Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Lung-resident macrophages and...
3. Macrophage immunometabolism...
4. Therapeutic targeting of...
5. Conclusion and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(15):6580-6598. doi:10.7150/ijbs.123492 This issue Cite
Review
Macrophage Immunometabolism in Pulmonary Homeostasis and Chronic Lung Diseases
Cong Xie1,2#, Maimaititusun Yalikun1,3#, Zhenhui Ruan1,4#, Hang Yu1,5, Xi Huang1,6, Huahe Zhu1,7, Wenglam Choi1,8, Qingli Luo1,8, Zhen Gao1,8 ![]() , Jingcheng Dong1,3,4,8
, Jingcheng Dong1,3,4,8 ![]()
1. Institutes of Integrative Medicine, Fudan University, Shanghai 200032, China.
2. Yuquan Hospital, School of Clinical Medicine, Tsinghua University, Beijing 100040, China.
3. College of Traditional Chinese Medicine, Xinjiang Medical University, Urumqi 830017, China.
4. Shanghai Institute of Infectious Disease and Biosecurity, Fudan University Shanghai Medical College, Shanghai 200032, China.
5. Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin 10117, Germany.
6. Department of Biochemistry, Faculty of Medicine and Dentistry, University of Alberta, Edmonton T6G 2H7, Canada.
7. Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai 200433, China.
8. Department of Integrative Medicine, Huashan Hospital Affiliated to Fudan University, Shanghai 200040, China.
# These authors contributed equally to this work.
Received 2025-8-10; Accepted 2025-9-30; Published 2025-10-10
Abstract

Macrophages play a central role in maintaining pulmonary immune homeostasis and responding to injury. In the lung, alveolar macrophages modulate their metabolic profiles to support essential functions such as microbial clearance, inflammation resolution, and tissue repair. Recent studies have shown that these metabolic adaptations are not merely byproducts of activation but represent key regulators of macrophage behavior. In chronic lung diseases including asthma, chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF), macrophage metabolism is pathologically reprogrammed, contributing to persistent inflammation in asthma and COPD, or to unrestrained fibrotic remodeling in IPF, and ultimately leading to ongoing tissue damage. Specifically, in asthma, type 2 cytokine signaling promotes alternative macrophage activation, accompanied by increased fatty acid oxidation and disrupted lipid mediator profiles. COPD-associated macrophages exhibit mitochondrial dysfunction, enhanced glycolysis, and iron overload, impairing bacterial phagocytosis and amplifying oxidative stress. In IPF, macrophages simultaneously engage glycolytic and oxidative pathways while losing regulatory metabolites such as itaconate, supporting persistent fibrogenic signaling. These disease-specific metabolic features sustain maladaptive macrophage phenotypes and constitute promising targets for therapeutic intervention. This review outlines current knowledge of macrophage immunometabolism in the lung and its contribution to chronic respiratory diseases. It also discusses strategies to restore metabolic balance, including the use of antioxidants, metabolic modulators, and targeted drug delivery. Understanding macrophage metabolism may open new avenues for treating chronic lung diseases at the level of cellular function.
Keywords: macrophage immunometabolism, pulmonary homeostasis, chronic lung diseases, alveolar macrophages, metabolic reprogramming, therapeutic targets
1. Introduction
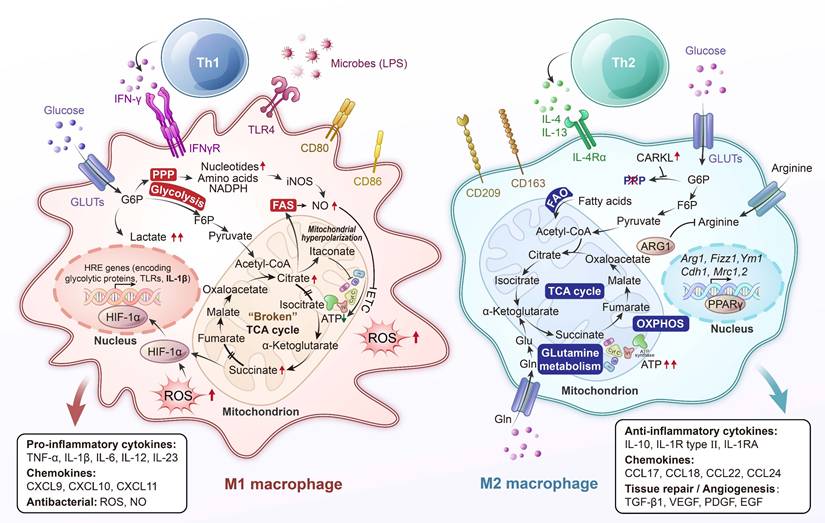
Lung-resident macrophages play a central role in pulmonary immune defense and the maintenance of tissue homeostasis [1]. Alveolar macrophages, which are the predominant immune cells in the healthy lung, act as frontline sentinels that continuously clear inhaled particles and microbes while regulating inflammation to preserve the integrity of the gas-exchange surface. A defining characteristic of macrophages is their immunometabolism, the close integration between cellular metabolic pathways and immune functions. Pro-inflammatory (“M1”) activation, such as stimulation by lipopolysaccharide (LPS), typically induces a shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, even in the presence of sufficient oxygen, a phenomenon reminiscent of the Warburg effect observed in cancer cells [2]. In contrast, exposure to anti-inflammatory (“M2”) signals like interleukin (IL)-4 promotes mitochondrial respiration and fatty acid oxidation (FAO) [3]. These metabolic programs are not just consequences of activation states but actively shape macrophage behavior, including cytokine production, antimicrobial capacity, and tissue-repair functions (Figure 1) [4].
Metabolic rearrangement in macrophage polarization to classically activated proinflammatory “M1” macrophages or alternatively activated anti-inflammatory “M2” macrophages. Pro-inflammatory stimuli induce the activation of specific pathways through the stabilization of transcription factors such as nuclear factor (NF)-κB and hypoxia-inducible factor (HIF)-1α, which trigger the expression of markers like inducible nitric oxide synthase (iNOS), CD80, and CD86 and the release of tumor necrosis factor (TNF)-α, IL-1β, IL-6, IL-12, and IL-23. Cells undergo a metabolic reprogramming toward glycolysis, the pentose-phosphate pathway, and fatty acid synthesis. This associates to interruption of tricarboxylic acid (TCA) cycle, ROS formation and efflux of citrate, which supports nicotinamide adenine dinucleotide phosphate (NADPH) and prostaglandin (PG) E2 synthesis, and succinate, which stabilizes HIF-1α, thereby promoting lipopolysaccharide (LPS)-induced expression of IL-1β. Alternatively, anti-inflammatory “M2” macrophages are induced by cytokines such as IL-4 and IL-13, leading to activation of transcription factors like STAT6 and peroxisome proliferator-activated receptor-γ (PPAR-γ). These cells are characterized by the expression of markers such as arginase-1 (Arg1), CD206 (mannose receptor), and Ym1, and the secretion of anti-inflammatory mediators like IL-10 and TGF-β. M2 polarization is associated with enhanced oxidative metabolism, including increased fatty acid oxidation (FAO) and mitochondrial oxidative phosphorylation (OXPHOS), along with an intact TCA cycle. This metabolic profile supports cellular functions such as tissue repair, matrix remodeling, efferocytosis (clearance of dead cells), and resolution of inflammation. Additionally, metabolic regulators like carbohydrate kinase-like protein (CARKL) and PPAR-γ coactivator 1 β (PGC-1β) contribute to maintaining redox balance and limiting pro-inflammatory responses. ATP, adenosine triphosphate; ETC, electron transport chain; FAS, fatty acid synthesis; F6P, fructose-6-phosphate; Gln, glutamine; Glu, glutamic acid; GLUT, glucose transporter; G6P, glucose-6-phosphate; HRE, hypoxia response element; IFN-γ, interferon-γ; PPP, pentose phosphate pathway; TLR4, toll-like receptor 4.
It is widely accepted that macrophage activation is driven by cytokines present in infected or damaged tissues [5]. However, recent findings suggest that metabolic adaptation itself also plays a critical and instructive role in determining macrophage functional identity [6]. Rather than being a passive byproduct, metabolic state can influence intracellular signaling, epigenetic remodeling, and effector differentiation. Within the lung, local environmental conditions such as high oxygen tension, limited glucose availability, and the presence of surfactant lipids impose unique metabolic constraints on resident macrophages [7, 8]. Under chronic disease conditions such as asthma, chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF), these macrophages undergo profound metabolic reprogramming as they adapt to inflammatory and remodeling signals in their microenvironment [9]. These metabolic changes can support protective functions such as microbial clearance and debris removal, but they may also contribute to pathological inflammation, tissue injury, and fibrosis [10].
Specific metabolic signaling pathways act as crucial bridges between cellular metabolic state and immune function. In M1 macrophages, for example, accumulation of the tricarboxylic acid (TCA) intermediate succinate stabilizes hypoxia-inducible factor (HIF)-1α, which shifts metabolism toward glycolysis and drives the production of IL-1β [11]. In parallel, the energy-sensing kinase AMP-activated protein kinase (AMPK) is activated by cellular stress or anti-inflammatory signals such as IL-10, promoting oxidative metabolism while inhibiting the mechanistic target of rapamycin (mTOR) pathway and thereby enhancing an anti-inflammatory macrophage phenotype [12]. By contrast, mTOR complex 1 (mTORC1) supports anabolic metabolism and facilitates translation of pro-inflammatory cytokines, including the optimal synthesis of IL-1β in classically activated (“M1”) macrophages [13]. Nutrient and oxygen sensors therefore tightly regulate macrophage polarization: HIF-1α favors a glycolytic, pro-inflammatory program [14], whereas peroxisome proliferator-activated receptor-γ (PPAR-γ) and AMPK tilt metabolism toward FAO and encourage anti-inflammatory, tissue-reparative functions [12]. Through these pathways, metabolic adaptation not only results from external activation signals but also feeds back to shape them, creating a bidirectional link between a macrophage's metabolic state and its immune effector profile.
In this review, we examine how macrophage immunometabolism is altered in chronic lung diseases and the implications of these changes for disease progression. We begin by describing the metabolic features of lung macrophages under homeostatic conditions, followed by a focused analysis of disease-specific metabolic reprogramming in asthma, COPD, and IPF. We also highlight emerging therapeutic strategies aimed at restoring healthy macrophage metabolism. A deeper understanding of these mechanisms may provide novel opportunities for targeted treatment of chronic respiratory diseases.
2. Lung-resident macrophages and their heterogeneity, functional plasticity, and metabolic phenotype at homeostasis
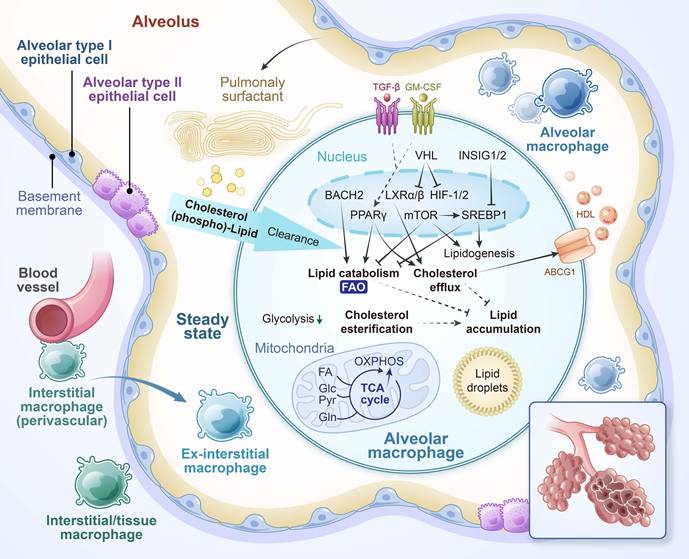
The lung harbors multiple populations of macrophages that differ in developmental origin, anatomical localization, and function [15]. Among them, alveolar macrophages (AMs) are located in the alveolar space, where they continuously clear inhaled particles, surfactant components, and apoptotic cells. Interstitial macrophages (IMs) are situated in the lung parenchyma, often adjacent to blood vessels and airways, and are more involved in modulating immune responses and maintaining tissue homeostasis (Figure 2) [16].
Functional and metabolic characteristics of alveolar macrophages (AMs) under homeostatic conditions. Under steady-state conditions, AMs maintain immune tolerance and regulate pulmonary surfactant—a lipoprotein complex produced by respiratory epithelial cells to lubricate the lungs and facilitate frictionless expansion/contraction—through anti-inflammatory signaling and lipolytic metabolism. Equipped with robust phagocytic capacity, AMs efficiently clear apoptotic/senescent cells and inhaled particles, serving as the first line of defense against airborne pathogens. Their energy metabolism primarily relies on oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO), a process governed by peroxisome proliferator-activated receptor-γ (PPARγ)-driven transcriptional programs and modulated by epithelial-derived signals such as granulocyte-macrophage colony-stimulating factor (GM-CSF). Impaired cholesterol handling, including deficiencies in liver X receptor (LXR) and its downstream transporter ABCG1 (ATP-binding cassette subfamily G member 1), can lead to pathological surfactant accumulation, as observed in pulmonary alveolar proteinosis. Furthermore, oxygen-sensing pathways such as the von Hippel-Lindau (VHL)-HIF axis shape AM identity and metabolic states, highlighting the critical role of tightly regulated lipid metabolism in maintaining pulmonary homeostasis. BACH2, BTB and CNC homology 2; HDL, high-density lipoprotein; INSIG, insulin-induced gene; SREBP1, sterol regulatory element-binding protein 1. This figure was substantially revised and repurposed based on Figure 2A from Wculek SK et al., Cell Mol Immunol. 2022;19:384-408.
Under physiological conditions, most AMs originate from embryonic precursors and maintain themselves through local proliferation [17, 18]. In fact, the majority of tissue-resident macrophages in adults are seeded before birth, arising from yolk sac- and fetal liver- derived progenitors during embryogenesis. Fate-mapping/lineage-tracing approaches, parabiosis experiments, and adoptive transfer studies have established that AMs sustain their population throughout life primarily via self-renewal, with minimal contribution from adult bone marrow-derived monocytes [19]. This long-lived, self-maintaining population is supported by factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and transforming growth factor (TGF)-β [20]. When the lung is injured or inflamed, circulating monocytes can be recruited to the alveoli and differentiate into monocyte-derived AMs (Mo-AMs) [21]. These newly differentiated cells initially exhibit transcriptional and metabolic profiles that differ from those of long-lived resident AMs [22-24]. For example, they tend to be more pro-inflammatory and rely more on glycolysis during early stages of adaptation [24, 25]. Over time, monocyte-derived macrophages may gradually adopt phenotypic features of resident AMs [26], although some differences can persist, particularly under chronic disease conditions [27].
The pulmonary environment imposes distinct metabolic demands on resident macrophages. The alveolar space is characterized by high oxygen levels, relatively low glucose availability, and abundant lipids derived from pulmonary surfactant [28]. In response to these conditions, AMs predominantly utilize FAO and OXPHOS to meet their energy requirements. At baseline, they exhibit low glycolytic activity and depend on mitochondrial respiration fueled by lipid catabolism. This metabolic profile is regulated by transcription factors such as PPAR-γ, which promotes lipid uptake and mitochondrial activity, and is induced by GM-CSF signaling [29]. These pathways support both surfactant clearance and the immunoregulatory phenotype of AMs. IMs, on the other hand, display more heterogeneity in both function and metabolism, reflecting their distribution across different interstitial niches [30]. Although less well-characterized, IMs are thought to engage more variable metabolic programs, including glycolysis and glutaminolysis, depending on local environmental cues.
Metabolism is not simply a reflection of macrophage activation; rather, it plays a directive role in shaping immune function (Table 1) [6]. Inflammatory stimuli such as LPS trigger a shift toward glycolysis and a disrupted TCA cycle to support cytokine production and antimicrobial activity. In contrast, anti-inflammatory signals like IL-4 promote a metabolic state characterized by intact TCA activity and enhanced FAO, which supports tissue repair and resolution of inflammation. Central regulators of these pathways include HIF-1α [31], which promotes glycolytic gene expression under hypoxic or inflammatory conditions, and AMPK, which favors catabolic metabolism and energy conservation during stress. The mTOR pathway, responsive to nutrient availability, supports anabolic metabolism and contributes to pro-inflammatory activation [32-34]. This metabolic flexibility allows lung macrophages to adjust their function according to the tissue environment. While such adaptability is essential for maintaining homeostasis, it also provides a basis for dysregulation in chronic disease, where persistent environmental stressors can lock macrophages into maladaptive metabolic and inflammatory states.
Functional plasticity and metabolic regulation in macrophage subsets.
| M1 [LPS(+IFN-γ)] | M2 [IL-4] | |
|---|---|---|
| Stimuli | Classical activation: LPS, IFN-γ, TNF-α, GM-CSF | Alternative activation: IL-4, IL-13, IL-10, TGF-β, IL-33, IL-21 |
| Markers | Surface: CD80/86, MHC-Ⅱ Intracellular: iNOS, IRF5, STAT1, NF-κB, HIF-1α | Surface: CD163, CD209, CXCR1, CXCR2, Dectin-1 Intracellular: Arginase 1, IRF4, STAT6, PPARγ |
| Function | Pro-inflammatory defense, Phagocytic, Microbial killing, Tissue damage, Anti-cancer immunity, Host defense | Anti-inflammatory, Wound healing, Efferocytosis, Hypersensitive response, Angiogenesis, Matric and tissue remodeling, Tumor progression and metastasis |
| Secretion | Cytokines: IL-1β, IL-6, IL-12, IL-23, TNF-α Chemokines: CXCL9, CXCL10, CXCL11, MIP-1α (CCL3) Free radicals: ROS, iNOS, NO | Cytokines: IL-10, IL-1RII, IL-1RA Chemokines: CCL17, CCL18, CCL22, CCL24 Growth factors: TGF-β1, VEGF, PDGF, EGF |
| Glycolysis | Increased glycolytic flux Lactate accumulation HIF-1α-induced production of pro-inflammatory cytokines (IL-1β, etc.) | Dispensable when OXPHOS is intact Glycolysis produces pyruvate to fuel TCA cycle |
| TCA cycle | Broken in two places: after citrate and after succinate | An intact TCA cycle Replenished with FAO and glutamine metabolism |
| OXPHOS | Dysfunctional OXPHOS and ETC Increased ROS generation | Increased mitochondrial biogenesis and respiratory capacity PGC1β-induced gene expression |
| PPP | Induced and required for ROS generation via NADPH oxidase, NO production, and nucleotide and protein synthesis | Not required/suppressed by the sedoheptulose kinase CARKL |
| Fatty acid metabolism | Increased lipid synthesis SREBP-induced gene expression | Increased fatty acid β-oxidation (FAO) STAT6- and PPARγ-induced gene expression |
| Amino acid metabolism | Arginine is converted to NO and citrulline by iNOS Glutamine metabolism regulates trained innate immunity | Arginase-1 metabolizes arginine to generate ornithine and urea Glutamine is essential for M2 polarization |
| Iron metabolism | Ferritin (iron storage) | Ferroportin (iron export) |
Abbreviations: M1, classically activated macrophages; M2, alternatively activated macrophages; LPS, lipopolysaccharide; TNF-α, Tumor necrosis factor-α; GM-CSF, granulocyte-macrophage colony-stimulating factor; TGF-β, transforming growth factor-β; MHC, major histocompatibility complex class; iNOS, inducible nitric oxide synthase; IRF, interferon regulatory factor; STAT, signal transducer and activator of transcription; NF-κB, nuclear factor-κB; PPAR, peroxisome proliferator activated receptor; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor; PDGF, platelet derived growth factor; EGF, epidermal growth factor; FAO, fatty acid β-oxidation; PGC-1β, PPARγ coactivator 1β; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation; ETC, electron transport chain; PPP, pentose phosphate pathway.
3. Macrophage immunometabolism in chronic lung diseases
Chronic lung diseases such as asthma, COPD, and IPF are associated with persistent immune activation and progressive tissue remodeling. In each of these conditions, macrophages exhibit distinct metabolic profiles that reflect their adaptation to the local microenvironment. These disease-specific metabolic programs influence macrophage function and, in many cases, contribute directly to the progression of inflammation, tissue damage, or fibrosis.
3.1 Asthma
Asthma is a chronic airway disease characterized by type 2 inflammation, airway hyperresponsiveness, mucus overproduction, and structural remodeling [35, 36]. In allergic asthma, AMs are exposed to a cytokine environment dominated by IL-4 and IL-13, which promote alternative (M2-like) activation. As a result, airway macrophages display increased expression of CD206 and other markers associated with IL-4/IL-13-mediated polarization, indicating a shift toward a tissue-repair and immunomodulatory phenotype [37]. Notably, in mild allergic asthma, M2-polarized AMs can help resolve inflammation by engulfing apoptotic eosinophils and producing anti-inflammatory cytokines such as IL-10 [38]. However, in more severe forms of asthma, this resolution program is impaired. AMs lose their ability to clear apoptotic cells effectively and show reduced IL-10 production, leading to persistent inflammation and airway remodeling, including subepithelial fibrosis and smooth muscle hyperplasia [39]. In severe asthma, pro-inflammatory macrophage subsets also emerge, such as those expressing interferon regulatory factor 5 (IRF5), suggesting considerable phenotypic heterogeneity within the macrophage pool [40]. This diversity reflects a functional plasticity shaped by the chronic inflammatory microenvironment.
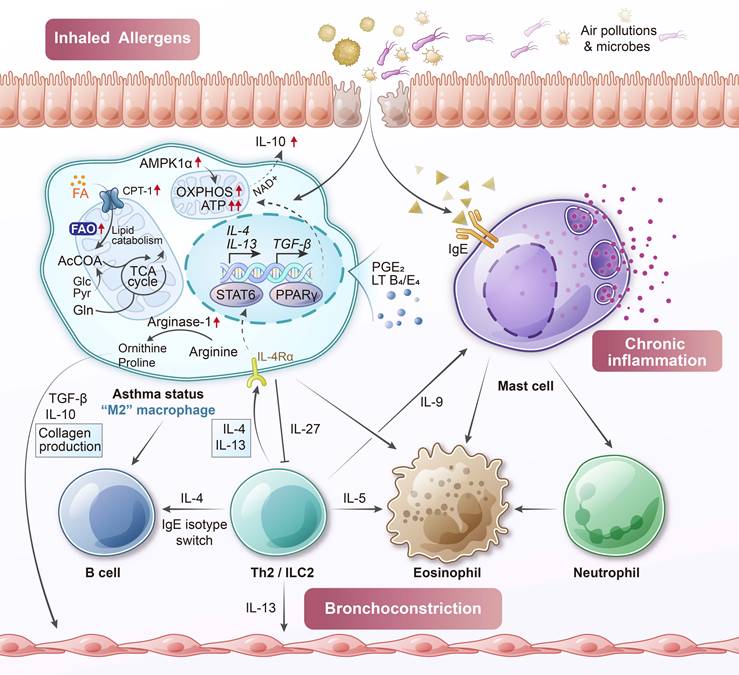
The metabolic programming of macrophages in asthma reflects this skewed activation state (Figure 3). Chronic exposure to allergens and type 2 cytokines enhances mitochondrial oxidative metabolism in AMs. These cells exhibit increased FAO, as evidenced by elevated expression of carnitine palmitoyl transferase 1 (CPT1) and other enzymes involved in mitochondrial fatty acid uptake and β-oxidation [41]. Such metabolic orientation supports the energy demands of M2-like functions, including matrix remodeling and secretion of fibrogenic mediators. Moreover, this FAO enhancement may represent a compensatory adaptation to the lipid-rich airway environment during inflammation [42, 43].
Allergic asthma-associated macrophage metabolism. In asthma, alveolar macrophages are exposed to a cytokine environment dominated by IL-4 and IL-13, which promote alternative (M2-like) activation and shift metabolic programming toward enhanced fatty acid oxidation. These macrophages express high levels of carnitine palmitoyl transferase 1 (CPT1) and arginase-1, supporting tissue repair functions but also contributing to subepithelial fibrosis and airway remodeling. Concurrently, elevated reactive oxygen species (ROS) production and disordered glycolytic activity reflect a broader metabolic imbalance. Asthmatic macrophages also display altered lipid mediator metabolism, producing excess leukotrienes while failing to generate sufficient pro-resolving eicosanoids such as 15-hydroxy eicosatetraenoic acid (15-HETE) and prostaglandin E2 (PGE2). These changes impair inflammation resolution, disrupt efferocytosis, and drive persistent airway inflammation. AcCoA, acetyl-coenzyme A; AMPK, adenosine monophosphate-activated protein kinase; NAD+, nicotinamide adenine dinucleotide (oxidized form).
At the same time, asthmatic macrophages demonstrate increased oxidative stress. Elevated production of reactive oxygen species (ROS), such as hydrogen peroxide and superoxide, has been detected in AMs from asthmatic patients [44]. This is accompanied by upregulation of oxidative stress response genes, including heme oxygenase-1 (HO-1), indicating sustained redox imbalance [45]. ROS overproduction can contribute to epithelial injury [46], promote pro-inflammatory signaling [47], and enhance airway hyperresponsiveness. Importantly, this oxidative burden coexists with metabolic dysfunction in glycolysis pathways, suggesting a broader metabolic imbalance rather than a simple shift toward FAO [48].
Asthma is also associated with disruptions in macrophage lipid mediator metabolism [49]. IL-4 and IL-13 stimulation enhances the production of pro-inflammatory eicosanoids, such as leukotriene B4 (LTB4), LTE4, and 15-hydroxyeicosatetraenoic acid (15-HETE) [50]. These mediators promote bronchoconstriction, leukocyte recruitment, and mucus secretion. In contrast, the generation of anti-inflammatory lipid mediators, including prostaglandin E2 (PGE2) and 15-HETE, is diminished in asthmatic macrophages [51]. This imbalance between pro-inflammatory and pro-resolving lipid mediators impairs efferocytosis and contributes to the perpetuation of airway inflammation [52]. Furthermore, the expression of arginase-1 (Arg1) is increased in asthmatic macrophages [53, 54]. This enzyme metabolizes L-arginine into ornithine and urea [55, 56], diverting substrate away from nitric oxide (NO) synthesis and instead facilitating polyamine and proline production [56], which support collagen synthesis and airway remodeling [57]. The upregulation of Arg1, together with increased FAO, reflects a metabolic environment that favors tissue repair and matrix deposition but may also contribute to fibrosis if left unchecked [58-60].
Although some of these M2-associated features serve to limit inflammation and promote resolution, their persistence or dysregulation in chronic disease may result in pathological tissue remodeling [61, 62]. In severe asthma, the regulatory capacity of AMs becomes compromised [39]. These cells lose their ability to effectively clear apoptotic cells and dampen excessive inflammation, leading to a mixed phenotype that combines elements of M2-like metabolism with elevated oxidative stress and persistent cytokine production [63-65]. Consistent with these alterations, biomarkers of macrophages activation are modified in asthmatic airway. AMs from asthmatic patients exhibit high pression of Arg1, reflecting an M2-skewed phenotype, and reduced production of PGE2. Additionally, levels of the chitinase-like protein YKL-40 (CHI3L1), secreted by macrophages, are elevated in both serum and bronchoalveolar lavage fluid of patients with severe asthma [66, 67].
The immunometabolic reprogramming of AMs in asthma involves enhanced FAO, increased ROS generation, and imbalanced eicosanoid synthesis. These changes promote both structural remodeling and chronic inflammation. The asthmatic environment pushes AMs into a metabolically active yet functionally imbalanced state. It is as if one foot is pressing the gas pedal, driven by increased ROS production and disordered glycolysis, while the other foot is on the brake, sustaining M2-like tissue-repair activity. This conflicting metabolic behavior prevents proper resolution of inflammation and contributes to ongoing tissue damage [63].
3.2 COPD
COPD is a chronic lung condition that is primarily caused by prolonged exposure to cigarette smoke [68]. It is characterized by persistent airway inflammation, progressive airflow limitation, and destruction of alveolar structures, leading to emphysema [69]. A hallmark of COPD is the accumulation of activated macrophages in the small airways and alveolar spaces. In smokers and individuals with COPD, the number of AMs in bronchoalveolar lavage fluid can increase by several folds [70]. These macrophages contribute to disease progression by releasing pro-inflammatory cytokines, chemokines, and proteases [71]. Among these, macrophage metalloelastases are directly implicated in elastin degradation and tissue destruction. Simultaneously, COPD AMs are functionally compromised in their host defense capacity. Multiple studies have demonstrated that COPD macrophages exhibit reduced phagocytic activity and impaired bacterial killing, which contributes to frequent microbial colonization and recurrent disease exacerbations [72].
A defining feature of the COPD lung microenvironment is the presence of high oxidative stress, which far exceeds that observed in asthma [73]. Cigarette smoke introduces large quantities of exogenous free radicals into the lungs and stimulates resident macrophages to produce additional ROS [74]. Compared to non-smokers, AMs from COPD patients release significantly higher levels of mitochondrial ROS, superoxide anion (O2⁻), and hydrogen peroxide (H2O2), indicating chronic oxidant exposure [75]. This oxidative imbalance is further aggravated by insufficient antioxidant defenses in COPD macrophages. For example, the expression of glutamate-cysteine ligase, the rate-limiting enzyme in glutathione biosynthesis, is downregulated in these cells [76]. As a result, the oxidant burden in the COPD lung remains high, not only damaging surrounding tissue structures but also impairing the function and viability of the macrophages themselves.
Mitochondrial dysfunction is a prominent feature of COPD AMs and is strongly linked to the sustained oxidative environment caused by cigarette smoke exposure [77]. Persistent ROS accumulation leads to mitochondrial membrane depolarization and disruption of the electron transport chain [78]. In COPD patient-derived AMs, studies have revealed a decreased mitochondrial membrane potential, along with significantly reduced maximal respiratory capacity and diminished spare respiratory reserve [74]. Bioenergetic profiling further shows that while baseline glycolysis rates remain similar between healthy individuals and those with COPD, COPD macrophages exhibit substantially lower OXPHOS efficiency and increased proton leak [79]. This indicates a population of mitochondria that are energetically inefficient and structurally compromised. Consequently, these cells cannot generate adenosine triphosphate (ATP) effectively through OXPHOS. Moreover, mitochondrial damage leads to greater leakage of ROS, forming a vicious cycle of oxidative stress. This bioenergetic dysfunction likely contributes to impaired phagocytosis. When mitochondria fail to provide sufficient ATP, or when excess mitochondrial ROS interferes with cellular signaling, macrophages become unable to efficiently engulf and eliminate bacteria. One study has demonstrated that the increase in mitochondrial ROS observed in COPD AMs is causally linked to their defective bacterial clearance [74].
In response to mitochondrial impairment, COPD macrophages may attempt to adapt their metabolism. Evidence supports a shift toward glycolytic metabolism, largely driven by HIF-1α [80, 81]. Transcriptomic analyses of COPD AMs show increased expression of HIF-1α and its downstream targets, including the adenosine A2B receptor, a gene often upregulated under hypoxic or glycolytic conditions [82]. These findings suggest that despite the presence of normal oxygen levels, COPD macrophages experience a “pseudo-hypoxic” state due to mitochondrial dysfunction and oxidative stress, which stabilizes HIF-1α and promotes a metabolic shift toward glycolysis. However, this glycolytic compensation may be insufficient to restore energy balance. The resulting accumulation of lactate and altered metabolite profiles could further contribute to inflammation and immune dysregulation.
In addition to these changes, COPD macrophages show abnormalities in nitrogen and arginine metabolism. Unlike AMs in healthy lungs, which produce minimal NO, COPD macrophages exhibit concurrent upregulation of inducible nitric oxide synthase (iNOS) and Arg1 [83, 84]. Elevated iNOS expression is particularly evident in patients with advanced disease or during exacerbations [85, 86]. Although this might be expected to increase NO production, levels of exhaled NO in stable COPD are not elevated. One likely explanation is the excessive level of superoxide in the COPD lung, which rapidly reacts with NO to form peroxynitrite (ONOO⁻), a highly reactive nitrogen species (RNS) [83]. Supporting this, increased nitrotyrosine staining, an indicator of peroxynitrite-mediated protein damage, has been detected in sputum macrophages from COPD patients and correlates with worsened lung function [87]. Meanwhile, Arg1 is also induced by cigarette smoke and depletes intracellular L-arginine, the common substrate for both iNOS and Arg1 [88]. When L-arginine levels become limiting, iNOS becomes “uncoupled,” shifting its activity from NO production to superoxide generation [84]. This favors additional peroxynitrite formation and intensifies nitrosative stress. Therefore, COPD macrophages experience combined oxidative and nitrosative stress, producing ONOO⁻ that damages proteins, lipids, and DNA, and contributing to sustained inflammation and cell death. The simultaneous expression of iNOS and Arg1 in these cells reflects a mixed M1/M2 polarization pattern, driven by the complex and dysregulated microenvironment in COPD lungs [83].
Iron metabolism adds another layer to the immunometabolic dysfunction of COPD macrophages [89]. Cigarette smoke contains iron-rich particles and promotes alveolar microhemorrhages that release heme, leading to iron loading in AMs [90, 91]. These macrophages often appear as hemosiderin-laden “siderophages” under histological analysis, indicating increased intracellular iron content [92]. Excess iron promotes the Fenton reaction, in which hydrogen peroxide is converted into highly reactive hydroxyl radicals, thereby exacerbating oxidative injury [93, 94]. Moreover, iron overload and the associated oxidative stress can trigger ferroptosis, a form of iron-dependent cell death, in lung cells. This is increasingly recognized in COPD and pulmonary fibrosis, as iron-catalyzed lipid peroxidation feeds into cell death and inflammation [95]. Iron-loaded macrophages further propagate inflammation and tissue damage [96]. In one study, reducing iron availability—either by iron chelation or dietary iron restriction—provided protection against cigarette smoke-induced lung injury in mice [97]. These findings support a direct pathological role for iron overload in driving oxidative stress and macrophage dysfunction in COPD.
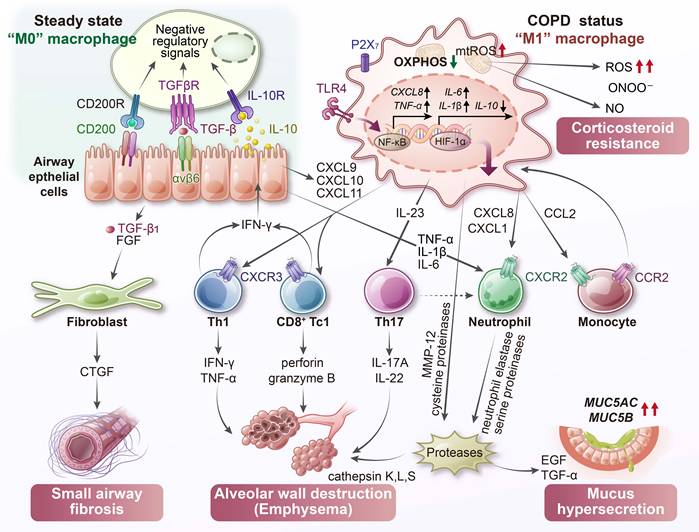
COPD induces profound metabolic disturbances in lung macrophages (Figure 4). Chronic smoke exposure and persistent ROS production lead to mitochondrial damage, disrupted energy metabolism, and imbalances in both reactive oxygen and nitrogen species. These stressors impair essential immune functions such as bacterial clearance and promote chronic inflammation and tissue destruction. Multiple inflammatory biomarkers reflect the macrophage-driven changes in COPD (Table 2). Sputum levels of IL-8 and matrix metalloproteinase (MMP)-9, partly derived from macrophages, are significantly elevated and correlate with neutrophilic inflammation as well as progressive decline in lung function [98]. Likewise, sputum macrophages in COPD exhibit increased levels of 3-nitrotyrosine, a marker of peroxynitrite-mediated oxidative damage, and these elevations show an inverse correlation with FEV1% [87]. Therapeutic strategies aimed at correcting macrophage metabolic dysfunction, such as restoring mitochondrial function or enhancing antioxidant capacity, hold promise for improving disease outcomes. Indeed, activation of the NRF2 (nuclear erythroid-related factor 2) antioxidant pathway in COPD macrophages has been shown to improve bacterial clearance [99], suggesting that immunometabolic correction can provide clinical benefit.
Metabolic dysfunction in COPD macrophages. In COPD, alveolar macrophages display mitochondrial impairment with reduced membrane potential and oxidative phosphorylation (OXPHOS) efficiency. Chronic exposure to cigarette smoke promotes iron accumulation and excessive ROS, fueling Fenton reactions and the formation of peroxynitrite (ONOO⁻). Concurrent stabilization of hypoxia-inducible factor-1α (HIF-1α), even under normoxic conditions, drives a metabolic shift toward glycolysis and enhances pro-inflammatory activity. These macrophages secrete a range of inflammatory mediators that orchestrate immune cell recruitment: CXCL9, CXCL10, and CXCL11 attract Th1 and Tc1 lymphocytes; CXCL8, CXCL1, and leukotriene B4 (LTB4) recruit neutrophils; and CCL2 mediates monocyte infiltration. The release of proteolytic enzymes, including matrix metalloproteinases (MMPs) and cathepsins, contributes to elastin degradation, synergizing with cytotoxic T cells to promote emphysema. Transforming growth factor-β1 (TGF-β1) released by macrophages also drives small airway fibrosis. Furthermore, the interaction between macrophage-derived ROS and nitric oxide (NO) leads to excessive ONOO⁻ generation, a process potentially linked to corticosteroid resistance in COPD. CTGF, connective tissue growth factor; FGF, fibroblast growth factor.
Representative biomarkers of macrophage activation and metabolic dysfunction in chronic lung diseases.
| Disease | Biomarker | Source | Macrophage-derived? | Clinical Relevance |
|---|---|---|---|---|
| Asthma | Arginase-1 | BALF macrophage | Yes (M2 macrophage) | Promotes collagen synthesis, airway fibrosis |
| YKL-40 (CHI3L1) [67] | Serum/BALF | Yes (macrophages, others) | Correlates with asthma severity and airway remodeling | |
| FeNO [160] | Breath | Partially (epithelium via iNOS; modulated by Arg1) | Elevated in allergic (type 2) asthma; indicates airway eosinophilic inflammation; influenced by macrophage arginase activity | |
| LTB4 | Sputum/BALF | Yes (macrophages, neutrophils) | Increased in asthmatic airways; contributes to bronchoconstriction and inflammation | |
| 15-HETE & Lipoxin A4 | BALF | Yes (macrophages) | Reduced in asthma; anti-inflammatory and pro-resolving lipid mediators | |
| CCL17 (TARC) | BALF | Yes | Macrophage-secreted chemokine; eosinophil/T cell recruitment | |
| Periostin | Serum | No | Biomarker of type 2 inflammation | |
| COPD | CXCL8 (IL-8) | Sputum | Yes (macrophages, epithelium) | Markedly elevated in COPD sputum; drives neutrophil recruitment |
| MMP-9, MMP-12 | BALF/Sputum | Yes (macrophages, neutrophils) | Promote elastin degradation and emphysema progression | |
| 3-Nitrotyrosine | Sputum cells | Yes (macrophages) | Reflects peroxynitrite (ONOO-)-mediated oxidative stress | |
| Hemosiderin [161] | BALF macrophages | Yes (indicative) | Indicates iron overload and oxidative injury | |
| SP-A, SP-D [162] | Serum | No | General marker of lung injury and exacerbations | |
| IPF | CCL18/PARC [163, 164] | Serum, BALF | Yes (M2 macrophages) | Reflects profibrotic M2 activation; predicts poor survival and faster disease progression |
| YKL-40 (CHI3L1) | Serum, BALF | Yes (macrophages, others) | Associated with fibrosis and matrix remodeling | |
| MMP-7 [165] | Serum | Partially (epithelium, macrophages) | Indicates active epithelial injury and remodeling | |
| KL-6 (MUC1) [164, 166, 167] | Serum | No (alveolar type Ⅱ cells) | Widely used diagnostic and prognostic biomarker for ILDs | |
| SP-D [164, 168] | Serum | No | Reflect epithelial injury and surfactant disturbance | |
| Reduced itaconate (ACOD1 product) [114] | BALF macrophages | Yes | Loss of anti-inflammatory metabolite regulation; linked to persistent activation | |
| CD44 [169] | BALF/lung tissue | Partially (macrophages, fibroblasts) | Regulates macrophage-fibroblast crosstalk; associated with fibrosis progression | |
| CX3CL1 (fractalkine) [170] | Serum, BALF | Yes (Endothelium, macrophages) | Potential diagnostic biomarker; correlates with IPF progression | |
| S100A8/A9/A12 (S100 family) [171] | Serum, BALF | Partially (macrophages, neutrophils) | Associated with inflammation, oxidative stress, and fibrogenesis |
Abbreviations: BALF, bronchoalveolar lavage fluid; FeNO, fractional exhaled nitric oxide; LTB4, leukotriene B4; 15-HETE, 15-hydroxyeicosatetraenoic acid; CCL17, chemokine (C-C motif) ligand 17; TARC, thymus and activation-regulated chemokine; MMP, matrix metalloproteinase; SP, surfactant protein; CCL18/PARC, pulmonary and activation-regulated chemokine; KL-6, Krebs von den Lungen-6, a mucin 1 glycoprotein; ILDs, interstitial lung diseases; ACOD1, aconitate decarboxylase 1.
3.3 IPF
IPF is a chronic and progressive interstitial lung disease characterized by excessive fibrotic remodeling [100]. This condition involves the accumulation of collagen and extracellular matrix within the lung parenchyma, which leads to architectural distortion and ultimately respiratory failure. The pathogenesis of IPF is believed to stem from aberrant wound-healing responses to unidentified lung injuries. These responses include repetitive alveolar epithelial cell damage and dysregulated activation of fibroblasts and myofibroblasts [101]. Importantly, unlike asthma and COPD, IPF is not primarily an inflammatory disorder; broad anti-inflammatory or immunosuppressive therapies have shown little benefit and may even be harmful [102]. Instead, the differentiation of fibroblasts into myofibroblasts is considered the central pathogenic event driving disease progression, as these cells directly mediate fibrotic remodeling of the lung. AMs play a critical supporting role by secreting pro-fibrotic mediators such as TGF-β and platelet-derived growth factor (PDGF), while also sustaining fibroblast activation; however, they are not the main effectors of scarring [103]. In IPF lungs, AMs accumulate in fibrotic foci and exhibit an “M2-like” activation profile associated with wound healing. However, under persistent stimulation, this phenotype becomes pathogenic. These macrophages release large amounts of TGF-β and ROS, both of which directly stimulate myofibroblasts to produce collagen and thereby perpetuate fibrogenesis.
Recent studies suggest that AMs in IPF undergo global metabolic activation and become hypermetabolic cells. In contrast to the metabolically restrained phenotype of homeostatic AMs, IPF macrophages upregulate both glycolytic and oxidative metabolic pathways. In animal models of pulmonary fibrosis, such as bleomycin-induced lung injury, AMs demonstrate increased glucose uptake and a shift toward aerobic glycolysis, as evidenced by enhanced expression of key glycolytic enzymes and glucose transporters [104, 105]. Consistent findings have been observed in humans: AMs from IPF patients display elevated expression of glucose transporter 1 (GLUT1) [105], indicating an increased capacity for glucose uptake [104]. This is somewhat analogous to cancer cells in a tumor microenvironment, in which glycolysis supports the rapid generation of ATP and biosynthetic intermediates required for high secretory activity [106, 107]. Moreover, glycolytic reprogramming can stabilize HIF-1α and enhance the production of pro-fibrotic cytokines like IL-1β in macrophages [13], further contributing to the fibrotic process.
In parallel, IPF AMs exhibit heightened FAO and increased mitochondrial respiration. Notably, studies in IPF patients have shown upregulated activity of the mitochondrial calcium uniporter and its downstream effector peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α), a central regulator of mitochondrial biogenesis and FAO [108]. Upregulation of CPT1, a key enzyme in FAO, has been observed in both IPF patient samples and bleomycin-induced fibrotic lungs, indicating a significant increase in FAO capacity [104, 108]. Thus, IPF macrophages display a hybrid metabolic state, engaging in both aerobic glycolysis and oxidative metabolism simultaneously (Figure 5) [109]. This state of enhanced metabolic flux likely reflects the energy demands of continuous secretion of growth factors, proteases, and ROS [110]. By utilizing both glucose and fatty acids as energy substrates, these cells maintain a robust supply of ATP and biosynthetic precursors necessary for their profibrotic activity [111]. However, this hypermetabolic state also leads to elevated mitochondrial ROS production, a known consequence of increased OXPHOS [112].
Immunometabolic profile of IPF macrophages. In IPF, alveolar macrophages exhibit distinct metabolic polarization that reflects the progression from early inflammation to late-stage fibrosis. Early in disease, macrophages may adopt a glycolysis-driven M1-like phenotype associated with pro-inflammatory cytokine production, including IL-1β. As fibrosis advances, these cells shift toward an M2-like metabolic profile characterized by enhanced fatty acid oxidation, upregulated PGC-1α signaling, and sustained TGF-β secretion. Concurrent mitochondrial calcium influx amplifies oxidative phosphorylation and ROS output. Suppression of the aconitate decarboxylase 1 (ACOD1)-itaconate pathway removes an anti-inflammatory regulatory node, enabling persistent macrophage activation. Together, these metabolic changes promote fibroblast activation, collagen deposition, and irreversible tissue remodeling. AEC, alveolar epithelial cell; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; HO-1, heme oxygenase 1.
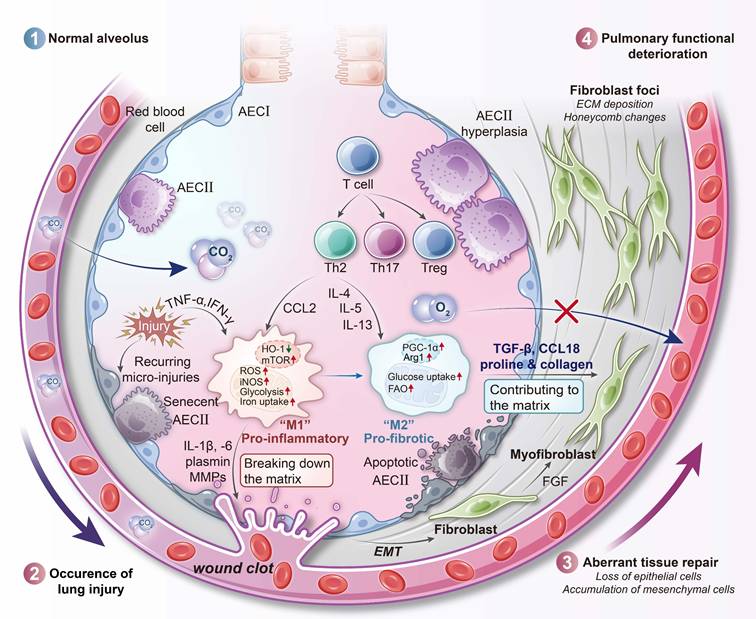
One metabolic checkpoint that appears disrupted in IPF macrophages is the itaconate pathway. Itaconate, produced by the mitochondrial enzyme immune-responsive gene 1 (IRG1, also known as ACOD1 [aconitate decarboxylase 1]), serves as a negative feedback regulator during macrophage activation [113]. It inhibits succinate dehydrogenase activity in the TCA cycle, activates the NRF2 pathway, and reduces IL-1β production, thereby exerting anti-inflammatory effects. In IPF, this protective mechanism is compromised. Alveolar lining fluid from IPF patients contains significantly reduced levels of itaconate, and macrophages from fibrotic lungs exhibit decreased expression of ACOD1 compared to healthy controls [114].
Functionally, this loss of itaconate signaling shifts macrophages toward a more pro-inflammatory and pro-fibrotic phenotype. In mouse models, genetic deletion of Acod1 exacerbates lung fibrosis after injury, while treatment with exogenous itaconate or derivatives such as 4-octyl itaconate reduces fibroblast activation and collagen deposition [114]. These findings suggest that the absence of endogenous itaconate removes an important regulatory brake on TGF-β signaling and extracellular matrix production in IPF. The loss of this single metabolite contributes significantly to macrophage-driven fibrogenesis.
Consistent with their hyperactivated metabolic profile, IPF macrophages also generate large amounts of ROS and RNS [115]. Elevated levels of Rac1, a small GTPase that activates NADPH oxidase, have been observed in IPF macrophages, driving superoxide production [116, 117]. In IPF lungs, the combined effects of NADPH oxidase-derived ROS and mitochondrial ROS from upregulated OXPHOS create a highly oxidizing milieu. Simultaneously, these macrophages express iNOS, and inflammatory stimuli in IPF are capable of inducing robust iNOS activity. As a result, NO and superoxide co-exist within the same microenvironment and react to form peroxynitrite (ONOO⁻). Elevated levels of ONOO⁻ and its oxidative byproducts, such as nitrotyrosine, have been detected in IPF patient-derived AMs [118]. In the bleomycin model of fibrosis, AMs show increased levels of superoxide, NO, and ONOO⁻ during the active fibrotic phase [119]. These reactive species can activate latent TGF-β and induce alveolar epithelial cell apoptosis, thereby driving further fibrosis.
Iron dysregulation in macrophages represents another immunometabolic abnormality in IPF. Similar to findings in COPD, IPF lungs frequently contain hemosiderin-laden AMs, suggesting chronic microhemorrhages or enhanced iron uptake. These iron-loaded macrophages contribute to fibrogenesis through several mechanisms [120]. Interestingly, the proportion of AMs expressing transferrin receptor (CD71) is reduced in IPF lungs, which may indicate a compensatory mechanism in response to intracellular iron overload [121]. The result is increased levels of extracellular, transferrin-bound iron in the fibrotic niche. Free iron can catalyze redox reactions that generate hydroxyl radicals via the Fenton reaction, promoting additional oxidative damage. Moreover, iron accumulation promotes macrophage polarization toward a pro-fibrotic phenotype [122]. Iron-rich AMs secrete ROS and pro-fibrotic cytokines and can directly stimulate fibroblast proliferation and myofibroblast differentiation [123]. Experimental reduction of iron availability through dietary restriction or iron chelation has been shown to ameliorate lung fibrosis in animal models [124, 125], underscoring the pathological relevance of iron-driven macrophage activation in IPF. Emerging evidence also links iron overload in IPF to ferroptosis [95, 126]. In late-stage fibrotic lungs, substantial iron deposition and oxidative lipid damage have been observed, which may promote death of alveolar epithelial cells and even macrophages via ferroptosis [124]. This ferroptotic injury is thought to further amplify profibrotic signaling by releasing damage-associated molecular patterns (DAMPs) and activating neighboring fibroblasts [95]. Thus, the dysregulated iron homeostasis in IPF not only accelerates ROS/RNS generation but also intersects with novel pathogenic mechanisms like ferroptosis, creating self-perpetuating cycles of injury and fibrosis.
AMs in IPF are metabolically reprogrammed into highly active, fibrosis-promoting cells. They simultaneously engage glycolysis and FAO, maintain high levels of ATP and ROS production, and lose regulatory mechanisms such as itaconate signaling. Iron accumulation and sustained oxidative/nitrosative stress further amplify their pathogenic activity. This metabolic polarization of macrophages plays a central role in IPF progression and represents a promising target for therapeutic intervention.
3.4 Comparative perspective
Despite the distinct metabolic fingerprints of asthma, COPD, and IPF, there are unifying themes in how macrophage metabolism is dysregulated. Each condition drives macrophages toward a particular metabolic bias that reinforces the predominant pathology. In asthma, a cytokine-rich, type 2 environment pushes macrophages toward oxidative metabolism and lipid utilization, resembling an M2-like profile that supports tissue remodeling but, when unchecked, contributes to fibrosis and impaired resolution. In COPD, chronic oxidative stress and hypoxia-inducible signals enforce a glycolytic, Warburg-like metabolism (akin to an M1 state) but in the context of mitochondrial dysfunction, resulting in energy failure, persistent inflammation, and tissue injury. IPF macrophages uniquely combine these metabolic programs, exhibiting concurrent glycolysis and OXPHOS to fuel relentless fibrogenesis (Table 3).
Comparative metabolic reprogramming of macrophages in chronic lung diseases.
| Feature | Asthma | COPD | IPF |
|---|---|---|---|
| Dominant phenotype | M2-like, but dysfunctional in severe disease | Mixed M1/M2, functionally impaired | Hypermetabolic, profibrotic |
| Primary metabolic shift | ↑ FAO, ↑ arginase activity, altered lipid mediator metabolism | Mitochondrial dysfunction → ↑ glycolysis (pseudo-hypoxia), ↓ OXPHOS | ↑ Glycolysis + ↑ FAO (hybrid state) |
| ROS/RNS balance | ↑ ROS, impaired antioxidant defenses | Excessive ROS + peroxynitrite (ONOO-) due to iNOS/Arg1 co-expression | ↑ ROS + RNS, mitochondrial and NADPH oxidase-derived |
| Iron metabolism | Mild changes, not central | Iron overload, siderophages, Fenton reaction | Iron overload, CD71low AMs, ferroptosis link |
| Regulatory metabolites | Imbalanced lipid mediators (↓PGE2, ↓ resolvins, ↑ leukotrienes) | ↓ Glutathione synthesis, impaired antioxidant capacity | ↓ Itaconate (ACOD1), loss of anti-inflammatory brake |
| Crosstalk with other cells | Eosinophils (impaired efferocytosis), epithelial cells | Neutrophils, T cells, epithelial damage | Fibroblasts/myofibroblasts (profibrotic loop) |
| Clinical implication | Contributes to airway remodeling, persistent inflammation | Drives impaired host defense, steroid resistance, emphysema | Sustains fibroblast activation, irreversible fibrosis |
The interplay between glycolysis and fatty acid oxidation is context-dependent. Inflammatory macrophages typically downregulate FAO as they upregulate glycolysis, as seen in acute M1 activation, whereas pro-resolving macrophages favor FAO and dampen glycolysis, consistent with classic M2 polarization. However, in chronic disease microenvironments, these pathways can co-exist or cycle dynamically, indicating that macrophages can flexibly rewire their metabolism when faced with complex stimuli. Key regulators such as HIF-1α and PPAR-γ act in opposition to fine-tune this balance: HIF-1α activation, even under normoxic conditions, promotes glycolysis and can inhibit mitochondrial respiration, whereas PPAR-γ, upregulated by signals like IL-4 and GM-CSF, enhances lipid uptake and oxidation while antagonizing inflammatory glycolysis. In diseases like IPF, both arms are engaged, suggesting that continuous injury signals override the conventional glycolysis-versus-FAO toggle.
Understanding these metabolic interactions is essential, as targeting a single pathway such as glycolysis may cause compensatory shifts in another (FAO), so successful therapies might need to address the metabolic network as a whole. In the next section, we discuss how these insights are being leveraged to design interventions that recalibrate macrophage metabolism across different lung diseases.
4. Therapeutic targeting of macrophage immunometabolism
Given the central role of macrophage metabolic reprogramming in chronic lung diseases, a number of therapeutic strategies are being explored with the aim of reconfiguring macrophage metabolism toward a more balanced and health-promoting state. These interventions seek to restore the physiological equilibrium between pro-inflammatory and reparative macrophage functions, or to enhance antimicrobial capacity through metabolic modulation. Several approaches have demonstrated encouraging results in preclinical or early-phase clinical studies.
4.1 Antioxidants and NRF2 activators
One potential therapeutic approach centers on reducing oxidative stress within macrophages, with the goal of alleviating tissue injury and restoring cellular function. N-acetylcysteine, a precursor of glutathione, has been tested as an oral antioxidant therapy in IPF, as exemplified by the PANTHER trial [127]. Although this trial did not demonstrate significant improvements in lung function, and the concomitant use of prednisone and azathioprine in that treatment arm actually worsened outcomes, N-acetylcysteine and other thiol-based antioxidants remain of interest as strategies to replenish intracellular glutathione in diseases such as COPD or cystic fibrosis, where they may mitigate oxidant-mediated macrophage dysfunction. More targeted strategies aim to activate the NRF2 pathway, a key regulator of antioxidant defense gene expression. Sulforaphane, a naturally occurring NRF2 agonist found in cruciferous vegetables, has been shown to enhance phagocytosis in AMs isolated from COPD patients by upregulating antioxidant responses [128]. These findings suggest that NRF2 activation can reverse certain functional deficits in COPD macrophages. Similarly, dimethyl fumarate, a pharmacologic NRF2 activator currently used for multiple sclerosis [129], has been reported to shift macrophages toward an anti-inflammatory phenotype [130]. Collectively, agents that enhance the expression of HO-1, glutathione biosynthesis enzymes, and other antioxidant mediators constitute a mechanistically grounded approach to suppressing the ROS/RNS-driven inflammatory cycle characteristic of chronic lung diseases.
4.2 Metabolic pathway inhibitors (glycolysis and FAO)
Because disease-associated macrophages in asthma and IPF exhibit concurrent upregulation of glycolysis and FAO, selective inhibition of these pathways may attenuate pathogenic macrophage activation [41, 131, 132]. 2-deoxy-D-glucose, a competitive inhibitor of glycolysis, has been shown in murine models of allergic asthma to prevent the pathological glycolytic shift in AMs [133]. Treatment with 2-deoxy-D-glucose reduced airway inflammation and hyperresponsiveness, suggesting that limiting aerobic glycolysis can downregulate cytokine production in asthma-associated AMs [134]. Conversely, etomoxir, an inhibitor of CPT1 that blocks mitochondrial FAO, has also shown beneficial effects in asthma models. Administration of etomoxir in mice reduced airway hyperreactivity, potentially by impairing the FAO-dependent alternative activation of AMs (“M2”) [41]. Similar logic may apply to IPF, where inhibition of glycolysis or FAO may disrupt the hypermetabolic and pro-fibrotic programming of macrophages [135]. However, systemic inhibition of core metabolic pathways poses significant risks, given their essential roles in all cell types. Therefore, careful control of drug dosage and delivery route is critical. Inhaled administration of low-dose 2-deoxy-D-glucose or etomoxir may enable local metabolic reprogramming of pulmonary macrophages with reduced systemic exposure and toxicity. Early studies support this idea, but clinical trials will be needed to confirm safety and efficacy in patients.
4.3 AMPK and mTOR modulators
The AMPK and mTOR pathways serve as central metabolic regulators that determine the balance between catabolic and anabolic processes. Activation of AMPK favors oxidative metabolism and promotes cellular resilience to stress, while mTOR activation supports glycolysis, protein synthesis, and pro-inflammatory cytokine production. Metformin, a well-characterized AMPK activator, has attracted attention for its potential to reprogram macrophage metabolism toward an anti-inflammatory state. In vitro, metformin enhances FAO and improves mitochondrial function in macrophages, which may support anti-inflammatory and autophagic phenotypes [136]. In models of lung fibrosis, metformin has been shown to activate AMPK and concurrently inhibit TGF-β signaling in both macrophages and myofibroblasts, thereby reducing collagen deposition [137, 138]. Nonetheless, translation to clinical benefit remains uncertain. A post hoc analysis of IPF patients receiving metformin did not demonstrate a clear therapeutic advantage, although patient heterogeneity and dosing variability could have influenced outcomes [139]. On the other hand, pharmacological inhibition of mTOR, such as with rapamycin, may suppress macrophage activation by reducing glycolytic flux and inflammatory cytokine production [140]. mTORC1 promotes protein synthesis and IL-1β translation in classically activated (“M1”) macrophages. Preclinical studies in COPD models have suggested that mTOR inhibition may reduce airway inflammation, but safety and long-term efficacy remain to be established [141, 142]. Altogether, precise tuning of macrophage metabolic sensors—enhancing AMPK activity while suppressing mTOR—offers a rational framework for therapeutic intervention but necessitates targeted approaches to avoid adverse systemic effects [3, 143].
4.4 Lipid mediator and iron homeostasis interventions
Another promising therapeutic strategy targets the metabolic outputs of activated macrophages, particularly the imbalance between pro-inflammatory and pro-resolving lipid mediators [144]. In asthma, overproduction of LTs by macrophages contributes to bronchoconstriction and airway inflammation [145]. Pharmacologic agents that block LT synthesis (e.g., 5-lipoxygenase inhibitors such as zileuton) or LT receptor signaling (e.g., montelukast, a CysLT1 antagonist) are already in clinical use for symptom control [145]. Beyond blocking harmful lipid mediators, supplementing beneficial, pro-resolving eicosanoids offers an alternative therapeutic angle. For example, 15-HETE, an anti-inflammatory lipid molecule, is deficient in asthmatic macrophages. In animal studies, exogenous delivery of 15-HETE inhibited LT production and improved airway responsiveness [146]. Likewise, specialized pro-resolving mediators such as lipoxin A4 and resolvin D1—both derived from Ω-3 fatty acids—have been shown to reduce neutrophilic inflammation and enhance clearance of cellular debris in models of asthma and cystic fibrosis [147, 148].
Iron homeostasis also plays a crucial role in macrophage immunometabolism, particularly in COPD and IPF, where macrophages often accumulate excess intracellular iron [89, 94]. Chelating agents such as deferoxamine and deferasirox can reduce iron availability and may be delivered via inhalation to selectively target iron-laden AMs [149]. In murine models, dietary iron restriction or iron chelation protected against cigarette smoke-induced emphysema, suggesting that limiting iron burden may reduce macrophage-driven oxidative damage [150]. Additional strategies include increasing macrophage expression of iron-export proteins such as ferroportin, or enhancing iron uptake via transferrin receptor modulation. These approaches aim to restore iron handling and mitigate iron-catalyzed oxidative injury [151]. By reducing iron-driven ROS production, such interventions could also lower the risk of ferroptosis in lung tissues, a mechanism implicated in both pulmonary fibrosis and emphysema [126].
4.5 Targeted drug delivery to lung macrophages
A recurring challenge in macrophage-targeted metabolic therapy is achieving cell-type specificity without systemic toxicity [152]. To this end, a variety of targeted delivery systems are under development that aim to direct therapeutic agents specifically to AMs. These systems leverage the phagocytic capacity of macrophages to preferentially uptake engineered carriers, such as nanoparticles or microspheres [153]. Inhalable formulations, including liposome-encapsulated drugs, have demonstrated preferential uptake by AMs [154]. For instance, in the context of tuberculosis, aerosolized rifampicin-loaded microspheres have been successfully used to deliver antibiotics directly into AMs [155]. Similar strategies may be adapted for metabolic modulation in chronic lung diseases. Nanoparticles carrying agents such as 2-deoxy-D-glucose, metformin, or antioxidants can be delivered via inhalation to achieve intracellular release within AMs. Surface modifications such as mannose coating further increase uptake efficiency by targeting the mannose receptor expressed on AMs [156].
Another promising modality involves inhaled nucleic acid therapeutics. Aerosolized small interfering RNAs (siRNAs) or antisense oligonucleotides can be designed to silence specific metabolic genes in AMs [157], such as HIF-1α or IRG1. This approach allows for selective attenuation of pathogenic signaling within pulmonary macrophages while minimizing effects on other cell types. In preclinical studies, combination therapies, such as co-delivery of iron chelators and nitrated fatty acids that activate anti-inflammatory pathways, have been shown to reprogram AMs toward a less injurious phenotype [153, 158].
Overall, these therapeutic strategies reflect a fundamental shift from broad immunosuppression (e.g., steroids) toward selective modulation of macrophage metabolic states. Their objective is no longer merely to suppress inflammation, but rather to restore physiological macrophage function by correcting underlying metabolic defects. This involves facilitating the resolution of inflammation, enhancing tissue repair, and improving pathogen elimination. While some strategies have demonstrated efficacy in animal models, their translation to human patients presents challenges. The redundancy of metabolic pathways means that macrophages might compensate for the inhibition of one pathway by upregulating others, and patient heterogeneity implies that a one-size approach may not fit all. Nevertheless, this customized regulation of macrophage immunometabolism holds great promise. It targets the core functional alterations of macrophages in chronic lung diseases, potentially restoring their capacity to promote homeostasis and eliminate pathogens, rather than simply shutting them down. Future inhaled therapies or smart drugs targeting macrophage metabolism could become adjuncts or alternatives to existing treatments for asthma, COPD, and IPF.
5. Conclusion and future directions
Over the past decade, significant advances have deepened our understanding of how metabolic adaptation shapes macrophage function in the lung. AMs exhibit remarkable metabolic plasticity: they can shift between OXPHOS, glycolysis, and lipid metabolism to meet context-dependent demands. In chronic lung diseases such as asthma, COPD, and IPF, this plasticity is pathologically reprogrammed, resulting in maladaptive phenotypes that perpetuate inflammation, tissue remodeling, or fibrosis. Each disease imposes a distinct metabolic signature on AMs. In asthma, macrophages are skewed toward fatty acid metabolism and lipid mediator imbalance. In COPD, iron overload, mitochondrial dysfunction, and HIF-1α-driven glycolysis impair microbial clearance while promoting oxidative stress. In IPF, macrophages display concurrent upregulation of glycolysis and FAO, coupled with loss of anti-inflammatory metabolites such as itaconate.
Despite these disease-specific profiles, a unifying theme is evident: chronic perturbations in immunometabolism underlie macrophage dysfunction and disease persistence. Targeting macrophage metabolism presents an opportunity to restore homeostatic function without broadly suppressing immune responses. Preclinical data suggest that modulating key metabolic pathways, such as glycolysis, FAO, antioxidant defense, and iron handling, can shift macrophages away from pro-inflammatory or pro-fibrotic states. Moreover, early efforts in drug delivery platforms, including inhalable nanoparticles and macrophage-specific carriers, have demonstrated potential for selective and localized intervention.
A major challenge lies in the heterogeneity of macrophage populations within the lung. Single-cell and spatial transcriptomic studies have begun to reveal distinct metabolic phenotypes among macrophage subsets in both health and disease [159]. To move beyond correlation, these insights must be integrated with direct metabolic measurements and functional models that capture human disease contexts. Lung organoids, precision-cut lung slices, and humanized mouse models may serve as critical platforms to test causal links between metabolic rewiring and disease progression.
Future therapies will require not only effective metabolic targets but also precise patient selection. Identifying metabolic biomarkers that reflect macrophage dysfunction could support personalized treatment strategies and improve clinical trial design. Ultimately, by restoring balanced immunometabolism, it may be possible to re-establish the macrophage's protective functions and interrupt the self-sustaining cycles of inflammation and injury that characterize chronic lung disease.
In conclusion, macrophage immunometabolism offers both a conceptual and mechanistic framework for comprehending chronic lung diseases at the cellular level. It also paves the way for a promising therapeutic approach that targets underlying functional impairments rather than merely addressing downstream symptoms. By correcting metabolic imbalance, it may be possible to restore macrophage homeostatic roles in host defense, resolution, and repair. Continued research at the intersection of immunology, metabolism, and respiratory biology will be key to turning this potential into clinical reality.
Abbreviations
AM: alveolar macrophage; AMPK: AMP-activated protein kinase; Arg1: arginase-1; ATP: adenosine triphosphate; CARKL: carbohydrate kinase-like protein; COPD: chronic obstructive pulmonary disease; CPT1: carnitine palmitoyl transferase 1; FAO: fatty acid oxidation; GLUT: glucose transporter; GM-CSF: granulocyte-macrophage colony-stimulating factor; HETE: hydroxy eicosatetraenoic acid; HIF: hypoxia-inducible factor; HO-1: heme oxygenase-1; IL: interleukin; IM: interstitial macrophage; iNOS: inducible nitric oxide synthase; IPF: idiopathic pulmonary fibrosis; IRF: interferon regulatory factor; IRG: immune-responsive gene; LPS: lipopolysaccharide; LT: leukotriene; mTOR: mechanistic target of rapamycin; NO: nitric oxide; NRF2: nuclear erythroid-related factor 2; OXPHOS: oxidative phosphorylation; PDGF: platelet-derived growth factor; PG: prostaglandin; PPAR-γ: peroxisome proliferator-activated receptor-γ; RNS: reactive nitrogen species; ROS: reactive oxygen species; TCA: tricarboxylic acid; TGF: transforming growth factor.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82474264, 82174170 to J.D.); the Shenzhen Municipal Government's “Three Famous Health Project” (to J.D.); and a horizontal collaboration with China Railway First Group Co., Ltd. (Xi'an, China), all undertaken under the auspices of the Institutes of Integrative Medicine at Fudan University.
Author contributions
J.D and Z.G developed the concept; C.X, M.Y, and Z.R drafted the manuscript; H.Y, X.H, H.Z, W.C, and Q.L edited the manuscript; C.X drawn the figures and tables. All authors reviewed and approved the final version before submission.
Competing interests
Jingcheng Dong's laboratory received research sponsorship from Sinopharm Taiji Group Co., Ltd. (Chongqing, China) and Inner Mongolia Otaqi Pharmaceutical Co., Ltd. (Ulanhot, China). The sponsors had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55:1564-80
2. Galvan-Pena S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420
3. Chen S, Saeed A, Liu Q, Jiang Q, Xu H, Xiao GG. et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8:207
4. Van den Bossche J, O'Neill LA, Menon D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017;38:395-406
5. Rodriguez-Morales P, Franklin RA. Macrophage phenotypes and functions: resolving inflammation and restoring homeostasis. Trends Immunol. 2023;44:986-98
6. Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol. 2022;19:384-408
7. Svedberg FR, Brown SL, Krauss MZ, Campbell L, Sharpe C, Clausen M. et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat Immunol. 2019;20:571-80
8. Sano H, Sohma H, Muta T, Nomura S, Voelker DR, Kuroki Y. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J Immunol. 1999;163:387-95
9. Ogger PP, Byrne AJ. Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol. 2021;14:282-95
10. Papaioannou O, Karampitsakos T, Barbayianni I, Chrysikos S, Xylourgidis N, Tzilas V. et al. Metabolic Disorders in Chronic Lung Diseases. Front Med (Lausanne). 2017;4:246
11. Sun JX, Xu XH, Jin L. Effects of Metabolism on Macrophage Polarization Under Different Disease Backgrounds. Front Immunol. 2022;13:880286
12. Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5'-monophosphate-activated protein kinase regulates IL-10-mediated anti-inflammatory signaling pathways in macrophages. J Immunol. 2015;194:584-94
13. Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771-84
14. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599:23-37
15. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70:1189-96
16. Hou F, Xiao K, Tang L, Xie L. Diversity of Macrophages in Lung Homeostasis and Diseases. Front Immunol. 2021;12:753940
17. Soucie EL, Weng Z, Geirsdottir L, Molawi K, Maurizio J, Fenouil R. et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science. 2016;351:aad5510
18. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M. et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792-804
19. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S. et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977-92
20. Shi T, Denney L, An H, Ho LP, Zheng Y. Alveolar and lung interstitial macrophages: Definitions, functions, and roles in lung fibrosis. J Leukoc Biol. 2021;110:107-14
21. Guilliams M, Svedberg FR. Does tissue imprinting restrict macrophage plasticity? Nat Immunol. 2021;22:118-27
22. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T. et al. Cell Origin Dictates Programming of Resident versus Recruited Macrophages during Acute Lung Injury. Am J Respir Cell Mol Biol. 2017;57:294-306
23. Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM. et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood. 2015;126:1357-66
24. Woods PS, Kimmig LM, Meliton AY, Sun KA, Tian Y, O'Leary EM. et al. Tissue-Resident Alveolar Macrophages Do Not Rely on Glycolysis for LPS-induced Inflammation. Am J Respir Cell Mol Biol. 2020;62:243-55
25. Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ. et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193:4245-53
26. Kulikauskaite J, Wack A. Teaching Old Dogs New Tricks? The Plasticity of Lung Alveolar Macrophage Subsets. Trends Immunol. 2020;41:864-77
27. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM. et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol. 2020;21:145-57
28. Bain CC, MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol. 2022;15:223-34
29. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 2014;15:1026-37
30. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019 363
31. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G. et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238-42
32. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD. et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834
33. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27:286-96
34. Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P. et al. mTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science. 2017;357:1014-21
35. Porsbjerg C, Melen E, Lehtimaki L, Shaw D. Asthma. Lancet. 2023;401:858-73
36. Xie C, Yang J, Gul A, Li Y, Zhang R, Yalikun M. et al. Immunologic aspects of asthma: from molecular mechanisms to disease pathophysiology and clinical translation. Front Immunol. 2024;15:1478624
37. Draijer C, Boorsma CE, Robbe P, Timens W, Hylkema MN, Ten Hacken NH. et al. Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J Allergy Clin Immunol. 2017;140:280-3 e3
38. Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl Res. 2018;191:1-14
39. Fricker M, Gibson PG. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J. 2017 50
40. Byrne AJ, Weiss M, Mathie SA, Walker SA, Eames HL, Saliba D. et al. A critical role for IRF5 in regulating allergic airway inflammation. Mucosal Immunol. 2017;10:716-26
41. Al-Khami AA, Ghonim MA, Del Valle L, Ibba SV, Zheng L, Pyakurel K. et al. Fuelling the mechanisms of asthma: Increased fatty acid oxidation in inflammatory immune cells may represent a novel therapeutic target. Clin Exp Allergy. 2017;47:1170-84
42. Stevenson ER, Smith LC, Wilkinson ML, Lee SJ, Gow AJ. Etiology of lipid-laden macrophages in the lung. Int Immunopharmacol. 2023;123:110719
43. Little I, Bersie S, Redente EF, McCubbrey AL, Tarling EJ. Alveolar macrophages: guardians of the alveolar lipid galaxy. Curr Opin Lipidol. 2025;36:153-62
44. Calhoun WJ, Bush RK. Enhanced reactive oxygen species metabolism of airspace cells and airway inflammation follow antigen challenge in human asthma. J Allergy Clin Immunol. 1990;86:306-13
45. Xia Z, Zhong W. Immune Regulation of Heme Oxygenase-1 in Allergic Airway Inflammation. Antioxidants (Basel). 2022 11
46. Park HS, Kim SR, Lee YC. Impact of oxidative stress on lung diseases. Respirology. 2009;14:27-38
47. Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84:581-90
48. Goretzki A, Lin YJ, Schulke S. Immune metabolism in allergies, does it matter?-A review of immune metabolic basics and adaptations associated with the activation of innate immune cells in allergy. Allergy. 2021;76:3314-31
49. Qin Z, Chen Y, Wang Y, Xu Y, Liu T, Mu Q. et al. Immunometabolism in the pathogenesis of asthma. Immunology. 2024;171:1-17
50. Damon M, Chavis C, Daures JP, Crastes de Paulet A, Michel FB, Godard P. Increased generation of the arachidonic metabolites LTB4 and 5-HETE by human alveolar macrophages in patients with asthma: effect in vitro of nedocromil sodium. Eur Respir J. 1989;2:202-9
51. Huynh ML, Malcolm KC, Kotaru C, Tilstra JA, Westcott JY, Fadok VA. et al. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med. 2005;172:972-9
52. Zheng W, Zhou Z, Guo X, Zuo X, Zhang J, An Y. et al. Efferocytosis and Respiratory Disease. Int J Mol Sci. 2023 24
53. Vercelli D. Arginase: marker, effector, or candidate gene for asthma? J Clin Invest. 2003;111:1815-7
54. Chang CI, Zoghi B, Liao JC, Kuo L. The involvement of tyrosine kinases, cyclic AMP/protein kinase A, and p38 mitogen-activated protein kinase in IL-13-mediated arginase I induction in macrophages: its implications in IL-13-inhibited nitric oxide production. J Immunol. 2000;165:2134-41
55. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front Immunol. 2014;5:532
56. Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol Rev. 2018;98:641-65
57. Ricciardolo FL, Zaagsma J, Meurs H. The therapeutic potential of drugs targeting the arginase pathway in asthma. Expert Opin Investig Drugs. 2005;14:1221-31
58. Guan F, Wang R, Yi Z, Luo P, Liu W, Xie Y. et al. Tissue macrophages: origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduct Target Ther. 2025;10:93
59. Ma H, Gao L, Chang R, Zhai L, Zhao Y. Crosstalk between macrophages and immunometabolism and their potential roles in tissue repair and regeneration. Heliyon. 2024;10:e38018
60. Hough KP, Curtiss ML, Blain TJ, Liu RM, Trevor J, Deshane JS. et al. Airway Remodeling in Asthma. Front Med (Lausanne). 2020;7:191
61. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. 2016;38:409-23
62. Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol. 2012;5:605-9
63. Liu Y, Zhang M, Wang T, Zhang J. Reactive Oxygen Species in Asthma: Regulators of Macrophage Polarization and Therapeutic Implications: A Narrative Review. J Asthma Allergy. 2025;18:1129-46
64. Perez S, Rius-Perez S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants (Basel). 2022 11
65. Jiang Z, Zhu L. Update on the role of alternatively activated macrophages in asthma. J Asthma Allergy. 2016;9:101-7
66. Specjalski K, Chelminska M, Jassem E. YKL-40 protein correlates with the phenotype of asthma. Lung. 2015;193:189-94
67. Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R. et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N Engl J Med. 2008;358:1682-91
68. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399:2227-42
69. Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA. et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076
70. Ni L, Dong C. Roles of Myeloid and Lymphoid Cells in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Front Immunol. 2018;9:1431
71. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138:16-27
72. Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest. 2012;141:1055-62
73. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144:266-73
74. Bewley MA, Preston JA, Mohasin M, Marriott HM, Budd RC, Swales J. et al. Impaired Mitochondrial Microbicidal Responses in Chronic Obstructive Pulmonary Disease Macrophages. Am J Respir Crit Care Med. 2017;196:845-55
75. McGuinness AJ, Sapey E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J Clin Med. 2017 6
76. Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16:534-54
77. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I. et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. 2019 54
78. Narala VR, Narala SR, Aiya Subramani P, Panati K, Kolliputi N. Role of mitochondria in inflammatory lung diseases. Front Pharmacol. 2024;15:1433961
79. O'Beirne SL, Kikkers SA, Oromendia C, Salit J, Rostmai MR, Ballman KV. et al. Alveolar Macrophage Immunometabolism and Lung Function Impairment in Smoking and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2020;201:735-9
80. Russell KE, Chung KF, Clarke CJ, Durham AL, Mallia P, Footitt J. et al. The MIF Antagonist ISO-1 Attenuates Corticosteroid-Insensitive Inflammation and Airways Hyperresponsiveness in an Ozone-Induced Model of COPD. PLoS One. 2016;11:e0146102
81. Chen X, Tang Y, Wang W, Zhu Y. [Heterogeneity of airway macrophage in different clinical phenotypes of COPD patients with frequent or infrequent exacerbations]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2019;44:1376-84
82. Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One. 2010;5:e9224
83. Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. 2014;5:435
84. Maarsingh H, Pera T, Meurs H. Arginase and pulmonary diseases. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:171-84
85. Bhowmik A, Seemungal TA, Donaldson GC, Wedzicha JA. Effects of exacerbations and seasonality on exhaled nitric oxide in COPD. Eur Respir J. 2005;26:1009-15
86. Agusti AG, Villaverde JM, Togores B, Bosch M. Serial measurements of exhaled nitric oxide during exacerbations of chronic obstructive pulmonary disease. Eur Respir J. 1999;14:523-8
87. Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med. 2000;162:701-6
88. Gebel S, Gerstmayer B, Kuhl P, Borlak J, Meurrens K, Muller T. The kinetics of transcriptomic changes induced by cigarette smoke in rat lungs reveals a specific program of defense, inflammation, and circadian clock gene expression. Toxicol Sci. 2006;93:422-31
89. Cloonan SM, Mumby S, Adcock IM, Choi AMK, Chung KF, Quinlan GJ. The "Iron"-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2017;196:1103-12
90. Baker JM, Hammond M, Dungwa J, Shah R, Montero-Fernandez A, Higham A. et al. Red Blood Cell-Derived Iron Alters Macrophage Function in COPD. Biomedicines. 2021 9
91. Zhang WZ, Butler JJ, Cloonan SM. Smoking-induced iron dysregulation in the lung. Free Radic Biol Med. 2019;133:238-47
92. Philippot Q, Deslee G, Adair-Kirk TL, Woods JC, Byers D, Conradi S. et al. Increased iron sequestration in alveolar macrophages in chronic obstructive pulmonary disease. PLoS One. 2014;9:e96285
93. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K. et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145
94. Ho T, Nichols M, Nair G, Radford K, Kjarsgaard M, Huang C. et al. Iron in airway macrophages and infective exacerbations of chronic obstructive pulmonary disease. Respir Res. 2022;23:8
95. Yao M, Liu Z, Zhao W, Song S, Huang X, Wang Y. Ferroptosis in idiopathic pulmonary fibrosis: mechanisms, impact, and therapeutic opportunities. Front Immunol. 2025;16:1567994
96. Wugk S, Cloonan SM. Iron-loaded alveolar macrophages alter alveolar type 2 epithelial cell function in COPD. ERJ Open Research. 11: TP237.
97. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA. et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22:163-74
98. Paone G, Conti V, Vestri A, Leone A, Puglisi G, Benassi F. et al. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis Markers. 2011;31:91-100
99. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33:101544
100. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941-52
101. Moss BJ, Ryter SW, Rosas IO. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu Rev Pathol. 2022;17:515-46
102. Idiopathic Pulmonary Fibrosis Clinical Research N, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968-77
103. Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res. 2018;19:170
104. Xie N, Cui H, Ge J, Banerjee S, Guo S, Dubey S. et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2017;313:L834-L44
105. El-Chemaly S, Malide D, Yao J, Nathan SD, Rosas IO, Gahl WA. et al. Glucose transporter-1 distribution in fibrotic lung disease: association with [(1)(8)F]-2-fluoro-2-deoxyglucose-PET scan uptake, inflammation, and neovascularization. Chest. 2013;143:1685-91
106. Pan BS, Hsu CC, Wu HE, Chen YR, Zhou X, Wang SC. et al. Glucose metabolism and its direct action in cancer and immune regulation: opportunities and challenges for metabolic targeting. J Biomed Sci. 2025;32:71
107. Tufail M, Jiang CH, Li N. Altered metabolism in cancer: insights into energy pathways and therapeutic targets. Mol Cancer. 2024;23:203
108. Gu L, Larson Casey JL, Andrabi SA, Lee JH, Meza-Perez S, Randall TD. et al. Mitochondrial calcium uniporter regulates PGC-1alpha expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019;26:101307
109. Rajesh R, Atallah R, Barnthaler T. Dysregulation of metabolic pathways in pulmonary fibrosis. Pharmacol Ther. 2023;246:108436
110. Pokhreal D, Crestani B, Helou DG. Macrophage Implication in IPF: Updates on Immune, Epigenetic, and Metabolic Pathways. Cells. 2023 12
111. Li J, Zhai X, Sun X, Cao S, Yuan Q, Wang J. Metabolic reprogramming of pulmonary fibrosis. Front Pharmacol. 2022;13:1031890
112. Bueno M, Calyeca J, Rojas M, Mora AL. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020;33:101509
113. Wu R, Chen F, Wang N, Tang D, Kang R. ACOD1 in immunometabolism and disease. Cell Mol Immunol. 2020;17:822-33
114. Ogger PP, Albers GJ, Hewitt RJ, O'Sullivan BJ, Powell JE, Calamita E. et al. Itaconate controls the severity of pulmonary fibrosis. Sci Immunol. 2020 5
115. Gonzalez-Gonzalez FJ, Chandel NS, Jain M, Budinger GRS. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl Res. 2017;190:61-8
116. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313
117. Murthy S, Adamcakova-Dodd A, Perry SS, Tephly LA, Keller RM, Metwali N. et al. Modulation of reactive oxygen species by Rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2009;297:L846-55
118. Saleh D, Barnes PJ, Giaid A. Increased production of the potent oxidant peroxynitrite in the lungs of patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:1763-9
119. Yamazaki C, Hoshino J, Sekiguchi T, Hori Y, Miyauchi S, Mizuno S. et al. Production of superoxide and nitric oxide by alveolar macrophages in the bleomycin-induced interstitial pneumonia mice model. Jpn J Pharmacol. 1998;78:69-73
120. Puxeddu E, Comandini A, Cavalli F, Pezzuto G, D'Ambrosio C, Senis L. et al. Iron laden macrophages in idiopathic pulmonary fibrosis: the telltale of occult alveolar hemorrhage? Pulm Pharmacol Ther. 2014;28:35-40
121. Allden SJ, Ogger PP, Ghai P, McErlean P, Hewitt R, Toshner R. et al. The Transferrin Receptor CD71 Delineates Functionally Distinct Airway Macrophage Subsets during Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019;200:209-19
122. Maus M, Lopez-Polo V, Mateo L, Lafarga M, Aguilera M, De Lama E. et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat Metab. 2023;5:2111-30
123. Lee J, Arisi I, Puxeddu E, Mramba LK, Amicosante M, Swaisgood CM. et al. Bronchoalveolar lavage (BAL) cells in idiopathic pulmonary fibrosis express a complex pro-inflammatory, pro-repair, angiogenic activation pattern, likely associated with macrophage iron accumulation. PLoS One. 2018;13:e0194803
124. Song L, Gao F, Man J. Ferroptosis: the potential key roles in idiopathic pulmonary fibrosis. Eur J Med Res. 2025;30:341
125. Zhu Y, Chang J, Tan K, Huang SK, Liu X, Wang X. et al. Clioquinol Attenuates Pulmonary Fibrosis through Inactivation of Fibroblasts via Iron Chelation. Am J Respir Cell Mol Biol. 2021;65:189-200
126. Jiang S, Shi X, Kong X, Chen Y, Xu W, Hao M. et al. Iron dysregulation and ferroptosis are associated with pulmonary fibrosis: Insight from idiopathic pulmonary fibrosis, systemic sclerosis, and COVID-19 patients. J Trace Elem Med Biol. 2025;91:127728
127. Oldham JM, Ma SF, Martinez FJ, Anstrom KJ, Raghu G, Schwartz DA. et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2015;192:1475-82
128. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH. et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3:78ra32
129. Carlstrom KE, Ewing E, Granqvist M, Gyllenberg A, Aeinehband S, Enoksson SL. et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat Commun. 2019;10:3081
130. Palsson-McDermott EM, O'Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300-14
131. Albers GJ, Michalaki C, Ogger PP, Lloyd AF, Causton B, Walker SA. et al. Airway macrophage glycolysis controls lung homeostasis and responses to aeroallergen. Mucosal Immunol. 2025;18:121-34
132. Geng J, Liu Y, Dai H, Wang C. Fatty Acid Metabolism and Idiopathic Pulmonary Fibrosis. Front Physiol. 2021;12:794629
133. Tian Z, Zheng H, Fan Y, Li B, Huang Z, Wang M. et al. Upregulated Hexokinase-2 in Airway Epithelium Regulates Apoptosis and Drives Inflammation in Asthma via Peptidylprolyl Isomerase F. Cells. 2025 14
134. Zhao Q, Chu Z, Zhu L, Yang T, Wang P, Liu F. et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front Immunol. 2017;8:637
135. Gu L, Surolia R, Larson-Casey JL, He C, Davis D, Kang J. et al. Targeting Cpt1a-Bcl-2 interaction modulates apoptosis resistance and fibrotic remodeling. Cell Death Differ. 2022;29:118-32
136. Septembre-Malaterre A, Boina C, Douanier A, Gasque P. Deciphering the Antifibrotic Property of Metformin. Cells. 2022 11
137. Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J. et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res. 2016;17:107
138. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K. et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med. 2018;24:1121-7
139. Spagnolo P, Kreuter M, Maher TM, Wuyts W, Bonella F, Corte TJ. et al. Metformin Does Not Affect Clinically Relevant Outcomes in Patients with Idiopathic Pulmonary Fibrosis. Respiration. 2018;96:314-22
140. Dong L, Wang Y, Chen H, Li Z, Xu X, Zhou J. et al. MTOR Suppresses Cigarette Smoke-Induced Airway Inflammation and MMP12 Expression in Macrophage in Chronic Obstructive Pulmonary Disease. Int J Chron Obstruct Pulmon Dis. 2024;19:269-79
141. Huckestein BR, Zeng K, Westcott R, Alder JK, Antos D, Kolls JK. et al. Mammalian Target of Rapamycin Complex 1 Activation in Macrophages Contributes to Persistent Lung Inflammation following Respiratory Tract Viral Infection. Am J Pathol. 2024;194:384-401
142. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L. et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018 3
143. Goyal A, Chopra V, Garg K, Sharma S. Mechanisms coupling the mTOR pathway to chronic obstructive pulmonary disease (COPD) pathogenesis. Cytokine Growth Factor Rev. 2025;82:55-69
144. Kytikova O, Novgorodtseva T, Denisenko Y, Antonyuk M, Gvozdenko T. Pro-Resolving Lipid Mediators in the Pathophysiology of Asthma. Medicina (Kaunas). 2019 55
145. Paggiaro P, Bacci E. Montelukast in asthma: a review of its efficacy and place in therapy. Ther Adv Chronic Dis. 2011;2:47-58
146. Lai CK, Phillips GD, Jenkins JR, Holgate ST. The effect of inhaled 15-(s)-hydroxyeicosatetraenoic acid (15-HETE) on airway calibre and non-specific responsiveness in normal and asthmatic human subjects. Eur Respir J. 1990;3:38-45
147. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349-61
148. Tian Y, Sun J, Jiao D, Zhang W. The potential role of n-3 fatty acids and their lipid mediators on asthmatic airway inflammation. Front Immunol. 2024;15:1488570
149. Chen WJ, Kung GP, Gnana-Prakasam JP. Role of Iron in Aging Related Diseases. Antioxidants (Basel). 2022 11
150. Meng D, Zhu C, Jia R, Li Z, Wang W, Song S. The molecular mechanism of ferroptosis and its role in COPD. Front Med (Lausanne). 2022;9:1052540
151. Ru Q, Li Y, Chen L, Wu Y, Min J, Wang F. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct Target Ther. 2024;9:271
152. Liu H, Lv H, Duan X, Du Y, Tang Y, Xu W. Advancements in Macrophage-Targeted Drug Delivery for Effective Disease Management. Int J Nanomedicine. 2023;18:6915-40
153. Hu G, Guo M, Xu J, Wu F, Fan J, Huang Q. et al. Nanoparticles Targeting Macrophages as Potential Clinical Therapeutic Agents Against Cancer and Inflammation. Front Immunol. 2019;10:1998
154. Parikh R, Dalwadi S, Aboti P, Patel L. Inhaled microparticles of antitubercular antibiotic for in vitro and in vivo alveolar macrophage targeting and activation of phagocytosis. J Antibiot (Tokyo). 2014;67:387-94
155. Garcia-Contreras L, Sethuraman V, Kazantseva M, Godfrey V, Hickey AJ. Evaluation of dosing regimen of respirable rifampicin biodegradable microspheres in the treatment of tuberculosis in the guinea pig. J Antimicrob Chemother. 2006;58:980-6
156. Costa A, Sarmento B, Seabra V. Mannose-functionalized solid lipid nanoparticles are effective in targeting alveolar macrophages. Eur J Pharm Sci. 2018;114:103-13
157. Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H. et al. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol Ther. 2010;18:993-1001
158. Zheng M, Zhu W, Gao F, Zhuo Y, Zheng M, Wu G. et al. Novel inhalation therapy in pulmonary fibrosis: principles, applications and prospects. J Nanobiotechnology. 2024;22:136
159. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163-72
160. Marcos MC, Cisneros Serrano C. What is the added value of FeNO as T2 biomarker? Front Allergy. 2022;3:957106
161. Mohan S, Ho T, Kjarsgaard M, Radford K, Borhan AS, Thabane L. et al. Hemosiderin in sputum macrophages may predict infective exacerbations of chronic obstructive pulmonary disease: a retrospective observational study. BMC Pulm Med. 2017;17:60
162. Lv MY, Qiang LX, Wang BC, Zhang YP, Li ZH, Li XS. et al. Complex Evaluation of Surfactant Protein A and D as Biomarkers for the Severity of COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:1537-52
163. Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR. et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:717-23
164. Elhai M, Hoffmann-Vold AM, Avouac J, Pezet S, Cauvet A, Leblond A. et al. Performance of Candidate Serum Biomarkers for Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2019;71:972-82
165. Bauer Y, White ES, de Bernard S, Cornelisse P, Leconte I, Morganti A. et al. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2017 3
166. Chung C, Kim J, Cho HS, Kim HC. Baseline serum Krebs von den Lungen-6 as a biomarker for the disease progression in idiopathic pulmonary fibrosis. Sci Rep. 2022;12:8564
167. d'Alessandro M, Conticini E, Bergantini L, Mazzei MA, Bellisai F, Selvi E. et al. Krebs von den Lungen-6 as biomarker of the new progressive fibrotic phenotype of interstitial lung disease. Tissue Cell. 2024;90:102516
168. He X, Ji J, Zheng D, Luo Z, Luo L, Guo L. Serum surfactant protein D as a significant biomarker for predicting occurrence, progression, acute exacerbation, and mortality in interstitial lung disease: a systematic review and meta-analysis. Front Immunol. 2025;16:1450798
169. Suchankova M, Zsemlye E, Urban J, Barath P, Kohutova L, Sivakova B. et al. The bronchoalveolar lavage fluid CD44 as a marker for pulmonary fibrosis in diffuse parenchymal lung diseases. Front Immunol. 2024;15:1479458
170. Urban J, Suchankova M, Ganovska M, Leksa V, Sandor F, Tedlova E. et al. The Role of CX3CL1 and ADAM17 in Pathogenesis of Diffuse Parenchymal Lung Diseases. Diagnostics (Basel). 2021 11
171. Todd JL, Neely ML, Overton R, Durham K, Gulati M, Huang H. et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir Res. 2019;20:227
Author contact
![]() Corresponding authors: E-mail: Jingcheng Dong (jcdong2004com) or Zhen Gao (gaozhenedu.cn).
Corresponding authors: E-mail: Jingcheng Dong (jcdong2004com) or Zhen Gao (gaozhenedu.cn).