Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Endometrial Senescence:...
3. Characteristics of Cellular...
4. Endometrial-Related Diseases...
5. Summary and Discussion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(15):6745-6758. doi:10.7150/ijbs.123036 This issue Cite
Review
Cellular Senescence in Endometrium: A Pivotal Regulator in Physiological Remodeling and Pathological Disorders
Zi-Yang Yan1,2, Wen-Jie Zhou3, Jiang-Feng Ye4, Feng Xie5, ![]() , Chun-Xue Zhang2,6,
, Chun-Xue Zhang2,6, ![]() , Ming-Qing Li1,2,
, Ming-Qing Li1,2, ![]()
1. Department of Reproductive Immunology, The International Peace Maternity and Child Health Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200030, China.
2. Shanghai Key Laboratory of Embryo Original Diseases, Shanghai 200030, China.
3. Reproductive Medical Center, Department of Obstetrics and Gynecology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
4. Institute for Molecular and Cell Biology, Agency for Science, Technology and Research, Singapore.
5. Medical Center of Diagnosis and Treatment for Cervical Diseases, Obstetrics and Gynecology Hospital of Fudan University, 200433, Shanghai, China.
6. Central Laboratory, The International Peace Maternity and Child Health Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 201112, China.
Received 2025-8-3; Accepted 2025-10-6; Published 2025-10-20
Abstract

As a highly dynamic tissue, the endometrium undergoes complex remodeling during the menstrual cycle and pregnancy. Recent studies have revealed that cellular senescence plays a pivotal role in both physiological renewal (e.g., menstrual shedding, decidualization) and pathological disorders (e.g., endometriosis, intrauterine adhesions, thin endometrium) of the endometrium. Under physiological conditions, senescent cells contribute to tissue repair and embryo implantation through precise regulation. However, pathological accumulation of senescent cells drives chronic inflammation, fibrosis, and reproductive dysfunction. Here we aim to summarize the mechanism indicating endometrial senescence and elucidating their pleiotropic roles in both physiological homeostasis and pathological progression, while discussing emerging therapeutic strategies for clinical translation—including senolytics and SASP inhibitors.
Keywords: senescence, endometrium, decidualization, endometriosis, intrauterine adhesions, thin endometrium, implantation failure, spontaneous abortion
1. Introduction
The endometrium is a multicellular mucosal tissue lining the uterine cavity, composed of epithelial cells, stromal cells, immune cells, and other cellular components. Functionally, the human endometrium comprises two distinct layers: the hormone-responsive functional layer that undergoes cyclical shedding, and the basalis layer responsible for physiological regeneration (1). Across the proliferative, secretory, and menstrual phases, estradiol drives epithelial and stromal proliferation, while progesterone induces decidualization of endometrial stromal cells (ESCs) in response to signals such as cAMP (2-4). Decidualization is strictly temporally regulated: during a brief proinflammatory phase, ESCs secrete chemokines and interleukins supports endometrial receptivity followed by a strictly time-locked 2-4-day implantation window (1,5-8), providing an appropriate niche for embryo implantation (9). Upon embryo implantation, this specialized tissue provides critical support for embryonic development while protecting maternal tissues from excessive trophoblast invasion (3). During this stage, local immunity shifts toward an anti-inflammatory, tolerogenic milieu (10-12). Missing the critical window significantly elevates the risk of adverse pregnancy outcomes (13). Thus, endometrium serves as the cornerstone of reproductive health while simultaneously representing a potential site for pathological development (14,15). Endometrial dysfunction can lead to various pathological conditions including endometriosis, implantation failure, and spontaneous abortion (16), significantly impacting both fertility and quality of life for numerous women of reproductive age. Physiologically, within these cyclical transitions, the tissue deploys a transient, rapidly cleared senescence program that facilitates decidualization, shedding, and repair; when clearance fails or pro-senescent cues persist, cellular senescence becomes persistent, promoting inflammation, fibrosis, and immune imbalance that link normal cycling to pathology.
By contrast, within the endometrium cellular senescence is not inherently detrimental. Cellular senescence represents an irreversible cell cycle arrest state that occurs in diverse physiological and pathological processes, including tissue remodeling, injury response, fibrosis, carcinogenesis, and maintenance of immune microenvironment homeostasis. Recent studies have demonstrated that the high turnover state of the endometrium is closely associated with dysregulation of cell cycle control, and cellular senescence may play a pivotal role in various endometrial-related pathologies. Nevertheless, current research is hampered by small sample sizes, reliance on in vitro systems, and limited in vivo or translational validation. Moreover, the senescence-associated secretory phenotype (SASP) mechanisms in endometrium remain incompletely defined, and standardized senescence markers in the endometrium are lacking, complicating cross-study comparisons. These gaps underscore the need for further investigation on endometrial senescence. Here, we review cellular senescence in endometrial diseases, highlighting the current understanding of senescence signatures and molecular mechanisms, and discuss the potential of emerging therapeutic strategies such as senolytics and SASP inhibitors for clinical translation.
2. Endometrial Senescence: Hallmarks and Endometrium-Specific Features
2.1 Hallmarks and Signaling of Cellular Senescence
In vitro cultured endometrial senescent cells (SNCs) typically exhibit characteristic morphological alterations, including cellular enlargement, flattened morphology, and cytoplasmic vacuolization(7,18). In contrast, in vivo senescent cells often maintain their normal morphology as determined by the surrounding tissue architecture. At the molecular level, characteristic senescence-associated alterations serve as definitive biomarkers for identifying cellular senescence (19). Senescent cells characteristically exhibit elevated expression of senescence-associated β-galactosidase (SA-β-gal), which is widely utilized for senescence identification. Additionally, molecular components of senescence-associated signaling pathways - particularly the gene and protein expression of p16 (CDKN2A) and p21 (CDKN1A) are considered classical biomarkers of cellular senescence. SNCs maintain active secretory capacity, releasing a spectrum of bioactive molecules including interleukins, chemokines, and growth factors that collectively constitute SASP. This SASP generates inflammation-related signals resembling immune responses (20) and mediates paracrine interactions with the extracellular microenvironment.
Multiple studies have demonstrated that SASP plays a dual role: Acute senescence transiently generates SASP components that recruit immune cells for rapid clearance of SNCs, thereby facilitating tissue remodeling processes such as embryonic development and wound healing (19). However, excessive cellular senescence or impaired clearance leads to chronic senescence (21,22), where in persistent SASP signaling establishes a proinflammatory microenvironment, promotes tissue fibrosis, and potentially facilitates tumorigenesis (22,23).The SASP amplifies local inflammatory responses through secreted cytokines such as IL-1β, IL-6, and IL-8, which activate NF-κB and JAK/STAT signaling pathways. This process may contribute to the establishment of a chronic inflammatory microenvironment and facilitate senescence propagation to neighboring cells (21).
Through the secretion of chemokines (e.g. CCL2 and CXCL1), SASP recruits immune cells such as macrophages, T cells, and NK cells. These recruited cells subsequently release additional inflammatory factors and reactive oxygen species (ROS), thereby exacerbating both local and systemic inflammatory responses (24). SASP also mediates extracellular matrix (ECM) remodeling through the secretion of proteases and matrix metalloproteinases (MMPs), thereby disrupting tissue integrity and promoting cancer metastasis (23). Furthermore, SASP derived profibrotic factors, including transforming growth factor-β (TGF-β) and plasminogen activator inhibitor-1 (PAI-1), directly drive fibrotic progression (25,26).
2.2 Endometrium-specific features and physiological roles
The endometrium, as a highly dynamic tissue, undergoes approximately 400 cyclical changes during a woman's reproductive lifespan (14). Its periodic regeneration and shedding rely on precisely regulated mechanisms of cellular proliferation, differentiation, and clearance. Recent studies have revealed that controlled cellular senescence occurs during endometrial tissue turnover, serves dual roles: maintaining tissue homeostasis and contributing to functional disorders such as infertility, recurrent implantation failure (RIF), and recurrent pregnancy loss (RPL) (1) (Figure 1). The unique physiological context of the endometrium imparts specific characteristics to its senescent cells. For instance, endometrial senescence is tightly coupled to steroid hormone fluctuations, particularly the withdrawal of progesterone, which acts as a key trigger for senescence-associated inflammatory responses during menstruation (15,27). Furthermore, the senescence program in decidualizing stromal cells is orchestrated by the FOXO1-DIO2 axis and is crucial for establishing endometrial receptivity and facilitating embryo implantation (7,28). Following the initiation of decidualization, transcription factors such as FOXO1 induce the expression of decidualization marker genes, including prolactin (PRL) and insulin-like growth factor binding protein 1 (IGFBP1) (1,29). The immune microenvironment of the endometrium, rich in specialized uterine natural killer (uNK) cells, is particularly adept at surveilling and clearing senescent cells, a process critical for cyclical renewal and pregnancy maintenance (7,16). Therefore, positioned at the interface between physiological renewal and pathological remodeling, endometrial senescence serves as a key mechanistic nexus connecting cellular senescence with both physiological and pathological processes in endometrium.
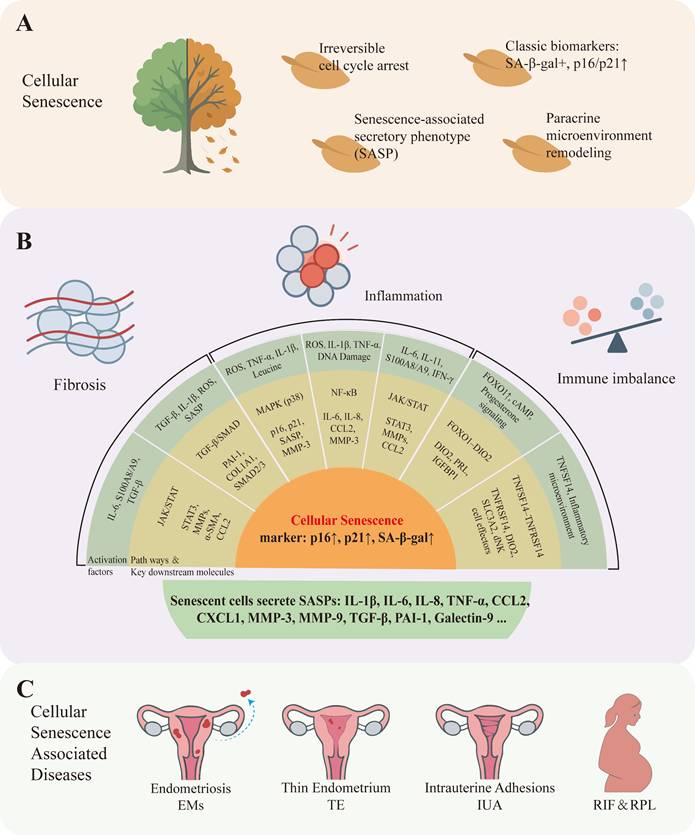
Cellular senescence markers in endometrium and endometrium-related disorders. A: According to existing research, cellular senescence has the following characteristics: Irreversible cell cycle arrest, Classic biomarkers such as SA-β-gal positive, p16/p21 upward, secrete a variety of cytokines collectively constitute the senescence-associated secretory phenotype (SASP), remodeling the cellular microenvironment through paracrine signaling. B: Upstream signaling pathways that drive cellular senescence in the endometrium can be grouped by functional role into three categories: (1) inflammation-related pathways, e.g., MAPK (mitogen-activated protein kinase), NF-κB (nuclear factor kappa-B), and JAK/STAT (Janus kinase / signal transducer and activator of transcription); (2) fibrosis-related pathways, e.g., TGF-β (transforming growth factor-β) and downstream SMAD signaling; and (3) immune-imbalance-related axes, e.g., the FOXO1-DIO2 axis and TNFSF14-TNFRSF14 signaling. Activation of these pathways promotes entry into a senescent state. Typical senescence phenotypes include increased expression of p16 INK4a (CDKN2A), p21 CIP1 (CDKN1A), and senescence-associated β-galactosidase (SA-β-gal), together with secretion of senescence-associated secretory phenotype (SASP) factors (for example IL-1β, IL-6, IL-8, CCL2) that mediate inflammation, extracellular matrix remodeling, and paracrine senescence. C: Numerous studies have shown that cellular senescence can cause many endometrial diseases such as endometriosis, thin endometrium, intrauterine adhesions, recurrent implantation failure and recurrent pregnancy loss.
3. Characteristics of Cellular Senescence in Physiological Endometrium
3.1 Menstrual Cycle Dynamics and Cellular Senescence
During the proliferative phase, rising estrogen levels drive endometrial proliferation, with mitotic activity being the most prominent feature of stromal and perivascular cells (27,30). Estrogen upregulates telomerase activity (31), thereby delaying cellular senescence. Upon entering the secretory phase, progesterone becomes the dominant regulatory hormone that drives endometrial differentiation to prepare for potential embryo implantation. The most prominent morphological change during this phase is the initiation of ESC decidualization. Following successful embryo implantation, cellular senescence within decidualized cells plays critical roles in modulating embryonic invasion depth and establishing local immune tolerance.
During the menstrual phase, endometrial cells progressively enter a senescent state accompanied by localized inflammatory responses and tissue remodeling, which collectively facilitate the shedding of the functional layer (27). Apoptosis plays a pivotal role in this phase by eliminating accumulated SNCs from the functional layer, thereby maintaining endometrial cellular homeostasis (32). Single-cell transcriptomic analyses have demonstrated that by day 7 post-luteinizing hormone surge (LH+7), corresponding to implantation window, ESCs exhibit significantly elevated senescence markers. This increase in senescence, coupled with moderate secretion of inflammatory factors, commonly prepare for the decidualization. By the premenstrual phase (LH+11), senescence signals intensify further and demonstrates a significant correlation with localized inflammation. (1,33). Progesterone withdrawal relieves the suppression of the NF-κB pathway (15), leading to upregulation of specific inflammatory factors by SNCs (2,14). These inflammatory mediators subsequently recruit macrophages and neutrophils, which in turn secrete MMPs (including MMP-9) to facilitate ECM degradation and subsequent shedding of the functional layer (34). Uterine natural killer cells (uNK) possess the capacity to recognize and eliminate senescent decidual cells, thereby fulfilling a critical immune surveillance function (7). This cyclical senescence process, coupled with subsequent inflammatory-repair mechanisms, forms an intrinsic regulatory circuit governing endometrial shedding and regeneration. This unique physiological program enables complete renewal of the functional layer, representing a distinct biological phenomenon compared to scar-forming repair processes characteristic of other tissues (35).
3.2 Decidualization and Cellular Senescence
During the mid-luteal phase, a series of programmed changes occur to facilitate embryo implantation and pregnancy maintenance, involving cellular differentiation, matrix remodeling, and immune regulation (7). Accumulating evidence indicates that programmed cellular senescence constitutes an integral component of normal decidualization (1). During decidualization, a subset of stromal cells exhibits classical senescence markers, including stabilized p53 expression, upregulated p16, and detectable SA-β-Gal activity. These cells are operationally defined as senescent-like decidual cells (snDCs) (1). Experimental evidence demonstrates that controlled cellular senescence is essential for successful decidualization: rapamycin-mediated inhibition of senescence signaling concomitantly suppresses expression of decidualization marker genes PRL and IGFBP1(18,29). Acute senescence of decidual cell subpopulations is induced by the FOXO1-DIO2 signaling axis (7,28). Knockdown of FOXO1 suppresses DIO2 expression and attenuates decidual senescence in ESCs, whereas FOXO1 activation upregulates DIO2 expression and promotes decidual senescence (28).
Furthermore, the orderly regulation of cellular senescence significantly impacts embryo implantation capacity. SnDCs exhibit a classic acute senescence phenotype (7), and their secreted SASP factors enhance the initial inflammatory response during decidualization, thereby inducing secondary senescence (36). On one hand, SASP factors promote moderate decidual expansion, induce the expression of implantation-essential factors, and recruit immune cells, thereby enhancing endometrial receptivity (1,37); On the other hand, SNCs alter the mechanical and immune properties of decidual tissue, providing the structural and spatial conditions required for embryo invasion (7). SnDCs recruit and activate decidual natural killer cells (dNKs), which eliminate SnDCs via exocytosis of perforin and granzymes. This process facilitates embryo penetration through structurally loosened regions of the decidual barrier, enabling successful invasion and implantation (38,39). Excessive suppression of cellular stress and senescent subpopulations during early decidualization may lead to over-stabilization of decidual tissue, resulting in insufficient tissue remodeling dynamics and ultimately impairing normal embryo implantation (18). However, excessive accumulation of uncleared SNCs in decidual tissue leads to overproduction of SASP factors, which triggers disproportionately strong inflammatory and matrix degradation responses (18). This subsequently attenuates decidual cell responsiveness to progesterone and elevates the risk of pregnancy failure (36,40).
In summary, cellular senescence-like changes during decidualization constitute a critical process for pregnancy establishment, where precise regulation is essential for successful embryo implantation and pregnancy maintenance (36). Dysregulated senescence may lead to decidualization defects, resulting in pregnancy complications such as implantation failure and recurrent miscarriage (28,41). Furthermore, the functional crosstalk between snDCs and dNK cells plays a pivotal role in maintaining immune homeostasis during early pregnancy (16).
4. Endometrial-Related Diseases and Senescence Characteristics in Pathological and Therapeutical Conditions
Under physiological conditions, cellular senescence participates in the cyclic regeneration of the endometrium and the regulation of embryo implantation. However, excessive accumulation or impaired clearance of SNCs may contribute to various endometrial disorders (Figure 2). In thin endometrium, senescent basal stem cells lead to diminished regenerative capacity. In intrauterine adhesions (IUA), senescent stromal cells secrete pro-fibrotic factors (e.g., TGF-β, CTGF), driving excessive collagen deposition and uterine cavity obliteration. In endometriosis (EMs), ectopic lesions exhibit SNCs that sustain chronic inflammation and angiogenesis through SASP components (e.g. IL-6, MMP-3) (Table 1).
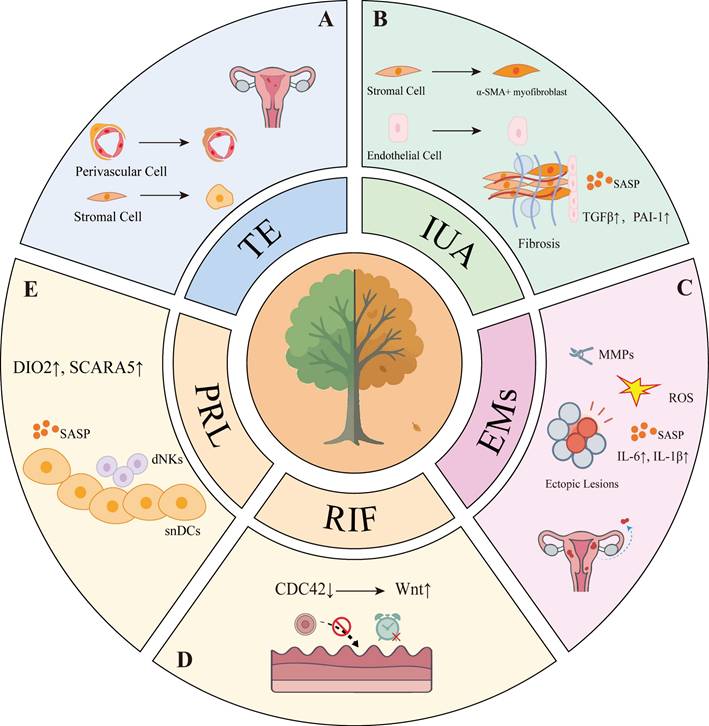
Abnormal cellular senescence and distinct pathological features in endometrium-related disorders. A: In TE, senescence of stromal cells and perivascular cells undergo aberrant senescence. B: In IUA, senescent endothelial cells release SASP factors, which collaborate with α-SMA-positive myofibroblasts to drive fibrosis. C: In EMs, senescent stromal cells sustain chronic inflammation through SASP components such as IL-6 and IL-1β, while ROS and MMPs further contribute to disease progression. D: In RIF, dysregulated temporal control of cellular senescence leads to impaired endometrial receptivity. E: In PRL, inadequate clearance of senescent decidual cells results in diminished endometrial receptivity.
Characteristics and clinical correlations of cellular senescence in endometrial disorders
| Disorder | Key Senescent Cell Types | Biomarkers | Pathological Consequences | Clinical Evidence |
|---|---|---|---|---|
| TE | Endometrial stem cells, Perivascular cells | p16↑, p21↑, DIO2↑ | Impaired regeneration, Reduced vascular density | scRNA-seq shows senescence gene enrichment (74) |
| IUA | Stromal cells, Endothelial cells | PAI-1↑, TGF-β↑, Galectin-9↑ | Fibrosis, Uterine cavity occlusion | EC senescence increases collagen deposition (73,90) |
| EMs | Ectopic lesion stromal cells | IL-6↑, MMP-3↑, ROS↑ | Chronic inflammation, Angiogenesis | Higher p16 in lesions than eutopic endometrium (48) |
| RIF | Decidual stromal cells | IL-6↓, DIO2↓, CDC42↓ | Decidualization defects, Impaired receptivity | Reduced p16+ cell proportion (101) |
| PRL | snDCs, Dysfunctional dNKs | DIO2↑, TNFSF14↓, SLC3A2↑ | Excessive inflammation, Embryo rejection | Leucine accumulation in decidua (93,96) |
Although manifestations vary across endometrium-related disorders, a common feedback loop is widely recognized: the accumulation of senescent cells and persistent inflammation mutually reinforce each other. Senescent endometrial cells, via the SASP, secrete inflammatory cytokines such as IL‑1β, IL‑6, and TNF‑α that activate NF‑κB and p38 MAPK signaling, leading to stromal cell migration, neovascularization, and immune cell infiltration. Inflammation in turn provokes DNA damage, hyperactivation of mTOR, and oxidative stress, which further drives cellular senescence and SASP - establishing the senescence-inflammation feedback loop. Notably, agents such as quercetin, metformin, and rapamycin - despite targeting distinct pathways - have all been shown to exhibit activity against the senescence-inflammation axis and therapeutic efficacy in multiple endometrial disorders.
These agents represent disparate drug classes—quercetin as a senolytic flavonoid, metformin as a metabolic modulator, and rapamycin as an mTOR inhibitor—they converge on the same core regulatory axis of endometrial pathology: the senescence-inflammation cycle. Mechanistically, all three target key nodes of this loop, notably the mTOR/AKT pathway, NF-κB signaling, AMPK activation, and the p53 network (42). Through this unified axis, they share the capacity to clear or modulate senescent cell populations, blunt the pro-inflammatory SASP, and reprogram the local immune-metabolic microenvironment of the endometrium (43,44). In concert, quercetin eliminates senescent cells, metformin suppresses NF-κB/SASP signaling via AMPK activation, and rapamycin inhibits mTOR, together disrupting the self-perpetuating senescence-inflammation loop that drives endometrial tissue dysfunction (43,44). These shared actions explain the broad therapeutic potential of these agents across pathologically distinct endometrial disorders despite the divergent etiologies of these conditions (44). Indeed, their convergent efficacy underscores the centrality of the senescence-inflammation axis in driving diverse endometrial pathologies and supports this cycle as a unified therapeutic target in endometrial disease (44).
Collectively, these findings indicate that eliminating senescent cells, inhibiting the SASP, and modulating key signaling pathways can effectively disrupt this self-perpetuating “senescence-inflammation” cycle. The combined or optimized use of these interventions serves as a common, foundational therapeutic strategy across various endometrial disorders. In the following sections, we will summarize these strategies in the context of specific endometrial pathologies, and discuss their applications and benefits for each condition.
4.1 Endometriosis
EMs is a prevalent chronic inflammatory and hormone-dependent disorder characterized by the ectopic growth of endometrial tissue. Clinically, EMs primarily manifests as pelvic pain and infertility, affecting 5-10 % of reproductive-aged women worldwide and significantly impairing their quality of life (45,46).
Recent studies have revealed the presence of canonical cellular senescence features in endometriotic lesions (47). Specifically, deep infiltrating endometriosis (DIE) lesions exhibit significantly higher p16 expression compared to matched eutopic endometrium, along with elevated IL-1β levels, while further analyses reveal that peritoneal fluid and the peri-lesional microenvironment in these patients contain multiple proinflammatory cytokines—including IL-6, IL-8, and IL-1β—showing substantial overlap with the SASP secretory profile (48). Furthermore, studies on ESCs demonstrate that ESCs from EMs patients exhibit significantly higher SA-β-Gal activity even without external stimulation, whereas minimal SA-β-Gal staining is detected in healthy controls (49).
IL-1β, as an early-phase SASP factor, initiates downstream NF-κB signaling and accelerates ESC senescence via the JNK pathway (50). This cascade induces production of major SASP components such as IL-6 and IL-8, establishing a positive feedback loop that amplifies inflammation and propagates senescence: SASP factors further enhance local inflammatory responses while inducing secondary senescence in neighboring cells through paracrine mechanisms. Under physiological conditions, NK cells and macrophages effectively identify and eliminate SNCs (51). However, in EMs patients, the aberrant pelvic immune microenvironment enables SNCs to evade immune clearance and accumulate locally (52). Concurrently, these SNCs frequently exhibit an anti-apoptotic phenotype, resisting programmed cell death activation (53).
Although the mechanistic role of cellular senescence in EMs pathogenesis remains incompletely understood, insights can be drawn from its established functions in tumor microenvironments. During tumor progression, senescent stromal cells establish an immunosuppressive microenvironment and drive tumorigenesis (54). These cells secrete proinflammatory SASP factors (e.g., IL-6, IL-1β) that sustain chronic inflammation (23), while simultaneously recruiting immunosuppressive cells (e.g., myeloid-derived suppressor cells, Tregs) to form an immunosuppressive microenvironment. This process not only inhibits NK cell function (55) but also enables tumor cells to evade immune surveillance. Concurrently, these senescent stromal cells release pro-angiogenic factors such as VEGF to support tumor vascularization (56). Furthermore, through the secretion of MMPs, they degrade the ECM, thereby facilitating tumor cell invasion and metastasis (57). As an invasive disorder, endometriotic lesions may concurrently harbor both proliferative cell populations and senescent yet metabolically active cells. The inflammatory mediators released by the latter could modulate the local immune milieu toward an immunosuppressive phenotype, thereby promoting cellular survival and contributing to lesion persistence and expansion. Accumulating evidence demonstrates that senolytic therapy can selectively eliminate pro-tumorigenic SNCs, with therapeutic benefits observed in diverse diseases including diabetes, Alzheimer's disease, and Parkinson's disease (58,59). As a classic senolytic cocktail, the combination of dasatinib and quercetin (D+Q) has shown efficacy in clearing SNCs and restoring tissue function across multiple disease models (60). In EMs, preliminary studies indicate that quercetin suppresses stromal cell proliferation while promoting normal differentiation in patient-derived cells, and reduces ectopic lesion size in animal models (61) (Table 2). Therefore, senolytic therapy holds potential to ameliorate the chronic inflammatory milieu in EMs by eliminating SNCs within ectopic lesions, which may partially restore healthy cellular function (see Figure 3). However, further experimental validation is required to assess its clinical translatability. Additionally, as SASP inhibitors, several anti-senescence drugs—including resveratrol, rapamycin, and metformin—have demonstrated therapeutic potential in inflammatory diseases. Future studies should explore their efficacy in alleviating endometriosis-associated pelvic inflammation (62-64).
Therapeutic effects of anti-senescence agents in murine models of endometrium-related disorders
| Drug/Therapy | Type | Anti-cellular senescence Mechanism | Indications as Anti-cellular Senescence Agent | Application in Endometrial Diseases | Ref. |
|---|---|---|---|---|---|
| Dasatinib & Quercetin | Senolytics | Inhibitor of BCL-2 and inhibitor of PI3Ks and serpins | Diabetic kidney disease, Idiopathic pulmonary | EMs, PRL | (44,60,61,109-113) |
| Metformin | SASP inhibition | Activates AMPK / inhibits NF‑κB / mTOR | T2DM, age-related disorders, inflammation | PRL, RIF, EMs | (41,64,93,109,114-116) |
| Rapamycin | SASP inhibition | Inhibits mTORC1 / reduces SASP | Age-related pathologies | EMs, RIF, RPL | (41,117-121) |
| Resveratrol | SASP inhibition | Activates SIRT1 / AMPK → NF‑κB suppression | NA | EMs, RIF | (108,122-124) |
| CAR-NK/T cell therapies | Senolytics | Immune-mediated clearance of senescent cells | Fibrosis, age-related disorders | NA | (125,126) |
NA = no direct evidence in endometrial diseases; extrapolated from other indications
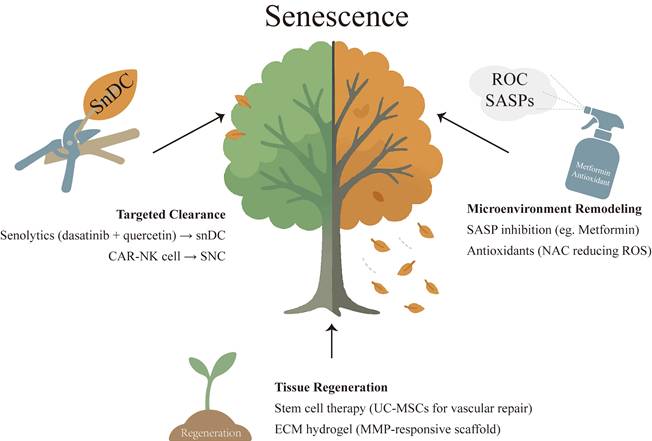
Potential therapeutic strategies targeting cellular senescence in endometrial-related disorders. Three main approaches: targeted clearance of senescent cells using senolytics (dasatinib and quercetin) to eliminate specific populations such as senescent dendritic cells; microenvironment remodeling via suppression of SASP and reduction of ROS with SASP inhibitors (metformin) and antioxidants (N-acetylcysteine), thereby attenuating chronic inflammation; and tissue regeneration by stem cell therapy (umbilical cord-derived mesenchymal stem cells) and ECM hydrogel (MMP-responsive scaffold).
ROS and iron overload also play significant roles in the progression of EMs. Compared to healthy women, EMs patients exhibit higher levels of iron in peritoneal fluid, may associated with retrograde menstruation and intra-lesional hemorrhage (65). These factors increase free iron levels, which through the Fenton reaction generate excessive ROS, further inducing oxidative stress damage (66). The accumulated ROS impose persistent oxidative stress on lesional cells, inducing senescence through multiple pathways including P38/MAPK and mTORC1 (67). Furthermore, ROS trigger NF-κB pathway activation in peritoneal macrophages, exacerbating chronic inflammation and promoting angiogenesis within endometriotic lesions (68,69). ROS not only induce cellular senescence in both eutopic endometrium and ectopic lesions but may also disrupt the reproductive microenvironment. For instance, oxidative stress can cause spindle damage and telomere depletion in oocytes—mechanisms that are recognized as pivotal to oocyte aging and may contribute to endometriosis-associated infertility (70). Iron overload also drives cellular senescence and oocyte damage in ovarian endometriosis-associated infertility (71). Notably, ROS-scavenging interventions and iron overload mitigation have demonstrated potential to alleviate EMs symptoms in preclinical studies (62,71).
4.2 Thin Endometrium and Intrauterine Adhesions
Thin endometrium (TE) and IUA are common endometrial disorders associated with female infertility, both of which are closely linked to impaired endometrial regeneration. Emerging evidence suggests that cellular senescence may represent a shared pathological mechanism (72-74): On the one hand, senescent cell accumulation directly suppresses endometrial regeneration; On the other hand, SASP drives fibrotic progression.
4.2.1 Thin Endometrium
TE is typically defined as a maximal endometrial thickness <7 mm during the proliferative phase, a condition associated with compromised endometrial receptivity, reduced implantation rates, and increased miscarriage risk (75). Single-cell RNA sequencing has revealed aberrant cellular senescence as a hallmark feature of TE (74). Specifically, the senescence of endometrial stem cells impairs endometrial proliferation, resulting in insufficient thickness to support embryo implantation (74). Furthermore, TE exhibits elevated p16 and p21 expression in epithelial cells, along with an increased proportion of senescent-associated elongated ciliated epithelial cells, indicating a pervasive senescent state within the endometrial epithelium (74,76). Endometrial mesenchymal stem cells (eMSCs), which reside in perivascular niches, normally possess high proliferative potential and multilineage differentiation capacity (77,78). However, in TE, perivascular cells demonstrate significant upregulation of senescence-related genes accompanied by impaired proliferation and differentiation, ultimately leading to reduced vascular density and compromised endometrial regeneration (30,74). Functional enrichment analysis further revealed that DIO2, a major key gene in the decidual senescence pathway, is significantly upregulated in thin endometrial stroma (30). Additionally, the abundance of uNK cells with senescent cell clearance capacity is markedly reduced in TE, leading to impaired senescent cell removal. This accumulation of SNCs perpetuates chronic senescence and excessive SASP production (79), which in turn promotes fibrotic progression. Recent findings further indicate that insufficient expansion or functional immaturity of cytolytic uNK cell subsets during the implantation window exacerbates the persistence of senescent stromal cells and local inflammation, thereby impairing endometrial receptivity and increasing the risk of implantation failure or pregnancy loss(80).
Although occurring in distinct tissues, chronic wounds and TE share remarkably similar "senescence-regenerative dysfunction" mechanisms. In chronic wounds, excessive accumulation of senescent fibroblasts together with insufficient immune clearance establishes a persistent inflammatory milieu that suppresses angiogenesis and impedes repair (81,82). Senolytic therapy to eliminate SNCs has been shown to promote wound healing (81), implying that reducing the senescent burden or enhancing its immune clearance may be therapeutically relevant to TE. By contrast, current TE management has largely emphasized stem cell-based approaches to increase endometrial thickness and improve pregnancy outcomes, while the contributions of cellular senescence and immune microenvironmental change remain underexplored. Mechanistically, both conditions converge on a senescence-amplified loop; interrupting this loop by targeting senescent cells and recalibrating the immune microenvironment constitutes a testable therapeutic strategy for TE.
4.2.2 Intrauterine Adhesions
IUA, also known as Asherman syndrome, refers to endometrial fibrosis following damage to the basal layer of the endometrium, resulting in partial or complete uterine cavity obliteration. This condition manifests clinically as menstrual abnormalities, infertility, and RPL. IUA is typically triggered by intrauterine procedures (83).
Studies have demonstrated that ESCs isolated from IUA patients exhibit significantly impaired colony-forming capacity, migratory/invasive potential, and angiogenic support compared to those from healthy women, displaying a premature senescence phenotype (84). Single-cell and transcriptomic analyses further reveal marked upregulation of senescence-associated genes and an increased proportion of SNCs in the proliferative-phase stromal compartment of IUA endometrium (72). Under stress conditions, ESCs undergo accelerated senescence and secrete abundant SASP factors, creating an immunosuppressive, pro-fibrotic inflammatory microenvironment (72,85). The SASP factor CCL2 is upregulated in IUA, recruiting immune cells including macrophages, a subset of which polarize into CD301+ pro-fibrotic phenotypes. These macrophages exacerbate endometrial fibrosis by activating the JAK/STAT signaling pathway (86). Concurrently, elevated levels of neutrophil-derived S100A8/A9 (a calcium-binding protein complex) promote ESC proliferation and differentiation into α-SMA-positive myofibroblasts via JAK2/STAT3 activation, driving excessive collagen and ECM deposition (87). Notably, galectin-9 (LGALS9), a lectin family protein highly expressed in senescent stromal cells of IUA, interacts with immune cell receptors to induce immunosuppressive effects, impairing the clearance of senescent and aberrant cells (72,88). Furthermore, stromal cell senescence may contribute to endometrial thinning, diminished ovarian hormone responsiveness, and reduced receptivity in patients (30,36). Collectively, the crosstalk between SNCs and inflammatory cells in IUA endometrium establishes a positive feedback loop, perpetuating a scarred microenvironment with impaired regenerative capacity.
Beyond ESCs, emerging evidence implicates endothelial cell (EC) senescence as a pivotal contributor to IUA pathogenesis. Endometrial cyclic regeneration critically depends on a robust microvascular network, which is disrupted in IUA due to basal layer damage and subsequent destruction of the decidua-associated capillary plexus, ultimately impairing tissue repair (89). At the molecular level, IUA patients exhibit significantly elevated expression of the senescence markers p16 and p21 in endometrial ECs compared to healthy controls (73). Single-cell sequencing and in vitro assays further confirm that microvascular ECs in IUA undergo pronounced senescence, characterized by diminished proliferative capacity and impaired angiogenic potential (73). These senescent ECs fail to adequately support endometrial revascularization, creating localized ischemia that activates stromal fibroblasts and promotes scar tissue deposition. Experimental studies using conditioned medium from senescent ECs to stimulate ESCs have demonstrated significantly elevated expression of fibrosis markers in the latter (73). Concurrently, ESCs from IUA patients overproduce PAI-1, which binds to the uPAR receptor on EC surfaces to induce endothelial senescence (73,90). Notably, TGF-β—already upregulated in early fibrosis stages—further amplifies PAI-1 production in ESCs via SMAD-dependent signaling, thereby establishing a self-reinforcing cycle (91,92). These findings position PAI-1 inhibition and other endothelial senescence-targeting strategies as promising therapeutic avenues for IUA prevention and treatment.
4.3 Recurrent Pregnancy Loss and Recurrent Implantation Failure
RPL and RIF are commonly associated with decidualization defects. Emerging evidence suggests that dysregulated temporal control of decidual senescence or impaired clearance of SNCs may compromise endometrial receptivity, ultimately contributing to these adverse reproductive outcomes (1,93,94). Mid-luteal phase endometrial biopsies from RPL patients reveal a "pro-senescent decidual response", characterized by upregulated expression of senescence-associated markers (e.g., DIO2, SCARA5) in the stroma. This excessive decidual senescence propensity correlates with elevated miscarriage risk (36). Decidual natural killer (dNK) cells—highly enriched during early pregnancy—normally surveil and eliminate aberrant cells. Studies demonstrate that dNK cells maintain decidual homeostasis by clearing SNCs, preventing their harmful accumulation (16). However, in RPL patients, insufficient dNK cell-mediated snDC clearance and impaired recognition of SNCs lead to excess snDC accumulation during decidualization, resulting in reduced endometrial receptivity (36,95). Endometrial tissues from RPL patients demonstrate elevated expression of DIO2 (a canonical snDC marker gene) alongside abnormal dNK cell accumulation, suggesting either expansion of decidual senescence phenotypes or a breakdown in senescence clearance homeostasis (96). In murine models, a high-leucine diet triggers extensive decidual cell senescence and subsequent embryonic loss (93). Branched-chain amino acids (e.g., leucine) accumulate in decidual stromal cells, activating the p38/MAPK pathway to drive cellular senescence (93,97). Conversely, TNFSF14-expressing dNK cells counteract leucine-induced senescence by engaging TNFRSF14 receptors on stromal cells (93). Notably, RPL patients exhibit enhanced decidual senescence, upregulated leucine transporter SLC3A2 while reduced TNFRSF14 expression. This pattern suggests coexisting leucine accumulation and defective anti-senescence regulation. Collectively, these findings demonstrate that dNK cells maintain decidual senescence homeostasis via the TNFSF14/TNFRSF14 axis.
In RIF patients, ESCs subjected to in vitro decidualization exhibit significantly lower levels of the classic SASP factor IL-6 and the snDC marker DIO2 compared to controls (98). Similarly, PAI-1—an established senescence marker (99)—shows reduced expression in the endometrium of RIF patients (100). Notably, the proportion of p16-positive endometrial cells is markedly decreased in RIF women relative to those with successful pregnancies, suggesting that insufficient cellular senescence may paradoxically impair endometrial receptivity (101). Furthermore, primary ESCs isolated from patients with impaired endometrial receptivity during the proliferative phase demonstrate significant upregulation of SA-β-Gal activity and CDKN2A (the gene encoding p16), indicating premature ESC senescence (102). Studies have revealed reduced CDC42 (a GTPase) expression in RIF patient-derived ESCs (103). In vitro knockdown of CDC42 activates Wnt signaling and upregulates p21, thereby inducing cellular senescence and impairing decidualization—ultimately compromising uterine receptivity (103).
Clinical studies in both RIF and RPL patients have consistently documented ESC senescence signatures and disrupted endometrial immune microenvironments (102,104). Animal model investigations further confirm the critical role of senescence-regulating factors in decidual function (93,105). These collective findings highlight the therapeutic potential of targeting senescence pathways—such as snDC-clearing agents or TNFSF14-TNFRSF14 axis modulators—for improving endometrial receptivity and reducing RIF/RPL risk, representing novel intervention strategies.
5. Summary and Discussion
Emerging evidence indicates that, despite distinct phenotypic manifestations—chronic inflammation in EMs, regenerative impairment in TE, fibrosis in IUA, and receptivity defects in RIF or RPL—these disorders converge on a core pathogenic axis: aberrant accumulation of senescent cells coupled with dysregulated/failed immune surveillance. This axis integrates the senescence-associated secretory phenotype with persistent inflammation, dysregulated matrix remodeling, angiogenic imbalance, and regenerative failure, providing a coherent mechanistic framework that spans these conditions.
The therapeutic approach to endometrial disorders is undergoing a paradigm shift—from morphological restoration to mechanism-guided regulation. Traditional clinical studies have disproportionately focused on singular endpoints (e.g., excision of endometriotic lesions, lysis of intrauterine adhesions, or increasing endometrial thickness in TE), while overlooking the shared pathological axis of cellular senescence and immune microenvironment dysregulation. This mechanistic commonality unveils opportunities for cross-disease anti-senescence therapies, yet necessitates disease-specific adaptations. For instance, senolytics (e.g., dasatinib + quercetin) may effectively eliminate pathological senescent cells in endometriotic lesions, but require cautious application in RIF to avoid disrupting physiological decidualization, given the demonstrated role of controlled senescence in embryo implantation. Current anti-senescence strategies (e.g., senolytics, SASP inhibitors) have demonstrated therapeutic efficacy across multiple diseases. In EMs, the senolytic quercetin has shown promising results in animal models, significantly reducing lesion size and improving tissue remodeling. However, the precise mechanisms—particularly how combinations such as dasatinib plus quercetin selectively clear senescent ESCs while preserving physiological functions—remain incompletely elucidated. Senescence-directed CAR-T/NK approaches that improved myocardial fibrosis (86,106) suggest potential applicability to fibrosis-associated TE and IUA. In terms of immunomodulation, metformin shows cross-disease promise, with a large randomized controlled trial on “Metformin Targeting Aging” underway (107); in mice, metformin reduces the risk of spontaneous abortion by antagonizing decidual stromal cell senescence, indicating potential clinical value for senescence-driven endometrial disorders. A central caveat is the senescence paradox: physiological senescence is indispensable for cyclic endometrial remodeling and embryo implantation, whereas pathological senescence drives disease progression. For example, resveratrol's anti-inflammatory and anti-senescence actions may benefit pelvic endometriosis and uterine leiomyomas, yet simultaneously diminish decidual senescence and suppress PRL and IGFBP1 expression, thereby altering decidual programming (108). To enable precise clinical application, priorities include: (1) establishing endometrium-specific senescence biomarkers to identify treatment-responsive populations; (2) optimizing drug-delivery technologies to enrich therapeutic agents in diseased endometrial tissues; and (3) translating advances from anti-senescence drug development in other disciplines to launch cross-disciplinary clinical trials.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFC2705403), the Major Research Program of National Natural Science Foundation of China (NSFC92478122, NSFC92357306, NSFC32370914, NSFC82371673, NSFC92057119, NSFC31970798), the Shanghai Natural Science Foundation (23ZR1408200), the Shanghai Oriental Talent Plan, the Open Fund for Shanghai Key Laboratory of Embryo Original Diseases (shelab2024ZD02).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deryabin P, Griukova A, Nikolsky N, Borodkina A. The link between endometrial stromal cell senescence and decidualization in female fertility: the art of balance. Cell Mol Life Sci. 2020Apr1;77(7):1357-70
2. Critchley HOD, Maybin JA, Armstrong GM, Williams ARW. Physiology of the Endometrium and Regulation of Menstruation. Physiol Rev. 2020 July 1;100(3):1149-79
3. Ticconi C, Di Simone N, Campagnolo L, Fazleabas A. Clinical consequences of defective decidualization. Tissue Cell. 2021Oct1;72:101586
4. Ng SW, Norwitz GA, Pavlicev M, Tilburgs T, Simón C, Norwitz ER. Endometrial Decidualization: The Primary Driver of Pregnancy Health. Int J Mol Sci. 2020Jan;21(11):4092
5. Lucas ES, Dyer NP, Murakami K, Hou Lee Y, Chan YW, Grimaldi G. et al. Loss of Endometrial Plasticity in Recurrent Pregnancy Loss. Stem Cells. 2016Feb1;34(2):346-56
6. Lucas ES, Dyer NP, Fishwick K, Ott S, Brosens JJ. Success after failure: the role of endometrial stem cells in recurrent miscarriage. Reproduction. 2016Nov;152(5):R159-66
7. Brighton PJ, Maruyama Y, Fishwick K, Vrljicak P, Tewary S, Fujihara R. et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. Wagner G, editor. eLife. 2017Dec11;6:e31274
8. Marquez CMD, Ibana JA, Velarde MC. The female reproduction and senescence nexus. Am J Reprod Immunol. 2017;77(5):e12646
9. Massimiani M, Lacconi V, La Civita F, Ticconi C, Rago R, Campagnolo L. Molecular Signaling Regulating Endometrium-Blastocyst Crosstalk. Int J Mol Sci. 2020Jan;21(1):23
10. The maternal-fetal interface at single-cell resolution. uncovering the cellular anatomy of the placenta and decidua. Am J Obstet Gynecol. 2025Apr1;232(4):S55-79
11. Salker MS, Nautiyal J, Steel JH, Webster Z, Šućurović S, Nicou M. et al. Disordered IL-33/ST2 Activation in Decidualizing Stromal Cells Prolongs Uterine Receptivity in Women with Recurrent Pregnancy Loss. PLOS ONE. 2012Dec27;7(12):e52252
12. Li Z, Si P, Meng T, Zhao X, Zhu C, Zhang D. et al. CCR8+ decidual regulatory T cells maintain maternal-fetal immune tolerance during early pregnancy. Sci Immunol. 2025Apr18;10(106):eado2463
13. Valdes CT, Schutt A, Simon C. Implantation failure of endometrial origin: it is not pathology, but our failure to synchronize the developing embryo with a receptive endometrium. Fertil Steril. 2017 July 1;108(1):15-8
14. Jabbour HN, Kelly RW, Fraser HM, Critchley HOD. Endocrine Regulation of Menstruation. Endocr Rev. 2006Feb1;27(1):17-46
15. Azlan A, Salamonsen LA, Hutchison J, Evans J. Endometrial inflammasome activation accompanies menstruation and may have implications for systemic inflammatory events of the menstrual cycle. Hum Reprod. 2020 June 1;35(6):1363-76
16. Zheng ZM, Shi JW, Wang L, Li MQ. NK cells: shielding senescence homeostasis in the decidua during early pregnancy. Semin Immunopathol. 2025Mar11;47(1):22
17. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular Senescence: Aging, Cancer, and Injury. Physiol Rev. 2019Apr;99(2):1047-78
18. Rawlings TM, Makwana K, Taylor DM, Molè MA, Fishwick KJ, Tryfonos M. et al. Modelling the impact of decidual senescence on embryo implantation in human endometrial assembloids. Spencer TE, Cooper JA, Spencer TE, Wagner G, Vankelecom H, Julie Kim JY, editors. eLife. 2021 Sept 6;10:e69603
19. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014 July;15(7):482-96
20. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020Jan16;18(1):e3000599
21. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013Aug;15(8):978-90
22. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020Dec1;34(23-24):1565-76
23. Coppé JP, Desprez PY, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol Mech Dis. 2010 Feb 28;5(Volume 5, 2010):99-118
24. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013 Sept 16;210(10):2057-69
25. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ. et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017Feb23;8(1):14532
26. Maus M, López-Polo V, Mateo L, Lafarga M, Aguilera M, De Lama E. et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat Metab. 2023;5(12):2111-30
27. Wang W, Vilella F, Alama P, Moreno I, Mignardi M, Isakova A. et al. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat Med. 2020Oct;26(10):1644-53
28. Kendirci-Katirci R, Sati L, Celik-Ozenci C. Deciphering the role of rapamycin in modulating decidual senescence: implications for decidual remodeling and implantation failure. J Assist Reprod Genet. 2024 Sept 1;41(9):2441-56
29. Endometrial stromal cells and decidualized stromal cells. Origins, transformation and functions. Gene. 2014Nov1;551(1):1-14
30. Lv H, Zhao G, Jiang P, Wang H, Wang Z, Yao S. et al. Deciphering the endometrial niche of human thin endometrium at single-cell resolution. Proc Natl Acad Sci U S A. 2022Feb22;119(8):e2115912119
31. Hapangama DK, Kamal A, Saretzki G. Implications of telomeres and telomerase in endometrial pathology. Hum Reprod Update. 2017Mar;23(2):166-87
32. Harada T, Kaponis A, Iwabe T, Taniguchi F, Makrydimas G, Sofikitis N. et al. Apoptosis in human endometrium and endometriosis. Hum Reprod Update. 2004Jan1;10(1):29-38
33. Cao D, Liu Y, Cheng Y, Wang J, Zhang B, Zhai Y. et al. Time-series single-cell transcriptomic profiling of luteal-phase endometrium uncovers dynamic characteristics and its dysregulation in recurrent implantation failures. Nat Commun. 2025Jan2;16(1):137
34. Vassilev V, Pretto CM, Cornet PB, Delvaux D, Eeckhout Y, Courtoy PJ. et al. Response of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases Messenger Ribonucleic Acids to Ovarian Steroids in Human Endometrial Explants Mimics Their Gene- and Phase-Specific Differential Control in Vivo. J Clin Endocrinol Metab. 2005Oct;90(10):5848-57
35. Maybin JA, Murray AA, Saunders PTK, Hirani N, Carmeliet P, Critchley HOD. Hypoxia and hypoxia inducible factor-1α are required for normal endometrial repair during menstruation. Nat Commun. 2018Jan23;9(1):295
36. Lucas ES, Vrljicak P, Muter J, Diniz-da-Costa MM, Brighton PJ, Kong CS. et al. Recurrent pregnancy loss is associated with a pro-senescent decidual response during the peri-implantation window. Commun Biol. 2020Jan21;3(1):1-14
37. Gong GS, Muyayalo KP, Zhang YJ, Lin XX, Liao AH. Flip a coin: cell senescence at the maternal-fetal interface†. Biol Reprod. 2023 Sept 1;109(3):244-55
38. Tsuru A, Yoshie M, Kojima J, Yonekawa R, Azumi M, Kusama K. et al. PGRMC1 Regulates Cellular Senescence via Modulating FOXO1 Expression in Decidualizing Endometrial Stromal Cells. Biomolecules. 2022 July 28;12(8):1046
39. Antonangeli F, Zingoni A, Soriani A, Santoni A. Senescent cells: Living or dying is a matter of NK cells. J Leukoc Biol. 2019May27;105(6):1275-83
40. Cloke B, Huhtinen K, Fusi L, Kajihara T, Yliheikkilä M, Ho KK. et al. The Androgen and Progesterone Receptors Regulate Distinct Gene Networks and Cellular Functions in Decidualizing Endometrium. Endocrinology. 2008 Sept;149(9):4462-74
41. Deryabin PI, Borodkina AV. Stromal cell senescence contributes to impaired endometrial decidualization and defective interaction with trophoblast cells. Hum Reprod. 2022 July 1;37(7):1505-24
42. Kobayashi H, Umetani M, Nishio M, Shigetomi H, Imanaka S, Hashimoto H. Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction. Cells. 2025Jan;14(12):858
43. Nie P, Wang M, Mo Y, Zhou H, Zha Q, Lash GE. et al. Metformin in gynecological disorders: pathogenic insights and therapeutic implications. Front Pharmacol [Internet]. 2025 Apr 22 [cited 2025 Sept 7];16. Available from: https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar. 2025 1526709/full
44. Delenko J, Hyman N, Chatterjee PK, Safaric Tepes P, Shih AJ, Xue X. et al. Targeting Cellular Senescence to Enhance Human Endometrial Stromal Cell Decidualization and Inhibit Their Migration. Biomolecules. 2025 June 16;15(6):873
45. Endometriosis is a chronic systemic disease. clinical challenges and novel innovations. The Lancet. 2021Feb27;397(10276):839-52
46. Symons LK, Miller JE, Kay VR, Marks RM, Liblik K, Koti M. et al. The Immunopathophysiology of Endometriosis. Trends Mol Med. 2018 Sept;24(9):748-62
47. Sonehara R, Nakamura T, Takeda T, Kaseki S, Seki T, Tanaka H. et al. A novel senotherapeutic strategy with azithromycin for preventing endometriosis progression. Reprod Biol Endocrinol RBE. 2025Mar26;23:47
48. Malvezzi H, Dobo C, Filippi RZ, Mendes do Nascimento H, Palmieri da Silva e Sousa L, Meola J. et al. Altered p16Ink4a, IL-1β, and Lamin b1 Protein Expression Suggest Cellular Senescence in Deep Endometriotic Lesions. Int J Mol Sci. 2022Feb24;23(5):2476
49. Malvezzi H, Cestari BA, Meola J, Podgaec S. Higher Oxidative Stress in Endometriotic Lesions Upregulates Senescence-Associated p16ink4a and β-Galactosidase in Stromal Cells. Int J Mol Sci. 2023Jan4;24(2):914
50. Taylor RN, Berga SL, Zou E, Washington J, Song S, Marzullo BJ. et al. Interleukin-1β induces and accelerates human endometrial stromal cell senescence and impairs decidualization via the c-Jun N-terminal kinase pathway. Cell Death Discov. 2024 June 15;10:288
51. Iannello A, Raulet DH. Immune Surveillance of Unhealthy Cells by Natural Killer Cells. Cold Spring Harb Symp Quant Biol. 2013Jan1;78:249-57
52. Palmieri L, Malvezzi H, Cestari B, Podgaec S. Colocalization of senescent biomarkers in deep, superficial, and ovarian endometriotic lesions: a pilot study. Sci Rep. 2022Oct14;12:17280
53. Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. 2019May1;117:15-22
54. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016 June 8;7(1):11762
55. Ye J, Baer JM, Faget DV, Morikis VA, Ren Q, Melam A. et al. Senescent CAFs mediate immunosuppression and drive breast cancer progression. Cancer Discov. 2024 July 1;14(7):1302-23
56. Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA. et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med. 2016Oct26;8(362):362ra144-362ra144
57. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019Aug;19(8):439-53
58. Schmitt CA, Wang B, Demaria M. Senescence and cancer — role and therapeutic opportunities. Nat Rev Clin Oncol. 2022Oct;19(10):619-36
59. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022Aug;28(8):1556-68
60. Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu Y. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest. 2022Aug1;132(15):e158450
61. Delenko J, Xue X, Chatterjee PK, Hyman N, Shih AJ, Adelson RP. et al. Quercetin enhances decidualization through AKT-ERK-p53 signaling and supports a role for senescence in endometriosis. Reprod Biol Endocrinol RBE. 2024Aug8;22:100
62. Anastasi E, Scaramuzzino S, Viscardi MF, Viggiani V, Piccioni MG, Cacciamani L. et al. Efficacy of N-Acetylcysteine on Endometriosis-Related Pain, Size Reduction of Ovarian Endometriomas, and Fertility Outcomes. Int J Environ Res Public Health. 2023Jan;20(6):4686
63. Christy B, Demaria M, Campisi J, Huang J, Jones D, Dodds SG. et al. p53 and rapamycin are additive. Oncotarget. 2015 June 23;6(18):15802-13
64. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE. et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013;12(3):489-98
65. Ni Z, Li Y, Song D, Ding J, Mei S, Sun S. et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. 2022 July 4;13(7):579
66. Defrère S, Lousse JC, González-Ramos R, Colette S, Donnez J, Van Langendonckt A. Potential involvement of iron in the pathogenesis of peritoneal endometriosis. Mol Hum Reprod. 2008 July 1;14(7):377-85
67. Nacarelli T, Azar A, Sell C. Mitochondrial stress induces cellular senescence in an mTORC1-dependent manner. Free Radic Biol Med. 2016 June 1;95:133-54
68. González-Ramos R, Langendonckt AV, Defrère S, Lousse JC, Colette S, Devoto L. et al. Involvement of the nuclear factor-κB pathway in the pathogenesis of endometriosis. Fertil Steril. 2010Nov1;94(6):1985-94
69. Papaconstantinou J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells. 2019Nov4;8(11):1383
70. Lin X, Dai Y, Tong X, Xu W, Huang Q, Jin X. et al. Excessive oxidative stress in cumulus granulosa cells induced cell senescence contributes to endometriosis-associated infertility. Redox Biol. 2020Jan12;30:101431
71. Li Y, Zhou W, Ding J, Song D, Cheng W, Yu J. et al. Integrative Single-Cell Analysis Reveals Iron Overload-Induced Senescence and Metabolic Reprogramming in Ovarian Endometriosis-Associated Infertility. Adv Sci. 2025;12:e17528
72. Deryabin PI, Borodkina AV. Endometrial Stromal Senescence Mediates the Progression of Intrauterine Adhesions. Int J Mol Sci. 2025Jan;26(9):4183
73. Wu J, Wang J, Pei Z, Zhu Y, Zhang X, Zhou Z. et al. Endothelial senescence induced by PAI-1 promotes endometrial fibrosis. Cell Death Discov. 2025Mar6;11:89
74. Zhang X, Lv H, Weng Q, Jiang P, Dai C, Zhao G. et al. “Thin endometrium” at single-cell resolution. Am J Obstet Gynecol. 2025Apr1;232(4):S135-48
75. Gharibeh N, Aghebati-Maleki L, Madani J, Pourakbari R, Yousefi M, Ahmadian Heris J. Cell-based therapy in thin endometrium and Asherman syndrome. Stem Cell Res Ther. 2022Jan28;13(1):33
76. Breslin L. et al. Ciliary abnormalities in senescent human fibroblasts impair proliferative capacity. Cell Cycle. 2014 Sept 2;13(17):2773-9
77. Kirkwood PM, Gibson DA, Smith JR, Wilson-Kanamori JR, Kelepouri O, Esnal-Zufiaurre A. et al. Single-cell RNA sequencing redefines the mesenchymal cell landscape of mouse endometrium. FASEB J. 2021;35(4):e21285
78. Ding L, Vezzani B, Khan N, Su J, Xu L, Yan G. et al. CD10 expression identifies a subset of human perivascular progenitor cells with high proliferation and calcification potentials. Stem Cells. 2020Feb1;38(2):261-75
79. Zhao G, Dai J, Hu Y. Development of regenerative therapies targeting fibrotic endometrium in intrauterine adhesion or thin endometrium to restore uterine function. Sci China Life Sci [Internet]. 2025 Apr 11 [cited. 2025 May 21]; Available from: https://doi.org/10.1007/s11427-024-2842-6
80. Muter J, Kong CS, Nebot MT, Tryfonos M, Vrljicak P, Brighton PJ. et al. Stalling of the endometrial decidual reaction determines the recurrence risk of miscarriage. Sci Adv. 2025 June 25;11(26):eadv1988
81. Samarawickrama PN, Zhang G, Zhu E, Dong X, Nisar A, Zhu H. et al. Clearance of senescent cells enhances skin wound healing in type 2 diabetic mice. Theranostics. 2024Aug26;14(14):5429-42
82. Wilkinson HN, Hardman MJ. Cellular Senescence in Acute and Chronic Wound Repair. Cold Spring Harb Perspect Biol. 2022Nov;14(11):a041221
83. Zhao G, Hu Y. Mechanistic insights into intrauterine adhesions. Semin Immunopathol. 2024Nov29;47(1):3
84. Min J, Lu N, Huang S, Chai X, Wang S, Peng L. et al. Phenotype and biological characteristics of endometrial mesenchymal stem/stromal cells: A comparison between intrauterine adhesion patients and healthy women. Am J Reprod Immunol. 2021;85(6):e13379
85. Alekseenko LL, Zemelko VI, Domnina AP, Lyublinskaya OG, Zenin VV, Pugovkina NA. et al. Sublethal heat shock induces premature senescence rather than apoptosis in human mesenchymal stem cells. Cell Stress Chaperones. 2014May;19(3):355-66
86. Yao S, Zhou Z, Wang L, Lv H, Liu D, Zhu Q. et al. Targeting endometrial inflammation in intrauterine adhesion ameliorates endometrial fibrosis by priming MSCs to secrete C1INH. iScience. 2023 June 24;26(7):107201
87. Xin X, Liu H, Zhang S, Li P, Zhao X, Zhang X. et al. S100A8/A9 promotes endometrial fibrosis via regulating RAGE/JAK2/STAT3 signaling pathway. Commun Biol. 2024Jan22;7(1):1-15
88. Lv Y, Ma X, Ma Y, Du Y, Feng J. A new emerging target in cancer immunotherapy: Galectin-9 (LGALS9). Genes Dis. 2022 June 4;10(6):2366-82
89. Chen Y, Chang Y, Yao S. Role of angiogenesis in endometrial repair of patients with severe intrauterine adhesion. Int J Clin Exp Pathol. 2013 June 15;6(7):1343-50
90. Zhang G, Kim H, Cai X, Lopez-Guisa JM, Carmeliet P, Eddy AA. Urokinase Receptor Modulates Cellular and Angiogenic Responses in Obstructive Nephropathy. J Am Soc Nephrol. 2003May;14(5):1234
91. Kawarada Y, Inoue Y, Kawasaki F, Fukuura K, Sato K, Tanaka T. et al. TGF-β induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci Rep. 2016Oct19;6:35483
92. MicroRNA-122-5p alleviates endometrial fibrosis via inhibiting the TGF-β/SMAD pathway in Asherman's syndrome. Reprod Biomed Online. 2023 Nov 1;47(5):103253.
93. Shi JW, Lai ZZ, Zhou WJ, Yang HL, Zhang T, Sun JS. et al. TNFSF14+ natural killer cells prevent spontaneous abortion by restricting leucine-mediated decidual stromal cell senescence. EMBO J. 2024Nov4;43(21):5018-36
94. Schatz F, Guzeloglu-Kayisli O, Arlier S, Kayisli UA, Lockwood CJ. The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding. Hum Reprod Update. 2016 June;22(4):497-515
95. Yamamoto M, Fukui A, Mai C, Saeki S, Takayama R, Wakimoto Y. et al. Evaluation of NKp46 expression and cytokine production of decidual NK cells in women with recurrent pregnancy loss. Reprod Med Biol. 2022 July 12;21(1):e12478
96. Tang AW, Alfirevic Z, Turner MA, Drury JA, Small R, Quenby S. A feasibility trial of screening women with idiopathic recurrent miscarriage for high uterine natural killer cell density and randomizing to prednisolone or placebo when pregnant. Hum Reprod. 2013 July 1;28(7):1743-52
97. He D, Wu H, Xiang J, Ruan X, Peng P, Ruan Y. et al. Gut stem cell aging is driven by mTORC1 via a p38 MAPK-p53 pathway. Nat Commun. 2020Jan2;11(1):37
98. Decreased PIBF1/IL6/p-STAT3 during the mid-secretory phase inhibits human endometrial stromal cell proliferation and decidualization. J Adv Res. 2021 May 1;30:15-25.
99. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler Thromb Vasc Biol. 2017Aug;37(8):1446-52
100. Up-regulation of miR-145 may contribute to repeated implantation failure after IVF-embryo transfer by targeting PAI-1. Reprod Biomed Online. 2020 May 1;40(5):627-36.
101. Parvanov D, Ganeva R, Vidolova N, Stamenov G. Decreased number of p16-positive senescent cells in human endometrium as a marker of miscarriage. J Assist Reprod Genet. 2021Aug1;38(8):2087-95
102. Tomari H, Kawamura T, Asanoma K, Egashira K, Kawamura K, Honjo K. et al. Contribution of senescence in human endometrial stromal cells during proliferative phase to embryo receptivity†. Biol Reprod. 2020 June 23;103(1):104-13
103. Tang X, Zhu Y, Cao Z, Wang X, Cai X, Tang Y. et al. CDC42 deficiency leads to endometrial stromal cell senescence in recurrent implantation failure. Hum Reprod. 2024Dec1;39(12):2768-84
104. Parvanov D, Ganeva R, Arsov K, Decheva I, Handzhiyska M, Ruseva M. et al. Association between endometrial senescent cells and immune cells in women with repeated implantation failure. J Assist Reprod Genet. 2023 July;40(7):1631-8
105. Sirohi VK, Medrano TI, Kannan A, Bagchi IC, Cooke PS. Uterine-specific Ezh2 deletion enhances stromal cell senescence and impairs placentation, resulting in pregnancy loss. iScience. 2023 June 8;26(7):107028
106. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J. et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016Feb;530(7589):184-9
107. American Federation for Aging Research [Internet]. [cited 2025 July 19]. TAME - Targeting Aging with Metformin. Available from: https://www.afar.org/tame-trial.
108. Ochiai A, Kuroda K, Ozaki R, Ikemoto Y, Murakami K, Muter J. et al. Resveratrol inhibits decidualization by accelerating downregulation of the CRABP2-RAR pathway in differentiating human endometrial stromal cells. Cell Death Dis. 2019Mar20;10(4):276
109. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK. et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019Feb;40:554-63
110. Kusama K, Yamauchi N, Yoshida K, Azumi M, Yoshie M, Tamura K. Senolytic treatment modulates decidualization in human endometrial stromal cells. Biochem Biophys Res Commun. 2021 Sept 24;571:174-80
111. Robbins PD, Jurk D, Khosla S, Kirkland JL, LeBrasseur NK, Miller JD. et al. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu Rev Pharmacol Toxicol. 2021 Jan 6;61(Volume 61, 2021):779-803
112. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK. et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019 Sept;47:446-56
113. Cavalcante MB, Saccon TD, Nunes ADC, Kirkland JL, Tchkonia T, Schneider A. et al. Dasatinib plus quercetin prevents uterine age-related dysfunction and fibrosis in mice. Aging. 2020Jan18;12(3):2711-22
114. Abdelgawad IY, Agostinucci K, Sadaf B, Grant MKO, Zordoky BN. Metformin mitigates SASP secretion and LPS-triggered hyper-inflammation in Doxorubicin-induced senescent endothelial cells. Front Aging. 2023Apr24;4:1170434
115. Cheng FF, Liu YL, Du J, Lin JT. Metformin's Mechanisms in Attenuating Hallmarks of Aging and Age-Related Disease. Aging Dis. 2022 July 11;13(4):970-86
116. Stochino-Loi E, Major AL, Gillon TER, Ayoubi JM, Feki A, Bouquet de Joliniere J. Metformin, the Rise of a New Medical Therapy for Endometriosis? A Systematic Review of the Literature. Front Med. 2021May11;8:581311
117. Zhang Y, Zhang J, Wang S. The Role of Rapamycin in Healthspan Extension via the Delay of Organ Aging. Ageing Res Rev. 2021 Sept 1;70:101376
118. Alqahtani S, Alqahtani T, Venkatesan K, Sivadasan D, Ahmed R, Sirag N. et al. SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights. Cells. 2025Jan;14(8):608
119. Hirota Y, Cha J, Yoshie M, Daikoku T, Dey SK. Heightened uterine mammalian target of rapamycin complex 1 (mTORC1) signaling provokes preterm birth in mice. Proc Natl Acad Sci U S A. 2011Nov1;108(44):18073-8
120. Kendirci-Katirci R, Sati L, Celik-Ozenci C. Deciphering the role of rapamycin in modulating decidual senescence: implications for decidual remodeling and implantation failure. J Assist Reprod Genet. 2024 Sept;41(9):2441-56
121. Lu H, Yang HL, Zhou WJ, Lai ZZ, Qiu XM, Fu Q. et al. Rapamycin prevents spontaneous abortion by triggering decidual stromal cell autophagy-mediated NK cell residence. Autophagy [Internet]. 2021 Sept 2 [cited 2025 July 30]; Available from: https://www.tandfonline.com/doi/abs/10.1080/15548627. 2020 1833515
122. Liu S, Zheng Z, Ji S, Liu T, Hou Y, Li S. et al. Resveratrol reduces senescence-associated secretory phenotype by SIRT1/NF-κB pathway in gut of the annual fish Nothobranchius guentheri. Fish Shellfish Immunol. 2018 Sept;80:473-9
123. Kolahdouz-Mohammadi R, Shidfar F, Khodaverdi S, Arablou T, Heidari S, Rashidi N. et al. Resveratrol treatment reduces expression of MCP-1, IL-6, IL-8 and RANTES in endometriotic stromal cells. J Cell Mol Med. 2021Jan;25(2):1116-27
124. Kuroda K, Ochiai A, Brosens JJ. The actions of resveratrol in decidualizing endometrium: acceleration or inhibition?†. Biol Reprod. 2020Dec1;103(6):1152-6
125. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020 July;583(7814):127-32
126. Yang D, Sun B, Li S, Wei W, Liu X, Cui X. et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci Transl Med. 2023Aug16;15(709):eadd1951
Author contact
![]() Corresponding authors: Ming-Qing Li, E-mail: mqliedu.cn; Chun-Xue Zhang, E-mail: zhangchunxueedu.cn; Feng Xie, E-mail: fengxie10edu.cn.
Corresponding authors: Ming-Qing Li, E-mail: mqliedu.cn; Chun-Xue Zhang, E-mail: zhangchunxueedu.cn; Feng Xie, E-mail: fengxie10edu.cn.