Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Pathophysiology of MC damage in...
Crosstalk between MCs and other...
Therapeutic implications of MCs...
Conclusion and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(11):4762-4781. doi:10.7150/ijbs.114907 This issue Cite
Review
Mesangial Cells in Diabetic Kidney Disease: From Mechanisms to Therapeutic Implications
Li Feng, Yu-ying Feng, Qian Ren, Ping Fu ![]() , Liang Ma
, Liang Ma ![]()
Department of Nephrology, Institute of Kidney Diseases, West China Hospital of Sichuan University, and National Key Laboratory of Kidney Diseases, Chengdu 610041, China.
Received 2025-4-2; Accepted 2025-7-1; Published 2025-7-24
Abstract

Diabetic kidney disease (DKD) is a serious diabetic complication, the morbidity and mortality of which has rapidly increased worldwide. As known, the pathogenesis of DKD is complex and includes mesangial cell (MC) proliferation and hypertrophy, mesangial expansion, glomerular basement membrane thickening, podocyte detachment, and tubulointerstitial damage. Thus, the role of MCs cannot be underestimated, as their exclusive positioning and diverse physiological functions are crucial for preserving the glomerular filtration membrane's composition and functionality. Considerable animal studies and clinical trials have elaborated that MCs are pivotal to the occurrence and progression of DKD. In this review, we summarize and outline the mechanisms of MC injury and the interactions of MCs with other cells and discuss the progress of MC-targeted therapeutics to provide a comprehensive perspective on the prevention and treatment of DKD.
Keywords: diabetic kidney disease, mesangial cells, pathophysiology, drug target.
Introduction
Diabetes is a global metabolic disease. More than 10.5% of adults worldwide have diabetes [1-6]. Epidemiological studies have demonstrated that approximately 40% of diabetic patients ultimately progress into diabetic kidney disease (DKD), with significant variations among different geographical regions and ethnic groups [7]. Notably, African-descent and Asian populations exhibit particularly elevated disease susceptibility, whereas low-income countries bear a disproportionately heavier disease burden [8]. Geographically, DKD has consistently remained the leading etiology of end-stage renal disease (ESRD) in developed Western countries. Concurrently, epidemiological data from China have revealed a steady upward trajectory in DKD incidence, establishing it as one of the principal causes of kidney failure (KF) [9]. As the incidence of diabetes has increased worldwide, DKD has become highly prevalent and is a major cause of heart disease, early death, and worldwide health care expenditures [10]. However, the etiology of DKD is complex, and various pathophysiological mechanisms might contribute to DKD, including abnormalities in glomerular hemodynamic function, oxidative stress, mitochondrial dysfunction, and the activation of proinflammatory and profibrotic factors [11]. Moreover, multiple cellular abnormalities are responsible for the development and progression of DKD.
Glomerular endothelial cell dysfunction, mesangial cell (MC) damage, mesangial matrix accumulation, podocyte shedding, and tubular epithelial injury are involved in the process of renal fibrosis, which eventually lead to the renal impairment in DKD patients [12, 13]. Here, the MC injury is a significant pathogenic process during the development of DKD. MCs are widely known to maintain homeostasis under healthy conditions, with dynamic balancing of the synthesis and degradation of mesangial matrix proteins. Under stable conditions, MCs preserve the normal structure of the surrounding capillary network and ensure that the glomerulus can perform its filtering function [14]. However, when exposed to certain pathological factors and injury-inducing stimulations, such as hyperglycemia, hypertension, hyperlipidemia, reactive oxygen species (ROS), inflammatory factors, and so on, MCs are activated and both proliferate and secrete cytokines and matrix proteins, ultimately leading to the loss of glomerular capillary loops and glomerular function. Aberrant proliferation of MCs and excessive matrix expansion have been identified as pivotal pathological features in diseases such as IgA nephropathy and lupus nephritis [15, 16]. These processes contribute to renal fibrosis progression by obstructing Bowman's capsule, disrupting the filtration barrier, and ultimately leading to glomerulosclerosis and loss of renal function.
Additionally, MCs could directly contact with endothelial cells and separate from podocytes via the glomerular basement membrane (GBM). This architectural framework supports the necessary conditions for physiological intercellular communication within the glomerulus. Emerging evidence has indicated that kidney cells and the interaction between kidney cells and immune cells play pivotal roles in triggering the course of DKD [17]. This complex interaction is notably visible in endothelial dysfunction, mesangial enlargement, podocyte injury, and progressive podocyte apoptosis, which can exacerbate glomerulosclerosis and functional degradation [18]. Accumulating, studies have highlighted that MCs not only maintain the stability of the capillary network, but also receive and release a range of growth factors and cytokines to play a signaling role [19-21]. Therefore, further exploration of the interactions between MCs and other cells is highly important.
In this review, we examine the molecular mechanisms of MC damage and create a web of cellular crosstalk focused on MCs to investigate their roles in DKD. Importantly, we also discuss the MC-targeted treatment strategies for DKD.
Pathophysiology of MC damage in DKD
The central axis of the glomerulus is made up of MCs and their mesangial stroma, which are spread throughout the capillary network and, along with endothelial cells and podocytes, maintain the normal glomerular structure. In their resting state, MCs exhibit a stellate morphology; however, upon activation, they transform into elongated spindle-shaped cells closely resembling fibroblasts. [22]. MCs have cellular protrusions that extend from the cell's center of the cell into the surrounding capillary lumen, allowing them to perform a range of physiological activities [23]. In DKD, MCs undergo a significant phenotypic shift from a quiescent state to a dedifferentiated state with elevated α-SMA expression. MCs undergoing phenotypic transition exhibit robust secretory capacity, releasing substantial quantities of inflammatory mediators and pro-fibrotic factors. These secretory products collectively contribute to renal pathology in DKD through both autonomous mechanisms and paracrine interactions with neighboring cell populations. [24]. However, much remains undiscovered in the context of MC damage in DKD. With the progression of biomedical research, substantial scientific evidence has elucidated the molecular mechanisms underlying MCs injury in DKD pathophysiology. The following part will focus on the molecular mechanisms underlying DKD MC failure in DKD from four perspectives.
Oxidative stress in MCs
Oxidative stress arises from a disruptive imbalance between pro-oxidant and antioxidant forces within biological systems, triggering the relentless buildup of reactive oxygen species (ROS) and reactive nitrogen intermediates. This biochemical cascade ultimately inflicts oxidative injury upon vital biomolecules, compromising DNA integrity and denaturing functional proteins. [25]. The pathogenesis of DKD is complex, among which oxidative stress is particularly important [26]. Many studies suggest that MC exposure to a hyperglycemic environment increases the levels of ROS and inflammatory mediators, which may be strongly related to DKD [27]. The accumulation of ROS can lead to mesangial dysfunction, an imbalance of mesangial matrix synthesis and degradation, and cause glomerulosclerosis and loss of filtration function. It has been revealed that Fyn, a member of the Src kinase, is engaged in the oxidative stress associated with of acute renal damage [28]. Knockdown of Fyn reduced high-glucose-induced ROS levels in MCs and DKD kidney tissue, improving renal fibrosis [29]. Another study reported that overexpression of the transcription factor FOXO1 reduced the ROS accumulation in MCs under high-glucose stimulation [30]. Nuclear factor erythroid 2-related factor (Nrf2) is the main regulator of cells maintaining redox homeostasis and plays an antioxidant role [31]. Studies have shown that the expression of Nrf2 and its target gene expression are downregulated in DKD, and that activation of Nrf2 could improve high-glucose triggered oxidative stress and inflammation in MCs [32]. Research has demonstrated that the transcription factor YY1 promotes renal fibrogenesis in DKD through activation of the HIF-1α/mROS signaling pathway. Mechanistically, under HG conditions, YY1 increases hypoxia-inducible factor-1α (HIF-1α) expression and activity in MCs, leading to excessive mitochondrial ROS (mROS) production. Furthermore, mROS reciprocally enhances YY1/HIF-1α signaling activation, establishing a positive feedback loop that ultimately drives MC hyperproliferation [33].
Mitochondrial dysfunction in MCs
The factors that cause the development of DKD are not clear, but numerous studies have shown that mitochondrial dysfunction is related to the progression of DKD [34]. Mitochondria are extremely important organelles in eukaryotic cells and exert have a wide range of biological functions. Mitochondria, the main site of glucose, lipid, and amino acid metabolism, supply the energy and nutrients required to adjust to different cellular conditions and warning signs. The kidney is an organ with high metabolism and high energy demand, and various biological processes require mitochondria to supply energy. Therefore, mitochondrial function is a necessary condition for maintaining normal kidney structure and function. MCs contain several mitochondria, and the mitochondrial dysfunction of MCs under high-glucose conditions leads to MC damage and promotes the progression of DKD.
Mitophagy
Autophagy is a conserved process in which for removing excess organelles and misfolded proteins are removed from the body [35]. Autophagosomes encapsulate proteins or organelles that need to be degraded and transport them to lysosomes, where multiple proteases degrade these materials. The degraded products are recycled and reused to synthesize new proteins and organelles [36]. Mitophagy is the process of targeting the transfer of damaged or dysfunctional mitochondria to autophagosomes for degradation through lysosomes [37]. In DKD, a large number of functionally impaired mitochondria accumulate in the kidney, suggesting that mitophagy function may be impaired [38]. Mitophagy is mainly mediated by the PINK1/Parkin pathway [39], which is also inhibited in DKD [36, 38]. A study reported that erythropoietin (EPO) could reverse high-glucose-induced inhibition of the PINK1/Parkin pathway in MCs, restore mitophagy in DKD, and ameliorate renal damage [40].
Mitochondrial fusion and fission
The process of uniting two mitochondria's inner and outer membranes to create a single mitochondrion is known as mitochondrial fusion, and mitochondrial fission involves the breakage of the inner and outer membranes of one mitochondrion into two mitochondria [41]. Fusion and fission are adaptive changes in mitochondria that are used to meet cellular energy needs [42]. Mitochondria repair their function by fusing with mitochondrial DNA or proteins, which then divide to form new functional mitochondria, while the remaining defective proteins are recycled via autophagy [43, 44]. In DKD, mitochondrial fusion and fission are increased, and mitophagy is impaired, resulting in many defective mitochondria that cannot be recycled and reused, leading to insufficient mitochondrial ATP [44, 45]. Dynamic GTPase dynamite-related protein 1 (Drp1) is a mitochondrial cleavage protein that is located in the cytoplasm, translocates to the outer mitochondrial membrane, and promotes mitochondrial fission in a GTP-dependent manner [34]. Studies have shown that Rho-associated coiled-coil kinase 1 (ROCK1) in DKD causes mitochondrial fission by promoting the aggregation of Drp1 in mitochondria. Mechanistically, the serine residue at position 600 of Drp1 is phosphorylated by ROCK1, thereby triggering the translocation of Drp1 [46]. Multiple studies have shown that inhibiting mitochondrial fission caused by Drp1 could improve renal damage in DKD [47]. Phosphodiesterase-4 (PDE4) is a member of the phosphodiesterase family and is involved in the progression of DKD [48]. PDE4 overexpression can increase the expression of Drp1 and reduce the phosphorylation of Drp1 at serine 637 by inhibiting protein kinase A (PKA), thereby promoting high-glucose-induced mitochondrial fission in MCs [49]. Single amino acid phosphorylation of Drp1 can regulate mitochondrial fission and DKD progression, but it seems that different phosphorylation sites play different roles. For example, in rat aortic smooth muscle cells, angiotensin II induces the phosphorylation of Drp1 serine residue 616 to promote mitochondrial fission [50]. Elucidating the signaling mechanisms associated with altered mitochondrial dynamics is essential for understanding the pathogenic mechanisms underlying mitochondrial dysregulation during the early stages of hyperglycemic conditions.
Mitochondrial biogenesis
Mitophagy, fusion, and fission are all necessary for improved mitochondrial biogenesis which is the process of maintaining the number of mitochondria [51]. Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) jointly regulate mitochondrial biogenesis. In DKD, the reduction in mitochondrial DNA amount, the impairation of mitochondrial biogenesis, the mitochondrial dysfunction, and insufficiency in energy supply altogether, ultimately lead to cell apoptosis. Multiple studies have shown that nuclear respiratory factor 1 (NRF1), peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), and AMP-activated protein kinase (AMPK) are major regulators of mitochondrial biogenesis [52-54]. Downregulation of PGC1-α and NRF1 promotes renal injury and dysfunction in DKD [55, 56]. The expression and activity of the transcription factor FOXO1 are downregulated in MCs and DKD kidneys stimulated by high glucose [57, 58]. Studies have shown that the decreased FOXO1 activity reduces the transcription of its target gene PGC1-α in MCs, thereby inhibiting mitochondrial biogenesis and promoting the progression of DKD [30]. Additionally, recent studies have shown that the glucagon-like peptide 1 receptor (GLP-1R) agonist exendin-4 improves DKD by increasing the expression of PGC1-α [59]. The natural polyphenol resveratrol was also found to improve the damage to MCs stimulated by high glucose, acting through the AMPK-SIRT1-PGC1α axis [50].
Lipid metabolism of MCs
Lipid metabolism is severely dysregulated in the early stages of DKD, which has sparked widespread interest. Accumulating evidence has demonstrated the dysfunction of lipid metabolism during DKD. These alterations include increased fatty acid intake, reduced fatty acid utilization, cholesterol accumulation, aberrant lipoprotein levels, and ectopic lipid deposition [60-63]. Mitochondrial dysfunction and dysregulated lipid metabolism form a self-perpetuating vicious cycle in DKD. Excessive FFAs induce mitochondrial reactive ROS overproduction and electron transport chain (ETC) impairment via β-oxidation, whereas dysfunctional mitochondria further suppress lipid catabolism due to insufficient ATP generation, exacerbating intracellular lipid accumulation.
Cluster of differentiation 36 (CD36) and FA-binding protein (FABP) increase the absorption of fatty acids, which are subsequently delivered to mitochondria by carnitine palmitoyltransferase (CPT), a rate-limiting enzyme for FA oxidation [64, 65]. The misregulation of fatty acid utilization is well-acknowledged as a primary cause of diabetic kidney injury [66]. The lipid metabolism of human MCs exposed to hyperglycemia is altered through the combined activation of inflammatory and proliferative pathways [67]. According to recent studies, fatty acid oxidation is reduced in DKD MCs and that genetic deletion of ROCK1 leads to the restoration of fatty acid oxidation and mitochondrial function, indicating the detrimental role of deficient fatty acid oxidation in MC damage in DKD [66].
Additionally, several free fatty acids (FFAs) and phospholipids are elevated in DKD and are commonly acknowledged as risk factors of the disease [68]. Palmitate, a common saturated fatty acid, caused considerable lipid accumulation, ROS generation, oxidative stress, and fibrosis in human MCs [69]. One of the main potential mechanisms is associated with connected to the FFA transporter protein CD36. By facilitating lipid deposition, CD36 significantly increases transient receptor potential canonical channel 6 (TRPC6)-induced activation of nuclear factor of activated T cell 2 (NFAT2), which is crucial for renal fibrosis. In contrast, lysophosphatidic acid (LPA) causes fibrosis in DKD MCs through increasing the expression of carbohydrate-responsive element-binding protein (ChREBP), which is linked to glycolysis and adipogenic metabolism [70]. Mechanistically, the binding of LPA to its receptor causes ROS production in cells, upregulates the E3 ubiquitin ligase Traf4, and promotes the ubiquitination of the HECT-type E3 ubiquitin ligase Smurf2, which inhibits ChREBP ubiquitination and increases its expression [70]. However, Yao et al. reported that caffeoylisocitric acid activated Nrf2 and inhibited MAPK signaling, which is widely recognized as a significant mediator of oxidative stress, ultimately leading to attenuation of high-glucose-induced oxidative stress in MCs and extracellular matrix accumulation [67].
Research indicates that renal accumulation of cholesterol is linked to an increased risk of DKD [71, 72]. Abnormalities in intracellular cholesterol synthesis, uptake, and export cause total cholesterol accumulation in DKD kidneys. DKD MCs present the downregulation of cholesterol transport proteins such as adenosine triphosphate binding cassette transporter A1 (ABCA1), G1 (ABCG1), and type I scavenger receptor class B (SR-BI) [73]. In addition, liver X receptor (LXR) agonists, which play crucial roles in the activation of cholesterol transport proteins, ameliorated renal cholesterol accumulation and inflammatory outbreaks via the delivery of synthetic high-density lipoprotein (sHDL) nanodiscs to MCs [74]. Apolipoprotein C3 (ApoC3) is a triglyceride regulator. Huan et al. [75] found that ApoC3 exacerbates renal inflammation through activation of the TLR2/NF-κB pathway, leading to the initial development of DKD, and that this effect is independent of the triglyceride-regulating effects of ApoC3. The NF-κB pathway is well known as an inflammatory signaling pathway, and a recent article reported that the accumulation of the inflammatory factor high-mobility group box 1 (Hmgb1) in MCs exacerbated DKD progression by accelerating the activation of the NF-κB signaling pathway through binding to IκBα [76]. Future research should focus on the regulatory mechanisms of lipid metabolism in MCs in order to identify and create more promising DKD therapeutics.
Epigenetics in MCs
Epigenetic modifications are heritable alterations in cellular phenotype that primarily include DNA methylation, histone modifications, and noncoding RNAs and are unaffected by DNA sequence changes. The amount of information demonstrating that epigenetic pathways exert crucial roles in the pathogenesis of diseases, including cancer, liver fibrosis, and osteoarthritis, has gained substantial attention [77-79]. However, there is still remains much to explore regarding epigenetic regulation that is needed in the field of kidney diseases, particularly DKD. Due to considerable research and advancements in epigenetic techniques, it is commonly recognized that abnormal gene expression caused by epigenetic modifications is commonly recognized to be associated with MC damage and the consequent loss of renal function in DKD. In the following paragraphs, we will discuss how epigenetic alterations influence MC inflammation, MC matrix secretion, and renal function in DKD.
DNA methylation
DNA methylation, a thoroughly researched epigenetic regulatory mechanism involving the transfer of methyl groups to the carbon atom at position 5 of cytosine, occurs predominantly in CpG-rich regions known as CpG islands, which are found in regulatory regions such as promoters or enhancers of genes [80]. Compelling evidence suggests that DNA methylation alters the stability, conformation, and chromatin structure of DNA as well as how DNA interacts with proteins to regulate the expression of specific genes [81]. Notably, it is difficult for heavily methylated promoter-area CpG islands to bind to transcription factors, resulting in transcriptional arrest; hence, DNA methylation is commonly regarded as a gene silencing mechanism. DNA methylation is strongly associated with DKD. Previous research has shown that C/EBP homologous protein (CHOP) promotes high-glucose-induced apoptosis in MCs [82]. Recent research has demonstrated that in DKD, decreased expression of the E3 ligase TRIM13, also known as RFP2, prevents ubiquitinated degradation of CHOP, which promotes mesangial collagen formation. Mechanistically, reduced TRIM13 expression in DKD is largely due to DNA methylation, since TRIM13 was considerably upregulated in MCs treated with a DNA methyltransferase inhibitor (5-Aza-CdR) [83]. It is well known that DNA methylation is a reversible process, and high methylation levels catalyzed by DNA methyltransferases (DNMTs) can undergo demethylation in specific cellular environments or diseased states, which makes DNA methylation an extremely promising therapeutic strategy [84-86].
Histone modifications
Large molecules of DNA and histone complexes make up the nucleosome, the basic structural unit of chromatin [87]. Enzymes can modify the free amino terminus of histones in a variety of ways, including acetylation, methylation, phosphorylation, and ubiquitination [88]. These modifications can alter chromatin function, such as by altering the affinity of histones for DNA double strands and affecting the binding of transcription factors to gene promoters, but do not change the DNA sequence [89]. Histone methylation is the addition of methyl groups to histone tails by histone methyltransferases (HMTs), typically at arginine or lysine residues. Histone methylation, like DNA methylation, is a dynamically reversible modification process because histone demethylases remove methyl groups from amino acid residues, making histone methylation an important regulator of gene expression. The degree of histone methylation and the location of modification affect whether gene transcription is inhibited or enabled. For example, histone H3 lysine 9 (H3K9) and H3K27 methylation are related to gene silencing, whereas H3K4 methylation is connected with gene activation, which greatly increases the complexity of gene expression regulation. Enhancer of zeste homolog-2 (EZH2), a histone methyltransferase that is overexpressed in the cortex of diabetic mouse kdineys and was able to methylates H3K27, resulting in the transcriptional inhibition of target genes [90]. Das et al. [91] discovered that the high-glucose-induced increase in EZH2 expression promoted MC hypertrophy and matrix expansion, and mechanistic studies revealed that this may be because EZH2 promotes lysine-27 trimethylation of histone H3 (H3K27Me3), which induces downregulation of the expression of deptor, a negative regulator of the mTOR complex, and consequently activates the mTORC1 and mTORC2 activities. In contrast, another study revealed that EZH2 expression was downregulated in MCs exposed to high glucose, whereas overexpression of O-linked N-acetylglucosaminyltransferase (OGT) increased EZH2 glycosylation and maintained EZH2 stability to limit DKD progression [92]. The differences in the results could be because of the use of different animal strains and cell lines. To summarize, the role of EZH2 in DKD remains contentious and will require further investigation in the future.
Histone acetyltransferase (HAT) and histone deacetylase (HDAC) regulate histone acetylation, which mostly takes place at the lysine residues of H3 and H4. Acetylation can influence gene transcription by changing the histone charge and interacting with proteins. In general, DNA is negatively charged, but histones are positively charged, causing DNA and histones to bind closely. HAT adds acetyl groups to lysine residues, neutralizing the positive charge of histones, resulting in weaker connections with DNA and promoting transcription [93]. Thus, it is widely acknowledged that histone acetylation is related to gene expression, whereas deacetylation is involved in the suppression of gene transcription [94]. Histone acetylation is strongly linked to the development of DKD. Vega et al. [95] discovered that lysine acetylation facilitated high-glucose-induced fibronectin (FN) assembly in MCs, as FN assembly increased considerably in MCs exposed to HDAC. However, other forms of histone modifications, such as ubiquitination and phosphorylation, have received less attention in DKD MCs. The prospective significance of histone modifications in DKD MCs is not fully understood.
Regulation of noncoding RNAs
Noncoding RNAs, or RNAs that cannot be translated into proteins, such as microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs), have been identified as epigenetic regulators and are involved in controlling a variety of biological processes in the development of DKD, including mesangial proliferation, fibrosis, and matrix secretion. The miRNAs are small, highly conserved RNAs composed of 18-25 nucleotides that regulate gene expression by binding to the untranslated regions (UTRs) of target mRNAs, resulting in RNA-induced silencing complexes (RISCs) that inhibit the translation of or degrade the target mRNAs. Some miRNA dysregulation is involved in the pathophysiological mechanisms of DKD. A transcriptomics study revealed that miR-15b-5p is responsible for apoptosis in mouse MCs, which promotes DKD progression and renal function deterioration. Furthermore, urinary levels of miR-15b-5p have a negative correlation with the estimated glomerular filtration rate (eGFR) and a positive correlation with the urinary albumin/creatinine ratio (uACR), indicating that it could be used as a predictor of DKD progression [96]. Chen et al. [97] reported that miR-216a-5p was highly expressed in HG-treated MCs and promoted cell apoptosis. miR-21 is involved in renal fibrogenesis [98-100]. Compared with healthy individuals, diabetic patients have greater concentrations of miR-21 in their blood and urine. A study revealed that miR-21 expression was considerably increased in STZ-induced DKD, which promoted podocyte motility and MC hypertrophy while also increasing the inflammatory response and fibrosis gene expression [101]. miR-21 overexpression contributed to podocyte dedifferentiation and MC activation through the Wnt/β-catenin and TGF-β1/Smad3 pathways [102]. Notably, the inhibition of several miRNAs also promotes fibrosis. For example, TGF-β1 inhibits miR-29 expression, leading to increased collagen expression and kidney fibrosis [103]. These findings suggest that targeting miRNAs with pharmaceuticals could represent a unique and promising therapeutic approach for slowing the progression and consequences of DKD.
LncRNAs are noncoding RNAs that include more than 200 nucleotides. The existing evidence suggests that dysregulation of lncRNAs is linked to the development of a range of disorders, including DKD [104-106]. On the one hand, lncRNAs can act through a variety of signaling mechanisms. For example, Chen et al. [107] discovered that the expression of the lncRNA SOX2OT was reduced in STZ-induced diabetic nephropathy mice and high-glucose-treated MCs, resulting in autophagy suppression, MC proliferation, and fibrosis-related gene expression via the Akt/mTOR pathway. Additionally, DANCR, a new lncRNA, inhibited TGF-β/Smad signaling in human renal MCs (HRMCs) [108]. On the other hand, lncRNAs can act as miRNA sponges and indirectly affect protein expression. The lncRNA MIAT played a role in DKD progression by modulating the miR-147a/E2F3 axis [109]. XIST, an lncRNA X inactivation-specific transcript, increases MC proliferation in DKD by inhibiting miR-423-5p and increasing HMGA2 expression [110]. Silencing the lncRNA SNHG14 decreases MC proliferation and ECM accumulation by increasing the level of miR-424-5p and decreasing the level of SOX4 [111]. In contrast, Tu et al. [112] found that LINC01232 attenuated MC proliferation and fibrosis in DKD, which was achieved by attenuating MSH2 inhibition by miR-1250-3p.
The circRNAs, which are noncoding RNAs with a circular structure, influence gene expression through the mechanism of competing endogenous RNAs (ceRNAs) and play significant biological roles in a variety of disorders, similar to lncRNAs. Several investigations have confirmed that circRNAs serve as molecular sponges for miRNAs in DKD [113, 114]. For example, the circRNA DLGAP4 sponges miR-143 and regulates the ERBB3/NF-κB/MMP-2 axis, which in turn leads to the proliferation and fibrosis of MCs [115]. Taken together, these findings indicate that noncoding RNAs play a variety of roles in DKD and are critical epigenetic regulatory molecules for early detection, targeted therapy, and prevention (Table 1) (Figure 1).
DKD related non-coding RNA
| non-coding RNA changes | Mediators or Related Pathways | Effects | Reference(s) |
|---|---|---|---|
| miR-15b-5p upregulation | Targeting BCL-2 | Promoting DKD progression and deterioration of renal function | [96] |
| miR-216a-5p upregulation | Inhibiting MAPK pathway | Promoting MC apoptosis | [97] |
| MiR-21 upregulation | Activating the TGF-β1/Smad3 pathway | Promoting MC hypertrophy | [101, 102] |
| miR-29 downregulation | Targeting ECM components | Promoting collagen expression and kidney fibrosis | [103] |
| LncRNA SOX2OT downregulation | Activating Akt/mTOR pathway | Promoting MC proliferation, and fibrosis gene expression | [107] |
| LncRNA DANCR downregulation | Inhibiting TGF-β/Smad signaling | Inducing extracellular matrix accumulation in HMCs | [108] |
| lncRNA MIAT upregulation | Modulating the miR-147a/E2F3 axis | Inducing mesangial cell proliferation and fibrosis | [109] |
| lncRNA XIST upregulation | Inhibiting miR-423-5p | Increasing MC proliferation in DKD | [110] |
| lncRNA SNHG14 upregulation | Inhibiting miR-424-5p | Promoting mesangial cell proliferation and ECM accumulation | [111] |
| LINC01232 upregulation | Attenuating miR-1250-3p | Attenuating MCs proliferation and fibrosis in DKD | [112] |
| CircRNA DLGAP4 upregulation | Inhibiting miR-143 | Leading to proliferation and fibrosis of MCs | [115] |
Mechanisms of MCs damage in DKD. During DKD, MCs undergo a range of pathological changes, including oxidative stress, mitochondrial dysfunction, abnormal lipid metabolism, and epigenetic dysregulation. As shown, Fyn expression was upregulated in DKD MCs, which inhibited the expression of antioxidant genes such as catalase through the Sirt1/FoxO3a pathway. In addition, Nrf2 was downregulated in HG-induced MCs, leading to ROS accumulation and inducing MCs injury. Dysfunctional mitochondria contribute to the progression of DKD. In DKD, impaired mitophagy, abnormal mitochondrial dynamics, and decreased mitochondrial production all contribute to MC destruction. It is worth noting that lipid metabolism undergoes substantial dysregulation in the early stages of DKD. Among other ways, poor fatty acid utilisation causes the production of lipid droplets, which has a negative impact on MCs. Finally, epigenetic alterations such as DNA methylation, histone modifications and non-coding RNA modifications are involved in MCs damage and subsequent dysfunction in DKD.
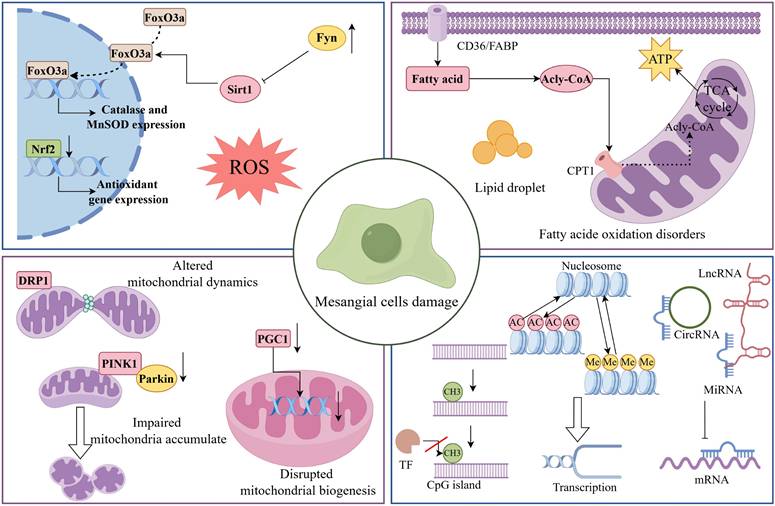
In addition to the characteristics discussed above, mechanical pulling is one of the mechanisms of early injury in MCs. Liu et al. reported that in DKD, glomeruli activate mechanosensitive transcriptional regulators, such as the transcription factor serum response factor (SRF) and coactivators myocardin-related transcription factor A and B (MRTFA/B) and yes-associated protein 1 (YAP1 or YAP) [116]. MRTF-SRF was specifically activated in DKD MCs. Furthermore, a recent study revealed that YAP and TAZ were activated in MCs cultivated with high glucose, which significantly increased extracellular matrix deposition in hyperproliferating MCs [117]. Activated YAP/TAZ bind to and increase the transcriptional activity of N-Myc proteins, which eventually leads to MC damage and DKD development. A groundbreaking study revealed a novel regulatory pathway underlying MC hyperproliferation in DKD. The research demonstrated that GABP is specifically overexpressed in DKD MCs, where it directly binds to the GLI1 gene promoter region to increase its transcription, thereby driving aberrant MC proliferation and ECM accumulation [118]. This discovery provides fresh insights into the mechanisms of renal fibrosis.
Crosstalk between MCs and other cells in DKD
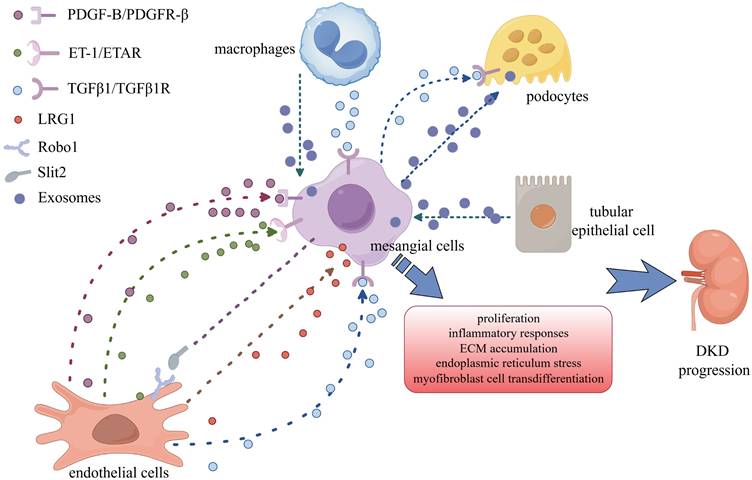
In recent years, there has been an endless stream of research on the role of cellular interactions in the occurrence and development of DKD. MCs possess secretory functions and can secrete large amounts of growth factors and cytokines in response to stimuli such as hyperglycemia and hemodynamic changes. These secretions act on surrounding cells, including podocytes and endothelial cells, through a paracrine mechanism and can also diffuse into the renal tubulointerstitium through the bulbotubular junctions, causing tubulointerstitial damage. In addition, MCs themselves can also receive signals transmitted by cytokines secreted by other cells. The unique positioning and physiological functions of MCs warrant exploration of their role in cellular interactions.
Crosstalk between MCs and podocytes
TGF-β1
TGF-β is upregulated in DKD and plays a role in matrix protein synthesis and fibrosis [119]. TGF-β1 is a common isoform of TGF-β and a biomarker for type 2 DKD [120]. Therefore, it is particularly important to study the role of TGF-β1 in DKD. Previous studies have reported that the interaction between MCs and podocytes plays an important role in IgA nephropathy [121]. A previous study revealed that HG can cause MCs to secrete exosomes, which was confirmed by detection of the exosome markers CD63 and TSG101 [122]. Coincubation of these exosomes with podocytes resulted in reduced expression of the podocyte-specific proteins nephrin, podocin, and WT-1 [122]. The level of TGF-β1 in exosomes from MCs treated with HG significantly increased and induced podocyte apoptosis through the TGFβ1/PI3K-AKT signaling pathway [122]. Inhibition of TGF-β1 in MCs significantly improved the damage to podocytes caused by exosomes from high-glucose-treated MCs [122].
Crosstalk between MCs and endothelial cells
Platelet-derived growth factor B (PDGF-B)/PDGFRβ
MCs are in direct contact with endothelial cells. MCs have contractile properties and maintain the structure of capillaries through contraction. MCs carry PDGFRβ during development [123]. During the development of human glomeruli, glomerular MCs are formed in the middle and late stages, and PDGF-B expressed by endothelial cells is key to the formation of MCs. It has been reported that mice lacking PDGF-B or PDGFRβ genes lack MCs in their glomeruli, indicating that the development and formation of glomerular MCs depend on PDGF-B and its receptor PDGFRβ [124, 125]. Surprisingly, in mice lacking these genes, the glomerular capillary network collapsed and was replaced by a single dilated capillary loop [124, 125]. Endothelial cells promote the development of MCs by secreting PDGF-B, which in turn supports and maintains the structure of the capillary network. Another study showed that the PDGF-B/PDGFRβ signaling pathway is activated in DKD, promoting the proliferation of MCs and aggravating the progression of DKD [17, 126]. Inhibiting PDGF-B improves STZ-induced MC dysfunction in DKD [127].
Integrin αvβ8 and its ligand transforming growth factor β (TGF-β)
Integrin αvβ8 is abundantly expressed in the kidney and is located mainly in MCs. According to research reports, mice lacking integrin αvβ8 develop proteinuria, azotemia, and endothelial cell apoptosis [128]. Latent TGF-β is the main ligand of integrin αvβ8. Integrin αvβ8 expressed by MCs binds and sequesters latent TGF-β, reduces the activation and release of TGF-β, and inhibits TGF-β-mediated damage in endothelial cells [128]. These findings suggest that MCs maintain the stability of glomerular endothelial cells by regulating the release of TGF-β through integrin αvβ8. In addition, another study revealed that exosomes from endothelial cells treated with high glucose were enriched in TGF-β1 compared with exosomes from endothelial cells treated with normal glucose [129]. Treating MCs with these exosomes activated TGFβ1/Smad3 signaling, mediating MC proliferation and increasing matrix protein production [130]. These studies provide a basis for the interaction between MCs and endothelial cells to maintain glomerular structure and function.
Endothelin-1 (ET-1)
ET-1 is one of the most potent vasoconstrictor factors. Earlier studies revealed that coculture of HMCs and human umbilical vein endothelial cells (HUVECs) significantly reduced the expression of ET-1 and endothelin-converting enzyme (ECE-1) compared with culturing HUVECs alone [131]. Studies have shown that endothelial cells treating with high glucose increases the secretion of ET-1, which activates the RhoA/ROCK signaling pathway by binding to the endothelin A receptor (ETAR) on MCs, thereby promoting MC proliferation and ECM accumulation [132-134]. Endothelin B receptor (ETBR)-deficient DKD mice exhibit aggravated renal injury [132]. Studies have shown that ET-1 combines with ETBR to inhibit the activity of NF-κB and reduce the secretion of ET-1 [132].
Leucine-rich α2-glycoprotein 1 (LRG1)
LRG1 was formerly assumed to be involved in ocular neovascularization, but more recent research has revealed links to psoriasis, rectal cancer, atherosclerosis, and diabetic nephropathy [135-138]. Previous research has indicated that LRG1 released by renal tubular epithelial cells enhances TGF-β-mediated Smad3 activation in renal fibroblasts, suggesting that LRG1 can mediate TGF-β signaling in a paracrine manner [139]. In 2023, a cohort study by Liu et al. [137] reported that urinary LRG1 was related to a rapid deterioration in renal function and the development of massive albuminuria, providing evidence that LRG1 may be an essential component driving the progression of DKD. Furthermore, single-cell RNA sequencing (scRNA-seq) research by Tsuruta et al. identified LRG1 as a marker of kidney damage linked to fructose overconsumption in DKD [140]. In DKD, LRG1 expression is increased in glomerular endothelial cells and is associated with glomerulosclerosis. The potential reason for this is that endothelial cells in a high-glucose environment synthesize and release secreted LRG1, which diffuses into the mesangial area and triggers TGF-β signaling [140, 141]. LGR1 may play a role in overactivating TGF-β signaling in DKD glomeruli, making it an attractive target for preventing and treating TGF-β-related renal disorders.
SLIT2/ROBO1
Slit2, a secreted protein, was initially discovered to play a functional role in axon rejection in the central nervous system. Recent evidence suggests that it can promote angiogenesis by binding to the Robo1 receptor [142, 143]. Notably, an intriguing study revealed that aberrant intraglomerular angiogenesis may be linked to Slit2/Robo1 signaling in the early stages of DKD. Slit2 was shown to be upregulated in HRMCs treated with HG. Additionally, Slit2/Robo1 signaling was triggered in HRGECs treated with HG-HRMC-CM. These findings indicate that Slit2 may facilitate crosstalk between HRMCs and HRGECs in a high-glucose environment and that the function of Slit2 is dependent on the receptor Robo1, as evidenced by the suppression of PI3K/Akt signaling activation in HG-HRMC-CM-treated HRGECs following Robo1 silencing [144].
VEGF-B/VEGFR1
A member of the VEGF family, vascular endothelial growth factor B (VEGF-B) has well-established metabolic regulatory roles [145]. Current evidence has demonstrated that systemic inhibition of VEGF-B signaling significantly reduces glomerular lipid accumulation and ameliorates diabetic nephropathy phenotypes in murine models [146]. However, the cell-specific expression patterns of VEGF-B in renal tissue, its precise molecular mechanisms in regulating renal lipid metabolism, and its impact on renal function in diabetic patients remain to be elucidated. Recent studies have elucidated a dual mechanistic role of VEGF-B in DKD: (1) MC-derived VEGF-B activates endothelial VEGFR1 through paracrine signaling, promoting fatty acid uptake and renal injury; (2) adipose tissue VEGF-B enhances lipolysis via hormone-sensitive lipase (HSL), increasing the number of circulating fatty acids that activate mesangial VEGF-B signaling, thereby establishing a pathological 'adipose-circulation-kidney' axis [147].
Crosstalk between MCs and tubular epithelial cells
miR-92a-1-5p
Renal tubular injury is also a pathological characteristic of DKD. Proximal tubular epithelial cell (PTEC) injury occurs in the early stage of DKD [148]. The interaction between glomeruli and tubules in DKD has been demonstrated multiple times [149, 150]. However, the interaction between PTECs and MCs needs to be further studied. One study revealed that supernatants or exosomes derived from HK-2 cells treated with HG can cause endoplasmic reticulum stress and myofibroblast cell transdifferentiation (MFT) in MCs [151]. Using RNA sequencing to analyze the miRNA profile of PTECs from normal individuals and type 2 DKD patients, we found that the level of miR-92a-1-5p was significantly increased in PTECs from DKD patients [151]. Moreover, the mimic of miR-92a-1-5p significantly induced the MFT of MCs. A potential target of miR-92a-1-5p may be RCN3 because HG-cultured HK-2-derived exosomes reduce the protein expression of RCN3 in MC, and significantly promote the MFT and endoplasmic reticulum stress in MCs when RCN3 is deleted [151]. The level of miR-92a-1-5p in the urine of DKD patients is positively correlated with the glomerular filtration rate and urinary protein level, which suggests that miR-92a-1-5p may be used as a biomarker of diabetic kidney damage and provide guidance for the clinical diagnosis and treatment of DKD patients.
Crosstalk between MCs and macrophages
TGF-β1
Hyperglycemia activates resident renal macrophages; leads to the production of many chemokines and inflammatory factors; induces the recruitment of circulating monocytes/macrophages and renal cell damage; and promotes renal inflammation, fibrosis, and apoptosis [152]. Coculture of macrophages and MCs under high-glucose conditions promotes the secretion of inflammatory factors and matrix proteins [152]. When TGF-β-activated kinase-1 (TAK1) inhibitors are used, the secretion of inflammatory factors and extracellular matrix components is reduced by inhibiting the nuclear translocation of NF-κB p65 [153]. Macrophages treated with high glucose secrete large amounts of exosomes, which act on MCs and promote MC proliferation and activation. Mechanistically, high glucose induces macrophages to secrete TGF-β1, which activates MCs to produce extracellular matrix through the TGF-β1/Smad3 signaling pathway, promoting the progression of DKD [154] (Table 2) (Figure 2).
Crosstalk between MCs and other cells in DKD
| Cell Types | Mediators or Related Pathways | Effects | Reference(s) |
|---|---|---|---|
| GECs → MCs | PDGF-B/PDGFR-β signaling pathways | Promoting the proliferation of MCs and aggravating the progression of DKD | [127] |
| GECs → MCs | TGFβ1/Smad3 signaling | Mediating MCs proliferation and increased matrix protein production | [130] |
| MCs→ GECs | Integrin αvβ8/TGF-β | Inhibiting TGF-β-mediated damage in endothelial cells | [128] |
| GECs → MCs | ET-1/ETAR | Promoting MCs proliferation and ECM accumulation | [132] |
| GECs → MCs | LRG1 | Overactivating TGF-β and driving the progression of DKD. | [141] |
| MCs→ GECs | SLIT2/ROBO1 | Promoting GECs aberrant intraglomerular angiogenesis | [144] |
| MCs→ podocytes | TGFβ1/PI3K-AKT signaling pathway | Induced podocytes apoptosis | [122] |
| MCs→ podocytes | Endoplasmic reticulum (ER)-associated degradation (ERAD) | Inhibiting of ERAD-related proteins, inducing podocyte apoptosis, and promoting the progression of DKD | [198] |
| PTECs → MCs | miR-92a-1-5p | Promoting endoplasmic reticulum stress and myofibroblast cell transdifferentiation (MFT) in MCs | [151] |
| Macrophages → MCs | TGF-β1/Smad3 signaling pathway | Promoting the proliferation of MCs and the progression of DKD | [154] |
| Macrophages → MCs | NLRP3 inflammasome | Activating of NLRP3 inflammasome and inhibiting MC autophagy | [158] |
Crosstalk between MCs and other cells in DKD. MCs communicate with podocytes, endothelial cells, renal tubular epithelial cells, and macrophages via a variety of signalling chemicals to accelerate DKD progression.
NLRP3 inflammasome
Activation of the NOD-like receptor 3 (NLRP3) inflammasome is involved in various kidney diseases through the regulation of proinflammatory cytokines such as IL-1β [155]. For example, inhibition of the NLRP3 inflammasome can improve renal injury in DKD model mice [156]. HG induces the activation of the NLRP3 inflammasome and NF-κB in HMCs, promotes the inflammatory response, and leads to the progression of DKD [157]. Recent studies have not only shown that HG induces the M1 polarization of macrophages and promotes their secretion of exosomes but also that MCs internalize these exosomes [158]. Exosomes from HG-treated macrophages promoted MC inflammatory responses and NLRP3 inflammasome activation and inhibited MC autophagy [158]. Exosomes derived from HG-treated macrophages damage the kidney and induce mesangial hyperplasia in vivo [158].
Therapeutic implications of MCs in DKD
Pharmacological targeting of oxidative stress and inflammation in MCs
Previously, DKD treatment consisted primarily of lifestyle management, glycemic control, blood pressure control, proteinuria reduction, and the management of associated complications. Recent research has revealed several new pharmacological classes that have been shown to enhance renal outcomes in patients with type 2 diabetes, particularly sodium‒glucose cotransporter protein-2 inhibitors, mineralocorticoid receptor antagonists (MRAs), and selective endothelin receptor antagonists. Nonetheless, a considerable proportion of patients with diabetes inevitably show progression to DKD, and these therapeutic measures have provided modest support, encouraging exploration of the therapeutic potential of targeting MCs.
Oxidative stress and inflammation have emerged as critical factors in renal MC injury during DKD, coordinating their actions through multiple pathways, particularly the NF-κB and Nrf2 pathways. In a mouse model of DKD, activation of NF-κB promoted immune cell infiltration, the MC inflammatory response and fibrosis, and accelerated renal fibrosis [159]. In OVE26 mice, Zhang et al. [160] found that pharmacological inhibition of PLK1 by the polo-like kinase 1 (PLK1) inhibitor BI-2536 lessened MC proliferation, alleviated proteinuria, and safeguarded renal function via NF-κB. Wang et al. [75] recently observed that ApoC3 can increase the phosphorylation of NF-κB and activate downstream inflammatory factors, including TNF-α, VCAM-1, and MCP-1, via TLR2 in MCs from T1DN mice, thereby exacerbating STZ-induced renal injury. Regrettably, ApoC3 deficiency did not seem to have a significant protective effect in STZ-induced DN mice, probably because there are already enough ligands for TLR2 in DN to exert an effect. In type 1 diabetic (T1DM) mice, dihydromyricetin (DHM), a flavonoid, inhibits NF-κB activation by binding to sphingosine kinase-1 (SphK1) and reducing its activity and protein expression, ultimately mitigating HG-induced expression of fibrosis and inflammatory molecules in GMCs [161]. Zheng et al. identified another flavonoid, wogonin, that ameliorates renal inflammation and fibrosis by blocking the NF-κB and TGF-β1/Smad3 signaling pathways in MCs in DN [162]. In addition, a cell-permeable peptide containing the kappa B kinase γ inhibitor /NF-κB essential regulatory factor-binding domain has been proven to ameliorate proteinuria and renal lesions [163]. In addition, several compounds, such as icariin, eucommia lignans, β-caryophyllene and ampelopsin, can decrease HG-induced oxidative stress in MCs by activating the Nrf2/HO-1 pathway, hence slowing the course of experimental diabetic nephropathy and further emphasizing the importance of Nrf2 in the treatment of DKD [164-167]. MCC950, an effective NLRP3 small molecule inhibitor, protects renal function by attenuating fibrosis in the HBZY-1 rat MCs line via inhibition of the NLPR3/caspase-1/IL-1β pathway [168]. These findings support the notion that addressing the inflammatory response of MCs with medicines is an exciting avenue for the treatment of renal disease (Table 3).
Pharmacological moderators of MCs in DKD
| Mediators or Agents | Mechanisms | Effects | Reference(s) |
|---|---|---|---|
| BI-2536 | Inhibiting PLK1 and NF-κB | Lessening MCs proliferation, alleviating proteinuria, and safeguarding renal function | [160] |
| ApoC3 | Increasing phosphorylation of NF-κB | Exacerbating STZ-induced renal injury | [75] |
| Dihydromyricetin | Inhibiting NF-κB activation by binding to sphingosine kinase-1 (SphK1) | Mitigating HG-induced expression of fibrosis and inflammatory molecules in GMCs | [161] |
| Wogonin | Blocking the TGF-β1/Smad3 signalling | Ameliorating renal inflammation and fibrosis | [162] |
| Cell-permeable peptide | Targeting NF-κB | Easing proteinuria and renal lesions | [163] |
| Icariin | Mediating p62-Dependent Keap1 Degradation and Nrf2 Activation | Decreasing extracellular matrix accumulation in MCs | [166] |
| Eucommia lignans | Mediating the AR/Nrf2/HO-1/AMPK axis | Alleviating the progression of diabetic nephropathy | [165] |
| β-Caryophyllene | Activating the Nrf2/HO-1 pathway | Decreasing HG-induced oxidative stress in MCs | [167] |
| Ampelopsin | Activating the Nrf2/HO-1 pathway | Inhibiting HG-induced extracellular matrix accumulation and oxidative stress in MCs | [164] |
| MCC950 | Inhibiting of the NLPR3/caspase-1/IL-1β pathway | Protecting renal function by attenuating rat MCs line HBZY-1 fibrosis | [168] |
| Epiberberine | Disrupting the Agt-TGF-β/Smad2 pathway | Limiting MCs proliferation and hypertrophy, and ultimately ameliorating kidney damage in db/db mice | [169] |
| Osthole | Reducing TGF-β1/Smads signaling pathway activation | Inhibiting HBZY-1 hypertrophy, ROS production and ECM deposition | [170] |
| EW-7197 | Inhibiting of TGF-β signaling | Attenuating fibrosis and inflammation in MCs | [171] |
| Liposomal puerarin | Reducing HG-induced TGF-β expression and Smad 2/3 nuclear translocation in RMCs | Protecting mesangial cells | [172] |
| Pyrrole-imidazole (PI) polyamides | Targeting TGF-β1 | Alleviating proteinuria and glomerular pathological changes | [173] |
| USF1 PI polyamide | Restricting the expression of TGF-β1 | Minimizing impaired renal function | [174] |
| Viral mimetic nanoparticles (NPs) encapsulated with cinaciguat | Regulating soluble guanylate cyclase (sGC) activity and 3′, 5′-cyclic guanosine monophosphate (cGMP) generation | Inhibiting MC proliferation and matrix accumulation | [176] |
| Resveratrol | Inhibiting PDE4D/PKA signaling pathway | Inhibiting oxidative stress and Drp1-mediated mitochondrial fission in diabetic renal MCs | [199] |
| Tilapia Skin Peptides | Activating the Bnip3/Nix signaling | Enhancing mitochondrial autophagic activity, scavenging damaged mitochondria in GMCs | [177] |
Pharmacological targeting of MC fibrosis
Considering that MC fibrosis and the secretion of large amounts of extracellular matrix components accelerate DKD renal damage, therapeutic strategies aimed at tackling the TGF-β pathway have aroused much attention. An isoquinoline alkaloid, epiberberine (EPI), was identified to ameliorate DKD by Xiao et al. Using protein-protein interaction (PPI) analysis and core gene screening, they found that EPI can dock with angiotensinogen (Agt) and disrupt the Agt-TGF-β/Smad2 pathway, thereby limiting MC proliferation and hypertrophy and ultimately ameliorating kidney damage in db/db mice [169]. Recently, Li et al. [170] have also identified a Chinese herb, osthole, reduces HBZY-1 cell hypertrophy, ROS production, TGF-β1/Smad signaling pathway activation and ECM deposition, hence alleviating pathological renal damage in an STZ/high-fat/high-sucrose diet-induced rat model of type 2 diabetes mellitus. Inhibition of TGF-β signaling with EW-7197 attenuated fibrosis and inflammation in MCs, thus delaying the progression of DKD. Moreover, this intervention also diminished ROS production, ameliorated endoplasmic reticulum stress, and strengthened mouse kidneys against HG-induced damage [171]. Barro et al. [172] synthesized a liposomal dosage form of puerarin to overcome its application limitations due to its water solubility. Liposomal puerarin reduced HG-induced TGF-β expression and Smad 2/3 nuclear translocation in RMCs. However, this impact has been confirmed only in vitro, and more research, including in vivo animal tests, is needed. Pyrrole-imidazole (PI) polyamides, a new biologic medicine, can block transcription by binding to the promoter regions of target genes. PI polyamides that target TGF-β1 have been developed to research chronic kidney disease, renal fibrosis, and DKD. In STZ-induced Wistar rats, PI polyamide treatment targeting TGF-β1 significantly alleviated proteinuria and pathological glomerular changes [173]. In another study, HG promoted the nuclear translocation of transcription factor upstream stimulatory factor 1 (USF1), while USF1 PI polyamide significantly restricted the expression of TGF-β1 mRNA and protein, markedly reduced the expression of osteopontin, and hindered the conversion of MCs from the contractile to the synthetic phenotypes in the HG milieu, minimizing impaired renal function [174]. Interestingly, viral mimetic nanoparticles (NPs) encapsulated with cinaciguat preferentially target MCs to regulate soluble guanylate cyclase (sGC) activity and 3′,5′-cyclic guanosine monophosphate (cGMP) generation. In injured kidneys, poor sGC function has been shown to promote MC proliferation and matrix accumulation [175, 176]. Identifying compounds that improve mitochondrial dynamics as potential DKD treatments has been an emerging topic of interest. Several agents targeting mitochondrial function have been explored. For example, resveratrol inhibited oxidative stress and Drp1-mediated mitochondrial fission in diabetic renal MCs through the PDE4D/PKA signaling pathway, improving mitochondrial function [49]. Furthermore, Lin et al. reported that tilapia skin peptides (TSP), a small-molecule mixture derived from tilapia skin, increases mitochondrial autophagic activity through the activation of Bnip3/Nix signaling, which lead to a reduction in mitochondrial ROS accumulation by scavenging damaged mitochondria and ultimately attenuates HG-induced fibrosis in GMCs [177].
Although the development of targeted anti-inflammatory and antifibrotic drugs remains in its early stages, clinical studies have demonstrated the protective effects of combined antioxidant therapy (N-acetylcysteine + taurine) in DKD. The trial data revealed that, compared with the placebo group, the treatment group presented an 18.6% reduction in the uACR, a 34.09% decrease in microalbuminuria, and a significantly greater improvement in the serum cystatin C level, suggesting that this regimen may delay DKD progression through multiple mechanisms [178].
Certain clinical medications targeting MCs
Antihyperglycemic agents have been employed in the management of patients with T2DM and its complications, including DKD, for the past several decades. Metformin, the recommended glycemic control medicine, is widely utilized in clinical practice because of its ability to reduce hepatic gluconeogenesis and intestinal glucose absorption. At the mechanistic level, metformin delayed renal progression in STZ/high-fat diet-induced type 2 diabetic rats by decreasing oxidative stress in HG-exposed RMCs via the AMPK/SIRT1-FoxO1 pathway, increasing autophagy and alleviating hyperproliferation [179]. Additionally, metformin partially reversed palmitate-mediated lipotoxicity-induced apoptosis in rat MCs. The underlying mechanism is likely that metformin stimulates GLP-1R expression, as agonists of GLP-1R alleviate the destruction of MCs under HG conditions; conversely, GLP-1R knockdown accelerates the apoptosis of MCs [180, 181]. A recent randomized controlled trial demonstrated that semaglutide treatment significantly reduced the risk of the primary composite kidney outcome (including kidney failure [defined as the need for chronic dialysis or kidney transplantation or a sustained eGFR <15 mL/min/1.73 m2], ≥50% decline in the eGFR, or kidney/cardiovascular-related death) by 24% (HR 0.76, 95% CI 0.66-0.88; P=0.0003) in patients with type 2 diabetes and CKD, with 331 versus 410 first events occurring in the semaglutide and placebo groups, respectively [182]. The abovementioned results not only emphasize the centrality of metformin in the treatment of DKD but also point out the utilization of GLP-1R agonists as an emerging therapeutic strategy in the prevention and treatment of DKD. Li and colleagues reported that GLP-1RA liraglutide treatment contributed to the shielding of MCs from hyperglycemia-mediated mitochondrial apoptosis, which had a beneficial influence on renal outcomes, supporting this assumption [183]. Additionally, in cultured RMCs, liraglutide suppresses HG-stimulated production of fibronectin and collagen by augmenting Wnt/β-catenin signaling, which alleviates glomerular matrix accumulation and renal pathology abnormalities in DKD [184]. Notably, several clinical trials have demonstrated that SGLT2 inhibitors may provide cardiovascular and renal benefits to patients with DKD in the form of a lower estimated GFR, a reduced risk of ESRD, and a lower rate of mortality due to cardiovascular effects [185-187]. Toshinobu et al. recently showed that low dosages of canagliflozin reduced aberrant ROS production in HG-stimulated MCs and renal mesangial dilatation in db/db mice via the PKC/NADPH oxidase pathway, regardless of its glucose-lowering action, indicating that it has the unique ability to target MCs [188]. Earlier studies have shown that SGLT2 inhibitors increase mitochondrial activity in diabetic vascular endothelial cells while decreasing oxidative stress injury, causing a reduction in vascular problems and an improved prognosis in type 2 diabetes patients [189]. SGLT2 inhibitors may help protect against diabetic nephropathy-related cell damage by inhibiting mitochondrial dysfunction. In addition, the DPP-4 inhibitor selegiline has been shown to alleviate DKD by inhibiting TGF-β1/Smad signaling and upregulating heme oxygenase-1 (HO-1) expression in MCs [190, 191]. Initial randomized controlled trials demonstrated that the DPP-4 inhibitor saxagliptin exerts potential renoprotective effects on DKD [192]. However, the recent CARMELINA trial, which enrolled patients with both normal renal function and varying degrees of renal impairment, revealed that while linagliptin did not significantly improve the primary composite cardiovascular and renal endpoint (HR 1.04; 95% CI 0.89-1.22) compared with placebo, it significantly attenuated albuminuria progression [193]. Prolyl hydroxylase structural domain (PHD) inhibitors have emerged as viable therapeutic agents for the treatment of chronic anemia in CKD because they activate HIF, which stimulates erythropoietin synthesis. When diabetic mice were given the PHD inhibitor enarodustat, proteinuria and glomerular damage were significantly reduced. The authors hypothesized that the renoprotective impact of enarodustat may be due to its ability to inhibit CCL2/MCP-1 production in MCs [194]. Hypertension and proteinuria are also risk factors for promoting ESRD in people with type 2 DKD; hence, RAS inhibitors with dual effects of lowering blood pressure and hyperglycemia are a popular first-line treatments for DKD patients. RAS blockers include ACE inhibitors and angiotensin receptor blockers (ARBs). Telmisartan, an angiotensin II type 1 receptor blocker, has been shown to reduce AGE-induced MCP-1 in MCs and govern HG-induced TGF-β1 expression in HRMCs, underscoring the protective effect of RASi in MCs [195, 196]. Furthermore, the SONAR trial, an international, multicenter, randomized controlled study involving 11,087 patients, demonstrated that the selective endothelin-A receptor antagonist atrasentan significantly reduced the risk of renal composite endpoints in patients with diabetes and CKD (HR 0.65; 95% CI 0.49-0.88) [197]. Further research is warranted to determine whether the therapeutic value of the abovementioned medications for MCs may be extended to clinical application (Table 4).
Clinical medications of MCs in DKD
| Mediators or Agents | Mechanisms | Effects | Reference(s) |
|---|---|---|---|
| Metformin | Activating AMPK/SIRT1-FoxO1 pathway; stimulating GLP-1R expression | Enhancing autophagy and slowed down abnormal cell proliferation in high glucose cultured RMCs | [179, 180] |
| Liraglutide | Activating the ERK‑Yap signaling pathway and upregulating Sirt3 expression | Contributing to the shielding of MCs from hyperglycaemia-mediated mitochondrial apoptosis | [183] |
| Liraglutide | Enhancing the Wnt/β-catenin signaling | Suppressing production of extracellular matrix proteins and ameliorating renal injury of diabetic nephropathy | [184] |
| cagliflozin | Inhibiting the PKC/NADPH oxidase pathway | Reducing aberrant ROS production in HG-stimulated MCs and renal mesangial dilatation in db/db mice | [188] |
| Selegiline | Inhibiting TGF-β1/Smad signaling; upregulating heme oxygenase-1 (HO-1) expression | Alleviating DKD | [190, 191] |
| Enarodustat | Inhibiting the CCL2/MCP-1 production | Reducing proteinuria and glomerular damage | [194] |
| Telmisartan | Reducing AGE-induced MCP-1;governing HG-induced TGFβ1 expression | Alleviating DKD | [195, 196] |
Conclusion and perspectives
MCs, a previously “forgotten” intrinsic glomerular cell, are receiving increasing attention due to their indispensable role in the course of DKD. Hyperglycemia in DKD causes damage to MCs via oxidative stress, mitochondrial failure, aberrant lipid metabolism, and epigenetic changes. These pathological changes accelerate the proliferation, hypertrophy, and phenotypic transformation of MCs in DKD, which in turn secrete a large number of cytokines, chemokines, and profibrotic factors, facilitating coordination and cooperation between MCs and other cells to promote DKD progression, ultimately leading to irreversible glomerulosclerosis and loss of renal function. Based on these pathological changes and damage phenotypes, it is clear that MCs will be the subject of extensive investigation in the future.
Although the mechanisms of harm to DKD MCs in DKD are classified into four types above, they are all intimately related and intersect with one another. In the pathological context of DKD, the hyperglycemic state and the progressive accumulation of advanced glycation end products (AGEs) jointly disrupt the redox homeostasis within MCs, leading to excessive generation of ROS and subsequently triggering oxidative stress. The overproduced ROS act as 'molecular scissors' that directly target mitochondria, resulting in an imbalance of mitochondrial dynamics and dysfunctional mitophagy. This severely disrupts the normal energy metabolism and quality control mechanisms of the cell. Moreover, MCs in patients with DKD commonly exhibit significant lipid metabolism dysregulation, characterized by markedly increased fatty acid uptake, excessive triglyceride accumulation, and disrupted cholesterol metabolic pathways. These aberrant lipid metabolites are far from 'inert bystanders'—instead, they drive alterations in epigenetic modifications through diverse molecular mechanisms. Notably, recent studies have revealed that sustained lipotoxic stimulation induces H3K9me3, an epigenetic marker that can be stably inherited during cell division, resulting in a 'metabolic memory' effect. Consequently, even after removal from the lipotoxic environment, cells retain dysregulated metabolic traits, persistently exacerbating DKD progression. These findings underscore the importance of comprehensively understanding the complex pathogenic mechanisms underlying MC injury in DKD. The ability of MCs to respond to innate immunity and stimuli by influencing immune cell recruitment via chemokine production highlights their role in numerous inflammatory responses and immunological-related disorders. Research has shown that MCs have significant phagocytic activity and can remove proteins that pass through the filtration membrane and collagen lost from the basement membrane, promoting basement membrane renewal and filtration barrier homeostasis. Currently, strategies for the prevention and treatment strategies of MCs damage in DKD involve two main pathways: anti-inflammatory and antifibrotic pathways. The most important signaling pathways are the NF-κB signaling and TGF-β pathways, which are activated by various stimuli in DKD, causing the proliferation and phenotypic transformation of MCs and the secretion of chemokines to mediate intercellular communication and the transmission of injury signals. In this review, various pharmacological molecules that act against MC inflammation and fibrosis are summarized, aiming to provide new perspectives for the clinical prevention and treatment of DKD. Furthermore, numerous commonly administered medicines, including the glucose-lowering drug metformin, GLP1R agonists, and SGLT2 inhibitors, have been demonstrated to improve mitochondrial performance in DKD MCs. Interestingly, the protective effect of SGLT2 inhibitors on thylakoid cells in DKD was not dependent on their hypoglycemic activity. This begs the question of whether there are other possible pathways for the positive effects of glucose-lowering medicines on MCs. Furthermore, drug safety concerns warrant equal attention. Although antioxidant therapy aims to mitigate oxidative stress by scavenging excessive ROS, overintervention in oxidative processes may disrupt the intrinsic redox homeostasis within cells, consequently leading to cellular dysfunction and even severe consequences such as increased tumorigenic risk. Therefore, in the development and application of therapies targeting MC injury in DKD, it is imperative to balance therapeutic efficacy with safety and comprehensively evaluate potential adverse effects.
At present, DKD is becoming increasingly common, and it is the leading cause of end-stage renal disease in developed countries. The complex pathogenesis of DKD is not favorable for its prevention and treatment. The main treatments for DKD include blood glucose lowering, proteinuria control, and renal function improvement. However, the incidence and mortality rates of diabetic nephropathy continue to increase. Therefore, in-depth study of the pathogenesis of diabetic nephropathy, the identification of new therapeutic targets, and the development of effective drugs are urgently needed for the current medical community to prevent and treat diabetic nephropathy. Future research is suggested to focus on two key directions: (1) elucidating novel mechanisms regulating MC proliferation, apoptosis, and ECM metabolism to uncover previously unidentified signaling pathways; and (2) utilizing single-cell multiomics technologies to decipher MC heterogeneity and identify critical pathogenic subpopulations, thereby providing new therapeutic targets for precision medicine approaches. In clinical research, optimizing existing therapeutic regimens and conducting novel drug trials are pivotal for improving outcomes in patients with DKD. Current pharmacological interventions targeting MCs, while demonstrating modest efficacy, remain limited in their therapeutic scope. Fundamentally, contemporary approaches to MC modulation persist at a 'broad-spectrum suppression' level—analogous to employing a fire extinguisher to quell an entire building when targeting a single flame. Future investigations must pioneer the development of 'molecular scalpels'—sophisticated modalities enabling single-cell precision targeting, spatiotemporally controlled drug delivery, and artificial intelligence-driven personalized therapeutic strategies to achieve precise regulation of pathogenic MCs.
Abbreviations
DKD: diabetic kidney disease; MC: mesangial cell; ESRD: end-stage renal disease; KF: kidney failure; ROS: oxygen species; GBM: glomerular basement membrane; Nrf2: nuclear factor erythroid 2-related factor; HIF-1α: hypoxia-inducible factor-1α; mROS: mitochondrial ROS; EPO: erythropoietin; Drp1: dynamite-related protein 1; ROCK1: Rho-associated coiled-coil kinase 1; PDE4: phosphodiesterase-4; PKA: protein kinase A; mtDNA: mitochondrial DNA; nDNA: nuclear DNA; AMPK: AMP-activated protein kinase; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1α; NRF1: nuclear respiratory factor 1; GLP-1R: glucagon-like peptide 1 receptor; CD36: cluster of differentiation 36; FABP: FA-binding protein; CPT: carnitine palmitoyltransferase; FFAs: free fatty acids; TRPC6: transient receptor potential canonical channel 6; NFAT2: nuclear factor of activated T cell 2; LPA: lysophosphatidic acid; ChREBP: carbohydrate-responsive element-binding protein; ABCA1: adenosine triphosphate binding cassette transporter A1; ABCG1: adenosine triphosphate binding cassette transporter G1; SR-BI: scavenger receptor class B; LXR: liver X receptor; sHDL: synthetic high-density lipoprotein; ApoC3: apolipoprotein C3; Hmgb1: high-mobility group box 1; CHOP: C/EBP homologous protein; DNMTs: DNA methyltransferases; HMT: histone methyltransferases; EZH2: zeste homolog-2; OGT: O-linked N-acetylglucosaminyltransferase; HAT: histone acetyltransferase; FN: fibronectin; miRNAs: microRNAs; lncRNAs: long non-coding RNAs; circRNAs: circular RNAs; UTRs: untranslated regions; RISCs: RNA-induced silencing complexes; ACR: albumin/creatinine ratio; eGFR: estimated glomerular filtration rate; HRMCs: human renal MCs; ceRNAs: competing endogenous RNAs; MRTFA/B: myocardin-related transcription factor A and B; YAP1: yes-associated protein 1; SRF: serum response factor; PDGF-B: platelet-derived growth factor B; TGF-β: transforming growth factor β; ET-1: endothelin-1; HUVEC: human umbilical vein endothelial cells; ECE-1: endothelin-converting enzyme; ETAR: endothelin A receptor; ETBR: endothelin B receptor; LRG1: leucine-rich α2-glycoprotein 1; scRNAseq: single-cell RNA sequencing; VEGF-B: vascular endothelial growth factor B; PTEC: proximal tubular epithelial cell; MFT: myofibroblast cell transdifferentiation; TAK1: TGF-β activated kinase-1; NLRP3: NOD-like receptor 3; MRA: mineralocorticoid receptor antagonist; PLK1: polo-like kinase 1; T1DM: type 1 diabetic; DHM: dihydromyricetin; SphK1: sphingosine kinase-1; EPI: epiberberine; Agt: angiotensinogen; PI: pyrrole-imidazole; USF1: upstream stimulatory factor 1; NPs: nanoparticles; sGC: soluble guanylate cyclase; cGMP: guanosine monophosphate; TSP: tilapia Skin Peptides; HO-1: oxygenase-1; PHD: prolyl hydroxylase structural domain; ARBs: angiotensin receptor blockers; AGEs: advanced glycation end products.
Acknowledgements
We would like to thank Figdraw for online figure generation.
Funding
This work was supported by the National Natural Science Foundation of China (82400795), the Science/Technology Project of Sichuan province (2024NSFSC1727, 2024NSFSC1501), the China Postdoctoral Science Foundation (2023M742479) and the 1.3.5 project for disciplines of excellence from West China Hospital of Sichuan University (ZYGD23015).
Author contributions
L. M, P. F. and L.F. designed the review and analyzed the references. L.F, L.M and Y.F. wrote and revised the manuscript. L.F, L.M and Q.R. supervised the study and contributed to the editing of the manuscript. All authors helped to interpret the results and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB. et al. Erratum to “IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045” [Diabetes Res. Clin. Pract. 183 (2022) 109119]. Diabetes research and clinical practice. 2023;204:110945
2. de Boer IH. Kidney disease and related findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes care. 2014;37:24-30
3. Afkarian M, Zelnick LR, Hall YN, Heagerty PJ, Tuttle K, Weiss NS. et al. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988-2014. Jama. 2016;316:602-10
4. Koye DN, Magliano DJ, Nelson RG, Pavkov ME. The Global Epidemiology of Diabetes and Kidney Disease. Advances in chronic kidney disease. 2018;25:121-32
5. Lai W, Wang B, Huang R, Zhang C, Fu P, Ma L. Ferroptosis in organ fibrosis: From mechanisms to therapeutic medicines. J Transl Int Med. 2024;12:22-34
6. Huang R, Fu P, Ma L. Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct Target Ther. 2023;8:129
7. Johansen KL, Chertow GM, Foley RN, Gilbertson DT, Herzog CA, Ishani A. et al. US Renal Data System 2020 Annual Data Report: Epidemiology of Kidney Disease in the United States. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2021;77:A7-a8
8. Sardar MB, Ahmed S, Ashraf H, Ashfaq H, Nadeem ZA, Babar M. et al. Temporal and regional trends in adults with diabetics kidney disease in the US from 1999 to 2020. Diabetes research and clinical practice. 2024;213:111729
9. Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clinical journal of the American Society of Nephrology: CJASN. 2017;12:2032-45
10. van Raalte DH, Bjornstad P, Cherney DZI, de Boer IH, Fioretto P, Gordin D. et al. Combination therapy for kidney disease in people with diabetes mellitus. Nature reviews Nephrology. 2024;20:433-46
11. Ricciardi CA, Gnudi L. Kidney disease in diabetes: From mechanisms to clinical presentation and treatment strategies. Metabolism: clinical and experimental. 2021;124:154890
12. Kriz W, Löwen J, Gröne HJ. The complex pathology of diabetic nephropathy in humans. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2023;38:2109-19
13. Yin T, Yang L, Tang L, Li J, Liu D, Guo F. et al. Podocyte FFAR4 deficiency aggravated glomerular diseases and aging. Molecular therapy: the journal of the American Society of Gene Therapy. 2025:S1525-0016(25)00406-X.
14. Marciano DK. Mesangial Cells: The Tuft Guys of Glomerular Development. Journal of the American Society of Nephrology: JASN. 2019;30:1551-3
15. Kim J, Lee JH, Jang SH, Lee EY, Lee M, Park S. et al. SBP1 contributes to mesangial proliferation and inflammation through mitochondrial respiration in glomerulus during IgA nephropathy. Free radical biology & medicine. 2024;225:711-25
16. Li W, Yao C, Guo H, Ni X, Zhu R, Wang Y. et al. Macrophages communicate with mesangial cells through the CXCL12/DPP4 axis in lupus nephritis pathogenesis. Cell death & disease. 2024;15:344
17. Zhang K, Fu Z, Zhang Y, Chen X, Cai G, Hong Q. The role of cellular crosstalk in the progression of diabetic nephropathy. Frontiers in endocrinology. 2023;14:1173933
18. Hu S, Hang X, Wei Y, Wang H, Zhang L, Zhao L. Crosstalk among podocytes, glomerular endothelial cells and mesangial cells in diabetic kidney disease: an updated review. Cell communication and signaling: CCS. 2024;22:136
19. Sakai T, Kriz W. The structural relationship between mesangial cells and basement membrane of the renal glomerulus. Anatomy and embryology. 1987;176:373-86
20. Zimmerman SE, Hiremath C, Tsunezumi J, Yang Z, Finney B, Marciano DK. Nephronectin Regulates Mesangial Cell Adhesion and Behavior in Glomeruli. Journal of the American Society of Nephrology: JASN. 2018;29:1128-40
21. Schlöndorff D. Roles of the mesangium in glomerular function. Kidney international. 1996;49:1583-5
22. Johnson RJ, Floege J, Yoshimura A, Iida H, Couser WG, Alpers CE. The activated mesangial cell: a glomerular “myofibroblast”? Journal of the American Society of Nephrology: JASN. 1992;2:S190-7
23. Latta H, Maunsbach AB, Madden SC. The centrolobular region of the renal glomerulus studied by electron microscopy. Journal of ultrastructure research. 1960;4:455-72
24. Sterzel RB, Schulze-Lohoff E, Marx M. Cytokines and mesangial cells. Kidney international Supplement. 1993;39:S26-31
25. Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nature reviews Drug discovery. 2021;20:689-709
26. Gong M, Guo Y, Dong H, Wu F, He Q, Gong J. et al. Modified Hu-lu-ba-wan protects diabetic glomerular podocytes via promoting PKM2-mediated mitochondrial dynamic homeostasis. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2024;123:155247
27. Wang X, Song R, Li Z. Salviolone protects against high glucose-induced proliferation, oxidative stress, inflammation, and fibrosis of human renal mesangial cells by upregulating membrane metalloendopeptidase expression. Chemical biology & drug design. 2023;101:819-28
28. Uddin MJ, Jeong J, Pak ES, Ha H. CO-Releasing Molecule-2 Prevents Acute Kidney Injury through Suppression of ROS-Fyn-ER Stress Signaling in Mouse Model. Oxidative medicine and cellular longevity. 2021;2021:9947772
29. Li S, Lin Z, Xiao H, Xu Z, Li C, Zeng J. et al. Fyn deficiency inhibits oxidative stress by decreasing c-Cbl-mediated ubiquitination of Sirt1 to attenuate diabetic renal fibrosis. Metabolism: clinical and experimental. 2023;139:155378
30. Wu L, Wang Q, Guo F, Zhou Y, Ji H, Liu F. et al. Activation of FoxO1/ PGC-1α prevents mitochondrial dysfunction and ameliorates mesangial cell injury in diabetic rats. Molecular and cellular endocrinology. 2015;413:1-12
31. Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney international. 2013;83:1029-41
32. Yao H, Kong M, Du D, Ai F, Li J, Li Y. Swinhoeic acid from Potentilla fragarioides ameliorates high glucose-induced oxidative stress and accumulation of ECM in mesangial cells via Keap1-dependent activation of Nrf2. Redox report: communications in free radical research. 2022;27:230-8
33. Yang TT, Shao YT, Cheng Q, He YT, Qiu Z, Pan DD. et al. YY1/HIF-1α/mROS positive-feedback loop exacerbates glomerular mesangial cell proliferation in mouse early diabetic kidney disease. Acta pharmacologica Sinica. 2025;46(7):1974-1989
34. Quiles JM, Gustafsson Å B. The role of mitochondrial fission in cardiovascular health and disease. Nature reviews Cardiology. 2022;19:723-36
35. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology. 2010;12:814-22
36. Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? British journal of pharmacology. 2014;171:1917-42
37. Lemasters JJ. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. Journal of gastroenterology and hepatology. 2007;22(Suppl 1):S31-7
38. Czajka A, Ajaz S, Gnudi L, Parsade CK, Jones P, Reid F. et al. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients With Diabetic Nephropathy. EBioMedicine. 2015;2:499-512
39. Zhu J, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy. 2013;9:1663-76
40. Yi X, Yan W, Guo T, Liu N, Wang Z, Shang J. et al. Erythropoietin Mitigates Diabetic Nephropathy by Restoring PINK1/Parkin-Mediated Mitophagy. Frontiers in pharmacology. 2022;13:883057
41. Liu S, Yuan Y, Xue Y, Xing C, Zhang B. Podocyte Injury in Diabetic Kidney Disease: A Focus on Mitochondrial Dysfunction. Frontiers in cell and developmental biology. 2022;10:832887
42. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA. et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell metabolism. 2018;27:869-85.e6
43. Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nature reviews Nephrology. 2018;14:291-312
44. Takabatake Y, Yamamoto T, Isaka Y. Stagnation of autophagy: A novel mechanism of renal lipotoxicity. Autophagy. 2017;13:775-6
45. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney international. 2013;83:568-81
46. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P. et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell metabolism. 2012;15:186-200
47. Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT. et al. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. Journal of the American Society of Nephrology: JASN. 2016;27:2733-47
48. Ookawara M, Nio Y. Phosphodiesterase 4 inhibitors in diabetic nephropathy. Cellular signalling. 2022;90:110185
49. Zhu X, Deng Z, Cao Y, Zhou Z, Sun W, Liu C. et al. Resveratrol prevents Drp1-mediated mitochondrial fission in the diabetic kidney through the PDE4D/PKA pathway. Phytotherapy research: PTR. 2023;37(12):5916-5931
50. Wang L, Wu L, Du Y, Wang X, Yang B, Guo S. et al. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) drives angiotensin II-induced vascular remodeling through regulating mitochondrial fragmentation. Redox biology. 2023;67:102893
51. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiological reviews. 2008;88:611-38
52. Zhang Z, Yuan Y, Zhang X, Gu L, Tang Y, Zhao Y. et al. GPR39 Agonist TC-G 1008 Promoted Mitochondrial Biogenesis and Improved Antioxidative Capability via CREB/PGC-1α Pathway Following Intracerebral Hemorrhage in Mice. Translational stroke research. 2025;16:625-644
53. Yin H, Li X, Wang C, Li X, Liu J. Nickel induces mitochondrial damage in renal cells in vitro and in vivo through its effects on mitochondrial biogenesis, fusion, and fission. Chemico-biological interactions. 2024;394:110975
54. Salagre D, Navarro-Alarcón M, Villalón-Mir M, Alcázar-Navarrete B, Gómez-Moreno G, Tamimi F. et al. Chronic melatonin treatment improves obesity by inducing uncoupling of skeletal muscle SERCA-SLN mediated by CaMKII/AMPK/PGC1α pathway and mitochondrial biogenesis in female and male Zücker diabetic fatty rats. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2024;172:116314
55. Ye S, Zhang M, Tang SCW, Li B, Chen W. PGC1-α in diabetic kidney disease: unraveling renoprotection and molecular mechanisms. Molecular biology reports. 2024;51:304
56. Song Y, Yu H, Sun Q, Pei F, Xia Q, Gao Z. et al. Grape seed proanthocyanidin extract targets p66Shc to regulate mitochondrial biogenesis and dynamics in diabetic kidney disease. Frontiers in pharmacology. 2022;13:1035755
57. Liu F, Ma XJ, Wang QZ, Zhao YY, Wu LN, Qin GJ. The effect of FoxO1 on the proliferation of rat mesangial cells under high glucose conditions. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2014;29:1879-87
58. Wu L, Zhang Y, Ma X, Zhang N, Qin G. The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Molecular biology reports. 2012;39:9085-93
59. Fang S, Cai Y, Lyu F, Zhang H, Wu C, Zeng Y. et al. Exendin-4 Improves Diabetic Kidney Disease in C57BL/6 Mice Independent of Brown Adipose Tissue Activation. Journal of diabetes research. 2020;2020:9084567
60. Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z. et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nature reviews Nephrology. 2017;13:769-81
61. Liu X, Ducasa GM, Mallela SK, Kim JJ, Molina J, Mitrofanova A. et al. Sterol-O-acyltransferase-1 has a role in kidney disease associated with diabetes and Alport syndrome. Kidney international. 2020;98:1275-85
62. Zhang J, Wu Y, Zhang J, Zhang R, Wang Y, Liu F. ABCA1 deficiency-mediated glomerular cholesterol accumulation exacerbates glomerular endothelial injury and dysfunction in diabetic kidney disease. Metabolism: clinical and experimental. 2023;139:155377
63. Wu M, Yang Z, Zhang C, Shi Y, Han W, Song S. et al. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism: clinical and experimental. 2021;118:154748
64. Susztak K, Ciccone E, McCue P, Sharma K, Böttinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS medicine. 2005;2:e45
65. Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Annals of medicine. 2011;43:198-211
66. Nagai Y, Matoba K, Takeda Y, Yako H, Akamine T, Sekiguchi K. et al. Rho-associated, coiled-coil-containing protein kinase 1 regulates development of diabetic kidney disease via modulation of fatty acid metabolism. Kidney international. 2022;102:536-45
67. Yao H, Zhang W, Yang F, Ai F, Du D, Li Y. Discovery of caffeoylisocitric acid as a Keap1-dependent Nrf2 activator and its effects in mesangial cells under high glucose. Journal of enzyme inhibition and medicinal chemistry. 2022;37:178-88
68. Ly LD, Xu S, Choi SK, Ha CM, Thoudam T, Cha SK. et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Experimental & molecular medicine. 2017;49:e291
69. Su Y, Chen Q, Ju Y, Li W, Li W. Palmitate induces human glomerular mesangial cells fibrosis through CD36-mediated transient receptor potential canonical channel 6/nuclear factor of activated T cell 2 activation. Biochimica et biophysica acta Molecular and cell biology of lipids. 2020;1865:158793
70. Kim D, Nam GY, Seo E, Jun HS. Inhibition of ChREBP ubiquitination via the ROS/Akt-dependent downregulation of Smurf2 contributes to lysophosphatidic acid-induced fibrosis in renal mesangial cells. Journal of biomedical science. 2022;29:31
71. Tufro A. Cholesterol accumulation in podocytes: a potential novel targetable pathway in diabetic nephropathy. Diabetes. 2013;62:3661-2
72. Ducasa GM, Mitrofanova A, Fornoni A. Crosstalk Between Lipids and Mitochondria in Diabetic Kidney Disease. Current diabetes reports. 2019;19:144
73. Tsun JG, Yung S, Chau MK, Shiu SW, Chan TM, Tan KC. Cellular cholesterol transport proteins in diabetic nephropathy. PloS one. 2014;9:e105787
74. He H, Halseth TA, Mei L, Shen C, Liu L, Schwendeman A. Nanodisc delivery of liver X receptor agonist for the treatment of diabetic nephropathy. Journal of controlled release: official journal of the Controlled Release Society. 2022;348:1016-27
75. Wang H, Huang X, Xu P, Liu X, Zhou Z, Wang F. et al. Apolipoprotein C3 aggravates diabetic nephropathy in type 1 diabetes by activating the renal TLR2/NF-κB pathway. Metabolism: clinical and experimental. 2021;119:154740
76. Wu K, Zha H, Wu T, Liu H, Peng R, Lin Z. et al. Cytosolic Hmgb1 accumulation in mesangial cells aggravates diabetic kidney disease progression via NFκB signaling pathway. Cellular and molecular life sciences: CMLS. 2024;81:408
77. Zhu C, Zhang L, Ding X, Wu W, Zou J. Non-coding RNAs as regulators of autophagy in chondrocytes: Mechanisms and implications for osteoarthritis. Ageing research reviews. 2024;99:102404
78. Dong QQ, Yang Y, Tao H, Lu C, Yang JJ. m6A epitranscriptomic and epigenetic crosstalk in liver fibrosis: Special emphasis on DNA methylation and non-coding RNAs. Cellular signalling. 2024;122:111302
79. Wang D, Zhang Y, Li Q, Li Y, Li W, Zhang A. et al. Epigenetics: Mechanisms, potential roles, and therapeutic strategies in cancer progression. Genes & diseases. 2024;11:101020
80. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature reviews Genetics. 2012;13:484-92
81. Mattei AL, Bailly N, Meissner A. DNA methylation: a historical perspective. Trends in genetics: TIG. 2022;38:676-707
82. Shao D, Ni J, Shen Y, Liu J, Zhou L, Xue H. et al. CHOP mediates XBP1S-induced renal mesangial cell necrosis following high glucose treatment. European journal of pharmacology. 2015;758:89-96
83. Li Y, Ren D, Shen Y, Zheng X, Xu G. Altered DNA methylation of TRIM13 in diabetic nephropathy suppresses mesangial collagen synthesis by promoting ubiquitination of CHOP. EBioMedicine. 2020;51:102582
84. Liang G, Weisenberger DJ. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics. 2017;12:416-32
85. Kakani P, Dhamdhere SG, Pant D, Joshi R, Mishra J, Samaiya A. et al. Hypoxia-induced CTCF promotes EMT in breast cancer. Cell reports. 2024;43:114367
86. Ren L, Chang YF, Jiang SH, Li XH, Cheng HP. DNA methylation modification in Idiopathic pulmonary fibrosis. Frontiers in cell and developmental biology. 2024;12:1416325
87. Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285-94
88. Wang S, Zhang X, Wang Q, Wang R. Histone modification in podocyte injury of diabetic nephropathy. Journal of molecular medicine (Berlin, Germany). 2022;100:1373-86
89. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41-5
90. Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Current opinion in genetics & development. 2004;14:155-64
91. Das F, Bera A, Ghosh-Choudhury N, Sataranatarajan K, Kamat A, Kasinath BS. et al. High glucose-stimulated enhancer of zeste homolog-2 (EZH2) forces suppression of deptor to cause glomerular mesangial cell pathology. Cellular signalling. 2021;86:110072
92. Fang N, Li P. O-linked N-acetylglucosaminyltransferase OGT inhibits diabetic nephropathy by stabilizing histone methyltransferases EZH2 via the HES1/PTEN axis. Life sciences. 2021;274:119226
93. Millán-Zambrano G, Burton A, Bannister AJ, Schneider R. Histone post-translational modifications - cause and consequence of genome function. Nature reviews Genetics. 2022;23:563-80
94. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell research. 2011;21:381-95
95. Vega ME, Kastberger B, Wehrle-Haller B, Schwarzbauer JE. Stimulation of Fibronectin Matrix Assembly by Lysine Acetylation. Cells. 2020;9:655
96. Tsai YC, Kuo MC, Hung WW, Wu LY, Wu PH, Chang WA. et al. High Glucose Induces Mesangial Cell Apoptosis through miR-15b-5p and Promotes Diabetic Nephropathy by Extracellular Vesicle Delivery. Molecular therapy: the journal of the American Society of Gene Therapy. 2020;28:963-74
97. Chen Y, Peng Y, Li P, Jiang Y, Song D. Ginsenoside Rg3 induces mesangial cells proliferation and attenuates apoptosis by miR-216a-5p/MAPK pathway in diabetic kidney disease. Aging. 2024;16:9933-43
98. McClelland AD, Herman-Edelstein M, Komers R, Jha JC, Winbanks CE, Hagiwara S. et al. miR-21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clinical science (London, England: 1979). 2015;129:1237-49
99. Loboda A, Sobczak M, Jozkowicz A, Dulak J. TGF-β1/Smads and miR-21 in Renal Fibrosis and Inflammation. Mediators of inflammation. 2016;2016:8319283
100. Wang JY, Gao YB, Zhang N, Zou DW, Xu LP, Zhu ZY. et al. Tongxinluo ameliorates renal structure and function by regulating miR-21-induced epithelial-to-mesenchymal transition in diabetic nephropathy. American journal of physiology Renal physiology. 2014;306:F486-95
101. Kölling M, Kaucsar T, Schauerte C, Hübner A, Dettling A, Park JK. et al. Therapeutic miR-21 Silencing Ameliorates Diabetic Kidney Disease in Mice. Molecular therapy: the journal of the American Society of Gene Therapy. 2017;25:165-80
102. Wang X, Gao Y, Tian N, Zou D, Shi Y, Zhang N. Astragaloside IV improves renal function and fibrosis via inhibition of miR-21-induced podocyte dedifferentiation and mesangial cell activation in diabetic mice. Drug design, development and therapy. 2018;12:2431-42
103. Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M. et al. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. Journal of the American Society of Nephrology: JASN. 2012;23:252-65
104. Wei Q, Huang J, Livingston MJ, Wang S, Dong G, Xu H. et al. Pseudogene GSTM3P1 derived long non-coding RNA promotes ischemic acute kidney injury by target directed microRNA degradation of kidney-protective mir-668. Kidney international. 2024;106:640-657
105. Gan T, Liu W, Wang Y, Huang D, Hu J, Wang Y. et al. LncRNA MAAMT facilitates macrophage recruitment and proinflammatory activation and exacerbates autoimmune myocarditis through the SRSF1/NF-κB axis. International journal of biological macromolecules. 2024;278:134193
106. Gao J, Wang W, Wang F, Guo C. LncRNA-NR_033515 promotes proliferation, fibrogenesis and epithelial-to-mesenchymal transition by targeting miR-743b-5p in diabetic nephropathy. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2018;106:543-52
107. Chen K, Yu B, Liao J. LncRNA SOX2OT alleviates mesangial cell proliferation and fibrosis in diabetic nephropathy via Akt/mTOR-mediated autophagy. Molecular medicine (Cambridge, Mass). 2021;27:71
108. Liu F, Cao Y, Zhang C, Su H. Decreased DANCR contributes to high glucose-induced extracellular matrix accumulation in human renal mesangial cell via regulating the TGF-β/Smad signaling. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2023;37:e22926
109. Ji TT, Qi YH, Li XY, Tang B, Wang YK, Zheng PX. et al. Loss of lncRNA MIAT ameliorates proliferation and fibrosis of diabetic nephropathy through reducing E2F3 expression. Journal of cellular and molecular medicine. 2020;24:13314-23
110. Chen H, Guo Y, Cheng X. Long non-cording RNA XIST promoted cell proliferation and suppressed apoptosis by miR-423-5p/HMGA2 axis in diabetic nephropathy. Molecular and cellular biochemistry. 2021;476:4517-28
111. Wang Y, Yang J, Wu C, Guo Y, Ding Y, Zou X. LncRNA SNHG14 silencing attenuates the progression of diabetic nephropathy via the miR-30e-5p/SOX4 axis. Journal of diabetes. 2024;16:e13565
112. Tu X, Zhang H, Ren H. LINC01232 targeting miR-1250-3p/MSH2 axis attenuates mesangial cell proliferation and fibrosis in diabetic nephropathy. Molecular and cellular biochemistry. 2024;479:2093-2103
113. Peng F, Gong W, Li S, Yin B, Zhao C, Liu W. et al. circRNA_010383 Acts as a Sponge for miR-135a, and Its Downregulated Expression Contributes to Renal Fibrosis in Diabetic Nephropathy. Diabetes. 2021;70:603-15
114. Tang B, Li W, Ji TT, Li XY, Qu X, Feng L. et al. Circ-AKT3 inhibits the accumulation of extracellular matrix of mesangial cells in diabetic nephropathy via modulating miR-296-3p/E-cadherin signals. Journal of cellular and molecular medicine. 2020;24:8779-88
115. Bai S, Xiong X, Tang B, Ji T, Li X, Qu X. et al. Exosomal circ_DLGAP4 promotes diabetic kidney disease progression by sponging miR-143 and targeting ERBB3/NF-κB/MMP-2 axis. Cell death & disease. 2020;11:1008
116. Liu S, Zhao Y, Lu S, Zhang T, Lindenmeyer MT, Nair V. et al. Single-cell transcriptomics reveals a mechanosensitive injury signaling pathway in early diabetic nephropathy. Genome medicine. 2023;15:2
117. Choi S, Hong SP, Bae JH, Suh SH, Bae H, Kang KP. et al. Hyperactivation of YAP/TAZ Drives Alterations in Mesangial Cells through Stabilization of N-Myc in Diabetic Nephropathy. Journal of the American Society of Nephrology: JASN. 2023;34:809-28
118. Du L, Liu S, Lu Y, Ren D, Yu X, Hu Y. et al. GABP Promotes Mesangial Cell Proliferation and Renal Fibrosis Through GLI1 in Diabetic Nephropathy. Advanced science (Weinheim, Baden-Wurttemberg, Germany). 2025;12:e2407462
119. Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M. et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:8015-20
120. Mou X, Zhou DY, Zhou DY, Ma JR, Liu YH, Chen HP. et al. Serum TGF-β1 as a Biomarker for Type 2 Diabetic Nephropathy: A Meta-Analysis of Randomized Controlled Trials. PloS one. 2016;11:e0149513
121. Leung JC, Chan LY, Saleem MA, Mathieson PW, Tang SC, Lai KN. Combined blockade of angiotensin II and prorenin receptors ameliorates podocytic apoptosis induced by IgA-activated mesangial cells. Apoptosis: an international journal on programmed cell death. 2015;20:907-20
122. Wang YY, Tang LQ, Wei W. Berberine attenuates podocytes injury caused by exosomes derived from high glucose-induced mesangial cells through TGFβ1-PI3K/AKT pathway. European journal of pharmacology. 2018;824:185-92
123. Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF. Developmental patterns of PDGF B-chain, PDGF-receptor, and alpha-actin expression in human glomerulogenesis. Kidney international. 1992;42:390-9
124. Levéen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes & development. 1994;8:1875-87
125. Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes & development. 1994;8:1888-96
126. Fagerudd JA, Groop PH, Honkanen E, Teppo AM, Grönhagen-Riska C. Urinary excretion of TGF-beta 1, PDGF-BB and fibronectin in insulin-dependent diabetes mellitus patients. Kidney international Supplement. 1997;63:S195-7
127. Sohn E, Kim J, Kim CS, Jo K, Lee YM, Kim JS. Root of Polygonum cuspidatum extract reduces progression of diabetes-induced mesangial cell dysfunction via inhibition of platelet-derived growth factor-BB (PDGF-BB) and interaction with its receptor in streptozotocin-induced diabetic rats. BMC complementary and alternative medicine. 2014;14:477
128. Khan S, Lakhe-Reddy S, McCarty JH, Sorenson CM, Sheibani N, Reichardt LF. et al. Mesangial cell integrin αvβ8 provides glomerular endothelial cell cytoprotection by sequestering TGF-β and regulating PECAM-1. The American journal of pathology. 2011;178:609-20
129. Zhang Y, Zhu Z, Wang T, Dong Y, Fan Y, Sun D. TGF-β1-containing exosomes from cardiac microvascular endothelial cells mediate cardiac fibroblast activation under high glucose conditions. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2021;99:693-9
130. Wu XM, Gao YB, Cui FQ, Zhang N. Exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells to promote renal fibrosis. Biology open. 2016;5:484-91
131. López-Ongil S, Díez-Marqués ML, Griera M, Rodríguez-Puyol M, Rodríguez-Puyol D. Crosstalk between mesangial and endothelial cells: angiotensin II down-regulates endothelin-converting enzyme 1. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2005;15:135-44
132. Zou HH, Wang L, Zheng XX, Xu GS, Shen Y. Endothelial cells secreted endothelin-1 augments diabetic nephropathy via inducing extracellular matrix accumulation of mesangial cells in ETBR(-/-) mice. Aging. 2019;11:1804-20
133. Peng F, Wu D, Gao B, Ingram AJ, Zhang B, Chorneyko K. et al. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes. 2008;57:1683-92
134. Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes. 2008;57:714-23
135. Jiang W, Zhang T, Qiu Y, Liu Q, Chen X, Wang Q. et al. Keratinocyte-to-macrophage communication exacerbate psoriasiform dermatitis via LRG1-enriched extracellular vesicles. Theranostics. 2024;14:1049-64
136. Zhu Z, Guo Y, Liu Y, Ding R, Huang Z, Yu W. et al. ELK4 Promotes Colorectal Cancer Progression by Activating the Neoangiogenic Factor LRG1 in a Noncanonical SP1/3-Dependent Manner. Advanced science (Weinheim, Baden-Wurttemberg, Germany). 2023;10:e2303378
137. Liu JJ, Liu S, Wang J, Pek SLT, Lee J, Gurung RL. et al. Urine Leucine-Rich α-2 Glycoprotein 1 (LRG1) Predicts the Risk of Progression to End-Stage Kidney Disease in Patients With Type 2 Diabetes. Diabetes care. 2023;46:408-15
138. Grzesiak L, Amaya-Garrido A, Feuillet G, Malet N, Swiader A, Sarthou MK. et al. Leucine-Rich Alpha-2 Glycoprotein 1 Accumulates in Complicated Atherosclerosis and Promotes Calcification. International journal of molecular sciences. 2023;24:16537
139. Hong Q, Cai H, Zhang L, Li Z, Zhong F, Ni Z. et al. Modulation of transforming growth factor-β-induced kidney fibrosis by leucine-rich ⍺-2 glycoprotein-1. Kidney international. 2022;101:299-314
140. Tsuruta H, Yasuda-Yamahara M, Yoshibayashi M, Kuwagata S, Yamahara K, Tanaka-Sasaki Y. et al. Fructose overconsumption accelerates renal dysfunction with aberrant glomerular endothelial-mesangial cell interactions in db/db mice. Biochimica et biophysica acta Molecular basis of disease. 2024;1870:167074
141. Wang X, Sun Z, Fu J, Fang Z, Zhang W, He JC. et al. LRG1 loss effectively restrains glomerular TGF-β signaling to attenuate diabetic kidney disease. Molecular therapy: the journal of the American Society of Gene Therapy. 2024;32:3177-3193
142. Coll M, Ariño S, Martínez-Sánchez C, Garcia-Pras E, Gallego J, Moles A. et al. Ductular reaction promotes intrahepatic angiogenesis through Slit2-Roundabout 1 signaling. Hepatology (Baltimore, Md). 2022;75:353-68
143. Rama N, Dubrac A, Mathivet T, RA NC, Genet G, Cristofaro B. et al. Slit2 signaling through Robo1 and Robo2 is required for retinal neovascularization. Nature medicine. 2015;21:483-91
144. Liu J, Hou W, Guan T, Tang L, Zhu X, Li Y. et al. Slit2/Robo1 signaling is involved in angiogenesis of glomerular endothelial cells exposed to a diabetic-like environment. Angiogenesis. 2018;21:237-49
145. Bry M, Kivelä R, Leppänen VM, Alitalo K. Vascular endothelial growth factor-B in physiology and disease. Physiological reviews. 2014;94:779-94
146. Falkevall A, Mehlem A, Palombo I, Heller Sahlgren B, Ebarasi L, He L. et al. Reducing VEGF-B Signaling Ameliorates Renal Lipotoxicity and Protects against Diabetic Kidney Disease. Cell metabolism. 2017;25:713-26
147. Folestad E, Mehlem A, Ning FC, Oosterveld T, Palombo I, Singh J. et al. Vascular endothelial growth factor B-mediated fatty acid flux in the adipose-kidney axis contributes to lipotoxicity in diabetic kidney disease. Kidney international. 2025;107:492-507
148. Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. American journal of physiology Regulatory, integrative and comparative physiology. 2011;300:R1009-22
149. Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K. et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nature medicine. 2013;19:1496-504
150. Nihalani D, Susztak K. Sirt1-Claudin-1 crosstalk regulates renal function. Nature medicine. 2013;19:1371-2
151. Tsai YC, Kuo MC, Hung WW, Wu PH, Chang WA, Wu LY. et al. Proximal tubule-derived exosomes contribute to mesangial cell injury in diabetic nephropathy via miR-92a-1-5p transfer. Cell communication and signaling: CCS. 2023;21:10
152. Ma T, Li X, Zhu Y, Yu S, Liu T, Zhang X. et al. Excessive Activation of Notch Signaling in Macrophages Promote Kidney Inflammation, Fibrosis, and Necroptosis. Frontiers in immunology. 2022;13:835879
153. Fan Z, Xu X, Qi X, Wu Y. Role of TGF-β activated kinase-1 inhibitor on the interaction between macrophages and mesangial cells on the condition of high glucose. Immunological investigations. 2018;47:303-14
154. Zhu QJ, Zhu M, Xu XX, Meng XM, Wu YG. Exosomes from high glucose-treated macrophages activate glomerular mesangial cells via TGF-β1/Smad3 pathway in vivo and in vitro. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2019;33:9279-90
155. Wang L, Hauenstein AV. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Molecular aspects of medicine. 2020;76:100889
156. Yang M, Zhao L. The Selective NLRP3-Inflammasome Inhibitor CY-09 Ameliorates Kidney Injury in Diabetic Nephropathy by Inhibiting NLRP3- inflammasome Activation. Current medicinal chemistry. 2023;30:3261-70
157. Tang ZZ, Zhang YM, Zheng T, Huang TT, Ma TF, Liu YW. Sarsasapogenin alleviates diabetic nephropathy through suppression of chronic inflammation by down-regulating PAR-1: In vivo and in vitro study. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2020;78:153314
158. Liu Y, Li X, Zhao M, Wu Y, Xu Y, Li X. et al. Macrophage-derived exosomes promote activation of NLRP3 inflammasome and autophagy deficiency of mesangial cells in diabetic nephropathy. Life sciences. 2023;330:121991
159. Impellizzeri D, Esposito E, Attley J, Cuzzocrea S. Targeting inflammation: new therapeutic approaches in chronic kidney disease (CKD). Pharmacological research. 2014;81:91-102
160. Zhang L, Wang Z, Liu R, Li Z, Lin J, Wojciechowicz ML. et al. Connectivity Mapping Identifies BI-2536 as a Potential Drug to Treat Diabetic Kidney Disease. Diabetes. 2021;70:589-602
161. Wen M, Sun X, Pan L, Jing S, Zhang X, Liang L. et al. Dihydromyricetin ameliorates diabetic renal fibrosis via regulating SphK1 to suppress the activation of NF-κB pathway. European journal of pharmacology. 2024;978:176799
162. Zheng ZC, Zhu W, Lei L, Liu XQ, Wu YG. Wogonin Ameliorates Renal Inflammation and Fibrosis by Inhibiting NF-κB and TGF-β1/Smad3 Signaling Pathways in Diabetic Nephropathy. Drug design, development and therapy. 2020;14:4135-48
163. Opazo-Ríos L, Plaza A, Sánchez Matus Y, Bernal S, Lopez-Sanz L, Jimenez-Castilla L. et al. Targeting NF-κB by the Cell-Permeable NEMO-Binding Domain Peptide Improves Albuminuria and Renal Lesions in an Experimental Model of Type 2 Diabetic Nephropathy. International journal of molecular sciences. 2020;21:4225
164. Dong C, Wu G, Li H, Qiao Y, Gao S. Ampelopsin inhibits high glucose-induced extracellular matrix accumulation and oxidative stress in mesangial cells through activating the Nrf2/HO-1 pathway. Phytotherapy research: PTR. 2020;34:2044-52
165. Huang Q, Zhang Y, Jiang Y, Huang L, Liu Q, Ouyang D. Eucommia lignans alleviate the progression of diabetic nephropathy through mediating the AR/Nrf2/HO-1/AMPK axis in vivo and in vitro. Chinese journal of natural medicines. 2023;21:516-26
166. Wang K, Zheng X, Pan Z, Yao W, Gao X, Wang X. et al. Icariin Prevents Extracellular Matrix Accumulation and Ameliorates Experimental Diabetic Kidney Disease by Inhibiting Oxidative Stress via GPER Mediated p62-Dependent Keap1 Degradation and Nrf2 Activation. Frontiers in cell and developmental biology. 2020;8:559
167. Li H, Wang D, Chen Y, Yang M. β-Caryophyllene inhibits high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation in mesangial cells. International immunopharmacology. 2020;84:106556
168. Zhang C, Zhu X, Li L, Ma T, Shi M, Yang Y. et al. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes, metabolic syndrome and obesity: targets and therapy. 2019;12:1297-309
169. Xiao Y, Deng J, Li C, Gong X, Gui Z, Huang J. et al. Epiberberine ameliorated diabetic nephropathy by inactivating the angiotensinogen (Agt) to repress TGFβ/Smad2 pathway. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2021;83:153488
170. Li Q, Wang Y, Yan J, Yuan R, Zhang J, Guo X. et al. Osthole ameliorates early diabetic kidney damage by suppressing oxidative stress, inflammation and inhibiting TGF-β1/Smads signaling pathway. International immunopharmacology. 2024;133:112131
171. Ha KB, Sangartit W, Jeong AR, Lee ES, Kim HM, Shim S. et al. EW-7197 Attenuates the Progression of Diabetic Nephropathy in db/db Mice through Suppression of Fibrogenesis and Inflammation. Endocrinology and metabolism (Seoul, Korea). 2022;37:96-111
172. Barro L, Hsiao JT, Chen CY, Chang YL, Hsieh MF. Cytoprotective Effect of Liposomal Puerarin on High Glucose-Induced Injury in Rat Mesangial Cells. Antioxidants (Basel, Switzerland). 2021;10:1177
173. Horikoshi S, Fukuda N, Tsunemi A, Okamura M, Otsuki M, Endo M. et al. Contribution of TGF-β1 and Effects of Gene Silencer Pyrrole-Imidazole Polyamides Targeting TGF-β1 in Diabetic Nephropathy. Molecules (Basel, Switzerland). 2020;25:950
174. Okamura M, Fukuda N, Horikoshi S, Kobayashi H, Tsunemi A, Akiya Y. et al. Transcriptional Suppression of Diabetic Nephropathy with Novel Gene Silencer Pyrrole-Imidazole Polyamides Preventing USF1 Binding to the TGF-β1 Promoter. International journal of molecular sciences. 2021;22:4741
175. Shen K, Johnson DW, Gobe GC. The role of cGMP and its signaling pathways in kidney disease. American journal of physiology Renal physiology. 2016;311:F671-f81
176. Fleischmann D, Harloff M, Maslanka Figueroa S, Schlossmann J, Goepferich A. Targeted Delivery of Soluble Guanylate Cyclase (sGC) Activator Cinaciguat to Renal Mesangial Cells via Virus-Mimetic Nanoparticles Potentiates Anti-Fibrotic Effects by cGMP-Mediated Suppression of the TGF-β Pathway. International journal of molecular sciences. 2021;22:2557
177. Jin L, Zheng D, Yang G, Li W, Yang H, Jiang Q. et al. Tilapia Skin Peptides Ameliorate Diabetic Nephropathy in STZ-Induced Diabetic Rats and HG-Induced GMCs by Improving Mitochondrial Dysfunction. Marine drugs. 2020;18:363
178. Verma H, Bhalla AK, Kumar S, Sheth G, Kumar B, Mondal R. WCN25-2546 A Prospective, Multi-Centric, Randomized, Double Blind, Placebo-Controlled Study to Evaluate the Effectiveness of Supplementation of N-Acetylcysteine& Taurine in Preventing Progression of Chronic Kidney Disease. Kidney International Reports. 2025;10:S269-S70
179. Ren H, Shao Y, Wu C, Ma X, Lv C, Wang Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Molecular and cellular endocrinology. 2020;500:110628
180. Kim DI, Park MJ, Heo YR, Park SH. Metformin ameliorates lipotoxicity-induced mesangial cell apoptosis partly via upregulation of glucagon like peptide-1 receptor (GLP-1R). Archives of biochemistry and biophysics. 2015;584:90-7
181. Xu WW, Guan MP, Zheng ZJ, Gao F, Zeng YM, Qin Y. et al. Exendin-4 alleviates high glucose-induced rat mesangial cell dysfunction through the AMPK pathway. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2014;33:423-32
182. Perkovic V, Tuttle KR, Rossing P, Mahaffey KW, Mann JFE, Bakris G. et al. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes. The New England journal of medicine. 2024;391:109-21
183. Li J, Li N, Yan S, Lu Y, Miao X, Gu Z. et al. Liraglutide protects renal mesangial cells against hyperglycemia-mediated mitochondrial apoptosis by activating the ERK-Yap signaling pathway and upregulating Sirt3 expression. Molecular medicine reports. 2019;19:2849-60
184. Huang L, Lin T, Shi M, Chen X, Wu P. Liraglutide suppresses production of extracellular matrix proteins and ameliorates renal injury of diabetic nephropathy by enhancing Wnt/β-catenin signaling. American journal of physiology Renal physiology. 2020;319:F458-f68
185. Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF. et al. Dapagliflozin in Patients with Chronic Kidney Disease. The New England journal of medicine. 2020;383:1436-46
186. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N. et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. The New England journal of medicine. 2017;377:644-57
187. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M. et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. The New England journal of medicine. 2016;375:323-34
188. Maki T, Maeno S, Maeda Y, Yamato M, Sonoda N, Ogawa Y. et al. Amelioration of diabetic nephropathy by SGLT2 inhibitors independent of its glucose-lowering effect: A possible role of SGLT2 in mesangial cells. Scientific reports. 2019;9:4703
189. Luna-Marco C, Iannantuoni F, Hermo-Argibay A, Devos D, Salazar JD, Víctor VM. et al. Cardiovascular benefits of SGLT2 inhibitors and GLP-1 receptor agonists through effects on mitochondrial function and oxidative stress. Free radical biology & medicine. 2024;213:19-35
190. Wang J, Hu L, Chen Y, Fu T, Jiang T, Jiang A. et al. Sitagliptin improves renal function in diabetic nephropathy in male Sprague Dawley rats through upregulating heme oxygenase-1 expression. Endocrine. 2019;63:70-8
191. Wang D, Zhang G, Chen X, Wei T, Liu C, Chen C. et al. Sitagliptin ameliorates diabetic nephropathy by blocking TGF-β1/Smad signaling pathway. International journal of molecular medicine. 2018;41:2784-92
192. Mosenzon O, Leibowitz G, Bhatt DL, Cahn A, Hirshberg B, Wei C. et al. Effect of Saxagliptin on Renal Outcomes in the SAVOR-TIMI 53 Trial. Diabetes care. 2017;40:69-76
193. Perkovic V, Toto R, Cooper ME, Mann JFE, Rosenstock J, McGuire DK. et al. Effects of Linagliptin on Cardiovascular and Kidney Outcomes in People With Normal and Reduced Kidney Function: Secondary Analysis of the CARMELINA Randomized Trial. Diabetes care. 2020;43:1803-12
194. Sugahara M, Tanaka S, Tanaka T, Saito H, Ishimoto Y, Wakashima T. et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. Journal of the American Society of Nephrology: JASN. 2020;31:560-77
195. Wei X, Ma Y, Li Y, Zhang W, Zhong Y, Yu Y. et al. Anti-Apoptosis of Podocytes and Pro-Apoptosis of Mesangial Cells for Telmisartan in Alleviating Diabetic Kidney Injury. Frontiers in pharmacology. 2022;13:876469
196. Matsui T, Yamagishi S, Ueda S, Nakamura K, Imaizumi T, Takeuchi M. et al. Telmisartan, an angiotensin II type 1 receptor blocker, inhibits advanced glycation end-product (AGE)-induced monocyte chemoattractant protein-1 expression in mesangial cells through downregulation of receptor for AGEs via peroxisome proliferator-activated receptor-gamma activation. The Journal of international medical research. 2007;35:482-9
197. Heerspink HJL, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou FF. et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet (London, England). 2019;393:1937-47
198. Fujimoto D, Kuwabara T, Hata Y, Umemoto S, Kanki T, Nishiguchi Y. et al. Suppressed ER-associated degradation by intraglomerular cross talk between mesangial cells and podocytes causes podocyte injury in diabetic kidney disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2020;34:15577-90
199. Zhu X, Deng Z, Cao Y, Zhou Z, Sun W, Liu C. et al. Resveratrol prevents Drp1-mediated mitochondrial fission in the diabetic kidney through the PDE4D/PKA pathway. Phytotherapy research: PTR. 2023;37:5916-31
Author contact
![]() Corresponding authors: Ping Fu, and Liang Ma. Email: fupinghxedu.cn (P Fu) and liang_medu.cn (L Ma).
Corresponding authors: Ping Fu, and Liang Ma. Email: fupinghxedu.cn (P Fu) and liang_medu.cn (L Ma).