Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structure and signaling cascade...
GPCRs in MSCs
GPCRs in osteoblast and osteocyte
GPCRs in macrophage and...
GPCRs in chondrocyte
GPCRs in other cells
Therapeutic applications of...
Emerging concepts in GPCR...
Perspective and Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(11):4736-4761. doi:10.7150/ijbs.113585 This issue Cite
Review
The function of GPCRs in different bone cells
Yan Zhang1,2*, Nai-Ning Wang1,3*, Zi-Han Qiu1, Jia-Hao Wang1, Wen-Na An1, Li-Dan Shi1, Fei Chen1, Da-Jin Zhang1, Si-Yue Wang1, Tie-Lin Yang1,3, Shou-Ye Hu2 ![]() , Yan Guo1,2
, Yan Guo1,2 ![]()
1. Key Laboratory of Biomedical Information Engineering of Ministry of Education, Key Laboratory of Biology Multiomics and Diseases in Shaanxi Province Higher Education Institutions, and Biomedical Informatics & Genomics Center, School of Life Science and Technology, Xi'an Jiaotong University, Xi'an, Shaanxi, 710049, China.
2. Department of Joint Surgery, Honghui Hospital, Xi'an Jiaotong University, Xi'an, Shaanxi, 710054, China.
3. Department of Orthopedics, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi, 710061, China.
* These authors contributed equally to this study.
Received 2025-3-11; Accepted 2025-6-28; Published 2025-7-24
Abstract

G protein-coupled receptors (GPCRs) are recognized as critical therapeutic targets in bone disorders, owing to their multifaceted regulatory roles across diverse bone cell lineages. This review systematically catalogs GPCR expression and functional heterogeneity in key bone cells: 12 GPCRs in mesenchymal stem cells (MSCs) orchestrate lineage specification; 21 GPCRs in osteoblasts/osteocytes mediate matrix mineralization and mechanotransduction; 23 GPCRs in macrophages/osteoclasts regulate inflammatory bone resorption; 31 GPCRs in chondrocytes govern endochondral ossification and osteoarthritis pathogenesis; and 8 GPCRs in other cell types modulate bone-related physiological processes. By integrating canonical signaling axes—cAMP/PKA-dependent transcriptional networks, PLC-β/IP3-driven calcium signaling, and NF-κB-modulated immuno-skeletal interactions—we elucidate how GPCRs dynamically coordinate cellular plasticity to maintain skeletal homeostasis. This work establishes a multidimensional research framework integrating historical context, molecular mechanisms, and cutting-edge methodologies to advance GPCR-targeted therapies for bone-related diseases. Moreover, this review provides insights for clinical translation, including biased agonism and allosteric modulation precision strategies to restore skeletal equilibrium in osteoporosis, arthritis, and regenerative medicine.
Keywords: GPCR, MSC, osteoblast, osteoclast, chondrocyte
Introduction
Bone modeling is a dynamic and continuous process fundamental for maintaining skeletal integrity and facilitating repair throughout an individual's lifetime. This intricate process hinges on a delicate interplay among multiple cells, governed by a sophisticated network of molecular interactions. Among the pivotal regulators of this process are G protein-coupled receptors (GPCRs), a family of transmembrane proteins that act as critical sensors of extracellular cues and translators of intracellular responses [1-3]. GPCRs play indispensable roles in transducing diverse extracellular signals into intracellular responses across various physiological systems.
Ubiquitously expressed across tissues and cells, GPCRs have long been key targets for drug development, accounting for approximately 33% of currently marketed drugs [4, 5]. Within skeletal biology, GPCRs directly modulate the functions of mesenchymal stem cells (MSCs), osteoblasts, osteocytes, osteoclasts, chondrocytes and other bone cells, thereby influencing bone metabolism and homeostasis. They have emerged as a significant target family for the treatment of bone-related diseases. Notable examples include the Parathyroid hormone (PTH) receptor, whose agonist abaloparatide promotes osteoblast differentiation while suppressing osteoclast activity, and teriparatide (a recombinant human PTH 1-34 fragment), which inhibits bone resorption, stimulates bone formation, and is approved for treating osteoporosis in postmenopausal women, old men, and glucocorticoid-induced cases [6, 7]. Additionally, the Calcium-sensing receptor (CaSR) and its agonists (e.g., calcimimetics) enhance extracellular calcium sensitivity, mitigating hypercalcemia-related complications like osteoporosis and extending therapeutic potential to fracture healing and bone tumors [6]. These applications demonstrate a pivotal role of GPCRs in skeletal biology and the regulatory mechanisms of bone modeling, and underscore GPCRs' translational relevance in addressing skeletal disorders.
Skeletal homeostasis emerges from the coordinated activities of diverse bone cell populations, with GPCRs orchestrating specialized functions through context-dependent signaling [1, 2, 7]. GPCR activity is influenced by age, genetic, and environmental factors, with functional changes directly contributing to bone metabolic imbalances during development, aging, and disease progression. In MSCs, GPCRs couple to G protein subtypes (e.g., Gs, Gq, Gi) to direct lineage commitment toward osteoblasts or chondrocytes. Osteoblasts utilize GPCRs to regulate matrix synthesis and mineralization while secreting paracrine signals that dampen osteoclastogenesis. Conversely, osteoclast-surface GPCRs integrate hormonal and local cues to modulate resorptive activity, whereas osteocytes, as mechanosensors, employ GPCRs to translate mechanical stimuli into adaptive remodeling signals. Macrophages and chondrocytes also leverage GPCRs to mediate inflammatory responses and joint cartilage metabolism, respectively. Dysregulation of these pathways—such as aberrant GPCR signaling—contributes to pathologies like osteoporosis, osteoarthritis, and rheumatoid arthritis. Emerging study highlights GPCRs as both biomarkers and therapeutic targets, with their functional plasticity offering opportunities for cells interventions [3, 5, 8]. Understanding the molecular functions and mechanisms underlying GPCR-mediated regulation in different bone cells has profound implications for the development of novel drug targets and therapeutic strategies for bone-related diseases.
Here, we comprehensively summarize the crucial roles and mechanisms of GPCRs in different bone cells, based on emerging evidence from numerous studies. We also establish a multidimensional framework that integrates historical context, molecular mechanisms, and cutting-edge methodologies to advance GPCR research in bone biology. In conclusion, our review consolidates the current understanding of GPCRs in various bone cells and paves the way for the development of novel drug targets and therapeutic strategies for bone-related diseases. As the field of GPCR research continues to evolve, we anticipate that future studies will further elucidate the functional nuances of these receptors in bone metabolism and uncover new therapeutic opportunities for addressing bone disorders.
Structure and signaling cascade of GPCR
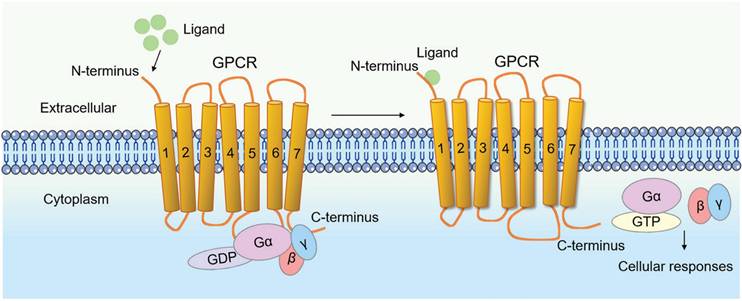
GPCRs constitute a large and diverse superfamily of transmembrane proteins, widely distributed and serving as crucial membrane protein receptors [4, 5, 9]. The hallmark of GPCRs is their characteristic seven-transmembrane α-helical structure. The binding site for the G protein (guanylate-binding protein) is located at the C-terminus of the polypeptide chain and on the third intracellular loop between the fifth and sixth transmembrane helices, as counted from the N-terminus (Fig. 1).
Schematic representation of the G protein-coupled receptor (GPCR) model. The hallmark of GPCR receptors is their characteristic seven-transmembrane α-helical structure. The binding site for the G protein (guanylate-binding protein) is located at the C-terminus of the polypeptide chain and on the third intracellular loop between the fifth and sixth transmembrane helices, as counted from the N-terminus. Their activation by external signals triggers a series of biochemical reactions through interactions with different G proteins or arrestins, thereby regulating diverse physiological processes.
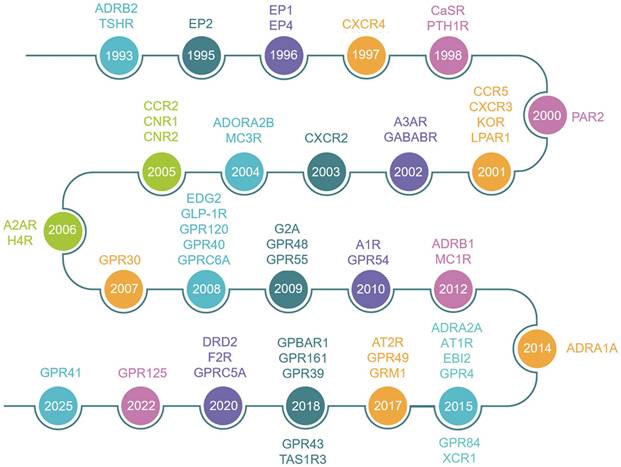
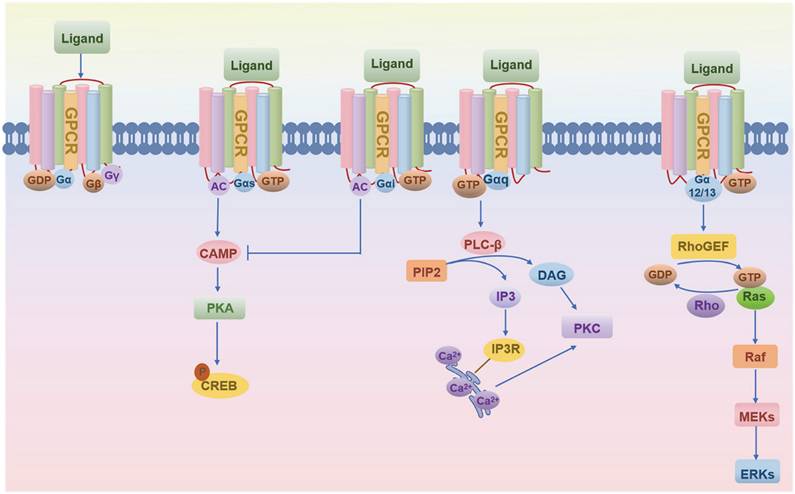
Since 1993, the skeletal research field has witnessed the identification of over 56 pivotal GPCRs as critical regulators of bone cell physiology (Fig. 2). Activation of these GPCRs by external signals triggers a series of biochemical reactions through interactions with distinct G proteins or arrestins, thereby regulating diverse physiological processes (Fig. 1 and Fig. 3) [4, 9]. GPCRs mediate rapid signal transmission through G-protein-dependent pathways. The signaling cascade of GPCRs comprises four key stages: ligand-receptor binding, G protein activation, downstream pathway initiation, and cell-specific functional regulation [10, 11]. GPCR signaling is initiated by extracellular ligands, including: hormones (e.g., PTH), ions (e.g., Calcium ions, Ca²⁺), lipid molecules (e.g., Lysophosphatidic acid, LPA; Sphingosine-1-phosphate, S1P), and chemokines (e.g., Stromal cell-derived factor-1, SDF-1) [10, 11]. Ligand binding induces conformational changes in GPCRs, triggering G protein activation. GPCRs couple to heterotrimeric G proteins composed of α, β, and γ subunits [10, 11]. At inactive state, Gα binds GDP and associates with Gβγ [10, 11]. At activation process, ligand binding catalyzes GDP-to-GTP exchange on Gα. Then GTP-bound Gα dissociates from Gβγ, forming Gα-GTP and free Gβγ. After that both subunits activate downstream effectors, initiating distinct pathways. Functional specialization of G protein subtypes mainly includes Gs-type (e.g., Gαs) activates Adenylyl cyclase (AC), increasing cAMP. On the contrary, Gi inhibits the formation of cAMP catalyzed by AC. Gq-type (e.g., Gαq) activates Phospholipase C (PLC), hydrolyzing Phosphatidylinositol 4,5-bisphosphate (PIP2) into Diacylglycerol (DAG) and Inositol triphosphate (IP3). Gi-type (e.g., Gαi) inhibits AC, reducing cAMP. Gβγ complex independently activates pathways like Ras-MAPK. G protein subunits regulate cellular behavior via three core pathways, including cAMP- protein kinase A (PKA)-CREB pathway (Gs-dominant), PLC-protein kinase C (PKC)/Ca²⁺ pathway (Gq-dominant), and Ras-MAPK Pathway (Gβγ- or Gα12/13-dominant) (Fig. 3) [3, 10, 11]. This pathway drives immediate physiological responses (e.g., metabolic regulation) and terminates signals via GTP hydrolysis and β-arrestin-mediated desensitization, enabling precise spatiotemporal control. Besides, GPCRs mediate prolonged signaling regulation through G-protein-independent pathways. Receptor phosphorylation recruits β-arrestin, forming complexes that activate MAPKs (e.g., ERK, JNK), Src family kinases, or NF-κB to regulate cell proliferation, stress responses, and gene expression. Signals persist longer in this pathway and may extend to perinuclear regions via endosomal trafficking, influencing nuclear functions. Biased ligands selectively targeting this pathway offer novel therapeutic opportunities for disease [4, 9]. The diversity of GPCR signaling pathways also lays a good foundation for analyzing the function of GPCR in bone homeostasis, including the effect of Gαs/Gαi mediated on the function of osteoblasts/osteoclasts, etc.
Historical timeline of the major GPCRs discovered in bone biology.
The schematic of GPCR-ligand binding and downstream G-protein signaling.
The dawn of the 21st century ushered in a structural biology revolution propelled by advancements in cryo-electron microscopy (cryo-EM) and X-ray crystallography, which fundamentally reshaped GPCR research paradigms. In bone biology, this structural elucidation provided unprecedented mechanistic insights into how GPCRs integrate skeletal signals - from hormonal cues (e.g., PTH) to mechanical stimuli to modulate osteoblastogenesis, osteoclastogenesis, and chondrogenesis. The post-2000 era saw exponential acceleration in GPCR research velocity (Fig. 2). This convergent evolution of structural biology, pharmacology, and skeletal medicine positions GPCRs as central nodes in the quest for precision osteotherapeutics.
GPCRs in MSCs
MSCs, known for their self-renewal capability and multilineage differentiation potential, are key players in tissue regeneration and repair, particularly in the bone marrow niche [12]. These cells express a wide array of GPCRs that respond to a multitude of ligands, including hormones, neurotransmitters, growth factors, and small molecules (Table 1). The interaction between these ligands and their cognate GPCRs orchestrates a complex signaling network that fine-tunes the behavior of MSCs, thereby regulating bone homeostasis and repair.
The functions and mechanisms of GPCR in mesenchymal stem cells (MSCs)
| GPCR Name | GRAFS Classification | Ligand | Coupled G Protein Subtype | Signaling Pathway | Functional/Phenotypic Changes and references |
|---|---|---|---|---|---|
| A1R (ADORA1) | Rhodopsin family - α subgroup (Amines) | Adenosine | Gαi | WNT signaling | Stimulates osteogenic differentiation in dental pulp stem cells [17]. |
| A2AR (ADORA2A) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | cAMP/PKA | Increases proliferation and differentiation of bone marrow MSCs [18]. |
| ADORA2B (A2BAR) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | Osteoblast differentiation gene regulation | Deletion lowers bone mineral density and reduces mineralization in BMSCs [19]. |
| ADRB2 (β-AR) | Rhodopsin family - α subgroup (Adrenergic) | Norepinephrine, Isoproterenol, Epinephrine | Gαs | cAMP/PKA | Agonists inhibit mineralization and osteogenesis; antagonists promote mineralization [22, 23]. |
| CaSR | Glutamate family | Extracellular Ca²⁺ | Gαq/Gαi | Calcium sensing → PTHrP/PTH1R inhibition | Maintains osteogenic potential; knockdown reduces bone formation capacity [15]. |
| EP1 | Rhodopsin family | PGE2 | Gαq/11 | ↑ PLCβ → IP₃/DAG → Ca²⁺ release, PKC activation | Inhibits osteoblast migration, exacerbates osteoarthritis [20, 21]. |
| EP2/EP4 (PTGER) | Rhodopsin family | PGE2 | Gαs | FAK/ERK1/2 → PGE2 release | Enhances MSC migration; boosts immunosuppressive effects on RA T cells [27, 28]. |
| GPR161 | Other 7TM receptors | / | Gαs | Mechanical force → cAMP signaling | Knockdown impairs mechanotransduction and reduces osteogenic marker expression [29]. |
| GPRC6A | Glutamate family | cation, amino acid, and testosterone | Gαq (putative) | Extracellular calcium → ERK activation | Promotes osteogenic marker expression and mineralization; knockout reduces bone mineral density [16]. |
| LGR5 (GPR49) | Adhesion family | R-spondin | Gαs (putative) | Wnt/β-catenin, ERK, mitochondrial dynamics | Promotes osteogenic differentiation of MSCs; overexpression accelerates fracture healing [14]. |
| LPAR1 (EDG2/GPCR26) | Rhodopsin family - δ subgroup (Lipids) | Lysophosphatidic acid, LPA | Gα12/Gα13 (major), Gαq/Gαi (minor) | Caspase-3 inhibition → anti-apoptosis | Protects MSCs from apoptosis; promotes proliferation [26]. |
| PTH1R | Secretin family | PTH/ PTHrP | Gαs | cAMP/PKA → Runx2/osteocalcin | Enhances osteogenic differentiation and bone formation; inhibits osteoclastogenesis [6, 15]. |
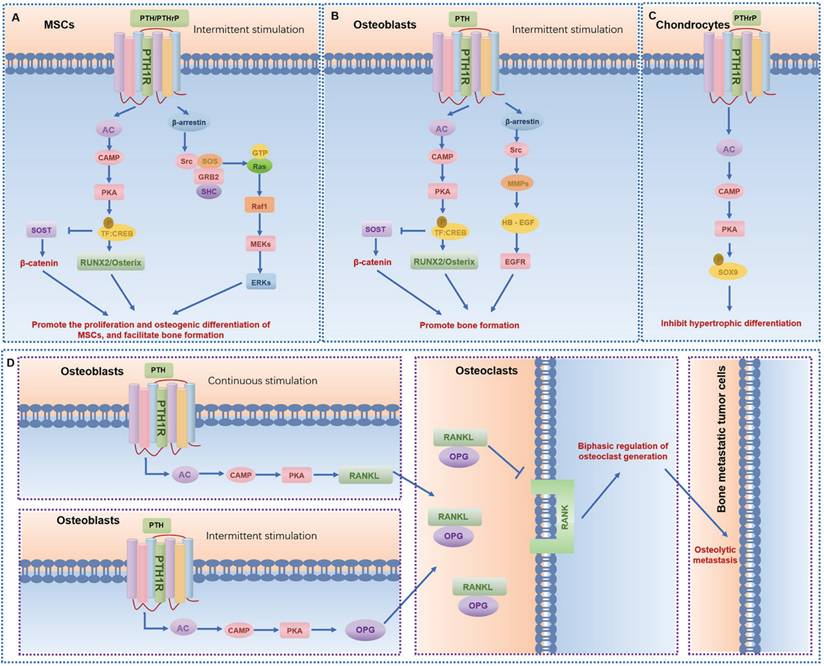
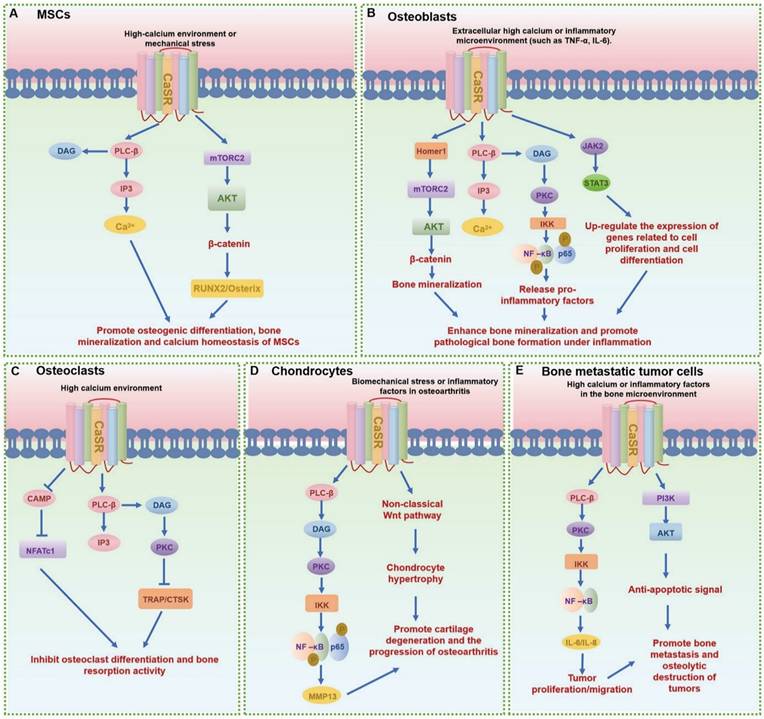
GPCRs regulate MSCs differentiation and mineralization, with specific pathways promoting osteogenic differentiation (Table 1). Leucine-rich repeat containing G protein-coupled receptor 5 (LGR5), also known as G protein-coupled receptor 49 (GPR49), a vital member of the rhodopsin family of GPCRs and a Wnt target gene, plays a significant role in osteogenic differentiation [13]. Lgr5 knockdown suppresses osteogenic differentiation via dysregulation of Wnt/ERK signaling and impaired mitochondrial dynamics, which are critical for MSC lineage commitment. Conversely, overexpression of Lgr5 accelerates fracture healing through enhanced osteogenesis and angiogenesis [14]. Another crucial GPCR subset in MSCs is the Parathyroid hormone receptor 1 (PTH1R), a member of the class B GPCR family. PTH1R regulates MSCs functions through multiple signaling cascades: upon ligand binding (PTH or PTHrP), it activates Gαs protein to stimulate adenylate cyclase (AC), generating cAMP that activates protein kinase A (PKA) to upregulate osteogenic genes (e.g., Runx2, Osterix) and drive osteoblast differentiation; concurrently, PTH-PTH1R interaction recruits β-arrestin, forming a signaling complex that activates extracellular signal-regulated kinase 1/2 (ERK1/2), which promotes MSC proliferation and integrates survival signals through the MAPK pathway, collectively modulating skeletal homeostasis and cellular fate decisions (Fig. 4A) [6, 15]. Moreover, CaSR, a class C GPCR, is vital for calcium homeostasis and bone turnover (Fig. 5A) [6, 15]. Knockdown of CaSR (using shRNA-CaSR) decreases the bone formation potential of MSCs. CaSR signaling counteracts PTH1R signaling by downregulating PTH1R via inhibition of PTHrP expression [15]. G protein-coupled receptor family C group 6 member A (GPRC6A), another GPCR, has been associated with human spine bone mineral density (BMD) [16]. GPRC6A knockout mice exhibit lower BMD and a suppressed response to extracellular calcium-stimulated ERK activation, leading to attenuated osteogenic marker gene expression and mineralization in bone marrow mesenchymal stem cells (BMSCs) [16].
The signaling pathways and functions of PTH1R in different bone-related cells. (A-C) The main signaling pathways of PTH1R in mesenchymal stem cells (MSCs) (A), osteoblasts (B), and chondrocytes (C), respectively. (D) Osteoclasts are indirectly regulated by PTH1R in osteoblasts, leading to osteolytic metastasis and regulating the dynamics of other bone-related cells.
The signaling pathways and functions of CaSR in different bone-related cells. (A-E) The main signaling pathways of CaSR in mesenchymal stem cells (MSCs) (A), osteoblasts (B), osteoclasts (C), chondrocytes (D) and other bone-related cells (E), respectively.
Adenosine receptors also play crucial roles in regulating MSC function. Adenosine A1 receptor (A1R) promotes osteogenic differentiation of human dental pulp stem cells (DPSCs) via Wnt signaling [17]. Adenosine A2A receptor (A2AR) increases the proliferation and differentiation of MSCs from mouse bone marrow [18]. The deletion of Adenosine receptor A2b (ADORA2B/A2BAR) results in lower BMD in mice, with decreased expression of osteoblast differentiation genes and fewer mineralized nodules in BMSCs [19]. Prostaglandin E receptors (Prostaglandin E receptor 1, EP1/PTGER1; Prostaglandin E receptor 2, EP2/PTGER2; and Prostaglandin E receptor 4, EP4/PTGER4) also influence MSC function. Deletion of mice in enhanced fracture healing, stronger cortical bones, and higher trabecular bone volume [20, 21]. This is due to inactivation of Hif1α, leading to increased oxygen consumption rate and promotion of osteogenic differentiation and bone formation [20]. GPCR signaling also plays a regulatory role in the adipogenic differentiation of MSCs. Adrenoceptor β2 (ADRB2, also known as β-AR) agonists, on the other hand, suppress MSC mineralization in a dose- and time-dependent manner and inhibit adipogenesis and osteogenesis via the cAMP/PKA signaling pathway. ADRB2 antagonists have the opposite effect, increasing calcium mineralization and adipogenesis in MSCs [22, 23].
GPCRs assume a pivotal role in influencing over various cellular functions of MSCs, including proliferation and migration. Inhibition of CaSR disturbs the proliferation and migration of human BM-MSCs [24]. CaSR in MSCs utilizes the G protein-dependent Gq/11-PLC-IP3 pathway: upon binding to Gq/11 proteins, CaSR activates PLC, catalyzing the hydrolysis of PIP2 into inositol IP3 and DAG. IP3 induces Ca²⁺ release from the endoplasmic reticulum, elevating cytoplasmic Ca²⁺ levels to activate calcium-dependent pathways (e.g., CaMK and PKC), thereby regulating MSCs proliferation and differentiation (Fig. 5A). In osteoarthritis, MSCs can serve as a substitute for chondrocytes and support cartilage regeneration. Norepinephrine, acting via ADRB2, suppresses the proliferation of BMSCs, thereby reducing their regenerative capacity. This suggests that targeting ADRB2 signaling may provide a novel therapeutic option for osteoarthritis [25]. Lysophosphatidic acid (LPA) protects human umbilical cord MSCs (UC-MSCs) from LPS-induced apoptosis by inhibiting caspase-3 activation through Lysophosphatidic acid receptor 1 (LPAR1, also known as Endothelial differentiation gene 2, EDG2; or G protein-coupled receptor 26, GPCR26) coupled with a G protein. LPAR1 regulates LPA-induced proliferation of UC-MSCs, enhancing their survival without affecting differentiation [26]. EP2 enhances the migration of MSCs by activating FAK and ERK1/2 pathways, without affecting osteogenic differentiation [27].
MSCs have immune regulatory functions, and GPCR signaling can affect the immune regulatory properties of MSCs. MSCs co-cultured with rheumatoid arthritis CD4+ T cells show that EP2/EP4-stressed MSCs have a better inhibitory effect on rheumatoid arthritis (RA) T cells by releasing PGE2, indicating that induction of EP2/EP4 stress can enhance the immunosuppressive effect of MSCs [28]. GPCRs responsive to adrenergic ligands, such as β-adrenergic receptors, play significant roles in regulating MSC function under stress conditions. Activation of these receptors by catecholamines like adrenaline and noradrenaline stimulates cAMP production, leading to increased MSC migration and proliferation in response to injury or inflammation. However, chronic adrenergic stimulation can negatively impact osteogenic differentiation, highlighting the GPCR signaling in MSCs.
GPCRs are not only targets for exogenous ligands but also sensors of the mechanical microenvironment. Primary cilia, an important organelle in bone mechanobiology and mechanical transduction, harbor GPCRs that respond to mechanical stimuli. G protein-coupled receptor 161 (GPR161), a mechanically responsive orphan GPCR located in the cilium, is crucial for fluid shear-induced cAMP signaling in MSCs [29]. The absence of GPR161 inhibits mechanical transduction, leading to decreased expression of osteogenic marker genes downstream [29]. This mechanosensitive is pivotal in bone adaptation to mechanical loading and in the pathogenesis of bone diseases such as osteoporosis.
In summary, 12 GPCRs orchestrate MSC functions, including differentiation, proliferation, migration, and immune regulation (Table 1). Key receptors include LGR5, which promotes osteogenesis via Wnt/ERK signaling and mitochondrial dynamics, accelerating fracture healing. PTH1R stimulates osteoblastogenesis through cAMP/PKA-mediated Runx2 and osteocalcin expression, enhancing bone formation (Fig. 4A). CaSR maintains calcium homeostasis (Fig. 5A). Adenosine receptors (A1R, A2AR, A2BAR) modulate osteogenic differentiation and proliferation. Prostaglandin E receptors (EP2/EP4) enhance MSCs migration and fracture repair via FAK/ERK pathways, while suppressing adipogenesis. ADRB2 regulate cAMP/PKA signaling, impacting MSC mineralization. Mechanically responsive GPCR like GPR161 mediates osteogenic responses to fluid shear stress and matrix stiffness. Understanding the intricate GPCR signaling networks in MSCs will pave the way for the development of novel therapeutic strategies to treat bone diseases, enhance bone regeneration, and improve the efficacy of MSC-based therapies.
GPCRs in osteoblast and osteocyte
In bone tissue, osteoblasts and osteocytes collaborate to maintain skeletal homeostasis. Osteoblasts, responsible for bone formation, synthesize and mineralize the organic matrix to generate new bone, while osteocytes, embedded within the mineralized matrix, orchestrate bone remodeling through mechanotransduction and intercellular signaling. GPCRs emerge as critical regulators of these processes, mediating diverse signaling pathways that govern osteoblast differentiation, osteocyte function, and overall bone metabolism (Table 2).
The functions and mechanisms of GPCR in osteoblasts and osteocytes
| GPCR Name | GRAFS Classification | Ligand | Coupled G Protein Subtype | Signaling Pathway | Functional/Phenotypic Changes and references |
|---|---|---|---|---|---|
| A1R (ADORA1) | Rhodopsin family - α subgroup (Amines) | Adenosine | Gαi | Adipogenic signaling | Promotes adipogenesis over osteoblast differentiation [50]. |
| A2AR (ADORA2A) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | cAMP/PKA | Stimulates osteoblast activity and suppresses osteoclast activity; promotes bone regeneration [50, 52]. |
| A3AR (ADORA3) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαi | Anti-inflammatory signaling | Reduces inflammatory cytokines (e.g., TNF-α); promotes bone repair in inflammatory conditions [53]. |
| ADORA2B | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | cAMP/PKA | Inhibits adipogenesis and stimulates osteoblast differentiation [51]. |
| ADRA1A (α1-AR) | Rhodopsin family - α subgroup (Adrenergic) | Epinephrine, Norepinephrine, Phentolamine | Gαq | / | Negatively regulates Bmp4 expression by up-regulating Nfil3/E4BP4 in osteoblasts [41]. |
| ADRA2A (α2A-AR) | Rhodopsin family - α subgroup (Adrenergic) | Epinephrine, Norepinephrine, Phentolamine | Gαi | Neuro-endocrine signaling | SNP rs553668 and rs1800544 locate in gene; alters mRNA stability and BMD [42]. |
| ADRB1 | Rhodopsin family - α subgroup (Adrenergic) | Norepinephrine, isoproterenol | Gαs | cAMP/PKA | Inhibits disuse-induced bone loss by reducing osteocyte apoptosis [37]. |
| ADRB2 | Rhodopsin family - α subgroup (Adrenergic) | Norepinephrine, Isoproterenol, Epinephrine | Gαs | cAMP/PKA → RANKL upregulation | Deficiency increases bone mass [38-40]. |
| CaSR | Glutamate family | Extracellular Ca²⁺ | Gαq/Gαi | mTORC2/AKT-β-catenin, NF-κB/JAK-Stat3 | Promotes bone formation (via β-catenin); drives pathological bone formation in inflammatory states [34]. |
| CNR2 (CB2) | Rhodopsin family - α subgroup (Cannabinoid) | Endocannabinoids, CB2 agonists | Gαi | Osteoclast survival suppression | Agonists promote osteoblast differentiation and protect against ovariectomy-induced bone loss [55, 56]. |
| EP2 | Rhodopsin family | PGE2 | Gαs | cAMP/PKA | Enhances osteoblast differentiation and mineralization [49]. |
| GABABR | Glutamate family | GABA | Gαi | cAMP suppression → BMP2/RANKL downregulation | Inhibits osteoblastogenesis; knockout increases BMP2 expression but reduces BMD [45]. |
| GPR161 | Other 7TM receptors | / | Gαi (putative) | Sonic Hedgehog (Shh) pathway suppression | Inhibits intramembranous bone formation; knockout completely blocks osteoblastogenesis [29, 46]. |
| GPR39 | Other 7TM receptors | Zinc | Gαq | Unknown (matrix deposition regulation) | Knockout causes disordered matrix deposition (low collagen, high mineral content) [36]. |
| GPR48 (LGR4) | Adhesion family | R-spondin | Gαs | cAMP-PKA-Atf4 | Regulates embryonic osteoblast differentiation and mineralization; knockout delays bone formation [31]. |
| GPRC6A | Glutamate family | Calcium/Amino acids | Gαq | ERK activation | Suppresses osteoblast differentiation and ALP activity; knockout reduces bone formation [16]. |
| GRM1 (mGluR1) | Glutamate family | Glutamate | Gαq | / | Knockout causes premature growth plate fusion and osteoblast dysfunction [48]. |
| LGR5 (GPR49) | Adhesion family | R-spondin | Gαs | Wnt/β-catenin activation | Promotes osteoblast differentiation and bone formation [30]. |
| LPAR1 (EDG2/GPCR26) | Rhodopsin family - δ subgroup (Lipids) | Lysophosphatidic acid (LPA) | Gα12/13 | RANKL→MAPK/AKT-NF-κB | Knockout reduces bone mineralization, increases osteocyte apoptosis, and bone porosity [47]. |
| PTH1R | Secretin family | PTH/ PTHrP | Gαs | cAMP/PKA | Activation (e.g., by teriparatide) stimulates bone formation; key therapeutic target for osteoporosis [6, 7]. |
| XCR1 | Other 7TM receptors | XCL1 | Gαi (putative) | / | Promotes osteoblast differentiation and enhances bone formation [54]. |
GPCRs exert bidirectional control over osteoblast activity, influencing both pro-osteogenic and anti-osteogenic pathways. Pro-osteogenic signaling pathways include the activation of the Wnt/β-catenin pathway by receptors such as LGR5 and Leucine-rich repeat-containing G protein-coupled receptor 4 (LGR4, also known as G protein-coupled receptor 48, GPR48), which promote osteoblast differentiation [30] [31]. LGR4 deficiency, for instance, delays osteoblast mineralization during embryonic bone development, yet does not impair chondrocyte maturation, highlighting its specificity for osteogenic signaling [31]. CaSR and PTH1R are pivotal for calcium homeostasis, with CaSR alleles associating with BMD and osteoporosis risk, and PTH1R agonists (e.g., teriparatide) serving as therapeutic agents for osteoporosis by enhancing bone formation (Fig. 4B,D and Fig. 5B) [32-35]. Additionally, LGR4 regulates osteoblast differentiation via the cAMP-PKA-ATF4 pathway, underscoring its role in early bone development [31]. G protein-coupled receptor 39 (GPR39), another critical GPCR, is involved in bone matrix deposition, with Gpr39-/- mice exhibiting disordered matrix deposition characterized by abnormally low collagen and high mineral contents in osteoblasts [36].
GPCRs also mediate anti-osteogenic and modulatory signaling. Adrenergic receptors exemplify this duality: Adrenoceptor β1 (ADRB1) agonists mitigate disuse-induced bone loss by reducing osteocyte apoptosis [37], while ADRB2 deficiency increases bone mass, and ADRB2 agonists stimulate RANKL expression in osteoblasts, promoting osteoclastogenesis and bone resorption [38-40]. ADRA1A (α1-adrenergic receptor, α1-AR) signaling upregulates the transcriptional repressor Nfil3, inhibiting BMP4 expression and establishing a circadian regulatory loop [41], and ADRA2A (α2A-adrenergic receptors, α2A-AR) polymorphisms correlate with altered bone resorption markers (e.g., CTX, Cathepsin K) and osteoporosis risk [42]. The overexpression of GRK2, a kinase that terminates GPCR signaling, suppresses osteoblast numbers and bone formation by attenuating Wnt/β-catenin activity, leading to low bone turnover [43, 44]. GABAB receptor (GABABR) deficiency increases ALP activity and BMP2/Osterix expression in osteoblasts, disrupting osteoclastogenesis via RANKL downregulation [45]. GPR161, a cilium-localized GPCR, is essential for intramembranous bone formation, with Gpr161 knockout mice lacking forelimbs due to hyperactive Sonic Hedgehog (Shh) signaling and blocked osteoblastogenesis [29, 46]. GPRC6A deficiency suppresses calvarial-derived osteoblast differentiation and Alkaline phosphatase (ALP) activity, with siRNA-mediated knockdown of Gprc6a in MC3T3-E1 osteoblasts restraining extracellular calcium-stimulated ERK activities [16].
The roles of GPCRs in bone metabolism are context-dependent. LPAR1 deficiency impairs bone mineralization and cortical thickness, accompanied by osteocyte apoptosis and lacunar defects [47]. Metabotropic glutamate receptor 1 (GRM1, also known as mGluR1 or mGlu1) knockout mice exhibit premature growth plate fusion and osteoblast dysfunction, linking glutamate signaling to skeletal maturation [48]. EP2A enhances osteoblastic differentiation and mineralization [49], while adenosine receptors, including A1R and ADORA2B, play roles in osteoblast and adipocyte lineage determination [50, 51], with A2AR agonists promoting new bone formation by increasing osteoblast activity and reducing osteoclast activity [50, 52].
Osteocytes, the most abundant bone cells, utilize GPCRs to maintain mechanical integrity and coordinate remodeling. ADRA2A (α2A-adrenergic receptors, α2A-AR) in osteoblasts and lining cells mediates neuroendocrine inputs, with single nucleotide polymorphisms (SNP rs553668 and rs1800544) affecting bone resorption markers [42]. CaSR signaling in osteoblasts is modulated by inflammatory cytokines (e.g., NF-κB/JAK-STAT3), influencing pathological bone formation in ankylosing spondylitis (Fig. 5B) [34]. Homer1 mediates CaSR signaling via mTORC2 in osteoblasts to enhance AKT-dependent β-catenin stabilization, while systemic inhibition of CaSR represses pathological new bone formation in animal models of ankylosing spondylitis [33]. In the inflammatory immune responses of osteoblasts, pulsed electromagnetic fields promote the anti-inflammatory effect of A2A and Adenosine A3 receptor (A3AR) in human hFOB 1.19 osteoblasts [53].
Genetic and therapeutic insights further highlight the importance of GPCRs in bone biology. An intergenic susceptibility SNP rs4683184, influences transcription factor RUNX2 binding and mediates long-range chromatin interactions with X-C chemokine receptor 1 (XCR1) [54]. XCR1, also named as G protein-coupled receptor 5 (GPR5), promotes osteoblast differentiation, and the bone-targeting adeno-associated virus targeting Xcr1 enhances bone formation in osteoporotic mice [54]. Therapeutically, PTH1R agonists (teriparatide, abaloparatide) are first-line treatments for osteoporosis, while Cannabinoid receptor 2 (CNR2/CB2) agonists show promise in preventing ovariectomy-induced bone loss by stimulating osteogenesis [55, 56].
In conclusion, 21 GPCRs form a complex regulatory network in bone biology, integrating hormonal, mechanical, and metabolic cues to fine-tune osteoblast and osteocyte activity (Table 2). LGR5, PTH1R, CNR2, and EP2A play important roles in promoting bone formation. On the contrary, GPRC6A, and ADRB2 agonists mainly inhibit bone formation. In terms of bone resorption regulation, ADRB2 and A2AR have become key driving factors. CaSR and A2AR/A3AR play crucial roles in inflammation and immune regulation. LGR4, GPR39 and A1R also affect development and metabolic regulation. Understanding the functional roles and signaling cascades of these receptors will pave the way for the development of novel therapeutic strategies to treat bone diseases and enhance bone regeneration.
GPCRs in macrophage and osteoclast
GPCRs play pivotal roles in orchestrating the migration, differentiation, and activation of macrophages and osteoclasts, thereby profoundly influencing bone mass, microstructure, and strength (Table 3). These receptors mediate complex signaling networks that either promote or inhibit osteoclastogenesis and macrophage polarization, with implications for skeletal homeostasis and disease.
The functions and mechanisms of GPCR in osteoclasts
| GPCR Name | GRAFS Classification | Ligand | Coupled G Protein Subtype | Signaling Pathway | Functional/Phenotypic Changes and references |
|---|---|---|---|---|---|
| A1R (ADORA1) | Rhodopsin family - α subgroup (Amines) | Adenosine | Gαi | Pro-osteoclast signaling | Promotes osteoclast formation and bone loss [74]. |
| A2AR (ADORA2A) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | cAMP/PKA | Inhibits osteoclast differentiation; agonists promote bone regeneration [74]. |
| ADRB2 | Rhodopsin family - α subgroup (Adrenergic) | Norepinephrine, Isoproterenol, Epinephrine | Gαs | RANKL upregulation | Enhances osteoclastogenesis and bone resorption [38-40]. |
| CaSR | Glutamate family | Extracellular Ca²⁺ | Gαq/Gαi | NF-κB, Akt | High Ca²⁺ inhibits osteoclast resorption; osteoblast knockout increases RANKL-driven resorption [67-71]. |
| CNR1 (CB1) | Rhodopsin family - α subgroup (Cannabinoid) | Endocannabinoids, e.g., anandamide | Gαi | Apoptosis induction | Antagonists increase bone mass by promoting osteoclast apoptosis [78]. |
| CNR2 (CB2) | Rhodopsin family - α subgroup (Cannabinoid) | Endocannabinoids, CB2 agonists | Gαi | Osteoclast survival suppression | Agonists promote osteoclast formation; antagonists inhibit bone loss [81]. |
| DRD2 (D2DR) | Rhodopsin family - α subgroup (Dopamine) | Dopamine | Gαi | NF-κB suppression | Inhibits M1 macrophage polarization; restricts inflammatory osteolysis [82]. |
| EBI2 (GPR183) | Rhodopsin family - γ subgroup (Chemokine) | 7α,25-OHC | Gαi | 7α,25-OHC → OCP migration/fusion | Promotes osteoclast precursor migration; defective signaling increases bone mass [58]. |
| EP4 (PTGER4) | Rhodopsin family | PGE2 | Gαs | PGE2 → cytokine-driven resorption | Enhances osteoclast formation in inflammation; overexpression inhibits resorption [85-87]. |
| F2r (Thrombin Receptor) | Rhodopsin family - δ subgroup (Thrombin) | Thrombin | Gαq/11 | Inhibiting Akt-GSK3β-NFATc1 and suppressing NF-κB signaling | Inhibits osteoclast formation and bone resorption [60]. |
| GABABR | Glutamate family | GABA | Gαi | cAMP suppression → BMP2/RANKL downregulation | Knockout elevates ALP levels and BMP2/Osterix expression, which inhibits osteoclast formation by reducing RANKL production [45]. |
| GPR120 (FFAR4) | Rhodopsin family - α subgroup (Fatty Acid) | Long-chain fatty acids | Gαq/Gαq11 | ROS suppression → antioxidant activation | Inhibits osteoclast formation/resorption; reduces ROS production [65]. |
| GPR125 | Adhesion family | / | Gαq/12/13 (putative) | RANKL → MAPK/AKT-NF-κB | Promotes osteoclast differentiation/activation; knockdown reduces signaling [84]. |
| GPR132 (G2A) | Other 7TM receptors | Lysophosphatidic acid, LPA | Gα12/13 | Macrophage polarization | Reduces M1-like macrophage infiltration; shifts to M2 polarization [64]. |
| GPR30 | Other 7TM receptors | Estrogen | Gαi (putative) | Membrane estrogen signaling | Inhibits osteoclastogenesis via non-nuclear pathways [61]. |
| GPR48 (LGR4) | Adhesion family | R-spondin | Gαs (putative) | cAMP-PKA-CREB → Atf4 | Delays embryonic osteoblast differentiation; postnatal knockout increases osteoclast activity [62, 63]. |
| GPR54 | Other 7TM receptors | Kisspeptin | Gαq (putative) | Kp-10 → Dusp18/Src dephosphorylation | Suppresses osteoclast activity; prevents bone loss [66]. |
| GPR55 | Other 7TM receptors | Lysophosphatidic acid, LPA | Gα12/13 (putative) | RANKL → NFATc1 activation | Enhances osteoclast maturation; inhibition reduces bone resorption [59]. |
| GPRC6A | Glutamate family | Calcium/Amino acids | Gαq (putative) | ucOCN-mediated inhibition | Inhibits early osteoclast differentiation and resorption [72]. |
| H4R (GPCR105) | Rhodopsin family - α subgroup (Histamine) | Histamine | Gαi (putative) | RANKL upregulation | Promotes RA-associated osteoclastogenesis; antagonists reduce bone destruction [88]. |
| LPAR1 (EDG2/GPCR26) | Rhodopsin family - δ subgroup (Lipids) | Lysophosphatidic acid, LPA | Gα12/13 | RANKL → MAPK/AKT-NF-κB | Essential for osteoclast differentiation; antagonists inhibit resorption [83]. |
| TAS1R3 | Glutamate family | Sweet tastants | Gαi (putative) | Nutrient sensing | Increases cortical bone mass; reduces osteoclast activity [73]. |
| TSHR (LGR3) | Rhodopsin family - δ subgroup (Glycoprotein) | TSH | Gαs | TSH → cAMP/PKA | Inhibits osteoclastogenesis; knockout reduces bone strength [57]. |
As a rhodopsin-family GPCR, Thyroid-stimulating hormone receptor (TSHR/LGR3) suppresses osteoclast activity by inhibiting RANKL-induced differentiation, as evidenced by increased bone resorption in Tshr-/- mice and reduced TRAP-positive osteoclasts following TSH treatment [57]. EBV-induced G protein-coupled receptor 2 (EBI2, also known as GPR183) and its ligand 7α,25-dihydroxycholesterol (7α,25-OHC), secreted by osteoblasts, guide osteoclast precursor (OCP) migration to bone surfaces, with EBI2 deficiency enhancing bone mass by disrupting OCP homing [58]. G protein-coupled receptor 55 (GPR55) modulates osteoclastogenesis by attenuating RANKL-stimulated transcription of osteoclast markers, while its inhibition via peptide P1 blocks osteoclast maturation [59]. Similarly, Coagulation factor II thrombin receptor (F2r) restrains osteoclast formation and function by attenuating RANKL-induced signaling through the Akt and NF-κB pathways [60].
In addition, quercetin, acting through G protein-coupled receptor 30 (GPR30) rather than nuclear estrogen receptors, inhibits osteoclastogenesis, highlighting crosstalk between cytokine and GPCR pathways [61]. LGR4 competes with RANK for RANKL binding, initiating cAMP-PKA-CREB signaling that upregulates Atf4 in osteoblasts and counteracts RANK-mediated osteoclast activation [31]. Postnatal Lgr4 deficiency increases osteoclast activity, underscoring its role in fine-tuning bone remodeling [62, 63]. G protein-coupled receptor G2A (GPR132) plays a role in macrophage migration and polarization during inflammation. In G2A-deficient mice, there is reduced M1-like macrophage infiltration at the site of inflammation and a shift towards M2-like polarization, highlighting the importance of GPCRs in regulating macrophage function during immune responses [64]. G protein-coupled receptor 120 (GPR120, also known as Free fatty acid receptor 4, FFAR4) activation by TUG-891 inhibits osteoclast formation and resorption in RAW264.7 macrophages by reducing ROS and upregulating antioxidant proteins [65]. Recently, the study reveals that G protein-coupled receptor 54 (GPR54), activated by Kisspeptin-10 (Kp-10), recruits Dusp18 phosphatase to dephosphorylate Src at Tyr416. Knockout of Kiss1, Gpr54, or Dusp18 in mice results in osteoclast hyperactivation and bone loss [66]. Kp-10 treatment suppresses osteoclast activity and bone loss in vivo [66]. Thus, the Kp-10/Gpr54 pathway represents a potential therapeutic target for bone resorption via Dusp18-mediated Src dephosphorylation [66].
CaSR, expressed in mature osteoclasts, inhibits bone resorption in response to high extracellular Ca²⁺ or agonists, with osteoblast-specific CaSR knockout upregulating RANKL and increasing osteoclast activity (Fig. 5C) [67-71]. GABABR in osteoblasts suppresses cAMP formation, ALP activity, and osteogenic genes (e.g., BMP2, Osterix), thereby reducing osteoblastogenesis and indirectly modulating osteoclastogenesis via RANKL [45]. Undercarboxylated osteocalcin (ucOCN) inhibits early osteoclast differentiation through GPRC6A [72], while Taste 1 receptor member 3 (Tas1R3) deficiency enhances cortical bone mass by uncoupling bone remodeling and reducing osteoclast function [73].
Adenosine receptors exhibit opposing effects. A1R promotes osteoclastogenesis, whereas A2AR inhibits differentiation and function [74]. A2AR agonists enhance bone regeneration by increasing osteoblasts and decreasing osteoclasts in skull defects, while Adora2b deficiency reduces bone mass and trabecular number [52, 75]. ADRB2 signaling in periodontal ligament cells (PDLCs) stimulates osteoclastogenesis and accelerates orthodontic tooth movement via RANK-L upregulation, with noradrenaline and selective agonists enhancing osteoclast multinuclearity without directly affecting osteoblasts [76, 77].
Moreover, cannabinoid receptors also regulate bone turnover. Cannabinoid receptor 1 (CNR1) antagonism increases bone mass by promoting osteoclast apoptosis [78], while combined Cnr1/Cnr2 deficiency protects against age-related and ovariectomy-induced bone loss despite reduced osteoblast function [78, 79]. In a mouse model of diet-induced obesity, treatment with the Cannabinoid receptor 1 (CB1) antagonist AM251 resulted in weight loss and reduced inflammation [80]. CB2 (CNR2) agonists stimulate osteoclastogenesis, with CB2-/- mice exhibiting blunted ovariectomy-induced bone loss, suggesting therapeutic potential for CB2 antagonists [81]. Additionally, Dopamine receptor D2 (DRD2) suppresses M1 macrophage polarization and NF-κB/NLRP3 inflammasome activation [82], while LPAR1/EDG2/GPCR26 deficiency impairs osteoclastogenesis and prevents ovariectomy-induced bone loss [83]. Recently, the study found that G protein-coupled receptor 125 (GPR125) is highly expressed in osteoclasts and positively regulates their differentiation and activation [84]. Additionally, GPR125 knockdown reduced the expression of phosphorylated MAPK (p-ERK and p-p38) and AKT-NF-κB (p-AKT and p-IKBα) signaling pathway proteins in response to RANKL stimulation [84].
PGE2 receptors exhibit context-dependent roles. EP4 downregulation in osteoclasts prevents PGE2-mediated inhibition of bone resorption, yet EP4 on osteoblasts is critical for osteoclast formation induced by inflammatory cytokines [85-87]. Besides, Histamine H4 receptor (H4R), also known as GPCR105, blockade reduces RANKL expression and osteoclastogenesis in rheumatoid arthritis, where synovial histamine levels correlate with disease severity [88].
Collectively, these evidences emphasize the crucial role of 23 GPCRs in regulating macrophage and osteoclast function (Table 3). A1R, ADRB2, LPAR1, GPR55, H4R promote osteoclastogenesis, while GPR120, GPR54, A2AR, CNR1, GPRC6A inhibit osteoclastogenesis. The regulation of bone resorption is regulated by CaSR, PTH1R, F2r, and GIT1. Immune and inflammatory regulation are also regulated by GPR132, EP4, and H4R. In terms of metabolism and nutrient sensing, the role of TASR3 and GPR120 cannot be ignored. Their diverse roles in osteoclastogenesis, macrophage polarization, and osteoblast-osteoclast crosstalk underscore their therapeutic potential in osteoporosis, inflammatory arthritis, and metabolic bone diseases. Targeting these receptors offers innovative strategies to modulate bone resorption and formation, with implications for regenerative medicine and anti-resorptive therapies.
GPCRs in chondrocyte
GPCRs serve as master regulators of chondrocyte biology, orchestrating responses to growth factors, cytokines, mechanical cues, and inflammatory mediators. These receptors govern critical processes such as chondrocyte proliferation, differentiation, extracellular matrix (ECM) synthesis, and survival, thereby maintaining cartilage integrity and modulating the pathogenesis of osteoarthritis (Table 4). Dysregulation of GPCR signaling contributes to cartilage degradation, synovial inflammation, and subchondral bone remodeling, highlighting their therapeutic potential in osteoarthritis management.
The functions and mechanisms of GPCR in chondrocytes
| GPCR Name | GRAFS Classification | Ligand | Coupled G Protein Subtype | Signaling Pathway | Functional/Phenotypic Changes and references |
|---|---|---|---|---|---|
| A2AR | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | FoxO/autophagy activation | Stimulation improves cartilage function; enhances autophagy and reduces inflammation [114]. |
| A3AR (ADORA3) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαi | Suppression of RUNX2/CaMKII | Agonists inhibit matrix degradation and cartilage hypertrophy in OA [108]. |
| ADORA2B | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαs | cAMP/PKA | Activation inhibits chondrogenic differentiation in MSCs by downregulating SOX9 and COL2A1 [50]. |
| ADRA1A (α1-AR) | Rhodopsin family - α subgroup (Adrenergic) | Epinephrine, Norepinephrine, Phentolamine | Gαq | ERK/PKA | Low-dose NE induces apoptosis via α1-AR; accelerates OA pathogenesis [113]. |
| ADRA2A (α2A-AR) | Rhodopsin family - α subgroup (Adrenergic) | Epinephrine, Norepinephrine, Phentolamine | Gαi | ERK1/2-PKA/cGMP | Activation ↑ MMPs and RANKL, causing cartilage degeneration; antagonists as enhancers of chondrogenesis and suppressors of hypertrophy, agonists induced detrimental hypertrophy [111]. |
| ADRB2 (β-AR) | Rhodopsin family - α subgroup (Adrenergic) | Norepinephrine, Isoproterenol, Epinephrine | Gαs | ERK1/2-PKA/Jun-B | Agonists inhibit chondrocyte differentiation markers; high-dose NE reverses IL-1β damage [109, 110]. |
| AT1R | Rhodopsin family | Angiotensin II | Gαq | ERK/PKA | High AT1R/AT2R ratio impedes chondrocyte proliferation under stress; inhibition promotes survival [115]. |
| AT2R | Rhodopsin family | Angiotensin II | Gαi (putative) | Counteracts AT1R | Enhanced expression reduces apoptosis in stressed chondrocytes [115]. |
| CaSR | Glutamate family | Extracellular Ca²⁺ | Gαq | Pro-differentiation signaling | Biomechanical stress ↑ CaSR expression, accelerating OA; calcilytics block cartilage degradation [15, 24, 33]. |
| CNR1 (CB1) | Rhodopsin family - α subgroup (Cannabinoid) | Endocannabinoids, e.g., anandamide | Gαi | SIRT1 activation | Agonists protect against IL-1β-induced senescence and cell cycle arrest [117]. |
| CNR2 (CB2) | Rhodopsin family - α subgroup (Cannabinoid) | Endocannabinoids, CB2 agonists | Gαi | Anti-inflammatory | Deficiency worsens OA; agonists reduce OA severity and enhance proteoglycan synthesis [117]. |
| CCR2 | Rhodopsin family - γ subgroup (Chemokine) | CCL2 | Gαi | NF-κB/MAPK | Drives macrophage recruitment and cartilage erosion in OA; antagonism reduces synovitis and damage [93]. |
| CCR5 | Rhodopsin family - γ subgroup (Chemokine) | CCL3/CCL4/CCL5 | Gαi | / | Deficiency protects against cartilage degeneration in OA [92]. |
| CXCR2 | Rhodopsin family | CXCL1/CXCL8 | Gαi | AKT signaling | Maintains chondrocyte homeostasis; knockout increases osteoarthritis severity (↑ apoptosis, ↓ ECM) [90]. |
| CXCR3 | Rhodopsin family | CXCL9/CXCL10/CXCL11 | Gαi | ER stress (CHOP/GRP78) | Elevated in OA; siRNA knockdown reduces nitrate-induced chondrocyte apoptosis [91]. |
| CXCR4 | Rhodopsin family | CXCL12 | Gαi | SDF-1/CXCR4-Runx2 feedback loop | Promotes chondrocyte hypertrophy; blocking CXCR4 inhibits hypertrophy and delays growth plate closure [89]. |
| EP1 | Rhodopsin family | PGE2 | Gαq | PGE2 signaling | Inhibits fracture healing; EP1 knockout accelerates bone repair [21]. |
| EP2 | Rhodopsin family | PGE2 | Gαs | cAMP/PKA | Suppresses MMP-13 (anti-catabolic); combined EP2/EP4 activation mimics PGE2-induced collagen synthesis [100]. |
| EP4 | Rhodopsin family | PGE2 | Gαs | cAMP/PKA | Cooperates with EP2 to regulate chondrocyte differentiation and matrix synthesis [21, 99]. |
| GLP-1R | Secretin family | GLP-1 | Gαs | PI3K/Akt/NF-κB | Activation reduces ER stress, apoptosis, and inflammation; attenuates OA cartilage degeneration [132, 167]. |
| GPR120 | Rhodopsin family - α subgroup (Fatty Acid) | Long-chain fatty acids | Gαq | SOX9-mediated ECM protection | Agonists rescue type II collagen and aggrecan expression; suppresses IL-1β-induced ECM loss [106]. |
| GPBAR1 | Rhodopsin family | Bile acids | Gαs | Anti-senescence | Protects chondrocytes from IL-1β-induced senescence; activation reduces β-galactosidase activity [104]. |
| GPR4 | Rhodopsin family | Protons | Gα12/13 or Gαq | NF-κB/MAPK | Drives OA progression; knockout or inhibition attenuates cartilage degradation [105]. |
| GPR40 | Rhodopsin family | Medium/long-chain fatty acids | Gαq (putative) | NF-κB inhibition | Agonists reduce matrix-degrading enzymes and inflammation; slows OA progression [102]. |
| GPR43 | Rhodopsin family | Short-chain fatty acids, e.g., propionate | Gαi | Anti-inflammatory signaling | Activated by butyrate; mitigates IL-1β-induced MMPs and collagen degradation [103]. |
| GPR84 | Rhodopsin family | Medium-chain fatty acids | Gαi | NF-κB inhibition | Deficiency ↑ cartilage catabolism; activation blocks IL-1β-induced OA pathogenesis [107]. |
| H4R (GPCR105) | Rhodopsin family - α subgroup (Histamine) | Histamine | Gαi | cAMP↓, MAPK↑ | Linked to hypertrophic chondrocyte differentiation (co-expressed with COLX) [98]. |
| KOR (OPRK1) | Rhodopsin family | Dynorphin | Gαi | cAMP/CREB | Protects cartilage via ↑ anabolic enzymes and ↓ catabolism; agonists may treat early OA [101]. |
| MC1R | Rhodopsin family - α subgroup (Melanocortin) | α-MSH | Gαs | cAMP/PKA | Reduces inflammatory cytokines and cartilage-degrading enzymes; enhances chondroprotective factors [97]. |
| MC3R | Rhodopsin family - α subgroup (Melanocortin) | α-MSH | Gαs | cAMP/PKA | Synergizes with MC1R to suppress cartilage degradation in OA [97]. |
| PAR2 | Rhodopsin family | Proteases, e.g., trypsin | Gαq | NF-κB/ERK | Promotes OA inflammation and cartilage damage; PAR2 antagonists reduce joint swelling and senescence [94-96]. |
Chemokine receptors play crucial roles in regulating chondrocyte biology and osteoarthritis pathogenesis. During endochondral ossification, C-X-C motif chemokine receptor 4 (CXCR4) is highly expressed in hypertrophic chondrocytes at the chondro-osseous junction [89]. Here, CXCR4 binds stromal cell-derived factor 1 (SDF-1) secreted by adjacent osteoblasts and marrow stromal cells, forming a positive feedback loop with RUNX2 to amplify hypertrophic differentiation and type X collagen expression [89]. In rabbit models, SDF-1 infiltration accelerates growth plate hypertrophy and premature physeal closure, underscoring CXCR4's role in skeletal maturation [89]. Conversely, C-X-C motif chemokine receptor 2 (CXCR2) deficiency exacerbates osteoarthritis pathology by reducing ECM production (e.g., aggrecan, type II collagen) and increasing chondrocyte apoptosis via attenuated AKT signaling [90]. C-X-C motif chemokine receptor 3 (CXCR3) expression was significantly elevated in osteoarthritis patients [91]. By using siRNA to downregulate CXCR3 in chondrocyte models induced by IL-1β and sodium nitroprusside, it was found that CXCR3 reduction had no effect on IL-1β-induced chondrocyte apoptosis but significantly decreased nitrate levels [91]. However, it markedly reduced nitrate levels and alleviated sodium nitroprusside-induced chondrocyte apoptosis [91]. The UPR pathway factors CHOP and GRP78 were involved in this process, suggesting that CXCR3 modulates chondrocyte apoptosis via the ER stress signaling pathway [91].
The C-C chemokine receptor family also plays pivotal roles in osteoarthritis pathogenesis. C-C motif chemokine receptor 5 (CCR5) ablation protects against cartilage degeneration in post-traumatic osteoarthritis models, independent of synovial or bone changes, suggesting a cartilage-specific protective role [92]. Interestingly, another study found that mice lacking C-C motif chemokine ligand 2 (CCL2) or C-C motif chemokine receptor 2 (CCR2), but not CCL5 or CCR5, were protected against osteoarthritis with reduced monocyte/macrophage recruitment [93]. Elevated levels of CCR2 ligands were found in synovial fluids from osteoarthritis patients [93]. CCR2+ macrophages were abundant in human osteoarthritis synovium and associated with cartilage erosion [93]. Blocking CCL2/CCR2 signaling significantly reduced macrophage accumulation, synovitis, and cartilage damage in mouse osteoarthritis, suggesting that selective targeting of the CCL2/CCR2 system may be a very promising therapeutic approach for osteoarthritis [93].
Emerging evidence reveals additional GPCRs as modulators of chondrocyte stress responses. Proteinase-activated receptor 2 (PAR2) emerges as a key mediator of osteoarthritis inflammation and structural damage. Studies employing PAR2 knockout mouse models have demonstrated a reduction in knee swelling and cartilage damage severity compared to wild-type mice, thereby highlighting the involvement of PAR2 in structural alterations within osteoarthritic joints [94]. Specifically, the PAR2 antagonist AZ3451 has been shown to alleviate inflammation, cartilage degradation, and cellular senescence in chondrocytes, while promoting autophagy to decrease apoptosis [95]. Notably, PAR2 deficiency in mice also leads to decreased osteophyte formation and absence of osteosclerosis, indicating its role in bone changes associated with osteoarthritis [96]. These findings collectively underscore the therapeutic potential of PAR2 antagonists in treating osteoarthritis by addressing multiple disease facets, including pain perception and bone pathology.
Melanocortin receptors MC1R/MC3R (Melanocortin 1 receptor/ Melanocortin 3 receptor) exert chondroprotective effects by suppressing inflammatory cytokines and matrix-degrading enzymes while promoting anti-inflammatory mediators [97]. H4R expression correlates with type X collagen (COLX)-positive hypertrophic chondrocytes, suggesting involvement in terminal differentiation [98]. Prostaglandin E2 (PGE2) receptors exhibit subtype-specific functions [99]. EP1 negatively regulates bone formation, while combined EP2/EP4 stimulation is required for type II collagen upregulation [21]. EP2 agonists suppress MMP-13 expression via cAMP-PKA signaling, demonstrating anti-catabolic effects without compromising cell viability [100].
Multiple GPCRs exhibit protective profiles in cartilage biology. Opioid receptor kappa 1 (OPRK1/KOR) signaling enhances anabolic activity through cAMP/CREB pathways [101], while G protein-coupled receptor 40 (GPR40) agonists reduce metalloproteinase expression and NF-κB activation [102]. GPR43 activation by butyrate mitigates IL-1β-induced inflammation [103], and GPBAR1 activation protects against senescence [104]. GPR4 promotes osteoarthritis progression through NF-κB/MAPK signaling, making its inhibition a therapeutic target [105]. GPR120 agonists preserve matrix components via SOX9-mediated pathways [106], and GPR84 deficiency exacerbates cartilage catabolism through impaired NF-κB regulation [107].
Adrenergic signaling demonstrates contextual regulation [108]. β-AR (also known as ADRB2) activation inhibits chondrogenic differentiation through ERK1/2-mediated AP-1 signaling [109, 110], while α2A-AR stimulation drives matrix degradation via ERK1/2/PKA pathways [111, 112]. Sympathetic nerve-derived norepinephrine exhibits dose-dependent effects—low concentrations promote proliferation/apoptosis through α1-AR [113], while high concentrations maintain phenotypic stability via β-AR [113]. Adenosine receptors modulate cartilage integrity through distinct mechanisms: A3AR agonists suppress RUNX2/CaMKII to prevent matrix degradation [108], while A2AR stimulation enhances autophagy via FOXO activation [114]. Besides, local injection of adrenoreceptor antagonists or agonists showed that α2A-AR activation in chondrocytes leads to cartilages degeneration and subchondral bone loss by suppressing aggrecan expression and stimulating MMP-3, MMP-13, and RANKL production via ERK1/2 and PKA pathways [111]. Inhibiting α2A-AR attenuated degenerative changes, while activating it intensified them [111]. Thus, α2A-adrenergic signal activation in chondrocytes accelerates temporomandibular joint degenerative remodeling [111]. Moreover, Bai's team developed a cartilaginous organoid system from hEPSCs with dual reporters to monitor chondrogenesis and hypertrophy, identifying α-adrenergic receptor antagonists (e.g., phentolamine) as enhancers of chondrogenesis and suppressors of hypertrophy, while α2-AR agonists induced detrimental hypertrophy [112]. Mechanistically, α2-AR signaling drives hypertrophic degeneration via cGMP-dependent SLPI production, and targeting this pathway, including SLPI inhibition, shows therapeutic potential for regenerating hyaline cartilage and repairing defects without fibrosis [112].
Angiotensin receptors demonstrate balanced regulation, with Angiotensin II receptor type 1 (AT1R) inhibition promoting proliferation and Angiotensin II receptor type 2 (AT2R) activation mitigating apoptosis under oxidative stress [115]. Cannabinoid receptors CB1/CB2 regulate skeletal growth and osteoarthritis progression [116]. CB1 deficiency causes femoral elongation defects, while CB2 activation protects against osteoarthritis through SIRT1 upregulation and senescence inhibition [117].
In the pathophysiology of cartilage and osteoarthritis, PTH1R mediates anabolic responses to intermittent PTH administration, promoting bone formation and subchondral bone integrity [118]. After activation of PTH1R, it upregulates the level of cAMP, thereby activating PKA and regulating gene expression in chondrocytes (Fig. 4C). Moreover, PTH1R activation modulates the subchondral bone microenvironment by suppressing aberrant bone remodeling, reducing sensory nerve innervation and vascular invasion, thereby decreasing inflammatory mediators like PGE2 [119]. This process not only alleviates pain but also slows osteoarthritis progression through preservation of Nestin+ mesenchymal stem cell-driven bone remodeling [7]. Conversely, CaSR, activated by abnormal biomechanical stimuli (e.g., fluid shear stress) in osteoarthritis, induces endoplasmic reticulum calcium overload, accelerating chondrocyte hypertrophy and matrix degradation [15, 24, 33]. Abnormal fluid shear stress activates CaSR and promotes chondrocyte hypertrophy and the expression of stromal degradation enzymes (such as MMP-13) through the MAPK/NF-κB pathway (Fig. 5D). CaSR inhibition emerges as a therapeutic strategy to mitigate these pathological processes [120]. Additionally, CaSR contributes to subchondral bone metabolic dysregulation, with its hyperactivation exacerbating osteoarthritis progression through aberrant bone remodeling. Collectively, PTH1R and CaSR represent opposing regulators in osteoarthritis: PTH1R exerts protective effects via bone-cartilage crosstalk, while CaSR drives pathological differentiation and matrix breakdown, highlighting their therapeutic potential as targets for osteoarthritis management.
Overall, these findings underscore the 31 GPCRs in regulating chondrocyte activity and differentiation, offering potential therapeutic targets for the treatment of bone and cartilage-related disorders such as fracture healing and osteoarthritis (Table 4). Such as promoting cartilage protection, MC1R/MC3R, GPR120, KOR, GPR40, A3AR, A2AR are indispensable. CCR2, GPR4, α2A-AR, and CaSR will accelerate the progression of osteoarthritis. EP2/EP4, CXCR2, and GPR43 mainly play a role in regulating ECM. PAR2, GPR43, A2AR, CB1/CB2 are related to inflammation regulation. CXCR4, H4R, and EP1 play important roles in the direction of chondrocyte hypertrophy and differentiation. This comprehensive GPCR network integrates diverse signals to maintain cartilage integrity. Targeting these receptors offers opportunities to modulate inflammation, matrix turnover, and cellular senescence—key pathological drivers in osteoarthritis.
GPCRs in other cells
GPCRs also play pivotal roles in other bone-related cells, such as synovial fibroblasts, immune cells, adipocytes, muscle cells and tumor cells, exerting significant influences on a multitude of physiological and pathological processes [121-123]. For instance, they impact tumor bone metastasis, a critical aspect in the progression of certain cancers where the interaction between GPCRs and bone cells can facilitate the metastatic spread of tumor cells to bone tissues (Table 5). Calcium, via overexpressed CaSR, promotes the migration and proliferation of bone-metastasizing renal cell carcinoma (RCC) cells by activating downstream pathways (Fig. 5D). Thus, CaSR could serve as a novel prognostic marker for RCC bone metastasis [122]. A3AR, which shares similar biological properties with TMIGD3 (Transmembrane and immunoglobulin domain containing 3) isoform 1, also functions as a suppressor of osteosarcoma cell aggressiveness by inhibiting the PKA-Akt-NF-κB signaling axis [124].
The functions and mechanisms of GPCR in other cells
| GPCR Name | GRAFS Classification | Ligand | Coupled G Protein Subtype | Signaling Pathway | Functional/Phenotypic Changes and references |
|---|---|---|---|---|---|
| A3AR (ADORA3) | Rhodopsin family - α subgroup (Adenosine) | Adenosine | Gαi | PKA-Akt-NF-κB inhibition | Suppresses osteosarcoma cell aggressiveness; inhibits tumor progression [124]. |
| CaSR | Glutamate family | Extracellular Ca²⁺ | Gαq | Calcium signaling/NF-κB | Promotes migration and proliferation of bone-metastasizing renal cell carcinoma (RCC) cells; potential prognostic marker for RCC bone metastasis [122]. |
| CB1 | Rhodopsin family - α subgroup (Cannabinoid) | Endocannabinoids, e.g., anandamide | Gαi | Pro-inflammatory cytokine signaling | Knockdown in Kupffer cells improves insulin sensitivity and reduces hepatic insulin resistance in obesity [125]. |
| LPAR1 (EDG2/GPCR26) | Rhodopsin family - δ subgroup (Lipids) | Lysophosphatidic acid, LPA | Gα12/13 | NF-κB/MMP activation | SNP in EDG2 promoter enhances inflammatory cytokine and MMP expression; contributes to osteoarthritis pathogenesis [126]. |
| GPR41 (FFAR3) | Rhodopsin family | Short-chain fatty acids, e.g., propionate | Gαi/Gαq (putative) | Calcium signaling | Enhances glucose uptake in muscle cells; improves insulin sensitivity and glucose tolerance in diabetic models [123]. |
| GPR43 | Rhodopsin family | Short-chain fatty acids, e.g., propionate | Gαi | Anti-inflammatory signaling pathways | Reduce osteoarthritis cartilage inflammation and matrix destruction [103, 128]. |
| GPRC5A | Glutamatefamily | Tretinoin | / | STAT3-dependent signaling | Knockout prevents bone metastasis in prostate cancer; correlates with Gleason score and metastasis in patients [127]. |
| CXCR4 | Rhodopsin family | CXCL12 | Gαi | CXCL12/CXCR4-EMT axis | Promotes EMT-like changes and osteotropism in neuroendocrine tumor cells; CXCR4 silencing abrogates migration and metastasis [129]. |
Furthermore, GPCRs are involved in regulating insulin sensitivity, which is crucial for maintaining metabolic homeostasis and preventing conditions such as diabetes. In diabetic mouse models induced by streptozotocin or a high-fat diet, AR420626 elevated plasma insulin levels, increased skeletal muscle glycogen content, and improved glucose tolerance [123]. Activation of G protein-coupled receptor 41 (GPR41) with AR420626 enhanced glucose uptake in muscle cells by boosting calcium signaling [123]. These findings indicate that GPR41 is a promising target for diabetes treatment, as it can enhance insulin sensitivity and glucose regulation. Selectively knocking down CB1R in Kupffer cells improves glucose tolerance and insulin sensitivities in obese mice without influencing hepatic lipid contents or body weight. This effect is associated with a shift to an anti-inflammatory cytokine profile and enhanced insulin signaling, indicating that CB1R in Kupffer cells plays a crucial role in obesity-related hepatic insulin resistance through a pro-inflammatory mechanism [125]. In diabetic mouse models, GPR41/FFAR3 activation by AR420626 improved glucose tolerance via enhanced calcium signaling, elevated insulin levels, and increased muscle glycogen content [123]. These findings indicate that GPR41 is a promising target for diabetes treatment, as it can enhance insulin sensitivity and glucose regulation.
Additionally, they modulate immune responses and inflammatory pathways, thereby influencing the body's defense mechanisms and its ability to manage inflammation, which are particularly relevant in bone health and disease. Osteoimmunology is receiving increasing attention, as there exist numerous shared molecules between the immune system and the skeletal system, including members of the GPCR family. These GPCRs mediate bone-immune crosstalk, providing critical insights into bone diseases and immune disorders. Persistent inflammation in impaired joints leads to metabolic dysregulation in the synovial microenvironment, altering cell activity and contributing to rheumatoid arthritis pathogenesis. Recent research highlights the role of metabolite-sensing GPCRs in rheumatoid arthritis related inflammatory immune responses. Some GPCRs influence RA progression by modulating immune cell behavior [121]. Additionally, a study identified a significant association between a SNP in the promoter region of the EDG2 gene, which encodes an LPA receptor, and knee osteoarthritis in two independent populations [126]. The susceptibility allele of this SNP enhances transcriptional activities and DNA binding in synovial cells, leading to increased expression of inflammatory cytokines and matrix metalloproteases [126]. These findings indicate that the LPA-EDG2 signal contributes to the pathogenesis of osteoarthritis through catabolic processes [126].
Moreover, GPCRs assume a pivotal function in the course of bone metastasis. G protein-coupled receptor class C group 5 member A (GPRC5A) knockout PC3 cells fail to establish bone metastasis in mice [127]. The expression of GPRC5A correlates with bone metastasis and the Gleason score in prostate cancer patients, suggesting its potential as a therapeutic target and prognostic marker for advanced prostate cancer [127]. LSSIG, a novel murine leukocyte-specific GPCR induced by STAT3 activation, has high homology to human GPR43. The expression of LSSIG is induced in M1 leukemia cells during differentiation to macrophages in a STAT3-dependent manner [128]. Similarly, GPR43 expression is induced during the differentiation of HL-60 and U937 leukemia cells to monocytes [128]. Both LSSIG and GPR43 are highly restricted to hematopoietic tissues and are induced by cytokine stimulation in bone marrow cells, monocytes, and neutrophils [128]. These findings indicate that LSSIG and GPR43 may play vital roles in the differentiation and immune response of monocytes and granulocytes [128]. In addition, CXCL12 promotes EMT-like changes and osteotropism in CXCR4 high/CXCL12 low neuroendocrine tumor (NET) cells via CXCR4. Silencing CXCR4 abrogates CXCL12-induced EMT, migration, and mesenchymal transcriptional patterns [129]. The subcellular localization of CXCR4 may suggest unique functions, hinting at potential relevance for future in vivo studies [129]. Notably, a CXCR4 agonist pepducin, a synthetic molecule that combines a peptide from CXCR4's intracellular loop with a lipid tether, has been found to mobilize bone marrow hematopoietic cells [130].
These studies emphasize the crucial roles of 8 GPCRs in processes such as tumor cell behavior, insulin resistance, immune response, fracture healing, and bone metastasis (Table 5). These findings enhance our understanding of various biological processes and diseases, suggesting GPCRs as potential therapeutic targets and prognostic markers.
Therapeutic applications of GPCR-targeting drugs in bone disorders
The development of GPCR-targeted therapeutics has maintained a pioneering role in biomedical innovation. Currently, therapeutic agents engaging these receptors represent a critical component of modern clinical practice, with ongoing expansion observed in both drug discovery pipelines and experimental therapeutic programs (Table 6). This progression reflects the established importance of GPCR modulation across multiple therapeutic domains. The evolving landscape of GPCR-based interventions underscores their enduring relevance as drug development targets. Building on mechanistic insights into GPCR-orchestrated cellular processes in bone biology—including osteoblast differentiation, osteoclast activity modulation, and paracrine signaling during remodeling—the therapeutic rationale for targeting these receptors has gained substantial traction. Dysregulation of GPCR signaling pathways, manifesting through aberrant ligand interactions, receptor desensitization, or imbalanced downstream effector cascades, forms the pathophysiological basis for skeletal disorders such as osteoporosis, rheumatoid arthritis, and osteogenesis imperfecta. GPCRs represent a critical class of therapeutic targets in bone disorders, offering significant clinical value and research potential due to their widespread tissue distribution and diverse signal transduction mechanisms (Fig. 6). These receptors enable precise modulation of bone metabolism through multiple intervention points, forming a continuum of drug development from approved medications to innovative investigational agents.
Drugs targeting GPCRs in clinical trials
| Targeted GPCR | Drug Name | Indications | Stage (Year) | Mechanism | References |
|---|---|---|---|---|---|
| Dopamine D2 Receptor (DRD2) | Chlorpromazine | Schizophrenia | Approved (1957) | D2 antagonist | [168] |
| β1/β2-Adrenergic Receptor (ADRB1/2) | Propranolol | Hypertension, Angina, Arrhythmias | Approved (1964) | β1/β2 antagonist | [169] |
| β2-Adrenergic Receptor (ADRB2) | Salbutamol | Acute Asthma | Approved (1969) | β2 agonist | [170] |
| H1 Receptor (HRH1) | Loratadine | Allergic Rhinitis | Approved (1993) | H1 antagonist | [171] |
| Calcium-Sensing Receptor (CaSR) | Cinacalcet | Hyperparathyroidism | Approved (2004) | CaSR positive allosteric modulator | [160] |
| S1P Receptor | Fingolimod | Multiple Sclerosis | Approved (2010) | S1P1 functional antagonist | [172] |
| GLP-1 Receptor (GLP1R) | Semaglutide | T2DM, Obesity | Approved (2017) | GLP-1R agonist | [173] |
| CGRP Receptor | Erenumab (Aimovig) | Migraine Prevention | Approved (2018) | CGRP receptor antagonist (mAb) | [174] |
| GPRC5D Receptor | Teclistamab | Relapsed/Refractory Multiple Myeloma | Approved (2022) | GPRC5D antagonist (bispecific antibody) | [175] |
| 5-HT1A Receptor (HTR1A) | Gepirone | Major Depressive Disorder | Approved (2023) | 5-HT1A partial agonist | [176] |
| Muscarinic M1/M4 Receptor (CHRM1/4) | KarXT (Xanomeline) | Schizophrenia | Approved (2024) | M1/M4 agonist, peripheral antagonist | [177] |
| EP4 Receptor (PTGER4) | YY001 (ECNU) | Advanced Solid Tumors | Phase II (2021) | EP4 antagonist | [178] |
| CCR8 Receptor | HBM1022 (Harbour BioMed) | Solid Tumors | Phase I (2023) | CCR8 antagonist (mAb) | [3] |
| PTH1 Receptor (PTH1R) | SEP-786 (Septerna) | Hypoparathyroidism | Phase II (2023) | PTH1R oral allosteric agonist | [166] |
| GLP-1R/GCGR/GIPR | Retatrutide (Lilly) | Obesity | Phase III (2024) | Triple agonist | [179] |
| ADGRG2 Receptor | Nb23-bi (SDU) | Orchitis/Neuroinflammation | Preclinical (2025) | Allosteric nanobody (w/DHEA) | [180] |
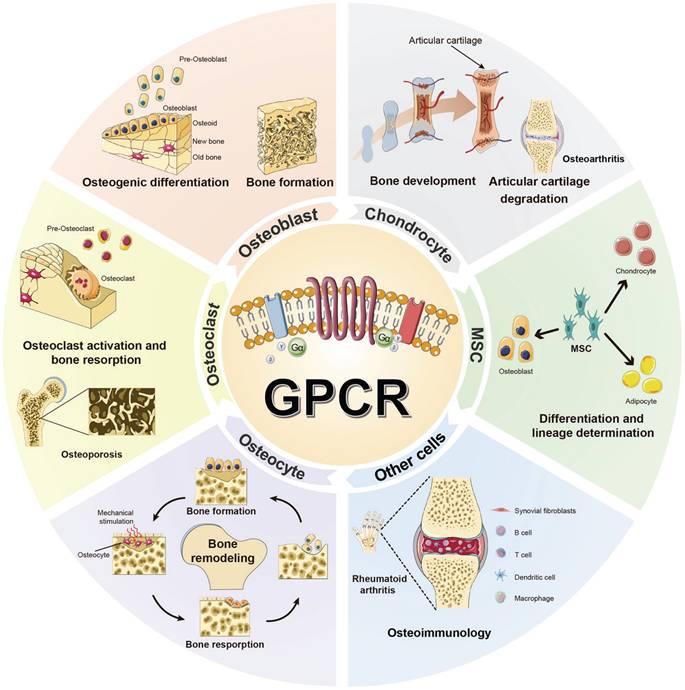
The function of GPCRs in different bone cells.
Among FDA-approved therapies, PTH1R agonists have established a paradigm shift in anabolic bone treatment [4, 7]. Teriparatide (PTH1-34) exemplifies this class through non-selective activation of Gαs/cAMP and Gαq pathways, enhancing osteoblast activity while necessitating careful monitoring for osteosarcoma risks. This challenge has driven the development of next-generation agents like abaloparatide, a biased agonist that preferentially activates Gαs-mediated osteoanabolic signals, maintaining efficacy with improved safety profiles [55]. Both medications hold FDA approval for managing severe osteoporosis in high-fracture-risk populations.
CaSR modulators constitute another therapeutic pillar. Calcimimetics such as cinacalcet and evocalcet enhance CaSR sensitivity to extracellular calcium, effectively suppressing PTH hypersecretion and stabilizing bone turnover in secondary hyperparathyroidism and chronic kidney disease-related bone disorders [6, 24]. Notably, evocalcet demonstrates reduced gastrointestinal adverse effects through enhanced receptor selectivity. Emerging as a complementary approach, calcilytics (CaSR antagonists) are being investigated as intermittent endogenous PTH stimulators with unique bone-forming potential in preclinical models [6, 15].
Exploration of novel targets has revealed multidimensional regulatory roles for GPCRs in bone metabolism. CB2 agonists exhibit dual regulatory capacity, inhibiting osteoclastogenesis while promoting osteogenesis in osteoporosis models and protecting against cancer-induced bone destruction in metastatic settings [81, 117]. Adenosine receptor modulation demonstrates therapeutic synergy, with A2A agonists simultaneously enhancing osteoblast differentiation and suppressing osteoclast formation, while A2B signaling directly augments bone matrix mineralization. Pulsed electromagnetic fields further amplify this system's therapeutic potential by upregulating A2A/A3 receptor expression, creating synergistic bone regeneration effects [53].
Precision signaling modulation has become the vanguard of next-generation drug design. Biased PTH1R agonists exemplify this strategy by selectively activating anabolic pathways while minimizing unwanted bone resorption signals [7]. Calcium-sensing receptor allosteric modulators are engineered for skeletal-specific responses, and β-adrenergic receptor regulators are being optimized to modulate osteoblast-osteoclast communication with maximal target specificity [4, 5]. These approaches reflect a paradigm shift from traditional orthosteric agonists to more nuanced signal control mechanisms.
Therapeutic boundaries continue expanding across disease spectra. CXCR4 antagonism, already validated for stem cell mobilization, demonstrates preclinical efficacy in blocking tumor cell bone homing through CXCL12-CXCR4 axis disruption [129]. GPR40/GPR120 agonists exhibit chondroprotective and anti-inflammatory effects in osteoarthritis models [102, 106], while GPR41/GPR43 activation mediates immunometabolic regulation with dual benefits in metabolic bone diseases [131]. Notably, GLP-1R/GCGR dual agonists achieve synergistic osteoanabolic and metabolic control in diabetes-associated osteoporosis [132].
Technological advancements are accelerating therapeutic innovation. Cryo-electron microscopy structures of B-class GPCR-G protein complexes provide atomic-level templates for biased ligand design. Single-cell transcriptomics uncovers skeletal cell heterogeneity, enabling targeted drug delivery systems. AI-driven platforms expedite the discovery of multifunctional molecules, though challenges persist in managing signaling cross-talk, ensuring long-term safety, and optimizing precision strategies for age-specific populations.
As the field progresses, GPCR modulation is transitioning from single-target interventions to network-based precision therapies. This evolution holds transformative potential across osteoporosis, osteoarthritis, skeletal metastases, and inflammatory bone diseases, heralding a new era of skeletal medicine grounded in molecular precision and therapeutic innovation.
Emerging concepts in GPCR research
The evolution of GPCR research has profoundly impacted bone biology, offering transformative insights into skeletal physiology and therapeutic strategies. Recent advancements in structural biology, signaling modulation, and spatial omics are reshaping our understanding of GPCRs as key regulators of bone metabolism, with direct implications for diseases like osteoporosis and osteoarthritis. This review highlights five pivotal concepts bridging GPCR innovation to bone health, emphasizing translational relevance.
Structural revelations via Cryo-EM: unlocking class B/C GPCR mechanisms
Cryo-EM has revolutionized structural elucidation of bone-relevant GPCRs. For class B receptors, the high-resolution structure of PTH1R - a master regulator of calcium homeostasis - revealed how PTH stabilizes transmembrane helix 6 to activate Gs proteins, providing a molecular template for osteoporosis drug design [133, 134]. Similarly, class C GABAB receptor heterodimer structures demonstrated asymmetric activation: GB1 subunit binds ligands while GB2 couples to Gi proteins, offering mechanistic insights into pathways disrupted in skeletal dysplasias [135-137]. These structures not only clarify ligand-receptor interactions but also enable rational design of biased agonists and allosteric modulators that selectively target bone-specific GPCR conformations.
Biased signaling and allosteric modulation: precision control of bone remodeling
Allosteric modulators, binding outside orthosteric sites, introduce therapeutic advantages by fine-tuning receptor activity without desensitization [138, 139]. In bone, PCO371 exemplifies this paradigm: by stabilizing PTH1R's intracellular transducer pocket, it activates Gs signaling independent of extracellular ligand binding, bypassing traditional desensitization pathways [139-141]. Such biased allosteric modulators direct signaling toward G protein-mediated anabolic pathways (e.g., osteoblast stimulation) while avoiding β-arrestin-driven catabolic effects (e.g., osteoclast activation) [142-145]. This precision is critical for osteoporosis, where maintaining balanced bone turnover remains challenging.
GPCR heterodimerization: emerging regulatory paradigms in bone cells
Heterodimerization expands GPCR functional diversity beyond monomeric paradigms [146-148]. The GABAB receptor heterodimer exemplifies this principle, where subunit cooperation enables nuanced signaling control. In bone, heterodimers like CXCR4-CXCR7 may integrate chemokine gradients to regulate osteoclast migration, while CaSR dimers modulate osteoblast differentiation. Targeting these complexes offers dual therapeutic angles: enhancing protective heterodimers or disrupting pathogenic ones. For instance, stabilizing osteoprotective mGluR2-mGlu4 heterodimers could mitigate aberrant resorption in rheumatoid arthritis [143, 149].
Endosomal signaling: subcellular control of bone homeostasis
Traditional views of plasma membrane-restricted GPCR signaling are evolving with discoveries of endosomal persistence [150-152]. The PTH1R-β-arrestin1 complex, for example, sustains Gs signaling from endosomes to regulate calcium fluxes long after surface receptor internalization [153-155]. This spatial redistribution explains PTH's dual anabolic (acute) and catabolic (chronic) effects in bone, where prolonged endosomal signaling may drive pathological remodeling. Therapeutically, manipulating endosomal trafficking - via dynamin inhibitors or lysosomal escape modulators - could optimize PTH analogs for sustained anabolic benefits in osteoporosis [156-160].
Single-cell GPCR atlas: mapping heterogeneity in skeletal niches
Single-cell RNA sequencing (scRNA-seq) is resolving GPCR expression heterogeneity across bone cell lineages. Preliminary studies in immune cells highlight how neuroinflammatory conditions alter GPCR profiles in osteoclast precursors, but bone-centric analyses remain sparse [161]. Future efforts should profile osteoblasts, osteocytes, and osteoclasts under pathological states (e.g., aging, diabetes) to identify disease-specific GPCR signatures. Such atlases would enable targeted delivery of GPCR modulators - such as CX3CR1 agonists to polarize osteoclasts toward anti-inflammatory phenotypes - while sparing off-target tissues.
Modern GPCR research converges on bone biology through structural precision, signaling refinement, and spatial omics. Cryo-EM-guided drug design, biased allosteric modulation, heterodimer targeting, endosomal signaling manipulation, and single-cell GPCR mapping collectively address unmet needs in skeletal medicine. As these technologies mature, the vision of precision-engineered GPCR therapeutics - from PTH1R-biased agonists to osteocyte-specific allosteric modulators - moves closer to clinical reality, offering new hope for fractured bones and fragile lives.
Perspective and Conclusion
GPCRs represent highly promising targets for pharmacological intervention due to their widespread distribution and involvement in numerous physiological processes [9, 121]. This review systematically consolidates the roles of GPCRs across bone cell types: 12 GPCRs in MSCs, 21 in osteoblasts and osteocytes, 23 in macrophages and osteoclasts, 31 in chondrocytes, and 8 in other bone-related cells (Tables 1-5).
These GPCR receptors are functionally categorized by their coupled Gα subunits (Fig. 3). Gαs-coupled receptors (e.g., PTH1R, A2AR, MC1R/MC3R) promote osteogenesis or suppress osteoclast activity via cAMP/PKA signaling, as exemplified by PTH1R-driven RUNX2 activation in osteoblasts, a mechanism harnessed clinically by the osteoporosis drug teriparatide. Conversely, ADRB2 in chondrocytes paradoxically inhibits differentiation markers through ERK1/2-PKA, underscoring cell-type specificity. Gαi/o-coupled receptors (e.g., GPR120, A1R, CNR2) primarily inhibit osteoclastogenesis or inflammation, with GPR41 enhancing insulin sensitivity in diabetic models via calcium signaling. Gαq-coupled receptors (e.g., CaSR, PAR2, EDG2) regulate bone metabolism and pathologies such as tumor metastasis through PLC/PKC or NF-κB pathways—renal cancer cells overexpressing CaSR, for instance, exhibit heightened bone metastatic potential. Gα12/13-coupled GPCRs like GPR55 drive osteoclastogenesis via Rho/ROCK, highlighting their role in bone resorption (Table 1-5).
GPCR activity is influenced by age, genetic, and environmental factors, with functional changes directly contributing to bone metabolic imbalances during development, aging, and disease progression. The functional diversity of GPCRs arises from pathway and cell-type specificity (Table 1-5). For example, ADRB2 enhances RANKL expression in osteoblasts to amplify osteoclast activity, yet suppresses chondrocyte differentiation markers, illustrating context-dependent roles. In osteoblasts, core pathways include Gαs/cAMP-PKA and Gαq/PLC-PKC: PTH1R activates RUNX2 and Osterix to stimulate bone formation, while CaSR boosts mineralization via AKT-β-catenin (Fig. 4 and Fig. 5). Osteoclasts rely on Gαq/PLC-IP3-Ca²⁺ and Gαi/EBI2-RhoA pathways, where CaSR activates NFATc1 to upregulate bone-resorbing enzymes (TRAP, CTSK), and EBI2 directs precursor migration to bone surfaces. MSCs balance osteogenic and adipogenic differentiation through Gαs/Wnt-β-catenin and Gβγ/MAPK signaling—LGR5 promotes osteogenesis, whereas ADRB2 suppresses differentiation via cAMP. Chondrocyte homeostasis hinges on Gαi/CXCR4-ERK and Gαq/GPR120-NF-κB pathways: CXCR4 drives hypertrophic differentiation, while GPR120 preserves cartilage matrix by inhibiting IL-1β-induced collagen degradation.
Therapeutically, GPCRs offer versatile targets. Gαs agonists (e.g., A2AR agonists, PTH1R activators) enhance bone regeneration, while Gαi agonists like GPR120 ligands suppress osteoclast activity in inflammatory bone loss. Gαq antagonists such as the PAR2 inhibitor AZ3451 mitigate osteoarthritis progression by blocking cartilage degradation. In cancer, targeting GPRC5A or CaSR may inhibit bone metastasis, whereas GPR41 activation improves glucose metabolism in metabolic disorders. Beyond single-cell functions, GPCRs integrate mechanical, metabolic, and immune signals: mechanical sensors like Gpr161 and Piezo1 guide MSC differentiation under stress; metabolic GPCRs (e.g., GPR41) link energy status to bone remodeling; and immune-bone crosstalk involves EP2/EP4-PGE2 (anti-inflammatory) and H4R (pro-osteoclastogenic in rheumatoid arthritis). For instance, CXCR4/SDF-1 and PAR2/NF-κB pathways exacerbate osteoarthritis through cartilage hypertrophy and matrix degradation, while MC1R/MC3R and GPR120/SOX9 counteract inflammation to preserve cartilage.
Despite their therapeutic promise, critical gaps persist. Most studies focus on GPCR roles in isolated cell types, neglecting systemic coordination across osteoblasts, osteoclasts, and immune cells (Fig. 6). For example, bone formation-resorption equilibrium likely requires GPCR-mediated dialogue between these lineages, yet such networks remain poorly mapped. Additionally, disease-stage-specific GPCR dynamics—such as CaSR's dual roles in early inflammation versus late fibrosis in osteoarthritis—are underexplored, obscuring precise therapeutic windows. Overcoming these challenges demands longitudinal studies of GPCR crosstalk and temporal regulation, leveraging advances in single-cell omics and biased ligand design. By bridging these gaps, GPCR-targeted therapies could revolutionize treatment for osteoporosis, osteoarthritis, and metastatic bone diseases, transforming mechanistic insights into clinical breakthroughs.
Moreover, the journey from bench to bedside of GPCR targeted therapies is fraught with complexity. The translation of GPCRs research into effective clinical treatments for bone diseases presents several challenges [4, 5].
Complexity of GPCR signaling
GPCRs can activate multiple signaling pathways, which can have diverse and sometimes opposing effects on bone metabolism. This signaling complexity can cause unpredictable outcomes when GPCRs are targeted therapeutically. For example, while one pathway may promote bone formation, another might simultaneously enhance bone resorption. Dissecting these pathways for selective targeting is a major challenge. To address the complexity of GPCR signaling in therapeutic development, integrated strategies should combine pathway-selective modulation with emerging technologies. First, biased agonists that preferentially activate G protein- versus β-arrestin-dependent pathways could decouple therapeutic effects from adverse liabilities, as demonstrated by μ-opioid receptor ligands separating analgesia from respiratory depression [3]. Complementary approaches include allosteric modulators and nanobody-based tools to stabilize functional receptor conformations or disrupt pathological oligomerization complexes, enabling spatial control over signaling microenvironments. Temporal regulation may be achieved through targeted protein degradation systems like PROTACs, which dynamically adjust receptor surface density to mitigate desensitization in chronic disorders, or CRISPR-based epigenome editing to permanently correct transcriptional imbalances in genetic conditions [162, 163]. Multi-omics integration offers another dimension, with single-cell analytics and machine learning identifying patient-specific signaling signatures to guide precision ligand combinations. For localized interventions, optogenetic GPCR variants provide non-invasive [164], spatiotemporally precise control over bone remodeling processes, enabling circadian rhythm restoration in osteoblast-osteoclast coupling or acute suppression of inflammatory cascades in arthritic joints.
Pleiotropic effects
Many GPCRs play roles in multiple physiological processes beyond bone metabolism [1, 7]. Targeting these receptors can therefore lead to unintended side effects in other tissues or systems. For instance, the cannabinoid receptors CB1 and CB2, which influence bone metabolism, are also involved in neurological and immune system functions [125]. This pleiotropy complicates the development of therapies that are both effective and safe.
Drug specificity and selectivity
Developing drugs that specifically target bone-related GPCRs without affecting similar receptors in other tissues is challenging. The high degree of homology among GPCR family members complicates the creation of highly selective ligands [4, 5]. Non-specific activation or inhibition of GPCRs can lead to off-target effects, reducing the therapeutic utility of a potential drug.
Limitations in models
In vitro experiments are usually carried out in cell lines or purified receptor systems, lacking the complex physiological environment in vivo, including interactions between cells, tissue specificity, and various regulatory factors in the body. In addition, in the heterologous expression system, the expression level of GPCRs may differ from that under physiological conditions in vivo. This may lead to changes in the aggregation state of the receptors, ligand-binding characteristics, and signal transduction functions.
When studying the structure and function of GPCRs in vivo, due to the lack of effective real-time monitoring techniques, it is difficult to directly observe the dynamic changes of the receptors under physiological conditions. Moreover, the genetic background of animal models may affect the observation and analysis of GPCR-related phenotypes. Some unexpected genetic changes may be introduced during the gene editing process, thus interfering with the accurate evaluation of the functions of GPCRs.
Pharmacological desensitization and tachyphylaxis
Repeated stimulation of GPCRs can lead to desensitization, where the receptor becomes less responsive to its ligand. This phenomenon can undermine the long-term effectiveness of GPCR-targeted therapies, as seen with certain therapies for osteoporosis that use PTH analogs like teriparatide, abaloparatide and romosozumab [7]. Managing receptor desensitization and developing strategies to overcome it are significant hurdles.
Despite these challenges, the therapeutic potential of targeting GPCRs in bone diseases remains significant. Advances in molecular biology, Structural biology, pharmacology, and drug design are gradually overcoming these hurdles. The development of Cryo-EM has provided us with a clearer understanding of the molecular structure of GPCRs, laying the foundation for the study of their more complex molecular functions [165, 166]. Based on this, research on biased agonists may bring breakthroughs in the treatment of precisely targeting GPCR signaling pathways. In addition, emerging concepts such as GPCR heterodimers and GPCR spatio-temporal signaling may lead to the discovery of more GPCR drug targets. Research on GPCR expression in single - cell transcriptomes of the skeletal niches may provide new perspectives for the study of the synergistic effects of GPCR signaling among different bone cells. Continued research, interdisciplinary collaboration, and innovative approaches are essential to harness the full therapeutic potential of GPCRs in treating bone diseases. Future research should focus on elucidating the cross-talk between different GPCR signaling pathways and their integration with other signaling cascades to fully harness the potential of GPCRs in modulating bone cells behavior and bone health.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32300486, 82170896, and 32170616), China Postdoctoral Science Foundation (2021M692582), Science Fund for Distinguished Young Scholars of Shaanxi Province (2025JC-JCQN-054), Innovation Capability Support Program of Shaanxi Province (2022TD-44), Fundamental Research Funds for the Central Universities (xtr052022013, xzy012022032).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Luo J, Sun P, Siwko S, Liu M, Xiao J. The role of GPCRs in bone diseases and dysfunctions. Bone Res. 2019;7:19
2. Wang F, Liu M, Wang N, Luo J. G Protein-Coupled Receptors in Osteoarthritis. Front Endocrinol (Lausanne). 2021;12:808835
3. Lorente JS, Sokolov AV, Ferguson G, Schiöth HB, Hauser AS, Gloriam DE. GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2025;24:458-79
4. Zhang M, Chen T, Lu X, Lan X, Chen Z, Lu S. G protein-coupled receptors (GPCRs): advances in structures, mechanisms and drug discovery. Signal Transduct Target Ther. 2024;9:88
5. Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829-42
6. White AD, Vilardaga J-P. Chapter 10 - GPCR Signaling in Ca2+ Homeostasis: The PTH Type 1 and Calcium-Sensing Receptors. In: Ulloa-Aguirre A, Tao Y-X, editors. Cellular Endocrinology in Health and Disease (Second Edition). Boston: Academic Press. 2021 p. 219-27
7. Marino S, Bellido T. PTH receptor signalling, osteocytes and bone disease induced by diabetes mellitus. Nat Rev Endocrinol. 2024;20:661-72
8. Kalogriopoulos NA, Tei R, Yan Y, Klein PM, Ravalin M, Cai B. et al. Synthetic GPCRs for programmable sensing and control of cell behaviour. Nature. 2025;637:230-9
9. Cheng L, Xia F, Li Z, Shen C, Yang Z, Hou H. et al. Structure, function and drug discovery of GPCR signaling. Mol Biomed. 2023;4:46
10. Davies A, Tomas A. Chapter Six - Appreciating the potential for GPCR crosstalk with ion channels. In: Shukla AK, editor. Progress in Molecular Biology and Translational Science: Academic Press. 2023 p. 101-20
11. Luo Y, Sun L, Peng Y. The structural basis of the G protein-coupled receptor and ion channel axis. Curr Res Struct Biol. 2025;9:100165
12. Aghebati-Maleki L, Dolati S, Zandi R, Fotouhi A, Ahmadi M, Aghebati A. et al. Prospect of mesenchymal stem cells in therapy of osteoporosis: A review. J Cell Physiol. 2019;234:8570-8
13. Park S, Wu L, Tu J, Yu W, Toh Y, Carmon KS. et al. Unlike LGR4, LGR5 potentiates Wnt-β-catenin signaling without sequestering E3 ligases. Sci Signal. 2020;13:eaaz4051
14. Lin W, Xu L, Pan Q, Lin S, Feng L, Wang B. et al. Lgr5-overexpressing mesenchymal stem cells augment fracture healing through regulation of Wnt/ERK signaling pathways and mitochondrial dynamics. FASEB J. 2019;33:8565-77
15. Sarem M, Heizmann M, Barbero A, Martin I, Shastri VP. Hyperstimulation of CaSR in human MSCs by biomimetic apatite inhibits endochondral ossification via temporal down-regulation of PTH1R. Proc Natl Acad Sci U S A. 2018;115:E6135-e44
16. Pi M, Zhang L, Lei SF, Huang MZ, Zhu W, Zhang J. et al. Impaired osteoblast function in GPRC6A null mice. J Bone Miner Res. 2010;25:1092-102
17. D'Alimonte I, Nargi E, Lannutti A, Marchisio M, Pierdomenico L, Costanzo G. et al. Adenosine A1 receptor stimulation enhances osteogenic differentiation of human dental pulp-derived mesenchymal stem cells via WNT signaling. Stem Cell Res. 2013;11:611-24
18. Katebi M, Soleimani M, Cronstein BN. Adenosine A2A receptors play an active role in mouse bone marrow-derived mesenchymal stem cell development. J Leukoc Biol. 2009;85:438-44
19. Carroll SH, Wigner NA, Kulkarni N, Johnston-Cox H, Gerstenfeld LC, Ravid K. A2B adenosine receptor promotes mesenchymal stem cell differentiation to osteoblasts and bone formation in vivo. J Biol Chem. 2012;287:15718-27
20. Feigenson M, Eliseev RA, Jonason JH, Mills BN, O'Keefe RJ. PGE2 Receptor Subtype 1 (EP1) Regulates Mesenchymal Stromal Cell Osteogenic Differentiation by Modulating Cellular Energy Metabolism. J Cell Biochem. 2017;118:4383-93
21. Zhang M, Ho HC, Sheu TJ, Breyer MD, Flick LM, Jonason JH. et al. EP1(-/-) mice have enhanced osteoblast differentiation and accelerated fracture repair. J Bone Miner Res. 2011;26:792-802
22. Li H, Fong C, Chen Y, Cai G, Yang M. beta2- and beta3-, but not beta1-adrenergic receptors are involved in osteogenesis of mouse mesenchymal stem cells via cAMP/PKA signaling. Arch Biochem Biophys. 2010;496:77-83
23. Li H, Fong C, Chen Y, Cai G, Yang M. Beta-adrenergic signals regulate adipogenesis of mouse mesenchymal stem cells via cAMP/PKA pathway. Mol Cell Endocrinol. 2010;323:201-7
24. Cho H, Lee J, Jang S, Lee J, Oh TI, Son Y. et al. CaSR-Mediated hBMSCs Activity Modulation: Additional Coupling Mechanism in Bone Remodeling Compartment. Int J Mol Sci. 2020;22:325
25. Hedderich J, El Bagdadi K, Angele P, Grässel S, Meurer A, Straub RH. et al. Norepinephrine Inhibits the Proliferation of Human Bone Marrow-Derived Mesenchymal Stem Cells via β2-Adrenoceptor-Mediated ERK1/2 and PKA Phosphorylation. Int J Mol Sci. 2020;21:3924
26. Li N, Yan YL, Fu S, Li RJ, Zhao PF, Xu XY. et al. Lysophosphatidic acid enhances human umbilical cord mesenchymal stem cell viability without differentiation via LPA receptor mediating manner. Apoptosis. 2017;22:1296-309
27. Lu X, Han J, Xu X, Xu J, Liu L, Huang Y. et al. PGE2 Promotes the Migration of Mesenchymal Stem Cells through the Activation of FAK and ERK1/2 Pathway. Stem Cells Int. 2017;2017:8178643
28. Wei J, Ouyang X, Tang Y, Li H, Wang B, Ye Y. et al. ER-stressed MSC displayed more effective immunomodulation in RA CD4(+)CXCR5(+)ICOS(+) follicular helper-like T cells through higher PGE2 binding with EP2/EP4. Mod Rheumatol. 2020;30:509-16
29. Johnson GP, Fair S, Hoey DA. Primary cilium-mediated MSC mechanotransduction is dependent on Gpr161 regulation of hedgehog signalling. Bone. 2021;145:115846
30. Yu W, Xie CR, Chen FC, Cheng P, Yang L, Pan XY. LGR5 enhances the osteoblastic differentiation of MC3T3-E1 cells through the Wnt/β-catenin pathway. Exp Ther Med. 2021;22:889
31. Luo J, Zhou W, Zhou X, Li D, Weng J, Yi Z. et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development. 2009;136:2747-56
32. Lorentzon M, Lorentzon R, Lerner UH, Nordström P. Calcium sensing receptor gene polymorphism, circulating calcium concentrations and bone mineral density in healthy adolescent girls. Eur J Endocrinol. 2001;144:257-61
33. Rybchyn MS, Islam KS, Brennan-Speranza TC, Cheng Z, Brennan SC, Chang W. et al. Homer1 mediates CaSR-dependent activation of mTOR complex 2 and initiates a novel pathway for AKT-dependent β-catenin stabilization in osteoblasts. J Biol Chem. 2019;294:16337-50
34. Li X, Chen S, Hu Z, Chen D, Wang J, Li Z. et al. Aberrant upregulation of CaSR promotes pathological new bone formation in ankylosing spondylitis. EMBO Mol Med. 2020;12:e12109
35. Dong B, Endo I, Ohnishi Y, Kondo T, Hasegawa T, Amizuka N. et al. Calcilytic Ameliorates Abnormalities of Mutant Calcium-Sensing Receptor (CaSR) Knock-In Mice Mimicking Autosomal Dominant Hypocalcemia (ADH). J Bone Miner Res. 2015;30:1980-93
36. Jovanovic M, Schmidt FN, Guterman-Ram G, Khayyeri H, Hiram-Bab S, Orenbuch A. et al. Perturbed bone composition and integrity with disorganized osteoblast function in zinc receptor/Gpr39-deficient mice. FASEB J. 2018;32:2507-18
37. Swift JM, Swift SN, Allen MR, Bloomfield SA. Beta-1 adrenergic agonist treatment mitigates negative changes in cancellous bone microarchitecture and inhibits osteocyte apoptosis during disuse. PLoS One. 2014;9:e106904
38. Bonnet N, Pierroz DD, Ferrari SL. Adrenergic control of bone remodeling and its implications for the treatment of osteoporosis. J Musculoskelet Neuronal Interact. 2008;8:94-104
39. Mulcrone PL, Campbell JP, Clément-Demange L, Anbinder AL, Merkel AR, Brekken RA. et al. Skeletal Colonization by Breast Cancer Cells Is Stimulated by an Osteoblast and β2AR-Dependent Neo-Angiogenic Switch. J Bone Miner Res. 2017;32:1442-54
40. Huang HH, Brennan TC, Muir MM, Mason RS. Functional alpha1- and beta2-adrenergic receptors in human osteoblasts. J Cell Physiol. 2009;220:267-75
41. Hirai T, Tanaka K, Togari A. α1-adrenergic receptor signaling in osteoblasts regulates clock genes and bone morphogenetic protein 4 expression through up-regulation of the transcriptional factor nuclear factor IL-3 (Nfil3)/E4 promoter-binding protein 4 (E4BP4). J Biol Chem. 2014;289:17174-83
42. Mlakar V, Jurkovic Mlakar S, Zupan J, Komadina R, Prezelj J, Marc J. ADRA2A is involved in neuro-endocrine regulation of bone resorption. J Cell Mol Med. 2015;19:1520-9
43. Wang L, Liu S, Quarles LD, Spurney RF. Targeted overexpression of G protein-coupled receptor kinase-2 in osteoblasts promotes bone loss. Am J Physiol Endocrinol Metab. 2005;288:E826-34
44. Wang L, Gesty-Palmer D, Fields TA, Spurney RF. Inhibition of WNT signaling by G protein-coupled receptor (GPCR) kinase 2 (GRK2). Mol Endocrinol. 2009;23:1455-65
45. Takahata Y, Takarada T, Hinoi E, Nakamura Y, Fujita H, Yoneda Y. Osteoblastic γ-aminobutyric acid, type B receptors negatively regulate osteoblastogenesis toward disturbance of osteoclastogenesis mediated by receptor activator of nuclear factor κB ligand in mouse bone. J Biol Chem. 2011;286:32906-17
46. Hwang SH, White KA, Somatilaka BN, Shelton JM, Richardson JA, Mukhopadhyay S. The G protein-coupled receptor Gpr161 regulates forelimb formation, limb patterning and skeletal morphogenesis in a primary cilium-dependent manner. Development. 2018;145:dev154054
47. Alioli CA, Demesmay L, Laurencin-Dalacieux S, Beton N, Farlay D, Follet H. et al. Expression of the type 1 lysophosphatidic acid receptor in osteoblastic cell lineage controls both bone mineralization and osteocyte specification. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158715
48. Musante I, Mattinzoli D, Otescu LA, Bossi S, Ikehata M, Gentili C. et al. Phenotypic characterization of Grm1(crv4) mice reveals a functional role for the type 1 metabotropic glutamate receptor in bone mineralization. Bone. 2017;94:114-23
49. Choudhary S, Alander C, Zhan P, Gao Q, Pilbeam C, Raisz L. Effect of deletion of the prostaglandin EP2 receptor on the anabolic response to prostaglandin E2 and a selective EP2 receptor agonist. Prostaglandins Other Lipid Mediat. 2008;86:35-40
50. Gharibi B, Abraham AA, Ham J, Evans BA. Contrasting effects of A1 and A2b adenosine receptors on adipogenesis. Int J Obes (Lond). 2012;36:397-406
51. Corciulo C, Wilder T, Cronstein BN. Adenosine A2B receptors play an important role in bone homeostasis. Purinergic Signal. 2016;12:537-47
52. Mediero A, Wilder T, Perez-Aso M, Cronstein BN. Direct or indirect stimulation of adenosine A2A receptors enhances bone regeneration as well as bone morphogenetic protein-2. FASEB J. 2015;29:1577-90
53. Vincenzi F, Targa M, Corciulo C, Gessi S, Merighi S, Setti S. et al. Pulsed electromagnetic fields increased the anti-inflammatory effect of A₂A and A₃ adenosine receptors in human T/C-28a2 chondrocytes and hFOB 1.19 osteoblasts. PLoS One. 2013;8:e65561
54. Zhang Y, Li XH, Peng P, Qiu ZH, Di CX, Chen XF. et al. RUNX2 Phase Separation Mediates Long-Range Regulation Between Osteoporosis-Susceptibility Variant and XCR1 to Promote Osteoblast Differentiation. Adv Sci (Weinh). 2024;12:e2413561
55. Ehrenmann J, Schöppe J, Klenk C, Rappas M, Kummer L, Doré AS. et al. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat Struct Mol Biol. 2018;25:1086-92
56. Sophocleous A, Landao-Bassonga E, Van't Hof RJ, Idris AI, Ralston SH. The type 2 cannabinoid receptor regulates bone mass and ovariectomy-induced bone loss by affecting osteoblast differentiation and bone formation. Endocrinology. 2011;152:2141-9
57. Zhang W, Zhang Y, Liu Y, Wang J, Gao L, Yu C. et al. Thyroid-stimulating Hormone Maintains Bone Mass and Strength by Suppressing Osteoclast Differentiation. J Biomech. 2014;47:1307-14
58. Nevius E, Pinho F, Dhodapkar M, Jin H, Nadrah K, Horowitz MC. et al. Oxysterols and EBI2 promote osteoclast precursor migration to bone surfaces and regulate bone mass homeostasis. J Exp Med. 2015;212:1931-46
59. Mosca MG, Mangini M, Cioffi S, Barba P, Mariggiò S. Peptide targeting of lysophosphatidylinositol-sensing GPR55 for osteoclastogenesis tuning. Cell Commun Signal. 2021;19:48
60. Zhang Y, Wang H, Zhu G, Qian A, Chen W. F2r negatively regulates osteoclastogenesis through inhibiting the Akt and NFκB signaling pathways. Int J Biol Sci. 2020;16:1629-39
61. Masuhara M, Tsukahara T, Tomita K, Furukawa M, Miyawaki S, Sato T. A relation between osteoclastogenesis inhibition and membrane-type estrogen receptor GPR30. Biochem Biophys Rep. 2016;8:389-94
62. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G. et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med. 2016;22:539-46
63. Zaidi M, Iqbal J. Closing the loop on the bone-resorbing osteoclast. Nat Med. 2016;22:460-1
64. Kern K, Schäfer SMG, Cohnen J, Pierre S, Osthues T, Tarighi N. et al. The G2A Receptor Controls Polarization of Macrophage by Determining Their Localization Within the Inflamed Tissue. Front Immunol. 2018;9:2261
65. Sithole C, Pieterse C, Howard K, Kasonga A. GPR120 Inhibits RANKL-Induced Osteoclast Formation and Resorption by Attenuating Reactive Oxygen Species Production in RAW264.7 Murine Macrophages. Int J Mol Sci. 2021;22:10544
66. Li Z, Yang X, Fu R, Wu Z, Xu S, Jiao J. et al. Kisspeptin-10 binding to Gpr54 in osteoclasts prevents bone loss by activating Dusp18-mediated dephosphorylation of Src. Nat Commun. 2024;15:1300
67. Goltzman D, Hendy GN. The calcium-sensing receptor in bone-mechanistic and therapeutic insights. Nat Rev Endocrinol. 2015;11:298-307
68. Kameda T, Mano H, Yamada Y, Takai H, Amizuka N, Kobori M. et al. Calcium-Sensing Receptor in Mature Osteoclasts, Which Are Bone Resorbing Cells. Biochem Biophys Res Comm. 1998;245:419-22
69. Chang W, Tu C, Chen T-H, Bikle D, Shoback D. The Extracellular Calcium-Sensing Receptor (CaSR) Is a Critical Modulator of Skeletal Development. Sci Signal. 2008;1:ra1-ra
70. Dvorak-Ewell MM, Chen TH, Liang N, Garvey C, Liu B, Tu C. et al. Osteoblast extracellular Ca2+-sensing receptor regulates bone development, mineralization, and turnover. J Bone Miner Res. 2011;26:2935-47
71. Okada H, Okabe K, Tanaka S. Finely-Tuned Calcium Oscillations in Osteoclast Differentiation and Bone Resorption. Int J Mol Sci. 2021;22:180
72. Wang H, Li J, Xu Z, Wu F, Zhang H, Yang C. et al. Undercarboxylated osteocalcin inhibits the early differentiation of osteoclast mediated by Gprc6a. PeerJ. 2021;9:e10898
73. Eaton MS, Weinstein N, Newby JB, Plattes MM, Foster HE, Arthur JW. et al. Loss of the nutrient sensor TAS1R3 leads to reduced bone resorption. J Physiol Biochem. 2018;74:3-8
74. Kara FM, Chitu V, Sloane J, Axelrod M, Fredholm BB, Stanley ER. et al. Adenosine A1 receptors (A1Rs) play a critical role in osteoclast formation and function. FASEB J. 2010;24:2325-33
75. Mediero A, Kara FM, Wilder T, Cronstein BN. Adenosine A(2A) receptor ligation inhibits osteoclast formation. Am J Pathol. 2012;180:775-86
76. Cao H, Kou X, Yang R, Liu D, Wang X, Song Y. et al. Force-induced Adrb2 in periodontal ligament cells promotes tooth movement. J Dent Res. 2014;93:1163-9
77. Aitken SJ, Landao-Bassonga E, Ralston SH, Idris AI. Beta2-adrenoreceptor ligands regulate osteoclast differentiation in vitro by direct and indirect mechanisms. Arch Biochem Biophys. 2009;482:96-103
78. Idris AI, van 't Hof RJ, Greig IR, Ridge SA, Baker D, Ross RA. et al. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med. 2005;11:774-9
79. Sophocleous A, Marino S, Kabir D, Ralston SH, Idris AI. Combined deficiency of the Cnr1 and Cnr2 receptors protects against age-related bone loss by osteoclast inhibition. Aging Cell. 2017;16:1051-61
80. Miranda K, Mehrpouya-Bahrami P, Nagarkatti PS, Nagarkatti M. Cannabinoid Receptor 1 Blockade Attenuates Obesity and Adipose Tissue Type 1 Inflammation Through miR-30e-5p Regulation of Delta-Like-4 in Macrophages and Consequently Downregulation of Th1 Cells. Front Immunol. 2019;10:1049
81. Idris AI, Sophocleous A, Landao-Bassonga E, van't Hof RJ, Ralston SH. Regulation of bone mass, osteoclast function, and ovariectomy-induced bone loss by the type 2 cannabinoid receptor. Endocrinology. 2008;149:5619-26
82. Tan Y, Sun R, Liu L, Yang D, Xiang Q, Li L. et al. Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-κB signaling to trigger pyroptosis in breast cancer. Theranostics. 2021;11:5214-31
83. David M, Machuca-Gayet I, Kikuta J, Ottewell P, Mima F, Leblanc R. et al. Lysophosphatidic acid receptor type 1 (LPA1) plays a functional role in osteoclast differentiation and bone resorption activity. J Biol Chem. 2014;289:6551-64
84. Tang CY, Wang H, Zhang Y, Wang Z, Zhu G, McVicar A. et al. GPR125 positively regulates osteoclastogenesis potentially through AKT-NF-κB and MAPK signaling pathways. Int J Biol Sci. 2022;18:2392-405
85. Kobayashi Y, Take I, Yamashita T, Mizoguchi T, Ninomiya T, Hattori T. et al. Prostaglandin E2 receptors EP2 and EP4 are down-regulated during differentiation of mouse osteoclasts from their precursors. J Biol Chem. 2005;280:24035-42
86. Take I, Kobayashi Y, Yamamoto Y, Tsuboi H, Ochi T, Uematsu S. et al. Prostaglandin E2 strongly inhibits human osteoclast formation. Endocrinology. 2005;146:5204-14
87. Sakuma Y, Tanaka K, Suda M, Yasoda A, Natsui K, Tanaka I. et al. Crucial involvement of the EP4 subtype of prostaglandin E receptor in osteoclast formation by proinflammatory cytokines and lipopolysaccharide. J Bone Miner Res. 2000;15:218-27
88. Kim KW, Kim BM, Lee KA, Lee SH, Firestein GS, Kim HR. Histamine and Histamine H4 Receptor Promotes Osteoclastogenesis in Rheumatoid Arthritis. Sci Rep. 2017;7:1197
89. Wei L, Kanbe K, Lee M, Wei X, Pei M, Sun X. et al. Stimulation of chondrocyte hypertrophy by chemokine stromal cell-derived factor 1 in the chondro-osseous junction during endochondral bone formation. Dev Biol. 2010;341:236-45
90. Sherwood J, Bertrand J, Nalesso G, Poulet B, Pitsillides A, Brandolini L. et al. A homeostatic function of CXCR2 signalling in articular cartilage. Ann Rheum Dis. 2015;74:2207-15
91. Yin ZC, Xiong WH, Pang QJ. CXCR3 mediates chondrocyte injury through regulating nitric oxide. Eur Rev Med Pharmacol Sci. 2018;22:2454-60
92. Takebe K, Rai MF, Schmidt EJ, Sandell LJ. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthritis Cartilage. 2015;23:454-61
93. Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F. et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis. 2017;76:914-22
94. Amiable N, Martel-Pelletier J, Lussier B, Kwan Tat S, Pelletier JP, Boileau C. Proteinase-activated receptor-2 gene disruption limits the effect of osteoarthritis on cartilage in mice: a novel target in joint degradation. J Rheumatol. 2011;38:911-20
95. Huang X, Ni B, Xi Y, Chu X, Zhang R, You H. Protease-activated receptor 2 (PAR-2) antagonist AZ3451 as a novel therapeutic agent for osteoarthritis. Aging (Albany NY). 2019;11:12532-45
96. Huesa C, Ortiz AC, Dunning L, McGavin L, Bennett L, McIntosh K. et al. Proteinase-activated receptor 2 modulates OA-related pain, cartilage and bone pathology. Ann Rheum Dis. 2016;75:1989-97
97. Kaneva MK, Kerrigan MJ, Grieco P, Curley GP, Locke IC, Getting SJ. Chondroprotective and anti-inflammatory role of melanocortin peptides in TNF-α activated human C-20/A4 chondrocytes. Br J Pharmacol. 2012;167:67-79
98. Yamaura K, Akiyama S, Ueno K. Increased expression of the histamine H4 receptor subtype in hypertrophic differentiation of chondrogenic ATDC5 cells. J Cell Biochem. 2012;113:1054-60
99. Miyamoto M, Ito H, Mukai S, Kobayashi T, Yamamoto H, Kobayashi M. et al. Simultaneous stimulation of EP2 and EP4 is essential to the effect of prostaglandin E2 in chondrocyte differentiation. Osteoarthritis Cartilage. 2003;11:644-52
100. Sato T, Konomi K, Fujii R, Aono H, Aratani S, Yagishita N. et al. Prostaglandin EP2 receptor signalling inhibits the expression of matrix metalloproteinase 13 in human osteoarthritic chondrocytes. Ann Rheum Dis. 2011;70:221-6
101. Wu L, Zhang S, Shkhyan R, Lee S, Gullo F, Eliasberg CD. et al. Kappa opioid receptor signaling protects cartilage tissue against posttraumatic degeneration. JCI Insight. 2017;2:e88553
102. Gu J, Lin H, Zhang Y, Xu T, Wang T, Xue X. et al. Activation of GPR40 Suppresses AGE-Induced Reduction of Type II Collagen and Aggrecan in Human SW1353 Chondrocytes. Drug Des Devel Ther. 2020;14:2371-9
103. Pirozzi C, Francisco V, Guida FD, Gómez R, Lago F, Pino J. et al. Butyrate Modulates Inflammation in Chondrocytes via GPR43 Receptor. Cell Physiol Biochem. 2018;51:228-43
104. Huang H, Lei H, Yang F, Fan X, Dang Q, Li Y. Activation of the bile acid receptor GPBAR1 (TGR5) ameliorates interleukin-1β (IL-1β)- induced chondrocytes senescence. Biomed Pharmacother. 2018;106:1713-9
105. Li R, Guan Z, Bi S, Wang F, He L, Niu X. et al. The proton-activated G protein-coupled receptor GPR4 regulates the development of osteoarthritis via modulating CXCL12/CXCR7 signaling. Cell Death Dis. 2022;13:152
106. Xu Z, Ke T, Zhang Y, Fu C, He W. Agonism of GPR120 prevented IL-1β-induced reduction of extracellular matrix through SOX-9. Aging (Albany NY). 2020;12:12074-85
107. Wang F, Ma L, Ding Y, He L, Chang M, Shan Y. et al. Fatty acid sensing GPCR (GPR84) signaling safeguards cartilage homeostasis and protects against osteoarthritis. Pharmacol Res. 2021;164:105406
108. Shkhyan R, Lee S, Gullo F, Li L, Peleli M, Carlstrom M. et al. Genetic ablation of adenosine receptor A3 results in articular cartilage degeneration. J Mol Med (Berl). 2018;96:1049-60
109. Mitchell J, Lai LP, Peralta F, Xu Y, Sugamori K. β2-adrenergic receptors inhibit the expression of collagen type II in growth plate chondrocytes by stimulating the AP-1 factor Jun-B. Am J Physiol Endocrinol Metab. 2011;300:E633-9
110. Lai LP, Mitchell J. Beta2-adrenergic receptors expressed on murine chondrocytes stimulate cellular growth and inhibit the expression of Indian hedgehog and collagen type X. J Cell Biochem. 2008;104:545-53
111. Jiao K, Zeng G, Niu LN, Yang HX, Ren GT, Xu XY. et al. Activation of α2A-adrenergic signal transduction in chondrocytes promotes degenerative remodelling of temporomandibular joint. Sci Rep. 2016;6:30085
112. Wei X, Qiu J, Lai R, Wei T, Lin Z, Huang S. et al. A human organoid drug screen identifies α2-adrenergic receptor signaling as a therapeutic target for cartilage regeneration. Cell Stem Cell. 2024;31:1813-30.e8
113. Lorenz J, Schäfer N, Bauer R, Jenei-Lanzl Z, Springorum RH, Grässel S. Norepinephrine modulates osteoarthritic chondrocyte metabolism and inflammatory responses. Osteoarthritis Cartilage. 2016;24:325-34
114. Friedman B, Corciulo C, Castro CM, Cronstein BN. Adenosine A2A receptor signaling promotes FoxO associated autophagy in chondrocytes. Sci Rep. 2021;11:968
115. Cai HQ, Miao MY, Zhang WL. AT1/2R affects the proliferation and apoptosis of chondrocytes induced by oxygen-glucose deprivation. Bratisl Lek Listy. 2020;121:584-8
116. Wasserman E, Tam J, Mechoulam R, Zimmer A, Maor G, Bab I. CB1 cannabinoid receptors mediate endochondral skeletal growth attenuation by Δ9-tetrahydrocannabinol. Ann N Y Acad Sci. 2015;1335:110-9
117. Sophocleous A, Börjesson AE, Salter DM, Ralston SH. The type 2 cannabinoid receptor regulates susceptibility to osteoarthritis in mice. Osteoarthritis Cartilage. 2015;23:1586-94
118. Chu TL, Chen P, Yu AX, Kong M, Tan Z, Tsang KY. et al. MMP14 cleaves PTH1R in the chondrocyte-derived osteoblast lineage, curbing signaling intensity for proper bone anabolism. Elife. 2023;12:e82142
119. Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell. 2002;3:183-94
120. Zhang M, Yang H, Wan X, Lu L, Zhang J, Zhang H. et al. Prevention of Injury-Induced Osteoarthritis in Rodent Temporomandibular Joint by Targeting Chondrocyte CaSR. J Bone Miner Res. 2019;34:726-38
121. Yang X, Zhang W, Wang L, Zhao Y, Wei W. Metabolite-sensing GPCRs in rheumatoid arthritis. Trends Pharmacol Sci. 2024;45:118-33
122. Joeckel E, Haber T, Prawitt D, Junker K, Hampel C, Thüroff JW. et al. High calcium concentration in bones promotes bone metastasis in renal cell carcinomas expressing calcium-sensing receptor. Mol Cancer. 2014;13:42
123. Lee DH, Heo KS, Myung CS. Gα(i) -coupled GPR41 activation increases Ca(2+) influx in C2C12 cells and shows a therapeutic effect in diabetic animals. Obesity (Silver Spring). 2023;31:1871-83
124. Iyer SV, Ranjan A, Elias HK, Parrales A, Sasaki H, Roy BC. et al. Genome-wide RNAi screening identifies TMIGD3 isoform1 as a suppressor of NF-κB and osteosarcoma progression. Nat Commun. 2016;7:13561
125. Jourdan T, Nicoloro SM, Zhou Z, Shen Y, Liu J, Coffey NJ. et al. Decreasing CB(1) receptor signaling in Kupffer cells improves insulin sensitivity in obese mice. Mol Metab. 2017;6:1517-28
126. Mototani H, Iida A, Nakajima M, Furuichi T, Miyamoto Y, Tsunoda T. et al. A functional SNP in EDG2 increases susceptibility to knee osteoarthritis in Japanese. Hum Mol Genet. 2008;17:1790-7
127. Sawada Y, Kikugawa T, Iio H, Sakakibara I, Yoshida S, Ikedo A. et al. GPRC5A facilitates cell proliferation through cell cycle regulation and correlates with bone metastasis in prostate cancer. Int J Cancer. 2020;146:1369-82
128. Senga T, Iwamoto S, Yoshida T, Yokota T, Adachi K, Azuma E. et al. LSSIG is a novel murine leukocyte-specific GPCR that is induced by the activation of STAT3. Blood. 2003;101:1185-7
129. Cives M, Quaresmini D, Rizzo FM, Felici C, D'Oronzo S, Simone V. et al. Osteotropism of neuroendocrine tumors: role of the CXCL12/ CXCR4 pathway in promoting EMT in vitro. Oncotarget. 2017;8:22534-49
130. Tchernychev B, Ren Y, Sachdev P, Janz JM, Haggis L, O'Shea A. et al. Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc Natl Acad Sci U S A. 2010;107:22255-9
131. Lee D-H, Kim M-T, Han J-H. GPR41 and GPR43: From development to metabolic regulation. Biomed Pharmacother. 2024;175:116735
132. Roseweir AK, Millar RP. Kisspeptin antagonists. Adv Exp Med Biol. 2013;784:159-86
133. Zhao LH, Ma S, Sutkeviciute I, Shen DD, Zhou XE, de Waal PW. et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science. 2019;364:148-53
134. Kobayashi K, Kawakami K, Kusakizako T, Miyauchi H, Tomita A, Kobayashi K. et al. Endogenous ligand recognition and structural transition of a human PTH receptor. Mol Cell. 2022;82:3468-83.e5
135. Mao C, Shen C, Li C, Shen DD, Xu C, Zhang S. et al. Cryo-EM structures of inactive and active GABA(B) receptor. Cell Res. 2020;30:564-73
136. Park J, Fu Z, Frangaj A, Liu J, Mosyak L, Shen T. et al. Structure of human GABA(B) receptor in an inactive state. Nature. 2020;584:304-9
137. Shen C, Mao C, Xu C, Jin N, Zhang H, Shen DD. et al. Structural basis of GABA(B) receptor-G(i) protein coupling. Nature. 2021;594:594-8
138. Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41-54
139. Krumm BE, Roth BL. Intracellular GPCR modulators enable precision pharmacology. NPJ Drug Discov. 2025;2:8
140. Zhao LH, He Q, Yuan Q, Gu Y, He X, Shan H. et al. Conserved class B GPCR activation by a biased intracellular agonist. Nature. 2023;621:635-41
141. Kobayashi K, Kawakami K, Kusakizako T, Tomita A, Nishimura M, Sawada K. et al. Class B1 GPCR activation by an intracellular agonist. Nature. 2023;618:1085-93
142. Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205-16
143. Huang W, Jin N, Guo J, Shen C, Xu C, Xi K. et al. Structural basis of orientated asymmetry in a mGlu heterodimer. Nat Commun. 2024;15:10345
144. Slosky LM, Caron MG, Barak LS. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol Sci. 2021;42:283-99
145. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17:243-60
146. Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697-700
147. Serafini RA, Zachariou V. Opioid-galanin receptor heteromers differentiate the dopaminergic effects of morphine and methadone. J Clin Invest. 2019;129:2653-4
148. Song Y, Xu C, Liu J, Li Y, Wang H, Shan D. et al. Heterodimerization With 5-HT(2B)R Is Indispensable for beta(2)AR-Mediated Cardioprotection. Circ Res. 2021;128:262-77
149. Wang X, Wang M, Xu T, Feng Y, Shao Q, Han S. et al. Structural insights into dimerization and activation of the mGlu2-mGlu3 and mGlu2-mGlu4 heterodimers. Cell Res. 2023;33:762-74
150. Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276:19452-60
151. Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248-57
152. Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201-10
153. Yang F, Yu X, Liu C, Qu CX, Gong Z, Liu HD. et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun. 2015;6:8202
154. Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG. et al. The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci U S A. 1999;96:3712-7
155. Goodman OB Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW. et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447-50
156. Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM. et al. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907-19
157. Flores-Espinoza E, Thomsen ARB. Beneath the surface: endosomal GPCR signaling. Trends Biochem Sci. 2024;49:520-31
158. White AD, Pena KA, Clark LJ, Maria CS, Liu S, Jean-Alphonse FG. et al. Spatial bias in cAMP generation determines biological responses to PTH type 1 receptor activation. Sci Signal. 2021;14:eabc5944
159. Sutkeviciute I, Clark LJ, White AD, Gardella TJ, Vilardaga JP. PTH/PTHrP Receptor Signaling, Allostery, and Structures. Trends Endocrinol Metab. 2019;30:860-74
160. Horwitz MJ, Tedesco MB, Sereika SM, Syed MA, Garcia-Ocana A, Bisello A. et al. Continuous PTH and PTHrP infusion causes suppression of bone formation and discordant effects on 1,25(OH)2 vitamin D. J Bone Miner Res. 2005;20:1792-803
161. Tischner D, Grimm M, Kaur H, Staudenraus D, Carvalho J, Looso M. et al. Single-cell profiling reveals GPCR heterogeneity and functional patterning during neuroinflammation. JCI Insight. 2017;2:e95063
162. Li Z, Lin Y, Song H, Qin X, Yu Z, Zhang Z. et al. First small-molecule PROTACs for G protein-coupled receptors: inducing α1A-adrenergic receptor degradation. Acta Pharma Sin B. 2020;10:1669-79
163. Nakamura M, Gao Y, Dominguez AA, Qi LS. CRISPR technologies for precise epigenome editing. Nat Cell Biol. 2021;23:11-22
164. Wietek J, Nozownik A, Pulin M, Saraf-Sinik I, Matosevich N, Gowrishankar R. et al. A bistable inhibitory optoGPCR for multiplexed optogenetic control of neural circuits. Nat Methods. 2024;21:1275-87
165. Papasergi-Scott MM, Pérez-Hernández G, Batebi H, Gao Y, Eskici G, Seven AB. et al. Time-resolved cryo-EM of G-protein activation by a GPCR. Nature. 2024;629:1182-91
166. Duan J, He X-H, Li S-J, Xu HE. Cryo-electron microscopy for GPCR research and drug discovery in endocrinology and metabolism. Nat Rev Endocrinol. 2024;20:349-65
167. Meurot C, Jacques C, Martin C, Sudre L, Breton J, Rattenbach R. et al. Targeting the GLP-1/GLP-1R axis to treat osteoarthritis: A new opportunity? J Orthop Translat. 2022;32:121-9
168. Lehmann HE, Ban TA. The history of the psychopharmacology of schizophrenia. Can J Psychiatry. 1997;42:152-62
169. Black JW, Crowther AF, Shanks RG, Smith LH, Dornhorst AC. A New Adrenergic Betareceptor Antagonist. Lancet. 1964;1:1080-1
170. Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ. et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science. 2017;357:891-8
171. Clissold SP, Sorkin EM, Goa KL. Loratadine. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1989;37:42-57
172. Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P. et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387-401
173. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA. et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375:1834-44
174. Goadsby PJ, Reuter U, Hallstrom Y, Broessner G, Bonner JH, Zhang F. et al. A Controlled Trial of Erenumab for Episodic Migraine. N Engl J Med. 2017;377:2123-32
175. Moreau P, Garfall AL, van de Donk N, Nahi H, San-Miguel JF, Oriol A. et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2022;387:495-505
176. Kishi T, Meltzer HY, Matsuda Y, Iwata N. Azapirone 5-HT1A receptor partial agonist treatment for major depressive disorder: systematic review and meta-analysis. Psychol Med. 2014;44:2255-69
177. Kaul I, Sawchak S, Correll CU, Kakar R, Breier A, Zhu H. et al. Efficacy and safety of the muscarinic receptor agonist KarXT (xanomeline-trospium) in schizophrenia (EMERGENT-2) in the USA: results from a randomised, double-blind, placebo-controlled, flexible-dose phase 3 trial. Lancet. 2024;403:160-70
178. Peng S, Hu P, Xiao YT, Lu W, Guo D, Hu S. et al. Single-Cell Analysis Reveals EP4 as a Target for Restoring T-Cell Infiltration and Sensitizing Prostate Cancer to Immunotherapy. Clin Cancer Res. 2022;28:552-67
179. Bisson A, Fauchier G, Fauchier L. Triple-Hormone-Receptor Agonist Retatrutide for Obesity. N Engl J Med. 2023;389:1628
180. Zheng Y, Jiang D, Lu Y, Zhang C, Huang SM, Lin H. et al. Development of an allosteric adhesion GPCR nanobody with therapeutic potential. Nat Chem Biol. 2025
Author contact
![]() Corresponding authors: Yan Guo (guoyan253edu.cn), Shou-Ye Hu (hushouye2021com).
Corresponding authors: Yan Guo (guoyan253edu.cn), Shou-Ye Hu (hushouye2021com).