Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(4):1009-1023. doi:10.7150/ijbs.73570 This issue Cite
Research Paper
N6-Methyladenosine Modification of ANLN Enhances Hepatocellular Carcinoma Bone Metastasis
Hao Zheng1,2,3*, Zhang-Jun Cheng4*, Bo Liang5*, Zhen-Guang Wang1,2,3*, Yuan-Ping Tao2,3*, Sheng-Yu Huang6, Jun-sheng Ni1,2,3, Hui-Fen Li2,3, Le Yang2,3, Sheng-Xian Yuan1,2,3, Jennifer Wu7, Takumi Kawaguchi8, Hrishikesh Samant9, Wei-Ping Zhou1,2,3 ![]() , Dai-Min Xiang10
, Dai-Min Xiang10 ![]() , Yuan Yang1,2,3
, Yuan Yang1,2,3 ![]()
1. Third Department of Hepatic Surgery, Third Affiliated Hospital, Naval Medical University, Shanghai 200438, China.
2. Key Laboratory of Signaling Regulation and Targeting Therapy of Liver Cancer (SMMU), Ministry of Education, Shanghai 200438, China.
3. Department of Organization Sample Bank, Shanghai Key Laboratory of Hepatobiliary Tumor Biology (EHBH), Shanghai 200438, China.
4. Department of Hepato-Pancreato-Biliary Centers, Zhong Da Hospital, School of Medicine, Southeast University, Nanjing 210009, China.
5. Department of General Surgery, The Second Affiliated Hospital of Nanchang University, Nanchang, 330000, China.
6. Department of Hepatobiliary and Pancreatic Surgery, The 10th People's Hospital, Tongji University, Shanghai 200433, China.
7. Division of Hematology and Medical Oncology, Perlmutter Cancer Center, NYU Langone Health, New York, NY, USA.
8. Division of Gastroenterology, Department of Medicine, Kurume University School of Medicine, Kurume, Japan.
9. Hrishikesh Samant, Division of Gastroenterology and Hepatology, LSU Health Science Center, Shreveport, LA, USA.
10. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200032, China.
*Equal contributions to this work.
Received 2022-4-3; Accepted 2022-12-28; Published 2023-1-22
Abstract

Bones are categorized as the second most prevalent location of extra-hepatic metastasis in Hepatocellular Carcinoma (HCC), which is linked to an extremely poor prognosis due to limited therapeutic options. N6-methyladenosine (m6A) is a prominent modification involved in HCC, but the exact mechanisms on how m6A modifications induce HCC bone metastases (BM) remain unclear. The key modulators responsible for the abundant m6A RNA modification-induced HCC BM was found to be the METTL3 and YTHDF1. The expression of Anillin actin-binding protein (ANLN) was dramatically higher in HCC with BM tissues, and its messenger RNA (mRNA) stability was enhanced via m6A epitranscriptomic regulation by METTL3 and YTHDF1. High METTL3 and YTHDF1 expression along with nuclear ANLN protein was clinically correlated with BM in HCC patients. Furthermore, HCC BM was attributed to over-expression of nuclear ANLN forming a transcriptional complex with SP1 which enhanced KIF2C transcriptional activity to activate the mTORC1 pathway, therefore increased the expression of RANKL and disproportionated RANKL-OPG expression in bone microenvironment leading to malignant neoplasms invade bone tissue. In addition, inhibition of ANLN m6A modification by DZNeP attenuated HCC BM. This data provides meaningful understanding of the modulation and association of m6A epitranscriptomic-regulated BM in HCC, and moreover, defines potentially valuable therapeutic targets.
Keywords: hepatocellular carcinoma, bone metastasis, N6-methyladenosine, anillin actin binding protein, DZNeP
Background
Many cancer types exhibit preferential metastasis to the bone niche, which contributes significantly to both morbidity and mortality [1]. Bone metastasis (BM) has been observed in approximately 38.5 percent of individuals with extra-hepatic metastases in hepatocellular carcinoma (HCC) [2, 3], representing 11.7 percent of HCC patients who get curative resections [4]. HCC patients with BM also have intense pain, pathological fractures, and other nerve compression disorders [5], with an average survival time of 7.4 months [6]. Hence, gaining a better knowledge of the clinical and genetic processes driving HCC's metastatic growth to the bone is likely to facilitate improvements in patient prognosis and management.
N6-methyladenosine (m6A) methylation is discovered as the most common and ubiquitous modification in eukaryotic messenger RNAs (mRNAs) [7]. In mammalian cells, the m6A modification is dynamic and reversible, and it regulates mRNA stability, splicing, transport, localization, and translation, as well as RNA-protein interactions [8]. The m6A methyltransferases (also known as writers: METTL3, METTL14, and WTAP) install the m6A modifications and m6A demethylases (erasers: FTO and ALKBH5) remove them. Moreover, specific RNA-binding proteins (readers: YTHDF1/2/3, eIF3, IGF2BP1/2/3, and HNRNPA2B1) directly or indirectly bind to the m6A motif to influence RNA function [9]. From the perspective of cancer, multiple m6A regulators are reportedly dysregulated, functioning as oncogenes or tumor suppressors as per the context [10]. Cancer stem cell generation, epithelial-mesenchymal transition (EMT), cancer metabolism, and signal transduction through regulation of mRNA stability or protein translation of downstream effectors have all been linked to dysregulation of m6A modifications or m6A regulators [11]. The importance of m6A modification in HCC initiation and progression is being increasingly recognized with collective research efforts starting to clarify the complex functions played by m6A modifications and the dysregulation of m6A regulators [12, 13]. However, detailed investigations focusing on their role in HCC BM are still lacking.
Anillin actin-binding protein (ANLN) is a highly conserved actin binding protein playing a crucial part in cell mitosis and functions as a positive regulator of cell division and growth [14]. More recently, ANLN is found to be increasingly expressed in multiple malignancies and is strongly associated to tumor initiation and progression [15-20] and notably in HCC it is overexpressed and associated with poor prognosis [21]. Furthermore, knockdown of ANLN inhibits liver tumorigenesis and HCC growth [20, 22-24]. Nevertheless, the role of ANLN in HCC BM has remained obscure. The physiological function of m6A modification of ANLN in HCC BM was demonstrated in this study, and it was postulated that ANLN could be a new predictive biological marker and therapeutic target for HCC BM.
Materials and Methods
Clinical samples
The Institutional Review Board at Third Affiliated Hospital, Naval Medical University (Shanghai, China) approved this study, and it was executed in conformity with the Declaration of Helsinki (as revised in 2013). Before the trial began, the patients signed a written informed consent form. Supplementary Table 1 and 2 lists the clinical features of each HCC group.
Cell cultures
The HCC cell lines (Huh-7, HCCLM3, PLC, Hep3B, and Bel-7402) and human umbilical vein endothelial cells (HUVEC) cells and RAW264.7 were purchase from the American Type Culture Collection (ATCC; Manassas, VA, USA) and kept in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS; both Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Short tandem repeat (STR) sequencing indicated all cell lines were not contaminated by other cells such as HeLa (Biowing Applied Biotechnology Co., Ltd., Shanghai, China). The cells were kept in humidified incubators with 5% CO2 at 37 °C. For treatment, indicated HCC cells were stimulated with Wnt3a (2 ng/mL). As per the defined conditions, indicated cells were pre-treated with XAV939 (Wnt inhibitor, 10 μM) and INK-128 (mTORC1 inhibitor, 5 µM). The reagents mentioned above were all obtained from Sigma-Aldrich (St. Louis, MO, USA).
Animal models
All the mouse experiments which were performed according to the Naval Medical University's approved guidelines. The investigation complied with all applicable ethical regulations related to animal research. Using a 100 µl Hamilton Microliter syringe, all the groups of 1 × 107 luciferase-labeled Huh-7 cells in 50 µl 1 x phosphate-buffered saline (PBS) was injected intracardially in the left ventricle of nu/nu, female 4-6-week-old nude mice [25]. After injecting 4.0 mg luciferin (Gold Biotech, St. Louis, MO, USA) in 50 microliters of saline intraperitonially, the IVIS@ Lumina II system (Caliper Life Sciences, Hopkinton, MA, USA) was utilized for monitoring bone metastases for 10 minutes. The organs of the hindlimbs were excised and preserved for histological examination.
Statistical Analysis
Statistical analysis was performed in collaboration with the bioinformatics department. Each figure and figure legend shows the sample size (n), as well as the statistical test employed and the associated p-values. The data analysis did not exclude any samples or animals. GraphPad Prism Software (GraphPad Software, San Diego, CA, USA) and SPSS version 19.0 were employed for all statistical analyses (SPSS Inc., Chicago, IL, USA). P<0.05 was deemed significant. By using three independent biological repeats, all the quantitative experiments were repeated.
Other Methods
The material and methods are listed in detail in the Supplementary material.
Results
Abnormally increased m6A modification is associated with HCC BM
To determine if m6A modified RNAs were associated with BM, firstly m6A RNA levels in 6 samples of HCC with bone metastasis (BM), 13 HCC with metastasis (non-bone), and 10 advanced stage HCC without metastasis were compared via liquid chromatography-tandem mass spectrometry (LC-MS/MS). Interestingly, there was a significant increase in the overall m6A RNA levels in HCC patient samples with BM in comparison with samples with non-bone metastasis or no metastasis; while no significant differences were recorded between non-bone metastasis and no metastasis HCC cases (Fig. 1A and Supplementary Table 3). Then, by employing quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis, the mRNA levels of significant m6A writers (METTL3, METTL14, KIAA1429, and WTAP), erasers (ALKBH5 and FTO), and readers (YTHDF1, YTHDF2, and YTHDF3) were compared across all samples. We found that only the core methyltransferase writer METTL3 and reader YTHDF1 were considerably increased in HCC patient samples with BM in comparison with samples from non-bone metastasis or without metastasis (Fig. 1A and Supplementary Table 3). Again, no significant differences were found between non-bone metastasis and no metastasis cases. Then it was analyzed whether correlations existed between METTL3 and YTHDF1 protein expression and metastasis to different organs in 265 HCC cases. Notably, significantly higher expression of METTL3/YTHDF1 was found in HCC tumors with BM compared to HCC tumors without BM. In contrast, no differences were detected between HCC tumors with and without metastasis of the lung, lymph node, adrenal gland, or brain (Fig. 1B & C), therefore we focus METTL3 and YTHDF1 on HCC bone metastasis.
Abnormally increased METTL3 and YTHDF1-induced m6A modifications are associated with HCC BM. A. Total m6A levels of total mRNA (Upper panel), METTL3 (Middle panel) and YTHDF1(Down panel) transcript levels in 6 HCC with BM (BM HCC), 13 HCC with metastasis (non-bone), and 10 advanced stage HCC, as assessed using LC-MS/MS and qRT-PCR analysis. B. Representative IHC staining of METTL3 and YTHDF1 in matched HCC and adjacent normal tissues. Scale bars = 25 µm. C. IHC H-Scores from 265 HCC cases comparing the relative staining of METTL3 and YTHDF1. Of these 265 patients, 45 acquired BM, 53 had lung metastasis, 40 developed LNM, 10 suffered from adrenal gland metastasis, and 9 developed brain metastases. Note: In every eligible organ group, multiple organ metastases were included. D. Representative BLI images (Left panel), H&E-stained images of bone metastatic loci (Middle panel, scale bars, Magnification × 20), Osteoclast TRAP staining (Right panel, Magnification × 20) from each group (n =5 mice per group). M: marrow; B: bone; T: tumor. Note: H&E-stained and TRAP staining images from the same nude mouse. E. Osteoclast differentiation assays were co-cultured with conditioned media from METTL3/YTHDF1 knockdown and control HCC cells supplemented with M-CSF (50 ng/ml) and RANKL (100 ng/mL), magnification × 200. F. Concentration of RANKL in the culture medium measured by ELISA from METTL3/YTHDF1 knockdown and control HCC cell clones. G. Osteoclastogenesis differentiation-related factors mRNA level of RAW264.7 cells with conditioned medium from (G) was measured by qRT-PCR treated. H. The METTL3 overexpression and control HCC cells were infected with YTHDF1-KD or Control-KD. The in vitro osteoclast differentiation assays were performed, magnification × 200. I. Concentration of RANKL in the culture medium measured by ELISA from METTL3 overexpression and control HCC cells were infected with YTHDF1-KD or Control-KD HCC cell clones. J. Osteoclastogenesis differentiation-related factors mRNA level of RAW264.7 cells with conditioned medium from (J) was measured by qRT-PCR treated. For(A&C) Unpaired Student's t-test, (E-J) one-way ANOVA. Error bars shows mean ± SD derived from n = 3 independent experiments, *p < 0.05; **p < 0.01; ***p < 0.001, NS p>0.05. m6A, N6-methyladenosine; HCC BM, hepatocellular carcinoma bone metastasis; eHCC, early-stage HCC; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; LC-MS/MS, liquid chromatography tandem mass spectrometry; IHC, immunohistochemical; mRNA, messenger RNA; HUVECs, human umbilical vein endothelial cells; M-CSDF, macrophage colony-stimulating factor; BLI, bioluminescence imaging; H&E, hematoxylin and eosin; TRAP, tartrate resistant acid phosphatase positive; ANOVA, analysis of variance; SD, Standard Deviation; LNM, lymph node metastasis; NS, not significant.
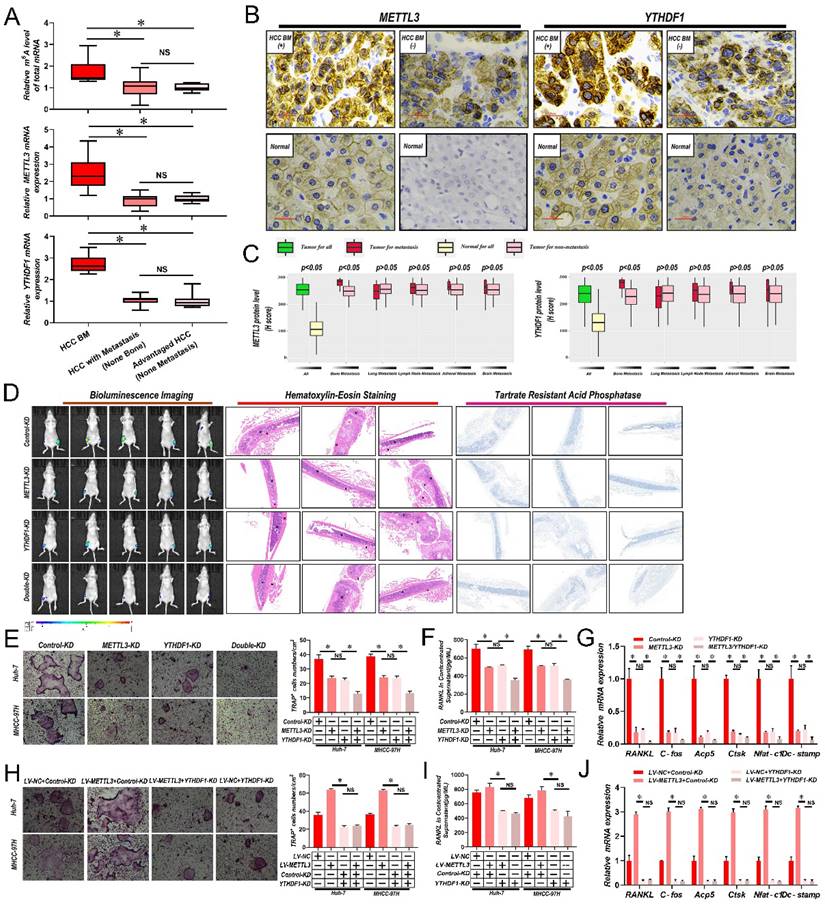
METTL3 and YTHDF1 depletion impairs HCC BM
Considerably greater levels of METTL3 and YTHDF1 were discovered, in cell lines with higher invasive and metastatic abilities, such as MHCC-97H, HCCLM3, and Huh-7, in comparison to cell lines with lesser metastatic potential, such as PLC, Hep3B, and Bel-7402 (Supplementary Fig. 1A). Then METTL3 and YTHDF1 were knocked down in MHCC-97H and Huh-7 HCC cells (Supplementary Fig. 1B and C). As shown in Fig. 1D, lower luciferase signals were detected in nude mice inoculated with METTL3/YTHDF1-KD Huh-7 cells compared with mice bearing control Huh-7 cells. Histological staining of tumor sections with hematoxylin and eosin (H&E) showed that the tumor lesion area in mouse limbs was greatly reduced by knockdown of METTL3 and YTHDF1 along with reduced tartrate resistant acid phosphatase (TRAP), indicative of clear decreases in osteoclast activity. Since osteoclastogenesis is recognized as essential for the establishment of BM, it was next examined how METTL3/YTHDF1 expression in HCC contributes to osteoclast differentiation. Indeed, fewer TRAP+ multinucleated cells were observed when bone marrow cells (BMCs) were cultured with conditioned medium from METTL3/YTHDF1-KD HCC cells compared with control cells (Fig. 1E). Moreover, the levels of RANKL which promote osteoclastogenesis decreased, while OPG which is a decoy receptor for RANKL that prevents osteoclastogenesis increased in conditioned medium after METTL3/YTHDF1 knockdown (Fig. 1F & Supplementary Fig. 1D). Additionally, culturing RAW246.7 cells with HCC-conditioned medium from METTL3/YTHDF1-KD HCC cells revealed significantly decreased expression of osteoclast differentiation/activation markers including RNAKL, C-fos, Acp5, Ctsk, Nfat-c1, and Dc-stamp (Fig. 1G and Supplementary Fig. 1E). Following, overexpression of METTL3 enhanced osteoclast differentiation; however; knockdown of YTHDF1 impaired these effects (Supplementary Fig. 1F-I and Fig. 1H-J). Lastly, supporting these findings, investigation of HCC in The Cancer Genome Atlas (TCGA) database using gene expression profiling interactive analysis (GEPIA) indicated that the expression of METTL3 and YTHDF1 were positively associated with several known factors related to osteoclastogenesis (Supplementary Fig. 1J). Collectively, these data suggested that the METTL3 and YTHDF1 function to promote HCC BM.
Hypermethylated m6A correlates with oncogene over-expression in HCC BM
To map the transcriptome-wide m6A modifications in HCC BM, m6A-seq was performed for 5 HCC primary focus (P-HCC) and 5 HCC BM focus (B-HCC) tissues. We found that the majority of the m6A sites (approximately 35%) were present in 3′ untranslated region (UTR) (Supplementary Fig. 2A), identifying 16,968-32,626 m6A sites in 8,618-12,309 mRNAs for each sample (Supplementary Fig. 2B). The m6A sites were enriched within the coding and 3'UTR sequence, and intriguingly, referentially accumulated in the CDS regions immediately downstream of translation initiation sites (Supplementary Fig. 2C & D). Consistent with previous reports [26], a proportion of the m6A sites conformed to the mammalian DRACH consensus motif (Supplementary Fig. 2E). Further analysis of our m6A-seq data revealed 6,681 differentially regulated m6A sites between P-HCC focus and B-HCC focus tissues (Fig. 2A). Among these, 5,994 m6A sites in 4,430 mRNAs were hypermethylated and 687 m6A sites in 547 mRNAs were hypomethylated in BM focus versus primary focus tissues (Supplementary Fig. 2F), indicating that m6A sites are frequently hypermethylated in HCC BM. The frequent m6A hypermethylation in BM HCC led us to categorize the function of the m6A-hypermethylated mRNAs. As shown in Fig. 2B and Supplementary Fig. 2G, the hypermethylated mRNAs were mainly enriched in several cancer-related pathways including the Wnt, Hippo, Notch, transforming growth factor-β (TGF-β), and nuclear factor-κß (NF-κß) pathway. Together, these data indicate that m6A hypermethylation is a frequent event in BM HCC and that the hypermethylated mRNAs are enriched in known oncogenic pathways.
METTL3 and YTHDF1 induces m6A modification of ANLN 3′-UTR to enhance its mRNA stability in HCC BM. A. Circos plot demonstrating m6A peaks on human chromosomes. The human genome's chromosome map is presented as the circos plot's outermost layer. Red or green bars (5 B-HCC vs. 5 P-HCC) indicate increased or decreased m6A peaks, respectively. All target m6A found by sequencing is shown by the second outermost circle. B. Heatmap showing the m6A hypermethylation tendency in B-HCC for 35 representative genes involved in key cancer-related pathways. The hypermethylated m6A sites were detected using the m6A-seq data for 5 B-HCC and 5 P-HCC. m6A-seq was performed once for each sample. Colored from blue to red to indicate low to high H-score values, respectively. C. ANLN m6A and transcript levels in 6 HCC with BM (BM HCC), 13 HCC with metastasis (non-bone), and 10 advanced stage HCC, as assessed using MeRIP-PCR and qRT-PCR analysis. D. qRT-PCR analysis of ANLN mRNA in Huh-7 and MHCC-97H cells after knockdown METTL3 or/and YTHDF1. E. The ANLN protein levels in Huh-7 and MHCC-97H cells infected with METTL3/YTHDF1-KD measured by western blotting. F. Coverage plots of m6A peaks in ANLN gene comparing matched B-HCC (n=5 independent biological samples) versus P-HCC (n=5 independent biological samples) by m6A-seq. Plotted coverages are the median of the n replicates presented. Eight DRACH motif sites were identified in ANLN 3′-UTR. The inset presents bp 242-696-bp of ANLN 3′-UTR region, with the DRACH motif highlighted by red text. G. METTL3-mediated ANLN m6A modifications were demonstrated using MeRIP-qPCR analysis. The m6A modification of ANLN was enhanced when METTL3 was up-regulated, but it was reduced when METTL3 was knocked out. H. qRT-PCR analysis of ANLN mRNA in Huh-7 and MHCC-97H cells infected with/without METTL3/YTHDF1-KD following Actinomycin D treatment for 0-24 h. I. ANLN mRNA was quantified by qRT-PCR as the percentage of input and graphed as fold enrichment relative to control. Immunoblot analysis of FLAG-YTHDF1 in the input and IP is shown on bottom panels. For (G, I) Paired Student's t-test, (C&D) one-way ANOVA, (H) Repeated-measures analysis of variance analysis. Error bars indicate mean ± SD were derived from n = 3 independent experiments, *p < 0.05; **p < 0.01; ***p < 0.001, NS p > 0.05. m6A, N6-methyladenosine; HCC BM, hepatocellular carcinoma bone metastasis; UTR, untranslated region; B-HCC, HCC BM focus; P-HCC, HCC primary focus; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; mRNA, messenger RNA; MeRIP-qPCR, methylated RNA immunoprecipitation-qPCR; ANOVA, analysis of variance; SD, Standard Deviation
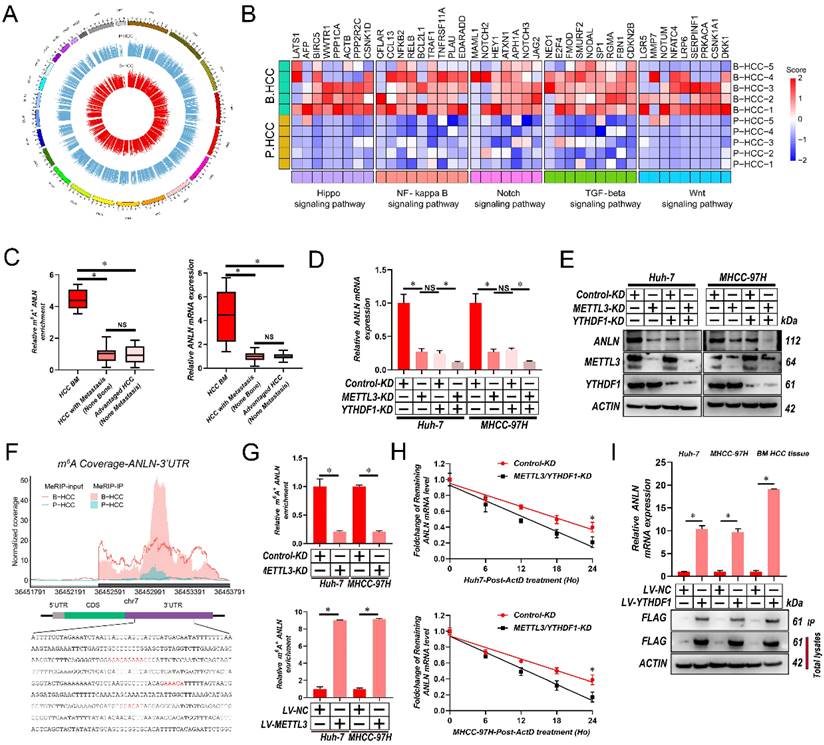
To better establish the association between m6A modifications and gene expression in HCC BM, we performed RNA sequencing (RNA-Seq) on the same cohort of 5 P-HCC and 5 B-HCC tissues. Analysis of these data identified 1,665 up- and 1,317 down-regulated mRNAs in B-HCC compared to P-HCC tissues (Supplementary Fig. 2H). Intersecting the m6A-Seq and RNA-Seq further revealed that expression and methylation of 68 mRNAs (up-regulated: Fold-Change>16, p-value<0.0001; and hypermethylated: Fold-Change>10, p-value<0.05) were positively correlated (Supplementary Table 4). Further interrogation of these 68 genes via MeRIP-qPCR and qRT-PCR assays in cohort (Fig. 1A) highlighted ANLN displayed strong correlations between the extent of m6A modification, mRNA and HCC BM (Fig. 2C and Supplementary Table 5). These data suggest that the m6A RNA modification enhances the mRNA expression of ANLN associated with HCC BM.
METTL3 and YTHDF1 Induces m6A Modification of ANLN 3'-UTR to Enhance its mRNA stability
Next, we validated the mRNA and protein levels ANLN were reduced by METTL3/YTHDF1 knockdown in HCC cells (Fig. 2D and E). Our m6A-seq results revealed that 4 DRACH motifs and m6A peaks within the 244-800-bp region of the 3′-UTR of ANLN were selectively increased in HCC BM focus tissues (Fig. 2F). In further analyses, MeRIP-qPCR assays were utilized for validating the m6A modification of ANLN mRNA in HCC cells and for confirming the function of METTL3 in the m6A modification of ANLN mRNA (Fig. 2G). Taking into consideration the positive regulation of mRNA levels of ANLN by m6A modification, it was investigated then, if the m6A modification influences the stability of ANLN mRNA. The HCC cells were treated with actinomycin D, an inhibitor of transcription, for the indicated times. As shown in Fig. 2H, knockdown of METTL3 and YTHDF1 reduced the half-life of ANLN mRNA in HCC cells. Moreover, in HCC cells and B-HCC tissue, an RNA immunoprecipitation (RIP) assay suggests that YTHDF1 recognizes and binds with the ANLN mRNA (Fig. 2I). Notably, examination of ANLN in HCC using GEPIA database indicated ANLN expression was significantly associated with the METTL3 and YTHDF1 expression (Supplementary Fig. 2I). Collectively the findings suggest that ANLN expression is maintained by METTL3-mediated m6A modification via YTHDF1-dependent ANLN mRNA stability.
High METTL3, YTHDF1 expression and nuclear ANLN levels are associated with poor survival and BM in HCC
ANLN is a highly dynamic protein, with notable variations in its expression and localization during various phases of the cell cycle [27]. Consequently, the nuclear and cytoplasmic distribution of ANLN in 265 HCC cases by immunohistochemical (IHC) analysis was evaluated, respectively. As shown in Supplementary Fig. 3A and B, both the nuclear and cytoplasmic staining of ANLN were increased in 265 HCC tissues in comparison to the neighboring normal tissues. Whether correlations existed between ANLN nuclear/cytoplasmic staining and metastasis to different organs was then analyzed. As expected, higher nuclear ANLN protein expression was found in HCC tumors with BM compared to HCC tumors without BM although no correlations existed with metastases to other sites including lung, lymph node, adrenal gland, and brain. Moreover, higher cytoplasmic stain for ANLN was not correlated with metastasis to different organs including bone, lung, lymph node, adrenal gland, and brain (Supplementary Fig. 3B). Furthermore, Kaplan-Meier survival curves presented that the HCC cases with elevated METTL3, YTHDF1, and nuclear ANLN expression had poorer overall survival (OS) and recurrence-free survival (RFS), respectively (Supplementary Fig. 3C-E). In contrast, cytoplasm ANLN staining exhibited no correlation with survival in 265 HCC cases (Supplementary Fig. 3F). Altogether, these outcomes suggest that a direct correlation of elevated METTL3, YTHDF1, and nucleus ANLN expression is associated with BM and low survival probability in HCC.
The METTL3 and YTHDF1-ANLN axis promotes HCC BM
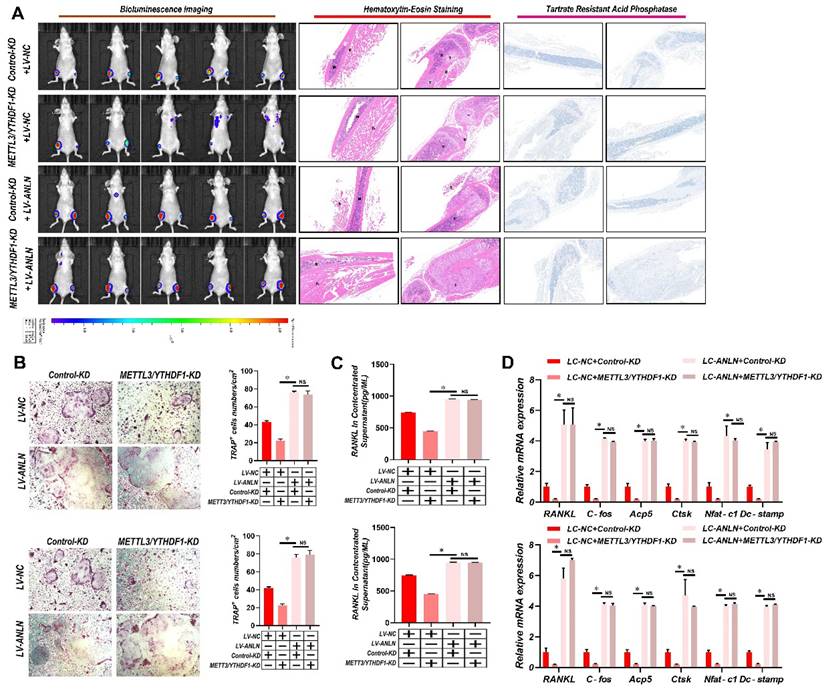
To learn more about ANLN's oncogenic function in HCC, further comparisons were made between ANLN knockdown and overexpression HCC cells (Supplementary Fig. 4A-D). Notably, knockdown of ANLN markedly suppressed HCC BM in vivo and osteoclast differentiation activities (Supplementary Fig. 4E & F and Supplementary Fig. 4H-J). Conversely, ectopic ANLN expression significantly enhanced cell osteoclast differentiation (Supplementary Fig. 4G and Supplementary Fig. 4K-M). Furthermore, the reintroduction of ANLN in METTL3/YTHDF1 knockdown HCC cells restored BM capabilities in vivo (Fig. 3A) along with osteoclast differentiation (Fig. 3B-D). Taken together, these findings indicated that METTL3 and YTHDF1 promote HCC BM through the upregulation of ANLN expression.
METTL3 and YTHDF1 -ANLN Axis Promotes HCC BM. A. Representative BLI images (Left panel), H&E-stained images of BM loci (Middle panel, Magnification × 20), Osteoclast TRAP staining (Right panel, Magnification ×20) from each group (n = 5 mice per group). M: marrow; B: bone; T: tumor. Note: H&E-stained and TRAP staining images from the same nude mouse. B. The METTL3/YTHDF1-KD and control HCC cells were infected with LV-NC or LV-ANLN. The in vitro migration osteoclast differentiation assays were performed, magnification × 200. C. Concentration of RANKL in the culture medium measured by ELISA from METTL3/YTHDF1-KD and control HCC cells clones. D. Osteoclastogenesis differentiation-related factors mRNA level of RAW264.7 cells with conditioned medium from (B) was measured by qRT-PCR treated. For (B-D) one-way ANOVA. Error bars indicate mean ± SD were derived from n = 3 independent experiments, *p < 0.05; **p < 0.01; ***p < 0.001, NS p > 0.05. HCC BM, hepatocellular carcinoma bone metastasis; BLI, bioluminescence imaging; H&E, hematoxylin and eosin; TRAP, tartrate resistant acid phosphatase positive; ANOVA, analysis of variance; SD, Standard Deviation; NS, not significant.
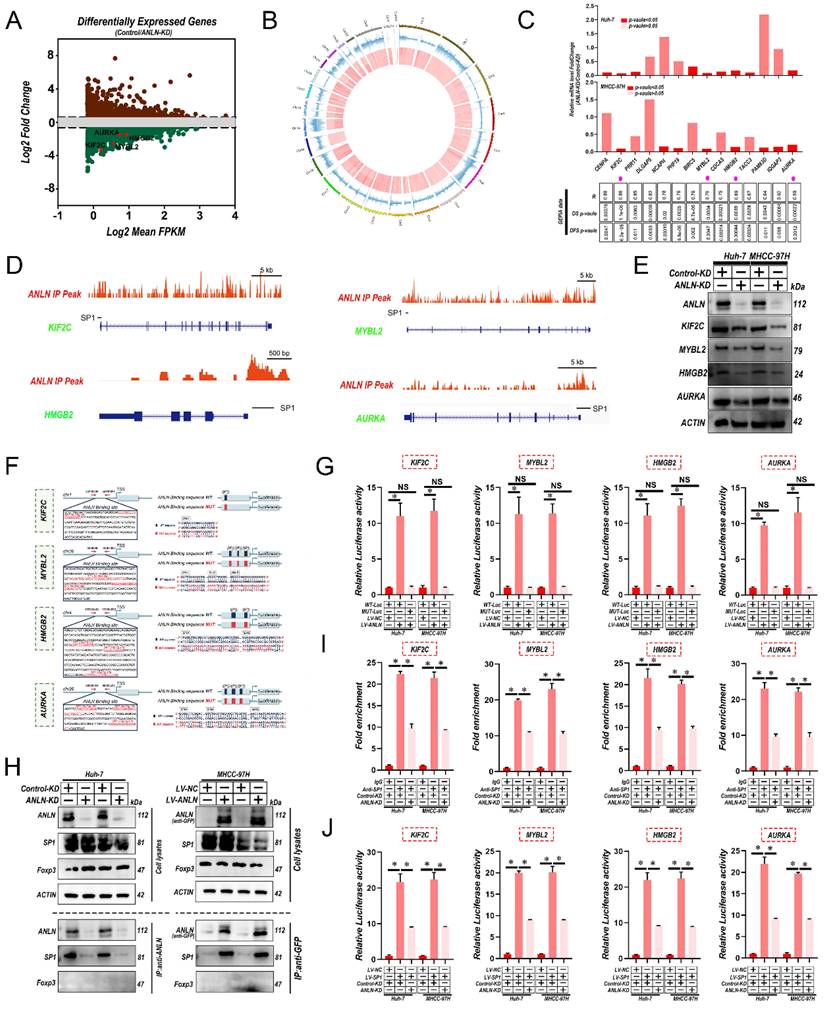
Interactions between ANLN and SP1 are indispensable for SP1-mediated oncogenic transcription
To further identify the potential substrates of ANLN, firstly RNA sequencing was utilized for comparing the expression profiles between Huh-7-control and ANLN-KD cells. Among ANLN-regulated genes, total 578 genes were greatly up-regulated, and 1,012 genes were down-regulated in Huh-7-ANLN-KD cells (Fig. 4A). As mentioned above, the nuclear localization of ANLN was associated with HCC BM. Combined with ANLN as an essential regulator of nucleoli biogenesis [28], a Cleavage Under Targets and Tagmentation (CUT&Tag) assessment on ANLN was conducted to specify its targets. Global mapping analysis demonstrated that the reads were most distributed amongst promoter regions (~39.23%) (Fig. 4B and Supplementary Fig. 5A & B), implying that ANLN may have a new role in regulating gene transcription in HCC. Combinatorial analysis of CUT&Tag versus RNA-seq data revealed that 238 of the differentially up-regulated genes were regulated by ANLN and exhibited ANLN binding at their respective promoters (Supplementary Fig. 5C). Thereafter, 14 genes showing upregulated expression levels in HCC (fold-change>2, p-value <0.01) and positive correlations with ANLN (r > 0.55, p-value <0.05) in the GEPIA database were chosen for further study (Fig. 4C & D, Supplementary Fig. 5D & E and Supplementary Table 6 & 7). Subsequent qRT-PCR analysis confirmed that 4 genes: KIF2C, MYBL2, HMGB2, and AURKA, were all downregulated after knockdown of ANLN in HCC cells (Fig. 4C, upper panel). Western blot analysis revealed their protein levels were also downregulated (Fig. 4E). Consistently, IHC analysis of the 265 HCC cases suggested that protein levels of KIF2C, MYBL2, HMGB2, and AURKA were positively correlated with the levels of nuclear but not cytoplasmic ANLN levels (Supplementary Fig. 5F & G and Supplementary Table 8). Instructively, activation of the KIF2C, MYBL2, HMGB2, and AURKA promoters was enhanced by ANLN overexpression in HCC cells (Fig. 4F & G) while chromatin immunoprecipitation (ChIP)-PCR assays further confirmed the direct binding of ANLN to their respective promoter regions (Supplementary Fig. 5H). It was recently reported that ANLN binds to particular transcription factors (TFs) in the nuclear context [29] so the potential functional collaborations between ANLN and TFs in HCC was investigated. Immunoprecipitation-mass spectrometry (IP-MS) analysis identified that ANLN bound to the well-known transcription factor SP1, and this interaction was further confirmed in co-immunoprecipitation (co-IP) assays (Fig. 4H). Consistently, interrogation of the JASPAR database (http://jaspar.genereg.net/) indicated that several SP1 binding sites were located within the ANLN-CUT-Tag peak of the KIF2C, MYBL2, HMGB2, and AURKA promoters (Fig. 4D & F). In addition, the mutation of SP1 binding site within the KIF2C, MYBL2, HMGB2, and AURKA promoter regions diminished the distinct activation differences in the KIF2C, MYBL2, HMGB2, and AURKA promoters between ANLN overexpression and control cells (Fig. 4G). Consistently, the ability of SP1 to bind to the promoter regions of KIF12C, MYBL2, HMGB2, and AURKA was significantly reduced after ANLN knockdown (Fig. 4I & J and Supplementary Fig. 5I). Lastly, analysis of HCC samples in the GEPIA database demonstrated that SP1 was positively correlated with KIF2C, MYBL2, HMGB2, and AURKA in HCC (Supplementary Fig. 5J). These results confirm that nuclear ANLN protein cooperatively combines with SP1 to enhance KIF2C, MYBL2, HMGB2, and AURKA expression in HCC.
Interaction between ANLN and SP1 is indispensable for SP1-mediated Oncogenic transcription. A. MA plot showing the mean log2expression against the log2-fold change of the RNA-seq data obtained from control and ANLN-KD Huh-7 cell lines. Green and brown depicts decreased and increased gene expression in the ANLN-KD group, respectively. B. Circos plot showing ANLN-binding peaks on human chromosomes. The outermost layer of the circos plot is a chromosome map of the human genome. The second outermost circle represents all target ANLN-binding peaks detected by sequencing. C. qRT-PCR confirmed that the mRNA level of 14 genes with the highest correlation with ANLN in HCC (base on GEPIA database) with ANLN-KD. The data in the grid below are R (coefficient of correlation, Pearson test); DFS value (disease-free survival, log-rank test); OS value (Overall survival, the log-rank test). D. Genome browser screenshot ANLN peaks (Red, based on CUT-Tag seq) at the KIF2C, MYBL2, HMGB2 and AURKA gene prompter loci, respectively. The thick black line is the predicted binding site of SP1 (Transcription Factor SP1 ChIP-seq Clusters from ENCODE 3 Source data version: ENCODE 3 Nov 2018). E. Western blot analysis of the level of KIF2C, MYBL2, HMGB2, and AURKA protein level in ANLN knockdown and control HCC cells. F. Schematic representation of the ANLN binding region of KIF2C, MYBL2, HMGB2, and AURKA, respectively. Luciferase reporter plasmids that included the WT or MUT (ANLN binding site sequence of KIF2C, MYBL2, HMGB2, and AURKA) were constructed. G. Luciferase activity in stable HCC cell clones transfected with luciferase reporters containing WT-ANLN-binding site or MUT-ANLN-binding site. Data are presented as the relative ratio of firefly luciferase activity to Renilla luciferase activity. H. The indicated HCC cell lysates were prepared and immunoprecipitated (IP) with anti-GFP (for GFP-tagged ANLN), or anti-ANLN (for endogenous ANLN) antibodies. IP and cell lysates were analyzed by western blotting. Foxp3 served as negative control. I. HCC cells (control-KD or ANLN-KD) were collected and subjected to ChIP assay using anti-SP1 antibody, followed by qPCR analysis. Graphs show ChIP-qPCR measurement of SP1 enrichment at the KIF2C, MYBL2, HMGB2, and AURKA promoter regions. J. Luciferase activity in stable HCC cell clones co-transfected with luciferase reporters containing WT-ANLN-binding site and ANLN-KD. Data are presented as the relative ratio of firefly luciferase activity to Renilla luciferase activity. For (C) Paired Student's t-test, (G, I and J) one-way ANOVA. Error bars indicate mean ± SD were derived from n = 3 independent experiments, *p < 0.05; **p < 0.01; ***p < 0.001, NS p > 0.05. HCC BM, hepatocellular carcinoma bone metastasis; BLI, bioluminescence imaging; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; ChIP, chromatin immunoprecipitation; SEM, standard error of the mean; ANOVA, analysis of variance; SD, Standard Deviation; NS, not significant.
ANLN facilitates HCC BM through KIF2C/mTORC1 signaling
Previous studies have indicated that KIF2C enhanced mammalian target of rapamycin complex1 (mTORC1) signal transduction in HCC [30], while recent studies have reported that activation of mTORC1 signaling promotes osteoclast formation [31, 32].
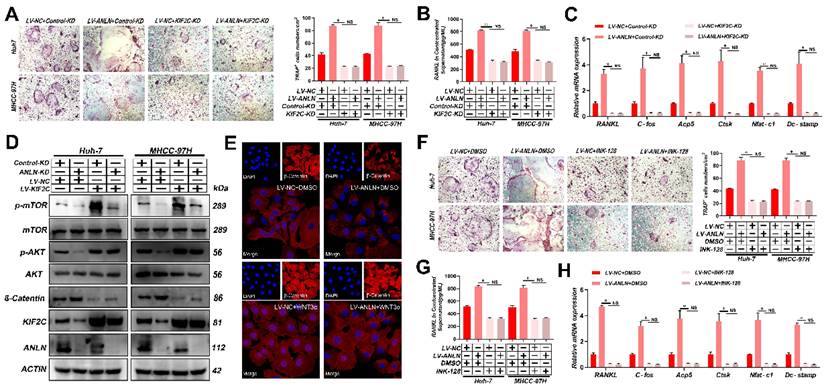
Notably, knockdown of KIF2C in HCC cells significantly reduced osteoclast differentiation activity promoted by ectopic ANLN expression (Fig. 5A-C and Supplementary Fig. 6A-E). Thus, it was further explored whether the ANLN-KIF2C axis enhances HCC BM via mTORC1 signaling. Indeed, mTOR phosphorylation was downregulated in ANLN-KD HCC cells and this could be partially recovered by ectopic KIF2C (Fig. 5D), indicating that the ANLN-KIF2C axis likely activates mTORC1 signaling. Activation of mTORC1 promotes osteoclast formation by regulating RANKL/OPG expression via activating the Akt pathway and negatively regulating β-catenin [31, 32]. Consistently, decreased phosphorylation of AKT and increased β-catenin were observed in ANLN-KD HCC cells that could be partially recovered by ectopic expression KIF2C (Fig. 5D). Moreover, subcellular fraction and immunofluorescence analysis indicated that overexpression of ANLN impaired the nuclear accumulation of β-catenin (Fig. 5E and Supplementary Fig. 6F). Furthermore, the specific mTORC1 inhibitor INK-128 abolished the osteoclast differentiation ability differences measured between ANLN overexpression and control HCC cells (Fig. 5F-H and Supplementary Fig. 6G & H). Importantly, KIF2C depletion or INK-128 treatment both abrogated the ANLN-enhanced BM of HCC cells in vivo (Fig. 5I). Lastly, analysis of patient-derived HCC tissues indicated that ANLN, KIF2C and mTORC1 pathway-related genes were increased in BM-HCC tissues compared to non-BM-HCC tissues, while p-AKT and β-catenin pathway related genes were decreased (Fig. 5J). Together, these outcomes suggest that the KIF2C/mTORC1 pathway is required for ANLN-enhanced HCC BM.
ANLN Facilitates HCC BM Through KIF2C/mTORC1 Signaling. A. Osteoclast differentiation assays were co-cultured with conditioned media from ANLN overexpression and control HCC cells were infected with KIF2C knockdown group supplemented with M-CSF (25 ng/mL) and RANKL (100 ng/ml), magnification × 200. B. Concentration of RANKL in the culture medium measured by ELISA from (A). C. Osteoclastogenesis differentiation-related factors mRNA level of RAW264.7 cells with conditioned medium from (A) was measured by qRT-PCR treated. D. Western blot analysis of the level of ANLN, KIF2C, and the phosphorylation of mTOR protein in indicated treated cells. E. Immunofluorescence staining for nuclear β-catenin after applying the LV-NC or LV-ANLN in MHCC-97H cells with or without Wnt3a treatment (2 ng/mL) for 8 h. β-Catenin is labelled in red. F. ANLN overexpression and control HCC cells were treated with or without INK-128, in vitro osteoclast differentiation assays supplemented with M-CSF (25 ng/mL) and RANKL (100 ng/ml), magnification × 200. G. Concentration of RANKL in the culture medium measured by ELISA from (F). H. Osteoclastogenesis differentiation-related factors mRNA level of RAW264.7 cells with conditioned medium from (F) was measured by qRT-PCR treated. I. Representative BLI images (Left panel), H&E-stained images of bone metastatic loci (Middle panel, scale bars, 500 µm), Osteoclast TRAP staining (Right panel, scale bars, 500 µm) from each group (n =3 mice per group). M: marrow; B: bone; T: tumor. J. Western blot analysis of the level of indicated protein level in BM-HCC (n=4) and no-BM-HCC (n=4) group. For (A-C, F-H) one-way ANOVA. Error bars indicate mean ± SD were derived from n = 3 independent experiments, *p < 0.05; **p < 0.01; ***p < 0.001, NS p>0.05. HCC BM, hepatocellular carcinoma bone metastasis; mRNA, messenger RNA; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; HUVEC, human umbilical vein endothelial cell; BLI, bioluminescence imaging; TRAP, tartrate resistant acid phosphatase positive; H&E, hematoxylin and eosin; ANOVA, analysis of variance; SEM, standard error of the mean; SD, Standard Deviation; NS, not significant.
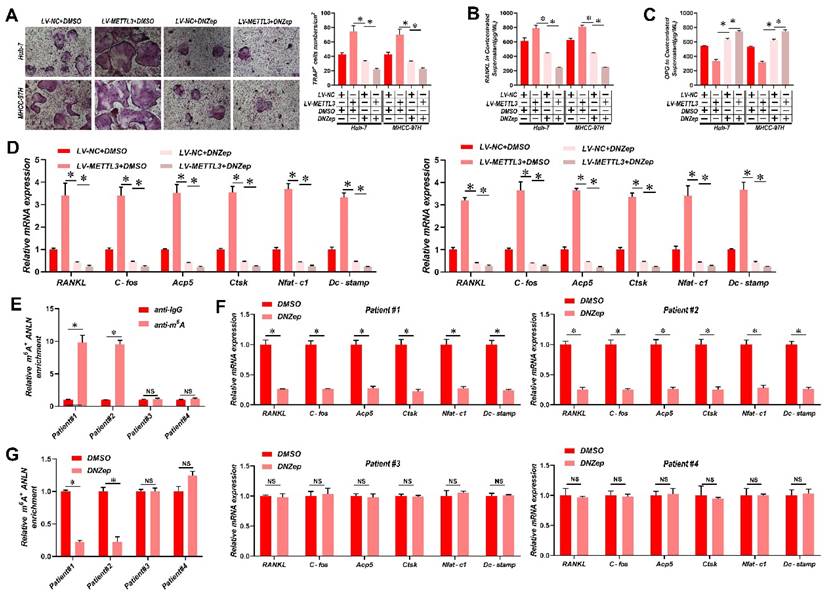
Suppression of ANLN m6A modification attenuates HCC BM
It has been reported that 3-deazaneplanocin A (DZNeP) inhibits RNA methylation [33]. DZNeP significantly reduced overall mRNA m6A methylation and the m6A modification of ANLN mRNA in HCC cells, as revealed in Supplementary Fig. 7A & B. Moreover, both the ANLN mRNA and protein levels were down-regulated in DZNeP-treated HCC cells (Supplementary Fig. 7C & D). In addition, DZNeP treatment abolished m6A hypermethylated ANLN and increased ANLN mRNA levels in HCC cells with LV-METTL3 (Supplementary Fig. 7E & F), also diminishing the differences in osteoclast differentiation and BM in vivo between control and METTL3-overexpressing HCC cells (Fig. 6A-E). A further observation worth noting is that DNZep treatment also inhibited HCC proliferation (Supplementary Fig. 7G). HCC patient-derived tumor xenograft (PDX) models were also established from four HCC patient tissues, two with high and two with low ANLN m6A modifications, respectively, and employed these to assess the clinical potential of using DZNeP to target m6A modified ANLN (Fig. 6F). These results showed that DZNeP treatment significantly suppressed m6A modified ANLN mRNA levels and also mRNAs of genes associated with osteoclast differentiation (Fig. 6G and H). Furthermore, DZNeP treatment significantly inhibited the tumor growth of the high ANLN m6A-modified HCC PDX models, while these effects were not evident in the two low ANLN m6A-modified HCC PDX lines (Supplementary Fig. 7H). Assessing the in vivo cytotoxicity of these treatments, the vital organs were collected after DZNep treatment but found no obvious side effects (Supplementary Fig. 7I). Altogether these outcomes revealed that antagonizing increased levels of m6A modified ANLN in HCC significantly inhibits BM.
Suppression of ANLN m6A modification attenuates HCC BM. A. Osteoclast differentiation assays were co-cultured with conditioned media from Huh-7 and MHCC-97H cell lines infected with LV-METTL3 or LV-NC were treated with vehicle (DMSO) or DNZep (10nM) supplemented with M-CSF (25 ng/mL) and RANKL (100 ng/ml), magnification × 200. B&C. Concentration of RANKL (B) and OPG (C) in the culture medium measured by ELISA from Huh-7 and MHCC-97H cell lines infected with LV-METTL3 or LV-NC were treated with vehicle (DMSO) or DNZep (10nM). D. Osteoclastogenesis differentiation-related factors mRNA level of RAW264.7 cells with conditioned medium from (A) was measured by qRT-PCR treated. E. MeRIP-qPCR analysis was employed to assessment the ANLN m6A modifications in the HCC tissues from patient#1-4. F. MeRIP-qPCR analysis was employed to assessment the ANLN m6A modifications from PDX model treated with Vehicle or DZNep (8 mg/kg per mouse). G. Osteoclastogenesis differentiation-related factors mRNA level was measured by qRT-PCR from PDX model treated with Vehicle or DZNep (8 mg/kg per mouse). For (A - D) one-way ANOVA, (E - G) Paired Student's t-test. Error bars indicate mean ± SD were derived from n = 3 independent experiments, *p < 0.05; **p < 0.01; ***p < 0.001, NS p > 0.05. m6A, N6-methyladenosine; HCC BM, hepatocellular carcinoma bone metastasis; BLI, bioluminescence imaging; H&E, hematoxylin and eosin; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; MeRIP-qPCR, methylated RNA immunoprecipitation-qPCR; mRNA, messenger RNA; TRAP, tartrate resistant acid phosphatase positive; DMSO, dimethylsulfoxide; PDX, patient-derived xenograft ANOVA, analysis of variance; SD, Standard Deviation; NS, not significant.
Discussion
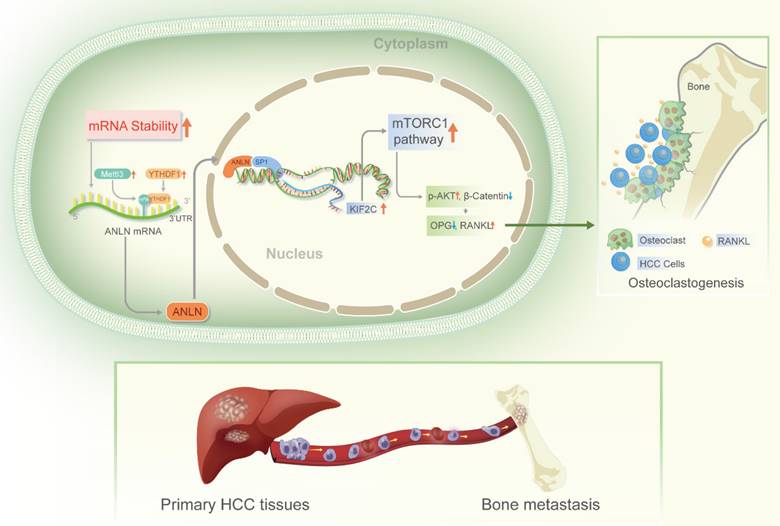
In advanced malignancies, BM is a common occurrence, and once established, patient prognosis is extremely poor [34]. However, the understanding of molecular mechanisms in BM from HCC is limited. The m6A is the most common RNA modification in both mRNA and non-coding RNA [9], and is involved in various human malignant tumors, including HCC [13]. According to Wen et al. [35] Long non-coding RNA NEAT1 increases prostate cancer bone metastases via N6-methyladenosine. Nevertheless, links between m6A RNA modifications and HCC BM are currently understudied. Here, the m6A levels of mRNA were demonstrated to be greatly increased in HCC BM tissues because of the upregulation of the methyltransferase METTL3 and reader YTHDF1. Mechanistically, it was deduced that upregulation of METTL3 stimulates m6A modified ANLN mRNA levels, with YTHDF1 directly binding to m6A sites in ANLN mRNA to maintain ANLN mRNA stability. Nuclear ANLN/SP1 complexes to the KIF2C promoters serve to promote the development of HCC BM through activating mTORC1 pathways (Fig. 7).
Schematic illustration of the METTL3 and YTHDF1 enhanced ANLN/KIF2C/mTRORC1 axis in driving HCC BM. Abbreviations: HCC BM, hepatocellular carcinoma bone metastasis.
A growing body of evidence suggests that dysregulation of m6A modification is linked to tumor metastasis [11]. Among the m6A modulators, the m6A writer METTL3 or reader YTHDF1 have been reported to either facilitate or impair carcinogenesis in different cancer types [36]. However, detailed investigations about their role in site-specific metastases to bone, especially in HCC, remains unclear. In the present research, it was found for the first time that METTL3 and YTHDF1 induced increased m6A modifications associated with HCC BM. Furthermore, METTL3 and YTHDF1 overexpression in HCC cells were required for the BM process, thus highlighting METTL3 and YTHDF1 as potential predictive biomarkers and therapeutic targets for HCC. By employing RNA-seq and m6A-seq, promising findings were discovered related to ANLN being a significant downstream target of METTL3 and YTHDF1 associated with HCC BM. Earlier research has revealed that ANLN exhibits an important part in a variety of cancer related mechanisms, which includes cancer initiation, growth, angiogenesis, and metastasis [14]. In HCC it was reported that ANLN promotes hepatic carcinogenesis as well as cancer cell proliferation [20, 22-24]. Regardless of these reported results, the function of ANLN in HCC BM remains unclear. ANLN expression and localization have been shown to be dynamic [27] and ANLN is primarily nuclear in cancer cells, with only a weak cytoplasmic staining observed, as reported by most of the published clinical investigations [14]. Consistently high nuclear expression of ANLN was found in HCC, also finding this distribution acts as an important prognostic factor for poor survival. Instructively, the increased nuclear expression of ANLN was shown to be associated with HCC BM, for the first time. While the activities of nuclear ANLN in tumorigenesis have been unexpectedly overlooked, the nuclear localization of ANLN in many cancer cells has been thoroughly described. Through CUT & Tag and RNA-seq, KIF2C was identified as a key target gene of ANLN. Furthermore, IP-MS analysis showed that nucleus ANLN and SP1 enhance KIF2C transcriptional activity via formation of transcription complex to enhance HCC BM via the mTORC1 pathway. The identification of this functional mechanism enriches the understanding of how HCC BM is promoted while also providing a new direction for the clinical diagnosis and treatment of HCC.
Bone metastasis is a complicated process involving the interaction among the tumor cells and the bone milieu. Elucidating the constitution of the bone milieu and how it interacts with tumor cells assists in understanding the potential mechanism of metastatic organ tendency. The bone microenvironment of RANKL/OPG ratio is a key in regulating osteoclast formation and bone resorption [37,38]. Increasing of RANKL and/or decreasing of OPG are sufficient to encourage the generation of osteoclasts and trigger the bone dissolution process [38]. RANK is one of the surface receptors of tumor necrosis factor (TNF) family [38]. This receptor modulates calcium metabolism and is required for the development, activation, and functionality of osteoclasts [37]. Recent research has found that RANK is expressed in tumor cells, insinuating its role in tumor metastasis [37]. The level of formation of osteoclasts is largely determined by the balance of RANKL and OPG activities, with elevated levels of OPG leading to a reduction in bone resorption [37]. OPG is the soluble receptor of RANKL and belongs to the small integrin binding ligand N-linked glycoprotein family [37,38]. OPG competes with RANK for RANKL in this manner, affecting osteoclast generation and ensuing bone resorption by preventing RANKL-RANK communication on the osteoclast membrane. Mammalian rapamycin target (mTOR) is considered to be a key cellular factor regulating most basic cellular functions [39]. In addition, mTOR has a crucial function in determining intraosseous homeostasis. It is reported that mTOR in osteoclasts regulates osteoclast differentiation, while rapamycin mTOR signal in osteoclasts can eliminate osteoclasts through RANKL/OPG pathway [39]. These results showed that important participants of m6A modifications (METTL3 and YTHDF1) enhance the stability of ANLN mRNA through m6A epitope transcriptional regulation, in turn with nuclear ANLN forming a transcriptional complex with SP1, which enhances the transcriptional activity of KIF2C, and activates the mTORC1 pathway. This increases the expression of RANKL and breaks the balance of RANKL-OPG expression in bone microenvironment leading to malignant neoplasms invade bone tissue. This study explored the relationship between m6A methylation and the microenvironment of hepatocellular carcinoma, enriched the understanding of the role of abnormal m6A methylation in promoting bone metastasis of hepatocellular carcinoma, and provided new ideas for the clinical diagnosis and treatment of bone metastasis of advanced hepatocellular carcinoma.
The bone marrow has a diverse composition of immune cells and may provide an immune-privileged habitat for disseminated tumor cells [40]. Bone metastasis involves many different immune cells [41] and takes place in an individual immunological environment. It is thought that m6A methylation influences the control of the tumor immune milieu, impacting the metastatic processes of a variety of malignancies [41]. m6A regulates the differentiation of T cells, the stabilization of Tregs, the maturation of Dendritic Cells (DCs), the polarization of macrophages and the functional modulation of myeloid-derived suppressor cells (MDSCs) [41]. m6A modulators are thus involved in tumor immunity and relevant to immunotherapy. Abnormal expression of m6A regulators has the potential to influence anticancer immune functions and modulate tumor metastasis [42]. The current research focused on the relationship between the m6A-ANLN axis and the equilibrium of RANKL-OPG expression in the bone microenvironment, immune microenvironment in bone metastasis not be accommodated and should be investigated in greater depth in the future.
Currently, effective therapeutic targets for HCC patients with BM are still not available [34]. Therefore, identifying novel therapeutic targets towards improving for HCC patient survival remains an unmet need. The ex vivo analysis of patient tissues revealed that BM HCC cases with high m6A modifications of ANLN mRNA, highlighting the potential therapeutic value of targeting the m6A modification in ANLN in HCC. Indeed, these findings using the RNA methylation inhibitor DZNeP [33], showed that DZNeP inhibits ANLN m6A methylation and effectively attenuates the malignant phenotype and BM in vivo of HCC cells exhibiting ANLN overexpression. The correlation between ANLN m6A methylation levels and DZNeP responses using PDX models was further validated. The results showed that PDXs with high m6A ANLN methylation level displayed significant growth inhibition upon DZNeP treatment, while PDXs with low m6A ANLN methylation level expression did not respond to DZNeP, implying that employing DZNeP to inhibit the m6A-ANLN axis may be a promising therapeutic option for HCC BM patients.
Conclusion
In summary, this work uncovered the functions of the m6A-ANLN axis induced by METTL3/YTHDF1 in HCC BM, and provided compelling evidence that multiple phases of the HCC BM cascade require m6A-ANLN upregulation. Furthermore, these findings provide understanding of the modulation and linkage of the m6A epitranscriptome in regulating BM in the HCC-specific context, also proposing new therapeutic targets for bone metastasis. Finally, the limitations of this study must be acknowledged. The sample size of the cohort needs to be increased, especially the patient subgroups with specific organ metastases. Future studies would benefit from expanding sample sizes in a multicenter environment to establish a clearer relationship between the expression of METTL3, YTHDF1, ANLN and bone metastasis. In addition, as with all experimental models, this in vivo bone metastasis model may not fully reflect the disease in patients, so further verification with other models would be beneficial to substantiate both the phenotype and molecular mechanisms of the m6A-METTL3/YTHDF1-ANLN axis in driving HCC BM.
Abbreviations
m6A: N6-methyladenosine; HCC: hepatocellular carcinoma; BM: bone metastasis; ANLN: Anillin actin-binding protein; HCC: early advantage HCC; IHC: immunohistochemical; TMAs: tissue microarrays; OS: overall survival; RFS: recurrence-free survival; MIH: metastasis-inclined HCC; MAH: metastasis-averse HCC; qRT-PCR: quantitative reverse transcription PCR; UTR: untranslated region; mTOR: mammalian target of rapamycin; SP1: specificity protein 1; mRNA: messenger RNA; TSS: transcription start site; WTAP: Wilm's tumor 1-associating protein; FTO: fat mass and obesity-associated protein; LC-MS/MS: liquid chromatography-tandem mass spectrometry; EMT: epithelial-mesenchymal transition; RANKL: receptor activator of NF-κB ligand; OPG: osteoprotegerin; IP-MIS: immunoprecipitation-mass spectrometry.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
We are grateful to Edit Springs (https://www.editsprings.com/) for their professional linguistic assistance and appreciate the academic support from the AME Hepatocellular Carcinoma Collaborative Group and bioinformation analysis from Shanghai Newcore Biotech Biotechnology Co., Ltd (http://www.rnastar.com/).
Funding
This work was supported by the following institutes: National Natural Science Foundation of China (No. 82160578, No. 81760438, No. 81871988). The Jiangsu Province Key Research and Development Program (BE2019747); Science Fund for Creative Research Groups, NSFC, China (81521091, 82073031). Clinical Research Plan of SHDC (No. SHDC2020CR5007, SHDC22020213).
Ethical approval and consent to participate
The Institutional Review Board at Naval Medical University (Shanghai, China) approved this study, and it was executed in conformity with the Declaration of Helsinki (as revised in 2013). Before the trial began, the patients signed a written informed consent form.
Author contributions
WP Z, DM X, and Y Y designed and conceived this project. H Z, SY H, L Y and HF L performed experiments and generated data. H Z, D-M X, and W-P Z developed methodology. H Z and DM X wrote the draft. All authors revised and finalized the manuscript. All authors contributed to and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fukutomi M, Yokota M, Chuman H. et al. Increased incidence of bone metastases in hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2001;13:1083-8
2. Natsuizaka M, Omura T, Akaike T. et al. Clinical features of hepatocellular carcinoma with extrahepatic metastases. J Gastroenterol Hepatol. 2005;20:1781-7
3. Aino H, Sumie S, Niizeki T. et al. Clinical characteristics and prognostic factors for advanced hepatocellular carcinoma with extrahepatic metastasis. Mol Clin Oncol. 2014;2:393-8
4. Xiang ZL, Zeng ZC, Tang ZY. et al. Potential prognostic biomarkers for bone metastasis from hepatocellular carcinoma. Oncologist. 2011;16:1028-39
5. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-64
6. He J, Zeng ZC, Tang ZY. et al. Clinical features and prognostic factors in patients with bone metastases from hepatocellular carcinoma receiving external beam radiotherapy. Cancer. 2009;115:2710-20
7. Roundtree IA, Evans ME, Pan T. et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187-200
8. Geula S, Moshitch-Moshkovitz S, Dominissini D. et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002-6
9. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608-24
10. Wang T, Kong S, Tao M. et al. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19:88
11. Deng X, Su R, Weng H. et al. RNA N (6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507-17
12. Lu Y, Xu W, Ji J. et al. Alternative splicing of the cell fate determinant Numb in hepatocellular carcinoma. Hepatology. 2015;62:1122-31
13. Chen M, Wong CM. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol Cancer. 2020;19:44
14. Naydenov NG, Koblinski JE, Ivanov AI. Anillin is an emerging regulator of tumorigenesis, acting as a cortical cytoskeletal scaffold and a nuclear modulator of cancer cell differentiation. Cell Mol Life Sci. 2021;78:621-33
15. Wang G, Shen W, Cui L. et al. Overexpression of Anillin (ANLN) is correlated with colorectal cancer progression and poor prognosis. Cancer Biomark. 2016;16:459-65
16. Zeng S, Yu X, Ma C. et al. Transcriptome sequencing identifies ANLN as a promising prognostic biomarker in bladder urothelial carcinoma. Sci Rep. 2017;7:3151
17. Olakowski M, Tyszkiewicz T, Jarzab M. et al. NBL1 and anillin (ANLN) genes over-expression in pancreatic carcinoma. Folia Histochem Cytobiol. 2009;47:249-55
18. Wang Z, Chen J, Zhong MZ. et al. Overexpression of ANLN contributed to poor prognosis of anthracycline-based chemotherapy in breast cancer patients. Cancer Chemother Pharmacol. 2017;79:535-43
19. Nie Y, Zhao Z, Chen M. et al. Anillin is a prognostic factor and is correlated with genovariation in pancreatic cancer based on databases analysis. Oncol Lett. 2021;21:107
20. Xiao JX, Xu W, Fei X. et al. Anillin facilitates cell proliferation and induces tumor growth of hepatocellular carcinoma via miR-138/SOX4 axis regulation. Transl Oncol. 2020;13:100815
21. Zhang LH, Wang D, Li Z. et al. Overexpression of anillin is related to poor prognosis in patients with hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2020
22. Lian YF, Huang YL, Wang JL. et al. Anillin is required for tumor growth and regulated by miR-15a/miR-16-1 in HBV-related hepatocellular carcinoma. Aging (Albany NY). 2018;10:1884-901
23. Zhang S, Nguyen LH, Zhou K. et al. Knockdown of Anillin Actin Binding Protein Blocks Cytokinesis in Hepatocytes and Reduces Liver Tumor Development in Mice Without Affecting Regeneration. Gastroenterology. 2018;154:1421-34
24. Li L, Huang K, Zhao H. et al. CDK1-PLK1/SGOL2/ANLN pathway mediating abnormal cell division in cell cycle may be a critical process in hepatocellular carcinoma. Cell Cycle. 2020;19:1236-52
25. Li C, Wang S, Xing Z. et al. A ROR1-HER3-lncRNA signalling axis modulates the Hippo-YAP pathway to regulate bone metastasis. Nat Cell Biol. 2017;19:106-19
26. Dominissini D, Moshitch-Moshkovitz S, Schwartz S. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201-6
27. Avino PP. How to scaffold the contractile ring for a safe cytokinesis - lessons from Anillin-related proteins. J Cell Sci. 2009;122:1071-9
28. Farley-Barnes KI, McCann KL, Ogawa LM. et al. Diverse Regulators of Human Ribosome Biogenesis Discovered by Changes in Nucleolar Number. Cell Rep. 2018;22:1923-34
29. Zheng B, Han M, Bernier M. et al. Nuclear actin and actin-binding proteins in the regulation of transcription and gene expression. FEBS J. 2009;276:2669-85
30. Wei S, Dai M, Zhang C. et al. KIF2C: a novel link between Wnt/β-catenin and mTORC1 signaling in the pathogenesis of hepatocellular carcinoma. Protein Cell. 2020
31. Dodd KM, Yang J, Shen MH. et al. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. 2015;34:2239-50
32. Xu S, Zhang Y, Liu B. et al. Activation of mTORC1 in B Lymphocytes Promotes Osteoclast Formation via Regulation of β-Catenin and RANKL/OPG. J Bone Miner Res. 2016;31:1320-33
33. Fustin JM, Doi M, Yamaguchi Y. et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793-806
34. Yang Y, Ma Y, Sheng J. et al. A multicenter, retrospective epidemiologic survey of the clinical features and management of bone metastatic disease in China. Chin J Cancer. 2016;35:40
35. Wen S, Wei Y, Zen C. et al. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol Cancer. 2020;19:171
36. He L, Li H, Wu A. et al. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176
37. Wang M, Xia F, Wei Y. et al. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 2020Jul29;8(1):30
38. Okamoto K. Role of RANKL in cancer development and metastasis. J Bone Miner Metab. 2021Jan;39(1):71-81
39. Tian Y, Chen J, Yan X. et al. Overloaded Orthopedic Force Induces Condylar Subchondral Bone Absorption by Stimulating Rat Mesenchymal Stem Cells Differentiating into Osteoclasts via mTOR-Regulated RANKL/OPG Secretion in Osteoblasts. Stem Cells Dev. 2021Jan1;30(1):29-38
40. Xiang L, Gilkes DM. The Contribution of the Immune System in Bone Metastasis Pathogenesis. Int J Mol Sci. 2019Feb25;20(4):999
41. D'Amico L, Roato I. The Impact of Immune System in Regulating Bone Metastasis Formation by Osteotropic Tumors. J Immunol Res. 2015;2015:143526
42. Guo L, Yang H, Zhou C. et al. N6-Methyladenosine RNA Modification in the Tumor Immune Microenvironment: Novel Implications for Immunotherapy. Front Immunol. 2021Dec9;12:773570
Author contact
![]() Corresponding authors: Yuan Yang, Third Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 225 Changhai Road, Shanghai, 200438 China. E-mail: yangyuanedu.cn; Dai-Min Xiang, State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 25 Lane 2200 Xie Tu Road, Shanghai, 200032, China. E-mail: xdm20079com; Wei-Ping Zhou, Third Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 225 Changhai Road, Shanghai, 200438 China. E-mail: ehphwpcom.
Corresponding authors: Yuan Yang, Third Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 225 Changhai Road, Shanghai, 200438 China. E-mail: yangyuanedu.cn; Dai-Min Xiang, State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 25 Lane 2200 Xie Tu Road, Shanghai, 200032, China. E-mail: xdm20079com; Wei-Ping Zhou, Third Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 225 Changhai Road, Shanghai, 200438 China. E-mail: ehphwpcom.