Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and methods
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(8):3379-3396. doi:10.7150/ijbs.106219 This issue Cite
Research Paper
Pterostilbene Induces Apoptosis in Awakening Quiescent Prostate Cancer Cells by Upregulating C/EBP-β-Mediated SOD2 Transcription
Zhichao Xi1,2†, Mengfan Liu1,2†, Xue Jiang1,2, Jiling Feng1,2,3, Rongchen Dai1,2, Wan Najbah Nik Nabil1,2,4, Xueyang Sun1,2, Jiayi Chen1,2, Hangui Ren1,2, Juan Zhang5, Qihan Dong6,7, Man Yuan1,2 ![]() , Yang Li1,2
, Yang Li1,2 ![]() , Hongxi Xu1,2
, Hongxi Xu1,2 ![]()
1. School of Pharmacy, Shanghai University of Traditional Chinese Medicine, No. 1200, Cailun Road, Shanghai 201203, China
2. Engineering Research Center of Shanghai Colleges for TCM New Drug Discovery, No. 1200, Cailun Road, Shanghai 201203, China
3. Precision Research Center for Refractory Diseases, Institute for Clinical Research, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 201620, China
4. National Pharmaceutical Regulatory Agency, Ministry of Health, Lot 36, Jalan Universiti, Petaling Jaya, Selangor 46200, Malaysia
5. School of Chinese Medicine, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong SAR 999077, China
6. Chinese Medicine Anti-Cancer Evaluation Program, Greg Brown Laboratory, Central Clinical School and Charles Perkins Centre, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia
7. Department of Endocrinology, Royal Prince Alfred Hospital, Sydney, NSW 2050, Australia
†These authors contributed equally to this work.
Received 2024-11-2; Accepted 2025-4-23; Published 2025-5-7
Abstract

Quiescent cancer cells (QCCs) are known to resist chemoradiotherapy, evade immune surveillance and have the potential to drive recurrence years after initial treatment. However, the key regulators of QCC survival during reactivation remain unclear. This study revealed that superoxide dismutase 2 (SOD2) levels are significantly greater in quiescent prostate cancer (PCa) cells than in proliferative cells. SOD2 overexpression induces apoptosis in awakening quiescent PCa cells, whereas its knockdown promotes reactivation. Elevated SOD2 also suppresses recurrent tumor growth by quiescent PCa cells and prolongs survival. Pterostilbene (PTE), a natural compound, preferentially induces apoptosis in quiescent PCa cells during awakening and reduces their long-term proliferative capacity by upregulating SOD2. Additionally, PTE inhibits tumorigenesis and significantly reduces the growth of quiescent PCa cells without apparent toxicity. Further mechanistic studies revealed that CCAAT/enhancer-binding protein beta (C/EBP-β) is critical for PTE-mediated SOD2 upregulation by enhancing SOD2 transcription. C/EBP-β knockdown significantly reduces PTE-induced apoptosis in awakening quiescent PCa cells. Clinical analysis revealed a positive correlation between CEBPB and SOD2, with low C/EBP-β expression linked to poor prognosis. Overall, the C/EBP-β-SOD2 pathway is crucial for eliminating awakening quiescent PCa cells and highlights PTE as a promising agent for preventing PCa recurrence.
Keywords: Quiescent cancer cells, Prostate cancer recurrence, SOD2, C/EBP-β, Pterostilbene, Apoptosis
Introduction
Despite considerable advances in primary cancer therapies, a significant proportion of patients still experience late recurrences following prolonged, disease-free intervals [1]. Quiescent cancer cells (QCCs) experience a reversible arrest in the G0 phase of the cell cycle and pose a significant therapeutic challenge, as they are clinically undetectable, resistant to chemotherapy and immune privileged, allowing them to survive for years or even decades [2, 3]. When QCCs exit the G0 phase, their cell cycle reentry (also termed “awakening”) restores proliferative capacity, thereby driving tumor recurrence [4]. Several signaling pathways have been implicated in the transition between quiescence and proliferation in cancer cells. For example, the FBXW7-c-Myc axis and fibroblasts-derived collagen-I promote cancer cells exit from quiescence [5-7], whereas factors such as DYRK1B, Axl tyrosine kinase, and integrins inhibit QCC reactivation and enhance chemotherapy sensitivity [8-10]. Moreover, STING suppresses QCC-driven metastatic recurrence by establishing an immunosuppressive microenvironment [11]. Despite these investigations, the key regulators that sustain QCC survival during reawakening remain elusive.
Our previous proteomic and transcriptomic analyses identified superoxide dismutase 2 (SOD2) as the most upregulated molecule in quiescent prostate cancer (PCa) cells. Clinical data further reveal that reduced SOD2 expression correlates with poorer survival and higher recurrence rates in PCa patients, emphasizing its significance as a key regulator of QCC reactivation. In quiescent cells, elevated SOD2 facilitate the transition of mouse embryonic fibroblasts into quiescence, a process associated with SOD2-mediated alterations in glucose metabolism [12-14]. Moreover, CCAAT/enhancer-binding protein beta (C/EBP-β), one of the primary transcription factors regulating SOD2 expression [15-17], is essential for maintaining the noncycling state of satellite cells and fibroblasts, with its loss promoting their exit from quiescence [18, 19]. Although current studies implicate both C/EBP-β and SOD2 in the transition between quiescence and proliferation, the precise mechanism in the reactivation process of QCCs remains to be fully elucidated.
In this study, we demonstrated the detrimental role of SOD2 in awakening process. Notably, forced elevation of SOD2 induces apoptosis in awakening QCCs, while its knockdown accelerates cell cycle reentry. Pterostilbene (PTE) is a potent SOD2 activator that is commonly found in blueberries, grapes and herbal medicines [20, 21]. It significantly induces apoptosis in awakening PCa cells and suppresses recurrent tumor growth without apparent toxicity. Further mechanistic studies revealed that C/EBP-β plays a pivotal role in PTE-mediated SOD2 upregulation by increasing its transcription. The present study provides new insights into the role of the C/EBP-β-SOD2 axis in eradicating awakening quiescent PCa cells and highlights PTE as a promising agent for preventing PCa recurrence.
Results
PTE eradicates awakening quiescent PCa cells by inducing apoptosis
Experimental quiescence was induced in DU145 and LNCaP cells by withdrawing serum for 7 days. Subsequently, reintroducing serum triggered cell cycle re-entry and the reactivation of QCCs [5, 22]. DNA resynthesis is a hallmark of quiescent cancer cells resuming proliferation [23]. To assess changes in DNA content during the awakening of quiescent PCa cells with or without PTE, we performed a SYBR Green assay. Seventy-two hours after serum replenishment, PTE significantly reduced the DNA content in awakening PCa cells in a dose-dependent manner compared with the DMSO control group (Fig. 1A, B). Notably, following treatment with 75 μM PTE, the DNA content was even lower than that at baseline, suggesting that PTE induced death in quiescent PCa cells during reawakening. The inhibitory concentration (IC) values of PTE (IC25, IC50, IC75 and IC90) in awakening quiescent PCa cells were calculated (Table 1).
PTE eradicates awakening quiescent PCa cells by inducing apoptosis. A, B Quiescent DU145 (A) and LNCaP (B) cells were induced to re-enter the cell cycle, then exposed to various concentrations of PTE for 72 h. DNA content was subsequently measured using a SYBR Green assay. C, D Flow cytometric analysis with PI staining was performed to evaluate the cell cycle phase distributions in DU145 (C) and LNCaP (D) cells in the proliferative, quiescent, and cell cycle reentry phases, with or without PTE (IC90), at the indicated time points. E, F Quiescent DU145 and LNCaP cells were induced to re-enter the cell cycle with DMSO, PTE (IC50 and IC90) or 50 μM Z-VAD-FMK, either alone or in combination, for 48 h. Representative images (E) and quantification data of the percentage of apoptotic cells (F) were determined by flow cytometry with Annexin V-FITC/PI staining. G, H Western blotting was used to assess the protein expression levels of (cleaved-)caspase-3, (cleaved-)caspase-9, and (cleaved-)PARP in DU145 (G) and LNCaP (H) cells after PTE (IC90) treatment at the indicated times, with β-actin serving as the loading control. I, J Quiescent DU145 and LNCaP cells were induced to resume proliferation with or without PTE (IC25, IC50 and IC75) for 24, 48 and 72 h. Afterward, cells were cultured in PTE-free complete medium for 14 days and then stained with crystal violet. Representative images and colony quantification data of DU145 (I) and LNCaP (J) cells are presented. 'Cont' refers to proliferative control cells, and 'Qsct' indicates quiescent cells. The data are presented as the means ± standard deviations from three biological replicates. One-way ANOVA was applied to panels A and B; two-way ANOVA to panels C, D, I, and J; and a t‑test to panel F. *P < 0.05, **P < 0.01, ***P < 0.001 versus the indicated groups.
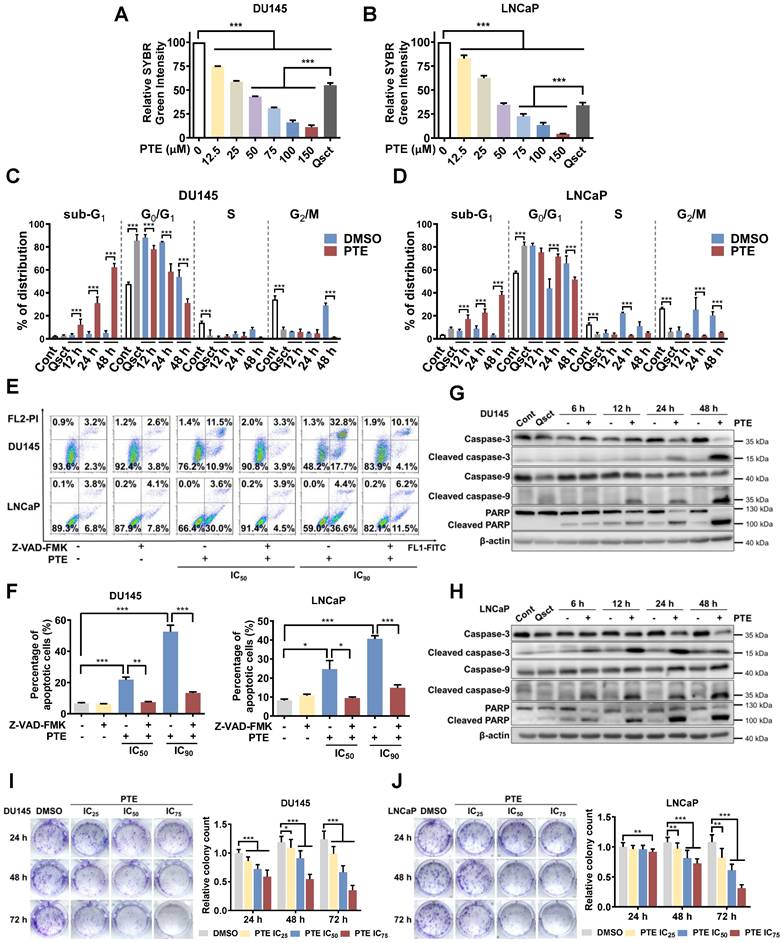
IC values of PTE in quiescent PCa cells
| IC% | DU145 (μM) | LNCaP (μM) |
|---|---|---|
| IC25 | 17.51 ± 0.51 | 19.73 ± 1.21 |
| IC50 | 34.45 ± 0.74 | 34.31 ± 1.44 |
| IC75 | 67.79 ± 2.42 | 59.73 ± 2.43 |
| IC90 | 133.47 ± 7.58 | 104.04 ± 5.98 |
The IC values of PTE were calculated based on the results of the SYBR Green assay (Fig. 1A, B) using GraphPad Prism 10. The IC values of each cell line are shown as the mean ± standard errors.
We further examined the cell cycle distribution during reactivation by employing PI-stained cells and flow cytometry. After 7 days of serum withdrawal, 85% of the DU145 cells and 81% of the LNCaP cells were in the G0/G1 phase, whereas 47% and 57% were in complete medium, respectively (Fig. 1C, D). Co-staining with Hoechst 33258 and Pyronin Y confirmed a pronounced increase in G0-phase cells in both PCa cell lines during quiescence (Fig. S1A, B). Twenty-four hours after release from the quiescent stage, PCa cells in the control group successfully re-entered the S and G2/M phases without significant accumulation in sub-G1. However, PTE significantly elevated the sub-G1 proportion within 48 h of reactivation compared with DMSO treatment (Fig. 1C, D; Fig. S1C). Annexin V-FITC/PI staining further revealed that PTE induced apoptosis in a dose-dependent manner, an effect that was substantially reversed by the pancaspase inhibitor Z-VAD-FMK (Fig. 1E, F). PTE activated caspase-3, caspase-9 and PARP in awakening quiescent PCa cells (Fig. 1G, H). Additionally, PTE treatment significantly diminished the mitochondrial membrane potential in these cells, corroborating the impairment of mitochondrial function (Fig. S1D). Notably, the same PTE treatment for 48 h resulted in less than 10% apoptosis in proliferative PCa cells (Fig. S1E).
To evaluate PTE's long-term inhibitory effect on awakening PCa cells, quiescent DU145 and LNCaP cells were treated with DMSO or PTE at the IC25, IC50 and IC75 concentrations for 24, 48 and 72 h during reactivation, followed by an additional 14 days of culture without PTE. Compared with the control, PTE markedly decreased colony formation in a dose- and time-dependent manner (Fig. 1I, J). Overall, these findings suggest that PTE preferentially induces apoptosis in PCa cells during their awakening from quiescence and reduces their long-term reproliferative ability.
SOD2 overexpression induces apoptosis in awakening quiescent PCa cells
Our previous multi-omics analyses identified SOD2 as the most upregulated molecule in quiescent PCa cells, we verified its expression during the reactivation process. In quiescent PCa cells, SOD2 protein and mRNA levels were significantly elevated than those in proliferative cancer cells, and these levels decreased during the awakening process (Fig. S2A-D). Interestingly, PTE treatment hindered the decrease in SOD2 protein levels in awakening PCa cells during cell cycle reentry (Fig. 2A). These observations indicate that SOD2 may have a regulatory function in the reactivation of quiescent PCa cells. Additionally, increased levels of phospho-Rb (Ser807/811) and decreased levels of p27 indicate progression of cell cycle reactivation, confirming the successful establishment of the quiescent model [5, 24] (Fig. S2A, B).
SOD2 overexpression induces apoptosis in awakening quiescent PCa cells. A The levels of SOD2 protein in PTE (IC90)-treated DU145 and LNCaP cells during cell cycle reentry at the indicated time points. B, C Protein (B) and mRNA (C) expression levels of SOD2 in DU145 and LNCaP cells stably transfected with either a SOD2-overexpression plasmid (SOD2) or a parental empty vector (EV). GAPDH and TBP were used as loading controls for western blotting and RT‒PCR, respectively. D, E Quiescent EV and SOD2-overexpressing DU145 and LNCaP cells were induced to re-enter the cell cycle. Cell viability (D) and cell cycle distribution (E) were assessed by flow cytometry using PI staining at the indicated intervals. F, G Following release from quiescence, EV and SOD2-overexpressing PCa cells were treated with or without Z-VAD-FMK (50 μM) for 12 or 24 h. Apoptotic cells (F) and the mitochondrial membrane potential (G) were measured by flow cytometry with Annexin V-FITC/PI and JC-1 staining, respectively. H, I Expression of apoptosis-related proteins in quiescent EV and SOD2-overexpressing DU145 (H) and LNCaP (I) cells during cell cycle reentry, with β-actin used as the loading control. J, K Apoptosis in SOD2-overexpressing DU145 (J) and LNCaP (K) cells, along with their EV control cells, was assessed by flow cytometry with Annexin V-FITC/PI staining after 48 h of PTE (IC90) treatment during cell cycle reentry. 'Cont' refers to proliferative control cells, and 'Qsct' indicates quiescent cells. The data are presented as means ± standard deviations from three individual experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the indicated groups.
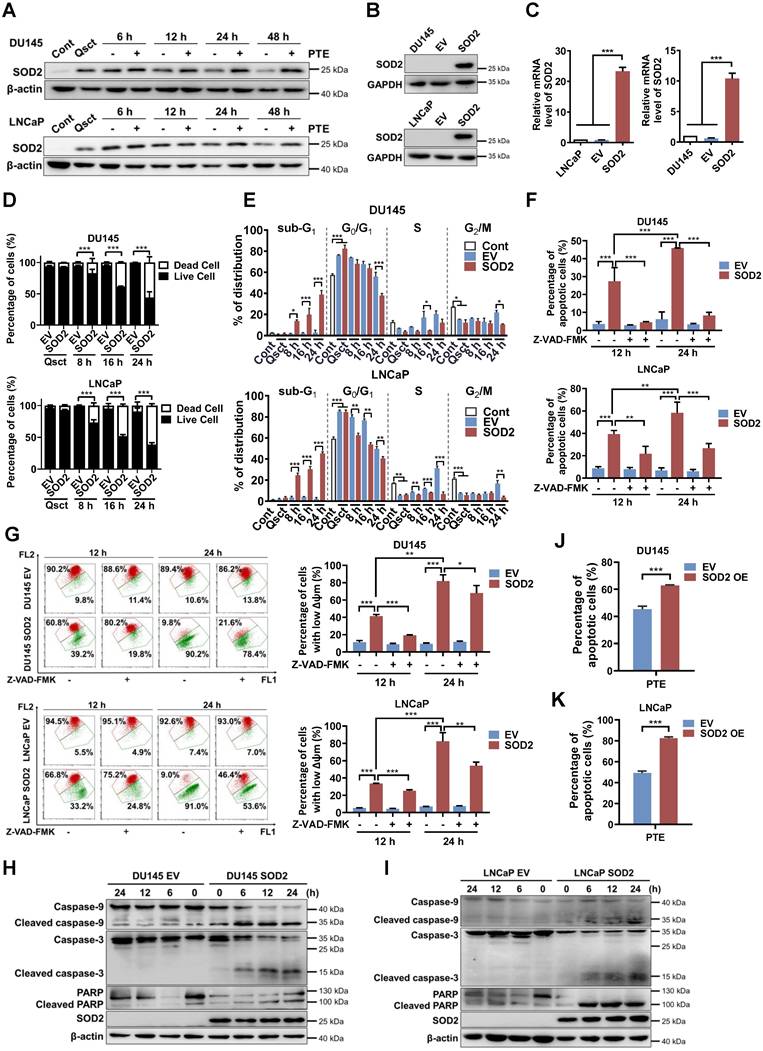
The primary function of SOD2 is to scavenge superoxide anion radicals and reduce mitochondrial ROS, thereby protecting against oxidative-induced cell death [25, 26]. We first investigated the changes in mitochondrial ROS levels in response to PTE-induced SOD2 upregulation and apoptosis. Interestingly, we observed that the elevated SOD2 levels in quiescent PCa cells did not result in lower mitochondrial ROS levels than those in proliferative cells (Fig. S3A, B). PTE treatment increased mitochondrial ROS levels in quiescent PCa cells as they re-entered the cell cycle (Fig. S3C, D). Notably, the mitochondrial ROS scavenger, mitoTEMPO, neither reduced ROS levels nor prevented PTE-induced apoptosis. These findings suggest that PTE-induced SOD2 promotes apoptosis independently of mitochondrial ROS accumulation (Fig. S3C-F).
We then investigated whether SOD2 overexpression could inhibit the awakening of quiescent PCa cells. DU145 and LNCaP cells were stably transfected with a full-length SOD2 cDNA construct or an empty vector (EV). This transfection significantly elevated SOD2 protein and mRNA levels (Fig. 2B, C). Notably, 90.7% of EV PCa cells remained viable 24 h after reentry into the cell cycle, whereas SOD2 overexpression induced approximately 58.5% and 61.4% cell death in reactivated DU145 cells and LNCaP cells, respectively (Fig. 2D). Consistently, SOD2 overexpression increased the sub-G1 population as early as 8 h after reactivation in awakening PCa cells compared with EV PCa cells (Fig. 2E).
Further analysis using Annexin V-FITC/PI staining revealed that SOD2 overexpression significantly induced apoptosis during the reactivation of quiescent DU145 and LNCaP cells, and this effect was significantly reversed by Z-VAD-FMK treatment (Fig. 2F, Fig. S4A, B). Additionally, SOD2-overexpressing PCa cells presented a loss of mitochondrial membrane potential following cell cycle reentry, which was mitigated by Z-VAD-FMK, indicating the occurrence of mitochondria-dependent apoptosis (Fig. 2G). Caspase-3, caspase-9 and PARP were activated in SOD2-overexpressing PCa cells upon exiting quiescence (Fig. 2H, I). Importantly, the combination of SOD2 overexpression and PTE treatment further increased apoptosis in awakening quiescent PCa cells (Fig. 2J, K; Fig. S4C). These findings suggest that SOD2 is integral to the reactivation process and could be a promising therapeutic target for preventing PCa recurrence.
SOD2 overexpression suppresses the growth of recurrent tumors from quiescent DU145 cells and prolongs mouse survival
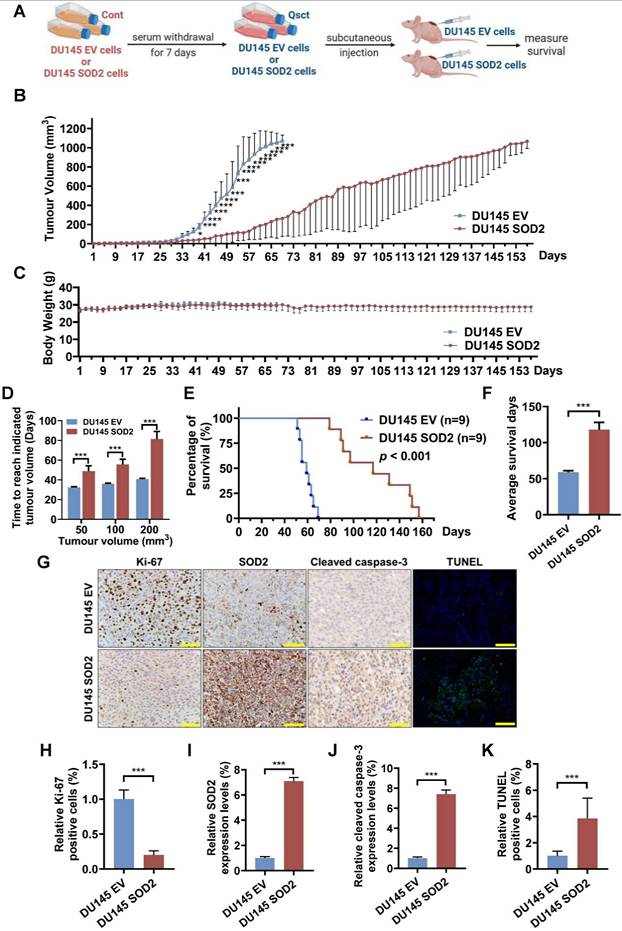
To evaluate the effect of SOD2 overexpression on the growth of recurrent tumors from quiescent PCa cells in vivo, quiescent control (DU145 EV) cells and SOD2-overexpressing (DU145 SOD2) cells were inoculated subcutaneously into nude mice (Fig. 3A). The growth rate of recurrent tumors in the SOD2-overexpressing group was significantly slower than that in the EV control group (Fig. 3B). No notable differences in body weight were observed between the two groups (Fig. 3C). The average times required to reach tumor volumes of 50, 100 and 200 mm³ were substantially prolonged in the SOD2-overexpressing group (Fig. 3D). Survival analysis revealed that SOD2 overexpression nearly doubled survival days relative to EV control group (Fig. 3E, F). Additionally, tumors from SOD2 overexpression group maintained high SOD2 levels, exhibited fewer Ki-67-positive cells and higher amount of cleaved caspase-3- and TUNEL-positive cells (Fig. 3G-K). These in vivo results demonstrate that elevated SOD2 expression suppresses the growth of recurrent tumors from quiescent PCa cells and prolongs survival.
SOD2 overexpression suppresses the tumorigenicity of quiescent DU145 cells and prolongs the mouse survival. A Schematic diagram of the mouse model for recurrent PCa using quiescent DU145 EV or DU145 SOD2 cells. B, C Tumor volumes (B) and mouse body weights (C) were measured every other day, with euthanasia performed when tumor size reached 1000 mm3. D The average times for tumors in the DU145 EV and DU145 SOD2 groups to reach volumes of 50, 100, and 200 mm3. E, F Survival curve analysis (E) and quantification data (F) showing the overall survival probability of the mice in each group. G-K For immunohistochemical analysis, three randomly selected views from each section (n = 3 per group) were quantitatively analyzed using ImageJ software. A representative image is shown in panel G, and quantification data for Ki‑67, SOD2, cleaved caspase‑3, and TUNEL staining in resected tumors are provided in panels H-K. Scale bar: 50 μm. The data are presented as the means ± standard deviations. *P < 0.05, ***P < 0.001 versus the control group or indicated group.
Knockdown of SOD2 promotes the awakening of quiescent PCa cells and reduces the apoptotic effects of PTE
To further explore the regulatory role of SOD2 in the reactivation of quiescent PCa cells, we established stable cell lines with doxycycline (DOX)-inducible SOD2 shRNA (DOX-shSOD2). PCa cells stably transfected with a parental empty vector (DOX-shCon) were used as controls. DOX treatment effectively reduced both SOD2 protein and mRNA expression levels in DU145 shSOD2 (Fig. 4A, C) and LNCaP shSOD2 cells (Fig. 4B, D). PCa cells, including nontransfected, DOX-shCon and DOX-shSOD2 cells, were treated with or without DOX on Day 5 of 7-day serum withdrawal and then reactivated into the cell cycle. SOD2 knockdown significantly increased the number of quiescent DU145 and LNCaP cells 48 h after serum reintroduction, indicating enhanced reproliferative capacity (Fig. 4E, F). In contrast, DOX treatment had no effect on the awakening of quiescent nontransfected or DOX-shCon PCa cells. Further analysis revealed that, compared with non-DOX-treated cells, DOX-treated shSOD2 PCa cells exhibited an accelerated shift from the G0/G1 to S and G2/M phases at 24 and 32 h (Fig. 4G, H). These findings further highlight the importance of SOD2 in quiescent PCa cell reactivation.
Knockdown of SOD2 promotes cell cycle reentry in quiescent PCa cells. A-D SOD2 protein and mRNA levels in nontransfected, DOX-inducible shCon or shSOD2 plasmid-transfected DU145 (A, C) and LNCaP (B, D) cells were analyzed by western blotting and RT‒PCR. E, F The reproliferative activity of the indicated cells during reactivation was assessed using a Trypan blue viability assay for 0‒120 h (normalized to 0 h). The cells were treated with DOX (4 μg/ml) for 48 h prior to reactivation. G, H Quiescent DU145 shSOD2 (G) and LNCaP shSOD2 (H) cells, with or without DOX treatment, were induced to re-enter the cell cycle, harvested at 24 and 32 h, and subjected to flow cytometry with PI staining for cell cycle distribution analysis. I, J Quiescent DU145 shSOD2 (I) and LNCaP shSOD2 (J) cells, in the presence or absence of DOX, were treated with either DMSO or PTE (IC50 and IC90) for 48 h. Apoptosis was assessed by flow cytometry with Annexin V-APC/PI staining. 'Cont' refers to proliferative control cells, and 'Qsct' indicates quiescent cells. The data are presented as the means ± standard deviations from three individual experiments. **P < 0.01, ***P < 0.001 versus the indicated groups.
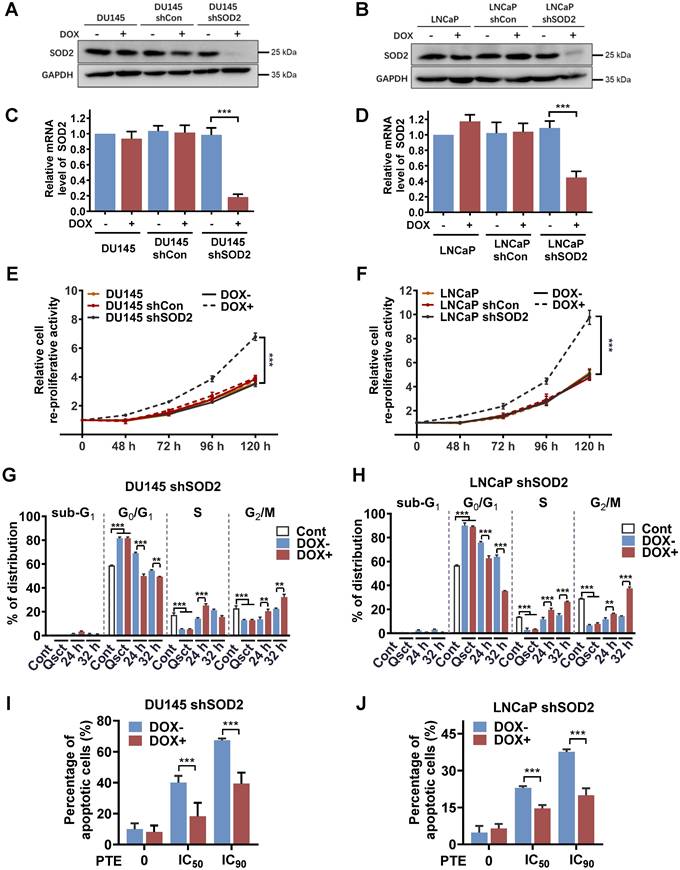
A rescue experiment was conducted to confirm the role of SOD2 in PTE-induced apoptosis during the awakening of quiescent PCa cells. DOX was administered to PCa shSOD2 cells 48 h before inducing cell cycle reentry. Subsequently, the cells were released from quiescence and exposed to PTE or control treatment for 48 h. SOD2 knockdown significantly reduced PTE-induced apoptosis in awakening quiescent DU145 and LNCaP cells. Notably, rescue percentages of 52.7% and 63.8% were achieved in PTE IC50- and IC90-treated DU145 shSOD2 cells, respectively (Fig. 4I, Fig. S5A). Similarly, rescue rates of 35.9% and 44.0% were observed in PTE IC50- and IC90-treated LNCaP shSOD2 cells, respectively (Fig. 4J, Fig. S5B). These results further confirm that PTE induces apoptosis during the awakening of quiescent PCa cells by increasing SOD2 expression.
PTE upregulates SOD2 at the transcriptional level via C/EBP-β
To further investigate the mechanism by which PTE upregulates SOD2, we first examined whether PTE treatment alters SOD2 transcription. The results demonstrated that SOD2 mRNA expression was greater in quiescent PCa cells than in proliferative cells. Upon release from quiescence, the mRNA expression of SOD2 decreased during the awakening of PCa cells, whereas PTE treatment significantly prevented this decrease in SOD2 mRNA expression (Fig. 5A), indicating PTE affects the transcription of SOD2. C/EBP-β, a transcription factor known to regulate SOD2 transcription [17, 27], was significantly upregulated by PTE treatment in a time- and dose-dependent manner after PCa cell reawakening (Fig. 5B, C). PTE markedly promoted the nuclear accumulation of C/EBP-β in awakening DU145 and LNCaP cells at 12 and 24 h after release from quiescence, accompanied by increased cytoplasmic SOD2 expression (Fig. 5D). To further confirm the transcriptional activity of C/EBP-β in regulating SOD2 expression after PTE treatment, ChIP analysis was performed. Compared with the control group, antibody against C/EBP-β significantly increased the interaction with SOD2 intronic enhancer following PTE treatment during the reactivation of quiescent PCa cells (Fig. 5E).
PTE upregulates SOD2 at the transcriptional level via C/EBP-β. A DU145 and LNCaP cells treated with PTE (IC90) during cell cycle reentry were analyzed for SOD2 mRNA levels by RT‒PCR, with values normalized to TBP. B Western blot analysis of C/EBP-β and SOD2 in awakening quiescent DU145 and LNCaP cells with PTE (IC90) treatment for the indicated intervals. GAPDH served as a loading control. C Western blotting was used to assess the expression of C/EBP-β and SOD2 in DU145 and LNCaP cells after PTE treatment at the indicated concentrations for 48 h. GAPDH was used as the loading control. D Protein expression levels of C/EBP-β and SOD2 in the nuclear and cytoplasmic extracts of awakening quiescent DU145 and LNCaP cells were analyzed using western blotting after PTE (IC90) treatment for 12 and 24 h. GAPDH and Lamin A/C were used as loading controls for cytoplasmic and nuclear extracts, respectively. E ChIP assay shows the enrichment of C/EBP-β on the SOD2 enhancer in awakening DU145 and LNCaP cells treated with either DMSO or PTE (IC90) for 24 h. F-H Western blotting (F) and RT‒PCR (G, H) analyses of C/EBP-β and SOD2 in quiescent DU145 and LNCaP cells transfected with the scramble and shC/EBP-β constructs. I, J Cell apoptosis was assessed by flow cytometry with Annexin V-FITC/PI staining (I), and apoptosis-related protein levels were detected by western blotting (J) in quiescent DU145 and LNCaP cells with scramble control or shC/EBP-β knockdown after 48 h of PTE (IC90) treatment. K C/EBP-β expression levels in the prostate tissues of patients with PRAD (n=492) and normal controls (n=152) were analyzed using the GEPIA database. L Kaplan‒Meier survival curves from the GEPIA database showing the correlation between C/EBP-β expression and overall survival in patients with PRAD. M TCGA data revealed a positive association between CEBPB and SOD2. The data are presented as the means ± standard deviations from three individual experiments. n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001 versus the indicated groups.
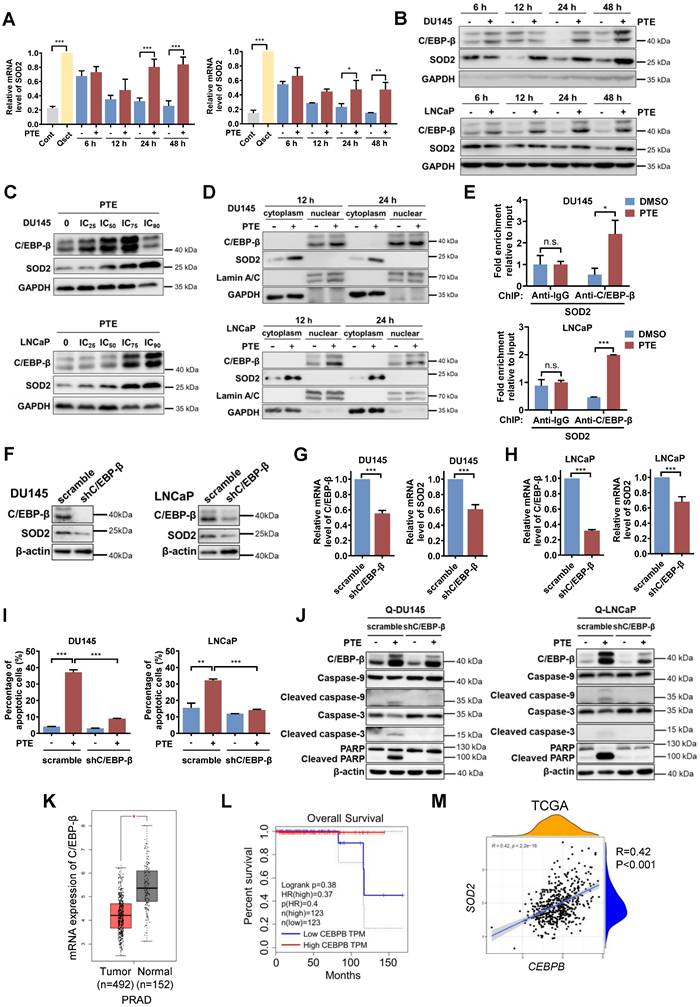
To determine whether C/EBP-β plays a crucial role in PTE-induced apoptosis in awakening PCa cells, we established stable C/EBP-β knockdown DU145 and LNCaP cell lines using lentiviral transfection of shC/EBP-β plasmids, with a nonsense plasmid (scramble) used as a control. Knockdown of C/EBP-β in quiescent PCa cells led to reductions in both C/EBP-β and SOD2 protein levels (Fig. 5F) as well as in C/EBP-β mRNA levels (Fig. 5G, H). C/EBP-β knockdown significantly reduced PTE-induced apoptosis in awakening quiescent PCa cells compared with control cells (Fig. 5I; Fig. S6C, D). Consistently, the PTE-induced increases in the expression of cleaved caspase-3, caspase-9 and PARP were diminished by C/EBP-β knockdown (Fig. 5J). Taken together, PTE upregulates SOD2 and induces apoptosis in awakening PCa cells, at least partially by enhancing the transcriptional activity of C/EBP-β on SOD2.
To further evaluate the clinical relevance of C/EBP-β in human PCa tissue, we examined its expression using the publicly available GEPIA database, which includes 492 samples from patients with PRAD and 152 normal human prostate samples (Fig. 5K). The results revealed that C/EBP-β mRNA expression was significantly lower in PRAD tissues than in normal prostate tissues. Kaplan‒Meier survival analysis revealed that reduced C/EBP-β in PRAD expression correlated with reduced overall survival in patients with PCa (Fig. 5L). Additionally, analysis of the TCGA database revealed a positive correlation between CEBPB and SOD2 (Fig. 5M), indicating that CEBPB tightly regulates SOD2 expression. Together, these data indicate that C/EBP-β plays a critical role in PTE-induced eradication of awakening PCa cells from quiescence by upregulating SOD2, highlighting its therapeutic potential for preventing PCa recurrence.
PTE suppresses the growth of recurrent tumors derived from quiescent PCa cells in vivo
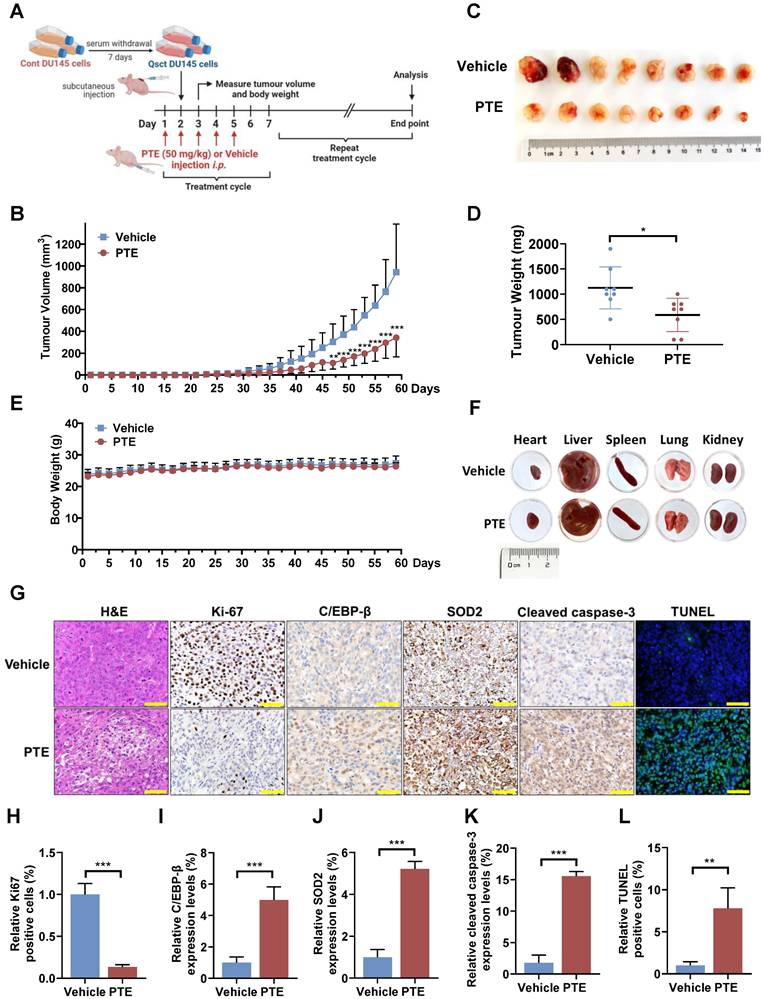
The reactivation of quiescent cancer cells is widely recognized as a key contributor to cancer recurrence [28, 29]. To further investigate the potential of PTE in preventing the reawakening of quiescent PCa cells in vivo, quiescent DU145 cells were subcutaneously implanted in mice to allow for recurrent tumor growth. The mice were administered PTE (50 mg/kg) or vehicle intraperitoneally starting the day before cell implantation and continuing weekly for five consecutive days over two months (Fig. 6A). In the vehicle-treated group, measurable tumors (100 mm³) began to appear by Day 39, with tumor growth progressing until the ethical endpoint (1000 mm³) was reached on Day 59 (Fig. 6B). In contrast, the PTE-treated group exhibited a significant delay in tumor regrowth, with measurable tumors appearing on Day 49. Compared with vehicle-treated mice, PTE-treated mice developed smaller tumors, with an average reduction in tumor weight of 65% after 59 days (Fig. 6C, D). Moreover, no significant changes in body weight or the gross anatomy of major organs were observed during PTE treatment (Fig. 6E, F). Histological analysis of tumors from PTE-treated mice revealed decreased cell density (H&E staining), a decreased number of Ki-67-positive cells, elevated protein levels of C/EBP-β, SOD2 and cleaved caspase-3, and an increased number of TUNEL-positive cells (Fig. 6G-L).
PTE suppresses the regrowth of quiescent PCa tumors in vivo. A Schematic diagram of quiescent PCa tumor regrowth in a PTE-treated xenograft model. i.p., intraperitoneally. B-E Tumor volumes (B) and mouse body weights (E) were measured every other day, and the mice were euthanized when the tumor volume reached 1000 mm3. On Day 59, the tumors were excised, photographed (C) and weighed (D). F Representative heart, liver, spleen, lung and kidney morphologies in mice treated with vehicle control or PTE (50 mg/kg) on the day of sacrifice. G-L Paraffin-embedded tumor tissues were subjected to H&E staining, TUNEL, and immunostaining with antibodies against Ki-67, C/EBP-β, SOD2 and cleaved caspase-3. Three randomly selected fields per section (n = 3 per group) were quantitatively analyzed using ImageJ software. Representative images (G) and quantification data (H-L) are presented. Scale bar: 50 μm. The data are presented as the means ± standard deviations. *P < 0.05, **P < 0.01, ***P < 0.001 versus the control group or indicated group.
Discussion
Androgen signaling, predominantly mediated through the androgen receptor (AR), is essential for the initiation and progression of PCa. Androgen-deprivation therapy (ADT), which inhibits androgen production or suppresses AR function, remains the first-line treatment for advanced and metastatic PCa [30, 31]. Although ADT initially results in disease regression, most patients experience only a transient response and ultimately develop to castration-resistant prostate cancer (CRPC) [32, 33]. Following treatments such as prostatectomy, radiotherapy, or ADT, a dormant phase may occur during which residual QCCs persist. These cells can later reactivate, ultimately leading to tumor recurrence [34]. PCa thus serves as an ideal model for investigating tumor dormancy, given that many patients often remain disease-free for over a decade post-prostatectomy before developing biochemical recurrence or metastasis [35, 36]. Therefore, strategies aimed at preventing the reactivation of QCCs hold significant promise for reducing recurrence and improving long-term patient outcomes. In our study, we demonstrate that SOD2 overexpression is critical for preventing QCC reactivation by inducing apoptosis, suppressing recurrent tumor growth, and prolonging survival in preclinical models. Moreover, our findings report for the first time that PTE effectively blocks the reawakening of QCCs through apoptosis induction, highlighting its potential to prevent PCa recurrence.
PTE, a natural compound found in herbal medicines and edible fruits such as blueberries and grapes, is typically produced through both biosynthesis and chemical synthesis. PTE has exhibited no significant toxicity in both preclinical and clinical trials [21, 37, 38], making it a promising and cost-effective drug candidate for long-term therapy. Notably, our results revealed a robust induction of apoptosis by PTE in both LNCaP (AR-positive) and DU145 (AR-negative) cells during awakening. This finding holds significant clinical implications for treating ADT-resistant PCa phenotypes such as CRPC. Developing clinically pertinent patient-derived xenograft (PDX) models, using serum prostate specific antigen (PSA) levels and tumor volume as key indicators, can better mimic ADT-induced dormancy in the clinic [32, 39, 40]. In parallel, it is imperative to establish in vitro models using media supplemented with charcoal-stripped FBS to simulate prolonged androgen-deprived conditions [41, 42], which are critical for studying the dormancy and subsequent reactivation of PCa cells. These experimental platforms can facilitate detailed mechanistic investigations into the role of PTE in androgen deprivation-induced PCa dormancy. Moreover, future research should elucidate whether AR plays a pivotal role in mediating PTE-induced apoptosis in quiescent PCa cells and explore the underlying molecular mechanisms. Furthermore, evaluating the potential synergistic effects of combining PTE with standard therapies in localized or advanced PCa may offer novel strategies for enhancing clinical outcomes.
In our study, SOD2 knockdown reduced PTE-induced apoptosis in awakening quiescent PCa cells, with rescue percentages of 63.8% and 44.0% observed in PTE IC90-treated DU145 and LNCaP shSOD2 cells, respectively (Fig. 4I, J). This partial rescue suggests that additional mechanisms may contribute to PTE-induced apoptosis. Studies in proliferating PCa cells have demonstrated that PTE triggers apoptosis through multiple pathways, including enhanced ROS production, downregulation of Akt and Bcl-2, activation of mitochondrial apoptotic cascades, and AMPK activation [43, 44]. Moreover, PTE has been reported to induce apoptosis in non‑small‑cell lung cancer cells by suppressing cyclooxygenase-2 expression and in gastric cancer cells by downregulating mitochondria‑related genes [45]. This downregulation leads to mitochondrial iron (II) accumulation and ROS‑mediated activation of HIF‑1α, which subsequently triggers endoplasmic reticulum stress and apoptosis [46, 47]. Further investigation is needed to determine their relative contributions in PTE-induced apoptosis in quiescent PCa cells.
Besides, genetic overexpression of SOD2 demonstrated more sustained tumor suppression than PTE, likely due to the inherent limitations of small-molecule therapeutics. PTE is rapidly metabolized by hepatic cytochrome P450 enzymes (CYP1A2, CYP2C9, and CYP3A4) [48], and the tumor microenvironment may further hinder its penetration and activity. In its native form, PTE may not fully meet the pharmacological demands in humans due to limited bioavailability; therefore, structural modifications (such as alterations to its carbon-carbon double bonds or benzene ring) have been developed to improve its chemopreventive and therapeutic potential [49]. Future strategies, including prodrug modifications, nanoparticle-based delivery, or alternative formulations, could enhance its clinical bioavailability by improving water solubility, enabling controlled drug release, and increasing stability under physiological conditions [48]. For its clinical advancement, future research should include comprehensive in vivo studies, such as PCa PDX models, to assess long-term efficacy and recurrence prevention. Detailed pharmacokinetic and toxicological evaluations in PCa animal models are also necessary to establish its safety profile. Additionally, Phase I/II clinical trials will be essential to confirm tolerability and to provide preliminary efficacy data in PCa patients.
Recent toxicity studies in both animals and humans support the safety of PTE at therapeutic doses. For example, dietary intake of up to 3000 mg/kg/day in Swiss mice increased red blood cell counts without significant alterations in serum proteins, electrolytes, or liver/kidney enzymes, and histopathological assessments of major organs revealed no toxicity-related changes [50]. Moreover, a clinical study in adults confirmed that daily doses of up to 250 mg of PTE are safe, with no significant adverse effects on liver, kidney, or glucose markers compared to placebo [38]. Therefore, although the applications of PTE in cancer therapy are still in its early stages, it represents a promising and effective agent that may enhance oncotherapy outcomes and serve as a valuable adjuvant therapy following chemotherapy.
Preventing cancer recurrence poses a significant therapeutic challenge, as the underlying mechanisms are complex and not yet fully understood. Our data reveal that SOD2 is overexpressed in quiescent PCa cells, which is in line with observations in other cancer types. For example, in head and neck squamous cell carcinoma, quiescent Cal27 and FaDu cells exhibited a three- to tenfold increase in SOD2 mRNA compared to proliferating cells [51]. Similarly, SOD2 was significantly upregulated by the kinase Mirk/Dyrk1B in quiescent pancreatic cancer cells [52]. Our findings demonstrate for the first time that overexpression of SOD2 is critical in preventing the reactivation of QCCs by inducing apoptosis, suppressing the growth of recurrent tumours from quiescent PCa cells, and prolonging mice survival. Although SOD2 is conventionally characterized as a protective antioxidant, catalyzing the conversion of mitochondrial superoxide radicals into hydrogen peroxide (H₂O₂) and oxygen (O₂) [53], emerging evidence indicates that its overexpression can also exert tumor-suppressive effects [54-58]. Moreover, the H₂O₂ generated by SOD2 can either be metabolized to water or participate in the Fenton reaction in the presence of iron (Fe2+ + H2O2 → Fe3++ ·OH + OH-), generating damaging hydroxyl radicals, thereby contributing to cell death [59].
Cancer dormancy is frequently characterized by substantial alterations in mitochondrial respiration and metabolic reprogramming [60]. Consequently, quiescent cells are less susceptible to apoptosis, as they typically employ differential anti-apoptotic mechanisms to ensure their survival [1, 61]. QCCs often display variable oxidative phosphorylation (OXPHOS) activity—enhanced OXPHOS can optimize ATP production under low proliferative demand, while reduced metabolic activity and OXPHOS may be linked to reduced proliferative signaling in QCCs [62-64]. Moreover, quiescent cancer stem-like cells are generally characterized by reduced metabolic activity, which minimizes ROS production [65, 66]. This distinct metabolic phenotype not only enables these cells to evade cytotoxic agents that target rapidly dividing cells but also is integral to their long-term survival [67, 68]. In our study, PTE treatment paradoxically increased mitochondrial ROS levels during cell cycle reentry; however, the mitochondrial ROS scavenger, mitoTEMPO, failed to mitigate PTE-induced apoptosis (Fig. S3). This suggests that the pro-apoptotic effects of PTE-mediated SOD2 upregulation are not solely due to changes in mitochondrial ROS levels. Instead, PTE may interfere with mitochondrial complex activities, alter SOD2 catalytic efficiency through acetylation of key lysine residues (K68 and K122) [69, 70], or promote the accumulation of an iron-bound SOD2 form (FeSOD2) with peroxidase activity [53, 71]. Additionally, the interplay between SOD2 and other antioxidant systems, such as catalase, glutathione peroxidase, and peroxiredoxin, adds further complexity to the regulation of ROS [72, 73].
Our study demonstrated that C/EBP-β significantly enhanced the association with SOD2 intronic enhancer following PTE treatment during the reactivation of quiescent PCa cells, thereby promoting SOD2 transcription. Similar mechanisms have been reported in previous studies. For example, in human endometrial stromal cells, cAMP-induced SOD2 expression is dependent on C/EBP-β binding with the enhancer region, and the full-length LAP isoform of C/EBP-β was identified as a critical activator of SOD2 transcription [27, 74]. Further investigation through site-directed mutagenesis of these binding sites could help delineate the specific role of C/EBP-β in PTE-treated awakening quiescent PCa cells. Notably, knockdown of C/EBP-β in quiescent PCa cells partially reduced SOD2 expression, suggesting that additional regulatory signaling pathways may be involved. It has been reported that NF‑κB can cooperate with C/EBP-β to regulate murine SOD2 via a complex intronic enhancer in response to TNF-α and IL-1β [75]. The regulatory mechanisms in quiescent cells warrant further investigation. Besides, C/EBP-β knockdown did not impair the ability of DU145 and LNCaP cells to enter or maintain quiescence (Fig. S6A, B), but it accelerated cell cycle reentry (Fig. S6E, F). Clinical data further corroborate these findings, as higher levels of C/EBP-β correlate with increased SOD2 expression and improved prognosis in PCa patients. Collectively, these results suggest that targeting C/EBP-β-SOD2 axis may be an effective strategy for eliminating awakening QCCs and preventing cancer recurrence.
In conclusion, our study demonstrates that PTE, a natural, low-toxicity compound with, effectively induces apoptosis in awakening quiescent PCa cells, suppresses recurrent tumor growth, and prolongs survival. The C/EBP-β-SOD2 signaling pathway is pivotal in PTE-mediated eradication of quiescent PCa cells during reactivation and represents a promising therapeutic target for preventing PCa recurrence.
Materials and methods
Chemicals and reagents
PTE (T2888, 99.71% purity) was obtained from TargetMol (Boston, MA, USA). It was dissolved in dimethyl sulfoxide (DMSO; 543900, Sigma‒Aldrich, Carlsbad, CA, USA), and stored at -20 °C. Z-VAD-FMK (C1202), RIPA lysis buffer (P0013C), crystal violet staining solution (C0121) and protease inhibitor cocktail (P1005) were obtained from the Beyotime Institute of Biotechnology (Shanghai, China). RNAiso Plus (TRIzol) and the PrimeScript RT Reagent Kit (RR037A) were purchased from Takara Biotechnology (Shiga, Japan). Additional reagents included SYBR Green Real-time PCR Master Mix (QPK-201, Toyobo Life Science, Osaka, Japan), propidium iodide (PI, P4170, Sigma‒Aldrich), SYBR Green (S-7563, Life Technologies, Carlsbad, CA, USA), an Annexin V-FITC/PI Apoptosis Kit (Meilun Biotechnology, Dalian, China) and an Annexin V-APC/PI Apoptosis Kit from Multi sciences Biotech (Hangzhou, China).
Cell culture
Human prostate cancer cell lines DU145 and LNCaP, along with the human embryonic kidney cell line HEK‑293T, were obtained from the American Type Culture Collection (Manassas, VA, USA). The PCa cell lines and HEK-293T cells were cultured in complete RPMI 1640 medium (MB4374) or DMEM (MA0212) from Meilun Biotechnology, respectively, with 10% fetal bovine serum (04-001-1 ACS, Biological Industries, Israel). The cells were maintained in a humidified atmosphere in a humidified atmosphere containing 5% CO₂ and 95% air. Experimental quiescence of PCa cells was achieved following established protocols [5]. Briefly, DU145 and LNCaP cells were cultured to 80% confluence, followed by serum deprivation for an additional 7 days. To induce cell cycle reentry, the cells were replated in the presence of serum. The cell culture materials were procured from NEST Biotechnology Co., Ltd. (Wuxi, China).
Western blotting
Cell pellets were lysed using ice-cold RIPA buffer supplemented with a protease inhibitor cocktail. Protein quantification, electrophoresis, transfer and immunoblotting were carried out following previously described methods [76]. The membranes were incubated with primary antibodies: anti-SOD2 (sc-137254), anti-p27 (sc-528) and anti-C/EBP-β (sc-7962) from Santa Cruz Biotechnology (Dallas, CA, USA); anti-caspase-3 (9662), anti-poly ADP-ribose polymerase (PARP, 9542), anti-caspase-9 (9502) and anti-phosphorylated retinoblastoma (phospho-Rb, Ser807/811, 9308) from Cell Signaling Technology (Danvers, MA, USA); β-actin (66009-1-lg) and GAPDH (60004-1-Ig) from Proteintech (Wuhan, China); and Lamin A/C (ab108595) from Abcam (Cambridge, United Kingdom).
For the subcellular fractionation assay, cytoplasmic and nuclear proteins were isolated using a nuclear and cytoplasmic protein extraction kit (Beyotime Biotechnology) and subsequently analyzed by western blotting. Lamin A/C and GAPDH served as controls for the nuclear and cytoplasmic fractions, respectively.
Real-time polymerase chain reaction (RT‒PCR)
Total RNA was extracted from cells using TRIzol reagent, and then quantified using a NanoDrop spectrophotometer (DeNovix, Wilmington, DE, USA); reverse transcription was conducted with a PrimeScript RT Reagent Kit. RT‒PCR was performed using the SYBR Green Real-time PCR Master Mix on a StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). The experimental protocol was consistent with a previous description, and normalization was performed relative to the expression level of TATA box-binding protein (TBP) [23]. Primer sequences were as follows: 5′-GAAC CACG GCAC TGAT TTTC-3′ (forward) and 5′-CCCC ACCA TGTT CTGA ATCT-3′ (reverse) for TBP; 5′-ATTT GTAA GTGT CCCC GTTC C-3′ (forward) and 5′-GTGG TGGT CATA TCAA TCAT AGC-3′ (reverse) for SOD2; 5'-CCCG CCCG TGGT GTTA TTTA-3' (forward) and 5'-CACG CGTT CAGC CATG TTTA-3' (reverse) for CEBPB. All expression levels were quantified using StepOne Software version 2.3 and analyzed with Microsoft Excel software.
Establishment of doxycycline (DOX)-inducible stable SOD2 shRNA and stable C/EBP-β shRNA cell lines
A lentiviral GFP-IRES-DOX-inducible FH1tUTG shRNA expression construct, generously provided by Dr. Marco Herold, has been previously characterized [59]. The SOD2 shRNA sequence, GGTG GTCA TATC AATC ATAG CTTC AAGA GAGC TATG ATTG ATAT GACC ACC, was introduced into the DOX-inducible GFP-IRES-shRNA FH1tUTG construct, whereas the unaltered FH1tUTG served as the empty vector (EV) control. The PGMLV-hU6-MCS-CMV-Puro-WPRE construct, procured from Genomeditech (Shanghai, China), was used to silence C/EBP-β. Lentiviral preparation was conducted following the established protocol [39]. The DOX-inducible EV shRNA, DOX-inducible SOD2 shRNA, scramble shRNA or C/EBP-β shRNA constructs were introduced into HEK-293T cells via transfection using a Lenti-PacTM HIV Expression Packaging Kit (GeneCopoeia, Changzhou, China). Viral supernatants were collected and used to infect DU145 and LNCaP cells in serum‑free medium. Cells expressing high levels of GFP were selected with 0.2 mg/ml puromycin (REVG1001; GeneChem, Shanghai, China). For in vitro induction of Dox-inducible shRNA expression, the cells were exposed to 4 µg/ml doxycycline (DOX, ST039A; Beyotime Institute of Biotechnology, Shanghai, China) for 48 h.
Establishment of stable SOD2-overexpressing PCa cell lines
PGMLV-CMV-Empty Vector-3×Flag-EF1-mCherry-T2A-Puromycin (EV) and PGMLV-CMV-SOD2-3×Flag-EF1-mCherry-T2A-Puromycin (SOD2) expression constructs were procured from Genomeditech (Shanghai, China) and subsequently transfected into HEK-293T cells using a Lenti-PacTM HIV Expression Packaging Kit (GeneCopoeia, Changzhou, China). The viral media were then harvested and stored at -80 °C. DU145 and LNCaP cells were infected with the lentiviral particles in serum-free growth medium. The culture medium was replaced with complete RPMI 1640 medium, followed by incubation and subsequent selection with 0.2 mg/ml puromycin.
Cell cycle analysis
Quiescent PCa cells or PCa cells transfected with various plasmids were stimulated to re-enter the cell cycle with or without PTE treatment for different durations. Cells were collected, fixed overnight in ice‑cold 70% ethanol prepared in phosphate‑buffered saline (PBS) and subsequently stored at 4 °C. Flow cytometry with propidium iodide (PI) staining was subsequently conducted according to previously established protocols [23]. The distribution of the cell cycle phases was analyzed to determine the proportion of cells in each phase using FlowJo software (version 7.6.1).
Cell viability assay
Cell viability was evaluated using a Trypan Blue Staining Assay Kit (C0011) from the Beyotime Institute of Biotechnology. Quiescent DU145 and LNCaP cells, along with DOX-inducible shCon/shSOD2 DU145 and LNCaP cells (2 × 104 cells/well), with or without DOX treatment, were seeded into 24-well plates and maintained in complete medium to induce proliferation for 48, 72, 96 and 120 h. The cells were collected at the indicated intervals, and 20 µl of the cell suspension was combined with an equal volume of Trypan blue staining solution. The viable cells were then analyzed and counted using a Countstar instrument (Alit Biotech, Shanghai, China).
A PI exclusion assay was also employed to assess cell viability. Quiescent EV and SOD2-overexpressing DU145 and LNCaP cells were plated in 6 cm dishes and activated to proliferate for 8, 16 and 24 h. The cells were collected, suspended in PBS supplemented with 100 µg/ml RNase and stained with PI solution (20 µg/ml) for 5 min. Flow cytometry (FACSCalibur, BD Biosciences) was utilized to determine the percentage of viable and dead cells, with analysis by FlowJo software (version 7.6.1).
Mitochondrial membrane potential detection
Mitochondrial membrane potential was measured with a JC-1 staining assay kit (C2006, Beyotime), as previously described [77]. Quiescent EV- and SOD2-overexpressing DU145 and LNCaP cells were plated in 6 cm dishes and simulated to re-enter the cell cycle in the presence or absence of Z-VAD-FMK (50 μM) for 12 and 24 h. Subsequently, they were harvested and stained with JC-1 at 37 °C for 20 min, rinsed and analyzed by flow cytometry (FACSCalibur, BD Biosciences).
SYBR Green assay
Quiescent DU145 (8 × 103 cells per well) and LNCaP (1 × 104 cells per well) cells were stimulated to resume the cell cycle by culturing them in complete medium in 96-well plates, supplemented with specified concentrations of PTE for 72 h. After removing the culture medium, the cells were stored at -80 °C. An equivalent number of synchronized cells was retained as a baseline condition and stored at -80 °C for comparison. The SYBR Green assay was performed following a previously established protocol [22]. DNA quantification data were analyzed using Microsoft Excel, and the inhibitory concentrations (ICs) for the cells [25% (IC25), 50% (IC50), 75% (IC75) and 90% (IC90)] were calculated using the following formula: [1 - (DNA quantity in the PTE group)/(DNA quantity in the control group)] × 100%.
Colony formation assay
Quiescent DU145 (1500 cells/well) and LNCaP (800 cells/well) cells were stimulated to re-enter the cell cycle by culturing them in complete medium containing either DMSO or PTE (at IC25, IC50 and IC75 concentrations) in 6-well plates for 24, 48 and 72 h. Afterwards, the culture medium was replaced with PTE-free complete medium, and the cells were incubated for an additional 14 days to allow colony formation. Colonies were fixed in 95% ethanol, stained with a 1% crystal violet solution in PBS and then imaged. Colonies containing more than 50 cells were counted as clones.
Annexin V/PI double-staining assay
Cell cycle reentry was induced in quiescent PCa cells by seeding them into 6 cm dishes (1 × 106 cells) containing complete medium with either DMSO, PTE (at IC50 and IC90 concentrations) or Z-VAD-FMK (50 μM) for 24 and 48 h. The cells were then harvested, washed and stained with either an Annexin V-FITC/PI or an Annexin V-APC/PI Apoptosis Kit according to the manufacturer's protocols, with apoptotic cells quantified by flow cytometry (FACSCalibur, BD Biosciences), with Annexin V-positive cells considered indicative of apoptosis.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was conducted using a ChIP Assay Kit (P2087, Beyotime) following the manufacturer's protocol. Cells were fixed with 1% formaldehyde, and nuclei were subsequently extracted. The chromatin/DNA complex was fragmented using sonication. The lysates were clarified and incubated overnight at 4 °C with protein A+G agarose beads coupled with an anti-C/EBP-β antibody (23431-1-AP, Proteintech). Normal rabbit IgG (A7016, Beyotime) served as the negative control. The DNA was eluted and analyzed by qPCR. The primer sequences for the SOD2 enhancer region were as follows: 5′- AAGT GTGG TATT TTAG CATA GTTG TGTA-3′ (forward) and 5′- AGAG GAAA GTTG TCAG ATGT CACC-3′ (reverse).
Public dataset-based bioinformatic analysis
The Gene Expression Profiling Integrative Analysis (GEPIA) database was used to analyze expression profiles from The Cancer Genome Atlas database and the Genotype-Tissue Expression project. GEPIA also facilitated differential expression analysis of C/EBP-β in prostate adenocarcinoma (PRAD) tumor tissue versus normal tissue. The correlation between C/EBP-β expression and overall survival in patients with PRAD was analyzed using GEPIA with a Cox model [78]. Data were downloaded from the TCGA-PRAD database (https://portal.gdc.cancer.gov/), and Pearson's correlation between CEBPB and SOD2 expression levels was analyzed using the "ggpubr" R package. Correlation plots were generated to visualize the results based on the TCGA expression data.
Establishment of tumor xenografts in nude mice
All animal experiments were approved by the Shanghai University of Traditional Chinese Medicine Committee for the Use of Live Animals for Teaching and Research (approval numbers PZSHUTCM200717024 and PZSHUTCM201225005), and conducted in accordance with institutional guidelines. Four-week-old male BALB/c nude mice were obtained from the Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China) and housed in a sterile, pathogen-free environment. The mice were randomly divided into two groups of nine and subcutaneously injected in the right flanks with 4.3 × 106 quiescent EV DU145 or SOD2-overexpressing DU145 cells. Tumor volume was measured every other day using a digital caliper by recording the length (L), width (W) and height (H) and was calculated using the following formula: 3.14 × L × W × H/6). Mice were euthanized when tumor volumes reached 1000 mm³, and tumors were excised, weighed and photographed.
For the PTE treatment study, four-week-old male BALB/c nude mice were subcutaneously injected with 3.6 × 106 quiescent DU145 cells in the right flank and randomly divided into two groups (n = 8 per group). PTE (50 mg/kg) and a vehicle control (10% Cremophor EL, 10% absolute ethanol and 5% glucose in saline) were administered intraperitoneally one day before tumor cell implantation. PTE and the vehicle control were then administered from Days 1 to 5 of each week for two months, with tumor volume and body weight measured every other day. After 61 days, the mice were euthanized, and the tumors were excised, weighed and photographed.
Immunohistochemistry
Immediately after euthanasia, tumors were harvested and fixed in 4% neutral buffered paraformaldehyde. The samples were then embedded in paraffin and sectioned into 5 µm thick slices. Subsequently, the samples underwent hematoxylin and eosin (H&E) staining as well as TUNEL staining (MA0223, Meilun Biotechnology, Dalian, China), and staining with antibodies against Ki-67 (ab16667, Abcam), SOD2 (ab68155, Abcam), cleaved caspase-3 (#9661, Cell Signaling Technology) and C/EBP-β (23431-1-AP, Proteintech). The sections were prepared for histological analysis by mounting them with DPX. Three views were randomly selected from each section (n=3 for each group) and the area of each section was quantitatively measured using ImageJ software. The IHC quantification was conducted using ImageJ following a standardized protocol: each IHC-stained image was converted to an RGB stack, a threshold was applied to isolate the positive staining, and measurements were set to obtain the “% Area” values.
Statistical analysis
Statistical analysis was conducted based on experiments independently repeated at least three times, and data are presented as the mean ± standard deviations from three biological replicates. Data analysis was performed using GraphPad Prism 10 software and SPSS (version 23.0) statistical software. Comparisons between two groups were performed using Student's two-tailed t test, whereas comparisons among more than two groups were conducted using ANOVA with Fisher's least significant difference (LSD) multiple comparisons test. Survival analyses were conducted using the Kaplan‒Meier method in GraphPad Prism 10 software, and comparisons between survival curves were assessed using the two-sided log-rank test. The levels of statistical significance were denoted as *P < 0.05, **P < 0.01 and ***P < 0.001.
Abbreviations
ADT: androgen-deprivation therapy; AMPK: AMP-activated protein kinase; AR: androgen receptor; ATP: adenosine triphosphate; C/EBP-β: CCAAT/enhancer-binding protein beta; Cont: control cells (proliferative cells); CRPC: castration-resistant prostate cancer; ChIP: chromatin immunoprecipitation; DMSO: dimethyl sulfoxide; DOX: doxycycline; EV: empty vector; FBXW7: F-box and WD repeat domain containing 7; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; GEPIA: gene expression profiling integrative analysis; H₂O₂: hydrogen peroxide; H&E: hematoxylin and eosin; IC: inhibitory concentration; Mirk/Dyrk1B: minibrain-related kinase/dual-specificity tyrosine phosphorylation-regulated kinase 1B; NEPC: neuroendocrine prostate cancer; O2: oxygen; OXPHOS: oxidative phosphorylation; PARP: poly ADP-ribose polymerase; PBS: phosphate-buffered saline; PCa: prostate cancer; PI: propidium iodide; PDX: patient-derived xenograft; PRAD: prostate adenocarcinoma; PSA: prostate specific antigen; PTE: pterostilbene; QCCs: quiescent cancer cells; Qsct: quiescent cells; Rb: retinoblastoma; ROS: reactive oxygen species; RT‒PCR: real-time polymerase chain reaction; SOD2: superoxide dismutase 2; TCGA: The Cancer Genome Atlas; TBP: TATA box-binding protein.
Supplementary Material
Supplementary figures.
Acknowledgements
We are grateful to Boya Chen for helpful suggestions and assistance on the study.
Funding
This work was supported by National Natural Science Foundation of China (NO. 82204437, NO. 82404919); Shanghai Science and Technology Innovation Action Plan (NO. 22YF1445100); Shanghai University of Traditional Chinese Medicine Budget Project (NO. 23KFL041); Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (NO. GZC20231703); Traditional Chinese Medicine Research Project of the Shanghai Municipal Health Commission (NO. 2024QN029).
Author contributions
ZC Xi and MF Liu contributed to investigation, validation, data curation, writing-original draft and writing-review & editing. M Yuan, Y Li and HX Xu contributed to conceptualization and methodology. X Jiang, JL Feng and HG Ren helped data curation, formal analysis and investigation. RC Dai, WN Nik Nabil and J Zhang helped writing-review & editing and methodology. XY Sun and JY Chen helped data curation and validation. QH Dong and HX Xu contributed to supervision and project administration. Y Li, MF Liu and ZC Xi contributed to funding acquisition. All authors read and approved the final manuscript.
Ethics committee approval
Animal procedures were authorized under the approval of the Shanghai University of Traditional Chinese Medicine Committee on the Use of Live Animals for Teaching and Research (Approval Number PZSHUTCM200717024 and PZSHUTCM201225005), and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Data availability
All data relevant to the study are included in the article or uploaded as supplementary information.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yang S, Seo J, Choi J, Kim SH, Kuk Y, Park KC. et al. Towards understanding cancer dormancy over strategic hitching up mechanisms to technologies. Mol Cancer. 2025;24:47
2. Liu R, Zhao Y, Su S, Kwabil A, Njoku PC, Yu H. et al. Unveiling cancer dormancy: Intrinsic mechanisms and extrinsic forces. Cancer Lett. 2024;591:216899
3. Nik Nabil WN, Xi Z, Song Z, Jin L, Zhang XD, Zhou H. et al. Towards a Framework for Better Understanding of Quiescent Cancer Cells. Cells. 2021;10:562
4. D'Antonio C, Liguori GL. Dormancy and awakening of cancer cells: the extracellular vesicle-mediated cross-talk between Dr. Jekill and Mr. Hyde. Front Immunol. 2024;15:1441914
5. Xi Z, Yao M, Li Y, Xie C, Holst J, Liu T. et al. Guttiferone K impedes cell cycle re-entry of quiescent prostate cancer cells via stabilization of FBXW7 and subsequent c-MYC degradation. Cell Death Dis. 2016;7:e2252
6. Shimizu H, Takeishi S, Nakatsumi H, Nakayama KI. Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight. 2019;4:e125138
7. Cho J, Lee HJ, Hwang SJ, Min HY, Kang HN, Park AY. et al. The Interplay between Slow-Cycling, Chemoresistant Cancer Cells and Fibroblasts Creates a Proinflammatory Niche for Tumor Progression. Cancer Res. 2020;80:2257-72
8. Tamamouna V, Pavlou E, Neophytou CM, Papageorgis P, Costeas P. Regulation of Metastatic Tumor Dormancy and Emerging Opportunities for Therapeutic Intervention. Int J Mol Sci. 2022;23:13931
9. Achyut BR, Shankar A, Iskander AS, Ara R, Angara K, Zeng P. et al. Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks. Cancer Lett. 2015;369:416-26
10. Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K. et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349-55
11. Hu J, Sanchez-Rivera FJ, Wang Z, Johnson GN, Ho YJ, Ganesh K. et al. STING inhibits the reactivation of dormant metastasis in lung adenocarcinoma. Nature. 2023;616:806-13
12. Sarsour EH, Venkataraman S, Kalen AL, Oberley LW, Goswami PC. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell. 2008;7:405-17
13. Collins SJ, Tumpach C, Groveman BR, Drew SC, Haigh CL. Prion protein cleavage fragments regulate adult neural stem cell quiescence through redox modulation of mitochondrial fission and SOD2 expression. Cell Mol Life Sci. 2018;75:3231-49
14. Sarsour EH, Kalen AL, Xiao Z, Veenstra TD, Chaudhuri L, Venkataraman S. et al. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Res. 2012;72:3807-16
15. Winnik S, Gaul DS, Siciliani G, Lohmann C, Pasterk L, Calatayud N. et al. Mild endothelial dysfunction in Sirt3 knockout mice fed a high-cholesterol diet: protective role of a novel C/EBP-beta-dependent feedback regulation of SOD2. Basic Res Cardiol. 2016;111:33
16. Cesena TI, Cardinaux JR, Kwok R, Schwartz J. CCAAT/enhancer-binding protein (C/EBP) beta is acetylated at multiple lysines: acetylation of C/EBPbeta at lysine 39 modulates its ability to activate transcription. J Biol Chem. 2007;282:956-67
17. Miao L, St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009;47:344-56
18. Lala-Tabbert N, AlSudais H, Marchildon F, Fu D, Wiper-Bergeron N. CCAAT/enhancer-binding protein beta promotes muscle stem cell quiescence through regulation of quiescence-associated genes. Stem Cells. 2021;39:345-57
19. Saunders MA, Sansores-Garcia L, Gilroy DW, Wu KK. Selective suppression of CCAAT/enhancer-binding protein beta binding and cyclooxygenase-2 promoter activity by sodium salicylate in quiescent human fibroblasts. J Biol Chem. 2001;276:18897-904
20. Ma Z, Zhang X, Xu L, Liu D, Di S, Li W. et al. Pterostilbene: Mechanisms of its action as oncostatic agent in cell models and in vivo studies. Pharmacol Res. 2019;145:104265
21. Liu Y, You Y, Lu J, Chen X, Yang Z. Recent Advances in Synthesis, Bioactivity, and Pharmacokinetics of Pterostilbene, an Important Analog of Resveratrol. Molecules. 2020;25:5166
22. Feng J, Xi Z, Jiang X, Li Y, Nik Nabil WN, Liu M. et al. Saikosaponin A enhances Docetaxel efficacy by selectively inducing death of dormant prostate cancer cells through excessive autophagy. Cancer Lett. 2023;554:216011
23. Jiang X, Li Y, Feng JL, Nik Nabil WN, Wu R, Lu Y. et al. Safrana l Prevents Prostate Cancer Recurrence by Blocking the Re-activation of Quiescent Cancer Cells via Downregulation of S-Phase Kinase-Associated Protein 2. Front Cell Dev Biol. 2020;8:598620
24. Bi L, Xie C, Yao M, Thae Hnit SS, Vignarajan S, Wang Y. et al. The histone chaperone complex FACT promotes proliferative switch of G(0) cancer cells. Int J Cancer. 2019;145:164-78
25. Amos A, Jiang N, Zong D, Gu J, Zhou J, Yin L. et al. Depletion of SOD2 enhances nasopharyngeal carcinoma cell radiosensitivity via ferroptosis induction modulated by DHODH inhibition. BMC Cancer. 2023;23:117
26. Ruggeri P, Farina AR, Di Ianni N, Cappabianca L, Ragone M, Ianni G. et al. The TrkAIII oncoprotein inhibits mitochondrial free radical ROS-induced death of SH-SY5Y neuroblastoma cells by augmenting SOD2 expression and activity at the mitochondria, within the context of a tumour stem cell-like phenotype. PLoS One. 2014;9:e94568
27. Qiu X, Aiken KJ, Chokas AL, Beachy DE, Nick HS. Distinct functions of CCAAT enhancer-binding protein isoforms in the regulation of manganese superoxide dismutase during interleukin-1beta stimulation. J Biol Chem. 2008;283:25774-85
28. Gomatou G, Syrigos N, Vathiotis IA, Kotteas EA. Tumor Dormancy: Implications for Invasion and Metastasis. Int J Mol Sci. 2021;22:4862
29. Basu S, Dong Y, Kumar R, Jeter C, Tang DG. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin Cancer Biol. 2022;78:90-103
30. Fujita K, Nonomura N. Role of Androgen Receptor in Prostate Cancer: A Review. World J Mens Health. 2019;37:288-95
31. Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36:3-23
32. Dong X, Xue H, Mo F, Lin YY, Lin D, Wong NKY. et al. Modeling Androgen Deprivation Therapy-Induced Prostate Cancer Dormancy and Its Clinical Implications. Mol Cancer Res. 2022;20:782-93
33. Kang N, Xue H, Wong NKY, Lin YY, Classen A, Wu R. et al. Exploring B7-H4's role in prostate cancer dormancy post-androgen deprivation therapy: extracellular matrix interactions and therapeutic opportunities. Mol Cancer Res. 2025;23:327-38
34. Cackowski FC, Heath EI. Prostate cancer dormancy and recurrence. Cancer Lett. 2022;524:103-8
35. Crea F, Nur Saidy NR, Collins CC, Wang Y. The epigenetic/noncoding origin of tumor dormancy. Trends Mol Med. 2015;21:206-11
36. Ruppender NS, Morrissey C, Lange PH, Vessella RL. Dormancy in solid tumors: implications for prostate cancer. Cancer Metastasis Rev. 2013;32:501-9
37. Wang Z, Liu Z, Qu J, Sun Y, Zhou W. Role of natural products in tumor therapy from basic research and clinical perspectives. Acta Mater Med. 2024;3:163-206
38. Riche DM, McEwen CL, Riche KD, Sherman JJ, Wofford MR, Deschamp D. et al. Analysis of safety from a human clinical trial with pterostilbene. J Toxicol. 2013;2013:463595
39. Kang N, Xue H, Wong NKY, Lin YY, Classen A, Wu R. et al. Exploring B7-H4's Role in Prostate Cancer Dormancy after Androgen Deprivation Therapy: Extracellular Matrix Interactions and Therapeutic Opportunities. Mol Cancer Res. 2025;23:327-38
40. Kang N, Xue H, Lin YY, Dong X, Classen A, Wu R. et al. Influence of ADT on B7-H3 expression during CRPC progression from hormone-naive prostate cancer. Cancer Gene Ther. 2023;30:1382-9
41. Carpenter V, Saleh T, Min Lee S, Murray G, Reed J, Souers A. et al. Androgen-deprivation induced senescence in prostate cancer cells is permissive for the development of castration-resistance but susceptible to senolytic therapy. Biochem Pharmacol. 2021;193:114765
42. Yu J, Lim JE, Song W. Therapeutic Potential of Bipolar Androgen Therapy for Castration-Resistant Prostate Cancer: In vitro and In vivo Studies. Biomedicines. 2024;12:181
43. Chakraborty A, Gupta N, Ghosh K, Roy P. In vitro evaluation of the cytotoxic, anti-proliferative and anti-oxidant properties of pterostilbene isolated from Pterocarpus marsupium. Toxicol In vitro. 2010;24:1215-28
44. Lin VC, Tsai YC, Lin JN, Fan LL, Pan MH, Ho CT. et al. Activation of AMPK by pterostilbene suppresses lipogenesis and cell-cycle progression in p53 positive and negative human prostate cancer cells. J Agric Food Chem. 2012;60:6399-407
45. Wang Z, Wang T, Chen X, Cheng J, Wang L. Pterostilbene regulates cell proliferation and apoptosis in non-small-cell lung cancer via targeting COX-2. Biotechnol Appl Biochem. 2023;70:106-19
46. Huang CJ, Shieh PC, Yang JS, Li YC, Chiu YJ, Bau DT. et al. Pterostilbene Suppressed Cell Viability, Induced Apoptosis and Autophagy of Cisplatin-resistant Gastric Cancer Cells. Anticancer Res. 2025;45:511-23
47. Nishiguch Y, Fujiwara-Tani R, Nukaga S, Nishida R, Ikemoto A, Sasaki R. et al. Pterostilbene Induces Apoptosis from Endoplasmic Reticulum Stress Synergistically with Anticancer Drugs That Deposit Iron in Mitochondria. Int J Mol Sci. 2024;25:2611
48. Yu X, Xu M, Gao Z, Guan H, Zhu Q. Advances in antitumor effects of pterostilbene and its derivatives. Future Med Chem. 2025;17:109-24
49. Lin WS, Leland JV, Ho CT, Pan MH. Occurrence, Bioavailability, Anti-inflammatory, and Anticancer Effects of Pterostilbene. J Agric Food Chem. 2020;68:12788-99
50. Obrador E, Salvador-Palmer R, Jihad-Jebbar A, Lopez-Blanch R, Dellinger TH, Dellinger RW. et al. Pterostilbene in Cancer Therapy. Antioxidants (Basel). 2021;10:492
51. Eckers JC, Kalen AL, Sarsour EH, Tompkins VS, Janz S, Son JM. et al. Forkhead box M1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat Res. 2014;182:420-9
52. Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009;69:3317-24
53. Palma FR, He C, Danes JM, Paviani V, Coelho DR, Gantner BN. et al. Mitochondrial Superoxide Dismutase: What the Established, the Intriguing, and the Novel Reveal About a Key Cellular Redox Switch. Antioxid Redox Signal. 2020;32:701-14
54. Li S, Yan T, Yang JQ, Oberley TD, Oberley LW. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000;60:3927-39
55. Zhong W, Oberley LW, Oberley TD, Yan T, Domann FE, St Clair DK. Inhibition of cell growth and sensitization to oxidative damage by overexpression of manganese superoxide dismutase in rat glioma cells. Cell Growth Differ. 1996;7:1175-86
56. Jaramillo MC, Frye JB, Crapo JD, Briehl MM, Tome ME. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Res. 2009;69:5450-7
57. Hempel N, Melendez JA. Intracellular redox status controls membrane localization of pro- and anti-migratory signaling molecules. Redox Biol. 2014;2:245-50
58. Venkataraman S, Jiang X, Weydert C, Zhang Y, Zhang HJ, Goswami PC. et al. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene. 2005;24:77-89
59. Lyngsie G, Krumina L, Tunlid A, Persson P. Generation of hydroxyl radicals from reactions between a dimethoxyhydroquinone and iron oxide nanoparticles. Sci Rep. 2018;8:10834
60. Tau S, Miller TW. The role of cancer cell bioenergetics in dormancy and drug resistance. Cancer Metastasis Rev. 2023;42:87-98
61. Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A. et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell. 2020;37:104-22 e12
62. La T, Chen S, Guo T, Zhao XH, Teng L, Li D. et al. Visualization of endogenous p27 and Ki67 reveals the importance of a c-Myc-driven metabolic switch in promoting survival of quiescent cancer cells. Theranostics. 2021;11:9605-22
63. Qiu S, Sheth V, Yan C, Liu J, Chacko BK, Li H. et al. Metabolic adaptation to tyrosine kinase inhibition in leukemia stem cells. Blood. 2023;142:574-88
64. Ogata R, Mori S, Kishi S, Sasaki R, Iwata N, Ohmori H. et al. Linoleic Acid Upregulates Microrna-494 to Induce Quiescence in Colorectal Cancer. Int J Mol Sci. 2021;23:225
65. Zhang M, Peng R, Wang H, Yang Z, Zhang H, Zhang Y. et al. Nanog mediated by FAO/ACLY signaling induces cellular dormancy in colorectal cancer cells. Cell Death Dis. 2022;13:159
66. Chen K, Zhang C, Ling S, Wei R, Wang J, Xu X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis. 2021;12:835
67. Kusumbe AP, Bapat SA. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res. 2009;69:9245-53
68. Xie XP, Laks DR, Sun D, Ganbold M, Wang Z, Pedraza AM. et al. Quiescent human glioblastoma cancer stem cells drive tumor initiation, expansion, and recurrence following chemotherapy. Dev Cell. 2022;57:32-46 e8
69. Dhar SK, St Clair DK. Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med. 2012;52:2209-22
70. Liu M, Sun X, Chen B, Dai R, Xi Z, Xu H. Insights into Manganese Superoxide Dismutase and Human Diseases. Int J Mol Sci. 2022;23:15893
71. Wang Y, Branicky R, Noe A, Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. 2018;217:1915-28
72. Wei T, Thakur SS, Liu M, Wen J. Oral delivery of glutathione: antioxidant function, barriers and strategies. Acta Materia Medica. 2022;1:177-92
73. Cheung EC, Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. 2022;22:280-97
74. Tamura I, Sato S, Okada M, Tanabe M, Lee L, Maekawa R. et al. Importance of C/EBPbeta binding and histone acetylation status in the promoter regions for induction of IGFBP-1, PRL, and Mn-SOD by cAMP in human endometrial stromal cells. Endocrinology. 2014;155:275-86
75. Jones PL, Ping D, Boss JM. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Mol Cell Biol. 1997;17:6970-81
76. Wong M, Sun Y, Xi Z, Milazzo G, Poulos RC, Bartenhagen C. et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat Commun. 2019;10:3319
77. Nie S, Shi Z, Shi M, Li H, Qian X, Peng C. et al. PPARgamma/SOD2 Protects Against Mitochondrial ROS-Dependent Apoptosis via Inhibiting ATG4D-Mediated Mitophagy to Promote Pancreatic Cancer Proliferation. Front Cell Dev Biol. 2021;9:745554
78. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98-W102
Author contact
![]() Corresponding authors: peggyyuan1990com (Man Yuan), liyangsh1991com (Yang Li), xuhongxi88com (Hongxi Xu)
Corresponding authors: peggyyuan1990com (Man Yuan), liyangsh1991com (Yang Li), xuhongxi88com (Hongxi Xu)