Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(8):3726-3739. doi:10.7150/ijbs.110504 This issue Cite
Review
Regulatory Action of Calcium and Calcium Channels in Pain Pathways
Yan Zhang1, John Shannonhouse1, Hyeonwi Son1, Joon Tae Park3, Yu Shin Kim1,2 ![]()
1. Department of Oral and Maxillofacial Surgery, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA.
2. Programs in Integrated Biomedical Sciences, Translational Sciences, Biomedical Engineering, Radiological Sciences, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA.
3. Division of Life Sciences, College of Life Sciences and Bioengineering, Incheon National University, Incheon, South Korea.
Received 2025-1-15; Accepted 2025-4-1; Published 2025-5-31
Abstract

Calcium ions (Ca2+) and Ca2+ channels are pivotal in the regulation of pain pathways and serve as key regulators of neuronal excitability and neurotransmitter release. We review the different types of Ca2+ channels involved in pain processing, including voltage-gated Ca2+ channels (VGCCs), such as L-, N-, P/Q-, and T-type channels. Each subtype is intricately involved in different aspects of pain perception, from acute pain signaling to the development and maintenance of chronic pain states. In addition, the roles of transient receptor potential (TRP) channels, particularly TRPV1 and TRPA1, are discussed in the context of their contribution to chronic pain. Advances in Ca2+ imaging techniques, particularly through genetically encoded Ca2+ indicators (GECIs), such as GCaMPs, have provided unprecedented insight into the dynamic role of Ca2+ channels in pain pathways. These efforts have deepened our understanding of Ca2+ channels and suggest novel therapeutic targets for more effective pain management strategies within Ca2+ channels.
Keywords: Ca2+, Ca2+ channel, VGCC, TRP channel, GCaMP, GECI, pain, regulatory mechanism
Introduction
Pain is an important biological response that alerts the body to potential injury or harm. protects the body from injury or potential harm. Acute pain is usually temporary, but chronic pain may persist or recur for more than three months, often resulting in long-term physical and psychological problems [1]. At the forefront of pain detection are specialized sensory neurons called nociceptors, which act as primary sensors for noxious stimuli [2]. These cells initiate the complex pain process that ultimately leads to the conscious perception of pain. Central to this intricate system are Ca2+ channels, which play a pivotal role in pain signaling.
Ca2+ channels are essential components of the pain signaling apparatus, significantly affecting the transmission and perception of nociceptive stimuli [3]. Ca2+ channels are widely present throughout the nervous system and play a key role in regulating pain pathways [3]. Specifically, Ca2+ channels regulate neurotransmitter release, neuronal excitability, and signal transmission in the pain pathway. In peripheral nociceptors, Ca2+ channels contribute to the detection and initial encoding of noxious stimuli. In the dorsal root ganglia, they modulate the excitability of sensory neurons and influence pain signal processing. In the central terminals of the spinal cord, Ca2+ channels are important for neurotransmitter release and synaptic transmission of pain signals to second-order neurons. These diverse roles of Ca2+ channels in pain signaling make them particularly attractive targets for pain therapeutic strategies.
In vivo Ca2+ imaging techniques have been developed and have become invaluable tools for exploring the role of Ca2+ and Ca2+ channels in pain pathways. By allowing real-time observation of Ca2+ dynamics in living organisms, Ca2+ imaging provides insight into how Ca2+ and Ca2+ channels function within intact neural circuits during pain processing. These techniques also allow for direct observation of the role in regulating neuronal excitability and neurotransmitter release by visualizing the activation and modulation of Ca2+ in response to nociceptive stimuli. The ability to observe these processes in real-time is essential for identifying the specific contributions of different Ca2+ channel subtypes to pain signals and developing targeted therapeutic interventions. To date, in vivo Ca2+ imaging has been used effectively to study neuronal and non-neuronal cell populations in the brain [4, 5], spinal cord [6, 7], and primary sensory neurons [8, 9] in living mice. It also has been applied to a variety of pain models [10-12], providing a powerful tool for investigating the complex processes underlying pain perception and other sensory functions.
We discuss the regulatory role of Ca2+ and Ca2+ channels in pain pathways, highlighting how visualization of Ca2+ imaging has improved our understanding of these mechanisms. We examine the specific roles of Ca2+ channels in pain transmission and modulation, with the aim of elucidating their potential as therapeutic targets for the management of chronic pain. We also describe the critical role of advanced imaging techniques in unraveling the complex intricacies of pain biology, thereby providing insights that may lead to more effective treatments for chronic pain conditions.
Types of Ca2+ Channels in Pain Pathways
Voltage-Gated Ca2+ Channels (VGCCs)
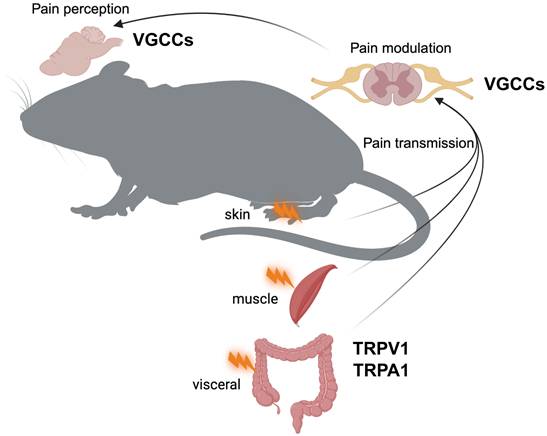
Neurons typically express several types of voltage-gated Ca2+ channels (VGCCs), which are classified as high-voltage-activated (HVA) and low-voltage-activated (LVA) channels based on their activation thresholds [13]. HVA channels require stronger depolarization to open and are typically activated at potentials more positive than -40 mV. HVA channels include L-type (Cav1.1-Cav1.4), P/Q-type (Cav2.1), N-type (Cav2.2), and R-type (Cav2.3) channels, each of which provide unique contributions to pain processing [14]. HVA channels are pharmacologically distinct and consist of heteromultimeric complexes containing a pore-forming α1 subunit, one of four types of β subunits, and one of four types of α2δ subunits [15]. In contrast, LVA channels, also known as T-type channels, are activated at more negative membrane potentials (approximately -70 to -60 mV). LVA channels include Cav3.1, Cav3.2, and Cav3.3. Unlike their HVA counterparts, LVA channels appear to function as monomers composed solely of α1 subunits. The functional importance of HVA and LVA channels in pain signal transduction becomes apparent at the synaptic level. When an action potential reaches the presynaptic terminal of a nociceptive neuron, membrane depolarization occurs. This depolarization opens VGCCs, allowing Ca2+ ions to enter the cell. The resulting influx of Ca2+ ions causes the release of neurotransmitters such as glutamate, calcitonin gene-related peptide (CGRP), and substance P into the synaptic cleft. These neurotransmitters then bind to receptors on the postsynaptic neuron, propagating the pain signal (Figure 1).
Depiction of the journey of pain signals from peripheral tissues to the brain, where they are ultimately perceived as pain. Pain signals are initiated in specialized sensory nerve endings called nociceptors, located in the skin, muscles, and organs (e.g., visceral tissues). In these nociceptors, Ca²⁺-permeable channels, TRPV1 and TRPA1, play a key role in detecting noxious stimuli, such as mechanical injury, temperature extremes (hot or cold), and chemical irritants. When these channels are activated, pain signals are initiated. Transmission of these pain signals to the central nervous system is facilitated by voltage-gated Ca²⁺ channels (VGCCs) in the presynaptic terminals of the spinal cord, which trigger the release of neurotransmitters and allow the continuation of the pain signal through the ascending pathways. VGCCs play a key role in pain modulation by affecting the intensity and duration of pain signals within the spinal cord and higher brain regions, ultimately influencing the perception and experience of pain.
HVA Ca2+ channels depend critically on their auxiliary α2δ subunit, which orchestrates multiple aspects of channel function despite not participating directly in ion conduction. Encoded by one of four genes (CACNA2D1-4) [16], with α2δ1 being the most prevalent in pain-processing neurons [17], this glycosylated subunit profoundly modulates the α1 subunit. The α2δ subunit fine-tunes Ca2+ channel biophysics by enhancing opening probability, shifting voltage-dependent activation toward more negative potentials, and accelerating both activation and inactivation kinetics [18, 19]. Beyond these electrophysiological effects, α2δ governs channel trafficking and membrane stabilization, facilitating movement from the endoplasmic reticulum to the plasma membrane and extending channel residence time at the cell surface [16, 20, 21]. In pain signaling, the α2δ1 subunit plays a crucial role, particularly in pathological conditions such as neuropathic pain [17]. Following nerve injury, α2δ1 expression undergoes marked upregulation in affected neural circuits [22, 23]. This molecular adaptation enhances calcium influx through both Cav2.2 and Cav1.2 channels in dorsal root ganglia neurons and their central projections in the spinal dorsal horn [24-26]. The resulting amplification of excitatory neurotransmitter release drives central sensitization, a hallmark of chronic pain.
L-type (Cav1.2 and Cav1.3)
Among the four subtypes of HVA channels, only L-type Cav1.2 and Cav1.3 channels are predominantly found in neurons, including medium-sized dorsal root ganglia (DRG) neurons as well as the cell bodies and dendrites of dorsal horn neurons [27]. Cav1.2 and Cav1.3 have distinct roles in pain transmission and the development of neuropathic pain. In the spinal dorsal horn, Cav1.3 channels are primarily involved in short-term sensitization during physiological pain transmission, whereas Cav1.2 channels play a key role in long-term plasticity associated with neuropathic pain [28, 29]. Peripheral nerve injury results in the accumulation of VGCC auxiliary units in primary afferent terminals, which increases the activity of Cav1.2 channels in the spinal dorsal horn and contributes to the maintenance of the neuropathic pain state [24]. Cav1.2 channels are also found in peripheral afferent neurons [30], but their role at this location is not considered significant in the development of neuropathic pain. Pharmacological interventions targeting these channels have shown nuanced results. L-channel blockers such as nitrendipine have been shown to reduce hyperalgesia (increased sensitivity to pain) and mechanical allodynia (pain evoked by normally non-painful stimuli) in animal models of neuropathic pain but are generally not considered as effective or well established as other Ca2+ channel modulators, such as gabapentin and pregabalin, which bind to the α2δ subunit for the management of neuropathic pain.
N-type (Cav2.2)
Cav2.2 channels are abundantly expressed in key nociceptive structures, including dorsal horn neurons and DRG neurons [3]. At the cellular level, Cav2.2 channels control Ca2+ influx at presynaptic terminals, thereby regulating the release of neurotransmitters involved in pain signaling [31]. Interestingly, Cav2.2 channels in peripheral nociceptors have been suggested to be required for thermal hypersensitivity but not for intradermal capsaicin-induced mechanical hypersensitivity [32]. During inflammation induced by complete Freund's adjuvant (CFA), Cav2.2 channels have been found to be upregulated in thermal-sensitive DRG neurons but not in mechanical-sensitive DRG neurons, and blocking Cav2.2 channels inhibited CFA-induced thermal hyperalgesia but not mechanical hyperalgesia [33]. These results suggest that Cav2.2 channels play a critical role in regulating thermal pain responses during acute and chronic inflammation. The importance of Cav2.2 channels in pain processing has been further elucidated by genetic studies. Mice deficient in N-type channels demonstrate reduced nociceptive responses and attenuated inflammatory and neuropathic pain-related behaviors [34-36], confirming the important role of Cav2.2 channels in nociceptive processing. Therefore, Cav2.2 channels are considered an important therapeutic target for pain management. Selective inhibition of these channels has shown promise in reducing pain responses in a variety of animal models. A notable example is the Cav2.2-specific peptide pore blocker ziconotide (Prialt™). Ziconotide is a synthetic version of the ω-conotoxin MVIIA and is clinically approved for the treatment of refractory chronic pain [37]. However, the clinical application of ziconotide is limited by the need for intrathecal administration and potential side effects, which limit its widespread use. Research efforts are underway to develop novel modulators of Cav2.2 channels. For example, the peptidomimetic compound CBD3063 has been shown to selectively modulate Cav2.2 activity and reduce pain behaviors without significant side effects [38]. Peptidomimetic modulators targeting the interaction of collapsin response mediator protein 2 (CRMP2) with Cav2.2 channels have shown promise in alleviating chronic pain in preclinical models and in reversing nociceptive behaviors in rodents without eliciting sensory, affective, or cognitive effects [39]. These results highlight the potential of targeted therapies that can modulate Cav2.2 function to achieve analgesia.
P/Q-type (Cav2.1)
P/Q-type Ca2+ channels are found primarily at presynaptic terminals in the dorsal horn; their distribution pattern is complementary to that of N-type channels [40]. Although their precise role in pain processing is not fully understood, P/Q-type channels are thought to modulate synaptic transmission, particularly in the context of postsynaptic nociceptive transmission. In terms of pain modulation, the involvement of P/Q-type channels appears to vary depending on the type of pain. For instance, in neuropathic pain models, blocking P/Q-type channels with ω-agatoxin IVA does not significantly affect mechanical allodynia [41]. However, studies in mice lacking the Cav2.1 α1 subunit have revealed a dual role for P/Q-type channels in pain modulation. They contribute to reducing sensitivity to non-injurious noxious thermal stimuli (antinociceptive role), while promoting pain sensitivity in inflammatory and neuropathic states (pronociceptive role) [42]. Mutations in the CACNA1A gene, which encodes the Cav2.1 α1 subunit, have been found in patients with trigeminal neuralgia, a debilitating facial pain disorder [43]. These mutations have also been associated with familial hemiplegic migraine, a severe form of migraine with aura [44]. Mice carrying gain-of-function mutations in this gene display a phenotype associated with trigeminal pain, providing a valuable model to study the genetic and physiological mechanisms underlying trigeminal neuralgia and familial hemiplegic migraine.
T-type (Cav3.1, Cav3.2, Cav3.3)
T-type Ca2+ channels exhibit a unique structural composition that sets them apart from HVA Ca2+ channels. While HVA channels are hetero-multimers composed of a pore-forming α1 subunit and auxiliary subunits (β, α2δ, γ), T-type channels are monomers composed solely of the α1 subunit [45]. This structural simplicity has profound implications for their function and regulation. The absence of auxiliary subunits in T-type channels contributes to their distinctive electrophysiological profile characterized by low-voltage activation and fast deactivation kinetics. These properties may contribute to important roles in neural functions, including generation of low-threshold Ca2+ spikes, facilitation of burst firing in diverse neuronal populations, and regulation of neurotransmitter release at dorsal horn synapses. T-type Ca2+ channels are also highly expressed in both peripheral nociceptive neurons and dorsal horn neurons [46-48]. In vitro electrophysiological recordings have shown that genetic deletion or pharmacological inhibition of these channels results in diminished action potential firing [49-51], highlighting the importance of these channels in neuronal excitability. Cav3.2 channels, a subtype of T-type Ca2+ channels, play an important role in mechanosensation and pain perception. In mechanosensation, Cav3.2 channels serve as selective markers for Aδ-LTMRs and C-LTMRs, two major types of low-threshold mechanoreceptors that innervate skin hair follicles [52]. Cav3.2 regulates light-touch perception and nociceptive sensation and is essential for the development of allodynia symptoms in neuropathic pain. Upregulation of Cav3.2 channels has been implicated in a variety of chronic pain conditions, including inflammatory pain [53-55], neuropathic pain (diabetes-induced [56, 57] and nerve injury-induced [58-62]), visceral pain [63, 64], bone cancer pain [65], and chemotherapy-induced peripheral neuropathy (e.g., paclitaxel-induced hypersensitivity [66]). In the trigeminal system, Cav3.2 channels can impact pain transmission and contribute to trigeminal-mediated neuropathic pain [67-69]. Mutations in the CACNA1H gene, which encodes the Cav3.2 α1 subunit, have been found in patients with trigeminal neuralgia. These mutations may alter the biophysical properties of these channels [70, 71] and potentially contribute to the pathophysiology of the condition.
Modulation of Cav3.2 channels offers a variety of avenues for therapeutic intervention for pain management, a particular focus of which has been the development of specific Cav3.2 channel blockers. The specific T-type Ca2+ channel blocker, TTA-P2, has been shown to reduce hypersensitivity in animal models of diabetic neuropathic pain [72], osteoarthritis pain [73], spinal cord injury-induced neuropathic pain [74], nerve-injury induced pain [75] and inflammatory pain [72]. Z944, a highly selective piperazine-based T-type channel blocker, has shown effectiveness in reducing pain behaviors in multiple rodent pain models, including neuropathic pain [76], inflammatory pain [77] and trigeminal pain [70]. Despite the promising preclinical results, no T-type Ca2+ blockers are currently available on the market for pain management.
Targeting posttranslational modifications such as glycosylation, phosphorylation, and ubiquitination offers promising avenues for developing novel pain management therapies by modulating T-type channel function and expression. Glycosylation of CaV3.2 channels involves the addition of carbohydrate groups (N-linked glycan chains) to asparagine residues on the channel protein, which can influence the channel's surface expression, stability, and gating properties [78, 79]. In animal models of painful peripheral diabetic neuropathy in both type 1 and type 2 diabetes, glycosylation of Cav3.2 channels in nociceptors contributes to increased neuronal excitability [80-82]. The increased presence and activity of these channels on the cell surface amplifies pain signaling, leading to pain hypersensitivity. In vitro, inhibition of the glycosylation process by de-glycosylating enzymes such as neuraminidase or PNGase-F reduces T-type current density in nociceptors and recombinant Cav3.2 channels, as shown in electrophysiological recordings [80, 81]. In vivo, selective reversal of Cav3.2 channel glycosylation can alleviate pain hypersensitivity in diabetic models [80-83], highlighting the importance of glycosylation in modulating Cav3.2 function and its role in diabetic pain pathways. Phosphorylation is another important and dynamic regulatory mechanism for Cav3.2 channels. Phosphorylation targets the intracellular links connecting the four domains of CaV3.2 channels, allowing for rapid and precise modulation of channel function. Several protein kinases, including calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA), and protein kinase C (PKC), are involved in this regulation. CaMKII primarily modulates T-type Ca2+ channel activation by enhancing their probability of opening through phosphorylation [84, 85]. PKA and PKC similarly modulate Cav3.2 channels, typically by phosphorylating specific residues to enhance activity and thus increase Ca2+ influx and neuronal excitability, which contribute to pain signaling [86]. Cyclin-dependent kinase 5 (Cdk5) is particularly notable for augmenting surface expression of Cav3.2 channels by targeting two specific residues, Ser561 and Ser1987 [61]. Increased Cdk5 levels in DRG after spinal nerve injury suggest a role in pain modulation. Pharmacological inhibition of Cdk5 with olomoucine has been shown to reduce compound action potentials and alleviate mechanical allodynia induced by spinal nerve ligation [61, 87]. Ubiquitination is an important mechanism for regulating the stability, trafficking, and function of Cav3.2 channels. The ubiquitinating enzyme USP5, which specifically binds to the intracellular domain III-IV linker region of the channels, removes ubiquitin from these channels. USP5 expression is upregulated in DRG and spinal cord in inflammatory and neuropathic pain states [88, 89]. Knockdown of USP5 via shRNA reduces Cav3.2 protein levels and attenuates Cav3.2 whole-cell currents [88], suggesting that the ubiquitinating activity of USP5 prevents degradation of Cav3.2 channels and maintains their functional presence at the cell surface. Disruption of the interaction between USP5 and Cav3.2 in vivo results in analgesic effects in both inflammatory and neuropathic pain models [88-91]. Furthermore, SUMOylation of USP5 appears to decrease its affinity for Cav3.2 channels, indicating a complex regulatory interplay that influences pain signaling pathways.
PKC modulates the activity of various Cav channels through phosphorylation at specific serine and threonine residues primarily located on the intracellular loops and C-terminal domains of the α1 subunit. In L-type channels, Ser1928 on Cav1.2 is a well-characterized PKC target that modulates channel activity in various tissues [92]. For N-type and P/Q-type channels, PKC enhances channel activity by phosphorylation of the α1 subunit, particularly at the domain I-II intracellular linker, which contributes to increased channel opening at presynaptic terminals [93]. Additionally, PKC targets the synprint site (a region critical for SNARE protein binding) at Ser774 and Ser898 of N-type channels, further modulating synaptic transmission [94]. In T-type channels, phosphorylation of Cav3.2 channels by Cdk5 and PKC has been linked to enhanced channel activity in neuropathic pain models [95, 96], suggesting a role for these kinases in pain sensitization.
Transient Receptor Potential (TRP) channels
Transient receptor potential (TRP) channels form a large family of ion channels that play a key role in various sensory processes, including pain perception [97]. These channels act as polymodal detectors that respond to a variety of physical and chemical stimuli. The TRP channel superfamily is divided into six subfamilies: TRPC (Canonical), TRPV (Vanilloid), TRPM (Melastatin), TRPA (Ankyrin), TRPP (Polycystin), and TRPML (Mucolipin). Among these subfamilies, TRPV1 and TRPA1 channels, which are also Ca2+-permeable channels, are particularly important in pain pathways (Figure 1). These two channels are extensively involved in nociception and play a key role in acute and chronic pain states. Their ability to respond to a wide range of noxious stimuli and their involvement in various pain modalities make them critical components of the pain signaling network and important targets for pain research and therapeutic development.
TRPV1
TRPV1 is a polymodal receptor activated by noxious heat, capsaicin, acidic pH, and endogenous ligands, which are often associated with inflammation. It is primarily expressed in nociceptors associated with unmyelinated C fibers and medium-diameter thinly myelinated Aδ fibers. When activated, TRPV1 promotes the influx of cations (Ca2+ and Na+) into the nociceptive neuron, causing depolarization and initiation of action potentials. This channel is involved in both peripheral and central sensitization processes, contributing to conditions such as thermal hyperalgesia and inflammatory pain [98, 99].
TRPV1 regulation in the pain pathways involves multiple intricate mechanisms. During inflammation, TRPV1 undergoes posttranslational modifications that significantly increase its sensitivity to activation stimuli. This process is mediated by the local release of various inflammatory mediators, such as substance P, CGRP, bradykinin, cytokines (e.g., TNF-α, IL-1β), nerve growth factor (NGF), and other factors [100]. These inflammatory agents sensitize TRPV1 by lowering its activation threshold and/or increasing its expression. Phosphorylation by kinases such as PKA, PKC, and Src often enhances TRPV1 sensitivity through various mechanisms, including increased channel activity, reduced desensitization, and increased membrane trafficking [101]. PKC-mediated phosphorylation of TRPV1 occurs at Ser 502, Tyr704, and Ser 800 [102]. Phosphorylation of Ser502 contributes to capsaicin-induced hypersensitivity following PKC activation but does not affect responses to heat or acid. Tyr704 is selectively involved in heat hypersensitivity. In contrast, Ser800 serves as a polymodal sensitization site, integrating multiple inflammatory signals in nociceptors and contributing to hypersensitivity across capsaicin, heat, and acid stimuli. Functional studies have shown that mutations at these sites abolish PKC-induced sensitization, underscoring their critical role in regulating TRPV1 activity [103]. Lipid regulation, particularly by phosphoinositides and endocannabinoids, can either sensitize or inhibit TRPV1 activity [104-106]. The function of the channel is further influenced by interactions with proteins such as calmodulin [107] and AKAP79/150 [108]. Environmental factors such as increased temperature [109] and acidic pH [110], common in inflamed tissues, directly activate TRPV1. This multifaceted modulation of TRPV1 highlights its central role in pain perception and its potential as a target for pain management strategies. Because of its central role in the pain pathway, TRPV1 is an important target for analgesic development, with both antagonists and agonists being explored for pain management. Although preclinical studies have established a critical role for TRPV1 in various pain conditions, clinical trials have encountered challenges, one of which, the induction of hyperthermia, has been a major issue [111].
TRPA1
TRPA1 is an important TRP channel in pain pathways, activated by a diverse range of chemical, noxious cold, and mechanical stimuli. TRPA1 is specialized for sensing endogenous disease-causing agents such as reactive oxygen species (ROS), reactive nitrogen species (RNS), and inflammatory mediators such as bradykinin and cytokines. These agents sensitize TRPA1 channels, thereby increasing nociceptor hypersensitivity and contributing to pain perception. TRPA1 is expressed on nociceptive neurons in peripheral tissues, where it detects harmful stimuli and contributes to the initiation of pain signals, especially in inflammatory conditions [112]. TRPA1 is also present on the central terminals of nociceptive neurons in the spinal dorsal horn, where it amplifies pain transmission and contributes to central sensitization [113]. When TRPA1 is activated, neuropeptides, including substance P and CGRP that mediate neurogenic inflammation and further sensitize nociceptors, are released. In neuropathic pain models, TRPA1 activation following nerve injury contributes to mechanical allodynia and cold hypersensitivity [114, 115], and in Schwann cells orchestrates neuroinflammation, ethanol and oxidative stress, sustaining neuropathic pain [116, 117]. During inflammation, TRPA1 activation by inflammatory agents leads to nociceptor hypersensitivity via pathways such as PLC/Ca2+ signaling [112]. This channel has also been implicated in cancer pain, osteoarthritis, chemotherapy-induced neuropathy, migraine, and postoperative pain [112]. In these conditions, TRPA1 activation or blockade increases or decreases pain behavior and hypersensitivity accordingly. The wide-ranging activation profile of TRPA1 and its role in diverse pain pathways make it an attractive target for the development of analgesic drugs aimed at treating various types of pain, including inflammatory, neuropathic, and migraine pain.
While TPRA1 shares several regulatory mechanisms with TRPV1, including phosphorylation by PKA and PKC, lipid regulation, and interactions with intracellular proteins, it also has unique regulatory features. Specific phosphorylation sites on TRPA1 have been identified that are important for its regulation by PKC. Notably, phosphorylation at Ser119, Thr281, and Thr529 play key roles in sensitizing the channel [118]. In addition to functioning as a pain sensor, TRPA1 also functions as a sensor of oxidative stress. It can be directly activated by ROS and RNS through oxidation of specific cysteine residues [119]. A unique characteristic of TRPA1 is its activation by covalent modifications, particularly by electrophilic agents such as mustard oil, which directly modify cysteine residues on the channel [120]. These distinct regulatory mechanisms highlight the special role of TRPA1 in sensing not only painful stimuli but also cellular stress and specific chemical agents, contributing to its importance in various pain and inflammatory conditions.
Calcium-Activated Ion Channels in Pain Pathways
In addition to VGCCs and TRP channels, calcium-activated ion channels play a significant role in the onset and maintenance of pain by modulating neuronal excitability and synaptic transmission in pain pathways. These channels are activated by intracellular Ca²⁺, often downstream of Ca2+ influx through VGCCs or TRP channels, and include calcium-dependent chloride channels, such as anoctamin 1 (ANO1), and calcium-dependent potassium channels, such as large-conductance (BKCa) and small-conductance (SKCa) channels.
Anoctamin 1 (ANO1)
ANO1, alternatively known as TMEM16A, is a calcium-activated chloride channel widely expressed in nociceptive DRG neurons and dorsal horn neurons [121-123]. ANO1 is functionally coupled with TRPV1 channels. When calcium enters through TRPV1, it triggers ANO1 activation, resulting in chloride efflux that depolarizes neurons and heightens their excitability [124, 125]. ANO1 enhances neuronal excitability in inflammatory and neuropathic pain, contributing to hyperalgesia, and pharmacological or genetic inhibition of ANO1 in nociceptive neurons attenuates pain behaviors [121, 122].
BKCa (large-conductance)
Large-conductance calcium-activated potassium (BKCa) channels, encoded by the KCNMA1 gene, are expressed in DRG neurons and spinal cord neurons and are activated by both Ca2+ and membrane depolarization [126]. BKCa channels contribute to the repolarization phase of action potentials and mediate the afterhyperpolarization (AHP), which regulates the firing frequency of neurons. In the context of pain, BKCa channels generally play an antinociceptive role by reducing neuronal excitability. BKCa channel activity is downregulated in DRG neurons following peripheral injury, leading to increased excitability and contributing to inflammatory or neuropathic pain [127-129]. Intrathecal delivery of viral vectors or pharmacological activation of BKCa channels with agonists like NS1619 reduces excitability in nociceptive neurons and attenuates mechanical and thermal hyperalgesia in animal models of neuropathic and inflammatory pain [127, 129-131]. Conversely, inhibition of BKCa channels with blockers like iberiotoxin enhances neuronal firing and exacerbates pain behaviors, underscoring their protective role [127, 132].
SKCa (small-conductance)
Small-conductance calcium-activated potassium (SKCa) channels, encoded by KCNN1-3 genes, are expressed in both peripheral nociceptors and spinal cord neurons and are activated solely by Ca2+ binding to calmodulin [133]. SKCa channels contribute to the medium AHP, which regulates spike frequency adaptation and neuronal excitability [134]. Silencing SK channels in Drosophila results in heightened pain sensitivity behaviors, suggesting these channels function to attenuate pain signaling pathways [135]. In injured human and mouse DRG, KCNN1(SK1) expression is downregulated, leading to increased DRG neuron excitability and contributing to the development of neuropathic pain [136-139]. Restoring KCNN1 expression in injured DRG, either through sensory neuron-specific long non-coding RNA modulation [136] or viral-mediated gene delivery [137], effectively reduces neuronal hyperexcitability and attenuates neuropathic pain behaviors in animal models.
Ca2+ Imaging in Pain Research
In vivo or ex vivo Ca2+ imaging has emerged as a powerful technique in pain research, offering significant advantages over traditional cultured neuron imaging. These methods employ Ca2+-sensitive dyes or other Ca2+ indicators, most typically, genetically encoded Ca2+ indicators (GECIs), to visualize and measure Ca2+ dynamics in neurons and other cells involved in the pain pathway. Key advantages of these techniques include the preservation of neurons in their natural context, the maintenance of native connectivity and microenvironmental factors; the capture of complex interactions between neurons and non-neuronal cells in intact networks; the observation of dynamic processes such as real-time responses to stimuli and pharmacological effects; and the capacity to study whole-system responses in mature neurons within intact tissues. These benefits provide a comprehensive and physiologically relevant view of neuronal function that is critical for understanding the intricate mechanisms underlying pain perception and processing.
Ca2+ indicators
Ca2+-sensitive dyes
Ca²⁺-sensitive dyes are designed to selectively bind to intracellular Ca²⁺ ions, causing a conformational change that alters the fluorescence properties of the dye. This change in fluorescence intensity correlates with the concentration of Ca2+ ions, allowing visualization and measurement of real-time Ca2+ dynamics. Ca²⁺-sensitive dyes are less favored for in vivo Ca2+ imaging because their use involves invasive procedures, as they require injection to introduce the dye into the target tissue. These procedures lack cell type specificity, involve time-consuming loading procedures, and are of limited suitability for long-term studies [140].
GECI
GECIs are generally preferred over Ca²⁺-sensitive dyes as they offer targeted expression, stable long-term imaging, and a less invasive introduction method. GECIs are typically based on fluorescent proteins linked to Ca2+ binding domains, such as calmodulin or troponin C. Upon binding to Ca2+, these proteins undergo a conformational change that either enhances or quenches fluorescence. This change in fluorescence intensity correlates with Ca2+ concentration, providing a proxy for neuronal activity [141].
GCaMPs were first introduced in 2001 and have become the most widely used Ca2+ probes for studying in vivo Ca2+ activity. These GECIs are based on circular permutations of GFP fused to calmodulin and the M13 peptide from myosin light chain [142]. The design of GCaMPs allows them to emit fluorescence in response to Ca2+ binding, providing a real-time readout of intracellular Ca2+ dynamics. Over the years, various GCaMP variants have been developed, offering different sensitivities and kinetics, applicable to diverse experimental needs. Early versions, GCaMP1 and GCaMP2, served as proof-of-concept but had limitations in sensitivity and dynamic range [142-145]. GCaMP3 showed significant improvements, offering better signal-to-noise ratio and enhanced sensitivity [8, 9]. The GCaMP5 series introduced various subtypes, such as GCaMP5A, GCaMP5G, and GCaMP5K, each optimized for different experimental conditions including kinetics and brightness as well as species compatibility [146]. The GCaMP6 series includes GCaMP6s (slow), GCaMP6m (medium), and GCaMP6f (fast), providing options for different signal kinetics and amplitudes [147]. The latest generation, jGCaMP8, offers improved performance characteristics including improved sensitivity and reduced photobleaching, making it the most advanced option for Ca2+ imaging to date [148]. Each generation of GCaMPs was built on the strengths of the previous generation and addresses its limitations, making these indicators invaluable tools for exploring Ca2+ signaling in various biological contexts, including pain research.
Despite the availability of other GECIs, such as Förster resonance energy transfer (FRET)-based GECIs like Twitch [149], which rely on energy transfer between two fluorescent proteins to report calcium levels, and red fluorescent GECIs like jRGECO1a [150] and RCaMP [151], which emit red fluorescence in response to calcium binding, GCaMP still remains the primary choice for in vivo Ca2+ imaging in pain studies. This preference is a consequence of several key advantages: GCaMPs provide superior signal strength and sensitivity, resulting in a higher signal-to-noise ratio that allows detection of subtle Ca2+ transients. They have a broad dynamic range allowing them to capture both small and large Ca2+ fluctuations, which is important for accurately monitoring pain-related neural activity. GCaMPs have become the preferred choice for Ca2+ imaging in pain research, in part, because the continuous optimization of GCaMPs has resulted in variants with improved performance characteristics such as faster kinetics and reduced baseline fluorescence. The increasing popularity of cell type-specific GCaMPs, such as those specifically expressed in nociceptors (Figure 2), has provided deeper insights into the molecular and cellular mechanisms underlying various pain conditions. The user-friendly nature, broad compatibility, and extensive validation of GCaMPs have established them as reliable and versatile tools, resulting in widespread adoption and a robust foundation of protocols and comparative data in the field.
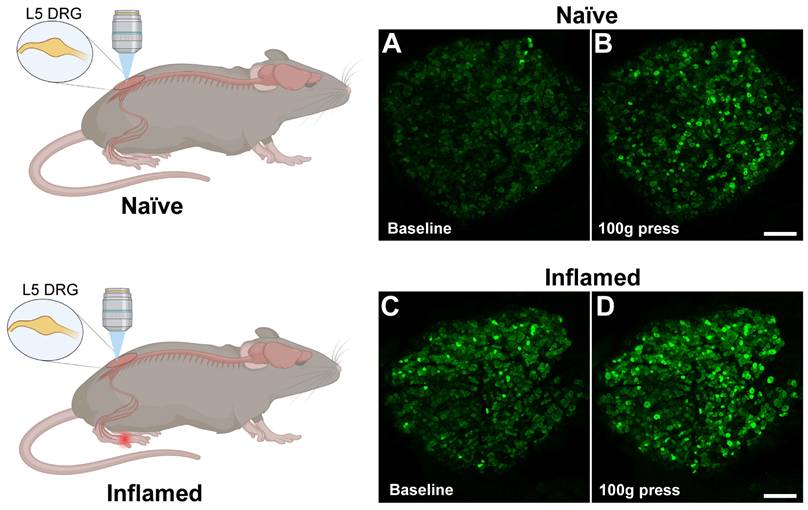
In vivo fluorescence imaging of dorsal root ganglion (DRG) during inflammation using Pirt-GCaMP3 mice. A. (top) Control mice received mock injection in right hindpaw; (bottom) Experimental mice received injection of complete Freund's adjuvant (CFA) into the right hindpaw to induce inflammation. For both mice, two days later, the DRG was surgically exposed at the right lumbar L5, which innervates the right hindpaw and examined by Ca2+ imaging. B. Representative confocal fluorescence images of the L5 DRG showing GCaMP3 expression in primary sensory neurons (green). At baseline, naïve mouse (panel a) showed few steady-state high Ca2+ level cells and Ca2+oscillated cells (spontaneous activity), whereas CFA-treated mouse (panel c) exhibited some steady-state high Ca2+ level cells and Ca2+oscillated cells (indicated by greener and brighter neurons). Among mice that received a 100-g mechanical press delivered to the right hindpaw (panel b, d), there was an increase in the number of activated DRG neurons (indicated by greener and brighter neurons), and the increase was much greater in the CFA-treated mice than in the control mice (compare panels b & d). Scale bar: 100 μm.
Applications in pain research
Unlike in vitro studies, in vivo imaging preserves the integrity of the cellular and tissue microenvironment, including the complex interactions between neuronal and non-neuronal cells. This preservation is essential for studying pain mechanisms, as it allows for a more accurate representation of how these cells communicate and function within their native context. In contrast, ex vivo imaging provides a more controlled setting for investigating pharmacological effects, especially when using perfusion techniques. In ex vivo setups, tissues can be exposed to precise concentrations of drug, ensuring consistent and uniform exposure while allowing direct observation of tissue responses without the confounding influence of the animal's physiological state. Thus, ex vivo imaging is particularly valuable for assessing the pharmacological effects of calcium channel blockers. By combining a variety of Ca2+ markers, in vivo and ex vivo imaging techniques have greatly improved our understanding of how different calcium channel subtypes affect neuronal excitability and synaptic transmission in the pain pathway, as summarized in Table 1.
Current in vivo and ex vivo imaging techniques use in studying the regulatory mechanisms of Ca2+ channels in pain transmission
| Ca2+ indicator | Species | Imaging method | Experimental approach | Regulatory mechanism(s) | Year |
|---|---|---|---|---|---|
| GCaMP6s | Transgenic mice (Trpv1-GaMP6s) | Ex vivo 2-photon imaging of L4 DRG neuron central terminals | Pharmacological blockage | N- and P/Q-type channels mediate the increased Ca2+ amplitude in central terminals of DRG neurons following spared nerve injury. | 2024 [152] |
| GCaMP6f | Transgenic mice (Advillin-GaMP6f) | Ex vivo imaging of spinal cord slices | Genetic ablation | TRPA1 is essential for the Ca2+ response in axonal terminals triggered by microRNA lethal-7 (let-7), which acts as a potent pain inducer. | 2024 [153] |
| GCaMP6m | Mice (AAV expression) | In vivo optical fiber photometer imaging of brain | Pharmacological blockage | The functional upregulation of T-type Ca2+ channels induced by remifentanil increases activity in the thalamocortical VPLGlu→S1HLGlu circuit, contributing to the development of secondary pain in mice. | 2022 [154] |
| GCaMP3 | Transgenic mice (Pirt-GCaMP3) | In vivo confocal imaging of trigeminal ganglia | Pharmacological blockage | Stress and/or pituitary adenylate cyclase-activating polypeptide-38 (PACAP38) elevation leads to nociceptor sensitization via TRPV1. | 2024 [155] |
| GCaMP3 | Transgenic mice (Pirt-GCaMP3) | In vivo confocal imaging of dorsal root ganglia | Agonist pretreatment | Desensitization of TRPV1 in nociceptors alleviate postoperative surgery pain by reducing neuronal hyperexcitability. | 2021 [156] |
| GCaMP3 | Transgenic mice (Pirt-GCaMP3) | In vivo confocal imaging of trigeminal ganglia | shRNA interference | Upregulated descending serotonergic input causes central terminal TRPV1 sensitization in the dorsal horn and contributes to chronic neuropathic pain. | 2014 [8] |
| GCaMP | Transgenic C. elegans | In vivo imaging of ASH neurons | Diet supplementation | Phosphoinositide lipids are negative modulators of TRPV1 in vivo. | 2021 [157] |
| FRET | Transgenic C. elegans | In vivo imaging of thermonociceptor | mutations | (1) N-type channels play a crucial role in thermal sensitivity. (2) L-type channels help maintain high sensory gain. (3) Neither channel type is essential for sustaining stable Ca2+ signaling during long-term stimulation. | 2019 [158] |
| Ca2+-sensitive dye | Mice (dye injection) | In vivo 2-photon imaging of spinal cord | Genetic ablation | (1) TRPM8-positive DRG neurons are responsible for spinal responses to mild cooling. (2) TRPV1-positive DRG neurons are responsible for spinal responses to heat and strong cold. | 2016 [159] |
Although many types of calcium channels are involved in pain transmission, studies of only a few have used in vivo Ca2+ imaging, and the in vivo regulatory mechanisms of most types remain largely unexplored. While in vivo Ca2+ imaging has provided important insights into many aspects of neuronal function, its application has been relatively constrained due to several technical challenges. (1) Achieving high spatial resolution with in vivo Ca2+ imaging can be challenging, especially in deep brain regions or densely packed areas like the spinal cord. This limitation hinders the precise localization of calcium channel activity within neurons and non-neuronal cells involved in pain pathways. (2) In vivo imaging is highly susceptible to motion artifacts due to freely moving animals or physiological processes such as breathing and heartbeat. These artifacts can complicate Ca2+ signal analysis and prevent accurate observations. (3) In vivo Ca2+ imaging typically reflects the overall Ca2+ dynamics within a cell or tissue. This makes it challenging to isolate the activity of specific Ca²⁺ channel subtypes involved in pain pathways, especially when multiple types of Ca²⁺ channels contribute to the observed Ca²⁺ transients. (4) Ca2+ imaging relies on changes in intracellular Ca2+ levels as a proxy for neuronal activity, but these changes are slower and more indirect than membrane potential changes captured by voltage imaging. Voltage imaging using genetically encoded voltage indicators (GEVIs) directly measures membrane potential fluctuations with millisecond precision, capturing fast electrical events like action potentials and subthreshold depolarizations that are critical in pain signaling [160]. This makes voltage imaging better suited for studying the temporarily precise timing and dynamics of nociceptive signaling, particularly in fast-spiking neurons involved in pain responses.
These challenges highlight the need for further technological advances and innovative approaches to overcome these limitations if we are to gain a comprehensive understanding of calcium channel regulation in pain pathways. Future developments in imaging technologies and genetic tools may help address these issues and may potentially lead to more precise and informative studies of calcium channel regulation in pain processing in vivo.
Future directions
The use of genetically modified animals (e.g., transgenic mice or mice with specific gene silencing) in combination with in vivo calcium imaging will provide a powerful approach for elucidating the role of specific calcium channel subtypes in pain pathways. This method will provide detailed insights into how genetic alterations affect neural activity and pain signaling within living animals. By applying this technique across various pain models, including inflammatory, neuropathic, and other chronic pain models, as well as disease-associated transgenic mutant mice, we can unravel the complex mechanisms underlying diverse pain states and elucidate the specific contributions of different calcium channel subtypes. This approach has the potential to identify new targets for pain management and to deepen our understanding of the molecular and cellular basis of pain perception and modulation.
Conclusion
Ca2+ and Ca2+ channels are fundamental to the regulation of pain pathways, with different subtypes contributing to different aspects of pain perception and modulation. Advances in in vivo Ca2+ imaging, particularly using GECIs such as GCaMP, have greatly improved our understanding of the neural and non-neuronal dynamics in pain processing. However, a notable gap persists in the field with respect to using in vivo Ca2+ imaging to fully elucidate the regulatory role of specific Ca2+ channel subtypes in pain pathways. Current research has yet to fully explore the complex interactions and precise roles of different Ca2+ channel subtypes in vivo, and bridging this gap will be critical to developing novel therapeutic approaches targeting specific Ca2+ channels, which may lead to more effective pain relief strategies, particularly for chronic pain conditions. Progress in this direction holds the promise of unveiling new insights into pain mechanisms and opening avenues for innovative pain management techniques.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R. et al. Chronic pain as a symptom or a disease: the IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain. 2019;160:19-27
2. Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest. 2010;120:3760-72
3. Park J, Luo ZD. Calcium channel functions in pain processing. Channels (Austin). 2010;4:510-7
4. Zong W, Obenhaus HA, Skytoen ER, Eneqvist H, de Jong NL, Vale R. et al. Large-scale two-photon calcium imaging in freely moving mice. Cell. 2022;185:1240-56 e30
5. Chen C, Niehaus JK, Dinc F, Huang KL, Barnette AL, Tassou A. et al. Neural circuit basis of placebo pain relief. Nature. 2024
6. Xu Q, Ford NC, He S, Huang Q, Anderson M, Chen Z. et al. Astrocytes contribute to pain gating in the spinal cord. Sci Adv. 2021;7:eabi6287
7. Sekiguchi KJ, Shekhtmeyster P, Merten K, Arena A, Cook D, Hoffman E. et al. Imaging large-scale cellular activity in spinal cord of freely behaving mice. Nat Commun. 2016;7:11450
8. Kim YS, Chu Y, Han L, Li M, Li Z, LaVinka PC. et al. Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron. 2014;81:873-87
9. Kim YS, Anderson M, Park K, Zheng Q, Agarwal A, Gong C. et al. Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron. 2016;91:1085-96
10. Bibineyshvili Y, Vajtay TJ, Salsabilian S, Fliss N, Suvarnakar A, Fang J. et al. Imaging the large-scale and cellular response to focal traumatic brain injury in mouse neocortex. bioRxiv. 2024
11. Shannonhouse J, Bernabucci M, Gomez R, Son H, Zhang Y, Ai CH. et al. Meclizine and Metabotropic Glutamate Receptor Agonists Attenuate Severe Pain and Ca(2+) Activity of Primary Sensory Neurons in Chemotherapy-Induced Peripheral Neuropathy. J Neurosci. 2022;42:6020-37
12. Son H, Zhang Y, Shannonhouse J, Ishida H, Gomez R, Kim YS. Mast-cell-specific receptor mediates alcohol-withdrawal-associated headache in male mice. Neuron. 2024;112:113-23 e4
13. Felix R, Calderon-Rivera A, Andrade A. Regulation of high-voltage-activated Ca(2+) channel function, trafficking, and membrane stability by auxiliary subunits. Wiley Interdiscip Rev Membr Transp Signal. 2013;2:207-20
14. Rajagopal S. Modulatory Action of Voltage-gated Ion Channels in Inflammation and Inflammatory Pain. The Open Neurology Journal. 2023;17:1874-205X /23
15. Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594:5369-90
16. Dolphin AC. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13:542-55
17. Cui W, Wu H, Yu X, Song T, Xu X, Xu F. The Calcium Channel alpha2delta1 Subunit: Interactional Targets in Primary Sensory Neurons and Role in Neuropathic Pain. Front Cell Neurosci. 2021;15:699731
18. Felix R, Gurnett CA, De Waard M, Campbell KP. Dissection of functional domains of the voltage-dependent Ca2+ channel alpha2delta subunit. J Neurosci. 1997;17:6884-91
19. Felix R. Voltage-dependent Ca2+ channel alpha2delta auxiliary subunit: structure, function and regulation. Recept Channels. 1999;6:351-62
20. Cassidy JS, Ferron L, Kadurin I, Pratt WS, Dolphin AC. Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary alpha2delta-1 subunits. Proc Natl Acad Sci U S A. 2014;111:8979-84
21. Bernstein GM, Jones OT. Kinetics of internalization and degradation of N-type voltage-gated calcium channels: role of the alpha2/delta subunit. Cell Calcium. 2007;41:27-40
22. Tachiya D, Sato T, Ichikawa H. Nerve Injury Increases the Expression of Alpha-2/Delta-1 Subunit of L-Type Calcium Channel in Sensory Neurons of Rat Spinal and Trigeminal Nerves. Ann Neurosci. 2018;24:191-200
23. Cui WQ, Chu YX, Xu F, Chen T, Gao L, Feng Y. et al. Calcium Channel alpha2delta1 Subunit Mediates Secondary Orofacial Hyperalgesia Through PKC-TRPA1/Gap Junction Signaling. J Pain. 2020;21:238-57
24. Alles SR, Garcia E, Balasubramanyan S, Jones K, Tyson JR, Joy T. et al. Peripheral nerve injury increases contribution of L-type calcium channels to synaptic transmission in spinal lamina II: Role of alpha2delta-1 subunits. Mol Pain. 2018;14:1744806918765806
25. Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME. et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868-75
26. Li CY, Zhang XL, Matthews EA, Li KW, Kurwa A, Boroujerdi A. et al. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain. 2006;125:20-34
27. Rangel-Galvan M, Rangel-Galvan V, Rangel-Huerta A. T-type calcium channel modulation by hydrogen sulfide in neuropathic pain conditions. Front Pharmacol. 2023;14:1212800
28. Fossat P, Dobremez E, Bouali-Benazzouz R, Favereaux A, Bertrand SS, Kilk K. et al. Knockdown of L calcium channel subtypes: differential effects in neuropathic pain. J Neurosci. 2010;30:1073-85
29. Radwani H, Lopez-Gonzalez MJ, Cattaert D, Roca-Lapirot O, Dobremez E, Bouali-Benazzouz R. et al. Cav1.2 and Cav1.3 L-type calcium channels independently control short- and long-term sensitization to pain. J Physiol. 2016;594:6607-26
30. Ramachandra R, Hassan B, McGrew SG, Dompor J, Farrag M, Ruiz-Velasco V. et al. Identification of CaV channel types expressed in muscle afferent neurons. J Neurophysiol. 2013;110:1535-43
31. Weber AM, Wong FK, Tufford AR, Schlichter LC, Matveev V, Stanley EF. N-type Ca2+ channels carry the largest current: implications for nanodomains and transmitter release. Nat Neurosci. 2010;13:1348-50
32. DuBreuil DM, Lopez Soto EJ, Daste S, Meir R, Li D, Wainger B. et al. Heat But Not Mechanical Hypersensitivity Depends on Voltage-Gated Ca(V)2.2 Calcium Channel Activity in Peripheral Axon Terminals Innervating Skin. J Neurosci. 2021;41:7546-60
33. Pitake S, Middleton LJ, Abdus-Saboor I, Mishra SK. Inflammation Induced Sensory Nerve Growth and Pain Hypersensitivity Requires the N-Type Calcium Channel Cav2.2. Front Neurosci. 2019;13:1009
34. Hatakeyama S, Wakamori M, Ino M, Miyamoto N, Takahashi E, Yoshinaga T. et al. Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca(2+) channels. Neuroreport. 2001;12:2423-7
35. Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T. et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 2001;20:2349-56
36. Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H. et al. Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol Cell Neurosci. 2001;18:235-45
37. Nair AS, Poornachand A, Kodisharapu PK. Ziconotide: Indications, Adverse Effects, and Limitations in Managing Refractory Chronic Pain. Indian J Palliat Care. 2018;24:118-9
38. Gomez K, Santiago U, Nelson TS, Allen HN, Calderon-Rivera A, Hestehave S. et al. A peptidomimetic modulator of the Ca(V)2.2 N-type calcium channel for chronic pain. Proc Natl Acad Sci U S A. 2023;120:e2305215120
39. Perez-Miller S, Gomez K, Khanna R. Peptide and Peptidomimetic Inhibitors Targeting the Interaction of Collapsin Response Mediator Protein 2 with the N-Type Calcium Channel for Pain Relief. ACS Pharmacol Transl Sci. 2024;7:1916-36
40. Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. 1998;18:6319-30
41. Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994;269:1117-23
42. Luvisetto S, Marinelli S, Panasiti MS, D'Amato FR, Fletcher CF, Pavone F. et al. Pain sensitivity in mice lacking the Ca(v)2.1alpha1 subunit of P/Q-type Ca2+ channels. Neuroscience. 2006;142:823-32
43. Gambeta E, Gandini MA, Souza IA, Ferron L, Zamponi GW. A CACNA1A variant associated with trigeminal neuralgia alters the gating of Cav2.1 channels. Mol Brain. 2021;14:4
44. Grangeon L, Lange KS, Waliszewska-Prosol M, Onan D, Marschollek K, Wiels W. et al. Genetics of migraine: where are we now? J Headache Pain. 2023;24:12
45. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411-25
46. Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895-911
47. Cai S, Gomez K, Moutal A, Khanna R. Targeting T-type/CaV3.2 channels for chronic pain. Transl Res. 2021;234:20-30
48. Jacus MO, Uebele VN, Renger JJ, Todorovic SM. Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci. 2012;32:9374-82
49. Wang R, Lewin GR. The Cav3.2 T-type calcium channel regulates temporal coding in mouse mechanoreceptors. J Physiol. 2011;589:2229-43
50. Wu J, Peng S, Xiao L, Cheng X, Kuang H, Zhu M. et al. Cell-Type Specific Distribution of T-Type Calcium Currents in Lamina II Neurons of the Rat Spinal Cord. Front Cell Neurosci. 2018;12:370
51. Liao YF, Tsai ML, Chen CC, Yen CT. Involvement of the Cav3.2 T-type calcium channel in thalamic neuron discharge patterns. Mol Pain. 2011;7:43
52. Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S. et al. The Low-Threshold Calcium Channel Cav3.2 Determines Low-Threshold Mechanoreceptor Function. Cell Rep. 2015;10:370-82
53. Gadotti VM, Huang S, Zamponi GW. The terpenes camphene and alpha-bisabolol inhibit inflammatory and neuropathic pain via Cav3.2 T-type calcium channels. Mol Brain. 2021;14:166
54. Watanabe M, Ueda T, Shibata Y, Kumamoto N, Shimada S, Ugawa S. Expression and Regulation of Cav3.2 T-Type Calcium Channels during Inflammatory Hyperalgesia in Mouse Dorsal Root Ganglion Neurons. PLoS One. 2015;10:e0127572
55. Lin SF, Yu XL, Wang B, Zhang YJ, Sun YG, Liu XJ. Colocalization of insulin-like growth factor-1 receptor and T type Cav3.2 channel in dorsal root ganglia in chronic inflammatory pain mouse model. Neuroreport. 2016;27:737-43
56. Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER. et al. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci. 2007;27:3305-16
57. Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ. et al. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain. 2009;145:184-95
58. Yue J, Liu L, Liu Z, Shu B, Zhang Y. Upregulation of T-type Ca2+ channels in primary sensory neurons in spinal nerve injury. Spine (Phila Pa 1976). 2013;38:463-70
59. Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O. et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315-24
60. Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK. et al. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol. 2008;99:3151-6
61. Gomez K, Calderon-Rivera A, Sandoval A, Gonzalez-Ramirez R, Vargas-Parada A, Ojeda-Alonso J. et al. Cdk5-Dependent Phosphorylation of Ca(V)3.2 T-Type Channels: Possible Role in Nerve Ligation-Induced Neuropathic Allodynia and the Compound Action Potential in Primary Afferent C Fibers. J Neurosci. 2020;40:283-96
62. Feng XJ, Ma LX, Jiao C, Kuang HX, Zeng F, Zhou XY. et al. Nerve injury elevates functional Cav3.2 channels in superficial spinal dorsal horn. Mol Pain. 2019;15:1744806919836569
63. Marger F, Gelot A, Alloui A, Matricon J, Ferrer JF, Barrere C. et al. T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc Natl Acad Sci U S A. 2011;108:11268-73
64. Tsubota M, Matsui K, Nakano M, Kajitani R, Ishii Y, Tomochika K. et al. Essential role of Ca(v)3.2 T-type calcium channels in butyrate-induced colonic pain and nociceptor hypersensitivity in mice. Eur J Pharmacol. 2020;887:173576
65. Liu Q, Lu Z, Ren H, Fu L, Wang Y, Bu H. et al. Cav3.2 T-Type calcium channels downregulation attenuates bone cancer pain induced by inhibiting IGF-1/HIF-1alpha signaling pathway in the rat spinal cord. J Bone Oncol. 2023;42:100495
66. Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM. et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain. 2017;158:417-29
67. Zhang Y, Wei Y, Zheng T, Tao Y, Sun Y, Jiang D. et al. Adiponectin receptor 1-mediated stimulation of Cav3.2 channels in trigeminal ganglion neurons induces nociceptive behaviors in mice. J Headache Pain. 2023;24:117
68. Qi R, Cao J, Sun Y, Li Y, Huang Z, Jiang D. et al. Histone methylation-mediated microRNA-32-5p down-regulation in sensory neurons regulates pain behaviors via targeting Cav3.2 channels. Proc Natl Acad Sci U S A. 2022;119:e2117209119
69. Wang H, Wei Y, Pu Y, Jiang D, Jiang X, Zhang Y. et al. Brain-derived neurotrophic factor stimulation of T-type Ca(2+) channels in sensory neurons contributes to increased peripheral pain sensitivity. Sci Signal. 2019 12
70. Gambeta E, Gandini MA, Souza IA, Zamponi GW. Ca V 3.2 calcium channels contribute to trigeminal neuralgia. Pain. 2022;163:2315-25
71. Mustafa ER, Gambeta E, Stringer RN, Souza IA, Zamponi GW, Weiss N. Electrophysiological and computational analysis of Ca(v)3.2 channel variants associated with familial trigeminal neuralgia. Mol Brain. 2022;15:91
72. Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R. et al. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol. 2011;80:900-10
73. Shin SM, Cai Y, Itson-Zoske B, Qiu C, Hao X, Xiang H. et al. Enhanced T-type calcium channel 3.2 activity in sensory neurons contributes to neuropathic-like pain of monosodium iodoacetate-induced knee osteoarthritis. Mol Pain. 2020;16:1744806920963807
74. Liu H, Lauzadis J, Gunaratna K, Sipple E, Kaczocha M, Puopolo M. Inhibition of T-Type Calcium Channels With TTA-P2 Reduces Chronic Neuropathic Pain Following Spinal Cord Injury in Rats. J Pain. 2023;24:1681-95
75. Chen W, Chi YN, Kang XJ, Liu QY, Zhang HL, Li ZH. et al. Accumulation of Ca(v)3.2 T-type Calcium Channels in the Uninjured Sural Nerve Contributes to Neuropathic Pain in Rats with Spared Nerve Injury. Front Mol Neurosci. 2018;11:24
76. Antunes FTT, Huang S, Chen L, Zamponi GW. Effect of ABT-639 on Cav3.2 channel activity and its analgesic actions in mouse models of inflammatory and neuropathic pain. Eur J Pharmacol. 2024;967:176416
77. Harding EK, Dedek A, Bonin RP, Salter MW, Snutch TP, Hildebrand ME. The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. Br J Pharmacol. 2021;178:3517-32
78. Ondacova K, Karmazinova M, Lazniewska J, Weiss N, Lacinova L. Modulation of Cav3.2 T-type calcium channel permeability by asparagine-linked glycosylation. Channels (Austin). 2016;10:175-84
79. Weiss N, Black SA, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013;465:1159-70
80. Joksimovic SL, Evans JG, McIntire WE, Orestes P, Barrett PQ, Jevtovic-Todorovic V. et al. Glycosylation of Ca(V)3.2 Channels Contributes to the Hyperalgesia in Peripheral Neuropathy of Type 1 Diabetes. Front Cell Neurosci. 2020;14:605312
81. Orestes P, Osuru HP, McIntire WE, Jacus MO, Salajegheh R, Jagodic MM. et al. Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes. 2013;62:3828-38
82. Hoffmann T, Kistner K, Joksimovic SLJ, Todorovic SM, Reeh PW, Sauer SK. Painful diabetic neuropathy leads to functional Ca(V)3.2 expression and spontaneous activity in skin nociceptors of mice. Exp Neurol. 2021;346:113838
83. Todorovic SM, Jevtovic-Todorovic V. Targeting of CaV3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflugers Arch. 2014;466:701-6
84. Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J Neurosci. 2003;23:10116-21
85. Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, Howell N. et al. Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. J Clin Invest. 2006;116:2403-12
86. Iftinca MC, Zamponi GW. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci. 2009;30:32-40
87. Gomez K, Vargas-Parada A, Duran P, Sandoval A, Delgado-Lezama R, Khanna R. et al. L5-6 Spinal Nerve Ligation-induced Neuropathy Changes the Location and Function of Ca(2+) Channels and Cdk5 and Affects the Compound Action Potential in Adjacent Intact L4 Afferent Fibers. Neuroscience. 2021;471:20-31
88. Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V. et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron. 2014;83:1144-58
89. Garcia-Caballero A, Gadotti VM, Chen L, Zamponi GW. A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Mol Pain. 2016 12
90. Stemkowski PL, Garcia-Caballero A, Gadotti VM, M'Dahoma S, Chen L, Souza IA. et al. Identification of interleukin-1 beta as a key mediator in the upregulation of Cav3.2-USP5 interactions in the pain pathway. Mol Pain. 2017;13:1744806917724698
91. Gadotti VM, Zamponi GW. Disrupting USP5/Cav3.2 interactions protects female mice from mechanical hypersensitivity during peripheral inflammation. Mol Brain. 2018;11:60
92. Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF. et al. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207-14
93. Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442-6
94. Yokoyama CT, Myers SJ, Fu J, Mockus SM, Scheuer T, Catterall WA. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28:1-17
95. Gomez K, Vallecillo TGM, Moutal A, Perez-Miller S, Delgado-Lezama R, Felix R. et al. The role of cyclin-dependent kinase 5 in neuropathic pain. Pain. 2020;161:2674-89
96. Gaifullina AS, Lazniewska J, Gerasimova EV, Burkhanova GF, Rzhepetskyy Y, Tomin A. et al. A potential role for T-type calcium channels in homocysteinemia-induced peripheral neuropathy. Pain. 2019;160:2798-810
97. Anand S, Rajagopal S. A Comprehensive Review on the Regulatory Action of TRP Channels: A Potential Therapeutic Target for Nociceptive Pain. Neurosci Insights. 2023;18:26331055231220340
98. Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P. et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183-7
99. Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR. et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306-13
100. Ma W, Quirion R. Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin Ther Targets. 2007;11:307-20
101. Shuba YM. Beyond Neuronal Heat Sensing: Diversity of TRPV1 Heat-Capsaicin Receptor-Channel Functions. Front Cell Neurosci. 2020;14:612480
102. Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ. et al. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc Natl Acad Sci U S A. 2003;100:12480-5
103. Wang S, Joseph J, Ro JY, Chung MK. Modality-specific mechanisms of protein kinase C-induced hypersensitivity of TRPV1: S800 is a polymodal sensitization site. Pain. 2015;156:931-41
104. Cao E, Cordero-Morales JF, Liu B, Qin F, Julius D. TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron. 2013;77:667-79
105. Lukacs V, Thyagarajan B, Varnai P, Balla A, Balla T, Rohacs T. Dual regulation of TRPV1 by phosphoinositides. J Neurosci. 2007;27:7070-80
106. Ryskamp DA, Redmon S, Jo AO, Krizaj D. TRPV1 and Endocannabinoids: Emerging Molecular Signals that Modulate Mammalian Vision. Cells. 2014;3:914-38
107. Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol. 2004;123:53-62
108. Zhang X, Li L, McNaughton PA. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron. 2008;59:450-61
109. Grandl J, Kim SE, Uzzell V, Bursulaya B, Petrus M, Bandell M. et al. Temperature-induced opening of TRPV1 ion channel is stabilized by the pore domain. Nat Neurosci. 2010;13:708-14
110. Dhaka A, Uzzell V, Dubin AE, Mathur J, Petrus M, Bandell M. et al. TRPV1 is activated by both acidic and basic pH. J Neurosci. 2009;29:153-8
111. Khairatkar-Joshi N, Szallasi A. TRPV1 antagonists: the challenges for therapeutic targeting. Trends Mol Med. 2009;15:14-22
112. Souza Monteiro de Araujo D, Nassini R, Geppetti P, De Logu F. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin Ther Targets. 2020;24:997-1008
113. Koivisto A, Chapman H, Jalava N, Korjamo T, Saarnilehto M, Lindstedt K. et al. TRPA1: a transducer and amplifier of pain and inflammation. Basic Clin Pharmacol Toxicol. 2014;114:50-5
114. Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y. et al. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393-401
115. Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S. et al. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain. 2008;4:48
116. De Logu F, Nassini R, Materazzi S, Carvalho Goncalves M, Nosi D, Rossi Degl'Innocenti D. et al. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat Commun. 2017;8:1887
117. De Logu F, Li Puma S, Landini L, Portelli F, Innocenti A, de Araujo DSM. et al. Schwann cells expressing nociceptive channel TRPA1 orchestrate ethanol-evoked neuropathic pain in mice. J Clin Invest. 2019;129:5424-41
118. Brackley AD, Gomez R, Guerrero KA, Akopian AN, Glucksman MJ, Du J. et al. A-Kinase Anchoring Protein 79/150 Scaffolds Transient Receptor Potential A 1 Phosphorylation and Sensitization by Metabotropic Glutamate Receptor Activation. Sci Rep. 2017;7:1842
119. Takahashi N, Mizuno Y, Kozai D, Yamamoto S, Kiyonaka S, Shibata T. et al. Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin). 2008;2:287-98
120. Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF. et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541-5
121. Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y. et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15:1015-21
122. Lee B, Cho H, Jung J, Yang YD, Yang DJ, Oh U. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol Pain. 2014;10:5
123. Pineda-Farias JB, Barragan-Iglesias P, Loeza-Alcocer E, Torres-Lopez JE, Rocha-Gonzalez HI, Perez-Severiano F. et al. Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Mol Pain. 2015;11:41
124. Takayama Y, Uta D, Furue H, Tominaga M. Pain-enhancing mechanism through interaction between TRPV1 and anoctamin 1 in sensory neurons. Proc Natl Acad Sci U S A. 2015;112:5213-8
125. Shah S, Carver CM, Mullen P, Milne S, Lukacs V, Shapiro MS. et al. Local Ca(2+) signals couple activation of TRPV1 and ANO1 sensory ion channels. Sci Signal. 2020 13
126. Echeverria F, Gonzalez-Sanabria N, Alvarado-Sanchez R, Fernandez M, Castillo K, Latorre R. Large conductance voltage-and calcium-activated K(+) (BK) channel in health and disease. Front Pharmacol. 2024;15:1373507
127. Chen SR, Cai YQ, Pan HL. Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J Neurochem. 2009;110:352-62
128. Cao XH, Chen SR, Li L, Pan HL. Nerve injury increases brain-derived neurotrophic factor levels to suppress BK channel activity in primary sensory neurons. J Neurochem. 2012;121:944-53
129. Lu R, Lukowski R, Sausbier M, Zhang DD, Sisignano M, Schuh CD. et al. BKCa channels expressed in sensory neurons modulate inflammatory pain in mice. Pain. 2014;155:556-65
130. Liu CY, Li N, Zhao YF, Ma B. [BK(Ca) channel agonist NS1619 and Kv channel antagonist 4-AP on the facial mechanical pain threshold in a rat model of chronic constriction injury of the infraorbital nerve]. Sheng Li Xue Bao. 2010;62:441-9
131. Zhang FX, Gadotti VM, Souza IA, Chen L, Zamponi GW. BK Potassium Channels Suppress Cavalpha2delta Subunit Function to Reduce Inflammatory and Neuropathic Pain. Cell Rep. 2018;22:1956-64
132. Zhang XF, Gopalakrishnan M, Shieh CC. Modulation of action potential firing by iberiotoxin and NS1619 in rat dorsal root ganglion neurons. Neuroscience. 2003;122:1003-11
133. DosSantos MF, Filha LGA, Verissimo CP, Sanz CK, Gazerani P. Presence of Small-Conductance Calcium-Activated Potassium (SK) Channels in the Central and Peripheral Nervous Systems and Their Role in Health and Disease. J Integr Neurosci. 2023;22:69
134. Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol. 2004;554:255-61
135. Walcott KCE, Mauthner SE, Tsubouchi A, Robertson J, Tracey WD. The Drosophila Small Conductance Calcium-Activated Potassium Channel Negatively Regulates Nociception. Cell Rep. 2018;24:3125-32 e3
136. Wang B, Ma L, Guo X, Du S, Feng X, Liang Y. et al. A sensory neuron-specific long non-coding RNA reduces neuropathic pain by rescuing KCNN1 expression. Brain. 2023;146:3866-84
137. Wang H, Zuo W, Feng X, Huo X, Liang Y, Wang B. et al. ESRRG-controlled downregulation of KCNN1 in primary sensory neurons is required for neuropathic pain. JCI Insight. 2024 9
138. Hall BE, Macdonald E, Cassidy M, Yun S, Sapio MR, Ray P. et al. Transcriptomic analysis of human sensory neurons in painful diabetic neuropathy reveals inflammation and neuronal loss. Sci Rep. 2022;12:4729
139. Boettger MK, Till S, Chen MX, Anand U, Otto WR, Plumpton C. et al. Calcium-activated potassium channel SK1- and IK1-like immunoreactivity in injured human sensory neurones and its regulation by neurotrophic factors. Brain. 2002;125:252-63
140. Li ES, Saha MS. Optimizing Calcium Detection Methods in Animal Systems: A Sandbox for Synthetic Biology. Biomolecules. 2021 11
141. Tian L, Hires SA, Looger LL. Imaging neuronal activity with genetically encoded calcium indicators. Cold Spring Harb Protoc. 2012;2012:647-56
142. Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137-41
143. Reiff DF, Ihring A, Guerrero G, Isacoff EY, Joesch M, Nakai J. et al. In vivo performance of genetically encoded indicators of neural activity in flies. J Neurosci. 2005;25:4766-78
144. Ohkura M, Matsuzaki M, Kasai H, Imoto K, Nakai J. Genetically encoded bright Ca2+ probe applicable for dynamic Ca2+ imaging of dendritic spines. Anal Chem. 2005;77:5861-9
145. Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R. et al. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci U S A. 2006;103:4753-8
146. Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S. et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819-40
147. Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295-300
148. Zhang Y, Rozsa M, Liang Y, Bushey D, Wei Z, Zheng J. et al. Fast and sensitive GCaMP calcium indicators for imaging neural populations. Nature. 2023;615:884-91
149. Thestrup T, Litzlbauer J, Bartholomaus I, Mues M, Russo L, Dana H. et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods. 2014;11:175-82
150. Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP. et al. Sensitive red protein calcium indicators for imaging neural activity. Elife. 2016 5
151. Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolo J. et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci. 2013;6:2
152. Ferron L, Harding EK, Gandini MA, Brideau C, Stys PK, Zamponi GW. Functional remodeling of presynaptic voltage-gated calcium channels in superficial layers of the dorsal horn during neuropathic pain. iScience. 2024;27:109973
153. Chen O, Jiang C, Berta T, Powell Gray B, Furutani K, Sullenger BA. et al. MicroRNA let-7b enhances spinal cord nociceptive synaptic transmission and induces acute and persistent pain through neuronal and microglial signaling. Pain. 2024;165:1824-39
154. Jin Y, Mao Y, Chen D, Tai Y, Hu R, Yang CL. et al. Thalamocortical circuits drive remifentanil-induced postoperative hyperalgesia. J Clin Invest. 2022 132
155. Son H, Zhang Y, Shannonhouse J, Gomez R, Kim YS. PACAP38/mast-cell-specific receptor axis mediates repetitive stress-induced headache in mice. J Headache Pain. 2024;25:87
156. Ishida H, Zhang Y, Gomez R, Shannonhouse J, Son H, Banik R. et al. In vivo Calcium Imaging Visualizes Incision-Induced Primary Afferent Sensitization and Its Amelioration by Capsaicin Pretreatment. J Neurosci. 2021;41:8494-507
157. Caires R, Bell B, Lee J, Romero LO, Vasquez V, Cordero-Morales JF. Deficiency of Inositol Monophosphatase Activity Decreases Phosphoinositide Lipids and Enhances TRPV1 Function In vivo. J Neurosci. 2021;41:408-23
158. Saro G, Lia AS, Thapliyal S, Marques F, Busch KE, Glauser DA. Specific Ion Channels Control Sensory Gain, Sensitivity, and Kinetics in a Tonic Thermonociceptor. Cell Rep. 2020;30:397-408 e4
159. Ran C, Hoon MA, Chen X. The coding of cutaneous temperature in the spinal cord. Nat Neurosci. 2016;19:1201-9
160. Zhang Y, Shannonhouse J, Gomez R, Son H, Ishida H, Stephen Evans GJ. et al. Imaging sensory transmission and neuronal plasticity in primary sensory neurons with genetically-encoded voltage indicators. bioRxiv. 2021
Author contact
![]() Corresponding author: Dr. Yu Shin Kim Email: kimy1edu.
Corresponding author: Dr. Yu Shin Kim Email: kimy1edu.