Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(11):4942-4960. doi:10.7150/ijbs.112133 This issue Cite
Research Paper
MALAT1 promotes malignancy of HBV-related hepatocellular carcinoma by regulating IGF2BP3-mediated nuclear-cytoplasmic shuttling
Ze-Bang Du1†, Xin-Mou Wu1†, Tun Han1†, Yu-Xin Cai1, Bo Qian1, Yu-Shi Shen1, Han-Yu Zhang1, Jia-Shen Wu1, Jie He1, Xiao-Xuan Chen1, Dong-Bei Guo1, Hang-Tian Zhong2, Xiong Li2, Lei Zhang1, Xiao-Ming Luo2 ![]() , Wen-Gang Li3
, Wen-Gang Li3 ![]() , Yu-Chun Lin1
, Yu-Chun Lin1 ![]() , Zhong-Ning Lin1
, Zhong-Ning Lin1 ![]()
1. State Key Laboratory of Vaccines for Infectious Diseases, Xiang An Biomedicine Laboratory, Xiang'an Hospital of Xiamen University, National Innovation Platform for Industry-Education Integration in Vaccine Research, School of Public Health, Xiamen University, Xiamen 361102, China
2. Department of Preventive Medicine, School of Public Health, Chengdu Medical College, Chengdu 610500, China.
3. Department of Hepatobiliary Surgery, Cancer Research Center, Xiang'an Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, Fujian 361102, China.
† These authors contributed equally to this work.
Received 2025-2-13; Accepted 2025-7-8; Published 2025-7-28
Abstract

Hepatitis B virus (HBV) X protein (HBx) plays a critical role in the progression of HBV-related hepatocellular carcinoma (HCC). Long non-coding RNAs (lncRNAs) regulate various biological processes and contribute to HCC development, with their therapeutic potential in disease progression recently gaining significant attention. However, the involvement of lncRNAs in HBx-related hepatocarcinogenesis and the underlying regulatory mechanisms remain unclear. In this study, we conducted a comprehensive analysis of multi-database sequencing data to identify metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) as an HBx-associated lncRNA and observed its upregulation in HBV-related HCC tissues and cells upon HBx expression. Additionally, high MALAT1 expression was correlated with poor prognosis and advanced HCC progression. MALAT1 overexpression significantly promoted the proliferation, migration, and invasion of HCC cells. Mechanistic investigations revealed that MALAT1 was transported to the cytoplasm and enhanced RNA stability in a N6-methyladenosine (m6A)-dependent manner through direct interaction with and recruitment of insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3). Targeting MALAT1 in vivo with antisense oligonucleotides (ASO)-MALAT1 treatment effectively suppressed the progression of xenograft tumors and orthotopic liver tumors in HBx-related HCC. Moreover, hydrodynamic-based gene delivery (HGD) was utilized to introduce anti-HBx transposon plasmids into murine hepatocytes, thereby suppressing MALAT1-m6A-mediated HBV-related hepatocarcinogenesis in HBx transgenic (HBx-Tg) mice. Overall, our findings shed novel light on the regulatory role of IGF2BP3-mediated MALAT1 nuclear-cytoplasmic shuttling and RNA stabilization via m6A modification during HCC progression. These results suggest that m6A-based MALAT1 expression serves as a novel diagnostic and prognostic biomarker for targeted epigenetic intervention in HBV-related HCC.
Keywords: HBV-related HCC, MALAT1, m6A modification, nuclear-cytoplasmic shuttling, IGF2BP3, anti-HBx transposon plasmids
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent and lethal malignancies worldwide, posing a significant challenge to public health with a growing incidence rate [1]. The primary risk factor for HCC development, identified in 80%-90% of cases, is chronic hepatitis B virus (HBV) infection, which often progresses to end-stage liver diseases such as cirrhosis and ultimately leads to HCC [2]. Chronic HBV infection contributes to hepatocarcinogenesis through both direct mechanisms, including gene integration, mutation-induced genomic instability, and activation of cancer-related signaling pathways, and indirect mechanisms involving viral proteins [3]. Among the viral proteins encoded by the HBV genome, the HBV X protein (HBx) is a multifunctional regulatory protein that plays a critical role in initiating and progressing HCC by activating various oncogenes [4, 5]. However, the epigenetic mechanisms by which HBx contributes to HCC pathogenesis remain poorly understood.
Accumulating evidence has demonstrated that HBx plays a critical role in the pathogenesis of HCC by inducing genetic alterations in tumor suppressor genes and oncogenes. Additionally, epigenetic aberrations are also pivotal in the pathogenesis of HCC [4, 6]. Recent studies have highlighted the dysregulation of long non-coding RNAs (lncRNAs) in HBV-related HCC and HBV/HBx-expressing cells. These lncRNAs, which are longer than 200 nucleotides and lack protein-coding capacity [7], are involved in a wide range of cellular processes, including chromatin modification, transcriptional and post-transcriptional regulation, and function as signals, decoys, guides, scaffolds, and competing endogenous RNAs (ceRNAs) [8, 9]. Dysregulated lncRNAs, such as HOTAIR, have been associated with diseases like liver fibrosis [10], liver cancer [11], and metastasis [12], and serve as early prognostic indicators for lower survival rates. These findings suggest that lncRNAs can act as molecular markers and prognostic indicators for HCC due to their abnormal expression and specific molecular functions. Importantly, emerging evidence indicates that lncRNAs mediate interactions between host cells and viruses, including HBV. For example, HULC enhances the stability of HBV covalently closed circular DNA (cccDNA) [13], while the HBx-bound lncRNA DLEU2 sustains the transcription of cccDNA and cancer-relatd genes [14]. Moreover, HBx-upregulated TRERNA1 has been shown to confer resistance to sorafenib in HCC cells [15]. Collectively, these studies imply that differential lncRNA expression is regulated by HBV infection and HBx expression, and that HBx significantly contributes to the lncRNA-associated epigenetic regulation of hepatocytes in HBV-related HCC. Therefore, identifying functional lncRNAs targeted by HBx is crucial for developing novel therapeutic strategies against HBV-related HCC.
N6-methyladenosine (m6A) modification, constituting 0.1%-0.4% of native cellular RNA adenosine residues, is a prevalent epigenetic mark across various RNA types [16]. This internal modification extensively regulates gene expression by modulating RNA processing, localization, translation, and decay. These processes are orchestrated by three categories of proteins: "writers", "erasers", and "readers". m6A modification is also widely present in lncRNAs and regulates their functions, including nuclear export, potential peptide translation, and stability. However, the role of m6A in regulating HBx-induced lncRNAs in HBV-related HCC remains elusive.
In this study, we identified metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) as an lncRNA that is upregulated in HBV/HBx-expressing cells, HBx transgenic (HBx-Tg) mice, and patients with HBV-related HCC. We further investigated the biological function of MALAT1 and its m6A regulation in hepatocarcinogenesis both in vitro and in vivo. Mechanistically, we explored the m6A methylation status of MALAT1 at specific sites (A6021 and A7265) and the role of insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3) in mediating MALAT1 nuclear-cytoplasmic shuttling and RNA stabilization. Our findings provide new insights into the mechanisms of m6A-based lncRNAs in HBV-related HCC and highlight the potential of m6A-modified lncRNAs as novel epigenetic biomarkers and theranostic targets.
Materials and Methods
HCC tissue specimens
Twelve human HCC tumor tissues and corresponding peritumor tissues were obtained from patients who underwent liver tumor resection at Xiang'an Hospital of Xiamen University (Xiamen, China). Immediately upon retrieval, all samples were frozen in liquid nitrogen and stored at -80 °C until use. The levels of MALAT1 were detected via RNA fluorescence in situ hybridization (FISH) assays. Some samples were fixed and paraffin-embedded for histological examination. Protein levels, including HBx, IGF2BP3, E-cadherin, and Vimentin, were detected by western blotting (WB) and immunohistochemistry (IHC) staining. Written informed consent was obtained from all HCC patients involved in this study, and the study was approved by the Ethics Committee of Xiamen University.
In vivo xenograft tumor study
An in vivo xenograft tumor study was conducted following a previously described protocol [17, 18]. To explore the effects of HBx expression and MALAT1 genetic targeting intervention on the progression of HCC cell xenograft tumors, BALB/c nude mice (4- to 6-week-old) were randomly divided into four groups (n = 8 per group) as follows: Ctrl (HepG2-pc3.1-NC), HBx (HepG2-pc3.1-HBx), HBx+ASO-NC, and HBx+ASO-MALAT1. After allowing the xenograft tumors to grow for one week, the HBx+ASO-NC and HBx+ASO-MALAT1 groups were injected with ASO-NC or ASO-MALAT1 (5 nmol per injection, once every 2 days, for a total of 7 injections) via the tail vein. The mice were sacrificed on the day of the last injection. Tumor width (W), tumor length (L), and body weight were measured. Tumor volume (V) was calculated using the formula: V= (W2×L)/2. Xenograft tumors were collected for RNA FISH, quantitative real-time PCR (qRT-PCR), WB, hematoxylin and eosin (HE) staining, and IHC assays. All animal experiments were approved by the Experimental Animal Ethics Committee of Xiamen University (Ethics Approval No. XMULAC20220282, dated 2022-03-12).
Orthotopic liver tumor model
To establish an orthotopic liver tumor model mimicking HBV-related HCC in vivo, luciferase-expressing HepG2.2.15 cells (HepG2.2.15-Luc) were used. Briefly, 14 male nude mice (7-8 weeks old, average weight 20 g) were anesthetized with 1.5-2% isoflurane. After cleaning the surgical area with iodine and alcohol, a 1-1.5 cm midline incision was made below the xiphoid process. A total of 50 µl of a suspension containing 5×105 HepG2.2.15-Luc cells mixed with Matrigel was slowly injected into the top lobe of the liver, which was gently exteriorized through the incision. The liver was then carefully replaced, and the wound was sutured. The mice were monitored until they fully recovered from anesthesia and were then returned to their cages. Humane endpoints were defined as 20% body weight loss, loss of mobility, or significant decrease in activity. However, no mice reached these endpoints during the study.
The tumor-bearing mice were randomly divided into two groups (n = 7 per group): ASO-NC and ASO-MALAT1. The ASO-MALAT1 group received intravenous injections of 10 nmol ASO-MALAT1 on days 1, 3, and 5, while the ASO-NC group received equivalent doses of ASO-NC as a negative control (NC). Tumor progression was monitored noninvasively using bioluminescence imaging (BLI) on days 7, 14, 21, and 28 post-implantation. Imaging was performed using an IVIS Spectrum system (PerkinElmer, Inc., Waltham, MA, USA) following intraperitoneal injection of D-luciferin (150 mg/kg). After 4 weeks, the animals were sacrificed, and their livers were harvested for macroscopic tumor assessment, histological examination, and molecular analysis. Intrahepatic tumor burden and abdominal dissemination were recorded.
HBx transgenic (HBx-Tg) mouse study
HBx-Tg mice were generated as previously described [19, 20]. For the RNA sequencing analysis, livers were collected from HBx-Tg mice (n = 4) and wild-type (WT) mice (n = 4). Eight cDNA libraries were constructed using 3 μg of RNA per liver sample. The quality of the libraries was assessed using the Agilent Bioanalyzer 2100 system. The libraries were sequenced at the Novogene Bioinformatics Institute (Beijing, China) on an Illumina HiSeq 4000 platform, generating 150 bp paired-end reads after clustering of the index-coded samples. Data were extracted from the resulting images using Agilent's Feature Extraction Software (Agilent Technologies; Santa Clara, CA, USA).
To suppress HBx expression in the livers of HBx-Tg mice, in vivo gene delivery was performed via intravenous injection of PB-20F3 transposon plasmids expressing the anti-HBx antibody (provided by Prof. Quan Yuan, Xiamen University) using TransIT-EE Hydrodynamic Delivery Solution (TaKaRa; Kyoto, Japan). Injections were administered through the tail vein at a constant speed and completed within 10 seconds. For long-term gene expression experiments, HBx-Tg mice were injected with 0.3 mL of TransIT-EE containing PB-20F3 (10 μg) and PB-Trans (5 μg) (a universal plasmid containing PBase transposase) once every 4 days for a total of 7 injections. After the final injection, both the WT and HBx-Tg mice were sacrificed, and their livers were collected for tissue lysate extraction and serial sectioning. All animal procedures were ethically approved by the Experimental Animal Ethics Committee of Xiamen University (Ethics Approval No. XMULAC20220282, dated 2022-03-12).
Other materials and methods are described in the Supplementary files.
Results
HBx expression-upregulated MALAT1 is positively correlated with poor prognosis in HCC patients
To identify differentially expressed lncRNAs associated with HBV-related HCC, we extracted three HCC gene expression datasets from the GEO database. Employing the criteria of P < 0.05 and |log2FC| ≥ 1.2, we identified 1043, 1219, and 3379 differentially expressed genes (DEGs) from GSE6764, GSE62232, and GSE98269, respectively, which included both HCC tumor tissues and healthy liver controls. Heatmaps and volcano plots were generated to visualize the DEGs of mRNAs and lncRNAs in patients with HCC (Fig. 1A-C). By intersecting the three cohorts, we identified 346 significantly altered DEGs. Subsequently, we screened 4 lncRNAs for survival analysis using the overall survival (OS) and disease-free survival (DFS) data from the TCGA-LIHC cohort. After survival analysis, we successfully identified 3 candidate lncRNAs (Fig. 1D).
HBx expression-upregulated MALAT1 is positively correlated with poor prognosis in HCC patients. A-C Three HCC gene expression datasets, including GSE6764 (n = 45), GSE62232 (n = 26), and GSE98269 (n = 6), were retrieved from the GEO database. The raw data were normalized, and the numbers of DEGs (P < 0.05 and |log2FC| ≥ 1.2) were 1043, 1219 and 3379, respectively, as shown in the heatmap and volcano plots. D Venn diagram showing the overlap between the DEGs of the three cohorts. E Heatmap of 10 HBx-related HCC-associated lncRNAs in HBx-Tg mice (n = 4) and WT mice (n = 4). F-G Relative expression of 7 candidate lncRNAs was detected via qRT-PCR in HBV/HBx-expressing HCC cells. H Relative expression of 7 candidate lncRNAs was detected via qRT-PCR in HBx-Tg mice livers. I Pan-cancer analysis of MALAT1 expression in the TCGA-LIHC cohort. J-K Quantification of MALAT1 expression in HBV-infected and uninfected HCC tissue and peritumor tissue samples. L GSEA of MALAT1 in the TCGA-LIHC cohort. NES, normalized enrichment score. M-N Paired tumor and adjacent peritumor liver tissues were collected from 12 patients with HBV-related HCC. M Relative expression of MALAT1 was detected by qRT-PCR. N Quantification of FISH results showing fluorescence intensity and percentage of MALAT1-positive area. O Kaplan-Meier analysis of the OS and DFS of patients with MALAT1, with or without hepatitis infection in the TCGA-LIHC cohort. HR, hazard ratio. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
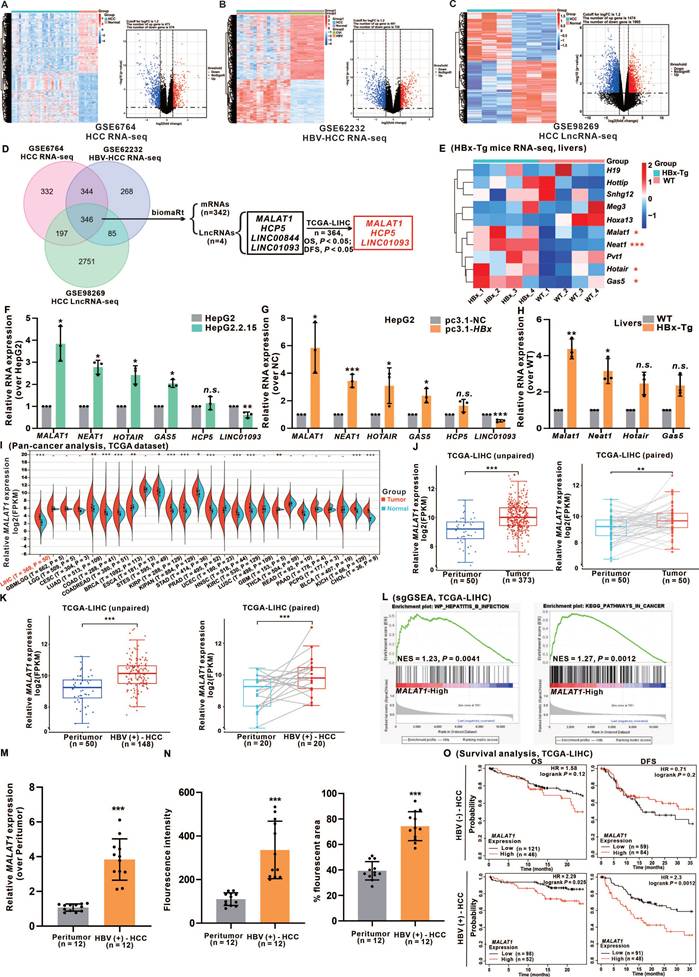
Furthermore, considering the deregulation of multiple lncRNAs in HBV infection and HBV-related HCC patients [21, 22], we evaluated 9 known HBV-related HCC-modulated lncRNAs in the livers of HBx-Tg and WT mice livers via RNA-seq analysis (Fig. 1E). Based on the above results, we successfully identified 6 HBx-associated candidate lncRNAs (MALAT1, NETA1, HOTAIR, GAS5, HCP5, and LINC01093). These lncRNAs were significantly differentially expressed between tumor and normal tissues and were significantly correlated with both the OS and DFS of HCC patients (Fig. S1A). Validation of these candidate lncRNAs' expression levels was performed in HBV/HBx-expressing HCC cells and the livers of HBx-Tg mice (Fig. 1F-H). Based on the expression data, MALAT1, which exhibited the highest abundance, displayed a dose- and time-dependent relationship with HBx expression, suggesting that further investigation is warranted (Fig. S1B-C). To further determine whether the induction of MALAT1 is uniquely driven by HBx rather than by other HBV components, we transfected HepG2 cells with a full-length HBV plasmid, an HBx-deficient (HBV-x-null) plasmid, and an HBx-expressing plasmid alone. Only the presence of HBx significantly upregulated MALAT1 expression, while the x-null construct had no effect (Fig. S1D). Additionally, individual overexpression of HBV-encoded proteins (HBs, HBc, HBp, and HBx) revealed that only HBx was capable of inducing MALAT1 upregulation, further confirming HBx as the sole viral component responsible for this effect (Fig. S1E-F). GO and KEGG enrichment analyses of RNA-Seq data from HBx-Tg mice livers revealed the activation of oncogenic pathways (Fig. S1G-H), which is consistent with the role of HBx in the carcinogenesis of HCC.
To elucidate the oncogenic relevance of MALAT1, a pan-cancer analysis was conducted, revealing that MALAT1 is significantly overexpressed across various tumor types compared with corresponding peritumor tissues (Fig. 1I). Examination of TCGA-LIHC RNA-seq data revealed MALAT1 upregulation in both unpaired and paired HCC tissues, particularly in association with HBV infection (Fig. 1J-K). Single-gene gene-set enrichment analysis (sgGSEA) further highlighted the strong association between MALAT1 and HBV infection, as well as with cancer-related signaling pathways (Fig. 1L). To further evaluate the specificity of MALAT1 in HBV-related HCC, we extended our analysis to publicly available GEO datasets covering multiple HCC etiologies. Specifically, MALAT1 expression was examined in 7 HBV-HCC datasets, 7 HCV-HCC datasets, 4 NAFLD-related HCC datasets, and 2 datasets from patients with alcoholic liver disease (ALD) (Fig. S2A-B). MALAT1 was consistently upregulated in 5 out of 7 HBV-HCC datasets (log2FC > 1), while no significant or consistent change was observed in HCV- or NAFLD-related HCC. Moreover, ALD patient samples showed no significant difference in MALAT1 expression compared with healthy controls (Fig. S2A-B), supporting the notion that MALAT1 upregulation is preferential to HBV-driven hepatocarcinogenesis. In addition to public datasets, we validated MALAT1 expression in an independent clinical cohort of 12 patients with HBV-related HCC from our collaborating hospital. qRT-PCR and FISH performed on paired tumor and adjacent liver tissues confirmed significantly elevated MALAT1 expression in tumor tissues (Fig. 1M-N, S2C), further reinforcing the robustness and translational relevance of our findings. Consistent patterns of upregulation were also observed in the livers of HBx-Tg mice (Fig. S2D), supporting the functional relevance of HBx in MALAT1 activation. Furthermore, survival analysis of the TCGA-LIHC cohort stratified by HBV status indicated that MALAT1 overexpression was significantly associated with reduced OS and DFS exclusively in HBV-positive patients (Fig. 1O). These prognostic implications were further corroborated by the predictive accuracy of MALAT1 in a nomogram-based model (Fig. S2E). Collectively, these results underscore the specificity and prognostic value of MALAT1 in HBV/HBx-related HCC and support its potential utility as a biomarker for early detection and risk stratification.
MALAT1 promotes the proliferation and metastasis of HBV/HBx-expressing HCC cells in vitro
The elevated MALAT1 expression in HCC patients prompted an investigation into its functional role in HBV-related HCC progression. Among the seven HBx-expressing HCC cell lines, HepG2 cells presented the most substantial increase in HBx-upregulated MALAT1 expression (Fig. S3A). Consequently, HepG2 and HepG2.2.15 cells, which harbor the HBV genome, were selected for further investigation. To achieve stable MALAT1 overexpression, two strategies were employed. The initial approach involved transient transfection of the pCDH-MALAT1 plasmid, resulting in a modest threefold increase (Fig. S3B). However, due to the large size of the full-length MALAT1 transcript (8779 bp), further enhancement of expression was limited. Subsequently, CRISPR/Cas9 technology was utilized to construct dCas9-MALAT1 plasmids for transfection into HBV/HBx-expressing HCC cells, ensuring stable and upregulated MALAT1 expression through the dCas9-SAM system (Fig. S3C) [23, 24]. This strategy, which is more effective for achieving high lncRNA expression levels and stable cell lines, circumvents limitations related to transcript length. For MALAT1 silence, ASOs and CRISPR/Cas9 technology were employed, both of which demonstrated efficient silence (Fig. S3D-E). Considering the potential off-target effects associated with Cas9-MALAT1, ASO-MALAT1 has emerged as the preferred approach for subsequent intervention, holding promise in contemporary drug development.
To elucidate the downstream molecular mechanisms governed by MALAT1 in HBV-related HCC, transcriptome profiling was performed in HepG2.2.15 cells following MALAT1 silence. Differential gene expression analysis (|log2FC| > 1, P < 0.05) confirmed successful silencing of MALAT1 (log2FC = -1.2, P < 0.05) and identified a set of significantly regulated genes (Fig. 2A). GO enrichment analysis revealed that these DEGs were predominantly associated with "Cell migration", "Angiogenesis", "Regulation of cell growth", "Epithelial to mesenchymal transition (EMT)", and "ncRNA processing" (Fig. 2B). KEGG analysis further indicated enrichment in the pathways such as "P53 signaling", "Hippo signaling", "VEGF signaling", and "Hepatitis B" (Fig. 2C). Moreover, GSEA demonstrated significant suppression of oncogenic processes such as "Viral hepatitis", "Cell growth regulation", "EMT", and "P53 signaling" in MALAT1-silenced cells (Fig. S3F-I). Collectively, these findings demonstrate that MALAT1 functions as a key oncogenic lncRNA in HBV-related HCC by orchestrating a broad network of cancer-associated signaling pathways. This regulatory role is consistent with MALAT1's known pro-metastatic functions and provides novel insights into its HBx-specific oncogenic mechanisms.
MALAT1 promotes the proliferation and metastasis of HBV/HBx-expressing HCC cells in vitro. A-C HepG2.2.15 cells were transfected with ASO-MALAT1 (100 nM, for 24 h) to construct MALAT1-silence cells, while ASO-NC was used as a negative control (NC). RNA sequencing was performed for transcriptomic profiling analysis in cells. A Volcano plot illustrating DEGs. B GO enrichment analysis of DEGs. C KEGG enrichment analysis of DEGs. D-I HepG2.2.15 and HBx-expressing HepG2 cells were transfected with ASO-MALAT1 (100 nM, for 24 h), while ASO-NC was used as a control. D Colony formation assays were performed to determine the clonogenicity of the cells. The relative number of colonies is shown. E Cell proliferation was detected using EdU staining (green). Nuclei were counterstained with Hoechst 33342 (blue). Scale bar: 100 μm. F-G The levels of E-cadherin and Vimentin were detected by WB (F) and IF staining (G). Scale bar: 20 μm. H Cell migration was measured by wound healing assays. I The migration and invasion of the cells were examined via Transwell assays. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
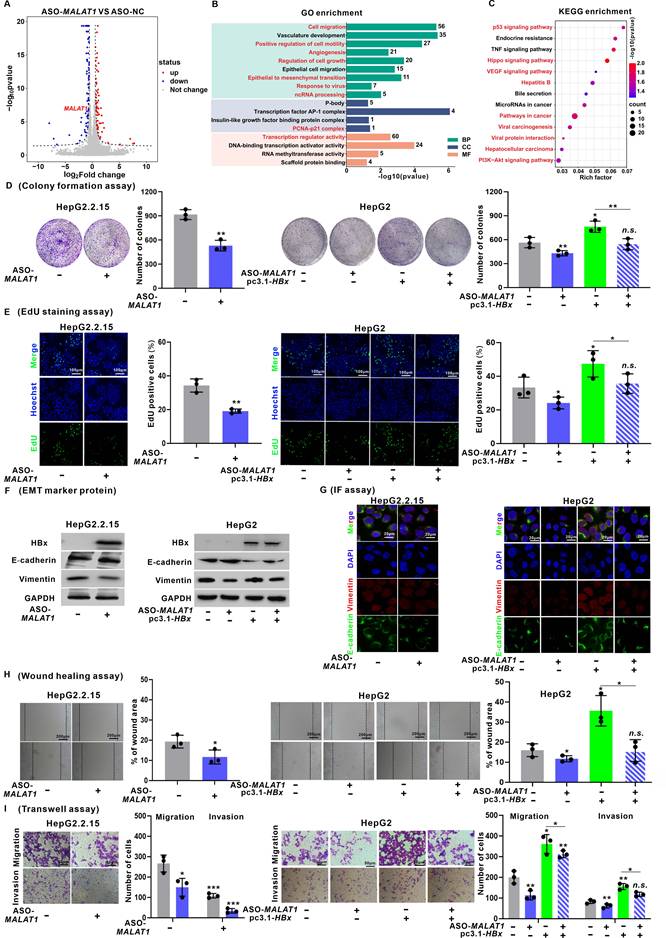
Consistent with these transcriptomic changes, cell viability assays revealed that MALAT1 silence suppressed proliferation, whereas MALAT1 overexpression promoted proliferation in HBV/HBx-expressing HCC cells (Fig. S3J-K). This observation was further corroborated by colony formation and EdU assays (Fig. 2D-E, Fig. S4A-B). Additionally, a marked decrease in the epithelial marker E-cadherin was detected upon MALAT1 silence, while an increase was observed with MALAT1 overexpression. Conversely, the expression of the mesenchymal marker Vimentin decreased with MALAT1 silence but increased with MALAT1 overexpression (Fig. 2F-G, Fig. S4C-D). Wound healing and Transwell assays demonstrated that MALAT1 silence notably reduced the migratory and invasive capacities, whereas MALAT1 overexpression enhanced these abilities in HBV/HBx-expressing HCC cells (Fig. 2H-I, Fig. S4E-F). These findings illustrate the oncogenic role of MALAT1 in promoting the proliferation and metastasis abilities of HBV-associated and HBx-expressing HCC cells.
Interestingly, beyond its impact on tumor progression, MALAT1 silencing also affected HBV replication. Quantification of virological parameters after ASO-MALAT1 treatment showed that while HBsAg and HBeAg levels remained largely unchanged (Fig. S5A-B), pgRNA, intracellular HBV DNA, and relaxed circular cccDNA (rcccDNA) levels were significantly reduced (Fig. S5C-E). These findings suggest a previously unrecognized role for MALAT1 in modulating HBV transcription and replication, possibly through regulation of viral RNA stability or chromatin-related transcriptional machinery. This expands the functional significance of MALAT1 beyond tumorigenesis, revealing its dual involvement in both hepatocarcinogenesis and viral persistence.
The m6A level of MALAT1 is upregulated by the recruitment of the m6A methyltransferase complex
Recent studies on tumor epigenetics have shed light on the impact of m6A modification on lncRNAs [25]. To investigate the potential influence of m6A modification on MALAT1 regulation in HCC, we analyzed transcriptomic data from HBV-related HCC patients and normal individuals from the GEO database (GSE94660) and identified 7 "writers", 2 "erasers", and 11 "readers" of m6A modification on the basis of prior research [26]. A heatmap revealed significant m6A methylation activation in the livers of patients with HBV-related HCC (Fig. 3A), with a positive correlation between the relative expression levels of these genes (Fig. 3B). Comparable results were observed in our transcriptome analysis of HBx-Tg mice livers (Fig. S6A-B). By demonstrating a significant increase in overall m6A levels in HBV/HBx-expressing HCC cells (Fig. 3C), we found that this increase in m6A was specifically induced by HBx and displayed a dose-dependent relationship with HBx expression (Fig. S6C-D). Additionally, by assessing the transcription levels of the seven "writers", we observed notable increases in the mRNA and protein expression levels of methyltransferase-like 3 (METTL3), METTL14, and Wilms tumor 1-associating protein (WTAP) in HepG2.2.15 cells and HBx-expressing HepG2 cells in an HBx-dependent manner (Fig. 3D-E, Fig. S6E). These findings suggested that HBx promotes m6A levels mainly by upregulating METTL3, METTL14, and WTAP.
The m6A level of MALAT1 is upregulated by the recruitment of the m6A methyltransferase complex. A Heatmap of 20 m6A-related genes in the livers of HBV-HCC patients (n = 21) and peritumor (n = 21) tissues. B Pearson's correlation analysis was used to determine the relationships between the mRNA levels of 20 m6A-related genes. C The overall m6A content in HepG2.2.15 and HepG2 cells was analyzed by m6A quantitation analysis. D Relative expression of the 7 m6A methyltransferase genes was detected by qRT-PCR in HepG2.2.15 and HepG2 cells. E The protein levels of METTL3, METTL14, and WTAP in HepG2.2.15 and HepG2 cells were detected by WB. F Venn diagram showing the overlap of METTL3-, METTL14-, and WTAP-bound transcripts revealed by CLIP-seq and the TCGA-LIHC cohort DEGs. G MeRIP of MALAT1 transcripts in HepG2.2.15 and HepG2 cells upon indicated knockdown. The abundance of MALAT1 among MeRIP with anti-m6A antibodies was measured via qPCR and normalized to that of IgG. H MeRIP of MALAT1 transcripts in HepG2.2.15 cells. I METTL3, METTL14, and WTAP were pulled down by biotin-labeled sense MALAT1 (S) but not MALAT1 antisense (AS) RNA in HepG2.2.15 and HepG2 cells. J METTL3, METTL14, and WTAP were pulled down in cytoplasmic and nuclear fractions of HepG2.2.15 and HepG2 cells. K MALAT1 secondary structure was predicted by lnCAR. L Scheme of full-length biotinylated-MALAT1 (S#2), antisense MALAT1 (S#1), and 8 truncated biotinylated-MALAT1 fragments based on the sub-structures: S#3(1-1097 nt), S#4 (1098-2195 nt), S#5 (2196-3293 nt), S#6 (3294-4391 nt), S#7 (4392-5489 nt), S#8 (5490-6587 nt), S#9 (6588-7685 nt) and S#10 (7686-8779 nt). The full-length biotinylated-MALAT1 and its truncations were separately transfected into HepG2.2.15 cells. M Dual-luciferase reporter assays were used to confirm the interaction between METTL3, METTL14, WTAP and 2 mutations (6021C and 7265C) of the MALAT1 m6A sites. N MeRIP of MALAT1 transcripts in HepG2.2.15 cells transfected with MALAT1 mutant plasmids. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
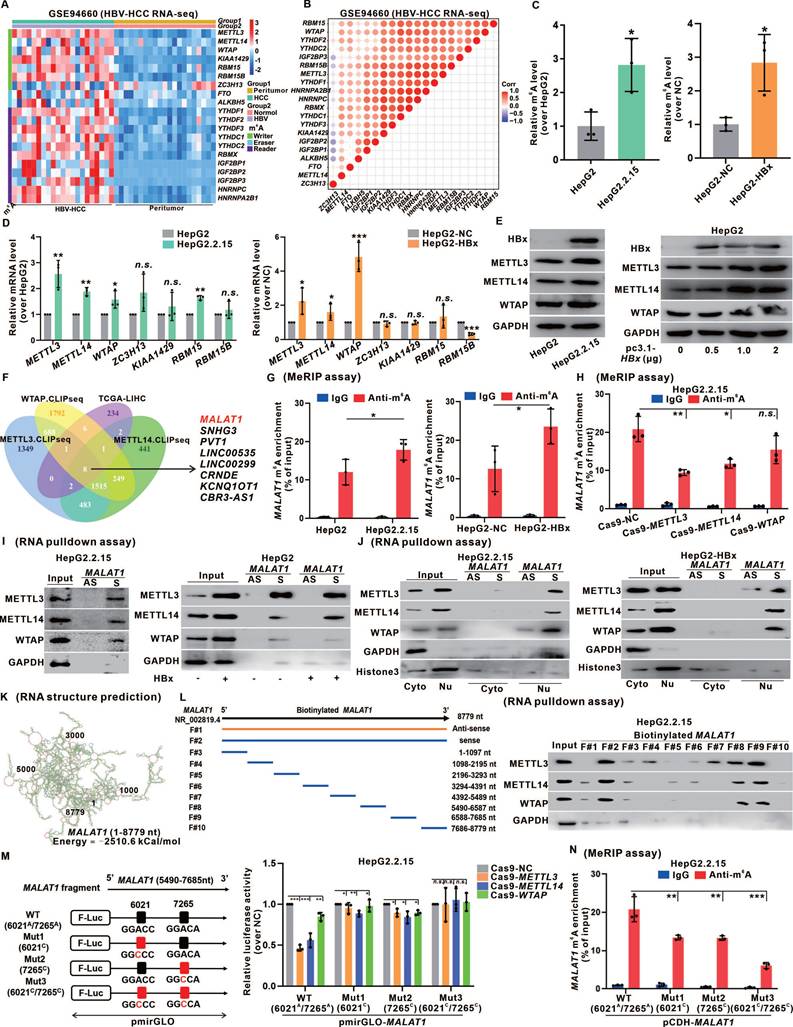
Analysis of the TCGA-LIHC cohort and the cross-linking and immunoprecipitation high-throughput sequencing (CLIP-seq) data reported previously [27] revealed that the METTL3, METTL14, and WTAP proteins potentially bind to MALAT1, which is linked to HCC pathogenesis (Fig.3F). MeRIP assays confirmed a significant increase in MALAT1 m6A levels in HBV/HBx-expressing HCC cells (Fig. 3G). To investigate the involvement of METTL3, METTL14, and WTAP in regulating MALAT1 m6A methylation, we constructed the corresponding Cas9 plasmids to knock down these "writers" (Fig. S6F). There was a significant decrease in MALAT1 m6A levels in HepG2.2.15 cells upon knockdown of METTL3 or METTL14 (Fig. 3H). We designed RNA pulldown biotinylated probes specific for MALAT1 and confirmed their effectiveness (Fig. S6G), validating the direct binding between MALAT1 and METTL3, METTL14, and WTAP in HepG2.2.15 cells and HBx-expressing HepG2 cells, with increased interactions in HBx-expressing cells (Fig. 3I). Moreover, subcellular fractionation in the RNA pulldown assays confirmed this binding regulation of HBV/HBx-associated MALAT1 m6A methylation, specifically within the nucleus (Fig. 3J). Given that METTL3 serves as the primary catalytic enzyme in the methylation system, we generated both METTL3 wild-type (WT) and its mutant (Mut) enzyme active site to confirm its influence on MALAT1 m6A regulation definitively, and MeRIP assays revealed a significant reduction in MALAT1 m6A levels in the METTL3-Mut group compared with those in the METTL3-WT group (Fig. S6H).
MALAT1 binding with METTL3, METTL14, and WTAP is regulated by its recognition of the m6A consensus motif with specific m6A sites. We utilized the lnCAR platform to analyze the secondary structure (maximum free energy was -2510 kCal/mol) of MALAT1 (Fig. 3K). Based on the predicted structure and full length (F#2, 1-8779 nt) of MALAT1, eight smaller fragments were transcribed (F#3-10) via in vitro transcription. These fragments were biotinylated and subjected to RNA pulldown assays. We found that METTL3, METTL14, and WTAP were simultaneously pulled down by F#8 and F#9, which correspond to the 5490 nt to 7685 nt region of MALAT1 (Fig. 3L), indicating that the potential regulatory domain for MALAT1 m6A modification might reside within this flanking region. We subsequently employed the common prediction tools SRAMP and BERMP to screen the m6A-targeting sites in this domain further. The intersection of the predictions revealed two m6A sites: the 6021A site in the eighth fragment (F#8) and the 7265A site in the ninth fragment (F#9) (Fig. S6I). Additionally, the m6A binding motif of MALAT1 was identified via the online platforms RMBase v2.0 and STAMP, which was consistent with 6021A (GGACC) and 7265A (GGACA) (Fig. S6J). To confirm our target screening results, dual-luciferase reporter gene plasmids based on pmirGLO that fused the linear sequence of the 5490 nt to 7685 nt fragment of MALAT1 and the mutants of the 6021C or/and 7265C sites were constructed. Dual-luciferase reporter assays confirmed that both 6021A and 7265A were the m6A-targeting modification sites regulated by the binding of MALAT1 to METTL3, METTL14, and WTAP in HepG2.2.15 cells (Fig. 3M). Moreover, site-directed mutagenesis plasmids with 6021A or/and 7265A sites based on pCDH-MALAT1 were generated (Fig. S6K), and the results of MeRIP assays revealed that both 6021A and 7265A sites were necessary for the upregulation of MALAT1 m6A modification in HepG2.2.15 cells (Fig. 3N). Additionally, the bacterial single-stranded RNase MazF assays identified the MALAT1 7265A site with m6A methylation in HBV-related HepG2.2.15 cells (Fig. S6L). These findings suggested that the MALAT1 m6A level was upregulated due to the recruitment of the METTL3/METTL14/WTAP methylation complex in HBV/HBx-related HCC cells, with specific binding at m6A sites identified as 6021A and 7265A.
IGF2BP3 interacts with and stabilizes MALAT1 in a m6A-dependent manner
Growing evidence indicates that many lncRNAs can function to regulate the expression of target genes through direct interactions with proteins [28, 29]. In the present study, we employed the starBase database to identify potential RNA-binding proteins (RBPs) that interact with MALAT1. By intersecting the results with those of the TCGA-LIHC and GEO HCC datasets, we identified six RBPs potentially interacting with MALAT1 (Fig. 4A). Subsequent assessment in HBx-expressing cells highlighted significant upregulation of IGF2BP3 among the candidate RBPs in response to HBV/HBx expression (Fig. 4B). Furthermore, a significant positive correlation between IGF2BP3 and MALAT1 expression in the TCGA-LIHC cohort was observed (Fig. 4C). Given these findings, we further investigated the role of IGF2BP3 in MALAT1 regulation in HBV/HBx-related HCC. Immunohistochemistry analysis demonstrated that HBx expression significantly induced the upregulation of IGF2BP3 in the livers of HBx-Tg mice and was associated with HBV-related HCC (Fig. S7A-B). Pan-cancer analysis revealed elevated IGF2BP3 expression across various cancer types, particularly in liver tumors, compared with that in peritumor tissues (Fig. S7C). Additionally, Kaplan-Meier analysis revealed an association between high IGF2BP3 expression and lower OS and DFS in the TCGA-LIHC cohort (Fig. S7D). These findings highlight the potential role of IGF2BP3 regulation in HBV/HBx-related HCC prognosis and its potential association with MALAT1.
IGF2BP3 interacts with and stabilizes MALAT1 in a m6A-dependent manner. A Venn diagram showing the overlap of DEGs from GSE6764, GSE94660, TCGA-LIHC cohort, and starBase database. B Relative expression of the six candidate RBPs was detected by qRT-PCR in HepG2.2.15 and HBx-expressing HepG2 cells. C Correlation analysis showing a positive relationship between IGF2BP3 and MALAT1 in the TCGA-LIHC cohort. D Binding ability of MALAT1 and IGF2BP3 was predicted by RPISeq. E Interaction between MALAT1 and IGF2BP3 was predicted by HDOCK. F Representative images showing the colocalization of MALAT1 (red) and IGF2BP3 (green) in HepG2.2.15 and HBx-expressing HepG2 cells. Scale bars: 20 μm. G RNA pulldown assays followed by silver staining of protein extracts from HBx-expressing HepG2 cells. H RNA pulldown assays were performed in HepG2.2.15 and HepG2 cells. I RIP assays were performed in HepG2.2.15 and HepG2 cells. J RIP assays were performed in HepG2.2.15 cells transfected with Cas9-METTL3, Cas9-METTL14, and Cas9-WTAP plasmids. K Dual-luciferase reporter assays were used to confirm the interaction between IGF2BP3 and two mutants (6021C and 7265C) of the MALAT1 m6A. L RIP assays were performed in HepG2.2.15 cells transfected with MALAT1 containing the indicated mutations. M Scheme of Flag-tagged full-length IGF2BP3 (F#6) and the five truncated mutants (F#1: 1-77aa; F#2: 1-156aa; F#3: 1-262aa; F#4:1-344aa; F#5:1-471aa) were constructed (left). In vitro binding assays showing the enriched MALAT1 in HepG2.2.15 cells detected by RT-PCR (right, upper panel) after incubation with full-length or truncated Flag-tagged IGF2BP3 protein validated by WB (right, lower panel). N RIP assays were performed in HepG2.2.15 cells transfected with plasmids containing the full-length or truncated constructs. O RNA pulldown assays were performed in HepG2.2.15 cells transfected with plasmids containing the indicated full-length or truncated constructs. P The RNA half-life of MALAT1 was measured by performing RNA decay assaysin HepG2.2.15 and HBx-expressing HepG2 cells with IGF2BP3 overexpression or knockdown. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
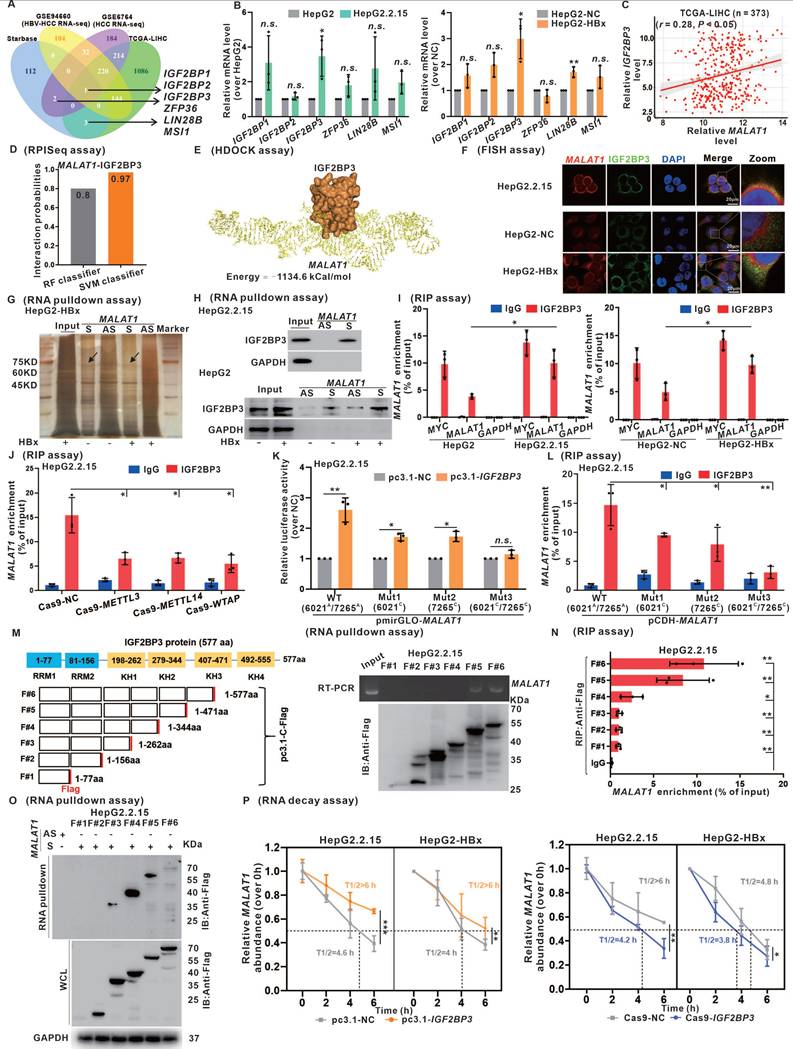
To investigate the interactive regulation between IGF2BP3 and MALAT1, we quantitatively assessed their binding potential via RPISeq (Fig. 4D) and established a molecular docking model via HDOCK, which illustrates their structural binding (Fig. 4E). Validation via RNA-FISH confirmed the IGF2BP3-MALAT1 interaction in HBV/HBx-expressing HCC cells, with stronger binding observed in the presence of HBx (Fig. 4F). Subsequent RNA pulldown for silver staining (Fig. 4G) and WB (Fig. 4H) confirmed the presence of IGF2BP3 (~75 kDa) in the MALAT1 fraction from HBx-expressing cells. In addition, RIP assays were utilized to validate the specific binding interaction between IGF2BP3 and MALAT1. Compared with the positive control MYC, which is known to bind with IGF2BP3 [31], HBV/HBx expression increased the interactive binding of MALAT1 with IGF2BP3 (Fig. 4I). An investigation of m6A-dependent regulation revealed that knockdown of METTL3, METTL14, or WTAP reduced the binding between IGF2BP3 and MALAT1 (Fig. 4J). Dual-luciferase reporter assays using MALAT1-WT and its m6A site mutant plasmids in IGF2BP3-expressing HepG2.2.15 cells revealed that mutations of the 6021C and 7265C sites of MALAT1 inhibited its binding activity with IGF2BP3 (Fig. 4K). Further RIP assays in cells transfected with MALAT1 mutants demonstrated attenuated binding between IGF2BP3 and MALAT1 upon mutation of these sites (Fig. 4L).
Considering the structure of IGF2BP3 protein (1-577 aa), which includes two RNA recognition motifs (RRMs) and four K homology (KH) domains, we constructed six flag-tagged recombinant plasmids containing overlapping truncated fragments (F#1-6) of IGF2BP3. In in vitro RNA binding assays, deletion of the KH3 domain (407-471 aa) of the flag-tagged IGF2BP3 protein significantly decreased, or even abolished, the binding interaction between IGF2BP3 and MALAT1 (Fig. 4M-O). These results indicate the crucial role of the KH3 domain in the HBV/HBx-related IGF2BP3-targeting binding interaction with MALAT1.
To assess the regulatory effect of IGF2BP3 on MALAT1 expression, our study revealed that MALAT1 expression did not affect IGF2BP3 mRNA or protein levels in HBV/HBx-expressing HCC cells, regardless of whether MALAT1 was overexpressed or knocked down (Fig. S7E-G). Conversely, in cells with IGF2BP3 overexpression or knockdown (Fig. S7H-I), overexpressing IGF2BP3 increased MALAT1 levels, whereas knocking down IGF2BP3 decreased MALAT1 expression in HBV/HBx-expressing HCC cells (Fig. S7J-K). Furthermore, RNA decay assays demonstrated that IGF2BP3 inhibited MALAT1 decay rates, suggesting that IGF2BP3 stabilizes MALAT1 in HBV/HBx-expressing HCC cells (Fig. 4P). These findings, along with the identification of IGF2BP3 as a newly characterized m6A lncRNA-binding protein and in conjunction with our earlier experiments shown in Fig. 3, suggest that IGF2BP3 might specifically interact with MALAT1 to increase its stability in a m6A site-dependent manner in HBV/HBx-related HCC.
IGF2BP3 promotes MALAT1 nuclear-cytoplasmic shuttling in a m6A‑dependent manner
MALAT1 is known to be highly abundant in the nucleus, specifically in nuclear speckles, which are nuclear compartments involved in pre-mRNA splicing and storage [32]. Upon performing FISH and RNA nucleocytoplasmic isolation assays of MALAT1, we discovered the abundant presence of MALAT1 in the cytoplasm in HBV/HBx-expressing HCC cells, challenging our original assumption that it was enriched primarily in the nucleus (Fig. 5A-B). Moreover, knockdown of METTL3, METTL14, or WTAP led to increased nuclear localization of MALAT1 in HBV/HBx-expressing HCC cells, indicating a link between the nucleocytoplasmic shuttling of MALAT1 and its m6A modification (Fig. 5C). Transfection of m6A site mutants (6021C and/or 7265C) of MALAT1 predominantly confined these overexpressed mutants to the nucleus of HBV/HBx-expressing HCC cells (Fig. 5D).
IGF2BP3 promotes MALAT1 nuclear-cytoplasmic shuttling in a m6A‑dependent manner. HepG2.2.15 cells and HBx-expressing HepG2 cells were used for experiments, whereas HepG2 cells and HepG2-NC cells served as controls. A Representative images of FISH assays identifying the subcellular location of MALAT1 (red) in the cells. Scale bars: 20 μm. B The subcellular distribution of MALAT1 was analyzed via qRT-PCR in the cells. U6 and GAPDH were used as nuclear and cytoplasmic markers, respectively. C-D The subcellular distribution of MALAT1 was analyzed via qRT-PCR in the cells transfected with the indicated plasmids. E sgGSEA of IGF2BP3 in the TCGA-LIHC cohort. NES, normalized enrichment score. F The protein level of IGF2BP3 was detected by WB in the cytoplasmic and nuclear fractions of the cells. G RNA pulldown assays were conducted, and IGF2BP3 was pulled down by biotin-labeled sense MALAT1 (S) but not by MALAT1 antisense (AS) RNA in the cytoplasmic and nuclear fractions of the cells. H The subcellular distribution of MALAT1 was analyzed via qRT-PCR in the cells transfected with the indicated plasmids. I-J RNA decay assays showing the effect of overexpressing and knocking down IGF2BP3 on MALAT1 RNA half-life in the cells.
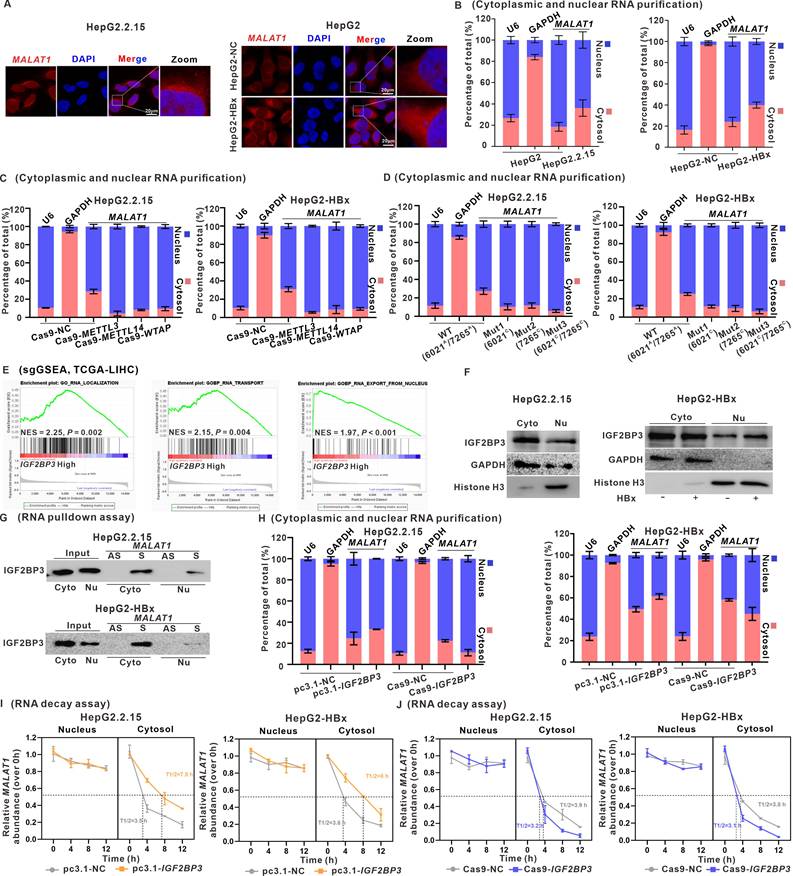
As shown in Fig. 4, we identified the role of IGF2BP3 in regulating the stability of MALAT1 through m6A modification, which led us to consider whether IGF2BP3 participated in the regulatory nucleocytoplasmic shuttling of MALAT1. sgGSEA identified the involvement of IGF2BP3 in RNA localization (NES = 2.25), RNA transport (NES = 2.15), and RNA export from the nucleus (NES = 1.97) (Fig. 5E). The results of the cytoplasmic and nuclear protein separation assays demonstrated the presence of IGF2BP3 in the nucleus, whereas its levels were increased in HBV/HBx-expressing HCC cells (Fig. 5F). Furthermore, RNA pulldown experiments showed that IGF2BP3 binds to MALAT1 in both the cytoplasm and nucleus (Fig. 5G). Moreover, IGF2BP3 knockdown resulted in the retention of MALAT1 in the nucleus, whereas its overexpression facilitated its cytoplasmic distribution in HBV/HBx-expressing cells (Fig. 5H). As shown in Fig. 4P, IGF2BP3 enhanced MALAT1 stability, prompting further investigation into whether this stability is linked to its cellular localization. Following the separation of cytoplasmic and nuclear RNA, RNA decay assays revealed that HBx primarily prevented MALAT1 degradation in the cytoplasm, with minimal degradation in the nucleus (Fig. S8A-B), possibly due to MALAT1 binding to multiple RBPs in the nucleolus. Additionally, IGF2BP3 also reduced MALAT1 decay rates in the cytoplasm (Fig. 5I-J). These findings indicate that IGF2BP3, which binds to m6A-modified MALAT1, facilitates the nuclear export of m6A-modified MALAT1 for its cytoplasmic localization and RNA stabilization.
Targeting MALAT1 in vivo with ASO-MALAT1 treatment effectively suppresses xenograft tumor progression in HBx-related HCC
To assess the effects of MALAT1 and its m6A methylation regulatory signaling axis on HBx-related HCC migration and metastasis, a nude mouse model harboring subcutaneous xenograft liver tumors was used (Fig. 6A). There were no observable changes in the body weights of the mice in the various groups (Fig. S9A). Compared with control tumors, xenograft tumors derived from HBx-expressing HepG2 cells were larger in size, heavier in weight, and grew faster (Fig. 6B-D). When MALAT1 was knocked down with ASO-MALAT1 in xenograft tumors derived from HBx-expressing HepG2 cells, significantly smaller tumor sizes, weights, and volumes were detected than in the HBx+ASO-NC group (Fig. 6B-D). The MALAT1 level and its distribution in xenograft tumor tissues were increased in the HBx group (Fig. 6E, Fig. S9B), whereas they were decreased in the ASO-MALAT1 group ((Fig. 6E, Fig. S9B). Moreover, the overall m6A level and its related METTL3, METTL14, and WTAP protein levels, as well as cytoplasmic MALAT1-exporting IGF2BP3 and its distribution in xenograft tumor tissues, were increased in the HBx group (Fig. 6F-G, Fig. S9C). During the malignant progression of HBx-related HCC, compared with those in the HBx group, the levels of HBx-induced Ki67 and Vimentin in xenograft tumor tissues were lower in the ASO-MALAT1 group (Fig. 6F-G), indicating that MALAT1 modulates HBx-driven tumor growth and metastatic potential through an m6A-dependent mechanism.
Targeting MALAT1 in vivo with ASO-MALAT1 treatment effectively suppresses xenograft tumor progression in HBx-related HCC. A-G BALB/c nude mice were used to construct the subcutaneous xenograft models with HBx-expressing HepG2 cells. The mice were divided into four groups: Ctrl (HepG2-pc3.1-NC), HBx (HepG2-pc3.1-HBx), HBx+ASO-NC, and HBx+ASO-MALAT1 (n = 8 per group). For MALAT1-targeting intervention, mice were treated with ASO-MALAT1(5 nmol per injection, once every 2 days, for a total of 7 injections), while control oligonucleotides (ASO-NC) served as a negative control. A Schematic diagram of the experimental design, where SPSS represents stroke-physiological saline solution. B Representative images showing the subcutaneous tumor xenografts. C Tumor weights of the xenografts were evaluated. D Tumor growth curves of the xenografts were plotted. E Representative images showing FISH assays of the RNA levels of MALAT1 (red) in the xenograft tumors. Nuclei were counterstained with DAPI (blue). Scale bars: 100 μm. F Levels of HBx, METTL3, METTL14, WTAP, IGF2BP3, E-cadherin, and Vimentin proteins in the xenograft tumors were detected by WB. G Representative images showing the HE staining, HBx, METTL3, METTL14, WTAP, IGF2BP3, Ki67, E-cadherin, and Vimentin immunostaining of the xenograft tumors. Scale bars: 100 μm. H-M An orthotopic liver tumor model was established in nude mice by injecting HepG2.2.15-Luc cells. Mice were then treated with ASO-MALAT1, while ASO-NC were used as a negative control (n = 7 per group). H Representative bioluminescence images of orthotopic liver tumors, with imaging performed on days 7, 14, 21, and 28 post-treatment. I-J Quantification of tumor burden based on total bioluminescence signal intensity (I) and tumor volume (J) over time. K Gross images of excised livers from each group on day 28. Intrahepatic orthotopic tumor nodules are outlined with dashed circles. L-M Quantification of tumor weight (L) and number of intrahepatic tumor nodules (M) is presented in dot plots. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
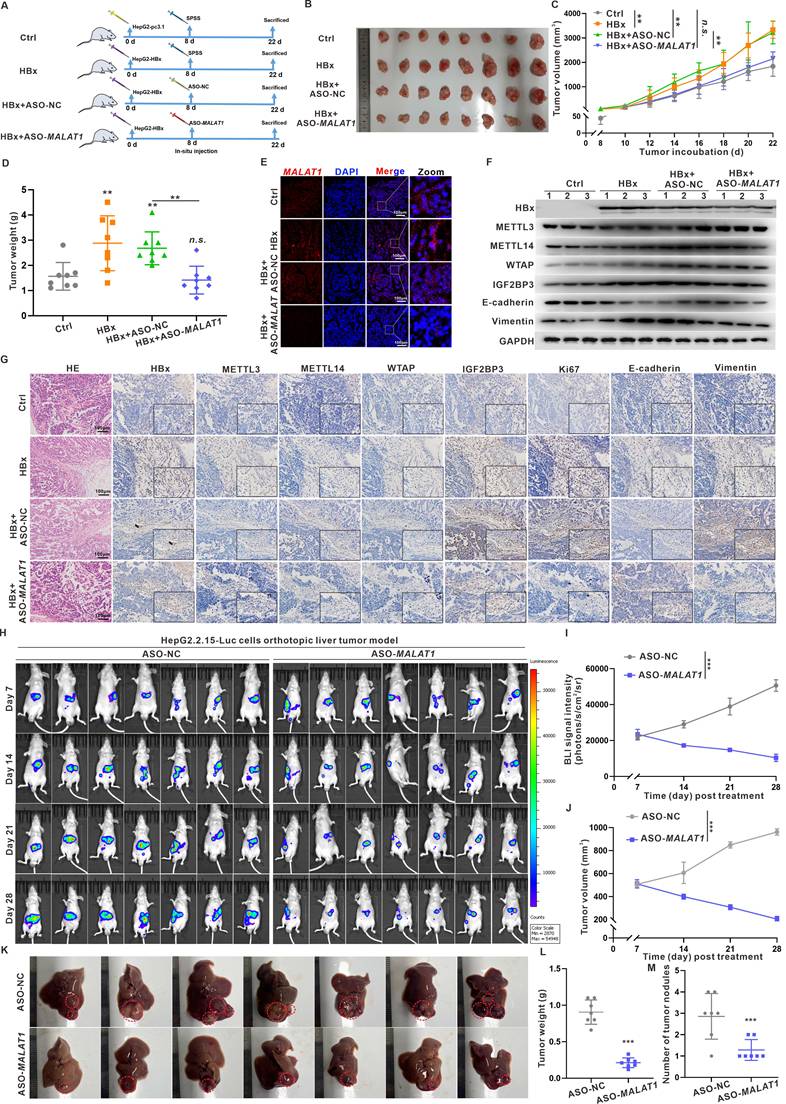
To further validate the metastatic function of MALAT1 in a physiologically relevant model, we established an orthotopic liver tumor model using luciferase-labeled HepG2.2.15 (HepG2.2.15-Luc) cells. There were no observable changes in the body weights of the mice in the various groups (Fig. S9D). Mice receiving ASO-MALAT1 treatment showed significantly reduced bioluminescent signal intensity over 28 days, suggesting suppressed tumor progression (Fig. 6H-J). At necropsy, tumors in the ASO-MALAT1 group were visibly smaller with fewer intrahepatic nodules, indicating local tumor suppression. Moreover, qRT-PCR analysis confirmed that ASO-MALAT1 treatment markedly reduced hepatic MALAT1 expression levels (Fig. S9E). Subsequent WB revealed increased E-cadherin and decreased Vimentin expression, indicating suppression of EMT following MALAT1 knockdown (Fig. S9F). Although no lung metastases were observed—likely due to the relatively short duration of the experiment—abdominal dissemination was detected in control animals but was notably reduced in the ASO-MALAT1 group (Fig. 6K-M). These observations provide functional evidence that MALAT1 contributes to both tumor growth and early metastatic dissemination in HBV-related HCC. Together, findings from the xenograft and orthotopic models underscore the critical role of MALAT1 in promoting HBx-mediated hepatocarcinogenesis and metastasis. Suppression of MALAT1 not only attenuates tumor proliferation but also limits intrahepatic expansion and peritoneal spread, supporting its therapeutic potential as a dual-target regulator of tumor progression and early dissemination.
Anti-HBx gene delivery via transposons suppresses MALAT1-m6A-initiated HBV-related hepatocarcinogenesis in vivo
Given the pivotal role of HBx in HBV-related hepatocarcinogenesis initiation and progression, HBx-targeting suppression represents a promising strategy to inhibit HBV replication and prevent HBV-related HCC [17, 20]. In previous studies, we successfully suppressed the biofunction of intercellular HBx expression in HCC cells via an anti-HBx monoclonal antibody (mAb) delivered through a recombinant anti-HBx plasmid (pTT5-anti-HBx) [20]. In this study, we further expanded our anti-HBx intervention to target the MALAT1-m6A-initiated pathway, thereby suppressing hepatocarcinogenesis phenotypes in HBx-Tg mice. We employed the PiggyBac system, a mammalian gene transfer system based on a transposon, to express the anti-HBx mAb and interfere with HBx expression in mice (Fig. 7A-B). Concurrently, hydrodynamic-based gene delivery (HGD) was utilized as a highly efficient technique to introduce the anti-HBx plasmid into murine hepatocytes [33]. HBx-Tg mice were administered an intravenous injection of a PB-20F3 transposon containing an anti-HBx mAb-encoding sequence (Fig. 7A-B), which was then delivered into the mouse liver via HGD to efficiently express the anti-HBx mAb, visualized with mRuby3 (Fig. 7C).
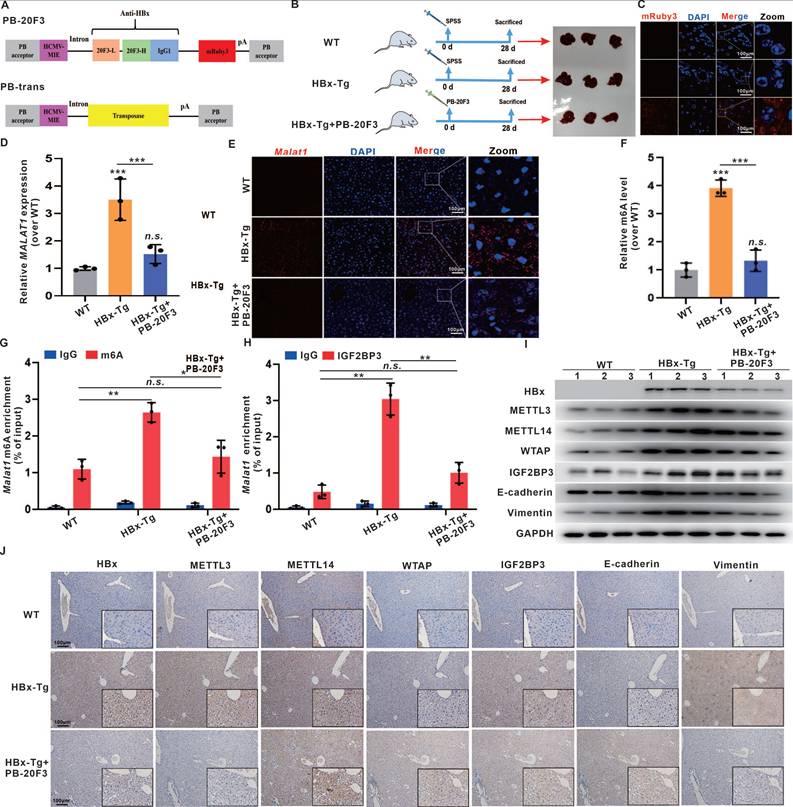
Anti-HBx gene delivery via transposons suppresses MALAT1-m6A-initiated HBV-related hepatocarcinogenesis in vivo. HBx-Tg mice were injected with 0.3 mL of TransIT-EE containing PB-20F3 (10 μg) and PB-Trans (5 μg) once every 4 days for a total of 7 injections. A Scheme of the plasmids PB-20F3, with SPSS used as a negative control. B Schematic diagram of the experimental design for HBx-targeting intervention in HBx-Tg mice. C Representative images showing fluorescence of mRuby3 in livers from the different groups, with nuclei counterstained with DAPI (blue). Scale bars: 100 μm. D The relative expressions of MALAT1 in the livers was detected by qRT-PCR. E Representative images showing FISH assays of the RNA levels of MALAT1 (red) in the livers, with nuclei counterstained with DAPI (blue). Scale bars: 100 μm. F The overall m6A content in the livers was analyzed by m6A quantitation analysis. G MeRIP assays were performed in the livers, while the abundance of MALAT1 with anti-m6A antibodies was measured by qRT-PCR and normalized to that of IgG. H RIP assays were performed with IGF2BP3 antibody in the livers, while the abundance of MALAT1 was measured by qRT-PCR. I The levels of HBx, METTL3, METTL14, WTAP, IGF2BP3, E-cadherin, and Vimentin proteins in the livers were detected by WB. J Representative images showing HBx, METTL3, METTL14, WTAP, IGF2BP3, E-cadherin, and Vimentin immunostaining of livers. Scale bars: 100 μm. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
Although the HBx-Tg mice did not develop overt liver tumors during the 6-month observation period, consistent with previous reports requiring a longer latency (14-18 months) for spontaneous HCC development [34, 35], histopathological examination revealed clear preneoplastic alterations. HE staining identified hepatocellular nuclear atypia (e.g., anisokaryosis, hyperchromasia, prominent nucleoli), while immunohistochemistry showed elevated expression of early HCC markers—GPC3, AFP, Ki67, and PCNA—and activation of EMT-associated markers (Fig. S10). These findings demonstrate that persistent HBx expression initiates early oncogenic events prior to macroscopic tumor formation.
Importantly, anti-HBx treatment via the PB-20F3 plasmid significantly reversed these molecular and histological alterations. The expression and hepatic distribution of Malat1 were elevated in HBx-Tg mice livers but reduced in the PB-20F3 group (Fig. 7D-E). The overall m6A level and related METTL3, METTL14, and WTAP protein levels, as well as their distribution in liver tissues, were increased in the HBx group (Fig. 7F-J), whereas these HBx-induced m6A regulators were downregulated in the PB-20F3 group (Fig. 7F-J). In particular, Malat1 m6A enrichment and Malat1 binding with IGF2BP3 were increased in the HBx group but downregulated in the PB-20F3 group (Fig. 7G-H). Compared with those in the HBx group, the levels of HBx-induced Vimentin in liver tissues were lower in the PB-20F3 group (Fig. 7I-J). Collectively, these data demonstrate that genetic intervention targeting HBx not only mitigates MALAT1 overexpression but also disrupts the associated m6A epitranscriptomic network that contributes to early hepatocarcinogenic signaling.
Discussion
HBx acts as an oncoprotein and plays a key role in HBV-related HCC by influencing epigenetic modifications and genetic regulation. Epigenetic alterations, including DNA methylation, histone acetylation, and microRNA (miRNA) regulation, have been reported as early events in tumorigenesis, contributing to the malignant transformation of hepatocytes [36]. Emerging evidence has implicated lncRNAs in HBV-induced carcinogenesis via interactions with epigenetic modification complexes. For example, HBx repressed LINC01431 transcription and decreased the stability of PRMT1, resulting in reduced PRMT1 enrichment on cccDNA and increased cccDNA accessibility and transcription [37]. Despite these findings, the precise role and mechanisms of lncRNAs in HBV-related HCC, particularly how HBx promotes carcinogenesis and tumorigenesis through lncRNAs, remain unclear and warrant further investigation. This study comprehensively analyzed differentially expressed lncRNAs in HBV-related HCC patients via multiple datasets and validated their significance in the liver transcriptomes of HBx-Tg mice. Our investigation revealed that HBx expression alters the lncRNA profile in HBx-expressing cells, indicating that a distinct lncRNA profile is associated with HBV-related HCC tumorigenesis. Notably, MALAT1 was significantly overexpressed in response to HBx across multiple datasets and in HBx-Tg mice, and this overexpression was correlated with poor prognosis in HBV-related HCC patients. MALAT1, an oncogenic lncRNA, promotes cancer progression through various mechanisms, such as miRNA sequestration, autophagy stimulation, and the induction of epithelial-mesenchymal transition [32]. However, the specific involvement of HBx in elevated MALAT1 levels in HCC and the underlying regulatory mechanisms have remained elusive. Our results revealed that HBx induced MALAT1 upregulation, which is correlated with poor prognosis in HBV-related HCC patients. Additionally, the knockdown of MALAT1 in HBx-expressing cells suppressed hepatocarcinogenesis phenotypes, and in vivo experiments demonstrated the inhibition of xenograft tumor growth upon MALAT1 knockdown (Fig. 8). These findings emphasize the significant role of MALAT1 in HBV-related HCC progression and prognosis, indicating that MALAT1 is a potential therapeutic target.
Schematic diagram illustrating the mechanisms of IGF2BP3-mediated m6A modification of MALAT1 in promoting the malignant progression of HBV/HBx-related HCC, as well as potential strategies for epigenetic or genetic intervention.
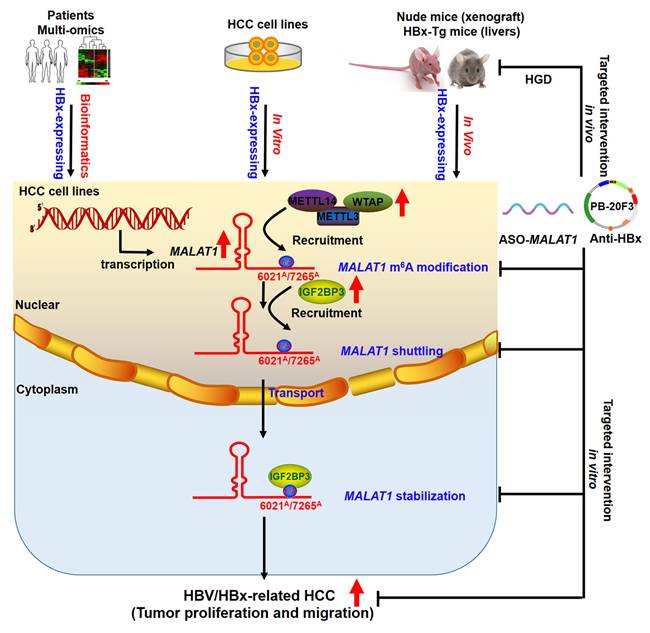
m6A modification governs various cellular and viral RNA functions [38-40]. Viral infections can induce cellular lncRNAs with antiviral effects, while viruses exploit lncRNAs to regulate metabolic networks for their survival [41]. HBx interacts with METTL3/14 enzymes, recruiting them to both viral cccDNA and host chromosomal loci, indicating that m6A is involved in HBx-driven hepatocarcinogenesis [42]. Mechanistically, our study revealed that HBx modulates the m6A epitranscriptomic landscape to enhance MALAT1 stability. HBx upregulated METTL3, METTL14, and WTAP expression, increasing m6A deposition on MALAT1, particularly at the 6021A and 7265A sites. MeRIP and pulldown assays confirmed MALAT1 binding to these m6A writers. IGF2BP3, a known m6A reader, was found to directly interact with and stabilize m6A-modified MALAT1 via its KH3 domain, as validated by RIP and RNA decay assays (Fig. 8). These findings reveal a novel HBx-mediated epigenetic regulatory axis involving the METTL3-m6A-MALAT1-IGF2BP3 cascade in HBV-related liver tumorigenesis.
Identifying targeted interventions for the specific functions of HBx presents a challenge. Given the crucial role of HBx in HBV replication and cell survival, targeting HBx is essential for preventing HBV-associated liver disease. Monoclonal antibodies have emerged as therapeutic options for infectious diseases and cancer. Previous studies have demonstrated that anti-HBx antibodies can reduce intracellular HBx levels through TRIM21-mediated protein degradation, while also activating the NF-κB, AP-1, and IFN-β pathways, leading to enhanced antiviral responses in host cells [43]. In our study, we utilized a hydrodynamic delivery system to express an intracellular anti-HBx mAb (PB-20F3) in HBx-Tg mice. Although this model did not develop macroscopic tumors within six months, consistent with prior findings, histological analysis revealed marked preneoplastic changes in HBx-Tg livers, including nuclear atypia, elevated GPC3, AFP, Ki67, and activation of EMT markers. Anti-HBx treatment effectively suppressed these changes, restored tissue architecture, and downregulated MALAT1, METTL3/14, and IGF2BP3 expression (Fig. 8). These findings demonstrate that anti-HBx intervention attenuates early oncogenic signaling and epigenetic remodeling driven by HBx.
Altogether, our data establish that HBx promotes hepatocarcinogenesis via MALAT1 upregulation and m6A-mediated stabilization, thereby facilitating tumor growth and metastasis. The orthotopic liver model, transcriptomic analysis, and HBx-Tg mouse interventions collectively elucidate the role of the HBx/MALAT1/IGF2BP3 axis in HBV-related HCC (Fig. 8). These findings provide mechanistic insights and experimental evidence supporting lncRNA-based therapeutic strategies in HBV-driven liver cancer, with MALAT1 serving as both a prognostic biomarker and a potential therapeutic target.
Abbreviations
HBV: Hepatitis B virus; HBx: Hepatitis B virus X protein; HCC: Hepatocellular carcinoma; lncRNA: Long non-coding RNA; MALAT1: Metastasis-associated lung adenocarcinoma transcript 1; m6A: N6-methyladenosine; IGF2BP3: Insulin like growth factor 2 mRNA-binding protein 3; ASO: antisense oligonucleotides; HGD: Hydrodynamics-based gene delivery; HBx-Tg: HBx transgenic; ceRNAs: Competing endogenous RNAs; cccDNA: Covalently closed circular DNA; FISH: Fluorescence in situ hybridization; WB: Western blotting; IHC: immunohistochemistry; qRT-PCR: Quantitative real-time PCR; H&E: Hematoxylin and eosin; NC: Negative control; BLI: Bioluminescence imaging; WT: Wild-type; DEGs: Differentially expressed genes; OS: Overall survival; DFS: Disease-free survival; sgGSEA: Single-gene gene-set enrichment analysis; ALD: Alcoholic liver disease; EMT: Epithelial to mesenchymal transition; rcccDNA: Relaxed circular cccDNA; METTL3/14: Methyltransferase-like 3/14; WTAP: Wilms tumor 1-associating protein; CLIP-seq: Cross-linking and immunoprecipitation high-throughput sequencing; RBP: RNA-binding protein; RRMs: RNA recognition motifs; mAb: Monoclonal antibody.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (Nos. 82273667, 82073588), the Industry-University-Research Cooperation Project of Fujian Science and Technology Plan (No. 2022Y4009), the Fundamental Research Funds for the Central Universities (No. 20720250004, 20720232001), XMU Undergraduate Innovation and Entrepreneurship Training Programs (Nos. S202110384378, 2021X1114, 2022X1162, 2023Y1104).
Data availability
The data supporting this study's findings are available within the article and its supplementary materials. Researchers and interested parties can access the relevant information and supporting evidence from the corresponding authors upon reasonable request.
Authors contributions
Ze-Bang Du, Xin-Mou Wu, and Tun Han designed and performed the experiments, prepared figures, and wrote the manuscript. Yu-Xin Cai, Bo Qian, Yu-Shi Shen, Han-Yu Zhang, Jia-Shen Wu, and Jie-He performed in vivo animal studies; Xiao-Xuan Chen, Dong-Bei Guo, Hang-Tian Zhong, Xiong Li, and Lei Zhang performed the experiments, analyzed a portion of the data; Xiao-Ming Luo and Wen-Gang Li designed the experiment, Resources; Yu-Chun Lin and Zhong-Ning Lin reviewed drafts of the paper, and approved final version of the manuscript for publication. All authors reviewed and approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huang DQ, Mathurin P, Cortez-Pinto H, Loomba R. Global epidemiology of alcohol-associated cirrhosis and HCC: trends, projections and risk factors. Nat Rev Gastroenterol Hepatol. 2023;20:37-49
2. Huang DQ, Hoang JK, Kamal R, Tsai PC, Toyoda H, Yeh ML. et al. Antiviral Therapy Utilization and 10-Year Outcomes in Resected Hepatitis B Virus- and Hepatitis C Virus-Related Hepatocellular Carcinoma. J Clin Oncol. 2024;42:790-9
3. Jiang Y, Han Q, Zhao H, Zhang J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J Hepatocell Carcinoma. 2021;8:435-50
4. Lin X, Li AM, Li YH, Luo RC, Zou YJ, Liu YY. et al. Silencing MYH9 blocks HBx-induced GSK3beta ubiquitination and degradation to inhibit tumor stemness in hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5:13
5. Ghosh A, Ghosh S, Dasgupta D, Ghosh A, Datta S, Sikdar N. et al. Hepatitis B Virus X Protein Upregulates hELG1/ ATAD5 Expression through E2F1 in Hepatocellular Carcinoma. Int J Biol Sci. 2016;12:30-41
6. Zoulim F, Chen PJ, Dandri M, Kennedy P, Seeger C. Hepatitis B Virus DNA integration: Implications for diagnostics, therapy, and outcome. J Hepatol. 2024;81:1087-1099
7. Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen LL. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. 2023;24:430-47
8. Nemeth K, Bayraktar R, Ferracin M, Calin GA. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat Rev Genet. 2024;25:211-32
9. Yuan D, Chen Y, Li X, Li J, Zhao Y, Shen J. et al. Long Non-Coding RNAs: Potential Biomarkers and Targets for Hepatocellular Carcinoma Therapy and Diagnosis. Int J Biol Sci. 2021;17:220-35
10. Wu M, Sun J, Wang L, Wang P, Xiao T, Wang S. et al. The lncRNA HOTAIR via miR-17-5p is involved in arsenite-induced hepatic fibrosis through regulation of Th17 cell differentiation. J Hazard Mater. 2023;443:130276
11. Yang L, Peng X, Li Y, Zhang X, Ma Y, Wu C. et al. Long non-coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol Cancer. 2019;18:78
12. Liu SJ, Dang HX, Lim DA, Feng FY, Maher CA. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21:446-60
13. Liu Y, Feng J, Sun M, Yang G, Yuan H, Wang Y. et al. Long non-coding RNA HULC activates HBV by modulating HBx/STAT3/miR-539/APOBEC3B signaling in HBV-related hepatocellular carcinoma. Cancer Lett. 2019;454:158-70
14. Salerno D, Chiodo L, Alfano V, Floriot O, Cottone G, Paturel A. et al. Hepatitis B protein HBx binds the DLEU2 lncRNA to sustain cccDNA and host cancer-related gene transcription. Gut. 2020;69:2016-24
15. Song W, Zheng C, Liu M, Xu Y, Qian Y, Zhang Z. et al. TRERNA1 upregulation mediated by HBx promotes sorafenib resistance and cell proliferation in HCC via targeting NRAS by sponging miR-22-3p. Mol Ther. 2021;29:2601-16
16. Deng X, Qing Y, Horne D, Huang H, Chen J. The roles and implications of RNA m(6)A modification in cancer. Nat Rev Clin Oncol. 2023;20:507-26
17. Chen YY, Wang WH, Che L, Lan Y, Zhang LY, Zhan DL. et al. BNIP3L-Dependent Mitophagy Promotes HBx-Induced Cancer Stemness of Hepatocellular Carcinoma Cells via Glycolysis Metabolism Reprogramming. Cancers (Basel). 2020;12:655
18. Che L, Wu JS, Du ZB, He YQ, Yang L, Lin JX. et al. Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma. Cancers (Basel). 2022;14:821
19. He C, Qiu Y, Han P, Chen Y, Zhang L, Yuan Q. et al. ER stress regulating protein phosphatase 2A-B56gamma, targeted by hepatitis B virus X protein, induces cell cycle arrest and apoptosis of hepatocytes. Cell Death Dis. 2018;9:762
20. Che L, Du ZB, Wang WH, Wu JS, Han T, Chen YY. et al. Intracellular antibody targeting HBx suppresses invasion and metastasis in hepatitis B virus-related hepatocarcinogenesis via protein phosphatase 2A-B56gamma-mediated dephosphorylation of protein kinase B. Cell Prolif. 2022;55:e13304
21. Wang X, Kang M, Liu C, Lin T, Han X, Jiang X. Current State and Progress of Research on the Role of lncRNA in HBV-Related Liver Cancer. Front Cell Infect Microbiol. 2021;11:714895
22. Zhao X, Fan H, Chen X, Zhao X, Wang X, Feng Y. et al. Hepatitis B Virus DNA Polymerase Restrains Viral Replication Through the CREB1/HOXA Distal Transcript Antisense RNA Homeobox A13 Axis. Hepatology. 2021;73:503-19
23. Rahman MM, Tollefsbol TO. Targeting cancer epigenetics with CRISPR-dCAS9: Principles and prospects. Methods. 2021;187:77-91
24. Anderson DA, Voigt CA. Competitive dCas9 binding as a mechanism for transcriptional control. Mol Syst Biol. 2021;17:e10512
25. Li W, Gao Y, Jin X, Wang H, Lan T, Wei M. et al. Comprehensive analysis of N6-methylandenosine regulators and m6A-related RNAs as prognosis factors in colorectal cancer. Mol Ther Nucleic Acids. 2022;27:598-610
26. Yang Z, Wang T, Wu D, Min Z, Tan J, Yu B. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J Exp Clin Cancer Res. 2020;39:203
27. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93-5
28. Zhang W, Zhao J, Deng L, Ishimwe N, Pauli J, Wu W. et al. INKILN is a Novel Long Noncoding RNA Promoting Vascular Smooth Muscle Inflammation via Scaffolding MKL1 and USP10. Circulation. 2023;148:47-67
29. Morelli E, Fulciniti M, Samur MK, Ribeiro CF, Wert-Lamas L, Henninger JE. et al. A MIR17HG-derived long noncoding RNA provides an essential chromatin scaffold for protein interaction and myeloma growth. Blood. 2023;141:391-405
30. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285-95
31. Zhu K, Gao T, Wang Z, Zhang L, Tan K, Lv Z. RNA N6-methyladenosine reader IGF2BP3 interacts with MYCN and facilitates neuroblastoma cell proliferation. Cell Death Discov. 2023;9:151
32. Goyal B, Yadav SRM, Awasthee N, Gupta S, Kunnumakkara AB, Gupta SC. Diagnostic, prognostic, and therapeutic significance of long non-coding RNA MALAT1 in cancer. Biochim Biophys Acta Rev Cancer. 2021;1875:188502
33. Nakamura S, Ishihara M, Watanabe S, Ando N, Ohtsuka M, Sato M. Intravenous Delivery of piggyBac Transposons as a Useful Tool for Liver-Specific Gene-Switching. Int J Mol Sci. 2018;19:3452
34. Ahodantin J, Bou-Nader M, Cordier C, Megret J, Soussan P, Desdouets C. et al. Hepatitis B virus X protein promotes DNA damage propagation through disruption of liver polyploidization and enhances hepatocellular carcinoma initiation. Oncogene. 2019;38:2645-57
35. McBrearty N, Arzumanyan A, Bichenkov E, Merali S, Merali C, Feitelson M. Short chain fatty acids delay the development of hepatocellular carcinoma in HBx transgenic mice. Neoplasia. 2021;23:529-38
36. Nagaraju GP, Dariya B, Kasa P, Peela S, El-Rayes BF. Epigenetics in hepatocellular carcinoma. Semin Cancer Biol. 2022;86:622-32
37. Sun Y, Teng Y, Wang L, Zhang Z, Chen C, Wang Y. et al. LINC01431 Promotes Histone H4R3 Methylation to Impede HBV Covalently Closed Circular DNA Transcription by Stabilizing PRMT1. Adv Sci (Weinh). 2022;9:e2103135
38. Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA. et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe. 2016;20:654-65
39. Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y. et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. 2016;1:16011
40. Imam H, Khan M, Gokhale NS, McIntyre ABR, Kim GW, Jang JY. et al. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc Natl Acad Sci U S A. 2018;115:8829-34
41. Gonzales-van Horn SR, Sarnow P. Making the Mark: The Role of Adenosine Modifications in the Life Cycle of RNA Viruses. Cell Host Microbe. 2017;21:661-9
42. Kim GW, Siddiqui A. Hepatitis B Virus X Protein Expression Is Tightly Regulated by N6-Methyladenosine Modification of Its mRNA. J Virol. 2022;96:e0165521
43. Zhang JF, Xiong HL, Cao JL, Wang SJ, Guo XR, Lin BY. et al. A cell-penetrating whole molecule antibody targeting intracellular HBx suppresses hepatitis B virus via TRIM21-dependent pathway. Theranostics. 2018;8:549-62
Author contact
![]() Corresponding authors: lxmedu.cn (X.-M. Luo); lwgangedu.cn (W.-G. Li); linychedu.cn (Y.-C. Lin); and linzhnedu.cn (Z.-N. Lin). Tel.: +86 592 2880615; Fax: +86 592 2881578.
Corresponding authors: lxmedu.cn (X.-M. Luo); lwgangedu.cn (W.-G. Li); linychedu.cn (Y.-C. Lin); and linzhnedu.cn (Z.-N. Lin). Tel.: +86 592 2880615; Fax: +86 592 2881578.