Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. Astrocyte and epilepsy
2. Astrocytes and AD
3. Astrocytes and POCD
4. Astrocytes and SAE
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(7):3122-3143. doi:10.7150/ijbs.109315 This issue Cite
Review
Astrocytes Lingering at a Crossroads: Neuroprotection and Neurodegeneration in Neurocognitive Dysfunction
Xiaoxia Xu1,2†, Bin Mei1†, Yue Yang3†, Junxiong Li4†, Juntao Weng1, Yueyue Yang1, Qianyun Zhu1, Honghai Zhang2,3 ![]() , Xuesheng Liu1
, Xuesheng Liu1 ![]()
1. Department of Anesthesiology, the First Affiliated Hospital of Anhui Medical University, Key Laboratory of Anesthesiology and Perioperative Medicine of Anhui Higher Education Institutes, Anhui Medical University, Hefei, Anhui Province, 230022, China.
2. Department of Anesthesiology, Affiliated Hangzhou First People's Hospital, Westlake University School of Medicine, Hangzhou, 310006, China.
3. Department of Anesthesiology, the Fourth Clinical School of Medicine, Zhejiang Chinese Medical University, Hangzhou, 310006, China.
4. Department of Orthodontics, Affiliated Stomatological Hospital of Kunming Medical University, Yunnan Key Laboratory of Stomatology, Kunming Medical University, Kunming Yunnan 650106, China.
†Equal contribution.
Received 2024-12-24; Accepted 2025-4-10; Published 2025-4-28
Abstract

Astrocytes, a major class of glial cells in the central nervous system, play a fundamental role in maintaining homeostasis, supporting neuronal function, and regulating synaptic activity. Recent studies have increasingly highlighted the pivotal role of astrocytes in the initiation and progression of neurocognitive dysfunction. Alterations in astrocytic morphology and functionality have been strongly associated with the onset of cognitive impairments, positioning astrocytes as key regulators in neurocognitive processes. Astrocytes influence neurocognitive function through their involvement in the uptake and release of gliotransmitters, modulation of inflammatory mediators, and metabolic regulation. These processes have been implicated in various neurodegenerative and neurocognitive disorders, including epilepsy, Alzheimer's disease, postoperative cognitive dysfunction, and sepsis-associated encephalopathy. Given the emerging role of astrocytes in these conditions, understanding the mechanisms by which they modulate neurocognitive function is essential for identifying potential therapeutic targets. This review provides an overview of the current understanding of astrocytic contributions to neurocognitive dysfunction and explores the therapeutic opportunities provided by the targeting of astrocyte-mediated pathways.
Keywords: astrocyte, neurocognitive dysfunction, epilepsy, neuroinflammation, Alzheimer's disease, postoperative cognitive dysfunction, sepsis-associated encephalopathy
Introduction
Due to the escalating global aging phenomenon, the number of patients with neurocognitive dysfunction caused by neurodegenerative diseases has increased dramatically[1-3]. Alzheimer's disease (AD), for example, is one of the leading causes of dementia worldwide and affects millions of people[4-6]. In addition, neurocognitive dysfunction stemming from conditions like epilepsy and sepsis has a serious impact on patients' quality of life and generates a socio-economic burden[7-9]. Postoperative cognitive dysfunction (POCD), which affects many elderly patients following surgery, is associated with prolonged hospitalization, long-term cognitive decline, and mortality[10-12]. Presently, the optimization of medical treatments to benefit such patients has become an important focus in both basic and clinical research. Astrocytes play a key role in multiple critical physiological functions such as brain homeostasis and memory[13-15]. This review thus summarizes current research regarding the roles of astrocytes in neurocognitive dysfunction associated with epilepsy, AD, POCD, and sepsis-associated encephalopathy (SAE) as well as the underlying mechanisms implicated therein and their potential as therapeutic targets.
1. Astrocyte and epilepsy
1.1 Mechanisms of epilepsy-induced neurocognitive dysfunction
1.1.1 Amyloid and tau protein
Amyloid-β (Aβ) accumulation and tau hyperphosphorylation are central to epilepsy-associated neurocognitive dysfunction[16-19]. Seizures drive Aβ deposition and tau phosphorylation, potentially via stress pathways like the c-Jun N-terminal kinase (JNK), mammalian target of rapamycin (mTOR)/p70S6K, and protein kinase R -like endoplasmic reticulum kinase/eukaryotic initiation factor 2α pathways, which are activated during epileptogenesis[16, 20]. For example, childhood seizures correlate with midlife Aβ accumulation, suggesting long-term neurodegenerative consequences[21, 22]. Critically, Aβ and tau synergistically exacerbate cognitive decline: high levels of both biomarkers predict severe memory impairment, as shown in the 11C-Pittsburgh Compound B and 18F-Flortaucipir maps[23, 24]. Mechanistically, Aβ hyperexcites hippocampal neurons, lowering seizure thresholds, while tau potentiates network hypersynchrony and hippocampal atrophy[25-27]. Tau pathology also recruits cytotoxic T cells, driving neuronal loss and synaptic dysfunction[28-30]. Moreover, Aβ and tau protein deposition in the brain may disrupt both anatomical and functional connectivity, leading to reduced attention, language impairment, and executive dysfunction[31, 32]. Therapeutic strategies targeting both proteins (e.g., tau ablation) restore excitation-inhibition balance, reduce seizure severity, and improve outcomes, highlighting their intertwined roles in epileptogenic cognitive decline[33-35].
1.1.2 Manganese-containing superoxide dismutase (MnSOD)
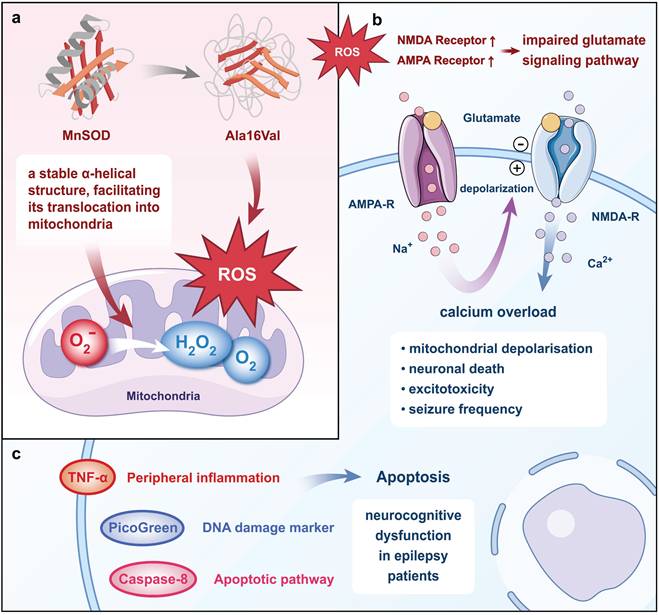
MnSOD, a nuclear-encoded antioxidant metalloenzyme, primarily functions to convert O2ˉ to H2O2 and O2[36]. In pathological conditions, a single nucleotide polymorphism in the MnSOD gene results in a change from Ala to Val at the 16th amino acid position of its primary structure, termed Ala16Val. This single nucleotide polymorphism is one of the most extensively studied polymorphisms in the MnSOD gene. Molecular dynamics simulation results revealed that wild-type MnSOD maintains a stable α-helical secondary structure, facilitating its translocation into mitochondria for the production of MnSOD, whereas the valine polymorphism rapidly disrupts this α-helical structure, leading to an increase in mitochondrial reactive oxygen[37]. Studies using electrophysiology and redox proteomics in cultured human pluripotent stem cell-derived cortical neurons revealed that the overproduction of mitochondrial reactive oxygen species (ROS) upregulates the expression of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and n-methyl-d-aspartate (NMDA) receptors containing glutamate receptor subunit 1 and NMDA receptor subunit 2B subunits, leading to impaired glutamate signaling, calcium overload, and excitotoxicity[38]. Inhibition of ROS reduces Ca2+ oscillation, mitochondrial depolarization, neuronal death, and seizure frequency in rats[39].
Moreover, evidence from other studies suggested that the Ala16Val polymorphism is implicated in various processes including caspase-3-induced activation of apoptotic pathways, elevated levels of DNA damage markers (PicoGreen), and increased peripheral inflammation in epilepsy patients, all of which may contribute to neurocognitive dysfunction[40]. While the specific cell types undergoing apoptosis were not directly identified in this study, caspase-3 activation is commonly associated with neuronal and glial cell apoptosis in epilepsy[41, 42]. Neuronal apoptosis, in particular, is a known consequence of excitotoxicity and chronic inflammation, hallmarks of epilepsy pathophysiology[43, 44]. Additionally, glial cells, such as astrocytes and microglia, may also undergo apoptosis due to sustained inflammatory responses and oxidative stress, further contributing to cognitive dysfunction[45, 46]. The genotypes of the MnSOD Ala16Val single nucleotide polymorphism were classified as Ala/Ala, Ala/Val, and Val/Val, with the Val/Val genotype associated with higher levels of tumor necrosis factor-α (TNF-α), caspase-8, and PicoGreen[47] (Fig. 1). Consequently, epileptic patients with the Val/Val genotype exhibit more severe inflammatory and apoptotic status[48]. Consistent with these findings, the Val/Val genotype has been closely linked to a variety of neurocognitive dysfunctions, including practice, perception, attention, language, executive function, long-term semantic memory, short-term visual memory, and total memory[47, 49]. Hence, the MnSOD Ala16Val polymorphism, particularly the Val/Val genotype, may play a role in the pathogenesis of epileptogenic neurocognitive dysfunction.
MnSOD Ala16Val polymorphism causes neurocognitive dysfunction in epilepsy. The Ala16Val polymorphism disrupts the α-helix structure and prevents the translocation of MnSOD precursor proteins to the mitochondria to exert antioxidant effects, leading to an increase in mitochondrial ROS. The overproduction of ROS increases the expression of the AMPA and NMDA receptors, causing calcium overload, mitochondrial depolarization, neuronal death, and an increase in seizure frequency. The Ala16Val polymorphism also induces neuronal apoptosis, DNA damage, and peripheral inflammation, ultimately leading to neurocognitive dysfunction in epilepsy.
1.1.3 Aquaporin 4 (AQP4)
AQP4 is predominantly expressed in the astrocytic endfeet around cerebral blood vessels and regulates ion and water homeostasis in the central nervous system (CNS)[50]. As a key component of the glymphatic system, AQP4 supports the brain-wide clearance of interstitial solutes, including Aβ and other neurotoxic substances[51, 52]. In epilepsy, AQP4 modulates the dynamics of water and ion balance in the brain, which can influence neuronal excitability and seizure susceptibility[53, 54]. The highly polarized expression of AQP4 water channels on astroglial endfeet is essential for efficient glymphatic function. Polarity refers to the distribution of AQP4 immunoreactivity, with higher polarity indicating a greater concentration of AQP4 in perivascular regions and lower polarity reflecting a more even distribution between perivascular endfeet and the soma[55]. It was found that the transient upregulation of AQP4 expression and depolarization following epilepsy-induced brain edema in mice may disrupt the function of the glial lymphoid system, resulting in significantly impaired performance in the Morris water maze test, which was ameliorated upon downregulation of AQP4 in Trpm4-/- mice[53]. However, deficiency in AQP4 has been associated with an increase in the frequency and duration of spontaneous seizures, potentially compromising memory consolidation[54, 56]. Additionally, AQP4 deficiency also impairs the compensatory upregulation of glutamate transporter-1, leading to glutamate accumulation and excitatory neurotoxicity[57]. In vivo rat studies have revealed that excess glutamate can induce elevated electroencephalogram (EEG) spikes, intense epileptic discharges, epileptiform behavioral changes, and reduced neuronal counts, ultimately contributing to neurocognitive dysfunction[58-60]. However, blocking AQP4 in an in vitro model of 4-aminopyridine-induced epilepsy did not abolish rapid volumetric pulsations and sustained contraction of the extracellular space[61]. These findings collectively suggest that seizures may precipitate epilepsy-related memory impairment, with AQP4 potentially playing a significant role.
1.2 Role of astrocytes in epileptogenic neurocognitive dysfunction
Astrocytes dynamically interact with neurons by releasing signaling molecules such as ATP, glutamate, and other gliotransmitters that regulate synaptic plasticity, neuronal excitability, and network stability. Under pathological conditions, reactive astrocytes exhibit aberrant signaling, including excessive ATP release or impaired gliotransmitter uptake, which disrupts synaptic homeostasis and promotes hyperexcitability[62, 63]. For example, ATP released by astrocytes can activate neuronal purinergic receptors, enhance excitatory neurotransmission, and lower the seizure threshold[64, 65]. Conversely, glutamate uptake by astrocytes through glutamate transporter 1 (GLT-1) is essential for preventing excitotoxicity and reducing seizure susceptibility[66]. Dysregulation of these processes can lead to epileptogenesis and cognitive deficits, highlighting the importance of astrocyte-neuron crosstalk in health and disease[65, 67].
Another critical function of astrocytes is their regulation of extracellular ion concentrations, particularly K⁺ and Ca²⁺. By dynamically buffering K⁺ through the inward-rectifying potassium channel Kir4.1 and spatial redistribution, astrocytes prevent extracellular K⁺ accumulation, which can depolarize neurons and lower seizure thresholds[68]. Additionally, overexpression of Kir4.1 in astrocytes attenuates presynaptic Ca²⁺ entry and release probability at excitatory synapses[69]. Similarly, astrocytes modulate extracellular Ca²⁺ levels via transporters or channels (e.g., gamma-aminobutyric acid transporter 3), maintaining ionic balance critical for synaptic transmission[70]. Impaired Kir4.1 function or dysregulated Ca²⁺ signaling disrupts ion homeostasis, lowering seizure thresholds and propagating hyperexcitability[68, 69].
1.2.1 Astrocytes and amyloid and tau proteins
Astrocytes play a pivotal role in regulating the metabolism of amyloid and tau proteins. In epilepsy models, astrogliosis in the prefrontal cortex and hippocampus correlates with Aβ or tau accumulation[71, 72]. Activated astrocytes facilitate tau propagation, as evidenced by plasma glial fibrillary acidic protein (GFAP) serving as a biomarker for Aβ positivity and a predictor of cognitive decline[73, 74]. GFAP has emerged as a more accurate indicator of Aβ positivity compared with other glial markers, and it has been shown to predict longitudinal deposition in whole samples, regardless of the participants' cognitive status or the presence of AD or other neurodegenerative disorders[73]. Mechanistically, the transcription factor signal transducer and activator of the transcription 3 (STAT3) pathway drives astrocyte proliferation, and its conditional knockout reduces Aβ plaques and improves spatial learning and memory decline[72]. Paradoxically, inhibiting the Janus kinase 2 (JAK2)/STAT3 pathway in reactive astrocytes does not alter tau or Aβ pathology but modulates anxiety-related behaviors, indicating pathway-specific effects[75]. This duality indicates that astrocyte activation may engage distinct molecular mechanisms—some that promote Aβ clearance (e.g., via STAT3 suppression) and others that exacerbate neuroinflammation. The unresolved interplay between astrocytic pathways and protein aggregation underscores the need for targeted therapies that selectively modulate astrocyte functions, balancing neuroprotection and inflammation mitigation to address Aβ- or tau-driven cognitive decline.
1.2.2 Astrocytes and MnSOD
Astrocytes are implicated in MnSOD-mediated neurocognitive dysfunction. Zimmer et al. demonstrated that hippocampal astrocytes in patients with intractable temporal lobe epilepsy combined with hippocampal sclerosis exhibit pronounced upregulation of peroxiredoxin 6, suggesting that astrocytes play a predominant role in antioxidant gene expression in the hippocampus of epilepsy patients[76]. Critically, MnSOD deficiency in astrocytes exacerbates mitochondrial ROS accumulation, which directly contributes to epileptogenesis through multiple pathways. In vitro experiments demonstrated that exposure of human fetal astrocytes to graphene oxide induces a pro-inflammatory phenotype characterized by elevated expression of C3, exacerbating epileptic inflammatory processes and lowering seizure thresholds[76]. Downregulation of MnSOD enhances ROS production in primary astrocytes, leading to heightened cell death, whereas restoration of MnSOD levels protects astrocytes from these impairments and even reverses aggravated astrocyte damage[77]. MnSOD downregulation not only leads to elevated ROS levels but may also impair astrocytic ATP production, further destabilizing neuron-astrocyte metabolic coupling[78, 79]. Astrocyte injury is associated with elevated levels of S100 calcium-binding protein B and GFAP, along with decreased brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor. The levels of serum markers of astrocyte injury are correlated with insomnia severity and/or neurocognitive dysfunction[80]. Thus, MnSOD may mitigate astrocyte damage, thereby ameliorating neurocognitive dysfunction.
1.2.3 Astrocytes and AQP4
Astrocytes play an important role in AQP4-mediated neurocognitive dysfunction. AQP4 expression in astrocytes exhibits a selective distribution, with a notably higher density in the endfeet membrane than in the non-endfeet membrane. Disruption of AQP4 polarization impairs astrocyte-mediated K⁺ buffering and water transport[81], and this impaired water transport potentially causes water uptake and swelling by astrocytes[82]. Additionally, the mislocalization of AQP4 disrupts the spatial buffering of K+, leading to increased extracellular K+ levels[83]. Research has shown that AQP4 depolarization-induced dysregulation of water and K+ homeostasis may contribute to neurocognitive dysfunction and epileptic propensities[81]. For example, AQP4-knockout mice exhibit impaired synaptic plasticity (e.g., deficits in long-term potentiation and depression) and worsened seizure outcomes due to reduced astrocyte scar formation and dysfunctional glutamate clearance[81, 84, 85]. Astrocytes regulate glutamate via GLT-1, a glutamate transporter, and GLT-1 deficiency in zebrafish models elevates extracellular glutamate, lowering seizure thresholds[86]. Conversely, astrocytic GLT-1 achieved overexpression via the AAV-Gfa2-GLT-1-cHA vector reduces seizure duration and severity in epileptic mice by restoring glutamate homeostasis[87].
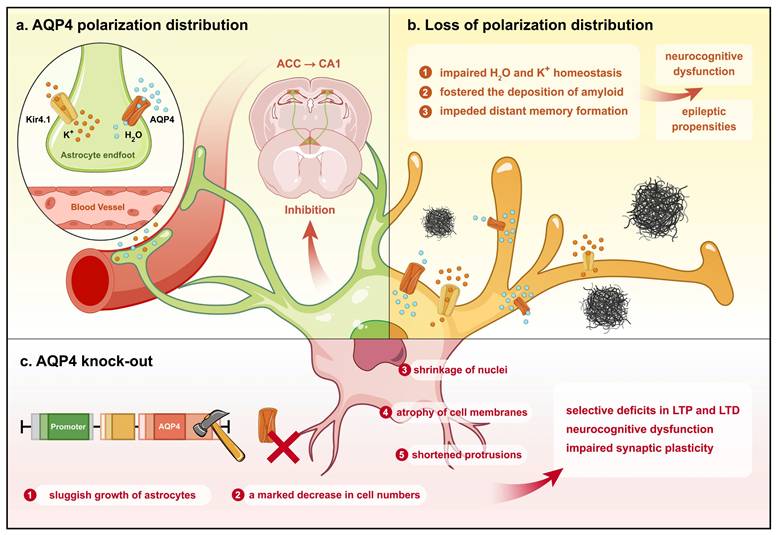
In vitro experiments further demonstrated that AQP4 gene silencing leads to impaired astrocyte growth with reduced cell numbers, diminished nuclei, shrunken cytosol, and shortened protrusions[88]. During memory acquisition, the anterior cingulate cortex (ACC) projects to the hippocampal CA1 region, prompting massive neuronal recruitment accompanied by activation of ACC neurons. This process is essential for distant memory formation and is inhibited by astrocytes[15]. AQP4 deficiency, then, may impede distant memory formation by impairing astrocytic function (Fig. 2). Additionally, in mice with intrahippocampal kainic acid-induced epilepsy, the neuregulin (NRG)/ErbB4 signaling pathway is upregulated in astrocytes, along with reductions in amino acid carrier 1 protein and NeuN-positive neurons, but exogenous NRG-1 treatment increases NRG/ErbB4 activity, upregulating glutamine synthetase and amino acid carrier 1 to restore glutamate homeostasis[89].
Different conditions in which AQP4 affects neurocognition by regulating astrocyte function. Under physiological circumstances, AQP4 exhibits a polarized distribution on astrocytes, thereby preserving physiological water and K+ homeostasis and restraining ACC projections to CA1 in the hippocampus for memory formation. Conversely, the depolarized distribution of AQP4 in astrocytes impairs water and K+ homeostasis and fosters the deposition of amyloid, inducing neurocognitive dysfunction. Knockout of AQP4 culminates in slowed astrocyte growth, markedly decreased cell numbers, shrinkage of nuclei, atrophy of cell membranes, and shortened protrusions, thereby impairing synaptic plasticity and neurocognitive function.
Astrocytic pH and Ca²⁺ dynamics also modulate neuronal activity. During seizure intensification, a transient alkaline shift in astrocytes is followed by an abrupt intracellular acidic shift, which potentially stimulates gliotransmitter release and exacerbates epilepsy[90]. Given that all cellular enzymatic reactions are sensitive to Ca2+ and pH changes, modulation of pH and/or Ca2+ levels in astrocytes emerged as a potential but challenging therapeutic target for the treatment of epilepsy or the prevention of adverse plasticity associated with epileptogenesis[90]. While these intracellular changes could influence a wide range of cellular enzymes and processes, targeted approaches that selectively regulate astrocytic pH and Ca²⁺ dynamics, without broadly affecting other cell types or systemic processes, may offer a pathway to mitigate epileptogenesis with reduced off-target effects.
1.3 Summary
A bidirectional feed-forward relationship exists between epilepsy and amyloid and tau protein levels, wherein epilepsy triggers the accumulation of amyloid and tau proteins, and conversely, amyloid and tau proteins exacerbate seizure activity, thereby contributing to neurocognitive dysfunction. The presence of amyloid and tau proteins may indeed exacerbate neurocognitive dysfunction, potentially facilitated by astrocytic hyperplasia. Astrocytes serve as crucial intermediaries in this interplay, and amyloid deposition triggers astrocyte activation, characterized by reactive astrogliosis—a process involving morphological changes, increased expression of GFAP, and the release of pro-inflammatory cytokines. This activation of astrocytes contributes to subsequent tau protein accumulation. However, no significant impact of the astrocyte-activating JAK2/STAT pathway on amyloid and tau deposition has been observed.
Moreover, the MnSOD Ala16Val polymorphism is linked to oxidative stress, peripheral inflammation, apoptosis, and DNA damage in patients with epilepsy. These associations may explain its potential correlation with various neurocognitive dysfunctions, particularly evident in individuals with the ValVal genotype. Given that MnSOD itself exerts a protective effect on astrocytes, targeting the 16th amino acid shift could potentially serve as a strategy for mitigating epilepsy-induced neurocognitive dysfunction. While inherited mutations pose challenges for direct correction, therapeutic approaches such as gene editing technologies (e.g., CRISPR-Cas9) or pharmacological modulation of downstream pathways affected by the mutation could be explored as strategies to mitigate the functional consequences of the amino acid shift. Further studies are needed to develop clinically feasible strategies for targeting this specific mutation.
Additionally, research has demonstrated that seizures impede memory consolidation and deficiency in AQP4 increases the frequency and duration of spontaneous seizures while elevating glutamate levels, triggering CNS neuroexcitotoxicity[91]. AQP4 plays a pivotal role in regulating synaptic plasticity, astrocytic growth and function, glutamate clearance, and water and K+ homeostasis, as well as in influencing amyloid deposition. Either knockout or silencing of AQP4 has been associated with varying degrees of neurocognitive dysfunction. Hence, targeting AQP4 may represent a viable approach for treating neurocognitive dysfunction arising from epilepsy. Therefore, astrocytes play an important role in epilepsy, and interventions aimed at ameliorating neurocognitive dysfunction through astrocyte modulation could yield favorable outcomes in epilepsy treatment.
2. Astrocytes and AD
2.1 Mechanisms of neurocognitive dysfunction in AD
2.1.1 Amyloid
AD is a neurodegenerative disease associated with aging. The predominant pathological features of AD include amyloid plaques and neurofibrillary tangles in the brain[92, 93]. These histopathological features impair neuronal circuit function and alter the behavioral response by affecting the activation of neurotransmitter receptors[94, 95]. In primary neural cultures, exposure to Aβ was shown to result in a 33% decrease in cell survival and a 38% reduction in neurite extensions among surviving cortical neurons[73]. Furthermore, studies have demonstrated that elevated levels of Aβ, amyloid precursor protein (APP), or APP C-terminal fragments increase the expression of adenosine A2A receptors in the hippocampal and neocortical astrocytes of aging mice. This A2A receptor up-regulation may contribute to memory impairment in patients with AD[96].
Then how does Aβ contribute to neurocognitive dysfunction? A study comparing levels of protein kinase C alpha (PKCα) protein in the frontal cortex of post-mortem human brains from individuals with AD and age-matched controls via immunoblotting revealed upregulation of PKCα in AD brains[97]. To investigate whether enhanced PKCα signaling output was associated with AD, the researchers studied mice harboring the PKCα M489V mutation, which is associated with increased constitutive PKCα activity. In these mice, the mutation was found to alter the brain phosphoproteome, reduce the spine density in hippocampal neurons, enhance Aβ-induced synaptic inhibition, and ultimately impair spatial learning memory in the Barnes maze test without affecting Aβ levels in the brain. These findings suggest that Aβ may inhibit synapses by enhancing PKCα activity, leading to cognitive decline[97]. Triggering receptor expressed on myeloid cells 2 (TREM2), a receptor on microglia, plays a critical role in regulating microglial migration and phagocytosis of oligomeric Aβ and amyloid plaques[98-100]. In a study using the 5XFAD mouse model of AD, TREM2 was activated by a TREM2 agonistic antibody tetra-variable domain immuno-globulin (Ab18 TVD-Ig), which increased TREM2 activation by 100-fold compared to its bivalent form. This enhanced activation significantly reduced amyloid burden, increased microglial migration toward and phagocytosis of amyloid plaques, and decreased the presence of lysosome-associated membrane protein 1, a marker of dystrophic neurites surrounding plaques[101]. Additionally, the treatment enhanced the expression of synaptophysin (a presynaptic marker) and postsynaptic markers in the hippocampus and cortex, indicating improved synaptic function. Behavioral tests revealed that the treatment reduced anxiety-like behaviors in the open arms of the elevated plus maze test and improved hippocampal-dependent contextual memory during the fear conditioning test, indicating enhanced cognitive function[101].
Are reductions in Aβ content protective against neurocognitive dysfunction? A study using a mouse model that altered GPR3 (an orphan G protein-coupled receptor implicated in AD) to be G protein-biased (engineered to favor G protein signaling over β-arrestin pathways by mutation of phosphorylation sites in its C-terminus) demonstrated that this change led to reduced endogenous Aβ40 and Aβ42 production and decreased amyloid plaque area[102]. Reducing amyloid deposition has been shown to reverse memory deficits and anxiety symptoms, thereby improving cognitive performance in mice[103, 104]. However, reducing the accumulation of amyloidogenic APP C-terminal fragments, soluble Aβ, Aβ oligomers, and amyloid deposits was not found to ameliorate neural network dysfunction and behavioral abnormalities[105]. Thus, amyloid deposition is implicated in a myriad of neurocognitive deficits, and its removal may ameliorate only some aspects of AD neuropathology.
2.1.2 Tau protein
The accumulation of tau protein tangles represents a pivotal component of AD-related neurocognitive dysfunction[106]. Levels of plasma biomarkers, including GFAP, t-tau, p-tau181, and p-tau231 are elevated in patients with preclinical AD, with GFAP and p-tau181 increasing over time. These observations emphasize the diagnostic and longitudinal monitoring potential of plasma GFAP and p-tau isoforms in preclinical AD[107]. From comparisons of various plasma biomarkers in early-stage AD cohorts, longitudinal increases in p-tau217 are specifically linked to lower Mini-Mental State Examination scores and brain atrophy, and in a delayed recall memory test, only the slope of p-tau217 is significantly associated with cognitive decline[108]. Symptomatic younger AD patients show stronger tau positron emission tomography signals in the frontoparietal hub, a region critical for AD-related cognition, which is highly connected to other brain regions. These independently verified findings suggest that symptomatic episodes in younger AD patients correlate with heightened tau pathology in the brain hub, accelerating tau propagation across interconnected brain regions and cognitive decline[109]. Plasma levels of p-tau217 increase in early preclinical AD, preceding tau-positron emission tomography positivity[110].
In the brains of AD patients, pathological tau protein accumulation leads to neuronal loss, synaptic dysfunction, and subsequent neurocognitive dysfunction[92, 111]. BIN1, a susceptibility gene for AD, has multiple isoforms, with 10 isoforms identified as being expressed in the CNS. The peptide encoded by exon 7 of the BIN1 N-BAR structural domain exhibits a significant inverse association with AD-related traits, particularly tau protein tangle accumulation, and reduced expression of the BIN1 isoform containing exon 7 correlates with increased tau protein tangle deposition and subsequent neurocognitive dysfunction[112]. Aβ facilitates the accumulation of pathogenic tau, while TREM2 can slow AD progression and reduce tau-driven neurodegeneration by limiting this process[113]. Genome-wide miRNA screening in Drosophila identified inhibitors of tau-mediated AD pathology, pinpointing the miR-9 family as potent inhibitors of human tau overexpression, and overexpression of CG11070, a target gene of Drosophila miR-9a, or its mammalian homolog UBE4B, increased tau ubiquitination and degradation, leading to reduced total and phosphorylated tau levels, thereby suggesting a potential innovative therapeutic strategy for AD[114]. Thus, the tau protein tangles play an important role in AD-induced neurocognitive dysfunction.
2.1.3 P2Y1 purinoreceptor (P2Y1R)
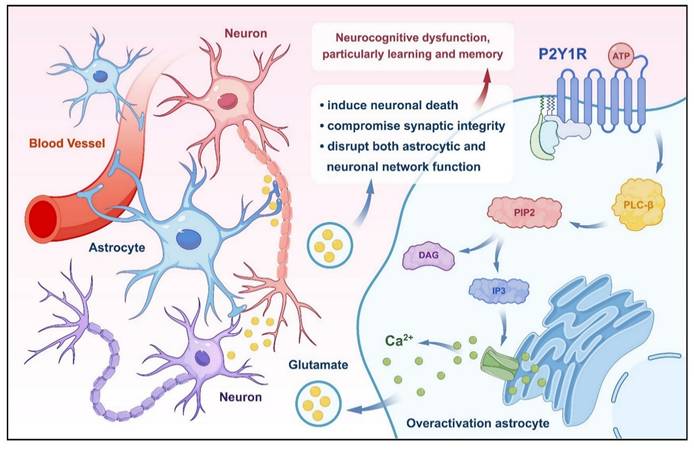
The P2YR purinoceptors, including P2Y1R, are metabotropic ATP receptors. In the context of AD, P2Y1R plays a significant role in neurocognitive dysfunction. Research indicates that excessive activation of astrocytes is an important factor in neuronal-glial network dysfunction in AD, with P2Y1R-mediated signaling pathways implicated in driving astrocyte overactivation[115]. Specifically, selective activation of P2Y1R in cultured astrocytes has been shown to induce massive glutamate release, a phenomenon associated with neuronal death in the rat hippocampus[116, 117]. Activation of P2Y1R in the medial prefrontal cortex disrupts the inhibitory control and behavioral flexibility mediated by increased mesocorticolimbic activity and local disinhibition, suggesting a role for P2Y1R in modulating cognitive-behavioral responses in the prefrontal cortex[118]. Furthermore, extracellular ATP regulates neuronal damage and memory impairment primarily through activation of P2Y1R, and antagonizing P2Y1R protects against memory deficits[119]. Restoration of inhibitory homeostasis, a process crucial for resilience to Aβ accumulation, structural preservation, and synaptic hyperinhibition, can be achieved by blocking P2Y1R in calreticulin interneurons, which is selectively dysfunctional during AD pathogenesis[120] (Fig. 3). Hence, antagonizing P2Y1R presents a potential therapeutic approach for ameliorating the neurocognitive dysfunction caused by AD.
P2Y1R improves neurocognitive function by regulating glutamate release from astrocytes. Activation of P2Y1R by ATP leads to overactivation of astrocytes, triggering the release of glutamate. Excess glutamate levels induce neuronal death, compromise synaptic integrity, and disrupt both astrocytic and neuronal network function. These pathological processes collectively contribute to neurocognitive dysfunction, particularly impairing learning and memory.
2.2 Role of astrocytes in AD-induced neurocognitive dysfunction
2.2.1 Astrocytes and amyloid
In the context of AD, a reciprocal regulatory relationship exists between astrocytes and Aβ. Neuroinflammation, driven by reactive astrocytes and microglia, facilitates the development of neurodegenerative diseases and cognitive decline[121-123]. Astrocytes transition from a homeostatic state to a reactive state in response to Aβ aggregates, releasing pro-inflammatory cytokines, chemokines, and synaptotoxic secretions[124, 125]. This inflammatory environment perpetuates neuronal damage, disrupted synaptic plasticity, and accelerated Aβ aggregation, forming a vicious cycle that drives disease progression[126]. For example, Aβ plaques activate astrocytes via Toll-like receptor 4, triggering nuclear factor- kappa B (NF-κB)-dependent transcription of pro-inflammatory mediators[127]. These reactive astrocytes upregulate Na+/H+ exchanger isoform 1, enhancing Aβ accumulation[128].
Despite this detrimental feedback loop, astrocytes also exhibit protective roles in modulating Aβ pathology[129]. Astrocytes differentiate into two distinct C3-positive and C3-negative reactive populations, in which soluble adenylyl cyclase and compartmented, nuclear- and cytoplasmic-localized cyclic adenosine monophosphate in reactive astrocytes act as a molecular switch for neuroprotective astrocyte reactivity[130]. In neurodegenerative diseases like AD, an imbalance between neurotoxic and neuroprotective astrocytes has been observed in animal models and human patients. For example, in a mouse model of neurodegeneration, neuroprotective astrocytes initially emerge in an intermediate state but then shift to a neurotoxic phenotype under sustained neuroinflammation, and targeting mTOR in astrocytes can alleviate astrocyte neurotoxicity[131].
Alterations in amyloid pathology are accompanied by the activation and hypertrophy of microglia and astrocytes, which may limit Aβ progression under specific conditions. While the role of glial activation in AD is complex and context-dependent, one study found that the observed glial response appears to be protective, as these glial changes could limit the progression of amyloid pathology in G protein-biased GPR3 AD mice[102]. Mechanistically, astrocytes are recruited to Aβ plaques and enhance autophagosome generation, reducing soluble and insoluble Aβ levels in hippocampal and cortical tissues[96]. This process may involve signaling pathways such as the phosphatidylinositide 3-kinase/protein kinase B/actin, Sirtuin 1/AMP-activated protein kinase/sterol response element binding protein 2, and chemerin/chemokine-like receptor 1/stimulator and deactivation of ephrin receptor A4/c-Abl pathways[132, 133]. Furthermore, astrocytes regulate Aβ clearance through cholesterol homeostasis: maintaining low neuronal cholesterol levels inhibits Aβ accumulation, while astrocytic dysfunction impairs Aβ clearance via NF-κB inflammatory signaling[124, 134].
Astrocytes also secrete clusterin, a synaptogenic factor with anti-amyloid properties, which enhances synaptic transmission and reduces Aβ levels[135, 136]. Conversely, Aβ itself can subvert these protective mechanisms. For instance, Aβ downregulates phagocytic receptors (e.g., Mertk, Megf10) in astrocytes, directly impairing their ability to clear oligomeric Aβ and encapsulate dystrophic synapses near plaques[137]. This bidirectional interaction underscores the duality of astrocyte function: while they possess intrinsic capacities to mitigate Aβ pathology (e.g., via autophagy, clusterin secretion), chronic Aβ exposure disrupts these mechanisms, exacerbating neuroinflammation and neurodegeneration.
2.2.2 Astrocytes and apolipoprotein E (ApoE)
Astrocytes regulate tau protein aggregation and phosphorylation through the influence of ApoE4, the mutation of which is currently the strongest genetic risk factor associated with late-onset AD. Notably, tau protein accumulation has been observed in astrocytes within the dentate gyrus of AD patients. In mice, overexpression of 3R tau protein (contains three microtubule-binding repeats in the carboxy-terminal) in dentate gyrus astrocytes alters mitochondrial dynamic and function, resulting in impaired spatial memory capacity[138]. ApoEε4, one of the high-frequency alleles of ApoE4 and a major genetic risk factor for AD, enhanced tau protein aggregation and phosphorylation in neurons when expressed in astrocytes[139]. In vivo and in vitro investigations have demonstrated that phosphatidylinositol proteoglycan 4, secreted by astrocytes, strongly interacts with APOE4 and exacerbates APOE4-induced diffusion and hyperphosphorylation of tau[140]. Utilizing brain organoid models derived from induced pluripotent stem cells, a study revealed that homozygous conversion of ApoE4 to ApoE3 attenuates the ApoE4-associated phenotype in brain organoid cells from AD patients[72]. Removal of astrocyte ApoE4 alleviates tau-induced synaptic deficits and reduces phagocytosis of synaptic components by microglia[141]. Hippocampal synaptic dysfunction and dendritic deficits associated with microglial activation may underlie memory deficits[142]. These findings collectively indicate that ApoE4 may drive memory deficits in AD through its effects on astrocytes. APOE4 disrupts the neuronal microenvironment by impairing the cholesterol metabolism balance in astrocytes, activating inflammatory signaling pathways, and causing dysregulation of extracellular matrix production. These disruptions lead to synaptic dysfunction and reduce the efficiency of neural networks, ultimately contributing to AD-related memory deficits[143]. Specifically, APOE4-induced cholesterol dysregulation in astrocytes inhibits the myelination of oligodendrocytes, further exacerbating neuronal dysfunction and impairing memory formation[144]. Mechanistically, astrocytes regulate tau accumulation through the histone deacetylase 7/transcriptional factor EB lysosomal and NF-κB/chemokine (C-C motif ligand 2, C-X-C motif ligand 1, and interleukin [IL]-6) signaling pathways[145, 146]. Thus, APOE4 exerts its detrimental effects on memory by causing a combination of cholesterol metabolism disruption, inflammation, and synaptic dysfunction, highlighting the central role of astrocytes in AD pathogenesis.
2.2.3 Astrocytes and P2Y1R
P2Y1R exerts significant effects on astrocytes, particularly in the context of AD. Research has shown that long-term intracerebroventricular infusion of P2Y1R inhibitors, monitored through the vivo two-photon microscopy, preserves normal astrocytic and neuronal network function, bolsters synaptic structure integrity, and safeguards hippocampal long-term potentiation (LTP). In the early stages of AD, P2Y1R mediates a prolonged duration of the response to ATP-induced [Ca2+]i[147]. Notably, inhibition of astrocytic Ca2+-dependent overexcitatory purinergic signaling, primarily mediated by P2Y1R, rescues theta burst-induced LTP, suggesting the critical role of spontaneous astrocytic Ca2+ signaling in maintaining CA3-CA1 synaptic plasticity[148]. Long-term administration of P2Y1R antagonists in AD mice, or the genetic deletion of astrocyte-specific P2Y1R downstream signaling pathways, prevents the decline in spatial learning and memory in these mouse models[115]. Therefore, activation of P2Y1R in astrocytes may be an important mechanism underlying the neurocognitive dysfunction in AD.
2.3 Summary
In summary, Aβ deposition leads to impaired neuronal circuit function and altered behavioral response. Clearance of amyloid improves memory deficits, anxiety symptoms, and cognitive performance, although it does not address network dysfunction and behavioral abnormalities. Astrocytes are able to clear Aβ via autophagy, but Aβ itself impairs the ability of astrocytes to clear Aβ by downregulating the expression of astrocyte phagocytic receptors. Augmentation of clusterin levels in astrocytes has shown promise for reducing Aβ levels and ameliorating the associated neurocognitive dysfunction.
Accumulation of tau protein also causes cognitive and memory impairments. Astrocytic expression of ApoE4 exacerbates tau protein accumulation, and removal of astrocytic ApoE4 appears to mitigate synaptic loss and phagocytosis, potentially attenuating memory deficits. Activation of P2Y1R not only triggers glutamate release, leading to hippocampal neuronal death, but also disrupts the inhibitory control and behavioral flexibility mediated via increased mesocortical limbic activity and local disinhibition, ultimately leading to neuronal damage and memory impairment. Conversely, inhibition of P2Y1R normalizes astrocytic and neuronal network function, enhances synaptic structure integrity, and preserves hippocampal LTP. In conclusion, astrocytes play an important role in AD, and interventions targeting astrocytic function hold promise for improving AD-related neurocognitive dysfunction, thus bearing important implications for clinical treatment strategies.
3. Astrocytes and POCD
3.1 Mechanism of POCD
3.1.1 Connexin-43 (Cx43)
Cx43 expression may be involved in the progression of POCD. Sevoflurane, a volatile anaesthetic, increases the expression of Cx43 and induces neuronal apoptosis by activating the JNK signaling pathway in the rat hippocampus on postnatal day 7, which can lead to neurocognitive dysfunction during puberty[149]. Cx43 is the predominant gap junction (GJ) protein in the nervous system, serving vital physiological functions including regulation of ion transport, regulation of substrate exchange, and facilitation of intercellular signaling[150, 151]. A study investigating long-term isoflurane-induced perioperative neurocognitive dysfunction showed that prolonged exposure to isoflurane results in the uncoupling of Cx43-based GJs without altering the total expression of Cx43, which increases oxidative stress and neuroinflammation in the hippocampus and primary astrocytes, leading to neurocognitive dysfunction, as evidenced by the results of Y-maze and fear conditioning tests, while increased Cx43-based GJ formation ameliorates these effects[152]. Notably, the Cx43 protein can form both GJ channels and hemichannels[153]. After long-term isoflurane anesthesia, enhancement of the total Cx43 hemichannels increases astrocyte permeability, neuronal excitability, and neuronal toxicity[154]. Research has indicated that internal fixation of tibial fractures induces the opening of astrocytic Cx43 hemichannels, triggering an inflammatory response in the hippocampus and resulting in neurocognitive dysfunction in aged mice[155]. These findings suggest that Cx43 contributes to POCD not only in surgical procedures (e.g., orthopedic surgery) but also in models of anesthesia induction (e.g., prolonged exposure to isoflurane or sevoflurane). The dual role of Cx43 (through gastrojunctional uncoupling and hemichannel activation) highlights its widespread involvement in neuroinflammation and synaptic disruption in response to various perioperative stressors, further supporting its potential as a mechanistic target in the pathogenesis of POCD.
3.1.2 MicroRNA-146a (miR-146a)
MiR-146a is a small non-coding RNA that plays a key role in the negative regulation of the innate immune response. MiR-146a dysregulation is associated with a variety of inflammatory diseases[156, 157]. MiR-146a may negatively regulate surgery-induced hippocampal neuroinflammation and ameliorate cognitive decline through inhibition of the IL receptor associated kinase 1 (IRAK1)/tumor necrosis factor receptor associated factor 6/NF-κB pathway. Thus, overexpression of miR-146a attenuates early postoperative neurocognitive dysfunction by suppressing surgery-induced hippocampal neuroinflammation in mice. Downregulation of miR-146a may exacerbate hippocampal-dependent learning memory deficits and hippocampal inflammation in POCD mice[158]. A study on the neuroinflammatory response mediated by Escherichia coli infection suggested that inhibition of Escherichia coli-induced miR-146a in vivo may further promote the production of inflammatory cytokines, exacerbate astrocyte and microglial reactivity, and shorten the survival time of mice via the toll-like receptor/NF-κB or epidermal growth factor receptor/NF-κB signaling pathways[159]. A reduction in lysosomal activity (Lyso Tracker and BECN1 gene) and enhanced expression of synaptic/axonal transport-related genes (DLG4, KIF5B, and DYNC1H1) in motor neurons occurs after co-culture with astrocytes with altered miR-146a expression[160]. In addition, research showed that expression of B-cell translocation gene 2 (BTG2), a tumor suppressor gene[161], is increased in a mouse model of POCD, and inhibition of BTG2 reduces neuronal apoptosis, necrosis, and brain damage, thereby attenuating neurocognitive dysfunction. MiR-146a negatively regulates the expression of BTG2[162], suggesting that increasing the level of miR-146a could potentially prevent and treat POCD by inhibiting BTG2.
3.1.3 Abnormal activation of N-methyl-D-aspartate receptor (NMDAR)
Aberrant activation of NMDAR may be involved in the onset and development of POCD. The BDNF/tropomyosin-related kinase receptor B (TrkB) signaling pathway is considered an important mechanism in POCD. BDNF is known to activate the high-affinity TrkB, including the full-length (TrkB-FL) and truncated (TrkB-T) isoforms[163]. Anesthesia- and surgery-induced neuroinflammation excessively activates NMDAR, which in turn triggers excessive activation of calpain, causing TrkB-FL truncation, dysregulation of the BDNF/TrkB signaling pathway, loss of dendritic spines, and apoptosis, resulting in neurocognitive dysfunction in aged mice. Notably, treatment with the NMDAR antagonist memantine or the calpain inhibitor MDL-28170 can attenuate these abnormalities, suggesting that inhibition of abnormal activation of NMDAR or TrkB-FL truncation may be a strategy for treating POCD[11].
Memantine is commonly used in the treatment of AD. Experiments in POCD mice have shown a potential protective effect of memantine on memory, depression-like behavior, and social novelty preferences[164]. Isoflurane, another volatile anesthetic, may induce reversible neurocognitive dysfunction, and sustained upregulation of the extra-synaptic NMDAR 2B subunit after isoflurane anesthesia may suppress synaptic transmission and thus contribute to neurocognitive dysfunction[165]. Therefore, upregulation of the extra-synaptic NMDAR 2B subunit could be another potential mechanism of POCD induction by NMDAR overactivation.
3.2 Role of astrocytes in POCD
3.2.1 Astrocytes and Cx43
Cx43 plays an important regulatory role in astrocytes. Astrocytes form astrocyte networks through connections of GJs, which are mainly composed of Cx43, forming astrocyte networks[166-168]. In long-term isoflurane anesthesia-induced POCD, a significant reduction in Cx43 formation occurs in the hippocampus and primary mouse astrocytes along with impaired function of the astrocyte network and elevated levels of IL-1β and IL-6 in the hippocampus and astrocytes, and the mice exhibit severe neurocognitive dysfunction[169]. Deletion of Cx30 and Cx43 impairs astrocyte GJ coupling, leading to activation of astrocytes, enhancement of excitability and excitatory synaptic transmission in hippocampal CA1 neurons, and reduced LTP, ultimately resulting in deficits in sensorimotor performance and spatial learning and memory[170]. Abnormal over-opening of Cx43 hemichannels on reactive astrocytes exacerbates inflammatory response and cell death in CNS pathology[171]. Cx43-specific deletion in astrocytes improves neurocognitive dysfunction by reducing astrocyte proliferation and increasing synaptic function[172]. Moreover, Cx43 deletion before and after birth did not impair the laminar distribution of neocortical excitatory neurons. Consequently, mice deficient in Cx43 during the early postnatal period show normal anxiety-like behavior, depression-related behavior, and learning and memory-related behavior in adolescence[173]. Therefore, inhibition of Cx43 hemichannels in astrocytes may be a potential target for the treatment of POCD.
3.2.2 Astrocytes and miR-146a
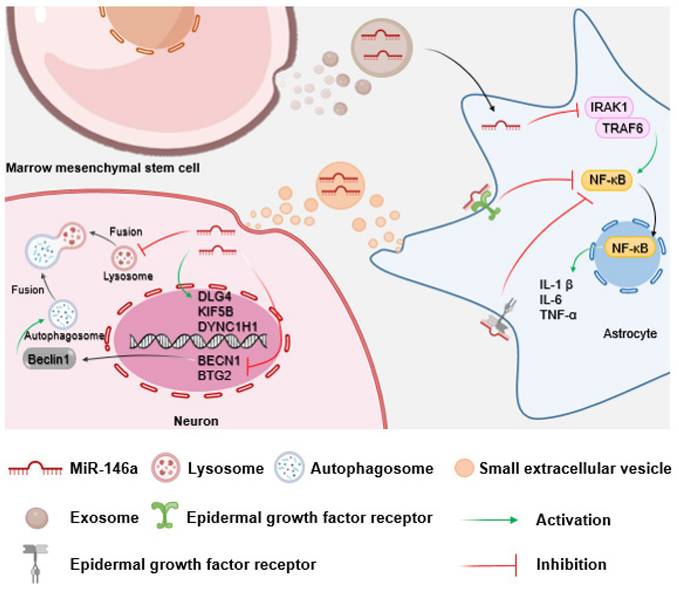
MiR-146a exhibits critical functions in the modulation of astrocyte function. In vitro experiments have shown that exosomal miR-146a secreted by bone marrow mesenchymal stem cells is taken up by astrocytes, where it suppresses NF-κB signaling and inflammation[174]. This aligns with the view that miR-146a is a negative feedback regulator of the astrocyte-mediated inflammatory response[175]. Therefore, astrocyte-specific miR-146a overexpression may attenuate neuroinflammation, ameliorate neuritis pathology, and prevent neuronal loss, thereby reducing neurocognitive dysfunction in learning and memory[176, 177]. Given that astrocytes play a pivotal role in synapse formation, restoration of astrocyte function could facilitate the correction of aberrant synapse formation and improve neurocognitive dysfunction[174]. Thus, miR-146a may reduce neuroinflammation by regulating astrocyte function, thereby improving neurocognitive dysfunction (Fig. 4).
Schematic of the impact of miR-146a on neurons mediated by astrocytes. Bone marrow mesenchymal stem cells secrete miR-146a-containing exosomes to inhibit neuroinflammation and protect neurocognitive function by suppressing the IRAK1/TRAF6/NF-κB pathway in astrocytes. Furthermore, astrocytes can regulate neuronal autophagy and axonal transport function through the paracrine pathway.
3.2.3 Astrocytes and NMDAR
Neurons and astrocytes regulate each other reciprocally via neuronal NMDAR. Evidence suggests an inter-cellular signaling mechanism between neurons and astrocytes, wherein neuronal NMDAR activation promotes neuroglial cell divisions during postnatal development or as a major component of neuroinflammation after mild brain injury[178]. Astrocytes release D-serine to stimulate NMDAR, causing glutamatergic synapses to undergo short-term enhancement and promoting water-seeking memory and water intake[179]. Hippocampal astrocytes modulate NMDAR via the glutamate-permeable anion channel Bestrophin1-mediated co-release of D-serine and glutamate, fostering heterosynaptic long-term depression and metaplasticity, thereby contributing to cognitive flexibility[180]. However, under pathological conditions, hyperactivated astrocytes release excessive levels of excitatory transmitters such as glutamate and D-serine, inducing overactivation of the NMDAR in neurons, calcium overload, and ultimately apoptotic neuronal death due to excitotoxicity[181, 182]. Mechanistically, astrocytic dysfunction contributes to POCD by impairing synaptic plasticity via the Sirtuin 1/BDNF and BDNF/tropomyosin receptor kinase B signaling pathways[183-185]. L-serine, a precursor of D-serine, acts as a co-agonist of synaptic NMDAR and is required for synaptic plasticity[186, 187]. Dietary supplementation with L-serine can prevent synaptic and behavioral deficits in mice[188]. Therefore, avoiding excessive activation of NMDAR caused by astrocyte hyperexcitability may be beneficial in preventing and treating POCD.
3.3 Summary
Both surgery and anesthesia can contribute to POCD[189]. Cx43 forms GJ channels and hemichannels. Uncoupling of Cx43-based GJs increases oxidative stress and neuroinflammation levels, leading to neurocognitive dysfunction, while increased Cx43-based GJs coupling ameliorates these effects. Elevation of Cx43 hemichannels contributes to neurocognitive dysfunction by promoting the inflammatory response, and inhibition of Cx43 expression improves learning and memory function. Astrocyte-specific deletion of Cx43 improves neurocognitive dysfunction by reducing astroglial proliferation and increasing synaptic function. At the molecular level, miR-146a blunts hippocampal neuroinflammation to minimize neurocognitive dysfunction. In addition, miR-146a inhibits neuronal apoptosis, necrosis, and brain damage by negatively regulating BTG2 expression, ultimately improving neurocognitive dysfunction. Increased miR-146a expression in astrocytes reduces neuroinflammation and thus improves neurocognitive dysfunction. Overactivation of NMDAR and sustained upregulation of the extrasynaptic NMDAR 2B subunit can lead to neurocognitive dysfunction. Under pathological conditions, hyperactive astrocytes release excessive amounts of excitatory transmitters, resulting in neuronal apoptosis due to excitotoxicity. Thus, astrocytes play a crucial role in POCD, and modulation of astrocytes may be an important approach in improving POCD-induced neurocognitive dysfunction.
4. Astrocytes and SAE
4.1 Mechanisms of neurocognitive dysfunction caused by SAE
4.1.1 Nucleotide-binding oligomerization domain (NOD) like receptor protein 3/cysteine aspartate protease 1 (NLRP3/caspase-1)
NLRP3 and caspase-1, inflammation-associated proteins, are pivotal players in the pathogenesis of SAE and contribute significantly to neurocognitive dysfunction. Studies in cecal ligation and puncture (CLP)-induced models demonstrated hippocampal-dependent memory deficits coinciding with elevated NLRP3- and caspase-1-positive neurons in the hippocampal CA1 region as well as increased hippocampal contents of NLRP3/caspase-1 signaling, gasdermin-D, and pro-inflammatory cytokines within the hippocampus, suggesting that the NLRP3/caspase-1 signaling pathway may play an important role in neuronal apoptosis and the resultant neurocognitive dysfunction during SAE progression[190]. Moreover, inhibition of NLRP3 or caspase-1 was shown to reverse neurobehavioral abnormalities[190, 191]. The NLRP-caspase-1 complex was shown to direct the localization of caspase-1 activity[192], while inhibition of NLRP3 mitigates microglia activation and neurocognitive dysfunction, thereby attenuating CLP-induced neurocognitive dysfunction[193]. Similarly, inhibition of caspase-1 can prevent neurocognitive dysfunction by reducing expression of IL-1β, monocyte chemotactic protein-1, and TNF-α in serum and brain tissue in septic mice, resulting in the inhibition of microglial activation, reduced blood-brain barrier (BBB) disruption and ultrastructural damage to brain tissue, protection of synaptic plasticity, and preservation of LTP[194]. Behavioral assessments in mouse models of lipopolysaccharide (LPS)-induced SAE, utilizing novel object recognition, elevated plus maze, and tail suspension tests, highlighted the efficacy of pharmacologically inhibiting the NLRP3 inflammasome for alleviating cognitive deficits, mood disturbances, inflammation, and BBB dysfunction in SAE[195]. Specifically, inhibition of NLRP3/caspase-1 signaling with NU9056 inhibits the classical pathway of gasdermin-D-mediated pyroptosis and attenuates BBB injury, further ameliorating SAE pathology[196]. Moreover, inhibition of NLRP3/caspase-1/toll-like receptor 4 signaling attenuates neuroinflammation, NLRP3 inflammasome activation, neuronal damage, and mitochondrial damage in young rats[197]. These findings underscore the therapeutic potential of targeting the NLRP3/caspase-1 pathway as an approach to mitigate the neurocognitive dysfunction associated with SAE.
4.1.2 Neuronal damage
Neuronal damage is the primary driver of SAE-associated neurocognitive dysfunction. In rodent models, SAE induces hippocampal iron overload, activating apoptotic pathways linked to neurocognitive deficits[198]. Iron chelation with deferoxamine mitigates neuronal apoptosis by reducing iron deposition and activating the nuclear factor E2-related factor 2 (Nrf2)/glutathione peroxidase 4 antioxidant pathway, thereby preserving synaptic integrity and improving cognitive outcomes[199]. Electroacupuncture further confers neuroprotection by ameliorating neuronal damage, synaptic injury, and oxidative stress through Nrf-2/heme oxygenase-1 (HO-1) signaling[200]. Additionally, CLP and low-dose isoflurane (0.7%) increase hippocampal HO-1, reducing neuronal injury and SAE severity[201]. These findings highlight how oxidative stress and ferroptosis offer critical targets for intervention, emphasizing the potential of combining antioxidant and iron-chelating therapies to improve neurocognitive recovery in sepsis survivors.
4.1.3 BBB damage
BBB damage is another critical mechanism underlying SAE. Sepsis induces substantial systemic inflammation, neuroinflammation, BBB leakage, and neurocognitive dysfunctions[205]. Exposure of mice to LPS activated mitochondrial localization of dynamin-related protein 1, provoking oxidative stress, upregulating the expression of vascular permeability modulators, and inducing mitochondrial defects in primary microvascular endothelial cells, consequently heightening BBB permeability[202]. However, inhalation of 2% H2 was found to reduce BBB permeability through Nrf2-induced gene expression, thereby alleviating SAE-associated neurocognitive dysfunction[203]. Moreover, subcutaneous injection of recombinant human brain natriuretic peptide has demonstrated efficacy in reducing BBB damage and inhibiting neuronal apoptosis, and recombinant human brain natriuretic peptide treatment also reduces hippocampal inflammatory cytokine levels by suppressing the toll-like receptor 4/NF-κB pathway, resulting in improved 14-day survival rates and neurocognitive dysfunction in septic mice[204]. Systemic administration of dexmedetomidine, an α2-adrenoceptor agonist with sedative, anxiolytic, and analgesic properties, also was shown to attenuate the negative neurological effects of sepsis[205]. Further studies have revealed that the neuroprotective effects of dexmedetomidine are mediated by astrocyte α2A-adrenergic receptors[205, 206]. Therefore, ameliorating BBB injury is an important measure for managing the neurocognitive dysfunction caused by SAE.
4.2 Role of astrocytes in SAE
4.2.1 Astrocytes and NLRP3/caspase-1
Bidirectional functional interaction occurs between astrocytes and NLRP3/caspase-1 signaling. First, in vitro studies have shown that exposure to LPS induces inflammatory vesicle activation and apoptosis in astrocytes[207]. In vivo, LPS treatment promotes caspase-1 immunoreactivity in astrocytes and triggers the release of the IL-1β and IL-18 pro-inflammatory cytokines[208]. Second, activation of NLRP3/caspase-1 inflammatory vesicles upregulates astrocyte IL-1β expression and effectively inhibits LPS-induced IL-1β expression by suppressing the expression of the astrocytic NLRP3/caspase-1 inflammasome[209]. In addition, treatment of astrocytes with lysophosphatidylcholine, a molecule associated with neurodegeneration and demyelination, induces the activation of the NLRP3 and NLRC4 inflammatory vesicles, which are central players in neuroinflammation and mediators of astrocyte proliferation[210]. Thus, astrocytes and NLRP3/caspase-1 signaling interact, each influencing the function of the other.
4.2.2 Astrocytes and neuronal damage
Astrocytes play a crucial role in neuronal protection. As previously described, LPS treatment in rats promotes caspase-1 immunoreactivity in astrocytes and increases the release of IL-1β and IL-18 pro-inflammatory cytokines, leading to neuronal injury, which can be attenuated by dexmedetomidine. Research has suggested that dexmedetomidine protects glial cells and subsequently safeguards neurons by reducing apoptosis, which may preserve brain function and ultimately improve outcomes in patients with sepsis[162]. Furthermore, astrocyte-derived exosomes accumulate predominantly around neurons and were found to mitigate neuronal damage by inhibiting autophagy in vivo[211, 212]. Additionally, significant upregulation of 17β-estradiol 2 synthase aromatase expression occurs in astrocytes after brain injury. To investigate the precise role of astrocyte-derived estradiol 2 in the damaged brain, a GFAP promoter-driven aromatase knockout mouse model was established to deplete astrocyte-derived estradiol 2 in the brain, and the GFAP promoter-driven aromatase knockout mice showed intense neuronal damage, microglia activation, and neurocognitive dysfunction[213]. These findings underscore the multifaceted ways through which astrocytes protect neurons.
4.2.3 Astrocytes and BBB damage
Astrocytes serve as crucial components of the BBB, actively contributing to its maintenance and regulation. Astrocytes express the glucagon-like peptide-1 receptor (GLP-1R), and activation of GLP-1R not only protects the BBB and reduces ischemia-induced neuroinflammation but also reduces the release of vascular endothelial growth factor A, matrix metalloproteinase 9 (MMP9), monocyte chemotactic protein-1 and chemokine ligand 1 (C-X-C motif). GLP-1R also increases the expression of tight junction proteins in brain endothelial cells, thereby improving BBB integrity[214]. Astrocytes can reduce neuron-derived C-C motif chemokine ligand 2 expression and protect BBB integrity by secreting fatty-acid-binding protein 7[215]. In mice deficient in pH-sensitive Na+/H+ exchanger 1 (NHE1), astrocytes release Wnt7a to activate the Wnt/β-catenin signaling pathway in cerebral vessels to protect BBB integrity[216]. The effect may be related to the control of caveolin-1 expression, vesicle abundance, and end-foot integrity in the neurovascular unit after activation of Wnt/β-catenin[217]. Astrocyte activation of pSTAT3 maintains cerebral vascular coverage through a process termed “endfoot plasticity,” which involves replacing nearby cells during the initial stages of astrocyte pedicle retraction, and the significant loss of endfoot coverage by astrocytes did not necessarily disrupt BBB integrity[218].
However, under certain conditions, astrocytes can also contribute to BBB damage. Reactive astrocytes have been shown to disrupt BBB integrity in both in vivo and in vitro experiments[205, 219]. Oncostatin M secretion by reactive astrocytes induces BBB damage during neuroinflammation, which induces permeability and recruits T helper 17 cells through enhancing endothelial C-C motif chemokine ligand 20 secretion and integrin activation[220]. In response to inflammation, astrocytes contribute to BBB dysfunction through activation of STAT3 and increased expression of SERPINA3, which encodes α1-antichymotrypsin[221]. ApoE4 production by astrocytes leads to BBB leakage, increased MMP9, impaired tight junctions, and reduced vascular coverage of astrocyte terminals[222]. Under pathological conditions, N-myc downstream-regulated gene 2 binds directly to protein phosphatase and Mg2+/Mn2+-dependent 1A, increasing MMP9 expression in reactive astrocytes[223]. The activation of astrocytic p75 neurotrophin receptor further activates NF-κB and hypoxia inducible factor-1α signaling, upregulates MMP9 and vascular endothelial growth factor expression, and subsequently leads to post-ischemic tight junction degradation[224]. Therefore, astrocytes play an important role in both the onset of and recovery from BBB injury.
4.3 Summary
Indeed, the involvement of the NLRP3/caspase-1 complex in sepsis-induced hippocampal-dependent memory deficits underscores the critical role of inflammatory pathways in neurocognitive dysfunction. Inhibition of NLRP3 or caspase-1 not only reverses neurobehavioral abnormalities but also attenuates inflammatory responses, microglial activation, BBB disruption, and ultrastructural damage to brain tissue, protecting synaptic plasticity and thereby ameliorating neurocognitive dysfunction. The inflammatory response in astrocytes during SAE highlights the intricate interplay between astrocytes and inflammatory cascades. Regulation of astrocytic NLRP3/caspase-1 inflammatory vesicles can modulate the release of inflammatory factors, potentially influencing astrocyte proliferation and neuroinflammation. Additionally, reducing septic neuronal damage, possibly through astrocyte-mediated mechanisms like autophagy inhibition via released exosomes, holds promise for improving neurocognitive function in SAE. As astrocytes are important components of the BBB, they can resist BBB disruption. We have enumerated some of the receptors and pathways in astrocytes that positively regulate the BBB. However, under pathological conditions, astrocyte activation can lead to the formation of reactive astrocytes, which contribute to BBB damage and exacerbate neuroinflammation. In addition, dexmedetomidine can improve BBB leakage and neurocognitive dysfunction via the astrocyte α2A-adrenergic receptor. Overall, the emerging prominence of astrocytes in the context of SAE suggests that astrocyte inhibition may be a promising intervention to improve the neurocognitive dysfunction caused by SAE.
In summary, neurocognitive dysfunction can arise from a variety of conditions, including AD, epilepsy, exposure to surgery and anesthesia, and septicemic encephalopathy. These conditions impair cognitive function through different mechanisms, with astrocytes playing central and multifaceted roles in their pathogenesis. Astrocytes are involved in regulating synaptic activity, modulating inflammatory responses, and maintaining metabolic balance, all of which are critical for proper cognitive function (Fig. 5). Given their pivotal role in these processes, targeting astrocytes presents a promising therapeutic strategy for both preventing and treating neurocognitive dysfunction. By focusing on astrocytic pathways—such as modulating gliotransmitter release, controlling neuroinflammation, and enhancing synaptic plasticity—it may be possible to mitigate cognitive impairments across a range of neurological conditions. Thus, astrocytes offer viable targets for future research and clinical interventions aimed at improving cognitive outcomes in patients suffering from neurodegenerative diseases, post-operative complications, and other neurological disorders.
Schematic overview of astrocyte involvement in neurocognitive dysfunction. Under pathological conditions such as AD, epilepsy, POCD, and SAE, dysfunction of astrocytes leads to impaired neuronal signaling, increased oxidative stress, and altered synaptic activity, contributing to cognitive decline. The figure illustrates how astrocytes interact with neuroinflammatory pathways, modulate glutamate clearance, and regulate amyloid and tau protein deposition, all of which exacerbate neurocognitive dysfunction. Targeting astrocytic pathways may offer therapeutic opportunities for improving cognitive outcomes in various neurological disorders.
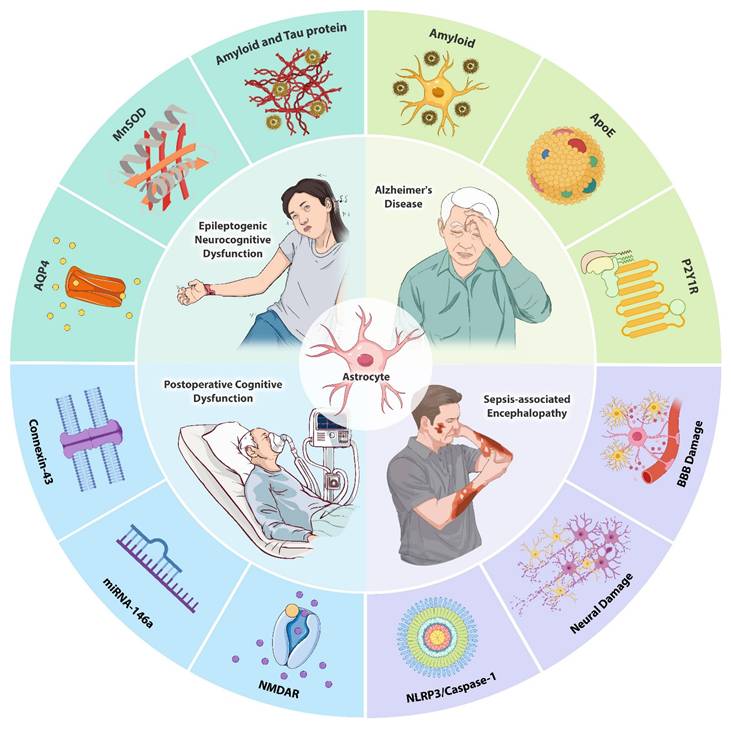
Abbreviations
AD: Alzheimer's disease; POCD: postoperative cognitive dysfunction; SAE: sepsis-associated encephalopathy; JNK: c-Jun N-terminal kinase; mTOR: mammalian target of rapamycin; Aβ: amyloid-beta; MnSOD: manganese-containing superoxide dismutase; ROS: reactive oxygen species; NMDA: n-methyl-d-aspartate; AQP4: aquaporin 4; GLT-1: glutamate transporter 1; CNS: central nervous system; GFAP: glial fibrillary acidic protein; STAT3: transcription 3; JAK2: Janus kinase 2; BDNF: brain-derived neurotrophic factor; ACC: anterior cingulate cortex; NRG: neuregulin; APP: amyloid precursor protein; PKCα: protein kinase C alpha; TREM2: trigger receptor 2; P2Y1R: P2Y1 purinoreceptor; ApoE: apolipoprotein E; LTP: long-term potentiation; Cx43: connexin-43; GJ: gap junction; IRAK1: IL receptor associated kinase 1; miR-146a: MicroRNA-146a; NF-κB: nuclear factor kappa B; BTG2: B-cell translocation gene 2; NMDAR: N-methyl-D-aspartate receptor; TrkB: tropomyosin-related kinase receptor B; TrkB-FL: tropomyosin-related kinase receptor B-full-length; TrkB-T: tropomyosin-related kinase receptor B- truncated; IL: interleukin; NLRP3: Nucleotide-binding oligomerization domain (NOD)-like receptor protein 3; CLP: cecal ligation and puncture; LPS: lipopolysaccharide; Nrf2: nuclear factor-E2-related factor 2; HO-1: heme oxygenase-1; GLP-1R: glucagon-like peptide-1 receptor; MMP9: matrix metalloproteinase 9; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid.
Acknowledgements
Funding
This work was supported by the National Nature Science Foundation of China (Grant No. 82172136) and the Natural Science Foundation of Zhejiang Province (LZ25H090003).
Author contributions
Xiaoxia Xu: conceptualization, investigation, and writing (original draft, review and editing). Bin Mei: writing (original draft, review and editing). Yue Yang: writing and graphics (original draft, review and editing). Junxiong Li: writing (original draft, review and editing). Juntao Weng, Yueyue Yang, and Qianyun Zhu: writing original draft. Honghai Zhang and Xuesheng Liu: writing-reviewing and editing, supervision, validation, funding acquisition.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mai T, Flerchinger C. Prevalence of cognitive impairments in acute nursing care-Analysis and comparison of routine data. Z Gerontol Geriatr. 2021;54:264-71
2. Ganz AB, Beker N, Hulsman M, Sikkes S, Netherlands Brain B, Scheltens P. et al. Neuropathology and cognitive performance in self-reported cognitively healthy centenarians. Acta Neuropathol Commun. 2018;6:64
3. Levin OS. Predementia neurocognitive impairment in the elderly. Zh Nevrol Psikhiatr Im S S Korsakova. 2019;119:10-7
4. H. M, Z. P, CA. B, Victor MB, Leary N, Babu S, et al. Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer's disease pathology. Cell. 2023;186:4365-85
5. 2024 Alzheimer's disease facts and figures. Alzheimers Dement. 2024;20:3708-821
6. Qiu S, Miller MI, Joshi PS, Lee JC, Xue C, Ni Y. et al. Multimodal deep learning for Alzheimer's disease dementia assessment. Nat Commun. 2022;13:3404
7. Kim H, Lee S, Ku BD, Ham SG, Park WS. Associated factors for cognitive impairment in the rural highly elderly. Brain Behav. 2019;9:e01203
8. Tedrus G, Limongi JMJ, Zuntini JVR. Resilience, quality of life, and clinical aspects of patients with epilepsy. Epilepsy Behav. 2020;103:106398
9. Nannan Panday RS, Minderhoud TC, Chantalou DS, Alam N, Nanayakkara PWB. Health related quality of life in sepsis survivors from the Prehospital Antibiotics Against Sepsis (PHANTASi) trial. PLoS One. 2019;14:e0222450
10. Evered LA, Chan MTV, Han R, Chu MHM, Cheng BP, Scott DA. et al. Anaesthetic depth and delirium after major surgery: a randomised clinical trial. Br J Anaesth. 2021;127:704-12
11. Qiu LL, Pan W, Luo D, Zhang GF, Zhou ZQ, Sun XY. et al. Dysregulation of BDNF/TrkB signaling mediated by NMDAR/Ca(2+)/calpain might contribute to postoperative cognitive dysfunction in aging mice. J Neuroinflammation. 2020;17:23
12. Xin J, Shan W, Li J, Yu H, Zuo Z. Activation of the Lateral Habenula-Ventral Tegmental Area Neural Circuit Contributes to Postoperative Cognitive Dysfunction in Mice. Adv Sci (Weinh). 2022;9:e2202228
13. Zhou Z, Okamoto K, Onodera J, Hiragi T, Andoh M, Ikawa M. et al. Astrocytic cAMP modulates memory via synaptic plasticity. Proc Natl Acad Sci U S A. 2021;118:e2016584118
14. Jimenez OA NS, Barbian HJ, Albalawi YA, Seaton MS, Robinson KF, Al-Harthi L. β-Catenin Restricts Zika Virus Internalization by Downregulating Axl. Virol. 2021;95:e0070521
15. Kol A, Adamsky A, Groysman M, Kreisel T, London M, Goshen I. Astrocytes contribute to remote memory formation by modulating hippocampal-cortical communication during learning. Nat Neurosci. 2020;23:1229-39
16. Gourmaud S, Shou H, Irwin DJ, Sansalone K, Jacobs LM, Lucas TH. et al. Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain. 2020;143:191-209
17. Alves M, Kenny A, de Leo G, Beamer EH, Engel T. Tau Phosphorylation in a Mouse Model of Temporal Lobe Epilepsy. Front Aging Neurosci. 2019;11:308
18. Yang SH, Lee DK, Shin J, Lee S, Baek S, Kim J. et al. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med. 2017;9:61-77
19. Kiss E, Kins S, Zoller Y, Schilling S, Gorgas K, Gross D. et al. Artesunate restores the levels of inhibitory synapse proteins and reduces amyloid-beta and C-terminal fragments (CTFs) of the amyloid precursor protein in an AD-mouse model. Mol Cell Neurosci. 2021;113:103624
20. Canet G, Zub E, Zussy C, Hernandez C, Blaquiere M, Garcia V. et al. Seizure activity triggers tau hyperphosphorylation and amyloidogenic pathways. Epilepsia. 2022;63:919-35
21. Joutsa J, Rinne JO, Hermann B, Karrasch M, Anttinen A, Shinnar S. et al. Association Between Childhood-Onset Epilepsy and Amyloid Burden 5 Decades Later. JAMA Neurol. 2017;74:583-90
22. Joutsa J, Rinne JO, Karrasch M, Hermann B, Johansson J, Anttinen A. et al. Brain glucose metabolism and its relation to amyloid load in middle-aged adults with childhood-onset epilepsy. Epilepsy Res. 2017;137:69-72
23. Sperling RA, Mormino EC, Schultz AP, Betensky RA, Papp KV, Amariglio RE. et al. The impact of amyloid-beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85:181-93
24. Zhang Q, Wang J, Zhu L, Jiang SJ, Liu J, Wang LX. et al. Ligustrazine Attenuates Hyperhomocysteinemia-induced Alzheimer-like Pathologies in Rats. Curr Med Sci. 2021;41:548-54
25. Costa C, Parnetti L, D'Amelio M, Tozzi A, Tantucci M, Romigi A. et al. Epilepsy, amyloid-beta, and D1 dopamine receptors: a possible pathogenetic link? Neurobiol Aging. 2016;48:161-71
26. Toscano ECB, Vieira ELM, Grinberg LT, Rocha NP, Brant JAS, Paradela RS. et al. Hyperphosphorylated Tau in Mesial Temporal Lobe Epilepsy: a Neuropathological and Cognitive Study. Mol Neurobiol. 2023;60:2174-85
27. Chojdak-Łukasiewicz J M-MM, Zimny A, Noga L, Paradowski B. Plasma tau protein and Aβ42 level as markers of cognitive impairment in patients with Parkinson's disease. Adv Clin Exp Med. 2020;29:115-21
28. Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E. et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:668-77
29. Song JX, Malampati S, Zeng Y, Durairajan SSK, Yang CB, Tong BC. et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer's disease models. Aging Cell. 2020;19:e13069
30. Foo H, Mak E, Yong TT, Wen MC, Chander RJ, Au WL. et al. Progression of subcortical atrophy in mild Parkinson's disease and its impact on cognition. Eur J Neurol. 2017;24:341-8
31. Pereira JB, Ossenkoppele R, Palmqvist S, Strandberg TO, Smith R, Westman E. et al. Amyloid and tau accumulate across distinct spatial networks and are differentially associated with brain connectivity. Elife. 2019;8:e50830
32. Qin Q, Tang Y, Dou X, Qu Y, Xing Y, Yang J. et al. Default mode network integrity changes contribute to cognitive deficits in subcortical vascular cognitive impairment, no dementia. Brain Imaging Behav. 2021;15:255-65
33. Badawi GA, Shokr MM, Zaki HF, Mohamed AF. Pentoxifylline prevents epileptic seizure via modulating HMGB1/RAGE/TLR4 signalling pathway and improves memory in pentylenetetrazol kindling rats. Clin Exp Pharmacol Physiol. 2021;48:1111-24
34. Shao E, Chang CW, Li Z, Yu X, Ho K, Zhang M. et al. TAU ablation in excitatory neurons and postnatal TAU knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model. Sci Transl Med. 2022;14:eabm5527
35. Chang CW, Evans MD, Yu X, Yu GQ, Mucke L. Tau reduction affects excitatory and inhibitory neurons differently, reduces excitation/inhibition ratios, and counteracts network hypersynchrony. Cell Rep. 2021;37:109855
36. Sirisena D, Perera NCN, Godahewa GI, Kwon H, Yang H, Nam BH. et al. A manganese superoxide dismutase (MnSOD) from red lip mullet, Liza haematocheila: Evaluation of molecular structure, immune response, and antioxidant function. Fish Shellfish Immunol. 2019;84:73-82
37. Broz M, Furlan V, Lesnik S, Jukic M, Bren U. The Effect of the Ala16Val Mutation on the Secondary Structure of the Manganese Superoxide Dismutase Mitochondrial Targeting Sequence. Antioxidants (Basel). 2022;11:2348
38. Esteras N, Kopach O, Maiolino M, Lariccia V, Amoroso S, Qamar S. et al. Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimers Dement. 2022;18:318-38
39. Singh PK, Saadi A, Sheeni Y, Shekh-Ahmad T. Specific inhibition of NADPH oxidase 2 modifies chronic epilepsy. Redox Biol. 2022;58:102549
40. Arend J, Kegler A, Caprara ALF, Almeida C, Gabbi P, Pascotini ET. et al. Depressive, inflammatory, and metabolic factors associated with cognitive impairment in patients with epilepsy. Epilepsy Behav. 2018;86:49-57
41. Kim JE, Park H, Kim TH, Kang TC. LONP1 Regulates Mitochondrial Accumulations of HMGB1 and Caspase-3 in CA1 and PV Neurons Following Status Epilepticus. Int J Mol Sci. 2021;22:2275
42. Tzeng TT TH, Chang L, Hsu CL, Lai TH, Huang FL, Shiao YJ. Caspase 3 involves in neuroplasticity, microglial activation and neurogenesis in the mice hippocampus after intracerebral injection of kainic acid. J Biomed Sci. 2013;6:1
43. Jiang J, Yu Y, Kinjo ER, Du Y, Nguyen HP, Dingledine R. Suppressing pro-inflammatory prostaglandin signaling attenuates excitotoxicity-associated neuronal inflammation and injury. Neuropharmacology. 2019;149:149-60
44. Cao J, Yu X, Liu J, Fu J, Wang B, Wu C. et al. Ruxolitinib improves the inflammatory microenvironment, restores glutamate homeostasis, and promotes functional recovery after spinal cord injury. Neural Regen Res. 2024;19:2499-512
45. Kim J, Lim J, Yoo ID, Park S, Moon JS. TXNIP contributes to induction of pro-inflammatory phenotype and caspase-3 activation in astrocytes during Alzheimer's diseases. Redox Biol. 2023;63:102735
46. Socodato R, Rodrigues-Santos A, Tedim-Moreira J, Almeida TO, Canedo T, Portugal CC. et al. RhoA balances microglial reactivity and survival during neuroinflammation. Cell Death Dis. 2023;14:690
47. Kegler A, Cardoso AS, Caprara ALF, Pascotini ET, Arend J, Gabbi P. et al. Involvement of MnSOD Ala16Val polymorphism in epilepsy: A relationship with seizure type, inflammation, and metabolic syndrome. Gene. 2019;711:143924
48. Kegler A, Caprara ALF, Pascotini ET, Arend J, Gabbi P, Duarte M. et al. Apoptotic Markers Are Increased in Epilepsy Patients: A Relation with Manganese Superoxide Dismutase Ala16Val Polymorphism and Seizure Type through IL-1beta and IL-6 Pathways. Biomed Res Int. 2020;2020:6250429
49. Arend J, Kegler A, Caprara ALF, Gabbi P, Pascotini ET, de Freitas LAV. et al. MnSOD Ala16Val polymorphism in cognitive dysfunction in patients with epilepsy: A relationship with oxidative and inflammatory markers. Epilepsy Behav. 2020;112:107346
50. Rosu GC, Catalin B, Balseanu TA, Laurentiu M, Claudiu M, Kumar-Singh S. et al. Inhibition of Aquaporin 4 Decreases Amyloid Abeta40 Drainage Around Cerebral Vessels. Mol Neurobiol. 2020;57:4720-34
51. Salman MM, Kitchen P, Halsey A, Wang MX, Törnroth-Horsefield S, Conner AC. et al. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain. 2022;145:64-75
52. Feng S, Wu C, Zou P, Deng Q, Chen Z, Li M. et al. High-intensity interval training ameliorates Alzheimer's disease-like pathology by regulating astrocyte phenotype-associated AQP4 polarization. Theranostics. 2023;13:3434-50
53. Liu K, Zhu J, Chang Y, Lin Z, Shi Z, Li X. et al. Attenuation of cerebral edema facilitates recovery of glymphatic system function after status epilepticus. JCI Insight. 2021;6:e151835
54. Szu JI, Patel DD, Chaturvedi S, Lovelace JW, Binder DK. Modulation of posttraumatic epileptogenesis in aquaporin-4 knockout mice. Epilepsia. 2020;61:1503-14
55. Wang M, Iliff JJ, Liao Y, Chen MJ, Shinseki MS, Venkataraman A. et al. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J Neurosci. 2012;32:17948-60
56. Lambert I, Tramoni-Negre E, Lagarde S, Roehri N, Giusiano B, Trebuchon-Da Fonseca A. et al. Hippocampal Interictal Spikes during Sleep Impact Long-Term Memory Consolidation. Ann Neurol. 2020;87:976-87
57. Wang C, Wu Q, Wang Z, Hu L, Marshall C, Xiao M. Aquaporin 4 knockout increases complete freund's adjuvant-induced spinal central sensitization. Brain Res Bull. 2020;156:58-66
58. Beraldo Neto E, Freitas LA, Pimenta DC, Lebrun I, Nencioni ALA. Tb1, a Neurotoxin from Tityus bahiensis Scorpion Venom, Induces Epileptic Seizures by Increasing Glutamate Release. Toxins (Basel). 2020;12:65
59. Reddy DS, Thompson W, Calderara G. Does Stress Trigger Seizures? Evidence from Experimental Models. Curr Top Behav Neurosci. 2022;55:41-64
60. Tan YX, Liu GC, Chen HL, Lu MN, Chen B, Hu T. et al. Exercise-Induced Cognitive Improvement Is Associated with Sodium Channel-Mediated Excitability in APP/PS1 Mice. Neural Plast. 2020;2020:9132720
61. Colbourn R, Hrabe J, Nicholson C, Perkins M, Goodman JH, Hrabetova S. Rapid volume pulsation of the extracellular space coincides with epileptiform activity in mice and depends on the NBCe1 transporter. J Physiol. 2021;599:3195-220
62. Perez-Nunez R, Chamorro A, Gonzalez MF, Contreras P, Artigas R, Corvalan AH. et al. Protein kinase B (AKT) upregulation and Thy-1-alpha(v)beta(3) integrin-induced phosphorylation of Connexin43 by activated AKT in astrogliosis. J Neuroinflammation. 2023;20:5
63. Li Puma DD, Ripoli C, Puliatti G, Pastore F, Lazzarino G, Tavazzi B. et al. Extracellular tau oligomers affect extracellular glutamate handling by astrocytes through downregulation of GLT-1 expression and impairment of NKA1A2 function. Neuropathol Appl Neurobiol. 2022;48:e12811
64. Nikolic L, Shen W, Nobili P, Virenque A, Ulmann L, Audinat E. Blocking TNFalpha-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia. 2018;66:2673-83
65. Fukuyama K, Motomura E, Okada M. Age-Dependent Activation of Purinergic Transmission Contributes to the Development of Epileptogenesis in ADSHE Model Rats. Biomolecules. 2024;14:204
66. Qu Q, Zhang W, Wang J, Mai D, Ren S, Qu S. et al. Functional investigation of SLC1A2 variants associated with epilepsy. Cell Death Dis. 2022;13:1063
67. Ma L, Wu Q, You Y, Zhang P, Tan D, Liang M. et al. Neuronal small extracellular vesicles carrying miR-181c-5p contribute to the pathogenesis of epilepsy by regulating the protein kinase C-delta/glutamate transporter-1 axis in astrocytes. Glia. 2024;72:1082-95
68. Jia X, Xu J, Zhang Y, Kong S, Cheng X, Wu N. et al. Elevated Astrocytic NFAT5 of the hippocampus increases epilepsy susceptibility in hypoxic-ischemic mice. Epilepsia. 2025;66:817-832
69. Tyurikova O, Kopach O, Zheng K, Rathore D, Codadu N, Wu S-Y. et al. Astrocyte Kir4.1 expression level territorially controls excitatory transmission in the brain. Cell Reports. 2025;44:115299
70. Shen W, Chen F, Tang Y, Zhou W, Zhao Y, Li X. et al. Astrocytic GAT-3 Regulates Synaptic Transmission and Memory Formation in the Dentate Gyrus. Glia. 2025;73:788-804
71. Munger EL, Edler MK, Hopkins WD, Ely JJ, Erwin JM, Perl DP. et al. Astrocytic changes with aging and Alzheimer's disease-type pathology in chimpanzees. J Comp Neurol. 2019;527:1179-95
72. Reichenbach N, Delekate A, Plescher M, Schmitt F, Krauss S, Blank N. et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer's disease model. EMBO Mol Med. 2019;11:e9665
73. Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE. et al. Plasma GFAP is an early marker of amyloid-beta but not tau pathology in Alzheimer's disease. Brain. 2021;144:3505-16
74. Lee Y, Miller MR, Fernandez MA, Berg EL, Prada AM, Ouyang Q. et al. Early lysosome defects precede neurodegeneration with amyloid-beta and tau aggregation in NHE6-null rat brain. Brain. 2022;145:3187-202
75. Guillemaud O, Ceyzériat K, Saint-Georges T, Cambon K, Petit F, Ben Haim L. et al. Complex roles for reactive astrocytes in the triple transgenic mouse model of Alzheimer disease. Neurobiology of Aging. 2020;90:135-46
76. Zimmer TS, David B, Broekaart DWM, Schidlowski M, Ruffolo G, Korotkov A. et al. Seizure-mediated iron accumulation and dysregulated iron metabolism after status epilepticus and in temporal lobe epilepsy. Acta Neuropathol. 2021;142:729-59
77. Xiang J, Zhang J, Cai X, Yang F, Zhu W, Zhang W. et al. Bilobalide protects astrocytes from oxygen and glucose deprivation-induced oxidative injury by upregulating manganese superoxide dismutase. Phytother Res. 2019;33:2329-36
78. Dong YX, Li TH, Wang SS, Hu YH, Liu Y, Zhang F. et al. Bu zhong Yiqi Decoction ameliorates mild cognitive impairment by improving mitochondrial oxidative stress damage via the SIRT3/MnSOD/OGG1 pathway. J Ethnopharmacol. 2024;331:118237
79. Mongin AA, Kimelberg HK. ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol. 2002;283:C569-78
80. Zhang P, Li YX, Zhang ZZ, Yang Y, Rao JX, Xia L. et al. Astroglial Mechanisms Underlying Chronic Insomnia Disorder: A Clinical Study. Nat Sci Sleep. 2020;12:693-704
81. Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LM, Nilsson LN. et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer's disease. J Alzheimers Dis. 2011;27:711-22
82. Zhu DD, Yang G, Huang YL, Zhang T, Sui AR, Li N. et al. AQP4-A25Q Point Mutation in Mice Depolymerizes Orthogonal Arrays of Particles and Decreases Polarized Expression of AQP4 Protein in Astrocytic Endfeet at the Blood-Brain Barrier. J Neurosci. 2022;42:8169-83
83. Amiry-Moghaddam M WA, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100:13615-20
84. Skucas VA, Mathews IB, Yang J, Cheng Q, Treister A, Duffy AM. et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J Neurosci. 2011;31:6392-7
85. Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT. Aquaporin-4 Reduces Post-Traumatic Seizure Susceptibility by Promoting Astrocytic Glial Scar Formation in Mice. J Neurotrauma. 2021;38:1193-201
86. Hotz AL, Jamali A, Rieser NN, Niklaus S, Aydin E, Myren-Svelstad S. et al. Loss of glutamate transporter eaat2a leads to aberrant neuronal excitability, recurrent epileptic seizures, and basal hypoactivity. Glia. 2021;70:196-214
87. Peterson AR, Garcia TA, Cullion K, Tiwari-Woodruff SK, Pedapati EV, Binder DK. Targeted overexpression of glutamate transporter-1 reduces seizures and attenuates pathological changes in a mouse model of epilepsy. Neurobiol Dis. 2021;157:105443
88. Zhang Y, Sun L, Liu E, Wang A, Yan J. The olfactory stimulation slows down the substance clearance in the extracellular space of the hippocampus in rat brain. Biochem Biophys Res Commun. 2019;515:429-35
89. Peterson AR, Garcia TA, Ford BD, Binder DK. Regulation of NRG-1-ErbB4 signaling and neuroprotection by exogenous neuregulin-1 in a mouse model of epilepsy. Neurobiol Dis. 2021;161:105545
90. Ikoma Y, Sasaki D, Matsui K. Local brain environment changes associated with epileptogenesis. Brain. 2023;146:576-86
91. Yang L, Chen Z, Wan X, Liu M, Wu J, Chen Y. et al. Angiotensin II type 1 receptor deficiency protects against the impairment of blood-brain barrier in a mouse model of traumatic brain injury. Int J Neurosci. 2023;133:604-11
92. Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S. et al. Acetylation changes tau interactome to degrade tau in Alzheimer's disease animal and organoid models. Aging Cell. 2020;19:e13081
93. Giovinazzo D, Bursac B, Sbodio JI, Nalluru S, Vignane T, Snowman AM. et al. Hydrogen sulfide is neuroprotective in Alzheimer's disease by sulfhydrating GSK3beta and inhibiting Tau hyperphosphorylation. Proc Natl Acad Sci U S A. 2021;118:e2017225118
94. Karthick C, Nithiyanandan S, Essa MM, Guillemin GJ, Jayachandran SK, Anusuyadevi M. Time-dependent effect of oligomeric amyloid-beta (1-42)-induced hippocampal neurodegeneration in rat model of Alzheimer's disease. Neurol Res. 2019;41:139-50
95. Gauthier-Umana C, Munoz-Cabrera J, Valderrama M, Munera A, Nava-Mesa MO. Acute Effects of Two Different Species of Amyloid-beta on Oscillatory Activity and Synaptic Plasticity in the Commissural CA3-CA1 Circuit of the Hippocampus. Neural Plast. 2020;2020:8869526
96. Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W. et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis. 2018;110:29-36
97. Lorden G, Wozniak JM, Dore K, Dozier LE, Cates-Gatto C, Patrick GN. et al. Enhanced activity of Alzheimer disease-associated variant of protein kinase Calpha drives cognitive decline in a mouse model. Nat Commun. 2022;13:7200
98. Wang X, Xie Y, Fan X, Wu X, Wang D, Zhu L. Intermittent hypoxia training enhances Abeta endocytosis by plaque associated microglia via VPS35-dependent TREM2 recycling in murine Alzheimer's disease. Alzheimers Res Ther. 2024;16:121
99. Yang Y, Shi G, Ge Y, Huang S, Cui N, Tan L. et al. Accumulated BCAAs and BCKAs contribute to the HFD-induced deterioration of Alzheimer's disease via a dysfunctional TREM2-related reduction in microglial beta-amyloid clearance. J Neuroinflammation. 2024;21:327
100. Park JC, Han JW, Lee W, Kim J, Lee SE, Lee D. et al. Microglia Gravitate toward Amyloid Plaques Surrounded by Externalized Phosphatidylserine via TREM2. Adv Sci (Weinh). 2024;11:e2400064
101. Zhao P, Xu Y, Jiang L, Fan X, Li L, Li X. et al. A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer's disease. Science Translational Medicine. 2022;14:eabq0095
102. Huang Y, Rafael Guimaraes T, Todd N, Ferguson C, Weiss KM, Stauffer FR. et al. G protein-biased GPR3 signaling ameliorates amyloid pathology in a preclinical Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2022;119:e2204828119
103. Luo R, Su LY, Li G, Yang J, Liu Q, Yang LX. et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy. 2020;16:52-69
104. He Z, Yang Y, Xing Z, Zuo Z, Wang R, Gu H. et al. Intraperitoneal injection of IFN-gamma restores microglial autophagy, promotes amyloid-beta clearance and improves cognition in APP/PS1 mice. Cell Death Dis. 2020;11:440
105. Johnson ECB, Ho K, Yu GQ, Das M, Sanchez PE, Djukic B. et al. Behavioral and neural network abnormalities in human APP transgenic mice resemble those of App knock-in mice and are modulated by familial Alzheimer's disease mutations but not by inhibition of BACE1. Mol Neurodegener. 2020;15:53
106. Wen Q, Risacher SL, Xie L, Li J, Harezlak J, Farlow MR. et al. Tau-related white-matter alterations along spatially selective pathways. Neuroimage. 2021;226:117560
107. Chatterjee P, Pedrini S, Ashton NJ, Tegg M, Goozee K, Singh AK. et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer's disease. Alzheimers Dement. 2022;18:1141-54
108. Ashton NJ, Janelidze S, Mattsson-Carlgren N, Binette AP, Strandberg O, Brum WS. et al. Differential roles of Abeta42/40, p-tau231 and p-tau217 for Alzheimer's trial selection and disease monitoring. Nat Med. 2022;28:2555-62
109. Frontzkowski L, Ewers M, Brendel M, Biel D, Ossenkoppele R, Hager P. et al. Earlier Alzheimer's disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat Commun. 2022;13:4899
110. Janelidze S, Berron D, Smith R, Strandberg O, Proctor NK, Dage JL. et al. Associations of Plasma Phospho-Tau217 Levels With Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021;78:149-56
111. Aillaud I, Kaniyappan S, Chandupatla RR, Ramirez LM, Alkhashrom S, Eichler J. et al. A novel D-amino acid peptide with therapeutic potential (ISAD1) inhibits aggregation of neurotoxic disease-relevant mutant Tau and prevents Tau toxicity in vitro. Alzheimers Res Ther. 2022;14:15
112. Taga M, Petyuk VA, White C, Marsh G, Ma Y, Klein HU. et al. BIN1 protein isoforms are differentially expressed in astrocytes, neurons, and microglia: neuronal and astrocyte BIN1 are implicated in tau pathology. Mol Neurodegener. 2020;15:44
113. Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH. et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by beta-amyloid pathology. Neuron. 2021;109:1283-301 e6
114. Subramanian M, Hyeon SJ, Das T, Suh YS, Kim YK, Lee JS. et al. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat Commun. 2021;12:3291
115. Reichenbach N, Delekate A, Breithausen B, Keppler K, Poll S, Schulte T. et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer's disease model. J Exp Med. 2018;215:1649-63
116. Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem. 2006;281:30684-96
117. Simoes AP, Silva CG, Marques JM, Pochmann D, Porciuncula LO, Ferreira S. et al. Glutamate-induced and NMDA receptor-mediated neurodegeneration entails P2Y1 receptor activation. Cell Death Dis. 2018;9:297
118. Koch H, Bespalov A, Drescher K, Franke H, Krugel U. Impaired cognition after stimulation of P2Y1 receptors in the rat medial prefrontal cortex. Neuropsychopharmacology. 2015;40:305-14
119. Carmo MR, Simoes AP, Fonteles AA, Souza CM, Cunha RA, Andrade GM. ATP P2Y1 receptors control cognitive deficits and neurotoxicity but not glial modifications induced by brain ischemia in mice. Eur J Neurosci. 2014;39:614-22
120. Shi A, Petrache AL, Shi J, Ali AB. Preserved Calretinin Interneurons in an App Model of Alzheimer's Disease Disrupt Hippocampal Inhibition via Upregulated P2Y1 Purinoreceptors. Cereb Cortex. 2020;30:1272-90
121. Caneque-Rufo H, Fontan-Baselga T, Galan-Llario M, Zuccaro A, Sanchez-Alonso MG, Gramage E. et al. Pleiotrophin deletion prevents high-fat diet-induced cognitive impairment, glial responses, and alterations of the perineuronal nets in the hippocampus. Neurobiol Dis. 2025;205:106776
122. Qiao CM, Tan LL, Ma XY, Xia YM, Li T, Li MA. et al. Mechanism of S100A9-mediated astrocyte activation via TLR4/NF-kappaB in Parkinson's disease. Int Immunopharmacol. 2025;146:113938
123. Wijesinghe SS, Rowe JB, Mason HD, Allinson KSJ, Thomas R, Vontobel DS. et al. Post-mortem validation of in vivo TSPO PET as a microglial biomarker. Brain. 2025: awaf078.
124. Wirth S, Schlosser A, Beiersdorfer A, Schweizer M, Woo MS, Friese MA. et al. Astrocytic uptake of posttranslationally modified amyloid-beta leads to endolysosomal system disruption and induction of pro-inflammatory signaling. Glia. 2024;72:1451-68
125. Perez-Nievas BG, Johnson L, Beltran-Lobo P, Hughes MM, Gammallieri L, Tarsitano F. et al. Astrocytic C-X-C motif chemokine ligand-1 mediates beta-amyloid-induced synaptotoxicity. J Neuroinflammation. 2021;18:306
126. Tang C, Gao J, Li S, Cheng H, Peng YY, Ding Y. et al. Chlorogenic acid improves SPS-induced PTSD-like behaviors in rats by regulating the crosstalk between Nrf2 and NF-kappaB signaling pathway. Free Radic Biol Med. 2025;231:136-52
127. Sanjay Sood R, Jaiswal V Kang SU, Park M Lee HJ. Nobiletin regulates intracellular Ca(2+) levels via IP(3)R and ameliorates neuroinflammation in Abeta42-induced astrocytes. Redox Biol. 2024;73:103197
128. Collier JM, Metwally S, McFarland M, Krishna S, Kurella P, Fiesler V. et al. Upregulation of Na/H Exchanger in Astrogliosis and Early Alzheimer's Disease Pathogenesis. Aging Dis. 2025
129. Du S, Jin F, Maneix L, Gedam M, Xu Y, Catic A. et al. FoxO3 deficiency in cortical astrocytes leads to impaired lipid metabolism and aggravated amyloid pathology. Aging Cell. 2021;20:e13432
130. Cameron EG, Nahmou M, Toth AB, Heo L, Tanasa B, Dalal R. et al. A molecular switch for neuroprotective astrocyte reactivity. Nature. 2024;626:574-82
131. Zhang L, Xu Z, Jia Z, Cai S, Wu Q, Liu X. et al. Modulating mTOR-dependent astrocyte substate transitions to alleviate neurodegeneration. Nat Aging. 2025;5:468-475
132. Sun X PS, Li D, Su M, Zheng H, Zhang YW, Li Y. Cell adhesion molecule protocadherin-γC5 ameliorates Aβ plaque pathogenesis by modulating astrocyte function in Alzheimer's disease. The Journal of neuroscience. 2025
133. Zhong M, Xu QQ, Hu Z, Yang W, Lin ZX, Xian YF. Tianma-Gouteng pair ameliorates the cognitive deficits on two transgenic mouse models of Alzheimer's disease. J Ethnopharmacol. 2024;328:118113
134. Wang H, Kulas JA, Wang C, Holtzman DM, Ferris HA, Hansen SB. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc Natl Acad Sci U S A. 2021;118:e2102191118
135. Chen F, Swartzlander DB, Ghosh A, Fryer JD, Wang B, Zheng H. Clusterin secreted from astrocyte promotes excitatory synaptic transmission and ameliorates Alzheimer's disease neuropathology. Mol Neurodegener. 2021;16:5
136. Wojtas AM, Sens JP, Kang SS, Baker KE, Berry TJ, Kurti A. et al. Astrocyte-derived clusterin suppresses amyloid formation in vivo. Mol Neurodegener. 2020;15:71
137. Sanchez-Mico MV, Jimenez S, Gomez-Arboledas A, Munoz-Castro C, Romero-Molina C, Navarro V. et al. Amyloid-beta impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer's disease. Glia. 2021;69:997-1011
138. Richetin K, Steullet P, Pachoud M, Perbet R, Parietti E, Maheswaran M. et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer's disease. Nat Neurosci. 2020;23:1567-79
139. Jablonski AM, Warren L, Usenovic M, Zhou H, Sugam J, Parmentier-Batteur S. et al. Astrocytic expression of the Alzheimer's disease risk allele, ApoEepsilon4, potentiates neuronal tau pathology in multiple preclinical models. Sci Rep. 2021;11:3438
140. Saroja SR, Gorbachev K, Julia T, Goate AM, Pereira AC. Astrocyte-secreted glypican-4 drives APOE4-dependent tau hyperphosphorylation. Proc Natl Acad Sci U S A. 2022;119:e2108870119
141. Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS. et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021;109:1657-74 e7
142. Bourel J, Planche V, Dubourdieu N, Oliveira A, Sere A, Ducourneau EG. et al. Complement C3 mediates early hippocampal neurodegeneration and memory impairment in experimental multiple sclerosis. Neurobiol Dis. 2021;160:105533
143. Tcw J, Qian L, Pipalia NH, Chao MJ, Liang SA, Shi Y. et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell. 2022;185:2213-33 e25
144. Blanchard JW, Akay LA, Davila-Velderrain J, von Maydell D, Mathys H, Davidson SM. et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature. 2022;611:769-79
145. Ye J, Zhong S, Wan H, Guo X, Yao X, Liu Q. et al. Upregulated astrocyte HDAC7 induces Alzheimer-like tau pathologies via deacetylating transcription factor-EB and inhibiting lysosome biogenesis. Mol Neurodegener. 2025;20:5
146. Beltran-Lobo P, Hughes MM, Troakes C, Croft CL, Rupawala H, Jutzi D. et al. P2X(7)R influences tau aggregate burden in human tauopathies and shows distinct signalling in microglia and astrocytes. Brain Behav Immun. 2023;114:414-29
147. Dias L, Madeira D, Dias R, Tome AR, Cunha RA, Agostinho P. Abeta(1-42) peptides blunt the adenosine A(2A) receptor-mediated control of the interplay between P(2)X(7) and P(2)Y(1) receptors mediated calcium responses in astrocytes. Cell Mol Life Sci. 2022;79:457
148. Martorell A, Wellmann M, Guiffa F, Fuenzalida M, Bonansco C. P2Y1 receptor inhibition rescues impaired synaptic plasticity and astroglial Ca2+-dependent activity in the epileptic hippocampus. Neurobiology of Disease. 2020;146:105132
149. Bi C, Cai Q, Shan Y, Yang F, Sun S, Wu X. et al. Sevoflurane induces neurotoxicity in the developing rat hippocampus by upregulating connexin 43 via the JNK/c-Jun/AP-1 pathway. Biomed Pharmacother. 2018;108:1469-76
150. Wang X, Feng L, Xin M, Hao Y, Wang X, Shang P. et al. Mechanisms underlying astrocytic connexin-43 autophagy degradation during cerebral ischemia injury and the effect on neuroinflammation and cell apoptosis. Biomed Pharmacother. 2020;127:110125
151. Hyland C, Mfarej M, Hiotis G, Lancaster S, Novak N, Iovine MK. et al. Impaired Cx43 gap junction endocytosis causes morphological and functional defects in zebrafish. Mol Biol Cell. 2021;32:ar13
152. Dong R, Lv P, Han Y, Jiang L, Wang Z, Peng L. et al. Enhancement of astrocytic gap junctions Connexin43 coupling can improve long-term isoflurane anesthesia-mediated brain network abnormalities and cognitive impairment. CNS Neurosci Ther. 2022;28:2281-97
153. Lee HJ, Cha HJ, Jeong H, Lee SN, Lee CW, Kim M. et al. Conformational changes in the human Cx43/GJA1 gap junction channel visualized using cryo-EM. Nat Commun. 2023;14:931
154. Almad AA, Taga A, Joseph J, Gross SK, Welsh C, Patankar A. et al. Cx43 hemichannels contribute to astrocyte-mediated toxicity in sporadic and familial ALS. Proc Natl Acad Sci U S A. 2022;119:e2107391119
155. Ju H WY, Shi Q, Zhou Y, Ma R, Wu P, Fang H. Inhibition of connexin 43 hemichannels improves postoperative cognitive function in aged mice. Am J Transl Res. 2019;11:2280-7
156. Ghotloo S, Motedayyen H, Amani D, Saffari M, Sattari M. Assessment of microRNA-146a in generalized aggressive periodontitis and its association with disease severity. J Periodontal Res. 2019;54:27-32
157. Liu GJ, Zhang QR, Gao X, Wang H, Tao T, Gao YY. et al. MiR-146a Ameliorates Hemoglobin-Induced Microglial Inflammatory Response via TLR4/IRAK1/TRAF6 Associated Pathways. Front Neurosci. 2020;14:311
158. Chen L, Dong R, Lu Y, Zhou Y, Li K, Zhang Z. et al. MicroRNA-146a protects against cognitive decline induced by surgical trauma by suppressing hippocampal neuroinflammation in mice. Brain Behav Immun. 2019;78:188-201
159. Yang B, Yang R, Xu B, Fu J, Qu X, Li L. et al. miR-155 and miR-146a collectively regulate meningitic Escherichia coli infection-mediated neuroinflammatory responses. J Neuroinflammation. 2021;18:114
160. Gomes C, Sequeira C, Likhite S, Dennys CN, Kolb SJ, Shaw PJ. et al. Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes. Cells. 2022;11:1186
161. Lim SK, Choi YW, Lim IK, Park TJ. BTG2 suppresses cancer cell migration through inhibition of Src-FAK signaling by downregulation of reactive oxygen species generation in mitochondria. Clin Exp Metastasis. 2012;29:901-13
162. Mao L, Zeng Q, Su W, Song M, Li J, Xie M. Elevation of miR-146a Inhibits BTG2/BAX Expression to Ameliorate Postoperative Cognitive Dysfunction Following Probiotics (VSL#3) Treatment. Mol Neurobiol. 2021;58:3457-70
163. Aroeira RI, Sebastiao AM, Valente CA. BDNF, via truncated TrkB receptor, modulates GlyT1 and GlyT2 in astrocytes. Glia. 2015;63:2181-97
164. Almahozi A, Radhi M, Alzayer S, Kamal A. Effects of Memantine in a Mouse Model of Postoperative Cognitive Dysfunction. Behav Sci (Basel). 2019;9:24
165. Li L, Li Z, Cao Y, Fan D, Chui D, Guo X. Increased extrasynaptic GluN2B expression is involved in cognitive impairment after isoflurane anesthesia. Exp Ther Med. 2016;12:161-8
166. Mishchenko TA, Yarkov RS, Saviuk MO, Krivonosov MI, Perenkov AD, Gudkov SV. et al. Unravelling Contributions of Astrocytic Connexin 43 to the Functional Activity of Brain Neuron-Glial Networks under Hypoxic State In Vitro. Membranes (Basel). 2022;12:948
167. McCutcheon S, Spray DC. Glioblastoma-Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol Cancer Res. 2022;20:319-31
168. Hastings N, Yu YL, Huang B, Middya S, Inaoka M, Erkamp NA. et al. Electrophysiological In Vitro Study of Long-Range Signal Transmission by Astrocytic Networks. Adv Sci (Weinh). 2023: e2301756.
169. Dong R, Han Y, Jiang L, Liu S, Zhang F, Peng L. et al. Connexin 43 gap junction-mediated astrocytic network reconstruction attenuates isoflurane-induced cognitive dysfunction in mice. J Neuroinflammation. 2022;19:64
170. Hosli L, Binini N, Ferrari KD, Thieren L, Looser ZJ, Zuend M. et al. Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep. 2022;38:110484
171. Yu H, Cao X, Li W, Liu P, Zhao Y, Song L. et al. Targeting connexin 43 provides anti-inflammatory effects after intracerebral hemorrhage injury by regulating YAP signaling. J Neuroinflammation. 2020;17:322
172. Ren R, Zhang L, Wang M. Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sci. 2018;208:175-91
173. Wang M, Chen JJ, Huang Q, Su X, Yu YC, Liu LY. Connexin43 in neonatal excitatory neurons is important for short-term motor learning. Brain Res. 2019;1720:146287
174. Nakano M, Kubota K, Kobayashi E, Chikenji TS, Saito Y, Konari N. et al. Bone marrow-derived mesenchymal stem cells improve cognitive impairment in an Alzheimer's disease model by increasing the expression of microRNA-146a in hippocampus. Sci Rep. 2020;10:10772
175. Iyer A, Zurolo E, Prabowo A, Fluiter K, Spliet WG, van Rijen PC. et al. MicroRNA-146a: a key regulator of astrocyte-mediated inflammatory response. PLoS One. 2012;7:e44789
176. Liang C, Zou T, Zhang M, Fan W, Zhang T, Jiang Y. et al. MicroRNA-146a switches microglial phenotypes to resist the pathological processes and cognitive degradation of Alzheimer's disease. Theranostics. 2021;11:4103-21
177. Zhang Z ZX, Zhang R, Xie Y, Feng Z, Li F, Han J, Sun H, Ouyang Q, Hua S, Lv B, Hua T, Liu Z, Cai Y, Zou Y, Tang Y, Jiang X. Human umbilical cord mesenchymal stem cell-derived exosomal miR-146a-5p reduces microglial-mediated neuroinflammation via suppression of the IRAK1TRAF6 signaling pathway after ischemic stroke. Aging (Albany NY). 2021;13:3060-79
178. Zhou J, Geng Y, Su T, Wang Q, Ren Y, Zhao J. et al. NMDA receptor-dependent prostaglandin-endoperoxide synthase 2 induction in neurons promotes glial proliferation during brain development and injury. Cell Rep. 2022;38:110557
179. Park A, Croset V, Otto N, Agarwal D, Treiber CD, Meschi E. et al. Gliotransmission of D-serine promotes thirst-directed behaviors in Drosophila. Curr Biol. 2022;32:3952-70 e8
180. Koh W, Park M, Chun YE, Lee J, Shim HS, Park MG. et al. Astrocytes Render Memory Flexible by Releasing D-Serine and Regulating NMDA Receptor Tone in the Hippocampus. Biol Psychiatry. 2022;91:740-52
181. Sun Y, Yang J, Hu X, Gao X, Li Y, Yu M. et al. Conditioned medium from overly excitatory primary astrocytes induced by La(3+) increases apoptosis in primary neurons via upregulating the expression of NMDA receptors. Metallomics. 2018;10:1016-28
182. Green MV, West AE. TRPing into excitotoxic neuronal death. Cell Calcium. 2021;93:102331
183. Qiu LL TX, Yang JJ, Zhang H, Xu N, Zhao C, Sun J. Lactate improves postoperative cognitive function through attenuating oxidative stress and neuroinflammation in aged mice via activating the SIRT1 pathway. Experimental neurology. 2025;385:115136
184. Lu X, Xiong W, Chen Z, Li Y, Xu F, Yang X. et al. Exercise-conditioned plasma ameliorates postoperative cognitive dysfunction by activating hippocampal cholinergic circuit and enhancing BDNF/TrkB signaling. Cell Commun Signal. 2024;22:551
185. Wu WF, Chen C, Lin JT, Jiao XH, Dong W, Wan J. et al. Impaired synaptic plasticity and decreased glutamatergic neuron excitability induced by SIRT1/BDNF downregulation in the hippocampal CA1 region are involved in postoperative cognitive dysfunction. Cell Mol Biol Lett. 2024;29:79
186. Nuzzo T, Punzo D, Devoto P, Rosini E, Paciotti S, Sacchi S. et al. The levels of the NMDA receptor co-agonist D-serine are reduced in the substantia nigra of MPTP-lesioned macaques and in the cerebrospinal fluid of Parkinson's disease patients. Sci Rep. 2019;9:8898
187. Bodner O, Radzishevsky I, Foltyn VN, Touitou A, Valenta AC, Rangel IF. et al. D-Serine Signaling and NMDAR-Mediated Synaptic Plasticity Are Regulated by System A-Type of Glutamine/D-Serine Dual Transporters. J Neurosci. 2020;40:6489-502
188. Le Douce J, Maugard M, Veran J, Matos M, Jego P, Vigneron PA. et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer's Disease. Cell Metab. 2020;31:503-17 e8
189. Luo T, Lin D, Hao Y, Shi R, Wei C, Shen W. et al. Ginkgolide B-mediated therapeutic effects on perioperative neurocognitive dysfunction are associated with the inhibition of iNOS-mediated production of NO. Mol Med Rep. 2021;24:537
190. Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q. et al. NLRP3/Caspase-1 Pathway-Induced Pyroptosis Mediated Cognitive Deficits in a Mouse Model of Sepsis-Associated Encephalopathy. Inflammation. 2019;42:306-18
191. Zhou R, Yang X, Li X, Qu Y, Huang Q, Sun X. et al. Recombinant CC16 inhibits NLRP3/caspase-1-induced pyroptosis through p38 MAPK and ERK signaling pathways in the brain of a neonatal rat model with sepsis. J Neuroinflammation. 2019;16:239
192. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL. et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215:827-40
193. Zhong X, Xie L, Yang X, Liang F, Yang Y, Tong J. et al. Ethyl pyruvate protects against sepsis-associated encephalopathy through inhibiting the NLRP3 inflammasome. Mol Med. 2020;26:55
194. Xu XE, Liu L, Wang YC, Wang CT, Zheng Q, Liu QX. et al. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav Immun. 2019;80:859-70
195. Chen L, Qing W, Yi Z, Lin G, Peng Q, Zhou F. NU9056, a KAT 5 Inhibitor, Treatment Alleviates Brain Dysfunction by Inhibiting NLRP3 Inflammasome Activation, Affecting Gut Microbiota, and Derived Metabolites in LPS-Treated Mice. Front Nutr. 2021;8:701760
196. Zhou S, Li Y, Hong Y, Zhong Z, Zhao M. Puerarin protects against sepsis-associated encephalopathy by inhibiting NLRP3/Caspase-1/GSDMD pyroptosis pathway and reducing blood-brain barrier damage. Eur J Pharmacol. 2023;945:175616
197. Dumbuya JS, Chen X, Du J, Li S, Liang L, Xie H. et al. Hydrogen-rich saline regulates NLRP3 inflammasome activation in sepsis-associated encephalopathy rat model. International Immunopharmacology. 2023;123:110758
198. Wei XB, Jiang WQ, Zeng JH, Huang LQ, Ding HG, Jing YW. et al. Exosome-Derived lncRNA NEAT1 Exacerbates Sepsis-Associated Encephalopathy by Promoting Ferroptosis Through Regulating miR-9-5p/TFRC and GOT1 Axis. Mol Neurobiol. 2022;59:1954-69
199. Yao P, Chen Y, Li Y, Zhang Y, Xu W. Hippocampal neuronal ferroptosis involved in cognitive dysfunction in rats with sepsis-related encephalopathy through the Nrf2/GPX4 signaling pathway. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2019;31:1389-94
200. Li C, Yu TY, Zhang Y, Wei LP, Dong SA, Shi J. et al. Electroacupuncture Improves Cognition in Rats With Sepsis-Associated Encephalopathy. J Surg Res. 2020;256:258-66
201. Zhang L, Zhang X, Wu T, Pan X, Wang Z. Isoflurane reduces septic neuron injury by HO-1-mediated abatement of inflammation and apoptosis. Mol Med Rep. 2021;23:155
202. Haileselassie B, Joshi AU, Minhas PS, Mukherjee R, Andreasson KI, Mochly-Rosen D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J Neuroinflammation. 2020;17:36
203. Yu Y, Feng J, Lian N, Yang M, Xie K, Wang G. et al. Hydrogen gas alleviates blood-brain barrier impairment and cognitive dysfunction of septic mice in an Nrf2-dependent pathway. Int Immunopharmacol. 2020;85:106585
204. Li N, Zhang EF, Zhang J, Zhang L, Liu YE, Jin HX. et al. Therapeutic effects of recombinant human brain natriuretic peptide on sepsis-associated encephalopathy in mice. Int Immunopharmacol. 2020;81:106280
205. Mei B, Li J, Zuo Z. Dexmedetomidine attenuates sepsis-associated inflammation and encephalopathy via central alpha2A adrenoceptor. Brain Behav Immun. 2021;91:296-314
206. Mei B, Xu X, Weng J, Yang Y, Wang P, Qiu G. et al. Activating astrocytic alpha2A adrenoceptors in hippocampus reduces glutamate toxicity to attenuate sepsis-associated encephalopathy in mice. Brain Behav Immun. 2024;117:376-98
207. Li L, Shu MQ, Chen J. CYLD deficiency exacerbates lipopolysaccharide (LPS)-induced pyroptosis in astrocytes of mice with sepsis. Biochem Biophys Res Commun. 2019;514:1066-73
208. Sun YB, Zhao H, Mu DL, Zhang W, Cui J, Wu L. et al. Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 2019;10:167
209. Hong Y, Liu Y, Yu D, Wang M, Hou Y. The neuroprotection of progesterone against Abeta-induced NLRP3-Caspase-1 inflammasome activation via enhancing autophagy in astrocytes. Int Immunopharmacol. 2019;74:105669
210. Mi J, Yang Y, Yao H, Huan Z, Xu C, Ren Z. et al. Inhibition of heat shock protein family A member 8 attenuates spinal cord ischemia-reperfusion injury via astrocyte NF-kappaB/NLRP3 inflammasome pathway: HSPA8 inhibition protects spinal ischemia-reperfusion injury. J Neuroinflammation. 2021;18:170
211. Venturini A, Passalacqua M, Pelassa S, Pastorino F, Tedesco M, Cortese K. et al. Exosomes From Astrocyte Processes: Signaling to Neurons. Front Pharmacol. 2019;10:1452
212. Pei X, Li Y, Zhu L, Zhou Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp Cell Res. 2019;382:111474
213. Wang J, Sareddy GR, Lu Y, Pratap UP, Tang F, Greene KM. et al. Astrocyte-Derived Estrogen Regulates Reactive Astrogliosis and is Neuroprotective following Ischemic Brain Injury. J Neurosci. 2020;40:9751-71
214. Shan Y, Tan S, Lin Y, Liao S, Zhang B, Chen X. et al. The glucagon-like peptide-1 receptor agonist reduces inflammation and blood-brain barrier breakdown in an astrocyte-dependent manner in experimental stroke. J Neuroinflammation. 2019;16:242
215. Zhu Y, Yan P, Wang R, Lai J, Tang H, Xiao X. et al. Opioid-induced fragile-like regulatory T cells contribute to withdrawal. Cell. 2023;186:591-606 e23
216. Song S, Huang H, Guan X, Fiesler V, Bhuiyan MIH, Liu R. et al. Activation of endothelial Wnt/beta-catenin signaling by protective astrocytes repairs BBB damage in ischemic stroke. Prog Neurobiol. 2021;199:101963
217. Guerit S, Fidan E, Macas J, Czupalla CJ, Figueiredo R, Vijikumar A. et al. Astrocyte-derived Wnt growth factors are required for endothelial blood-brain barrier maintenance. Prog Neurobiol. 2021;199:101937
218. Mills WA 3rd, Woo AM, Jiang S, Martin J, Surendran D, Bergstresser M. et al. Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat Commun. 2022;13:1794
219. Zhang Q, Liu C, Shi R, Zhou S, Shan H, Deng L. et al. Blocking C3d(+)/GFAP(+) A1 Astrocyte Conversion with Semaglutide Attenuates Blood-Brain Barrier Disruption in Mice after Ischemic Stroke. Aging Dis. 2022;13:943-59
220. Hermans D, Houben E, Baeten P, Slaets H, Janssens K, Hoeks C. et al. Oncostatin M triggers brain inflammation by compromising blood-brain barrier integrity. Acta Neuropathol. 2022;144:259-81
221. Kim H LK, Park J, Sorets AG, Kim S, Shostak A. et al. Reactive astrocytes transduce inflammation in a blood-brain barrier model through a TNF-STAT3 signaling axis and secretion of alpha 1-antichymotrypsin. Nat Commun. 2022;13:6581
222. Jackson RJ, Meltzer JC, Nguyen H, Commins C, Bennett RE, Hudry E. et al. APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain. 2022;145:3582-93
223. Feng D ZJ, Liu H, Wu X, Li F, Zhao J. et al. Astrocytic NDRG2-PPM1A interaction exacerbates blood-brain barrier disruption after subarachnoid hemorrhage. Sci Adv. 2022;8:eabq2423
224. Qin X WJ, Chen S, Liu G, Wu C, Lv Q. et al. Astrocytic p75(NTR) expression provoked by ischemic stroke exacerbates the blood-brain barrier disruption. Glia. 2022;70:892-912
Author contact
![]() Corresponding authors: Honghai Zhang, E-mail: zhanghonghai_0902com; Xuesheng Liu, E-mail: liuxueshengedu.cn.
Corresponding authors: Honghai Zhang, E-mail: zhanghonghai_0902com; Xuesheng Liu, E-mail: liuxueshengedu.cn.