Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structure and molecular...
Key physiological functions of...
The role of MAM in DKD
Outlook
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(14):4427-4441. doi:10.7150/ijbs.86608 This issue Cite
Review
Broadening horizons: the contribution of mitochondria-associated endoplasmic reticulum membrane (MAM) dysfunction in diabetic kidney disease
Yong Liu1,2,3,4, Yingjin Qiao5, Shaokang Pan1,2,3,4, Jingfang Chen1,2,3,4, Zihui Mao1,2,3,4, Kaidi Ren6, Yang Yang7, Qi Feng1,2,3,4 ![]() , Dongwei Liu1,2,3,4
, Dongwei Liu1,2,3,4 ![]() , Zhangsuo Liu1,2,3,4
, Zhangsuo Liu1,2,3,4 ![]()
1. Research Institute of Nephrology, Zhengzhou University, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, P. R. China.
2. Traditional Chinese Medicine Integrated Department of Nephrology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, P. R. China.
3. Henan Province Research Center for Kidney Disease, Zhengzhou 450052, P. R. China.
4. Key Laboratory of Precision Diagnosis and Treatment for Chronic Kidney Disease in Henan Province, Zhengzhou 450052, P. R. China
5. Blood Purification Center, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, P. R. China.
6. Department of Pharmacy, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, P. R. China.
7. Clinical Systems Biology Laboratories, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, P. R. China.
Received 2023-5-29; Accepted 2023-8-15; Published 2023-8-21
Abstract

Diabetic kidney disease (DKD) is a global health issue that presents a complex pathogenesis and limited treatment options. To provide guidance for precise therapies, it is crucial to accurately identify the pathogenesis of DKD. Several studies have recognized that mitochondrial and endoplasmic reticulum (ER) dysfunction are key drivers of the pathogenesis of DKD. The mitochondria-associated ER membrane (MAM) is a dynamic membrane contact site (MSC) that connects the ER and mitochondria and is essential in maintaining the normal function of the two organelles. MAM is involved in various cellular processes, including lipid synthesis and transport, calcium homeostasis, mitochondrial fusion and fission, and ER stress. Meanwhile, recent studies confirm that MAM plays a significant role in the pathogenesis of DKD by regulating glucose metabolism, lipid metabolism, inflammation, ER stress, mitochondrial fission and fusion, and autophagy. Herein, this review aims to provide a comprehensive summary of the physiological function of MAMs and their impact on the progression of DKD. Subsequently, we discuss the trend of pharmaceutical studies that target MAM resident proteins for treating DKD. Furthermore, we also explore the future development prospects of MAM in DKD research, thereby providing a new perspective for basic studies and clinical treatment of DKD.
Keywords: Mitochondria-associated endoplasmic reticulum membrane (MAM), diabetic kidney disease (DKD), mitochondrial physiology, calcium homeostasis, lipid metabolism
Introduction
Diabetes mellitus (DM) is a prevalent global health problem with approximately 10% morbidity [1]. Epidemiological statistics show that the number of diabetic patients is projected to reach 537 million by 2021 and is expected to increase to 783 million by 2045[2, 3]. Diabetic kidney disease (DKD), a chronic renal microvascular disease caused by DM, has been considered the major cause of end-stage renal disease (ESRD) [4]. It is estimated that approximately 30-40% of patients with DM will develop DKD [5], and the prevention and treatment of DKD has become a global health problem [6]. DKD is characterized by a decrease in the glomerular filtration rate (GFR), fusion of podocytic foot processes, mesentery proliferation, and thickening of the glomerular basement membrane. It is also accompanied by albuminuria, renal inflammation, and tubulointerstitial fibrosis. The pathogenesis of DKD is very complex, and only limited therapeutic options are available [7]. Although the progression of DKD can be delayed by inhibiting the continuous rise of blood sugar, blood pressure and lipid levels, some DKD patients still experience severe and progressive renal injury due to either untimely treatment or insensitivity to medication, which may ultimately lead to ESRD [8-10]. Therefore, there is a pressing need to expand our understanding of the pathogenesis and identify promising targets for the prevention and treatment of DKD. Renal hemodynamic changes, glucose and lipid metabolism disorders, inflammatory responses, and oxidative stress (including mitochondrial damage and endoplasmic reticulum stress) are generally considered the primary causes of DKD [11, 12].
During the past few years, the important role of organelles in human diseases has been widely studied. Maintaining organelle homeostasis is crucial for living cells, but organelles are not isolated structures. Organelles facilitate material exchange and signal transduction among each other through membrane junctions and vesicle transport, which can have synergistic or antagonistic effects. With the in-depth study of organelles, many studies have shown that mitochondrial dysfunction and endoplasmic reticulum (ER) stress play key roles in the development of DKD [13, 14]. The mitochondrial-associated endoplasmic reticulum (MAM) is a dynamic membrane contact site (MCS) between the mitochondria and ER that helps maintain their normal function [15], and this crucial connection has been found to play a pivotal role in the development of DKD [16].
Intercellular communication plays a crucial role in maintaining cellular homeostasis and overall organism health. Both direct communication of MCSs and vesicle transport are essential for this process. MCS refers to areas where intracellular membrane compartments are in close proximity, but membrane fusion does not occur [17]. In these regions, the protein frenulum facilitates tight fitting of the membranes of the two organelles, enabling fast, direct, and mutual signaling between them [18]. MAM is the first discovered physical link between two intracellular organelles [19, 20]. The intricate relationship between MAM and cellular function has been the subject of extensive research in recent years, with new insights emerging into their role in various physiological and pathological processes. Recent research has revealed that MAM plays crucial roles in various cellular processes, including Ca2+ signal transduction, inflammation, lipid metabolism, ER stress, autophagy, and apoptosis [15].
Structure and molecular composition of MAM
The ER is tightly attached to 5-20% of the mitochondrial surface, forming a specialized ER domain known as the MAM [21] (Figure 1). The close membrane association between the ER and mitochondria was initially observed using electron microscopy in the 1950s [22]. With the advent of advanced imaging technology, the existence of MAMs has been confirmed using various techniques, such as real-time imaging, new electron microscopy methods, and subcellular isolation [23]. MAM can be isolated through proteolytic hydrolysis, revealing a protein bridge that connects the outer mitochondrial membrane (OMM) and the ER. Electron microscopy studies have confirmed the existence of this bridge [24]. This close association between the two organelles facilitates the exchange of lipids, calcium ions, and other signaling molecules. For an MAM to function properly, it is important to maintain a distance between the ER and OMM of no less than 10 nm and no more than 30 nm [25]. This distance is necessary for efficient protein interaction and material exchange. If the distance between the mitochondria and ER is too close, less than 7 nm, or too wide, greater than 50 nm, it can lead to dysfunction in the MAM [26].
The structure and components of MAM. The physical connection region between the ER and mitochondria is called the MAM, which is composed of the mitochondrial outer membrane and the endoplasmic reticulum membrane. This structure is rich in various proteins and is connected by tethering proteins, which include MFN2-MFN1/2, VAPB-PTPIP51, WASF3-ATAD3, IP3R-GRP75-VDAC1, BAP31-FIS1, OPR5/8-PTPIP51, MFN2-MITOL, MOSPD2-PTPIP51, WASF3-GRP78 and FUNDC1-IP3R2. MFN1/2, mitofusin 1/2; VAPB, vesicle-associated membrane-protein-associated protein B; PTPIP51, protein tyrosine phosphatase-interacting protein 51; WASF3, Wiskott-Aldrich syndrome protein family member 3; ATAD3, ATPase family AAA Domain-containing protein 3; IP3R, inositol 1,4,5-trisphosphate receptor; GRP75, glucose regulate protein 75; VDAC1, voltage-dependent anion channel 1; BAP31, B-cell receptor-associated protein 31; FIS1, mitochondrial fission 1 protein; OPR5/8, oxysterol-binding protein related-protein 5 and 8; MITOL, mitochondrial ubiquitin ligase; MOSPD2, motile sperm domain-containing protein 2; GRP78, glucose regulate protein 78; FUNDC1, FUN14 domain-containing protein 1.
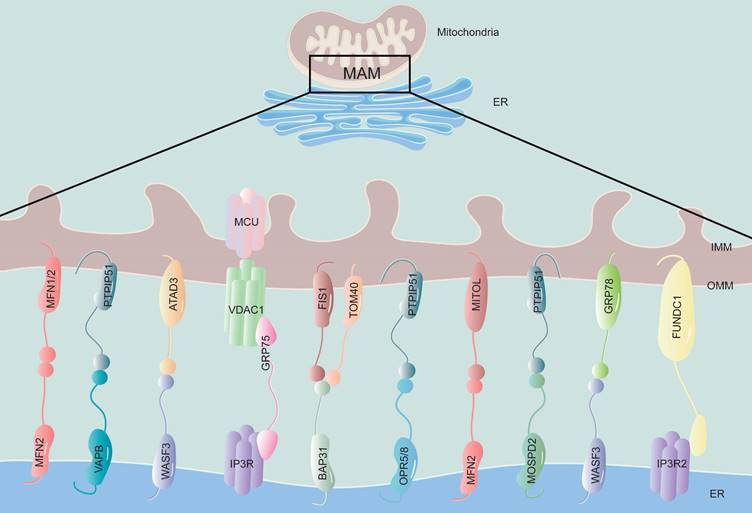
The MAM acts as a bridge between the ER and mitochondria and relies mainly on proteins to carry out its functions. Proteomics technology has identified hundreds to thousands of proteins in MAMs across various species and tissues, with highly conserved components [27-29]. A recent proteome analysis of MAM in diabetic and nondiabetic mouse brains revealed significant changes in 144 proteins due to DM [30], indicating that components of MAM are altered during the process of disease. Studies have increasingly found newly discovered MAM-related proteins, and their functions are being gradually revealed. The flexibility of MAM protein assemblies allows for the recruitment of different signaling molecules based on specific cell states [31, 32]. Resident proteins on the MAM are classified according to their different functions, for example, Ca2+ transport-related protein: inositol 1,4,5-trisphosphate receptor (IP3R) [33]; lipid synthesis and transfer-associated protein: phosphatidylserine synthase 1 and 2 (PSS1/2) [34]; mitochondrial dynamic regulatory proteins: mitofusin 2 (MFN2), dynamin-related protein 1 (DRP1) [35]; insulin signaling-related protein: protein kinase B (PKB), mammalian target of rapamycin complex (mTORC) [36]; and so on. The presence of multifunctional proteomes in MAM indicates that they exert crucial roles in regulating cellular homeostasis and biological processes (Table 1).
Structure and function-related proteins in MAM
| Function types | Proteins | Abbreviation | Relevant functions in MAM |
|---|---|---|---|
| Lipid metabolism | Fatty acid CoA ligase 4 | FACL4 | Immobilize fatty acids on CoA [37] |
| Acy1-Coenzyme A-cholesterol acyltransferase | ACAT | Synthesize cholesteryl esters [38] | |
| Phosphatidylethanolamine N-methyltransferase 2 | PEMT2 | Convert PE to PC in ER [39] | |
| Mitofusin-2 | MFN2 | Transfer PS to mitochondria from ER [40] | |
| Oxysterol-binding protein-related protein 5 | ORP5/8 | Mediating the transfer of PS from ER to mitochondria [41, 42] | |
| Protein tyrosine phosphatase-interacting protein 51 | PTPIP51 | Regulates the transfer of PA in MAM [43] | |
| Diacylglycerol O-acyltransferase 2 | DGAT2 | Catalyzes triglyceride synthesis [44] | |
| Phosphatidylserine synthase 1 and 2 | PSS1/2 | Synthesize PS [34] | |
| Caveolin-1 | CAV1 | Regulate cholesterol efflux [45] | |
| Ca2+ hemostasis | Inositol1,4,5-trisphosphate receptor | IP3Rs | Major calcium channels in ER [46] |
| Voltage-dependent anion channel 1 | VDAC1 | Calcium uptake channels in mitochondria [47] | |
| Glucose regulated protein 75 | GRP75 | Connects IP3R and VDAC to form VDAC1/GRP75/IP3R1 channel complex [47] | |
| Cyclophilin D | CYPD | A partner of the IP3R1-GRP75-VDAC1 complex and changes the MAM spatial structure [48] | |
| Sarco/endoplasmic reticulum Ca2+ ATPase | SERCA2b | Acts as an important pump involved in Ca2+ transport into ER [46] | |
| Mitochondrial dynamics | Dynamin-related protein 1 | DRP1 | Control mitochondrial fusion [49] |
| Inverted formin 2 | INF2 | Driving initial mitochondrial constriction [50] | |
| Mitofusin-2 | MFN2 | Mediate mitochondrial fusion [51] | |
| Optic atrophy 1 | OPA1 | Mediate mitochondrial fusion [51] | |
| PTEN-induced putative kinase 1 | PINK1 | Mediates mitophagy [52] | |
| FUN14 domain-containing protein 1 | FUNDC1 | Mediates mitophagy [53] | |
| Insulin signaling | Protein kinase B | PKB | Maintains insulin signal transduction [36] |
| mammalian target of rapamycin complex (mTORC) | mTORC | Maintains insulin signal transduction [36] | |
| PTEN-induced putative kinase 1 | PINK1 | Maintains insulin signal transduction [54] |
Key physiological functions of MAMs
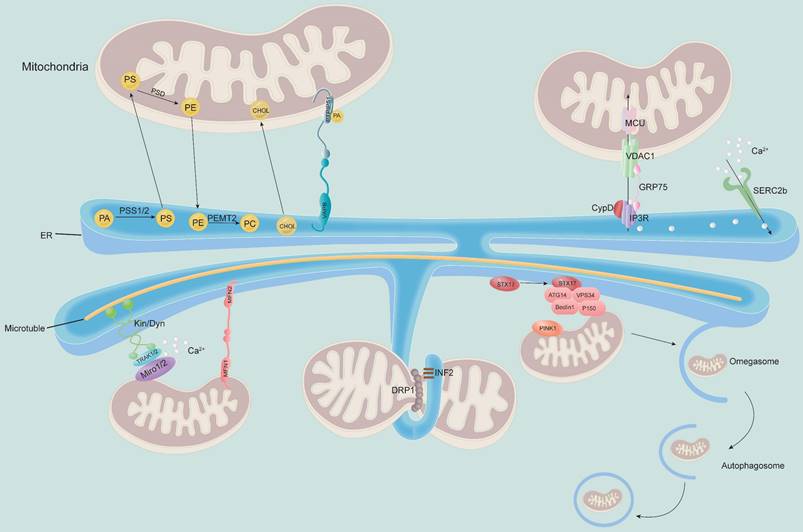
MAM plays a key role in multiple physiological activities of cells, participating in lipid synthesis and transport, maintaining Ca2+ homeostasis, regulating mitochondrial fission and fusion, apoptosis and autophagy and other cellular processes (Figure 2).
MAM regulates the physiological function of mitochondria. MAM regulates cellular lipid metabolism (top left) and calcium homeostasis (top right). The MAM is a key physical connection region for lipid metabolism, including phosphatidylcholine (PC) synthesis and cholesterol transport. In addition, MAM also participates in regulating calcium homeostasis through the IP3R-GRP75-VDAC1-MCU axis, which is an important channel for calcium transport from the ER to mitochondria (bottom). Meanwhile, MAM is considered a regulator of mitochondrial physiology: mitochondrial contraction occurs near the site of contact with the ER, which determines where mitochondrial fission occurs. Other proteins localized to MAM, such as STX17 and INF2, are involved in this process. Moreover, the ER remains attached to the mitochondria and travels along microtubules in the cell. MAM-resident STX17 binds ATG14 and recruits the class III PI3K complex to MAM, which contributes to the formation of autophagosomes.
Lipid synthesis and transport
Accumulating evidence indicates that one of the major functions of MAMs is the regulation of biosynthesis and intermembrane transport of phospholipids [55]. MAM forms a hydrophilic environment between the ER and mitochondria, which is conducive to the bidirectional noncystic transfer of lipids [38]. Phosphatidylethanolamine (PE) and cardiolipin (CL) are essential phospholipids for mitochondrial respiratory function and can be synthesized in the mitochondria; however, their precursors and most of the mitochondrial phospholipids need to be synthesized in the ER before being transported to the mitochondria [56]. The precursor of phosphatidylserine (PS), which is required for PE synthesis, is synthesized by phosphatidylserine synthase (PSS) in MAMs [34]. In the study of Yang et. al, it was found that when PSS1 and PSS2 were knocked down, the content of PS was decreases, subsequently led to a change in the steady state of phosphatidylserine in the ER and diversion of lipid synthesis to triacylglycerol and diacylglycerol synthesis, thereby resulting in lipid accumulation [57]. María Isabel et. al confirmed that MFN2 could selectively bind to PS and transfer it to mitochondria. Knockout of MFN2 reduced the expression of PSS1 and decreased the transfer of PS from the ER to the mitochondria, resulting in impaired phospholipid synthesis and ER stress [40]. Moreover, overexpression of PSS1 promotes PS synthesis but cannot rescue the synthesis of PE [40]. Additionally, Oxysterol-binding protein-related protein 5 (ORP5) and ORP8 also localize to the ER-mitochondria contact and interact with the intermembrane space bridging (MIB)/mitochondrial contact site and junctional junction organizing system (MICOS) complex and the OMM protein tyrosine phosphatase-interacting protein 51 (PTPIP51), mediating the transfer of PS from ER to mitochondria, and their depletion leads to defects in mitochondrial morphology and respiratory function [41, 42]. Therefore, it is assumed that these functions of PS may be of great significance to explore the ectopic lipid deposition of kidney in DKD. However, whether the change of PSS itself at MAM directly triggers MAM dysfunction remains unclear, and it is also worth exploring in the future. CL is synthesized through a series of modifications of phosphatidic acid (PA), and recent studies have shown that the VAPB-PTPIP51 complex regulates the transfer of PA in MAMs [43]. Moreover, many proteins related to lipid metabolism are enriched in MAMs, including diacylglycerol O-acyltransferase 2 (DGAT2), phosphatidylethanolamine N-methyltransferase 2 (PEMT2), fatty acid CoA ligase 4 (FACL4), cholesterol acyltransferase/sterol O-acyltransferase 1 (ACAT1/SOAT1), and PSS1/2 [34, 58]. Cholesterol likely reaches mitochondria through specialized MAM [59]. The MAM-associated protein caveolin-1 (CAV1) has recently been shown to regulate ER-mitochondrial cholesterol transfer [45], and it regulates cholesterol efflux by binding to VDAC2. Inhibition of CAV1 is associated with abnormal intracellular accumulation of free cholesterol and reduced MAM physical extension and integrity [60].
Ca2+ homeostasis
Intracellular Ca2+ homeostasis is mainly characterized by the balance and exchange of Ca2+ between the ER and mitochondria to maintain normal cell function [61]. MAMs are hubs for Ca2+ signaling. The ER and mitochondria are important Ca2+ storage organelles, and the transfer of Ca2+ from the ER to the mitochondria relies on MAM-mediated physical contact sites between these two organelles [62]. The ER is the largest intracellular Ca2+ pool [63], which performs its physiological function mainly through SERCA uptake and Ca2+ release by IP3R [46]. The OMM protein voltage-dependent anion channel 1 (VDAC1) and the IMM protein mitochondrial calcium uniporter (MCU) are important proteins that take Ca2+ from the MAM gap into the mitochondrial matrix [64]. The physical junction between the ER and mitochondria has a high concentration of Ca2+ microregions conducive to Ca2+ signal transduction [65]. Various proteins in MAMs are related to Ca2+ signal transduction. IP3R⁃GRP75⁃VDAC1 is the major protein complex responsible for Ca2+ signaling between the ER and mitochondria [66]. After activation, IP3R on the ER membrane releases Ca2+ from the ER lumen. With the assistance of GRP75, it is taken up by VDAC1 on the OMM, enters the mitochondrial matrix through the MCU of the IMM, and forms a Ca2+ signal flow in MAMs to play a biological role. In addition to Grp75, recent studies have shed light on other molecular partners at MAMs, such as cyclophilin D (CypD), which regulates ER-mitochondria crosstalk. CypD is a mitochondrial chaperone foldase and is sensitive to Ca2+ that interacts with the VDAC1-GRP75-IP3R complex at the interface of MAM and transfers Ca2+ from the ER to mitochondria in the heart and liver through IP3R1 and IP3R2 [48, 67]. Recent studies have shown that activation of CypD by high Ca2+ concentrations is one of the causes of oxidative stress and kidney injury in DKD [68], and a lack of CypD appears to exacerbate kidney injury in STZ-induced diabetic mice [69]. However, whether MAM has an effect on this process needs to be addressed in future studies.
Mitochondrial dynamics
Mitochondria are double-membrane organelles that regulate cellular metabolism and overall function. Mitochondrial dysfunction is recognized as an important contributor to DKD [70]. Mitochondrial dynamics include mitochondrial fission, fusion and motility and mitophagy [71].
Mitochondrial fission is controlled by a group of motility-associated GTPase proteins, including DRP1 and its outer membrane receptors fission-1 (Fis1), mitochondrial fission factor (MFF), and mitochondrial dynamics proteins 49 and 59 (MiD49 and MiD59) [49]. DRP1 possesses membrane contractile and severing capabilities and is the main driving force for performing mitochondrial and peroxisome fission [72]; it increases ER-mitochondria interactions by promoting tubule formation in the ER [73]. Binding of DRP1 to F-actin activates DRP1 GTPase activity, leading to the recruitment of DRP1 to preconstricted mitochondria to stimulate their fission [74]. In the process of mitochondrial fission, DRP1 is recruited to the OMM, forming oligomeric complexes that surround, constrict and divide mitochondria [75]. As DRP1 is normally cytosolic and lacks a domain that directly binds to membrane phospholipids, a collection of MFFs, MiD49 and MiD51, are recruited to the OMM through the receptor protein Fis1 [76]. These proteins are known to localize at the ER‐mitochondria interface. Several other MAM-associated molecules are also involved in mitochondrial fission. The ER-anchored isoform of the formin protein inverted formin 2 (INF2) mediates actin polymerization and facilitates mitochondrial DRP1 recruitment at ER-mitochondria intersections [77]. It is a key step in mitochondrial fission, resulting in increased ER-mitochondria contacts, mitochondrial Ca2+ uptake, and mitochondrial fission [78]. Mitochondrial Ca2+ uptake decreases after knocking out MCU, which leads to a decrease in mitochondrial fission [79]. Overexpression of phosphofurin acidic cluster sorting protein 2 (PACS2) also suppresses mitochondrial fission and was reported to alleviate excessive mitochondrial fission by blocking mitochondrial recruitment of DRP1 in HK-2 cells cultured with high glucose [80]. In addition, FUN14 domain-containing protein 1 (FUNDC1) aggregates in large quantities at the MAM during anoxia and promotes mitochondrial fission by binding to calcinetin (CANX) in response to anoxia [81].
The mitochondrial fusion process is divided into OMM fusion and IMM fusion [82]. The OMM proteins MFN1 and MFN2, as well as the OMM protein optic atrophy 1 (OPA1), play major roles in mitochondrial fusion [51]. Whereas MFN2 on the surface of the ER regulates mitochondrial connections to maintain the whole mitochondrial network, MFN1 on the surface of mitochondria plays a critical role in mitochondrial docking and fusion. The two proteins generate dimers and drive fusion of the OMM [83].
Mitochondrial motility is defined as transport along microtubules by opposing kinesin and dynein motors to regulate the distribution of mitochondria in the cytoplasm, which is essential to maintain the normal function of cells. Kinesin and dynein do not directly associate with the OMM but bind indirectly to the OMM proteins Miro1 and Miro2 via TRAK1 and TRAK2 [59]. Current studies have shown that the role of Miro1/2 in mitochondrial movement is closely related to MAMs. It was reported that the yeast ortholog of Miro1 [60], whereas loss of Miro1/2 alters mitochondria-ER communication [61]. However, this binding requires a higher Ca2+ concentration. Due to the relatively low calcium affinity of Miro1/2, MAM is ideal for mitochondrial motility [60, 61].
Mitophagy is a selective autophagy process used to eliminate damaged mitochondria [84]. The key regulators of mitophagy, PTEN-induced putative kinase 1 (PINK1) and Beclin1, both relocalize to MAMs during autophagy, which promotes the enhancement of ER-mitochondrial contact sites and the formation of autophagosome probes [52]. Another mitophagy-associated protein, FUNDC1, was also shown to accumulate at the ER-mitochondrion interface during mitophagy by binding to ER-resident IP3R2 [53]. It was reported that MAM-resident syntaxin 17 can bind to the autophagosome to label autophagy related 14 and accumulate in the MAM until autophagosome formation is complete [85].
The role of MAM in DKD
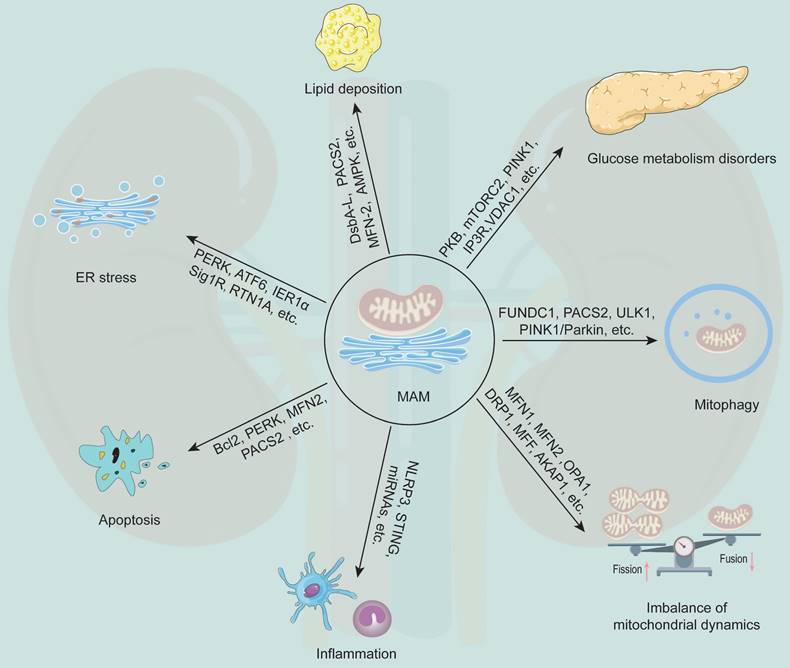
MAM plays a generalist role in the occurrence and development of DKD (Figure 3). Current studies have revealed that MFN2, DRP1, PACS2, PINK1, disulfide-bond A oxidoreductase-like protein (DsbA-L), nucleotide binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) and other MAM-related proteins are involved in regulating the initiation and development of various cellular processes, such as lipid metabolism, cell apoptosis, mitochondrial fission and fusion, and mitophagy (Table 2).
The multifunction of MAM in DKD. MAM is a special membrane contact site between the ER and mitochondria, and MAM-resident proteins play key roles in regulating various cellular processes that associated with the development of DKD, including glucose metabolism disorders, lipid deposition, ER stress, apoptosis, inflammation, mitochondrial dynamics imbalance, and mitophagy.
MAM-associated key components participate in DKD
| Functions in DKD | Proteins in MAMs |
|---|---|
| Regulate glucose metabolism | PINK1[54], mTORC1[86], PP2A[86] |
| Regulate lipid metabolism | DsbA-L, PACS2, MFN2[93], AMPK [95] |
| Regulate inflammation | STING[87], NLRP3[88] |
| Regulate ER stress | PERK, MFN2[89], RTN1A, VDAC1[16] |
| Regulate renal cell apoptosis | Bcl2[90], PERK, MFN2[79], PACS2[91] |
| Regulate mitochondrial fission and fusion | MFN1, MFN2, OPA1, DRP1[136], MFF[92], AKAP1[93] |
| Regulate mitophagy | FUNDC1[94], PINK1/Parkin[95], ULK1[96] |
Regulation of glucose metabolism
The main cause of T1D is the destruction of pancreatic β-cells, leading to deficient insulin production and secretion [97]. Disruption of Ca2+ signaling at the MAM interface may be a key feature of glucotoxicity-mediated β-cell dysfunction [98]. T2D is characterized by insulin resistance and compensatory hyperinsulinemia, which results in the development of microvascular complications, including DKD [99]. Podocytes are insulin-sensitive renal cells, and thus, insulin resistance is more likely to cause kidney damage [100]. Insulin-stimulated podocytes rapidly transport glucose to the cytoplasm to provide energy for maintaining their actin skeleton and normal filtration function. A growing body of evidence has supported that MAM integrity is required for insulin signaling [101]. It may mediate insulin resistance by regulating Ca2+ transduction, lipid metabolism, mitochondrial function, ER stress, etc. [102]. The proteins mTORC1 and protein phosphatase 2A (PP2A) located in MAMs play core roles in regulating podocyte insulin resistance [86]. Another MAM protein associated with insulin resistance, PINK, has been found to be deficient, significantly increasing albumin permeability and hindering glucose uptake in podocytes, which supports the crucial role of PINK1 in maintaining insulin signal transduction and podocyte permeability [54]. Therefore, MAM might participate in regulating insulin resistance in DKD; however, further studies are needed to confirm its potential.
Regulation of lipid metabolism
Lipid metabolism disorder is a key pathogenic feature of DKD [103]. High glucose induces lipid deposition in the kidney, and lipid deposition in nonadipose tissues such as the liver and kidney are called ectopic fat deposition (EFD) [104]. Lipid deposition has been found to be mainly distributed in tubules and glomeruli in animal models of DKD [105, 106]. EFD in renal tissue is one of the main factors leading to renal fibrosis and CKD [107]. It was reported that MAMs were significantly reduced in renal tissues of DKD patients, and the expression of MAM-related proteins such as DsbA-L, PACS2 and MFN2 was reduced and inversely correlated with blood lipid levels and the degree of renal lipid deposition, suggesting that a disruption of MAM integrity may lead to renal lipid deposition in DKD [108]. AMP-activated protein kinase (AMPK) is an important protein involved in the formation of MAMs [109]. The study by Chen et al. found that inhibition of DsbA-L expression aggravated lipid deposition in DKD, and activation of the AMPK pathway is a potential mechanism by which DsbA-L plays a role in renal lipid deposition [110]. Zhao et al. found that diabetic mice with proximal tubule PACS2-specific knockout had more severe tubular damage and proteinuria than controls, which was accompanied by increased lipid synthesis, decreased cholesterol uptake and efflux, and lipid deposition in renal tubules [111], indicating that PACS2 is a major regulator in lipid-related kidney lesions in DKD. These studies suggested that MAM-related proteins played key roles in regulating lipid deposition in DKD, but more research is needed to elucidate the molecular mechanism of MAM involvement in EFD in DKD.
Regulation of inflammation
Persistent inflammation in the circulatory system and kidney tissue is an important pathophysiological basis in the development of DKD [112]. The role of MAMMAMs in inflammation is related to NLRP3 inflammasome assembly and activation [113]. Inactivated NLRP3 is located in the ER membrane and cytoplasm, while activated NLRP3 can combine with apoptosis-associated speck-like protein containing a CARD (ASC) in MAM to form inflammasomes [114, 115]. Recently, Yang et al. found that DsbA-L attenuated NLRP3-mediated renal inflammatory injury by promoting AMPK phosphorylation in DKD [88], which suggested that MAM acted as an inflammatory regulator through NLRP3 in DKD. In addition, as a key mediator of innate immunity, stimulator of interferon genes (STING), which can recognize exogenous and endogenous DNA in cells, has been shown to be present in the ER and MAM [116]. A new discovery by Feng et al. showed that the enhanced inflammatory response caused by HIV virus infection in podocytes might be a key accelerator for the progression of DKD [117]. Coincidentally, STING is known to bind mitochondrial antiviral signaling protein (MAVS) on MAM, thereby increasing the interferon response to viral infection [118]. Depending on the unique localization of STING on the MAM, it has been suggested that STING-MAM crosstalk has a nonnegligible effect on the immune response [119]. Additionally, as a key player in metabolic inflammation, STING induces podocyte injury in db/db mice [87]. Therefore, STING may regulate the inflammation progression of DKD through MAMs. In addition, it has also been found that MAMs are subcellular sites of specific miRNAs [120]. In this study, Wang et al. found that MAMs are rich in inflammatory response miRNAs in human and rat brains, including miR-146a, miR-142-3p and miR-142-5p [120]. Coincidentally, miR-146, miR-142-3p, and miR-142-5p have been shown to play a regulatory role in the progression of DKD inflammation [121-123]. Therefore, there is a possibility that MAMs participate in the inflammatory response of DKD through miRNAs. However, due to the highly conserved nature of MAMs, more studies are needed to confirm whether MAMs enrich the expression levels of inflammatory-responsive miRNAs in DKD.
Regulation of ER stress
The ER is a major site for protein synthesis, folding, processing, and quality control [124]. The imbalance between ER protein-folding load and capacity under various physiological and pathological conditions results in ER stress [125]. However, sustained stress stimulation promotes the switch of the adaptive unfolded protein response (UPR) into proapoptotic signals [123]. ER stress is mediated by ER-localized sensor protein kinases RNA-dependent protein kinase (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1α (IRE1α), which are retained in their inactive states by interacting with GRP78 [126]. When ER stress occurs, they are separated from GRP78 to activate downstream signaling pathways and then restore ER homeostasis by reducing protein translation and promoting chaperone production [127]. Various ER partners involved in protein folding, including Bid, calnexin, calreticulin and sigma-1 receptor (Sig1R), are located in the MAM [128]. IRE1α was reported to be expressed in the MAM and to associate with Sig1R during ER stress [129]. PERK is a sensor for the UPR and has been identified as a key MAM component [130]. MFN2 is an upstream modulator of PERK [131]. It was found that reduced MFN2-PERK interaction was accompanied by decreased MFN2 expression and activation of all three UPR pathways in DKD [89]. The ER-resident protein reticulon-1A (RTN1A) is known to mediate podocyte and tubular cell injury in DKD by modulating ER stress [132, 133]. A recent study found that overexpression of RTN1A exacerbated ER stress in DKD by modulating MAM [16]. This is mainly because RTN1A interferes with the interaction of mitochondrial hexokinase-1 and VDAC1, leading to the activation of apoptosis and inflammatory pathways [16]. In summary, the involvement of MAM in DKD is substantial, as it contributes to ER stress, and PERK, MFN2, Sig1R, and RTN1A are known to be integral to this process.
Regulation of cell apoptosis
Apoptosis is believed to be an important cause of DKD [134]. Correspondingly, this process is closely related to MAM-mediated Ca2+ regulation. B-cell lymphoma 2 (Bcl2) family proteins are key members in mediating Ca2+ transport from the ER to mitochondria and regulating apoptosis [135]. Bcl2 is predominantly located in the resting ER and translocates to the MAM and mitochondria upon induction of apoptosis [136]. As an extensively studied anti-apoptotic protein, Bcl2 is often used as a marker to detect apoptosis in DKD. Its expression was significantly decreased in DKD patients [90]. Recent studies have shown that TOM20 promotes the transfer of Bcl2 from the ER to the MAM and mitochondria during apoptosis induction [136]. Many multifunctional MAM-associated proteins have been found to play an antiapoptotic role in DKD. MFN2 exerts mitochondrial protective and antiapoptotic effects by maintaining the number and integrity of MAMs and inhibiting the activation of the PERK pathway [89]. Meanwhile, several MAM-associated proteins are also involved in apoptosis by interacting with MFN2. For example, DsbA-L plays an antiapoptotic role in DKD by promoting the expression of MFN2 and maintaining the integrity of MAMs [137]. However, these beneficial effects were partially blocked by overexpression of FATE-1, a MAM uncoupling protein [137]. PACS2 is also a key molecule required to maintain MAM homeostasis in diabetic tubular injury. Xue et al. observed that PACS2 deficiency reduced the integrity of MAMs and exacerbated renal cell apoptosis in diabetic mice [91]. Taken together, these studies strongly support that MAM plays an important role in regulating cell apoptosis in DKD.
Regulation of mitochondrial fission and fusion
As an energy-intensive organ, the kidney is the second-largest relative to the heart in mitochondrial abundance [138]. Mitochondrial dysfunction has a serious impact on kidney function. Furthermore, changes in the regulation of mitochondrial dynamics and ultrastructure precede the development of albuminuria and renal histological changes in diabetes, and these mitochondrial changes evolve as DKD progresses [139]. Therefore, it is of great significance to study the markers related to mitochondrial dysfunction in DKD. Mitochondria are highly dynamic organelles that continually maintain cell survival and bioenergetics through fission, fusion, mitophagy and other mitochondrial quality control processes [140]. The balance of mitochondrial fusion and fission is necessary to maintain the normal shape and function of mitochondria under physiological conditions [141]. It has been reported that MAM-related proteins, such as MFN1, MFN2, OPA1 and DRP1, are involved in regulating mitochondrial fission and fusion. For example, under high glucose condition, the expression of the mitochondrial fusion markers MFN1, MFN2 and OPA1 was decreased, while the activity of the fission marker DRP1 was increased in human podocytes [142]. Xiao et al. found that the mitochondrial fission of podocytes increased under high glucose conditions, while the expression of MFF was upregulated. After MFF expression inhibition, the podocyte survival rate was significantly reduced [92]. It is suggested that the increase in mitochondrial number caused by MFF-mediated mitochondrial fission may be a response mechanism to mitochondrial overload and protect podocytes in the short term. Li et al. reported that the translocation of A-kinase-anchored protein 1 (AKAP1) to MAMs was increased, which promoted podocyte mitochondrial fission by regulating DRP1 phosphorylation and its subsequent mitochondrial translocation [93].
Regulation of autophagy
Autophagy is considered an evolutionarily conserved cellular process responsible for digesting or recycling organelles and long-lived proteins to maintain cellular homeostasis [143]. Accumulation of fragmented mitochondria has been found in the kidney in both humans and animals with DKD [144, 145], suggesting that the mitochondrial clearance machinery may be impaired as the kinetics shift. The OMM protein FUNDC1, a novel MAM protein, is enriched at MAM through interaction with the ER-resident protein CANX under hypoxic conditions. During mitochondrial phagocytosis, it dissociates from CANX and preferably recruits DNM1L/DRP1 to drive mitochondrial fission in response to hypoxic stress [81]. FUNDC1 has been shown to be involved in podocyte mitophagy in DKD [94]. Furthermore, Li et al. revealed that the overexpression of PACS2 in HK-2 cells blocked mitochondrial recruitment of DRP1 and alleviated excessive mitochondrial fission induced by high glucose condition, subsequently restoring MAM integrity and enhancing mitophagy [80]. PINK1/Parkin and ULK1 are considered mitophagy-related genes in DKD [95, 96] and have been reported to localize to MAMs during mitophagy [52, 146].
Potential role of MAM in DKD
In view of the important role of MAM in kidney injury, it is possible to develop MAM-related proteins as therapeutic targets for DKD. New research has found that AMPK, MFN2, PACS2 and DsbA-L play essential roles in regulating glucose and lipid metabolism and thus can be used as promising targets for the treatment of DKD. For example, curcumin inhibited renal lipid accumulation and oxidative stress through the AMPK and Nrf2 signaling pathways in DKD mice [147].
As an important protein mediating ER stress in DKD, targeting PERK for DKD has been extensively studied. Yuan et al. found that resveratrol treatment reduced the phosphorylation of PERK and regulated ER stress in the kidneys of diabetic rats [148]. Additionally, Tian et al. found that emodin attenuates ER stress-induced podocyte apoptosis by inhibiting the PERK signaling pathway in DKD [149]. In addition, NLRP3 has attracted wide attention as a target for inflammation regulation. Curcumin, the major bioactive compound of turmeric, exerts antifibrotic effects in DKD by inhibiting the NLRP3 inflammasome [146]. Dapagliflozin, an SGLT2 inhibitor, has been shown to reduce renal injury even in the absence of diabetes through inhibition of the NLRP3 inflammasome, protecting against kidney fibrosis [150]. In addition, Bcl2, MFN2, PACS2 and Beclin1 are potential targets for modulating apoptosis. It was reported that wogonin protects glomerular podocytes by targeting Bcl2-mediated autophagy and apoptosis in DKD [90]. Curcumin has also been shown to regulate apoptosis in DKD. It inhibited podocyte apoptosis and accelerated autophagy in diabetic nephropathy by regulating Beclin1/UVRAG/Bcl2[151]. FUNDC1 and Parkin are potential targets for regulating mitophagy in DKD. Metformin is a classic antidiabetic drug that inhibits the expression of Parkin and mitophagy by activating PP2A and inhibiting NF-κB, which protects renal epithelial cells from high glucose stimulation in vitro [152].
Due to the imbalance of mitochondrial division and fusion in DKD, inhibition of DRP1 function may be a new therapy for diabetic albuminuria [153]. A novel mitochondrial target peptide, SS31, which targets mitochondrial fission and fusion, has been found to reduce mitochondrial fragmentation by inhibiting the expression of DRP1 and increasing the expression of MFN1, thus preventing STZ-induced kidney injury in mice [154]. In addition, a recent study developed a covalent compound, mitochondrial division inhibitor (MIDI), which functionally mimics DRP1 knockdown to inhibit mitochondrial fission but prevents the recruitment of DRP1 to mitochondria, thereby preventing mitochondrial fission [155].
Outlook
The interactions between organelles are essential for eukaryotes to maintain normal physiological functions. It is widely acknowledged that the mitochondria and ER are significant players in the development of DKD [13, 14]. The MAM serves as a platform for facilitating communication between the ER and mitochondria, enabling the rapid exchange of biomolecules to maintain cellular health and function [57]. MAM dysfunction leads to the development of various diseases through multiple pathways. Studies have demonstrated that MAM plays a crucial role in maintaining Ca2+ homeostasis, lipid synthesis and transport, and metabolism, as well as regulating autophagy [156] and monitoring the morphology and function of the ER and mitochondria. In addition, as a dynamic structure, MAM also provides a platform for numerous enzymes to operate, and miRNAs are also highly concentrated in MAM [125].
Recent studies have revealed that MAM plays a crucial role in several processes related to DKD, such as Ca2+ overload, lipid metabolism, apoptosis, mitochondrial fission and fusion, and mitophagy [80, 104, 137]. Based on the present studies, we speculated that MAM contributed to the progression of DKD via insulin resistance and inflammation [52, 54, 88, 113]. Several MAM-related proteins have been deeply investigated in DKD, such as DsbA-L, PACS2, DRP1, and MFN2 [91, 93]. All of these proteins play crucial roles in different aspects of cell function and are considered promising targets for DKD treatment. Numerous drugs have been discovered to target MAM, which are beneficial in DKD treatment. For example, curcumin targets AMPK, NLRP3, and Bcl2 to lower ER stress, inflammation and apoptosis in DKD [146, 147, 151]. Metformin, dapagliflozin, wogonin, resveratrol, and SS31 are also effective in treating DKD by targeting MAM [90, 150, 152, 154]. However, targeting MAM-related proteins does not necessarily alter the MAM itself. For example, a study by Maya et al. showed that although metformin treatment improves blood glucose levels and insulin sensitivity to some extent in diabetic mice, it does not prevent the changes in MAM Ca2+ coupling in cardiac muscle cells and the resulting heart dysfunction induced by T2D [157]. On the other hand, Wei et al. found that activation of transient receptor potential cation channel subfamily V member 1 (TRPV1) by capsaicin alleviated mitochondrial dysfunction caused by high glucose in podocytes and was accompanied by a reduction in MAM formation and decreased Ca2+ transport from the ER to mitochondria [158]. This is because the transient influx of Ca2+ mediated by TRPV1 reduced the transcription of the key molecule FUNDC1, which was crucial for MAM formation [158]. These findings strongly support MAM as a potential target for the treatment of DKD.
Although some MAM-related metabolic regulators and signaling pathways are involved in regulating DKD, there remain several unclear questions. What are the essential elements for preserving MAM structure and function? What is the precise regulatory mechanism by which MAM contributes to DKD? Notably, there is still no specific molecules to dynamically trace ER-mitochondria interaction, which makes it difficult to conduct in-depth study on the key role of MAM in DKD. In addition, the kidney is composed of various cell types, including podocytes, mesangial cells, endothelial cells, and renal tubular cells, which may have distinct functions associated with MAM. Therefore, investigating the MAM of different cell lines is crucial to understanding its role in various diseases. It is noteworthy that MAM is a transient structure, and its state may vary during different stages of disease. Therefore, further extensive research is necessary to fully explain the multifunctional attributes of MAM dysfunction in DKD.
Data availability
The data used to support the findings of this study are included within the paper.
Author Contributions
Yong Liu, Qi Feng and Zhangsuo Liu designed the manuscript. Yong Liu, Qi Feng, Dongwei Liu and Zhangsuo Liu contributed to writing the manuscript. Yong Liu, Yingjin Qiao, Dongwei Liu, Shaokang Pan, Jingfang Chen, Zihui Mao, Kaidi Ren, Yang Yang, Qi Feng and Zhangsuo Liu reviewed and revised the manuscript. All authors have seen and approved the final version of the manuscript being submitted.
Acknowledgements
This work was financially supported by the National Natural Science Young Scientists Foundation of China (No.82200796, No.81900624, No.31900502, No.82204389,), the China Postdoctoral Science Foundation (No.2022M722901), the National Natural Science Foundation of China Joint Project (No. U21A20348), the Medical Science and Technology Research Project of Henan Province (SBGJ202102145, SBGJ202103079), and the General Program of the National Natural Science Foundation of China General Project (No.81970633). We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service.
Abbreviations
MAM: Mitochondria-associated endoplasmic reticulum membrane
ER: Endoplasmic reticulum
MSC: Membrane contact site
DKD: Diabetic kidney disease
DM: Diabetes mellitus
T1D: Type 1 diabetes
T2D: Type 2 diabetes
ESRD: End-stage renal disease
GFR: Glomerular filtration rate
DGAT2: Diacylglycerol O-acyltransferase 2
PEMT2: Phosphatidylethanolamine N-methyltransferase 2
FACL4: Fatty acid CoA ligase 4
ACAT1/SOAT1: Cholesterol acyltransferase/sterol O-acyltransferase 1
PSS1/2: Phosphatidylserine synthase 1/2
PS: Phosphatidylserine
PE: Phosphatidylethanolamine
PA: Phosphatidic acid
CL: Cardiolipin
OMM: Outer mitochondrial membrane
IMM: Inner mitochondrial membrane
PC: Phosphatidylcholine
PEMT2: Phosphatidylethanolamine N-methyltransferase 2
PTPIP51: Protein tyrosine phosphatase-interacting protein 51
CAV1: Caveolin-1
IP3R: Inositol 1,4,5-trisphosphate receptor
GRP75: Glucose regulated protein 75
VDAC1: Voltage-dependent anion channel 1
MCU: Mitochondrial calcium uniporter
CypD: Cyclophilin D
CANX: Calcinetin
MFN2: Mitofusin 2
PKB: Protein kinase B
mTORC: Mammalian target of rapamycin complex
DRP1: Dynamin-related protein 1
Fis1: Fission-1
MFF: Mitochondrial fission factor
MID: mitochondrial dynamics proteins 49
OPA1: Optic atrophy 1
INF2: Inverted formin 2
PACS2: Phosphofurin acidic cluster sorting protein 2
PP2A: Protein phosphatase 2A
PINK1: PTEN-induced putative kinase 1
DsbA-L: Disulfide-bond A oxidoreductase-like protein
FUNDC1: FUN14 domain-containing protein 1
EFD: Ectopic fat deposition
NLRP3: Nucleotide binding and oligomerization domain-like receptor family pyrin domain-containing 3
ASC: Apoptosis-associated speck-like protein containing a CARD
MAVS: Mitochondrial antiviral signaling protein
STING: Stimulator of interferon genes
AMPK: AMP-activated protein kinase
UPR: Unfolded protein response
Sig1R: Sigma-1 receptor
PERK: RNA-dependent protein kinase (PKR)-like ER kinase
ATF6: Activating transcription factor 6
IRE1α: Inositol-requiring enzyme 1α
RTN1A: Reticulon 1A
AKAP1: A-kinase anchoring protein 1
Bcl2: B-cell lymphoma 2
MIDI: Mitochondrial division inhibitor
WASF3: Wiskott-Aldrich syndrome protein family member 3
ATAD3: ATPase family AAA Domain-containing protein 3
MITOL: Mitochondrial ubiquitin ligase
MOSPD2: Motile sperm domain-containing protein 2
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yao W, Liao H, Pang M, Pan L, Guan Y, Huang X. et al. Inhibition of the NADPH Oxidase Pathway Reduces Ferroptosis during Septic Renal Injury in Diabetic Mice. Oxid Med Cell Longev. 2022;2022:1193734
2. Zhou M, Huang R. Associations of Serum Total 25OHD, 25OHD3, and epi-25OHD3 with Insulin Resistance: Cross-Sectional Analysis of the National Health and Nutrition Examination Survey, 2011-2016. Nutrients. 2022 14
3. Zhu R, Zhou S, Xia L, Bao X. Incidence, Morbidity and years Lived with Disability due to Type 2 Diabetes Mellitus in 204 Countries and Territories: Trends From 1990 to 2019. Frontiers in endocrinology. 2022;13:905538
4. Wang N, Lu Z, Zhang W, Bai Y, Pei D, Li L. Serum Cystatin C Trajectory Is a Marker Associated with Diabetic Kidney Disease. Front Endocrinol (Lausanne). 2022;13:824279
5. Rico-Fontalvo J, Aroca G, Cabrales J, Daza-Arnedo R, Yánez-Rodríguez T, Martínez-Ávila MC. et al. Molecular Mechanisms of Diabetic Kidney Disease. Int J Mol Sci. 2022 23
6. Yang M, Wang X, Han Y, Li C, Wei L, Yang J. et al. Targeting the NLRP3 Inflammasome in Diabetic Nephropathy. Current medicinal chemistry. 2021;28:8810-24
7. Said SM, Nasr SH. Silent diabetic nephropathy. Kidney international. 2016;90:24-6
8. Zac-Varghese S, Winocour P. Managing diabetic kidney disease. British Medical Bulletin. 2017;125:55-66
9. Lazar AG, Vlad ML, Manea A, Simionescu M, Manea SA. Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney. Antioxidants (Basel). 2021 10
10. Ma T, Li X, Zhu Y, Yu S, Liu T, Zhang X. et al. Excessive Activation of Notch Signaling in Macrophages Promote Kidney Inflammation, Fibrosis, and Necroptosis. Front Immunol. 2022;13:835879
11. Tang J, Yao D, Yan H, Chen X, Wang L, Zhan H. The Role of MicroRNAs in the Pathogenesis of Diabetic Nephropathy. International Journal of Endocrinology. 2019;2019:8719060
12. Samsu N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Research International. 2021;2021:1497449
13. Saxena S, Mathur A, Kakkar P. Critical role of mitochondrial dysfunction and impaired mitophagy in diabetic nephropathy. Journal of cellular physiology. 2019;234:19223-36
14. Ni L, Yuan C, Wu X. Endoplasmic Reticulum Stress in Diabetic Nephrology: Regulation, Pathological Role, and Therapeutic Potential. Oxidative medicine and cellular longevity. 2021;2021:7277966
15. Luan Y, Luan Y, Yuan R-X, Feng Q, Chen X, Yang Y. Structure and Function of Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Role in Cardiovascular Diseases. Oxidative Medicine and Cellular Longevity. 2021;2021:4578809
16. Xie Y, E J, Cai H, Zhong F, Xiao W, Gordon RE. et al. Reticulon-1A mediates diabetic kidney disease progression through endoplasmic reticulum-mitochondrial contacts in tubular epithelial cells. Kidney international. 2022;102:293-306
17. Li S, Yan R, Xu J, Zhao S, Ma X, Sun Q. et al. A new type of ERGIC-ERES membrane contact mediated by TMED9 and SEC12 is required for autophagosome biogenesis. Cell Research. 2022;32:119-38
18. Krols M, van Isterdael G, Asselbergh B, Kremer A, Lippens S, Timmerman V. et al. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathologica. 2016;131:505-23
19. Perrone M, Caroccia N, Genovese I, Missiroli S, Modesti L, Pedriali G. et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int Rev Cell Mol Biol. 2020;350:119-96
20. Perrone M, Caroccia N, Genovese I, Missiroli S, Modesti L, Pedriali G. et al. Chapter Four - The role of mitochondria-associated membranes in cellular homeostasis and diseases. In: Kepp O, Galluzzi L, editors. International Review of Cell and Molecular Biology: Academic Press. 2020 p. 119-96
21. Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ. et al. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017;134:129-49
22. Bernhard W, Rouiller C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol. 1956;2:73-8
23. Barazzuol L, Giamogante F, Calì T. Mitochondria Associated Membranes (MAMs): Architecture and physiopathological role. Cell Calcium. 2021;94:102343
24. Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF. et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915-21
25. Delprat B, Maurice T, Delettre C. Wolfram syndrome: MAMs' connection? Cell Death & Disease. 2018;9:364
26. Giacomello M, Pellegrini L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 2016;23:1417-27
27. Wang X, Wen Y, Dong J, Cao C, Yuan S. Systematic In-Depth Proteomic Analysis of Mitochondria-Associated Endoplasmic Reticulum Membranes in Mouse and Human Testes. Proteomics. 2018;18:e1700478
28. Lu X, Gong Y, Hu W, Mao Y, Wang T, Sun Z. et al. Ultrastructural and proteomic profiling of mitochondria-associated endoplasmic reticulum membranes reveal aging signatures in striated muscle. Cell Death & Disease. 2022;13:296
29. Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteomics. 2013;79:219-30
30. Ma JH, Shen S, Wang JJ, He Z, Poon A, Li J. et al. Comparative Proteomic Analysis of the Mitochondria-associated ER Membrane (MAM) in a Long-term Type 2 Diabetic Rodent Model. Scientific Reports. 2017;7:2062
31. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843:2253-62
32. Liu J, Yang J. Mitochondria-associated membranes: A hub for neurodegenerative diseases. Biomedicine & Pharmacotherapy. 2022;149:112890
33. Peng J, Peng C, Wang L, Cao H, Xing C, Li G. et al. Endoplasmic reticulum-mitochondria coupling attenuates vanadium-induced apoptosis via IP 3 R in duck renal tubular epithelial cells. Journal of inorganic biochemistry. 2022;232:111809
34. Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. 2000;275:34534-40
35. Adachi Y, Kato T, Yamada T, Murata D, Arai K, Stahelin RV. et al. Drp1 Tubulates the ER in a GTPase-Independent Manner. Molecular cell. 2020;80:621-32.e6
36. Rieusset J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death & Disease. 2018;9:388
37. Teodoro BG, Sampaio IH, Bomfim LH, Queiroz AL, Silveira LR, Souza AO. et al. Long-chain acyl-CoA synthetase 6 regulates lipid synthesis and mitochondrial oxidative capacity in human and rat skeletal muscle. J Physiol. 2017;595:677-93
38. Rusiñol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269:27494-502
39. Shen W, Gao C, Cueto R, Liu L, Fu H, Shao Y. et al. Homocysteine-methionine cycle is a metabolic sensor system controlling methylation-regulated pathological signaling. Redox Biol. 2020;28:101322
40. Hernández-Alvarez MI, Sebastián D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P. et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell. 2019;177:881-95.e17
41. Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO reports. 2016;17:800-10
42. Monteiro-Cardoso VF, Rochin L, Arora A, Houcine A, Jääskeläinen E, Kivelä AM. et al. ORP5/8 and MIB/MICOS link ER-mitochondria and intra-mitochondrial contacts for non-vesicular transport of phosphatidylserine. Cell reports. 2022;40:111364
43. Yeo HK, Park TH, Kim HY, Jang H, Lee J, Hwang G-S. et al. Phospholipid transfer function of PTPIP51 at mitochondria-associated ER membranes. EMBO Rep. 2021;22:e51323
44. Jin Y, McFie PJ, Banman SL, Brandt C, Stone SJ. Diacylglycerol acyltransferase-2 (DGAT2) and monoacylglycerol acyltransferase-2 (MGAT2) interact to promote triacylglycerol synthesis. The Journal of biological chemistry. 2014;289:28237-48
45. Sala-Vila A, Navarro-Lérida I, Sánchez-Alvarez M, Bosch M, Calvo C, López JA. et al. Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci Rep. 2016;6:27351
46. Son SM, Byun J, Roh SE, Kim SJ, Mook-Jung I. Reduced IRE1α mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis. 2014;5:e1188
47. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D. et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901-11
48. Rieusset J, Fauconnier J, Paillard M, Belaidi E, Tubbs E, Chauvin M-A. et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia. 2016;59:614-23
49. Patten D, Harper M-E, Boardman N. Harnessing the protective role of OPA1 in diabetic cardiomyopathy. Acta Physiologica. 2020;229:e13466
50. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358-62
51. Nolden KA, Egner JM, Collier JJ, Russell OM, Alston CL, Harwig MC. et al. Novel <i>DNM1L</i> variants impair mitochondrial dynamics through divergent mechanisms. Life Sci Alliance. 2022 5
52. Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M. et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy. 2017;13:654-69
53. Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X. et al. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in vivo. Circulation. 2017;136:2248-66
54. Audzeyenka I, Rachubik P, Typiak M, Kulesza T, Kalkowska D, Rogacka D. et al. PTEN-induced kinase 1 deficiency alters albumin permeability and insulin signaling in podocytes. Journal of Molecular Medicine. 2022;100:903-15
55. Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2014;1841:595-609
56. Horvath SE, Daum G. Lipids of mitochondria. Progress in Lipid Research. 2013;52:590-614
57. Yang X, Liang J, Ding L, Li X, Lam S-M, Shui G. et al. Phosphatidylserine synthase regulates cellular homeostasis through distinct metabolic mechanisms. PLOS Genetics. 2019;15:e1008548
58. Anastasia I, Ilacqua N, Raimondi A, Lemieux P, Ghandehari-Alavijeh R, Faure G. et al. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 2021;34:108873
59. Bosch M, Marí M, Herms A, Fernández A, Fajardo A, Kassan A. et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr Biol. 2011;21:681-6
60. Fu Y, Hoang A, Escher G, Parton RG, Krozowski Z, Sviridov D. Expression of caveolin-1 enhances cholesterol efflux in hepatic cells. The Journal of biological chemistry. 2004;279:14140-6
61. Theobald SJ, Gräb J, Fritsch M, Suárez I, Eisfeld HS, Winter S. et al. Gasdermin D mediates host cell death but not interleukin-1β secretion in Mycobacterium tuberculosis-infected macrophages. Cell Death Discov. 2021;7:327
62. More J, Galusso N, Veloso P, Montecinos L, Finkelstein JP, Sanchez G. et al. N-Acetylcysteine Prevents the Spatial Memory Deficits and the Redox-Dependent RyR2 Decrease Displayed by an Alzheimer's Disease Rat Model. Front Aging Neurosci. 2018;10:399
63. Davis LC, Morgan AJ, Galione A. NAADP-regulated two-pore channels drive phagocytosis through endo-lysosomal Ca(2+) nanodomains, calcineurin and dynamin. Embo j. 2020;39:e104058
64. Ligeza J, Marona P, Gach N, Lipert B, Miekus K, Wilk W. et al. MCPIP1 contributes to clear cell renal cell carcinomas development. Angiogenesis. 2017;20:325-40
65. Kerkhofs M, Bittremieux M, Morciano G, Giorgi C, Pinton P, Parys JB. et al. Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018;9:334
66. Lin H, Peng Y, Li J, Wang Z, Chen S, Qing X. et al. Reactive Oxygen Species Regulate Endoplasmic Reticulum Stress and ER-Mitochondrial Ca(2+) Crosstalk to Promote Programmed Necrosis of Rat Nucleus Pulposus Cells under Compression. Oxid Med Cell Longev. 2021;2021:8810698
67. Paillard M, Tubbs E, Thiebaut P-A, Gomez L, Fauconnier J, Silva CCD. et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes From Lethal Hypoxia-Reoxygenation Injury. Circulation. 2013;128:1555-65
68. John ASP, Kundu S, Pushpakumar S, Amin M, Tyagi SC, Sen U. Hydrogen sulfide inhibits Ca2+-induced mitochondrial permeability transition pore opening in type-1 diabetes. American Journal of Physiology-Endocrinology and Metabolism. 2019;317:E269-E83
69. Lindblom Runa SJ, Higgins Gavin C, Nguyen T-V, Arnstein M, Henstridge Darren C, Granata C. et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clinical Science. 2020;134:239-59
70. Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nature Reviews Nephrology. 2018;14:291-312
71. Che Y, Chen Y, Wang Z, Zheng S, Xing K, Yuan S. et al. The Combination of Rhodosin and MMF Prolongs Cardiac Allograft Survival by Inhibiting DC Maturation by Promoting Mitochondrial Fusion. Oxid Med Cell Longev. 2022;2022:7260305
72. Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, Ryan MT. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nature Communications. 2018;9:5239
73. Mao H, Chen W, Chen L, Li L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochemical pharmacology. 2022;199:115011
74. Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659-67
75. Plewes MR, Hou X, Talbott HA, Zhang P, Wood JR, Cupp AS. et al. Luteinizing hormone regulates the phosphorylation and localization of the mitochondrial effector dynamin-related protein-1 (DRP1) and steroidogenesis in the bovine corpus luteum. Faseb j. 2020;34:5299-316
76. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. Journal of Cell Biology. 2016;212:379-87
77. Jin X, Wang J, Gao K, Zhang P, Yao L, Tang Y. et al. Dysregulation of INF2-mediated mitochondrial fission in SPOP-mutated prostate cancer. PLoS Genet. 2017;13:e1006748
78. Steffen J, Koehler CM. ER-mitochondria contacts: Actin dynamics at the ER control mitochondrial fission via calcium release. J Cell Biol. 2018;217:15-7
79. Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol. 2018;217:251-68
80. Li C, Li L, Yang M, Yang J, Zhao C, Han Y. et al. PACS-2 Ameliorates Tubular Injury by Facilitating Endoplasmic Reticulum-Mitochondria Contact and Mitophagy in Diabetic Nephropathy. Diabetes. 2022;71:1034-50
81. Wu W, Li W, Chen H, Jiang L, Zhu R, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 2016;12:1675-6
82. Wang A, Zhang D, Liu J, Yan H, Zhang P, Yuan H. et al. Guanxinning Injection Combined with Ischemic Postconditioning Attenuate Myocardial Ischemic Reperfusion Injury in Chronic Renal Failure Rats by Modulating Mitochondrial Dynamics. Front Cardiovasc Med. 2022;9:905254
83. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189-200
84. Wang FX, Luo YM, Ye ZQ, Cao X, Liang JN, Wang Q. et al. iTRAQ-based proteomics analysis of autophagy-mediated immune responses against the vascular fungal pathogen Verticillium dahliae in Arabidopsis. Autophagy. 2018;14:598-618
85. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N. et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389-93
86. Kumar S, Tikoo K. Independent role of PP2A and mTORc1 in palmitate induced podocyte death. Biochimie. 2015;112:73-84
87. Zang N, Cui C, Guo X, Song J, Hu H, Yang M. et al. cGAS-STING activation contributes to podocyte injury in diabetic kidney disease. iScience. 2022;25:105145
88. Yang M, Luo S, Jiang N, Wang X, Han Y, Zhao H. et al. DsbA-L Ameliorates Renal Injury Through the AMPK/NLRP3 Inflammasome Signaling Pathway in Diabetic Nephropathy. Frontiers in Physiology. 2021 12
89. Cao Y, Chen Z, Hu J, Feng J, Zhu Z, Fan Y. et al. Mfn2 Regulates High Glucose-Induced MAMs Dysfunction and Apoptosis in Podocytes via PERK Pathway. Frontiers in Cell and Developmental Biology. 2021 9
90. Liu X-Q, Jiang L, Li Y-Y, Huang Y-B, Hu X-R, Zhu W. et al. Wogonin protects glomerular podocytes by targeting Bcl-2-mediated autophagy and apoptosis in diabetic kidney disease. Acta pharmacologica Sinica. 2022;43:96-110
91. Xue M, Fang T, Sun H, Cheng Y, Li T, Xu C. et al. PACS-2 attenuates diabetic kidney disease via the enhancement of mitochondria-associated endoplasmic reticulum membrane formation. Cell Death & Disease. 2021;12:1107
92. Xiao M, Kong Z-L, Che K, Hu J-X, Li Y, Huang Y-J. et al. The role of mitochondrial fission factor in podocyte injury in diabetic nephropathy. Biochemical and Biophysical Research Communications. 2022;624:40-6
93. Li X, Yang Q, Liu S, Song S, Wang C. Mitochondria-associated endoplasmic reticulum membranes promote mitochondrial fission through AKAP1-Drp1 pathway in podocytes under high glucose conditions. Experimental cell research. 2023;424:113512
94. Zheng T, Wang H-y, Chen Y, Chen X, Wu Z-l, Hu Q-y. et al. Src Activation Aggravates Podocyte Injury in Diabetic Nephropathy via Suppression of FUNDC1-Mediated Mitophagy. Frontiers in Pharmacology. 2022 13
95. Yi X, Yan W, Guo T, Liu N, Wang Z, Shang J. et al. Erythropoietin Mitigates Diabetic Nephropathy by Restoring PINK1/Parkin-Mediated Mitophagy. Frontiers in Pharmacology. 2022 13
96. Yang Y-Y, Gao Z-X, Mao Z-H, Liu D-W, Liu Z-S, Wu P. Identification of ULK1 as a novel mitophagy-related gene in diabetic nephropathy. Frontiers in endocrinology. 2022;13:1079465
97. Zayed AE, Saleh A, Gomaa AMS, Abd-Elkareem M, Anwar MM, Hassanein KMA. et al. Protective Effect of Ginkgo biloba and Magnetized Water on Nephropathy in Induced Type 2 Diabetes in Rat. Oxid Med Cell Longev. 2018;2018:1785614
98. Dingreville F, Panthu B, Thivolet C, Ducreux S, Gouriou Y, Pesenti S. et al. Differential Effect of Glucose on ER-Mitochondria Ca2+ Exchange Participates in Insulin Secretion and Glucotoxicity-Mediated Dysfunction of β-Cells. Diabetes. 2019;68:1778-94
99. Purvis GSD, Collino M, Loiola RA, Baragetti A, Chiazza F, Brovelli M. et al. Identification of AnnexinA1 as an Endogenous Regulator of RhoA, and Its Role in the Pathophysiology and Experimental Therapy of Type-2 Diabetes. Front Immunol. 2019;10:571
100. Wu Z, Yu S, Kang X, Liu Y, Xu Z, Li Z. et al. Association of visceral adiposity index with incident nephropathy and retinopathy: a cohort study in the diabetic population. Cardiovasc Diabetol. 2022;21:32
101. Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin M-A. et al. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Is Required for Insulin Signaling and Is Implicated in Hepatic Insulin Resistance. Diabetes. 2014;63:3279-94
102. Cheng H, Gang X, He G, Liu Y, Wang Y, Zhao X. et al. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Frontiers in Endocrinology. 2020 11
103. Yang M, Luo S, Yang J, Chen W, He L, Liu D. et al. Lipid droplet - mitochondria coupling: A novel lipid metabolism regulatory hub in diabetic nephropathy. Frontiers in Endocrinology. 2022 13
104. Yang M, Han Y, Luo S, Xiong X, Zhu X, Zhao H. et al. MAMs Protect Against Ectopic Fat Deposition and Lipid-Related Kidney Damage in DN Patients. Front Endocrinol (Lausanne). 2021;12:609580
105. Thongnak L, Pongchaidecha A, Lungkaphin A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am J Med Sci. 2020;359:84-99
106. Schelling JR. The Contribution of Lipotoxicity to Diabetic Kidney Disease. Cells. 2022;11:3236
107. Gu W, Yang L, Wang X, Geng J, Li X, Zheng K. et al. Pterostilbene, a Resveratrol Derivative, Improves Ectopic Lipid Deposition in the Kidneys of Mice Induced by a High-Fat Diet. Kidney and Blood Pressure Research. 2022;47:514-22
108. Yang M, Han Y, Luo S, Xiong X, Zhu X, Zhao H. et al. MAMs Protect Against Ectopic Fat Deposition and Lipid-Related Kidney Damage in DN Patients. Frontiers in Endocrinology. 2021 12
109. Hu Y, Chen H, Zhang L, Lin X, Li X, Zhuang H. et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2021;17:1142-56
110. Chen X, Han Y, Gao P, Yang M, Xiao L, Xiong X. et al. Disulfide-bond A oxidoreductase-like protein protects against ectopic fat deposition and lipid-related kidney damage in diabetic nephropathy. Kidney Int. 2019;95:880-95
111. Zhao C, Li L, Li C, Tang C, Cai J, Liu Y. et al. PACS-2 deficiency in tubular cells aggravates lipid-related kidney injury in diabetic kidney disease. Molecular Medicine. 2022;28:117
112. Qiu YY, Tang LQ. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol Res. 2016;114:251-64
113. Pereira AC, De Pascale J, Resende R, Cardoso S, Ferreira I, Neves BM. et al. ER-mitochondria communication is involved in NLRP3 inflammasome activation under stress conditions in the innate immune system. Cellular and Molecular Life Sciences. 2022;79:213
114. Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M. et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death & Disease. 2018;9:329
115. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221-5
116. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788-92
117. Feng J, Bao L, Wang X, Li H, Chen Y, Xiao W. et al. Low expression of HIV genes in podocytes accelerates the progression of diabetic kidney disease in mice. Kidney International. 2021;99:914-25
118. Ishikawa H, Barber GN. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell Mol Life Sci. 2011;68:1157-65
119. Xue C, Dong N, Shan A. Putative role of STING-mitochondria associated membrane crosstalk in immunity. Trends in immunology. 2022;43:513-22
120. Wang W-X, Prajapati P, Nelson PT, Springer JE. The Mitochondria-Associated ER Membranes Are Novel Subcellular Locations Enriched for Inflammatory-Responsive MicroRNAs. Molecular Neurobiology. 2020;57:2996-3013
121. Li X, Venkatesh I, Villanueva V, Wei H, Geraghty T, Rajagopalan A. et al. Podocyte-specific deletion of miR-146a increases podocyte injury and diabetic kidney disease. Frontiers in medicine. 2022;9:897188
122. Zhao N, Luo Q, Lin R, Li Q, Ma P. MiR-142-3p ameliorates high glucose-induced renal tubular epithelial cell injury by targeting BOD1. Clinical and experimental nephrology. 2021;25:1182-92
123. Chen FY, Huang MY, Lin YM, Ho CH, Lin SY, Chen HY. et al. BIK ubiquitination by the E3 ligase Cul5-ASB11 determines cell fate during cellular stress. J Cell Biol. 2019;218:3002-18
124. Binder P, Wang S, Radu M, Zin M, Collins L, Khan S. et al. Pak2 as a Novel Therapeutic Target for Cardioprotective Endoplasmic Reticulum Stress Response. Circ Res. 2019;124:696-711
125. Riess C, Del Moral K, Fiebig A, Kaps P, Linke C, Hinz B. et al. Implementation of a combined CDK inhibition and arginine-deprivation approach to target arginine-auxotrophic glioblastoma multiforme cells. Cell Death Dis. 2022;13:555
126. Bae H, Lee W, Song J, Hong T, Kim MH, Ham J. et al. Polydatin Counteracts 5-Fluorouracil Resistance by Enhancing Apoptosis via Calcium Influx in Colon Cancer. Antioxidants (Basel). 2021 10
127. Yi S, Chen K, Zhang L, Shi W, Zhang Y, Niu S. et al. Endoplasmic Reticulum Stress Is Involved in Stress-Induced Hypothalamic Neuronal Injury in Rats via the PERK-ATF4-CHOP and IRE1-ASK1-JNK Pathways. Front Cell Neurosci. 2019;13:190
128. Knoell J, Chillappagari S, Knudsen L, Korfei M, Dartsch R, Jonigk D. et al. PACS2-TRPV1 axis is required for ER-mitochondrial tethering during ER stress and lung fibrosis. Cell Mol Life Sci. 2022;79:151
129. Mori T, Hayashi T, Hayashi E, Su TP. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One. 2013;8:e76941
130. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP. et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880-91
131. Muñoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A. et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. Embo j. 2013;32:2348-61
132. Fan Y, Zhang J, Xiao W, Lee K, Li Z, Wen J. et al. Rtn1a-Mediated Endoplasmic Reticulum Stress in Podocyte Injury and Diabetic Nephropathy. Scientific Reports. 2017;7:323
133. Xiao W, Fan Y, Wang N, Chuang PY, Lee K, He JC. Knockdown of RTN1A attenuates ER stress and kidney injury in albumin overload-induced nephropathy. American Journal of Physiology-Renal Physiology. 2016;310:F409-F15
134. Shen S, Ji C, Wei K. Cellular Senescence and Regulated Cell Death of Tubular Epithelial Cells in Diabetic Kidney Disease. Frontiers in Endocrinology. 2022 13
135. Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T. et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135-9
136. Lalier L, Mignard V, Joalland M-P, Lanoé D, Cartron P-F, Manon S. et al. TOM20-mediated transfer of Bcl2 from ER to MAM and mitochondria upon induction of apoptosis. Cell Death & Disease. 2021;12:182
137. Yang M, Zhao L, Gao P, Zhu X, Han Y, Chen X. et al. DsbA-L ameliorates high glucose induced tubular damage through maintaining MAM integrity. EBioMedicine. 2019;43:607-19
138. Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg. 2018;1859:940-50
139. Coughlan MT, Nguyen TV, Penfold SA, Higgins GC, Thallas-Bonke V, Tan SM. et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci (Lond). 2016;130:711-20
140. Kim MJ, Kim HJ, Jang B, Kim HJ, Mostafa MN, Park SJ. et al. Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration. Cells. 2022 11
141. Wang X, Xie D, Dai H, Ye J, Liu Y, Fei A. Clemastine protects against sepsis-induced myocardial injury in vivo and in vitro. Bioengineered. 2022;13:7134-46
142. Audzeyenka I, Rachubik P, Typiak M, Kulesza T, Topolewska A, Rogacka D. et al. Hyperglycemia alters mitochondrial respiration efficiency and mitophagy in human podocytes. Experimental Cell Research. 2021;407:112758
143. Wu QY, Cheng Z, Zhou YZ, Zhao Y, Li JM, Zhou XM. et al. A novel STAT3 inhibitor attenuates angiotensin II-induced abdominal aortic aneurysm progression in mice through modulating vascular inflammation and autophagy. Cell Death Dis. 2020;11:131
144. Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT. et al. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. Journal of the American Society of Nephrology. 2016;27:2733-47
145. Ma Y, Chen Z, Tao Y, Zhu J, Yang H, Liang W. et al. Increased mitochondrial fission of glomerular podocytes in diabetic nephropathy. Endocrine Connections. 2019;8:1206-12
146. Lu M, Yin N, Liu W, Cui X, Chen S, Wang E. Curcumin Ameliorates Diabetic Nephropathy by Suppressing NLRP3 Inflammasome Signaling. BioMed research international. 2017;2017:1516985
147. Kim BH, Lee ES, Choi R, Nawaboot J, Lee MY, Lee EY. et al. Protective Effects of Curcumin on Renal Oxidative Stress and Lipid Metabolism in a Rat Model of Type 2 Diabetic Nephropathy. Yonsei medical journal. 2016;57:664-73
148. Yuan D, Liu X-M, Fang Z, Du L-L, Chang J, Lin S-H. Protective effect of resveratrol on kidney in rats with diabetic nephropathy and its effect on endoplasmic reticulum stress. European review for medical and pharmacological sciences. 2018;22:1485-93
149. Tian N, Gao Y, Wang X, Wu X, Zou D, Zhu Z. et al. Emodin mitigates podocytes apoptosis induced by endoplasmic reticulum stress through the inhibition of the PERK pathway in diabetic nephropathy. Drug design, development and therapy. 2018;12:2195-211
150. Ke Q, Shi C, Lv Y, Wang L, Luo J, Jiang L. et al. SGLT2 inhibitor counteracts NLRP3 inflammasome via tubular metabolite itaconate in fibrosis kidney. Faseb j. 2022;36:e22078
151. Zhang P, Fang J, Zhang J, Ding S, Gan D. Curcumin Inhibited Podocyte Cell Apoptosis and Accelerated Cell Autophagy in Diabetic Nephropathy via Regulating Beclin1/UVRAG/Bcl2. Diabetes, metabolic syndrome and obesity: targets and therapy. 2020;13:641-52
152. Zhao Y, Sun M. Metformin rescues Parkin protein expression and mitophagy in high glucose-challenged human renal epithelial cells by inhibiting NF-κB via PP2A activation. Life Sci. 2020;246:117382
153. Tagaya M, Kume S, Yasuda-Yamahara M, Kuwagata S, Yamahara K, Takeda N. et al. Inhibition of mitochondrial fission protects podocytes from albumin-induced cell damage in diabetic kidney disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2022;1868:166368
154. Yang S-k, Li A-m, Han Y-c, Peng C-h, Song N, Yang M. et al. Mitochondria-Targeted Peptide SS31 Attenuates Renal Tubulointerstitial Injury via Inhibiting Mitochondrial Fission in Diabetic Mice. Oxidative Medicine and Cellular Longevity. 2019;2019:2346580
155. Yang J, Chen P, Cao Y, Liu S, Wang W, Li L. et al. Chemical inhibition of mitochondrial fission via targeting the DRP1-receptor interaction. Cell Chemical Biology. 2023;30:278-94.e11
156. Li M-D, Fu L, Lv B-B, Xiang Y, Xiang H-X, Xu D-X. et al. Arsenic induces ferroptosis and acute lung injury through mtROS-mediated mitochondria-associated endoplasmic reticulum membrane dysfunction. Ecotoxicology and Environmental Safety. 2022;238:113595
157. Dia M, Leon C, Chanon S, Bendridi N, Gomez L, Rieusset J. et al. Effect of Metformin on T2D-Induced MAM Ca2+ Uncoupling and Contractile Dysfunction in an Early Mouse Model of Diabetic HFpEF. International Journal of Molecular Sciences. 2022;23:3569
158. Wei X, Wei X, Lu Z, Li L, Hu Y, Sun F. et al. Activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metabolism. 2020;105:154182
Author contact
![]() Corresponding authors: Qi Feng, Dongwei Liu and Zhangsuo Liu, Research Institute of Nephrology, Zhengzhou University, Traditional Chinese Medicine Integrated Department of Nephrology, Henan Province Research Center for Kidney Disease, Key Laboratory of Precision Diagnosis and Treatment for Chronic Kidney Disease in Henan Province, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. Email: fengqi2019edu.cn; liu-dongweiedu.cn; zhangsuoliuedu.cn.
Corresponding authors: Qi Feng, Dongwei Liu and Zhangsuo Liu, Research Institute of Nephrology, Zhengzhou University, Traditional Chinese Medicine Integrated Department of Nephrology, Henan Province Research Center for Kidney Disease, Key Laboratory of Precision Diagnosis and Treatment for Chronic Kidney Disease in Henan Province, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. Email: fengqi2019edu.cn; liu-dongweiedu.cn; zhangsuoliuedu.cn.