrounding normal cells [17] and to generate resistance to chemotherapy [18]. Potent OXPHOS inhibitors induce mitochondria stress from a mild state to a severe lethal energetic crisis [19].
Mitochondria generate ATP through OXPHOS mainly by using pyruvate derived from glycolysis. In the absence of glucose, ATP will be produced via the degradation of fatty acids and proteins. Ketone bodies (3-β-hydroxybutyrate, acetoacetate, acetone) will be produced from fatty acid oxidation (FAO) during fasting or prolonged exercise when the glucose is insufficient. Ketone bodies are generated in the mitochondrial matrix of liver cells and are subsequently exported via the blood to other organs to cover the energy demands [20]. When the glucose transporters or ETC complexes deficiency happens, the glucose metabolism and the oxidation of pyruvate in mitochondria are bypassed, and cellular energy supply will be shifted from glucose to ketone bodies [20].
Mitochondrial function in lipid metabolism
Mitochondria consist of around 1000 proteins and more than 400 lipids [21]. Mitochondrial membrane lipids including phosphatidylethanolamine and cardiolipin are synthesized within mitochondria while other lipids are synthesized in the endoplasmic reticulum (ER) [22]. Mitochondria lipid homeostasis and trafficking among different organelles inside the cell are critical to mitochondrial function [23]. Mitochondria can synthesize phospholipids, phosphatidylethanolamine (PE), cardiolipin (CL) and the latter two are essential phospholipids for mitochondria respiratory functions [24, 25]. Therefore, mitochondria maintain a highly active exchange of phospholipids with other cellular compartments [26].
The history of the relation between mitochondria and lipid synthesis and metabolism can be dated back to 1959 [27]. After lipolysis (fat mobilization), fat is decomposed into fatty acid and glycerol, then fatty acids are transferred from the cytosol to mitochondria by carnitine, which is called carnitine shuttle, fulfilled by palmitoylcarnitine transferase 1 and 2, SLC25A20 in the inner mitochondrial membrane. The β-oxidation of fatty acids takes place in mitochondria and peroxisomes [28]. Free fatty acids alter respiration by providing fuel for β-oxidation, acting as weak uncouplers and activating UCP1 [29].
Mitochondria associate with lipid droplets (LDs) in fat-oxidizing tissues with fatty acid storage and oxidation capacity [30], mitochondria-LD association enhances LD biogenesis and thereby protects mitochondria from lipotoxicity [31]. LDs combined with mitochondria participate in metabolic regulation to mediate cellular function [32]. Dgat1-dependent LD biogenesis protects mitochondrial function during starvation-induced autophagy [31, 33]. mitochondria bounding with LDs are named peridroplet mitochondria (PDM) [34], it can be assessed by several quantifiable parameters such as membrane contact with an LD and adherence to an LD even after cell disruption or in restrictive purification conditions [35-37]. Mitochondria in brown adipocytes continuously engage in fusion and fission activities [38]. In the fission process, this PDM were segregated from cytoplasmic mitochondria and promoted triacylglyceride (TAG) synthesis [34]. The segregation of PDM is achieved by keeping mitochondrial fusion proteins intact which are quite different from that in regular mitochondria whose segregation leads to disparate cristae structures, proteomes, and super-complexes. By anchoring LDs, the PDM's motility was reduced, PDM have distinct proteome and metabolic capabilities and are demonstrated as a novel node of control for lipid metabolism [39], separation of PDM from cytoplasmic mitochondria requires a modified approach. PDM can function in LDs accumulation and FAO. They are playing different roles in different physiological conditions. For example, in brown adipose tissue in mice, both the Mito-LD interaction and LD mass will be increased while FAO will be lower under the condition of thermoneutrality compared to cold exposure, thus the PDM contributes to the lipid synthesis under this condition [34, 37]. In the human vastus lateralis muscle, Mito-LD interaction, LDs and FAO are higher after endurance training, thus the PDM contributes to both LDs synthesis and FAO metabolism [36]. Mass spectrometry results indicate similar mitochondrial protein in CM and PDM, similar membrane potential, but the PDM have increased respiratory capacity, higher proton efflux rate and enhanced OXPHOS capacity [34]. The mitochondria-lipid droplet association promotes triacyl glyceride synthesis and the peridroplet mitochondria have a unique structure, fusion dynamics, movement but reduced DPR1 recruitment and OPA1 processing. But the peridroplet mitochondria have no relation with FAO. LDs can exist in all different cell types and tissues. LDs play an important role in carcinogenesis, malignant development of cancers, and immune cells.
In addition, side products of respiratory chain such as ROS attack unsaturated lipids thus lead to lipid peroxidation (LPO), which will initiate secondary cellular responses. LPO is one of the markers for oxidative stress [40]. LPO damage DNA, proteins and enzyme activity, it activates signaling pathways initiating cell death [41]. Polyunsaturated fatty acids (PUFA) in the glycolipids, phospholipids and cholesterols are often the targets for PLO, among them, PUFA is one of the major targets of oxidative stress via LPO in the cellular system [42]. The cardiolipin oxidation is catalyzed by cytochrome c in mitochondria, in this process, cytochrome c interacts with cardiolipin and gains peroxidase function instead of participation in electron transfer. The complex lead to cardiolipin oxidation, which is pivotal for the release of pro-apoptotic factors including cytochromes [43]. Different mitochondria-targeted inhibitors of the cardiolipin oxidation pathway have been developed based on the inhibition of hydrogen peroxide or peroxidase activity of cytochrome c in this complex [44].
Mitochondrial function in nucleic acid metabolism
Purine and pyrimidine nucleotides are required for the synthesis of RNA and DNA. The purine and pyrimidine bases are from various nonessential amino acids and methyl groups donated from the one-carbon or folate pool. DNA replication and RNA production are essential processes in cell division and proliferation, their biosynthesis is upregulated in the late G1 phase [45, 46]. Nucleic acid biosynthesis is an energy-intensive process which is involved multiple metabolic pathways [47]. In order to maintain sufficient energy and precursors for RNA biosynthesis, a high ATP/ADP ratio must be kept, the ATP concentration is usually stable as above 1 mM [48, 49], if the ATP level is lower than a required level, corresponding cell functions may cease or even result in necrotic cell death [50, 51]. In some special stages or some high-energy requested cells such as skeletal muscle cells and cardiac myocytes, a higher ATP level (> 5mM) should be maintained to supply the materials for nucleic acid synthesis [51, 52]. Cells have to adjust energy metabolism and the nucleotide biosynthetic pathways to produce enough nucleic acids for cell proliferation. Most cells synthesize nucleic acids de novo and mainly from glucose, glutamine and CO2 [53]. In the TCA cycle, oxaloacetate generated from malic acid is transaminated to aspartate under the catalysis of transaminase and aspartate is the intermediate for synthesizing both purine and pyrimidine bases. Nucleotide biosynthesis can be a target for cancers too [54].
Small metabolites or molecules are transported to the matrix of mitochondria by the mitochondrial carrier family (MCF) [55]. Nucleotides are one of the largest solutes to cross the inner membrane, thus the transporting process requests conformational changes of the MCF proteins [56]. The ADP/ATP carrier proteins located in the inner membrane of mitochondria are the most studied MCFs [57], they transport ADP into the mitochondrial matrix for ATP synthesis and ATP out to reach high cytosolic ATP concentrations for energy-requiring reactions [56, 58]. Carboxyatractyloside (CATR), a membrane-impermeable toxic inhibitor, blocks the nucleotide translocation from the inner membrane space to the matrix, the CATR and ADP binding sites from the inner membrane space partially overlap. The detailed transport mechanisms for adenine nucleotides need to be further explored [59].
The interplay between mitochondria and glycolysis
Cancer cells exhibit heterogeneity in energy resource, some rely more on OXPHOS and some others rely on more on glycolysis. The energy resource of cancer cells may shift their reliance from one to the other. For example, loss of LUC7L2 and U1 snRNP subunits shifts the energy metabolism from glycolysis to OXPHOS [60]. The recent study also demonstrated that cancer cells relying on more OPXHOS are highly expressing DNMT1 and lowly expressing NNMT, by genetically manipulating the expression of NNMT and DNMT1, the sensitivity to OXPHOS inhibitors are changed [61]. Overexpression of NNMT and knockdown of DNMT1 change OXPHOS-inhibition sensitive cell line to be resistant ones, indicating their energy resource reliance shifted from OXPHOS to glycolysis [61]. In the process of energy metabolic switch, the M2 isoform of pyruvate kinase (PKM2, a rate-limiting enzyme in glycolysis) interacts with the key regulator of mitochondrial fusion protein MFN2 to promote the mitochondria fusion and OXPHOS, meanwhile attenuate glycolysis [62], consistently, the activation of MFN2 interacting with PFK1 mediate PFK1 degradation and therefore suppresses glycolysis [63]. In Hepatocellular carcinoma (HCC), inhibition of lysophosphatidic acid receptor 6 (LPAR6) overcomes sorafenib resistance by switching glycolysis into OXPHOS [64]. Moreover, the cancer suppressor gene p53 suppresses glycolysis and promotes mitochondria oxidative phosphorylation by a series of downstream targets against the Warburg effect [65]. In addition, the modification of RNA also contributes to the energy switch in cancer metastasis. In human oral cancer, the RNA modifications-5-methylcytosine (m5C)-deficient cancer cells exhibit increased glycolysis and decreased mitochondria function without affecting cell viability or primary tumor growth [66].
Function of mitochondrial metabolism in cancer
Mitochondrial metabolism plays an essential role in a variety of physiological and pathological processes (Figure 3). It is crucial for tumor proliferation, survival, metastasis and drug resistance [67, 68].
Mitochondria function in cancer metabolism. 1) Mutation of enzymes in TCA cycles such as mutation of IDH, SDH leads to high concentrations of D2HG, fumarate and succinate accumulation promoting tumor cell proliferation and progression. These enzymes also serve as the driver of human cancer. The metabolic dysregulation is not only a consequence of oncogenic transformation but also that it drives cancer. 2) Acetyl-CoA which is the most important substrates for acetylation modification, can be derived from glucose (pyruvate oxidation), fatty acid, and amino acid catabolism, it is the key indicator of cell metabolism and links metabolism, signaling, and epigenetics. 3) Succinate exists intracellular (cytosol and mitochondria) and extracellular, its efflux from mitochondria to the cytosol relies on SLC25 and VDAC.
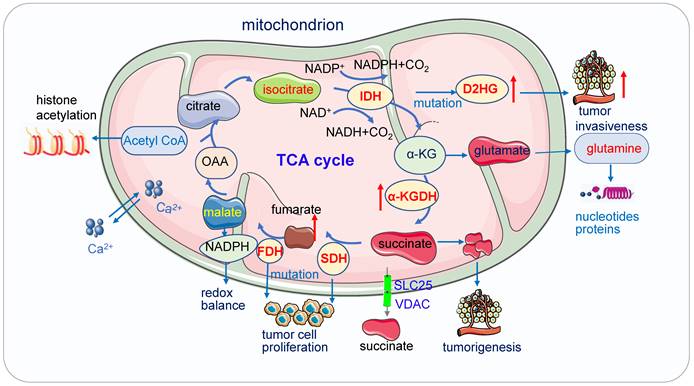
Acetyl-coenzyme A
Acetyl-coenzyme A (Acetyl-CoA), the most important substrates for acetylation modification, can be derived from glucose (pyruvate oxidation), fatty acid, and amino acid catabolism, it is the key indicator of cell metabolism and links metabolism, signaling, and epigenetics [69-71]. Fluctuations of Acetyl-CoA have been demonstrated to be closely associated with changes in overall histone acetylation and gene expression [70, 72]. Under the physiological stage, dynamic histone modifications have important roles in Maternal-to-zygotic transition (MZT). In mouse embryos, H3K4me3 and H3K27ac module MZT process [73]. In pigs, Acetyl-CoA synthases are essential for maintaining ATP and histone acetylation in porcine early embryos under metabolic stress during Zygotic genome activation (ZGA) [74]. During early embryogenesis, H4K16ac is maintained from oocytes to fertilized embryos in Drosophila and mammals, providing an instructive function to the offspring by priming gene activation [75]. The level of Acetyl-CoA and histone acetylation also controls the early differentiation of embryonic stem cells, inhibition of acetyl-CoA or its upstream causes differentiation of pluripotent cells, while inhibition of its downstream delays differentiation [76, 77]. Cooperation between lncRNA DIGIT and BRD3 at the regions of H3K18ac regulates the transcription factors which drive endoderm differentiation [78]. In embryonic neurol stem cell (NSC), histone acetylation plays important roles in NSC differentiation, TP53 inducible glycolysis and apoptosis regulator (TIGAR) promotes NSC differentiation through upregulating Acetyl-CoA [79]. Under the pathological state, acetyl-CoA play important roles in tumor migration. For example, in glioblastoma, it promotes the expression of cell migration and adhesion-related genes by controlling Ca2+ and nuclear factor of activated T called signaling [72, 80]. In pancreatic carcinogenesis, histone acetylation facilitates cell plasticity and proliferation [81]. And in the non-small-cell lung cancer (NSCLC), acetyl-CoA carboxylase (ACCS) catalyzes the ATP-dependent carboxylation of acetyl-CoA to form malonyl-CoA, it is required for de novo fatty acid synthesis needed for tumor growth and viability. Inhibition of ACCS suppresses fatty acid synthesis and thus suppress tumor growth [82], ACCS is a potential cancer target for cancer therapy. Acetyl-CoA synthetase 2 (ACSS2) contributes to cancer cell growth under low-oxygen and lipid-depleted conditions by promoting acetate utilization [83]. In hepatocellular carcinoma (HCC) metastasis, acyl-CoA thioesterase 12 (ACOT12) links the alteration of acetyl-CoA with HCC metastasis by suppressing HCC metastasis both in vitro and in vivo [84].
Citrate
Citrate in mitochondria is oxidized in the TCA cycle to produce ATP, it is important for lipid synthesis and epigenetic regulation; cancer cells import extracellular citrate to support their growth [85]. Citrate can also be transported to the cytosol and cleaved by citrate lyase to regenerate acetyl-CoA and oxaloacetate [86, 87]. Manipulating citrate level affects both cancer and immune cells thus modulating cancer cell apoptosis and immune responses [85]. Citrate level keeps low in highly proliferative cancer cells, it is an activator of gluconeogenesis and an inhibitor of glycolysis [88, 89]. For cancer cells relying more on glycolysis, this low level of citrate help to sustain the Warburg effect, but for others relying more on OXPHOS, more citrate is produced from FAO but it is converted to acetyl-CoA and oxaloacetate quickly [87]. Elevation of intracellular citrate level arrests glycolysis, proliferation, dedifferentiation, and aggressiveness of cancer cells [90]. In prostate cancer, citrate activates the autophagic death of cancer cells by downregulating CaMKII/AKT/mTOR pathway [91]. In addition, citrate inhibits the proliferation of multiple cultured cancer cells such as ovarian, mesothelioma, pancreas, lung, stomach, and melanoma, indicating that citrate might be a promising anti-tumor target.
Isocitrate
Isocitrate is the product of aconitase and the substrate of isocitrate dehydrogenase, it is an intermediate metabolite in the citric acid cycle found both inside the mitochondria and outside in the cytosolic shunt. Treatment of isocitrate may enhance breathing and render it more resistant to hypoxic insult and increase the amplitude of ventilation in vivo in normoxia, indicating isocitrate is helpful in avoiding the failure of gasping generation and autoresuscitation in pathological conditions. The application of isocitrate showed a significant and persistent attenuation for anemia of inflammation in a mouse acute and severe anemia of inflammation (AI) and in a rat arthritis model of moderate chronic AI [92, 93].
Isocitrate is oxidized by the enzyme IDH, where carbon dioxide is liberated and 2-oxoglutarate (2OG, also known as α-ketoglutarate) is the oxidized product, it converts isocitrate to α-KG. IDH can be inside mitochondria in a nicotinamide adenine dinucleotide (NAD)+-dependent manner and nicotinamide adenine dinucleotide phosphate (NADP)+-dependent manner, it can also be formed in the cytosol in an NADP+-dependent manner [94]. IDH1 and IDH2 impair the production of 2OG and reduce NADPH from isocitrate, meanwhile they oxidize NADP+ and promote 2OG to D-2-hydroxyglutarate (D-2HG) [95]. The elevated d-2HG is a biomarker for many cancers [96]. IDH1 is the most frequently mutated gene in cancer. In low-grade glioblastoma (LGG), more than 80% of IDH mutations occur in the IDH1 gene, being dominated by R132H IDH1 [97].
Moreover, IDH mutations are associated with the altered IL-1β responses in acute myeloid leukemia (AML) [98]. Mutations in IDH1 and IDH2 induce epigenetic and transcriptional reprogramming, and differentiation bias and promote the development of a number of malignancies [99, 100] including but not limited to chondrosarcoma, AML [101, 102], cholangiocarcinoma [103], prostate cancers [104], angioimmunoblastic T-cell lymphomas [105], myeloid neoplasia [106]. Thus, inhibition of IDH1 and IDH2 variants is being pursued as a medicinal chemistry target [107].
α-ketoglutarate
α-KG is a membrane-impermeable and key endogenous metabolite in the TCA cycle, its level changes upon fasting, exercise and aging [108]. Level of α-KG affects cell metabolism, collagen synthesis, epigenetic regulation, stem cell proliferation and immune response [109]. Intracellular α-KG inhibits starvation-induced autophagy and it has no direct respiration-inhibitory effect [110]. α-ketoglutarate can extend lifespan in mice by suppressing chronic inflammation via the induction of IL-10 by dietary [108].
α-KG is an important cofactor for histone demethylase including Jumonji C (JmjC) and TET, it plays important roles in methylation modification [111, 112]. In age-related osteoporosis, α-KG decreases H3K9me3 and H3K27me3 and upregulates BMP signaling and Nanog expression, resulting in rejuvenating mesenchymal stromal/stem cells (MSCs) and ameliorating age-related osteoporosis [113]. α-KG directly regulates signal pathway, in colorectal cancer (CRC), α-KG attenuates the Wnt signaling pathway by promoting hypomethylation of DNA and histone H3K4me3 and drives differentiation of CRC cells, identifying α-KG as a potent antineoplastic metabolite for potential differentiation therapy for CRC patients [114]. In p53-deficient pancreatic ductal adenocarcinoma (PDAC), the addition of cell-permeable α-KG increased cell differentiation and decreased tumor-cell fitness, indicating the role of α-KG in linking p53 to cell fate during tumor suppression [115]. α-KG also plays important roles in the immune system, α-KG suppresses M1 macrophage activation but promotes M2 macrophage activation to exhibit anti-inflammatory effects by mediating metabolic and epigenetic reprogramming [116].
α-KG is catalyzed by α-ketoglutarate dehydrogenase (α-KGDH) and metabolized into succinyl-CoA, this GDH-mediated α-KG can inhibit IKKβ activation and block nuclear factor κB (NF-κB) activation, promote glucose update and cell survival by upregulating GLUT1, thereby accelerating gliomagenesis [117]. α-KGDH inhibition increases α-KG levels and leads to DNA demethylation and finally impairs cell migration in breast cancer-associated lung metastasis [118].
Succinate
Succinate is a key modulator of cell metabolism in hypoxic response, an important player in tumorigenesis, protein succinylation and inflammatory signal [119]. Succinate exists intracellular (cytosol and mitochondria) and extracellular, its efflux from mitochondria to the cytosol relies on solute carrier family 25 (SLC25) and the voltage-dependent anion channel (VDAC) [120]. Succinate can be generated via three pathways: the reductive branch of the TCA cycle, the glyoxylate pathway, and the oxidative TCA cycle.
Succinate also acts as an inflammation signal [119, 121], a key player in macrophage activation [122, 123]. It inhibits transcription factor hypoxia-inducible factor-1α (HIF-1α) prolyl hydroxylases in the cytosol, leading to stabilization and activation of HIF-1α via succinate receptor in specific tumors and in activated macrophages and stimulates dendritic cells [121, 124]. Succinate is an extracellular metabolic stress signal sensed by the mainly Gi-coupled succinate receptor SUCNR1, regulating the transcription of human M2 macrophages [125]. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8+ T cells [126].
Succinate and other metabolites with similar structures, such as D-2HG and fumarate, are considered oncometabolites [127]. Accumulation of succinate promotes immune function and tumorigenesis [128], circulating succinate has emerged as a promising biomarker in chronic metabolic diseases. Elevated succinate levels within the gut lumen association with microbiome disturbances (dysbiosis) and in patients with inflammatory bowel disease (IBD) and animal models of intestinal inflammation [120]. Physiologically, succinate activates the receptor GPR91 identified in the bladder and it is essential to bladder structure and contraction [129].
SDH, also known as mitochondrial Complex II, is a mitochondrial enzyme participating in both the citric acid cycle and the electron transport chain. It oxidizes succinate to fumarate in the TCA cycle and oxidizes ubiquinone to ubiquinol in the mitochondrial ETC [130]. Localized in the inner membrane of mitochondria, Complex II holoenzyme consists of four subunits, SDHA, SDHB, SDHC, and SDHD, and two assembly factors, SDHF1 and SDHF2 [131].
SDH has now been defined as a tumor suppressor and succinate an oncometabolite [132]. SDH deficiency leads to the accumulation of succinate [133, 134], which inhibits α-KG dependent dioxygenase family enzymes. These enzymes include around 60 members and regulate key aspects of tumorigenesis such as DNA and histone demethylation, hypoxia responses, and m6A mRNA modification [127]. SDH deficiency changes cellular metabolism, such as the demand for extracellular pyruvate and the biosynthesis of aspartate [135, 136]. SDH-deficient cells also increase activities in glycolysis, lactate production, and pentose phosphate pathways [137]. Dysfunction of the SDH impairs mitochondrial activity, ATP generation and energy hemostasis. Increased mitochondrial oxidation of succinate by SDH and an elevation of mitochondrial membrane potential (MMP) induces mitochondrial ROS production [122]. Functional SDH deficiency is a common adverse feature of clear cell renal cell carcinoma (ccRCC) [138]. SDH gene germline mutations lead to SDH-deficient renal cell carcinoma [139]. Moreover, SDH acts as a key regulator in neurodegenerative disorders [140], neuroendocrine tumors [141], Chronic obstructive pulmonary disease [142]. In addition, SDH is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation, SDH deficiency induced a proinflammatory gene signature in T cells and promoted T helper 1 and T helper 17 lineage differentiation [143].
Fumarate
Fumarate is generated in the TCA cycle by the SDH-catalyzed dehydrogenation of succinate, acting as a bona fide oncogenic molecule and a key activator of a variety of oncogenic cascades [144]. Fumarate will be accumulated when FH is deficient, which leads to high concentration fumarate in multiple subcellular compartments and the extracellular microenvironment, affecting the balance of multiple enzymatic reactions and induction of tumorigenesis. The loss of FH and the accumulation of fumarate elicit the pro-oncogenic signals which contribute to the transformation of normal cells into tumor cells.
FH is distributed in both cytosol and mitochondria; the mitochondrial FH is part of the TCA cycle and it catalyzes the reversible hydration of fumarate to malate. The mutation of FH has been identified in a variety of cancers. FH-deficient cells respond to mitochondrial impairment by a series of compensatory metabolic changes: they increase their glycolytic pathway with the increase of glycolytic-related gene transcription and the inhibition of pyruvate dehydrogenase (PDH); the glutamine is converted to α-KG and eventually to citrate via reductive carboxylation. FH-deficient cells harbor lower fumarate and preserve Aconitase 2 (ACO2) function to promote tumor progression.
The FH deficiency also affects epigenetics. The loss of FH causes hypermethylation in the promoter of the tumor suppressor cyclin-dependent kinase inhibitor 2A (CDKN2A), the hypermethylation of CDKN2A is a predictive factor for unfavorable prognosis of various cancers [145]. It also causes the hypermethylation and suppression of MIR200 and leads to an epithelial-to-mesenchymal transition (EMT), which promotes tumor metastasis [146]. The accumulation of fumarate inhibits JmjC-KDMs and increases the methylation of H3K4, H3K27 and H3K79, which promote gene transcription [147].
Malate
Malate is synthesized from fumarate by fumarase and further oxidized to oxaloacetate by malate dehydrogenase (MDH) with the accompanying reduction of NAD+. The metabolic imbalance results in the impairment of either mitochondrial and/or cytoplasmic metabolism. The oxidation of cytosolic NADH by the mitochondria requires malate-aspartate shuttle [148], by which cells maintain a balance of metabolic intermediates between the cytosol and the mitochondria to enable lactate metabolism to continue. This shuttle controls NAD+/NADH homeostasis to maintain the activity of mitochondrial MDH and promote L-lactate oxidation in mitochondria [149]. Inhibition of the malate-aspartate shuttle leads to the decrease of ATP production in glioma cells [150]. The major function of the shuttle in cancer cells is to maintain glycolysis, instead of increasing mitochondrial energy metabolism.
MDH reversibly converts malate to oxaloacetate using NADH and serves as important oxidoreductase in several metabolic pathways. MDH has two isoenzymes, MDH1 in cytosol and MDH2 in mitochondria. Both are closely associated with cancers. The mutation of MDH2 destroys the TCA cycle and affects energy metabolism, leading to mitochondria dysfunctional diseases such as epilepsy, hypotension etc. The upregulation of MDH2 increase energy metabolism and the deficiency of MDH2 reduces the production of ATP and the augment of ROS. MDH2 is highly expressed in many types of cancers, MDH2 concentration was higher in early-stage NSCLC patients compared with that in controls, therefore, MDH2 can be a potential biomarker for early detection of NSCLC [151].
Oxaloacetate
Oxaloacetate can be generated from malate, pyruvate, phosphoenolpyruvate and aspartate. It plays important roles in cancers and other diseases by modulating cell metabolism. OAA is a competitive inhibitor of LDHA, high pyruvate kinase M2 (PKM2) activity reduces LDHA activity via upregulating cytosolic OAA in cancer cells, the elevated PKM2 increase the de novo synthesis of OA and inhibit LDHA in cancer cells [152]. In KRAS-mutated cancer cells, inhibition of glutamate oxaloacetate transaminase 1 (GOT1) increases the sensitivity to glucose deprivation, GOT1 is critical to provide oxaloacetate at low glucose to maintain the redox homeostasis [153]. Furthermore, treatment of oxaloacetate in human hepatocellular liver carcinoma cell inhibit the glycolysis and enhance the OXPHOS, leads to cancer cell apoptosis [154]. In addition, oxaloacetate has been demonstrated playing a neuroprotective role for acute ischemic stroke since it reduces the brain and blood glutamate levels when the blood-resident enzyme glutamate-oxaloacetate transaminase is activated [155]. It also protects liver from warm ischemia injury by improving cellular energy metabolism [156].
Mitochondrial function in cancer stem cells
Cancer stem cells (CSC) or tumor-initiating cells, characterized by the ability to self-renew, are a small subset of cancer cells responsible for tumor initiation, maintenance, growth, metastasis, recurrence and drug resistance. Mitochondria play crucial roles in cancer stem cells in different aspects such as cell metabolism, drug resistance, apoptosis etc. Metabolic reprogramming through the regulation of mitochondrial activities is a key feature of CSCs [157]. Inhibition of mitochondrial translation and mitochondrial fission, targeting the OXPHOS pathway or the related key genes are potent therapeutic strategies in cancer stem cells [158]. Therefore, mitochondria have been seen as a new therapeutic target for eradicating cancer stem cells [159].
CSCs rely on more on OXPHOS
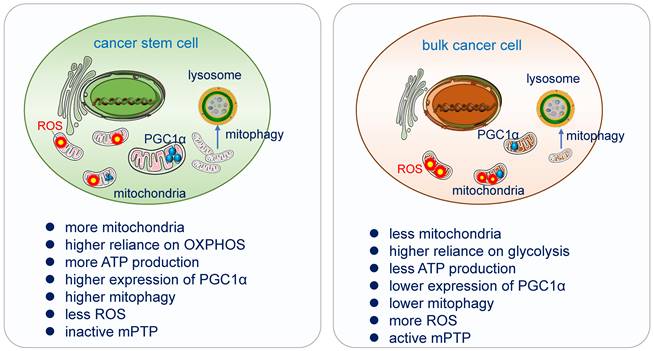
Increasing evidence had demonstrated that a myriad of CSCs relies highly on OXPHOS for obtaining energy [160]. They are less glycolytic, consuming less glucose and producing less lactate while maintaining higher ATP levels than their differentiated version and surrounding bulk tumor cells [161]. CSCs possess high mitochondrial membrane potential and elevated mitochondrial gene expression, over-expressed PGC-1α [162]. In pancreatic ductal adenocarcinoma (PDAC) cancer stem cells, the utilization of OXPHOS increased tumorigenic potential and pluripotency gene expression, enhanced invasiveness and upregulated immune evasion properties [163].
Inhibition of OXPHOS in CSCs induces cell death, in chronic myeloid leukemia (CML), the combination treatment with imatinib (a tyrosine kinase inhibitor) and tigecycline (an antibiotic) that inhibits mitochondrial protein translation, selectively eradicates CSCs both in vitro and in a xenotransplantation model of human CML [164]. In AML, it also promotes cell differentiation [164-166]. Various OXPHOS inhibitors have been demonstrated as cancer stem cell killers. For instance, Targeting Co-enzyme Q10 of mitochondrial complex III by atovaquone [167], targeting complex I by Rotenone [159], and targeting complex V by Gboxin [13] eradicate CSC effectively.
CSCs possess higher PGC-1α
PGC1-α, the transcription co-activator peroxisome proliferator-activated receptor gamma co-activator 1α, acts as a master regulator of mitochondria de novo synthesis, and regulates different energy-producing metabolic processes including OXPHOS. It has been demonstrated to be overexpressed in circulating tumor cells. High expression of PGC-1α enhances mitochondrial biogenesis, strong antioxidant activity and fewer ROS, increase OXPHOS and promote cancer cells' invasion and metastasis [168, 169]. Suppression of MYC and subsequent increase of PGC-1α were identified as key determinants for the OXPHOS dependency of CSCs, which was abolished in resistant CSC clones [170].
CSCs exhibit lower ROS
ROS as the side product of the respiratory chain has been applied in radiation therapy, which causes DNA damage directly by ionization or indirectly through the generation of ROS to destroy cancer cells [171]. CSCs possess a more powerful antioxidant defense system which can counteract and scavenge ROS to keep their low ROS level and maintain their stemness and tumorigenesis [172]. In glioblastoma stem cells, the generation of ROS in mitochondria can be suppressed by the combination of Prohibitin and PRDX3, which lead to the degradation by the ubiquitin-proteinase system [173].
Differences of mitochondrial functions in cancer stem cell and bulk cancer cell. Cancer stem cells acquire their energy more from OXPHOS. High expression of PGC-1α enhances mitochondrial biogenesis, strong antioxidant activity and fewer ROS, increases OXPHOS and promotes cancer cells' invasion and metastasis. CSCs possess a more powerful antioxidant defense system which can counteract and scavenge ROS to keep their low ROS level and maintain their stemness and tumorigenesis. Mitophagy is essential in the differentiation and in the acquisition of “stemness” in cancer stem cells, it promotes CSC plasticity for better adaption in TME. CSCs have inactive mPTP and bulk tumor cells have active mPTP [13].
Mitochondria fission and fusion affect CSCs' cell fate
Mitochondria are dynamic organelles frequently undergoing fission and fusion to maintain their morphology and regulate their function. Mitochondrial morphology and function contribute to a stem-like phenotype of cancer cells [174], implying that mitochondrial fission and fusion are critical players in CSC behavior. Mitochondrial fission may play a role in the removal of damaged mitochondria. High levels of mitochondrial fission activity are associated with high proliferation and invasiveness in some cancer cells and with self-renewal and resistance to differentiation in some stem cells [175]. Mechanistically, the FOXO and Notch signaling converge on the regulation of mitochondrial fission and in turn, the mitochondrial fission provokes the CSC differentiation into the bulk cells, thus the mitochondria morphology affecting cell fate of CSCs [176].
The exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate the AML resistance to OXPHOS inhibition [177]. The processes controlling mitochondria dynamics are mediated by specific proteins such as mitochondrial out membrane fusion protein MitoPLD, inner membrane fusion protein OPA1, mitochondrial fission protein Drp1 and its receptors FIS1, MID49, MID51, etc. [6, 178]. The mitochondria fission factor FIS1 augments mitophagy in lung CSCs to maintain their stemness [179]. Mitochondria fission has been treated as a driver of stemness in tumor cells. Drp1, the essential mediator of mitochondrial fission, has been found to be hyperactivated in brain tumor-initiating cells and its phosphorylation regulates mitochondrial morphology and stem cell marker expression [180]. mDIVI1, an inhibitor of the mitochondrial fission protein DRP1, inhibits 3D tumor sphere-forming capacity, cell migration and stemness-related signaling in breast cancer cells via inducing the loss of oxidative phosphorylation [181].
By asymmetric division, stem cells generate two daughter cells in different traits. Daughter cells that received fewer old mitochondria maintain stem cell traits. The failure to asymmetrically apportion old mitochondria in a single division can cause a persistent loss of stemness in stem cells and cancer stem cells [182].
Mitophagy in CSCs
Mitophagy is an evolutionarily conserved cellular process, it is critical in maintaining mitochondrial integrity and cell homeostasis by removing damaged or surplus mitochondrial fragments to the lysosome for degradation [183]. Mitophagy is essential in the differentiation and in the acquisition of “stemness” in cancer stem cells [184, 185]. It is balanced with mitochondrial biogenesis to maintain mitochondrial homeostasis in CSCs and promote CSCs plasticity for better adaption in tumor microenvironment (TME) [186, 187]. Mitophagy also contributes the energetic and metabolic shift from glycolytic phenotype into an OXPHOS state of CSCs via their dynamic adjustment [183, 184]. PINK1-dependent mitophagy driving mitochondrial rejuvenation affects pluripotency and differentiation state in stem cells [188]. In Pancreatic cancer stem cells (PaCSC), ISG15 and ISGylation are required for mitophagy and metabolic plasticity, loss of ISG15 lead to increased accumulation of dysfunctional mitochondria, reduced OXPHOS and impaired mitophagy and finally negatively impacts PaCSC stemness [189]. In HCC, mitophagy induction increases the hepatic CSC population while its inhibition decreases it [190], mitophagy promotes transcriptional activation of NANOG to maintain the self-renewal and cell stemness [190, 191]. Mitophagy is also required for self-renewal of human acute myeloid leukemia stem cells [192]. In glioblastoma CSCs, the platelet-derived growth factor (PDGF) signaling inhibits the activation of mitophagy and promotes cancer stem cell maintenance by inducing N6-methyladenosine (m6A) accumulation [193]. Furthermore, CSCs may also reply on mitophagy to keep low ROS levels and prevent the activation of programmed cell death [194]. However, an excessive rate of mitophagy flux can also confer chemoresistance [195]. Therefore, the role of mitophagy in CSCs may be more important than previously recognized.
Mitochondria in the tumor microenvironment (TME)
TME comprises various cell types including tumor cells, immune cells and the surrounding environment of tumors. TME plays pivotal roles in tumor imitation, progression and metastasis [196]. The heterogeneous TME requests different oxygen supply which leads to the heterogeneity of mitochondrial distribution [68]. T cell infiltrating tumors exhibit decreases in mitochondrial function and mass, this TME represses T cell mitochondrial biogenesis and T cell oxidative metabolism [197]. The hypoxia microenvironment of tumors requests mitochondria to adjust ETC activity, ROS production and reduce the TCA cycle to survive and proliferate in oxygen insufficiency.
ROS in cancer cell metabolism
Mitochondria generate ROS from complex I and complex III at the inner mitochondrial membrane during oxidative phosphorylation, this can be identified by inhibitors of the mitochondrial respiratory chain such as Rotenone and Antimycin A as well as the substrates for various complexes such as pyruvate or succinate [198]. Excessive ROS quantity induces mitochondria dysfunction by inducing a significant decrease in mtDNA-encoded gene transcripts for respiratory complexes I, III and IV. mtDNA is sensitive to oxidative damage due to its low repair capacity and close relation with the electron transfer chain, it has been one of the main targets of ROS [199]. mitoTEMPO, a mitochondria-targeted superoxide dismutase mimetic, decreases mitochondrial ROS production and reduces hypertension [200]. Mitoquinone, another mitochondrial-targeted antioxidant, can improve mitochondrial function and attenuate redox-related cardiomyopathies, but in cancer cells, it could also lead to ROS production, membrane depolarization, and apoptosis [201].
Mitochondrial ROS also oxidizes proteins in the ETC and leads to a decrease in mitochondrial respiration and finally causes related signaling pathway activation [202, 203]. ROS concentration inside the cell is modulated and tightly controlled to remain in a concentration range compatible with cell proliferation and to avoid ROS overproduction, provoking damage to mitochondria and extra-mitochondrial macromolecules and eliciting cell death [10, 204]. Cancer cells are able to reduce ROS quantity to survive and invade surroundings. Antioxidant proteins such as peroxiredoxins were specifically upregulated in lung metastatic breast cancer cells [205], PRDX2 had been demonstrated to protect metastatic cancer cells from oxidative stress [206].
The interaction of ROS and autophagy play important roles in both cell damage and cell survival. ROS may regulate autophagy at transcriptional and post-transcriptional levels via various signal pathways such as OS-FOXO3-LC3/BNIP3-autophagy, ROS-NRF2-P62-autophagy, ROS-HIF1-BNIP3/NIX-autophagy and ROS-TIGAR-autophagy [207]. By contrast, in tumor cells, autophagy can reduce the excessive production of mitochondrial or cytosolic ROS [208]. Oxidative damage leads to the production of ROS, but meanwhile also initiates autophagy to clear the increased ROS.
ROS can modify cellular functions via interacting with HIF-1α and NFκB [209]. ROS are directly involved in HIF-1a stabilization under hypoxia. ROS signaling via HIF-1a is a key process that is critical in cellular proliferation and angiogenesis [210].
Mitochondria function in hypoxia
Hypoxia has been regarded as a hallmark of the tumor microenvironment, it happened due to increased oxygen consumption and inadequate oxygen supply in the majority of tumors [211]. Hypoxia is associated with poor prognosis and has been demonstrated as a target for cancer therapy [212, 213]. It induces changes in gene expression of tumor cells for better adaption to a low oxygen environment. High-proliferation tumor cells rely on glycolysis to obtain ATP for survival. But slowly dividing cells such as cancer stem cells in hypoxic regions are more relying on OXPHOS to obtain ATP and they usually escape from the chemo- and radiotherapy. Hypoxia induces reduced energy production through decreased mitochondrial metabolic activity or altered hypoxia-inducible factor-1 (HIF-1) and peroxisome proliferator-activated receptor gamma coactivator 1(PGC1) dependent mitochondrial biogenesis. Mitochondria are the main consumer of oxygen, in response to hypoxia, mitochondria will moderate their respiratory chain to adjust metabolism, especially in decreasing the activity of TCA cycle [214]. By contrast, hypoxia alters mitochondrial fusion and fission, mitophagy and OXPHOS. Super-complexes were formed among complexes I, III, and IV of the ETC to produce minimum superoxide and improve the efficiency of oxygen consumption in hypoxia.
Hypoxia activates mitophagy [215]. Chronic hypoxic conditions can deplete the mitochondrial mass by autophagy, resulting in selective clearance of damaged mitochondria in cells [216]. The outer mitochondrial membrane protein FUNDC1, a receptor for hypoxia-induced mitophagy, recruits DNM1L/DRP1 to drive mitochondrial fission in response to hypoxic stress to enhance mitophagy [217]. The mitochondrial phosphatase PGAM5 dephosphorylates FUNDC1 to activate mitophagy [218]. FUNDC1 interacted with LC3 via LC3-binding motif Y(18)xxL(21), and mutation of the LC3-interaction region impaired its interaction with LC3 and the subsequent induction of mitophagy[219]. Knocking down of FUNDC1 prevents autophagy in hypoxia [220]. ULK1, one of the core human autophagy-related genes, plays a specific role in mitophagy. ULK1 interacts with FUNDC1, phosphorylating it at serine 17, which enhances FUNDC1 binding to LC3 [221]. ULK1 deficiency induces an invasive phenotype of breast cancer cells under hypoxia and increases osteolytic bone metastasis. ULK1 depletion attenuates mitophagy ability during hypoxia. phosphorylation of ULK1 by MAPK1/ERK2-MAPK3/ERK1 kinase triggers its interaction with BTRC and subsequent K48-linked ubiquitination and proteasome degradation [222]. In addition, Hypoxia can also activate the PINK1/Parkin-mediated mitophagy pathway, induce the excessive proliferation of PASMCs and lead to Pulmonary Artery Hypertension (PAH) [223].
HIF is a master regulator for adaptation to low oxygen content and HIF proteins are key determinants of cellular response to hypoxia [224]. They increase the expression of glycolysis and decrease oxygen-dependent ETC complex activity in hypoxia [214]. HIF mediates adaptive responses to oxidative stress by nuclear translocation and regulation of gene expression. A small fraction of HIF-1α will translocate to the mitochondria after exposure to hypoxia or H2O2 treatment. The mitochondrial HIF-1α protects against oxidative stress induced apoptosis and reduces ROS production to maintain mitochondrial membrane potential and reduce the expression of mitochondrial DNA-encoded mRNA in response to hypoxia [225].
Hypoxia also impacts the mitochondrial morphology including cristae structure. More mitochondria are in the fission process to promote mitophagy and keep ROS production in a comparatively low level and maintain decreased respiratory activity [226]. The impaired mitochondrial function caused by hypoxia also exacerbates the inflammatory response via metabolic perturbation. Hypoxia induces immune cell dysfunction and leads to an altered metabolic profile [227]. Mitochondrial respiration defects in cancer cells cause an increase of NADH, and activation of Akt, thus cancer cells obtain drug resistance and survival advantage in hypoxia. In tumor cells, hypoxia-induced autophagy could reduce oxidative damage and promote cell survival.
Targeting mitochondria for cancer therapy
Mitochondria dysfunction and oxidative stresses have been found in many diseases including cancers, neurodegenerative disorders, pathogen infections, etc. The mtDNA decrease, OXPHOS dysfunction, and the upregulation of ROS are found in various tumors. The OXPHOS downregulation is associated with poor clinical outcome across most of the cancer types and correlates with a gene signature characteristic of invasive and metastatic tumors. Targeting mitochondria has been reviewed a lot and various inhibitors have been developed (Table 1). Some of the inhibitors described above have moved on to clinical trials due to their relatively stable therapeutic effects.
Inhibitors of mitochondria
| Inhibitors | Target | References |
|---|---|---|
| Metformin | Complex I | [228] |
| IACS-010759 | Complex I | [166] |
| Phenformin | Complex I | [242] |
| ME344 | Complex I | [243] |
| Fenofibrate | Complex I | [244] |
| mIBG | Complex I | [245] |
| Pyrvinium | Complex I | [246] |
| Canagliflozin | Complex I | [247] |
| Pioglitazone | Complex I | [248] |
| Rosiglitazone | Complex I | [249] |
| Amobarbital | Complex I | [250] |
| Nefazodone | Complex I | [251] |
| Piericidin A | Complex I | [252] |
| mdivi-1 | Complex I | [253, 254] |
| ginsenoside Rb1 | Complex I | [255] |
| ASP4132 | Complex I | [256] |
| Authipyrin | Complex I | [257] |
| αTOS | Complex II | [258] |
| Lonidamine | Complex II | [259] |
| Malonate | Complex II | [260] |
| benzenesulfonamide | Complex II | [261] |
| Atpenin A5 | Complex II | [262] |
| 2 - alkyl - 4, 6 - dinitrophenols | Complex II | [263] |
| Napyradiomycin A1 | Complex II | [264] |
| Linalool | Complex II | [265] |
| picolinamide | Complex III | [266] |
| Antimycin A | Complex III | [267] |
| Azoxystrobin | Complex III | [268] |
| Ametoctradin | Complex III | [269] |
| Atovaquone | Complex III | [233] |
| Myxothiazol | Complex III | [270] |
| Stigmatellin | Complex III | [271] |
| mIBG | Complex III | [245] |
| Acremonium exuviarum | Complex III | [272] |
| Itaconic acid | Complex IV | [273] |
| Arsenic trioxide | Complex IV | [274] |
| Hydrocortisone | Complex IV | [275] |
| 6 - hydroxydopamine | Complex IV | [276] |
| Cyanide | Complex IV | [277] |
| Azide | Complex IV | [278] |
| Chrysin | Complex V | [279] |
| Oligomycin | Complex V | [280] |
| Paroxetine | Complex V | [281] |
| Chlorpromazine | Complex V | [282] |
| Gboxin | Complex V | [13] |
| Baicalein | mPTP | [5] |
Metformin
Metformin has been used as a treatment for diabetes for the past 100 years [228]. In recent years, it has been shown in clinical trials to be effective against cancer as well [229]. In breast cancer, Metformin reduces tumor cell proliferation by decreasing insulin level since insulin promotes breast cancer cell proliferation, meanwhile, it suppresses tumor progression via inhibiting complex I and PI3K pathway [230]. Moreover, Metformin has similar effect in the clinical trial of colorectal cancer, it inhibits mitochondrial complex I and activating AMPK and LKB1 pathways to suppress tumor growth [231]. Therefore, Metformin is considered to be a valuable multi-target therapy agent.
IACS-010759
IACS-010759 is a clinical-grade small-molecule inhibitor of complex I of the mitochondrial electron transport chain. Treatment with IACS-010759 robustly inhibited proliferation and induced apoptosis in models of brain tumors and AML reliant on OXPHOS. IACS-010759 is currently being evaluated in phase 2 clinical trials in relapsed/refractory AML and solid tumors [166].
Gboxin
Gboxin, a small molecule, emerges as a novel OXPHOS inhibitor that targeting the activity of F0F1 ATP synthase in mitochondria [13]. Gboxin inhibits the glioblastoma cancer stem cell growth without affecting the growth of normal cells. In liver cancer, Gboxin inhibits cancer cell ATP production and migration via disrupting the interaction between TOMM34 and ATP5B, it shows synergistic effect with Metformin in liver cancer treatment [232].
Atovaquone
Atovaquone, an approved antimicrobial drug, has demonstrated anti-cancer potential and ability in clinical trial in treating ovarian cancer recently, it inhibits tumor cell proliferation by reducing ATP production by inhibiting mitochondrial complex III and increasing ROS levels [233]. In NSCLC, Atovaquone reduces tumor cells' drug resistance, it inhibits complex III and activates AMPK, leading to cancer cell death [234].
Dichloroacetic acid
Dichloroacetic acid, a novel anticancer drug, has been proved to have synergistic and inhibitory effects on liver cancer cells in clinical studies [235]. Its main target is PDH [236]. It inhibits liver cancer cell growth by inhibiting TCA cycle to stimulate AMPK pathway and induce oxidative stress. It has similar effect in glioblastoma [237]. In HCC mouse models, it was demonstrated that the combination of Metformin and Dichloroacetic acid had a synergistic effect, Metformin inhibit OXPHOS and upregulates glycolysis in HCC and increases the sensitivity to the PDH kinase inhibitor [238].
CPI-613
CPI-613 is a potent mitochondria metabolism inhibitor [239]. ACC, a key enzyme modulating lipid metabolism, has been demonstrated as a vital target of CPI-613. CPI-613 exhibits anticancer activity in pancreatic cancer cells by triggering ROS-associated apoptosis, accompanied by increased autophagy and repressed lipid metabolism through activating the AMPK signaling [240]. Devimistat (CPI-613®) has achieved promising result in Phase I and Phase II study and right now in Phase III study [241].
Conclusion and perspective
Mitochondrial metabolism is necessary for cancer cell proliferation, tumorigenesis and metastasis. Mitochondria execute mitophagy to clear damaged mitochondria and their fusion and fission dynamics may support tumor stemness and growth. Mutations of mtDNA are commonly found in various tumors, eliminated mutated mtDNA limits tumorigenesis. mtDNA escaped from stressed mitochondria by mPTP- and VDAC-dependent channels to activate the cGAS-STING signaling and gave rise to pro-inflammatory extracellular DNA [283]. Mutations of enzymes and metabolites in TCA cycles such as FH and SDH in mitochondria play important roles in tumor invasiveness and metastasis [284]. The intermediates in TCA cycles such as succinate, citrate, and NAD+ have been demonstrated to possess signaling capacity and to influence immunity [285].
In multiple cancers, the majority of tumor cells especially slow-dividing cancer cells generate ATP via mitochondrial OXPHOS. CSCs are resistant to regular chemo- and radiotherapy, but a great number of them rely on OXPHOS for energy supply, plenty of data highlight the existing connection between CSCs fate and mitochondrial metabolism and lots of inhibitors have been developed to target the complexes or proteins in OXPHOS to reduce CSC viability.
Targeting specific players including both TCA enzymes and OXPHOS-related-genes in mitochondria or mitochondria-related pathways may serve as novel and promising therapeutic approaches for cancers due to the multiple functions of mitochondria in cancer cells. However, the obstacles are there too, mitochondria are not only working in cancer cells but also in normal cells, how to selectively eliminate cancer cells without affecting healthy cells is the biggest challenge. The novel inhibitor Gboxin is one of the trials since its function relies on the high gradient in mitochondria of CSCs. Further investigations are required on the difference of mitochondria between cancer cells and normal ones so that targeting this difference will be a potential selective therapy.
Acknowledgements
Funding
Funding for this project includes National Natural Science Foundation of China (32100615, W.F.), (82073274, Y.S.), Science Technology Commission of Shanghai Municipality (20S11900700, Y.S.).
Author Contributions
Conceptualization and modification: Prof. Yufeng Shi, Y.L;
Writing review: Y.L, Y.S, X.S;
Revision: Y.L, Y.G;
Literature checking: W.F, J.Z, Y.W, X.C, L.W, S.Y.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH. et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell. 2019;176:1325-39 e22
2. Vasan K, Werner M, Chandel NS. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metabolism. 2020;32:341-52
3. Bennett CF, Latorre-Muro P, Puigserver P. Mechanisms of mitochondrial respiratory adaptation. Nat Rev Mol Cell Biol. 2022;23:817-35
4. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clinical Cancer Research. 2018;24:2482-90
5. Huang S, Liu Y, Zhang Y, Zhang R, Zhu C, Fan L. et al. Baicalein inhibits SARS-CoV-2/VSV replication with interfering mitochondrial oxidative phosphorylation in a mPTP dependent manner. Signal Transduct Target Ther. 2020;5:266
6. Yoo SM, Jung YK. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol Cells. 2018;41:18-26
7. Song CK, Pan SZ, Zhang JJ, Li N, Geng Q. Mitophagy: A novel perspective for insighting into cancer and cancer treatment. Cell Proliferat. 2022
8. Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. 2018;293:12649-52
9. Man Z, Guo J, Zhang Y, Cai Z. Regulation of intracellular ATP supply and its application in industrial biotechnology. Crit Rev Biotechnol. 2020;40:1151-62
10. Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updat. 2018;38:1-11
11. Griguer CE, Oliva CR, Gillespie GY. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J Neurooncol. 2005;74:123-33
12. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628-32
13. Shi YF, Lim SK, Liang QR, Iyer SV, Wang HY, Wang ZL. et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature. 2019;567:341 -+
14. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504-7
15. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9-15
16. Hooda J, Cadinu D, Alam MM, Shah A, Cao TM, Sullivan LA. et al. Enhanced heme function and mitochondrial respiration promote the progression of lung cancer cells. PLoS One. 2013;8:e63402
17. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017 168
18. Vashisht Gopal YN, Gammon S, Prasad R, Knighton B, Pisaneschi F, Roszik J. et al. A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin Cancer Res. 2019;25:6429-42
19. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T. et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013 9
20. Vidali S, Aminzadeh S, Lambert B, Rutherford T, Sperl W, Kofler B. et al. Mitochondria: The ketogenic diet-A metabolism-based therapy. Int J Biochem Cell Biol. 2015;63:55-9
21. Bird SS, Marur VR, Stavrovskaya IG, Kristal BS. Qualitative characterization of the rat liver mitochondrial lipidome using LC-MS profiling and high energy collisional dissociation (HCD) all ion fragmentation. Metabolomics. 2013;9:S67-S83
22. Tatsuta T, Scharwey M, Langer T. Mitochondrial lipid trafficking. Trends Cell Biol. 2014;24:44-52
23. Tamura Y, Kawano S, Endo T. Lipid homeostasis in mitochondria. Biol Chem. 2020;401:821-33
24. Baker CD, Basu Ball W, Pryce EN, Gohil VM. Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol Biol Cell. 2016;27:2161-71
25. Calzada E, Avery E, Sam PN, Modak A, Wang CY, McCaffery JM. et al. Phosphatidylethanolamine made in the inner mitochondrial membrane is essential for yeast cytochrome bc(1) complex function. Nat Commun. 2019 10
26. Dimmer KS, Rapaport D. Mitochondrial contact sites as platforms for phospholipid exchange. Bba-Mol Cell Biol L. 2017;1862:69-80
27. Klein HP, Greenfield S. Effects of mitochondria on lipid synthesis in yeast homogenates. Exp Cell Res. 1959;17:185-8
28. Nalecz KA, Nalecz MJ. [Carnitine - mitochondria and beyond]. Postepy Biochem. 2016;62:85-93
29. Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. 2012;151:400-13
30. Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R. et al. Mfn2 is critical for brown adipose tissue thermogenic function. Embo J. 2017;36:1543-58
31. Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R. et al. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev Cell. 2017;42:9-21 e5
32. Welte MA, Gould AP. Lipid droplet functions beyond energy storage. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:1260-72
33. Klecker T, Braun RJ, Westermann B. Lipid Droplets Guard Mitochondria during Autophagy. Dev Cell. 2017;42:1-2
34. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA. et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metabolism. 2018;27:869 -+
35. Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, Ruperez C. et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun. 2015;6:7176
36. Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak-Tarnopolsky SN, Devries MC, Hamadeh MJ. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol-Reg I. 2007;292:R1271-R8
37. Yu J, Zhang S, Cui L, Wang W, Na H, Zhu X. et al. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim Biophys Acta. 2015;1853:918-28
38. Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si YG, Las G. et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. Embo J. 2014;33:418-36
39. Benador IY, Veliova M, Liesa M, Shirihai OS. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019;29:827-35
40. Angelova PR, Esteras N, Abramov AY. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med Res Rev. 2021;41:770-84
41. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D. et al. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843
42. Anthonymuthu TS, Kenny EM, Bayir H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016;1640:57-76
43. Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA. et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature Neuroscience. 2012;15:1407-13
44. Kagan VE, Wipf P, Stoyanovsky D, Greenberger JS, Borisenko G, Belikova NA. et al. Mitochondrial targeting of electron scavenging antioxidants: Regulation of selective oxidation vs random chain reactions. Adv Drug Deliv Rev. 2009;61:1375-85
45. Fridman A, Saha A, Chan A, Casteel DE, Pilz RB, Boss GR. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem J. 2013;454:91-9
46. Sigoillot FD, Berkowski JA, Sigoillot SM, Kotsis DH, Guy HI. Cell cycle-dependent regulation of pyrimidine biosynthesis. J Biol Chem. 2003;278:3403-9
47. Peng T, Zhou LX, Zuo L, Luan YX. miR-506 functions as a tumor suppressor in glioma by targeting STAT3 (Retraction of Vol 35, Pg 1057, 2016). Oncology Reports. 2022 47
48. Gadian DG, Gadian DG. NMR and its applications to living systems. 2nd ed. Oxford; New York: Oxford University Press. 1995
49. Manfredi G, Yang L, Gajewski CD, Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317-26
50. Zong WX, Thompson CB. Necrotic death as a cell fate. Gene Dev. 2006;20:1-15
51. MacIntosh BR, Holash RJ, Renaud JM. Skeletal muscle fatigue-regulation of excitation-contraction coupling to avoid metabolic catastrophe. J Cell Sci. 2012;125:2105-14
52. Tatsumi T, Shiraishi J, Keira N, Akashi K, Mano A, Yamanaka S. et al. Intracellular ATP is required for mitochondrial apoptotic pathways in isolated hypoxic rat cardiac myocytes. Cardiovascular Research. 2003;59:428-40
53. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
54. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671-84
55. Walker JE, Runswick MJ. The mitochondrial transport protein superfamily. J Bioenerg Biomembr. 1993;25:435-46
56. Ruprecht JJ, King MS, Zogg T, Aleksandrova AA, Pardon E, Crichton PG. et al. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell. 2019;176:435-47 e15
57. Klingenberg M. Molecular aspects of the adenine nucleotide carrier from mitochondria. Arch Biochem Biophys. 1989;270:1-14
58. Kunji ER, Aleksandrova A, King MS, Majd H, Ashton VL, Cerson E. et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim Biophys Acta. 2016;1863:2379-93
59. Pebay-Peyroula E, Brandolin G. Nucleotide exchange in mitochondria: insight at a molecular level. Curr Opin Struc Biol. 2004;14:420-5
60. Jourdain AA, Begg BE, Mick E, Shah H, Calvo SE, Skinner OS. et al. Loss of LUC7L2 and U1 snRNP subunits shifts energy metabolism from glycolysis to OXPHOS. Mol Cell. 2021;81:1905-19 e12
61. Wu C, Liu Y, Liu W, Zou T, Lu S, Zhu C. et al. NNMT-DNMT1 Axis is Essential for Maintaining Cancer Cell Sensitivity to Oxidative Phosphorylation Inhibition. Adv Sci (Weinh). 2022: e2202642.
62. Li T, Han J, Jia L, Hu X, Chen L, Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. 2019;10:583-94
63. Tang Y, Jia Y, Fan L, Liu H, Zhou Y, Wang M. et al. MFN2 Prevents Neointimal Hyperplasia in Vein Grafts via Destabilizing PFK1. Circ Res. 2022;130:e26-e43
64. Gnocchi D, Kurzyk A, Mintrone A, Lentini G, Sabba C, Mazzocca A. Inhibition of LPAR6 overcomes sorafenib resistance by switching glycolysis into oxidative phosphorylation in hepatocellular carcinoma. Biochimie. 2022;202:180-9
65. Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol. 2022;85:4-32
66. Delaunay S, Pascual G, Feng B, Klann K, Behm M, Hotz-Wagenblatt A. et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature. 2022;607:593-603
67. Bosc C, Selak MA, Sarry JE. Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment. Cell Metabolism. 2017;26:705-7
68. Liu YE, Shi YF. Mitochondria as a target in cancer treatment. Medcomm. 2020;1:129-39
69. Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. 2015;33:125-31
70. Lozoya OA, Wang T, Grenet D, Wolfgang TC, Sobhany M, Ganini da Silva D. et al. Mitochondrial acetyl-CoA reversibly regulates locus-specific histone acetylation and gene expression. Life Sci Alliance. 2019 2
71. Liu Y, Chen C, Wang X, Sun Y, Zhang J, Chen J. et al. An Epigenetic Role of Mitochondria in Cancer. Cells. 2022 11
72. Martinez-Reyes I, Chandel NS. Acetyl-CoA-directed gene transcription in cancer cells. Genes Dev. 2018;32:463-5
73. Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M. et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature. 2016;537:548-52
74. Zhou W, Nie ZW, Zhou DJ, Cui XS. Acetyl-CoA synthases are essential for maintaining histone acetylation under metabolic stress during zygotic genome activation in pigs. J Cell Physiol. 2021;236:6948-62
75. Samata M, Alexiadis A, Richard G, Georgiev P, Nuebler J, Kulkarni T. et al. Intergenerationally Maintained Histone H4 Lysine 16 Acetylation Is Instructive for Future Gene Activation. Cell. 2020;182:127-44 e23
76. Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D. et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21:392-402
77. Shyh-Chang N, Daley GQ. Metabolic switches linked to pluripotency and embryonic stem cell differentiation. Cell Metab. 2015;21:349-50
78. Daneshvar K, Ardehali MB, Klein IA, Hsieh FK, Kratkiewicz AJ, Mahpour A. et al. lncRNA DIGIT and BRD3 protein form phase-separated condensates to regulate endoderm differentiation. Nat Cell Biol. 2020;22:1211-22
79. Zhou WJ, Zhao TT, Du JY, Ji GY, Li XY, Ji SF. et al. TIGAR promotes neural stem cell differentiation through acetyl-CoA-mediated histone acetylation. Cell Death & Disease. 2019 10
80. Lee JV, Berry CT, Kim K, Sen P, Kim T, Carrer A. et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca2+-NFAT signaling. Gene Dev. 2018;32:497-511
81. Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC. et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019;9:416-35
82. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN. et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22:1108-19
83. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57-71
84. Cui S, Zhang K, Li C, Chen J, Pan Y, Feng B. et al. Methylation-associated silencing of microRNA-129-3p promotes epithelial-mesenchymal transition, invasion and metastasis of hepatocelluar cancer by targeting Aurora-A. Oncotarget. 2016;7:78009-28
85. Huang L, Wang CD, Xu HX, Peng GY. Targeting citrate as a novel therapeutic strategy in cancer treatment. Bba-Rev Cancer. 2020 1873
86. Srere PA. The citrate cleavage enzyme. I. Distribution and purification. J Biol Chem. 1959;234:2544-7
87. Icard P, Coquerel A, Wu Z, Gligorov J, Fuks D, Fournel L. et al. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int J Mol Sci. 2021 22
88. Crochet RB, Kim JD, Lee H, Yim YS, Kim SG, Neau D. et al. Crystal structure of heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) and the inhibitory influence of citrate on substrate binding. Proteins. 2017;85:117-24
89. Icard P, Wu ZR, Alifano M, Fournel L. Gluconeogenesis of Cancer Cells Is Disrupted by Citrate. Trends Cancer. 2019;5:265-6
90. Icard P, Lincet H. The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target (vol 29, pg 47, 2016). Drug Resist Update. 2017;30:63 -
91. Fan X, Zhou J, Yan X, Bi X, Liang J, Lu S. et al. Citrate activates autophagic death of prostate cancer cells via downregulation CaMKII/AKT/mTOR pathway. Life Sci. 2021;275:119355
92. Richardson CL, Delehanty LL, Bullock GC, Rival CM, Tung KS, Kimpel DL. et al. Isocitrate ameliorates anemia by suppressing the erythroid iron restriction response. J Clin Invest. 2013;123:3614-23
93. Kim A, Fung E, Parikh SG, Gabayan V, Nemeth E, Ganz T. Isocitrate treatment of acute anemia of inflammation in a mouse model. Blood Cells Mol Dis. 2016;56:31-6
94. Larsen T. Fluorometric determination of free and total isocitrate in bovine milk. J Dairy Sci. 2014;97:7498-504
95. Liu S, Cadoux-Hudson T, Schofield CJ. Isocitrate dehydrogenase variants in cancer - Cellular consequences and therapeutic opportunities. Curr Opin Chem Biol. 2020;57:122-34
96. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44
97. Yan D, Li W, Liu Q, Yang K. Advances in Immune Microenvironment and Immunotherapy of Isocitrate Dehydrogenase Mutated Glioma. Front Immunol. 2022;13:914618
98. Sunthankar KI, Jenkins MT, Cote CH, Patel SB, Welner RS, Ferrell PB. Isocitrate dehydrogenase mutations are associated with altered IL-1beta responses in acute myeloid leukemia. Leukemia. 2022;36:923-34
99. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730-41
100. Stuani L, Sabatier M, Saland E, Cognet G, Poupin N, Bosc C. et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J Exp Med. 2021 218
101. Chaturvedi A, Cruz MMA, Jyotsana N, Sharma A, Yun HY, Gorlich K. et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122:2877-87
102. Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L. et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636-43
103. Wu MJ, Shi L, Merritt J, Zhu AX, Bardeesy N. Biology of IDH mutant cholangiocarcinoma. Hepatology. 2022;75:1322-37
104. Ghiam AF, Cairns RA, Thoms J, Dal Pra A, Ahmed O, Meng A. et al. IDH mutation status in prostate cancer. Oncogene. 2012;31:3826 -
105. Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP. et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901-3
106. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599-612
107. Waitkus MS, Diplas BH, Yan H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell. 2018;34:186-95
108. Asadi Shahmirzadi A, Edgar D, Liao CY, Hsu YM, Lucanic M, Asadi Shahmirzadi A. et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020;32:447-56 e6
109. Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413-6
110. Baracco EE, Castoldi F, Durand S, Enot DP, Tadic J, Kainz K. et al. alpha-Ketoglutarate inhibits autophagy. Aging (Albany NY). 2019;11:3418-31
111. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811-6
112. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5
113. Wang Y, Deng P, Liu Y, Wu Y, Chen Y, Guo Y. et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat Commun. 2020;11:5596
114. Tran TQ, Hanse EA, Habowski AN, Li H, Ishak Gabra MB, Yang Y. et al. alpha-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat Cancer. 2020;1:345-58
115. Morris JPt, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC. et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature. 2019;573:595-9
116. Liu S, Yang J, Wu Z. The Regulatory Role of alpha-Ketoglutarate Metabolism in Macrophages. Mediators Inflamm. 2021;2021:5577577
117. Wang X, Liu R, Qu X, Yu H, Chu H, Zhang Y. et al. alpha-Ketoglutarate-Activated NF-kappaB Signaling Promotes Compensatory Glucose Uptake and Brain Tumor Development. Mol Cell. 2019;76:148-62 e7
118. Atlante S, Visintin A, Marini E, Savoia M, Dianzani C, Giorgis M. et al. alpha-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell Death Dis. 2018;9:756
119. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G. et al. Succinate is an inflammatory signal that induces IL-1 beta through HIF-1 alpha. Nature. 2013;496:238 -+
120. Connors J, Dawe N, Van Limbergen J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients. 2018 11
121. Mills E, O'Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 2014;24:313-20
122. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE. et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167:457-70 e13
123. Keiran N, Ceperuelo-Mallafre V, Calvo E, Hernandez-Alvarez MI, Ejarque M, Nunez-Roa C. et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol. 2019;20:581-92
124. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD. et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77-85
125. Trauelsen M, Hiron TK, Lin D, Petersen JE, Breton B, Husted AS. et al. Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated Gq signaling. Cell Rep. 2021 35
126. Elia I, Rowe JH, Johnson S, Joshi S, Notarangelo G, Kurmi K. et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab. 2022;34:1137-50 e6
127. Zhao Y, Feng F, Guo QH, Wang YP, Zhao R. Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors. World J Gastroenterol. 2020;26:5074-89
128. Jiang S, Yan W. Succinate in the cancer-immune cycle. Cancer Lett. 2017;390:45-7
129. Mossa A, Velasquez-Flores M, Cammisotto PG, Campeau L. Receptor GPR91 contributes to voiding function and detrusor relaxation mediated by succinate. Neurourol Urodyn. 2021;40:120-30
130. Huang S, Millar AH. Succinate dehydrogenase: the complex roles of a simple enzyme. Curr Opin Plant Biol. 2013;16:344-9
131. Van Vranken JG, Na U, Winge DR, Rutter J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit Rev Biochem Mol Biol. 2015;50:168-80
132. Dalla Pozza E, Dando I, Pacchiana R, Liboi E, Scupoli MT, Donadelli M. et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol. 2020;98:4-14
133. van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM. et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764-71
134. Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC. et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231-9
135. Cardaci S, Zheng L, MacKay G, van den Broek NJ, MacKenzie ED, Nixon C. et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol. 2015;17:1317-26
136. Lussey-Lepoutre C, Hollinshead KE, Ludwig C, Menara M, Morin A, Castro-Vega LJ. et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun. 2015;6:8784
137. Aspuria PP, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA. et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014;2:21
138. Aggarwal RK, Luchtel RA, Machha V, Tischer A, Zou YY, Pradhan K. et al. Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. P Natl Acad Sci USA. 2021 118
139. Tsai TH, Lee WY. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma. Arch Pathol Lab Med. 2019;143:643-7
140. Jodeiri Farshbaf M, Kiani-Esfahani A. Succinate dehydrogenase: Prospect for neurodegenerative diseases. Mitochondrion. 2018;42:77-83
141. Dona M, Neijman K, Timmers H. MITOCHONDRIA: Succinate dehydrogenase subunit B-associated phaeochromocytoma and paraganglioma. Int J Biochem Cell Biol. 2021;134:105949
142. Schumacker PT. Mitochondrial Succinate Dehydrogenase in Chronic Obstructive Pulmonary Disease: Is Complex II Too Complex? Am J Resp Cell Mol. 2021;65:231-2
143. Chen X, Sunkel B, Wang M, Kang S, Wang T, Gnanaprakasam JNR. et al. Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation. Sci Immunol. 2022;7:eabm8161
144. Schmidt C, Sciacovelli M, Frezza C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin Cell Dev Biol. 2020;98:15-25
145. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223-33
146. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488-94
147. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H. et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326-38
148. Borst P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. Iubmb Life. 2020;72:2241-59
149. Altinok O, Poggio JL, Stein DE, Bowne WB, Shieh AC, Snyder NW. et al. Malate-aspartate shuttle promotes l-lactate oxidation in mitochondria. J Cell Physiol. 2020;235:2569-81
150. Wang C, Chen H, Zhang M, Zhang J, Wei X, Ying W. Malate-aspartate shuttle inhibitor aminooxyacetic acid leads to decreased intracellular ATP levels and altered cell cycle of C6 glioma cells by inhibiting glycolysis. Cancer Lett. 2016;378:1-7
151. Ma YC, Tian PF, Chen ZP, Yue DS, Liu CC, Li CG. et al. Urinary malate dehydrogenase 2 is a new biomarker for early detection of non-small-cell lung cancer. Cancer Sci. 2021;112:2349-60
152. Wiese EK, Hitosugi S, Loa ST, Sreedhar A, Andres-Beck LG, Kurmi K. et al. Enzymatic activation of pyruvate kinase increases cytosolic oxaloacetate to inhibit the Warburg effect. Nat Metab. 2021;3:954-68
153. Zhou X, Curbo S, Li F, Krishnan S, Karlsson A. Inhibition of glutamate oxaloacetate transaminase 1 in cancer cell lines results in altered metabolism with increased dependency of glucose. BMC Cancer. 2018;18:559
154. Kuang Y, Han X, Xu M, Yang Q. Oxaloacetate induces apoptosis in HepG2 cells via inhibition of glycolysis. Cancer Med. 2018;7:1416-29
155. Campos F, Sobrino T, Ramos-Cabrer P, Castillo J. Oxaloacetate: a novel neuroprotective for acute ischemic stroke. Int J Biochem Cell Biol. 2012;44:262-5
156. Merlen G, Raymond VA, Cassim S, Lapierre P, Bilodeau M. Oxaloacetate Protects Rat Liver From Experimental Warm Ischemia/Reperfusion Injury by Improving Cellular Energy Metabolism. Liver Transpl. 2019;25:627-39
157. Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019;26:690-702
158. Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z. et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2011;20:674-88
159. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014;5:11029-37
160. Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front Oncol. 2018;8:203
161. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M. et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell. 2013;12:329-41
162. Raggi C, Taddei ML, Sacco E, Navari N, Correnti M, Piombanti B. et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J Hepatol. 2021;74:1373-85
163. Valle S, Alcala S, Martin-Hijano L, Cabezas-Sainz P, Navarro D, Munoz ER. et al. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat Commun. 2020;11:5265
164. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL. et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234-40
165. Jones CL, Stevens BM, D'Alessandro A, Reisz JA, Culp-Hill R, Nemkov T. et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell. 2018;34:724-40 e4
166. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C. et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036-46
167. Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR. et al. Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016;7:34084-99
168. Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K. et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287-301
169. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC. et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992-1003 1-15
170. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M. et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015;22:590-605
171. Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH. et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017;16:10
172. Wang X, Chen Y, Wang X, Tian H, Wang Y, Jin J. et al. Stem Cell Factor SOX2 Confers Ferroptosis Resistance in Lung Cancer via Upregulation of SLC7A11. Cancer Res. 2021;81:5217-29
173. Huang H, Zhang S, Li Y, Liu Z, Mi L, Cai Y. et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat Commun. 2021;12:3720
174. Feng X, Zhang W, Yin W, Kang YJ. The involvement of mitochondrial fission in maintenance of the stemness of bone marrow mesenchymal stem cells. Exp Biol Med (Maywood). 2019;244:64-72
175. Chen H, Chan DC. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017;26:39-48
176. Ludikhuize MC, Meerlo M, Gallego MP, Xanthakis D, Burgaya Julia M, Nguyen NTB. et al. Mitochondria Define Intestinal Stem Cell Differentiation Downstream of a FOXO/Notch Axis. Cell Metab. 2020;32:889-900 e7
177. Saito K, Zhang Q, Yang H, Yamatani K, Ai T, Ruvolo V. et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv. 2021;5:4233-55
178. Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27:105-17
179. Liu D, Sun Z, Ye T, Li J, Zeng B, Zhao Q. et al. The mitochondrial fission factor FIS1 promotes stemness of human lung cancer stem cells via mitophagy. FEBS Open Bio. 2021;11:1997-2007
180. Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W. et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501-10
181. Peiris-Pages M, Bonuccelli G, Sotgia F, Lisanti MP. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget. 2018;9:13254-75
182. Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S. et al. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340-3
183. Feng X, Yin W, Wang J, Feng L, Kang YJ. Mitophagy promotes the stemness of bone marrow-derived mesenchymal stem cells. Exp Biol Med (Maywood). 2021;246:97-105
184. Naik PP, Birbrair A, Bhutia SK. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell Mol Life Sci. 2019;76:27-43
185. Lin QY, Chen JQ, Gu LF, Dan XG, Zhang C, Yang YZ. New insights into mitophagy and stem cells. Stem Cell Research & Therapy. 2021 12
186. Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Patra S, Behera BP. et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020;66:45-58
187. Praharaj PP, Patro BS, Bhutia SK. Dysregulation of mitophagy and mitochondrial homeostasis in cancer stem cells: Novel mechanism for anti-cancer stem cell-targeted cancer therapy. Br J Pharmacol. 2022;179:5015-35
188. Vazquez-Martin A, Van den Haute C, Cufi S, Corominas-Faja B, Cuyas E, Lopez-Bonet E. et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging (Albany NY). 2016;8:1330-52
189. Alcala S, Sancho P, Martinelli P, Navarro D, Pedrero C, Martin-Hijano L. et al. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat Commun. 2020 11
190. Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST. et al. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell. 2017;68:281-92 e5
191. Lin R, Iacovitti L. Classic and novel stem cell niches in brain homeostasis and repair. Brain Res. 2015;1628:327-42
192. Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC. et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell. 2018;23:86 -+
193. Lv DG, Gimple RC, Zhong CQ, Wu QL, Yang KL, Prager BC. et al. PDGF signaling inhibits mitophagy in glioblastoma stem cells through N-6-methyladenosine. Developmental Cell. 2022;57:1466 -+
194. Held NM, Houtkooper RH. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays. 2015;37:867-76
195. Yan C, Luo L, Guo CY, Goto S, Urata Y, Shao JH. et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017;388:34-42
196. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61-8
197. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC. et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016;45:374-88
198. Mazat JP, Devin A, Ransac S. Modelling mitochondrial ROS production by the respiratory chain. Cell Mol Life Sci. 2020;77:455-65
199. Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J Am Coll Cardiol. 2017;70:230-51
200. Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC. et al. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med. 2016;90:12-23
201. Pokrzywinski KL, Biel TG, Kryndushkin D, Rao VA. Therapeutic Targeting of the Mitochondria Initiates Excessive Superoxide Production and Mitochondrial Depolarization Causing Decreased mtDNA Integrity. PLoS One. 2016;11:e0168283
202. Bugger H, Pfeil K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165768
203. Rababa'h AM, Guillory AN, Mustafa R, Hijjawi T. Oxidative Stress and Cardiac Remodeling: An Updated Edge. Curr Cardiol Rev. 2018;14:53-9
204. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560-9
205. Espana L, Martin B, Aragues R, Chiva C, Oliva B, Andreu D. et al. Bcl-x(L)-mediated changes in metabolic pathways of breast cancer cells: from survival in the blood stream to organ-specific metastasis. Am J Pathol. 2005;167:1125-37
206. Stresing V, Baltziskueta E, Rubio N, Blanco J, Arriba MC, Valls J. et al. Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene. 2013;32:724-35
207. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol. 2015;35:615-21
208. Giordano S, Darley-Usmar V, Zhang JH. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biology. 2014;2:82-90
209. Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M. et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409-14
210. Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem. 2015;116:696-703
211. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H. et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18:157
212. Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
213. Koukourakis MI, Giatromanolaki A, Skarlatos J, Corti L, Blandamura S, Piazza M. et al. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res. 2001;61:1830-2
214. Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208-15
215. Wu H, Chen Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid Redox Signal. 2015;22:1032-46
216. Fuhrmann DC, Wittig I, Heide H, Dehne N, Brune B. Chronic hypoxia alters mitochondrial composition in human macrophages. Biochim Biophys Acta. 2013;1834:2750-60
217. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H. et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362-77
218. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24:787-95
219. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P. et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177-85
220. Wu WX, Li W, Chen H, Jiang L, Zhu RZ, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 2016;12:1675-6
221. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W. et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. Embo Rep. 2014;15:566-75
222. Deng R, Zhang HL, Huang JH, Cai RZ, Wang Y, Chen YH. et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy. 2021;17:3011-29
223. Linqing L, Yuhan Q, Erfei L, Yong Q, Dong W, Chengchun T. et al. Hypoxia-induced PINK1/Parkin-mediated mitophagy promotes pulmonary vascular remodeling. Biochem Biophys Res Commun. 2021;534:568-75
224. Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207-17
225. Li HS, Zhou YN, Li L, Li SF, Long D, Chen XL. et al. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019;25:101109
226. Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biology. 2017;12:208-15
227. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12:385-97
228. Triggle CR, Mohammed I, Bshesh K, Marei I, Ye K, Ding H. et al. Metformin: Is it a drug for all reasons and diseases? Metabolism. 2022;133:155223
229. De A, Kuppusamy G. Metformin in breast cancer: preclinical and clinical evidence. Curr Probl Cancer. 2020;44:100488
230. Kawakita E, Yang F, Kumagai A, Takagaki Y, Kitada M, Yoshitomi Y. et al. Metformin Mitigates DPP-4 Inhibitor-Induced Breast Cancer Metastasis via Suppression of mTOR Signaling. Mol Cancer Res. 2021;19:61-73
231. Kamarudin MNA, Sarker MMR, Zhou JR, Parhar I. Metformin in colorectal cancer: molecular mechanism, preclinical and clinical aspects. J Exp Clin Cancer Res. 2019;38:491
232. Jin P, Jiang J, Zhou L, Huang Z, Qin S, Chen HN. et al. Disrupting metformin adaptation of liver cancer cells by targeting the TOMM34/ATP5B axis. Embo Mol Med. 2022: e16082.
233. Guo Y, Hu B, Fu BB, Zhu HY. Atovaquone at clinically relevant concentration overcomes chemoresistance in ovarian cancer via inhibiting mitochondrial respiration. Pathol Res Pract. 2021 224
234. Xie F, Gong J, Tan H, Zhang H, Ma J. Preclinical evidence of synergism between atovaquone and chemotherapy by AMPK-dependent mitochondrial dysfunction. Eur J Pharmacol. 2021;907:174256
235. Meng G, Li B, Chen A, Zheng M, Xu T, Zhang H. et al. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br J Cancer. 2020;122:111-20
236. Buchsteiner M, Quek LE, Gray P, Nielsen LK. Improving culture performance and antibody production in CHO cell culture processes by reducing the Warburg effect. Biotechnol Bioeng. 2018;115:2315-27
237. Albayrak G, Konac E, Dere UA, Emmez H. Targeting Cancer Cell Metabolism with Metformin, Dichloroacetate and Memantine in Glioblastoma (GBM). Turk Neurosurg. 2021;31:233-7
238. Kim TS, Lee M, Park M, Kim SY, Shim MS, Lee CY. et al. Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1. Int J Mol Sci. 2021 22
239. Egawa Y, Saigo C, Kito Y, Moriki T, Takeuchi T. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS One. 2018;13:e0198940
240. Gao L, Xu Z, Huang Z, Tang Y, Yang D, Huang J. et al. CPI-613 rewires lipid metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC signaling. J Exp Clin Cancer Res. 2020;39:73
241. Philip PA, Buyse ME, Alistar AT, Rocha Lima CM, Luther S, Pardee TS. et al. A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol. 2019;15:3189-96
242. Di Magno L, Manni S, Di Pastena F, Coni S, Macone A, Cairoli S. et al. Phenformin Inhibits Hedgehog-Dependent Tumor Growth through a Complex I-Independent Redox/Corepressor Module. Cell Rep. 2020;30:1735-52 e7
243. Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mouron S. et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep. 2016;15:2705-18
244. Stalinska J, Zimolag E, Pianovich NA, Zapata A, Lassak A, Rak M. et al. Chemically Modified Variants of Fenofibrate with Antiglioblastoma Potential. Transl Oncol. 2019;12:895-907
245. Cornelissen J, Wanders RJ, Van Gennip AH, Van den Bogert C, Voute PA, Van Kuilenburg AB. Meta-iodobenzylguanidine inhibits complex I and III of the respiratory chain in the human cell line Molt-4. Biochem Pharmacol. 1995;49:471-7
246. Momtazi-Borojeni AA, Abdollahi E, Ghasemi F, Caraglia M, Sahebkar A. The novel role of pyrvinium in cancer therapy. J Cell Physiol. 2018;233:2871-81
247. Papadopoli D, Uchenunu O, Palia R, Chekkal N, Hulea L, Topisirovic I. et al. Perturbations of cancer cell metabolism by the antidiabetic drug canagliflozin. Neoplasia. 2021;23:391-9
248. Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M. et al. PPARgamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018 10
249. Peng J, Wang K, Xiang W, Li Y, Hao Y, Guan Y. Rosiglitazone polarizes microglia and protects against pilocarpine-induced status epilepticus. CNS Neurosci Ther. 2019;25:1363-72
250. Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ. Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J Cardiovasc Pharmacol. 2012;59:101-8
251. Davis R, Whittington R, Bryson HM. Nefazodone. A review of its pharmacology and clinical efficacy in the management of major depression. Drugs. 1997;53:608-36
252. Hall C, Wu M, Crane FL, Takahashi H, Tamura S, Folkers K. Piericidin A: a new inhibitor of mitochondrial electron transport. Biochem Biophys Res Commun. 1966;25:373-7
253. Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V. et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Developmental Cell. 2017;40:583 -+
254. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T. et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193-204
255. Jiang L, Yin X, Chen YH, Chen Y, Jiang W, Zheng H. et al. Proteomic analysis reveals ginsenoside Rb1 attenuates myocardial ischemia/reperfusion injury through inhibiting ROS production from mitochondrial complex I. Theranostics. 2021;11:1703-20
256. Janku F, LoRusso P, Mansfield AS, Nanda R, Spira A, Wang T. et al. First-in-human evaluation of the novel mitochondrial complex I inhibitor ASP4132 for treatment of cancer. Invest New Drugs. 2021;39:1348-56
257. Kaiser N, Corkery D, Wu Y, Laraia L, Waldmann H. Modulation of autophagy by the novel mitochondrial complex I inhibitor Authipyrin. Bioorg Med Chem. 2019;27:2444-8
258. Angulo-Molina A, Reyes-Leyva J, Lopez-Malo A, Hernandez J. The role of alpha tocopheryl succinate (alpha-TOS) as a potential anticancer agent. Nutr Cancer. 2014;66:167-76
259. Nath K, Guo L, Nancolas B, Nelson DS, Shestov AA, Lee SC. et al. Mechanism of antineoplastic activity of lonidamine. Biochim Biophys Acta. 2016;1866:151-62
260. Kim YS. Malonate metabolism: biochemistry, molecular biology, physiology, and industrial application. J Biochem Mol Biol. 2002;35:443-51
261. Cheng H, Liu HF, Yang L, Zhang R, Chen C, Wu Y. et al. N-(3,5-Dichloro-4-(2,4,6-trichlorophenoxy)phenyl)benzenesulfonamide: A new dual-target inhibitor of mitochondrial complex II and complex III via structural simplification. Bioorg Med Chem. 2020;28:115299
262. Wojtovich AP, Nehrke KW, Brookes PS. The mitochondrial complex II and ATP-sensitive potassium channel interaction: quantitation of the channel in heart mitochondria. Acta Biochim Pol. 2010;57:431-4
263. Yankovskaya V, Sablin SO, Ramsay RR, Singer TP, Ackrell BA, Cecchini G. et al. Inhibitor probes of the quinone binding sites of mammalian complex II and Escherichia coli fumarate reductase. J Biol Chem. 1996;271:21020-4
264. Yamamoto K, Tashiro E, Motohashi K, Seto H, Imoto M. Napyradiomycin A1, an inhibitor of mitochondrial complexes I and II. J Antibiot (Tokyo). 2012;65:211-4
265. Usta J, Kreydiyyeh S, Knio K, Barnabe P, Bou-Moughlabay Y, Dagher S. Linalool decreases HepG2 viability by inhibiting mitochondrial complexes I and II, increasing reactive oxygen species and decreasing ATP and GSH levels. Chem Biol Interact. 2009;180:39-46
266. Cheng H, Yang L, Liu HF, Zhang R, Chen C, Wu Y. et al. N-(4-(2-chloro-4-(trifluoromethyl)phenoxy)phenyl)picolinamide as a new inhibitor of mitochondrial complex III: Synthesis, biological evaluation and computational simulations. Bioorg Med Chem Lett. 2020;30:127302
267. James J, Valuparampil Varghese M, Vasilyev M, Langlais PR, Tofovic SP, Rafikova O. et al. Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape. Int J Mol Sci. 2020 21
268. Gao AH, Fu YY, Zhang KZ, Zhang M, Jiang HW, Fan LX. et al. Azoxystrobin, a mitochondrial complex III Qo site inhibitor, exerts beneficial metabolic effects in vivo and in vitro. Biochim Biophys Acta. 2014;1840:2212-21
269. Zhu X, Zhang M, Liu J, Ge J, Yang G. Ametoctradin is a potent Qo site inhibitor of the mitochondrial respiration complex III. J Agric Food Chem. 2015;63:3377-86
270. Parker WD Jr, Frerman F, Haas R, Parks JK. Myxothiazol resistance in human mitochondria. Biochim Biophys Acta. 1988;936:133-8
271. Gurung B, Yu L, Yu CA. Stigmatellin induces reduction of iron-sulfur protein in the oxidized cytochrome bc1 complex. J Biol Chem. 2008;283:28087-94
272. Kruglov AG, Andersson MA, Mikkola R, Roivainen M, Kredics L, Saris NEL. et al. Novel Mycotoxin from Acremonium exuviarum Is a Powerful Inhibitor of the Mitochondrial Respiratory Chain Complex III. Chem Res Toxicol. 2009;22:565-73
273. Belosludtsev KN, Belosludtseva NV, Kosareva EA, Talanov EY, Gudkov SV, Dubinin MV. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie. 2020;176:150-7
274. Medda N, Patra R, Ghosh TK, Maiti S. Neurotoxic Mechanism of Arsenic: Synergistic Effect of Mitochondrial Instability, Oxidative Stress, and Hormonal-Neurotransmitter Impairment. Biol Trace Elem Res. 2020;198:8-15
275. Gallagher CH. Effect of hydrocortisone on mitochondrial membrane permeability. Nature. 1958;182:1315-6
276. Glinka YY, Youdim MB. Inhibition of mitochondrial complexes I and IV by 6-hydroxydopamine. Eur J Pharmacol. 1995;292:329-32
277. Way JL, Leung P, Cannon E, Morgan R, Tamulinas C, Leongway J. et al. The Mechanism of Cyanide Intoxication and Its Antagonism. Ciba F Symp. 1988;140:232-48
278. Zvyagilskaya RA, Bogucka K, Wojtczak L. Accumulation of azide in mitochondria and the effect of azide on energy metabolism. Acta Biochim Pol. 1969;16:163-73
279. Salimi A, Roudkenar MH, Seydi E, Sadeghi L, Mohseni A, Pirahmadi N. et al. Chrysin as an Anti-Cancer Agent Exerts Selective Toxicity by Directly Inhibiting Mitochondrial Complex II and V in CLL B-lymphocytes. Cancer Investigation. 2017;35:174-86
280. Vaamonde-Garcia C, Loureiro J, Valcarcel-Ares MN, Riveiro-Naveira RR, Ramil-Gomez O, Hermida-Carballo L. et al. The mitochondrial inhibitor oligomycin induces an inflammatory response in the rat knee joint. BMC Musculoskelet Disord. 2017;18:254
281. Steiner JP, Bachani M, Wolfson-Stofko B, Lee MH, Wang T, Li G. et al. Interaction of paroxetine with mitochondrial proteins mediates neuroprotection. Neurotherapeutics. 2015;12:200-16
282. Berger M, Strecker HJ, Waelsch H. Action of chlorpromazine on oxidative phosphorylation of liver and brain mitochondria. Nature. 1956;177:1234-5
283. Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H. et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity. 2022;55:1370-85 e8
284. Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT. et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell. 2016;61:199-209
285. Wang ZH, Chen L, Li W, Chen L, Wang YP. Mitochondria transfer and transplantation in human health and diseases. Mitochondrion. 2022;65:80-7
Author contact
![]() Corresponding authors: E-mail: yshiedu.cn (leading contact); E-mail: yueliuedu.cn.
Corresponding authors: E-mail: yshiedu.cn (leading contact); E-mail: yueliuedu.cn.