Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(12):5514-5530. doi:10.7150/ijbs.113890 This issue Cite
Review
FAM134B in Cellular Homeostasis: Bridging Endoplasmic Reticulum-Phagy to Human Diseases
Ning Chen1, Jia-Qi Yang2, Sen Tong1, Lu Xu1, Ning Dong1, Yao Wu1, Yu-Xuan Li3 ![]() , Ren-Qi Yao3
, Ren-Qi Yao3 ![]() , Yong-Ming Yao1
, Yong-Ming Yao1 ![]()
1. Medical Innovation Research Division and Fourth Medical Center, Chinese PLA General Hospital, Beijing 100853, China.
2. Department of Acute Abdominal Surgery, Beijing Chao-Yang Hospital, Capital Medical University, Beijing 100020, China.
3. Department of General Surgery, First Medical Center of the Chinese PLA General Hospital, Beijing 100853, China.
Ning Chen and Jia-Qi Yang have contributed equally to this work.
Received 2025-3-17; Accepted 2025-8-8; Published 2025-8-30
Abstract

FAM134B, originally characterized as an oncogene in esophageal squamous carcinoma, has also been implicated in the pathogenesis of hereditary sensory and autonomic neuropathy type IIB (HSAN2B). It is recognized as the inaugural endoplasmic reticulum (ER)-phagy receptor in mammals containing an LC3-interacting region, which facilitates its interaction with LC3 and GABARAP proteins in the autophagosome. ER-phagy, a critical process involved in ER quality control, selectively degrades superfluous or damaged ER fragments in lysosomes, thereby maintaining ER and protein homeostasis. This review offers an in-depth analysis of FAM134B's structure, function, and regulation, emphasizing its role in infectious diseases, neuropathies, cancer, metabolic disorders, degenerative conditions, and cardiovascular diseases. The evidence presented highlights the need for further research on FAM134B as a potential therapeutic target in human diseases.
Keywords: FAM134B, ER-phagy, cellular homeostasis, human diseases
Introduction
Autophagy, or macroautophagy, is a conserved cellular process in eukaryotes that employs lysosomes to degrade cytoplasmic proteins and damaged organelles. Autophagy-related genes regulate the formation of autophagic vesicles for substrate sequestration and autolysosomes for degradation. Under normal conditions, autophagy operates at a baseline level, however, it undergoes significant alterations in response to cellular stress [1]. This dynamic process is integral to the synthesis, breakdown, and recycling of cellular components, thereby sustaining homeostasis within the cell. However, excessive autophagy can lead to metabolic stress, cellular degradation, and cell death [2]. Recent studies have shown that autophagy, once considered a non-selective process, can specifically target certain cellular components such as impaired organelles, aggregated proteins, and invasive bacteria [3, 4]. ER-phagy, a form of selective autophagy, targets excess or damaged ER for degradation within lysosomes or vacuoles. The ER, a single-membrane intracellular system, is essential for protein synthesis, calcium regulation, and lipid metabolism. Thus, ER-phagy is essential for ER quality control and overall cellular homeostasis. Three distinct ER-phagy pathways have been identified: macro-ER-phagy, micro-ER-phagy, and LC3-dependent vesicular transport. To date, reported ER-phagy receptors include membrane-bound ER-phagy receptors (FAM134 protein family, RTN3L, SEC62, CCPG1, TEX264, atlastin (ATL) GTPases, STING) and cytoplasmic ER-phagy receptors (CALCOCO1, C53, p62) [5]. Notably, FAM134B was the first ER-phagy receptor discovered in mammals, exhibiting functional homology with Atg40, an ER receptor concurrently reported in yeast [6].
During ER-phagy, FAM134B strongly interacts with MAP1LC3B and GABARAP-L2. Enrichment analysis has revealed that FAM134B interacts with a diverse array of proteins involved in critical cellular functions, including ER function, ATP synthesis, translocon complexes, ER morphology, vesicle formation, Ca2+ signaling, and cytoskeletal microfilaments. Additionally, FAM134B is linked to various organelles, including ubiquitin (Ub)-proteasome system, mitochondria, and Golgi apparatus [7]. Downregulation of FAM134B leads to ER and Golgi expansion, while its overexpression leads to ER fragmentation and lysosomal degradation in human cells [8]. Beyond its role in selective ER-phagy, FAM134B also facilitates LC3B-dependent vesicle translocation to degrade misfolded polypeptide aggregates. We undertook a thorough search and review of the published literature to elucidate the significant biological functions of FAM134B and to offer theoretical guidance for future research endeavors.
Review Methodology
A literature search on PubMed and Web of Science covered original research in peer-reviewed journals from 2010 to 2024. Studies were chosen for their relevance to FAM134B mechanisms or diseases, excluding conference abstracts and non-peer-reviewed works. The search terms applied in our study included "FAM134B" or "RETREG1" as well as "ER-Phagy" or "reticulophagy".
Structure and expression patterns of FAM134B proteins
The FAM134 family, comprising FAM134A/RETREG2, FAM134B/RETREG1, and FAM134C/RETREG3, is crucial for ER fragmentation and lysosomal degradation during ER stress [9]. The FAM134B gene (also known as RETREG1 or JK-1) is located on chromosome 5p15.1 and spans approximately 144 kb. FAM134B is a 497-amino acid protein with a molecular weight of 54 kDa, primarily located in the ER sheet and Golgi apparatus. Structurally, FAM134B comprises three domains (Fig. 1): a N-terminal disordered domain (~80 amino acids), a reticulon homology domain (RHD), and a C-terminal disordered domain (~240 amino acids) containing an LC3-interacting region (LIR) motif [6, 10-12]. The LIR motif-mediated interaction between FAM134B and LC3/GABARAP is essential for targeting ER fragments to autophagosomes, leading to subsequent lysosomal degradation [8]. The RHD of FAM134B plays a fundamental role in ER membrane remodeling and consists of four key structural components: TM1-TM2, AHL, TM3-TM4, and AHC [13, 14]. TM1-TM2 and TM3-TM4 are two helical hairpins, each containing two transmembrane (TM) helices linked by a polar luminal loop, which is crucial for membrane deformation and protein clustering. AHL and AHc, two cytoplasmic amphipathic helices, flank TM3-TM4, linking TM1-TM2 with TM3-TM4 and TM3-TM4 with the C-terminus of FAM134B, respectively. These interactions are essential for membrane shaping. The asymmetric wedge structure of RETREG1 RHD facilitates ER membrane bending, whereas the aggregation of six RHDs forms an inverted pyramid structure, further enhancing membrane curvature. During ER-phagy, the clustering of FAM134B RHDs, combined with the pulling force exerted by the C-terminal LIR motif binding to LC3 on the phagophore, promotes vesicle budding, ER pinching off, and subsequent lysosomal degradation [15].
Detailed structural composition of FAM134B. FAM134B is an ER-phagy receptor found in Homo sapiens and other mammals. The structure of FAM134B consists of three main components: the N-terminal region, reticulon homology domain (RHD), and C-terminal region. The RHD contains TM1-2, AHL, TM3-4, and AHC, which are primarily responsible for detecting and promoting membrane curvature and scission. The C-terminus contains an LC3-interacting region (LIR) motif.
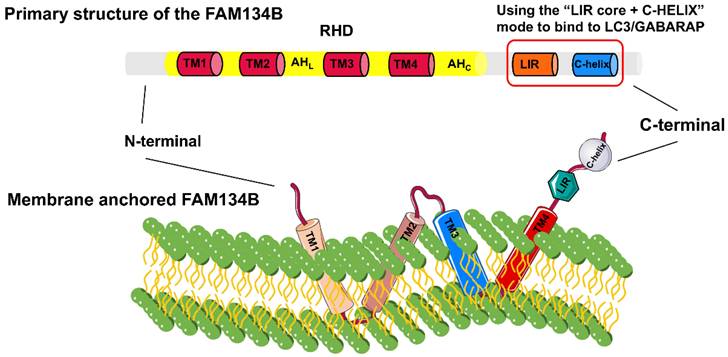
FAM134B-2, a truncated isoform of FAM134B, partially lacks the RHD and consists of six exons encoding a 356-amino acid protein. Starvation leads to an increase in FAM134B-2 expression rather than FAM134B-1 in the liver. Starvation activates the C/EBPβ-FAM134B-2 axis, initiating ER-phagy and lysosomal degradation of FAM134B-2. The ER secretory protein ApoCIII interacts with FAM134B-2 and is degraded through FAM134B-2-mediated ER-phagy, without affecting overall ER turnover [16]. Moreover, amino acid deprivation (AAD) in HeLa cells activates the transcription factors MEF2D and NR4A1, which bind upstream of the FAM134B-2 promoter to enhance mRNA expression and facilitate FAM134B-2-mediated ER-phagy, thereby maintaining amino acid homeostasis. MEF2D functions upstream of NR4A1, and the MEF2D-NR4A1-FAM134B2-mediated ER-phagy cascade is Ca2+- and PKC-dependent; however, the precise PKC-dependent mechanisms remain to be elucidated [17]. Within the FAM134 family, the lysosomal flux of FAM134B shows a greater increase in BafA-treated U2OS cells compared to FAM134A and FAM134C [18]. The wedge-shaped RHD of FAM134B allows for quicker ER vesicle formation compared to FAM134A and FAM134C, enhancing ER membrane remodeling efficiency [9].
Biological functions and regulatory mechanisms of FAM134B
FAM134B and ER-phagy
Under stressful conditions, misfolded protein accumulation within the ER disrupts cellular function. To restore cellular homeostasis, cells initiate ER-phagy, a selective autophagic process that eliminates excess or misfolded proteins and damaged ER membranes. ER-phagy is driven by specific receptors and ultimately integrates into different stages of classical autophagy (recognition, encapsulation, fusion, and degradation) to achieve targeted degradation of abnormal ER components. So far, the dual-fluorescence reporter system (e.g., ssRFP-GFP-KDEL or mCherry-GFP-FAM134B) is the primary method for monitoring ER-phagy flux, leveraging GFP quenching in lysosomal acidity while RFP/mCherry remains stable, allowing for quantification via fluorescence co-localization or ratio changes [19]. FAM134B is critical for maintaining physiological homeostasis and modulating pathological conditions by regulating ER dynamics [8]. During ER-phagy, FAM134B's LIR domain interacts with LC3-like proteins within the autophagosome and is subsequently transported to the lysosome for degradation through core autophagy machinery. Disruptions in FAM134B function result in significant ER expansion, whereas upregulated FAM134B leads to ER fragmentation.
FAM134B and ER-mitochondria interaction
Calcium homeostasis is maintained through the interaction between the ER and mitochondria. Excessive mitochondrial calcium accumulation triggers mitochondrial dysfunction and neuronal apoptosis, representing a significant pathogenic mechanism in acquired epilepsy. The FAM134B-mediated ER-phagy could mitigate mitochondrial calcium overload by diminishing specific receptor IP3R levels on the mitochondrial-associated ER membranes. This action provides a protective effect on mitochondria and reduces aberrant neuronal cell death in the context of acquired epilepsy [20, 21]. Beyond its role in Ca²⁺ regulation, FAM134B is involved in the turnover of dual organelles, the ER and mitochondria, to maintain cellular homeostasis. Elevated cytosolic Ca2+ levels induce the upregulation of E3 Ub ligase autocrine motility factor receptor (AMFR), which facilitates the proteasomal degradation of mitofusin on the outer mitochondrial membrane under stress conditions. This process results in the formation of unstable mitoplasts, exposing the inner mitochondrial membrane (IMM) and promoting closer ER-mitochondria interactions. The interaction between FAM134B and optic atrophy 1, a mitochondrial dynamin-like GTPase located in the IMM, triggers “reticulo-mito-phagy”, wherein autophagic vesicles encapsulate AMFR and IMM components, targeting them for lysosomal degradation. Reticulo-mito-phagy serves as a protective mechanism that regulates ER-mitochondria crosstalk and restores cellular homeostasis [22]. Additionally, FAM134B facilitates adipocyte differentiation by promoting mitophagy and reducing mitochondrial numbers, further underscoring its role in cellular metabolism [23].
FAM134B mediated ERLAD
In contrast to the double-membrane autophagosomes in ER-phagy, ER-to-lysosome-associated degradation (ERLAD) involves the transport of a single-membrane vesicle from ER to lysosome. The process of ERLAD requires the interaction between FAM134B and LC3, along with LC3 lipidation, which is essential for vesicle trafficking. Proteasome-resistant polymers of ATZ, a mutant alpha-1-antitrypsin (AAT) protein, accumulate in the ER lumen and are segregated by calnexin (CNX) into a FAM134B-enriched ER subdomain. Subsequently, FAM134B facilitates LC3II-dependent vesicle translocation, encapsulating CNX-FAM134B-LC3II complexes containing ATZNNN polymers within SNARE STX17 ER vesicles. These vesicles then migrate to LAMP1/RAB7-positive endolysosomes, where they facilitate ATZ polymer degradation [24]. Misfolded proteins within the ER are degraded via mannose and glucose processing of N-glycans, which activate either the ER-associated degradation (ERAD) pathway or the ERLAD pathway, respectively. In parallel with classical ER-phagy, FAM134B can mediate ERLAD independently of autophagic vesicle formation. Proteasome-resistant ATZ polymers, which undergo N-glycan processing, generate ERAD-resistant ATZNNN polymers that interact with CNX and are selectively segregated into the FAM134B-enriched ER subdomain. This process is independent of ER-phagy, but the LIR function of FAM134B and CNX's glucose-binding activity are essential for endolysosomal clearance [24, 25].
Regulatory mechanisms of FAM134B expression
Transcription factors, including TFEB/TFE3, activate FAM134B-mediated ER-phagy by inducing FAM134B expression during prolonged starvation. Dephosphorylated TFEB/TFE3 translocates to the nucleus and binds specific DNA-binding site to enhance FAM134B transcription [26, 27]. In maintaining ER morphology, the ATL protein family—consisting of ATL1, ATL2, and ATL3—likely exerts complementary functions in regulating ER-phagy. Their GTPase domains aid in ER membrane scission for lysosomal engulfment. FAM134B upregulation results in decreased ATL2 levels, whereas ATL2 downregulation diminishes FAM134B-mediated ER-phagy triggered by FAM134B overexpression. Therefore, ATL2 operates downstream of FAM134B, serving as a FAM134B-mediated ER-phagy effector and cargo [28]. Similarly, DDRGK domain-containing 1 acts as a cargo in FAM134B-mediated ER-phagy, with its depletion hindering ER-phagy triggered by FAM134B overexpression, regardless of starvation conditions [29].
The nucleotide exchange factor SIL1, a co-chaperone of BiP, enhances BiP's ATPase activity and energy exchange efficiency in molecular chaperone dynamics. Pathogenic SIL1 mutations impair BiP-mediated protein folding, significantly reducing FAM134B expression. Notably, SIL1 and FAM134B colocalize in the Golgi apparatus [30]. Additionally, N6-methyladenosine (m6A) mRNA modification regulates FAM134B expression and affects fat formation in porcine adipocytes. YTH m6A RNA-binding protein 2 (YTHDF2) binds to the m6A site of FAM134B, promoting mRNA degradation and lowering FAM134B levels. A mutation at the m6A binding site prevents mRNA degradation, leading to increased levels of downstream proteins, which boost fat deposition [31].
Post-translational modifications of autophagy-related factors can either activate or inhibit autophagy, thereby influencing disease progression and drug efficacy (Table 1) [32]. FAM134B and ER-phagy are regulated by phosphorylation, ubiquitination, O-GlcNAcylation, and acetylation (Fig. 2). Phosphorylation of FAM134B sequentially induces oligomerization, ER fragmentation, and ER-phagy. During ER stress, CAMK2B phosphorylation modifies the S151 site of the RHD in FAM134B, promoting FAM134B-mediated ER-phagy to maintain cellular homeostasis [33, 34]. Similarly, mTOR inhibition triggers casein kinase 2 (CK2) to phosphorylate FAM134B and FAM134C, leading to their CK2-dependent ubiquitination. Phosphorylation promotes the growth and densification of FAM134 clusters, triggering ER-phagy once these high-density nanoscale clusters reach 90-100 nm. Notably, FAM134C functions as a substrate and an enhancer of FAM134B-mediated ER-phagy [35].
FAM134B regulatory factors, mechanisms, and physiological roles.
| Pathway/Key Factors | Effect on FAM134B | Pathophysiological model | Outcome | References |
|---|---|---|---|---|
| TFEB/TFE3 | Upregulation | Prolonged starvation | Activates ER-phagy, maintains cellular homeostasis | [26] |
| YTHDF2 | Downregulation | Porcine adipocytes | Reduces fat deposition | [31] |
| CAMK2 | S141 phosphorylation enhances oligomerization. | ER stress | Maintain cellular homeostasis | [33] |
| CK2 | Activating phosphorylation-based ubiquitination of FAM134B and FAM134C | mTOR inhibition | Triggers ER-phagy via FAM134B/FAM134C nanocluster formation | [35] |
| AMFR | Ubiquitination of FAM134B and ARL6IP1 enhances oligomerization and membrane curvature | HeLa | Boosts FAM134B-LC3B interaction and ER-phagy; | [37-38] |
| USP20 | Stabilizes FAM134B by deubiquitination | Starvation | Enhances FAM134B-LC3B interaction and recruits WIPI2 to promote autophagy initiation | [39] |
| OGT | O-GlcNAcylation of FAM134B | Nutrient-deprived NP cells | Suppresses apoptosis/senescence and enhances ER-phagy | [40] |
| CBP/SIRT7 | K160 acetylation/deacetylation of FAM134B | ER stress | Enhances ER-phagy/Prevents excessive ER degradation | [41-42] |
| AT-1 | Acetylation ATG9A inhibit FAM134B/SEC62-LC3B axis | AT-1-overexpressing mice | ATG9A acetylation blocks ER-phagy, causing progeria-like symptoms; ATase inhibition rescues aging defects | [43-44] |
Post-translational modifications of FAM134B promote or inhibit ER-phagy. (a) Phosphorylation of FAM134B enhances ER-phagy by promoting its oligomerization and ER fragmentation. FAM134C acts as a substrate and an enhancer for FAM134B-mediated ER-phagy. (b) Ubiquitination of FAM134B and ARL6IP1 by AMFR enhances FAM134B oligomerization and facilitates the formation of FAM134B/ARL6IP1 heterodimers, thereby promoting ER-phagy. (c) USP20 specifically deubiquitinated FAM134B by K48/K63-linked polyubiquitination, activating ER-phagy during starvation. USP20 also interacts with VAPs, facilitating the recruitment of autophagy initiation proteins, such as WIPI2. (d) Under nutrient deprivation conditions, the O-GlcNAcylation of FAM134B by OGT facilitates ER-phagy and cell survival. (e) The acetylated ATG-9A regulates FAM134B-mediated ER-phagy. AT-1/SLC33A1 mediated acetylation of ATG-9A enhances its interaction with the chaperones HSPB1 (cytoplasmic) and CALR (ER lumen), preventing its binding to FAM134B and SEC62, thereby inhibiting ER-phagy. Conversely, AT-1/SLC33A1 knockdown enhances ER-phagy. The acetyltransferase CBP and deacetylase SIRT7 are critically involved in modulating the acetylation status of FAM134B, with acetylated FAM134B facilitating CAMK2-mediated phosphorylation, leading to increased ER-phagy.
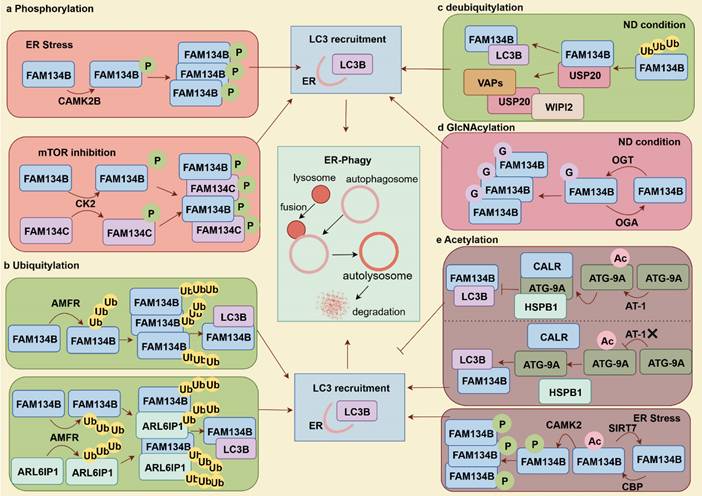
Protein ubiquitination is the tagging of target proteins for degradation, involving ubiquitin activation (E1), conjugation (E2) and ligation (E3) [36]. AMFR, an E3 ligase catalyzes the FAM134B's RHD ubiquitination, promoting FAM134B oligomerization and inducing membrane curvature, which enhances ER-phagy flux by facilitating interactions between FAM134B and LC3B. Additionally, ubiquitinated RHDs strengthen the interactions between adjacent RHDs, forming dense FAM134B clusters that contribute to extensive membrane remodeling [37]. Beyond the FAM134B, AMFR also mediates the ubiquitination of the RHD of ARL6IP1, which does not directly interact with LC3. Instead, ARL6IP1 forms a heterodimer with FAM134B, allowing AMFR-mediated ubiquitination of ARL6IP1 to enhance the FAM134B-LC3B interaction, thereby promoting ER-phagy and improving cellular fitness. Defects in ARL6IP1 are associated with sensory nerve loss, abnormal ER morphology, reduced ER stress resistance, impaired ER-phagy, and decreased cellular adaptability [38]. Deubiquitination, the reverse of ubiquitination, is mediated by deubiquitinating enzymes that remove Ub molecules from substrate proteins, thereby protecting them from proteasomal degradation. USP20 specifically cleaves K48/K63-linked Ub chains from FAM134B, stabilizing it and enhancing FAM134B-LC3B interactions to facilitate ER-phagy under starvation conditions. Moreover, USP20 interacts with ER TM proteins (VAMP-associated proteins [VAPs]), which recruit autophagy initiation proteins like WD repeat domain and phosphoinositide interacting 2 (WIPI2), enhancing ER-phagy [39].
O-GlcNAcylation, facilitated by O-GlcNAc transferase (OGT), transfers a single N-Acetyl glucosamine to serine or threonine residues, and can be reversed by O-GlcNAc glycosylase (OGA) for de-O-GlcNAcylation. Upregulated O-GlcNAcylation mitigate apoptosis and senescence in nucleus pulposus (NP) cells deprived of nutrients. This protective effect is mediated through the interaction between OGT and FAM134B, which stabilizes FAM134B, reduces FAM134B ubiquitination and degradation, and enhances ER-phagy, thereby facilitating cellular adaptation and survival under nutrient-deficient conditions [40]. Nε-lysine acetylation within the ER is another key mechanism regulating ER homeostasis and protein secretion. CBP acetylates and SIRT7 deacetylates FAM134B at K160 site, respectively, creating a regulatory circuit for ER homeostasis. ER stress-induced acetylation of FAM134B by CBP promotes FAM134B oligomerization, ER membrane fragmentation, and ER-phagy. Then, acetylated FAM134B enhances CAMKII-mediated phosphorylation, inducing mild and sustained ER-phagy. Conversely, SIRT7 deacetylates FAM134B to prevent excessive ER degradation. Interestingly, the nuclear acetyltransferase CBP, traditionally recognized as a transcriptional regulator, can relocate to the cytosol to modulate ER stress responses by regulating ER-phagy [41, 42]. AT1/SLC33A1 facilitates cytosol-to-ER acetyl-CoA flux, negatively regulates ER-phagy levels by suppressing the ATG9A-FAM134B/SEC62-LC3 axis. Acetylation of ATG9A disrupts its interaction with FAM134B and SEC62, thereby inhibiting ER-phagy. In this process, the chaperone protein CALR binds to acetylated ATG9A within the ER lumen, whereas HSPB1 undergoes a conformational change to bind to ATG9A, further preventing its interaction with FAM134B and SEC62 and ultimately inhibiting ER-phagy. AT-1/SLC33A1 overexpression in mice results in a progeria-like phenotype, characterized by aberrant metabolism and systemic inflammation [43, 44].
FAM134B in inflammatory and immune diseases
Sepsis
Sepsis is a critical syndrome marked by multiorgan dysfunction due to an abnormal host response to infection. Myocardial injury is a frequent complication of sepsis and septic shock, occurring in up to 40% of cases, and is a significant contributor to poor prognosis. In sepsis, FAM134B and ER-phagy is upregulated to serve a protective role by attenuating myocardial injury through the suppression of inflammatory responses and apoptosis. FAM134B overexpression or rapamycin (RAP, an autophagy inducer) treatment significantly reduces myocardial lesions in septic mice, whereas FAM134B knockout or treatment with 3-methyladenine (3-MA, an autophagy inhibitor) reverses these protective effects [45].
Viral infectious
The ER provides membranes for virus-replicating organelles, facilitating viral assembly and maturation. Studies have established FAM134B's key role in viral infection pathogenesis (Fig. 3). Early in flaviviral infections, FAM134B's antiviral effect against dengue virus (DENV) and Zika viruses (ZIKV), linked to reduced viral RNA production, was confirmed through FAM134B knockdown experiments. The flaviviral protease NS2B3 enhances viral replication by manipulating FAM134B-mediated ER-phagy. Specifically, NS2B3 cleaves the RHD of FAM134B, disrupting its oligomerization and membrane bending, thereby inhibiting ER-phagy and promoting replication vesicle formation [46]. The depletion of the flavivirus positive regulator BPI fold-containing family B member 3 (BPIFB3) is hypothesized to increase ER sheet turnover and promote FAM134B-mediated ER-phagy [47]. FAM134B contributes to the inhibition of Ebola virus replication, specifically in the Makona and Mayinga strains. Increased levels of virus-associated glycoprotein (GP) and VP40, along with the accumulation of nucleocapsid lattices, were observed in FAM134B-/- cells compared with FAM134B+/+ cells [48]. Whole-exome sequencing revealed three single nucleotide variants of CCRL2 (rs3204849), RETREG1/FAM134B (rs61733811), and YWHAH (rs73884247) in linked to pediatric HIV associated neurocognitive disorder. FAM134B knockdown exacerbates the IL-1β release, and suppresses autophagy in microglia cells upon HIV ssRNA40 exposure [49].
FAM134B-mediated ER-phagy in viral infectious diseases. During DENV and ZIKV infection, the cleavage of FAM134B by NS2B at the RHD site disrupts FAM134B oligomerization, impairing ER-phagy and enhancing viral replication. BPIFB3 is an autophagy regulator that positively regulates DENV and ZIKV replication. Inhibition of FAM134B-mediated ER-phagy by BPIFB3 results in increased viral replication. In Ebola virus infection, FAM134B-mediated ER-phagy exerts antiviral effects by suppressing the expression of viral replication-associated proteins GP and VP and reducing the accumulation of nucleocapsid lattices. During COVID-19 infection, SARS-CoV-2 ORF3a manipulates ER-phagy by interacting with HMGB1, facilitating FAM134B-mediated ER-phagy via the HMGB1-BECN1 pathway, leading to ER stress, inflammation, cell death, and viral replication. SARS-CoV-2 ORF8 undergoes phase separation and interacts with p62 to form condensates. These ORF8/p62 condensates hijack FAM134B/ATL, preventing its interaction with LC3 and impairing ER-phagy. This disruption results in increased viral DMV formation and severe ER stress.
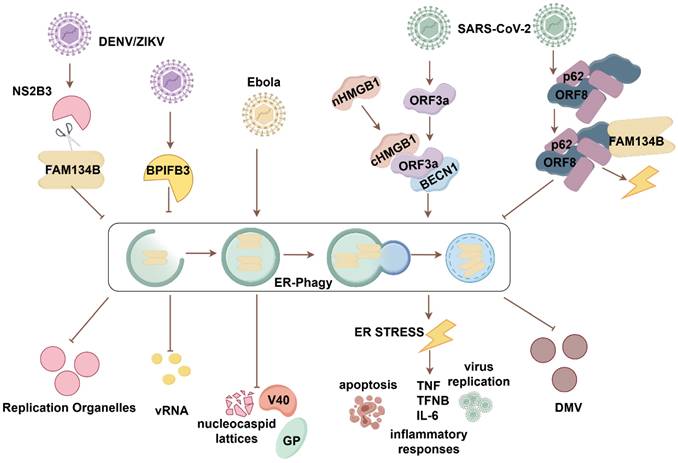
The impairment of FAM134B-mediated ER-phagy has been contributed to the pathogenesis of SARS-CoV-2 infection. In particular, ORF3a protein is capable of subverting FAM134B-mediated ER-phagy, thereby facilitating its own replication and propagation. ORF3a targets the ER, promoting HMGB1 movement from the nucleus to the cytoplasm and its recruitment to the ER. ORF3a interacts with HMGB1 and promotes FAM134B-mediated ER-phagy via the HMGB1-BECN1 pathway. Sustained ER-phagy exacerbates ER stress, early apoptosis, inflammatory cytokine storms, and viral replication. Therefore, proteins involved in ER-phagy and ER stress pathways could be potential therapeutic targets for SARS-CoV-2 infection [50]. Accompanied by hijacking FAM134B and ATL3, ORF8 interacts with P62 to undergo an ORF8/P62 condensates. It subsequently attenuates the antiviral efficacy of FAM134B-mediated ER-phagy, thereby facilitating the formation of double-membrane vesicles (DMVs) that are conducive to viral replication. Importantly, ORF8 homodimer formation is essential for the hijack of FAM134B and ATL3[51]. Blocking ORF8 homodimerization may serve as an effective antiviral strategy to inhibit viral replication.
Allergic rhinitis
Genetic analysis identified that the single nucleotide polymorphism (SNP) rs7071836 in the CD39 gene and rs257174 in the FAM134B gene are associated with heightened inflammation and an elevated risk of allergic rhinitis patients. Subsequent analysis demonstrated that reduced CD39 in regulatory T cells impairs the hydrolysis of extracellular ATP. Concurrently, the rs257174 SNP enhances FAM134B expression and further leads aberrant immune response in monocytes [52].
FAM134B in neurodegenerative diseases
Hereditary neuropathies
HSAN2 is a rare autosomal recessive disorder resulting from specific genetic mutations. It is characterized by early-onset impairment of sensory and autonomic nerves, with a predominant impact on distal sensory functions. HSAN2 patients frequently complicated with changes in skin nutrition, recurrent ulcers, soft tissue infections, and osteomyelitis, potentially leading to distal amputation. HSAN2 has four subtypes, with numerous reports linking FAM134B mutations to subtype HSAN2B (Table 2) [53-58]. FAM134B knockdown leads to changes in Golgi apparatus structure and size, along with increased neuronal apoptosis. Notably, the homozygous FAM134BW107X mutation led to chronic renal failure with HSAN2B, indicating that renal failure might be an advanced stage of FAM134B-related diseases. The FAM134B mutation likely causes pathological progression due to autoinflammation from ER-to-Golgi axis disruption [59]. Excessive cell death in dorsal root ganglion sensory neurons, caused by the FAM134BG216R mutation, could be attenuated by modulating FAM134B oligomerization levels, highlighting a potential therapeutic target for HSAN2 [33].
Mutations in FAM134B causing HSAN2B
| Patient | Exon/Intron | cDNA change | Amino acid change | Mutation type | Zygosity | Reference |
|---|---|---|---|---|---|---|
| 1 | EX2 | c.471C > T | p.Q145X | Nonsense | Hom | [58] |
| 2 | EX6 | c.926C > G | p.S309X | Nonsense | Hom | [56] |
| 3 | EX2 | c.433C > T | p.Q145X | Nonsense | Hom | |
| 4 | EX1 | c.17-18delCT | p.P7GfsX133 | Frameshift | Hom | |
| 5 | Intron7 | c.873+2T > C | / | Splice site | Hom | |
| 6 | EX4 | c.646G > A | p.G216R | SNP | Hom | [54] |
| 7 | EX1 | c.321G > A | p.W107X | Nonsense | Hom | [59] |
| 8 | EX5 | c.826delA | p.W276Vfs*8 | Deletion | Hom | [55] |
| 9 | EX6 | c.896-897delAA | p.K299Rfs*6 | Deletion | Hom | [53] |
| 10 | EX7 | c.1426del | p.Q476Rfs*57 | Deletion | Hom | |
| 11 | EX7 | c.1009C > C/T | p.R337* | Nonsense | Het | [57] |
| EX1 | c.259_266del | p.L87Efs*51 | Frameshift | Het |
* EX: exon; Hom: homozygous; Het: heterozygous.
Epidermal growth factor transactivates NTRK2/TrkB, promoting its transport to the cell surface and augmenting its responsiveness to brain-derived neurotrophic factors, a process essential for the proper development of the cortical plate. ER-phagy degrades NTRK2/TrkB to restrict cell membrane trafficking. CNX directs NTRK2/TrkB to FAM134B, where it undergoes lysosomal degradation. Phosphorylation of CNX releases NTRK2/TrkB from the ER, enabling its cell surface transport. Strikingly, CNX determines whether NTRK2/TrkB is transported to the cell surface or targeted for degradation via autophagy [60-62]. In a rat model of prolonged cervical cord compression, FAM134B levels increased following compression and were further elevated in neuronal cells after melatonin treatment. Furthermore, glutamate-induced neurotoxicity resulted in increased FAM134B expression in neuronal cells [63].
Protein aggregation disorders
FAM134B-mediated ER-phagy helps degrade the Niemann-Pick type C1 (NPC1) mutant. The NPC1I1061T mutation causes NPC disease, a fatal progressive neurodegenerative disorder. Misfolded NPC1 is also degraded via the E3 ligase MARCH6-dependent ERAD, contributing to the complementary regulation of protein turnover [64]. In Parkinson's disease, α-synuclein accumulation is linked to a reduction in dopaminergic neurons and motor dysfunction. Accumulated α-synuclein in the ER binds to CNX and is targeted for clearance via FAM134B-mediated ER-phagy, alleviating ER dysfunction and protecting dopaminergic neurons [65].
Age-related degeneration
Accumulated advanced glycation end products disrupts mitochondrial and ER homeostasis by generating ROS. Then, activated ROS pathway upregulates senescence and apoptosis-related proteins (p53, p16, and caspase-3), ultimately contributing to intervertebral disc degeneration (IDD). FAM134B-mediated ER-phagy exerts a protective effect by modulating ROS signaling, thereby reducing apoptosis and senescence in NP cells [66]. Moreover, nutrient-deprived IDD tissues and cells exhibit increased proteins associated with apoptosis and senescence, including p53, p21, p16, caspase-3, and Bax. OGT interacts with FAM134B to stabilize the protein, preventing ubiquitination and degradation. OGT overexpression or O-GlcNAcase inhibition enhances FAM134B and ER-phagy, promoting NP cell survival and delaying IDD progression under nutrient-deprived conditions. Therefore, O-GlcNAcylation may represent a potential therapeutic target for IDD disease [40]. Caloric restriction has been demonstrated to enhance skeletal muscle health in the elderly population. GEO analysis revealed increased FAM134B expression in aged skeletal muscles subjected to caloric restriction, suggesting that FAM134B-mediated ER-phagy contributes to muscle maintenance and longevity [67].
FAM134B in cancer
Tumor-Suppressive Roles
Reduced FAM134B expression promotes colorectal adenoma transition toward colorectal adenocarcinoma, enhances tumor recurrence and metastasis, and reduces patient survival rates [68, 69]. Mutations in FAM134B induce structural and functional abnormalities, with the most common mutation sites identified in colorectal cancer (CRC) tissues and cells being p.Glu354Glu, p.Val342Ala, p.Thr359Thr, p.Ser276Cys, and p.Asp349ArgfsX13 [70]. Moreover, copy number variations in FAM134B are associated with cancer progression [71]. FAM134B expression is progressively downregulated as the disease advances, and its inhibition enhances colon cancer cell proliferation and colony formation. A xenograft model revealed increased tumor formation in FAM134B-deficient mice, further supporting its tumor-suppressive role [72]. FAM134B interacts with several proteins in colon cancer cells, including end-binding protein (EB1), adenylyl cyclase-associated protein 1, peptidyl-prolyl cis-trans isomerase B, and ER protein retention receptor 2 (KDELR2). These interactions influence colon carcinogenesis and subcellular structure regulation. FAM134B downregulation decreases KDELR2 expression while increasing EB1 expression. As KDELR2 is potentially involved in protein secretion pathways, its downregulation disrupts cellular homeostasis, contributing to tumorigenesis. EB1 interacts with adenomatous polyposis coli (APC) and aurora kinase B, modulating microtubule-associated processes that promote tumor cell growth. FAM134B knockdown upregulates EB1 expression, which activates the WNT/β-catenin pathway, thereby promoting adenoma-to-adenocarcinoma progression in CRC [73]. FAM134B also stabilizes APC and p53 via the WNT/β-catenin pathway and interacts with MIF and p53 to control mitochondrial metabolism and the cell cycle. Its overexpression leads to G1-phase arrest as well as reduced apoptosis and mitochondrial respiration, thereby limiting tumor cell growth [74]. FAM134B promoter hypermethylation negatively correlated with FAM134B expression, in good agreement with tumor metastasis and poor prognosis in CRC. In addition, microRNA-186-5p inversely affects FAM134B levels, promoting tumor growth and worsening prognosis. Therefore, FAM134B promoter methylation and microRNA-186-5p retain a promising potential for CRC therapies [75, 76]. In addition, FAM134B might suppress tumors in malignant mesothelioma and is considered a potential antigen for an mRNA vaccine, especially aiding patients with the TM2 immune subtype [77].
Oncogenic Roles
FAM134B demonstrates a pro-carcinogenic function in esophageal squamous cell carcinoma, as its inhibition leads to a reduction in migration, and invasion [78]. Copy number variations and 37 mutation sites of FAM134B have been identified in cancerous tissues, and mutations in FAM134B have been associated with metastatic lymph nodes [79]. FAM134B-mediated ER-phagy is also implicated in melanoma tumor stem cell regulation. In M14-SE cells, FAM134B-mediated ER-phagy promotes tumor formation and the tumor stem cell markers, such as Aldh1, CD133, Nanog, Oct4, and Sox2. In addition, SEC23a suppresses tumor stemness gene expression and self-renewal by inhibiting ER stress and FAM134B-mediated ER-phagy [80]. FAM134B has been identified as a pivotal gene in the integrated diagnostic network used for breast cancer detection [81]. Notably, hypoxia represents a major challenge in cancer therapy. Hypoxia-induced ER stress induce misfolded protein accumulation, activating the UPR. BiP chaperones detach to identify these proteins and interact with FAM134B, thereby triggering ER-phagy to clear damaged ER, reduce stress, and maintain homeostasis. Silencing FAM134B increases ER stress, while BiP depletion hinders ER-phagy and cancer cell growth, positioning ER-phagy targeting as a breast cancer therapeutic strategy (Fig. 4) [82].
Signaling pathways of FAM134B in tumor-related diseases. FAM134B suppresses colorectal cancer by inhibiting EB1 and upregulating KDELR2 expression, which in turn activates the WNT/β-catenin pathway. FAM134B also upregulates MIF and p53 levels via the WNT/β-catenin pathway, influencing cell cycle regulation and mitochondrial function. Conversely, FAM134B acts as an oncogene in HCC, promoting tumorigenesis, EMT, and tumor metastasis through the AKT signaling pathway. Downregulation of FAM134B in HCC contributes to radiotherapy resistance by modulating the JAK/STAT3 pathway. Moreover, FAM134B suppresses the expression of ER stress-related degradation factors such as DERL2, EDEM1, SEL1L, and HDR1, thereby accelerating HCC progression.
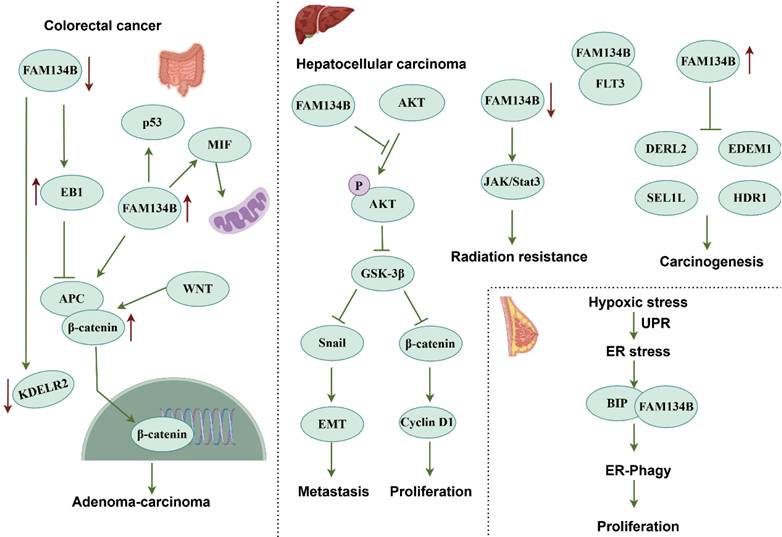
In hepatocellular carcinoma (HCC), FAM134B exerts oncogenic properties by promoting tumorigenesis, epithelial-mesenchymal transition (EMT), and metastasis through AKT signaling pathway. Specifically, FAM134B-induced activation of AKT phosphorylated and inhibited GSK-3β, resulting in β-catenin accumulation, which enhances HCC cell proliferation. FAM134B stabilizes Snail expression, further facilitating EMT [83]. The downregulation of FAM134B impairs cell proliferation and triggers apoptosis, suggesting its involvement in HCC progression. Mechanically, FAM134B promotes HCC progression by inhibiting DERL2, EDEM1, SEL1L, and HRD1 [84].
Interestingly, FAM134B interacts with FMS-related receptor tyrosine kinase 3 (FLT3) to activate the JAK2/STAT3 pathway, enhancing radiotherapy sensitivity, suggesting its potential as a therapeutic target [85]. Moreover, ferroptosis enhancement in HCC is a promising strategy against treatment resistance. The ferroptosis inducer sorafenib activates FAM134B-mediated ER-phagy, whereas inhibition of FAM134B enhances sorafenib-induced ferroptosis in HCC cells. In vivo, sorafenib reduced tumor growth and downregulated FAM134B and GPX4, aligning with in vitro results. The RNA-binding protein PABPC1 binds and upregulates FAM134B, indicating that PABPC1-FAM134B-ER-phagy pathway targeting might be a viable HCC treatment strategy [86]. In Ewing sarcoma, FAM134B-mediated ER-phagy supports dormant cell survival. When the oncogenic fusion gene EWSR1/FLI1 is inhibited, approximately 1% of the cells enter dormancy, depending on this pathway for persistence [87].
FAM134B in metabolic diseases
In an in vitro model of diabetes, high glucose levels triggered ER stress and ER-phagy, and apoptosis, which may contribute to the diabetic cardiomyopathy (DCM) progression (Fig. 5). In HepG2 cells, high glucose exposure induced the upregulation of ER stress and upstream regulatory proteins (GRP78, PERK, IRE1α, ATF6, αPDI, and ERO1α). Concurrently, it also elevated the ER-phagy proteins (Sec62 and RTN3), whereas FAM134B expression was notably decreased. Moreover, CNX and apoptosis-related proteins (CHOP and caspase-12) were upregulated. In vivo model, structural abnormalities were observed in the myocardium, and BNP, a marker of myocardial damage, was elevated. In the above model, chlorogenic acid reduces ER stress and cell death by boosting FAM134B to ER-phagy [88]. In non-alcoholic fatty liver disease (NAFLD) rats, high-fat diet increased p62 and FAM134B levels and disrupted the ER-phagy, prolonged ER stress, and CHOP, GRP78, and TNF-α upregulation, thereby contributing to precancerous lesions. Notably, the hepatic metabolic enzyme inducer phenobarbital (PB) inhibits precancerous lesion formation by activating ER-phagy and reducing ER stress [89]. NAFLD is another common complication of T2DM, characterized by the upregulation of ER stress proteins (GRP78 and ATF6) and downregulation of ER-phagy markers (FAM134B, p62, Beclin-1, and LC3II/I). Chinese herbs 'Jianpi Xiaozhi formula' shows protective effect of liver injury in T2DM rats by modulating ER stress and ER-phagy [90]. In a streptozotocin-induced model of diabetic kidney disease (DKD), renal tubular cell dysfunction significantly disrupted cellular homeostasis, leading to inflammatory cell infiltration and interstitial fibrosis. This was mainly linked to decreased PACS-2 expression, which activates the TFEB-FAM134B axis that alleviates inflammatory and fibrotic responses [91]. Collectively, boosting FAM134B-mediated ER-phagy in diabetic patients could protect the heart, liver, and kidney, offering a new treatment approach for metabolic diseases.
Regulation of FAM134B in diabetes and proinsulin clearance. High glucose levels in diabetes induce ER stress, activating ER-phagy pathways associated with RTN3L and SEC62 while simultaneously inhibiting FAM14B, leading to cell death and potential DCM. Chlorogenic acid has been shown to reduce ER stress and cell death in DCM models by upregulating FAM134B expression. In DKD, a reduction in ER-phagy in renal tubular cells is observed, primarily as a result of decreased levels of PACS-2 and FAM134B. PACS-2 plays a crucial role in TFEB nuclear translocation, which subsequently enhances FAM134B expression. The absence of PACS-2 exacerbates kidney damage in DKD by impairing ER-phagy via the TFEB/FAM134B pathway. UVRAG plays a role in proinsulin aggregate clearance, modulating ER-phagy through interactions with cargo receptors, including FAM134B, ATL3, and RTN3L. This process facilitates Atg8 protein recruitment and promotes receptor oligomerization, enhancing the efficient assembly of ER-phagy sites.
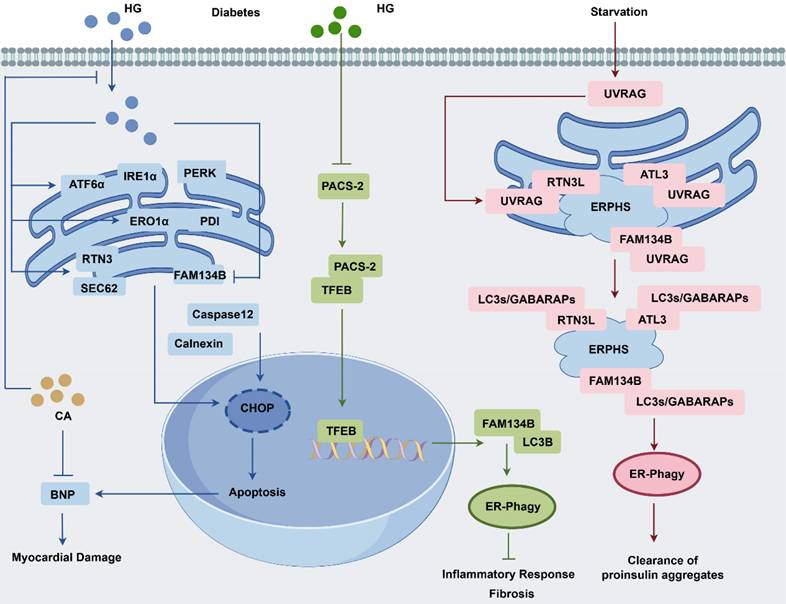
Ultraviolet (UV) resistance-associated gene (UVRAG), a protein integral to UV radiation resistance, interacts with FAM134B, ATL3, and RTN3L via its N-terminal PR structural domain. The interaction is critical for initiating autophagosome formation at ER-phagy sites, autophagosome maturation and lysosomal fusion. Notably, UVRAG enhances ER-phagy by promoting ER-phagy receptor oligomerization, potentially reducing the cellular accumulation of pathogenic proinsulin [92]. Hepatic steatosis is more vulnerable to ischemia/reperfusion injury, which triggers oxidative stress and inflammatory responses in hepatocytes. Interestingly, berberine exerts a protective effect against hepatic steatosis by inhibiting FAM134B-mediated ER-phagy [93]. In hepatocytes, dithiothreitol (DDT) induces ER stress, characterized by a temporal increase in CHOP, GPR78, and CNX expression, leading to mitochondrial calcium destabilization and apoptosis. However, DDT-induced ER stress and apoptosis could be reversed by FAM134B-mediated ER-phagy, providing a protective mechanism against cellular damage [94].
FAM134B positively regulated the lipid metabolism and subcutaneous lipid deposition in pigs, largely owing to activating lipogenic genes, including fatty acid synthetase and acetyl-CoA carboxylase, while suppressing adipose triglyceride lipase and hormone-sensitive lipase. Notably, adipocyte differentiation is inhibited in cells treated with pFAM134B siRNA, suggesting that targeting FAM134B may offer a novel approach for controlling adiposity and managing metabolic diseases [95]. In acute pancreatitis, ER stress-related proteins like BiP, p-PERK, and p-eIF2α increase, while ATF6 degrades more. High cleaved-caspase-3, p-MLKL, and Rip3 levels lead persistent ER stress and progressed to apoptosis and necrosis. Early on, CCPG1- and FAM134B-mediated ER-phagy boosts pancreatic cell survival, but feedback between ER-phagy and ER stress eventually hinders ER clearance, worsening the condition [96]. FAM134B-mediated ER-phagy is essential for maintaining in skeletal muscle homeostasis during acute exercise. Acute exercise reduces SR/ER calcium ATPase activity, causing cytoplasmic Ca2+ accumulation and subsequent muscle dysfunction. This is accompanied by increased ER stress proteins (PDI, GRP78, and CRT) and ER-phagy markers (FAM134B and LC3B), which gradually stabilize throughout exercise [97].
FAM134B in cardiovascular diseases
The rare SNP of FAM134B (rs78314670) has been regarded as a potential genetic site of thromboembolism susceptibility. This SNP of FAM134B correlates with reduced levels of the anticoagulant glucosylceramide, thereby affecting protein C activation and influencing sphingolipid metabolism [98]. A strong epistatic effect between FAM134B and TNFRSF19 has also been observed, highlighting a significant link to vascular dementia predisposition [99]. In a doxorubicin-induced cardiotoxicity model, the activation of FAM134B-mediated ER-phagy was enhanced via the caspase-11/GSDMD signaling pathway, leading to increased apoptosis and aggravated myocardial damage. The formation of GSDMD-induced pores in the ER membrane triggered ER stress, which in turn activated general autophagy and FAM134B-mediated ER-phagy, ultimately culminating in cardiomyocyte apoptosis and substantial myocardial injury. Selective silencing of GSDMD in cardiomyocytes preserved cardiac function and alleviated doxorubicin-induced cardiotoxicity by reducing FAM134B-mediated ER-phagy [100]. Apelin-13, an endogenous APJ receptor ligand, induces myocardial hypertrophy, characterized by increased cardiomyocyte size, volume, and protein content. The hypertrophic response is mediated by activation of the pannexin-1/eATP/purinergic ligand-gated ion channel 7 (P2X7) axis and FAM134B-mediated ER-phagy. Specifically, apelin-13 stimulates pannexin-1 hemichannels, leading to ATP release and subsequent eATP-mediated P2X7 receptor activation. This process further activates FAM134B-mediated ER-phagy, thereby promoting cardiac hypertrophy [101].
FAM134B as a biomarker and potential therapeutic target in diseases
The modulation of FAM134B expression and ER-phagy represents a promising therapeutic avenue for disease treatment and pharmacological innovation (Table 3 and Table 4). Vitexin, a potential inhibitor of breast cancer progression, has been found to inhibit BiP-FAM134B complex formation, suppress BiP-dependent UPR and ER-phagy, and reduce neoplastic cell proliferation in mouse breast cancer xenograft models [82]. Investigators have developed an electrochemical approach to quantify FAM134B mRNA levels in patients with esophageal cancer. This method involves magnetic isolation of the target mRNA, adsorption onto SPE-Au, and quantification via differential pulse voltammetry using a [Fe (CN)6] 4-/3- redox system. Notably, this PCR-free assay has exhibited high sensitivity in detecting tumor-specific mRNA in esophageal cancer, making it a promising tool for early cancer diagnosis [102]. LOP complexes upregulate ATF4, which subsequently induces FAM134B- and TEX264-mediated ER-phagy and autophagic cell death in glioblastoma cells [103]. Brefeldin A (BFA), an inhibitor of protein secretion from the ER to the Golgi apparatus, is encapsulated in mesoporous silica nanoparticles (MSNs) to create MSNs-BFA. The combination of MSNs with the autophagy-inducing peptide TAT-B enhances BFA translocation to the perinuclear region, ensuring controlled release. This strategic localization induces a perinuclear ER stress response, leading to ER expansion. BFA deactivates the AKT/TSC/mTOR pathway to increases the LC3II/LC3I ratio and non-selective autophagy. However, it also suppresses the transcriptional regulation of FAM134B, thereby inhibiting ER-phagy. Ideally, MSN-BFA could be optimized to induce non-selective autophagy while preserving ER integrity, preventing ER fragmentation and enhancing cell viability [104].
FAM134B as a biomarker and potential treatment target for diseases
| Reagent | Targeted protein or pathway | Desired outcome | Target indication | Effect on FAM134B | Reference |
|---|---|---|---|---|---|
| Vitexin | Inhibits BiP-FAM134B complexes and UPR | Reduces cell proliferation; tumor suppression | Breast cancer | Suppresses | [82] |
| Phenobarbital | Relieves ER Stress | Inhibits precancerous lesion formation | NAFLD | Induces | [89] |
| Sorafenib | PABPC1-FAM134B-ER-phagy | Reduces tumor weight and growth; tumor suppression | Hepatocellular carcinoma | Induces | [86] |
| Chlorogenic acid | PERK, IRE1α, ATF6α, PDI, ERO1α, BNP | Reduces ER stress and cell death | T2DM | Induces | [88] |
| LOP complexes | Upregulates ATF4, DDIT3, HSPA5, and p-EIF2A | Induces autophagic cell death | Glioblastoma | Induces | [103] |
| MSNs-BFA | Induces ER stress, inactivates AKT/TSC/mTOR, and enhances macroautophagy | Promotes cell survival | U2OS and A549 cells | Suppresses | [104] |
| CdTe-QDs | Activates the PERK-ATF4 pathway | Decreases cell viability | Renal dysfunction | Induces | [106] |
| Palmitate (nutrient-rich) | p-PERK-ATF4 pathway | Induces ER stress | Hypothalamus | Induces | [107] |
| Palmitate (starvation) | Enhances Bcl-2 expression and inhibits ER stress | Relieves cellular stress | Suppresses | ||
| Z36 | PERK-ATF4-CHOP/IRE1 pathway | Induces cell death | Hela cells | Induces | [108] |
The pathological role and targeting of FAM134B-mediated ER-Phagy in diseases
| Disease Category | Disease/Condition | Primary Organ/System | Key Cell Type(s) | Animal/Experimental Model | References |
|---|---|---|---|---|---|
| Sepsis | Septic myocardial injury | Myocardium | Cardiomyocytes | Lipopolysaccharide (LPS)-induced cardiomyocytes/Cecal ligation and puncture (CLP) treatment | [45] |
| Viral Infections | Flaviviruses | Systemic infection | Human brain microvascular endothelial cells | DENV/ZIKV infection | [46-47] |
| Ebola virus (EBOV) | Systemic infection | Mouse embryonic fibroblasts | EBOV-infected Vero E6 cells | [48] | |
| SARS-CoV-2 (COVID-19) | Respiratory system | Lung epithelial cells (A549) | A549; HEK293T | [50] | |
| SARS-CoV-2 | Respiratory system | Vero E6 cell | Vero-E6; HeLa, HEK293T; A549 | [51] | |
| Neurodegenerative Diseases | HSAN2B | Autonomic nervous system | Sensory and autonomic ganglion neurons | Fam134b KO mice | [54,57] |
| Niemann-Pick type C disease | Cerebellum, liver | Primary human fibroblasts | Npc1-I1061T mice | [64] | |
| Parkinson's disease | Autonomic system | Dopaminergic neurons | AAV-ER-α-syn mouse model | [65] | |
| Age-Related Degeneration | Intervertebral disc degeneration | Spine system | Nucleus pulposus (NP) cells | Annulus Fibrosus Injury rat | [40] |
| Cancer | Colon cancer | Digestive system | SW-480, SW-48, HCT116 | Severe combined immunodeficiency (SCID) mice | [73] |
| Breast cancer | Breast cancer tissue | MCF-7 Cells | Mouse xenograft (hypoxia model) | [82] | |
| Hepatocellular carcinoma | liver | HCC tissue | HCC xenograft mice | [83] | |
| Metabolic Diseases | Diabetic cardiomyopathy (DCM) | heart | Cardiomyocytes | HFFD-induced rats + STZ | [88] |
| Non-alcoholic fatty liver | liver | Hepatocytes | HFD-fed rat | [89] | |
| Diabetic kidney disease | Kidney (proximal tubule) | Tubular epithelial cells | STZ-induced mice | [91] | |
| Acute pancreatitis (AP) | Pancreas | Pancreatic acinar cells | L-arginine-induced mice | [96] | |
| Cardiovascular Disease | Doxorubicin-Induced Cardiotoxicity (DIC) | Heart | Cardiomyocytes | Mice (WT; GSDMD-KO; GSDMD-CKO) | [100] |
| Cardiac hypertrophy | Heart | Cardiomyocytes | HL-1 and H9c2 cell line | [101] |
Regarding disease diagnosis, FAM134B serves as an exosome-specific marker for the quantitative detection of colon cancer. Serum exosomes are first captured using CD9 or CD63 magnetic beads and subsequently identified using FAM134B functionalized with CdSe quantum dot. After magnetic elution and purification, electrochemical methods are employed to measure the amplified Cd2+ quantum dot signals, thereby quantifying the exosome contents. This method achieves a detection sensitivity of 100 exosomes/µL. Quantum dots are gaining attention for their applications in bioimaging, medical diagnostics, and therapy; however, biosafety remains a major concern within the scientific community [105]. CdTe quantum dots enter renal cells via clathrin-dependent endocytosis, leading to ER swelling and vacuolization. They trigger FAM134B-mediated ER-phagy through the PERK-ATF4 pathway, resulting in reduced cell viability and impaired renal function. Inhibiting the UPR and silencing FAM13B has been shown to mitigate CdTe-induced cytotoxicity [106]. In a nutrient-rich environment, hypothalamic cells treated with acute palmitate shows increased ER stress and FAM134B-mediated ER-phagy via the p-PERK-ATF4 pathway. However, chronic palmitate treatment disrupts autophagosome maturation, exacerbating ER stress and impairing ER-phagy. Palmitate shields hypothalamic cells from stress by inhibiting ER-phagy under starvation [107]. Similarly, Z36 exposure of Hela cells boosts FAM134B, LC3, and Atg9, causing excessive ER-phagy and subsequent cell death. Moreover, Z36-induced ER stress and UPR activate the PERK-ATF4-CHOP pathway, promoting cell death, whereas the IRE1 pathway contributes to cell survival. Thus, modulating FAM134B expression via Z36 to promote cancer cell death represents a promising strategy for cancer therapy [108].
Conclusions and prospects
The RHD and LIR motifs of FAM134B are crucial for ER-phagy, with RHD involved in membrane remodeling and LIR in LC3 binding. The LIR motif has co-evolved with autophagy proteins like LC3 to enhance ER-phagy efficiency. Despite variations in the disordered domain, these functions are conserved, coordinating with RHD to adapt to the regulatory needs of different species, e.g., phosphorylation site-related differences. [13]. In contrast, the C-terminal region of FAM134B binds the GABARAP subfamily in a “LIR core + C-helix” mode with approximately 10-fold higher affinity than the LC3 subfamily [109]. FAM134B collaborates with other ER-phagy receptors to maintain ER homeostasis, with its RHD bending sheet-like ER but the RHD of RTN3L fitting the tubular ER curvature [92]. RTN3L and FAM134B can partially compensate functionally during starvation [7]. During prolonged ER stress, FAM134B and SEC62 activate at different stages [5]. FAM134B2, a truncated variant of FAM134B, helps maintain amino acid balance and cell survival during AAD by the MEF2D-NR4A1-FAM134B2 pathway. Moreover, the regulatory network of transcription factors BACH1 and ZBTB10 with FAM134B2 can be further explored via molecular interaction or functional rescue assessment [17].
FAM134B-mediated ER-phagy exerts distinct effects depending on the specific pathophysiological state. For example, ZIKV blocks ER-phagy to form replication compartments, whereas SARS-CoV-2 disrupts it to create double-membrane replication vesicles [46, 51]. It suppresses CRC via the WNT pathway and enhances tumor cell survival in HCC through the AKT pathway. In HSAN2B, FAM134B mutations impair ER-Golgi trafficking, causing cell death, while in melanoma stem cells, FAM134B-mediated ER-phagy boosts stemness markers, aiding tumor survival. FAM134B supports nociceptor and neuron survival within the autonomic ganglia. However, excessive ER-phagy might induce neuronal cell death. In the heart, FAM134B-mediated ER-phagy protects against septic myocardial injury by reducing apoptosis and inflammation [45], although it also contributes to apelin13-induced cardiomyocyte hypertrophy [101]. While no clinical trials specifically target FAM134, adjusting its expression holds promising potential for future research. In ER storage disorders, the XBP1-Sestrin2-TFEB/FAM134B pathway facilitates ER-phagy to remove misfolded proteins. The drugs Fluphenazine (FPZ) and Tetrandrine (TET) boost ER-phagy by inhibiting mTORC1 localization to the lysosomes and activating TFEB/FAM134B, without inducing ER stress [110]. Thiamet G (TMG), an O-GlcNAcase inhibitor, boosts FAM134B expression both in vitro and in vivo, helping to alleviate IDD [40]. Notably, TMG also reduces pathological tau aggregation in Alzheimer's disease [111].
FAM134B is particularly implicated in inter-organelle crosstalk, especially in Ca2+ exchange between the ER and mitochondria[112]. ER-Phagy is crucial for ER quality control, as it removes damaged ER to maintain cellular balance. As technology advances and we understand better FAM134B-mediated ER-phagy, its therapeutic potential will become clearer.
Abbreviations
AAT: alpha-1-antitrypsin; AAD: amino acid deprivation; AMFR: autocrine motility factor receptor; ATL: atlastin; APC: adenomatous polyposis coli; BPIFB3: BPI fold-containing family B member 3; BFA: brefeldin A; CNX: calnexin; CK2: casein kinase 2; CRC: colorectal cancer; DCM: diabetic cardiomyopathy; DDT: dithiothreitol; DENV: dengue virus; DKD: diabetic kidney disease; DMV: double-membrane vesicle; ERLAD: ER-to-lysosome-associated degradation; ERAD: ER-associated degradation; EB1: end-binding protein; EMT: epithelial-mesenchymal transition; FLT3: FMS-related receptor tyrosine kinase 3; GP: glycoprotein; HSAN2B: hereditary sensory and autonomic neuropathy type IIB; HCC: hepatocellular carcinoma; IMM: inner mitochondrial membrane; IDD: intervertebral disc degeneration; KDELR2: ER protein retention receptor 2; LIR: LC3-interacting region; MSNs: mesoporous silica nanoparticles; NPC1: Niemann-Pick type C1; m6A: N6-methyladenosine; NP: nucleus pulposus; NCI: neurocognitive impairment; NAFLD: non-alcoholic fatty liver disease; OGT: O-GlcNAc transferase; OGA: O-GlcNAc glycosylase; P2X7: purinergic ligand-gated ion channel 7; RAP: rapamycin; RHD: reticulon homology domain; SNP: single nucleotide polymorphism; TM: transmembrane; T2DM: type 2 diabetes mellitus; Ub: ubiquitin; UV: ultraviolet; UVRAG: UV resistance-associated gene; VAPs: VAMP-associated proteins; WIPI2: WD repeat domain and phosphoinositide interacting 2; YTHDF2: YTH m6A RNA-binding protein 2; ZIKV: Zika virus; 3-MA: 3-methyladenine.
Acknowledgements
We thank Figdraw 2.0 for their support in drawing.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2022YFA1104600), the National Natural Science Foundation of China (82241062, 82130062), and the Beijing Natural Science Foundation (7244296).
Author contributions
N.C. drafted the original manuscript. N.C. and J.Y. drew the figures. S.T., L.X., N.D., and Y.W. gathered information. Y.L., R.Y., and Y.Y revised and approved the manuscript. All authors have read and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176:11-42
2. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349-64
3. Jin M, Liu X, Klionsky DJ. SnapShot: Selective autophagy. Cell. 2013;152:368-e2
4. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24:167-85
5. Reggiori F, Molinari M. ER-phagy: mechanisms, regulation, and diseases connected to the lysosomal clearance of the endoplasmic reticulum. Physiological reviews. 2022;102:1393-448
6. Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y. et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359-62
7. Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R. et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife. 2017 6
8. Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M. et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354-8
9. Reggio A, Buonomo V, Berkane R, Bhaskara RM, Tellechea M, Peluso I. et al. Role of FAM134 paralogues in endoplasmic reticulum remodeling, ER-phagy, and Collagen quality control. EMBO reports. 2021 22
10. Rubinsztein DC. Cell biology: Receptors for selective recycling. Nature. 2015;522:291-2
11. Song S, Tan J, Miao Y, Zhang Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J Cell Physiol. 2018;233:3867-74
12. Gubas A, Dikic I. ER remodeling via ER-phagy. Mol Cell. 2022;82:1492-500
13. Bhaskara RM, Grumati P, Garcia-Pardo J, Kalayil S, Covarrubias-Pinto A, Chen W. et al. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat Commun. 2019;10:2370
14. Siggel M, Bhaskara RM, Moesser MK, I DI, Hummer G. FAM134B-RHD Protein Clustering Drives Spontaneous Budding of Asymmetric Membranes. J Phys Chem Lett. 2021;12:1926-31
15. Popelka H, Klionsky DJ. Molecular dynamics simulations reveal how the reticulon-homology domain of the autophagy receptor RETREG1/FAM134B remodels membranes for efficient selective reticulophagy. Autophagy. 2020;16:585-8
16. Kohno S, Shiozaki Y, Keenan AL, Miyazaki-Anzai S, Miyazaki M. An N-terminal-truncated isoform of FAM134B (FAM134B-2) regulates starvation-induced hepatic selective ER-phagy. Life Sci Alliance. 2019 2
17. Shiozaki Y, Miyazaki-Anzai S, Keenan AL, Miyazaki M. MEF2D-NR4A1-FAM134B2-mediated reticulophagy contributes to amino acid homeostasis. Autophagy. 2022;18:1049-61
18. Di Lorenzo G, Iavarone F, Maddaluno M, Plata-Gómez AB, Aureli S, Quezada Meza CP. et al. Phosphorylation of FAM134C by CK2 controls starvation-induced ER-phagy. Science advances. 2022;8:eabo1215
19. Sang Y, Li B, Su T, Zhan H, Xiong Y, Huang Z. et al. Visualizing ER-phagy and ER architecture in vivo. J Cell Biol. 2024 223
20. Wang C, Li Y, Li Y, Du L, Zhang J, Li N. et al. FAM134B-Mediated ER-Phagy in Mg(2+)-Free Solution-Induced Mitochondrial Calcium Homeostasis and Cell Death in Epileptic Hippocampal Neurons. Neurochem Res. 2021;46:2485-94
21. Chen W, Ouyang X, Chen L, Li L. FAM134B-mediated ER-phagy regulates ER-mitochondria interaction through MAMs. Acta Biochim Biophys Sin (Shanghai). 2022;54:412-4
22. Mookherjee D, Das S, Mukherjee R, Bera M, Jana SC, Chakrabarti S. et al. RETREG1/FAM134B mediated autophagosomal degradation of AMFR/GP78 and OPA1 -a dual organellar turnover mechanism. Autophagy. 2021;17:1729-52
23. Cai M, Zhao J, Liu Q, Wang X, Wang Y. FAM134B improves preadipocytes differentiation by enhancing mitophagy. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:158508
24. Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Solda T. et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018 37
25. Fregno I, Fasana E, Solda T, Galli C, Molinari M. N-glycan processing selects ERAD-resistant misfolded proteins for ER-to-lysosome-associated degradation. EMBO J. 2021;40:e107240
26. Cinque L, De Leonibus C, Iavazzo M, Krahmer N, Intartaglia D, Salierno FG. et al. MiT/TFE factors control ER-phagy via transcriptional regulation of FAM134B. EMBO J. 2020;39:e105696
27. Fraiberg M, Elazar Z. Selective autophagy bears bone. Embo j. 2020;39:e105965
28. Liang JR, Lingeman E, Ahmed S, Corn JE. Atlastins remodel the endoplasmic reticulum for selective autophagy. J Cell Biol. 2018;217:3354-67
29. Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T. et al. A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell. 2020;180:1160-77.e20
30. Gatz C, Hathazi D, Munchberg U, Buchkremer S, Labisch T, Munro B. et al. Identification of Cellular Pathogenicity Markers for SIL1 Mutations Linked to Marinesco-Sjogren Syndrome. Front Neurol. 2019;10:562
31. Cai M, Liu Q, Jiang Q, Wu R, Wang X, Wang Y. Loss of m(6) A on FAM134B promotes adipogenesis in porcine adipocytes through m(6) A-YTHDF2-dependent way. IUBMB Life. 2019;71:580-6
32. Shu F, Xiao H, Li Q-N, Ren X-S, Liu Z-G, Hu B-W. et al. Epigenetic and post-translational modifications in autophagy: biological functions and therapeutic targets. Signal Transduction and Targeted Therapy. 2023 8
33. Jiang X, Wang X, Ding X, Du M, Li B, Weng X. et al. FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. EMBO J. 2020;39:e102608
34. De Leonibus C, Cinque L, Settembre C. Beating the ER: novel insights into FAM134B function and regulation. The EMBO Journal. 2020 39
35. Berkane R, Ho-Xuan H, Glogger M, Sanz-Martinez P, Brunello L, Glaesner T. et al. The function of ER-phagy receptors is regulated through phosphorylation-dependent ubiquitination pathways. Nature Communications. 2023 14
36. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nature medicine. 2014;20:1242-53
37. González A, Covarrubias-Pinto A, Bhaskara RM, Glogger M, Kuncha SK, Xavier A. et al. Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature. 2023;618:394-401
38. Foronda H, Fu Y, Covarrubias-Pinto A, Bocker HT, González A, Seemann E. et al. Heteromeric clusters of ubiquitinated ER-shaping proteins drive ER-phagy. Nature. 2023;618:402-10
39. Zhang M, Wang Z, Zhao Q, Yang Q, Bai J, Yang C. et al. USP20 deubiquitinates and stabilizes the reticulophagy receptor RETREG1/FAM134B to drive reticulophagy. Autophagy. 2024;20:1780-97
40. Luo R, Li G, Zhang W, Liang H, Lu S, Cheung JPY. et al. O-GlcNAc transferase regulates intervertebral disc degeneration by targeting FAM134B-mediated ER-phagy. Experimental & molecular medicine. 2022;54:1472-85
41. Wang X, Jiang X, Li B, Zheng J, Guo J, Gao L. et al. A regulatory circuit comprising the CBP and SIRT7 regulates FAM134B-mediated ER-phagy. Journal of Cell Biology. 2023 222
42. Wang X, Li B, Sun Q. The spatiotemporal control of ER membrane fragmentation during reticulophagy. Autophagy. 2023;20:210-1
43. Peng Y, Shapiro SL, Banduseela VC, Dieterich IA, Hewitt KJ, Bresnick EH. et al. Increased transport of acetyl-CoA into the endoplasmic reticulum causes a progeria-like phenotype. Aging Cell. 2018;17:e12820
44. Sheehan BK, Orefice NS, Peng Y, Shapiro SL, Puglielli L. ATG9A regulates proteostasis through reticulophagy receptors FAM134B and SEC62 and folding chaperones CALR and HSPB1. iScience. 2021;24:102315
45. Li T, Chen Y, Li Y, Yao Z, Liu W. FAM134B-mediated endoplasmic reticulum autophagy protects against sepsis myocardial injury in mice. Aging (Albany NY). 2021;13:13535-47
46. Lennemann NJ, Coyne CB. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13:322-32
47. Evans AS, Lennemann NJ, Coyne CB. BPIFB3 Regulates Endoplasmic Reticulum Morphology to Facilitate Flavivirus Replication. Journal of virology. 2020 94
48. Chiramel AI, Dougherty JD, Nair V, Robertson SJ, Best SM. FAM134B, the Selective Autophagy Receptor for Endoplasmic Reticulum Turnover, Inhibits Replication of Ebola Virus Strains Makona and Mayinga. J Infect Dis. 2016;214:S319-S25
49. Rawat P, Brummel SS, Singh KK, Kim J, Frazer KA, Nichols S. et al. Genomics Links Inflammation with Neurocognitive Impairment in Children Living with Human Immunodeficiency Virus Type-1. J Infect Dis. 2021;224:870-80
50. Zhang X, Yang Z, Pan T, Long X, Sun Q, Wang PH. et al. SARS-CoV-2 ORF3a induces RETREG1/FAM134B-dependent reticulophagy and triggers sequential ER stress and inflammatory responses during SARS-CoV-2 infection. Autophagy. 2022:1-17
51. Tan X, Cai K, Li J, Yuan Z, Chen R, Xiao H. et al. Coronavirus subverts ER-phagy by hijacking FAM134B and ATL3 into p62 condensates to facilitate viral replication. Cell reports. 2023;42:112286
52. Melchiotti R, Puan KJ, Andiappan AK, Poh TY, Starke M, Zhuang L. et al. Genetic analysis of an allergic rhinitis cohort reveals an intercellular epistasis between FAM134B and CD39. BMC medical genetics. 2014;15:73
53. Falcao de Campos C, Vidailhet M, Toutain A, de Becdelievre A, Funalot B, Bonello-Palot N. et al. Hereditary sensory autonomic neuropathy type II: Report of two novel mutations in the FAM134B gene. J Peripher Nerv Syst. 2019;24:354-8
54. Davidson G, Murphy S, Polke J, Laura M, Salih M, Muntoni F. et al. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol. 2012;259:1673-85
55. Elif Ilgaz Aydinlar M, Arndt Rolfs M, Mustafa Serteser M, Yesim Parman M. MUTATION IN FAM134B CAUSING HEREDITARY SENSORY NEUROPATHY WITH SPASTICITY IN A TURKISH FAMILY. Muscle Nerve. 2014;49:774-5
56. Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A. et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179-81
57. Luo ZY, Wang HJ, Zhao YK, Liu JH, Chen YM, Lin ZM. et al. Late-onset hereditary sensory and autonomic neuropathy type 2B caused by novel compound heterozygous mutations in FAM134B presenting as chronic recurrent ulcers on the soles. Indian J Dermatol Venereol Leprol. 2021;87:455
58. Murphy SM, Davidson GL, Brandner S, Houlden H, Reilly MM. Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry. 2012;83:119-20
59. Tasdelen E, Calame DG, Akay G, Mitani T, Fatih JM, Herman I. et al. Novel RETREG1 (FAM134B) founder allele is linked to HSAN2B and renal disease in a Turkish family. Am J Med Genet A. 2022;188:2153-61
60. Lüningschrör P, Andreska T, Veh A, Wolf D, Giridhar NJ, Moradi M. et al. Calnexin controls TrkB cell surface transport and ER-phagy in mouse cerebral cortex development. Developmental Cell. 2023;58:1733-47.e6
61. Lüningschrör P, Sendtner M. Connecting reticulophagy and neuronal NTRK2/TrkB signaling. Autophagy. 2023:1-2
62. Hoyer MJ, Capitanio C, Smith IR, Paoli JC, Bieber A, Jiang Y. et al. Combinatorial selective ER-phagy remodels the ER during neurogenesis. Nat Cell Biol. 2024;26:378-92
63. Yao M, Pu Pm, Li Zy, Zhu K, Zhou Ly, Sun YL. et al. Melatonin restores endoplasmic reticulum homeostasis to protect injured neurons in a rat model of chronic cervical cord compression. Journal of Pineal Research. 2023 74
64. Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L. et al. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun. 2018;9:3671
65. Kim DY, Shin JY, Lee JE, Kim HN, Chung SJ, Yoo HS. et al. A selective ER-phagy exerts neuroprotective effects via modulation of α-synuclein clearance in parkinsonian models. Proceedings of the National Academy of Sciences of the United States of America. 2023;120:e2221929120
66. Luo R, Li S, Li G, Lu S, Zhang W, Liu H. et al. FAM134B-Mediated ER-phagy Upregulation Attenuates AGEs-Induced Apoptosis and Senescence in Human Nucleus Pulposus Cells. Oxid Med Cell Longev. 2021;2021:3843145
67. Murphy A, Vyavahare S, Kumar S, Lee TJ, Sharma A, Adusumilli S. et al. Dietary interventions and molecular mechanisms for healthy musculoskeletal aging. Biogerontology. 2022;23:681-98
68. Kasem K, Gopalan V, Salajegheh A, Lu CT, Smith RA, Lam AK. The roles of JK-1 (FAM134B) expressions in colorectal cancer. Exp Cell Res. 2014;326:166-73
69. Kasem K, Sullivan E, Gopalan V, Salajegheh A, Smith RA, Lam AK. JK1 (FAM134B) represses cell migration in colon cancer: a functional study of a novel gene. Exp Mol Pathol. 2014;97:99-104
70. Islam F, Gopalan V, Wahab R, Lee KT, Haque MH, Mamoori A. et al. Novel FAM134B mutations and their clinicopathological significance in colorectal cancer. Hum Genet. 2017;136:321-37
71. Kasem K, Gopalan V, Salajegheh A, Lu CT, Smith RA, Lam AK. JK1 (FAM134B) gene and colorectal cancer: a pilot study on the gene copy number alterations and correlations with clinicopathological parameters. Exp Mol Pathol. 2014;97:31-6
72. Islam F, Gopalan V, Wahab R, Smith RA, Qiao B, Lam AK. Stage dependent expression and tumor suppressive function of FAM134B (JK1) in colon cancer. Mol Carcinog. 2017;56:238-49
73. Islam F, Chaousis S, Wahab R, Gopalan V, Lam AK. Protein interactions of FAM134B with EB1 and APC/beta-catenin in vitro in colon carcinoma. Mol Carcinog. 2018;57:1480-91
74. Lee KT, Islam F, Vider J, Martin J, Chruscik A, Lu CT. et al. Overexpression of family with sequence similarity 134, member B (FAM134B) in colon cancers and its tumor suppressive properties in vitro. Cancer Biol Ther. 2020;21:954-62
75. Islam F, Gopalan V, Pillai S, Lu CT, Kasem K, Lam AK. Promoter hypermethylation inactivate tumor suppressor FAM134B and is associated with poor prognosis in colorectal cancer. Genes Chromosomes Cancer. 2018;57:240-51
76. Islam F, Gopalan V, Vider J, Wahab R, Ebrahimi F, Lu CT. et al. MicroRNA-186-5p overexpression modulates colon cancer growth by repressing the expression of the FAM134B tumour inhibitor. Exp Cell Res. 2017;357:260-70
77. Wang S, Yang Y, Li L, Ma P, Jiang Y, Ge M. et al. Identification of Tumor Antigens and Immune Subtypes of Malignant Mesothelioma for mRNA Vaccine Development. Vaccines. 2022 10
78. Islam F, Gopalan V, Law S, Tang JC-o, Lam AK-y. FAM134B promotes esophageal squamous cell carcinoma in vitro and its correlations with clinicopathologic features. Human Pathology. 2019;87:1-10
79. Haque MH, Gopalan V, Chan K-w, Shiddiky MJA, Smith RA, Lam AK-y. Identification of Novel FAM134B (JK1) Mutations in Oesophageal Squamous Cell Carcinoma. Scientific Reports. 2016 6
80. Sun Z, Liu D, Zeng B, Zhao Q, Li X, Chen H. et al. Sec23a inhibits the self-renewal of melanoma cancer stem cells via inactivation of ER-phagy. Cell Commun Signal. 2022;20:22
81. Dai X, Hua T, Hong T. Integrated diagnostic network construction reveals a 4-gene panel and 5 cancer hallmarks driving breast cancer heterogeneity. Scientific Reports. 2017 7
82. Chipurupalli S, Ganesan R, Martini G, Mele L, Reggio A, Esposito M. et al. Cancer cells adapt FAM134B/BiP mediated ER-phagy to survive hypoxic stress. Cell Death Dis. 2022;13:357
83. Zhang ZQ, Chen J, Huang WQ, Ning D, Liu QM, Wang C. et al. FAM134B induces tumorigenesis and epithelial-to-mesenchymal transition via Akt signaling in hepatocellular carcinoma. Mol Oncol. 2019;13:792-810
84. Wang H, Liu L, Gong H, Li H. Upregulation of FAM134B inhibits endoplasmic reticulum stress-related degradation protein expression and promotes hepatocellular carcinogenesis. Journal of Cellular and Molecular Medicine. 2023
85. Xie B, Xie Y, Fang C, Zhong B, Ye R, Zhang J. et al. Elevated FAM134B expression induces radiation-sensitive in hepatocellular carcinoma. BMC Cancer. 2023 23
86. Liu Z, Ma C, Wang Q, Yang H, Lu Z, Bi T. et al. Targeting FAM134B-mediated reticulophagy activates sorafenib-induced ferroptosis in hepatocellular carcinoma. Biochemical and biophysical research communications. 2022;589:247-53
87. Khoogar R, Li F, Chen Y, Ignatius M, Lawlor ER, Kitagawa K. et al. Single-cell RNA profiling identifies diverse cellular responses to EWSR1/FLI1 downregulation in Ewing sarcoma cells. Cell Oncol (Dordr). 2022;45:19-40
88. Preetha Rani MR, Salin Raj P, Nair A, Ranjith S, Rajankutty K, Raghu KG. In vitro and in vivo studies reveal the beneficial effects of chlorogenic acid against ER stress mediated ER-phagy and associated apoptosis in the heart of diabetic rat. Chemico-Biological Interactions. 2022 351
89. Uomoto S, Takesue K, Shimizu S, Maeda N, Oshima K, Hara E. et al. Phenobarbital, a hepatic metabolic enzyme inducer, inhibits preneoplastic hepatic lesions with expression of selective autophagy receptor p62 and ER-phagy receptor FAM134B in high-fat diet-fed rats through the inhibition of ER stress. Food Chem Toxicol. 2023;173:113607
90. Yuanyuan Z, Huaizhen L. Mechanistic Research into the Effects of the Jianpi Xiaozhi Formula on Liver Injury in Diabetic Rats. Evidence-based complementary and alternative medicine: eCAM. 2022;2022:7490747
91. Yang J, Li L, Li C, Chen W, Liu Y, Luo S. et al. PACS-2 deficiency aggravates tubular injury in diabetic kidney disease by inhibiting ER-phagy. Cell Death & Disease. 2023 14
92. Qian X, He L, Yang J, Sun J, Peng X, Zhang Y. et al. UVRAG cooperates with cargo receptors to assemble the ER-phagy site. The EMBO Journal. 2023 42
93. Zhang N, Sheng M, Wu M, Zhang X, Ding Y, Lin Y. et al. Berberine protects steatotic donor undergoing liver transplantation via inhibiting endoplasmic reticulum stress-mediated reticulophagy. Experimental biology and medicine (Maywood, NJ). 2019;244:1695-704
94. Guo YX, Han B, Yang T, Chen YS, Yang Y, Li JY. et al. Family with sequence similarity 134 member B-mediated reticulophagy ameliorates hepatocyte apoptosis induced by dithiothreitol. World journal of gastroenterology. 2022;28:2569-81
95. Yuan Z, Song D, Wang Y. The novel gene pFAM134B positively regulates fat deposition in the subcutaneous fat of Sus scrofa. Biochemical and biophysical research communications. 2014;454:554-9
96. Huang W, Zhang J, Jin W, Yang J, Yu G, Shi H. et al. Piperine alleviates acute pancreatitis: A possible role for FAM134B and CCPG1 dependent ER-phagy. Phytomedicine. 2022;105:154361
97. Jin S, Li L, Ren Z, Li J, Wang R, Ding H. FAM134B-mediated ER-phagy alleviates endoplasmic reticulum stress of rat soleus muscle in response to acute exercise. General physiology and biophysics. 2022;41:71-8
98. Deguchi H, Shukla M, Hayat M, Torkamani A, Elias DJ, Griffin JH. Novel exomic rare variants associated with venous thrombosis. Br J Haematol. 2020;190:783-6
99. Kong M, Kim Y, Lee C. A strong synergistic epistasis between FAM134B and TNFRSF19 on the susceptibility to vascular dementia. Psychiatric genetics. 2011;21:37-41
100. Qu Y, Gao R, Wei X, Sun X, Yang K, Shi H. et al. Gasdermin D mediates endoplasmic reticulum stress via FAM134B to regulate cardiomyocyte autophagy and apoptosis in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2022;13:901
101. Yang Y, Zhang K, Huang S, Chen W, Mao H, Ouyang X. et al. Apelin-13/APJ induces cardiomyocyte hypertrophy by activating the Pannexin-1/P2X7 axis and FAM134B-dependent reticulophagy. J Cell Physiol. 2022;237:2230-48
102. Islam MN, Gopalan V, Haque MH, Masud Mostafa K, Hossain MSA, Yamauchi Y. et al. A PCR-free electrochemical method for messenger RNA detection in cancer tissue samples. Biosensors and Bioelectronics. 2017;98:227-33
103. Zielke S, Kardo S, Zein L, Mari M, Covarrubias-Pinto A, Kinzler MN. et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy. 2021;17:2432-48
104. Wang Y, Zhao Z, Wei F, Luo Z, Duan Y. Combining autophagy-inducing peptides and brefeldin A delivered by perinuclear-localized mesoporous silica nanoparticles: a manipulation strategy for ER-phagy. Nanoscale. 2018;10:8796-805
105. Boriachek K, Islam MN, Gopalan V, Lam AK, Nguyen NT, Shiddiky MJA. Quantum dot-based sensitive detection of disease specific exosome in serum. Analyst. 2017;142:2211-9
106. Jiang S, Lin Y, Yao H, Yang C, Zhang L, Luo B. et al. The role of unfolded protein response and ER-phagy in quantum dots-induced nephrotoxicity: an in vitro and in vivo study. Arch Toxicol. 2018;92:1421-34
107. Lim Y, Kim S, Kim EK. Palmitate reduces starvation-induced ER stress by inhibiting ER-phagy in hypothalamic cells. Mol Brain. 2021;14:65
108. Liao Y, Duan B, Zhang Y, Zhang X, Xia B. Excessive ER-phagy mediated by the autophagy receptor FAM134B results in ER stress, the unfolded protein response, and cell death in HeLa cells. The Journal of biological chemistry. 2019;294:20009-23
109. Zhao J, Li Z, Li J. The crystal structure of the FAM134B-GABARAP complex provides mechanistic insights into the selective binding of FAM134 to the GABARAP subfamily. FEBS Open Bio. 2022;12:320-31
110. De Leonibus C, Maddaluno M, Ferriero R, Besio R, Cinque L, Lim PJ. et al. Sestrin2 drives ER-phagy in response to protein misfolding. Dev Cell. 2024;59:2035-52.e10
111. Hastings NB, Wang X, Song L, Butts BD, Grotz D, Hargreaves R. et al. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol Neurodegener. 2017;12:39
112. Ge WD, Du TT, Wang CY, Sun LN, Wang YQ. Calcium signaling crosstalk between the endoplasmic reticulum and mitochondria, a new drug development strategies of kidney diseases. Biochem Pharmacol. 2024;225:116278
Author contact
![]() Corresponding authors: Yong-ming Yao, MD, PhD, Medical Innovation Research Division and Fourth Medical Center, Chinese PLA General Hospital, 28 Fuxing Road, Haidian District, Beijing 100853, People's Republic of China. Tel: (+86) 1066867394; Fax: (+86)1068989955; E-mail: c_ffcom. Ren-Qi Yao, MD, PhD, Department of General Surgery, First Medical Center of the Chinese PLA General Hospital, 28 Fuxing Road, Haidian District, Beijing 100853, People's Republic of China. E-mail: yaorenqixx1995com. Yu-Xuan Li, MD, PhD, Department of General Surgery, First Medical Center of the Chinese PLA General Hospital, 28 Fuxing Road, Haidian District, Beijing 100853, People's Republic of China. E-mail: liyuxuan301com.
Corresponding authors: Yong-ming Yao, MD, PhD, Medical Innovation Research Division and Fourth Medical Center, Chinese PLA General Hospital, 28 Fuxing Road, Haidian District, Beijing 100853, People's Republic of China. Tel: (+86) 1066867394; Fax: (+86)1068989955; E-mail: c_ffcom. Ren-Qi Yao, MD, PhD, Department of General Surgery, First Medical Center of the Chinese PLA General Hospital, 28 Fuxing Road, Haidian District, Beijing 100853, People's Republic of China. E-mail: yaorenqixx1995com. Yu-Xuan Li, MD, PhD, Department of General Surgery, First Medical Center of the Chinese PLA General Hospital, 28 Fuxing Road, Haidian District, Beijing 100853, People's Republic of China. E-mail: liyuxuan301com.