Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Myeloid Cell and Regulation of...
Lymphocyte-Mediated Immune...
Platelets in MASLD-HCC
Gut Microbiota Modulates Immune...
Current Dilemma and Potential...
Conclusion and Future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(13):5666-5690. doi:10.7150/ijbs.117394 This issue Cite
Review
Immunotherapy resistance in MASLD-related hepatocellular carcinoma: special immune microenvironment and gut microbiota
Jie Jin1*, Kun Cheng1*, Mingrui Chen1*, Huifang Liang1,2,3 ![]() , Wanguang Zhang1,2,3,4
, Wanguang Zhang1,2,3,4 ![]()
1. Hepatic Surgery Centre, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, People's Republic of China.
2. Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Wuhan, Hubei 430030, People's Republic of China.
3. Clinical Medicine Research Centre for Hepatic Surgery of Hubei Province, Wuhan, Hubei 430030, People's Republic of China.
4. Key Laboratory of Organ Transplantation, Ministry of Education; NHC Key Laboratory of Organ Transplantation; Key Laboratory of Organ Transplantation, Chinese Academy of Medical Sciences, People's Republic of China.
* These authors contribute equally.
Received 2025-5-13; Accepted 2025-8-8; Published 2025-9-3
Abstract

Obesity represents a major global public health challenge. Consequently, metabolic dysfunction-associated steatotic liver disease (MASLD) has become the primary driver of chronic liver disease globally and is currently the most rapidly accelerating factor contributing to hepatocellular carcinoma (HCC). However, current evidence indicates that immunotherapy, a cornerstone of HCC management, yields suboptimal results specifically in MASLD-related HCC (MASLD-HCC) cases. Various immune components constitute a special immune microenvironment in MASLD-HCC, including heterogeneous myeloid cells, lymphocytes and platelets. Furthermore, disruptions in the intestinal barrier, along with the ectopic presence of intestinal flora and metabolites, also influence the immune microenvironment in MASLD-HCC. Elucidating immune cells functions and their interplay with gut microbiota is critical to deciphering MASLD progression to carcinogenesis and immunotherapy resistance. This review synthesizes current insights into the immune microenvironment and gut microbiome in MASLD-HCC, identifies factors influencing the efficacy of immunotherapy, and summarizes potential therapeutic targets to provide detailed guidance for developing effective immunotherapy strategies for MASLD-HCC.
Keywords: metabolic dysfunction-associated steatotic liver disease, hepatocellular carcinoma, immune microenvironment, gut microbiota, immunotherapy
Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD), the successor diagnosis to non-alcoholic fatty liver disease (NAFLD), currently impacts an estimated 25% of people globally. Fueled by growing epidemics of obesity and metabolic syndrome, this condition has become the primary cause of chronic liver disease worldwide [1-3]. Although the term NAFLD, first introduced by Ludwig et al. in 1980, has been used for nearly half a century, it was formally replaced by MASLD in June 2023. This change, based on a consensus document jointly released by international liver societies, addresses concerns regarding the term's ambiguous exclusionary diagnostic criteria and its stigmatizing nature [4]. MASLD encompasses both a relatively benign, non-progressive phenotype characterized by ≥5% hepatic steatosis, and progressive metabolic dysfunction-associated steatohepatitis (MASH). Steatosis, lobular inflammation, hepatocellular ballooning, and fibrosis represent the defining histological features of MASH, a condition with potential for progression to cirrhosis and hepatocellular carcinoma (HCC) [5, 6]. HCC comprises the majority of primary liver cancers, stands as the sixth most prevalent cancer diagnosis, and is the third most common cause of cancer-related death [7]. It is well known that hepatitis B virus (HBV) and hepatitis C virus (HCV) infections act as the most important drivers of primary liver cancer [8, 9]. The role of viral hepatitis in HCC etiology has declined ascribed to the greater vaccination coverage and antiviral medications, which can suppress viral effectivity. Accumulating epidemiological evidence reveals that MASLD is the fastest-growing etiological driver of HCC incidence globally [10, 11]. The underlying mechanisms of MASLD-related HCC (MASLD-HCC) include excessive lipid accumulation and lipid-induced hepatic insulin resistance (IR), liver cell damage caused by dysregulated metabolism, unique host genetic variants, gut microbiota and their metabolic products, and chronic inflammation-induced immune response [12, 13].
Contemporary HCC management encompasses surgical options (resection, transplantation), ablation, transarterial approaches, radiotherapy, and systemic therapies consisting of tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) [7, 14, 15]. Due to the confounding effects of subcutaneous adipose tissue and hepatic steatosis on ultrasonographic accuracy, coupled with the recognition that not all MASLD patients progressing to HCC traverse a cirrhotic pathway, MASLD-HCC is frequently diagnosed at advanced stages [16-18]. Mounting clinical evidence reveals an etiological stratification in HCC responsiveness to immunotherapy. Two meta-analyses, respectively including 8 trials with 3739 patients and 3 trials with 1656 patients, revealed significantly greater efficacy of ICIs in viral-related HCC compared to nonviral HCC. In contrast, the efficacy of TKIs showed no etiological dependence [19, 20]. Crucially, MASLD-HCC exhibits profound immunotherapy resistance, evidenced by two independent cohorts reporting significantly reduced median overall survival compared to other etiologies. This consistent survival disadvantage delineates a distinct resistance phenotype inherent to metabolic dysfunction-driven hepatocarcinogenesis [19].
The efficacy of ICIs, including anti-programmed death receptor-1 (PD-1), anti-programmed death-ligand 1 (PD-L1), and anti-cytotoxic T-lymphocyte antigen 4 (CTLA4) mAbs, is largely determined by the composition and state of the tumor immune microenvironment. MASLD-HCC exhibits a unique immunometabolic microenvironment. Lipid-laden macrophages promote hepatocyte lipid accumulation, while deficiencies in nuclear receptor coactivator 5 (NCOA5) or neuregulin 4 (NRG4) drive macrophages polarization towards a tumor-associated macrophages (TAMs)-like phenotype, accelerating MASLD-driven hepatocarcinogenesis [21-23]. Accumulated polyunsaturated fatty acids (PUFAs) foster neutrophil extracellular traps (NETs) generation that shifts naïve CD4+ T cells differentiation toward regulatory T cells (Tregs) [24]. Concurrently, CD8+ T cells display functional impairment, increased exhaustion, and reduced motility [19, 25]. The lipid-enriched milieu also depletes CD4+ T cells and induces linoleic acid-mediated oxidative damage, further promoting tumorigenesis [26]. Immunosuppressive IgA+ B cells accumulate in MASLD-HCC, impairing antitumor immunity by inhibiting cytotoxic CD8+ T cells [27]. Diverse immune cells coordinate an immunosuppressive milieu conducive to tumorigenesis, significantly exacerbating MASLD-HCC transition dynamics, though their precise individual roles warrant further elucidation. Pathogenic alterations in these bidirectional signaling pathways trigger a sequence of pathological events that culminate in metabolic diseases, with MASLD being a prominent example [28]. Gut dysbiosis contributes to MASLD pathogenesis by compromising intestinal barrier integrity, thereby facilitating the translocation of microbiota-derived factors and microbial-associated molecular patterns (MAMPs) to the liver. Engagement of hepatic pattern recognition receptors (PRRs), notably Toll-like receptors (TLRs), by these molecules initiate potent pro-inflammatory pathways, driving increased hepatic inflammation and fibrogenesis [12]. This review also delineates how gut microbiota-derived signals modulate the hepatic immune landscape, offering novel perspectives on immunological perturbations in MASLD progression.
Elucidating the changes in the immune system during MASLD-HCC progression can provide crucial insights into the potential mechanisms behind the reduced effectiveness of immunotherapy in MASLD-HCC patients. In this review, we discuss the innate and adaptive immune responses, alongside gut microbiota and metabolite-mediated immunological shifts in MASLD-HCC pathogenesis. Accumulating a deeper understanding of these immune mechanisms may provide new insights into MASLD-HCC development and help improve the efficacy of prevention and immunotherapy strategies for MASLD-HCC.
Myeloid Cell and Regulation of the MASLD-HCC Immune Microenvironment
Macrophages
Macrophages, present as abundant resident cells throughout the body's organs, are integral to tissue development and homeostasis while potentially playing a role in diverse ailments pathogenesis [29, 30], including MASLD (Figure 1). Macrophages can be categorized by origin into embryo-derived Kupffer cells (EmKC) and bone marrow/monocyte-derived macrophages [30, 31]. EmKCs form the predominant resident macrophage subset. Their functional repertoire, encompassing the secretion of anti-inflammatory mediators and proficient phagocytosis of particulates arriving through the portal circulation, is essential for sustaining liver immune equilibrium [32, 33]. Bone marrow/monocyte-derived macrophages, however, infiltrate liver tissue during liver injury or inflammation, exhibiting proinflammatory features [34, 35]. Macrophages can also be divided into inflammatory, lipid- and scar-associated, and restorative macrophages based on their different functions [30, 36, 37].
Innate immune modulation and platelets in MASLD-HCC pathogenesis. CCL2 secreted by steatotic hepatocytes recruits monocyte-derived macrophages, which then differentiate into anti-inflammatory TREM2+ macrophages and proinflammatory CX3CR1+CCR2+ macrophages. CX3CR1+CCR2+ macrophages participate in the formation of hepatic crown-like structures. EmKCs are also activated, which secrete IL-1β and TNF-α to induce TERM2 shedding in TREM2+ macrophages and aggravate hepatocyte steatosis. Neutrophils produce MPO, NE, and ROS, increasing oxidative stress and hepatocyte damage. NETs form and recruit naïve CD4+ T cells, driving TLR4-dependent Tregs differentiation. Platelets are increased and activated during the progression. pEVs containing impaired mitochondria are transferred to hepatocytes, causing excess LD buildup, heightened mitochondrial ROS, and apoptosis. The α and δ granules secreted by platelets release particles laden with pro-aggregatory molecules, inflammatory cytokines, chemokines, and growth factors, potentiating inflammatory responses. Platelets recruit CD8+ T cells and NKT cells by hyaluronan-CD44 binding with Kupffer cells in a platelet membrane GPIbα-dependent manner. HSCs are activated by TGF-β from macrophages, MPO from neutrophils, and PDGF-β or PDGF-AA from platelets, finally causing fibrogenesis. Abbreviations: CCL, C-C motif chemokine ligand; CX3CR1, C-X3-C motif chemokine receptor 1; EmKC, Embryonic Kupffer cell; GPIbα, glycoprotein Ibα; HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells; IL-1β, interleukin 1 beta; pEVs, platelet-derived extracellular vesicles; PDGF, platelet-derived growth factor; LD, lipid droplet; MPO, myeloperoxidase; MASH, metabolic dysfunction associated steatohepatitis; MASLD-HCC, metabolic dysfunction associated steatotic liver disease-related hepatocellular carcinoma; NE, neutrophil elastase; ROS, reactive oxygen species; NETs, neutrophil extracellular traps; NKT, natural killer T cells; TGF-β, transforming growth factor beta; TLR4, toll-like receptor 4; TNF-α, tumor necrosis factor alpha; Tregs, regulatory T cells; TREM2, triggering receptor expressed on myeloid cells 2.
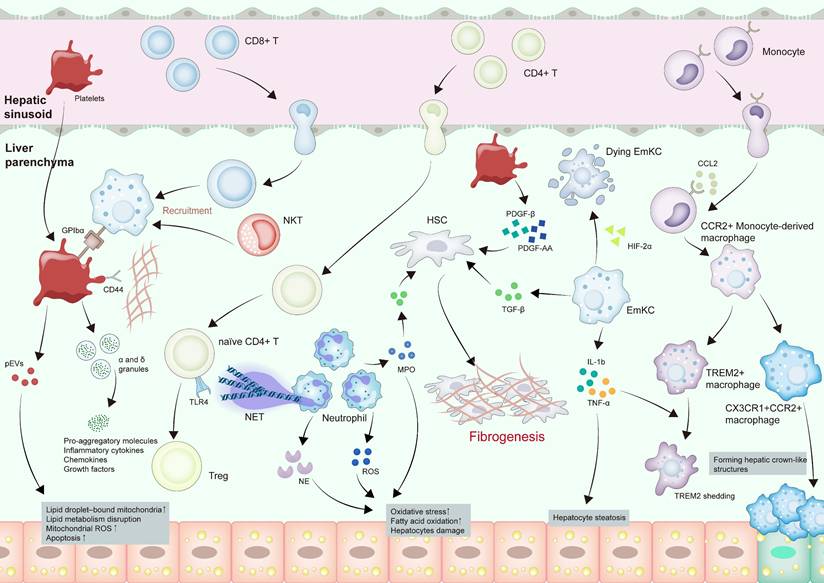
The occurrence and progression of MASLD are inseparable from significant macrophages involvement [32, 38]. During hepatic steatosis onset, the deposition of excess fatty acids places the liver in a state of stress, cholesterol, chemokine C-C motif chemokine ligand 2 (CCL2) and C-X-C motif chemokine ligand 10 (CXCL10) secreted by steatotic hepatocytes activate macrophages [39, 40]. Activated Kupffer cells can inhibit hepatocyte lipid metabolism through paracrine release of interleukin 1 beta (IL-1β) and tumor necrosis factor (TNF) α, ultimately encouraging hepatocyte steatosis [41]. In the pro-inflammatory hepatic milieu, EmKCs exhibit compromised self-renewal potential [42, 43]. The recruitment of C-C motif chemokine receptor 2 (CCR2) + monocyte-derived macrophages into the liver suppresses intrahepatic hepatic triglyceride (TG) retention and drives disease evolution toward steatohepatitis [43]. This process is further amplified by paracrine signaling between macrophages and hepatic stellate cells (HSCs), wherein transforming growth factor beta (TGF-β) acts as a key effector to stimulate pro-fibrotic HSC activation, thereby facilitating MASH development [44, 45].
In MASH progression, lipid-associated macrophages, a monocyte-derived subset, comprise two phenotypes: transitional CX3CR1+CCR2+ macrophages and classic triggering receptor expressed on myeloid cells 2 (TREM2) + macrophages [46]. TREM2+ macrophages exhibit anti-inflammatory effects by modulating lipid uptake, thereby mitigating MASH progression, while CX3CR1+CCR2+ macrophages contribute to hepatic crown-like structures formed by macrophages encircling lipid-laden hepatocytes [46]. Hepatic crown-like structures serve as histological markers of advanced disease, with their density closely tracking the severity of liver fibrosis [47]. In MASH, IL-1β and TNF-α induce TREM2 shedding via a disintegrin and metalloproteinase 17 (ADAM17)-mediated proteolytic cleavage, leading to abnormal accumulation of dying hepatocytes, which exacerbates proinflammatory cytokine production and drives disease progression [48]. Hypoxia-inducible factor (HIF) has emerged as a pivotal regulator of immune function and inflammatory pathways [49]. Within macrophages in MASH mouse models, elevated HIF-1α levels impair autophagic flux while promoting IL-1β secretion. Concurrently, HIF-1α-driven NF-κB activation upregulates monocyte chemoattractant protein-1 (MCP-1), with both cytokines exacerbating hepatic steatosis and inflammatory responses [50]. HIF-2α exerts cell type-specific effects in liver macrophages: it compromises EmKC homeostasis by aggravating lysosomal stress, resulting in diminished proliferation and phagocytosis. In contrast, bone marrow/monocyte-derived macrophages undergo HIF-2α-driven pro-inflammatory polarization via mitochondrial ROS amplification and coordinated upregulation of inflammasome-related genes [51].
The role played by macrophages in MASLD-HCC is incompletely understood. Some studies have discovered that myeloid differentiation primary response 88 (MyD88) in myoblasts enhances MASLD-HCC development by promoting M2 macrophage polarization [52]. NCOA5 deficiency in macrophages was also identified as a key factor in the transition [22, 53]. Previous studies identified NRG4 as a regulatory checkpoint suppressing tumor-permissive liver microenvironments. Loss of NRG4 promotes macrophages with TAM-like properties and drives cytotoxic CD8+ T cells exhaustion in MASLD-HCC [21]. A study demonstrated a significant upregulation of the YT521-B homology (YTH) m6A RNA-binding protein 1 (YTHDF1) in MASLD-HCC compared to peri-tumor regions. Upregulated YTHDF1 promotes MASLD-associated carcinogenesis through EZH2-IL6 pathway stimulation. This signaling cascade recruits and activates myeloid-derived suppressor cells (MDSCs), ultimately suppressing CD8+ T cells cytotoxicity [54]. Besides, former studies indicated that tumor-activated monocytes exhibit robust PD-L1 surface expression, which potently suppresses T cells function and accelerates HCC progression [55, 56].
During MASLD progression to HCC, distinct macrophage subsets drive lipid accumulation and inflammation through cytokine secretion. Critically, macrophage dysfunction—manifested by MyD88-dependent M2 polarization, NCOA5 deficiency, NRG4 loss, and YTHDF1-EZH2-IL6-mediated MDSCs recruitment—establishes a profoundly immunosuppressive microenvironment. This axis may play a pivotal role in both MASLD-HCC development and resistance to immunotherapy.
Neutrophils
Circulating neutrophils, being the most numerous white blood cells, execute frontline protective functions within the innate immune framework [57]. However, abnormally activated neutrophils are associated with certain inflammation-related diseases [58, 59]. Studies have shown that neutrophils infiltration is frequently noted in MASLD patients and correlates with disease progression [60, 61]. Neutrophils drive MASLD-HCC pathogenesis through ROS generation, protease secretion, and NETs formation [62, 63] (Figure 1).
In MASLD liver, upregulated CXCL1 expression recruits neutrophils, leading to ROS production, which promotes the transition from steatosis to steatohepatitis by inducing oxidative stress and activating related signaling pathways [64, 65]. IL-8 also contributes to the recruitment of neutrophils to the liver and promotes MASH by overexpressing CXCL1 and inducing mitochondrial oxidative stress [66]. IL-22, on the other hand, can upregulate hepatic antioxidant enzymes, metallothionein (MT) 1 and MT2, to impede neutrophils recruitment, thereby alleviating MASH development [67].
The proteolytic enzymes released by neutrophils—notably myeloperoxidase (MPO), neutrophil elastase (NE), and human neutrophil peptides (HNPs) may substantially contribute to MASLD development. MASH is characterized by increased MPO levels compared with simple steatosis [67]. In the MASLD mouse model, MPO catalyzes HOCl formation from H₂O₂, which damages hepatocytes and upregulates TGF-β, activating HSCs to drive fibrosis [68]. NE has multiple roles, including pro-inflammatory and pro-cancer effects [69, 70]. It has been demonstrated that NE promotes inflammation and insulin resistance by modulating the AMPK pathway and fatty acid oxidation [71, 72]. HNPs also promote the transition of MASH to fibrosis by stimulating HSCs proliferation [73].
NETs represent extracellular chromatin networks where unpacked DNA scaffolds embed neutrophil-derived granular enzymes and cytosolic proteins [74, 75]. Studies have indicated that the accumulation of PUFAs drives NETs formation in MASH progression [76]. In MASLD-HCC, NETs regulate the mitochondrial oxidative phosphorylation (OXPHOS) of naïve CD4+ T cells, driving their differentiation into Tregs in a TLR4-dependent manner. This process establishes an immunosuppressive microenvironment, promoting HCC development in MASH [24, 60].
Intercellular crosstalk critically modulates MASH progression. Within the MASLD-HCC tumor microenvironment (TME), CXCR2-expressing neutrophils demonstrate significant spatial enrichment, secreting diverse protumor mediators [77]. This phenotypic profile confers upon them the capacity to orchestrate a cascade of immunomodulatory events, including MDSC activation, the inhibition of dendritic cells maturation and function, and the promotion of tumor progression [77]. Lipocalin (LCN)-2 secreted by neutrophils also upregulates CXCR2 to facilitate the recruitment and proliferation of pro-inflammatory macrophages via NF-κB signaling [78]. Besides, NETs may also attract monocyte-derived macrophages to infiltrate the liver by releasing certain signaling molecules or altering the local microenvironment [60]. Additionally, microRNA-223 can be taken up by hepatocytes through the binding of low-density lipoprotein receptor (LDLR) and apolipoprotein E (APOE), thereby inhibiting MASH progression [79].
Taken together, neutrophils drive MASLD progression, fibrogenesis, and HCC pathogenesis by recruiting CXCR2+ neutrophils, producing ROS, proteases, LCN-2, and microRNA-223, and forming NETs.
Dendritic cells
Positioned at the innate-adaptive interface, dendritic cells (DCs) coordinate initial defense reactions while instigating antigen-driven lymphocyte activation [80, 81]. DCs differentiate into three principal subsets based on ontogeny: conventional DCs (cDCs), plasmacytoid DCs (pDCs), and Langerhans cells (LCs) [80, 82]. cDCs comprise two principal subtypes: conventional type I dendritic cells (cDC1) and conventional type II dendritic cells (cDC2) [83].
Previous studies have reported a decline in CD8+ pDCs and CD11c+CD8+ α-DCs during MASLD, concomitant with an increase in CD11b+CD8- pDCs [84]. CD103+ cDC1s and CD11b+ cDC2s also accumulate in the MASLD process [85, 86]. CD130+ DCs were discovered to serve as hepatoprotective agents in MASLD by regulating the immune response, limiting inflammatory cell activation, and potentially removing cell debris, thereby mitigating steatosis in MASLD [85, 87]. In contrast, chemokine X-C receptor 1 (XCR1) + cDC1s accumulate in MASH-affected livers across species, with their density positively correlating with histological severity [88, 89]. Mechanistically, liver pathology results from an excess of cDC1s, generated by enhanced proliferation of their bone marrow precursors. These amplified cDC1 populations drive inflammation by activating and reprogramming pro-inflammatory T cells [88]. However, some studies have yielded different conclusions. Batf3-deficient mice, which lack cDC1s, exhibit significant inhibition of the transition from steatosis to steatohepatitis [90]. The observed discrepancies may stem from the fact that the deletion of Batf3 may also impacts other immune cells. Furthermore, preclinical investigations indicated that co-blockade of PD-1 and CXCR2 significantly augments the XCR1+ cDC1s population, which promotes CD8+ T cells recruitment, thereby enhancing the therapeutic efficacy of the combination regimen [77].
DCs exhibit dichotomous roles in MASLD-HCC. Protective subsets like CD130+ DCs mitigate steatosis by regulating inflammation and clearing debris. Conversely, pathogenic XCR1+ cDC1s orchestrate pathology by activating pro-inflammatory T cells. Therapeutically, augmenting specific DC subsets (e.g., XCR1+ cDCs via anti-PD-1/CXCR2 inhibition) represents a therapeutic strategy to amplify CD8+ T cells infiltration and potentiate treatment efficacy.
Lymphocyte-Mediated Immune Response in MASLD-HCC
CD8+ T cells
The pathogen-clearing function of CD8+ T cells—mediating long-lasting protective immunity and homeostatic balance—demonstrates a significant association with improved patient survival metrics in HCC patients [91-93]. Depleting CD8+ T cells or administering anti-CD8α treatment promotes MASLD-HCC development [27, 77]. ICIs-based immunotherapy is also implemented based on their characteristics. However, some studies have revealed that CD8+ T cells in MASLD-HCC fail to exert anti-tumor effects and may even promote MASLD-HCC progression [19, 94]. Many studies have dedicated efforts to elucidating this intriguing but paradoxical phenomenon.
Quantitatively, despite an elevated systemic frequency, CD8+ T cells often exhibit impaired tumor infiltration, compromising their anti-tumor efficacy. During MASH progression, CD8+ T cells migrate to the liver via antigen presentation or cytokine signaling, increasing their overall population [95-98]. However, hepatic CD8+ T cells infiltration is primarily hindered by excessive collagen fiber deposition at the tumor margin [99]. Recent advances in spatial transcriptomics have also revealed that immune cells are predominantly enriched in adjacent normal tissues but markedly diminished within tumor regions [100]. This spatially marginal distribution pattern and diminished infiltration capacity of CD8+ T cells constrain their antitumor efficacy, resulting in immunotherapy being predominantly effective at tumor margins rather than within the tumor core, ultimately contributing to MASLD-HCC development and immunotherapy inefficiency.
Functionally, CD8+ T cells shift from naïve or effector states to dysfunctional or exhausted states, characterized by expanding intrahepatic CD8+PD-1+ T cells expressing genes linked to exhaustion, tissue residency, and impaired effector functions [19, 94, 96]. Compared to healthy individuals, MASLD-HCC patients exhibit higher rates of catenin beta 1 (CTNNB1) mutations, which elevate tumor necrosis factor receptor superfamily 19 (TNFRSF19) levels and suppress the secretion of senescence-associated secretory phenotype (SASP)-like cytokines, such as IL-6 and CXCL8, fostering an immune-excluded 'cold' TME that exacerbates CD8+ T cells dysfunction [101, 102]. Besides, overexpressed YTHDF1 has been implicated in suppressing cytotoxic CD8+ T cells function by enhancing IL-6 secretion [54]. Cholesterol accumulation dysregulates CD8+ T cells cytotoxicity through suppressed granzyme B (GZMB) and interferon gamma (IFN-γ) secretion [103]. Intratumoral CD8+ T cells in MASH-bearing mice additionally exhibit impaired motility—evidenced by reduced migration velocities and shortened displacement lengths—collectively diminishing antitumor capacity [25, 94]. Altered hepatic lipid metabolism likely drives CD8+ T cells metabolic reprogramming in MASH pathogenesis. Supporting in vitro data reveal that MASH impairs tumor-infiltrating CD8+ T cells motility independently of chemokine signaling or adhesion molecule interactions. Metabolic profiling of CD8+ T cells derived from NASH mice reveals dysregulated glycolysis, fatty acid oxidation, and mitochondrial respiration, substantiating their functional impairment. This metabolic impairment is further evidenced by marked mitochondrial depolarization and diminished mitochondrial mass [25, 94, 96]. Therefore, despite the increase in CD8+ T cells, their functionality is predominantly impaired, rendering them unable to exert anti-tumor effects, contributing to MASLD-HCC development and immunotherapy inefficiency.
A unique CXCR6+CD8+ T cells subset that exerts distinct roles compared with other CD8+ T cells subsets has been identified. CXCR6+CD8+ T cells maintenance depends on CCR7+ DCs that express the cognate ligand CXCL16 and provide IL-15 cytokine signaling [104]. These T cells, characterized by low Forkhead Box O1 (FOXO1) activity, are detected to accumulate in MASH mice fed a choline-deficient, high-fat diet (CD-HFD) or a western diet (WD). Mechanistically, IL-15-mediated FOXO1 suppression coupled with CXCR6 induction metabolically sensitizes CXCR6+CD8+ T cells. This reprogramming enables aberrant recognition of acetate/ATP signals, provoking auto-aggressive cytolysis through factor associated with suicide (Fas) / Fas ligand (FasL) interaction [105]. Strikingly, CXCR6+CD8+ T cells exhibit heightened migratory velocity when activated by local tissue signals [94] (Figure 2).
Characteristic changes of T cells in MASLD-HCC pathogenesis. In CD8+ T cells, IL-15 populated the CXCR6+PD-1+CD8+ T cells and CD8+ Trm cells, which cause HSCs apoptosis through Fas/FasL interaction respectively. Some CD8+ T cells also exhibit lower velocities and shorter displacement lengths, thus reducing their motility and antitumor effect. In CD4+ T cells, high levels of ROS make CD4+ T cells much more vulnerable to exposure to lipid metabolites, finally causing CD4+ T cells apoptosis. α4β7+ CD4 T cells are recruited by increased MAdCAM-1 in the liver and colon, which exacerbates inflammation and fibrosis. CXCR3+Th17 (ihTh17) is also increased to contribute to the development of MASH through activating the CXCR3-CXCL9/10 axis and reprogramming the metabolic and proinflammatory phenotype. The formation of NETs regulates the mitochondrial OXPHOS of naïve CD4+ T cells and drives their differentiation into Tregs in a TLR4-dependent manner. Abbreviations: CXCR, C-X-C motif chemokine receptor; CXC, C-X-C motif chemokine ligand; Fas, factor associated with suicide; FasL, Fas ligand; HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells; IL-15, interleukin 15; MAdCAM-1, mucosal addressin cell adhesion molecule-1; MASH, metabolic dysfunction associated steatohepatitis; MASLD-HCC, metabolic dysfunction associated steatotic liver disease-related hepatocellular carcinoma; OXPHOS, oxidative phosphorylation; NETs, neutrophil extracellular traps; PD-1, programmed death receptor 1; ROS, reactive oxygen species; TLR4, Toll-like receptor 4; Tregs, regulatory T cells; Trm, tissue-resident memory T cells.
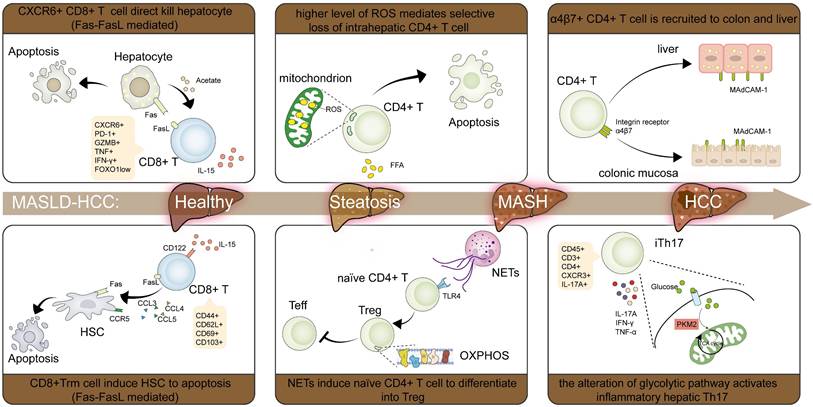
Therefore, immunotherapy resistance in MASLD may arise from: reduced tumor CD8+ T cells infiltration, functionally exhausted T cells with impaired motility, and pathological accumulation of CXCR6+ or PD-1+ CD8+ T cells subset in the liver. Cell metabolism is widely recognized as a factor affecting T cells function and migration. Given that MASLD is characterized by metabolic dysregulation, alterations in the MASLD-HCC TME may induce metabolic disturbances in CD8+ T cells, potentially exacerbating hepatic damage and promoting MASLD-HCC.
CD4+ T cells
As master regulators of adaptive immunity, CD4+ T cells represent a fundamental lymphocyte subpopulation. These cells segregate into two functional lineages: helper T cells (Th) and Tregs [106, 107]. Th cells undergo further specialization into distinct subsets (Th1, Th2, Th17, Th22) defined by unique transcriptional programs and cytokine signatures [107, 108]. CD4+ T cells subsets exert their respective effects, promoting or inhibiting disease progression, thus forming an immune regulatory network. Progression of MASH is accelerated through IFN-γ/TNF-α secretion from Th1 cells, mediating hepatocyte cytotoxicity and inflammation potentiation [109, 110]. Th2 cells, by secreting IL-13, promote HSC activation, leading to liver fibrosis [111]. Th17 cells accelerate MASH progression by secreting IL-17, which promotes hepatocellular injury and inflammatory responses [112, 113]. IL-17 also promotes hepatic fibrosis by up-regulating TGF-βRII on HSCs surfaces, which enhances their response to TGF-β [114]. In contrast, Th22 cells have a protective effect by secreting IL-22, attenuating hepatocytes injury and inflammatory response [115, 116].
In methionine-choline-deficient (MCD) or choline-deficient and amino acid-defined (CDAA) diet-induced MASH models, intrahepatic CD4+ T cells depletion occurs as lipid-rich microenvironments upregulate CPT expression. This amplifies mitochondrial biogenesis and reactive oxygen species generation, enhancing susceptibility to cytotoxic lipid metabolites like linoleic acid [26, 117]. Reports indicate a decrease in total hepatic CD4+ T cells, yet some subpopulations increase in MASH. Expanded central and effector memory CD4+ T cells drive liver inflammation and fibrosis [118]. Th17 cells are also demonstrated to be increased [119, 120]. A significant study uncovered the presence of a unique pathogenic subpopulation of liver Th17 cells, inflammatory hepatic CXCR3+ Th17 (ihTh17), which is sufficient to contribute to MASLD development through activating the CXCR3-CXCL9/10 axis and reprogramming cells toward a metabolic and proinflammatory phenotype [121]. Besides, enhanced hepatic and intestinal mucosal addressin cell adhesion molecule-1 (MAdCAM-1) expression in WD-fed mice facilitated α4β7+ CD4+ T cells recruitment, directly aggravating inflammatory responses and extracellular matrix deposition in the liver [122].
Tregs, a highly immunosuppressive subset of CD4+ T cells characterized by CD4+FOXP3+CD25+ expression, are essential for maintaining an immunosuppressive microenvironment [123]. Emerging evidence reveals a biphasic role of Tregs in MASLD pathogenesis [124]. During early steatosis, obesity and insulin resistance suppress Tregs differentiation and impair their functional capacity [125, 126]. Concurrently, oxidative stress triggers Tregs apoptosis and activates the TNF-α signaling pathway, collectively driving progressive hepatic injury [127]. As disease advances to MASH, substantial Tregs expansion occurs despite this initial suppression. NETs reprogram mitochondrial OXPHOS in naïve CD4+ T cells via TLR4 signaling, promoting their differentiation toward a regulatory phenotype over an effector phenotype [24]. During chronic liver injury, these elevated Tregs produce amphiregulin, which engages epidermal growth factor receptor (EGFR) on HSCs. This interaction directly promotes hepatic fibrogenesis and concurrently stimulates HSCs to secrete IL-6. The resulting IL-6 contributes to glucose intolerance, thereby establishing a vicious cycle that further drives MASH progression [128]. Gut dysbiosis represents a well-recognized pathological feature of MASLD. Microbial-derived short-chain fatty acids (SCFAs) amplify Tregs responses by enhancing IL-10-secreting Tregs abundance and expanding specialized effector Tregs populations [129]. As a microbial membrane constituent, lipoteichoic acid (LTA) translocates from gut to liver parenchyma, directly driving senescence programming in HSCs with consequent SASP factor secretion. Critically, bioactivation of IL-33 occurs through chymotrypsin-like elastase family member 1 (CELA1)-mediated proteolytic cleavage of its full-length precursor. Senescent HSCs export this mature IL-33 via gasdermin D (GSDMD) amino-terminal domain-mediated pore formation. The liberated cytokine then engages ST2+ Tregs (where ST2 functions as the IL-33 receptor), driving obesity-promoted hepatocarcinogenesis [130].
Herein, the metabolic reprogramming in CD4+ T cells is evident in MASLD-HCC pathogenesis. These cells exhibit increased mitochondrial mass and elevated mitochondrial OXPHOS activity. Heightened mitochondrially derived ROS instigates oxidative stress-mediated depletion of intrahepatic CD4+ T cells, accelerating hepatocarcinogenesis. Besides, enhanced OXPHOS activity directs naïve CD4+ T cells commitment to the Treg lineage, sustaining an immunosuppressive microenvironment (Figure 2).
B cells
B lymphocytes contribute significantly to MASLD-HCC pathogenesis due to their ability to secrete antibodies and various pro- and anti-inflammatory cytokines [131, 132] (Figure 3). B cells heterogeneity is principally defined by surface marker expression, distinguishing B1 and B2 lymphocyte subsets [133, 134]. B1 cells are generated from the fetal liver and produce IgM natural antibodies, participating in the innate immune response. Bone marrow-derived B2 precursors differentiate into antibody-secreting plasma cells via Th cell-mediated pathways, producing high-affinity antigen-specific immunoglobulins [131, 135]. An additional population of B cells characterized by CD5+CD1d high, known as regulatory B cells (Bregs), produce inhibitory cytokines, such as IL-10, or secrete inhibitory antibodies to affect the function of other immune cells, thus creating an immunosuppressive microenvironment [136, 137].
Dynamic changes of B cells in MASLD-HCC pathogenesis. B cells were activated by antigens derived from the gut and bacterial metabolites draining into the liver through MyD88-dependent or BCR signaling. The activated B cells are associated with increased inflammation and fibrosis, as well as BAFF, which forces B cell survival and maturation. IL-6 and TNF-α derived from intrahepatic B cells activate CD8+ T cells and CD4+ T cells, facilitating their secretion of IFN-γ. B cells also activate HSCs and promote fibrosis dependent on TNF signaling. IgA+ B cells are accumulated in the liver, which suppress CD8+ T cells by upregulating the expression of PD-L1 on the cell surface and producing the immunosuppressive cytokines. In the gut, IgA activates monocyte-derived macrophages in FcR-signalling. NcDase is significantly increased in the intestinal brush border of the small intestine and induces IgA-bound Desulfovibrio, which contributes to up-regulating SCD 1 expression with an increase of MUFAs, further facilitating the development of liver fibrosis. IgG antibodies are also elevated to be against OSEs. This anti-OSE IgG is connected to the differentiation of liver B2 cells to plasma cells. On the contrary, anti-OSE IgM is decreased in the process, suggesting a protective role. Abbreviations: BAFF, B cell-activating factor; HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells; IgA, immunoglobulin-A; IgG, Immunoglobulin-G; IgM, immunoglobulin-M; IFN-γ, interferon gamma; IL-6, interleukin 6; PD-L1, programmed death ligand 1; MyD88, myeloid differentiation primary response 88; MUFAs, monounsaturated fatty acids; NcDase, Neutral ceramidase; MASH, metabolic dysfunction associated steatohepatitis; MASLD-HCC, metabolic dysfunction associated steatotic liver disease-related hepatocellular carcinoma; OSEs, oxidative-stress-derived epitopes; SCD, stearoyl-CoA desaturase; TNF-α, tumor necrosis factor alpha.
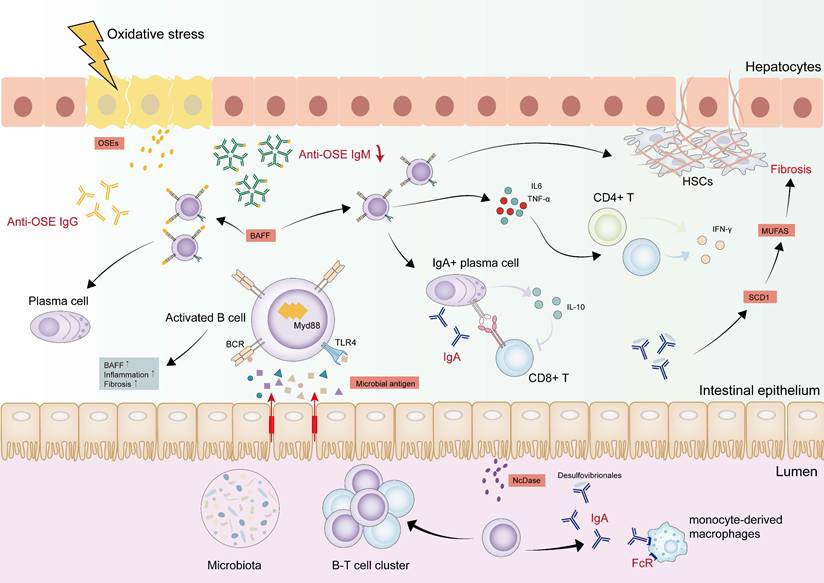
Activation of B cells in MASLD-HCC
Hepatic B cells accumulation occurs alongside an activated, pro-inflammatory phenotype linked to disease severity in MASLD [138-140]. MyD88 triggers B cell activation, and its B cell-specific deletion ameliorates inflammation and fibrosis [141]. The B cell activation cascade initiates before T cells engagement and features B cell-activating factor (BAFF) overexpression. This cytokine critically sustains B cells survival and developmental progression [138]. Using BAFF-neutralizing monoclonal antibodies or BAFF-/- mice can dramatically ameliorate steatohepatitis and reduce liver weight [138, 142]. Another potential factor is that gut-derived antigens and bacterial metabolites may drive intrahepatic B cells toward an inflammatory phenotype via MyD88-dependent or BCR signaling pathways. Fecal microbiota transplantation from MASLD patients augments intrahepatic B cells accumulation and hastens disease progression in recipient mice [141]. The roles of B cells are discussed from three critical perspectives: antigen presentation, pro-inflammatory cytokine secretion, and the generation of pathogenic antibodies in the progression of MASLD.
Antigen presentation
In MASLD, B cells display increased expression of cell surface major histocompatibility complex (MHC)-I and MHC-II, as well as CD86, suggesting enhanced antigen-presenting capability [138, 141]. Sometimes intestinal B cells induce T cells hyperactivation that does not rely on their traditional antigen presentation ability but on direct cell-cell interaction via intercellular cell adhesion molecule (ICAM)-1 and leukocyte function-associated antigen (LFA)-1 [143]. Hepatic B cell-derived cytokines orchestrate pro-inflammatory responses while modulating adjacent T cells activity. Specifically, intrahepatic B cell-secreted IL-6 and TNF-α activate CD4+ T cells and drive their Th1 polarization in MASH pathogenesis [141, 144]. B cells also have a prominent role in activating HSCs and promoting fibrosis via TNF signaling [145]. Bregs have been shown to promote HCC development, but a subset of Bregs expressing IL-10 has a protective effect against MASH progression [146, 147].
Secretion of pro-inflammatory cytokines
Evidence indicates that B cell-derived antibodies are involved in MASLD-HCC pathogenesis. In MASLD patients, elevated serum IgA levels are observed, which activate monocyte-derived macrophages via FcR signaling to promote hepatic fibrosis [143, 148]. The IL-21R-STAT1-c-Jun/c-Fos-IgA regulatory pathway is activated during MASLD-HCC, which leads to immunosuppressive IgA+ cells induction [149]. These cells can suppress CD8+ T cells by upregulating PD-L1 surface expression and producing the immunosuppressive cytokine IL-10, impacting tumor immune surveillance function in MASLD-HCC [27]. MASH models demonstrate elevated neutral ceramidase (NcDase) expression in the small intestinal brush border. NcDase acts as a regulator of gut B cells that induce IgA-bound Desulfovibrio and might contribute to up-regulating stearoyl-CoA desaturase (SCD) 1 expression and an increase in monounsaturated fatty acids (MUFAs). The increased SCD1/MUFAs activate Wnt/β-catenin signaling, further facilitating liver fibrosis [150].
Generation of pathogenic antibodies
Increased serum IgG2c levels point to a key role for secreted antibodies in MASH pathogenesis. Elevated IgG antibodies against oxidative-stress-derived epitopes (OSEs) have been demonstrated to drive lobular inflammation severity, fibrosis, and increased risk of MASH [138, 151]. Plasma cells differentiation from hepatic B2 precursors coincides with rising anti-OSE IgG titers [138, 152]. Conversely, there is a significant decline in IgM+B220+ hepatic B cells in MCD mice, paralleled by diminished anti-OSE IgM titers in MASLD patients compared to healthy controls [153]. Immunizing low-density lipoprotein receptor knock-out mice (Ldlr-/-) mice with heat-inactivated pneumococci to induce anti-OxLDL (oxidized low-density lipoproteins) IgM reduces liver inflammation under a high-fat, high-cholesterol diet, highlighting IgM's protective role [154]. The potential opposing effects of anti-OSE IgG and IgM demonstrate that the B1 and B2 cells may exert different roles in MASH, creating opportunities for novel therapies targeting specific B cells subsets or antibodies.
In summary, the inflammatory phenotype, pro-inflammatory cytokines, and pathogenic antibodies all play important roles in MASLD development. In other cancer types, substantial evidence supports a correlation between immunotherapy efficacy and B cells infiltration and tertiary lymphoid structure (TLS) formation, which enhance B cell-mediated antitumor immunity [155-157]. However, TLS formation may be rare in MASLD-HCC, and this lack might alter B cells function and subsequently impede immunotherapy response. Moreover, given that the generation of antibodies by B cells correlates with response to ICIs in mouse models of triple-negative breast cancer and the abnormal accumulation of antibodies in MASH, these antibodies likely also influence immune responses in MASLD-HCC [158].
Platelets in MASLD-HCC
While platelets are essential for hemostasis and wound repair, their extended functions now include significant contributions to hepatic inflammation and liver disease pathogenesis [159-161]. Hepatic physiology centrally governs platelet biogenesis and elimination. Reciprocally, platelets modulate liver functions through α-granule and dense granule exocytosis, releasing bioactive growth factors and immunoregulatory molecules [162]. Typically, patients with MASLD commonly display elevated platelet counts along with increases in mean platelet volume (MPV) and platelet distribution width (PDW) compared to healthy individuals [163, 164]. Platelet aggregation is induced by elevated leptin levels, a consequence of adipose tissue accumulation [165, 166]. However, a pronounced decrease in platelet counts becomes evident as the disease advances from MASH to hepatic fibrosis. This progressive thrombocytopenia demonstrates utility as a predictor of advancing fibrosis [167]. In biopsy-confirmed MASLD cohorts, lower baseline platelet counts correlate with elevated HCC incidence [163]. Preclinical studies indicate that simple steatosis and insulin resistance alone fail to elicit increased intrahepatic platelet numbers. This phenomenon manifests only upon progression to MASH, characterized by intrahepatic platelet accumulation, aggregation, and activation [168]. Upon activation, platelets shed platelet-derived extracellular vesicles (pEVs) carrying mitochondria with compromised function—evidenced by diminished fatty acid β-oxidation, acetyl-CoA carboxylase 2 (ACC2) inactivation, and defective OXPHOS activity [169]. Such mitochondria can transfer to hepatocytes via pEVs, increasing the number of faulty lipid droplet (LD)—bound mitochondria, which disrupts hepatocyte lipid metabolism, causes excess LD buildup, heightened mitochondrial ROS, and apoptosis, and finally aggravates MASH [169]. These platelets can also release the α and δ granules laden with myriad molecules including pro-aggregatory molecules such as ADP, serotonin, and thrombin, as well as inflammatory cytokines, chemokines, and growth factors that can directly potentiate inflammatory responses [170, 171]. Platelet-derived microparticles (PMPs) also participate in this process [172, 173]. Notably, platelet-derived growth factor (PDGF)-β and PDGF-AA drive HSC activation and contribute to biliary fibrosis progression. In contrast, adenosine 5'-triphosphate (ATP) released from platelets suppresses the activation of human HSCs, revealing a complex, multifaceted role for platelets in modulating the fibrotic microenvironment [174-176].
Platelet involvement in MASH progression to HCC exhibits context-dependent complexity, with reports suggesting both pro-tumorigenic and anti-tumor functions. Experimental evidence from CD-HFD mouse models implicates Kupffer cell-mediated platelet recruitment in the liver, facilitated by CD44-hyaluronan binding, as a driver of MASH progression. Critically, platelet-derived glycoprotein Ibα (GPIbα) has been demonstrated as essential for the development of MASH and subsequent HCC in this setting [168]. Conversely, studies employing orthotopic implantation of established HCC tumors or carcinogen/oncogene-driven HCC models reveal a protective role for platelets. In these models, platelets upregulate intrahepatic CD8+ T cells accumulation via CD40L release. This platelet-CD40L axis mediates robust anti-tumor immunity in a P2Y12 receptor-dependent manner, thereby inhibiting HCC growth and metastasis [177].
These contradictory findings are likely attributable to distinct experimental models—with the former focusing on platelet involvement in MASLD-driven hepatocarcinogenesis, while the latter specifically examines platelet-mediated modulation of established tumor progression within the MASLD microenvironment. Nevertheless, they converge to suggest that platelets play a pivotal role throughout MASLD pathogenesis.
Gut Microbiota Modulates Immune Microenvironment, Immunotherapy and MASLD-HCC
The portal vein delivers gut-derived microbial metabolites and microbiota components to the liver, establishing bidirectional gut-liver crosstalk that modulates hepatic physiology [178, 179]. Intestinal microbiota regulates liver homeostasis but can also produce damaging molecules and promote pathogenic overgrowth that compromises hepatic integrity [180-182]. Germ-free mice are effectively protected from obesity, whereas fecal microbiota transfer from obese mice promotes higher fat accumulation than transfers from lean counterparts [183, 184]. Gut microbiota also affects fat storage and fatty liver disease [185]. Typically, intestinal epithelial cells sustain gut barrier integrity primarily through tight junction complexes [186, 187]. However, during MASLD development and progression, the gut microbiota experiences a decline in diversity, which becomes more significant as the disease advances [188]. The abundance of certain microbiota, such as Streptococcus and gram-negative microbes, tends to increase [189]. These changes can damage the tight junctions between cells, impair gut barrier function, and facilitate portal vein translocation of microbiota and metabolites to the liver [189].
Translocation of microbial components
Compromised intestinal barrier function enables the pathological transfer of MAMPs, including lipopolysaccharide (LPS) and LTA, to the liver [185]. Hepatic TLR4 recognition of gram-negative bacterial LPS initiates NF-κB/MAPK signaling, driving inflammatory cytokine release and potentiating hepatic inflammation [190, 191]. The depletion of Akkermansia muciniphila (A. muciniphila) - a bacterium crucial for intestinal barrier integrity - is observed in fatty liver disease [192, 193]. Mechanistically, A. muciniphila prevents MASLD-HCC by downregulating γδT cells, upregulating CXCR6+ natural killer T cells (NKT), and inhibiting M1 macrophages polarization through the reduction of hepatic TLR2 expression [192, 194]. LTA, as previously indicated, ligation on HSCs orchestrates the activation of ST2-positive Tregs [130].
Bacterial metabolites
Microbial metabolites, including SCFAs, bile acids, and trimethylamine, may affect the immune system and contribute to MASH and MASLD-HCC [195-197]. In MASLD, bacterial metabolites drain into the liver via the portal vein, promoting intrahepatic B cells toward an inflammatory phenotype via MyD88-dependent or BCR signaling, and enhancing antigen presentation and costimulatory molecules expression [141]. SCFAs, including butyrate, propionate, and acetate, are enriched in MASLD-HCC patients and drive MASLD progression by regulating hepatic lipogenesis [198-202]. Acetate can promote tumor cell proliferation and HCC progression through upregulation of O-GlcNAcylation [181]. SCFAs also impact immune cells. Th1 cells exposure to SCFAs activates both signal transducer and activator of transcription 3 (STAT3) and mammalian target of rapamycin (mTOR) pathways, elevating B lymphocyte-induced maturation protein 1 (Blimp-1) expression. This potentiates IL-10 production, mediating anti-inflammatory effects [156]. However, some investigators have found that increased circulating SCFA levels protect against inflammation by promoting IL-22 production by CD4+ T cells [157]. On the other hand, increased SCFAs can lead to an immunosuppressed response by increasing Tregs and attenuating CD8+ T cells responses [129]. Through free fatty acid receptor (FFAR)2 signaling, SCFAs modulate colonic Tregs population dynamics and function while exerting protective effects against colitis [203].
Primary bile acids synthesized in the liver undergo extensive microbial transformation within the intestinal tract. This series of enzymatic reactions—including deconjugation, epimerization, 7-dehydroxylation, reconjugation, 3-acylation, 3-sulfation, and 3-glucosylation—converts them into significant microbiota-derived metabolites that critically influence the progression of MASLD [204]. Typically, bile acids act as endocrine mediators that critically maintain glucose and lipid balance via engagement of the G-protein-coupled bile acid receptor 5 (TGR5) and the nuclear farnesoid X receptor (FXR) [205, 206]. FXR activation reduces lipid synthesis and glucose levels by modulating SREBP-1C and gluconeogenesis-related genes [207]. Similarly, TGR5 helps maintain glucose homeostasis, alleviates hepatic steatosis, and suppresses inflammation [208]. Primary bile acids preferentially target FXR, while secondary bile acids prefer to combine with TGR5 [209]. However, patients with MASH often exhibit elevated total primary bile acids with concurrent reductions in secondary bile acids and 3-indole propionic acid (IPA) [210]. During MASLD-HCC, elevated steroidogenic acute regulatory protein 1 (STARD1) expression drives primary bile acid biosynthesis through mitochondrial cholesterol transport. This metabolic reprogramming potentiates cancer stem cell self-renewal, enhances stem-like properties, and amplifies pro-inflammatory signaling in tumor-initiating cells [211]. Treatment with anti-cholesterol drugs and manipulation of gut microbiota can completely prevent MASLD-HCC formation [212]. Mechanically, the gut microbiome modulates liver CXCL16 expression via bile acids, impacting CXCR6+ NKT cell dynamics. Specifically, primary bile acids boost CXCL16 levels on sinusoidal endothelial cells, whereas secondary bile acids diminish them. This upregulation of CXCL16, the major ligand for CXCR6, drives CXCR6+ NKT cells accumulation in the liver. These accumulated cells are phenotypically activated and secrete elevated IFN-γ upon antigen encounter [213]. These findings suggest that primary and secondary bile acids may play distinct roles in MASLD-HCC.
Changes in intestinal fungi
Studies focusing on intestinal fungi in MASLD mice models are sparse. Previous works demonstrates that mice fed a high-fat diet (HFD) exhibit reduced fungal diversity and constitutional changes [214, 215]. This mouse model showed markedly decreased populations of specific fungal taxa, including the genera Alternaria, Saccharomyces, Septoriella, and Tilletiopsis, along with the species Saccharomyces cerevisiae and Tilletiopsis washingtonensis [214]. Distinct fecal mycobiome profiles distinguish early-stage MASLD patients from advanced-stage counterparts, particularly in non-obese individuals. This dysbiosis correlates with heightened systemic reactivity to Candida albicans, evidenced by elevated anti-C. albicans IgG titers [216]. Furthermore, transferring feces from patients with MASH into a WD-fed gnotobiotic mice model and treating them with antifungal amphotericin B showed reduced liver damage, suggesting that targeting intestinal fungi may be a potential therapy to ameliorate MASH [216].
Collectively, gut barrier dysfunction, translocation of microbial components, and dysregulated bacterial metabolite abundance orchestrate MASLD-HCC pathogenesis by affecting the immune system to varying degrees (Figure 4).
Dynamic changes of gut microbiota in MASLD-HCC. Microbial metabolism, bacterial metabolites and antigens are draining into the liver, altering the environment. Bacteria and antigens activate B cells through MyD88-dependent or BCR signaling pathways, which finally induce B cells to differentiate into plasma cells or inflammatory phenotype. LPS binds to TLR4 in the liver and activates the downstream NF-κB and MAPK signaling pathways, leading to the secretion of inflammatory cytokines and promoting liver inflammation. LTA induces a senescent phenotype in HSCs, leading to the release of SASP factors. IL-33 is subsequently exported from senescent HSCs activating Tregs. SCFAs and primary bile acids tend to increase while 3-IPA and secondary bile acids tend to decrease. SCFAs activate STAT3 and mTOR in Th1 cells and upregulate transcription factor Blimp-1 consequently, thus promoting the secretion of IL-10 by Th1 cells. SCFAs also result in an immunosuppressed response by increasing Tregs and attenuating CD8+ T cells responses. Bile acids could bind to the TGR5 and FXR, thereby reducing lipid synthesis and glucose levels. Primary bile acids are used by the gut microbiome to upregulate the expression level of CXCL16 to mediate the accumulation of CXCR6+ NKT cells, which are activated and produce more IFN-γ upon antigen stimulation. Secondary bile acids had a negative effect on CXCL16 expression, causing an opposite result. Abbreviations: Blimp-1, B lymphocyte-induced maturation protein 1; CXCL16, C-X-C motif chemokine ligand 16; CXCR6, C-X-C motif chemokine receptor 6; FXR, farnesoid X receptor; HSC, hepatic stellate cell; IFN-γ, interferon gamma; IL-10, interleukin 10; IL-33, interleukin 33; IPA, indole propionic acid; LPS, lipopolysaccharide; LTA, lipoteichoic acid; mTOR, mammalian target of rapamycin; MyD88, myeloid differentiation primary response 88; MASH, metabolic dysfunction associated steatohepatitis; MASLD-HCC, metabolic dysfunction associated steatotic liver disease-related hepatocellular carcinoma; NKT, natural killer T cells; SCFAs, short-chain fatty acids; STAT3, signal transducer and activator of transcription 3; SASP, senescence-associated secretory phenotype; TGR5, G-protein-coupled bile acid receptor 5; Th, T helper cells; TLR4, Toll-like receptor 4; Tregs, regulatory T cells.
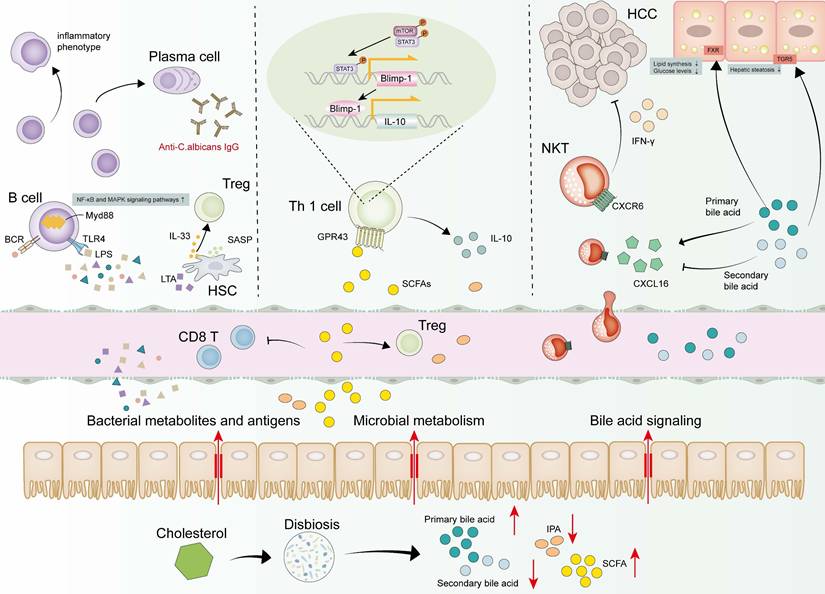
Current Dilemma and Potential Therapeutic Strategies in MASLD-HCC Immunotherapy
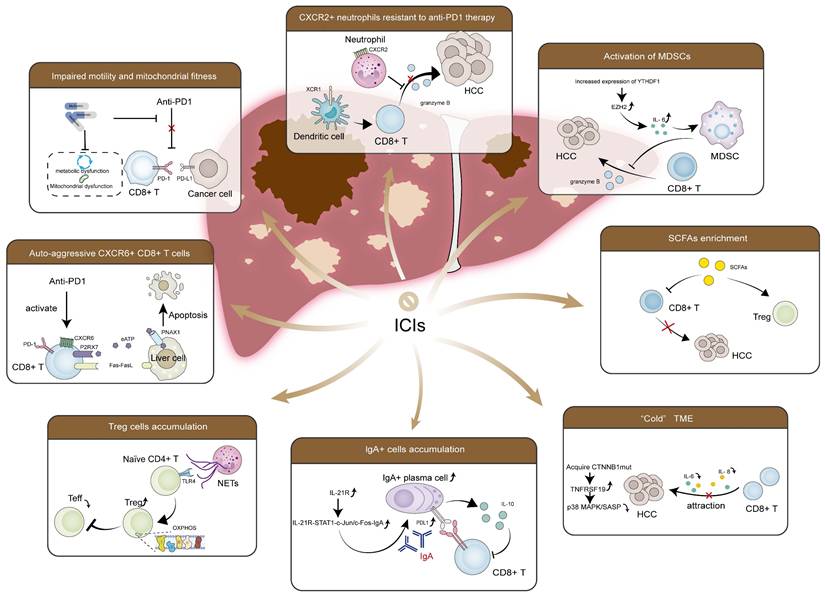
Currently, HCC treatment options encompass surgical interventions (resection/transplantation), ablation, intra-arterial therapies, radiotherapy, and systemic therapies. But clinicians choose treatment methods based on disease grading rather than etiology. In MASLD-HCC, excessive lipid accumulation and lipid-induced hepatic insulin resistance, dysregulated metabolism, the gut microbiota and its metabolic products, unique host genetic variants, and chronic inflammation-induced immune response collectively create a complex microenvironment, influencing therapy effectiveness. Current clinical trials focusing on MASLD-HCC remain scarce, most of which have investigated MASLD-HCC as part of non-viral HCC [217]. Despite their lack of specificity, these studies' results are still informative. Additionally, meta-analyses are attempting to determine the differences in therapeutic efficacy between MASLD-HCC and other etiologies. As mentioned previously and in former reviews [16], current research suggests that TKIs likely have comparable effectiveness, whereas ICIs might exhibit reduced efficacy in MASLD-HCC compared to viral HCC. Most studies on combination therapy with ICIs also report similar results [217, 218]. Mechanically, the distinct TME characteristic of MASLD-HCC, as summarized above-including activation of MDSCs, enriched CXCR2+ neutrophils, Treg cells and IgA+ cells, impaired CD8+ T cells recruitment and effector function, increased specific pro-cancerous CD8+ T cells subsets, accumulated CTNNB1 mutations and elevated SCFAs-may contribute to this phenomenon (Figure 5).
Key factors contribute to the immunotherapy resistance in MASLD-HCC. RNA m6A reader protein YTHDF1 is increased, which recruits and activates MDSCs to cause cytotoxic CD8+ T cells dysfunction. CXCR2+ neutrophils secrete pro-tumorigenic cytokines and immunosuppressive molecules, inhibiting the function of CD8+ T cells. The formation of NETs recruits naïve CD4+ T cells and drives their differentiation into Tregs in a TLR4-dependent manner, establishing an immunosuppressive microenvironment. Enriched SCFAs increase Tregs and attenuate CD8+ T cells response, resulting in an immunosuppressed response. IL-21R-STAT1-c-Jun/c-Fos-IgA regulatory pathway is also activated, which leads to the induction of immunosuppressive IgA+ cells. These cells suppress CD8+ T cells by upregulating the expression of PD-L1 on the cell surface and producing the immunosuppressive cytokine IL-10, impairing tumor surveillance function. Accumulated CTNNB1 mutations elevate TNFRSF19 levels, suppressing the secretion of SASP-like cytokines, such as IL-6 and CXCL8, inhibiting the effect of CD8+ T cells. In addition, impaired motility and mitochondrial fitness in CD8+ T cells are observed, along with a low infiltration rate. CXCR6+PD-1+CD8+ T cells are also increased, which causes hepatocyte apoptosis through Fas/FasL interaction, generating an adverse effect in MASLD-HCC immunotherapy. Abbreviations: CXCR, C-X-C motif chemokine receptor; CXCL8, C-X-C motif chemokine ligand 8; CTNNB1, catenin beta 1; Fas, factor associated with suicide; FasL, Fas ligand; HCC, hepatocellular carcinoma; ICIs, immune checkpoint inhibitors; IL-6, interleukin 6; IL-10, interleukin 10; PD-1, programmed death receptor 1; PD-L1, programmed death ligand 1; m6A, N6-methyladenosine; MDSCs, myeloid-derived suppressor cells; MASLD-HCC, metabolic dysfunction associated steatotic liver disease-related hepatocellular carcinoma; NETs, neutrophil extracellular traps; SASP, senescence-associated secretory phenotype; SCFAs, short-chain fatty acids; TNFRSF19, tumor necrosis factor receptor superfamily 19; Tregs, regulatory T cells; YTHDF1, YTH N6-methyladenosine RNA binding protein F1.
Given the continuum of MASLD progression to HCC, therapeutic interventions targeting early-stage disease may effectively halt hepatocarcinogenesis. Many studies and therapeutic interventions have focused on preventing the progression of MASLD by effectively managing and improving the underlying disease pathology through targeted therapy. Resmetirom currently represents the sole US Food and Drug Administration (FDA)-approved pharmacotherapy for MASLD, exerting therapeutic effects via selective thyroid hormone receptor activation [219]. Here, we summarize current therapeutic drugs targeting MASLD and MASLD progression, aiming to reveal the potential strategies for preventing MASLD-HCC (Table 1).
Current therapy drugs and treatment targeting MASLD progression
| Treatment | Mechanism | Drug name | Other interventions | Study phase | Clinical trial number or reference | Results |
|---|---|---|---|---|---|---|
| PPAR (PPARα/PPARγ/PPARδ) agonist | Promote the polarization of macrophages towards M2 type and reduce inflammatory response | Rosiglitazone | NA | II | NCT00492700,[236] | Insulin sensitivity and ALT levels are elevated, liver fat deposition is reduced |
| Pioglitazone | NA | IV | NCT00227110 | Liver fat deposition and fibrosis are reduced | ||
| Pioglitazone | Metformin | IV | NCT03796975 | Liver fat deposition and fibrosis are reduced | ||
| Pioglitazone | Empagliflozin | IV | NCT03646292 | Ongoing | ||
| Pioglitazone | Dapagliflozin | NA | NCT06649162 | Ongoing | ||
| GW501516 | NA | Preclinical | [237, 238] | Liver fat deposition and fibrosis are reduced | ||
| Lanifibranor | NA | III | NCT04849728 | Liver fibrosis is reduced | ||
| Lanifibranor | Empagliflozin | II | NCT05232071 | Ongoing | ||
| Elafibranor | NA | II | NCT01694849 | Liver fibrosis is reduced | ||
| CCR2/CCR5 antagonist | Block the CCR5/CCR2 and reduce hepatic infiltration of monocytes/macrophages | Cenicriviroc | NA | II | NCT02217475 | Liver fibrosis is reduced |
| Cenicriviroc | Tropifexor | II | NCT03517540 | Liver fat deposition and fibrosis are reduced | ||
| Maraviroc | NA | Preclinical | [239] | Liver fat deposition and fibrosis are reduced | ||
| CXCR2 antagonist | Block CXCL1-CXCR2 axis and inhibit neutrophils and macrophages recruitment | RS10289 | NA | Preclinical | [240] | Inflammatory responses, liver steatosis and liver damage are reduced |
| RS504393 | NA | Preclinical | [241] | Inflammatory responses, liver steatosis and liver damage are reduced | ||
| THR-β agonist | Promote fatty acid oxidation and reduce liver fat accumulation by activating THR-β | Resmetirom | NA | III | NCT05500222 | Liver fat deposition and fibrosis are reduced |
| ASC41 | NA | I | NCT04686994 | The drug was well-tolerated and no safety concerns were found | ||
| TERN-501 | NA | II | NCT05415722 | Liver fat deposition and fibrosis are reduced | ||
| TERN-501 | TERN-101 | II | NCT05415722 | Liver fat deposition and fibrosis are reduced | ||
| HSK31679 | NA | II | NCT06168383 | Ongoing | ||
| VK2809 | NA | II | NCT02927184 | Cholesterol and liver fibrosis are reduced | ||
| Kylo-0603 | NA | I | NCT06365580 | Ongoing | ||
| ECC4703 | NA | I | NCT05552274 | Ongoing | ||
| ALG-055009 | NA | I | NCT05090111 | Ongoing | ||
| IL-6 receptor antagonist | Inhibit IL-6-mediated signalling and attenuate inflammatory response and injury in hepatocytes by blocking the IL-6 receptor | Tocilizumab | NA | Preclinical | [242] | Liver fat deposition and fibrosis are reduced |
| NE inhibitor | Reduce hepatic inflammation and early hepatic fibrosis by inhibiting NE activity and ameliorate early inflammation in MASLD-HCC | Sivelestat | NA | Preclinical | [71, 223] | Insulin sensitivity is improved, inflammatory responses, liver steatosis and liver damage are reduced |
| Angiotensin II receptor antagonist | Inhibit TGF-β signalling pathway and reduce hepatic fibrosis, thereby ameliorating hepatic infiltration of CD8+ T cells | Losartan | NA | II | NCT00699036 | Ongoing |
| mTOR inhibitor | Regulate Th17/Treg cells balance and enhance the immunosuppressive function of Treg cells, thereby attenuating MASH-associated inflammation and fibrosis | Rapamycin | NA | Preclinical | [243] | Liver fat deposition and fibrosis are reduced |
| AZD2014 | NA | Preclinical | [244, 245] | Liver fat deposition is reduced, insulin sensitivity is improved, but there are some side effects (hyperglycemia and hyperlipidaemia) | ||
| Everolimus | NA | Preclinical | [246] | Adipose synthesis and inflammatory responses are reduced | ||
| Antiplatelet drug | Inhibit platelet activation and aggregation by irreversibly inhibiting cyclooxygenase (COX-1) and reduce thromboxane A2 (TXA2) production | Aspirin | NA | I/II | NCT04031729 | Liver fat deposition is reduced |
| A phosphodiesterase inhibitor that works by inhibiting platelet aggregation and promoting blood vessel dilation | Cilostazol | NA | I/II | NCT04761848 | Ongoing | |
| Inhibit P2Y12 receptor on platelets and block ADP-mediated platelet activation | Clopidogrel | NA | Preclinical | [247] | Platelet activation and hepatocyte inflammation are reduced | |
| FXR/TGR5 agonist | Activate FXR, regulate the synthesis, secretion and reabsorption of bile acids, which helps to reduce liver fat accumulation | Obeticholic Acid | NA | II | NCT01265498 | Liver fibrosis is reduced |
| Obeticholic Acid | Atorvastatin | II | NCT02633956 | LDLc and liver fibrosis are reduced | ||
| Cilofexor | NA | II | NCT03987074 | Liver fibrosis is reduced | ||
| Cilofexor | Semaglutide& Firsocostat | II | NCT03987074 | Liver fibrosis is reduced | ||
| EDP-305 | NA | II | NCT03421431, [248] | ALT level is elevated, and liver fat deposition is reduced | ||
| Tropifexor | NA | Preclinical | [249] | ALT level is elevated, and liver fat deposition is reduced | ||
| Tropifexor | Cenicriviroc | II | NCT03517540 | Liver fat deposition and fibrosis are reduced | ||
| MET409 | NA | II | NCT04702490 | Liver fat deposition is reduced | ||
| TERN-101 | NA | II | NCT04328077 | Liver fat deposition is reduced | ||
| TERN-101 | TERN-501 | II | NCT05415722 | Liver fat deposition and fibrosis are reduced | ||
| Activate TGR5, regulate the synthesis, secretion and reabsorption of bile acids, which helps to reduce liver fat accumulation | INT-777 | NA | Preclinical | [250] | Inflammatory responses, liver steatosis and liver damage are reduced | |
| FXR/TGR5 double agonist | INT-767 | NA | Preclinical | [250] | Inflammatory responses, liver steatosis and liver damage are reduced | |
| Probiotics/prebiotics/synbiotics | Regulate intestinal flora, reduce the abundance of harmful bacteria, strengthen intestinal barrier function, and reduce inflammatory response | Lactobacillus | Bifidobacterium | NA | NCT03467282,[251] | Ongoing, but former study has shown reduced ALT and AST levels and improved liver steatosis |
| Bifidobacterium | Lactobacillus | NA | NCT03467282,[252] | Ongoing | ||
| VSL#3 | NA | I/II | NCT03511365 | Obvious benefit was not observed | ||
| Oligofructose | NA | NA | NCT02568605 | Liver fibrosis is reduced | ||
| Oligofructose-enriched inuli | NA | NA | NCT03184376 | Liver fibrosis is reduced | ||
| GLP-1 agonist/GLP-1 receptor agonist | Activate PI3K/Akt signalling pathway and inhibit NF-κB signalling pathway, improve insulin sensitivity and prevent hepatocytes apoptosis | Liraglutide | NA | II | NCT01237119 | ALT level is elevated, and liver fat deposition is reduced |
| Tirzepatide | NA | II | NCT04166773 | Liver fibrosis is reduced | ||
| Retatrutide | NA | [253] | Liver fat deposition is reduced | |||
| Exenatide | NA | II/III | NCT00650546 | Liver fat deposition is reduced | ||
| Efinopegdutide | NA | I | NCT06052566 | Ongoing | ||
| Semaglutide | NA | II | NCT02970942 | Liver fibrosis is reduced | ||
| Semaglutide | Cilofexor | II | NCT04971785 | Liver fibrosis is reduced | ||
| Semaglutide | Firsocostat | II | NCT04971785 | Liver fibrosis is reduced | ||
| Semaglutide | Firsocostat&Cilofexor | II | NCT03987074 | Liver fibrosis is reduced | ||
| Semaglutide | NNC0194-0499 | I | NCT05766709 | Ongoing | ||
| Cotadutide | NA | II | NCT04019561 | Liver fat deposition is reduced | ||
| HM15211 | NA | I | NCT03744182 | The drug was well-tolerated and no safety concerns were found | ||
| ACC inhibitor | Inhibit acetyl coenzyme A carboxylase (ACC) and reduce fat synthesis | Firsocostat | NA | II | NCT02856555 | Liver fat deposition is reduced |
| Firsocostat | Semaglutide | II | NCT04971785 | Liver fat deposition and fibrosis are reduced | ||
| Firsocostat | Semaglutide&Cilofexor | II | NCT03987074 | Liver fat deposition and fibrosis are reduced | ||
| PF-05221304 | NA | II | NCT03248882 | Liver fat deposition is reduced | ||
| PF-05221304 | PF-06865571 | II | NCT03776175 | Liver fat deposition is reduced | ||
| WZ66 | NA | Preclinical | [254] | Liver fat deposition is reduced | ||
| FASN inhibitors | Inhibit fat synthase (FASN) and reduce Th17 cells differentiation, fat synthesis and pro-inflammatory effects | Denifanstat (TVB-2640) | NA | III | NCT04906421 | Liver fibrosis is reduced |
| TVB-3664 | NA | Preclinical | [255] | Triglyceride levels and liver fat are decreased | ||
| FT-4101 | NA | Preclinical | [255] | Liver fat deposition and fibrosis are reduced | ||
| Caspase inhibitor | Inhibit the activity of caspase enzyme, reduce apoptosis and inflammation | Emricasan | NA | II | NCT02686762 | Fail to improve liver histology in patients with MASH fibrosis despite target engagement and may have worsened fibrosis and ballooning |
| GS-9450 | NA | II | NCT00740610 | Prevents apoptosis | ||
| VX-166 | NA | Preclinical | [256] | Rate of apoptosis and inflammatory factor levels in hepatocytes are reduced | ||
| Gal-3 inhibitors | Inhibit galactoglucan-3 (Gal-3) and reduce inflammation and fibrosis | Belapectin (GR-MD-02) | NA | II/III | NCT04365868 | Liver histology is improved, and oesophageal varices are prevented |
| GM-CT-01 | NA | Preclinical | [257] | Inflammatory responses and liver fibrosis are reduced | ||
| GB1211 | NA | I | NCT03809052 | The drug was well tolerated and no safety concerns were found | ||
| LOXL2 inhibitor | Inhibit lysyl oxidase-like protein-2 (LOXL2) and reduce collagen cross-linking and fibrosis | Simtuzumab | NA | II | NCT01672866 | Failed to achieve the desired effect |
| Solithromycin | NA | II | NCT02510599 | Liver fibrosis is reduced | ||
| ASK1 inhibitor | Inhibit apoptosis signaling regulation kinase 1 (ASK1) and reduce apoptosis and fibrosis | Selonsertib | NA | III | NCT03053050 | Liver fibrosis is reduced |
| SRT-015 | NA | I | NCT04887038 | Ongoing | ||
| DGAT2 inhibitor | Inhibit diacylglycerol acyltransferase 2 (DGAT2) and reduce triacylglycerol incorporation in the liver | ION224 | NA | II | NCT04932512 | Liver fat deposition and fibrosis are reduced |
The ongoing elucidation of innate and adaptive immune dynamics within the MASLD-HCC microenvironment provides a mechanistic rationale for modulating these alterations to mitigate hepatocarcinogenesis and enhance immunotherapy efficacy. CXCR2 inhibitors can effectively improve the response of MASLD-HCC to PD-1 therapy by reducing neutrophil infiltration and ROS production [77, 220]. Anti-CD122 antibody treatment can decrease CD44+CXCR6+PD-1+CD8+ T cells, thus restoring CD8+ T cells function in MASLD and preventing HCC progression [221]. CXCR6+CD8+ T cells activity and function could be modulated therapeutically by targeting IL-15 or FOXO1 [94]. In the context of MASLD-HCC immunotherapy, metformin enhances CD8+ T cells activity and motility, likely by augmenting mitochondrial mass and promoting mitochondrial activation [94]. Furthermore, targeted therapy against activated macrophage subpopulations represents a potential strategy. In murine HCC models, targeting CCR2 effectively suppresses tumor growth and metastasis through limiting TAM infiltration and enhancing CD8+ T cell-mediated antitumor response [222]. Besides, targeting the YTHDF1-EZH2-IL-6 signaling axis prevents the recruitment and activation of MDSCs, which could potentially enhance anti-PD-1 efficacy [77]. Degradation of NETs by inhibiting their formation or function, e.g., using PAD4 inhibitors or DNase, can help reduce Tregs [223]. Employing mitochondria-targeted antioxidants or mitochondrial biogenesis promoters could also help restore the normal metabolic state of CD4+ T cells, thereby reducing their pro-inflammatory effects and preventing tumor progression [224, 225]. In addition, targeting the Wnt/β-catenin pathway with ICG001—a small-molecule inhibitor-reversed immune—excluded TME phenotypes in CTNNB1-mutant MASLD-HCC models. This intervention promoted robust CD8+ T cells infiltration and elevated M1/M2 macrophage ratios, indicating restored anti-tumor immunity. Therefore, using ICG001 to reprogram the immune microenvironment toward a pro-inflammatory phenotype may effectively improve the anti-tumor effect of ICIs in MASLD-HCC [101]. Targeting IL-21R signaling also has therapeutic potential by reducing the generation of IgA+ cells [149]. Since administration of A. muciniphila has been shown to decrease body weight, ameliorate IR in obese individuals and restore the efficacy of PD-1-based immunotherapy in cancer patients by increasing CD4+ T cells infiltration in tumors, it is reasonable to speculate that A. muciniphila could be used to improve the effect of immune therapy in MASLD-HCC [226, 227]. While Tregs promote immunotolerance and thus SCFAs might be considered a potential negative factor in cancer immunotherapy, SCFAs have been shown to enhance the anti-tumor activity of CTLs and the efficacy of CAR T cells in syngeneic murine melanoma and pancreatic cancer models [101]. Additional studies are required to elucidate how gut microbiota metabolite-regulated immune microenvironments influence immunotherapy efficacy (Table 2).
Potential therapeutic targets/strategies addressing the mechanisms of immunotherapy resistance in MASLD-HCC
| Potential therapeutic targets/strategy | Mechanism | Cancer type | Reference | Specific Drugs |
|---|---|---|---|---|
| Akkermansia muciniphila | Increase the infiltration of CD4+ T cells | MASLD-HCC | [226, 227] | NA |
| Anti-CD122 antibody | Decrease the amount of CD44+CXCR6+PD-1+CD8+ T cells | MASLD-HCC | [221] | NA |
| CCR2 inhibitor | Reduce the infiltration of TAMs and reinvigorate the antitumor activity of CD8+ T cells | HCC | [222] | NA |
| CXCR2 inhibitor | Reducing CXCR2+ neutrophils infiltration and ROS production | MASLD-HCC | [77, 220] | AZD5069 |
| SCFAs | Decrease the amount of SCFAs to reduce Tregs and improve CD8+ T cells responses | MASLD-HCC | NA | NA |
| IL-21R signalling blockade | Decrease the amount of IgA+ cells and improve CD8+ T cells responses | MASLD-HCC | [149] | NA |
| Modulate IL-15 or FOXO1 | Alter the activity and function of CXCR6+CD8+ T cells | MASLD-HCC | [94] | NA |
| PAD4 inhibitor/Dnase | Degradate of NETs by inhibiting their formation or function and reducing Tregs | MASLD-HCC | [223] | NA |
| CTNNB1 mutation | Inhibit Wnt/β-catenin pathway and reprogram the immune microenvironment towards a pro-inflammatory phenotype | HCC | [100] | ICG001 |
| YTHDF1-EZH2-IL-6 signaling axis | Decrease the expression of YTHDF and recruitment of MDSCs | MASLD-HCC | [54] | LNP-siRNA |
| Metformin | Increase mitochondrial mass and activation of CD8+ T cells | MASLD-HCC | [94] | Metformin |
Currently, despite some success achieved in preclinical studies targeting specific mechanisms of MASLD-HCC, evaluating specific drugs in clinical trials remains limited. Some drugs are undergoing animal testing. Combining the small molecule CXCR2 inhibitor AZD5069 with anti-PD-1 monoclonal antibody therapy significantly reduces tumor burden and extends survival in a MASLD-HCC mouse model [77]. Lipid nanoparticles (LNP)-encapsulated siRNA therapy is an FDA-approved approach for clinical use, and LNP-siRNA or YTHDF1 knockdown in combination with anti-PD-1 therapy has been proven to significantly increase the sensitivity of MASLD-HCC tumors to immunotherapy in mice models [54]. Combined metformin and anti-PD-1 therapy also demonstrated good efficacy against MASLD-HCC [94]. The efficacy of antiplatelet agents, such as aspirin, and certain antifibrotic drugs has been demonstrated in HCC of other etiologies, including viral-related HCC. However, the therapeutic outcomes of these agents in MASLD-HCC remain inconclusive and warrant further investigation. Consequently, therapeutic development targeting these pathways exhibits significant promise in both preclinical and clinical settings. Prioritizing investigation of the aforementioned targets and agents represents a strategic approach to enhance immunotherapy efficacy and advance curative strategies for MASLD-HCC.
Conclusion and Future Perspectives
MASLD critically drives HCC development by progressively remodeling the hepatic immune microenvironment. This remodeling occurs throughout the disease spectrum, from steatosis to steatohepatitis, fibrosis, and ultimately HCC, where shifts in the metabolic landscape alter immune cell phenotypes/function and gut microbial communities. These changes, in turn, influence MASLD progression, HCC development, and immunotherapy efficacy. The metabolic alterations in MASLD are multifaceted, extending beyond the widely recognized dysregulated lipid metabolism to encompass pivotal alterations in ammonia and glutamine handling [228]. Clinical evidence indicates elevated systemic ammonia levels and progressive downregulation of glutamine synthetase in MASH patients compared to simple steatosis [228]. Notably, enhanced glutamine catabolism—a hallmark metabolic adaptation in cancer—manifests in HCC through overexpression of glutaminase 1 (GLS1), which catalyzes the conversion of glutamine to glutamate. GLS1 inhibition attenuates tumor proliferation and suppresses epithelial-mesenchymal transition (EMT) [229]. Critically, GLS1 upregulation correlates with advanced clinicopathological features and stemness phenotypes, mechanistically driven by ROS-mediated activation of Wnt/β-catenin signaling that sustains cancer stemness [230]. Parallel investigations reveal that sustained hyperammonemia promotes HSC activation and fibrogenesis in MASLD models. Aberrant GLS1 induction exacerbates oxidative stress, impairs very-low-density lipoprotein (VLDL) particle assembly, and ultimately potentiates hepatocyte lipid accumulation and MASH progression [231]. Novel diagnostic strategies employing dynamic monitoring of glutamine flux through GLS expression patterns establish its potential as a noninvasive biomarker for detecting hepatic malignancies [232].
This review synthesizes alterations in immune cell dynamics and gut microbiota composition during MASH and MASLD-HCC pathogenesis. Notably, mechanistic insights into the MASH-HCC transition and immunotherapy response patterns in this patient population remain inadequately explored. First, existing MASLD models in mice cannot effectively mimic the pathophysiological signature of human MASLD. Second, consistent conclusions are difficult to obtain using the numerous different rodent experimental models of MASLD and various HCC models in the MASH context (Table 3). Comparative analysis of MASLD rodent models is complicated by their differential recapitulation of human disease pathophysiology [233]. Furthermore, not all cases of MASH progress to liver tumors, and many studies on MASH and MASLD do not adequately address pathogenesis and treatment of MASLD-HCC. This lack of focus limits our understanding of the immune microenvironment and the treatment options for MASLD-HCC. Therefore, it is essential to conduct more studies and develop more relevant animal models for MASH progression to HCC in the future.
Roles of different immune cells in different MASLD-HCC models
| Cell subset | Mouse Models | Mechanism | Reference |
|---|---|---|---|
| Macrophages | DEN + HFD | MyD88 in myoblasts enhances MASLD-HCC development by promoting M2 macrophages polarization | [52] |
| Myeloid-Lineage-Specific Heterozygous Deletion of Ncoa5 mice | NCOA5 deficiency in macrophages as a key factor in the transition of MASH to HCC | [22, 53] | |
| DEN + CDA-HFD | The loss of NRG4 induces TAM-like macrophages and exhausted cytotoxic CD8+ T cells in MASLD-HCC | [21] | |
| Neutrophils | Streptozotocin + HFD | NETs regulate the OXPHOS of naïve CD4+ T cells, drive their differentiation into Tregs | [24, 60] |
| Tumor cells + WD or DEN + ALIOS | Pro-tumorigenic cytokines and immunosuppressive molecules secreted by CXCR2+ neutrophils | [77] | |
| Dendritic cells | Tumor cells + WD or DEN + ALIOS | XCR1+ cDC1 mediate cDC1 and CD8 T cells interactions | [77] |
| CD8+ cytotoxic T cells | Spatial transcriptomics | The infiltration is diminished within tumor regions | [100] |
| CD-HFD | Induce liver damage, upregulate exhausted markers and promoting HCC, auto-aggressive killing hepatocytes | [19, 94, 96] | |
| DEN + CD-HFD or DEN + HFHC | YTHDF1 suppresses cytotoxic CD8+ T cells function by enhancing the secretion of IL-6 | [54] | |
| Spatial transcriptomics | CD8+PD-1+ T cells inducible T cells ICOS+, MDSCs, and tumor-TAMs | [100] | |
| MCD/CDAA/WD | Reduce cell motility, impaire metabolic fitness | [25, 94] | |
| B cells | HFD-fed MUP-uPA mice | IgA+ cells suppress CD8+ T cells, produce the immunosuppressive cytokine IL-10 | [27] |
| Platelets | CD-HFD | Recruit of CD8+ T cells and NKT cells, drive HCC | [168] |
| Tumor cells + MCD or DEN + CDAA or CCl4 + WD | Upregulate the accumulation of CD8+ T cells, inhibiting the growth and metastasis of HCC in MASH | [177] |
The response rate to immunotherapy is poor in MASLD-HCC. ICIs therapy seems to have encountered a significant setback in treating MASLD-HCC patients. Activation of MDSCs, enriched CXCR2+ neutrophils, Tregs, and IgA+ cells, diminished CD8+ T cells recruitment and functional impairment, increased specific pro-cancerous CD8+ T cells subsets, accumulated CTNNB1 mutations and elevated SCFAs may collectively contribute to this phenomenon. Research on gut dysbiosis-driven MASLD pathogenesis has elucidated key mechanisms centered on intestinal barrier compromise, which permits hepatic translocation of microbial components and metabolites (e.g., SCFAs, bile acids). These hepatotropic signals orchestrate immunometabolic reprogramming via upregulation of CXCR6+ NKT cells, suppressing M1 macrophages polarization, expanding Tregs, and attenuating CD8+ T cells effector functions. Compelling preclinical evidence demonstrates that microbiota—directed interventions-including fecal microbiota transplantation, probiotics, prebiotics, and synthetic biotics—effectively restore enteric homeostasis and ameliorate metabolic dysregulation and inflammation in MASLD models.
Advances in multi-omics sequencing have propelled tumor precision medicine into clinical focus, utilizing molecular and genetic profiling to tailor cancer therapies based on individual tumor characteristics. Methodologically, integrative analysis of tumor transcriptomes and patient prognoses has yielded the SAHR (Score of Aggregated Hazard Ratio) model—a universal quantitative metric for assessing clinical aggressiveness. Applying this framework to HCC revealed three molecular subtypes with distinct prognostic outcomes among Asian populations, each exhibiting multifaceted molecular disparities [234]. Notably in HBV-related HCC, proteogenomic profiling of 159 patients through integrated multi-omics analysis (encompassing somatic mutations, copy number alterations, transcriptomics, proteomics, and phosphoproteomics) delineated three tumor subclusters with characteristic pathway activation patterns. This approach further identified pyrroline-5-carboxylate reductase 2 (PYCR2) and alcohol dehydrogenase 1A (ADH1A) as robust prognostic biomarkers, with mechanistic studies confirming their roles in modulating pro-tumorigenic metabolic pathways [235]. However, such comprehensive multi-omics research on MASLD-HCC remains scarce. Spatial mapping at single-cell resolution reveals a previously unappreciated heterogeneity in immune cell distribution within MASLD-HCC. Contrary to prior models, immune cells are most abundant in adjacent non-tumor tissue, diminishing progressively towards the tumor core. Furthermore, spatial interactions shift from T cell networks towards immunosuppressive connections involving MDSCs and TAMs, potentially disrupting antitumor immunity [100]. Hence, further exploration of this heterogeneity in the distribution and composition of immune cells is warranted and may provide new directions for understanding immunotherapy resistance in MASLD-HCC. Every new mechanistic discovery may become the key to unlocking potential solutions to complex problems.
Elucidating dynamic alterations in immune cell populations and gut microbiota during MASLD-HCC progression not only enhances our understanding of the disease process but also provides a foundation for solving therapeutic dilemma and identifying new drugs and targets. Nowadays, with the underlying reason for immunotherapy resistance and immunometabolic changes in MASLD-HCC gradually being revealed, new targets have already been floated. However, specific drugs undergoing evaluation in clinical trials and validated in robust animal models are still limited. The lack of etiology-based classification of treatments also contributes to the scarcity of targeted research on MASLD-HCC. Future studies must focus on addressing existing research gaps in these areas, exploring additional reasons for immunotherapy resistance, identifying novel targets, and developing effective drugs to achieve a cure for MASLD-HCC ultimately. With the rising prevalence of MASLD-HCC continuing unabated, clarifying the mechanisms linking the metabolic microenvironment and immune responses in MASLD-HCC will be crucial not only to enhance treatment efficacy but also to minimize side effects and enable personalized treatment for patients with this condition.
Abbreviations
ACC2: acetyl-CoA carboxylase 2; ADAM17: a disintegrin and metalloproteinase 17; ADH1A: alcohol dehydrogenase 1A; APOE: apolipoprotein E; ATP: adenosine 5'-triphosphate; BAFF: B cell-activating factor; Blimp-1: B lymphocyte-induced maturation protein 1; Bregs: regulatory B cells; CCL2: C-C motif chemokine ligand 2; CCR2: C-C motif chemokine receptor 2; CDAA: choline-deficient and amino acid-defined diet; cDCs: conventional dendritic cells; cDC1: conventional type I dendritic cells; cDC2: conventional type II dendritic cells; CD-HFD: choline-deficient, high-fat diet; CPT: carnitine palmitoyltransferase; CELA1: chymotrypsin-like elastase family member 1; CTLA4: anti-cytotoxic T-lymphocyte antigen 4; CTNNB1: catenin beta 1; CXCL10: C-X-C motif chemokine ligand 10; CX3CR1: C-X3-C motif chemokine receptor 1; CXCR6: C-X-C motif chemokine receptor 6; DCs: dendritic cells; EmKC: embryo-derived Kupffer cells; EMT: epithelial-mesenchymal transition; Fas: factor associated with suicide; FasL: Fas ligand; FFAR: free fatty acid receptor; FOXO1: Forkhead Box O1; FXR: farnesoid X receptor; GPIbα: glycoprotein Ibα; GZMB: granzyme B; GSDMD: gasdermin D; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; HFD: high-fat diet; HIF: hypoxia-inducible factor; HNPs: human neutrophil peptides; HSCs: hepatic stellate cells; ihTh17: inflammatory hepatic CXCR3+ Th17; ICAM: intercellular cell adhesion molecule; ICIs: immune checkpoint inhibitors; ICOS: inducible T cell costimulator; IFN-γ: interferon gamma; IgA: immunoglobulin-A; IgG: immunoglobulin-G; IgM: immunoglobulin-M; IR: insulin resistance; IL-1β: interleukin 1 beta; IPA: indole propionic acid; LCs: Langerhans cells; LCN: lipocalin; LD: lipid droplet; LDLR: low density lipoprotein receptor; LFA: leukocyte function-associated antigen; LNP: lipid nanoparticles; LPS: lipopolysaccharide; LTA: lipoteichoic acid; mTOR: mammalian target of rapamycin; m6A: N6-methyladenosine; MAdCAM-1: mucosal addressin cell adhesion molecule-1; MASH: metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease; MCP-1: monocyte chemoattractant protein-1; MCD: methionine-choline-deficient diet; MDSCs: myeloid-derived suppressor cells; MHC: major histocompatibility complex; MPO: myeloperoxidase; MPV: mean platelet volumes; MR: magnetic resonance; MT: metallothionein; MUFAs: monounsaturated fatty acids; MyD88: myeloid differentiation primary response 88; NAFLD: non-alcoholic fatty liver disease; MASLD-HCC: metabolic dysfunction-associated steatotic liver disease-related hepatocellular carcinoma; MAMPs: microbial-associated molecular patterns; NcDase: neutral ceramidase; NCOA5: nuclear receptor coactivator 5; NKT: natural killer T cells; NRG4: neuregulin 4; NE: neutrophil elastase; NETs: neutrophil extracellular traps; OSE: oxidative-stress-derived epitopes; OxLDL: oxidized low-density lipoproteins; OXPHOS: oxidative phosphorylation; pDCs: plasmacytoid dendritic cells; pEVs: platelet-derived extracellular vesicles; PD-1: programmed death receptor-1; PD-L1: programmed death-ligand 1; PUFAs: polyunsaturated fatty acids; PRRs: pattern recognition receptors; PDGF: platelet-derived growth factor; PDW: platelet distribution width; PMPs: platelet-derived microparticles; PYCR2: pyrroline-5-carboxylate reductase 2; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; SAHR: Score of Aggregated Hazard Ratio; SCD: stearoyl-CoA desaturase; SCFAs: short-chain fatty acids; STARD1: steroidogenic acute regulatory protein 1; STAT3: signal transducer and activator of transcription 3; TAMs: tumor-associated macrophages; TG: triglyceride; TGF-β: transforming growth factor beta; TGR5: G-protein-coupled bile acid receptor 5; Th: T helper; Tregs: regulatory T cells; TREM2: triggering receptor expressed on myeloid cells 2; Trm: tissue-resident memory T cells; TKIs: tyrosine kinase inhibitors; TLRs: Toll-like receptors; TLS: tertiary lymphoid structures; TME: tumor microenvironment; TNF: tumor necrosis factor; TNFRSF19: tumor necrosis factor receptor superfamily 19; XCR1: X-C receptor 1; YTHDF1: YT521-B homology (YTH) m6A RNA-binding protein 1 (YTHDF1); WD: western diet; VLDL: very-low-density lipoprotein.
Acknowledgements
We thank several professional English editors (American and Chinese Journal Experts) for assistance in improving the quality of the language. We thank Professor Huifang Liang and Wanguang Zhang for their valuable scientific advice and substantial support.
Funding
This work was supported by The National Natural Science Foundation of China (No. 82373054 and 82173319 to Wanguang Zhang).
Author contributions
JJ, KC, and MRC gathered the relevant papers and made significant contributions to the manuscript. JJ and MRC created the figures and tables. KC wrote the initial draft of the paper and made critical revisions for important intellectual content. HFL and WGZ initiated the study and revised the manuscript. WGZ provided the final approval.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73-84
2. Yip TC, Lee HW, Chan WK, Wong GL, Wong VW. Asian perspective on NAFLD-associated HCC. J Hepatol. 2022;76:726-34
3. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18:223-38
4. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F. et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79:1542-56
5. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519-23
6. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62:S47-64
7. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301-14
8. Collaborators GBDHB. Global, regional, and national burden of hepatitis B, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol Hepatol. 2022;7:796-829
9. Maucort-Boulch D, de Martel C, Franceschi S, Plummer M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int J Cancer. 2018;142:2471-7
10. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J. et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 2018;69:896-904
11. Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, Duseja A. et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol. 2019;17:748-55 e3
12. Yahoo N, Dudek M, Knolle P, Heikenwalder M. Role of immune responses in the development of NAFLD-associated liver cancer and prospects for therapeutic modulation. J Hepatol. 2023;79:538-51
13. Llovet JM, Willoughby CE, Singal AG, Greten TF, Heikenwalder M, El-Serag HB. et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat Rev Gastroenterol Hepatol. 2023;20:487-503
14. Yang C, Zhang H, Zhang L, Zhu AX, Bernards R, Qin W. et al. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2023;20:203-22
15. Weber M, Lam M, Chiesa C, Konijnenberg M, Cremonesi M, Flamen P. et al. EANM procedure guideline for the treatment of liver cancer and liver metastases with intra-arterial radioactive compounds. Eur J Nucl Med Mol Imaging. 2022;49:1682-99
16. Foerster F, Gairing SJ, Muller L, Galle PR. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J Hepatol. 2022;76:446-57
17. Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA. et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology. 2018;155:1828-37.e2
18. Mohamad B, Shah V, Onyshchenko M, Elshamy M, Aucejo F, Lopez R. et al. Characterization of hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease (NAFLD) patients without cirrhosis. Hepatol Int. 2016;10:632-9
19. Pfister D, Nunez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M. et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450-6
20. Haber PK, Puigvehi M, Castet F, Lourdusamy V, Montal R, Tabrizian P. et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002-2020). Gastroenterology. 2021;161:879-98
21. Zhang P, Chen Z, Kuang H, Liu T, Zhu J, Zhou L. et al. Neuregulin 4 suppresses NASH-HCC development by restraining tumor-prone liver microenvironment. Cell Metabolism. 2022;34:1359-76.e7
22. Zhang Y, Luo Y, Liu X, Kiupel M, Li A, Wang H. et al. NCOA5 Haploinsufficiency in Myeloid-Lineage Cells Sufficiently Causes Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol. 2024;17:1-27
23. Kohlhepp MS, Liu H, Tacke F, Guillot A. The contradictory roles of macrophages in non-alcoholic fatty liver disease and primary liver cancer-Challenges and opportunities. Front Mol Biosci. 2023;10:1129831
24. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z. et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. 2021;75:1271-83
25. Lujambio A, Sarobe P. Metformin keeps CD8(+) T cells active and moving in NASH-HCC immunotherapy. J Hepatol. 2022;77:593-5
26. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P. et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253-7
27. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017;551:340-5
28. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. 2016;13:412-25
29. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. 2022;55:1515-29
30. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306-21
31. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090-6
32. Vonderlin J, Chavakis T, Sieweke M, Tacke F. The Multifaceted Roles of Macrophages in NAFLD Pathogenesis. Cell Mol Gastroenterol Hepatol. 2023;15:1311-24
33. Sasaki K, Rooge S, Gunewardena S, Hintz JA, Ghosh P, Pulido Ruiz IA. et al. Kupffer cell diversity maintains liver function in alcohol-associated liver disease. Hepatology. 2025;81:870-87
34. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J. et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16:145-59
35. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H. et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity. 2020;52:1057-74 e7
36. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512-8
37. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450-62
38. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M. et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67:1270-83
39. Horn CL, Morales AL, Savard C, Farrell GC, Ioannou GN. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol Commun. 2022;6:12-35
40. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313-26
41. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem. 2012;287:40161-72
42. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547-51
43. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gelineau A. et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity. 2020;53:627-40 e5
44. Wang Q, Zhou H, Bu Q, Wei S, Li L, Zhou J. et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J Hepatol. 2022;77:312-25
45. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z. et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020;31:406-21 e7
46. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW. et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021;34:108626
47. Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S. et al. Hepatic crown-like structure: a unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS One. 2013;8:e82163
48. Wang X, He Q, Zhou C, Xu Y, Liu D, Fujiwara N. et al. Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity. 2023;56:58-77 e11
49. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9-22
50. Wang X, de Carvalho Ribeiro M, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A. et al. Macrophage-Specific Hypoxia-Inducible Factor-1alpha Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology. 2019;69:545-63
51. Jeelani I, Moon JS, da Cunha FF, Nasamran CA, Jeon S, Zhang X. et al. HIF-2α drives hepatic Kupffer cell death and proinflammatory recruited macrophage activation in nonalcoholic steatohepatitis. Sci Transl Med. 2024;16:eadi0284
52. Liu Y, Chen H, Yan X, Zhang J, Deng Z, Huang M. et al. MyD88 in myofibroblasts enhances nonalcoholic fatty liver disease-related hepatocarcinogenesis via promoting macrophage M2 polarization. Cell Commun Signal. 2024;22:86
53. Liu Y, Feng GS. NCOA5 Deficiency in Macrophages Provokes NASH and HCC. Cell Mol Gastroenterol Hepatol. 2024;17:171-2
54. Wang L, Zhu L, Liang C, Huang X, Liu Z, Huo J. et al. Targeting N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J Hepatol. 2023;79:1185-200
55. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327-37
56. Cheng K, Cai N, Zhu J, Yang X, Liang H, Zhang W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun (Lond). 2022;42:1112-40
57. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459-89
58. Liew PX, Kubes P. The Neutrophil's Role During Health and Disease. Physiol Rev. 2019;99:1223-48
59. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159-75
60. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO. et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. 2018;68:1347-60
61. Cai J, Zhang XJ, Li H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology. 2019;70:1026-37
62. Nati M, Haddad D, Birkenfeld AL, Koch CA, Chavakis T, Chatzigeorgiou A. The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH). Rev Endocr Metab Disord. 2016;17:29-39
63. Hwang S, Yun H, Moon S, Cho YE, Gao B. Role of Neutrophils in the Pathogenesis of Nonalcoholic Steatohepatitis. Front Endocrinol (Lausanne). 2021;12:751802
64. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A. et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898-910
65. Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J. et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology. 2020;72:412-29
66. Cho YE, Kim Y, Kim SJ, Lee H, Hwang S. Overexpression of Interleukin-8 Promotes the Progression of Fatty Liver to Nonalcoholic Steatohepatitis in Mice. Int J Mol Sci. 2023 24
67. Liu J, Yu X, Ting HJ, Wang X, Xu S, Wang Y. et al. Myeloperoxidase-Sensitive T(1) and T(2) Switchable MR Imaging for Diagnosis of Nonalcoholic Steatohepatitis. ACS Nano. 2023;17:3324-33
68. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ. et al. Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid Redox Signal. 2015;23:1255-69
69. Jia W, Mao Y, Luo Q, Wu J, Guan Q. Targeting neutrophil elastase is a promising direction for future cancer treatment. Discov Oncol. 2024;15:167
70. Sun Z, Yang P. Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression. Lancet Oncol. 2004;5:182-90
71. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J. et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407-12
72. Mansuy-Aubert V, Zhou QL, Xie X, Gong Z, Huang JY, Khan AR. et al. Imbalance between neutrophil elastase and its inhibitor alpha1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013;17:534-48
73. Ibusuki R, Uto H, Arima S, Mawatari S, Setoguchi Y, Iwashita Y. et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int. 2013;33:1549-56
74. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS. et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-5
75. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134-47
76. Wu J, Zhang C, He T, Zhang S, Wang Y, Xie Z. et al. Polyunsaturated fatty acids drive neutrophil extracellular trap formation in nonalcoholic steatohepatitis. Eur J Pharmacol. 2023;945:175618
77. Leslie J, Mackey JBG, Jamieson T, Ramon-Gil E, Drake TM, Fercoq F. et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut. 2022;71:2093-106
78. Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y. et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol. 2016;65:988-97
79. He Y, Rodrigues RM, Wang X, Seo W, Ma J, Hwang S. et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J Clin Invest. 2021 131
80. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245-52
81. Howard CJ, Charleston B, Stephens SA, Sopp P, Hope JC. The role of dendritic cells in shaping the immune response. Anim Health Res Rev. 2004;5:191-5
82. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563-604
83. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU. et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14:571-8
84. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A. et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology. 2013;58:589-602
85. Mori T, Yoshio S, Kakazu E, Kanto T. Active role of the immune system in metabolic dysfunction-associated steatotic liver disease. Gastroenterol Rep (Oxf). 2024 12
86. Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, Fleury S. et al. Transcriptional Network Analysis Implicates Altered Hepatic Immune Function in NASH development and resolution. Nat Metab. 2019;1:604-14
87. Peiseler M, Tacke F. Inflammatory Mechanisms Underlying Nonalcoholic Steatohepatitis and the Transition to Hepatocellular Carcinoma. Cancers (Basel). 2021 13
88. Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B. et al. XCR1+ type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med. 2021;27:1043-54
89. Hess A, Gentile SD, Ben Saad A, Rahman RU, Habboub T, Pratt DS. et al. Single-cell transcriptomics stratifies organoid models of metabolic dysfunction-associated steatotic liver disease. EMBO J. 2023;42:e113898
90. Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T. et al. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol. 2017;66:1241-50
91. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ. et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550-7
92. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
93. Reig M, Forner A, Rimola J, Ferrer-Fabrega J, Burrel M, Garcia-Criado A. et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J Hepatol. 2022;76:681-93
94. Wabitsch S, McCallen JD, Kamenyeva O, Ruf B, McVey JC, Kabat J. et al. Metformin treatment rescues CD8(+) T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J Hepatol. 2022;77:748-60
95. Adams VR, Collins LB, Williams TI, Holmes J, Hess P, Atkins HM. et al. Myeloid cell MHC I expression drives CD8(+) T cell activation in nonalcoholic steatohepatitis. Front Immunol. 2023;14:1302006
96. Zhong X, Lv M, Ma M, Huang Q, Hu R, Li J. et al. State of CD8(+) T cells in progression from nonalcoholic steatohepatitis to hepatocellular carcinoma: From pathogenesis to immunotherapy. Biomed Pharmacother. 2023;165:115131
97. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K. et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549-64
98. Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P. et al. Hepatic Natural Killer T-cell and CD8+ T-cell Signatures in Mice with Nonalcoholic Steatohepatitis. Hepatol Commun. 2017;1:299-310
99. Gu L, Zhu Y, Lee M, Nguyen A, Ryujin NT, Huang JY. et al. Angiotensin II receptor inhibition ameliorates liver fibrosis and enhances hepatocellular carcinoma infiltration by effector T cells. Proc Natl Acad Sci U S A. 2023;120:e2300706120
100. Li M, Wang L, Cong L, Wong CC, Zhang X, Chen H. et al. Spatial proteomics of immune microenvironment in nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Hepatology. 2024;79:560-74
101. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A. et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat Commun. 2021;12:4077
102. McVey JC, Green BL, Ruf B, McCallen JD, Wabitsch S, Subramanyam V. et al. NAFLD indirectly impairs antigen-specific CD8(+) T cell immunity against liver cancer in mice. iScience. 2022;25:103847
103. Pan Y, Chen H, Zhang X, Liu W, Ding Y, Huang D. et al. METTL3 drives NAFLD-related hepatocellular carcinoma and is a therapeutic target for boosting immunotherapy. Cell Rep Med. 2023;4:101144
104. Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL. et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. 2021;184:4512-30 e22
105. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M. et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592:444-9
106. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557-69
107. Speiser DE, Chijioke O, Schaeuble K, Munz C. CD4(+) T cells in cancer. Nat Cancer. 2023;4:317-29
108. Richardson JR, Schollhorn A, Gouttefangeas C, Schuhmacher J. CD4+ T Cells: Multitasking Cells in the Duty of Cancer Immunotherapy. Cancers (Basel). 2021 13
109. Luo XY, Takahara T, Kawai K, Fujino M, Sugiyama T, Tsuneyama K. et al. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol. 2013;305:G891-9
110. Lu S, Wang Y, Liu J. Tumor necrosis factor-alpha signaling in nonalcoholic steatohepatitis and targeted therapies. J Genet Genomics. 2022;49:269-78
111. Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, Puri RK. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J Immunol. 2008;181:4656-65
112. Harley IT, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M. et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 2014;59:1830-9
113. Nie YJ, Wu SH, Xuan YH, Yan G. Role of IL-17 family cytokines in the progression of IPF from inflammation to fibrosis. Mil Med Res. 2022;9:21
114. Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-beta receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol. 2014;193:3925-33
115. Li J, Wu Z, Wu Y, Hu X, Yang J, Zhu D. et al. IL-22, a vital cytokine in autoimmune diseases. Clin Exp Immunol. 2024;218:242-63
116. Keir M, Yi Y, Lu T, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med. 2020;217:e20192195
117. Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, Luo J. et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis. 2018;9:620
118. Her Z, Tan JHL, Lim YS, Tan SY, Chan XY, Tan WWS. et al. CD4(+) T Cells Mediate the Development of Liver Fibrosis in High Fat Diet-Induced NAFLD in Humanized Mice. Front Immunol. 2020;11:580968
119. Rau M, Schilling A-K, Meertens J, Hering I, Weiss J, Jurowich C. et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. The Journal of Immunology. 2016;196:97-105
120. Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q. et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. 2011;166:281-90
121. Moreno-Fernandez ME, Giles DA, Oates JR, Chan CC, Damen M, Doll JR. et al. PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. 2021;33:1187-204 e9
122. Rai RP, Liu Y, Iyer SS, Liu S, Gupta B, Desai C. et al. Blocking integrin alpha(4)beta(7)-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J Hepatol. 2020;73:1013-22
123. Iglesias-Escudero M, Arias-Gonzalez N, Martinez-Caceres E. Regulatory cells and the effect of cancer immunotherapy. Mol Cancer. 2023;22:26
124. Riaz F, Wei P, Pan F. Fine-tuning of regulatory T cells is indispensable for the metabolic steatosis-related hepatocellular carcinoma: A review. Front Cell Dev Biol. 2022;10:949603
125. Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S. et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity (Silver Spring). 2013;21:461-8
126. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930-9
127. Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. 2007;46:1519-29
128. Savage TM, Fortson KT, de Los Santos-Alexis K, Oliveras-Alsina A, Rouanne M, Rae SS. et al. Amphiregulin from regulatory T cells promotes liver fibrosis and insulin resistance in non-alcoholic steatohepatitis. Immunity. 2024;57:303-18 e6
129. Behary J, Amorim N, Jiang XT, Raposo A, Gong L, McGovern E. et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. 2021;12:187
130. Yamagishi R, Kamachi F, Nakamura M, Yamazaki S, Kamiya T, Takasugi M. et al. Gasdermin D-mediated release of IL-33 from senescent hepatic stellate cells promotes obesity-associated hepatocellular carcinoma. Sci Immunol. 2022;7:eabl7209
131. Barrow F, Khan S, Wang H, Revelo XS. The Emerging Role of B Cells in the Pathogenesis of NAFLD. Hepatology. 2021;74:2277-86
132. Deng CJ, Lo TH, Chan KY, Li X, Wu MY, Xiang Z. et al. Role of B Lymphocytes in the Pathogenesis of NAFLD: A 2022 Update. Int J Mol Sci. 2022 23
133. R R Hardy KH. B cell development pathways. Annu Rev Immunol. 2001;19:595-621
134. Ma J, Wu Y, Ma L, Yang X, Zhang T, Song G. et al. A blueprint for tumor-infiltrating B cells across human cancers. Science. 2024;384:eadj4857
135. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol. 2020;17:81-92
136. Rosser EC, Mauri C. The emerging field of regulatory B cell immunometabolism. Cell Metab. 2021;33:1088-97
137. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607-12
138. Bruzzi S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A. et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med. 2018;124:249-59
139. van den Hoek AM, Verschuren L, Worms N, van Nieuwkoop A, de Ruiter C, Attema J. et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells. 2020 9
140. Kanemitsu-Okada K, Abe M, Nakamura Y, Miyake T, Watanabe T, Yoshida O. et al. Role of B Cell-Activating Factor in Fibrosis Progression in a Murine Model of Non-Alcoholic Steatohepatitis. Int J Mol Sci. 2023 24
141. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P. et al. Microbiota-Driven Activation of Intrahepatic B Cells Aggravates NASH Through Innate and Adaptive Signaling. Hepatology. 2021;74:704-22
142. Nakamura Y, Abe M, Kawasaki K, Miyake T, Watanabe T, Yoshida O. et al. Depletion of B cell-activating factor attenuates hepatic fat accumulation in a murine model of nonalcoholic fatty liver disease. Sci Rep. 2019;9:977
143. Kotsiliti E, Leone V, Schuehle S, Govaere O, Li H, Wolf MJ. et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J Hepatol. 2023;79:296-313
144. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, Chi YJ. et al. Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis. 2016;17:464-74
145. Thapa M, Chinnadurai R, Velazquez VM, Tedesco D, Elrod E, Han JH. et al. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology. 2015;61:2067-79
146. Shao Y, Lo CM, Ling CC, Liu XB, Ng KT, Chu AC. et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014;355:264-72
147. Karl M, Hasselwander S, Zhou Y, Reifenberg G, Kim YO, Park KS. et al. Dual roles of B lymphocytes in mouse models of diet-induced nonalcoholic fatty liver disease. Hepatology. 2022;76:1135-49
148. McPherson S, Henderson E, Burt AD, Day CP, Anstee QM. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J Hepatol. 2014;60:1055-62
149. Xie Y, Huang Y, Li ZY, Jiang W, Shi NX, Lu Y. et al. Interleukin-21 receptor signaling promotes metabolic dysfunction-associated steatohepatitis-driven hepatocellular carcinoma by inducing immunosuppressive IgA(+) B cells. Mol Cancer. 2024;23:95
150. Gu X, Sun R, Chen L, Chu S, Doll MA, Li X. et al. Neutral Ceramidase Mediates Nonalcoholic Steatohepatitis by Regulating Monounsaturated Fatty Acids and Gut IgA(+) B Cells. Hepatology. 2021;73:901-19
151. Albano E, Mottaran E, Vidali M, Reale E, Saksena S, Occhino G. et al. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. 2005;54:987-93
152. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C. et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886-97
153. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16:485-97
154. Nobili V, Parola M, Alisi A, Marra F, Piemonte F, Mombello C. et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int J Mol Med. 2010;26:471-6
155. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561-5
156. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549-55
157. Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556-60
158. Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR. et al. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell. 2019;179:1191-206 e21
159. Chauhan A, Adams DH, Watson SP, Lalor PF. Platelets: No longer bystanders in liver disease. Hepatology. 2016;64:1774-84
160. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264-74
161. Kurokawa T, Ohkohchi N. Platelets in liver disease, cancer and regeneration. World J Gastroenterol. 2017;23:3228-39
162. Mussbacher M, Brunnthaler L, Panhuber A, Starlinger P, Assinger A. Till Death Do Us Part-The Multifaceted Role of Platelets in Liver Diseases. Int J Mol Sci. 2021 22
163. Pal Chaudhary S, Reyes S, Chase ML, Govindan A, Zhao L, Luther J. et al. Resection of NAFLD/NASH-related Hepatocellular Carcinoma (HCC): Clinical Features and Outcomes Compared with HCC Due to Other Etiologies. Oncologist. 2023;28:341-50
164. Boccatonda A, Del Cane L, Marola L, D'Ardes D, Lessiani G, di Gregorio N. et al. Platelet, Antiplatelet Therapy and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Narrative Review. Life (Basel). 2024 14
165. Rotundo L, Persaud A, Feurdean M, Ahlawat S, Kim HS. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin Mol Hepatol. 2018;24:392-401
166. Corsonello A, Perticone F, Malara A, De Domenico D, Loddo S, Buemi M. et al. Leptin-dependent platelet aggregation in healthy, overweight and obese subjects. Int J Obes Relat Metab Disord. 2003;27:566-73
167. Zijlstra MK, Gampa A, Joseph N, Sonnenberg A, Fimmel CJ. Progressive changes in platelet counts and Fib-4 scores precede the diagnosis of advanced fibrosis in NASH patients. World J Hepatol. 2023;15:225-36
168. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E. et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25:641-55
169. Liao TL, Chen DY, Hsieh SL, Yang YY, Chen YM, Tang KT. et al. Platelet-derived mitochondria regulate lipid metabolism in nonalcoholic steatohepatitis through extracellular vesicles. Hepatology. 2024
170. Duran-Bertran J, Rusu EC, Barrientos-Riosalido A, Bertran L, Mahmoudian R, Aguilar C. et al. Platelet-associated biomarkers in nonalcoholic steatohepatitis: Insights from a female cohort with obesity. European Journal of Clinical Investigation. 2023 54
171. Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules.. or not? J Thromb Haemost. 2015;13:2141-51
172. Burnouf T, Goubran HA, Chou ML, Devos D, Radosevic M. Platelet microparticles: detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014;28:155-66
173. Taus F, Meneguzzi A, Castelli M, Minuz P. Platelet-Derived Extracellular Vesicles as Target of Antiplatelet Agents. What Is the Evidence? Front Pharmacol. 2019;10:1256
174. Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY. et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology. 2014;147:1378-92
175. Ye Q, Liu Y, Zhang G, Deng H, Wang X, Tuo L. et al. Deficiency of gluconeogenic enzyme PCK1 promotes metabolic-associated fatty liver disease through PI3K/AKT/PDGF axis activation in male mice. Nat Commun. 2023;14:1402
176. Ikeda N, Murata S, Maruyama T, Tamura T, Nozaki R, Kawasaki T. et al. Platelet-derived adenosine 5′-triphosphate suppresses activation of human hepatic stellate cell: In vitro study. Hepatology Research. 2011;42:91-102
177. Ma C, Fu Q, Diggs LP, McVey JC, McCallen J, Wabitsch S. et al. Platelets control liver tumor growth through CTNNB1-dependent CD40L release in NAFLD. Cancer Cell. 2022;40:986-98 e5
178. Pabst O, Hornef MW, Schaap FG, Cerovic V, Clavel T, Bruns T. Gut-liver axis: barriers and functional circuits. Nature Reviews Gastroenterology & Hepatology. 2023;20:447-61
179. Ohtani N, Kamiya T, Kawada N. Recent updates on the role of the gut-liver axis in the pathogenesis of NAFLD/NASH, HCC, and beyond. Hepatol Commun. 2023 7
180. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. 2020;72:558-77
181. Lang S, Schnabl B. Microbiota and Fatty Liver Disease-the Known, the Unknown, and the Future. Cell Host Microbe. 2020;28:233-44
182. Chopyk DM, Grakoui A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology. 2020;159:849-63
183. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A. et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718-23
184. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027-31
185. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J. et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol. 2020;17:279-97
186. Gierynska M, Szulc-Dabrowska L, Struzik J, Mielcarska MB, Gregorczyk-Zboroch KP. Integrity of the Intestinal Barrier: The Involvement of Epithelial Cells and Microbiota-A Mutual Relationship. Animals (Basel). 2022 12
187. Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L. et al. The Liver May Act as a Firewall Mediating Mutualism Between the Host and Its Gut Commensal Microbiota. Sci Transl Med. 2014;6:237ra66
188. Cai W, Qiu T, Hu W, Fang T. Changes in the intestinal microbiota of individuals with non-alcoholic fatty liver disease based on sequencing: An updated systematic review and meta-analysis. PLoS One. 2024;19:e0299946
189. Adorini L, Trauner M. FXR agonists in NASH treatment. J Hepatol. 2023;79:1317-31
190. Zou A, Magee N, Deng F, Lehn S, Zhong C, Zhang Y. Hepatocyte nuclear receptor SHP suppresses inflammation and fibrosis in a mouse model of nonalcoholic steatohepatitis. J Biol Chem. 2018;293:8656-71
191. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol Commun. 2020;4:478-92
192. Li T, Lin X, Shen B, Zhang W, Liu Y, Liu H. et al. Corrigendum: Akkermansia muciniphila suppressing nonalcoholic steatohepatitis associated tumorigenesis through CXCR6(+) natural killer T cells. Front Immunol. 2023;14:1297103
193. Niu H, Zhou M, Zogona D, Xing Z, Wu T, Chen R. et al. Akkermansia muciniphila: a potential candidate for ameliorating metabolic diseases. Front Immunol. 2024;15:1370658
194. Han Y, Ling Q, Wu L, Wang X, Wang Z, Chen J. et al. Akkermansia muciniphila inhibits nonalcoholic steatohepatitis by orchestrating TLR2-activated gammadeltaT17 cell and macrophage polarization. Gut Microbes. 2023;15:2221485
195. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K. et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225-35
196. Chu H, Duan Y, Yang L, Schnabl B. Small metabolites, possible big changes: a microbiota-centered view of non-alcoholic fatty liver disease. Gut. 2019;68:359-70
197. Golubeva JA, Sheptulina AF, Elkina AY, Liusina EO, Kiselev AR, Drapkina OM. Which Comes First, Nonalcoholic Fatty Liver Disease or Arterial Hypertension? Biomedicines. 2023 11
198. Rau M, Rehman A, Dittrich M, Groen AK, Hermanns HM, Seyfried F. et al. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United European Gastroenterol J. 2018;6:1496-507
199. Leonov GE, Varaeva YR, Livantsova EN, Starodubova AV. The Complicated Relationship of Short-Chain Fatty Acids and Oral Microbiome: A Narrative Review. Biomedicines. 2023 11
200. Dupraz L, Magniez A, Rolhion N, Richard ML, Da Costa G, Touch S. et al. Gut microbiota-derived short-chain fatty acids regulate IL-17 production by mouse and human intestinal gammadelta T cells. Cell Rep. 2021;36:109332
201. Liu W, Luo X, Tang J, Mo Q, Zhong H, Zhang H. et al. A bridge for short-chain fatty acids to affect inflammatory bowel disease, type 1 diabetes, and non-alcoholic fatty liver disease positively: by changing gut barrier. Eur J Nutr. 2021;60:2317-30
202. Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L. et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. 2020;579:586-91
203. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569-73
204. Luo X, Wang K, Jiang C. Gut microbial enzymes and metabolic dysfunction-associated steatohepatitis: Function, mechanism, and therapeutic prospects. Cell Host Microbe. 2025;33:836-53
205. Alonso-Pena M, Espinosa-Escudero R, Hermanns HM, Briz O, Herranz JM, Garcia-Ruiz C. et al. Impact of Liver Inflammation on Bile Acid Side Chain Shortening and Amidation. Cells. 2022 11
206. Gruner N, Mattner J. Bile Acids and Microbiota: Multifaceted and Versatile Regulators of the Liver-Gut Axis. Int J Mol Sci. 2021 22
207. Yang ZX, Shen W, Sun H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol Int. 2010;4:741-8
208. Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis. 2011;29:37-44
209. Baffy G, Portincasa P. Gut Microbiota and Sinusoidal Vasoregulation in MASLD: A Portal Perspective. Metabolites. 2024 14
210. Gottlieb A, Bechmann L, Canbay A. The Presence and Severity of Nonalcoholic Steatohepatitis Is Associated with Specific Changes in Circulating Bile Acids. Ann Hepatol. 2018;17:340-1
211. Conde de la Rosa L, Garcia-Ruiz C, Vallejo C, Baulies A, Nunez S, Monte MJ. et al. STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway. J Hepatol. 2021;74:1429-41
212. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX. et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2021;70:761-74
213. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018 360
214. Heisel T, Montassier E, Johnson A, Al-Ghalith G, Lin YW, Wei LN. et al. High-Fat Diet Changes Fungal Microbiomes and Interkingdom Relationships in the Murine Gut. mSphere. 2017 2
215. van der Merwe M, Sharma S, Caldwell JL, Smith NJ, Gomes CK, Bloomer RJ. et al. Time of Feeding Alters Obesity-Associated Parameters and Gut Bacterial Communities, but Not Fungal Populations, in C57BL/6 Male Mice. Curr Dev Nutr. 2020;4:nzz145
216. Demir M, Lang S, Hartmann P, Duan Y, Martin A, Miyamoto Y. et al. The fecal mycobiome in non-alcoholic fatty liver disease. J Hepatol. 2022;76:788-99
217. Yau T, Park J-W, Finn RS, Cheng A-L, Mathurin P, Edeline J. et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. The Lancet Oncology. 2022;23:77-90
218. Kelley RK, Rimassa L, Cheng AL, Kaseb A, Qin S, Zhu AX. et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022;23:995-1008
219. Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R. et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N Engl J Med. 2024;390:497-509
220. Dudek M, Tacke F. Immature neutrophils bring anti-PD-1 therapy in NASH-HCC to maturity. Gut. 2022
221. Lacotte S, Slits F, Moeckli B, Peloso A, Koenig S, Tihy M. et al. Anti-CD122 antibody restores specific CD8(+) T cell response in nonalcoholic steatohepatitis and prevents hepatocellular carcinoma growth. Oncoimmunology. 2023;12:2184991
222. Li X, Yao W, Yuan Y, Chen P, Li B, Li J. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157-67
223. Sounbuli K, Mironova N, Alekseeva L. Diverse Neutrophil Functions in Cancer and Promising Neutrophil-Based Cancer Therapies. Int J Mol Sci. 2022 23
224. Zhang A, Fan T, Liu Y, Yu G, Li C, Jiang Z. Regulatory T cells in immune checkpoint blockade antitumor therapy. Mol Cancer. 2024;23:251
225. Zhang L, Zhang W, Li Z, Lin S, Zheng T, Hao B. et al. Mitochondria dysfunction in CD8+ T cells as an important contributing factor for cancer development and a potential target for cancer treatment: a review. J Exp Clin Cancer Res. 2022;41:227
226. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91-7
227. Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S. et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. 2019;25:1096-103
228. Njei B, Al-Ajlouni YA, Ameyaw P, Njei LP, Boateng S. Role of ammonia and glutamine in the pathogenesis and progression of metabolic dysfunction-associated steatotic liver disease: A systematic review. J Gastroenterol Hepatol. 2024;39:1788-808
229. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S. et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12:558-80
230. Li B, Cao Y, Meng G, Qian L, Xu T, Yan C. et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine. 2019;39:239-54
231. Simon J, Nunez-Garcia M, Fernandez-Tussy P, Barbier-Torres L, Fernandez-Ramos D, Gomez-Santos B. et al. Targeting Hepatic Glutaminase 1 Ameliorates Non-alcoholic Steatohepatitis by Restoring Very-Low-Density Lipoprotein Triglyceride Assembly. Cell Metab. 2020;31:605-22 e10
232. Hu D, Wang W, Zhao X, An Y, Liu X, Yang M. et al. Expression pattern of glutaminase informs the dynamics of glutamine metabolism. The Innovation Life. 2025 3
233. Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019;29:18-26
234. Hu D, Zhang Z, Liu X, Wu Y, An Y, Wang W. et al. Generalizable transcriptome-based tumor malignant level evaluation and molecular subtyping towards precision oncology. J Transl Med. 2024;22:512
235. Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z. et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell. 2019;179:561-77 e22
236. Qiu YY, Zhang J, Zeng FY, Zhu YZ. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol Res. 2023;192:106786
237. Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K. et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182-91
238. Feng H, Yin Y, Zheng R, Kang J. Rosiglitazone ameliorated airway inflammation induced by cigarette smoke via inhibiting the M1 macrophage polarization by activating PPARgamma and RXRalpha. Int Immunopharmacol. 2021;97:107809
239. Bradshaw D, Abramowicz I, Bremner S, Verma S, Gilleece Y, Kirk S. et al. Hepmarc: A 96 week randomised controlled feasibility trial of add-on maraviroc in people with HIV and non-alcoholic fatty liver disease. PLoS One. 2023;18:e0288598
240. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310-21
241. Yang SJ, IglayReger HB, Kadouh HC, Bodary PF. Inhibition of the chemokine (C-C motif) ligand 2/chemokine (C-C motif) receptor 2 pathway attenuates hyperglycaemia and inflammation in a mouse model of hepatic steatosis and lipoatrophy. Diabetologia. 2009;52:972-81
242. Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H. et al. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab Invest. 2010;90:1169-78
243. Arora M, Kutinova Canova N, Farghali H. mTOR as an eligible molecular target for possible pharmacological treatment of nonalcoholic steatohepatitis. Eur J Pharmacol. 2022;921:174857
244. Basu B, Dean E, Puglisi M, Greystoke A, Ong M, Burke W. et al. First-in-Human Pharmacokinetic and Pharmacodynamic Study of the Dual m-TORC 1/2 Inhibitor AZD2014. Clin Cancer Res. 2015;21:3412-9
245. Powles T, Wheater M, Din O, Geldart T, Boleti E, Stockdale A. et al. A Randomised Phase 2 Study of AZD2014 Versus Everolimus in Patients with VEGF-Refractory Metastatic Clear Cell Renal Cancer. Eur Urol. 2016;69:450-6
246. Love S, Mudasir MA, Bhardwaj SC, Singh G, Tasduq SA. Long-term administration of tacrolimus and everolimus prevents high cholesterol-high fructose-induced steatosis in C57BL/6J mice by inhibiting de-novo lipogenesis. Oncotarget. 2017;8:113403-17
247. Fujita K, Nozaki Y, Wada K, Yoneda M, Endo H, Takahashi H. et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut. 2008;57:1583-91
248. Tian SY, Chen SM, Pan CX, Li Y. FXR: structures, biology, and drug development for NASH and fibrosis diseases. Acta Pharmacol Sin. 2022;43:1120-32
249. Sanyal AJ, Lopez P, Lawitz EJ, Lucas KJ, Loeffler J, Kim W. et al. Tropifexor for nonalcoholic steatohepatitis: an adaptive, randomized, placebo-controlled phase 2a/b trial. Nat Med. 2023;29:392-400
250. Iracheta-Vellve A, Calenda CD, Petrasek J, Ambade A, Kodys K, Adorini L. et al. FXR and TGR5 Agonists Ameliorate Liver Injury, Steatosis, and Inflammation After Binge or Prolonged Alcohol Feeding in Mice. Hepatol Commun. 2018;2:1379-91
251. Kessoku T, Kobayashi T, Imajo K, Tanaka K, Yamamoto A, Takahashi K. et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front Endocrinol (Lausanne). 2021;12:770986
252. Xiang H, Sun D, Liu X, She ZG, Chen Y. The Role of the Intestinal Microbiota in Nonalcoholic Steatohepatitis. Front Endocrinol (Lausanne). 2022;13:812610
253. Zheng Z, Zong Y, Ma Y, Tian Y, Pang Y, Zhang C. et al. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:234
254. Gao YS, Qian MY, Wei QQ, Duan XB, Wang SL, Hu HY. et al. WZ66, a novel acetyl-CoA carboxylase inhibitor, alleviates nonalcoholic steatohepatitis (NASH) in mice. Acta Pharmacol Sin. 2020;41:336-47
255. O'Farrell M, Duke G, Crowley R, Buckley D, Martins EB, Bhattacharya D. et al. FASN inhibition targets multiple drivers of NASH by reducing steatosis, inflammation and fibrosis in preclinical models. Sci Rep. 2022;12:15661
256. Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. 2012;92:713-23
257. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K. et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46:141-52
Author contact
![]() Corresponding authors: Huifang Liang, Ph.D., Professor, Email: lianghuifang1997com, Orcid ID: https://orcid.org/0000-0001-6874-3634; Wanguang Zhang, Ph.D., Professor, Email: wgzhangtjmu.edu.cn. Orcid ID: https://orcid.org/0000-0003-3184-9907.
Corresponding authors: Huifang Liang, Ph.D., Professor, Email: lianghuifang1997com, Orcid ID: https://orcid.org/0000-0001-6874-3634; Wanguang Zhang, Ph.D., Professor, Email: wgzhangtjmu.edu.cn. Orcid ID: https://orcid.org/0000-0003-3184-9907.