Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Cytoplasmic Metabolic...
3. Mitochondrial Metabolic...
4. Potential Metabolic Regulators
5. Future Prospective and...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(11):5056-5078. doi:10.7150/ijbs.114010 This issue Cite
Review
Innate Immunity Reimagined: Metabolic Reprogramming as a Gateway to Novel Therapeutics
Chuan-Han Deng1,#, Chen-Tong Wang1,#, Xin Zhou2, Xin Chen1,3,4, Ying Wang1,3,4,5 ![]()
1. Institute of Chinese Medical Sciences and State Key Laboratory of Mechanism and Quality of Chinese Medicine (University of Macau) (MQCM), University of Macau, Avenida da Universidade, Taipa, Macao SAR, China.
2. Longhua Hospital Shanghai University of Traditional Chinese Medicine, Shanghai, 200032, China.
3. Department of Pharmaceutical Sciences, Faculty of Health Science, University of Macau, Avenida da Universidade, Taipa, Macao SAR, China.
4. MoE Frontiers Science Center for Precision Oncology, University of Macau, Avenida da Universidade, Taipa, Macao SAR, China.
5. Minister Of Education Key Laboratory of Tumor Molecular Biology, Jinan University, Guangzhou 510632, China.
# These authors contribute equally.
Received 2025-3-19; Accepted 2025-6-25; Published 2025-7-28
Abstract

The interplay between cellular metabolism and innate immunity critically shapes the body's ability to fight infections, repair tissue, and manage stress. Metabolic reprogramming not only drives innate immune activation but also regulates the resolution of inflammation. Phenotypes of immune cell are closely linked to metabolic shifts that adapt to varying energy demands. However, the precise relationship between perturbations in the cellular respiratory-metabolic axis and the inflammatory response remains a critical field of investigation. In depth understanding of key metabolic pathways, such as glycolysis, NADPH oxidase activity, mitochondrial ROS production, TCA cycle metabolites, and cGAS-STING/AIM2 inflammasome activation, is essential to unravel the complexities of innate immunity. This article highlights the central role of metabolic reprogramming mainly in innate immunity and explores its potential as a therapeutic target for modulating inflammatory response.
Keywords: metabolism programming, cellular respiration, innate immune response, inflammation, mitochondria
1. Introduction
Innate immunity serves as the first line of defense against pathogen infection and tissue injury, mobilizing a suite of immune cells, such as dendritic cells, macrophages, and T cells, to engage in a rapid, non-specific response. These cells experience profound transcriptional and translational modifications, with a concurrent metabolic shift to sustain the immediate demands of the immune response. Typically, proinflammatory cells shift from oxidative phosphorylation (OXPHOS) to glycolysis, a metabolic alteration that provides both energy and biosynthetic precursors. The generation of reactive oxygen species (ROS) and mitochondrial signaling pathways are crucial determinants of the inflammatory response and immune cell function. Sustained activation of the innate immune response result in precipitate deleterious conditions, such as cytokine storms or autoimmune diseases [1]. Targeting metabolic reprogramming offers a promising strategy for developing novel therapies for inflammatory and autoimmune diseases. This review delineates the intricate steps and pivotal molecules in metabolic programming and sheds light on emerging therapeutic strategies aimed at their regulation.
2. Cytoplasmic Metabolic Signaling Hubs
2.1. Glycolysis Enzymes
Under normoxic conditions, the Warburg effect induces a switch of metabolism from OXPHOS to glycolysis, favoring aerobic glycolysis for ATP generation. Immune cells resort to Warburg metabolism upon encountering inflammatory stimuli, a strategy that underpins their resistance to lactate-mediated suppression and supports cellular proliferation [2]. Dendritic cells, for instance, augment glucose consumption and engage in Warburg metabolism following Toll-like receptor (TLR) activation [3]. Glycolysis and the pentose pathway become the predominant source for ATP production in T cells and M1 macrophages, sidelining the tricarboxylic acid (TCA) cycle [4]. The accumulation of lactic acid not only aids in restoring metabolic equilibrium but also can induce a phenotypic switch in immune cells towards a quiescent state, which marks the cessation of the immune response [5].
Glycolysis is an intricately controlled sequence of biochemical reactions (Figure 1). The rate of cellular glucose absorption is largely governed by glucose transporters (GLUT). Activation of T cells necessitates a prompt and robust upregulation of GLUT1, a requirement not shared by quiescent peripheral T cells for their survival [6]. During Streptococcus pneumoniae infection, the AIM2 inflammasome is triggered by GLUT1-mediated glycolysis, thereby intensifying pulmonary fibrosis [7]. Sepsis-induced Warburg effect via GLUT1 can lead to the apoptotic demise of CD4+ T cells, precipitating a collapse of immune function [8]. In models of encephalomyelitis and autoimmune colitis, glucose uptake via GLUT3 modulates glucose oxidation and ATP-citrate lyase-dependent acetyl-CoA synthesis in the mitochondria, influencing the epigenetic reprogramming of inflammatory genes in T helper (Th)17 cells [9].
The enzymes involved in glycolysis play important role in innate immune response, either directly or indirectly. Certain compounds have anti-inflammatory effects by inhibiting enzyme activity or altering enzyme conformation. Red arrows indicate pro-inflammatory effects; blue arrows indicate anti-inflammatory effects.
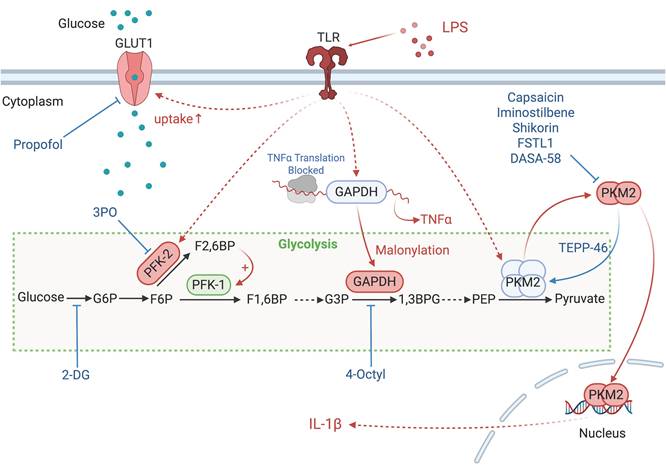
The enzymatic transformation of glucose into glucose-6-phosphate, catalyzed by hexokinase (HK) 1 to 4, marks the onset of aerobic glycolysis. The dissociation of HK1 from mitochondria and its binding to S100A8/A9 promotes iNOS-dependent nitrosylation and GAPDH inactivation. This redirects glycolytic flux to the pentose phosphate pathway and enhances nitric oxide signaling. This metabolic shift leads to oxidative stress and low-grade chronic inflammation that contribute to tissue damage in diabetic neuropathy and aging [10]. In LPS-primed macrophages, HK1 detects cytosolic N-acetylglucosamine, a peptidoglycan metabolite which triggers the dissociation of HK1 from the mitochondria. This inhibits enzyme activity of HK1 and leads to elevated ROS, which act as Signal 2. Signal 2 then drives the assembly of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, resulting in inflammation [11]. HK2 expression alteration is notably significant in activated T cells. The inhibition of HK2 by bacterial peptidoglycan-derived N-acetyl glucosamine results in its detachment from the mitochondrial outer membrane and the subsequent assembly of the NLRP3 inflammasome. Interfering with glycolysis via the addition of glucose-6-phosphate, the enzymatic product of HK2, nullifies its pattern recognition receptor function [11].
Glucokinase, a hexokinase isozyme with lower affinity for glucose, converts glucose to glucose-6-phosphate primarily in hepatocytes and pancreatic beta cells. The initiation of glucokinase-mediated glycolysis leads to its interaction with actin, which promotes cytoskeletal reorganization and the migration of T regulatory cells. This migratory response is driven by glucokinase expression upregulation via the phosphoinositide 3-kinase (PI3K)-mammalian target of rapamycin (mTOR) complex 2 signaling axis [12].
The third step of glycolysis is catalyzed by phosphofructokinase-2 (PFK-2), which facilitates conversion of fructose-6-phosphate to fructose-2,6-bisphosphate (F2,6BP). PFK-1 activity is allosterically upregulated by F2,6BP [13], which is synthesized from fructose-6-phosphate by fructose-6-phosphate-2-kinase (PFK2/PFKFB3). Genetic variants such as rs646564 in the PFKFB3 gene reduce glycolytic ATP production, resulting in impaired generation of ROS outburst. This leads to defective phagocytosis and poor fungal clearance in human macrophages [14].
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) undertakes the sixth step of glycolysis to catalyze the oxidation of glyceraldehyde-3-phosphate to 1,3-biphosphoglycerate. Malonylation at Lys213 on GAPDH disrupts its interaction with AU-rich mRNA elements, such as TNFα, resulting in the release of these transcripts for translation and the subsequent activation of pro-inflammatory signaling pathways. Concurrently, this modification impairs GAPDH's glycolytic enzymatic activity, and reprograms glycolysis to meet the energy demands of the macrophages during inflammation [15]. GAPDH also plays a role in T cell activation and glycolysis, with its direct interaction with the AU-rich element in the 3' untranslated region on interferon (IFN)-γ mRNA [16].
Pyruvate kinase isoezymes M2 (PKM2) orchestrates the final and rate-limiting step of glycolysis by catalyzing the transformation of phosphoenolpyruvate to pyruvate when in its active tetrameric state. In its dimeric configuration, PKM2 translocate to the nucleus to activate transcription factor 2, thereby enhancing LPS-induced pyroptosis in microglia [17]. Additionally, when associated with HIF-1α in the nucleus, PKM2 initiates the transcription of interleukin (IL)-1β, inhibits glycolysis and shifts macrophages towards the M2 phenotype under LPS and Salmonella typhimurium exposure [18]. PKM2-mediated glycolysis also facilitates the phosphorylation of eukaryotic translation initiation factor 2-alpha kinase 2, activating the AIM2 and NLRP3 inflammasomes in macrophages, a critical process in lethal endotoxemia and polymicrobial sepsis [19]. Pharmacological intervention that activates PKM2 to its tetrameric state impedes its nuclear translocation and subsequent transcription of pro-inflammatory genes. The allosteric PKM2 activator TEPP-46 mitigates CD4+ T cell-driven autoimmune and inflammatory responses in autoimmune encephalomyelitis models [20]. In contrast, sulfenylation of PKM2 impedes its tetramerization and reduces its enzymatic activity, which in turn augments glycolytic flux and the accumulation of harmful glucose metabolites [21].
Lactate dehydrogenase A (LDHA) catalyzes the conversion of pyruvate to lactate, a process that succeeds aerobic glycolysis. Elevated LDHA expression favors aerobic glycolysis, sustaining acetyl-coenzyme A concentrations necessary for histone acetylation, which in turn modulates epigenetic control of IFN-γ production in T cells upon activation. A deficiency in LDHA, however, can lead to PI3K-mediated dephosphorylation of Akt, reducing T cell-mediated immunity in mice challenged with bacterium Listeria monocytogenes [22].
2.2. Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase
The NADPH oxidase (NOX) family, along with the mitochondrial electron transport chain (ETC), are primary sources of reactive oxygen species (ROS) and directly generate ROS such as superoxide and hydrogen peroxide. To date, the NOX family has been expands to encompass seven isoforms, NOX1 to NOX5, along with dual oxidase (DUOX)1-2, each with unique tissue distribution and physiological functions [23].
The NOX2 complex, along with its regulatory subunits p40phox, p47phox, and p67phox, was initially characterized as the primary component of the phagocyte oxidative burst [24]. Upon infection, these regulatory subunits translocate to the membrane, and form the active oxidase complex together with gp91phox and p22phox. NOX2 facilitates the generation of superoxide via a biphasic electron transfer process, essential for pathogen eradication [24]. This activation promotes a significant upsurge in both OXPHOS and glycolysis [24]. A missense mutation in the neutrophil cytosolic factor 2 gene, which encodes p67phox, has been linked to early-onset IBD [25]. Conversely, a deficiency in NOX2 predisposes individuals to autoimmunity and elevate systemic lupus erythematosus risk [26]. On the other hand, hyper activation of NOX2 can lead to oxidative stress, contributing to chronic inflammation and tissue damage. Targeted reversible inhibitors that hinder p47phox and p22phox interactions effectively mitigate NOX2-induced oxidative stress [27].
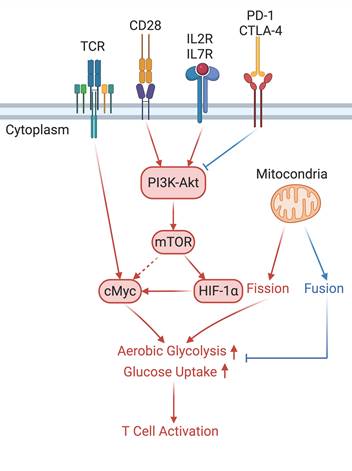
In the T cell activation cascade, the T cell receptor (TCR), along with co-stimulatory and co-receptor molecules like CD28, IL2R, and IL7R, orchestrates activation via the PI3K-Akt-cMyc axis. In contrast, programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) serve as inhibitory checkpoints, dampening T cell activation by modulating the PI3K-Akt pathway. The diagrammatic representation employs red arrows to denote the pathways promoting T cell activation, while blue arrows indicate the pathways conferring inhibitory signals that antagonize activation.
Compounds like LDC7559 and its more efficacious derivative, NA-11, target the AMP/ADP allosteric site on phosphofructokinase-1 liver type (PFKL). This interaction inhibits glycolysis and the subsequent pentose phosphate pathway, diminishing the NOX2-dependent oxidative burst and the defense capability of neutrophils, thereby curtailing tissue damage [28]. Additionally, during Staphylococcus aureus infection, NOX2 generates ROS, which alkalinizes phagosomes by consuming protons during the conversion of oxygen to superoxide (O₂⁻). This counteracts the acidifying activity of the V-ATPase proton pump. However, caspase-1 subsequently cleaves subunits of the NOX2 complex, reduces ROS production and allowing V-ATPase-driven acidification to proceed. This enhances the killing of Gram-positive bacteria via lysosomal enzymes [29].
Inhibition of NOX4 bolsters the endothelial cell barrier in sepsis and mitigate acute lung injury [30]. GKT137831, a NOX4 inhibitor, is undergoing clinical evaluation for idiopathic pulmonary fibrosis (Clinical trial No. NCT03865927), type 2 diabetes (NCT02010242), and primary biliary cholangitis (NCT03226067). While ablation of NOX4 enhances liver regeneration in mice [31], yet it appears to confer protection against tissue damage due to fibrogensis in chronic intestinal inflammation [32]. Given NOX4's multifaceted roles in different disease states, pharmacological targeting requires precise tailoring to minimize off-target effects.
NOX5, an oxidase primarily expressed during monocytes differentiation to dendritic cells and implicated in vascular remodeling and calcification [33]. NOX5 expression in podocytes is linked to the heightened ROS and pro-inflammatory cytokine production via activation of IL-1R-associated kinases (IRAK)-1, IRAK-4 [34], and protein kinase C-α signaling [35]. The broad-spectrum NOX inhibitor APX-115 enhances pancreatic beta-cell functionality and mitigates diabetic nephropathy in NOX5 overexpressing transgenic mice [36].
DUOX1 and DUOX2, initially identified in the thyroid, are crucial for thyroid hormone biosynthesis. In the lungs, IL-1β and ROS, generated by DUOX1, constitute a unified epithelial response to microbial infections [37]. DUOX2's primary function is to protect against pathogenic gut microbiota by producing hydrogen peroxide [38]. Notably, a monoallelic exonic variant of DUOX2 correlates with very early-onset IBD [39], and mutations in DUOX2 are associated with increased colonic IL-17C levels and risk of IBD [40].
2.3. Hypoxia-Inducing Factor-1α (HIF-1α)
HIF-1α transcription is principally regulated by nuclear factor kappa B (NF-κB) pathways in response to hypoxia [41]. The stability and subsequent nuclear translocation of HIF-1α are crucial in redirecting cellular metabolism towards glycolysis. During neutrophil-mediated oxidative burst, the glycerol 3-phosphate pathway is essential in preserving mitochondrial integrity and supporting glycolysis, thus facilitating HIF-1α stabilization [42]. The use of FG-4592 to stabilize HIF-1α diminishes both glycolytic metabolites and cytokine production in alveolar macrophages during acute lung injury [43]. Conversely, HIF-1α genetic ablation reduces glycolysis and curtails pro-inflammatory mediator production in macrophages, which has implications in systemic lupus erythematosus [44]. Additionally, Wnt ligand stimulation enhances the interaction between β-catenin and HIF-1α, leading to a surge in HIF-1α levels and a subsequent pro-inflammatory response in macrophages from patients with COVID-19 [45].
2.4. PI3K-Akt Signaling in Metabolic Regulation and Immune Cell Activation
The activation of naïve T cells is precipitated by the engagement of T cell receptor (TCR) complexes with co-stimulatory signals such as CD28, IL-2, and IL-7. The TCR complex influences the Myc pathway and the PI3K-Akt signaling signaling cascade [46]. Myc is indispensable for the metabolic reprogramming of T cells, with its absence impeding the induction of glycolytic flux upon T cell activation. Proteins associated with glycolysis and oxidative metabolism are markedly increased during the initial activation of naïve T cells [47]. For example, GLUT1 expression in activated T cells, regulated by PI3K-Akt signaling, corresponds with an adaptive increase in glucose metabolism. The reactivation of memory T cells similarly relies on CD28-mediated PI3K-Akt signaling for GLUT1 expression, which is critical for their metabolic demands [48].
3. Mitochondrial Metabolic Signaling Hubs
3.1. Mitochondria Generated ROS
Mitochondria act as the principal loci for aerobic respiration and serve as the energy biosynthesis powerhouses within eukaryotic cells. The ETC hosts the OXPHOS process that facilitates ATP synthesis, the predominant energy molecule. Electron transit through the ETC establishes a proton gradient across the inner mitochondrial membrane, which, upon reacting with oxygen, generates ROS within the ETC.
Electrons from nicotinamide adenine dinucleotide (NADH) enter the ETC at mitochondrial complex I. An elevated NADH/NAD+ ratio within the mitochondrial matrix allows for the interaction of molecular oxygen with reduced flavin mononucleotide (FMN), resulting in the production of the superoxide anion (O2-), which is liberated into the mitochondrial matrix. Subsequently, the reduction of ubiquinone and alterations in mitochondrial membrane proton concentration induce a reverse electron transport chain (RET), driving electrons back towards complex I and fostering additional O2- generation [49]. Complex I impairment impairs NADPH production and enhances the inflammatory response due to ROS accumulation [50].
Complex III is another significant contributor to mitochondrial ROS production. The O2- generated by complex III primarily enters the inner mitochondrial membrane space, while the H2O2 formed post-disproportionation permeates the matrix [51]. A deficit in complex III function leads to heightened DNA methylation and suppresses the expression of genes critical for the immunosuppressive function of regulatory T (Treg) cells without compromising Treg cell proliferation or viability [52]. ROS originating from complex III also precipitate the depletion of NAD+ levels and intensify DNA damage, processes crucial` for macrophage activation [53].
Mitochondria and NOX2 complexes are responsible for the generation and release of ROS into the cytoplasm. Mitochondrial Complexes I to III together with NOX2 complex generates O2-, a precursor to a multitude of ROS.
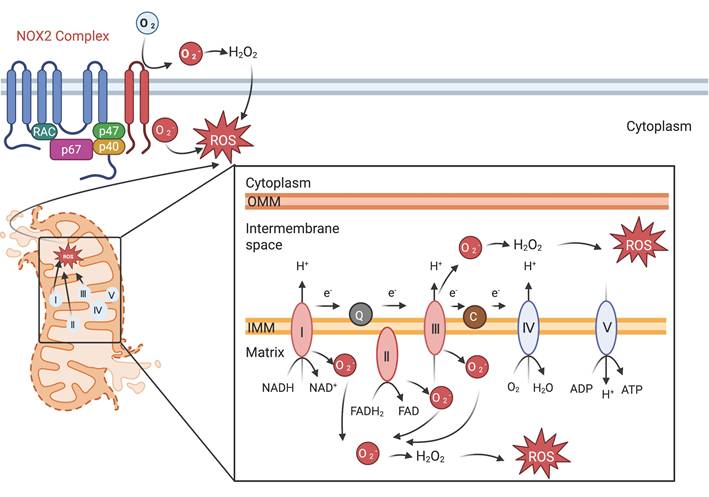
When both complexes I and complex III are inhibited, complex II becomes the predominant source of ROS [54]. Upon LPS stimulation, macrophages exhibit an increase in mitochondrial succinic acid oxidation and membrane potential due to complex II-mediated elevated mitochondrial ROS production. Targeting succinate dehydrogenase within complex II can mitigate ROS generation, dampen the macrophage inflammatory response, and reduce LPS-induced lethality in mice [55]. Itaconate, a metabolite produced by activated macrophages, acts as an inhibitor of succinate oxidation at complex II, modulating macrophage metabolism and attenuating inflammation in models of ischemia-reperfusion injury in Irg1-/- mice [56]. Cardiolipin biogenesis impedes the assembly of complex II, triggering lysosome-mediated degradation of this complex following LPS exposure in macrophages [57].
Mitochondrial-derived ROS play a pivotal role in modulating the innate immune response [58]. They facilitate the relocation of NLRP3 to the mitochondria-associated ER membrane, where it attracts both apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase-1, leading to inflammasome activation. Mitochondrial ROS (mtROS) trigger cleavage and oligomerization of the N-terminal domain of gasdermin D, enabling its insertion into the mitochondrial membrane to form pores. These pores amplify mtROS release, which then activates the RIPK1/RIPK3/MLKL necroptotic pathway and drive necroptotic cell death during Pseudomonas entomophila infection [58]. Excessive mtROS stemming from dysfunctional mitochondria instigate the assembly of the NLRP3 inflammasome. TLR7/8 agonists, such as imiquimod and CL097, impede the activity of quinone oxidoreductases NQO2 and complex I of the mitochondria, thereby heightening intracellular ROS triggering NLRP3 inflammasome activation independent of K+ efflux [59]. Conversely, curtailing mitochondrial ATP synthesis and DNA replication can avert NLRP3 inflammasome initiation in alveolar macrophages in acute respiratory distress syndrome induced by LPS or SARS-CoV-2 infection [60]. While obstructing the mitochondrial ETC can diminish the NLRP3-driven inflammatory cascade, the mitochondrial metabolite phosphocreatine activates the NLRP3 inflammasome in an ATP-dependent manner, irrespective of ROS generation [61].
3.2. Mitochondrial Dynamics in Immune Cell Fate and Inflammation
Mitochondrial dynamics is one of the key determent factors in metabolic programming and T cell destiny. Mitochondrial fusion proteins like OPA1 enhance OXPHOS in memory T cells by promoting fused mitochondrial networks and remodeling cristae structure, which optimizes ETC efficiency. This tight ETC coupling sustains high ATP production, supporting the metabolic demands and longevity of memory T cells [62].
Mitophagy, a specialized autophagic mechanism, selectively eliminates malfunctioning or surplus mitochondria and is instrumental in modulating inflammatory responses. FUNDC1, a receptor essential for mitophagy, ensures mitochondrial quality control under normal conditions, and its disruption worsens diet-triggered obesity and metabolic dysfunction [63]. Mitophagy prevents NLRC4 activation during Pseudomonas aeruginosa infection by removing mitochondria damaged by the type III secretion system (T3SS), thereby reducing mtROS and oxidized mtDNA release. This blocks the cytosolic accumulation of oxidized mtDNA, which is required for NLRC4 inflammasome oligomerization and activation, excessive ROS generation, mitochondrial DNA (mtDNA) release, and subsequent activation of the NLRC4 inflammasome in macrophages [64]. In intestinal macrophages, the deletion of IL-10 or its receptor prolongs mTOR pathway signaling, exacerbating inflammasome activity and intensifying intestinal inflammation [65].
The strategic induction of mitophagy through small-molecule agents presents great potential in regulating inflammatory response. Compounds such as rapamycin and resveratrol mitigate NLRC4 inflammasome activation by facilitating mitophagy, thereby clearing damaged mitochondria in mouse bone marrow-derived macrophages (BMDMs) [64]. Similarly, andrographolide, the main active substance first isolated from Andrographis paniculata, obstructs the advancement of colitis and associated cancers by inhibiting the NLRP3 inflammasome via mitophagy in mouse models [66].
3.3. TCA Cycle Metabolites
Mitochondrial metabolism plays a pivotal role in immune regulation, particularly through the tricarboxylic acid (TCA) cycle. The TCA cycle, or termed as the Krebs cycle, represents a fundamental process in biosynthesis and cellular energy production. In immune cells, intermediates of the TCA cycle serve a dual function: they are vital for ATP generation and act as signaling molecules that influence immune responses. For example, a low α-ketoglutarate/succinate ratio leads to proinflammatory state of macrophages, whereas a high α-ketoglutarate/succinate ratio facilitates the tissue repair phenotype of macrophages [67]. The inhibition of succinate oxidation by dimethyl malonate, in turn, drives the proinflammatory phenotype of macrophages [55].
One noteworthy endogenous metabolite derived from the TCA cycle-derived is itaconate, which is generated through the decarboxylation of cis-aconitate. Itaconate suppresses inflammatory responses by inhibiting activity of succinate dehydrogenase [56, 68] and to interact directly with Cys151, 257, 288, 273 and 297 on KEAP1 [69]. The covalent binding of itaconate and KEAP1 then enables increased expression of nuclear factor erythroid 2-related factor 2 (Nrf2) downstream anti-oxidant and anti-inflammatory genes [69]. Moreover, the cell permeable derivative of itaconate, 4-octyl itaconate, offers protection against lethality and systemic inflammation induced by LPS [69]. Treatment with glucocorticoids facilitates the interaction of glucocorticoid receptor with pyruvate dehydrogenase complex, and then elevates the TCA cycle-dependent production of itaconate and interfere with the production of proinflammatory cytokines [70]. This illustrates how metabolic pathways can directly influence immune cell behavior and responses, emphasizing the complex interconnection between metabolism and immunity.
3.4. Mitochondrial DNA (mtDNA) Triggered Inflammatory Response
mtDNA, positioned in proximity to the ETC, acts as a principal source of mitochondrial reactive oxygen species (mtROS). mtROS, such as hydroxyl radicals, oxidize mtDNA, generating strand breaks and 8-oxoguanine adducts that structurally mimic pathogen-derived DNA. When released into the cytoplasm, oxidized mtDNA is recognized by cGAS/STING as a "non-self" danger signal and leads to subsequent activation of innate immune pathways and conferring immunogenicity [71]. mtDNA is rich in hypomethylated CpG motifs, identifiable by pattern recognition receptors (PRRs) like cGAS-stimulator of interferon genes (STING), TLR9, and the AIM2 inflammasome [72, 73]. Experimental intra-articular injection of mtDNA in mice provokes a pro-inflammatory response [74]. Mitochondrial ROS can also impair mtDNA synthesis by diminishing the level of mitochondrial transcription factor A, intensifying the severity of ischemic acute kidney injury [75].
cGAS is activated by mtDNA, thereby catalyzing the formation of cGAMP. cGAMP binds to STING and promotes its transfer from the endoplasmic reticulum to the Golgi apparatus, subsequently activating downstream inflammatory pathways.
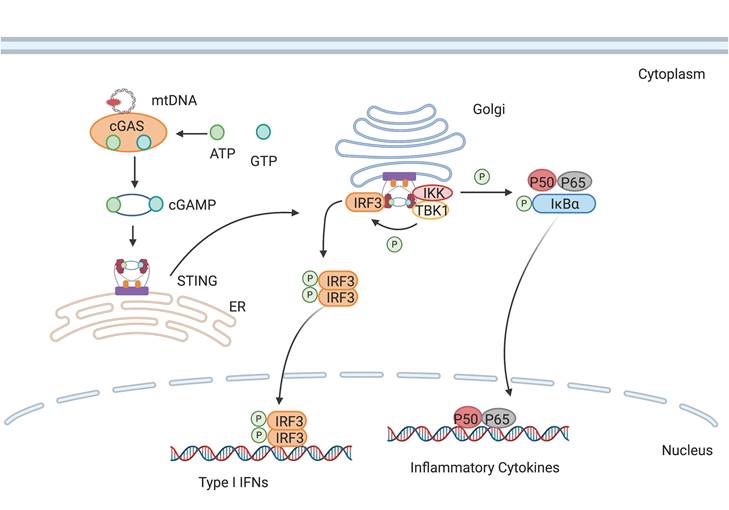
The release of mtDNA from damaged mitochondria into the cytoplasm stimulates the activation of AIM2 inflammasome. Caspase-1 subsequently cleaves gasdermin D to generate N-terminal fragments that assemble into membrane pores, while also maturing pro-IL-1β and pro-IL-18 for release through these channels.
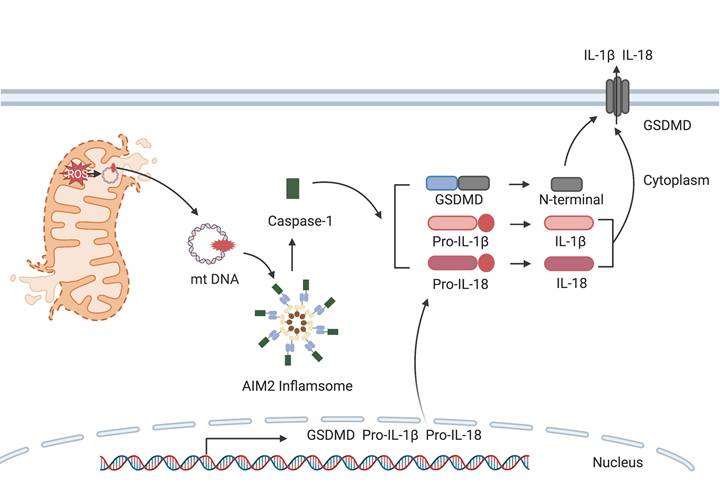
Cells expel defective mitochondria through mitophagy under normal conditions [76]. However, cells prioritize aerobic glycolysis over OXPHOS during immune activation, which leads to a surge in mtROS levels. This increase can induce mitochondrial damage and the subsequent mtDNA release, which then amplifies inflammatory responses [77].
3.4.1. cGAS-STING
The cGAS-STING (cyclic GMP-AMP synthase-stimulator of interferon genes) is a cytoplasmic DNA-sensing pathway that triggered by type I IFN production [78]. Upon binding to double-stranded DNA (dsDNA), cGAS utilizes the formation of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) using ATP and GTP, which subsequently binds and activates STING [79]. Dysregulated cGAS-STING signaling links to a spectrum of inflammatory diseases [80]. Therapeutic intervention targeting the cGAS-STING pathway, such as the inhibition of its substrate or catalytic sites, could potentially ameliorate autoimmune disorders [81]. STING antagonists can act by occupying its cyclic dinucleotide binding site [82] or by binding covalently to cysteine residue 91 to prevent STING palmitoylation, an essential posttranslational modification for its activity [83].
3.4.2. AIM2 Inflammasome
The AIM2 (absent in melanoma 2) inflammasome functions as a sensor for cytosolic double-stranded DNA that activates inflammatory caspases, engaging the adaptor protein ASC and procaspase-1 to facilitate its assembly. This complex initiates the cleavage and subsequent translocation of gasdermin D to cell membrane [84]. The activation of the AIM2 inflammasome is intricately linked to the metabolic reprogramming of immune cells. For example, in septic mice induced by LPS, PKM2-driven glycolysis leads to the phosphorylation of eukaryotic translation initiation factor 2-α kinase 2, which mediates the activation of NLRP3 and AIM2 inflammasome [19]. Heightened mtROS levels prompts the assembly of the AIM2 inflammasome, thus the cleavage of procaspase-1 and subsequent cleavage of Parkin, a negative regulator of mitophagy, thereby impeding mtROS clearance and enhancing mitochondrial damage [85]. Additionally, AIM2 inflammasome is suppressed in LPS-primed macrophages when the synthesis of 25-hydroxycholesterol is upregulated through cholesterol biosynthesis [86].
The role of the AIM2 inflammasome is context-dependent, varying with cell type and disease. In systemic lupus erythematosus, AIM2 expression is markedly upregulated in leukocytes and macrophages, though not in kidney tissue [87]. Conversely, activation of the AIM2 inflammasome aggravates atherosclerosis in individuals with clonal hematopoiesis [88]. In contrast, Akt interacts with AIM2 to inhibit the Akt/mTOR/Myc, thus promotes lipid oxidation in mitochondria. This enhances the stability of Treg cells in response to inflammatory stimuli, thereby limiting the development of autoimmunity in experimental autoimmune encephalomyelitis [89].
4. Potential Metabolic Regulators
For numerous immunologists and pharmacologists, the most urgent inquiry pertains to the potential for introducing a regulatory layer to oversee cellular metabolic programming, with the objective of controlling innate immune responses. The regulation of cellular metabolic programming has the potential to be a highly efficacious therapeutic intervention, as well as to provide crucial insights into the fundamental relationship between metabolic alterations and the signaling control of innate immunity.
4.1. Glycolysis Inhibitors
Small molecules can modulate glycolysis at various enzymatic steps. The compound 2-deoxyglucose (2-DG), a structural analog of glucose, competitively inhibits phosphoglucose isomerase, thereby curbing the formation of glucose-6-phosphate, a critical early intermediate in glycolysis [90]. The use of 2-DG and the fatty acid synthase inhibitor C75 has been shown to forestall the activation of DCs by disrupting the glycolysis-driven de novo synthesis of fatty acids [91]. Therapeutically, 2-DG administration attenuates oxidative stress and the systemic inflammatory response in murine models of acute lung injury and septic shock-induced kidney injury [92]. Furthermore, a recent Phase II clinical trial has reported that 2-DG, administered at a dose of 90 mg/kg/day in conjunction with the standard of care, provides additional benefit to patients with COVID-19, compared to the standard treatment alone [93].
Compound 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one, a potent inhibitor of PFKFB3, mitigates endothelial inflammation in LPS-induced acute lung injury mice [94]. The cell-permeable itaconate derivative 4-octyl itaconate covalently modifies the Cys22 residue on GAPDH to inhibit its glycolytic activity, thus resulting in the amelioration of the inflammatory response within macrophages [95].
Capsaicin interacts directly with Cys424 on PKM2, thereby inhibiting the enzyme's facilitation of the Warburg effect. Treatment with capsaicin, at a dosage of 1 mg/kg, mitigates systemic inflammation and multiple organ dysfunction in a murine model of septic shock induced by LPS [96]. The modulation of PKM2-mediated glycolytic metabolism through agents such as iminostilbene or shikonin associates with reduced inflammatory response in macrophages during myocardial ischemia-reperfusion injury [97, 98], and in Th17 cells in the context of non-alcoholic fatty liver disease [99]. In collagen-induced arthritis mice, Panax notoginseng saponins obstructs STAT3 phosphorylation by preventing nuclear translocation of PKM2, which in turn decreases differentiation of Th17 cells [100]. Conversely, enhancing PKM2 metabolic function with the allosteric activator TEPP-46 restricts Th17 cells maturation, thereby potentially reducing autoimmunity in models of experimental autoimmune encephalomyelitis and multiple sclerosis [20]. DASA-58, another well-characterized PKM2 activator [18], impedes glycolysis and the inflammatory response in macrophages triggered by LPS and follistatin-like protein [101]. Additionally, a series of coxylanolactone derivatives have been synthesized, among which the compound D5 is identified as a PKM2 activator. D5 inhibits Th17 cell differentiation, restoring the Th17/Treg cell balance and ameliorating symptoms of colitis in mice models induced by sodium glucan sulfate and 2,4,6-tritrobenzene sulfonic acid [102].
4.2. NADPH Oxidase Inhibitors
NADPH oxidase is pivotal in catalyzing the reduction of oxygen to superoxide anion, a reaction essential for the oxidative bursts that are a key component of the immune defense system [24]. Consequently, the development of NADPH oxidase inhibitors has become an area of intense research focus.
Apocynin, first isolated from the root of Apocynum cannabinum in 1908 and subsequently from Picrorhiz kurroa in 1971 , was later identified as a selective inhibitor of NADPH oxidase [103]. Apocynin alleviates corneal injury and inflammatory response in corneal neovascularization by its ROS scavenging activity [104]. Interestingly, apocynin also diminished neutrophil survival by modulating ERK1/2 phosphorylation induced by granulocyte-macrophage colony-stimulating factor (GM-CSF), independent of its inhibitory activity on NADPH oxidase [105]. The small molecule LDC7559 and its derivative NA-11 target PFKL, and selectively attenuate NOX-2-dependent oxidative bursts, effectively moderating excessive inflammation in human neutrophils [28].
The NOX1/4 inhibitor GKT137831, also known as setanaxib, potentiates immune activity in CD8+ T cells, enhancing their infiltration into cancer-associated fibroblasts and potentially reversing resistance to programmed cell death protein 1 (PD-1)/PD-ligand 1 immunotherapy [106]. GKT137831 is currently undergoing Phase IIb/III clinical trials for primary biliary cholangitis and hepatitis steatosis, as well as a Phase II clinical trial for squamous cell carcinoma of the head and neck (Clinical trial No. NCT05014672, NCT05323656).
The NOX5 specific inhibitor ML090 significantly reduces edema and cerebral induced cerebral ischemic injury when administered as a pretreatment, suggesting its potential as a preventative strategy in combination with thrombolytic drugs [107].
4.3. ROS Scavengers
The strategic removal of surplus mtROS relies on the development of specialized chemical scavengers. MitoQ, a ubiquinone-derived compound conjugated with a triphenylphosphonium cation, is a lipophilic cation engineered to cross biological membranes and accumulate in the mitochondrial inner membrane, leveraging the mitochondrial membrane potential [108]. Clinical studies have demonstrated the efficacy of MitoQ in a range of conditions, including Parkinson's disease [109], neuroinflammation [110], mtDNA damage associated with high-intensity exercise [111], and the enhancement of vascular function in healthy older adults [112].
Tiron, or sodium 4,5-dihydroxybenzene-1,3-disulfonate, represents another mitochondria-targeted antioxidant. It has shown promise in improving airway inflammation in a chronic asthma model in mice, displaying effectiveness comparable to the clinically prescribed corticosteroid, dexamethasone [113]. Moreover, Tiron inhibits the activation of the NLRP3 inflammasome in endothelin-1-induced models of erectile dysfunction [114], and offers superior protection against oxidative damage from hydroperoxide and UV radiation in the 315-400 nm range in human skin fibroblasts when compared to MitoQ [115].
Another notable compound is mito2HOBA ((4-(4-aminomethyl)-3-hydroxyphenoxy)butyl)-triphenylphosphonium), a mitochondria-targeted scavenger synthesized by conjugating 2-hydroxybenzylamine with the lipophilic cation triphenylphosphonium. Mito2HOBA significantly diminishes systemic inflammation in LPS-induced septic shock in mice [116].
Augmentation with key NAD+ precursors, such as nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), may activate enzymes critical for NAD biosynthesis. Deficits in NMN and NAD+ correlate with metabolic impairments and the enhanced presence of CD38 in immune cells, a phenomenon often observed with aging [117]. Long-term supplementation with NMN and NR is linked to a reduction in age-related inflammation and oxidative stress in murine models [117]. Clinical trials reveal that NMN can substantially improve insulin sensitivity and signaling in prediabetic women following a daily intake of 250 mg for a duration as brief as ten weeks [118].
However, it is imperative to consider treatment duration, dosing regimens, and potential long-term adverse effects. Current human studies typically use doses of up to 500 mg in the above mentioned studies, which are significantly lower than the doses used in mouse models, where approximately 300 mg per kilogram is common - equivalent to approximately 22.5 g for a 75 kg individual. This striking difference underscores the need for cautious dose extrapolation between species. Experts agree that further research using high-throughput methods is essential to elucidate the effects of NAD+ and its precursors on the epigenome, transcriptome, proteome, and metabolome. In addition, long-term administration of NMN or nicotinamide riboside and its effects on healthy individuals warrant rigorous investigation to ensure safety and efficacy.
4.4. cGAS Inhibitors
The inhibition of cGAS focuses on attenuating its catalytic function. PF-06928215, the inaugural cGAS inhibitor, was discovered via a fluorescence polarization assay, exhibits high affinity (kD=200 nM) and potency [81]. Enhanced derivatives, including compounds 18, S2, and S3, target the catalytic domain of cGAS and demonstrate superior inhibition, as confirmed through a pyrophosphate (PPi)-coupled assay and computational screening [119].
Another class of cGAS inhibitors emerged from a screen for compounds that hinder synthesis of cGAMP. RU.521, notable for its potency, binds to Arg364 and Tyr421 of cGAS, engaging the phthalide ring's aldehyde group and forming hydrogen bonds with Gly290 and Lys350 of murine cGAS. Ru.521 uniquely attenuates dsDNA-stimulated type I IFN expression in BMDMs isolated from mice with Aicardi-Goutieres syndrome [120].
Lama et al. introduced the small molecules G108 and G150 as human cGAS inhibitors, leveraging an ATP-dependent, luminescence-based high-throughput screen. These compounds target the cGAS active site, selectively reducing dsDNA-induced IFN response in human THP-1 cells and primary macrophages [121]. Similarly, Cu-32, Cu-76, and related molecules disrupt the dimer interface of human cGAS, specifically targeting cytosolic DNA-triggered, but not RNA-induced, IFN response [122].
Beyond these targeted molecules, certain approved drugs also exhibit inhibition to cGAS. Antimalarial agents, such as hydroxychloroquine sulfate, chloroquine, and quinine, along with the aminoacridine derivative X6, diminish IFN-β production by blocking cGAS-dsDNA interactions [123, 124]. Suramin, an established therapy for parasitic diseases, also inhibits cGAS activity without impacting TLR1/TLR2 or TLR4 pathways [125, 126]. Currently, suramin is undergoing Phase II trials for acute kidney injury (clinical trial No. NCT04496596), autism, and several types of cancer [127, 128]. Additionally, aspirin acetylates cGAS at Lys384, Lys394, or Lys414, and its administration at 50 mg/kg mitigates autoimmunity in models of Aicardi-Goutieres syndrome and in patient-derived peripheral blood mononuclear cells [129].
4.5. STING Agonists
Targeting the palmitoylation of STING offers therapeutic promise [130]. Nitrofuran derivatives C-170, C-171, C-176, C-178, and the indoles derivative H-151 irreversibly inhibit multimeric STING complexes assembly at the Golgi, curtailing downstream signaling [83]. C-176 specifically mitigates STING-associated inflammatory osteolysis [131]. Building on this, Liu et al. subsequently developed SP23, a STING-targeting proteolysis-targeting, from C-170, effectively dampening inflammation in a murine model of cisplatin-induced acute kidney injury [132]. Nitro-fatty acids (NO2-FAs), endogenously lipid, alkylate STING at Cys88, Cys91, and His16, impeding its palmitoylation [133].
Electrophilic acrylamide, BPK-21 and BPK-25, binds to Cys91 residue of STING, precluding its signaling activation in human primary T cells. Notably, BPK-25 also inhibits cGAMP-induced STING activation in peripheral blood mononuclear cells [134]. Tetrahydroisoquinolone derivative 18 interacts with Thr263 of STING, fostering an inactive conformation and obstructing cGAMP-mediated cytokine production [135]. Astin C, a cyclic peptide, competitively occupies cyclic dinucleotide binding sites, thwarting cGAS-STING signalosome assembly in inflammatory responses in Trex1-/- mice [82]. Further, compound 13, a butenolide heterodimer-based inhibitor, selectively inhibits the cGAS-STING pathway, reducing IFN-β and viral dsDNA-induced gene expression in THP-1 cells [136]. These findings indicate that the blockade of the downstream signal pathway is a more efficacious approach to controlling the metabolic changes-induced inflammatory response.
4.6. AIM2 Inflammasome Inhibitors
The activation of the AIM2 inflammasome consistently occurs concomitantly with the activation of other inflammasomes during viral infections, thus AIM2 inhibitors are anticipated to be used in combination with other agents that modulate the immune response. Several compounds have been identified to repress the AIM2 inflammasome-mediated immune response, including CRID3 [137], shikonin [138], compound J114 [139], and the bisphenol compound obovatol [140]. Thus far, no AIM2-specific inhibitors have been reported.
4.7. Metabolic Checkpoint Inhibitors
Metabolic checkpoints play a pivotal role in the regulation of the innate immune response, thereby ensuring the optimal functioning of immune cells such as macrophages and dendritic cells. Principal metabolic regulators include AMP-activated protein kinase (AMPK) and mTOR, which are capable of sensing cellular energy status and nutrient availability [141]. AMPK activation facilitates the catabolic pathways that generate ATP, thereby supporting the survival and function of innate immune cells under conditions of low energy. Conversely, mTOR stimulates anabolic processes, promoting cell growth, proliferation, and effector functions in response to nutrient abundance. The nuclear factor of activated T-cells (NFAT), which is activated by calcineurin, plays a role in the innate immune response by regulating the production of pro-inflammatory cytokines [142]. Immunosuppressive agents such as rapamycin inhibit the activity of mTOR, which in turn reduces the inflammatory activity of innate immune cells [143]. This can be beneficial in conditions such as sepsis, organ transplantation, and chronic inflammation. The modulation of these metabolic checkpoints by pharmacological agents demonstrates the intricate interplay between metabolism and innate immune regulation, thereby providing potential therapeutic strategies for inflammatory and immune-mediated diseases.
Inhibitors that control metabolic signaling.
| Inhibitor | Chemical Structure | Mode of Regulation | Disease Types | References |
|---|---|---|---|---|
| Glycolysis inhibitors | ||||
| 2-DG | 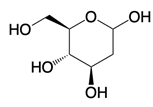 | Competitively inhibits the production of glucose-6-phosphate | Acute lung and kidney injury in mice; COVID-19 and herpes simplex virus infected patients | [91-93] |
| 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one | 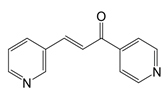 | Binds to PFKFB3 in a dose-dependent manner | Acute lung injury mice model | [88] |
| 4-Octyl itaconate | 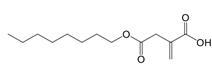 | Binds to Cys22 of GAPDH | Endotoxaemia mice model | [95] |
| Capsaicin | 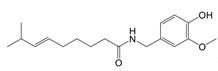 | Binds to Cys424 of PKM2 | Septic shock mice model; neuropathic pain and amyotrophic lateral sclerosis patients | [96] |
| iminostilbene or shikonin |  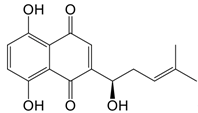 | Binds to PKM2 in a dose-dependent manner | Myocardial ischemia-reperfusion injury and non-alcoholic fatty liver disease mice models | [97, 99] |
| TEPP-46 | 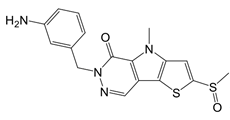 | Promotes PKM2 tetramer formation | Encephalomyelitis and multiple sclerosis mice models | [20] |
| DASA-58 | 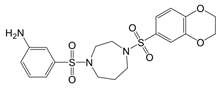 | Promotes PKM2 tetramer formation | Hepatic fibrosis mice model | [18, 101] |
| D5 | 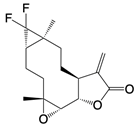 | Promotes PKM2 tetramer formation | Ulcerative colitis mice model | [102] |
| NADPH oxidase inhibitors | ||||
| Apocynin | 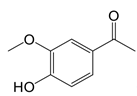 | Blocks p47phox membrane translocation | Corneal alkali burn mice model; sodium-induced declines in cutaneous microvascular function bronchial asthma patients | [104, 105, 148] |
| LDC7559 /NA-11 | 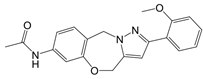 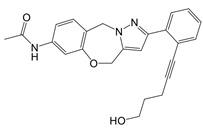 | Binds to the AMP/ADP allosteric activation site | Neutrophil-induced bronchial epithelial damage mice model | [28] |
| GKT137831 (Setanaxib) | 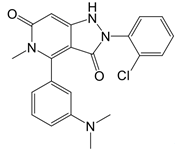 | Direct inhibitor of NOX1 and NOX4 | Type 2 diabetes and primary biliary cholangitis patients | Clinical trial No. NCT02010242, NCT03226067 |
| ML090 | 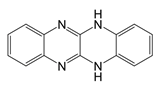 | NOX5 inhibitor | Stroke patients | [107] |
| ROS Scavenger | ||||
| MitoQ | 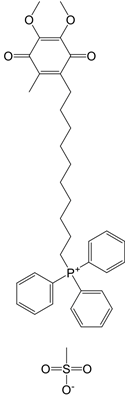 | A mitochondria-targeted antioxidant | Tissue hypoxia induced by neurological deficits in mice; improve vascular function Parkinson's disease exercise-induced mitochondrial DNA damage in patients | [108-112] |
| Tiron (sodium 4,5-dihydroxybenzene-1,3-disulfonate) | 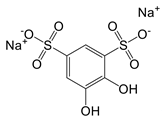 | A mitochondria-targeted antioxidant | Airway remodeling and erectile dysfunction in mice | [113-115] |
| mito2HOBA (4-(4-aminomethyl)-3-hydroxyphenoxy)butyl)-triphenylphosphonium) | 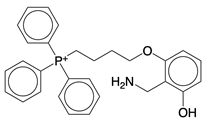 | A mitochondria-targeted isolevuglandins scavenger | N/A | [116] |
| nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) | 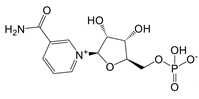 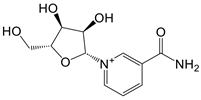 | Activate enzymes control biosynthesis of NAD | Aged mice; and prediabetes patients | [117, 118] |
| Itaconate derivative | ||||
| 4-Octyl itaconate | 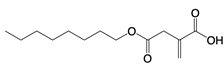 | Protects against lethality and systemic inflammation induced by LPS | Septic shock mice model | [69] |
| cGAS inhibitors | ||||
| PF-06928215 | 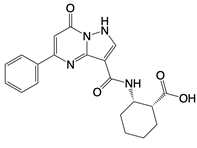 | Binds to the catalytic domain of cGAS | Aged mice and high fat diet-induced cardiac anomalies in mice | [81] |
| Compounds 18, S2, and S3 | 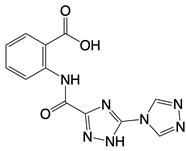 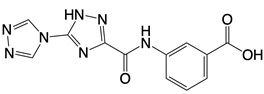 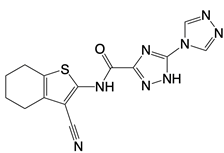 | Binds to the catalytic domain of cGAS | N/A | [119] |
| RU.521 | 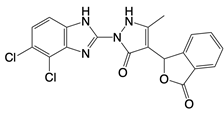 | Binds to residues Arg364 and Tyr421 of cGAS | Subarachnoid hemorrhage-induced brain injury, cerebral venous sinus thrombosis, acute liver injury, rheumatoid arthritis, and postoperative cognitive dysfunction models in mice | [120] |
| G108 and G150 | 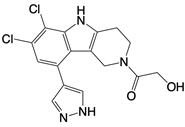 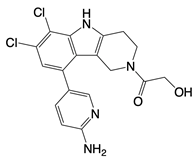 | Binds to the active site of cGAS | N/A | [121] |
| Cu-32, Cu-76, and their analogs | 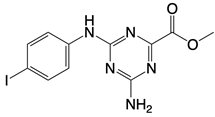 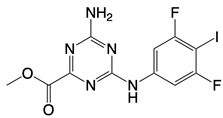 | Binds to the dimer interface of human cGAS | N/A | [122] |
| hydroxychloroquine sulfate, chloroquine, and quinine | 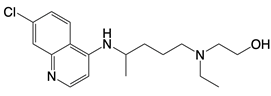 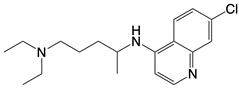 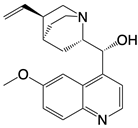 | Inhibits cGAS upon dsDNA stimulation of | Antineoplastic effects in mice; rheumatoid arthritis, systemic lupus rythematosus, and SARS-CoV-2 infection in patients | [123] |
| X6 | 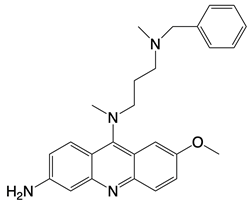 | Blocks interaction of cGAS to dsDNA | N/A | [124] |
| Suramin | 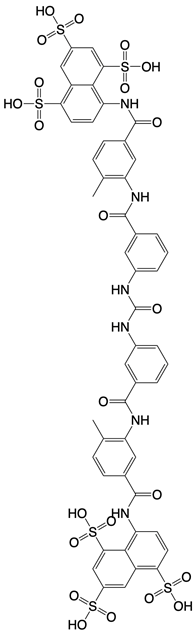 | Inhibits the enzymatic activity of cGAS | Osteoarthritis, chikungunya virus infection, and diabetic nephropathy in mice | [127, 128], Clinical trial No. NCT04496596 |
| Aspirin | 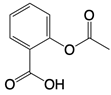 | Binds to residues Lys384, Lys394, or Lys414 of cGAS | Atherosclerotic cardiovascular disease, diabetes, and periodontitis in mice; patients with atherosclerotic cardiovascular disease | [129] |
| STING inhibitor | ||||
| Nitrofuran derivative C-170, C-171, C-176, C-178, and the indoles derivative H-151 | 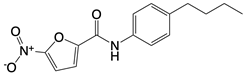 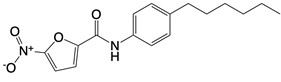 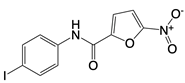 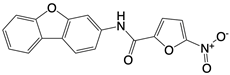 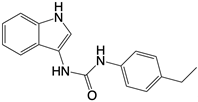 | Disrupt assembly of the multimeric STING complexes | N/A | [83, 131] |
| SP23 | 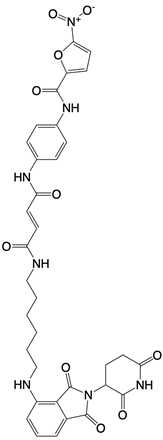 | Blocks assembly of the multimeric STING complexes | Mice renal cell carcinoma model | [132] |
| Nitro-fatty acids (NO2-FAs) | Not disclosed | Binds to residues Cys88, Cys91, and His16 of STING | Mice myocardial fibrosis model | [133] |
| BPK-21 and BPK-25 | 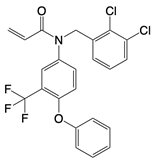 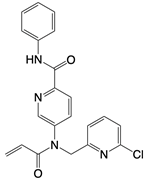 | Binds to residue Cys 91 of STING | N/A | [134] |
| Compound 18 | 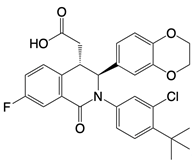 | Binds to residues Thr263 and Thr267 of STING | N/A | [135] |
| Astin C | 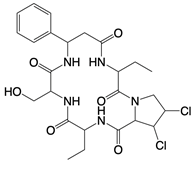 | Binds to the cyclic dinucleotide sites of STING | Colitis and cardiac anomalies in mice | [82] |
| Compound 13 | 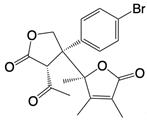 | A pathway-specific antagonists of cyclic GMP-AMP synthase | N/A | [136] |
| AIM2 inhibitors | ||||
| CRID3 | 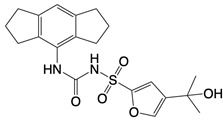 | Inhibits formation of ASC complexes | Spinal cord injury in mice | [137] |
| Shikonin | 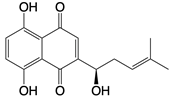 | Dampens formation of ASC specks and directly inhibit caspase-1 enzymatic activity | Acute liver injury, ovarian cancer, skin diseases, wound healing, and lung cancer in mice | [138] |
| J114 | 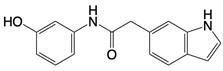 | Blocks interaction between AIM2 and ASC and inhibit ASC oligomerization | Mice keratitis model | [139] |
| Obovatol | 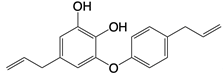 | Inhibits formation of ASC pyroptosome | Mice Alzheimer's disease, colorectal cancer, bone disorders, and hepatocellular carcinoma models | [140] |
| mTOR inhibitor | ||||
| Rapamycin | 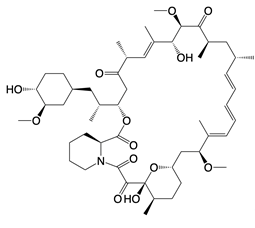 | Inhibits PI3K-Akt signaling, AMPK and mTOR activity | Glaucoma, lung injury, and aging in mice; tuberous sclerosis complex-associated tumors in patients | [141, 149] |
4.8. Combination Therapies
Recent studies highlight the promise of combining metabolic modulators with immune-targeted therapies to counteract pathological metabolic adaptations in immune cells. In cancer immunotherapy, glycolysis inhibitors (e.g., 2-DG) or monocarboxylate transporter 1 (MCT1) inhibitors (AR-C155858, MCT1i) synergize with anti-PD-1 antibodies to alleviate lactate-driven immunosuppression and reverse T cell exhaustion, enhancing antitumor responses [144, 145]. Similarly, MCT1 inhibitors like AZD3965 improve chimeric antigen receptor T-cell efficacy in B-cell malignancies by mitigating metabolic competition in the tumor microenvironment [146]. In psoriasis, the combination of IL-17 antibodies with soraphen A, an acetyl-CoA carboxylase (ACC) inhibitor, targets the metabolic reprogramming of γδT17 cells. These cells shift toward aerobic glycolysis and ATP-citrate synthase-dependent fatty acid synthesis under inflammatory conditions. The blocking of ACC by soraphen A has been shown to disrupt fatty acid synthesis, deplete lipid stores, and suppress IL-17A production in γδT17 cells. This, in turn, has been demonstrated to potentiate the therapeutic effect of IL-17 inhibition [147]. These examples underscore the importance of multi-pathway engagement to overcome metabolic plasticity in immune cells.
5. Future Prospective and Challenges
In future research, several key areas are likely to significantly impact the therapeutic approaches targeting cellular metabolic programming. Firstly, a comprehensive analysis of the intricate interconnections between pathways such as glycolysis, lipid metabolism, and the pentose phosphate pathway will provide a more nuanced understanding of metabolic programming, which in turn will inform the development of more practical therapeutic strategies. Computational modeling and artificial intelligence are emerging as powerful tools to decipher complex metabolic networks and predict therapeutic outcomes, enabling researchers to identify critical nodes for intervention. Secondly, the relationship between mitochondrial function and metabolic reprogramming needs to be thoroughly explored, which is of particular importance in the context of modulating innate immune responses and facilitate the development of more efficacious treatments.
Another significant challenge in the field is the need to account for interspecies differences in immune regulation, particularly between murine models and human physiology, to enhance the translational potential of preclinical findings. Challenges include metabolic redundancy, off-target effects (e.g., NOX inhibitors affecting non-immune cells), and interspecies variability limiting translational potential. Personalized approaches may be needed to account for patient-specific metabolic profiles. The advent of personalized medicine approaches, meticulously tailored to individual metabolic and immune profiles, plays a pivotal role in optimizing therapeutic efficacy and minimizing adverse effects. This assertion is particularly pronounced in the context of immune-mediated diseases, given their inherent heterogeneity. A paucity of human data exists regarding DUOX isoforms in IBD, as well as the role of TCA metabolites in chronic inflammation. Moreover, the majority of combination therapies are still in the preclinical stage, emphasizing the necessity for clinical validation.
In conclusion, the advent of new technologies and applications provides researchers with powerful tools for advancing metabolic reprogramming research. The integration of immunology, metabolism, bioinformatics, and clinical medicine can facilitate a comprehensive understanding of metabolic reprogramming. Techniques such as single-cell sequencing, mass spectrometry, and CRISPR gene editing are of pivotal importance for the uncovering of detailed molecular mechanisms and the identification of potential therapeutic targets. Continued research, coupled with innovative technologies and interdisciplinary collaboration, demonstrates considerable potential for translating metabolic reprogramming into groundbreaking therapies for immune-related diseases.
Abbreviations
2-DG: 2-deoxyglucose; ACC: acetyl-CoA carboxylase; AIM2: absent in melanoma 2; Akt: serine/threonine kinase 1; AMPK: AMP-activated protein kinase; ASC: PYD and CARD domain containing; BMDM: bone marrow-derived macrophage; CD28: cluster of differentiation 28; cGAMP: cyclic guanosine monophosphate-adenosine monophosphate; cGAS: cyclic GMP-AMP synthase; DCs: dendritic cells; dsDNA: double-stranded DNA; DUOX: dual oxidase; ETC: electron transport chain; F2,6BP: fructose-2,6-bisphosphate; F6P: fructose 6-phosphate; FADH2: flavin adenine dinucleotide; FUNDC1: fun14 domain-containing protein 1; G3P: glycerol-3-phosphate; G6P: glucose 6-phosphate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GLUT: glucose transporters; GM-CSF: granulocyte-macrophage colony stimulating factor; GSDMD: gasdermin d; GTP: guanosine-5'-triphosphate; HIF-1α: hypoxia-inducible factor-1α; HK: hexokinase; IBD: inflammatory bowel disease; IFN: interferon; IKK: inhibitor of nuclear factor κ B kinase; IL: interleukin; IMM: inner mitochondrial membrane; iNOS: inducible nitric oxide synthase; IRAK: IL-1R-associated kinases; IRF: interferon regulatory factor; IκB: inhibitor of nuclear factor κ B; LC3: microtubule-associated protein 1A/1B-light chain 3; LDHA: lactate dehydrogenase A; LPS: lipopolysaccharide; MCT1: monocarboxylate transporter 1; MitoQ: mitoquinol mesylate; mtDNA: mitochondrial DNA; mTOR: mammalian target of rapamycin; mtROS: mitochondrial reactive oxygen species; NAD: nicotinamide adenine dinucleotide; NADH: nicotinamide adenine dinucleotide+hydrogen; NADPH: triphosphopyridine nucleotide hydrogen; NF-κB: nuclear factor κ B; NLRC4: NOD-like receptor family card domain containing 4; NLRP3: NOD-like receptor family pyrin domain containing 3; NMN: nicotinamide mononucleotide; NOX: NADPH oxidase; NQO: quinone oxidoreductases; NR: nicotinamide riboside; OMM: outer mitochondrial membrane; OXPHOS: oxidative phosphorylation; PD-1: programmed cell death protein 1; PEP: 2-phosphoenolpyruvate; PFK1: 6-phosphofructokinase-1; PFK2: 6-phosphofructo-2-kinase; PFKFB3: fructose-2,6-biphosphatase 3; PFKL: phosphofructokinase-1 liver type; PI3K: phosphoinositide 3-kinase; PKM2: pyruvate kinase isozymes M2; RAC: Ras-related C3 botulinum toxin substrate; RET: reverse electron transport chain; ROS: reactive oxygen species; SARS: severe acute respiratory syndrome; siRNA: small interfering RNA; STING: stimulator of interferon genes; TBK: tank-binding kinase; TCA cycle: tricarboxylic acid cycle; TCA: tricarboxylic acid; TCR: T cell receptor; Th cell: T helper cell; TLR: Toll-like receptor; TNF-α: tumor necrosis factor-α; Treg cell: regulatory T cell.
Acknowledgements
We apologize to those whose work we could not include due to space limitation. We are also grateful to all members of the Wang laboratory and our collaborators for stimulating discussions and for testing the boundaries of knowledge. This work was partially supported by Macau Science and Technology Development Fund 0131/2022/A3, 0069/2023/RIB3, 005/2023/SKL.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work the authors used DeepL Write in order to check grammar and typographic errors. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Raffin C, Vo LT, Bluestone JA. T(reg) cell-based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20(3):158-72
2. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH. et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017;25(6):1282-93 e7
3. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ. et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742-9
4. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017;26(1):49-70
5. Ivashkiv LB. The hypoxia-lactate axis tempers inflammation. Nat Rev Immunol. 2020;20(2):85-6
6. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D. et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61-72
7. Cho SJ, Moon JS, Nikahira K, Yun HS, Harris R, Hong KS. et al. GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax. 2020;75(3):227-36
8. Huang L, Zhang X, Fan J, Liu X, Luo S, Cao D. et al. EGFR promotes the apoptosis of CD4(+) T lymphocytes through TBK1/Glut1 induced Warburg effect in sepsis. J Adv Res. 2023;44:4439-51
9. Hochrein SM, Wu H, Eckstein M, Arrigoni L, Herman JS, Schumacher F. et al. The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming. Cell Metab. 2022;34(4):516-32 e11
10. De Jesus A, Keyhani-Nejad F, Pusec CM, Goodman L, Geier JA, Stoolman JS. et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol Cell. 2022;82(7):1261-77 e9
11. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML. et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. 2016;166(3):624-36
12. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D. et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity. 2017;47(5):875-89 e10
13. Soto-Heredero G, Gomez de Las Heras MM, Gabande-Rodriguez E, Oller J, Mittelbrunn M. Glycolysis - a key player in the inflammatory response. FEBS J. 2020;287(16):3350-69
14. Goncalves SM, Antunes D, Leite L, Mercier T, Horst RT, Vieira J. et al. Genetic Variation in PFKFB3 Impairs Antifungal Immunometabolic Responses and Predisposes to Invasive Pulmonary Aspergillosis. mBio. 2021;12(3):e0036921
15. Galvan-Pena S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK. et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat Commun. 2019;10(1):338
16. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239-51
17. Li M, Lu H, Wang X, Duan C, Zhu X, Zhang Y. et al. Pyruvate kinase M2 (PKM2) interacts with activating transcription factor 2 (ATF2) to bridge glycolysis and pyroptosis in microglia. Mol Immunol. 2021:140250-66
18. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE. et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21(1):65-80
19. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L. et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016 713280
20. Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N. et al. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4(+) T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab. 2020;31(2):391-405 e8
21. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T. et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. 2017;23(6):753-62
22. Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P. et al. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science. 2021;371(6527):405-10
23. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. 2020;17(3):170-94
24. Graham DB, Becker CE, Doan A, Goel G, Villablanca EJ, Knights D. et al. Functional genomics identifies negative regulatory nodes controlling phagocyte oxidative burst. Nat Commun. 2015 67838
25. Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, Fattouh R. et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 2012;61(7):1028-35
26. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4(157):157ra41
27. Solbak SMO, Zang J, Narayanan D, Hoj LJ, Bucciarelli S, Softley C. et al. Developing Inhibitors of the p47phox-p22phox Protein-Protein Interaction by Fragment-Based Drug Discovery. J Med Chem. 2020;63(3):1156-77
28. Amara N, Cooper MP, Voronkova MA, Webb BA, Lynch EM, Kollman JM. et al. Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell. 2021;184(17):4480-94 e15
29. Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N. et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol. 2013;14(6):543-53
30. Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020 36101638
31. Herranz-Iturbide M, Lopez-Luque J, Gonzalez-Sanchez E, Caballero-Diaz D, Crosas-Molist E, Martin-Mur B. et al. NADPH oxidase 4 (Nox4) deletion accelerates liver regeneration in mice. Redox Biol. 2021 40101841
32. Stenke E, Aviello G, Singh A, Martin S, Winter D, Sweeney B. et al. NADPH oxidase 4 is protective and not fibrogenic in intestinal inflammation. Redox Biol. 2020 37101752
33. Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H. et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ Res. 2020;127(7):911-27
34. Holterman CE, Boisvert NC, Thibodeau JF, Kamto E, Novakovic M, Abd-Elrahman KS. et al. Podocyte NADPH Oxidase 5 Promotes Renal Inflammation Regulated by the Toll-Like Receptor Pathway. Antioxid Redox Signal. 2019;30(15):1817-30
35. Jha JC, Dai A, Garzarella J, Charlton A, Urner S, Ostergaard JA. et al. Independent of Renox, NOX5 Promotes Renal Inflammation and Fibrosis in Diabetes by Activating ROS-Sensitive Pathways. Diabetes. 2022;71(6):1282-98
36. Lee ES, Kim HM, Lee SH, Ha KB, Bae YS, Lee SJ. et al. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic Biol Med. 2020:16192-101
37. Sarr D, Gingerich AD, Asthiwi NM, Almutairi F, Sautto GA, Ecker J. et al. Dual oxidase 1 promotes antiviral innate immunity. Proc Natl Acad Sci U S A. 2021 118(26)
38. Sommer F, Backhed F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol. 2015;8(2):372-9
39. Parlato M, Charbit-Henrion F, Hayes P, Tiberti A, Aloi M, Cucchiara S. et al. First Identification of Biallelic Inherited DUOX2 Inactivating Mutations as a Cause of Very Early Onset Inflammatory Bowel Disease. Gastroenterology. 2017;153(2):609-11 e3
40. Viladomiu M, Longman RS. Decoding the matrix: multiomics reveals host-microbe biomarker for inflammatory bowel disease. J Clin Invest. 2021 131(9)
41. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V. et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807-11
42. Willson JA, Arienti S, Sadiku P, Reyes L, Coelho P, Morrison T. et al. Neutrophil HIF-1alpha stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood. 2022;139(2):281-6
43. Woods PS, Kimmig LM, Sun KA, Meliton AY, Shamaa OR, Tian Y. et al. HIF-1alpha induces glycolytic reprograming in tissue-resident alveolar macrophages to promote cell survival during acute lung injury. Elife. 2022 11
44. Jing C, Castro-Dopico T, Richoz N, Tuong ZK, Ferdinand JR, Lok LSC. et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc Natl Acad Sci U S A. 2020;117(26):15160-71
45. Zhu B, Wu Y, Huang S, Zhang R, Son YM, Li C. et al. Uncoupling of macrophage inflammation from self-renewal modulates host recovery from respiratory viral infection. Immunity. 2021;54(6):1200-18 e9
46. Shyer JA, Flavell RA, Bailis W. Metabolic signaling in T cells. Cell Res. 2020;30(8):649-59
47. Levine LS, Hiam-Galvez KJ, Marquez DM, Tenvooren I, Madden MZ, Contreras DC. et al. Single-cell analysis by mass cytometry reveals metabolic states of early-activated CD8(+) T cells during the primary immune response. Immunity. 2021;54(4):829-44 e5
48. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G. et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14(10):1064-72
49. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res. 2018;122(6):877-902
50. Balsa E, Perry EA, Bennett CF, Jedrychowski M, Gygi SP, Doench JG. et al. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat Commun. 2020;11(1):2714
51. Bleier L, Drose S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta. 2013;1827(11-12):1320-31
52. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I. et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565(7740):495-9
53. Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ. et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat Immunol. 2019;20(4):420-32
54. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287(32):27255-64
55. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE. et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167(2):457-70 e13
56. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E. et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016;24(1):158-66
57. Reynolds MB, Hong HS, Michmerhuizen BC, Lawrence AE, Zhang L, Knight JS. et al. Cardiolipin coordinates inflammatory metabolic reprogramming through regulation of Complex II disassembly and degradation. Sci Adv. 2023;9(5):eade8701
58. Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ. et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185(17):3214-31 e23
59. Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC. et al. K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity. 2016;45(4):761-73
60. Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG. et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity. 2021;54(7):1463-77 e11
61. Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M. et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. 2022;23(5):692-704
62. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE. et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell. 2016;166(1):63-76
63. Wu H, Wang Y, Li W, Chen H, Du L, Liu D. et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019;15(11):1882-98
64. Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D. et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. 2015;11(1):166-82
65. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513-9
66. Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P. et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10(6):972-85
67. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S. et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985-94
68. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G. et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496(7444):238-42
69. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z. et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113-7
70. Auger JP, Zimmermann M, Faas M, Stifel U, Chambers D, Krishnacoumar B. et al. Metabolic rewiring promotes anti-inflammatory effects of glucocorticoids. Nature. 2024;629(8010):184-92
71. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21(4):e49799
72. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42(3):406-17
73. Xu L, Zhou J, Che J, Wang H, Yang W, Zhou W. et al. Mitochondrial DNA enables AIM2 inflammasome activation and hepatocyte pyroptosis in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2021;320(6):G1034-G44
74. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75(6):995-1000
75. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y. et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021;11(4):1845-63
76. Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol. 2023;24(3):186-203
77. Tan HWS, Lu G, Dong H, Cho YL, Natalia A, Wang L. et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat Commun. 2022;13(1):3720
78. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I. et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380-4
79. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786-91
80. Woo MS, Mayer C, Binkle-Ladisch L, Sonner JK, Rosenkranz SC, Shaposhnykov A. et al. STING orchestrates the neuronal inflammatory stress response in multiple sclerosis. Cell. 2024;187(15):4043-60 e30
81. Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D. et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12(9):e0184843
82. Li S, Hong Z, Wang Z, Li F, Mei J, Huang L. et al. The Cyclopeptide Astin C Specifically Inhibits the Innate Immune CDN Sensor STING. Cell Rep. 2018;25(12):3405-21 e7
83. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A. et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269-73
84. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509-13
85. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM. et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111(43):15514-9
86. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171(5):1057-71 e11
87. Yang CA, Huang ST, Chiang BL. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology (Oxford). 2015;54(2):324-31
88. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T. et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296-301
89. Chou WC, Guo Z, Guo H, Chen L, Zhang G, Liang K. et al. AIM2 in regulatory T cells restrains autoimmune diseases. Nature. 2021;591(7849):300-5
90. Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer. 2002;87(7):805-12
91. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323-32
92. Tan C, Gu J, Li T, Chen H, Liu K, Liu M. et al. Inhibition of aerobic glycolysis alleviates sepsis-induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK-regulated autophagy. Int J Mol Med. 2021 47(3)
93. Bhatt AN, Shenoy S, Munjal S, Chinnadurai V, Agarwal A, Vinoth Kumar A. et al. 2-deoxy-D-glucose as an adjunct to standard of care in the medical management of COVID-19: a proof-of-concept and dose-ranging randomised phase II clinical trial. BMC Infect Dis. 2022;22(1):669
94. Li J, Zhou Y, Eelen G, Zhou QT, Feng WB, Labroska V. et al. A high-throughput screening campaign against PFKFB3 identified potential inhibitors with novel scaffolds. Acta Pharmacol Sin. 2023;44(3):680-92
95. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat Commun. 2019;10(1):5091
96. Zhang Q, Luo P, Xia F, Tang H, Chen J, Zhang J. et al. Capsaicin ameliorates inflammation in a TRPV1-independent mechanism by inhibiting PKM2-LDHA-mediated Warburg effect in sepsis. Cell Chem Biol. 2022;29(8):1248-59 e6
97. Lu S, Tian Y, Luo Y, Xu X, Ge W, Sun G. et al. Iminostilbene, a novel small-molecule modulator of PKM2, suppresses macrophage inflammation in myocardial ischemia-reperfusion injury. J Adv Res. 2021:2983-94
98. Rihan M, Sharma SS. Inhibition of Pyruvate kinase M2 (PKM2) by shikonin attenuates isoproterenol-induced acute myocardial infarction via reduction in inflammation, hypoxia, apoptosis, and fibrosis. Naunyn-Schmiedeberg's Archives of Pharmacology. 2023;397(1):145-59
99. Moreno-Fernandez ME, Giles DA, Oates JR, Chan CC, Damen M, Doll JR. et al. PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. 2021;33(6):1187-204 e9
100. Shen MY, Di YX, Wang X, Tian FX, Zhang MF, Qian FY. et al. Panax notoginseng saponins (PNS) attenuate Th17 cell differentiation in CIA mice via inhibition of nuclear PKM2-mediated STAT3 phosphorylation. Pharm Biol. 2023;61(1):459-72
101. Rao J, Wang H, Ni M, Wang Z, Wang Z, Wei S. et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut. 2022;71(12):2539-50
102. Wang P, Yang H, Lin W, Zhou J, Liu Y, Ma L. et al. Discovery of Novel Sesquiterpene Lactone Derivatives as Potent PKM2 Activators for the Treatment of Ulcerative Colitis. J Med Chem. 2023;66(8):5500-23
103. Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. American Journal of Respiratory Cell and Molecular Biology. 1994;11(1):95-102
104. Zhang K, Guo MY, Li QG, Wang XH, Wan YY, Yang ZJ. et al. Drp1-dependent mitochondrial fission mediates corneal injury induced by alkali burn. Free Radic Biol Med. 2021:176149-61
105. Pintard C, Ben Khemis M, Liu D, Dang PM, Hurtado-Nedelec M, El-Benna J. Apocynin prevents GM-CSF-induced-ERK1/2 activation and -neutrophil survival independently of its inhibitory effect on the phagocyte NADPH oxidase NOX2. Biochem Pharmacol. 2020 177113950
106. Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P. et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020;80(9):1846-60
107. Casas AI, Kleikers PW, Geuss E, Langhauser F, Adler T, Busch DH. et al. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J Clin Invest. 2019;129(4):1772-8
108. James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA. et al. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007;282(20):14708-18
109. Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V. et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord. 2010;25(11):1670-4
110. Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C. et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol. 2013;74(6):815-25
111. Williamson J, Hughes CM, Cobley JN, Davison GW. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox Biol. 2020 36101673
112. Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL. et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension. 2018;71(6):1056-63
113. El-Sherbeeny NA, Hassan ZA, Ateyya H. Tiron ameliorates oxidative stress and inflammation in a murine model of airway remodeling. Int Immunopharmacol. 2016:39172-80
114. Sobrano Fais R, Menezes da Costa R, Carvalho Mendes A, Mestriner F, Comerma-Steffensen SG, Tostes RC. et al. NLRP3 activation contributes to endothelin-1-induced erectile dysfunction. J Cell Mol Med. 2023;27(1):1-14
115. Oyewole AO, Wilmot MC, Fowler M, Birch-Machin MA. Comparing the effects of mitochondrial targeted and localized antioxidants with cellular antioxidants in human skin cells exposed to UVA and hydrogen peroxide. FASEB J. 2014;28(1):485-94
116. Mayorov V, Uchakin P, Amarnath V, Panov AV, Bridges CC, Uzhachenko R. et al. Targeting of reactive isolevuglandins in mitochondrial dysfunction and inflammation. Redox Biol. 2019 26101300
117. Chini CCS, Peclat TR, Warner GM, Kashyap S, Espindola-Netto JM, de Oliveira GC. et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat Metab. 2020;2(11):1284-304
118. Yoshino M, Yoshino J, Kayser BD, Patti GJ, Franczyk MP, Mills KF. et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science. 2021;372(6547):1224-9
119. Zhao W, Xiong M, Yuan X, Li M, Sun H, Xu Y. In Silico Screening-Based Discovery of Novel Inhibitors of Human Cyclic GMP-AMP Synthase: A Cross-Validation Study of Molecular Docking and Experimental Testing. J Chem Inf Model. 2020;60(6):3265-76
120. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y. et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8(1):750
121. Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V. et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun. 2019;10(1):2261
122. Padilla-Salinas R, Sun L, Anderson R, Yang X, Zhang S, Chen ZJ. et al. Discovery of Small-Molecule Cyclic GMP-AMP Synthase Inhibitors. J Org Chem. 2020;85(3):1579-600
123. An J, Minie M, Sasaki T, Woodward JJ, Elkon KB. Antimalarial Drugs as Immune Modulators: New Mechanisms for Old Drugs. Annu Rev Med. 2017:68317-30
124. An J, Woodward JJ, Lai W, Minie M, Sun X, Tanaka L. et al. Inhibition of Cyclic GMP-AMP Synthase Using a Novel Antimalarial Drug Derivative in Trex1-Deficient Mice. Arthritis Rheumatol. 2018;70(11):1807-19
125. Van den Enden E, Vlieghe E, Demeester R, Ieven G, Jansens H, Van den Hauwe L. A traveler with neurobrucellosis. Travel Med Infect Dis. 2009;7(4):215-8
126. Wang M, Sooreshjani MA, Mikek C, Opoku-Temeng C, Sintim HO. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-beta levels. Future Med Chem. 2018;10(11):1301-17
127. Naviaux RK, Curtis B, Li K, Naviaux JC, Bright AT, Reiner GE. et al. Low-dose suramin in autism spectrum disorder: a small, phase I/II, randomized clinical trial. Ann Clin Transl Neurol. 2017;4(7):491-505
128. George S, Dreicer R, Au JJ, Shen T, Rini BI, Roman S. et al. Phase I/II trial of 5-fluorouracil and a noncytotoxic dose level of suramin in patients with metastatic renal cell carcinoma. Clin Genitourin Cancer. 2008;6(2):79-85
129. Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS. et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell. 2019;176(6):1447-60 e14
130. Hansen AL, Mukai K, Schopfer FJ, Taguchi T, Holm CK. STING palmitoylation as a therapeutic target. Cell Mol Immunol. 2019;16(3):236-41
131. Yu ZC, Fu R, Li Y, Zhao DY, Jiang H, Han D. The STING inhibitor C-176 attenuates osteoclast-related osteolytic diseases by inhibiting osteoclast differentiation. FASEB J. 2023;37(4):e22867
132. Liu J, Yuan L, Ruan Y, Deng B, Yang Z, Ren Y. et al. Novel CRBN-Recruiting Proteolysis-Targeting Chimeras as Degraders of Stimulator of Interferon Genes with In Vivo Anti-Inflammatory Efficacy. J Med Chem. 2022;65(9):6593-611
133. Hansen AL, Buchan GJ, Ruhl M, Mukai K, Salvatore SR, Ogawa E. et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci U S A. 2018;115(33):E7768-E75
134. Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y. et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell. 2020;182(4):1009-26 e29
135. Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H. et al. Discovery of a Novel cGAMP Competitive Ligand of the Inactive Form of STING. ACS Med Chem Lett. 2019;10(1):92-7
136. Huffman BJ, Chen S, Schwarz JL, Plata RE, Chin EN, Lairson LL. et al. Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists. Nat Chem. 2020;12(3):310-7
137. Coll RC, Robertson A, Butler M, Cooper M, O'Neill LA. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS One. 2011;6(12):e29539
138. Zorman J, Susjan P, Hafner-Bratkovic I. Shikonin Suppresses NLRP3 and AIM2 Inflammasomes by Direct Inhibition of Caspase-1. PLoS One. 2016;11(7):e0159826
139. Jiao Y, Nan J, Mu B, Zhang Y, Zhou N, Yang S. et al. Discovery of a novel and potent inhibitor with differential species-specific effects against NLRP3 and AIM2 inflammasome-dependent pyroptosis. Eur J Med Chem. 2022 232114194
140. Kim J, Ahn H, Han BC, Shin H, Kim JC, Jung EM. et al. Obovatol inhibits NLRP3, AIM2, and non-canonical inflammasome activation. Phytomedicine. 2019 63153019
141. Gao J, Liu CF, Liu PP, Wang XW. Double-stranded RNA induces antiviral transcriptional response through the Dicer-2/Ampk/FoxO axis in an arthropod. Proc Natl Acad Sci U S A. 2024;121(31):e2409233121
142. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M. et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity. 2017;46(4):675-89
143. Barros RDS, Queiroz LAD, de Assis JB, Pantoja KC, Bustia SX, de Sousa ESA. et al. Effects of low-dose rapamycin on lymphoid organs of mice prone and resistant to accelerated senescence. Front Immunol. 2024 151310505
144. Wartewig T, Daniels J, Schulz M, Hameister E, Joshi A, Park J. et al. PD-1 instructs a tumor-suppressive metabolic program that restricts glycolysis and restrains AP-1 activity in T cell lymphoma. Nat Cancer. 2023;4(10):1508-25
145. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y. et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40(2):201-18 e9
146. Lopez E, Karattil R, Nannini F, Weng-Kit Cheung G, Denzler L, Galvez-Cancino F. et al. Inhibition of lactate transport by MCT-1 blockade improves chimeric antigen receptor T-cell therapy against B-cell malignancies. J Immunother Cancer. 2023 11(6)
147. Kao YS, Lauterbach M, Lopez Krol A, Distler U, Godoy GJ, Klein M. et al. Metabolic reprogramming of interleukin-17-producing gammadelta T cells promotes ACC1-mediated de novo lipogenesis under psoriatic conditions. Nat Metab. 2025
148. Satriano J, Schlondorff D. Activation and attenuation of transcription factor NF-kB in mouse glomerular mesangial cells in response to tumor necrosis factor-alpha, immunoglobulin G, and adenosine 3':5'-cyclic monophosphate. Evidence for involvement of reactive oxygen species. J Clin Invest. 1994;94(4):1629-36
149. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960-76
Author contact
![]() Corresponding author: emilyywangedu.mo (Y. Wang), ORCID: 0000-0002-1407-2668.
Corresponding author: emilyywangedu.mo (Y. Wang), ORCID: 0000-0002-1407-2668.