Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
General characteristics and...
PPARα
PPARβ/δ
PPARγ
LXR
FXR
REV-ERB and HNF4α
Concluding remarks and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(1):113-126. doi:10.7150/ijbs.87305 This issue Cite
Review
The Role of Nuclear Receptors in the Pathogenesis and Treatment of Non-alcoholic Fatty Liver Disease
Zhenhua Yang1,2, Awang Danzeng1,2, Qiumeng Liu1,2, Chenglong Zeng1,2, Lei Xu1,2, Jie Mo1,2, Ciren Pingcuo1,2, Xiaojing Wang3, Chao Wang1,2 ![]() , Bixiang Zhang1,2
, Bixiang Zhang1,2 ![]() , Binhao Zhang1,2
, Binhao Zhang1,2 ![]()
1. Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China.
2. Clinical Medicine Research Center for Hepatic Surgery of Hubei Province, Wuhan 430030, Hubei Province, China.
3. Department and Institute of Infectious Disease, Tongji Hospital, Tongji Medical College and State Key Laboratory for Diagnosis and Treatment of Severe Zoonostic Infectious Disease, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China.
Received 2023-6-19; Accepted 2023-9-21; Published 2024-1-1
Abstract

Non-alcoholic fatty liver disease (NAFLD) is a global health burden closely linked to insulin resistance, obesity, and type 2 diabetes. The complex pathophysiology of NAFLD involves multiple cellular pathways and molecular factors. Nuclear receptors (NRs) have emerged as crucial regulators of lipid metabolism and inflammation in NAFLD, offering potential therapeutic targets for NAFLD. Targeting PPARs and FXRs has shown promise in ameliorating NAFLD symptoms and halting disease progression. However, further investigation is needed to address side effects and personalize therapy approaches. This review summarizes the current understanding of the involvement of NRs in the pathogenesis of NAFLD and explores their therapeutic potential. We discuss the role of several NRs in modulating lipid homeostasis in the liver, including peroxisome proliferator-activated receptors (PPARs), liver X receptors (LXRs), farnesoid X receptors (FXRs), REV-ERB, hepatocyte nuclear factor 4α (HNF4α), constitutive androstane receptor (CAR) and pregnane X receptor (PXR).The expanding knowledge of NRs in NAFLD offers new avenues for targeted therapies, necessitating exploration of novel treatment strategies and optimization of existing approaches to combat this increasingly prevalent disease.
Keywords: NAFLD, NRs, PPARs, FXR, LXR
Introduction
Non-alcoholic fatty liver disease (NAFLD), recently proposed by scholars to be renamed metabolic dysfunction-associated steatotic liver disease (MASLD)[1], has emerged as one of the most prevalent and widespread liver disorders worldwide[2, 3]. Recent epidemiological data have revealed that the incidence of NAFLD has risen to an alarming 32.4% in 2022[4], surpassing previous estimates and showing a significant increase from a baseline of 25.24% in 2016[5]. Furthermore, NAFLD is projected to become the primary cause of cirrhosis necessitating liver transplantation over the next decade[6-8]. Due to the lack of early warning signs, NAFLD can lead to substantial healthcare expenses, economic losses and a significant reduction in health-related quality of life[9, 10]. At present, NAFLD lacks an approved therapy, although numerous drugs are progressing in advanced stages of development and researchers remain optimistic about the potential benefits of these therapies[11-13]. In light of the soaring incidence of NAFLD, there is an urgent need to accelerate research and development efforts to identify safe and effective therapies for this condition.
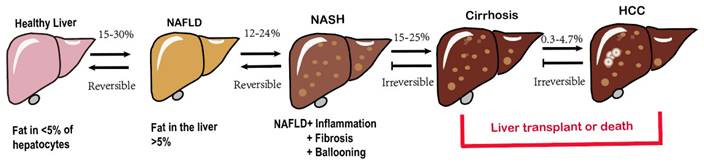
The hallmark characteristic of NAFLD is macrovescular steatosis, which is defined by the presence of lipid droplets in at least 5% of hepatocytes, without any secondary causes for hepatic fat accumulation, such as clinically significant alcohol consumption[14]. NAFLD, represented the hepatic manifestation of metabolic syndrome, demonstrates a bidirectional association with obesity[15], T2DM, elevated serum liver enzymes, poor physical condition and hypertension [13, 16-18]. Among these factors, T2DM poses a particularly severe threat as it significantly increases the risk of cirrhosis and its associated complications[19-21]. Fatty liver disease encompasses a variety of pathological conditions, ranging from lipid accumulation in liver cells (steatosis) to the development of superimposed inflammation (steatohepatitis), characterized by necrotizing inflammation and faster fibrosis progression compared to nonalcoholic liver disease[22], ultimately culminating in cirrhosis[23], even hepatocellular carcinoma(HCC) (Fig. 1) [24, 25]. The initiation and development of NAFLD are subject to an assortment of factors. Obesity and T2DM closely contribute to the increasing incidence of NAFLD and are directly linked to it[26]. Genetic makeup also has a significant impact on how diseases develop[27]. Additionally, complex interactions between environmental and genetic factors, particularly dietary factors, affect the progression of NAFLD[28, 29]. More recently, the gut microbiota has emerged as a significant player in the pathogenesis of NAFLD[30, 31]. The mechanisms underlying the development of NAFLD and its complications are intricate and not fully understood[32]. As mentioned earlier, numerous factors work together or synergistically contribute to the onset and progression of NAFLD to non-alcoholic steatohepatitis (NASH) giving rise to the multiple parallel hit hypothesis regarding NAFLD advancement[33, 34].
Spectrum of NAFLD.
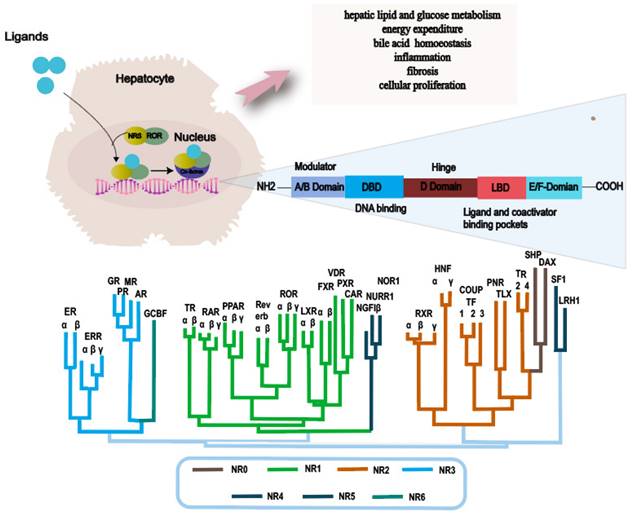
NRs are a superfamily of transcription factors that are regulated by a myriad of ligands and play an essential role in various physiological processes such as metabolism, immunity and development[35]. The human genome encodes 48 NRs that have been classified into 7 subfamilies, designated as NR0-NR6[36, 37]. A typical nuclear receptor consists of five structurally ordered regions for functionality: a variable N-terminal region that is commonly known to possess hormone-independent transactivation function, a conserved DNA binding domain that features two zinc-finger structures, a variable short hinge region that acts as a pivot for flexibility, a conserved ligand-binding domain that modulates interactions between the receptor and ligand, and a variable C-terminal region that contributes to the receptor's stability and specificity[38-40] (Fig. 2).
Main functions of nuclear receptor related to NAFLD and NR phylogenetic tree. Abbreviations: NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; ER: Estrogen Receptor; ERR: Estrogen-Related Receptor; GR: Glucocorticoid Receptor PR: Progesterone Receptor; MR: Mineralocorticoid Receptor; AR: Androgen Receptor; GCBF: Glucocorticoid-Induced TNFR-Related Protein; TR: Thyroid Hormone Receptor; RAR: Retinoic Acid Receptor; PPAR: Peroxisome Proliferator-Activated Receptor; ROR: Retinoic Acid Receptor-Related Orphan Receptor; LXR: Liver X Receptor; FXR: Farnesoid X Receptor; VDR: Vitamin D Receptor; PXR: Pregnane X Receptor; CAR: Constitutive Androstane Receptor; NGFIβ: Nerve Growth Factor-Inducible Protein β; NURR: Nuclear Receptor Subfamily 4 Group A; NOR1: Neuron-Derived Orphan Receptor 1; RXR: Retinoid X Receptor; HNF: Hepatocyte Nuclear Factor; COUPTF: Chicken Ovalbumin Upstream Promoter-Transcription Factor; PNR: Photoreceptor-Specific Nuclear Receptor; TLX: Tailless Homolog; TR: Thyroid Receptor; SHP: Small Heterodimer Partner; DAX: Dosage-Sensitive Sex Reversal, Adrenal Hypoplasia Critical Region, on Chromosome X; SF1: Steroidogenic Factor 1; LRH1: Liver Receptor Homolog 1.
NRs are mostly activated by chemically diverse small lipophilic ligands, several endogenous and exogenous lipids, such as steroids, retinoids, and phospholipids[41-43]. Upon ligands banding, NRs translocate to the nucleus and undergo conformational changes. Subsequently, by binding to reactive elements in the target gene promoter region, it recruits co-regulatory factors to activate or repress target gene expression. Through this process, transcription factors regulate gene expression in response to hormonal and environmental signal[40]. NRs transcriptionally control critical metabolic processes, including liver lipid and glucose metabolism, energy consumption, bile acid (BA) homeostasis, inflammation, fibrosis, and cell proliferation[44, 45]. Disruption of these processes contributes to the development and progression of fatty liver disease through the gut-liver-adipose axis and inflammatory signaling pathways[46]. Consequently, NRs have remained prominent drug targets in the forefront of novel therapeutic strategies for NAFLD[47].
This review focuses on the advancement of the metabolism and agonists of NRs closely related to NAFLD, including PPARs, LXRs, FXRs, REV-ERB, HNF4α, CAR and PXR nuclear receptors. We provide an overview of their impact on the progress of NAFLD, highlighting the interconnected nature of these receptors and the signaling pathways they regulate. Furthermore, we have elucidated the current state of preclinical and clinical studies investigating the efficacy of pharmacological agents targeting NRs in treating NAFLD.
General characteristics and three subtypes of PPAR
The superfamily of ligand-activated transcription factors is known as steroid hormone receptors, which causes the proliferation of peroxisomes[48]. Later on, as key integrators of inflammatory and metabolic signaling[49], PPARs are the most extensively researched NRs associated with NAFLD[50]. The three PPAR isoforms (PPARα, PPARβ/δ, and PPARγ) exhibit different tissue distributions and play distinct roles in energy metabolism[51, 52]. PPARα is extensively expressed in liver, skeletal muscle, brown fat and cardiac tissue, and it regulates energy homeostasis[53]. PPARβ/δ is ubiquitously expressed and enhances fatty acid metabolism[54]. PPARγ is predominant in adipose tissue and causes insulin sensitization and enhances glucose metabolism[55] (Table 1). The majority of PPARs create heterodimers with retinoic X receptors (RXRs). When a ligand binds, they then attach to peroxisome proliferator response elements (PPREs) in the promoters of target genes, depending on whether co-repressors or co-activators are present[54, 56, 57]. PPARs play a crucial role in lipid and glucose metabolism, as well as the regulation of energy balance, inflammation, and fibrosis (Fig. 3). Consequently, PPARs represent promising therapeutic targets for a more integrated and coordinated approach to NAFLD treatment[58-60].
The main roles and agonists of PPARs
| Isotypes | PPARα | PPARβ | PPARγ | ||
|---|---|---|---|---|---|
| Tissue expression | Liver Skeletal muscle Brown fat Cardiac tissue | Ubiquitously expressed | Adipose tissue | ||
| Natural ligands | FA Eicosanoids Phospholipids | FA VLDL components | FA Arachidonic acid metabolites | ||
| biolaogical functions related to NAFLD | FA catabolism Ketogenesis FGF21 production Anti-Inflammatory | FA catabolism Lipoprotein metabolism Anti-Inflammatory Glucose utilization | Adipogenesis Adipose FA storage Adipokine secretion Anti-Inflammatory | ||
| Main single agonists | Fenofibrate (NCT02781584) | Pemafibrate (NCT03350165) | Seladelpar | MSDC-0602K (NCT02784444) | Pioglitazone (NCT00063622) |
| Effect | Plasma triglycerides↓[210] | MRE-based liver stiffness↓ | Improves insulin sensitivity and steatohepatitis | Improves liver steatosis | Improve liver histology |
| Clinical status | Phase Ⅱ | Phase Ⅱ | Pause | Phase Ⅱ | Phase Ⅱ |
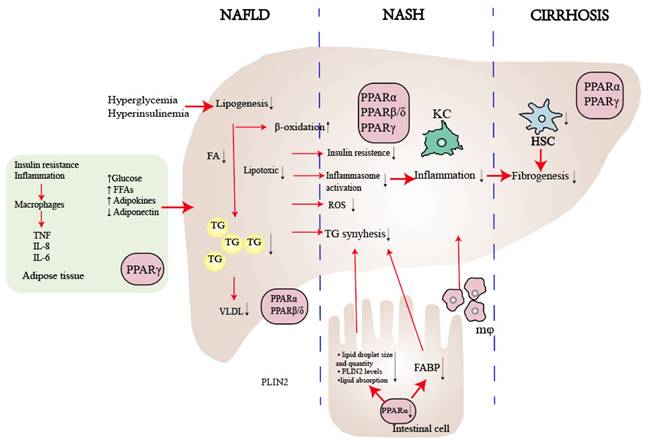
The role of PPARs in NAFLD. PPARα plays a crucial role in enhancing lipid metabolism by regulating the flow of lipids, controlling fatty acid transportation, and promoting β-oxidation. Moreover, it contributes to reducing inflammation by influencing liver cells, reducing visceral inflammation, and regulating intestinal permeability. On the other hand, PPARβ/δ suppresses the inflammatory phenotype in macrophages and facilitates the selective activation of a desired phenotype. As for PPARγ, it primarily regulates insulin sensitivity within adipose tissue and serves as the key regulator of hepatic stellate cell (HSC) fate. By preventing HSC activation, PPARγ plays a critical role in inhibiting fibrogenesis. Abbreviations: FFA: Free Fatty Acid; FA: Fatty Acid; TG: Triglyceride; ROS: Reactive Oxygen Species; KC: Kupffer Cell.
PPARα
PPARα, a nutrient sensor[61], plays a central role in metabolism and is expressed in tissues with high oxidative activity, such as the liver, skeletal muscle, brown fat, and cardiac tissue[62]. The fasting state stimulates the expression and activity of PPARα. In nocturnal rodents, the peak activity of liver PPARα occurs during the early night[63], and mice lacking PPARα can develop steatosis under during the fasting phase[64]. In NAFLD, hepatic PPARα expression is initially low but increases alongside histologic improvements following diet/exercise therapy[65]. A recent study found that eight weeks of aerobic training can reduce liver steatosis and inflammation by upregulating the AMP-activated protein kinase (AMPK)-PPARα pathway in the liver[66, 67]. In lipid metabolism, PPARα decreases liver fat synthesis through fatty acid transport, apolipoproteins production, genes expressed in β-oxidation [51, 68] or indirectly harmonizing via the LXR signaling pathway[68, 69]. In summary, the PPARα-mediated effect on lipid metabolism results in increased levels of serum high-density lipoprotein (HDL) and decreased levels of triglyceride-rich, lipoproteins and triglyceride accumulation in the liver[70]. In carbohydrate metabolism, PPARα regulates the expression of genes involved in gluconeogenesis, the process by which the liver produces glucose from non-carbohydrate sources[71]. PPARα also regulates the expression of genes involved glycogen breakdown and glucose release into the bloodstream [72]. Recently, gut-liver crosstalk has been recognized as playing a crucial role in regulating the progression of NAFLD[31, 73, 74]. A novel study demonstrates that the intestinal PPARα- Fatty acid binding protein 1(FABP1) axis is involved in controlling dietary fatty acid uptake, which in turn modulates obesity and NAFLD [75]. Another study has shown that deletion of intestinal PPARα leads to a reduction in both the size and quantity of lipid droplets, decreased fatty acid transport, and depletion of perilipin 2 (PLIN2), a critical regulator of lipid droplet formation in systemic lipid metabolism (Fig. 3)[76].
Current clinical trials targeting PPARα signaling for treatment
Both animal and in vivo models have shown the potential therapeutic benefits of PPAR agonists for NAFLD. Recent research has shown that fenofibrate, a PPARα agonist, reduces lipid levels in an mTOR-independent manner by activating autophagy and transcription factors E3 (TFE3) and EB (TFEB), which subsequently decreases hepatic fat accumulation[77]. Unfortunately, fenofibrate has minimal effect on insulin sensitivity or liver histology[78, 79]but exhibits better therapeutic advantages when combined with a PPARγ agonists rather than used alone[80]. Pemafibrate, a new and specific modulator of PPARα, has demonstrated advantageous effects on liver histology and liver enzymes in both preclinical NAFLD models and in individuals with diabetes and dyslipidemia[81]. In a double-blind, placebo-controlled, randomized multicenter phase Ⅱ trial, Pemafibrate did not decrease liver fat content but significantly reduced MRE-based liver stiffness[82]. Furthermore, it is important to note that species differences exist in cellular responses following PPARα activation between mice and humans[83]. This discrepancy could partially clarify why the effectiveness of isolated PPARα agonism shown in preclinical data has not been achieved in histological improvements in NAFLD patients[59].
PPARβ/δ
PPARβ/δ is also known to play a critical role in liver metabolism[57]. It is primarily expressed in hepatocytes, Kupffer cells, sinusoidal endothelial cells and hepatic stellate cells (HSCs) [84]. Nevertheless, it has been reported that the transcription and activation of PPARβ/δ are decreased in the livers of NAFLD patients in comparison to that of healthy individuals[85]. In lipid metabolism, activation of PPARβ/δ in the liver of mice can suppress the expression of sterol regulatory element binding protein-1c (SREBP-1c), which in turn reduces liver steatosis[86]. PPARβ/δ also inhibits hepatic steatosis and slows down NAFLD progression by regulating the very low-density lipoprotein receptor (VLDLR)[87]. Interestingly, the functions of PPARα and PPARβ/δ in the liver appear to be similar, implying that PPARβ/δ is the primary regulatory factor in liver intermediate metabolism[88]. However, PPARβ/δ cannot compensate for PPARα in PPARα deficient mice[64]. In addition, studies have shown that PPARβ/δ prevent fat toxicity by reducing levels of saturated fatty acids[89, 90]. Apart from its role in liver metabolism, PPARβ/δ also plays an important part in modulating inflammation. Ligands binding to PPARβ/δ are associated with the induction of anti-inflammatory signals and phenotypes in Kupffer cell[88], although the mechanism of its anti-inflammation role is not yet fully understood.
In conclusion, activation of PPARβ/δ results in decreased metabolic disturbances and insulin resistance in the liver, as well as the alternative activation of Kupffer cells with anti-inflammatory properties[91]. The selective PPARβ/δ agonist Seladelpar has shown improvements in insulin sensitivity and steatohepatitis in NASH patients[92]. However, CymaBay Therapeutics discontinued clinical trials of Seladelpar due to alarming results such as portal inflammation, along with plasma cells, interface hepatitis, and localized bile duct irregularities in initial end-of-treatment liver biopsies of NASH patients[93].
PPARγ
PPARγ performs specific functions in various tissues and cell type, with its primary role being in adipose tissue[94]. In individuals with NAFLD, the expression levels of PPARγ in the liver are significantly elevated[95]. PPARγ regulates a variety of target genes in adipocytes that are responsible for lipid uptake and storage, the production of inflammatory cytokines, and the secretion of adipokines that enhance insulin sensitivity[96]. In the liver, PPARγ stimulates the uptake of free fatty acids through FABP4-mediated fatty acid synthase (FASN) expression to increase triglyceride levels in hepatocytes. PPARγ also enhances the transcription of SREBP-1c, which in turn activates additional adipogenic genes and leads to the conversion of pyruvate into fatty acids[97, 98] In addition, PPARγ regulates various processes in hepatocytes, kupffer cells and HSCs, and the latter two play a pivotal role in the progression of hepatic inflammation, the development of fibrosis, and ultimately, patient outcome[99].
Interestingly, compared to traditional fatty liver models such as HFD feeding, chronic alcohol feeding, and leptin gene deletion, Gao et al. have developed a mouse model of acute steatohepatitis (HFD-plus-binge ethanol model) and successfully demonstrated hepatic PPARγ was found to induce steatosis through the upregulation of fat-specific protein 27 (Fsp27) and concurrently mitigate neutrophil infiltration by suppressing CXCL1, a chemokine involved in neutrophil recruitment[100].
Current clinical trials targeting PPARγ signaling for treatment
Approaches that reduce fat mass or shrink enlarged adipocytes (like weight loss) or improve insulin sensitivity in adipose tissue through medication (such as thiazolidinediones (TZDs)) are successful in treating NAFLD[101, 102]. This success is attributed to the reciprocal communication between the liver and adipose tissue, which adjusts to changes in energy requirements[59].A recently study found that GW9662, a PPARγ antagonist, attenuates NAFLD progression by reducing hepatic steatosis, inflammation, and oxidative stress[103]. In phase Ⅱb trials, pioglitazone notably improve liver histology liver histology features such as steatosis, inflammation, and ballooning, but not other histological features of NASH[7, 104, 105]. However, pioglitazone is particularly effective in patients with NASH and prediabetes or T2MD[101, 106]. The negative side effects associated with PPARγ activation, including weight gain, fluid retention, increased risk of cardiovascular incidents, and bone fractures, limit the widespread use of pioglitazone in treating NASH[107]. The recently created dual agonists for PPARα/γ, G4 and G5, reduce known side effects while improving systemic glucose metabolism, hyperlipidemia, and markers of liver injury in rats with insulin resistance induced by a high-fructose diet[108]. Saroglitazar, a different dual PPARα/γ agonist, improved steatosis, lobular inflammation, hepatocellular ballooning, and fibrosis levels in an animal model of NASH [109] and it was found to ameliorate ALT, liver fat content, insulin resistance, and atherogenic dyslipidemia in NASH patients, along with positive histological indications[110]. MSDC-0602K was intentionally engineered to diminish direct binding to PPARγ[111], yet it maintains its ability to inhibit the mitochondrial pyruvate carrier (MPC), which likely contributes to its beneficial effects on energy metabolism and glucose uptake [112]. Phase II clinical trials of the MSDC-0602K drug have shown promising results in obese individuals, including reduced glucose and insulin levels, as well as improved liver steatosis, without any adverse side effects[113, 114]. Preliminary data also indicates that MSDC-0602 might not possess the risk of bone loss associated with direct PPARγ agonists[115, 116]. Comparing with effects of the single or dual PPAR agonists, Lanifibranor, PPAR pan-agonist, improves all histological features of steatohepatitis in mice model of NASH, including liver fibrosis[60, 117, 118]. Likewise, in a phase Ⅱ clinical trial of Lanifibranor, there is evidence supporting its potential to provide benefits regarding numerous secondary endpoints, including hepatic fibrosis, lipid profile, and glycemic control[119].
LXR
LXR, the potential glucose sensor[120], comprises two isotypes, LXRα and LXRβ. It functions as nuclear receptors with crucial roles in lipid metabolism[69, 121, 122], regulating immunity[123], and exhibiting anti-inflammatory activity[124]. LXRα is predominantly found in metabolically active tissues and cells such as the liver, intestine, adipose tissue, and macrophages, while LXRβ is more universally expressed[125-127].
In hepatic metabolism, LXR serves a dual role. On the one hand is LXR directly increases SREBP-1c, FASN, stearoyl-coenzyme A desaturase 1 (SCD-1) and acetyl-CoA carboxylase (ACC), resulting in detrimental liver lipid deposition and hypertriglyceridemia[128]. An increase in LXR expression has been demonstrated to correlate with the worsening of NASH[127, 129]. Thus SR9238, an LXR inverse agonist, decreases the expression of genes encoding DNL enzymes, hepatic steatosis[130], and plasma liver enzymes in NAFLD mice model. It is worth noting that SR9238 treatment significantly suppressed hepatic inflammation and decreased hepatic fibrosis [127, 131]. On the other hand, LXRs regulate reverse cholesterol transport (RCT), which eliminates excess cholesterol through bile and feces after reaching the liver[126]. This process is facilitated by ATP Binding Cassette Subfamily A Member 1(ABCA1) and ATP binding cassette subfamily G member 1(ABCG1) in macrophages, both of which are directly targeted by LXR [132, 133]. Intriguingly, pharmacological activation of LXR increases cholesterol removal through feces, regulates cholesterol balance, produces anti-inflammatory effects, and improves insulin sensitivity by upregulating ABCG5/G8[69, 134]. However, the opposing pharmacodynamic effects of LXR in the treatment of NAFLD make it difficult to develop targeted drugs[127].
Current clinical trials targeting LXR signaling for treatment
A recent study has shown that inhibiting phosphorylation at Ser196 in LXRa can retard the progression of NAFLD in mice that are fed a high-fat and high-cholesterol (HFHC) diet[135]. Additionally, intranuclear MiR-552-3p has been found to suppress metabolic gene expression in vitro and exhibit positive impacts on glycolipid metabolism in vivo by modulating LXRα[136]. Although various selective LXR agonists, such as desmosterol, GW6340, and the LXRβ agonist LXR-623, have shown good tolerability, they are less commonly used for NAFLD treatment[137, 138]. Further data is necessary to assess the safety and effectiveness of LXR agonists in NASH therapy.
FXR
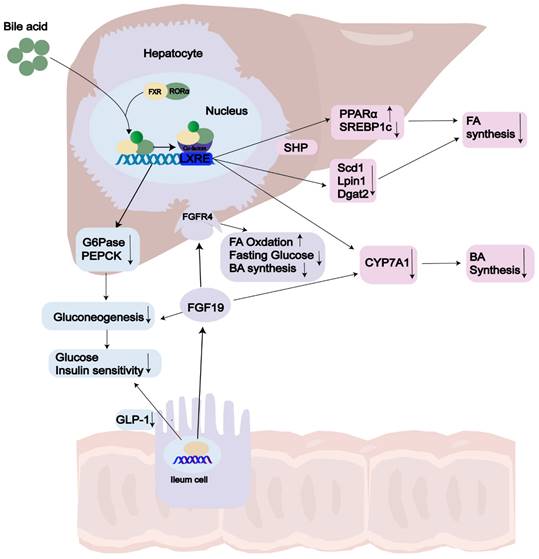
FXR, the primary controller of BA synthesis[139], is predominantly expressed in the liver and intestines, with lower levels found in the kidneys, adipose tissue, and adrenal glands[140, 141]. Chenodeoxycholic acid (CDCA) and cholic acid (CA) serve as endogenous ligands for FXR[142]. Recent studies have revealed that FXR acts as an enterohepatic regulator, controlling BA balance, lipid and glucose metabolism, and inflammation[143-146]. BA synthesis, which accounts for approximately 90% of daily cholesterol output in the body[147], has gained growing interest as a predictive indicator in NASH due to the increased BA levels observed in this condition[148, 149]. In a negative feedback loop controls, FXR is the pathway connecting the liver and intestine and regulates BA synthesis through two main pathway: the hepatic FXR-small heterodimer partner (SHP) pathway and the intestinal FXR- FGF15/19 pathway(FGF15 in mice[150]; FGF19 in human)[151, 152]. On the one hand, activation of hepatic FXR through BAs mediates induction of SHP expression, which belongs to the atypical nuclear receptor family and promotes inhibition of SREBP1c, thus leading to reduced hepatic synthesis of triglycerides [144]. FXR can limit the build-up of fat in the liver by enhancing fatty acid oxidation through the activation of hepatic PPARα and by clearing plasma VLDL triglycerides [151, 153, 154]. On the other hand, upon FXR activation in the ileum, human FGF19 is secreted into the bloodstream. It exerts its inhibitory action on BA synthesis and gluconeogenesis by activation two different mechanisms. Firstly, intestinal FXR agonists decrease intestinal lipid absorption by relying on BAs. Secondly, they selectively reduce the synthesis of monounsaturated fatty acids in the liver by inhibiting the expression of three key lipid synthesis genes: Scd1, Lpin1, and Dgat2, however, they do not affect Shp and Srebp-1c[155]. Mice lacking FXR exhibit notably elevated levels of serum and hepatic triglyceride, cholesterol, and free FA levels [156]. The removal of serum lipoproteins is contingent upon the function of FXR and is a FGF receptor 4 (FGFR4) located on the hepatocyte surface[157, 158], ultimately reducing liver steatosis and insulin resistance[159] (Fig. 2). However, the FGF19 agonists are not recommended due to their association with HCC[160]. Clifford et al. have shown that FXR activation reduces liver triglycerides through mediated through the regulation of gene expression of several key targets, including VLDLR, scavenger receptor B1, Syndecan-1, ApoCII/III and FA translocase (FAT/CD36)[161].
Current clinical trials targeting FXR signaling for treatment
FXR has become a prominent area of research for NAFLD treatment, with studies exploring the potential benefits of FXR agonists on NAFLD in both animal models and in vivo[148, 153, 162]. Obese mice treated with the gut-specific FXR agonist, Feraxamine, demonstrated improvements in obesity, insulin resistance, and steatosis[163]. Similarly, another FXR agonist, WAY-362450, reduced liver fibrogenesis and inflammation without triglyceride enrichment in methionine-choline deficient (MCD) diet-fed mice[164]. The non-BA FXR agonist, Tropifexor, has shown favorable outcomes in various preclinical NASH models[165, 166]. Furthermore, a phase Ⅱ clinical study has revealed that Cilofexor, a small molecule FXR agonist, has the potential to decrease fat accumulation and fibrosis in patients with NASH[167, 168]. Obeticholic acid (OCA) is a potent and specific FXR agonist[169, 170]. Key studies examining OCA include the FLINT trial[171]and the REGENERATE trial[172, 173]. OCA is the first FXR agonist to complete phase Ⅲ clinical trials in NASH patients, although the primary goal of NASH resolution was not achieved[170, 174].Consequently, the FDA has still not approved OCA for NASH, owing to consistently increased pruritus[173] and levels of serum LDL and decreased levels of serum HDL[175] (Table 2).
Current clinical trials for treatment of targeting to NR signaling
| Pharmacologic Compound | Drug Targrt | Effects | Clinical Trials |
|---|---|---|---|
| Saroglitazar[110, 211] | PPAR α/γ | Improved glucose/FFA metabolism TG↓, HDL-C↑, neural effect on LDL-C, ALT↓ | NCT03061721 PhaseⅡ |
| Elafibranor | PPAR α/σ | Improved glucose/FFA metabolism | NCT02704403 Phase Ⅲ, trial has been discontinued |
| Lanifibranor | Pan PPAR | Improved glucose/FFA metabolism TG↓, HDL-C↑, neural effect on LDL-C, ALT↓ | NCT03008070 PhaseⅡ, trial has positive results on histology |
| Cilofexor | FXR | Serum BA↓ Hepatic Steatosis ↓ | NCT02854605 PhaseⅡ |
| Obeticholic acid | FXR | Fibrosis↓ Hepatic inflammation↓ | NCT01265498 (FLINT, PhaseⅡ) NCT02548351 (REGENERATE, Phase Ⅲ) |
REV-ERB and HNF4α
REV-ERB exists two isotypes, REV-ERBα (NR1D1) and REV-ERBβ (NR1D2), and it regulates central and peripheral circadian clocks, lipid and glucose metabolism[176], and inflammation in the development of NAFLD[177, 178]. Treatment with a REV-ERB agonist improves hepatic health by reducing fat mass, improving dyslipidaemia and hyperglycaemia and suppressing hepatic fibrosis and inflammatory response in diet-induced obese mice[176, 178]. Central and peripheral circadian clocks as a crucial role in maintaining metabolic homeostasis in the development of liver diseases [179]. In liver circadian rhythms, hepatocytes are the peripheral clock that negatively impacts metabolism and overall health[180]. However, the function of REV-ERB can help restore the synchronization of liver clocks, which are frequently disturbed in conditions such as NAFLD, NASH, and metabolic syndrome[181]. In contrast to other nuclear receptors, REV-ERBs function as transcriptional repressors, attracting corepressors when their natural ligand, the iron-centered porphyrin heme, is present[182, 183]. REV-ERB is a recognized inflammatory regulator that directly regulates IL-1β, IL-6, TNFα, and the NOD-like receptor protein 3 (NLRP3) inflammasome[181]. NLRP3 activation occurs in NAFLD and increases liver inflammation and fibrosis in mouse NASH model[184, 185].
The role of FXR in NAFLD. Activation of FXR by bile acid (BA) ligands leads to a reduction in bile acid production and an enhancement of lipid and glucose metabolism. In terms of the bile acid pathway, this activation results in the upregulation of FGF19 and the downregulation of CYP7A1, consequently leading to a decrease in bile acid synthesis. Additionally, the activation of FXR increases the levels of SHP, which subsequently lowers the expression of SREBP1c, PEPCK, and G6Pase. As a result, there is an increase in both lipid and glucose metabolism. Abbreviations: GLP-1: Glucagon-Like Peptide 1; FGF19: Fibroblast Growth Factor 19; FGFR: Fibroblast Growth Factor Receptor; FA: Fatty Acid; BA: Bile Acid; LXRE: Liver X Receptor Response Element; SHP: Small Heterodimer Partner; CYP7A1: Cholesterol 7 Alpha-Hydroxylase.
HNF4α is found in large quantities in the liver[186] and has been observed to be notably reduced in both individuals with NAFLD and mouse models of NASH[187]. Multiple pieces of evidence indicate that HNF4α plays a role in the development of NAFLD[188]. Genetic studies have identified single nucleotide polymorphisms (SNPs) in the HNF4α gene associated with an increased risk of developing NAFLD[186]. Overexpression of hepatocyte HNF4α promotes lipolysis, fatty acid oxidation and VLDL secretion[189] to reduce hepatic triglyceride accumulation[190], whereas hepatocyte-specific HNF4α-/- mice has opposite effects[186, 191, 192]. In addition, hepatic HNF4a is markedly repressed in NASH and liver fibrosis[193, 194]. The crucial role of HNF4α in regulating hepatic lipid metabolism and maintaining liver homeostasis makes it a promising therapeutic target for NAFLD. HNF4α regulates liver fat storage by inducing lipophagy, a process that breaks down lipid droplets, and this effect can be reversed with a potent HNF4α agonist[195]. AAV8-mediated overexpression of HNF4a has been shown to attenuate HFD-induced NAFLD and NASH[190]. Sirtuin2, an NAD+-dependent deacetylase, largely alleviates insulin resistance, hepatic steatosis, and systematic inflammation in HFD-fed mice by binding to and deacetylating HNF4α[196]. However, further research is needed to fully understand the safety and efficacy of therapies targeting HNF4α in humans.
CAR and PXR
CAR, a member of the NR1I3 family of nuclear receptors, is almost exclusively expressed in the liver and primarily functions as a xenobiotic nuclear receptor[197]. In comparison to the classical NRS, CAR can directly or indirectly activate ligand binding through nuclear translocation from the cytoplasm[198]. In carbohydrate metabolism, CAR activation decreases glucose production in the liver by suppressing the expression of the crucial gluconeogenic genes PEPCK and G6P[199]. The activation of CAR leads to a reduction in SREBP-1c levels by promoting the expression of insulin-induced gene 1 protein (INSIG-1), which hinders the proteolytic activation of SREBPs [200]. Additionally, CAR activation increases the expression of the phase II enzyme SULT2B1b, which leads to a decrease in SREBP-1c expression. SULT2B1b also plays a role in sulfating and inactivating oxysterol agonists for LXR[199]. Peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) acts as a bridge connecting PPARs and CAR, as a transcriptional coactivator interacting with nuclear receptor PPARs. CAR regulates the degradation of PGC-1α by recruiting E3 ligase targeting PGC1α and promoting its ubiquitination in the liver[201].
PXR, a member of the NR1I2 family of nuclear receptors, is predominantly expressed in the liver[202]. Initially, PXR was defined as the main regulatory factor for exogenous reactions, similar to CAR, and its function was expanded to include lipid and glucose metabolism during the past years[203]. However, the role of PXR in NAFLD is debated, and both preclinical and clinical studies have yielded controversial results. Activation of PXR transcriptional activity has opposite effects on gluconeogenesis in rodents and humans[204]. Despite the presence of opposing evidence, the preponderance of the available data indicates that activation of PXR in mice subjected to a HFHC diet elicited the characteristic features of NAFLD and NASH, including steatosis, inflammation, and lipotoxicity. Karpale et. al. have proposed that the phenomenon of pseudo-improvement in glucose tolerance, where PXR activation resulted in aggravation of liver steatosis without being reflected in systemic glucose tolerance. The phenomenon is explained by remodulation of glucose metabolism [205].
In conclusion, further research is necessary to elucidate the potential roles of PXR and CAR in the development and progression of NAFLD/NASH, as well as to determine their temporal functions in the various stages of this complex disease.
Nuclear receptor crosstalk
Current strategies relying solely on a 'single-drug' or 'single-target' approach are inadequate in addressing the challenges posed by nuclear receptor ligands in clinical settings. Various nuclear receptors may exhibit shared sets of target genes, indicating overlapping regulatory functions[206]. An example of this is PPARα, through crosstalk with LXR, indirectly regulates the transcription of SREBP1c. Additionally, LXR and PPARα agonists exhibit synergy with insulin in inducing the expression of lipogenic genes like FAS and acetyl-CoA carboxylase 1 (ACC1)[207]. The crosstalk between PPAR and LXR is known to share a considerable number of target genes as they recognize similar response elements[208]. Alternatively, these receptors can also govern distinct genes associated with a common downstream biological process or pathway [206]. In the context of lipid metabolism, hepatic FXR directly downregulate three key lipogenic genes, SCD1, LPIN1 and DGAT2[155]. Whereas, PPARα regulates the expression of lipogenic genes mainly through the ChREBP, SREBP1c and LXR pathways[209]. Understanding the intricacies of nuclear receptor crosstalk is essential for developing targeted therapies that can manipulate these signaling pathways for therapeutic purposes.
Concluding remarks and future perspectives
NAFLD poses a significant health challenge, and researchers have identified several potential molecular targets for its treatment. These targets primarily involve four pathways: hepatic lipid metabolism, inflammation, intestinal flora, and anti-liver fibrosis. In recent years, significant progress has been made in developing metabolic NR ligands and understanding their role in liver physiological regulation. However, despite these advances, there are still challenges in discovering effective new strategies for treating NAFLD. Further research is needed to unravel the complex relationships between different NRs, their regulatory pathways, and their interactions with other metabolic factors during the progression of NAFLD. Exploring new drugs targeting NRs, such as selective PPAR modulators and FXR agonists, may improve treatment efficacy, minimize side effects, and overcome the limitations of current NAFLD treatment. In addition, the recognition of gut-liver crosstalk's significance in NAFLD progression emphasizes the need to study the role of NRs in gut-liver interactions and develop targeted interventions based on these findings. The combination of different NR-targeted drugs or their integration with lifestyle interventions can provide a more comprehensive and effective approach for managing NAFLD.
Abbreviations
NAFLD: non-alcoholic fatty liver disease; NRs: nuclear receptors; PPARs: peroxisome proliferator-activated receptors; LXRs: liver X receptors; FXRs: farnesoid X receptors; HNF4α: hepatocyte nuclear factor 4α; CAR: constitutive androstane receptor; PXR: Pregnane X receptor; MASLD: metabolic dysfunction-associated steatotic liver disease; HCC: hepatocellular carcinoma; NASH: non-alcoholic steatohepatitis; BA: bile acid; RXRs: retinoic X receptors; PPRE: peroxisome proliferator response elements; AMPK: AMP-activated protein kinase; HDL: high-density lipoprotein; FABP1: Fatty acid binding protein1; PLIN2: perilipin 2; TFE3: transcription factors E3; TFEB: transcription factors EB; HSCs: hepatic stellate cells; Fsp27: fat-specific protein 27; SREBP-1c: sterol regulatory element binding protein-1c; VLDLR: very low-density lipoprotein receptor; FASN: fatty acid synthase; TZDs: thiazolidinediones; MPC: mitochondrial pyruvate carrier; SCD-1: stearoyl-coenzyme A desaturase 1; ACC: acetyl-CoA carboxylase; RCT: reverse cholesterol transport; FGF: fibroblast growth factors; ABCA1: ATP Binding Cassette Subfamily A Member 1; ABCG1: ATP binding cassette subfamily G member 1; HFHC: high-fat and high-cholesterol; CDCA: Chenodeoxycholic acid; CA: cholic acid; SNP: small heterodimer partner; FGFR4: fibroblast growth factors receptor 4; MCD: methionine-choline deficient; OCA: Obeticholic acid; NLRP3: NOD-like receptor protein 3; SNPs: single nucleotide polymorphisms; PGC-1α: Peroxisome proliferator-activated receptor-gamma coactivator 1α; ACC1: acetyl-CoA carboxylase 1.
Acknowledgements
Funding
This work was supported by Chen Xiao-ping Foundation for the Development of Science and Technology of Hubei Province (No. CXPJJH122005-20, No. CXPJJH121001-2021004 and No. CXPJJH11900001-2019325) to Dr. Binhao Zhang.
Author contributions
ZY conducted literature survey and wrote the paper. XJ, BZ, AD, QL, CZ, CP, LX and JM provide insightful discussions and edits on this manuscript. BZ and CW conceptualized and edit the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Song SJ, Che-To Lai J, Lai-Hung Wong G, Wai-Sun Wong V, Cheuk-Fung Yip T. Can we use old NAFLD data under the new MASLD definition? Journal of hepatology. 2023
2. Powell EE, Wong VW-S, Rinella M. Non-alcoholic fatty liver disease. The Lancet. 2021;397:2212-24
3. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11-20
4. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE. et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. The Lancet Gastroenterology & Hepatology. 2022;7:851-61
5. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73-84
6. Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N. et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. The American journal of gastroenterology. 2018;113:1649-59
7. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic Steatohepatitis: A Review. Jama. 2020;323:1175-83
8. Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nature reviews Drug discovery. 2016;15:249-74
9. Boursier J, Tsochatzis EA. Case-finding strategies in non-alcoholic fatty liver disease. JHEP reports: innovation in hepatology. 2021;3:100219
10. Lazarus JV, Mark HE, Anstee QM, Arab JP, Batterham RL, Castera L. et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol. 2022;19:60-78
11. Ratziu V, Francque S, Sanyal A. Breakthroughs in therapies for NASH and remaining challenges. Journal of hepatology. 2022;76:1263-78
12. Ji L, Li Q, He Y, Zhang X, Zhou Z, Gao Y. et al. Therapeutic potential of traditional Chinese medicine for the treatment of NAFLD: A promising drug Potentilla discolor Bunge. Acta pharmaceutica Sinica B. 2022;12:3529-47
13. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet (London, England). 2021;397:2212-24
14. EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Obesity facts. 2016; 9: 65-90.
15. Li L, Liu DW, Yan HY, Wang ZY, Zhao SH, Wang B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obesity reviews: an official journal of the International Association for the Study of Obesity. 2016;17:510-9
16. Paik JM, Golabi P, Younossi Y, Mishra A, Younossi ZM. Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology. 2020;72:1605-16
17. Mantovani A, Byrne CD, Targher G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: a systematic review. The lancet Gastroenterology & hepatology. 2022;7:367-78
18. Xie J, Huang H, Liu Z, Li Y, Yu C, Xu L. et al. The associations between modifiable risk factors and nonalcoholic fatty liver disease: A comprehensive Mendelian randomization study. Hepatology. 2023;77:949-64
19. Ferguson D, Finck BN. Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nat Rev Endocrinol. 2021;17:484-95
20. Ko E, Yoon EL, Jun DW. Risk factors in nonalcoholic fatty liver disease. Clinical and molecular hepatology. 2023;29:S79-s85
21. Adams LA, Anstee QM, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66:1138-53
22. Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A. et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113-21
23. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology. 2020;158:1913-28
24. Ertle J, Dechêne A, Sowa JP, Penndorf V, Herzer K, Kaiser G. et al. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. International journal of cancer. 2011;128:2436-43
25. Manne V, Handa P, Kowdley KV. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clinics in liver disease. 2018;22:23-37
26. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686-90
27. Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat Rev Gastroenterol Hepatol. 2020;17:40-52
28. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10:627-36
29. Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S. et al. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology. 2010;139:1567-76 76.e1-6
30. Chen J, Vitetta L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. International journal of molecular sciences. 2020 21
31. Xu X, Poulsen KL, Wu L, Liu S, Miyata T, Song Q. et al. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal transduction and targeted therapy. 2022;7:287
32. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism: clinical and experimental. 2016;65:1038-48
33. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836-46
34. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537-64
35. Burris TP, Solt LA, Wang Y, Crumbley C, Banerjee S, Griffett K. et al. Nuclear receptors and their selective pharmacologic modulators. Pharmacological reviews. 2013;65:710-78
36. Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255-66
37. Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annual review of physiology. 2010;72:247-72
38. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K. et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835-9
39. Gustafsson JA. Historical overview of nuclear receptors. The Journal of steroid biochemistry and molecular biology. 2016;157:3-6
40. Luan ZL, Zhang C, Ming WH, Huang YZ, Guan YF, Zhang XY. Nuclear receptors in renal health and disease. EBioMedicine. 2022;76:103855
41. Hong T, Chen Y, Li X, Lu Y. The Role and Mechanism of Oxidative Stress and Nuclear Receptors in the Development of NAFLD. Oxidative medicine and cellular longevity. 2021;2021:6889533
42. D'Auria MV, Sepe V, Zampella A. Natural ligands for nuclear receptors: biology and potential therapeutic applications. Current topics in medicinal chemistry. 2012;12:637-69
43. Weikum ER, Liu X, Ortlund EA. The nuclear receptor superfamily: A structural perspective. Protein science: a publication of the Protein Society. 2018;27:1876-92
44. Trauner M, Fuchs CD. Novel therapeutic targets for cholestatic and fatty liver disease. Gut. 2022;71:194-209
45. Lopez-Velazquez JA, Carrillo-Cordova LD, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. Nuclear receptors in nonalcoholic Fatty liver disease. J Lipids. 2012;2012:139875
46. Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacology & therapeutics. 2017;179:142-57
47. Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG. et al. A comprehensive map of molecular drug targets. Nature reviews Drug discovery. 2017;16:19-34
48. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645-50
49. Venteclef N, Jakobsson T, Steffensen KR, Treuter E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends in endocrinology and metabolism: TEM. 2011;22:333-43
50. Dubois V, Eeckhoute J, Lefebvre P, Staels B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. The Journal of clinical investigation. 2017;127:1202-14
51. Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13:36-49
52. Wang YX. PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell research. 2010;20:124-37
53. Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. The Journal of clinical investigation. 2006;116:571-80
54. Wagner KD, Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacology & therapeutics. 2010;125:423-35
55. Anghel SI, Wahli W. Fat poetry: a kingdom for PPAR gamma. Cell research. 2007;17:486-511
56. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470-7
57. Wagner N, Wagner KD. The Role of PPARs in Disease. Cells. 2020 9
58. Poulsen L, Siersbæk M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Seminars in cell & developmental biology. 2012;23:631-9
59. Francque S, Szabo G, Abdelmalek MF, Byrne CD, Cusi K, Dufour JF. et al. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat Rev Gastroenterol Hepatol. 2021;18:24-39
60. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A. et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. Journal of hepatology. 2020;73:757-70
61. Patsouris D, Reddy JK, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology. 2006;147:1508-16
62. Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354-66
63. Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F. et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65:1202-14
64. Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. The Journal of clinical investigation. 1999;103:1489-98
65. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B. et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. Journal of hepatology. 2015;63:164-73
66. Diniz TA, de Lima Junior EA, Teixeira AA, Biondo LA, da Rocha LAF, Valadão IC. et al. Aerobic training improves NAFLD markers and insulin resistance through AMPK-PPAR-α signaling in obese mice. Life sciences. 2021;266:118868
67. Nikroo H, Hosseini SRA, Fathi M, Sardar MA, Khazaei M. The effect of aerobic, resistance, and combined training on PPAR-α, SIRT1 gene expression, and insulin resistance in high-fat diet-induced NAFLD male rats. Physiology & behavior. 2020;227:113149
68. Puengel T, Liu H, Guillot A, Heymann F, Tacke F, Peiseler M. Nuclear Receptors Linking Metabolism, Inflammation, and Fibrosis in Nonalcoholic Fatty Liver Disease. International journal of molecular sciences. 2022 23
69. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. 2018;14:452-63
70. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B. et al. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocrine reviews. 2018;39:760-802
71. Kersten S. Integrated physiology and systems biology of PPARα. Molecular metabolism. 2014;3:354-71
72. Samuel VT, Shulman GI. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018;27:22-41
73. Jayakumar S, Loomba R. Review article: emerging role of the gut microbiome in the progression of nonalcoholic fatty liver disease and potential therapeutic implications. Alimentary pharmacology & therapeutics. 2019;50:144-58
74. Zhang L, Guo GL. Gut-specific inhibition of PPARalpha as a novel approach of NAFLD treatment. Hepatology. 2022
75. Yan T, Luo Y, Yan N, Hamada K, Zhao N, Xia Y. et al. Intestinal peroxisome proliferator-activated receptor alpha-fatty acid-binding protein 1 axis modulates nonalcoholic steatohepatitis. Hepatology. 2022
76. Stojanović O, Altirriba J, Rigo D, Spiljar M, Evrard E, Roska B. et al. Dietary excess regulates absorption and surface of gut epithelium through intestinal PPARα. Nature communications. 2021;12:7031
77. Yoo J, Jeong IK, Ahn KJ, Chung HY, Hwang YC. Fenofibrate, a PPARα agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism: clinical and experimental. 2021;120:154798
78. Fabbrini E, Mohammed BS, Korenblat KM, Magkos F, McCrea J, Patterson BW. et al. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. The Journal of clinical endocrinology and metabolism. 2010;95:2727-35
79. Fernández-Miranda C, Pérez-Carreras M, Colina F, López-Alonso G, Vargas C, Solís-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2008;40:200-5
80. Mahmoudi A, Jamialahmadi T, Johnston TP, Sahebkar A. Impact of fenofibrate on NAFLD/NASH: A genetic perspective. Drug discovery today. 2022;27:2363-72
81. Ginsberg HN, Hounslow NJ, Senko Y, Suganami H, Bogdanski P, Ceska R. et al. Efficacy and Safety of K-877 (Pemafibrate), a Selective PPARα Modulator, in European Patients on Statin Therapy. Diabetes care. 2022;45:898-908
82. Nakajima A, Eguchi Y, Yoneda M, Imajo K, Tamaki N, Suganami H. et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Alimentary pharmacology & therapeutics. 2021;54:1263-77
83. Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annual review of biochemistry. 2006;75:295-332
84. Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochimica et biophysica acta. 2012;1821:809-18
85. Zarei M, Aguilar-Recarte D, Palomer X, Vázquez-Carrera M. Revealing the role of peroxisome proliferator-activated receptor β/δ in nonalcoholic fatty liver disease. Metabolism: clinical and experimental. 2021;114:154342
86. Flowers MT, Ntambi JM. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Current opinion in lipidology. 2008;19:248-56
87. Zarei M, Barroso E, Palomer X, Dai J, Rada P, Quesada-López T. et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Molecular metabolism. 2018;8:117-31
88. Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiological genomics. 2010;41:42-52
89. Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A. et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. Journal of gastroenterology and hepatology. 2009;24:830-40
90. Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM. et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. The Journal of biological chemistry. 2011;286:1237-47
91. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH. et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496-507
92. Haczeyni F, Wang H, Barn V, Mridha AR, Yeh MM, Haigh WG. et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatology communications. 2017;1:663-74
93. Dhingra S, Mahadik JD, Tarabishy Y, May SB, Vierling JM. Prevalence and clinical significance of portal inflammation, portal plasma cells, interface hepatitis and biliary injury in liver biopsies from patients with non-alcoholic steatohepatitis. Pathology. 2022;54:686-93
94. Chen H, Tan H, Wan J, Zeng Y, Wang J, Wang H. et al. PPAR-γ signaling in nonalcoholic fatty liver disease: Pathogenesis and therapeutic targets. Pharmacology & therapeutics. 2023: 108391.
95. Skat-Rørdam J, Højland Ipsen D, Lykkesfeldt J, Tveden-Nyborg P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic & clinical pharmacology & toxicology. 2019;124:528-37
96. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annual review of biochemistry. 2008;77:289-312
97. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Scientific reports. 2017;7:44612
98. Magliano DC, Bargut TC, de Carvalho SN, Aguila MB, Mandarim-de-Lacerda CA, Souza-Mello V. Peroxisome proliferator-activated receptors-alpha and gamma are targets to treat offspring from maternal diet-induced obesity in mice. PloS one. 2013;8:e64258
99. Morán-Salvador E, Titos E, Rius B, González-Périz A, García-Alonso V, López-Vicario C. et al. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. Journal of hepatology. 2013;59:1045-53
100. Wang W, Xu MJ, Cai Y, Zhou Z, Cao H, Mukhopadhyay P. et al. Inflammation is independent of steatosis in a murine model of steatohepatitis. Hepatology. 2017;66:108-23
101. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C. et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Annals of internal medicine. 2016;165:305-15
102. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM. et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England journal of medicine. 2010;362:1675-85
103. Baumann A, Burger K, Brandt A, Staltner R, Jung F, Rajcic D. et al. GW9662, a peroxisome proliferator-activated receptor gamma antagonist, attenuates the development of non-alcoholic fatty liver disease. Metabolism: clinical and experimental. 2022;133:155233
104. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J. et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. The New England journal of medicine. 2006;355:2297-307
105. Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G. et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445-53
106. Lian J, Fu J. Pioglitazone for NAFLD Patients With Prediabetes or Type 2 Diabetes Mellitus: A Meta-Analysis. Frontiers in endocrinology. 2021;12:615409
107. Gilardi F, Giudici M, Mitro N, Maschi O, Guerrini U, Rando G. et al. LT175 is a novel PPARα/γ ligand with potent insulin-sensitizing effects and reduced adipogenic properties. The Journal of biological chemistry. 2014;289:6908-20
108. Guru B, Tamrakar AK, Manjula SN, Prashantha Kumar BR. Novel dual PPARα/γ agonists protect against liver steatosis and improve insulin sensitivity while avoiding side effects. Eur J Pharmacol. 2022;935:175322
109. Kumar DP, Caffrey R, Marioneaux J, Santhekadur PK, Bhat M, Alonso C. et al. The PPAR α/γ Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Scientific reports. 2020;10:9330
110. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K. et al. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology. 2021;74:1809-24
111. Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N. et al. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor γ-sparing thiazolidinedione. The Journal of biological chemistry. 2012;287:23537-48
112. Ferguson D, Eichler SJ, Yiew NKH, Colca JR, Cho K, Patti GJ. et al. Mitochondrial pyruvate carrier inhibition initiates metabolic crosstalk to stimulate branched chain amino acid catabolism. Molecular metabolism. 2023;70:101694
113. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR. et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. Journal of hepatology. 2020;72:613-26
114. Davison BA, Harrison SA, Cotter G, Alkhouri N, Sanyal A, Edwards C. et al. Suboptimal reliability of liver biopsy evaluation has implications for randomized clinical trials. Journal of hepatology. 2020;73:1322-32
115. Kamm DR, Pyles KD, Sharpe MC, Healy LN, Colca JR, McCommis KS. Novel insulin sensitizer MSDC-0602K improves insulinemia and fatty liver disease in mice, alone and in combination with liraglutide. The Journal of biological chemistry. 2021;296:100807
116. Benova A, Ferencakova M, Bardova K, Funda J, Prochazka J, Spoutil F. et al. Novel thiazolidinedione analog reduces a negative impact on bone and mesenchymal stem cell properties in obese mice compared to classical thiazolidinediones. Molecular metabolism. 2022;65:101598
117. Wettstein G, Luccarini JM, Poekes L, Faye P, Kupkowski F, Adarbes V. et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatology communications. 2017;1:524-37
118. Boyer-Diaz Z, Aristu-Zabalza P, Andrés-Rozas M, Robert C, Ortega-Ribera M, Fernández-Iglesias A. et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. Journal of hepatology. 2021;74:1188-99
119. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ. et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. The New England journal of medicine. 2021;385:1547-58
120. Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V. et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219-23
121. Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L. et al. Role of LXRs in control of lipogenesis. Genes & development. 2000;14:2831-8
122. Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728-31
123. Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC. et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell. 2018;172:825-40.e18
124. Choi JY, Seo JY, Yoon YS, Lee YJ, Kim HS, Kang JL. Mer signaling increases the abundance of the transcription factor LXR to promote the resolution of acute sterile inflammation. Science signaling. 2015;8:ra21
125. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I. et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes & development. 2000;14:2819-30
126. Cariello M, Piccinin E, Moschetta A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cellular and molecular gastroenterology and hepatology. 2021;11:1519-39
127. Ni M, Zhang B, Zhao J, Feng Q, Peng J, Hu Y. et al. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2019;113:108778
128. Liu Y, Qiu DK, Ma X. Liver X receptors bridge hepatic lipid metabolism and inflammation. Journal of digestive diseases. 2012;13:69-74
129. Ahn SB, Jang K, Jun DW, Lee BH, Shin KJ. Expression of liver X receptor correlates with intrahepatic inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Digestive diseases and sciences. 2014;59:2975-82
130. Griffett K, Welch RD, Flaveny CA, Kolar GR, Neuschwander-Tetri BA, Burris TP. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Molecular metabolism. 2015;4:353-7
131. Griffett K, Burris TP. Development of LXR inverse agonists to treat MAFLD, NASH, and other metabolic diseases. Frontiers in medicine. 2023;10:1102469
132. Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. The Journal of biological chemistry. 2000;275:28240-5
133. Schulman IG. Liver X receptors link lipid metabolism and inflammation. FEBS letters. 2017;591:2978-91
134. Wang JQ, Li LL, Hu A, Deng G, Wei J, Li YF. et al. Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature. 2022
135. Becares N, Gage MC, Voisin M, Shrestha E, Martin-Gutierrez L, Liang N. et al. Impaired LXRα Phosphorylation Attenuates Progression of Fatty Liver Disease. Cell reports. 2019;26:984-95.e6
136. Fan L, Lai R, Ma N, Dong Y, Li Y, Wu Q. et al. miR-552-3p modulates transcriptional activities of FXR and LXR to ameliorate hepatic glycolipid metabolism disorder. Journal of hepatology. 2021;74:8-19
137. Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nature reviews Drug discovery. 2014;13:433-44
138. Tice CM, Noto PB, Fan KY, Zhuang L, Lala DS, Singh SB. The medicinal chemistry of liver X receptor (LXR) modulators. Journal of medicinal chemistry. 2014;57:7182-205
139. Perino A, Demagny H, Velazquez-Villegas L, Schoonjans K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiological reviews. 2021;101:683-731
140. Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T. et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687-93
141. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A. et al. Identification of a nuclear receptor for bile acids. Science (New York, NY). 1999;284:1362-5
142. Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta pharmacologica Sinica. 2015;36:44-50
143. Han CY. Update on FXR Biology: Promising Therapeutic Target? International journal of molecular sciences. 2018 19
144. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA. et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. The Journal of clinical investigation. 2004;113:1408-18
145. Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. The Journal of clinical investigation. 2006;116:1102-9
146. Claudel T, Staels B, Kuipers F. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2020-30
147. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annual review of biochemistry. 2003;72:137-74
148. Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology. 2017;65:350-62
149. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55-67
150. Gadaleta RM, Moschetta A. Metabolic Messengers: fibroblast growth factor 15/19. Nature metabolism. 2019;1:588-94
151. Zhou S, You H, Qiu S, Yu D, Bai Y, He J. et al. A new perspective on NAFLD: Focusing on the crosstalk between peroxisome proliferator-activated receptor alpha (PPARα) and farnesoid X receptor (FXR). Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2022;154:113577
152. de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657-69
153. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. Journal of hepatology. 2018;68:280-95
154. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Molecular endocrinology (Baltimore, Md). 2003;17:259-72
155. Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE. et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671-84.e4
156. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ. et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1006-11
157. Kumar RS, Brannigan JA, Prabhune AA, Pundle AV, Dodson GG, Dodson EJ. et al. Structural and functional analysis of a conjugated bile salt hydrolase from Bifidobacterium longum reveals an evolutionary relationship with penicillin V acylase. The Journal of biological chemistry. 2006;281:32516-25
158. Kliewer SA, Mangelsdorf DJ. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Digestive diseases (Basel, Switzerland). 2015;33:327-31
159. Alvarez-Sola G, Uriarte I, Latasa MU, Fernandez-Barrena MG, Urtasun R, Elizalde M. et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut. 2017;66:1818-28
160. Uriarte I, Latasa MU, Carotti S, Fernandez-Barrena MG, Garcia-Irigoyen O, Elizalde M. et al. Ileal FGF15 contributes to fibrosis-associated hepatocellular carcinoma development. International journal of cancer. 2015;136:2469-75
161. Fuchs CD, Traussnigg SA, Trauner M. Nuclear Receptor Modulation for the Treatment of Nonalcoholic Fatty Liver Disease. Seminars in liver disease. 2016;36:69-86
162. Sun L, Cai J, Gonzalez FJ. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol. 2021;18:335-47
163. Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D. et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nature medicine. 2015;21:159-65
164. Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. Journal of hepatology. 2009;51:380-8
165. Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB. et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). Journal of medicinal chemistry. 2017;60:9960-73
166. Hernandez ED, Zheng L, Kim Y, Fang B, Liu B, Valdez RA. et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatology communications. 2019;3:1085-97
167. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE. et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology. 2020;72:58-71
168. Alkhouri N, Herring R, Kabler H, Kayali Z, Hassanein T, Kohli A. et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. Journal of hepatology. 2022;77:607-18
169. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M. et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574-82.e1
170. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z. et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (London, England). 2019;394:2184-96
171. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF. et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet (London, England). 2015;385:956-65
172. Ratziu V, Sanyal AJ, Loomba R, Rinella M, Harrison S, Anstee QM. et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemporary clinical trials. 2019;84:105803
173. Younossi ZM, Stepanova M, Nader F, Loomba R, Anstee QM, Ratziu V. et al. Obeticholic Acid Impact on Quality of Life in Patients With Nonalcoholic Steatohepatitis: REGENERATE 18-Month Interim Analysis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2022;20:2050-8.e12
174. Rinella ME, Dufour JF, Anstee QM, Goodman Z, Younossi Z, Harrison SA. et al. Non-invasive evaluation of response to obeticholic acid in patients with NASH: Results from the REGENERATE study. Journal of hepatology. 2022;76:536-48
175. Kremoser C. FXR agonists for NASH: How are they different and what difference do they make? Journal of hepatology. 2021;75:12-5
176. Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T. et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62-8
177. Pourcet B, Zecchin M, Ferri L, Beauchamp J, Sitaula S, Billon C. et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology. 2018;154:1449-64.e20
178. Griffett K, Bedia-Diaz G, Elgendy B, Burris TP. REV-ERB agonism improves liver pathology in a mouse model of NASH. PloS one. 2020;15:e0236000
179. Crespo M, Leiva M, Sabio G. Circadian Clock and Liver Cancer. Cancers. 2021 13
180. Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT. et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature. 2012;485:123-7
181. Griffett K, Hayes ME, Boeckman MP, Burris TP. The role of REV-ERB in NASH. Acta pharmacologica Sinica. 2022;43:1133-40
182. Kim YH, Marhon SA, Zhang Y, Steger DJ, Won KJ, Lazar MA. Rev-erbα dynamically modulates chromatin looping to control circadian gene transcription. Science (New York, NY). 2018;359:1274-7
183. Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB. et al. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nature structural & molecular biology. 2007;14:1207-13
184. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM. et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. Journal of hepatology. 2017;66:1037-46
185. Duez H, Pourcet B. Nuclear Receptors in the Control of the NLRP3 Inflammasome Pathway. Frontiers in endocrinology. 2021;12:630536
186. Pan X, Zhang Y. Hepatocyte nuclear factor 4α in the pathogenesis of non-alcoholic fatty liver disease. Chinese medical journal. 2022;135:1172-81
187. Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nature communications. 2015;6:7466
188. Gunewardena S, Huck I, Walesky C, Robarts D, Weinman S, Apte U. Progressive loss of hepatocyte nuclear factor 4 alpha activity in chronic liver diseases in humans. Hepatology. 2022;76:372-86
189. Yu D, Chen G, Pan M, Zhang J, He W, Liu Y. et al. High fat diet-induced oxidative stress blocks hepatocyte nuclear factor 4α and leads to hepatic steatosis in mice. Journal of cellular physiology. 2018;233:4770-82
190. Xu Y, Zhu Y, Hu S, Xu Y, Stroup D, Pan X. et al. Hepatocyte Nuclear Factor 4α Prevents the Steatosis-to-NASH Progression by Regulating p53 and Bile Acid Signaling (in mice). Hepatology. 2021;73:2251-65
191. Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Molecular and cellular biology. 2001;21:1393-403
192. Thymiakou E, Tzardi M, Kardassis D. Impaired hepatic glucose metabolism and liver-α-cell axis in mice with liver-specific ablation of the Hepatocyte Nuclear Factor 4α (Hnf4a) gene. Metabolism: clinical and experimental. 2023;139:155371
193. Xu Y, Hu S, Jadhav K, Zhu Y, Pan X, Bawa FC. et al. Hepatocytic Activating Transcription Factor 3 Protects Against Steatohepatitis via Hepatocyte Nuclear Factor 4α. Diabetes. 2021;70:2506-17
194. Yang T, Poenisch M, Khanal R, Hu Q, Dai Z, Li R. et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. Journal of hepatology. 2021;75:1420-33
195. Lee SH, Veeriah V, Levine F. Liver fat storage is controlled by HNF4alpha through induction of lipophagy and is reversed by a potent HNF4alpha agonist. Cell death & disease. 2021;12:603
196. Ren H, Hu F, Wang D, Kang X, Feng X, Zhang L. et al. Sirtuin 2 Prevents Liver Steatosis and Metabolic Disorders by Deacetylation of Hepatocyte Nuclear Factor 4α. Hepatology. 2021;74:723-40
197. Bae SDW, Nguyen R, Qiao L, George J. Role of the constitutive androstane receptor (CAR) in human liver cancer. Biochimica et biophysica acta Reviews on cancer. 2021;1875:188516
198. Yang H, Wang H. Signaling control of the constitutive androstane receptor (CAR). Protein & cell. 2014;5:113-23
199. Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, Wakil SJ. et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18831-6
200. Roth A, Looser R, Kaufmann M, Blättler SM, Rencurel F, Huang W. et al. Regulatory cross-talk between drug metabolism and lipid homeostasis: constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Molecular pharmacology. 2008;73:1282-9
201. Gao J, Yan J, Xu M, Ren S, Xie W. CAR Suppresses Hepatic Gluconeogenesis by Facilitating the Ubiquitination and Degradation of PGC1α. Molecular endocrinology (Baltimore, Md). 2015;29:1558-70
202. Xing Y, Yan J, Niu Y. PXR: a center of transcriptional regulation in cancer. Acta pharmaceutica Sinica B. 2020;10:197-206
203. Hakkola J, Rysä J, Hukkanen J. Regulation of hepatic energy metabolism by the nuclear receptor PXR. Biochimica et biophysica acta. 2016;1859:1072-82
204. Mackowiak B, Hodge J, Stern S, Wang H. The Roles of Xenobiotic Receptors: Beyond Chemical Disposition. Drug metabolism and disposition: the biological fate of chemicals. 2018;46:1361-71
205. Karpale M, Kummu O, Kärkkäinen O, Lehtonen M, Näpänkangas J, Herfurth UM. et al. Pregnane X receptor activation remodels glucose metabolism to promote NAFLD development in obese mice. Molecular metabolism. 2023: 101779.
206. De Bosscher K, Desmet SJ, Clarisse D, Estébanez-Perpiña E, Brunsveld L. Nuclear receptor crosstalk - defining the mechanisms for therapeutic innovation. Nat Rev Endocrinol. 2020;16:363-77
207. Fernández-Alvarez A, Alvarez MS, Gonzalez R, Cucarella C, Muntané J, Casado M. Human SREBP1c expression in liver is directly regulated by peroxisome proliferator-activated receptor alpha (PPARalpha). The Journal of biological chemistry. 2011;286:21466-77
208. Liu Y, Izem L, Morton RE. Identification of a hormone response element that mediates suppression of APOF by LXR and PPARα agonists. Biochimica et biophysica acta Molecular and cell biology of lipids. 2020;1865:158583
209. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nature reviews Molecular cell biology. 2015;16:678-89
210. Lawitz EJ, Bhandari BR, Ruane PJ, Kohli A, Harting E, Ding D. et al. Fenofibrate Mitigates Hypertriglyceridemia in Nonalcoholic Steatohepatitis Patients Treated With Cilofexor/Firsocostat. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2023;21:143-52.e3
211. Siddiqui MS, Parmar D, Sheikh F, Sarin SK, Cisneros L, Gawrieh S. et al. Saroglitazar, a Dual PPAR α/γ Agonist, Improves Atherogenic Dyslipidemia in Patients With Non-Cirrhotic Nonalcoholic Fatty Liver Disease: A Pooled Analysis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2023
Author contact
![]() Corresponding authors: Binhao Zhang, bhzhang8com (ORCID ID: 0000-0003-1983-3507); Bixiang Zhang, bixiangzhangcom; Chao Wang, chaowangtjmu.edu.cn.
Corresponding authors: Binhao Zhang, bhzhang8com (ORCID ID: 0000-0003-1983-3507); Bixiang Zhang, bixiangzhangcom; Chao Wang, chaowangtjmu.edu.cn.