Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
MicroRNA biogenesis and...
Pathogenesis of SLE
Extracellular microRNAs
Concluding remarks and future...
Author Contributions
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(8):2495-2514. doi:10.7150/ijbs.74315 This issue Cite
Review
Dysregulated MicroRNAs in the Pathogenesis of Systemic Lupus Erythematosus: A Comprehensive Review
Daeun Choi1,*, Jimin Kim1,*, Jae Won Yang2,*, Ji Hong Kim3, Seoyeon Park1, Jae Il Shin3, ![]()
1. Yonsei University College of Medicine, Seoul, Republic of Korea.
2. Department of Nephrology, Yonsei University Wonju College of Medicine, Wonju, Republic of Korea.
3. Department of Pediatrics, Yonsei University College of Medicine, Seoul, Republic of Korea.
*These authors contributed equally to this work
Received 2022-4-25; Accepted 2022-12-11; Published 2023-5-8
Abstract

Systemic lupus erythematosus is a chronic autoimmune disease of which clinical presentation is vastly heterogeneous, ranging from mild skin rashes to severe renal diseases. Treatment goal of this illness is to minimize disease activity and prevent further organ damage. In recent years, much research has been done on the epigenetic aspects of SLE pathogenesis, for among the various factors known to contribute to the pathogenic process, epigenetic factors, especially microRNAs, bear the most therapeutic potential that can be altered unlike congenital genetic factors. This article reviews and updates what has been discovered so far about the pathogenesis of lupus, while focusing on the dysregulation of microRNAs in lupus patients in comparison to healthy controls along with the potentially pathogenic roles of the microRNAs commonly reported to be either upregulated or downregulated. Furthermore, this review includes microRNAs of which results are controversial, suggesting possible explanations for such discrepancies and directions for future research. Moreover, we aimed to emphasize the point that had been overlooked so far in studies regarding microRNA expression levels; that is, which specimen was used to assess the dysregulation of microRNAs. To our surprise, a vast number of studies have not considered this factor and have analyzed the potential role of microRNAs in general. Despite extensive investigations done on microRNA levels, their significance and potential role remain a mystery, which calls for further studies on this particular subject in regard of which specimen is used for assessment.
Keywords: microRNA (miRNA), systemic lupus erythematosus (SLE), T lymphocyte, B lymphocyte, plasmacytoid dendritic cell (pDC)
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease predominantly affecting the female sex, starting frequently at reproductive age. The course of disease in most cases involves alternating periods of remission and relapse, resulting in a life-long inflammatory condition (1). Often referred to as the prototype of systemic autoimmune diseases, SLE is characterized by elevated levels of circulating autoantibodies and involvement of multiple organs, including the skin, joints, blood vessels, lungs, heart, brain, and kidneys (2). Clinical presentations are heterogeneous, ranging from mild skin rashes to life-threatening renal failures. Etiology is thought to be multifactorial and is associated with genetic predispositions and environmental triggers, including UV light, infection, smoking, and drugs (3).
Contribution of both genetic and environmental factors to the initiation of SLE has been continuously described in the literature. However, as the nonspecific environmental factors are commonly exposed to the general population and yet, only a small percentage of people present with symptoms of lupus (4), genetic contributions to the disease have been investigated extensively, especially the field of epigenetic modifications. Epigenetic modification is defined as changes made in the chromosome without altering DNA sequence, which allows the biological system to affect gene expression levels in response to environmental signals and eventually result in a stably heritable phenotype (5). Increasing studies of epigenetics have shed light on a new perspective to describe the insufficiently-understood mechanisms of genetic and environmental factors, as seen in the low penetrance of lupus in monozygotic twins (25-45%) (6).
Means of epigenetic alterations are frequently depicted by three principal categories: DNA methylations, histone modifications, and non-coding RNAs. Particularly, microRNAs (miRNAs), a typical example of a non-coding RNA, have increasingly gained awareness of its capability to control the other two principal means of epigenetic alterations, along with its direct implication in the pathogenesis of autoimmune diseases (7, 8). Moreover, unlike the irreversible character of gene mutations or chromosomal anomalies, the reversibility of epigenetic mechanisms holds therapeutic potential, which is another aspect why this particular field bears much significance (9).
In this review, we summarize the expression levels of dysregulated miRNAs in SLE patients with or without renal complications, taking into account the specific source of miRNA samples. We aimed to focus on the implication of these miRNAs in the pathogenesis of SLE.
MicroRNA biogenesis and mechanism of action
MicroRNAs (miRNAs) are a class of small (~22 nucleotides), non-coding regulatory RNAs that are highly conserved among different species and has a crucial role in regulating the expression of its target genes at a post-transcriptional level. According to miRNA target gene studies, a single miRNA may have multiple target genes and vice versa, thus contributing to the complex yet fine-tuned regulatory network (10).
The biogenesis pathway of miRNAs involves a stepwise cleavage of long primary transcripts into smaller RNAs. In the canonical pathway, primary transcripts of miRNA coding genes (pri-miRNAs; pri-mir) are typically generated by RNA polymerase II (11). Pri-miRNAs contain hairpin-like secondary structures, which are cleaved off and processed into ~70-nucleotide (nt) precursor miRNAs (pre-miRNAs; pre-mir) by the nuclear microprocessor complex, comprised of the nuclear RNase III enzyme Drosha and its dsRNA-binding cofactor DGCR8 (DiGeorge syndrome critical region gene 8) (12). Some miRNAs are produced through non-canonical pathways in which hairpin structures are cleaved to produce pre-miRNAs via spliceosome-dependent mechanisms, bypassing the microprocessor machinery.
After transportation to the cytoplasm via exportin-5 (XPO5), pre-miRNA hairpins are further cleaved by the cytoplasmic RNase III Dicer and its dsRNA-binding cofactor TRBP (TAR-RNA binding protein), consequently yielding ~22-nt double-stranded RNAs (13, 14). This dsRNA molecule was conventionally termed as the miRNA/miRNA* duplex, for the more abundant strand was known to function as a guide strand for target gene silencing (miRNA), while the other strand was known to be degraded and was considered merely as a passenger strand (miRNA*). However, recent studies have provided evidence for the abundance and potential regulatory role of some miRNA* sequences (15). This led to the application of the current miRNA nomenclature, miRNA-5p and -3p, each corresponding to the 5'- and 3'- arm of the pre-miRNA hairpin.
For exertion of its regulatory function, the single-stranded mature miRNA is finally incorporated into the RNA-induced silencing complex (RISC), which contains a member of the endonuclease Argonaute family (AGO. The mature miRNA strand plays a guiding role in the RISC, by recognizing their mRNA target sequences through complementary base pairing. RISC negatively regulates its target mRNA, via destabilization of the mRNA molecule, along with inhibition of the translation initiation process (16, 17) (Figure 1).
Biogenesis of microRNAs. (a) MicroRNA (miRNA) genes are reported to be encoded in intergenic, intronic, and exonic regions. In the canonical pathway of miRNA biogenesis, these miRNA genes are transcribed into primary microRNA transcripts (pri-miRNAs), which are cleaved by the microprocessor complex (nuclear RNAase III enzyme Drosha and its cofactor DGCR8) into hairpin-shaped RNA molecule pre-miRNA. After transportion to the cytoplasm via nuclear membrane portein exportin-5, pre-miRNA is cleaved by the cytoplasmic RNase III Dicer and its cofactor TRBP into the miRNA/miRNA* duplex, or more recently termed as miRNA-5p and miRNA-3p RNA strands. The resulting single-stranded mature miRNA is incorporated into the RNA-induced silencing complex (RISC) and complementarily binds to its target gene. Depending on the level of complementarity, binding of RISC leads to the translation repression or cleavage of the target gene. (b) Some miRNAs are produced bypassing the microprocessor machinery, among which the most well-known process involves the mirtron molecule; hairpin-shaped RNA molecules originating from the intronic regions spliced from mRNAs through spliceosome-dependent mechanisms. (c) It is still controversial whether extracellular miRNAs are merely byproducts resulting from cell death or molecules secreted through elaborate regulatory systems. Nonetheless, miRNAs in extracellular fluids are generally observed in two forms: exosomal miRNAs and AGO (Argonaute) protein bound miRNAs. Abbreviations: miRNA, microRNA; DGCR8, DiGeorge syndrome critical region gene 8; TRBP, TAR-RNA binding protein.
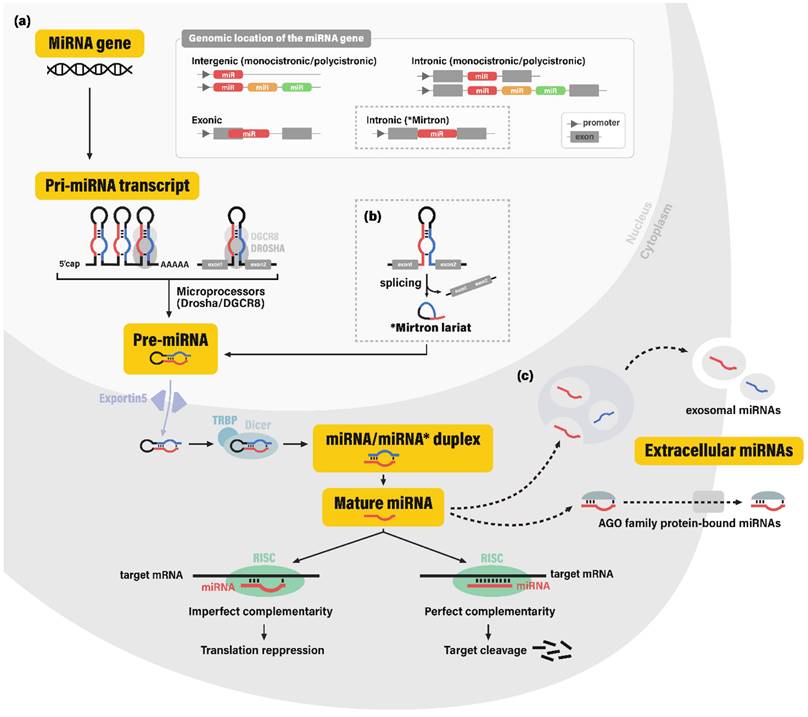
Pathogenesis of SLE
Overview
SLE is a multifactorial disease, in which both genetic and environmental factors are believed to contribute to disease progression. Epidemiologic studies have shown that environmental factors, such as ultraviolet (UV) light, cigarette smoking, and infection are associated with the development of lupus. These environmental risk factors are substances commonly found in everyday life but only a small portion of people who are exposed to these triggers present with symptoms of SLE, suggesting that people who develop SLE symptoms bear a predisposing factor, namely, genetic susceptibility. Indeed, there seems to be a strong link between genetics and the development of SLE, as demonstrated in the studies of different ancestries (18-22), siblings, and monozygotic/dizygotic twins. The complex interplay between genetic and environmental factors has not been fully elucidated, thus, we hereby present a brief summary of the pathogenesis of SLE about what has been discovered so far.
There are numerous aspects in the immune system which may contribute to loss of self-tolerance as the human body's immune reaction is very much complex and fine-tuned even to the slightest difference. Nevertheless, the fundamental phenomenon that underlies all hypotheses remains untouched: loss of tolerance to self-antigens and production of self-antigen-specific autoantibodies (23). The immune system becomes sensitized to self-antigens, such as nuclear proteins, cytoplasmic proteins, and membrane components of the body's own cell. Autoantibodies detected in SLE patients most commonly target antigens of nuclear origin, such as double-stranded DNA (dsDNA), histone proteins, Smith antigen (Sm), snRNP, and Sjögren's-syndrome-related antigen A and B (Ro/SS-A, and La/SS-B, respectively) (24, 25). These antinuclear antibodies (ANAs) are considered a serologic hallmark of SLE and are utilized as means of prognostic assessment of the disease (25, 26).
The initial contributing factor of this phenomenon of sensitization to self-antigens is an abnormal increase of exposure of self-antigens to the immune system, which would not have occurred as much in the absence of a pathologic condition. This feature is attributed to dysregulation of cell deaths, such as the classical apoptosis and neutrophil-specific death called NETosis, along with defective clearance of the remaining debris (27). With increased cell death and impaired clearance in SLE patients, the imbalance leads to accumulation of extracellular self-antigens, a state of chronic inflammation, and ultimately, stochastically generated autoreactive immune cells.
The process of sensitization begins as immature antigen presenting cells (APCs), such as immature macrophages or conventional dendritic cells (cDCs), recognize, engulf, and display self-antigens on their surfaces. The antigens are presented to naive T-helper (Th) cells in nearby lymph nodes, resulting in activation of the autoreactive Th-cell population into different types of Th cells. Amongst them, activated Th2 cells stimulate autoreactive B-cells, thus resulting in proliferation of plasma cells that produce pathogenic autoantibodies. In SLE patients, both T-cells and B-cells have been reported to show signs of hyperactivation: increased numbers of activated cells found in the peripheral blood, increased sensitivity to stimulatory signals, and skewing of T-cell response towards B-cell activation (28). Moreover, aberrant levels of cytokines, such as high levels of type 1 interferon, also promote T/B-cell activation and humoral immunity. Plasmacytoid dendritic cells are known to take part in this particular feature (Figure 2).
Pathogenesis of Systemic lupus erythematosus (SLE) and associated miRNAs upregulations/downregulations in SLE immune cells. Increased cell death via the classical apoptosis and the more recently investigated NETosis, along with defective clearance of the debris, lead to increased exposure of self-antigens to the immune system, and consequently, activation of autoreactive immune cells. SLE immune cells have been reported to show signs of hyperactivation, including increased numbers, increased sensitivity to stimulatory signals, and dysregulated secretion of chemokines/cytokines. Furthermore, altered ratio of T helper cell subtypes skew T-cell response towards B-cell activation. Moreover, type 1 interferon (IFN), a key immunoregulator mainly produced by plasmacytoid dendritic cell (pDCs), have also been observed to be upregulated in SLE patients. As a result, aberrant levels of autoantibodies and chemokine/cytokines lead to chronic inflammation, tissue damage, and end-organ injuries, clinically presenting as the well-known autoimmune disease.
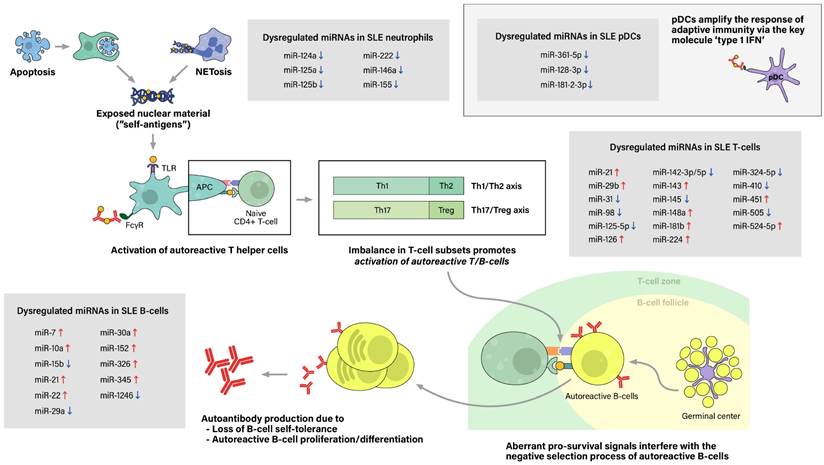
Step 1. Excessive exposure of self antigens to the immune system
Accumulation of extracellular self-antigens in SLE is attributable to increased cell deaths and defective clearance of their remnants, in which associated cell deaths include classical apoptosis and neutrophil-specific NETosis. The classical apoptosis process is triggered via the death-receptor-mediated (extrinsic) pathway or the mitochondrial-oxidative-stress-dependent (intrinsic) pathway, both pathways ultimately activating caspases and resulting in apoptotic blebbing of the cell (29). Apoptotic cells are recognized by phagocytic cells, such as macrophages, then are immediately engulfed and degraded to prevent the exposure of potential self-antigens to the immune system. Thus, in healthy individuals, apoptosis takes place in an immunologically silent manner (30). However, in vivo and in vitro studies have reported impairment of phagocytic functions in macrophages, monocytes, and neutrophils collected from SLE patients. Consequently, the uncleared apoptotic blebs lose their cellular integrity and undergo a necrotic phase called secondary necrosis, resulting in exposure of intracellular proteins to the immune system (31).
Another type of dysregulated cell death in SLE is NETosis. Traditionally, cell death of neutrophils has been referred to either programmed cell death (apoptosis) or non-programmed cell death (necrosis). Adding to this, another mean of cell death has been brought to light recently, called NETosis. NETosis is the process of neutrophils forming neutrophil extracellular traps (NETs), activated to trap and kill microbes when the pathogen outsizes the phagocytosis-possible capacity (32, 33). The neutrophil extracellular trap (NET) comprises decondensed chromatin fibers with histone proteins, cytoplasmic proteins, and contents of neutrophil granules. After NETosis, NETs are partially degraded by extracellular DNase 1, and these preprocessed NETs are recognized and cleared by macrophages with the assistance of C1q opsonization (34). While NET degradation is initiated promptly in healthy individuals, enabling a immunologically silent process, increased NETosis and inefficient clearance of its remnants observed in SLE patients result in increased exposure of NET components to the immune system.
Despite growing interest in NETosis, there is limited data on the epigenetic aspect of lupus neutrophils. Pérez-Sánchez et al. reported that miR-124a, 125a, 222, 125b, 146a, 155 are downregulated in neutrophils from SLE patients compared to neutrophils from healthy controls (35). However, the linkage between microRNA dysregulation and abnormal neutrophil function, particularly NETosis, has not been assessed, which calls for additional research to fill this gap.
Step 2. Exaggerated immune response to self antigens
In lupus patients, adding to excessive exposure of self-antigens to the immune system, T lymphocytes and B lymphocytes, both components of adaptive immunity, have been reported to show signs of hyperactivation. Aberrant T-cell signaling and imbalance in T-cell subsets lead to unrestricted hyperactivation of B-cells, along with the impaired development of naive B-cells in germinal centers (36). These aberrant activities of adaptive immunity lead to generation of excessive self-reactive immune cells, autoantibodies, inflammations, and consequently, end-organ damage of the disease.
Dysregulated miRNAs in T-cells and their potentially pathogenic roles
Overview of the mechanism of T-cell development
The role of CD8+ T-cells in the pathogenesis of SLE is not well-known (37). However, hyperactivation and increased levels of CD4+ T 'helper' cells have been observed in the peripheral blood of SLE patients. T helper cells are key regulators of the adaptive immune system. Not only do they help B-cells gain function to produce antibodies, but also shape the general landscape of the body's immune response to presented antigens. As a naïve CD4+ T-cell circulates within the lymph-vascular system, it encounters its cognate antigen displayed by antigen-presenting cells (APCs), especially dendritic cells (DCs) that have migrated from different tissues of the body. This interaction usually takes place in secondary lymphoid organs, such as the spleen, lymph nodes, and mucosal associated lymphoid tissues, consequently leading to alterations in the T-cell signaling pathway (38). A T-cell receptor (TCR) assembled with CD3 proteins (CD3δ, ε, γ, and ζ), recognizes its cognate antigenic peptide, and with the help of a secondary signal, initiates the intracellular signaling pathway in naive T-cells. Among the number of secondary signals that have been identified, CD28 on the T-cell binding to B7.1 (CD80) or B7.2 (CD86) on the APC is the key signal that specifically triggers proliferation of T-cells recognizing this particular antigen. Further differentiations into distinct lineages of effector CD4+ T-cells is determined by cytokine signaling.
Structural differences observed in lupus T-cells
Phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in the CD3ζ chain is one of the chief features of the T-cell activation process. Lck, a T-lymphocyte-specific tyrosine kinase, targets tyrosine residues of ITAMs in CD3ζ chains. These phosphorylated ITAMs act as binding sites for the 70-kDa zeta chain-associated protein (ZAP-70), another type of tyrosine kinase, and this CD3ζ-ZAP-70 interaction further leads to intracellular signaling cascades resulting in calcium influx into T-cells (39, 40). Thereafter, nuclear factor of activated T-cells (NFAT), a family of transcription factors known to be Ca-dependent, becomes dephosphorylated and activated via the Ca2+ mediated calmodulin-calcineurin pathway. NFAT then translocate into the nucleus, binds to the promoter of several genes and initiates their transcription, therefore inducing the production of cytokines essential for the proliferation and expansion of T-cells, such as IL-2, and promoting the expression of co-stimulatory molecules on the surface of T cells, such as CD40L. In lupus T-cells, however, expression of CD3ζ chains is significantly decreased and are replaced by the γ chain of high affinity IgE receptor (FcRγ), which is structurally homologous with the CD3ζ chain (41, 42). Thus, in lupus T-cells, the FcRγ chain binds with another type of tyrosine kinase Syk instead of ZAP-70. Signaling between FcRγ and Syk is known to be much stronger than it between CD3ζ and ZAP-70, causing hyperactivation of TCR signaling, thus resulting in increase of Ca2+ influx into SLE T-cells and elevation of NFAT level in the nucleus (41). However, consequences due to increased expression of NFAT are not as predictable; due to the overexpression of NFAT and its binding to its target promoters, CD40L gene is more easily activated, whereas production of IL-2 is observed to be lower in lupus T-cells. Several explanations have been suggested on this unexpected downregulation of IL-2 in lupus T-cells. First, since CD40L-CD40 signaling induces the differentiation of naive T-cells into Th17, it may contribute to the imbalance of Th cell subsets in SLE and dysregulation of cytokine levels. Second, decrease in IL-2 levels may be due to the increase of phosphorylated cAMP-responsive element modulator (p-CREM) due to Ca2+ /calmodulin-dependent kinase IV, which binds to the promoter of IL-2 gene and inhibits the production of IL-2 (43).
Another known feature of lupus T-cells involves their surface morphology during activation and entry into targeted inflamed tissues. Immune cells, specifically, leukocytes and lymphocytes, have a distinct function being able to quickly undergo multiple steps of polarization on activating cues, adhesion to endothelial tissues, and migration into sites of inflammation. Polarization of a lymphocyte refers to the morphological transition of a freely circulating cell to an endothelium-adhering cell with a specialized surface site called uropods. Various adhesion molecules, such as CD44, are concentrated in uropods, enabling them to stabilize migrating lymphocytes. The polarization and uropod formation process are known to be mainly regulated by ezrin, radixin, and moesin (ERM) proteins; serine/threonine kinases, such as Rho-associated protein kinase (ROCK) or protein kinase C (PKC) phosphorylates threonine residues of ERM proteins, then, phosphorylated ERM proteins set off a signaling cascade leading to alterations of cytoskeletal assembly (44, 45). In lupus T-cells, ERM proteins show increased levels of phosphorylation along with overexpression of surface molecule CD44 compared to T-cells of healthy controls, promoting T-cell adhesion and TCR-CD3 signaling in lupus patients (45). Furthermore, according to recent findings, ROCK has been found not only to regulate cytoskeletal assembly of lymphocytes, but also the effector function of CD4+ T-cells. Biswas et al reports that ROCK2, an isoform of Rho kinase, was activated in murine T-cells in Th17-cell dominant environments and phosphorylated interferon regulatory factor 4 (IRF4), a transcription factor involved in IL-17 and IL-21 gene expression. ROCK2-mediated activation of IRF4 leaded to IL-17 and IL-21 production, which are both key pathogenic cytokines known to contribute to autoimmune disorders, and Th17 cell differentiation from naïve T-cells.
Lastly, enhanced activity of phosphoinositide-3 kinase (PI3K), a key component in the PI3K/Akt/mTOR pathway, was observed in T cells of SLE patients. In the well-known PI3K/Akt/mTOR pathway, PI3K phosphorylates PIP2 (phosphatidylinositol 4,5-bisphosphate), then, phosphoinositide-dependent protein kinase 1 (PDK1) binds to the phosphorylated product, PIP3, and activates Akt, also known as protein kinase B (PKB). Akt, as the main molecule in the PI3K signaling pathway, has various downstream effects, amongst mammalian target of rapamycin (mTOR) is especially the molecule of interest in the field of autoimmunity. Phosphorylated Akt activates mTOR, which has been widely investigated for its role in naïve T-cell activation, differentiation into Th1, Th2, Th17 cell lineages, and inhibition of regulatory T-cells and T-cell anergy. Studies have shown that Akt levels are increased in T cells and B cells of SLE patients and that there lies a positive correlation between Akt/mTOR activation in B cells and disease severity (46, 47).
T-cell subset alterations observed in lupus patients
Upon stimulation by certain cytokines and membrane-bound molecules of APCs, the naïve T-cell undergoes clonal expansion and differentiation into functionally distinct CD4+ effector T-cell lineages (38). Different types of helper T-cells include the 'classical' effector T helper 1 cells (Th1 cells), T helper 2 cells (Th2 cells), T helper 17 cells (Th17 cells), the immunosuppressive regulatory T-cells (Treg cells), and lastly, T follicular helper cells (Tfh cells). The subsets can also be defined based on their cytokine secretion profiles: Th1 cells chiefly produce IFN-γ, IL-2, and tumor necrosis factor β (TNF-β), whereas Th2 cells produce IL-4, IL-5, IL-6, IL-10 and IL-13 (48). It has long been established that Th1 cells activate classical macrophages and amplify the body's immune response against intracellular pathogens, such as viruses, while Th2 cells stimulate mast cells and eosinophils, promote antibody production, and protect the body against extracellular pathogens, such as multicellular parasites. While the Th1/Th2 balance constitutes one axis of the immune homeostasis, another more recently recognized axis is the Th17/Treg balance. Th17 cells are characterized by the master transcription factor RAR-related organ receptor γ (RORγt) and are known to mainly produce IL-17, IL-21, IL-22, and IL-23, and plus, TNF-α and IL-6 when exposed to certain microenvironments (49, 50). It has been understood that Th17 cells play a major role in autoimmunity with their ability to recruit neutrophils and promote inflammatory responses. In contrast, Treg cells produce anti-inflammatory cytokines, such as IL-10 and transforming growth factor β (TGF-β), inhibiting a variety of immune cells and consequently inhibiting autoimmunity. IL-17, a key effector cytokine of Th17, are also known to promote B-cell differentiation and survival via upregulation of the B lymphocyte stimulator (BLyS) (36).
Imbalance in T-helper-cell subsets (Th1/Th2/Th17/Treg) has been suggested to contribute to the pathogenesis of SLE. However, it is still controversial which T-helper-cell subset and related cytokines mainly contribute to the pathogenic process (51). Regarding the Th1/Th2 balance axis, the traditional understanding was that function of Th2 cells are dominant in SLE patients compared to Th1 cells, promoting inappropriate development of autoantibodies in B-cells. However, recent studies report that Th1 cells may be involved in the pathogenic process as well, as exacerbation of lupus was observed after injection of IFN-γ in a lupus mouse model. Dolff et al has also reported the dominance of Th1 cells in the chronic stages of SLE, especially in patients with lupus nephritis IV (52). Meanwhile, regarding the more recently recognized Th17/Treg balance axis, it has been reported that T-helper-cells in lupus patients show skewing towards Th17 cells, as distinct cytokine profiles were observed: decreased IFN-γ, TGF-β and increased IL-17, each distinct cytokines of Th1 cells, Treg cells, and Th17 cells, respectively (51).
Expanded populations of Th17 cells have been reported in SLE patients, and the pathogenic role of these cells are well indicated in several studies. Increased levels of IL-17 and Th17 cells were found in the skin lesion biopsies of SLE patients and urinary sediment and kidney biopsies from patients with lupus nephritis. Treg cells, known to suppress and modulate the activity of other T-cells, have also been reported to play a key role in the pathogenesis of SLE as impaired function of Treg cells can result in generation of autoantibodies and loss of immune tolerance. Lower ratios of Treg/Th17 were observed in active SLE patients compared to healthy controls, and the deficiency of Treg cells were found to be correlated with SLE disease activity (49). Decreased levels and impaired function of Th17 cells in lupus patients have also been reported.
Dysregulated miRNAs and their potential contributions to aberrant T-cell activity
In regard of the aforementioned roles of T-cells in SLE pathogenesis, we ought to identify how dysregulated miRNAs in T-cells contribute to T-cell hyperactivation and the onset of the disease. According to Lu M et al, downregulation of miR-145 in lupus T-cells suppressed API5 expression that lead to T-cell activation-induced cell death, and upregulation of miR-224 enhanced expression of STAT-1 in SLE T-cells, causing lupus nephritis (53). miR-524-5p was also significantly overexpressed in SLE T-cells, increasing the production of IFN-γ in activated T-cells. It also diminished protein expression of Jagged-1 and Hes1 in T-cells, which are both related to Notch signaling pathway, resulting in increased SLE disease activity (54). Expression level of miR‐31 was decreased in SLE T-cells, leading to reduced expression of IL‐2 by suppressing NFAT and IL-2 promoter activity. RhoA expression was negatively correlated with miR-31 level, and therefore was upregulated in SLE T-cells (55). miR-410, which significantly suppressed the expression of IL-10 by targeting STAT3 activity, was decreased in SLE T cells. Furthermore, the level of miR-410 was also found lower in CD4+ T cells (56).
Pan et al reported that miR-21 and miR-148a were both overexpressed in CD4+ T cells. They were found to suppress DNMT1 by targeting RASGRP1 and its protein coding region respectively, promoting demethylation of CD4+ T cells and resulting in enhanced expression of methylation-sensitive autoimmune-associated genes such as CD70 and LFA-1 (57). Previous study has also shown that inhibition of BDH2, a direct target of miR-21-5p, contributes to DNA hypomethylation in CD4+ T cells by increasing intracellular iron concentration (58). It has been reported that PDCD4, another target of miR-21, might also play a central role in SLE (59). Up-regulation of miR-29b and miR-126 in CD4+ T cells also appeared to play an important role in SLE pathogenesis by suppressing DNMT1 and inducing hypomethylation, thereby enhancing expression of particular genes such as CD70 and CD11a that contributes to the autoreactive status of T cells (60, 61). miR-125a(-5p), which negatively regulates several effector T-cell factors including STAT3, IFN-γ and IL13, was downregulated in peripheral CD4+ T cells in SLE, leading to destabilization of Treg mediated immune homeostasis (62). miR‐142‐3p and miR‐142‐5p in CD4+ T cells of SLE patients were significantly downregulated due to DNA methylation of its promoter region, increasing translation level of its target proteins, SAP, CD84, and IL-10, and thereby causing overactivation of T cells and B cells (61, 63). Downregulation of miR-98 was found to contribute to SLE pathogenesis by inducing Fas-mediated apoptosis of CD4+ T cells (64). According to R Martinez-Ramos, levels of miR-224 and miR-143 were significantly overexpressed in CD4+ T cells of inactive, asymptomatic SLE patients. He suggested that overexpression of miR-224 could have induced IRF4 overexpression and increased secretion level of IL-21, causing hyper responsiveness of IL-21 dependent B cells (65). However, no correlation of target genes involved in autoimmune response with miR-143 has been found yet (65). miR-633 level was downregulated in SLE CD4+ T cells, contributing to SLE pathogenesis by targeting AKT1 and activating AKT1/mTOR pathway, which is known to play an important role in the activation of T cells (66). Additionally, Ming Zhao et al have shown that downregulation of miR-142-3p, miR-505, miR-324-5p, and upregulation of miR-126, miR-451, miR-181b were also observed in T cells of SLE patients regardless of their clinical phenotypes including skin lesions, chronic renal pathology, and polyarticular disease (67).
Number and function of Treg cells are known to be impaired in SLE and other autoimmune diseases (68). Sun et al reported that miR-326 expression level was significantly upregulated in Treg cells of new-onset SLE patients, positively correlated with CRP and anti-C1q antibody, and negatively correlated with Ets (69).
Dysregulated miRNAs in B-cells and their potentially pathogenic roles
Overview of the mechanism of B-cell development
Autoantibody production due to loss of B-cell self-tolerance and the alteration of B-cell differentiation is one of the major immunological aberrations in SLE pathogenesis. In lupus patients, B-cells are hyperactivated, increased in the peripheral blood, and are more prone to polyclonal activation by its specific antigens, resulting in the formation of a wide range of autoantibodies (3). B-cells are lymphocytes capable of secreting immunoglobulins, or antibodies, that specifically recognize proteins of potentially invasive pathogens, also called antigens. An immunoglobulin comprises two heavy chains and two light chains, both of which undergo gene rearrangements in the developmental stages, consequently generating an enormously diverse antibody repertoire in a single human being. The B-cell development process can be divided into two phases: the antigen-independent phase within the bone marrow (stem cells, pro-B cells, pre-B cells, and immature B-cells) and the antigen-dependent phase within secondary lymphoid tissues (naive mature B-cells, activated B-cells) (70). The intermediate phase of immature B-cells between the two phases is referred to as transitional B-cells, identified as CD19+ CD24high CD38high lymphocytes (71). These transitional B-cells further evolve into CD19+ CD24int CD38int naive mature B-cells, which remain quiescent and circulate through the peripheral blood and lymphatic system until they meet their specific antigens in secondary lymphoid organs (SLOs), such as lymph nodes, the spleen, and gut-associated lymphoid tissue (GALT) (72). Thereafter, an antigen-dependent process of B-cell expansion and differentiation takes place in order to yield high-affinity antibody-producing plasma cells and memory cells (70).
Throughout the early developmental stages of B-cells, immunoglobulin gene recombination takes place, and amongst them, malfunctioning or self-reactive combinations are eliminated through multiple immunological checkpoints. The resulting combinations of immunoglobulin heavy chains and light chains are assembled and presented on the cell membrane of immature B-cells, and are called B-cell receptors (BCRs). For maturation and proliferation, a developing B-cell requires pro-survival signals, including B-cell activating factor (BAFF, also known as BLyS), tonic BCR signaling, and certain cytokines, such as IL-7 (73). These signals synergize and promote the survival of B-cells at different stages of development. For example, while early developing B-cells (pro-B cells, pre-B cells) require signals through IL-7 receptors in order to survive, the impact of IL-7 on immature B-cells is minimal (74-76). Also, BAFF signaling is known to play its role starting from the transitional stage of B-cells, while in the survival of naive mature B-cells, it has been reported that both tonic BCR signaling and BAFF signaling are necessary. Of these three pro-survival signals, BAFF has been especially emphasized of its role in autoimmune diseases. Increased serum levels of BAFF and/or its homolog APRIL (a proliferation-inducing ligand) have been reported in lupus patients, some of which had also been found to be positively correlated with autoantibody titers (77, 78). Studies have discovered that excess BAFF promotes naive B-cell survival, interferes with the negative selection process of autoreactive B-cells within GCs, and facilitates immunoglobulin class switching, as a result, enhancing aberrant immune responses (79-82). There have also been promising results in murine studies regarding the therapeutic potential of BAFF/APRIL inhibitors in treating SLE, however, clinical trials of anti-BAFF antibodies have failed to show significant results so far (77, 78, 83). Nonetheless, BAFF/APRIL appears to play a key role in the pathogenesis of autoimmune diseases, including SLE, and many questions remain to be unsolved about its specific mechanism of action.
Adding to pro-survival signals, the fate of developing B-cells also depends on pathogen-associated molecules, especially in the antigen-dependent phase within SLOs. Naive mature B-cells express both IgM and IgD-BCR on their surfaces, and as they first encounter their cognate antigens presented by follicular dendritic cells (FDCs) in the B-cell follicles of SLOs, this leads to the internalization and presentation of the antigen on MHC class II molecules of B-cells. These antigen-presenting B-cells then migrate to the border of the T-cell zone and interact with CD4+ Th cells, which had already been primed by activated dendritic cells (70). Th cells further differentiate into T follicular helper cells (Tfh cells); a process driven by the upregulation of Tfh cell-associated transcription factors, including B-cell lymphoma 6 protein (BCL-6) (84). BCL-6 is a transcription factor that is crucial for the development of not only Tfh cells but also germinal center (GC) B-cells, playing a pivotal role in the germinal center reaction (85). Meanwhile, at the T-B border, stimulated B-cells differentiate into GC-independent memory B-cells, short-lived plasma cells, and GC B-cells, among which GC B-cells are the cells that participate in the GC reaction and generate high-affinity class-switched antibodies (70). Tfh cells, in combination with FDCs, play a key role in the formation of GCs, which are the very places where negative selection of autoreactive B-cells and positive selection of high-affinity B-cells take place, bearing the potential as culprit for self-reactive antibody generation in autoimmune diseases. According to multiple in vivo studies of lupus patients, increased levels of circulating Tfh cells were observed to be positively related with the disease activity and autoantibody titer of lupus (86, 87).
Although not fully understood, transitional B-cells seem to be closely related to IL-10-secreting regulatory B-cells (Breg cells), similarly secreting the anti-inflammatory cytokine IL-10. IL-10 suppresses the expansion and differentiation of Th1 and Th17 cells, thereby limiting production of pro-inflammatory cytokines (88-90). The general consensus is that transitional B-cells play a regulatory role in inflammatory conditions and prevent autoimmunity, as they are also known to take part in the differentiation of CD4+ FoxP3 Treg cells (88, 91). However, there have been some studies in which transitional B-cells were observed to secrete pro-inflammatory cytokines as well, for example, IL-6, which regulates the differentiation of Treg cells and promotes autoreactive responses of Th1 and Th17 cells (90, 92-95). Interestingly, despite the general understanding on transitional B-cells, studies on lupus patients have demonstrated elevated levels of CD19+ CD24high CD38high transitional B-cells (88, 92, 93, 96, 97) and decreased levels of the closely-related regulatory B-cells (98). In one of these studies, along with elevated levels of transitional B-cells, functional disabilities of these cells were also observed (88), providing a possible explanation for the conflicting results.
Dysregulated miRNAs and their potential contributions to aberrant B-cell activity
Regarding the previously mentioned roles of B-cells in the pathogenesis of SLE, we ought to identify how the dysregulated miRNAs in B-cells induce hyperactivation of B-cells and the onset of the disease. Increased expression of miR-7, miR-21, miR-22 in SLE B-cells were reported to play a critical role in B-cell hyper-responsiveness by down-regulating PTEN, an inhibitor of B cell signaling, and thereby enhancing BCR signaling. Moreover, IL-21, produced mainly by activated CD4+ T-cells, appeared to up-regulate miR-7 and miR-22 in both healthy and lupus B-cells, providing a positive feedback between activated T-cells and B-cells in lupus patients (99). However, IL-21 is also known to upregulate PTEN expression, making its relation between BCR signaling complex. A recent study has also revealed that the level of miR-30a in SLE B cells were significantly increased, negatively affecting the level of Lyn, a tyrosine kinase which negatively regulates BCR signaling by phosphorylating inhibitory receptors, which therefore leads to B-cell hyperactivation (100). MiRNA-152, which directly targets Kruppel-like factor 5 (KLF5) that binds to the promoter region of BAFF and inhibits its expression (101), was reported to be upregulated in B-cells of SLE patients, contributing to B-cell hyperactivation. Jin L et al. have shown that the upregulation of miR-326 also promotes B-cell hyperactivation by targeting Est-1, a negative regulator of B-cell differentiation (102). According to Martinez-Ramos R et al, miR-10a and miR-345 were overexpressed in B-cells from asymptomatic SLE patients. He expected that miR-10a has a possible inhibitory role on serum IL-8 level, which has been established to positively correlate with SLE disease activity, supporting the fact that SLE patients with high miR-10a in B-cells were asymptomatic and had low generation of autoantibodies from B-cells. Similarly, overexpressed miR-345 in inactive phase of SLE has a potential to inhibit IRF-8 in B-cells, a key transcription factor regulating B-cell differentiation (65).
MiR-15b, which directly targets Cyclin D3 (CCND3) that plays an important role in B-cell development and required for the transition of pro-B cell to pre-B cell, appeared to be downregulated in B cells. Ren D, et al. demonstrated that SLE patients showed significant upregulation of CCND3, due to the decrease of miR-15b in B-cells and the activation of TLR7, B-cell intrinsic receptor that senses single stranded RNA (103). Significant downregulation of miR-1246 was also reported in SLE B-cells compared to healthy controls. According to Luo et al, miR-1246 regulates the expression of Early B-cell factor 1(EBF1), which contributes to the proliferation of B-cells via the AKT signaling pathway. Therefore, decreased miR-1246 enhances EBF1 expression and B-cell activity. Also, activated B-cell's AKT-p53 pathway was reported to decrease the level of miR-1246, leading to further activation of B-cells (104). Level of miR-29a in lupus B-cells were downregulated compared to healthy controls. Et al. demonstrated that decreased miR-29a enhances the expression of its target gene, Crk-like protein (CRKL) in B-cells, resulting in increased secretion of IgG and activation of B-cells (105).
Role of plasmacytoid dendritic cells in systemic lupus erythematosus
Human dendritic cells are divided into two subsets: the 'classical' myeloid dendritic cells (mDC)s, and the plasmacytoid dendritic cells (pDCs) that are morphologically and functionally distinct from mDCs. On a molecular level, while mDCs express TLRs-1 to -6, -8 and -10 on their surface, pDCs have a less diverse profile of surface molecules, predominantly TLRs-7 and -9. Functionally, both dendritic cells engulf molecules of interest by endocytosis using their surface molecules, however, pDCs retain engulfed self-antigens for a longer time, enabling continuous production of type 1 IFN. Studies have shown that SLE patients have a normal or reduced frequency of mDCs compared to healthy controls. Studies regarding frequency of pDCs have not shown such consistent results, however recent research has shown that lupus pDCs exhibit more stimulatory activity and activated phenotypes (106). Although only recently recognized, the role of pDCs in the regulation of immune responses have been vigorously investigated throughout the years; while mDCs are professional antigen-presenting cells that links innate immunity to adaptive immunity, pDCs are “natural type 1 IFN-producers” that immune modulate and amplify the response of adaptive immunity via the key molecule 'type 1 IFN' (36, 106).
Formation and deposition of immune complexes containing autoantigens lead to endocytosis of pDCs and amplification of self-molecule directed immune responses. pDCs identify immune complexes by FcγRIIa, and the endosomal TLR7 and TLR9 plays a central role in the activation of pDCs, resulting in the production of type 1 IFN, a key cytokine of SLE pathogenesis (107). Upregulation of type 1 IFN arouses multiple effects that contribute to the activity of the disease, such as increased differentiation of precursor cells into mDCs, activation of Th cells, increased secretion of pro-inflammatory cytokines, upregulation of the B cell activating factor (BAFF), promotion of isotype switching in activated B-cells, and loss of immune regulatory function of Treg cells (36, 108). As a result, immune complexes lead to amplified inflammatory responses and complement activation, thereby injuring various systemic organs (23).
Despite the key role of type 1 IFN as master regulators in amplifying adaptive immunity and the relatively well accepted consensus that type 1 IFN levels are upregulated in lupus patients, not much research has been done on the epigenetic aspect of lupus pDCs. We have found one study that has investigated about dysregulation of miRNAs specifically in pDCs. It revealed that miR-361-5p, miR-128-3p, and miR-181-2-3p were significantly reduced in the pDCs of SLE patients, especially in those with high type 1 IFN levels (109). Moreover, the level of miR-361-5p was related with the disease activity (SLEDAI ≥4). Aberrations of miRNA expression in pDCs potentially may be able to explain the upregulated levels of type 1 IFN in SLE, which calls for additional investigations specifically on these master regulator immune cells.
Extracellular microRNAs
Differential in vivo expression levels of miRNAs have been widely investigated since microRNAs were recognized as important regulators of disease pathogenesis in various fields, notably autoimmunity (110, 111). Traditionally, the intracellular concentration of miRNAs was the topic of concern. However, a mounting number of studies have expanded their focuses to extracellular distributions of miRNAs (112, 113). Extracellular miRNA concentrations are measured in easily obtainable clinical samples, including blood plasma, serum, and urine (114-117). According to current understandings, miRNAs are able to endure the nuclease-rich environment outside cells by complexing with AGO family proteins, or being packaged into extracellular vesicles, such as exosomes, microvesicles, and apoptotic bodies. These circulating miRNAs are partly regarded simply as byproducts after cell death, while some suggest their significance as means of cell-to-cell communication (118, 119). In the latter point of view, miRNAs are secreted in a regulated process and function as autocrine, paracrine, or even endocrine messengers to modulate cellular activities of their recipient cells (120). Although further research is needed, it is regarded that both hypotheses are feasible, since a majority of extracellular miRNAs could be non-specific byproducts while some selected miRNAs engage in cell-cell signaling (118).
Despite lack of understanding on the physiology or function, dysregulated levels of extracellular miRNAs in plasma, serum, urine, etc., has been vigorously investigated in recent years. Here we update and summarize the pathogenic potentials of the most commonly studied miRNAs: miR-21, miR-146a, miR-155, and miR-181a.
MiR-21, which is significantly overexpressed in CD4+ T-cells and is associated with DNA hypomethylation, was shown to be upregulated in the plasma of SLE patients (121). According to Chen X et al, circulating miRNA are mostly derived from circulating blood cells (115), and this result proposes that the increased level of circulating miR-21 might be due to selective secretion of upregulated intracellular miR-21 in CD4+ T-cells and the exposure of CD4+ T-cells to circulating plasma in inflammatory conditions. However, the specific mechanism of selective secretion remains unclear. Moreover, circulating miR-21 was also reported to be increased in patients with RA, indicating that it might not be specific for SLE (121). Plasma miR-21 level was highly correlated with SLE disease activity score, and therefore may be used as a biomarker to distinguish the severence of SLE (122, 123). Its level was also negatively correlated with serum C3,4 levels and IL-2, and positively correlated with IL-10 expression (122-124).
According to Wang et al, the serum and plasma levels of miR-146a in SLE patients were down regulated compared to healthy controls and negatively correlated with SLEDAI (121, 125). Furthermore, serum exosomal miR-146a have shown to be decreased in SLE patients, and previous studies have reported that serum exosomal miR-146a negatively regulates senescence of SLE bone marrow mesenchymal stem cells by suppressing the TRAF6/NF-KB signaling pathway (182, 183). These are consistent with the report that miR-146a, which is known to contribute to the disease by regulating the type 1 IFN pathway, is significantly decreased in the PBMC of SLE patients (126). Urinary extracellular miR-146a level was notably higher in SLE patients and was correlated with estimated GFR, but its correlation between clinical measurements remains unclear (125, 127). Hernandez have also reported that in SLE patients, urinary miR-146a existed primarily in the form contained in exosomes and were increased in patients with active lupus nephritis (128).
The serum and plasma level of miR-155 in SLE have also been reported to be decreased in SLE patients, but its urinary level was significantly higher in lupus specimens and highly associated with SLEDAI and proteinuria (127). Divekar has reported that miR-155 level was upregulated in T cells in lupus, which may seem contradictory to its low circulating level in plasma (129). However, since several studies have shown that apoptotic cells can transfer its microparticles and apoptotic bodies to other cell types, increased transfer of miR-155 might have resulted in miRNAs' reduced circulating level (130).
Whether the circulating level of miR-181a increases is controversial. Carlsen A. et al and Motawi T. have reported that the plasma level of miR-181a was significantly increased in SLE patients and observed a correlation between SLEDAI score (122, 131). miR-181a was found to mediate inflammatory factors such as IL-1β, IL-6 (132), IL-8 (133), and TNF-α (134) and play a critical role in regulating T cell and B cell differentiation and innate immunity response (135). According to Li H, serum level of miR-181a was also increased in SLE patients and was positively correlated with ESR, CRP, anti-dsDNA, complement C4 and SLEDAI levels (136). However, several studies have observed downregulation of miR-181a in SLE patients. Its expression was found to be decreased in peripheral blood of Egyptian pediatric SLE patients, which may indicate that patterns of disease expression may differ in childhood onset and adult SLE patients (122, 137). In addition, Zhang H. et al has also observed a significant decrease of plasma miR-181a in SLE patients compared to healthy controls (138) (Table 1-3).
MiRNA profiles of systemic lupus erythematosus.
| Sample type | miRNAs | |
|---|---|---|
| Upregulated | Downregulated | |
| MiRNAs involved in SLE pathogenesis (SLE vs. HC, active SLE vs. inactive SLE, LNN vs. HC) | ||
| Renal tissue | miR-198 (147) miR-423-5p, miR-663a (148) | |
| Peripheral blood leukocytes | miR-146a (126) | |
| PBMC | miR-629, miR-525-5p, miR-516a-3p (149) miR-125, miR-1273h-5p (150) miR-16, miR-21, miR-155 (151) miR-155 (152) miR-199-3p (153) miR-27a-5p (154) miR-29b (155) miR-873 (156) | miR-125a (157) miR-125b (158) miR-141 (151) miR-146a (159) miR-155, miR-17, miR-181b (160) miR-155 (161) miR-17-5p (162) miR-181a-3p, miR-409-5p, miR-410, miR-485-3p, miR-543, miR-654-3p (163) miR-17-5p (164) miR-155 (165) |
| T-cell | miR-126, miR-451, miR-181b (163) miR-126 (61) miR-224, miR-143 (65) miR-224 (53) miR-148a, miR-21 (57) miR-21 (59) miR-21-5p (58) miR-29b (60) miR-326 (69) miR-449b, miR-524-5p (54) | miR-125a-5p (62) miR-142-3p, miR-505, miR-324-5p (67) miR-142-3p (61) miR-142-3p, miR-142-5p (63) miR-145 (53) miR-31 (55) miR-410 (56) miR-633 (66) miR-98 (64) |
| B-cell | miR-29b, miR-494, miR-9 (166) miR-10a, miR-345 (65) miR-326 (102) miR-152-3p (101) miR-21, miR-22, miR-7 (99) miR-30a (100) | miR-29c, miR-181c, miR-223, miR-324-5p, miR-328, miR-362-3p, miR-744, let-7d, miR-7e, miR-26a, miR-335, miR-532, miR-579, miR-629 (166) miR-15b (103) miR-1246 (104) |
| Monocyte | miR-146a, miR-155 (35) | miR-124a, miR-125a (35) miR-19b, miR-20a (167) miR-302d (168) |
| NK cell | miR-27a-5p (154) | |
| Neutrophil | miR-124a, miR-125a, miR-222, miR-125b, miR-146a, miR-155 (35) | |
| Extracellular sample (serum, plasma, urine) | miR-142-3p, miR-181a (131) miR-551b, miR-448 (169) miR-126, miR-16, miR-21, miR-223, miR-451 (121) miR-146a (128) miR-146a, miR-155 (127) miR-146a (125) miR-181a (136) miR-181a, miR-196a, miR-21 (122) miR-21 (124) miR-21 (123) miR-223-3p, miR-30e-5p, miR-92a-3p (170) miR-371-5p, miR-5100, miR-4642, miR-548a-5p, miR-518b, miR-4762-5p, miR-767-3p, miR-4708-3p (171) | miR-103, miR-150, miR-20a, miR-223, miR-27a, miR-15b, miR-16, miR-181a, miR-19b, miR-22, miR-23a, miR-25, miR-92a, miR-93 (138) miR-106a, miR-17, miR-20a, miR-203, miR-92a (131) miR-151a-3p, miR-148b-3p, miR-106b-3p, miR-28-5p (172) miR-124 (169) miR-125a-3p, miR-155, miR-146a (121) miR-126 (173) miR-200a, miR-200b, miR-200c, miR-429, miR-205, miR-192, miR-141 (174) miR-146a, miR-155 (125) miR-146a (146) miR-150-5p, miR-21-5p, miR-221-3p (175) miR-203a (136) miR-31 (123) miR-148b-3p, miR-146a-5p, miR-223-3p, miR-1246, miR-21-5p (171) |
| MiRNAs in involved in LN pathogenesis (LN vs. HC, active LN vs. inactive LN, LN vs. LNN) | ||
| Renal tissue | let-7a, let-7e, let-7i, let-7g (176) miR-130b (177) miR-198, miR-146a, miR-638, miR-198 (178) miR-150 (179) miR-146a (180) miR-422a, miR-21 (181) miR-423-5p, miR-663a (148) | miR-130b (182) miR-146a (183) miR-638 (178) miR-23b (180) miR-26a, miR-30b, miR-4286 (184) miR-371-5p (185) miR-26a, miR-133 (181) |
| PBMC | miR-21, miR-155, miR-16, miR-21 (151) | miR-141 (151) miR-146a (186) miR-146a (159) miR-5571-5p, miR-766-3p (187) |
| T-cell | miR-126, miR-451, miR-181b (67) | miR-142-3p, miR-505, miR-324-5p (67) |
| B-cell | miR-145 (166) | miR-29c, miR-345, miR-21, miR-18b, miR-365 (134) |
| Extracellular sample (serum, plasma, urine) | miR-125a-5p, miR-146a-5p, miR-155-5p (188) miR-204-5p, miR-30c-5p (189) miR-130b-3p (190) miR-148a-3p (191) miR-150, miR-21 (192) miR-21-5p, miR-423-3p (193) miR-182-5p (194)* miR-3135b, miR-654-5p, miR-146a-5p (195) | let-7a, miR-21 (196) miR-3201, miR-1273e (189) miR-130b-3p (190) miR-200b-5p, miR-141-5p, miR-200c-5p (197) miR-410, miR-29c (192) miR-26a, miR-30b (184) miR-29c (198) |
Abbreviations: RNA, ribonucleic acid; SLE, systemic lupus erythematosus; HC, healthy control; LNN, lupus nephritis-negative; PBMC, peripheral blood mononuclear cells; NK cell, natural killer cell.
*measured in a whole blood sample
Expression levels of dysregulated miRNAs in SLE patients (expressed in log2FC values).
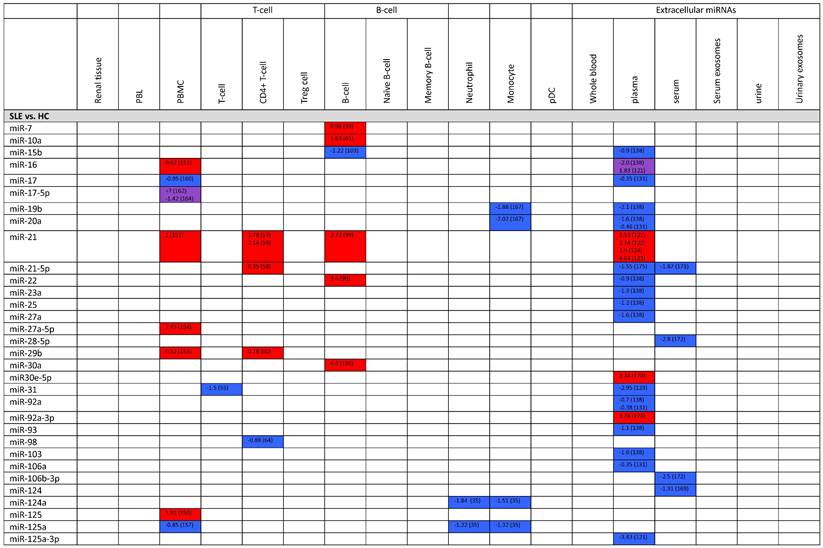
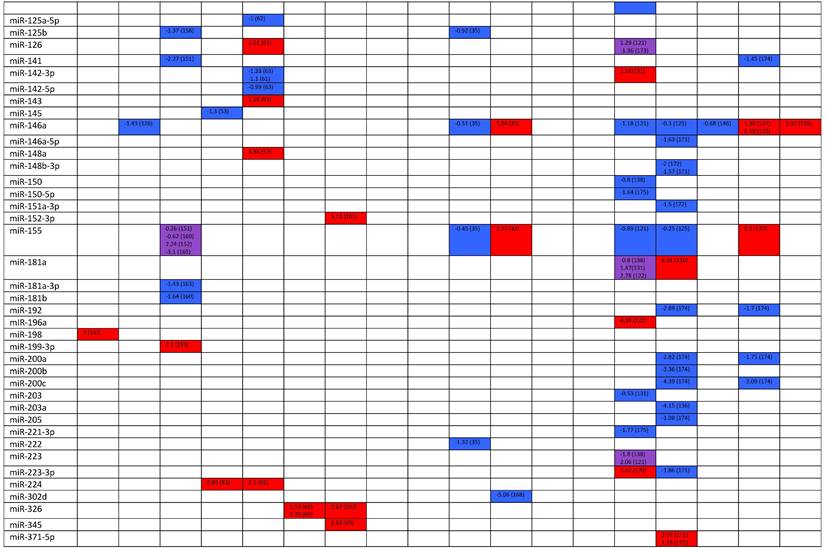
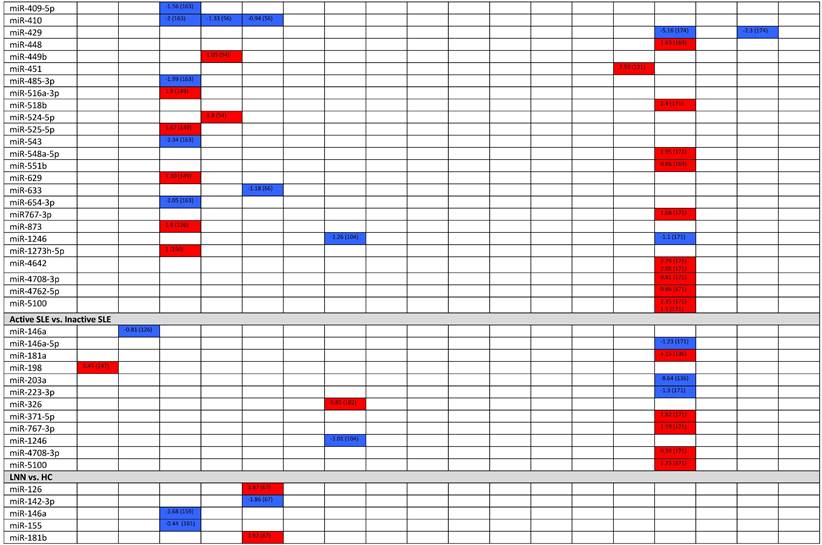
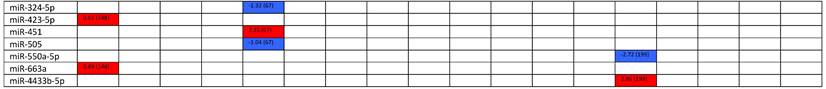
Abbreviations: FC, fold change; SLE, systemic lupus erythematosus; HC, healthy control; LNN, lupus nephritis-negative; PBL, peripheral blood leukocytes; PBMC, peripheral blood mononuclear cells; pDC, plasmacytoid dendritic cells.
Expression levels of dysregulated miRNAs in LN patients (expressed in log2FC values).
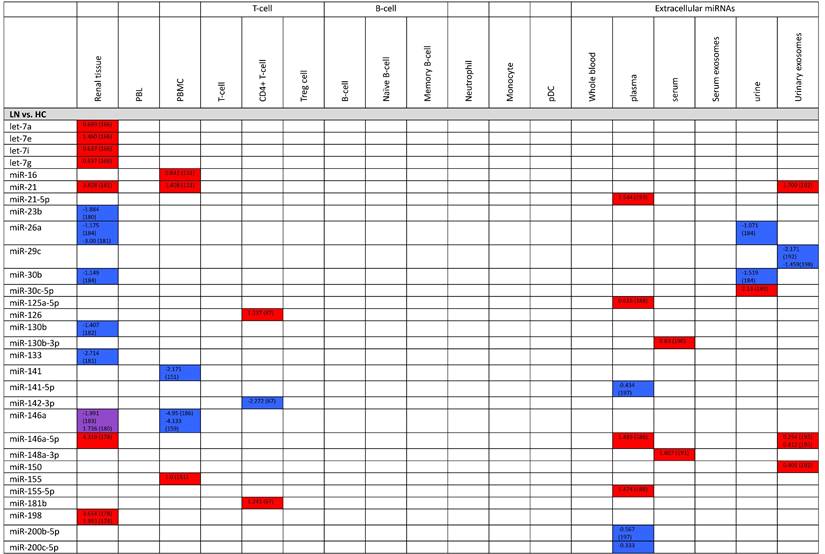
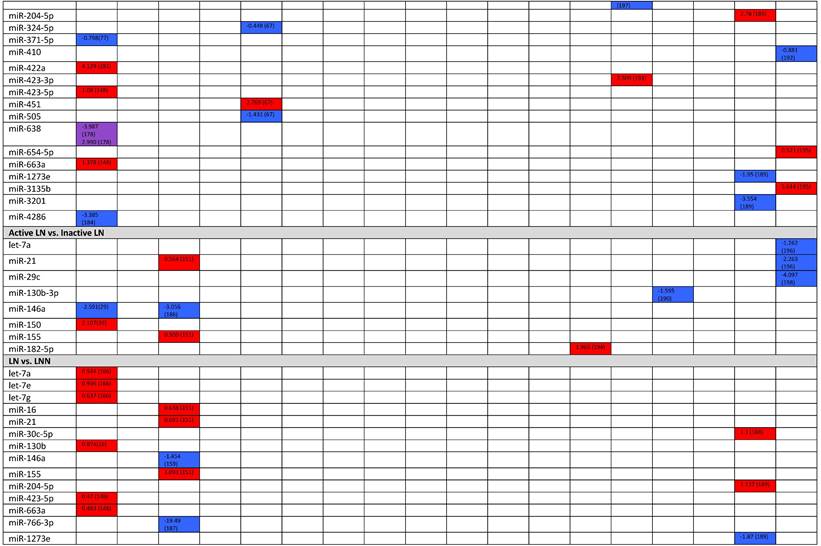

Abbreviations: FC, fold change; LN, lupus nephritis; HC, healthy control; LNN, lupus nephritis-negative; PBL, peripheral blood leukocytes; PBMC, peripheral blood mononuclear cells; pDC, plasmacytoid dendritic cells.
Concluding remarks and future perspectives
MiRNAs have been widely investigated in search for their roles in the pathogenesis of SLE. Many studies on dysregulation of miRNAs compare miRNA levels of lupus patients and healthy controls, then identify potentially pathogenic target genes using multiple databases that predict biological targets and functions of miRNAs (ex. TargetScan, miRDB). The most commonly studied miRNAs, also regarded as the most well-known contributors of autoimmune diseases, including lupus, are miR-146a, miR-155 and miR-181a (111). According to the databases used for identifying target genes, the potential targets of miR-146a are TRAF-6 and IRAK-1, both key components of the TLR4 signaling pathway, which takes part in innate immunity against extracellular pathogens, such as bacteria.
Studies have shown that miR-146a acts to downregulate this innate immune response against bacterial pathogens (139, 140). MiR-155, on the other hand, plays a key role in both innate and adaptive immunity, by regulating the germinal center response of B cells and Ig class switching of plasma cells (141, 142). Lastly, miR-181a is reported to take part in the development of bone marrow-derived B-cells and CD8+ T-cells (143).
In addition to the three miRNAs mentioned above, in the field of lupus pathogenesis, miR-21, miR-125b, miR-148a have also been studied extensively (144). Both miR-21 and miR-148a level are found to be increased in CD4+ T-cells of SLE patients. They are reported to function as suppressors of DNA methyltransferase 1 (DNMT1) in T-cells, promoting hypomethylation and overexpression of autoimmune-associated genes like CD70 in T-cells (57). MiR-21 is also known to be overexpressed in B-cells and plasma of lupus patients. Research has found that overexpressed miR-21 in B-cells suppress the expression of phosphatase and tensin homolog (PTEN), thereby inducing the generation and activation of self-reactive B-cells (99). Meanwhile, the specific mechanism leading to increased levels of miR-21 in the plasma of lupus patients remains unclear. Scholars predict that it might be due to the secretion from circulatory CD4+ T-cells with upregulated miR-21, which are overly exposed to circulatory plasma in inflammatory conditions such as SLE (121). Furthermore, miR-146a and miR-155 levels in plasma, serum, and neutrophils were decreased in SLE patients, whereas their urine level was increased (35, 121, 125-128, 145, 146). MiR-181a levels in plasma of SLE patients were controversial among studies, but most have reported that its level was increased, in correlation with SLEDAI score (122, 131, 136-138).
Although dysregulated miRNA levels measured in B-cells, T-cells, plasma, and urine each have very different meanings, the type of specimen used to estimate miRNA levels tend to be overlooked in the past studies on miRNAs. Moreover, there is growing interest in the dysregulation of extracellular miRNAs clinically obtained from plasma, serum, and urine, partly due to their easy accessibilities. However, its physiological function, whether it participates in cell-to-cell communication or is just a byproduct of circulatory cell death, and its specific mechanism of selective secretion from circulatory cells still remains unclear (118, 119, 121), which calls for further studies on the functions of extracellular miRNA itself.
Author Contributions
All authors made substantial contributions to all of the following: (1) conception and design of the study, data acquisition, or analysis and interpretation of data; (2) drafting or critical revision of the article for intellectual conduct; and (3) final approval of version to be submitted.
Competing Interests
The authors have declared that no competing interest exists.
References
1. D'Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet. 2007;369(9561):587-96
2. Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol. 2006;92:1-69
3. Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol. 2003;56(7):481-90
4. Carter EE, Barr SG, Clarke AE. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. 2016;12(10):605-20
5. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781-3
6. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J. et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20(2):170-9
7. Liu A, La Cava A. Epigenetic dysregulation in systemic lupus erythematosus. Autoimmunity. 2014;47(4):215-9
8. Foma AM, Aslani S, Karami J, Jamshidi A, Mahmoudi M. Epigenetic involvement in etiopathogenesis and implications in treatment of systemic lupus erythematous. Inflamm Res. 2017;66(12):1057-73
9. Ma X, Liu Q. MicroRNAs in the pathogenesis of systemic lupus erythematosus. Int J Rheum Dis. 2013;16(2):115-21
10. John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2(11):e363
11. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH. et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051-60
12. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415-9
13. Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24):3011-6
14. Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K. et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436(7051):740-4
15. Mah SM, Buske C, Humphries RK, Kuchenbauer F. miRNA*: a passenger stranded in RNA-induced silencing complex? Crit Rev Eukaryot Gene Expr. 2010;20(2):141-8
16. Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115(2):199-208
17. Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115(2):209-16
18. Harley JB, Kelly JA, Kaufman KM. Unraveling the genetics of systemic lupus erythematosus. Springer Semin Immunopathol. 2006;28(2):119-30
19. Ko K, Franek BS, Marion M, Kaufman KM, Langefeld CD, Harley JB. et al. Genetic ancestry, serum interferon-alpha activity, and autoantibodies in systemic lupus erythematosus. J Rheumatol. 2012;39(6):1238-40
20. Ko K, Koldobskaya Y, Rosenzweig E, Niewold TB. Activation of the Interferon Pathway is Dependent Upon Autoantibodies in African-American SLE Patients, but Not in European-American SLE Patients. Front Immunol. 2013;4:309
21. Sharma S, Jin Z, Rosenzweig E, Rao S, Ko K, Niewold TB. Widely divergent transcriptional patterns between SLE patients of different ancestral backgrounds in sorted immune cell populations. J Autoimmun. 2015;60:51-8
22. Sanchez E, Comeau ME, Freedman BI, Kelly JA, Kaufman KM, Langefeld CD. et al. Identification of novel genetic susceptibility loci in African American lupus patients in a candidate gene association study. Arthritis Rheum. 2011;63(11):3493-501
23. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716-30
24. Dema B, Charles N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies (Basel). 2016 5(1)
25. Cozzani E, Drosera M, Gasparini G, Parodi A. Serology of Lupus Erythematosus: Correlation between Immunopathological Features and Clinical Aspects. Autoimmune Dis. 2014;2014:321359
26. Kavanaugh A, Tomar R, Reveille J, Solomon DH, Homburger HA. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch Pathol Lab Med. 2000;124(1):71-81
27. Mahajan A, Herrmann M, Munoz LE. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol. 2016;7:35
28. Linker-Israeli M, Quismorio FP Jr, Horwitz DA. CD8+ lymphocytes from patients with systemic lupus erythematosus sustain, rather than suppress, spontaneous polyclonal IgG production and synergize with CD4+ cells to support autoantibody synthesis. Arthritis Rheum. 1990;33(8):1216-25
29. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516
30. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390(6658):350-1
31. Kaplan MJ. Apoptosis in systemic lupus erythematosus. Clin Immunol. 2004;112(3):210-8
32. Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30(11):513-21
33. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD. et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15(11):1017-25
34. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol. 2013;191(5):2647-56
35. Perez-Sanchez C, Aguirre MA, Ruiz-Limon P, Barbarroja N, Jimenez-Gomez Y, de la Rosa IA. et al. 'Atherothrombosis-associated microRNAs in Antiphospholipid syndrome and Systemic Lupus Erythematosus patients'. Sci Rep. 2016;6:31375
36. Pan L, Lu MP, Wang JH, Xu M, Yang SR. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J Pediatr. 2020;16(1):19-30
37. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6(6):317-25
38. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. 2019;16(7):634-43
39. Katsuyama T, Tsokos GC, Moulton VR. Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front Immunol. 2018;9:1088
40. Simeoni L. Lck activation: puzzling the pieces together. Oncotarget. 2017;8(61):102761-2
41. Tsokos GC, Nambiar MP, Tenbrock K, Juang YT. Rewiring the T-cell: signaling defects and novel prospects for the treatment of SLE. Trends Immunol. 2003;24(5):259-63
42. Liu CP, Lin WJ, Huang M, Kappler JW, Marrack P. Development and function of T cells in T cell antigen receptor/CD3 zeta knockout mice reconstituted with Fc epsilon RI gamma. Proc Natl Acad Sci U S A. 1997;94(2):616-21
43. Solomou EE, Juang YT, Gourley MF, Kammer GM, Tsokos GC. Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J Immunol. 2001;166(6):4216-22
44. Lee JH, Katakai T, Hara T, Gonda H, Sugai M, Shimizu A. Roles of p-ERM and Rho-ROCK signaling in lymphocyte polarity and uropod formation. J Cell Biol. 2004;167(2):327-37
45. Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M. et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007;178(3):1938-47
46. Tang H, Tan G, Guo Q, Pang R, Zeng F. Abnormal activation of the Akt-GSK3beta signaling pathway in peripheral blood T cells from patients with systemic lupus erythematosus. Cell Cycle. 2009;8(17):2789-93
47. Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H. et al. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J Clin Invest. 2007;117(8):2186-96
48. Viallard JF, Pellegrin JL, Ranchin V, Schaeverbeke T, Dehais J, Longy-Boursier M. et al. Th1 (IL-2, interferon-gamma (IFN-gamma)) and Th2 (IL-10, IL-4) cytokine production by peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE). Clin Exp Immunol. 1999;115(1):189-95
49. Kleczynska W, Jakiela B, Plutecka H, Milewski M, Sanak M, Musial J. Imbalance between Th17 and regulatory T-cells in systemic lupus erythematosus. Folia Histochem Cytobiol. 2011;49(4):646-53
50. Qu N, Xu M, Mizoguchi I, Furusawa J, Kaneko K, Watanabe K. et al. Pivotal roles of T-helper 17-related cytokines, IL-17, IL-22, and IL-23, in inflammatory diseases. Clin Dev Immunol. 2013;2013:968549
51. Talaat RM, Mohamed SF, Bassyouni IH, Raouf AA. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine. 2015;72(2):146-53
52. Dolff S, Bijl M, Huitema MG, Limburg PC, Kallenberg CG, Abdulahad WH. Disturbed Th1, Th2, Th17 and T(reg) balance in patients with systemic lupus erythematosus. Clin Immunol. 2011;141(2):197-204
53. Lu MC, Lai NS, Chen HC, Yu HC, Huang KY, Tung CH. et al. Decreased microRNA(miR)-145 and increased miR-224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin Exp Immunol. 2013;171(1):91-9
54. Lu MC, Yu CL, Chen HC, Yu HC, Huang HB, Lai NS. Aberrant T cell expression of Ca2+ influx-regulated miRNAs in patients with systemic lupus erythematosus promotes lupus pathogenesis. Rheumatology (Oxford). 2015;54(2):343-8
55. Fan W, Liang D, Tang Y, Qu B, Cui H, Luo X. et al. Identification of microRNA-31 as a novel regulator contributing to impaired interleukin-2 production in T cells from patients with systemic lupus erythematosus. Arthritis Rheum. 2012;64(11):3715-25
56. Liu D, Zhang N, Zhang X, Qin M, Dong Y, Jin L. MiR-410 Down-Regulates the Expression of Interleukin-10 by Targeting STAT3 in the Pathogenesis of Systemic Lupus Erythematosus. Cell Physiol Biochem. 2016;39(1):303-15
57. Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X. et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184(12):6773-81
58. Zhao M, Li MY, Gao XF, Jia SJ, Gao KQ, Zhou Y. et al. Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4(+) T cells of systemic lupus erythematosus. Clin Immunol. 2018;187:113-21
59. Suo QF, Sheng J, Qiang FY, Tang ZS, Yang YY. Association of long non-coding RNA GAS5 and miR-21 levels in CD4(+) T cells with clinical features of systemic lupus erythematosus. Exp Ther Med. 2018;15(1):345-50
60. Qin H, Zhu X, Liang J, Wu J, Yang Y, Wang S. et al. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. 2013;69(1):61-7
61. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S. et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011;63(5):1376-86
62. Pan W, Zhu S, Dai D, Liu Z, Li D, Li B. et al. MiR-125a targets effector programs to stabilize Treg-mediated immune homeostasis. Nat Commun. 2015;6:7096
63. Ding S, Liang Y, Zhao M, Liang G, Long H, Zhao S. et al. Decreased microRNA-142-3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. 2012;64(9):2953-63
64. Xie L, Xu J. Role of MiR-98 and Its Underlying Mechanisms in Systemic Lupus Erythematosus. J Rheumatol. 2018;45(10):1397-405
65. Martinez-Ramos R, Garcia-Lozano JR, Lucena JM, Castillo-Palma MJ, Garcia-Hernandez F, Rodriguez MC. et al. Differential expression pattern of microRNAs in CD4+ and CD19+ cells from asymptomatic patients with systemic lupus erythematosus. Lupus. 2014;23(4):353-9
66. Chen S, Wang Y, Qin H, Lin J, Xie L, Chen S. et al. Downregulation of miR-633 activated AKT/mTOR pathway by targeting AKT1 in lupus CD4(+) T cells. Lupus. 2019;28(4):510-9
67. Zhao M, Liu S, Luo S, Wu H, Tang M, Cheng W. et al. DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J Autoimmun. 2014;54:127-36
68. Yu N, Li X, Song W, Li D, Yu D, Zeng X. et al. CD4(+)CD25 (+)CD127 (low/-) T cells: a more specific Treg population in human peripheral blood. Inflammation. 2012;35(6):1773-80
69. Sun XG, Tao JH, Xiang N, Li XM, Wang GS, Fang X. et al. Negative Correlation Between miR-326 and Ets-1 in Regulatory T Cells from new-Onset SLE Patients. Inflammation. 2016;39(2):822-9
70. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol. 2020;20(4):229-38
71. Marie-Cardine A, Divay F, Dutot I, Green A, Perdrix A, Boyer O. et al. Transitional B cells in humans: characterization and insight from B lymphocyte reconstitution after hematopoietic stem cell transplantation. Clin Immunol. 2008;127(1):14-25
72. Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood. 2005;105(11):4390-8
73. Rowland SL, Leahy KF, Halverson R, Torres RM, Pelanda R. BAFF receptor signaling aids the differentiation of immature B cells into transitional B cells following tonic BCR signaling. J Immunol. 2010;185(8):4570-81
74. Milne CD, Fleming HE, Zhang Y, Paige CJ. Mechanisms of selection mediated by interleukin-7, the preBCR, and hemokinin-1 during B-cell development. Immunol Rev. 2004;197:75-88
75. Erlandsson L, Licence S, Gaspal F, Lane P, Corcoran AE, Martensson IL. Both the pre-BCR and the IL-7Ralpha are essential for expansion at the pre-BII cell stage in vivo. Eur J Immunol. 2005;35(6):1969-76
76. Milne CD, Paige CJ. IL-7: a key regulator of B lymphopoiesis. Semin Immunol. 2006;18(1):20-30
77. Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C. et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann N Y Acad Sci. 2005;1050:34-9
78. Davidson A. The rationale for BAFF inhibition in systemic lupus erythematosus. Curr Rheumatol Rep. 2012;14(4):295-302
79. Xu W, Santini PA, Matthews AJ, Chiu A, Plebani A, He B. et al. Viral double-stranded RNA triggers Ig class switching by activating upper respiratory mucosa B cells through an innate TLR3 pathway involving BAFF. J Immunol. 2008;181(1):276-87
80. He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004;173(7):4479-91
81. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F. et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20(6):785-98
82. Ota M, Duong BH, Torkamani A, Doyle CM, Gavin AL, Ota T. et al. Regulation of the B cell receptor repertoire and self-reactivity by BAFF. J Immunol. 2010;185(7):4128-36
83. Davidson A. Targeting BAFF in autoimmunity. Curr Opin Immunol. 2010;22(6):732-9
84. Qin L, Waseem TC, Sahoo A, Bieerkehazhi S, Zhou H, Galkina EV. et al. Insights Into the Molecular Mechanisms of T Follicular Helper-Mediated Immunity and Pathology. Front Immunol. 2018;9:1884
85. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T. et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34(6):961-72
86. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S. et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 2015;67(4):988-99
87. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N. et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013;39(4):770-81
88. Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR. et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32(1):129-40
89. Oleinika K, Mauri C, Salama AD. Effector and regulatory B cells in immune-mediated kidney disease. Nat Rev Nephrol. 2019;15(1):11-26
90. Hasan MM, Thompson-Snipes L, Klintmalm G, Demetris AJ, O'Leary J, Oh S. et al. CD24(hi)CD38(hi) and CD24(hi)CD27(+) Human Regulatory B Cells Display Common and Distinct Functional Characteristics. J Immunol. 2019;203(8):2110-20
91. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA. et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. 2013;5(173):173ra23
92. Taher TE, Ong VH, Bystrom J, Hillion S, Simon Q, Denton CP. et al. Association of Defective Regulation of Autoreactive Interleukin-6-Producing Transitional B Lymphocytes With Disease in Patients With Systemic Sclerosis. Arthritis Rheumatol. 2018;70(3):450-61
93. Piper CJM, Wilkinson MGL, Deakin CT, Otto GW, Dowle S, Duurland CL. et al. CD19(+)CD24(hi)CD38(hi) B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-alpha. Front Immunol. 2018;9:1372
94. Burton H, Dorling A. Transitional B cell subsets-a convincing predictive biomarker for allograft loss? Kidney Int. 2017;91(1):18-20
95. Liu M, Guo Q, Wu C, Sterlin D, Goswami S, Zhang Y. et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol Immunol. 2019;16(4):367-79
96. Simon Q, Pers JO, Cornec D, Le Pottier L, Mageed RA, Hillion S. In-depth characterization of CD24(high)CD38(high) transitional human B cells reveals different regulatory profiles. J Allergy Clin Immunol. 2016;137(5):1577-84 e10
97. Carvajal Alegria G, Gazeau P, Hillion S, Daien CI, Cornec DYK. Could Lymphocyte Profiling be Useful to Diagnose Systemic Autoimmune Diseases? Clin Rev Allergy Immunol. 2017;53(2):219-36
98. Wang L, Zhao P, Ma L, Shan Y, Jiang Z, Wang J. et al. Increased interleukin 21 and follicular helper T-like cells and reduced interleukin 10+ B cells in patients with new-onset systemic lupus erythematosus. J Rheumatol. 2014;41(9):1781-92
99. Wu XN, Ye YX, Niu JW, Li Y, Li X, You X. et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med. 2014;6(246):246ra99
100. Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y. et al. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum. 2013;65(6):1603-11
101. Luo S, Ding S, Liao J, Zhang P, Liu Y, Zhao M. et al. Excessive miR-152-3p Results in Increased BAFF Expression in SLE B-Cells by Inhibiting the KLF5 Expression. Front Immunol. 2019;10:1127
102. Jin L, Fang X, Dai C, Xiang N, Tao J, Sun X. et al. The potential role of Ets-1 and miR-326 in CD19(+)B cells in the pathogenesis of patients with systemic lupus erythematosus. Clin Rheumatol. 2019;38(4):1031-8
103. Ren D, Liu F, Dong G, You M, Ji J, Huang Y. et al. Activation of TLR7 increases CCND3 expression via the downregulation of miR-15b in B cells of systemic lupus erythematosus. Cell Mol Immunol. 2016;13(6):764-75
104. Luo S, Liu Y, Liang G, Zhao M, Wu H, Liang Y. et al. The role of microRNA-1246 in the regulation of B cell activation and the pathogenesis of systemic lupus erythematosus. Clin Epigenetics. 2015;7:24
105. Shi X, Ye L, Xu S, Guo G, Zuo Z, Ye M. et al. Downregulated miR29a promotes B cell overactivation by upregulating Crklike protein in systemic lupus erythematosus. Mol Med Rep. 2020;22(2):841-9
106. Chan VS, Nie YJ, Shen N, Yan S, Mok MY, Lau CS. Distinct roles of myeloid and plasmacytoid dendritic cells in systemic lupus erythematosus. Autoimmun Rev. 2012;11(12):890-7
107. Batteux F, Palmer P, Daeron M, Weill B, Lebon P. FCgammaRII (CD32)-dependent induction of interferon-alpha by serum from patients with lupus erythematosus. Eur Cytokine Netw. 1999;10(4):509-14
108. Liu Z, Davidson A. IFNalpha Inducible Models of Murine SLE. Front Immunol. 2013;4:306
109. van den Hoogen LL, Rossato M, Lopes AP, Pandit A, Bekker CPJ, Fritsch-Stork RDE. et al. microRNA downregulation in plasmacytoid dendritic cells in interferon-positive systemic lupus erythematosus and antiphospholipid syndrome. Rheumatology (Oxford). 2018;57(9):1669-74
110. Dai R, Ahmed SA. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res. 2011;157(4):163-79
111. Pauley KM, Cha S, Chan EK. MicroRNA in autoimmunity and autoimmune diseases. J Autoimmun. 2009;32(3-4):189-94
112. Chim SS, Shing TK, Hung EC, Leung TY, Lau TK, Chiu RW. et al. Detection and characterization of placental microRNAs in maternal plasma. Clin Chem. 2008;54(3):482-90
113. Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K. et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. 2008;141(5):672-5
114. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513-8
115. Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K. et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997-1006
116. Park NJ, Zhou H, Elashoff D, Henson BS, Kastratovic DA, Abemayor E. et al. Salivary microRNA: discovery, characterization, and clinical utility for oral cancer detection. Clin Cancer Res. 2009;15(17):5473-7
117. Hanke M, Hoefig K, Merz H, Feller AC, Kausch I, Jocham D. et al. A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol Oncol. 2010;28(6):655-61
118. Turchinovich A, Samatov TR, Tonevitsky AG, Burwinkel B. Circulating miRNAs: cell-cell communication function? Front Genet. 2013;4:119
119. Turchinovich A, Tonevitsky AG, Burwinkel B. Extracellular miRNA: A Collision of Two Paradigms. Trends Biochem Sci. 2016;41(10):883-92
120. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018;9:402
121. Wang H, Peng W, Ouyang X, Li W, Dai Y. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl Res. 2012;160(3):198-206
122. Motawi TK, Mohsen DA, El-Maraghy SA, Kortam MA. MicroRNA-21, microRNA-181a and microRNA-196a as potential biomarkers in adult Egyptian patients with systemic lupus erythematosus. Chem Biol Interact. 2016;260:110-6
123. Amr KS, Bayoumi FS, Elgengehy FT, Abdallah SO, Ahmed HH, Eissa E. The role of microRNA-31 and microRNA-21 as regulatory biomarkers in the activation of T lymphocytes of Egyptian lupus patients. Rheumatol Int. 2016;36(11):1617-25
124. Tang ZM, Fang M, Wang JP, Cai PC, Wang P, Hu LH. Clinical relevance of plasma miR-21 in new-onset systemic lupus erythematosus patients. J Clin Lab Anal. 2014;28(6):446-51
125. Wang G, Tam LS, Li EK, Kwan BC, Chow KM, Luk CC. et al. Serum and urinary cell-free MiR-146a and MiR-155 in patients with systemic lupus erythematosus. J Rheumatol. 2010;37(12):2516-22
126. Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y. et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60(4):1065-75
127. Wang G, Tam LS, Kwan BC, Li EK, Chow KM, Luk CC. et al. Expression of miR-146a and miR-155 in the urinary sediment of systemic lupus erythematosus. Clin Rheumatol. 2012;31(3):435-40
128. Perez-Hernandez J, Forner MJ, Pinto C, Chaves FJ, Cortes R, Redon J. Increased Urinary Exosomal MicroRNAs in Patients with Systemic Lupus Erythematosus. PLoS One. 2015;10(9):e0138618
129. Divekar AA, Dubey S, Gangalum PR, Singh RR. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. J Immunol. 2011;186(2):924-30
130. Prokopi M, Pula G, Mayr U, Devue C, Gallagher J, Xiao Q. et al. Proteomic analysis reveals presence of platelet microparticles in endothelial progenitor cell cultures. Blood. 2009;114(3):723-32
131. Carlsen AL, Schetter AJ, Nielsen CT, Lood C, Knudsen S, Voss A. et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013;65(5):1324-34
132. Xie W, Li Z, Li M, Xu N, Zhang Y. miR-181a and inflammation: miRNA homeostasis response to inflammatory stimuli in vivo. Biochem Biophys Res Commun. 2013;430(2):647-52
133. Galicia JC, Naqvi AR, Ko CC, Nares S, Khan AA. MiRNA-181a regulates Toll-like receptor agonist-induced inflammatory response in human fibroblasts. Genes Immun. 2014;15(5):333-7
134. Xie W, Li M, Xu N, Lv Q, Huang N, He J. et al. MiR-181a regulates inflammation responses in monocytes and macrophages. PLoS One. 2013;8(3):e58639
135. Ramkissoon SH, Mainwaring LA, Ogasawara Y, Keyvanfar K, McCoy JP Jr, Sloand EM. et al. Hematopoietic-specific microRNA expression in human cells. Leuk Res. 2006;30(5):643-7
136. Li HS, Ning Y, Li SB, Shao PY, Chen SJ, Ye Q. et al. Expression and clinical significance of miR-181a and miR-203 in systemic lupus erythematosus patients. Eur Rev Med Pharmacol Sci. 2017;21(21):4790-6
137. Lashine YA, Seoudi AM, Salah S, Abdelaziz AI. Expression signature of microRNA-181-a reveals its crucial role in the pathogenesis of paediatric systemic lupus erythematosus. Clin Exp Rheumatol. 2011;29(2):351-7
138. Zhang H, Huang X, Ye L, Guo G, Li X, Chen C. et al. B Cell-Related Circulating MicroRNAs With the Potential Value of Biomarkers in the Differential Diagnosis, and Distinguishment Between the Disease Activity and Lupus Nephritis for Systemic Lupus Erythematosus. Front Immunol. 2018;9:1473
139. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481-6
140. Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol. 2008;180(8):5689-98
141. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. 2018;392(10141):75-87
142. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S. et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27(6):847-59
143. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303(5654):83-6
144. Zhang L, Wu H, Zhao M, Lu Q. Identifying the differentially expressed microRNAs in autoimmunity: A systemic review and meta-analysis. Autoimmunity. 2020;53(3):122-36
145. Li X, Liu L, Meng D, Wang D, Zhang J, Shi D. et al. Enhanced apoptosis and senescence of bone-marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Stem Cells Dev. 2012;21(13):2387-94
146. Dong C, Zhou Q, Fu T, Zhao R, Yang J, Kong X. et al. Circulating Exosomes Derived-miR-146a from Systemic Lupus Erythematosus Patients Regulates Senescence of Mesenchymal Stem Cells. Biomed Res Int. 2019;2019:6071308
147. Cui D, Zhu D, Ren H, Lin J, Lai W, Huang Q. et al. MicroRNA198 contributes to lupus nephritis progression by inhibition of phosphatase and tensin homology deleted on chromosome ten expression. Mol Med Rep. 2017;16(5):7813-20
148. Wang W, Gao J, Wang F. MiR-663a/MiR-423-5p are involved in the pathogenesis of lupus nephritis via modulating the activation of NF-kappaB by targeting TNIP2. Am J Transl Res. 2017;9(8):3796-803
149. Zhu J, Huang X, Su G, Wang L, Wu F, Zhang T. et al. High expression levels of microRNA-629, microRNA-525-5p and microRNA-516a-3p in paediatric systemic lupus erythematosus. Clin Rheumatol. 2014;33(6):807-15
150. Guo G, Wang H, Shi X, Ye L, Wu K, Lin K. et al. NovelmiRNA-25 inhibits AMPD2 in peripheral blood mononuclear cells of patients with systemic lupus erythematosus and represents a promising novel biomarker. J Transl Med. 2018;16(1):370
151. Khoshmirsafa M, Kianmehr N, Falak R, Mowla SJ, Seif F, Mirzaei B. et al. Elevated expression of miR-21 and miR-155 in peripheral blood mononuclear cells as potential biomarkers for lupus nephritis. Int J Rheum Dis. 2019;22(3):458-67
152. Wen Z, Xu L, Chen X, Xu W, Yin Z, Gao X. et al. Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J Immunol. 2013;190(11):5411-22
153. Su X, Ye L, Chen X, Zhang H, Zhou Y, Ding X. et al. MiR-199-3p promotes ERK-mediated IL-10 production by targeting poly (ADP-ribose) Polymerase-1 in patients with systemic lupus erythematosus. Chem Biol Interact. 2019;306:110-6
154. Sourour SK, Aboelenein HR, Elemam NM, Abdelhamid AK, Salah S, Abdelaziz AI. Unraveling the expression of microRNA-27a* & NKG2D in peripheral blood mononuclear cells and natural killer cells of pediatric systemic lupus erythematosus patients. Int J Rheum Dis. 2017;20(9):1237-46
155. Wang X, Zhang C, Wu Z, Chen Y, Shi W. CircIBTK inhibits DNA demethylation and activation of AKT signaling pathway via miR-29b in peripheral blood mononuclear cells in systemic lupus erythematosus. Arthritis Res Ther. 2018;20(1):118
156. Liu L, Liu Y, Yuan M, Xu L, Sun H. Elevated expression of microRNA-873 facilitates Th17 differentiation by targeting forkhead box O1 (Foxo1) in the pathogenesis of systemic lupus erythematosus. Biochem Biophys Res Commun. 2017;492(3):453-60
157. Zhao X, Tang Y, Qu B, Cui H, Wang S, Wang L. et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 2010;62(11):3425-35
158. Cao W, Qian G, Luo W, Liu X, Pu Y, Hu G. et al. miR-125b is downregulated in systemic lupus erythematosus patients and inhibits autophagy by targeting UVRAG. Biomed Pharmacother. 2018;99:791-7
159. Hashad DI, Abdelmagid MH, Elsherif SH. microRNA146a expression in lupus patients with and without renal complications. J Clin Lab Anal. 2012;26(1):35-40
160. Kaga H, Komatsuda A, Omokawa A, Ito M, Teshima K, Tagawa H. et al. Downregulated expression of miR-155, miR-17, and miR-181b, and upregulated expression of activation-induced cytidine deaminase and interferon-alpha in PBMCs from patients with SLE. Mod Rheumatol. 2015;25(6):865-70
161. Rasmussen TK, Andersen T, Bak RO, Yiu G, Sorensen CM, Stengaard-Pedersen K. et al. Overexpression of microRNA-155 increases IL-21 mediated STAT3 signaling and IL-21 production in systemic lupus erythematosus. Arthritis Res Ther. 2015;17:154
162. Sarhan RA, Aboelenein HR, Sourour SK, Fawzy IO, Salah S, Abdelaziz AI. Targeting E2F1 and c-Myc expression by microRNA-17-5p represses interferon-stimulated gene MxA in peripheral blood mononuclear cells of pediatric systemic lupus erythematosus patients. Discov Med. 2015;19(107):419-25
163. Liu D, Zhao H, Zhao S, Wang X. MicroRNA expression profiles of peripheral blood mononuclear cells in patients with systemic lupus erythematosus. Acta Histochem. 2014;116(5):891-7
164. Aboelenein HR, Salah S, Lashine YA, Abdelaziz AI. Dual downregulation of microRNA 17-5p and E2F1 transcriptional factor in pediatric systemic lupus erythematosus patients. Rheumatol Int. 2013;33(5):1333-8
165. Lashine YA, Salah S, Aboelenein HR, Abdelaziz AI. Correcting the expression of miRNA-155 represses PP2Ac and enhances the release of IL-2 in PBMCs of juvenile SLE patients. Lupus. 2015;24(3):240-7
166. Duroux-Richard I, Cuenca J, Ponsolles C, Pineiro AB, Gonzalez F, Roubert C. et al. MicroRNA Profiling of B Cell Subsets from Systemic Lupus Erythematosus Patients Reveals Promising Novel Biomarkers. Int J Mol Sci. 2015;16(8):16953-65
167. Teruel R, Perez-Sanchez C, Corral J, Herranz MT, Perez-Andreu V, Saiz E. et al. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J Thromb Haemost. 2011;9(10):1985-92
168. Smith S, Fernando T, Wu PW, Seo J, Ni Gabhann J, Piskareva O. et al. MicroRNA-302d targets IRF9 to regulate the IFN-induced gene expression in SLE. J Autoimmun. 2017;79:105-11
169. Jin F, Hu H, Xu M, Zhan S, Wang Y, Zhang H. et al. Serum microRNA Profiles Serve as Novel Biomarkers for Autoimmune Diseases. Front Immunol. 2018;9:2381
170. Kim BS, Jung JY, Jeon JY, Kim HA, Suh CH. Circulating hsa-miR-30e-5p, hsa-miR-92a-3p, and hsa-miR-223-3p may be novel biomarkers in systemic lupus erythematosus. HLA. 2016;88(4):187-93
171. Zeng L, Wu JL, Liu LM, Jiang JQ, Wu HJ, Zhao M. et al. Serum miRNA-371b-5p and miRNA-5100 act as biomarkers for systemic lupus erythematosus. Clin Immunol. 2018;196:103-9
172. Perez-Sanchez C, Cecchi I, Barbarroja N, Patino-Trives AM, Luque-Tevar M, Perez-Sanchez L. et al. Early restoration of immune and vascular phenotypes in systemic lupus erythematosus and rheumatoid arthritis patients after B cell depletion. J Cell Mol Med. 2019;23(9):6308-18
173. Liu YJ, Fan WJ, Bai JZ. microRNA-126 expression and its mechanism of action in patients with systemic lupus erythematosus. Eur Rev Med Pharmacol Sci. 2015;19(20):3838-42
174. Wang G, Tam LS, Li EK, Kwan BC, Chow KM, Luk CC. et al. Serum and urinary free microRNA level in patients with systemic lupus erythematosus. Lupus. 2011;20(5):493-500
175. Su YJ, Tsai NW, Kung CT, Wang HC, Lin WC, Huang CC. et al. Investigation of MicroRNA in Mitochondrial Apoptotic Pathway in Systemic Lupus Erythematosus. Biomed Res Int. 2018;2018:9026357
176. Liu J, Zhu L, Xie GL, Bao JF, Yu Q. Let-7 miRNAs Modulate the Activation of NF-kappaB by Targeting TNFAIP3 and Are Involved in the Pathogenesis of Lupus Nephritis. PLoS One. 2015;10(6):e0121256
177. Wu S, Wang J, Li F. Dysregulation of PTEN caused by the underexpression of microRNA130b is associated with the severity of lupus nephritis. Mol Med Rep. 2018;17(6):7966-72
178. Lu J, Kwan BC, Lai FM, Tam LS, Li EK, Chow KM. et al. Glomerular and tubulointerstitial miR-638, miR-198 and miR-146a expression in lupus nephritis. Nephrology (Carlton). 2012;17(4):346-51
179. Zhou H, Hasni SA, Perez P, Tandon M, Jang SI, Zheng C. et al. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J Am Soc Nephrol. 2013;24(7):1073-87
180. Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y. et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-alpha. Nat Med. 2012;18(7):1077-86
181. Krasoudaki E, Banos A, Stagakis E, Loupasakis K, Drakos E, Sinatkas V. et al. Micro-RNA analysis of renal biopsies in human lupus nephritis demonstrates up-regulated miR-422a driving reduction of kallikrein-related peptidase 4. Nephrol Dial Transplant. 2016;31(10):1676-86
182. Han X, Wang Y, Zhang X, Qin Y, Qu B, Wu L. et al. MicroRNA-130b Ameliorates Murine Lupus Nephritis Through Targeting the Type I Interferon Pathway on Renal Mesangial Cells. Arthritis Rheumatol. 2016;68(9):2232-43
183. Zheng CZ, Shu YB, Luo YL, Luo J. The role of miR-146a in modulating TRAF6-induced inflammation during lupus nephritis. Eur Rev Med Pharmacol Sci. 2017;21(5):1041-8
184. Costa-Reis P, Russo PA, Zhang Z, Colonna L, Maurer K, Gallucci S. et al. The Role of MicroRNAs and Human Epidermal Growth Factor Receptor 2 in Proliferative Lupus Nephritis. Arthritis Rheumatol. 2015;67(9):2415-26
185. Yao F, Sun L, Fang W, Wang H, Yao D, Cui R. et al. HsamiR3715p inhibits human mesangial cell proliferation and promotes apoptosis in lupus nephritis by directly targeting hypoxiainducible factor 1alpha. Mol Med Rep. 2016;14(6):5693-8
186. Zhu Y, Xue Z, Di L. Regulation of MiR-146a and TRAF6 in the Diagnose of Lupus Nephritis. Med Sci Monit. 2017;23:2550-7
187. Jafari Ghods F, Topal Sarikaya A, Arda N, Hamuryudan V. MiRNA and mRNA Profiling in Systemic Lupus Reveals a Novel Set of Cytokine - Related miRNAs and their Target Genes in Cases With and Without Renal Involvement. Kidney Blood Press Res. 2017;42(6):1322-37
188. Zununi Vahed S, Nakhjavani M, Etemadi J, Jamshidi H, Jadidian N, Pourlak T. et al. Altered levels of immune-regulatory microRNAs in plasma samples of patients with lupus nephritis. Bioimpacts. 2018;8(3):177-83
189. Cardenas-Gonzalez M, Srivastava A, Pavkovic M, Bijol V, Rennke HG, Stillman IE. et al. Identification, Confirmation, and Replication of Novel Urinary MicroRNA Biomarkers in Lupus Nephritis and Diabetic Nephropathy. Clin Chem. 2017;63(9):1515-26
190. Wang W, Mou S, Wang L, Zhang M, Shao X, Fang W. et al. Up-regulation of Serum MiR-130b-3p Level is Associated with Renal Damage in Early Lupus Nephritis. Sci Rep. 2015;5:12644
191. Qingjuan L, Xiaojuan F, Wei Z, Chao W, Pengpeng K, Hongbo L. et al. miR-148a-3p overexpression contributes to glomerular cell proliferation by targeting PTEN in lupus nephritis. Am J Physiol Cell Physiol. 2016;310(6):C470-8
192. Sole C, Moline T, Vidal M, Ordi-Ros J, Cortes-Hernandez J. An Exosomal Urinary miRNA Signature for Early Diagnosis of Renal Fibrosis in Lupus Nephritis. Cells. 2019 8(8)
193. Nakhjavani M, Etemadi J, Pourlak T, Mirhosaini Z, Zununi Vahed S, Abediazar S. Plasma levels of miR-21, miR-150, miR-423 in patients with lupus nephritis. Iran J Kidney Dis. 2019;13(3):198-206
194. Wang X, Wang G, Zhang X, Dou Y, Dong Y, Liu D. et al. Inhibition of microRNA-182-5p contributes to attenuation of lupus nephritis via Foxo1 signaling. Exp Cell Res. 2018;373(1-2):91-8
195. Li Y, Xu X, Tang X, Bian X, Shen B, Zhao H. et al. MicroRNA expression profile of urinary exosomes in Type IV lupus nephritis complicated by cellular crescent. J Biol Res (Thessalon). 2018;25:16
196. Tangtanatakul P, Klinchanhom S, Sodsai P, Sutichet T, Promjeen C, Avihingsanon Y. et al. Down-regulation of let-7a and miR-21 in urine exosomes from lupus nephritis patients during disease flare. Asian Pac J Allergy Immunol. 2019;37(4):189-97
197. Zhang Y, Wang Y. The correlation of plasma microRNA-200 family expressions with risk and disease severity of lupus nephritis. Eur Rev Med Pharmacol Sci. 2018;22(10):3118-25
198. Sole C, Cortes-Hernandez J, Felip ML, Vidal M, Ordi-Ros J. miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol Dial Transplant. 2015;30(9):1488-96
199. Navarro-Quiroz E, Pacheco-Lugo L, Navarro-Quiroz R, Lorenzi H, Espana-Puccini P, Diaz-Olmos Y. et al. Profiling analysis of circulating microRNA in peripheral blood of patients with class IV lupus nephritis. PLoS One. 2017;12(11):e0187973
Author contact
![]() Corresponding author: Jae Il Shin, M.D., Ph.D. Address: 50 Yonsei-ro, Seodaemun-gu, C.P.O. Box 8044, Department of Pediatrics, Yonsei University College of Medicine, Seoul 03722, Republic of Korea. Tel: +82-2-2228-2050; Fax: +82-2-393-9118; E-mail: shinjiac.
Corresponding author: Jae Il Shin, M.D., Ph.D. Address: 50 Yonsei-ro, Seodaemun-gu, C.P.O. Box 8044, Department of Pediatrics, Yonsei University College of Medicine, Seoul 03722, Republic of Korea. Tel: +82-2-2228-2050; Fax: +82-2-393-9118; E-mail: shinjiac.