Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Undruggable targets in cancer
3.Targeted protein degradation
4. Targeting PPIs
5. Targeting IDPs
6. Targeting protein-DNA binding
7. Summary
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(11):3360-3382. doi:10.7150/ijbs.83026 This issue Cite
Review
Emerging Pharmacotherapeutic Strategies to Overcome Undruggable Proteins in Cancer
Yuqing Lu1#, Yuewen Yang1#, Guanghao Zhu2#, Hairong Zeng2, Yiming Fan3, Fujia Guo1, Dongshu Xu1, Boya Wang1, Dapeng Chen1 ![]() , Guangbo Ge2
, Guangbo Ge2 ![]()
1. Dalian Medical University, 116044 Dalian City, Liaoning Province, China.
2. Shanghai University of Traditional Chinese Medicine, 201203 Shanghai City, China.
3. Dalian Harmony Medical Testing Laboratory Co., Ltd, 116620 Dalian City, Liaoning Province, China.
#Equal contribution
Received 2023-1-30; Accepted 2023-6-13; Published 2023-6-26
Abstract

Targeted therapies in cancer treatment can improve in vivo efficacy and reduce adverse effects by altering the tissue exposure of specific biomolecules. However, there are still large number of target proteins in cancer are still undruggable, owing to the following factors including (1) lack of ligand-binding pockets, (2) function based on protein-protein interactions (PPIs), (3) the highly specific conserved active sites among protein family members, and (4) the variability of tertiary docking structures. The current status of undruggable targets proteins such as KRAS, TP53, C-MYC, PTP, are carefully introduced in this review. Some novel techniques and drug designing strategies have been applicated for overcoming these undruggable proteins, and the most classic and well-known technology is proteolysis targeting chimeras (PROTACs). In this review, the novel drug development strategies including targeting protein degradation, targeting PPI, targeting intrinsically disordered regions, as well as targeting protein-DNA binding are described, and we also discuss the potential of these strategies for overcoming the undruggable targets. Besides, intelligence-assisted technologies like Alpha-Fold help us a lot to predict the protein structure, which is beneficial for drug development. The discovery of new targets and the development of drugs targeting them, especially those undruggable targets, remain a huge challenge. New drug development strategies, better extraction processes that do not disrupt protein-protein interactions, and more precise artificial intelligence technologies may provide significant assistance in overcoming these undruggable targets.
Keywords: Cancer, Undruggable target, Protein-protein interaction, Intrinsically disordered protein, Targeted protein degradation, Proteolysis targeting chimera
1. Introduction
The incidence of cancer and associated mortality rates continue to increase globally. Cancer is the leading cause of death before the age of 70 years in 112 countries and either the third or fourth in 23 others. Cancer accounted for approximately 19.3 million new cases and nearly 10 million deaths worldwide in 2020 and 28.4 million new cases are anticipated in 2040[1]. At present, surgery, radiation therapy, and chemotherapy are the main strategies for cancer[2]. The success of surgical treatment is usually dependent on the absence of distant metastasis and local infiltration of tumor cells. Chemotherapy regimens use cytotoxic agents to prevent the proliferation, invasion, and metastasis of cancer cells. In 1949, the nitrogen mustard alkylating agent mechlorethamine was marketed as the first anti-cancer drug. Since then, the number of anti-cancer drugs entering the market has gradually increased[3]. Oncology remains the leading indication for Food and Drug Administration (FDA)-approved drugs in the past 2022[4]. Unfortunately, many promising and experimentally validated cancer targets are not within the scope of drug modifiability, such as the transcription factors (TFs). Besides cancer, in other diseases such as autoimmune diseases, neurodegenerative diseases, there are also some identified undruggable targets, and these identified targets are of great interest e.g., STAT3, lymphoid-specific tyrosine phosphatase, tau, alpha-synuclein, etc. Even though autoimmune diseases are considered rare compared to cancer, there are nearly 100 different autoimmune diseases that affect an additional 3% of the population[5]. Neurodegenerative diseases are diseases characterized by progressive impairment of motor and/or cognitive functions[6]. The number of cases worldwide is growing rapidly, especially in the context of the general trend of aging populations worldwide. However, their drug development is currently facing challenges [7, 8].
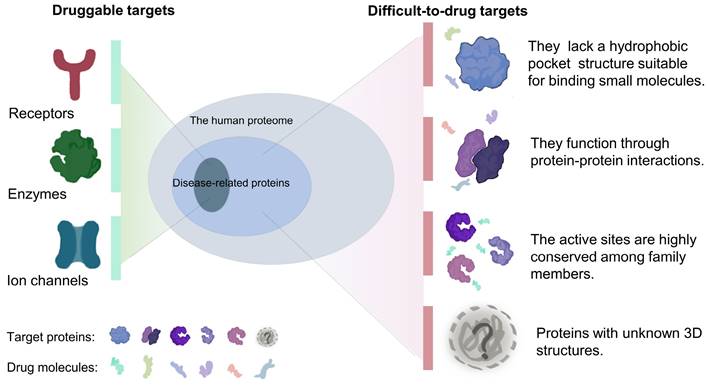
The therapeutic effects of drugs are traditionally determined by comparison of the efficacy and adverse reactions. Target-based drugs have gradually replaced traditional therapeutics as technologies have developed into the 21st century [9, 10]. Most targeted therapies aim at a specific ligand or modulates the function of a target protein. Cancers are usually caused by malfunction of various proteins, but only a relatively small fraction of these proteins can be targeted by small-molecule drugs or biologics[11]. Compared with traditional small molecule drugs, biologics, mainly monoclonal antibodies, have higher specificity and affinity[12, 13]. However, due to large molecular weights, most of biologics can only act on extracellular targets[13-15]. Nonetheless, a subset of targets is difficult to access by both small-molecule drugs and biologics. It is estimated that only 15% of drug targets (including enzymes, ion channels, and receptors) are considered druggable, while the remaining 85% are considered undruggable[16]. These so-called undruggable proteins tend to have several characteristics, including: (1) the lack of a hydrophobic pocket structure suitable for binding small molecules; (2) function via protein-protein interactions and formation of protein complexes; (3) highly conserved active sites that respond to specific inhibitors; and (4) intrinsically disordered or unknown tertiary structures (Figure 1). Based on the function of the target proteins in cancer, these undruggable targets can be classified as follows: (1) RAS family proteins. RAS proteins are small guanosine triphosphatases (GTPases) that play an important role in cell signaling transduction [17]. RAS is one of the most widely studied targets in cancer, and its mutations have a definite pathogenic role in many cancers, such as lung cancer and pancreatic cancer[18, 19]. The current status of its drug development has been dramatically transformed by relentless efforts. (2) Transcription factors, such as p53. Proteins involved in transcription mediate the expression of abnormal genes that lead to tumorigenesis and progression[20]. p53 is one of the mutated tumor suppressors in cancer. (3) Proteins involved in epigenetic regulation, such as protein tyrosine phosphatases (PTPs). PTPs and kinases together regulate tyrosine phosphorylation of proteins[21], and their family members play a dual role in carcinogenesis[22].
In fact, a few cancer-related targets are druggable, most are difficult to target, and often share some common characteristics.
Currently, researches try hard to overcome these undruggable targets owing to the new technology. With the continued advancements in drug development, various proteins are validated as potential drug targets. Most approaches to targeting proteins act by modulating the specific activity of the target protein, such as enzyme inhibition or ligand blocking. However, undruggable targets usually do not have enzymatic activity and an obvious active site. In such cases, a direct approach by selective degradation of the target protein is promising. TFs exert their biological activity through PPI and protein-DNA binding, thus facilitating the recruitment of other effectors to perform different functions, which are directly or indirectly involved in a variety of cancer-related gene expression and transcriptional abnormalities. Therefore, by blocking PPIs or protein-DNA interactions is another approach. In this process, the intervention of computer-aided drug development (CADD) and AI greatly facilitates the process of drug discovery and development. The purpose of this review is to provide an overview of the characteristics of undruggable targets in cancer. The drug development strategies to overcome the undruggable targets and current challenges in targeted therapies were also summarized. There are many undruggable targets in cancer[11, 23, 24], and the widely studied targets that have great therapeutic potential were selected as representative targets to introduce.
2. Undruggable targets in cancer
2.1. RAS proteins as “switches” for signal transduction
Mutations to RAS proteins are considered genetic drivers of multiple cancer types, and drug development is challenging. This is mainly because of the lack of pharmacologically actionable pockets for binding of small-molecule drugs. Further, drug development is complicated by the affinity of RAS proteins for guanosine-5'-triphosphate (GTP) combined with high intracellular GTP concentrations [25]. RAS proteins are small GTPases that regulate cell growth, differentiation, and apoptosis. There are three isoforms of Ras proteins: K-RAS, N-RAS, and H-RAS[26]. RAS mutations are among the most common drivers of human cancers [27]. K-RAS is the most mutated oncogene, occurring in 27%-39%, 40%-54%, and 86%-96% of lung, colorectal, and pancreatic cancers, respectively[28, 29]. Missense mutations are reported to increase the affinity of RAS proteins to GTP and lower the enzymatic activity of GTPase-activating proteins. This would leave KRAS in the “on” state and dysregulation of various signaling pathways that are dependent on active RAS regulation[30-32].
In the past decades, attempts to overcome KRAS have been made from different perspectives. More attention has been paid to indirect approaches, including (1) targeting upstream molecules; (2) targeting downstream effector molecules; (3) interfering with RAS mRNA expression; (4) targeting different metabolic processes associated with RAS mutations; and (5) screening of lethal synthetic interactors[17]. Small-molecule inhibitors (SMIs) that block RAS translocation to the cell membrane by targeting upstream molecules, such as PDEδ, SHP2, and STK19, include NHTD [33], JAB-3068 (ClinicalTrials.gov identifier: NCT03565003), and ZT-12-037-01 [34], among others. Inhibitors that block downstream adverse events by targeting downstream molecules, such as the RAF-MEK-ERK cascade and PI3K-AKT-mTOR signaling pathway, include RO5126766[35], BVD523[36], and NVP-BEZ235[37]. Inhibitors that impede the growth and metabolism of tumor cells by targeting metabolic processes associated with RAS mutations, such as autophagy, may have anti-tumor activities in combination with the above inhibitors[38]. Small interfering RNA exhibits anti-proliferative effects by interfering with RAS expression in tumors through delivery mediators[39, 40]. These methods have catalyzed many inhibitor molecules. However, many inhibitors have been difficult to translate to clinic applications due to safety concerns, limited anti-tumor activities, and other issues. In addition to SMIs, attempts have been made to develop degraders of KRAS. As G12C covalent inhibitors, ARS-1620 and Adagrasib were linked to E3 ligase to develop PROTACs. Experiments showed that KRASG12C degraders showed blockade of downstream signaling. In addition, they effectively inhibited the proliferation of tumor cells[41, 42]. Targeting protein degradation is a novel pathway to develop KRAS inhibitors.
Although drugs that directly target the RAS are considered elusive, the discovery of a new allosteric site in KRAS (G12C) has shown new hope for targeting KRAS. AMG510 (Sotorasib) and MRTX849 (Adagrasib) were recently approved for the treatment of non-small cell lung cancer (NSCLC) carrying the KRASG12C mutation[43], indicating that KRAS is no longer considered clinically undruggable. The KRASG12C mutation occurs in about 13% of lung adenocarcinomas and about 3% of colorectal adenocarcinomas[44, 45], and this mutant has a cysteine residue (glycine in position 12 becomes cysteine) that is used to design covalent inhibitors[46]. In cancer cells, KRAS G12C has been shown to cycle rapidly between an active GTP-bound state and an inactive GDP-bound state[47]. AMG510 is a selective inhibitor that binds to the cysteine residue in the Switch Ⅱ pocket of KRASG12C and irreversibly locks KRAS in an inactive state. Its binding and potency are enhanced by binding to a novel His groove on KRAS compared to previous preclinical attempts[44]. In the clinical phase I/II trial (ClinicalTrials.gov identifier: NCT03600883), AMG510 led to KRASG12C tumor regression and demonstrated a favorable safety profile. Treatment-related adverse effects included diarrhea, vomiting, elevated alanine aminotransferase levels, and elevated aspartate aminotransferase[48]. The phase II trial was evaluated in 126 patients with KRASG12C mutant NSCLC and the results were positive. Their objective response rate and disease control rate were 37.1% and 80.6%, respectively, with median remission duration and median progression-free survival of 10 months and 6.8 months, respectively[49]. The excellent trial results led to the accelerated approval of AMG510 by the FDA. Several clinical trials related to AMG510 combination therapy are still ongoing (Table 1). As the development of KRASG12C-targeted therapeutics has progressed at a rapid pace, there have been some notable achievements in the development of drugs for KRASG12D. In preclinical trials, MRTX1133, a KRASG12D inhibitor, mediated the apparent regression of KRASG12D mutant pancreatic ductal adenocarcinoma[50-52]. Currently, MRTX1133 is in a clinical trial to evaluate its safety, tolerability, and antitumor activity in patients with advanced solid tumor malignancies harboring KRASG12D mutations (ClinicalTrials.gov Identifier: NCT05737706).
Ongoing clinical trials of KRASG12C-targeted inhibitors and related combination therapy clinical trials.
| Drug | Treatment Strategy | ClinicalTrials.gov registration | Disease setting | Study phase | Recruitment Status |
|---|---|---|---|---|---|
| AMG510 (Sotorasib) | Monotherapy | NCT03600883 | Advanced solid tumors with KRAS G12C mutations | I/II | Active, not recruiting |
| Monotherapy | NCT04667234 | Advanced/unresectable/metastatic NSCLC with KRASG12C mutation | / | Available | |
| Monotherapy | NCT04380753 | Advanced/Metastatic Solid Tumors with KRASG12C Mutations | I | Active, not recruiting | |
| Monotherapy | NCT04625647 | Advanced NSCLC with KRAS G12C mutations | II | Recruiting | |
| Monotherapy | NCT04933695 | Stage IV NSCLC with KRAS G12C mutations without prior treatment | II | Active, not recruiting | |
| Monotherapy | NCT05400577 | Stage IV NSCLC with KRAS G12C mutations without prior treatment | II | Recruiting | |
| Monotherapy | NCT05398094 | Stage III unresectable NSCLC with KRAS G12C mutations | II | Recruiting | |
| Monotherapy | NCT05273047 | Metastatic NSCLC with KRAS G12C mutations | / | Recruiting | |
| Monotherapy | NCT05451056 | NSCLC with KRAS G12C mutations | II | Not yet recruiting | |
| Monotherapy vs. Docetaxel | NCT04303780 | Advanced NSCLC with KRAS G12C mutations | Ⅲ | Active, not recruiting | |
| Combined with Tarloxotinib | NCT05313009 | NSCLC with KRAS G12C mutations | I/II | Recruiting | |
| Combined with BBP-398 | NCT05480865 | Advanced solid tumors with KRAS G12C mutations | I | Recruiting | |
| Combined with VS-6766 | NCT05074810 | NSCLC with KRAS G12C mutations | I/II | Recruiting | |
| Combined with targeted therapy, chemotherapy, or immunotherapy | NCT04185883 | Advanced Solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| Combined with Panitumumab vs. Trifluridine and Tipiracil,or Regorafenib | NCT05198934 | CRC with KRAS G12C mutations | Ⅲ | Active, not recruiting | |
| Combined with MVASI | NCT05180422 | Advanced, unresectable or metastatic KRASG12C mutant NSCLC With asymptomatic brain metastasis | I/II | Recruiting | |
| Combined with Panitumumab | NCT05638295 | Advanced/Metastatic Solid Tumors With KRASG12C Mutation | II | Not yet recruiting | |
| Combined with RMC-4630 | NCT05054725 | NSCLC with KRAS G12C mutations | II | Recruiting | |
| Combined with cisplatin or carboplatin and pemetrexed | NCT05118854 | Stage IIA-IIIB resectable non-squamous NSCLC with KRAS G12C mutations | II | Recruiting | |
| MRTX849 (Adagrasib) | Monotherapy | NCT05162443 | Advanced solid tumors with KRAS G12C mutations | / | Available |
| Monotherapy | NCT05263986 | Advanced/Metastatic Solid Tumors With KRASG12C Mutations | I | Active, not recruiting | |
| Monotherapy | NCT05634525 | Metastatic pancreatic cancer with KRAS mutations | I | Not yet recruiting | |
| Monotherapy | NCT05673187 | Stage IV NSCLC with KRAS G12C mutations | II | Not yet recruiting | |
| Monotherapy or combined with Nivolumab | NCT05472623 | Resectable NSCLC with KRAS G12C mutations | II | Not yet recruiting | |
| Monotherapy or combined with Cetuximab or Pembrolizumab or Afatinib | NCT03785249 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| Monotherapy or combined with Pembrolizumab | NCT04613596 | Advanced or metastatic NSCLC with KRAS G12C mutations | II/Ⅲ | Recruiting | |
| Monotherapy vs. Docetaxel | NCT04685135 | Advanced or metastatic NSCLC with KRAS G12C mutations | Ⅲ | Recruiting | |
| Combined with Cetuximab vs. Chemotherapy | NCT04793958 | Advanced CRC with KRAS G12C mutations | Ⅲ | Recruiting | |
| Combined with BI 1701963 | NCT04975256 | Advanced solid tumors with KRAS G12C mutations | I | Completed | |
| Combined with Palbociclib | NCT05178888 | Advanced solid tumors with KRAS G12C mutations | I | Active, not recruiting | |
| Combined with TNO155 | NCT04330664 | Advanced solid tumors with KRAS G12C mutations | I/II | Active, not recruiting | |
| Combined with SAR442720 | NCT04418661 | NSCLC with KRAS G12C mutations | / | Active, not recruiting | |
| Combined with Pembrolizumab | NCT05609578 | Advanced NSCLC with KRAS G12C mutations | II | Recruiting | |
| Combined with Cetuximab and Irinotecan | NCT05722327 | CRC with KRAS G12C mutations | I | Not yet recruiting | |
| Combined with VS-6766 | NCT05375994 | NSCLC with KRAS G12C mutations | I/II | Recruiting | |
| JAB-21822 | Monotherapy | NCT05009329 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting |
| Monotherapy | NCT05276726 | Advanced or metastatic NSCLC with KRAS G12C mutations | I/II | Recruiting | |
| Monotherapy or combined with Cetuximab | NCT05002270 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| Combined with JAB-3312 | NCT05288205 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| Combined with Cetuximab | NCT05194995 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| JDQ443 | Monotherapy | NCT05329623 | Small Cell Lung Carcinoma | I | Suspended |
| Monotherapy | NCT05445843 | Advanced or metastatic NSCLC with KRAS G12C mutations | II | Recruiting | |
| Monotherapy | NCT05132075 | Advanced NSCLC with KRAS G12C mutations | Ⅲ | Recruiting | |
| Monotherapy or combined with TNO155 or tislelizumab or TNO155 + tislelizumab | NCT04699188 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| Combined with Trametinib or Ribociclib or cetuximab | NCT05358249 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting | |
| GDC-6036 | Monotherapy vs. Docetaxel | NCT03178552 | Advanced/unresectable/metastatic NSCLC | II/Ⅲ | Recruiting |
| Monotherapy or combined with chemotherapy, immunotherapy, etc. | NCT04449874 | Advanced or metastatic NSCLC with KRAS G12C mutations | I | Recruiting | |
| Combined with Pembrolizumab | NCT05789082 | Advanced or metastatic NSCLC with KRAS G12C mutations | I/II | Not yet recruiting | |
| Combined with Cetuximab | NCT04929223 | Metastatic CRC | II/Ⅲ | Recruiting | |
| D-1553 | Monotherapy | NCT05383898 | Advanced or metastatic NSCLC with KRAS G12C mutations | I/II | Recruiting |
| Monotherapy or combined with other therapies | NCT04585035 | Advanced or metastatic NSCLC with KRAS G12C mutations | I/II | Recruiting | |
| Combined with immunotherapy or targeted therapy | NCT05492045 | Advanced or metastatic NSCLC with KRAS G12C mutations | I/II | Not yet recruiting | |
| Combined with IN10018 | NCT05379946 | Advanced or metastatic NSCLC with KRAS G13C mutations | I/II | Not yet recruiting | |
| D3S-001 | Monotherapy | NCT05410145 | Advanced solid tumors with KRAS G12C mutations | I | Recruiting |
| GFH925 | Monotherapy | NCT05005234 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting |
| YL-15293 | Monotherapy | NCT05119933 | Advanced solid tumors with KRAS G12C mutations | I/II | Recruiting |
| JNJ-74699157 | Monotherapy | NCT04006301 | Advanced solid tumors with KRAS G12C mutations | I | Completed |
| RMC-6291 | Monotherapy | NCT05462717 | Advanced solid tumors with KRAS G12C mutations | I | Recruiting |
| HS-10370 | Monotherapy | NCT05367778 | Advanced solid tumors with KRAS G13C mutations | I/II | Not yet recruiting |
| MK-1084 | Monotherapy or combined with Pembrolizumab | NCT05067283 | Advanced solid tumors with KRAS G14C mutations | I | Recruiting |
| LY3537982 | Monotherapy or combined with targeted therapy, immunotherapy, etc. | NCT04956640 | Advanced solid tumors with KRAS G12C mutations | I | Recruiting |
However, acquired resistance ultimately occurs in most patients treated with monotherapy, and KRASG12C inhibitors do not appear to be an exception. Several possible mechanisms have been proposed to explain this phenomenon, including acquired mutations in KRAS[53, 54], activation of associated parallel signaling pathways[53], and phenotypic transformation[55]. It has been shown that KRASG12C inhibitors will lead to multiple different KRAS secondary mutations that can interfere with drug binding and have drug heterogeneity[56]. Bypass mechanisms of drug resistance include activation of upstream regulators, such as EGFR, MET, and RET[57, 58]; activation of downstream effectors, such as MEK and RAF[53, 59]; and activation of wild-type RAS [60]. Epithelial mesenchymal transition is also a mechanism of drug resistance[53]. It modulates and activates the PI3K pathway via the IGFR-IRS1 pathway, leading to endogenous and acquired drug resistance[61]. The mechanisms of acquired resistance to KRASG12C inhibitors are complex, and different mechanisms can coexist in the same patient. Combination therapy is an important approach to prevent or delay the onset of resistance, and many combination regimens are currently in clinical trials (Table 1), but attention needs to be paid to the occurrence of adverse events.
2.2. Undruggable proteins involved in transcription
TP53 is an important tumor suppressor gene that is mutated in more than half of all human cancers. However, drugs that act directly on this target are difficult to develop because p53 lacks deep pockets for binding SMIs, lacks enzymatic activity, and is in the nucleus. The p53 protein is a typical TF. Over the past three decades, the understanding of p53 has undergone three shifts: from an oncoprotein antigen, to an oncogene, to the “Guardian of the Genome” [62]. Activation of wild-type p53 in response to cellular damage caused by stressors, such as hypoxia and DNA damage, promotes cell cycle arrest, apoptosis, or senescence, thereby avoiding cellular carcinogenesis[63-66]. Therefore, inactivation of normal p53 function in cells often leads to carcinogenesis, and genetic mutation is the main mechanism underlying inactivation of p53. This leads to four possible consequences: loss of function, gain of function, dominant negative effects, or no effect on normal function[67]. Thus, mutated p53 may not only lose normal oncogenic function, but may also exhibit dominant negative effects and/or gain of function, thus acting as a cancer promoter. Although degradation of mutant p53 can be induced[68] or function can be restored with arsenic trioxide[69], the existence of different types of p53 mutants can inhibit application of this approach. In addition, various inhibitors of histone deacetylase 6 and HSP90 have been found to induce degradation of mutant p53 [70-72]. Specific deoxyribonucleases designed to target the mutation site of p53 can degrade mutant p53 transcripts[73, 74], thereby reducing protein expression. Drug development for wild-type p53 has focused on interfering with ubiquitination of p53 by MDM2, thereby stabilizing the p53 protein[75, 76]. To date, several small-molecule and natural drugs, such as nutlin-3a[77, 78], have been developed to inhibit interactions between p53 and MDM2.
Upregulation of MYC expression frequently occurs in cancers. A recent analysis of more than 9000 human cancers showed that MYC gene amplification occurs in approximately 28% of malignancies[79]. However, the design of MYC inhibitors is limited due to: (1) intrinsically disordered, but functionally important, domains and lack of enzymatically active sites; (2) high affinity interactions with MAX; (3) partial functional redundancy of family members; and (4) location mainly in the nucleus[80]. C-MYC, L-MYC, and N-MYC are TFs that encode MYC oncoproteins, which are known as “super” TFs that regulate the expression of genes involved in a variety of cellular processes[81-83]. Under normal conditions, the expression levels of C-MYC, L-MYC, and N-MYC are strictly limited by various mechanisms[84-86], but are often dysregulated in human cancers. Insertion of a retroviral promoter and chromosomal translocation/amplification can induce MYC overexpression[82]. Overall, N-MYC and L-MYC are reportedly amplified in less than 7% of cancers[79], whereas C-MYC is amplified in 21%[87]. Notably, even transient inactivation of MYC leads to tumor regression[88-90], suggesting that the modulation of oncogenic MYC is a feasible strategy for cancer treatment. However, the development of drugs directly targeting MYC has been challenging due to the lack of a specific active site for binding of small molecules and location, which is primarily in the nucleus. Therefore, compounds directly targeting MYC have not yet been tested in clinical trials. For these reasons, indirect methods to inhibit the oncogenic function of MYC, such as targeting transcription, translation, or the MYC-MAX complex, have been intensively investigated. The most extensively studied compound directly targeting MYC is Omomyc[91], which can potentially damage the MYC/MAX/MXD network and has been demonstrated to trigger tumor regression in a variety of cancer models [92-99]. Furthermore, the results of recent animal experiments suggest that the adverse side effects induced by the use of Omomyc are mild and reversible, and the therapeutic effect is improved in combination with paclitaxel [94]. Recently, GT19715, a novel dual C-MYC/GSPT1 degrader, was reported to effectively degrade C-MYC protein both in vivo and in vitro and to inhibit tumor growth at low doses in Acute Myeloid Leukemia and Lymphomas[100]. Although no therapies targeting MYC have been approved for clinical use, research conducted over the past 20 years has provided a solid foundation for the study of MYC-targeted inhibitors.
As a TF, STAT3 plays significant roles in a wide variety of biological processes. However, current drug development targeting STAT3 is limited due to: (1) the highly homologous SH2 structural domain shared by STAT family members[101]; and (2) the transcriptional activity of the monomeric STAT3 protein, which partially blocks the activities of inhibitors targeting the SH2 structural domain to prevent the formation of STAT3 dimers[102]. As a cytoplasmic TF, STAT3 modulates cell differentiation, proliferation, and apoptosis, in addition to angiogenesis, inflammation, and immune responses[103]. Among the seven conserved STAT family members, tumor cells often overexpress STAT3, which plays an essential role in antitumor immunity [104, 105]. STAT3 is a meeting point for a number of oncogenic pathways and is constitutively activated in both tumor cells and tumor-infiltrating immune cells. Overexpression of STAT3 impedes the antitumor immune response by inhibiting expression of mediators necessary for activation of the immune response against tumor cells[106, 107]. Besides, STAT3 can induce differentiation and proliferation of Th17 cells by enhancing expression of RAR-related orphan receptor gamma. It also suppresses the initial differentiation of regulatory T cells (Tregs) by suppressing expression of forkhead box P3, which plays vital roles in various autoimmune diseases[108]. Inhibition of STAT3 promotes the growth and differentiation of Tregs and regulates the balance of Tregs and Th17 cells, which can improve symptoms of autoimmune diseases[109-111]. Drug development against STAT3 can be broadly divided into two types: one targeting upstream molecules and the other directly targeting STAT3. Direct targeting involves inhibition of STAT3 phosphorylation, dimerization, nuclear translocation, and binding to DNA. Blocking of upstream molecules, such as JAK, can inhibit a variety of downstream pathways, resulting in undesirable consequences[112, 113]. Among the direct targeting strategies, the SH2 structural domain has been widely studied because of the key role in STAT3 activation. However, this strategy is limited because targeting the SH2 structural domain does not completely inhibit STAT3[102]. In recent years, degraders have become the focus of attention in drug development. Prof. Shaomeng Wang's team developed the small molecule SD-36, which is a selective degrader of STAT3[114]. In leukemia cell lines and lymphoma cell lines, SD-36 efficiently and selectively reduced STAT3 levels. In a mouse Molm-16 xenograft model, it achieved significant degradation of STAT3 and complete and durable tumor regression[114]. However, the development of PROTACs seems to be more challenging. To date, a number of STAT3 inhibitors have been investigated in clinical trials, but none are currently approved for clinical use, which has led to the development of more effective STAT3 inhibitors and further exploration of additional drug development strategies.
Variable splicing of pre-mRNA of the androgen receptor (AR) causes resistance of the AR-V7 splicing variant to AR signaling inhibitor therapy. In addition, the AR-V7 is considered difficult to target due to the lack of a ligand-binding domain for androgen and antagonists. AR signaling plays a non-negligible role in the development of prostate cancer[115]. As the mainstay treatment for prostate cancer, androgen deprivation therapy works by limiting the availability of androgens[116]. However, tumor cells are known to develop adaptive resistance to almost all targeted therapies and long-term treatment can eventually lead to castration-resistant prostate cancer (CRPC)[117-119]. The mechanism underlying treatment resistance is related to reactivation of AR signaling and formation of the AR-V7 splicing variant[120, 121]. In prostate cancer, cryptic exon 3 of intron 3 of the AR pre-mRNA sequence is selected by the spliceosome to replace the subsequent AR exon, resulting in the AR-V7 splicing variant[122, 123]. This in turn forms a heterodimer that can activate downstream target genes in the absence of androgens. Hence, next-generation drugs that directly or indirectly target AR-V7 signaling are urgently needed.
β-catenin is a classical oncogenic TF that is a key effector involved in the Wnt oncogenic pathway. Drug development targeting β-catenin is challenging due to: (1) the lack of deep pockets for binding SMIs; and (2) the tendency of β-catenin to bind to TCF-4 with low affinity, although the interaction surface is relatively large[124]. β-catenin is a multifunctional protein and a key transducer of the classical Wnt signaling pathway, which participates in the regulation of cell differentiation and proliferation [125]. Without Wnt ligands, β-catenin is recruited to a disruption complex composed of APC and AXIN that promotes phosphorylation of β-catenin, leading to ubiquitination and proteasomal degradation to maintain low expression levels in the cytoplasm[126]. When Wnt is activated or mutated, unphosphorylated β-catenin accumulates in the cytoplasm and subsequently migrates to the nucleus to interact with TCF/LEF and coactivators, resulting in transcription of specific target genes encoding oncoproteins[127, 128]. Therefore, targeting β-catenin presents a very attractive anticancer treatment strategy. However, β-catenin is rarely targeted[124] and no inhibitors targeting β-catenin to inhibit the Wnt signaling pathway have emerged.
Although HOXA9 and MEIS1 play synergistic and pathogenic roles in acute myeloid leukemia (AML), both molecules are considered difficult to drug due to the lack of deep pockets for binding SMIs. HOXA9 is a member of the HOX gene family of homologous TFs. AML is the most extensively studied disease involving dysregulation of HOX gene expression, as HOXA9 is overexpressed in about 50%[129]. Hence, HOXA9 is a potential target for the treatment of AML. Hematopoietic stem and progenitor cells normally express high levels of HOXA9, although expression levels are relatively decreased in mature cells[130]. Aberrant expression of HOXA9 is a salient feature of AML driven by multiple oncogenes. In current drug development, HOXA9 expression is often downregulated by indirect methods and thus is considered an undruggable target. Dysregulation of HOXA9 often occurs in conjunction with upstream genetic alterations, such as mixed-lineage leukemia (MLL) fusions, NUP98 fusions, nuclear translocation of phospholipid 1, and overexpression of CDX2, all of which can upregulate the expression of HOXA9[131, 132]. Many studies have proposed regulation of HOXA9 expression by targeting MLL fusion-related proteins, such as DOT1L and Menin[133]. MEIS1 is a member of the MEIS subfamily of TFs and plays important roles in leukemia and many solid tumors[134]. MEIS1 and HOXA9 act together to accelerate leukemogenesis by promoting cell proliferation and inhibiting apoptosis[134, 135]. In terms of drug development, like HOXA9, most strategies have focused on targeting MLL fusion-related proteins.
As an oncogenic TF, the EWS-FLI1 chimeric fusion protein is an attractive therapeutic target for Ewing sarcoma (ES). Although not present in normal cells, EWS-FLI1 is considered an intrinsically disordered protein (IDP) that is difficult to directly target[136]. ES is a malignant tumor of the bone and soft tissue and is the second most common primary bone malignancy in pediatric patients [137, 138]. EWS-FLI1, a major regulator of ES, is a clear target for the treatment of ES, as successful inhibition has led to tumor regression[139, 140]. As a potential therapeutic target, there are no drugs that act directly on the EWS-FLI1 fusion protein due to the lack of stable structures and enzymatic activities.
SMARCA2 is an ATPase subunit of the Switch/Sucrose Non-Fermentable chromatin remodeling complex. It is highly homologous to another subunit, SMARCA4, and together they regulate the repair of damaged DNA and DNA transcription[141]. In many cancers, especially NSCLC, SMARCA4 mutations result in expression deficiency[142]. Studies have shown a synthetic lethal effect of SMARCA2 with SMARCA4[143]. Synthetic lethality has been described as the interaction of two genes, where when one is repressed, the other can functionally compensate or replace the function of the first, while the loss of function of both is lethal to the cell, offering the possibility of indirect targeting of non-drug acting targets. Therefore, the strategy of targeting SMARCA2 to treat cancers with SMARCA4 mutations has attracted a lot of attention. However, due to the high similarity of SMARCA2 and SMARCA4 proteins, the selection of inhibitors is difficult to develop.
2.3. Undruggable proteins involved in epigenetic regulation
PTPs are considered difficult to target due to (1) the highly conserved active site of family members, (2) the active site of PTP is positively charged, thus screening against the active site often yields negatively charged phosphate analogs, in addition to poor cell permeability and pharmacokinetic properties, and (3) the side chains of catalytic cysteines that act as sulfate anions in the positively charged active site are major targets of various electrophiles, which can interfere with high-throughput screening (HTS)[144]. Tyrosine phosphorylation of intracellular proteins is regulated by the antagonistic activities of protein tyrosine kinases and PTPs, which remove phosphate groups from proteins by hydrolysis[21, 145]. Numerous studies have shown that disruption of tyrosine phosphorylation caused by dysregulation of PTP expression is involved in the pathogenesis of various cancers, autoimmune diseases, and diabetes[146]. The human genome encodes more than 100 PTPs[147], which are classified as a superfamily characterized by a conserved CX5R motif at the active site[148]. Numerous studies have shown that members of the PTP family play dual roles in oncogenesis and can therefore be classified as tumor suppressors or oncogenic PTPs[22]. In addition, PTPs are also involved in progression of autoimmune diseases. The single-nucleotide polymorphism c.1858C>T (rs2476601) of PTPN22, which encodes protein tyrosine phosphatase N22, is related to a variety of autoimmune diseases [149]. Although the roles of many PTPs have been well-documented in various diseases [150], no drugs targeting PTPs have yet been approved for clinical use.
Several drugs currently being tested in clinical and preclinical trials target epigenetic regulatory proteins, such as DNA methyltransferases and histone deacetylases, among others. In contrast, studies of histone acetyltransferases (HATs) as potential inhibitors are challenged by several factors, including: (1) the variety of cellular substrates ranging from histones and TFs to enzymes and nuclear receptors; and (2) the formation of multiprotein complexes that determine function, enzymatic activity, and substrate specificity[151, 152], which limit translation to cellular and in vivo experiments. Epigenetic modifications do not alter linear DNA sequences, but directly affect DNA conformation and gene activation or repression, and therefore have great therapeutic potential for treatment of human diseases[153]. Epigenetic regulatory proteins include a broad group of “writers,” “readers,” and “erasers,” which have distinctly different functions[154]. HATs are classified as “readers” that facilitate acetylation of lysine residues of cellular proteins. As compared to other family members, CBP/p300 has been more intensively studied in the field of cancer. It is unclear why both CBP/p300 deletion and overexpression can promote tumorigenesis[155]. However, several studies have shown that CBP/p300 inhibitors impede cancer cell survival, proliferation, and metastasis in a variety of cancer types[156-159]. HAT inhibitors are currently classified as dual-substrate inhibitors, natural product inhibitors, and synthetic SMIs[160]. Two CBP/p300 inhibitors are currently being tested in clinical trials as potential targets for treatment of cancer patients: CCS1477 (ClinicalTrials.gov identifier: NCT03568656) and FT-7051 (ClinicalTrials.gov identifier: NCT04575766). In addition, EP31670, a dual BET and CBP/p300 inhibitor, was recently approved for a phase 1 study of patients with advanced solid tumors (ClinicalTrials.gov identifier: NCT05488548). HATs were among the first epigenetic modifiers to be identified, but still no potent, selective drugs have been approved for clinical use due to the tertiary structures (Table 2).
Physiological functions and causes of abnormal expression patterns of potential protein targets in various diseases.
| Target protein | Physiological function | Abnormal expression | Disease |
|---|---|---|---|
| KRAS | Signal transduction | Mutation | Pancreatic cancer, colorectal cancer, non-small cell lung cancer, etc. |
| p53 | Transcription | Mutation | Lung cancer, stomach cancer, liver cancer, etc. |
| C-MYC | Transcription | Overexpression | Lung cancer, stomach cancer, breast cancer, etc. |
| STAT3 | Transcription | Overexpression | Rectal cancer, lung cancer, breast cancer, etc. |
| AR-V7 | Transcription | Overexpression | Metastatic castration-resistant prostate cancer |
| β-catenin | Transcription | Abnormal accumulation | Colon cancer, hepatocellular carcinoma, pancreatic cancer, etc. |
| HOXA9 | Transcription | Overexpression | Acute myeloid leukemia |
| MEIS1 | Transcription | Overexpression | Acute myeloid leukemia |
| EWS-FLI1 | / | Fusion | Ewing sarcoma |
| PTP | Post-translation modification | Elevated/reduced expression | Breast cancer, stomach cancer, prostate cancer, rheumatoid arthritis, systemic lupus erythematosus, etc. |
| HAT | Post-translation modification | Mutation | Leukemogenesis, Rubinstein-Taybi syndrome, etc. |
| SMARCA2 | Chromatin remodelling | / | Non-small cell lung cancer, etc. |
As mentioned above, many promising targets are facing difficulties in drug development. The following sections describe recent technological advances that have facilitated undruggable targets as promising therapeutics for treatment of different cancers.
3.Targeted protein degradation
The human body has a series of sophisticated systems, such as the ubiquitin-proteasome system (UPS) and the lysosomal system, that maintain protein homeostasis. TPD technology takes advantage of this natural mechanism and is able to directly degrade target proteins at the post-translational level with high selectivity and efficiency.
3.1. PROTACs
The term PROTAC was introduced in 2001 to describe a small bifunctional molecule. It can bind to both target proteins and E3 ubiquitin ligases, which leads to the ubiquitination and degradation of target proteins, and can catalyze the degradation of multiple target proteins. Following the synthesis of PROTAC-1 containing IκBα phosphopeptide and ovalicin[161], other small-molecule PROTACs have emerged based on MDM2 E3 ligase[162], IAP1 E3 ligase[163], VHL, and CRBN[164, 165]. In addition, Kelch-like ECH-associated protein 1-based PROTACs have emerged, including peptide-based and small molecule degraders[166, 167]. An increasing number of proteins are proving to be targets of PROTACs, AR, ER, STAT3, etc. As of 2019, two PROTACs have been tested in clinical trials conducted in the U.S. for the treatment of refractory prostate and breast cancers (ClinicalTrials.gov identifiers: NCT03888612 and NCT04072952). Currently, there are at least 20 PROTACs in clinical trials and the number will continue to increase. As compared to conventional drugs, the advantages of PROTACs include: (1) they can disrupt multiple functions of proteins; (2) they have more complete and longer-lasting therapeutic effects; (3) they require relatively lower affinity; and (4) they can prevent the development of adaptive drug resistance[168]. PROTACs use the cellular protein degradation machinery (i.e., UPS) to remove specific target proteins and thus have great potential for targeting undruggable proteins[101] (Table 3).
The PROTACs developed for the undruggable targets in cancers.
| Targets | PROTACS | Disease | Structure | Reference |
|---|---|---|---|---|
| KRAS | YF135 | Non-small cell lung cancer |  | [289] |
| KRAS | KP-14 | Non-small cell lung cancer |  | [290] |
| KRAS | LC-2 | Lung cancer, Pancreatic cancer |  | [41] |
| SHP2 | ZB-S-29 | Monocytic leukemia | 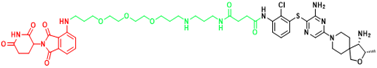 | [291] |
| SHP2 | SHP2-D26 | Esophageal cancer, Monocytic leukemia | 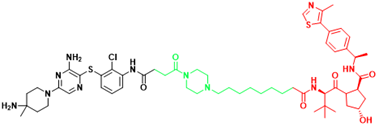 | [292] |
| SHP2 | SP4 | Cervical cancer | 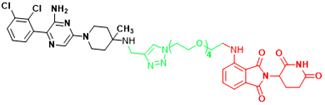 | [293] |
| SHP2 | R1-5C | Monocytic leukemia, Esophageal cancer, Acute myelogenous leukemia, Acute lymphoblastic leukemia | 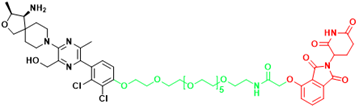 | [294] |
| BRD4(C-MYC) | ARV-825 | Burkitt's Lymphoma, B cell lymphoma | 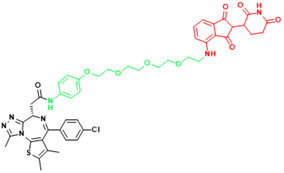 | [165] |
| STAT3 | SD-36 | Acute myelogenous leukemia, Anaplastic large cell lymphoma | 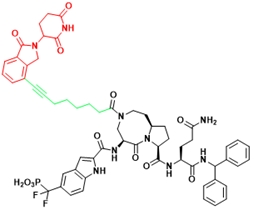 | [114] |
| STAT3 | SD-91 | Acute myelogenous leukemia | 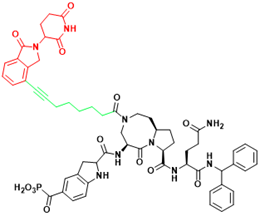 | [295] |
| AR-V7 | MTX-23 | Castration-resistant prostate cancer | 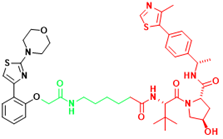 | [296] |
| AR-V7 | 6 | Castration-resistant prostate cancer | 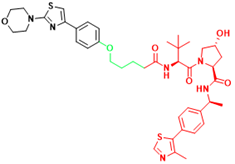 | [297] |
| β-catenin | xStAx-VHLL | Rectal cancer |  | [298] |
| p300/CBP | dCBP-1 | Multiple myeloma | 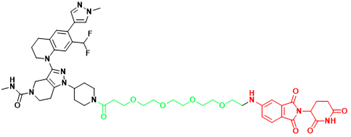 | [299] |
| EP300 | JQAD1 | Neuroblastoma | 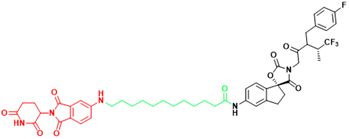 | [300] |
| SMARCA2 | A947 | Non-small cell lung cancer | 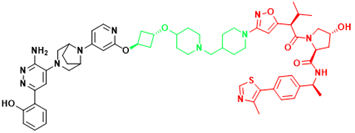 | [301] |
ARV-110 is an oral PROTAC developed by Arvinas that selectively targets and degrades AR and is proposed to be developed for the treatment of metastatic castration-resistant prostate cancer [169]. In populations with specific genetic mutations, ARV-110 reduced prostate-specific antigen levels by more than 50% in 40% of patients with metastatic desmoplastic resistant prostate cancer[169]. Based on safety, pharmacokinetics and efficacy, 420 mg QD was selected as the recommended clinical phase 2 dose (RP2D). Among the 113 subjects treated with RP2D, there were no grade ≥4 tx-related adverse events, of which nausea, vomiting, fatigue, and decreased appetite were common[170]. ARV-471 is a protein degrader that targets the estrogen receptor (ER) and is proposed for the treatment of ER+/HER2- locally advanced or metastatic breast cancer. Phase I clinical data for ARV-471 also showed that high levels of ER degradation (89%) were observed at dose levels of 30-700 mg with good safety and tolerability[171] In the phase Ⅱ clinical trial, 100% of 35 patients were previously treated with CDK4/6 inhibitors, 74% with fulvestrant, and 74% with chemotherapy. the clinical benefit rate (CBR) for ARV-471 200 mg QD was 37.1%, and the CBR for evaluable patients with mutant ESR1 (n=19) was 47.4%, and substantial on-treatment reductions in mutant ESR1 circulating tumor DNA levels were observed. The median progression-free survival was 3.5 months. Treatment-related adverse events were mainly fatigue, hot flashes, and nausea.ARV-110 and ARV-471 are currently being further evaluated in clinical trials and have the potential to be approved.
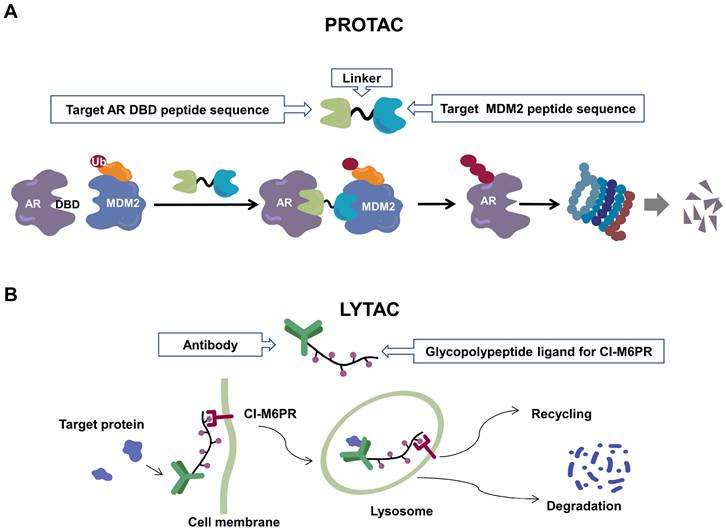
Recently, a group designed and synthesized Au-AR pep-PROTAC targeting AR-V7 by recruiting MDM2[172]. AR-V7 is often observed in CRPC. It is considered non-druggable due to the lack of ligand binding domain[120, 121]. In this study, they designed a novel peptide antagonist to target the DNA binding domain (DBD) of AR-V7 by artificial Inelligence (AI)-aided peptide drug design, which appears to have stronger affinity and specificity than small molecules. Due to the poor membrane permeability and short half-life of peptide PROTAC, an ultrasmall gold (Au)-peptide complex platform was developed for in vivo delivery of AR DBD PROTAC. It was shown that Au-AR pep-PROTAC induced degradation of AR and AR-V7 and inhibited cancer cell proliferation at both cellular and animal levels (Figure 2). KT-333 is a potent degradation agent that specifically degrades undruggable STAT3 in tumor cells. KT-333 causes a decrease in STAT3 levels in vitro and in vivo and induces tumor cell death[173]. KT-333 is in a Phase Ⅰ clinical trial (ClinicalTrials.gov Identifier: NCT05225584) to evaluate its safety, pharmacokinetics, pharmacodynamics, and clinical activity in adult patients with refractory lymphoma, large granular lymphocytic leukemia, solid tumors.
Schematic diagram of mechanism of PROTACs and Lysosome-Targeting Chimaeras (LYTACs). (A)An illustration of PROTAC (Au-AR pep-PROTAC) -mediated degradation of the AR and AR-V7. (B) An illustration of LYTAC-mediated degradation.
Three are still some challenges for us to apply PROTACs in clinic. First, the biological activity of PROTACs cannot be well predicted. Due to the different working principles, we cannot draw reliable conclusions based on the inhibitors of POI[174]. Second, the large molecular weight of PROTACs does not conform to Lipinski's Law of Five, which would affect their pharmacokinetic properties[175]. Thirdly, the degradation activity of PROTACs may be heterogeneous in different tissues or cells[176] due to the different expression of E3 in different tissues or cells[177]. Fourth, acquired resistance to PROTACs may emerge[178].
There are >600 E3 ubiquitin ligases in humans[179] and only a few are currently used for PROTAC development, including MDM2, CRBN, and IAP1. As the functions and tissue-specific expression of other E3 ligases are understood, more new E3 ligases such as RNF4[180], RNF114[181, 182] and KEAP1[167]will be used for the development of PROTACs in future [176]. A lot of effort and technology is required for the structural optimization of PROTACs drugs during the drug development process, which is costly and the results are very uncertain [183, 184]. The development of new technologies, such as NanoBiT system[185], enables the prediction and even real-time characterization of PROTACs-mediated degradation[186].
3.2. Molecular glue
The concept of molecular glue was first introduced in the 1990s[187]. Molecular glue "glues" together molecules that would not normally bind together by modifying the surface of the protein. The formation of a ternary complex facilitates the dimerization or colocalization of two proteins, thereby modulating their function. Cyclosporin A and FK506 were the first examples of molecular glue, followed by the discovery of rapamycin[187]. Like PROTACs, molecular glue degraders utilize UPS for the degradation of POIs. Unlike PROTACs, they do not bind both E3 ligase and target protein but one of the two. The interaction of the three is further induced or stabilized, subsequently leading to target protein ubiquitination and proteasome degradation. Since molecular glues do not have linkers, they have a smaller molecular weight similar to small molecules and therefore have better cell permeability[188]. A compelling example of glue degraders is thalidomide-like immunomodulatory drugs (IMiDs). They can function as protein degraders by binding to CRBN. It has been shown that Ikaros family zinc finger protein 1 (IKZF1) and IKZF3 can be ubiquitinated and degraded by forming complexes with IMiDs and CRBN[189]. This would be beneficial for the treatment of multiple myeloma and 5q-deletion-associated myelodysplastic syndrome, respectively[190, 191]. Iberdomide (CC-220) is a novel IKZF1/3 degradant. It has shown meaningful clinical efficacy in clinical trials (ClinicalTrials.gov NCT02773030) of patients with relapsed or refractory multiple myeloma. In the dose-escalation cohort (n=90), the overall response rate was 32%. In the dose-expansion cohort (n=107), the overall response rate was 26%. The most common adverse events of grade 3 or worse were neutropenia, anemia, infection, and thrombocytopenia. There was one treatment-related death, and 5 patients (5%) discontinued treatment because of adverse events[192]. Therefore, further evaluation of its efficacy in the treatment of myeloma is needed. It's important to note that the discovery of molecular glues is usually serendipitous and achieving their rational design requires overcoming many challenges.
3.3 LYTACs
Since the UPS is located intracellularly, PROTACs target intracellular proteins. Unlike PROTACs, LYTACs[193] mainly target extracellular and membrane-associated proteins, which comprise about 40% of the proteome and play key roles in disease progression[194]. LYTACs tagged with mannose-6-phosphonate link extracellular or membrane-bound proteins of interest to cation-independent mannose-6-phosphate receptors via the endosomal-lysosomal pathway to mediate protein endocytosis and lysosomal degradation[193, 195]. A recent study reported the development of molecular degraders of extracellular proteins through the asialoglycoprotein receptor that mediates the formation of ternary complexes through the same pathway, ultimately leading to endocytosis and degradation of target proteins [196] (Figure 2). Even though relatively few experimental studies have been conducted, the development of LYTACs has advanced therapeutic strategies targeting membrane and extracellular proteins.
Other protein degradation technologies include photodegradation-targeting chimeras[197], macroautophagy degradation targeting chimeras[198], autophagy-targeting chimera[199], autophagy-tethering compounds[200], and specific and non-genetic IAP-dependent protein erasers[201]. Targeted protein degradation strategies have gained considerable attention in the field of undruggable targets. Although these targeted degradation agents are reportedly effective, the results of experimental in vivo studies are insufficient and potential adverse events have not been addressed. In addition, the off-target toxicity, large molecular size, and high molecular complexity must be further optimized[202, 203].
4. Targeting PPIs
A single protein is often insufficient for most biological processes, thus most are conducted via PPIs. To date, more than 14,000 PPIs have been identified in humans[204]. These protein complexes are involved in many critical cellular functions, including cell growth, DNA replication, transcriptional activation, translation, and transmembrane signaling. In the rapidly advancing field of target-based drug discovery, inhibitors of PPIs have received increased attention. However, PPIs are considered undruggable targets due to the structural characteristics of the complex interfaces, such as large and highly hydrophobic interfacial regions, flat interfaces that are difficult to bind to inhibitor molecules, and interfaces with amino acid residues that bind to each other with high affinity and are difficult to target with SMIs[205].
4.1. SMIs
Accumulating evidence has challenged the traditional view that the structural characteristics of the PPI interface renders PPIs difficult to target with small-molecule drugs. The identification of “hot spots” has allowed the continued development of SMIs targeting PPIs[206]. Hot spots are key residues that are critical for high-affinity binding and significantly smaller in area than the interface. On average, hot spots account for 9.5% of interfacial residues[207, 208]. Competitive binding of a small-molecule ligand to a hot spot will block the original PPI[209]. Therefore, it is feasible to design SMIs to target the hot spots of PPIs. To date, several approaches have been employed for the development of SMIs targeting PPIs, including HTS, fragment screening, and virtual screening (VS). HTS, a major drug discovery paradigm, can rapidly and automatically screen hundreds of thousands of compounds for therapeutic applications over a relatively short period of time[210]. Applied techniques include fluorescence polarization-based drug screening[211], small-molecule microarray drug-screening platforms[212], surface plasmon resonance[213], MALDI-TOF mass spectrometry[214], and fluorescence resonance energy transfer (FRET)[215]. However, compound libraries are typically screened against traditional targets and, therefore, the rate of hits against targets of PPI is relatively low[216, 217]. Currently, the most successful examples of the application of HTS to identify targeted PPI inhibitors are the small molecules nutlins and benzodiazepinediones that target p53-MDM2 interactions[218, 219]. Fragment-based drug design (FBDD) is a method for developing effective compounds from fragments by random screening based on molecular structure[220] using highly sensitive screening techniques, such as nuclear magnetic resonance (NMR)[221] and X-ray crystallography[222], to identify suitable low molecular weight fragments with low affinity, followed by structural optimization to produce drug-like molecules[223]. FBDD is reportedly more effective than HTS for discovery of PPI inhibitors [224]. However, FBDD is limited by the difficulty in interpreting fragment hits to accurately identify drug candidates. The discovery of the novel antitumor drug ABT-263 (Navitoclax) [225], a Bcl-xL inhibitor, is considered a groundbreaking achievement for FBDD [226, 227]. VS is a complementary technique to aid HTS in drug development and can be divided into ligand- and structure-based VS[224], which has greater promise of success for PPI targets with well-defined hot spots. However, not all screened compounds can be successfully synthesized. In addition to the screening strategies described above, there are drug design strategies, including anchor-based PPI inhibitor design and design of small molecule mimics involved in PPI secondary structures. Anchors are hot residues of donor proteins and secondary structures that mediate most activities of PPIs[228]. Therefore, the use of small-molecule mimics of the key interactions of these components is an attractive strategy for modulation of PPIs[229]. The synthetic strategy is instead aimed at expanding the chemical space for screening of inhibitors[230].
4.2. Peptide-based drug discovery
In 2021, the number of peptides approved is at an all-time high[231]. Five peptides are approved for clinical use in 2022, representing 15% of the drugs approved by the FDA in 2022[4]. Peptide inhibitors are typically composed of less than 50 amino acids[232] and inhibit protein binding by modifying important residues at the interface of the original PPIs, such as hot spots[233]. Peptidomimetics are defined as peptide-like molecules containing amino acid analogs and other chemical moieties with specific pharmacophores[234] that bind competitively to PPI binding partners through similar structures, thereby blocking the original PPIs[235]. Peptides have higher selectivity and lower toxicity as compared to small-molecule drugs, and are cheaper to develop, more stable, and smaller with superior penetration than biologics[236, 237]. Therefore, peptides remain a promising class of drug candidates to target PPIs. Peptides have the combined advantages of small-molecule and protein drugs, but also some disadvantages, including low cell permeability, poor oral bioavailability, and short half-lives after administration, which pose great challenges for peptide-based drug discovery[237, 238]. Cyclization and modification of the backbone have emerged as two major strategies to address these issues[239]. General strategies for cyclization include hydrogen-bonded substitutes, stapling, and hairpins, which improve binding affinity, selectivity, and bioavailability by conformational restriction of the peptide[238]. A previous report proposed nucleophilic substitution of aromatic moieties for the synthesis of macrocyclic peptides by N-arylation[236]. This method was employed to obtain potent peptide inhibitors of p53-MDM2 with improved proteolytic stability and cell permeability[240]. Rational modification of the peptide backbone structure can reduces sensitivity to protein hydrolases and improve metabolic stability[239]. Display technologies, such as phage display, allow the creation of phage libraries through genetic modification for screening of peptide molecules that specifically bind to target molecules[241, 242]. In addition, integrated venomics allows bioinformatics analysis of genomic and transcriptomic data for screening of venom peptides[243] as specific therapeutic targets for the development of peptide-based drugs for undruggable targets. To date, more than 60 peptide-based drugs targeting PPIs have been developed, optimized, and approved for clinical application[244] (Figure 3).
Orthosteric inhibition of PPIs by SMIs and peptide inhibitors. The red dots indicate “hot spots”.
MDM2 is a negative regulator of P53 and mediates its degradation by binding to p53[245]. Numerous studies have shown that blocking the p53-MDM2 interaction can restore the tumor suppressor function of P53[246]. The key interface residues involved Phe19, Trp23 and Leu26 on P53[247]. This is an important structural basis for inhibitor development. Since the discovery of Nutlin, several new potential inhibitors of the p53-MDM2 interaction have emerged, some of which have shown some positive results in preclinical and clinical studies. Based on a full understanding of the structure of MDM2, dihydroisoquinolinone was selected as the scaffold of inhibitor by VS. A combination of X-ray crystallography, molecular modeling, and iterative medicinal chemistry stepwise optimization eventually led to the discovery of NVP-CG1097[248]. NVP-CG1097 is a specific and highly selective p53-MDM2 inhibitor. In preclinical studies, it induced an increase in p53 expression[249]. However, NVP-CGM097 did not appear to exhibit significant tumor regression and had some undesired hematologic toxicity in the phase Ⅰ clinical trial[250]. But there is no denying that this is a very meaningful attempt. In addition to SMIs, current peptide-based PPI inhibitors are also a promising class of drug candidates. β-peptides[251], peptoids[252], N-acylpolyamine[253] have previously been extensively studied to target the p53-MDM2 interaction. Currently, γ-AApeptide is receiving increasing attention as a novel peptidomimetic inhibitor that can disrupt the p53-MDM2 interaction[254]. To further enhance the inhibitory potency, sulfonamide groups were added to induce the scaffold to bend and fold[255]. Subsequently, from the addition of chiral side chains[255], development of D-sulfono-γ-AApeptide[256] to 1:1α/sulfono-γ-AApeptide[257], which showed a gradual increase in inhibitory potency and resistance to hydrolysis. This suggests that rational design of peptidomimetics is a promising way to block the p53-MDM2 interaction.
5. Targeting IDPs
Intrinsically disordered regions (IDRs) and IDPs lack stable secondary and/or tertiary structures[258], have low sequence complexity, lack large hydrophobic residues, are rich in charged and polar residues, and are highly flexible[259]. Over 30% of the eukaryotic proteins are IDPs and are involved in many important processes[260]. There is an association between dysregulation of IDPs and many human diseases, such as cancer, neurodegenerative diseases, and diabetes, among others [261]. Both p53 and C-MYC possess IDRs, which are major impediments to targeting with drugs. Currently, there are three main strategies to target IDPs: (1) stabilize disordered states to prevent formation of toxic polymers; (2) inhibit interactions with other proteins to prevent formation of protein complexes with unfavorable biological functions; and (3) induce allosteric inhibition[262]. Since IDPs are widely involved with PPIs, such as p53-MDM2 and C-MYC-MAX, the development of drugs to target IDPs involved in protein complexes employs some of the same strategies for targeting PPIs, such as FBDD. Commonly used drug discovery techniques include NMR, small-angle X-ray scattering, circular dichroism, FRET, and simulations of molecular dynamics (MDs). Among them, NMR is used to study the transient interactions of ligands with proteins and is arguably the best technique to observe and characterize the structural dynamics of IDRs[263], while small-angle X-ray scattering and FRET are highly complementary to NMR[264]. With different advantages and disadvantages, NMR revealed more detailed structural dynamics of C-MYC-MAX and smaller molecular weights of rigid proteins. For C-MYC-MAX, three NMR structures (PDB codes: 1A93, 1MV0, and 2A93) were identified[265, 266]. Although NMR provides snapshots of dynamic structures (20 models of 1MVO and 40 models of 2A93), the lengths of the three structures were less than 30 amino acids. For the X-ray determined structures of C-MYC-MAX (PDB codes: 1NKP, 6G6J, 6G6K, and 6G6L), the lengths of the structures were greater than 80 amino acids. In addition, the interactions between C-MYC-MAX and DNA (5'-CACGTG-3') were clearly revealed[267, 268]. The high-resolution structure of 1NKP explained how bHLHZ heterodimers can mediate specific and high-affinity binding to DNA, throwing light on the drug design targeting the C-MYC-MAX complex.
In contrast to the conformation-based approaches described above, the MD-based approach using MD simulations of the behavior of IDPs under physiological conditions in a solvent provide greater insight into protein dynamics[269]. The structure-based strategy was utilized to design efficient peptides to disrupt p53-MDM2/X interactions. In a prior study, the critical residues of validated inhibitors of MDM2/X were analyzed and computational HTS as well as MD simulations were conducted of two mutants, pDI (LTFEHYWAQLTS) and pDIQ (ETFEHWWSQLLS), which provided theoretical structural information of 27 native and mutants of p53-based inhibitory peptides[270]. A new Bayesian inference approach (MELD × MD) was used to simulate five peptides binding to MDM2, including the p53 epitope, pdiq, ATSP-7041, and two negative alanine-based peptides, which depicted the most likely bound conformations of MDM2 ligands[271]. Furthermore, physics-based atomic simulations can effectively overcome the uncertainties of disordered ensemble calculations and are expected to provide the rigorous thermodynamic ensembles required to reliably describe IDP-ligand interactions[264]. Even though several experimental and computational studies have offered insight into IDPs, additional experimental techniques are needed for drug discovery. Further understanding of the structure and function of IDPs is particularly important for treatment of neurodegenerative diseases associated with abnormal peptide aggregation. Moreover, IDPs are associated with the regulation of liquid-liquid phase separations in cells, which drives the formation of membrane-free cell organelles and the localization of biomolecules [272, 273], and may provide a novel approach to elucidate the roles of IDPs in tumors and neurological diseases.
6. Targeting protein-DNA binding
TFs are the most widely studied drug targets to block gene expression for cancer treatment. Although blocking protein-DNA interactions appears to be a more direct approach, studies have been limited. It is difficult to develop specific SMIs due to the large interface area of protein-DNA interactions, the large number of anchor points, the highly positive charge, and the fact that the DNA binding sites of related proteins are usually highly conserved [274, 275]. Despite these problems, the remarkable advances in CADD and experimental techniques have facilitated the development of protein-DNA inhibitors. Deep docking, an ultra-fast AI-based method, can significantly improve the efficiency of drug screening with significant runtime savings while handling large chemical libraries[276]. In addition, the most commonly used databases for AI-based prediction of binding affinity currently include the PDBbind database, Comparative Assessment of Scoring Functions benchmark dataset, and BindingDB, a web-accessible database of measured binding affinities [277-279]. The continued development of AI and deep learning has provided greater possibilities for protein structure prediction and structure-based drug design. Existing machine learning methods for prediction of binding sites can be divided into classical non-deep learning methods and modern deep learning methods, such as P2Rank [280] and DeepSurf [281]. In general, several computational tools, such as homology modeling[282] and ProDFace[282], can be used to predict druggable sites on the surface of DNA-binding domains, which are highly conserved and not prone to mutations, providing opportunities for drug development[283]. The DNA-binding interface of the AR was successfully identified using homology modeling and VS [284, 285]. InS3-54 is a STAT3-DNA binding inhibitor identified using VS and screened from 2 million compounds. InS3-54 has an IC50 of 13.8 ± 0.4 μM and binds selectively to STAT3. The main residues with which STAT3 interacts are Met331, Val343, Ile467, Met470, Lys 340 and Asn466. And InS3-54 does not bind to STAT1 due to the conflict of residues Pro326 and Thr327 with InS-54. In several lung and breast cancer cell lines, InS3-54 induced apoptosis to inhibit cancer cell growth and inhibited cancer cell migration and invasion[286]. Hence, CADD greatly improves the feasibility of targeting protein-DNA sites.
7. Summary
This review provides an overview of the current status of undruggable targets and potential solutions from several perspectives. Bifunctional small molecules enable targeted degradation of proteins of interest through various endogenous pathways, which can reduce the overall levels of disease-associated proteins and broaden the development of drugs for specific targets. At least 20 PROTACs against the targets mentioned above have been developed. Various drug entities and discovery techniques have its limitations. The permeability and bioavailability are poor due to its large molecular weight, and the degradation activity of target protein is difficult to evaluate. Also, the drug properties, toxicities and resistance of degraders need to be validated by adequate experiments. The stability of targeting PPI drugs like peptide should be further improved. High-throughput screening need to expand their screening libraries to improve hit rates, and the challenge for FBDD and virtual screening is to successfully translate screening hits into drug candidates.
Emerging technologies like excellent drug delivery system, conditional activated PROTACs (light-induced protein degradation), tissue specific E3 ligases and its ligands, high throughput screening method of active PROTACs may be helpful to overcome PROTACs limitations in the future. AI is also very helpful to discover and develop new target protein, obviously, which can also shed some lights into the overcome of the undruggable targets. AI technologies, such as DeepChem, DeepTox, and DeepNeuralNetQSAR, have been applied in virtual screening to predict the physicochemical properties, biological activities, and toxicities of drug molecules. AI can also help to predict the three-dimensional structures of target proteins and drug-protein interactions. AlphaFold significantly outperformed other protein structure prediction methods. This will help researchers better understand protein structures and interactions, which in turn will speed up the process of drug development. For example, using Alphafold's prediction results, candidate drug molecules that interact with target proteins can be quickly screened and their structures optimized for improved affinity and specificity. Alphafold also has some limitations, such as not being able to solve the long-standing protein folding problem; predicting a single ranked structure of a protein sequence, which cannot directly address the allosteric mechanism; and being less applicable to IDPs and IDRs, using low structural probabilities to describe them[287, 288]. Therefore, a combination of approaches is needed to address these targets. Moreover, further discovery of new drug targets, such as some proteins involved in cell metabolism, cell death pathways, seems to be beneficial for the treatment of diseases. Another approach is to develop activators of proteins against some targets, such as tumor suppressors activators, which seems to be another concise approach.
With the help of new drug-development strategies, some undruggable target proteins like RAS, HIF-2α, BCL-2, MDM2, and MLL are no longer considered undruggable right now. It remains a huge challenge to completely overcome the undruggable proteins, and new drug development strategies, better extraction processes that do not disrupt protein-protein interactions, and more precise AI are urgently needed. In any case, we believed that it is only a matter of time before undruggable targets become druggable.
Acknowledgements
Funding
This work was financially supported by the Basic scientific research project of Liaoning Province Education Department (No. LJKZ0832, China), the National Science Foundation of China (Nos. 81922070, 81973286, and 82141203, China), the Shanghai Science and Technology Innovation Action Plans of the Shanghai Science and Technology Committee (Nos. 20S21901500 and 20S21900900, China), the Innovation Team and Talents Cultivation Program of the National Administration of Traditional Chinese Medicine (No. ZYYCXTDD-202004, China), and the Three-year Action Plan of the Shanghai Traditional Chinese Medicine Development and Inheritance Program (No. ZY(2021-2023)-0401, China).
Author contributions
D.C., G.G. and Y.L.: Formulation of conception and design. Y.L., Y.Y., G.Z., Y.F., F.G. and D.X.: creation of the initial draft and revision of the draft. H.Z., Y.L. and B.W.: design figures and tables. D.C. and G.G.: oversight and critical revising.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Kifle ZD, Tadele M, Alemu E, Gedamu T, Ayele AG. A recent development of new therapeutic agents and novel drug targets for cancer treatment. SAGE Open Med. 2021;9:20503121211067083
3. Pourquier P. [Alkylating agents]. Bull Cancer. 2011;98:1237-51
4. de la Torre BG, Albericio F. The Pharmaceutical Industry in 2022: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules. 2023 28
5. Youinou P, Pers JO, Gershwin ME, Shoenfeld Y. Geo-epidemiology and autoimmunity. J Autoimmun. 2010;34:J163-7
6. Katsuno M, Sahashi K, Iguchi Y, Hashizume A. Preclinical progression of neurodegenerative diseases. Nagoya J Med Sci. 2018;80:289-98
7. Van Herle K, Behne JM, Van Herle A, Blaschke TF, Smith TJ, Yeaman MR. Integrative continuum: accelerating therapeutic advances in rare autoimmune diseases. Annu Rev Pharmacol Toxicol. 2012;52:523-47
8. Cuny GD. Foreword: neurodegenerative diseases: challenges and opportunities. Future Med Chem. 2012;4:1647-9
9. Berdigaliyev N, Aljofan M. An overview of drug discovery and development. Future Med Chem. 2020;12:939-47
10. Eder J, Herrling PL. Trends in Modern Drug Discovery. Handb Exp Pharmacol. 2016;232:3-22
11. Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol. 2016;56:23-40
12. Dimitrov DS, Marks JD. Therapeutic antibodies: current state and future trends-is a paradigm change coming soon? Methods Mol Biol. 2009;525:1-27 xiii
13. Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714-27
14. Baumann A. Early development of therapeutic biologics-pharmacokinetics. Curr Drug Metab. 2006;7:15-21
15. Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1-26
16. Spradlin JN, Zhang E, Nomura DK. Reimagining Druggability Using Chemoproteomic Platforms. Acc Chem Res. 2021;54:1801-13
17. Chen K, Zhang Y, Qian L, Wang P. Emerging strategies to target RAS signaling in human cancer therapy. J Hematol Oncol. 2021;14:116
18. Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non-small-cell lung cancer: recent progress and new approaches. Ann Oncol. 2021;32:1101-10
19. Luo J. KRAS mutation in pancreatic cancer. Semin Oncol. 2021;48:10-8
20. Liebl MC, Hofmann TG. The Role of p53 Signaling in Colorectal Cancer. Cancers (Basel). 2021 13
21. Hunter T. The genesis of tyrosine phosphorylation. Cold Spring Harb Perspect Biol. 2014;6:a020644
22. Bollu LR, Mazumdar A, Savage MI, Brown PH. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin Cancer Res. 2017;23:2136-42
23. Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the 'undruggable' cancer targets. Nat Rev Cancer. 2017;17:502-8
24. Zhuang JJ, Liu Q, Wu DL, Tie L. Current strategies and progress for targeting the "undruggable" transcription factors. Acta Pharmacol Sin. 2022;43:2474-81
25. Punekar SR, Velcheti V, Neel BG, Wong KK. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. 2022;19:637-55
26. Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004:RE13
27. Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020;80:2969-74
28. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M. et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116:15823-9
29. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457-67
30. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res. 2015;13:1325-35
31. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F. et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333-8
32. Krygowska AA, Castellano E. PI3K: A Crucial Piece in the RAS Signaling Puzzle. Cold Spring Harb Perspect Med. 2018 8
33. Leung EL, Luo LX, Li Y, Liu ZQ, Li LL, Shi DF. et al. Identification of a new inhibitor of KRAS-PDEdelta interaction targeting KRAS mutant nonsmall cell lung cancer. Int J Cancer. 2019;145:1334-45
34. Yin C, Zhu B, Zhang T, Liu T, Chen S, Liu Y. et al. Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell. 2019;176:1113-27 e16
35. Martinez-Garcia M, Banerji U, Albanell J, Bahleda R, Dolly S, Kraeber-Bodere F. et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res. 2012;18:4806-19
36. Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ. et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther. 2017;16:2351-63
37. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R. et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6
38. Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628-40
39. Xue W, Dahlman JE, Tammela T, Khan OF, Sood S, Dave A. et al. Small RNA combination therapy for lung cancer. Proc Natl Acad Sci U S A. 2014;111:E3553-61
40. Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T. et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther. 2014;13:2876-85
41. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent Sci. 2020;6:1367-75
42. Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ. et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem Biol. 2020;27:19-31 e6
43. Blair HA. Sotorasib: First Approval. Drugs. 2021;81:1573-9
44. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217-23
45. Kim D, Xue JY, Lito P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell. 2020;183:850-9
46. Kargbo RB. Inhibitors of G12C Mutant Ras Proteins for the Treatment of Cancers. ACS Med Chem Lett. 2019;10:10-1
47. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV. et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6:316-29
48. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI. et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383:1207-17
49. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J. et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384:2371-81
50. Ji X, Li Y, Kong X, Chen D, Lu J. Discovery of Prodrug of MRTX1133 as an Oral Therapy for Cancers with KRAS(G12D) Mutation. ACS Omega. 2023;8:7211-21
51. Kemp SB, Cheng N, Markosyan N, Sor R, Kim IK, Hallin J. et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023;13:298-311
52. Mahadevan KK, McAndrews KM, LeBleu VS, Yang S, Lyu H, Li B. et al. Oncogenic Kras (G12D) specific non-covalent inhibitor reprograms tumor microenvironment to prevent and reverse early pre-neoplastic pancreatic lesions and in combination with immunotherapy regresses advanced PDAC in a CD8 (+) T cells dependent manner. bioRxiv. 2023
53. Awad MM, Liu S, Rybkin, II, Arbour KC, Dilly J, Zhu VW. et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med. 2021;384:2382-93
54. Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT. et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599:679-83
55. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:386
56. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M. et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J Thorac Oncol. 2021;16:1321-32
57. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA. et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res. 2020;26:1633-43
58. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B. et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421-5
59. Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F. et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7:86-93
60. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA. et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021;11:1913-22
61. Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R. et al. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res. 2020;26:5962-73
62. Gomez-Lazaro M, Fernandez-Gomez FJ, Jordan J. p53: twenty five years understanding the mechanism of genome protection. J Physiol Biochem. 2004;60:287-307
63. Georgakilas AG, Martin OA, Bonner WM. p21: A Two-Faced Genome Guardian. Trends Mol Med. 2017;23:310-9
64. Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899-908
65. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
66. Faria MH, Patrocinio RM, Moraes Filho MO, Rabenhorst SH. Immunoexpression of tumor suppressor genes p53, p21 WAF1/CIP1 and p27 KIP1 in humam astrocystic tumors. Arq Neuropsiquiatr. 2007;65:1114-22
67. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008
68. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18:89-102
69. Chen S, Wu JL, Liang Y, Tang YG, Song HX, Wu LL. et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell. 2021;39:225-39 e8
70. Buyandelger B, Bar EE, Hung KS, Chen RM, Chiang YH, Liou JP. et al. Histone deacetylase inhibitor MPT0B291 suppresses Glioma Growth in vitro and in vivo partially through acetylation of p53. Int J Biol Sci. 2020;16:3184-99
71. Jhaveri K, Modi S. Ganetespib: research and clinical development. Onco Targets Ther. 2015;8:1849-58
72. Yu S, Cai X, Wu C, Liu Y, Zhang J, Gong X. et al. Targeting HSP90-HDAC6 Regulating Network Implicates Precision Treatment of Breast Cancer. Int J Biol Sci. 2017;13:505-17
73. Wang TH, Li WT, Yu SH, Au LC. The use of 10-23 DNAzyme to selectively destroy the allele of mRNA with a unique purine-pyrimidine dinucleotide. Oligonucleotides. 2008;18:295-9
74. Iyer SV, Parrales A, Begani P, Narkar A, Adhikari AS, Martinez LA. et al. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget. 2016;7:5401-15
75. Wang S, Zhao Y, Aguilar A, Bernard D, Yang CY. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb Perspect Med. 2017 7
76. Munisamy M, Mukherjee N, Thomas L, Pham AT, Shakeri A, Zhao Y. et al. Therapeutic opportunities in cancer therapy: targeting the p53-MDM2/MDMX interactions. Am J Cancer Res. 2021;11:5762-81
77. Secchiero P, Bosco R, Celeghini C, Zauli G. Recent advances in the therapeutic perspectives of Nutlin-3. Curr Pharm Des. 2011;17:569-77
78. Beloglazkina A, Zyk N, Majouga A, Beloglazkina E. Recent Small-Molecule Inhibitors of the p53-MDM2 Protein-Protein Interaction. Molecules. 2020 25
79. Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R. et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018;6:282-300 e2
80. Llombart V, Mansour MR. Therapeutic targeting of "undruggable" MYC. EBioMedicine. 2022;75:103756
81. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253-64
82. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976-90
83. Eilers M, Eisenman RN. Myc's broad reach. Genes Dev. 2008;22:2755-66
84. Brooks TA, Hurley LH. Targeting MYC Expression through G-Quadruplexes. Genes Cancer. 2010;1:641-9
85. Levens D. You Don't Muck with MYC. Genes Cancer. 2010;1:547-54
86. Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Drug targeting of the c-MYC promoter to repress gene expression via a G-quadruplex silencer element. Semin Oncol. 2006;33:498-512
87. Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D. et al. MYC Deregulation in Primary Human Cancers. Genes (Basel). 2017 8
88. Shachaf CM, Felsher DW. Tumor dormancy and MYC inactivation: pushing cancer to the brink of normalcy. Cancer Res. 2005;65:4471-4
89. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM. et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679-83
90. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313-7
91. Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene. 1998;17:2463-72
92. Jung LA, Gebhardt A, Koelmel W, Ade CP, Walz S, Kuper J. et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene. 2017;36:1911-24
93. Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN. et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504-13
94. Beaulieu ME, Jauset T, Masso-Valles D, Martinez-Martin S, Rahl P, Maltais L. et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med. 2019 11
95. Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I. et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun. 2014;5:4632
96. Fiorentino FP, Tokgun E, Sole-Sanchez S, Giampaolo S, Tokgun O, Jauset T. et al. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget. 2016;7:31014-28
97. Demma MJ, Mapelli C, Sun A, Bodea S, Ruprecht B, Javaid S. et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol Cell Biol. 2019 39
98. Savino M, Annibali D, Carucci N, Favuzzi E, Cole MD, Evan GI. et al. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS One. 2011;6:e22284
99. Soucek L, Nasi S, Evan GI. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 2004;11:1038-45
100. Nishida Y, Scruggs DA, Ayoub E, Patsilevas T, Ruvolo VR, Mak PY. et al. AML-147 C-MYC Targeting by Degradation: Novel Dual c-Myc/GSPT1 Degrader GT19715 Exerts Profound Cell Kill In Vitro and In Vivo in Acute Myeloid Leukemia and Lymphomas. Clinical Lymphoma Myeloma and Leukemia. 2022;22:S218
101. Samarasinghe KTG, Crews CM. Targeted protein degradation: A promise for undruggable proteins. Cell Chem Biol. 2021;28:934-51
102. Dong J, Cheng XD, Zhang WD, Qin JJ. Recent Update on Development of Small-Molecule STAT3 Inhibitors for Cancer Therapy: From Phosphorylation Inhibition to Protein Degradation. J Med Chem. 2021;64:8884-915
103. Lee H, Jeong AJ, Ye SK. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 2019;52:415-23
104. Chai EZ, Shanmugam MK, Arfuso F, Dharmarajan A, Wang C, Kumar AP. et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther. 2016;162:86-97
105. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer. 2020;19:145
106. Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018;415:117-28
107. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41-51
108. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016;37:11553-72
109. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L. et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605-15
110. Gharibi T, Hosseini A, Marofi F, Oraei M, Jahandideh S, Abdollahpour-Alitappeh M. et al. IL-21 and IL-21-producing T cells are involved in multiple sclerosis severity and progression. Immunol Lett. 2019;216:12-20
111. Egwuagu CE. STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine. 2009;47:149-56
112. Shah DR, Shah RR, Morganroth J. Tyrosine kinase inhibitors: their on-target toxicities as potential indicators of efficacy. Drug Saf. 2013;36:413-26
113. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem Biol. 2016;11:308-18
114. Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D. et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J Med Chem. 2019;62:11280-300
115. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C. et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401-6
116. Trump DL. Commentary on "Intermittent versus continuous androgen deprivation in prostate cancer." Hussain M, Tangen CM, Berry DL, Higano CS, Crawford ED, Liu G, Wilding G, Prescott S, Kanaga Sundaram S, Small EJ, Dawson NA, Donnelly BJ, Venner PM, Vaishampayan UN, Schellhammer PF, Quinn DI, Raghavan D, Ely B, Moinpour CM, Vogelzang NJ, Thompson IM Jr, University of Michigan, Division of Hematology/Oncology, 1500 E Medical Center Dr, 7314 CC, Ann Arbor, MI. N Engl J Med 2013;368(14):1314-25. Urol Oncol. 2013;31:1847
117. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS. et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424-33
118. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276-308
119. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180-92
120. Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN. et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. 2019;129:192-208
121. Uo T, Plymate SR, Sprenger CC. The potential of AR-V7 as a therapeutic target. Expert Opin Ther Targets. 2018;22:201-16
122. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E. et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16-22
123. Melnyk JE, Steri V, Nguyen HG, Hann B, Feng FY, Shokat KM. The splicing modulator sulfonamide indisulam reduces AR-V7 in prostate cancer cells. Bioorg Med Chem. 2020;28:115712
124. Cui C, Zhou X, Zhang W, Qu Y, Ke X. Is beta-Catenin a Druggable Target for Cancer Therapy? Trends Biochem Sci. 2018;43:623-34
125. Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169:985-99
126. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X. et al. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022;7:3
127. Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8:33972-89
128. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50-60
129. Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T. et al. C/EBPalpha is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci U S A. 2014;111:9899-904
130. Pineault N, Helgason CD, Lawrence HJ, Humphries RK. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp Hematol. 2002;30:49-57
131. Aryal S, Zhang Y, Wren S, Li C, Lu R. Molecular regulators of HOXA9 in acute myeloid leukemia. FEBS J. 2021
132. Collins CT, Hess JL. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr Opin Hematol. 2016;23:354-61
133. Lambert M, Alioui M, Jambon S, Depauw S, Van Seuningen I, David-Cordonnier MH. Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia. Cancers (Basel). 2019 11
134. Yao M, Gu Y, Yang Z, Zhong K, Chen Z. MEIS1 and its potential as a cancer therapeutic target (Review). Int J Mol Med. 2021 48
135. Mohr S, Doebele C, Comoglio F, Berg T, Beck J, Bohnenberger H. et al. Hoxa9 and Meis1 Cooperatively Induce Addiction to Syk Signaling by Suppressing miR-146a in Acute Myeloid Leukemia. Cancer Cell. 2017;31:549-62 e11
136. Hong SH, Youbi SE, Hong SP, Kallakury B, Monroe P, Erkizan HV. et al. Pharmacokinetic modeling optimizes inhibition of the 'undruggable' EWS-FLI1 transcription factor in Ewing Sarcoma. Oncotarget. 2014;5:338-50
137. Glass AG, Fraumeni JF Jr. Epidemiology of bone cancer in children. J Natl Cancer Inst. 1970;44:187-99
138. Ramachandran B, Rajkumar T, Gopisetty G. Challenges in modeling EWS-FLI1-driven transgenic mouse model for Ewing sarcoma. Am J Transl Res. 2021;13:12181-94
139. Jully B, Vijayalakshmi R, Gopal G, Sabitha K, Rajkumar T. Junction region of EWS-FLI1 fusion protein has a dominant negative effect in Ewing's sarcoma in vitro. BMC Cancer. 2012;12:513
140. Grohar PJ, Segars LE, Yeung C, Pommier Y, D'Incalci M, Mendoza A. et al. Dual targeting of EWS-FLI1 activity and the associated DNA damage response with trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growth. Clin Cancer Res. 2014;20:1190-203
141. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273-304
142. Schoenfeld AJ, Bandlamudi C, Lavery JA, Montecalvo J, Namakydoust A, Rizvi H. et al. The Genomic Landscape of SMARCA4 Alterations and Associations with Outcomes in Patients with Lung Cancer. Clin Cancer Res. 2020;26:5701-8
143. Thompson KW, Marquez SB, Reisman D. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1-letter. Cancer Res. 2014;74:4946-7
144. Zhang ZY. Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc Chem Res. 2017;50:122-9
145. Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140-6
146. He RJ, Yu ZH, Zhang RY, Zhang ZY. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin. 2014;35:1227-46
147. Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A. et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699-711
148. Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH. et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117-36
149. He Y, Liu S, Menon A, Stanford S, Oppong E, Gunawan AM. et al. A potent and selective small-molecule inhibitor for the lymphoid-specific tyrosine phosphatase (LYP), a target associated with autoimmune diseases. J Med Chem. 2013;56:4990-5008
150. Elhassan RM, Hou X, Fang H. Recent advances in the development of allosteric protein tyrosine phosphatase inhibitors for drug discovery. Med Res Rev. 2021
151. Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. 2016;8:59
152. Simon RP, Robaa D, Alhalabi Z, Sippl W, Jung M. KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J Med Chem. 2016;59:1249-70
153. Farria A, Li W, Dent SY. KATs in cancer: functions and therapies. Oncogene. 2015;34:4901-13
154. Baell JB, Miao W. Histone acetyltransferase inhibitors: where art thou? Future Med Chem. 2016;8:1525-8
155. Chen Q, Yang B, Liu X, Zhang XD, Zhang L, Liu T. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics. 2022;12:4935-48
156. Giotopoulos G, Chan WI, Horton SJ, Ruau D, Gallipoli P, Fowler A. et al. The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene. 2016;35:279-89
157. Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN. et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021;11:1118-37
158. Hogg SJ, Motorna O, Cluse LA, Johanson TM, Coughlan HD, Raviram R. et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell. 2021;81:2183-200 e13
159. Cai LY, Chen SJ, Xiao SH, Sun QJ, Ding CH, Zheng BN. et al. Targeting p300/CBP Attenuates Hepatocellular Carcinoma Progression through Epigenetic Regulation of Metabolism. Cancer Res. 2021;81:860-72
160. Manzo F, Tambaro FP, Mai A, Altucci L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin Ther Pat. 2009;19:761-74
161. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554-9
162. Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904-8
163. Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc. 2010;132:5820-6
164. Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10:1770-7
165. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K. et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755-63
166. Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y. et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251-9
167. Wei J, Meng F, Park KS, Yim H, Velez J, Kumar P. et al. Harnessing the E3 Ligase KEAP1 for Targeted Protein Degradation. J Am Chem Soc. 2021;143:15073-83
168. Burke MR, Smith AR, Zheng G. Overcoming Cancer Drug Resistance Utilizing PROTAC Technology. Front Cell Dev Biol. 2022;10:872729
169. Liu Z, Hu M, Yang Y, Du C, Zhou H, Liu C. et al. An overview of PROTACs: a promising drug discovery paradigm. Mol Biomed. 2022;3:46
170. Gao X, III HAB, Vuky J, Dreicer R, Sartor AO, Sternberg CN. et al. Phase 1/2 study of ARV-110, an androgen receptor (AR) PROTAC degrader, in metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2022;40:17 -
171. Qi SM, Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front Pharmacol. 2021;12:692574
172. Ma B, Fan Y, Zhang D, Wei Y, Jian Y, Liu D. et al. De Novo Design of an Androgen Receptor DNA Binding Domain-Targeted peptide PROTAC for Prostate Cancer Therapy. Adv Sci (Weinh). 2022;9:e2201859
173. Liu PCC, Dixit V, Mayo M, Dey J, Yuan K, Karnik R. et al. A First-in-Class STAT3 Degrader KT-333 in Development for Treatment of Hematologic Cancers. Blood. 2021;138:1865 -
174. Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S. et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol. 2018;25:78-87 e5
175. Matsson P, Doak BC, Over B, Kihlberg J. Cell permeability beyond the rule of 5. Adv Drug Deliv Rev. 2016;101:42-61
176. Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E. et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807-10
177. Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J. et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A. 2018;115:E7285-E92
178. Zhang L, Riley-Gillis B, Vijay P, Shen Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol Cancer Ther. 2019;18:1302-11
179. Ottis P, Toure M, Cromm PM, Ko E, Gustafson JL, Crews CM. Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem Biol. 2017;12:2570-8
180. Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K. et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem Biol. 2019;14:2430-40
181. Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L. et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol. 2019;15:747-55
182. Luo M, Spradlin JN, Boike L, Tong B, Brittain SM, McKenna JM. et al. Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem Biol. 2021;28:559-66 e15
183. Tamatam R, Shin D. Emerging Strategies in Proteolysis-Targeting Chimeras (PROTACs): Highlights from 2022. Int J Mol Sci. 2023 24
184. Moon Y, Jeon SI, Shim MK, Kim K. Cancer-Specific Delivery of Proteolysis-Targeting Chimeras (PROTACs) and Their Application to Cancer Immunotherapy. Pharmaceutics. 2023 15
185. Riching KM, Mahan S, Corona CR, McDougall M, Vasta JD, Robers MB. et al. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem Biol. 2018;13:2758-70
186. Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111-9
187. Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807-15
188. Dong G, Ding Y, He S, Sheng C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J Med Chem. 2021;64:10606-20
189. Asatsuma-Okumura T, Ito T, Handa H. Molecular mechanisms of cereblon-based drugs. Pharmacol Ther. 2019;202:132-9
190. Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M. et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301-5
191. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS. et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305-9
192. Lonial S, Popat R, Hulin C, Jagannath S, Oriol A, Richardson PG. et al. Iberdomide plus dexamethasone in heavily pretreated late-line relapsed or refractory multiple myeloma (CC-220-MM-001): a multicentre, multicohort, open-label, phase 1/2 trial. Lancet Haematol. 2022;9:e822-e32
193. Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584:291-7
194. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419
195. Gary-Bobo M, Nirde P, Jeanjean A, Morere A, Garcia M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945-53
196. Caianiello DF, Zhang M, Ray JD, Howell RA, Swartzel JC, Branham EMJ. et al. Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat Chem Biol. 2021;17:947-53
197. Liu S, Zhao X, Shui S, Wang B, Cui Y, Dong S. et al. PDTAC: Targeted Photodegradation of GPX4 Triggers Ferroptosis and Potent Antitumor Immunity. J Med Chem. 2022;65:12176-87
198. Alabi SB, Crews CM. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem. 2021;296:100647
199. Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A. et al. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol Cell. 2019;76:797-810 e10
200. Li Z, Zhu C, Ding Y, Fei Y, Lu B. ATTEC: a potential new approach to target proteinopathies. Autophagy. 2020;16:185-7
201. Naito M, Ohoka N, Shibata N. SNIPERs-Hijacking IAP activity to induce protein degradation. Drug Discov Today Technol. 2019;31:35-42
202. Hughes SJ, Testa A, Thompson N, Churcher I. The rise and rise of protein degradation: Opportunities and challenges ahead. Drug Discov Today. 2021;26:2889-97
203. Moreau K, Coen M, Zhang AX, Pachl F, Castaldi MP, Dahl G. et al. Proteolysis-targeting chimeras in drug development: A safety perspective. Br J Pharmacol. 2020;177:1709-18
204. Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N. et al. A proteome-scale map of the human interactome network. Cell. 2014;159:1212-26
205. Olah J, Szenasi T, Lehotzky A, Norris V, Ovadi J. Challenges in Discovering Drugs That Target the Protein-Protein Interactions of Disordered Proteins. Int J Mol Sci. 2022 23
206. Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383-6
207. Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1-9
208. Moreira IS, Fernandes PA, Ramos MJ. Hot spots-a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803-12
209. Zinzalla G, Thurston DE. Targeting protein-protein interactions for therapeutic intervention: a challenge for the future. Future Med Chem. 2009;1:65-93
210. Carnero A. High throughput screening in drug discovery. Clin Transl Oncol. 2006;8:482-90
211. Kiessling A, Sperl B, Hollis A, Eick D, Berg T. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem Biol. 2006;13:745-51
212. Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL. et al. Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem Biol. 2019;26:711-23 e14
213. Nguyen HH, Park J, Kang S, Kim M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (Basel). 2015;15:10481-510
214. Ritorto MS, Ewan R, Perez-Oliva AB, Knebel A, Buhrlage SJ, Wightman M. et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun. 2014;5:4763
215. Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629-33
216. Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727-30
217. Macarron R. Critical review of the role of HTS in drug discovery. Drug Discov Today. 2006;11:277-9
218. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8
219. Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK. et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005;48:909-12
220. Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6:211-9
221. Kang C. Applications of In-Cell NMR in Structural Biology and Drug Discovery. Int J Mol Sci. 2019 20
222. Schiebel J, Krimmer SG, Rower K, Knorlein A, Wang X, Park AY. et al. High-Throughput Crystallography: Reliable and Efficient Identification of Fragment Hits. Structure. 2016;24:1398-409
223. Li Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front Mol Biosci. 2020;7:180
224. Wu KJ, Lei PM, Liu H, Wu C, Leung CH, Ma DL. Mimicking Strategy for Protein-Protein Interaction Inhibitor Discovery by Virtual Screening. Molecules. 2019 24
225. Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A. et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008;51:6902-15
226. Pullarkat VA, Lacayo NJ, Jabbour E, Rubnitz JE, Bajel A, Laetsch TW. et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021;11:1440-53
227. Bertino EM, Gentzler RD, Clifford S, Kolesar J, Muzikansky A, Haura EB. et al. Phase IB Study of Osimertinib in Combination with Navitoclax in EGFR-mutant NSCLC Following Resistance to Initial EGFR Therapy (ETCTN 9903). Clin Cancer Res. 2021;27:1604-11
228. Koes D, Khoury K, Huang Y, Wang W, Bista M, Popowicz GM. et al. Enabling large-scale design, synthesis and validation of small molecule protein-protein antagonists. PLoS One. 2012;7:e32839
229. Dewal MB, Firestine SM. Non-peptidic alpha-helical mimetics as protein-protein interaction inhibitors. Curr Med Chem. 2011;18:2420-8
230. Sheng C, Dong G, Miao Z, Zhang W, Wang W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem Soc Rev. 2015;44:8238-59
231. de la Torre BG, Albericio F. The Pharmaceutical Industry in 2021. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules. 2022 27
232. Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur J Med Chem. 2015;94:459-70
233. Schatti J, Sezer U, Pedalino S, Cotter JP, Arndt M, Mayor M. et al. Tailoring the volatility and stability of oligopeptides. J Mass Spectrom. 2017;52:550-6
234. Lenci E, Trabocchi A. Peptidomimetic toolbox for drug discovery. Chem Soc Rev. 2020;49:3262-77
235. Wendt M, Bellavita R, Gerber A, Efrem NL, van Ramshorst T, Pearce NM. et al. Bicyclic beta-Sheet Mimetics that Target the Transcriptional Coactivator beta-Catenin and Inhibit Wnt Signaling. Angew Chem Int Ed Engl. 2021;60:13937-44
236. Ali AM, Atmaj J, Van Oosterwijk N, Groves MR, Domling A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput Struct Biotechnol J. 2019;17:263-81
237. Jing X, Jin K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med Res Rev. 2020;40:753-810
238. Wang X, Ni D, Liu Y, Lu S. Rational Design of Peptide-Based Inhibitors Disrupting Protein-Protein Interactions. Front Chem. 2021;9:682675
239. Wojcik P, Berlicki L. Peptide-based inhibitors of protein-protein interactions. Bioorg Med Chem Lett. 2016;26:707-13
240. Touti F, Gates ZP, Bandyopadhyay A, Lautrette G, Pentelute BL. In-solution enrichment identifies peptide inhibitors of protein-protein interactions. Nat Chem Biol. 2019;15:410-8
241. Ledsgaard L, Kilstrup M, Karatt-Vellatt A, McCafferty J, Laustsen AH. Basics of Antibody Phage Display Technology. Toxins (Basel). 2018 10
242. Li X, Craven TW, Levine PM. Cyclic Peptide Screening Methods for Preclinical Drug Discovery. J Med Chem. 2022;65:11913-26
243. Prashanth JR, Lewis RJ, Dutertre S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon. 2012;60:470-7
244. Lau JL, Dunn MK. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26:2700-7
245. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7
246. Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T. et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med. 2018 10
247. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ. et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948-53
248. Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S. et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J Med Chem. 2015;58:6348-58
249. Jeay S, Gaulis S, Ferretti S, Bitter H, Ito M, Valat T. et al. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife. 2015 4
250. Bauer S, Demetri GD, Halilovic E, Dummer R, Meille C, Tan DSW. et al. Pharmacokinetic-pharmacodynamic guided optimisation of dose and schedule of CGM097, an HDM2 inhibitor, in preclinical and clinical studies. Br J Cancer. 2021;125:687-98
251. Seebach D, Gardiner J. Beta-peptidic peptidomimetics. Acc Chem Res. 2008;41:1366-75
252. Estrada-Ortiz N, Neochoritis CG, Domling A. How To Design a Successful p53-MDM2/X Interaction Inhibitor: A Thorough Overview Based on Crystal Structures. ChemMedChem. 2016;11:757-72
253. Teveroni E, Luca R, Pellegrino M, Ciolli G, Pontecorvi A, Moretti F. Peptides and peptidomimetics in the p53/MDM2/MDM4 circuitry - a patent review. Expert Opin Ther Pat. 2016;26:1417-29
254. Teng P, Shi Y, Sang P, Cai J. gamma-AApeptides as a New Class of Peptidomimetics. Chemistry. 2016;22:5458-66
255. Sang P, Shi Y, Lu J, Chen L, Yang L, Borcherds W. et al. alpha-Helix-Mimicking Sulfono-gamma-AApeptide Inhibitors for p53-MDM2/MDMX Protein-Protein Interactions. J Med Chem. 2020;63:975-86
256. Sang P, Shi Y, Higbee P, Wang M, Abdulkadir S, Lu J. et al. Rational Design and Synthesis of Right-Handed d-Sulfono-gamma-AApeptide Helical Foldamers as Potent Inhibitors of Protein-Protein Interactions. J Org Chem. 2020;85:10552-60
257. Shi Y, Sang P, Lu J, Higbee P, Chen L, Yang L. et al. Rational Design of Right-Handed Heterogeneous Peptidomimetics as Inhibitors of Protein-Protein Interactions. J Med Chem. 2020;63:13187-96
258. Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol. 2011;21:419-25
259. Uversky VN. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2011;43:1090-103
260. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS. et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26-59
261. Martinelli AHS, Lopes FC, John EBO, Carlini CR, Ligabue-Braun R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int J Mol Sci. 2019 20
262. Joshi P, Vendruscolo M. Druggability of Intrinsically Disordered Proteins. Adv Exp Med Biol. 2015;870:383-400
263. Wojcik S, Birol M, Rhoades E, Miranker AD, Levine ZA. Targeting the Intrinsically Disordered Proteome Using Small-Molecule Ligands. Methods Enzymol. 2018;611:703-34
264. Chen J, Liu X, Chen J. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules. 2020 10
265. Lavigne P, Crump MP, Gagne SM, Hodges RS, Kay CM, Sykes BD. Insights into the mechanism of heterodimerization from the 1H-NMR solution structure of the c-Myc-Max heterodimeric leucine zipper. J Mol Biol. 1998;281:165-81
266. Pineda-Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R. et al. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J Mol Biol. 2005;351:182-94
267. Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193-205
268. Sammak S, Hamdani N, Gorrec F, Allen MD, Freund SMV, Bycroft M. et al. Crystal Structures and Nuclear Magnetic Resonance Studies of the Apo Form of the c-MYC:MAX bHLHZip Complex Reveal a Helical Basic Region in the Absence of DNA. Biochemistry. 2019;58:3144-54
269. Bhattacharya S, Lin X. Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins. Biomolecules. 2019 9
270. Rasafar N, Barzegar A, Mehdizadeh Aghdam E. Structure-based designing efficient peptides based on p53 binding site residues to disrupt p53-MDM2/X interaction. Sci Rep. 2020;10:11449
271. Lang L, Perez A. Binding Ensembles of p53-MDM2 Peptide Inhibitors by Combining Bayesian Inference and Atomistic Simulations. Molecules. 2021 26
272. Hosoya Y, Ohkanda J. Intrinsically Disordered Proteins as Regulators of Transient Biological Processes and as Untapped Drug Targets. Molecules. 2021 26
273. Biesaga M, Frigole-Vivas M, Salvatella X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr Opin Chem Biol. 2021;62:90-100
274. Luscombe NM, Thornton JM. Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. Journal of molecular biology. 2002;320:991-1009
275. Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer. 2019;19:611-24
276. Gentile F, Agrawal V, Hsing M, Ton AT, Ban F, Norinder U. et al. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent Sci. 2020;6:939-49
277. Dhakal A, McKay C, Tanner JJ, Cheng J. Artificial intelligence in the prediction of protein-ligand interactions: recent advances and future directions. Briefings in bioinformatics. 2022 23
278. Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic acids research. 2007;35:D198-201
279. Wang R, Fang X, Lu Y, Yang CY, Wang S. The PDBbind database: methodologies and updates. Journal of medicinal chemistry. 2005;48:4111-9
280. Krivák R, Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of cheminformatics. 2018;10:39
281. Mylonas SK, Axenopoulos A, Daras P. DeepSurf: A surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics (Oxford, England). 2021
282. Hameduh T, Haddad Y, Adam V, Heger Z. Homology modeling in the time of collective and artificial intelligence. Comput Struct Biotechnol J. 2020;18:3494-506
283. Radaeva M, Ton AT, Hsing M, Ban F, Cherkasov A. Drugging the 'undruggable'. Therapeutic targeting of protein-DNA interactions with the use of computer-aided drug discovery methods. Drug discovery today. 2021;26:2660-79
284. Li H, Ban F, Dalal K, Leblanc E, Frewin K, Ma D. et al. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. Journal of medicinal chemistry. 2014;57:6458-67
285. Matias-Barrios VM, Radaeva M, Song Y, Alperstein Z, Lee AR, Schmitt V. et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Frontiers in oncology. 2020;10:633142
286. Huang W, Dong Z, Wang F, Peng H, Liu JY, Zhang JT. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS chemical biology. 2014;9:1188-96
287. Nussinov R, Zhang M, Liu Y, Jang H. AlphaFold, Artificial Intelligence (AI), and Allostery. J Phys Chem B. 2022;126:6372-83
288. Ruff KM, Pappu RV. AlphaFold and Implications for Intrinsically Disordered Proteins. J Mol Biol. 2021;433:167208
289. Yang F, Wen Y, Wang C, Zhou Y, Zhou Y, Zhang ZM. et al. Efficient targeted oncogenic KRAS(G12C) degradation via first reversible-covalent PROTAC. Eur J Med Chem. 2022;230:114088
290. Li L, Wu Y, Yang Z, Xu C, Zhao H, Liu J. et al. Discovery of KRas G12C-IN-3 and Pomalidomide-based PROTACs as degraders of endogenous KRAS G12C with potent anticancer activity. Bioorg Chem. 2021;117:105447
291. Yang X, Wang Z, Pei Y, Song N, Xu L, Feng B. et al. Discovery of thalidomide-based PROTAC small molecules as the highly efficient SHP2 degraders. Eur J Med Chem. 2021;218:113341
292. Wang M, Lu J, Wang M, Yang CY, Wang S. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J Med Chem. 2020;63:7510-28
293. Zheng M, Liu Y, Wu C, Yang K, Wang Q, Zhou Y. et al. Novel PROTACs for degradation of SHP2 protein. Bioorg Chem. 2021;110:104788
294. Vemulapalli V, Donovan KA, Seegar TCM, Rogers JM, Bae M, Lumpkin RJ. et al. Targeted Degradation of the Oncogenic Phosphatase SHP2. Biochemistry. 2021;60:2593-609
295. Zhou H, Bai L, Xu R, McEachern D, Chinnaswamy K, Li R. et al. SD-91 as A Potent and Selective STAT3 Degrader Capable of Achieving Complete and Long-Lasting Tumor Regression. ACS Med Chem Lett. 2021;12:996-1004
296. Lee GT, Nagaya N, Desantis J, Madura K, Sabaawy HE, Kim WJ. et al. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Mol Cancer Ther. 2021;20:490-9
297. Bhumireddy A, Bandaru N, Raghurami Reddy B, Gore ST, Mukherjee S, Balasubramanian WR. et al. Design, synthesis, and biological evaluation of phenyl thiazole-based AR-V7 degraders. Bioorg Med Chem Lett. 2022;55:128448
298. Liao H, Li X, Zhao L, Wang Y, Wang X, Wu Y. et al. A PROTAC peptide induces durable beta-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020;6:35
299. Vannam R, Sayilgan J, Ojeda S, Karakyriakou B, Hu E, Kreuzer J. et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem Biol. 2021;28:503-14 e12
300. Durbin AD, Wang T, Wimalasena VK, Zimmerman MW, Li D, Dharia NV. et al. EP300 Selectively Controls the Enhancer Landscape of MYCN-Amplified Neuroblastoma. Cancer Discov. 2022;12:730-51
301. Cantley J, Ye X, Rousseau E, Januario T, Hamman BD, Rose CM. et al. Selective PROTAC-mediated degradation of SMARCA2 is efficacious in SMARCA4 mutant cancers. Nat Commun. 2022;13:6814
Author contact
![]() Corresponding authors: Dapeng Chen, Tel: +86 18940893462, E-mail addresses: friendchenedu.cn; Guangbo Ge, Tel: +86 13889602384, E-mail addresses: geguangboedu.cn.
Corresponding authors: Dapeng Chen, Tel: +86 18940893462, E-mail addresses: friendchenedu.cn; Guangbo Ge, Tel: +86 13889602384, E-mail addresses: geguangboedu.cn.