Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Components and structure of ECM
Biological functions of the...
Hidden connections: how ecm...
Applications of ECM in different...
Discussion
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(15):6808-6881. doi:10.7150/ijbs.119301 This issue Cite
Review
Extracellular matrix dysregulation in aging, calcification, and cancer diseases: insights into cellular senescence, inflammation, and novel therapeutic strategies
Diego Liviu Boaru1,2, Diego De Leon-Oliva1,2, Patricia De Castro-Martinez1,2, Cielo Garcia-Montero1,2, Oscar Fraile-Martinez1,2, Beatriz García-González1, Isabel Pérez-González1, Majd N. Michael Alhaddadin1, Silvestra Barrena-Blázquez2,3, Laura Lopez-Gonzalez2,4, Basilio de la Torre2,4, Leonel Pekarek1,2,5, Miguel A Saez1,2,6, Laura Ríos-Espinosa1, Tatiana Pekarek1,2, Roberto Fernández-Baillo Gallego de la Sacristana2,4, Mauricio Hernández-Fernández2,4, Carlos Casanova1,2, Ana Castel-Oñate4, Natalio Garcia-Honduvilla1,2, Julia Buján1,2, Raul Diaz-Pedrero2,4,7, Melchor Alvarez-Mon1,2,8, Miguel A Ortega1,2 ![]()
1. Department of Medicine and Medical Specialities, Faculty of Medicine and Health Sciences, Network Biomedical Research Center for Liver and Digestive Diseases (CIBEREHD), University of Alcalá, 28801 Alcala de Henares, Spain.
2. Ramón y Cajal Institute of Sanitary Research (IRYCIS), 28034 Madrid, Spain.
3. Department of Nursing and Physiotherapy, Faculty of Medicine and Health Sciences, University of Alcalá, 28801 Alcala de Henares, Spain.
4. Department of Surgery, Medical and Social Sciences, Faculty of Medicine and Health Sciences, University of Alcalá, 28801 Alcala de Henares, Spain.
5. Oncology Service, Guadalajara University Hospital, 19002 Guadalajara, Spain.
6. Pathological Anatomy Service, Central University Hospital of Defence-UAH Madrid, 28801 Alcala de Henares, Spain.
7. Department of General and Digestive Surgery, Príncipe de Asturias, University Hospital, 28805 Alcala de Henares, Spain.
8. Immune System Diseases-Rheumatology, Oncology Service an Internal Medicine (CIBEREHD), University Hospital Príncipe de Asturias, 28806 Alcala de Henares, Spain.
Received 2025-6-10; Accepted 2025-9-16; Published 2025-10-24
Abstract

This review underscores the dynamic role of the extracellular matrix (ECM) in regulating cellular behavior and maintaining tissue homeostasis, highlighting its pivotal involvement in aging, calcification, and cancer diseases. In healthy tissues, controlled ECM remodeling provides essential biochemical and mechanical cues, but dysregulation (driven by chronic inflammation, cellular senescence, and altered intercellular communication) leads to fibrosis, calcification, and the creation of a pro-tumorigenic microenvironment. Senescent cells contribute to these changes through senescence-associated secretory phenotype (SASP), which reinforces inflammation and matrix degradation, while extracellular vesicles (EVs) mediate intercellular signaling and further modulate ECM structure and function. In cancer, ECM remodeling not only facilitates tumor progression and metastasis by forming physical and biochemical barriers but also hinders the efficacy of conventional and immunotherapeutic interventions. Similarly, in cardiovascular diseases, aberrant ECM remodeling exacerbates tissue damage and impairs regenerative processes. Emerging therapeutic strategies aim to restore ECM homeostasis through targeted interventions, including ECM-normalizing agents, EV-based therapies, and stem cell approaches that modulate matrix composition to improve tissue repair. By elucidating the complex interplay between ECM dysfunction, cellular senescence, and chronic inflammation, this review highlights promising avenues for developing personalized treatments that address the underlying causes of age-related and tumorigenic pathologies, ultimately, the way to improved clinical outcomes.
Keywords: extracellular matrix, aging, calcification, cancer, cellular senescence, chronic inflammation, extracellular vesicles, tumor microenvironment, collagen, and metalloproteases
Introduction
The extracellular matrix (ECM) is a highly dynamic and complex network of proteins and other biomolecules that provides structural and biochemical support to cells and tissues. Beyond its fundamental role as a scaffold, the ECM actively regulates key cellular processes such as adhesion, migration, proliferation, differentiation, and apoptosis. It serves as a reservoir for growth factors and cytokines, mediates signal transduction through interactions with cell surface receptors (e.g., integrins), and contributes to tissue homeostasis, repair, and regeneration.
The mechanisms governing ECM secretion and assembly are highly conserved among eukaryotes, indicating their evolutionary origins long before the emergence of multicellular animals [1,2]. Many ECM proteins exhibit oligomeric structures, with the most common motifs being the GXY repeats in collagens, which facilitate the formation of a stable triple helix, and the α-helical heptad repeats, which promote oligomerization into trimers, tetramers, or pentamers [2]. These structural features are widely distributed across different biological domains, including viruses, suggesting an ancient and fundamental role for ECM components in cellular organization. Additionally, ECM proteins are characterized by the presence of repeated domains, such as the von Willebrand factor A (vWF-A) domain, the thrombospondin type 1 domain, and the epidermal growth factor (EGF) domain—all of which originated in early eukaryotic lineages and remain essential for ECM functionality [3,4]. The evolutionary conservation of these domains underscores the critical role of the ECM in the development and maintenance of complex multicellular life.
The ECM is essential for maintaining tissue integrity and function across various physiological contexts. In development, ECM components guide cellular differentiation and organogenesis, while in wound healing, they coordinate tissue repair through tightly regulated deposition and remodeling [5-7]. However, the dysregulation of ECM homeostasis contributes to a wide range of pathological conditions, particularly those associated with aging-related diseases, such as cancer, vascular calcification, fibrosis, and osteoarthritis [8-11].
As aging progresses, ECM components undergo biochemical and mechanical alterations, including increased collagen cross-linking, elastin degradation, and matrix stiffening [12]. These changes compromise tissue elasticity and impair cellular function, ultimately contributing to conditions such as fibrosis, osteoarthritis, and vascular diseases [13]. One prominent consequence of ECM dysfunction in aging is pathological calcification, characterized by the deposition of calcium phosphate crystals within the ECM [14]. This process disrupts tissue architecture and promotes dysfunction, playing a central role in disorders such as vascular calcification, valvular heart disease, and skeletal pathologies. Inflammatory and metabolic factors further influence ECM calcification, exacerbating disease progression. Within aging-related conditions, ECM remodeling is a key driver of tumor progression [15]. Aberrant ECM dynamics create a microenvironment that fosters cancer cell invasion, metastasis, and resistance to therapy. Increased ECM stiffness, excessive collagen deposition, and proteolytic ECM degradation facilitate epithelial-to-mesenchymal transition (EMT) and immune evasion, enabling malignant transformation and tumor expansion.
Given the critical role of ECM dysfunction in various diseases, extracellular vesicles (EVs) have emerged as promising therapeutic tools in regenerative medicine [16]. EVs—including exosomes and microvesicles—act as natural carriers of bioactive molecules such as proteins, nucleic acids, and lipids, enabling the modulation of ECM composition and function [17]. In degenerative diseases, EVs derived from mesenchymal stem cells (MSCs) have demonstrated the ability to restore ECM integrity by delivering reparative factors that stimulate fibroblasts, suppress inflammation, and enhance collagen synthesis [18]. Furthermore, in fibrosis and cancer, engineered EVs can transport anti-fibrotic agents, matrix metalloproteinases (MMPs) inhibitors, or RNA-based therapies to regulate ECM remodeling and prevent disease progression.
A particularly promising application of EVs lies in targeted drug delivery [19]. Functionalized EVs, designed with ECM-binding motifs, enable precise localization of therapeutic agents to affect tissues, thereby enhancing treatment efficacy while minimizing systemic toxicity. The emergence of EV-based therapies represents a paradigm shift in the management of ECM-related disorders, offering innovative strategies to restore tissue function and combat degenerative diseases [20].
This review provides a comprehensive analysis of the extracellular matrix (ECM), covering its biochemical composition, structural organization, and dynamic remodeling through synthesis, biogenesis, and enzymatic turnover. We explore its biological functions, including cell support, growth regulation, and its role as a signaling reservoir, as well as its involvement in Anoikis. In addition, we examine ECM interactions in pathological contexts such as cancer and aging, highlighting the role of extracellular vesicles (exosomes) in ECM modulation. We also review ongoing clinical trials and emerging ECM-targeted therapies, with a focus on regenerative medicine, oncology, and nanotechnology-based approaches to drug delivery and tissue engineering. This synthesis highlights the importance of ECM in health and disease and offers insights into its potential for therapeutic innovation.
Components and structure of ECM
Structure
As it is described above, ECM is important in regulating different cellular processes, such as adhesion, differentiation, proliferation, migration, and apoptosis [20]. This regulation is facilitated by the intricate network of matrix components and their interactions with each other, signaling factors, and membrane receptors [21]. ECM is an essential component of connective tissue, along with epithelial, muscle, and nerve tissue (the main tissue types in the human body) [22]. The ECM is a complex mixture of water, proteins, and polysaccharides, and the balance of these components varies depending on the tissue type, such as cartilage, bone, fat, or tendon [23,24]. As well as the tissue´s developmental stage and pathophysiological state. ECM components are synthesized and secreted locally by cells, primarily fibroblasts, which are the most numerous yet least specialized connective tissue cells [25]. The organization of the ECM structure is influenced by the arrangement and orientation of the intracellular cytoskeleton [26].
Although the basic organization of the ECM is consistent, there are two primary types distinguished by their location and composition: the interstitial matrix and the pericellular matrix. The interstitial matrix forms a three-dimensional porous network that surrounds cells, particularly in connective tissue. In contrast, the pericellular matrix is more compact and forms a layer adjacent to the cells [27].
The interstitial matrix, often referred to as the proper matrix, forms the structural scaffolding for cells. Its basic components are heterotypic fibrils primarily composed of type I collagen, with smaller amounts of type III and V collagens in varying proportions, all of which play an essential role in fibrillogenesis in cells [28]. These collagens are mostly secreted by fibroblasts. Additionally, important components of this amorphous three-dimensional gel include fibronectin and elastin, which are involved in the organization of the matrix structure [29].
A typical example of the pericellular matrix is the basement membrane, which is located at the interface between parenchyma and connective tissue. It provides a sheet-like anchoring layer that supports and stabilizes parenchymal cells, preventing them from tearing apart. Basement membranes are composed of collagen type IV, laminins, nidogens 1 and 2, and heparan sulfate proteoglycans (HSPGs) such as perlecan, agrin, collagen type XV, and collagen type XVIII [30-32]. Also, it contains matricellular proteins that do not contribute to its physical stability or structural integrity but have regulatory roles. These proteins interact with surface receptors, proteases, hormones, and other biologically active molecules and may be specific to certain tissues in terms of function and structure. Examples of these proteins include SPARC (secreted protein acidic and rich in cysteine, also known as osteonectin, primarily associated with mineralizing tissues like bone), thrombospondin-1 (which is abundant in platelet α-granules and, when secreted, activates TGF-β1, or transforming growth factor-beta 1), and tenascin-C (which is expressed during embryonic development but is minimally detectable in adult tissues, becoming more apparent during pathological processes) [33-38]. The basement membrane regulates tissue development, function, and regeneration by controlling cellular responses; it acts as a reservoir of growth factors, modulating their activity and concentration, and helps maintain the phenotype of surrounding cells [39]. The interstitial matrix and basement membrane are closely interconnected, working together to ensure tissue integrity [40].
On the other hand, the cells embedded within the ECM interact with this macromolecular network through surface receptors, including integrins, discoidin domain receptors (DDRs), cell surface proteoglycans (PGs), and the hyaluronan (HA) receptor CD44 [41]. These interactions allow cells to integrate signals from the ECM that influence their functions and behavior. All cell types synthesize and secrete ECM macromolecules in response to multiple signals, contributing to ECM formation [42]. Variations in ECM composition and structure affect both the architecture and biomechanical properties of the matrix, together with the signals transmitted to cells, thereby modulating their responses [43]. Growth factors, cytokines, and chemokines are deposited with the ECM through binding to specific ECM molecules. These factors can be released in a controlled manner, influencing development and physiological processes at appropriate times [44]. They are also actively involved in the reorganization of ECM. Besides synthesizing and secreting structural components, they also produce enzymes that degrade them. Remodeling processes are complex and must be tightly regulated to maintain environmental homeostasis [45].
Furthermore, ECM remodeling occurs under both physiological conditions and disease processes, influencing the structure and properties of ECMs in different ways [46]. For instance, proteolytic degradation mediated by enzymes such as matrix metalloproteinases (MMPs), disintegrin and metalloproteinases (ADAMTS), plasminogen activators, and the degradation of HSPGs chains by heparinase, releases heparin-binding growth factors that activate angiogenesis and cell growth, particularly during tumorigenesis [47]. During tumorigenesis, significant alterations in the ECM lead to the formation of fibrotic stroma characterized by increased stiffness, excessive deposition of ECM components, and the release of proteolytic enzymes that contribute to abnormal ECM remodeling [48]. Enhanced lysyl oxidase (LOX) activity promotes the cross-linking of collagen fibers with other ECM components, further increasing matrix stiffness. Various cell types within the tumor stroma, such as cancer-associated fibroblasts (CAFs), endothelial cells, immune cells, pericytes, and even the tumor cells themselves, contribute to the development of deregulated and disorganized ECMs that support and promote tumorigenesis [49].
ECM cycle
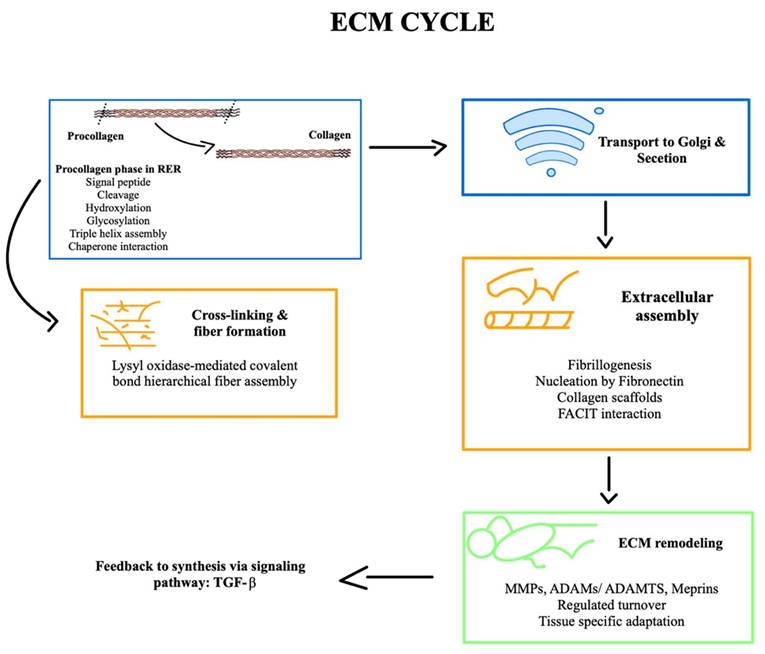
Extracellular matrix biogenesis: secretion and assembly
The biogenesis of the ECM is a fundamental, multi-step process essential for tissue development, maintenance, and repair. This process involves coordinated synthesis, secretion, and extracellular assembly and remodeling of a diverse set of macromolecules. Key structural ECM components include fibrous proteins such as collagens and elastins, proteoglycans, and adhesive glycoproteins like fibronectin and laminin [50]. Their precursors are synthesized primarily by fibroblasts, chondrocytes, and osteoblasts, although epithelial and other mesenchymal cells may contribute to tissue-specific contexts [51].
Intracellular synthesis of collagen begins in the rough endoplasmic reticulum (RER), where the initial translation product, known as preprocollagen, contains signal peptides and globular domains at the N- and C-termini. Although Porter et al initially proposed a model of direct cytoplasmic extrusion of collagen fibrils at the cell surface, later autoradiographic and immunohistochemical studies, notably those by Revel and Hay, demonstrated that collagen follows the canonical secretory pathway via the RER and Golgi apparatus [52]. These studies also revealed that newly synthesized collagen is typically deposited at some distance from the cell surface, suggesting that polymerization occurs extracellularly rather than directly on the plasmalemma [53]. Within the cisternae of the RER, a series of PTMs is initiated to transform preprocollagen into a functional precursor suitable for further processing and secretion:
1. Cleavage of the signal peptide: The N-terminal signal peptide of the preprocollagen is cleaved upon entry into the lumen of the RER, generating procollagen.
2. Hydroxylation of proline and lysine residues: Specific proline and lysine residues in procollagen chains undergo post-translational hydroxylation catalyzed by prolyl hydroxylases and lysyl hydroxylases, respectively. These enzymes require ascorbic acid (vitamin C) as an essential cofactor. Hydroxylation is critical for the formation of intramolecular hydrogen bonds that stabilize the collagen triple helix. Deficiencies in this modification, as observed in scurvy, compromise collagen stability, leading to impaired wound healing, defective bone formation, and increased tissue fragility. In addition to its structural role, hydroxylation of lysine residues—particularly in type IV and VI collagens—is followed by glycosylation, a modification essential for the proper secretion, assembly, and spatial distribution of these collagen types. These functions are especially relevant in basement membranes and muscle tissues, where these collagen isoforms are most abundant [54].
3. Glycosylation of hydroxylysine residues: Some hydroxylysine residues are glycosylated by the addition of galactose or glucosyl-galactose moieties. Additionally, N-linked oligosaccharides are added to the terminal regions. This glycosylation modulates collagen secretion, assembly, and interactions.
4. Formation of the C-terminal globular domain: The carboxy-terminal propeptide region of each α-chain folds into a globular domain stabilized by disulfide bonds. This domain serves as a nucleation site for the proper alignment and registration of the three α-chains.
5. Assembly of the triple helix: Starting from the C-terminal end, three α-chains associate and fold into a right-handed triple helix. The N- and C-terminal propeptides remain non-helical.
6. Formation of specific inter- and intrachain hydrogen bonds and disulfide bonds that contribute to structural stability.
7. Association with molecular chaperones: The triple-helical procollagen interacts with molecular chaperones such as HSP47, which prevent premature aggregation and ensure proper folding and stabilization of the collagen trimer within the RER.
8. Lateral association and transport to the Golgi: Next, properly folded procollagen molecules begin to associate laterally via interactions at their non-helical terminal regions. These assembled precursors are packaged into secretory vesicles for transport to the Golgi apparatus, which delivers ECM components to the plasma membrane for extracellular release.
The formation of mature collagen fibrils, or fibrillogenesis, is a highly orchestrated extracellular process critical for the structural integrity of connective tissues. Following secretion, ECM components self-assemble into supramolecular structures, often initiated by fibronectin, which nucleates collagen fibrillogenesis and guides the organization of matrix architecture [55]. The close association between the actin cytoskeleton and extracellular fibrils, observed in early electron microscopy (EM) studies and later confirmed by immunohistochemistry, highlights the dynamic interplay between cells and their surrounding matrix [56]. This interface is mediated by integrins—transmembrane receptors that couple the ECM to the cytoskeleton and transduce biochemical signals via focal adhesion complexes and intracellular kinase cascades.
Following their secretion from the cell, procollagen molecules undergo enzymatic processing by membrane-associated procollagen N- and C-proteinases, which cleave non-helical propeptides at both ends [57,58]. This proteolytic activation produces mature collagen monomers capable of assembling into higher-order structures. In particular, the serum concentration of the N-terminal propeptide of type I procollagen (PINP) serves as a clinically relevant biomarker for type I collagen synthesis and is often elevated in pathological states involving dysregulated extracellular matrix remodeling, such as bone metastasis in breast and prostate cancers [59].
Once activated, collagen molecules align in a row in the extracellular space and self-assemble in a longitudinal head-to-tail fashion [60]. This precise alignment is facilitated by specialized invaginations of the plasma membrane called “fibrillogenesis bays,” which serve as nucleation sites for collagen assembly [61]. These localized surface domains allow the accumulation and spatial coordination of secreted collagen molecules, promoting the formation of nascent fibrils with a defined orientation and periodicity.
The stability and mechanical properties of collagen fibrils are further enhanced by the enzymatic formation of covalent cross-links. These cross-links, mediated by lysyl oxidase, involve aldehyde groups generated from lysine and hydroxylysine residues and are essential for fibril maturation and resistance to tensile forces [62]. The assembled fibrils aggregate laterally to form larger collagen fibers. These supramolecular structures confer remarkable tensile strength to tissues, with a strength-to-weight ratio comparable to that of steel, underscoring the indispensable role of collagen in maintaining the architecture and function of the extracellular matrix. In addition to enzymatic cross-linking, proper collagen assembly and function depend on post-translational modifications, including glycosylation of hydroxylysine residues catalyzed by lysyl hydroxylase 3 (LH3). Recent findings demonstrate that LH3 requires intracellular trafficking via interaction with VIPAR and VPS33B proteins, with the assistance of RAB10 and RAB25, for delivery to specialized collagen IV carriers in epithelial cells [63].
Recent evidence suggests that the assembly of type I collagen fibrils is initiated by a fibrillar nucleation complex enriched in type V and XI collagens [64]. These molecules act as regulatory scaffolds that establish the initial fibril core, on which type I collagen molecules are subsequently deposited and polymerized. Type V and XI collagens play a critical role in controlling fibril diameter by limiting lateral growth once the desired thickness is reached.
In addition, mature collagen fibrils are often associated with the fibril-associated collagens with interrupted triple helices (FACIT) family, which includes collagens such as types IX and XII [65]. These molecules are located on the surface of fibrils and mediate interactions with other components of the ECM, thus contributing to matrix organization and tissue-specific architecture. In cartilage, for example, type IX collagen is found on the surface of type II fibrils, anchoring them to proteoglycans and other ECM components and reinforcing the mechanical integrity of the tissue [66].
Collagen molecules are synthesized by various cell types in both connective and epithelial tissue. In connective tissue, collagen production is mainly carried out by fibroblast-like cells, including tissue-specific analogues such as chondrocytes in cartilage, osteoblasts in bone and pericytes in blood vessel walls [67-69]. In addition, epithelial cells contribute to the synthesis of collagen components of basement membranes, underscoring the broad involvement of diverse cell types in collagen biogenesis [70].
The regulation of collagen synthesis is mediated by a complex network of signaling pathways involving growth factors, hormones and cytokines. For example, transforming growth factor beta (TGF-β) and platelet-derived growth factor (PDGF) are potent stimulators of collagen production by fibroblasts [70,71]. In contrast, glucocorticoid hormones exert an inhibitory effect, down-regulating collagen gene expression and synthesis [72]. These regulatory mechanisms are critical for maintaining tissue homeostasis and modulating extracellular matrix remodeling during development, repair, and disease.
In epithelial tissues, collagen fibrils are often arranged in highly ordered orthogonal lattices, such as those found in the dermis of aquatic vertebrates and in the corneal stroma of birds [73]. To explain the development of these specialized architectures, Porter and colleagues proposed the “shingle” or scindulene hypothesis, according to which narrow layers of collagen are inserted obliquely into the basal lamina and are sequentially displaced by newly synthesized layers [74]. This layered, anisotropic arrangement is thought to arise from tightly regulated interactions between secretory epithelial cells, the basal lamina, and the underlying ECM.
In connective tissues, the organization of collagen fibrils is tailored to the mechanical and functional demands of the tissue. In cartilage, type II collagen forms a dense three-dimensional network embedded in a hydrated matrix rich in proteoglycans, which confers tensile and compressive strength [75]. Minor fibrillar collagens, such as types IX and XI, copolymerize with type II and modulate fibril diameter, interfibrillar spacing and cross-linking, thus contributing to the biomechanical integrity of the tissue.
In bone, type I collagen fibrils constitute the primary organic scaffold for mineral deposition [76]. These fibrils exhibit a characteristic D-periodic banding pattern, reflecting the precise, staggered quarter alignment of tropocollagen molecules. This molecular organization is essential for the nucleation and orderly deposition of hydroxyapatite crystals, which endow the tissue with compressive strength. The hierarchical assembly of collagen in bone is orchestrated by osteoblasts and tightly regulated by local and systemic factors, such as mechanical stimuli, growth factors such as TGF-β and bone morphogenetic proteins (BMPs) [77]. Taken together, these examples underscore the structural versatility of collagen fibrils, whose tissue-specific arrangement is governed by both intrinsic molecular properties and extrinsic cellular and biomechanical factors.
Extracellular remodeling
The ECM is continuously remodeled through a dynamic balance between synthesis and degradation, essential for tissue homeostasis, development, repair, and adaptation to mechanical and biochemical stimuli. Collagen fibrils, despite their structural stability, are not static entities; they are subject to slow but continuous turnover, with half-lives ranging from a few days to several years depending on tissue type, e.g., skin vs. cartilage [78]. Degradation is initiated through mechanical stress, oxidative damage, or enzymatic cleavage and continues via proteolytic or phagocytic pathways [79,80].
Proteolytic degradation pathways
Proteolytic degradation occurs extracellularly, and the ECM can be cleaved by different families of proteases with overlapping substrate specificities (Table 1).
Enzymes involved in ECM proteolytic degradation pathways
| Enzyme family | Representative members | Substrates | Source cells | Mode of action |
|---|---|---|---|---|
| MMPs | MMP-1, MMP-2, MMP-9 | Collagens I-IV, elastin. Fibronectin | Fibroblasts, neutrophils | Extracellular cleavage |
| ADAMTS | ADAMTS2, ADAMTS4 | Procollagen, aggrecan | Chondrocytes fibroblasts | Secreted proteases |
| Meprins | Meprin-α, Meprin-β | Collagen IV, fibronectin | Kidney, intestine | Secreted/ membrane-bound |
| Cathepsins | Cathepsin K, L | Collagens, laminins | Macrophages, osteoclasts | Extracellular or lysosomal |
| Serine proteases | Plasmin, neutrophil elastase | Fibrin, fibronectin | Neutrophils epithelial cells | Pericellular/ solubleº |
| Heparanase | - | HSPGs | Endothelial cells, tumors | ECM-bound |
Matrix metalloproteinases. Matrix metalloproteinases (MMPs), a family of zinc-dependent endopeptidases secreted by fibroblasts, chondrocytes, macrophages, neutrophils, epithelial cells (e.g., keratinocytes), and cancer cells [81]. MMPs act on various components of the ECM: collagenases (e.g. MMP-1) degrade native fibrillar collagens (types I, II, III and X), gelatinases (MMP-2 and MMP-9) cleave denatured collagens (gelatins), laminin, fibronectin and elastin, stromelysins (e.g. MMP-3) degrade proteoglycans, fibronectin and non-helical collagens, matrilysins target basement membrane components such as type IV collagen and proteoglycans, membrane-type MMPs (MT-MMPs), produced mainly by tumor cells, exhibit potent pericellular proteolytic activity and macrophage metalloelastases (e.g. MMP-12) degrade elastin, type IV collagen and laminin [82-86]. Their expression and activity are typically low under physiological conditions but are markedly upregulated during tissue remodeling, inflammation, and pathological states such as cancer and fibrosis. MMPs are synthesized as inactive zymogens and are subsequently activated extracellularly through proteolytic cleavage by serine proteases or other MMPs, or via oxidative modification of a regulatory cysteine residue [87,88].
Adamalysins. This family includes disintegrinases and metalloproteinases (ADAMs) and ADAMs with thrombospondin motifs (ADAMTS). Of the 22 human ADAMs, 12 are catalytically active. ADAMs, often membrane-anchored, function as sheddases, cleaving ectodomains of membrane-bound proteins such as cytokines, growth factors, and receptors. Some ADAMs (e.g., ADAM10, ADAM12, ADAM15) can also degrade ECM proteins [89]. ADAMTS enzymes are secreted proteinases characterized by type I thrombospondin motifs. Several members of the ADAMTS family (e.g., ADAMTS4 and ADAMTS5) are aggrecanases, involved in proteoglycan degradation. Others (ADAMTS2, -3, -14) function as procollagen N-propeptidases, essential for the maturation and deposition of collagen fibrils [90]. ADAMTS13 cleaves von Willebrand factor and is essential for hemostasis [91].
Meprins. Meprins are zinc-dependent metalloproteinases belonging to the astacin family [92]. They are composed of α and β subunits that form homo- or hetero-oligomeric complexes. Meprin-α is secreted, whereas meprin-β is membrane-bound but can be shed via ADAM10 [93]. Meprins cleave several ECM components, including collagen IV, nidogen, and fibronectin. They also contribute to the processing of procollagen I and can regulate other metalloproteinases, such as promoting MMP-9 activation via MMP-3 [94].
Other enzymes are involved in ECM remodeling. On the one hand, serine proteases such as plasmin (activated by urokinase and tissue plasminogen activators) degrade fibrin, fibronectin, and laminin [95]. Neutrophil-derived elastase targets elastin and fibronectin, while matriptase, a serine protease bound to the epithelial membrane, maintains the integrity of the epithelial barrier [96,97]. On the other hand, cathepsins are lysosomal proteases with serine (cathepsins A and G), aspartic (cathepsins D and E), and cysteine (e.g., cathepsins B, K, L) variants [98,99]. They can function extracellularly or degrade ECM components after endocytic uptake and lysosomal processing. Finally, heparanase cleaves heparan sulfate proteoglycans (HSPG), altering ECM structure and releasing bound growth factors and cytokines [100]. Sulfatases 1 and 2 remove 6-O-sulfate groups from HSPGs, modulating the binding and signaling of factors such as FGF1 and VEGF [101].
Regulation of the proteolytic pathway
Endogenous inhibitors are essential for maintaining the integrity of the extracellular matrix (ECM) by tightly controlling proteolytic activity (Table 2). Tissue inhibitors of metalloproteinases (TIMPs)-TIMP1 to TIMP4- regulate the function of matrix metalloproteinases (MMPs), as well as members of the ADAM and ADAMTS families, with distinct binding specificities [102]. Among them, TIMP3 is sequestered in the ECM, whereas the others exist predominantly in soluble form. TIMP3 is also the main inhibitor of ADAM and ADAMTS [103]. Other regulatory molecules include RECK (reversion-inducing cysteine-rich protein with Kazal motifs), which modulates both MMP and ADAM activity, and other endogenous inhibitors such as cystatin C, elafin, and fetuin A, which have been shown to suppress meprin activity [104]. ECM turnover is governed by complex regulatory networks involving genetic, biochemical, and mechanistic signals. The balance between proteolytic enzymes and their inhibitors, exemplified by the MMP/TIMP ratio, determines the net proteolytic capacity within a given tissue and is finely tuned by transcriptional control, zymogen activation, and inhibitor availability [105]. Intact triple helix collagens are relatively resistant to proteolysis; however, once structurally compromised by mechanical stress or oxidative injury, they become more vulnerable, especially to gelatinases such as MMP2 and MMP9 [106]. Disruption of the delicate balance between MMPs and their inhibitors contributes to pathological degradation of the ECM, as seen in chronic inflammatory conditions, tumor invasion, and degenerative diseases such as rheumatoid arthritis and osteoporosis.
Endogenous inhibitors of ECM proteases
| Inhibitor | Target protease | Localization | Functional notes |
|---|---|---|---|
| TIMP-1 | MMPs (broad spectrum) | Soluble | Inhibits active MMPs |
| TIMP-3 | MMPs, ADAMs, ADAMTS | ECM-bound | Inhibits sheddases and aggrecanases |
| Serpins | Serine proteases (e.g., plasmin) | Plasma/ interstitial | Regulate coagulation and fibrinolysis |
| Cystatins | Cysteine cathepsins | Cytosol/ extracellular | Inhibit lysosomal enzymes |
| Sulfatases | Modulate HSPG sulfatation | ECM-associated | Influence FGF/VEGF signaling |
Phagocytic degradation pathway
Phagocytic degradation constitutes a crucial intracellular mechanism for the turnover of ECM components, particularly fibrillar collagens. In this pathway, relatively intact collagen fibers are recognized and internalized by fibroblasts or macrophages via receptor-mediated endocytosis, often facilitated by β1-integrins (e.g., α1β1, α10β1, α11β1) that bind to noncollagenous components such as fibronectin or proteoglycans decorating the collagen surface [107]. The formation of actin-rich pseudopodia allows engulfment of collagen segments in phagocytic vesicles [108]. Membrane matrix metalloproteinase type 1 (MT1-MMP), located on the cell surface, initiates partial cleavage of collagen prior to internalization [109]. Once inside the cell, the phagocytosed collagen is transported to the lysosomes, where it is degraded by lysosomal cysteine proteases, such as cathepsins. This pathway complements extracellular degradation by allowing the removal of ECM debris and promotes tissue remodeling under physiological conditions. In contrast to the rapid extracellular cleavage that predominates in pathological contexts, phagocytic degradation proceeds at a slower rate and is particularly important for the homeostatic maintenance of ECM integrity and the resolution of matrix turnover during tissue repair [110].
To summarize this explanation of the ECM cycle, a schematic graphic is provided to emphasize its key stages (Figure 1).
A schematic graphic of the development of the ECM cycle is presented to illustrate the dynamic processes involved in extracellular matrix remodelling and to provide a clear visual overview of its key stage and interactions.
Components
Collagen and elastic fibers
Collagen terms refer to a family of glycoproteins distinguished by three key features. The first is the repeating amino acid sequence [Gly-X-Y] n, which can include interruptions. The second feature is the common presence of proline and its hydroxylated form, hydroxyproline, in the X and Y positions, respectively. The third defining characteristic is the unique quaternary structure: a right-handed triple helix formed from three left-handed polyproline α-chains of identical length [111], which adopts a polyproline type II-type helical conformation and coils around each other. Interchain hydrogen bonds hold the α-chains together. The small hydrogen atom side chain of glycine in every third residue within the α-chains allows them to pack tightly together in a triple helix, with this residue in the interior of the helix and the rings of proline and 4-hydroxyproline pointing outward [112]. Collagens also have non-collagenous (NC) non-triple helical domains at both C- and N-termini, which are numbered from the C-terminus (NC1, NC2, NC3, etc.) [113].
It makes up to 30% of the total protein in humans. It is synthesized and secreted into ECMs primarily by fibroblasts. It is the most abundant fibrous protein within the interstitial ECMs of all animals and is also found in pericellular matrices such as the basement membrane [114]. The discovery of transmembrane collagens on the surface of various cell types containing bioactive peptides liberated upon degradation has amplified interest in collagen biology. By exerting tension on the matrix, fibroblasts organize collagen fibrils into sheets and cables, significantly influencing the alignment of collagen fibers [115].
It forms part of a superfamily of twenty-eight types, each formed by at least forty-six unique polypeptide chains (α chains) in vertebrates. Collagen type I, the most prevalent collagen, is widely expressed across various tissues. It forms ideal heterotrimeric triple helices that naturally assemble into fibrils and are a key structural component in tissues such as skin, bone, and tendons [116]. Collagen types vary significantly in their structure and functions. Some have breaks in their triple helices and do not self-assemble, while others, like transmembrane collagens, exhibit long interruptions and are crucial for cell signaling and adhesion [117].
Numerous mutations have been identified in collagen genes that can affect trimerization, collagen network formation, and propeptide cleavage. These mutations are associated with various clinical pathologies, such as Ehlers-Danlos syndrome (collagen types I, III, V) [118], osteogenesis imperfecta and osteoporosis (collagen type I) [119], osteoarthrosis (collagen type II, IX, X, XI) [120], chondrodysplasias (collagen types II, IX, X, XI) [121,122], arterial aneurysms (collagen type III) [123], Bethlem myopathy and Ullrich muscular dystrophy (collagen type VI) [124], epidermolysis bullosa acquisita (collagen type VII) [125], generalized atrophic epidermolysis bullosa (collagen type XII) [126], Fuchs endothelial corneal dystrophy (collagen type VIII) [127], Knobloch syndrome (collagen type XVIII) [128], Alport syndrome (collagen type IV) [129], and Schmid metaphyseal chondrodysplasia (collagen type X) [130].
Collagens are categorized into seven groups based on their domain homology and functions. These categories include:
- Fibrillar collagens: Including types I, II, III, V, XI, XXIV, and XXVII, are important for providing tensile strength to tissues. These collagens form 67-nm periodic fibrils through a regular staggered arrangement of triple helical molecules, with fibril diameters ranging from 12 nm to over 500nm, depending on development [131]. Their formation is influenced by other matrix macromolecules such as decorin and biglycan. Fibrillar collagens feature a long, uninterrupted Gly-X-Y domain and are flanked by N- and C-terminal propeptides. The α chains form homo- and heterotrimeric helices, with NC1 domains ensuring correct alignment for triple helix nucleation [132]. Post-translational modifications in the endoplasmic reticulum include hydroxylation, glycosylation, and disulfide bridge formation. Procollagen triple helices are cleaved by specific proteinases from tropocollagen, which then assemble into collagen microfibrils. LOX facilitates the formation of cross-links, stabilizing fibrils and contributing to their mechanical properties [133]. However, collagen types XXIV and XXVII exhibit imperfections in their Gly-X-Y sequences, indicating very short interruptions in their triple helical structure. Collagen fibrils found in the dermis, tendons, and other tissues are often composites of various collagen types, typically types I, III, and V [134]. These composite fibers are known as heterotypic fibrils, in contrast to homotypic fibrils, which consist of a single collagen type, such as collagen VII found in fibrils are the dermo-epidermal junction [134]. While collagen type I often predominated in a tissue, various types, and matrix macromolecules interact to impart specific structural and functional characteristics. Beyond their mechanical support role, collagen scaffolds also influence cell migration, adhesion, angiogenesis, tissue development, and repair [135].
- Network-forming collagens: Include types IV, VIII, and X, each playing distinct roles in various tissues. Collagen type IV, a key component of basement membranes, is essential for molecular filtration [136]. Collagen type VII is found in Descemet´s membrane and vascular sub-endothelial matrices [137], while collagen type X is present in the hypertrophic zone of growth plate cartilage [138]. These collagens feature interruptions in their triple helices, allowing flexibility and extensive network formation. Collagen type IV begins folding at the C-terminus and progresses towards the N-terminus. The NC1 domain facilitates the tail-to-tail association of trimeric molecules, forming hexamers stabilized by Met-Lys cross-links [139]. This assembly leads to the creation of a two-dimensional network. Similarly, collagen types VIII and X form polygonal lattices, with the NC1 domains for their supramolecular assembly. Additionally, these collagens interact with various ECM components to form complex multimolecular networks [140]. They offer structural support and attachment for cells and tissues, and function as a filtration barrier for macromolecules in organs like the kidneys [141].
- FACITs: encompass collagen types IX, XII, XIV, XVI, XIX, XX, XXI, and XXII. These collagens are relatively short and feature NC domains that interrupt their triple helical collagenous domains, granting them flexibility. FACITs interact with fibrillar collagens on their surfaces, connecting collagen fibers and other ECM molecules [142]. For instance, collagen type IX is covalently bound to the surface of collagen type II fibrils, with collagenous domains 1 and 2 positioned between NC1 and NC3 domains. The NC4 domain extends into the NC3 hinge region. Collagen type IX is essential for maintaining cartilage integrity. Collagen type XII associates with collagen type I and II fibrils, while collagen type XIV co-localizes with collagen type I indirectly through binding to DS chains of decorin, which in turn associates with collagen type I fibrils. In skin, collagen type XVI is incorporated into microfibrils, which are molecular composites, primarily consisting of collagen II and containing collagen type XI as a minor component [143].
- MACITs are type II transmembrane proteins characterized by a long extracellular C-terminal domain with collagenous segments interrupted by NC domains and a short cytoplasmic N-terminal domain. This group includes collagen types XIII, XVII, XXIII, and XXV, which are produced by various cells, including malignant ones, and different tissues. These proteins function as cell surface receptors, influencing cell adhesion. When proteolytically cleaved, they release from the cell surface into the extracellular matrix, creating soluble collagens. For instance, the ectodomain of collagen type XVII can be cleaved by ADAMs, thereby modulating cell motility [144].
- Anchoring fibrils: Collagen type VII is the primary component of anchoring fibrils located beneath the lamina densa of the basement membrane, linking it to the underlying stroma. This collagen type is created by the homotrimerization of α1 (VII), featuring a central triple helical collagenous domain interrupted by a short NC domain and flanked by N- and C-terminal NC domains. Two collagen type VII molecules dimerize and subsequently assemble into anchoring fibrils [145].
- Beaded-filament-forming collagens: Include types VI, XXVI, and XXVIII. Among these, collagen type VI is the most extensively studied. It is widely expressed in tissues, where it interacts with various ECM proteins, HA, PGs, and collagen type IV in basement membranes. Collagen-type VI molecules form antiparallel dimers through staggered alignments of monomers. These dimers are then associated laterally to create tetramers, which are stabilized by disulfide bonds. The tetramers connect at their globular ends, forming beaded filaments characterized by 25 nm beads spaced 100 nm apart [146].
- Multiplexin: Collagen type XV and XVIII fall into the category of Multiplexins/endostatin-producing collagens. These types are widely expressed across all vascular and epithelial basement membranes in human tissues. Collagen type XV connects adjacent collagen fibrils, forming various oligomeric assemblies that enhance the adhesion of basement membranes to the underlying connective tissue stroma [147]. Both collagen types XV and XVIII feature a central triple helical collagenous domain interrupted by multiple NC domains, with collagen XV carrying CS chains and collagen XVIII carrying HS chains. Collagen XVIII is a homotrimer made up of three α1 chains, containing ten interrupted collagenous domains flanked by eleven NC domains at their N- and C-termini [148]. It can carry three HS chains. Bot collagen types have a C-terminal NC domain that includes anti-angiogenic endostatin and endostatin-like modules. Cleavage of part of the NC1 domain releases endostatin, which interacts with several receptors such as integrins α5β1, αvβ5, and VEGFR2. These interactions disrupt the actin cytoskeleton, trigger a signaling network that downregulates the VEGF signaling pathway, and stimulate potent angiostatin components like TSPs, thereby significantly inhibiting angiogenesis [149].
All these collagen groups are summarized in the following Table 3.
All collagen types are divided into different groups that are classified. N.d. (Not defined).
| Collagen | ||||
|---|---|---|---|---|
| Groups | Collagen types | Molecular assembly | Localization | Function |
| Fibrillar collagens | I | [α1(I)]2 α2(I)] [α1(I)]3 | Dermis, tendons, and other tissues | Provide tensile strength to tissues; participate in cell migration, adhesion, angiogenesis, tissue development and repair |
| II | [α1(II)]3 | |||
| III | [α1(III)]3 | |||
| V | [α1(V)]2 α2(V) [α1(V) α2(V) α3(V)] [α1(V)]3 | |||
| XI | [α1(XI) α2(XI) αα3 (XI)] | |||
| XXIV | [α1 (XXIV)]3 | |||
| XXVII | [α1 (XXVII)]3 | |||
| Network-forming collagens | IV | [α1(IV)2 α2(IV)]; α3(IV), α4(IV), α5(IV), α6(IV) | Basement membranes | Formation of complex multimolecular networks from interaction with ECM components; structural support and attachment for cells and tissues; macromolecular filtration barrier |
| VII | [α1(VII)]3 | Descemet´s membrane and vascular sub-endothelial matrices | ||
| X | [α1(X)]3 | Hypertrophic zone of growth plate cartilage | ||
| FACITs | IX | [α1(IX) α2(IX) αα3(IX)] | Cartilage associated with type II collagen fibrils | Connecting collagen fibers with other components of the extracellular matrix to maintain tissue integrity |
| XII | [α1(XII)]3 | Collagen type I and II fibrils | ||
| XIV | [α1(XIV)]3 | n.d. | ||
| XVI | [α1(XVI)]3 | Microfibrils in skin | ||
| XIX | [α1(XIX)]3 | N.d. | ||
| XX | [α1(XX)]3 | N.d. | ||
| XXI | [α1(XXI)]3 | N.d. | ||
| XXII | [α1(XXII)]3 | N.d. | ||
| MACITs | XIII | [α1(XIII)]3 | Cell surface and extracellular matrix | Cell surface receptors; involved in cell adhesion and motility |
| XVII | [α1(XVII)]3 | |||
| XXIII | [α1(XXIII)]3 | |||
| XXV | [α1(XXV)]3 | |||
| Anchoring fibrils | VII | [α1(VII)]3 | Lamina densa of the basement membrane | Connects the basement membrane to the underlying stroma to provide structural stability |
| Beaded-filament-forming-collagens | VI | [α1(VI) α2(VI) αα3(VI)] | Widely expressed in different tissues | Interact with several ECM proteins, HA, PGs, in the tissues in which it is expressed, and type IV collagen in basement membranes |
| XXVI | [α1(XXIV)]3 | N.d. | N.d. | |
| XXVIII | [α1(XXVIII)]3 | N.d. | N.d. | |
| Multiplexin | XV | [α1(XV)]3 | Vascular and epithelial basement membranes in human tissues | Enhances the adhesion of basement membranes to the underlying connective tissue stroma by connecting adjacent collagen fibrils |
| XVIII | [α1(XVIII)]3 | Angiogenesis inhibition | ||
Elastic fibers are structural elements of the extracellular matrix, providing resilience to connective tissues. They are abundant in tissues that need mechanical flexibility, such as arteries, skin, lungs, and cartilage [150]. Elastic fibers consist of two primary components: elastin, a core-forming amorphous protein, and surrounding microfibrils composed of glycoproteins [151]. It is primarily produced from the ELN gene located on chromosome 7. The ELN gene is active during prenatal development and early life; it significantly decreases during adulthood. Mutations in the ELN gene can result in improper tropoelastin production or assembly, leading to diseases like supravalvular aortic stenosis (SVAS) and Williams-Beuren syndrome (WBS), where elastic fiber deficiency contributes to arterial stenosis [152,153].
The key precursor to elastin is tropoelastin, a protein with alternating hydrophobic and cross-link domains. Tropoelastin is secreted as a soluble polypeptide, with a molecular weight of around 70-72 kDa, into the extracellular matrix, where it self-assembles into globular aggregates through a process called coacervation, driven by its hydrophobic regions [154]. The polymerization of tropoelastin begins with the oxidative deamination of lysine reduced, catalyzed by LOX. These cross-links give rise to the durable elastic fibers that provide issues with their resilience [155]. The process also involved the formation of two amino acids, desmosine and isodesmosine, which further stabilize the elastic fibers through inter- and intra-molecular bonds. Tropoelastin deposition on microfibrils comprises glycoproteins such as fibrillin-1 and fibrillin-2, which are necessary to assemble mature fibers properly [156].
Beyond fibrillins, MAGP-1 (microfibril-associated glycoprotein-1) emerges as a significant structural component for microfibrils. Widely expressed in mesenchymal and connective tissues, MAGP-1 is associated with nearly all microfibrils [157]. It binds to the amino-terminal exons of fibrillin-1 through its carboxyl-terminal region, which contains common cysteine residues. This interaction is key to forming stable complexes between fibrillin-1 and other proteins, including decorin, biglycan, and tropoelastin, which all contribute to the structural assembly of elastic fibers [158].
Microfibrils guide the spatial arrangement of tropoelastin and promote the orderly cross-linking needed for fiber maturation. The final step involves extensive cross-linking to form a stable, hydrophobic, and insoluble polymer that resists proteolysis [159].
Elastic fibers, once formed, are stable. Elastin is so durable that it rarely breaks down or gets replaced throughout a person´s life [160]. This is one reason why elastin is so important for maintaining the structure and function of tissues, especially in organs that need to expand and contract, such as the lungs and heart [161,162]. Over time, however, changes in elastin, like reduced cross-linking or damage from environmental factors, can cause tissues to lose their elasticity.
This leads to common signs of aging, such as sagging skin or stiffening arteries. Elastin´s long-lasting maturity makes it a key focus in understanding how tissues age and how certain diseases related to elastic fiber defects, such as aortic stenosis and skin disorders, develop [163].
Multi-adhesive glycoproteins
Fibronectin (FN)
It is a glycoprotein that ranges in size from 230 to 270 kDa and exists as a dimer, with two subunits covalently bonded at their C-terminal by a pair of disulfide bonds [164]. Each monomer comprises three repeating units: 12 Type I, 2 Type II, and 15-17 Type III domains, which together constitute 90% of the FN sequence.
Similar structural domains (Type I, II, and III) are found in other biomolecules, suggesting FN evolved through exon shuffling [165]. While the conformations of type I and type II repeats are stabilized by pairs of intramodule disulfide bridges, the type III repeat forms a 7-stranded β-barrel structure that lacks disulfide bonds, allowing it to undergo conformational changes. Despite being long encoded by a single gene, FN exists in multiple variants due to extensive alternative splicing [166]. The domains or units of FN facilitate self-assembly and binding to ligands such as collagen/gelatin, integrins, heparin, fibronectin, and other extracellular molecules [167]. The 500 kDa FN dimer is created through a pair of antiparallel isoforms due to alternative splicing. FN exists in multiple isoforms due to alternative splicing. A single FN gene transcript encodes 20 isoforms in humans [168]. It is secreted as soluble inactive dimers with disulfide bonds, which must be activated by interaction with α5β1 and other integrins [169].
FN can be classified by its solubility into soluble plasma FN and cellular FN. Cellular FN is significantly more diverse because of splicing variations that are specific to different cell types and species [170].
According to its expression, it has an important role in embryos and adults, especially in areas of morphogenesis, cell migration, and inflammation. It is low in tumor cells, but it has a high expression in tissues undergoing repair [171]. FN facilitates the formation of fibrillar networks by binding to cell receptors like 5β1 integrin, organizing the actin cytoskeleton, and exposing additional binding sites for fibril formation. This process is essential for matrix assembly and the organization of other ECM proteins. FN´s ability to simultaneously bind to various ECM components makes it an organizer, particularly important for assembling a fibrillin-1 network [171].
Fibrinogen
It is an intricate fibrous glycoprotein composed of three pairs of polypeptide chains: Aα, Bβ, and γ. These chains are interconnected by 29 disulfide bonds. The fibrinogen molecules measure 45 nm in length and feature globular domains at both ends and, in the center, linked by α-helical coiled-coil rods, giving it a molecular weight of 340 kDa [172]. The E-region comprises the N-terminal ends of all six chains, while the D-regions consist of the C-terminal ends of the Bβ and γ chains along with part of the Aα chain, all connected by three-stranded α-helical coiled-coil regions. Both strongly and weakly bound calcium ions play roles in maintaining the structure and function of fibrinogen [173]. Its fibrinopeptides are cleaved by thrombin to convert soluble fibrinogen into soluble fibrin. This involves interactions between exposed knobs and holes, leading to the polymerization of fibrin monomers into protofibrils [174]. These protofibrils laterally aggregate to form fibers that branch into a three-dimensional fibrin clot essential for hemostasis [175]. Factor XIIIa further stabilizes the clot by linking fibrin molecules via isopeptide bonds [176].
The fibrinolytic system uses plasminogen, activated to plasmin, to degrade fibrin at specific lysine residues. Fibrinogen binds various proteins, influencing cardiovascular and ECM functions but not directly affecting important disorders discussed [177]. Research on dysfibrinogenemia and variant fibrinogen molecules has deepened the understanding of its functions [178]. Fibrinogen interacts with activated αIIbβ3 integrin on platelets, facilitating platelet aggregation for hemostasis, and it has adhesive and inflammatory roles through interactions with other cells [179].
Fibulins
Are a family of seven glycoproteins secreted by various cell types and tissues. They are intricately linked with basement membranes, elastic, fibers, and other extracellular ECM [180]. These proteins feature a fibulin module that allows them to release glycoproteins and possess a globular domain at their carboxy terminus. This domain is preceded by multiple tandem repeats of calcium-binding epithelial growth factor (cb-EGF) sequences. Research by Argraves has shown that fibulins are complex proteins with two distinct repeating motifs. One of these motifs shares similarities with anaphylatoxins C3a, C4a, and C5, as well as with the albumin gene family, while the other is like EGF [181].
The fibulin family is divided into classes based on length and domain configurations: Class I and Class II [182]. Class I is formed by fibulins 1, 2, and 6. Class II encompasses the shorter (50-60 kDa) fibulins 3, 4, 5, and 7. These shorter fibulins play a role in the formation of elastic fibers and are active during embryonic development in skeletal and cardiovascular tissues, with calcium ions aiding in this process [183].
Fibulin-2, part of the fibulin family, is recognized for its ability to engage with various ECM ligands and modulate the interaction between cells and their environment [184]. On the other hand, fibulin 3 is primarily found in mesenchyme that transforms into cartilage to bone, while fibulin 4 is notably present in heart muscle. Fibulin 5 is expressed in blood vessels, and fibulin 7 is abundant in teeth, placenta, hair follicles, and cartilage [185-188]. All fibulins have a C-terminal module with an elastic-binding domain, which is prominent in fibulin 5 [189].
Fibulins act not only as structural elements of the extracellular matrix but also as regulators of various cellular activities, including growth, differentiation, angiogenesis, and tumor development. They play a key role in modulating cellular behavior and function [190].
Fibrillins
They are a group of substantial extracellular glycoproteins (350 kDa) comprising three isoforms: Fibrillin-1, fibrillin-2, and fibrillin-3. These molecules feature 40-80 amino acid residues and multiple cbEGF domains interspersed with several motifs containing eight cysteines (TB motifs) that bind TGFβ. No other extracellular proteins contain as much cysteine as fibrillins [191]. While fibrillin-2 and fibrillin-3 are found in embryonic tissues, with some presence in peripheral nerves, skin, and tendons, fibrillin-1 is present in both embryonic and adult tissues [192].
They are the principal component of microfibrils in both elastic and non-elastic extracellular matrices, interacting closely with tropoelastin and integrins through direct binding. Microfibrils maintain structural integrity in specific organs and serve as scaffolds for elastogenesis in tissues like skin, lungs, and blood vessels [193]. Thus, fibrillin is used to incorporate elastin into elastic fibers. Various mutations, including those in the propeptide sequence encoded by the C-terminal domain of the fibrillin-1 gene, result in defective microfibril assembly in individuals with Marfan syndrome [194]. Besides fibrillins and elastin, numerous other proteins contribute to the makeup of microfibrils [195]. Fibronectin has a role in the process of binding a C-terminal fibrillin-1 region with the fibronectin gelatin-binding region. Homocysteinylation of fibronectin homocysteinylation reduces fibronectin dimers to monomers, impairing the assembly of fibrillin and microfibrils, like the effects of homocysteinylation of fibrillin-1 [196]. Fibrillins contain several TGF-binding motifs, making their structure and function akin to latent-TGF-binding proteins [197].
Laminins
Represent a fundamental group of large glycoproteins that function with the extracellular matrix, particularly in the basement membranes of tissue [198]. Their molecular weights typically range from 400 to 900 kDa, depending on the isoforms and subunit composition [199]. These molecules are composed of three distinct subunits: the first one, alpha (α), ranges between 160 and 400 kDa, the second one (β) from 120 to 210 kDa, and the third one, gamma (γ), from 150 to 200 kDa, which combine to form heterodimers. Out of the 11 identified in mammalian subunits, 16 different Laminin isoforms have been characterized [200]. Each isoform is designated by a code based on its subunit composition, enhancing its structural components and its function in various tissues, like Lm111 (α1β1γ1) or Lm211 (α2β1γ1). These subunits assemble into heterotrimers via a long coiled-coil region, forming laminins essential to the basement membrane's structure and function. The full hetero trimeric laminins vary in size but often measure around 800-900 kDa [201].
Laminin exhibits differences in its polymerization capabilities and interactions with cellular receptors, contributing to basement membranes' specific makeup across diverse tissues [202]. Most basement membranes contain laminin isoforms, suggesting that their combination contributes to the dynamic properties [203]. The structure of laminins varies, with some forms taking on a cross-like configuration. These forms possess three short arms, each capped by LN domains, and a longer arm, facilitating the interaction with nidogen and promoting polymerization [204]. Laminin with truncated subunits, such as those containing the α3A, α4, or γ4 chains, lacks the complete short arms and corresponding LN domains, thus precluding polymerization. These perform other functions related to signaling and structural organization without contributing to the polymer scaffold [205]. The long arms of laminin molecules extend from the subunit, terminating in a set of globular domains (LG). These domains interact with integrins, dystroglycan receptors, and other cell surface molecules such as sulfated glycolipids and heparan sulfate [206]. This interaction with cellular components enables laminins to regulate a range of biological processes, including cell adhesion, differentiation, and migration, which are essential for maintaining tissue integrity and function [207].
Laminin mutations, particularly in Lm332, can lead to severe diseases such as Herlitz-type junctional epidermolysis bullosa (JEB), underscoring the importance of these glycoproteins in tissue stability [208]. In contrast, the α3B splice variant is present in various developing tissues, including the brain, and it contributes to polymerizing laminins with stronger self-association capabilities, which may enhance their role in tissue development and repair [209].
Osteopontin
It is a bone matrix glycoprotein presented in bone and dental tissues by mediating the interactions between cells and minerals [210]. Osteopontin (OPN) is classified as a matricellular cytokine and functions in processes such as bone remodeling and bone formation under mechanical stress [211]. Structurally, OPN is an intrinsically disordered protein (IDP) enriched with acidic residues, particularly aspartic and glutamic acids, which constitute about 25% of its sequence [212]. The molecular weight of OPN typically ranges between 44 and 75 kDa, depending on its level of phosphorylation, which significantly influences its functionality [213]. Phosphorylation of OPN is a post-translational modification, regulating its interaction with calcium phosphate minerals. As a member of the SIBLING family (small integrin-binding ligand N-linked glycoproteins), OPN contains an ASARM motif known for inhibiting extracellular matrix mineralization by binding hydroxyapatite crystals. This inhibitory activity is controlled by the protease PHEX, which cleaves OPN into inactive fragments, thus modulating its regulatory role in mineralization [214].
ECM mineralization, such as hypophosphatemia, hyperphosphatemia, and hypophosphatasia [215-217]. In these models, full-length OPN was detected in bone extracts of Hyp and Fgf23 -/- mice, and its phosphorylation level was shown to decline. These conditions lead to an aging-like skeletal phenotype characterized by impaired mineralization and osteomalacia, where the ECM becomes compromised [218].
Nidogen
It is a sulfated glycoprotein consisting of three globular domains (G1, G2, and G3), a short linker, a rod-like domain, and is conserved across species [219]. These domains mediate carious interactions within the ECM, particularly with Collagen IV and Laminin, establishing a structural framework for tissue integrity [220]. Nidogen´s G2 domain binds to Collagen IV, while the G3 domain interacts with Laminin, facilitating the formation of stable ternary complexes [221]. These molecular interactions serve as a key element in maintaining the ECM´s architecture, especially at the basement membrane, contributing to cellular adhesion and tissue stability [222]. In addition to structural functions, Nidogen plays an important role in neuronal development, particularly in directing axon migration and forming neuromuscular junctions [223]. In Nidogen mutations, disruptions in nerve positioning and muscle connectivity result in motor and behavioral defects, further demonstrating its importance for proper ECM function in neural tissues [224]. Mice with Nidogen-1 mutations exhibit neurological abnormalities, including seizure-like symptoms and impaired muscle control [225]. Mutations in Nidogen have been linked to developmental disorders, such as Dandy-Walker malformation, where abnormalities in ECM integrity lead to defects in epithelial morphogenesis and neural development. Studies suggest that disruptions in the Nidogen-Laminin interaction contribute to a wide range of phenotypic variability, from subtle skeletal defects to severe neurological conditions [226,227].
Proteoglycans
Proteoglycans are complex molecules composed of glycosaminoglycan (GAG) chains covalently attached to a core protein, primarily located on the cell surface or within the extracellular matrix [228]. They maintain tissue hydration and act as molecular sieves in basement membranes. Their GAG chains, which contain repeating disaccharides of uronic acid and acetylated or sulfated hexosamines, vary in length based on available Ser-Gly sites on the protein core [229]. Proteoglycans include several types, such as heparan sulfate, chondroitin sulfate, and keratan sulfate, each with distinct disaccharide compositions. Classified into four main types, proteoglycans exhibit specific cellular distributions [230].
Serglycin is the only proteoglycan that forms part of the intracellular group, storing proteases with mast cell granules [231]. In contrast, heparan sulfate proteoglycans (HSPGs) are primarily associated with cell surfaces, where they support growth factor functions and interactions within the basement membrane [232]. HSPGs facilitate cellular communication and sustain morphogen gradients essential for development and regeneration by binding to growth factors like FGF and VEGF and structural ECM components [233]. Also, proteoglycans containing chondroitin and dermatan sulfate (CSPGs and DSPGs) become predominant [234]. These proteoglycans have an important role in the structure of complex matrices, including cartilage, brain, intervertebral discs, tendons, and corneas. Their functions include providing viscoelasticity, retaining water, maintaining osmotic pressure, ensuring organized collagen arrangement, and preserving corneal transparency [235]. Additionally, the ECM hosts the largest class of proteoglycans, small leucine-rich proteoglycans (SLRPs), which are abundant at the gene level. SLRPs act as structural elements and signaling molecules, especially during tissue remodeling associated with cancer, diabetes, inflammation, and atherosclerosis [236]. By interacting with receptor tyrosine kinases (RTKs) and toll-like receptors, SLRPs influence migration, proliferation, immune responses, apoptosis, autophagy, and angiogenesis [237].
Syndecan
It is a family that comprises transmembrane proteoglycans that connect cells to the ECM. Syndecans feature an ectodomain that binds GAG chains and a cytoplasmic domain with a PDZ-binding motif that anchors them to the cytoskeleton [238]. Syndecans are involved in a wide range of cellular functions, including growth factor binding, formation of morphogen gradients, endocytosis, and lipoprotein clearance, particularly through Syndecan-1 [239]. Proteolytic shedding of Syndecans, induced by cytokines and enzymes, regulates their presence on the cell surface and within the pericellular environment [240]. Shed syndecan-1, particularly in cancer, promotes tumor growth and metastasis, while syndecan-2 can inhibit angiogenesis by reducing endothelial cell migration [241,242]. A novel function of syndecan-1 includes nuclear translocation in cancer cells, where it affects gene transcription by modulating enzymes such as histone acetyltransferase (HAT), promoting tumorigenic gene expression [243].
Glypicans (GPCs)
HSPGs are attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor [244]. Six genes encode glypicans, divided into two groups with orthologs found across species. Glypicans have unique features, including GAG attachment sites close to the membrane. Allowing their HS chains to bind morphogens like Hedgehog (Hh), Wnt, and FGF and modulate cell signaling [245,246]. Glypicans undergo two types of processing; the first one is furin-like proteases that cleave to the ectodomain, creating two disulfide-linked subunits, and the second one, extracellular lipases, like Notum, release glypicans from the membrane, regulating morphogen gradients such as Wnt and BMP [247,248]. Functionally, glypicans regulate growth and angiogenesis and have implications for cancer. For instance, GPC3 mutations cause Simpson-Golabi-Behmel syndrome, which involves overgrowth and developmental abnormalities. While initially thought to inhibit IGF-II, GPC3 was later shown to suppress Hh signaling, binding Indian and Sonic Hh proteins and competing with the Patched receptor [249,250]. This suppression depends on HS chains and their sulfation. Glypicans ´complex regulation by proteases and lipases suggests evolving insights into their roles in biological processes [251].
Betaglycan
Also called the TGFβ type III receptor (TGFβ3), it is a transmembrane proteoglycan that serves as a c-receptor for the TGFβ superfamily, including growth factors such as activins and BMPs [252]. Betaglycan´s extracellular domain has multiple GAG attachment sites and protease-sensitive sequences, while its short intracellular domain is rich in serine and threonine residues, allowing for phosphorylation [253]. The ectodomain contains a unique zona pellucida (ZP) module that, unlike other ZP proteins, does not polymerize but instead facilitates TGFβ ligand binding [254]. Betaglycan enhances the affinity of TGFβ isoforms for their receptors and is essential for TGFβ2 signaling. It also acts as a suppressor in cancer, blocking NF-κB-mediated expression of matrix metalloproteinase 2 (MMP2), which limits tumor aggressiveness [255].
Perlecan
It is a large, modular heparan sulfate proteoglycan (HSPG) essential for various biological processes due to its structural complexity and widespread tissue distribution [256]. This 500 kDa core protein consists of five domains with homologies to several molecules, and it interacts with various receptors and ligands, making it integral to vascular and extracellular matrix biology [257]. Its N-terminal heparan sulfate chains promote angiogenesis by binding and presenting growth factors like VEGFA and FGF to their receptors, while protease cleavage can release pro-angiogenic factors, impacting blood vessel formation and repair. Perlecan´s C-terminal domain V, endorepellin, functions oppositely to its N-terminal proangiogenic region by inhibiting endothelial migration and angiogenesis [258]. Through dual receptor targeting of VEGFR2 and α2β1 integrin, endorepellin suppresses endothelial cell migration, induces autophagy, and alters cellular structures, impacting cancer, inflammation, and vascular pathologies [259].
Aggrecan
It is a vital structure proteoglycan in cartilage, forming large, resilient complexes with hyaluronan and link proteins that allow cartilage to withstand compression [260]. It includes multiple domains, each serving a specific function: the G1 and G2 domains stabilize aggrecan´s attachment to hyaluronan, forming robust networks with collagen that reinforce cartilage. The central GAG-rich domain, filled with negatively charged CS and KS chains, attracts water, providing hydration and compressive resistance to cartilage [261]. The G3 domain, with EGF-like and lectin elements, enables aggrecan to bind to other matrix proteins like tenascins and fibulins, enhancing structural support and mechanosensitivity [262]. Genetic defects in aggrecan, such as those seen in chondrodystrophies, weaken cartilage and associated health tissues, emphasizing its role in skeletal development [263]. Aggrecan also has functions in the brain, where it forms part of perineuronal nets around cortical interneurons. Here, it may aid neural maturation, stabilize synaptic connections, and protect neurons from oxidative stress, indicating roles beyond cartilage integrity in maintaining neural health [264].
Versican
The largest hyalecan family member has a central role in tissue organization and inflammation. Encoded by the VCAN gene, versican is structurally like aggrecan but includes unique features, such as an N-terminal IgG fold and link modules that bind hyaluronan with high affinity [265]. It contains central GAGα and GAGβ domains, which are variably spliced into isoforms V0, V1, V2, V3, and a newly identified V4 associated with breast cancer [266,267]. These isoforms exhibit tissue-specific expression, influencing cell adhesion and signaling, particularly in the heart, brain, and vascular tissue [268,269]. Versican´s C-terminal domain includes EGF-like and lectin motifs, connecting it with cell surface glycoproteins and stabilizing supramolecular structures at the plasma membrane. Versican supports inflammation by interacting with receptors like CD44 and Toll-like receptors, facilitating leukocyte migration during tissue repair [270]. Proteolytic processing by enzymes, such as MMPs and ADAMTs, modifies versican´s function, influencing cell adhesion and migration, and higher levels are linked to tumor growth and inflammation in diseases like leiomyosarcoma [271].
SLRPs
They are a large family of proteoglycans with 18 gene variants, known for their small protein cores and leucine-rich repeats (LRRs). They are broadly expressed in various extracellular matrices, especially around developing organs, where they shape and stabilize tissue structure [272]. SLRPs are categorized into five classes based on genetic similarities, with Classes I-III containing GAG chains that help them bind to collagen and stabilize collagen fibrils, thereby protecting them from enzymatic degradation [273]. The family´s best-known member, decorin, binds collagen type I and is essential for proper collagen fibrillogenesis. SLRPs also interact with various receptors and signaling pathways, including TGF-β, influencing cellular processes [274]. They show structural diversity in GAG chains, allowing them to support a wide range of functions across tissues, and may be transcriptionally co-regulated, indicating a coordinated role in development and tissue repair [273]. SLRPS are divided into five groups.
Class I SLRP
Decorin (PG40 or DSPG1), the most studied SLRP, binds collagen fibrils, stabilizing tissue structure and affecting collagen´s biomechanical properties, which is important for skin integrity and connective tissues [275]. Mice without decorin show weakened skin, like Ehlers-Danlos syndrome [276]. Decorin also has anti-cancer properties, acting as a growth suppressor by binding to growth receptors like EGFR and Met, inhibiting tumor progression and blood vessel formation [277,278].
Biglycan, decorin´s close relative, has pro-inflammatory roles through interactions with immune receptors, impacting immune response and tissue injury recovery [279].
Asporin, another SLRP, regulates bone formation and antagonizes cartilage formation via TGF-β signaling, with genetic variations linked to osteoarthritis severity [280].
Some SLRPs, including ECM2, are less understood but are thought to have distinct roles based on structural homology. SLRPs´ versatile interactions in collagen assembly, immune response, and disease progression [281].
Class II SLRP
This class consists of five distinct SLRPs, which can be further categorized into three subgroups based on protein homology: subgroup A (fibromodulin and lumican), subgroup B (PREPL and keratocan), and subgroup C (Osteoadherin) [282]. Despite their functional diversity, all Class II SLRPs share a conserved genomic structure, composed of three exons, with the largest exon encoding most of their leucine-rich repeats (LRRs). These proteins also contain a charged N-terminal region enriched in tyrosine sulfate residues, contributing to their anionic properties [283]. A defining feature of these SLPRs is their keratan sulfate and polylactosamine modifications, which contribute to their role in growth factor signaling. Notably, corneal keratan sulfate binds with high affinity to FGF2 and sonic hedgehog (SHH), suggesting its involvement in morphogen gradient formation [284]. Additionally, SLRPs interact with fibrillar collagens, reinforcing tissue structure and mechanical strength.
Fibromodulin was originally identified in cartilage. It is also notable for delaying collagen fibril formation and is necessary for maintaining ECM stability in tissues such as cartilage and tendons [285]. Its N-terminal domain contains tyrosine sulfate residues, enabling dual functions: collagen cross-linking and growth factor binding, including FGF, VEGF, and various cytokines [286]. It binds the same region of collagen I as lumican and is particularly important in regulating fibrillogenesis during development [287]. Fibromodulin also activates the classical complement pathway, supporting immune modulation and structural stability [288].
Lumican, also known for its role in maintaining corneal transparency, helps organize collagen fibrils, particularly in the cornea, where it preserves the spacing necessary for visual clarity [289]. Lumican also influences cancer and inflammation; in cancer, it is often upregulated in tumor stroma, especially in breast carcinomas and melanomas, and inhibits tumor progression [290]. It binds neutrophils during transmigration across endothelial barriers, enhancing neutrophil response in injury and wound healing [291].
PRELP (Proline/Arginine-Rich END Leucine-Rich Repeat Protein) primarily functions in connective tissues near basement membranes [292]. Its positively charged N-terminal domain binds heparin and heparan sulfate, creating structural links between ECM and basement membranes [293]. PRELP also inhibits NF-κB signaling, which reduces osteoclast formation, making it an effective antiresorptive molecule, as seen in osteoporosis models [294]. Additionally, PRELP prevents complement membrane attack complex formation, protecting vascularized tissues in inflammatory diseases [295].
Keratocan is essential for corneal transparency, and keratocan supports proper stromal collagen organization, which is necessary for corneal structure [296]. Although mice deficient in keratocan show normal corneal openness, they have irregular collagen fibril spacing, underscoring keratocan´s role in collagen packing [297]. Inflammation studies reveal that keratocan assists in chemokine gradient formation, promoting neutrophil recruitment during corneal injury and inflammation [298].
Osteoadherin (Osteomodulin) is highly expressed in mineralized tissue, and osteoadherin contributes to bone matrix organization [299]. With numerous tyrosine sulfates and acidic residues in its N-terminal domain, osteoadherin interacts with growth factors, mimicking heparin´s binding properties [300]. It facilitates osteoblast attachment, particularly via αvβe integrin, and shows a glycosylation pattern change during endochondral bone formation, suggesting a targeted role in mineralization and bone matrix integrity [301].
Class III SLRP
Class III consists of three closely related members: epiphycan, opticin, and osteoglycin. Unlike other SLRP classes, they have a distinct structure, containing only seven LRRs instead of the typical 10-12 found in other SLRP classes. Like Class II members, Class III SLPRs possess N-terminal consensus sequences for tyrosine sulfation, which likely serve as signals for keratan sulfate attachment during protein synthesis and post-translational modification [302].
Epiphycan is a glycoprotein found in epiphyseal cartilage and is the mammalian counterpart of avian PG-Lb [303]. It functions in chondrogenesis, distributed throughout the growth plate during cartilage development [304]. While mice with this proteoglycan show mild bone defects, epiphycan/biglycan double-knockout mice have shorter bones and develop osteoarthritis, suggesting a cooperative function [305]. It is also part of the collagen IX intercom, indicating a role in growth plate organization [306].
Osteoglycin, also known as mimecan, was first identified in bone and later as a keratan sulfate SLRP in the cornea [307]. Although multiple mRNA variants are produced from the Ogn genes, they generate a single protein core [308]. Besides, the inhibition of this gene in mice shows increased collagen fibril diameter in the cornea and dermis, like other SLRP-related phenotypes [309]. Osteoglycin undergoes proteolytic processing in vivo, particularly by BMP-1/Tolloid-like metalloproteinases, which enhances its role in collagen fibrillogenesis regulation. Nevertheless, this molecule is involved in myocardial integrity, cardiac remodeling, and injury response, interacting with ECM glycoproteins [310]. Additionally, osteoglycin functions as an anabolic bone factor secreted by muscle cells and may serve as a predictor of cardiovascular events [311].
Class IV SLRP
This is a non-canonical class of the SLPRs, and it includes the following chondroadherin: [312] and nyctalopin [313].
Chondroadherin is primarily found in cartilage, linking chondrocytes to the ECM through interactions with α2β1 integrin and heparan sulfate chains [314]. It also binds collagens II and VI, and mice knockout of this molecule exhibit cartilage and bone abnormalities, including disrupted collagen network assembly and weakened mechanical properties in cartilage [315,316].
Nyctalopin is unique among SLRPs as it is GPI-anchored to the plasma membrane, and X-linked- Mutations in NYX cause X-linked congenital stationary blindness, affecting night vision, myopia, and visual acuity [317]. In the retina, nyctalopin interacts with TRPM1 and mGluR6, forming a supramolecular complex for visual synapse signaling [318].
Class V SLRP
This lesser-known group of non-canonical SLRPs consists of podocan and podocan-like.
Podocan was first identified due to its elevated presence in podocytes from sclerotic glomeruli in HIV-associated kidney disease [319]. It is found in the glomerular basement membrane, proximal tubules, and aortic tissue, hinting at a role in vascular healing. It functions as a suppressor of smooth muscle cell migration and proliferation, potentially influencing atherosclerosis, much like biglycan [320]. In podocin-deficient cells, Wnt signaling activity is heightened, whereas cells with increased podocin expression show reduced Wnt signaling, a trait observed in other SLRPs [321]. Like decorin and biglycan, podocin interacts with collagen I and contributes to cell growth regulation through p21 WAF1 activation [322].
Metalloproteases
Matrix metalloproteinases (MMPs) are zinc-dependent endoproteases involved in ECM remodeling, which is involved in protein degradation, tissue homeostasis, and cellular signaling [323]. These enzymes regulate cell proliferation, migration, differentiation, apoptosis, and angiogenesis, influencing tissue repair and immune responses [324]. However, MMPs can modify bioactive molecules on the cell surface, affecting various signaling that govern cellular behavior [325]. While MMP expression and activity fluctuate in normal physiological processes, such as pregnancy and wound healing, their dysregulation has been linked to pathological conditions, including cardiovascular diseases, musculoskeletal disorders, and cancer [326]. In malignancies, MMPs contribute to tumor progression and invasiveness by degrading ECM barriers and facilitating cancer cell migration and metastasis [327].
Most of them are produced by a variety of tissues and cell types, including fibroblasts, osteoblasts, endothelial cells, vascular smooth muscle cells, macrophages, neutrophils, lymphocytes, and cytotrophoblasts [328]. Their activity is tightly controlled by tissue inhibitors of metalloproteinases (TIMPs), which regulate ECM turnover and prevent excessive degradation [329]. However, an imbalance between MMPs and TIMPs, either through increased MMP expression or reduced TIMP levels, can contribute to pathological conditions, including cardiovascular diseases, osteoarthritis, and cancer.
Collagenases
Collagenases (MMP-1, MMP-8, MMP-13, and MMP-18) degrade fibrillar collagens I, II, and III by first unwinding their triple-helical structure and then hydrolyzing peptide bonds [330]. Their hemopexin domain is necessary for cleaving intact collagen, while the catalytic domain processes non-collagen substrates. These enzymes are important for ECM remodeling, tissue repair, and homeostasis, but their dysregulation is linked to arthritis, fibrosis, and cancer [331].
MMP-1 is encoded in chromosome 11 and is involved in collagen degradation and inflammation [332]. Its expression in inflammatory conditions and autoimmune diseases contributes to delayed wound healing [333]. MMP-1 is also linked to cancer progression, with certain genetic variations associated with poor prognosis [334]. Anti-fibrotic agents like stratifin and kynurenic acid influence MMP-1 expression, enhancing tissue repair and reducing fibroids [335]. Overall, MMP-1 regulation affects both tissue remodeling and disease progression [336].
MMP-8, also known as collagenase-2 or neutrophil collagenase, is encoded in chromosome 11 and was first identified in a cDNA library from leukocytes of a leukemia patient [337]. It degrades interstitial collagens I, II, and III, with its activation regulated by other MMPs like MMP-3 and MMP-10. MMP-8 appears early in dermal wound healing, and its deficiency in mice leads to delayed healing and increased inflammation [338,339]. In periodontal disease, MMP-8 contributes to connective tissue breakdown, and its presence in saliva may serve as a diagnostic marker [340].
MMP-13, also known as collagenase-3, is encoded in chromosome 11 and efficiently degrades type II collagen [341]. It is overexpressed in osteoarthritic cartilage, contributing to disease progression, and is regulated by factors like miR-411 and dietary fatty acid ratios [342]. MMP-13 is implicated in lung diseases, brain astrocyte migration, and liver fibrosis, where targeted gene delivery has shown potential therapeutic effects [343]. It is also frequently overexpressed in tumors, promoting metastasis, particularly in nasopharyngeal cancer [344].
MMP-18, also known as collagenase-4, is encoded in chromosome 12 and shares structural similarities with MMP-1, -3, and -11. It has a unique dual-cleavage activation site and is expressed in various tissues but not in the brain, skeletal muscle, kidney, liver, or leukocytes [345]. MMP-18 is found in migrating macrophages and axonal growth, particularly in response to skin explants [346]. Its activity is linked to ECM breakdown, influencing nerve growth and regeneration [347].
Gelatinases
This group includes MMP-2 and MMP-9, which share structural similarities with other MMPs but have a unique collagen-binding domain essential for gelatin degradation. They are key enzymes in ECM breakdown and have a broad range of non-matrix protein targets [348]. Beyond their role in ECM remodeling, they are involved in embryonic development, angiogenesis, vascular diseases, inflammation, infections, neurodegenerative disorders, and cancer progression. Their diverse functions make them significant regulators in both normal and pathological processes [349].
MMP-2, also known as gelatinase-2, is on chromosome 16 and degrades collagen through a two-phase process [350]. It accumulates at the cell surface via the MT1-MMP/TIMP-2 complexes, contributing to localized collagen breakdown in angiogenesis, tissue repair, and inflammation [351]. MMP-2 is implicated in tumor invasion and metastasis, particularly in esophageal cancer, and targeting it with siRNA has shown promise in reducing lung cancer growth. In glioblastomas, MMP-2 interacts with α5β1 integrin, influencing IL-6/stat3 signaling and tumor progression [352].
MMP-9, or gelatinase-B, is a type IV collagenase located on chromosome 20 and is produced by various cell types, including epithelial cells, fibroblasts, and immune cells [353]. It acts in inflammatory cell migration and tissue remodeling [354]. In cholesteatoma, MMP-9 expression is elevated in tissues rather than serum, correlating with inflammatory severity [355]. Moreover, it is linked to esophageal cancer progression, with its activity correlating with vascular invasion [356].
Stromelysins
MMP-3, MMP-10, and MMP-11, also known as Stromelysins 1, 2, and 3, share the same domain structure as collagenases but do not cleave interstitial collagen [357]. MMP-3 and MMP-10 have similar structures and substrate specificities, actively degrading ECM components, particularly in proMMP activation [358]. In contrast, MMP-11 has weak ECM-degrading activity and is more distantly related. Unlike MMP-3 and MMP-10, which are secreted as inactive proenzymes, MMP-11 is activated intracellularly by furin and secreted in its active form [359].
MMP-3 is encoded on chromosome 11 and functions as a secretory endopeptidase that degrades ECM components, including various collagens, proteoglycans, elastin, and fibronectin [360,361]. It can activate other MMPs involved in tissue remodeling and has been detected in cell nuclei, suggesting a role in gene regulation and apoptosis. MMP-3 is implicated in post-traumatic osteoarthritis, where fibronectin fragments upregulate its expression, accelerating cartilage degradation [362]. Furthermore, it contributes to atherosclerosis, tumor growth, and metastasis [363]. It may interact with neurodegeneration by inducing mitochondrial oxidative stress and neuronal death [364].
MMP-10 is a secreted protein located on chromosome 11. It has multiple functions in various diseases, such as pulmonary fibrosis, where its serum levels correlate with lung function and disease severity [365]. MMP-10 is also implicated in respiratory syncytial virus infections, peripheral arterial disease, and wound healing [366]. It may contribute to tumor progression, metastasis, and liver regeneration, particularly in hepatocellular carcinoma [367].
MMP-11 is a protease with a gene locus on chromosome 22. It is secreted in its active form after intracellular activation, unlike other MMPs, which require extracellular activation [368]. MMP-11 acts in tissue remodeling during embryonic development, wound healing, and tumor invasion [369]. It has been linked to poor prognosis in various cancers, including breast, esophageal, and oral squamous cell carcinoma, with its elevated expression associated with tumor progression and metastasis [370,371]. However, some studies suggest it may inhibit metastasis in certain cancer models, indicating a complex role in cancer biology [372].
Matrilysins
MMP-7, or matrilysin-1, is a small protease located on chromosome 11. It appears in tissue remodeling, particularly in the uterus and following injury, by degrading ECM components and cleaving cell surface molecules like Fas-ligand and E-cadherin [373]. MMP-7 has dual effects on apoptosis, either inducing or inhibiting it, and is involved in immune response in the intestine. Studies suggest its involvement in chronic tonsillitis and idiopathic pulmonary fibrosis [374]. MMP-7 may also be a key factor in cancer progression, especially in oral squamous cell carcinoma [375].
MMP-26 (matrilysin-2), or endometase, is located on chromosome 11 and shares structural similarities with macrophage metalloelastase. It is primarily expressed in the placenta and uterus, but it is found in various tumor cell lines and malignant tumors, including those of epithelial origin [376]. MMP-26 exhibits broad proteolytic activity, targeting substrates like collagen, fibronectin, and gelatin, and is present in activating proMMP-9 [377]. As part of this, MMP-26 interacts in tissue remodeling, angiogenesis, and tumor progression, particularly in tumor invasion [378]. Expression of MMP-26 is linked to granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced tumor invasion and may serve as a marker for metastasis in pancreatic adenocarcinoma [379]. TIMP-2 and -4 regulate MMP-26, with TIMP-4 being a stronger inhibitor [380].
Membrane-type MMPs
This group consists of four transmembrane MMPs (MMP-14, -15, -16, and -24) and two GPI-anchored MMPs (MMP-17 and -25). These enzymes are activated intracellularly, with active forms expressed on the cell surface. MT-MMPs feature a membrane anchoring domain and display protease activity at the cell surface, making them efficient pericellular proteolytic machines. All MT-MMPs, except MMP-17, can be active proMMP-2. MMP-14 is also capable of activating proMMP-13 on the cell surface.
MMP-14 (MT1-MMP) is a transmembrane metalloproteinase encoded on chromosome 14, involved in extracellular matrix degradation and cell migration [381]. It activates proMMP-2, contributing to tumor invasion, metastasis, and tissue remodeling [382]. Elevated MT1-MMP levels correlate with aggressive cancer phenotypes, including head and neck squamous cell carcinoma and salivary gland carcinomas [383]. Deficiency in MT1-MMP leads to developmental abnormalities, emphasizing its role in skeletal and connective tissue integrity. Also, MT1-MMP is implicated in atherosclerotic plaque instability [384].
MMP-15 (MT2-MMP), encoded on chromosome 16, is a membrane-bound metalloproteinase implicated in extracellular matrix remodeling and MMP-2 activation, influencing tumor invasion [385]. It is indispensable for placental vasculogenesis, as its deficiency leads to embryonic lethality in mice. In colorectal cancer, MMP-15 is upregulated during early tumorigenesis, showing stromal localization [386]. However, in supraglottic carcinoma, MMP-14 appears more influential in tumor progression [387]. Elevated MT2-MMP expression correlates with increased invasion in laryngeal cancer [388].
MMP-16 (MT3-MMP) is located on chromosome 8 and is a membrane-bound protease that modulates MMP-2 and -9 activation, influencing tumor invasion and cell migration [389]. Its elevated expression in melanoma and bladder cancer correlates with adverse outcomes, while miR-146a and miR-155 regulate its activity in breast cancer and cardiac progenitor cells [390,391]. In neonatal lung development, MMP-16 polymorphisms impact susceptibility to bronchopulmonary dysplasia. Experimental inhibition of MMP-16 restricts cancer proliferation and enhances regenerative potential [392].
MMP-17 (MT4-MMP), encoded on chromosome 12, is a GPI-anchored metalloproteinase distinct from MT1-MMP, lacking a cytoplasmic tail and not activating proMMP-2 [393]. It features a unique nine-residue insertion necessary for furin-mediated activation. Expressed in multiple tissues, including leukocytes, its role may involve modulating growth factors and inflammatory mediators like TNF-α [394]. MMP-17 is overexpressed in various cancers, particularly breast cancer, and plays an important role in tumor progression [395].
MMP-24 (MT5-MMP), encoded on chromosome 20, is a membrane-associated metalloproteinase implicated in tumorigenesis and neural plasticity [396]. It facilitates proMMP-2 activation, contributing to malignancies such as glioblastoma. The brain regulates neural stem cell quiescence by cleaving N-cadherin and influences neuropathic pain by modulating sensory neuron plasticity [397]. Additionally, it mediated neuro-immune interactions in nociceptive signaling, affecting thermal pain responses. Elevated MMP-24 expression in breast cancer suggests its role in tumor progression, invasion, and angiogenesis [398].
MMP-25 (MT6-MMP), encoded on chromosome 16, is a GPI-anchored enzyme expressed in leukocytes and certain cancers [399]. It exists as a disulfide-linked homodimer, with its stability influenced by cysteine residues. MT6-MMP is localized in lipid rafts and translocates to the cell surface during neutrophil apoptosis, suggesting roles in immune response and IL-8 secretion [400]. It exhibits gelatinolytic activity and weak MMP-2 activation but does not promote cell migration. Elevated MMP-25 expression in glioblastomas, colon, urothelial, and prostate cancers implicates it in tumor progression [401].
Non-classified MMPs
MMP-12 is a macrophage metalloelastase located on chromosome 11 and interacts in immunity, inflammation, and tissue remodeling [402,403]. It facilitates interferon-α secretion, modulating antiviral responses, and contributing to asthma-related lung inflammation [404]. MMP-12 disrupts the blood-brain barrier in cerebral ischemia, exacerbating brain injury [405]. Its overexpression in head and neck squamous cell carcinoma correlates with extracapsular spread, making it an interest as a prognostic marker [406].
MMP-19, or stromelysin-4 or RASI-1, is an ECM proteinase located on chromosome 12 and is involved in tissue remodeling, fibrosis, and cancer. It regulates angiogenesis, inhibits pulmonary fibrosis, and contributes to liver fibrosis [407]. In cancer, MMP-19 promotes tumor progression in gallbladder carcinoma, colorectal cancer, and NSCLC but exhibits tumor-suppressive activity in nasopharyngeal carcinoma[408-410]. Genetic alterations in MMP-19 have been linked to congenital optic disc anomalies [411].
MMP-20, also known as enamelysin, is a tooth-specific MMP encoded by a gene on chromosome 11, clustered with several other MMP family members [412]. It degrades amelogenin and other enamel matrix proteins, having a role in enamel biomineralization [413]. Mutations in MMP-20 lead to amelogenesis imperfect, resulting in defective enamel formation [414,415]. MMP-20 also processes aggrecan and cartilage oligomeric matrix protein, contributing to tooth matrix remodeling [416].
MMP-21, an MMP with gelatinolytic activity located on chromosome 1, is involved in embryogenesis and cancer progression. It is highly expressed in invasive carcinomas and correlates with our prognosis in oral, esophageal, and colorectal cancers [417-419]. In Merkel cell carcinoma, MMP-21 is linked to less aggressive tumors, while MMP-10 is associated with disease progression [420]. In pancreatic cancer, MMP-21 marks differentiation rather than invasiveness and is upregulated by epidermal growth factor [421].
MMP-22, located on chromosome 1, is linked to tumor suppression and shares a locus with MMP-21. It contains domains similar to stromelysin-3 and features a Zn+2-binding region necessary for activation [422]. Unlike the MMPs, it lacks the “cysteine switch” for autocatalytic activation. MMP-22 produces multiple mRNA variants through alternative splicing [423].
MMP-23, located on chromosome 1, has a unique domain structure distinct from other MMPs. It lacks a conventional signal sequence, hemopexin-like repeats, and typical MPP subclass features. Instead, it contains cysteine-rich immunoglobulin-like domains and functions as a type II membrane protein [424]. MMP-23 is predominantly expressed in reproductive tissues. It is also upregulated in MDA-MB-231 breast cancer cells [425].
MMP-27, a stromelysin located on chromosome 11, is poorly secreted and primarily retained in the endoplasmic reticulum. It is expressed in B-lymphocytes, endometrial macrophages, and endometriotic lesions, with peak expression during menstruation [426]. In cancer, MMP-27 is more abundant in high-grade breast tumors, particularly in the MDA-MB-468 cell line. Its expression may contribute to breast cancer progression. Despite the structural resemblance to MT-MMPs, MMP-27 is not an integral membrane protein and remains stored until needed [427].
MMP-28 (epilysin) is a widely expressed MMP involved in tissue homeostasis, wound healing, and repair, located on chromosome 17 [428]. It is found in the epidermis and various organs and is linked with embryo implantation, cardiac remodeling, and periodontal health [429]. In myocardial infarction, MMP28 deficiency impairs healing, leading to cardiac rupture. Unlike other MMPs, it is downregulated in colon cancer, suggesting a protective role. And its expression remains elevated in osteoarthritis and rheumatoid arthritis [430,431].
All MMPs are summarized in Table 4.
All collagen types are divided into different groups that are classified. N.d. (Not defined).
| Metalloproteases | ||||
|---|---|---|---|---|
| Group of Metalloproteases | Subtype | Chromosome | Function | Implication |
| Collagenase | MMP-1 | 11 | Collagen degradation | Inflammation, autoimmune diseases |
| MMP-8 | 11 | Collagen type I, II and III degradation | Scarring and periodontal disease | |
| MMP-13 | 11 | Type II collagen degradation | Osteoarthritis, pulmonary disease, astrocyte migration, liver fibrosis, metastasis, nasopharyngeal cancer | |
| MMP-18 | 12 | ECM degradation | Macrophage migration, axonal growth | |
| Gelatinase | MMP-2 | 16 | Localized two-phase collagen degradation | Angiogenesis, tissue repair, inflammation, metastasis, esophageal and lung cancer |
| MMP-9 | 20 | ECM degradation | Inflammation, tissue remodeling, esophageal cancer | |
| Stromelysins | MMP-3 | 11 | Degradation of collagen, proteoglycans, elastin and fibronectin | Tissue remodeling, gene regulation, apoptosis, osteoarthritis, atherosclerosis, metastases |
| MMP-10 | 11 | N.d. | Pulmonary fibrosis, viral infections, arterial disease, scarring, metastasis, hepatocellular carcinoma | |
| MMP-11 | 22 | N.d. | Tissue remodeling in embryonic development, scarring, breast cancer, esophageal cancer, oral squamous cell carcinoma | |
| Mathylisins | MMP-7 | 11 | Fas and E-cadherin ligand cleavage | Tissue remodeling, dual involvement in apoptosis, chronic tonsillitis, idiopathic pulmonary fibrosis |
| MMP-26 | 11 | Degradation of collagen, fibronectin and gelatin. MMP-9 activation | Tissue remodeling, angiogenesis, tumor invasion, pancreatic adenocarcinoma | |
| Membrane type | MMP-14 | 14 | Modulates MMP-2 activation | Cell migration, tumor invasion, metastasis, aggressive head and neck carcinoma, salivary gland carcinoma |
| MMP-15 | 16 | Modulates MMP-2 activation | Placental vasculogenesis, colorectal cancer, supraglottic carcinoma, laryngeal cancer | |
| MMP-16 | 8 | Modulates activation of MMP-2 and MMP-9 | Melanoma, bladder cancer, breast cancer, bronchopulmonary dysplasia | |
| MMP-17 | 12 | Modulation of growth factors and inflammatory mediators | Breast cancer | |
| MMP-24 | 20 | Activation by MMP-2, N-cadherin cleavage | Tumorigenesis, neural plasticity, nociceptive signaling, glioblastoma, angiogenesis | |
| MMP-25 | 16 | Gelatinolytic, MMP-2 activation | Apoptosis, immune response, glioblastomas, colon, urothelial and prostate cancer | |
| Not classified | MMP-12 | 11 | Interferon-α secretion | Immunity, inflammation, cerebral ischemia, head and neck squamous cell carcinoma |
| MMP-19 | 12 | N.d. | Angiogenesis, fibrosis, colorectal cancer, lung cancer, gallbladder carcinoma, nasopharyngeal carcinoma | |
| MMP-20 | 11 | Degradation of amelogenin, processing of aggrecan and oligomeric protein | Enamel biomineralization | |
| MMP-21 | 1 | Gelatinolytic | Embryogenesis, invasive carcinomas, oral, esophageal and colorectal cancers, Merkel cell carcinoma, pancreatic cancer | |
| MMP-22 | 1 | N.d. | Tumor suppression | |
| MMP-23 | 1 | N.d. | Reproduction and breast cancer | |
| MMP-27 | 11 | N.d. | Endometriotic lesions, menstruation, breast cancer | |
| MMP-28 | 17 | N.d. | Tissue homeostasis, wound healing, repair, embryo implantation, cardiac remodeling, myocardial infarction, colon cancer, osteoarthritis, rheumatoid arthritis | |
Glycosaminoglycans
Glycosaminoglycans (GAGs) are negatively charged, linear polysaccharides composed of repeating disaccharide units, where hexosamines are linked to hexuronic acids or galactose via O-glycosidic bonds.
Based on disaccharide composition and sulfation patterns, GAGs are classified into four main types: chondroitin sulfate (CS) and dermatan sulfate (DS), heparin and heparan sulfate (HS), hyaluronan (HA), and keratan sulfate (KS). Most GAGs contain hexuronic acids, such as glucuronic acid (GlcA) in CH, HS, and HA, or iduronic acids (IdoA) in DS and heparin, while KS incorporates galactose instead. Hexosamine units vary, with N-acetylglucosamine (GlcNAc) found in HA, HS, and KS, and N-acetylgalactosamine (GalNAc) present in CS and DS.
Except for HA, all GAGs attach covalently to protein cores in proteoglycans (PGs), forming polymerized disaccharide chains, whereas HA is a free oligosaccharide binding noncovalently to PGs. HA is synthesized at the plasma membrane by hyaluronan synthases (HASes), while other GAGs undergo biosynthesis in the Golgi apparatus, where they attach to core proteins and undergo structural modifications.
- Chondroitin sulfate is a common component of the ECM and cell surfaces in animals. CS chains attach to core protein, forming proteoglycans involved in diverse biological processes through interactions with growth factors, morphogens, cytokines, and adhesion molecules [432]. Despite its simple disaccharide structure, CS undergoes extensive modifications, including sulfation and epimerization, leading to structural diversity [433]. CS and DS often exist as hybrid chains, influencing cellular functions through specific binding sites [434]. Structural studies have revealed CS-DS interactions with bioactive proteins, yet detailed saccharide sequences remain elusive [435].
- Dermatan sulfate, also known as chondroitin sulfate B, is a linear polysaccharide composed of disaccharide units containing GalNAc and GlcA or IdoA [436]. The presence of IdoA differentiates DS from other chondroitin sulfate and contributes to its unique protein-binding properties [437]. DS structure varies due to differences in chai length, sulfation patterns, and core proteins, influencing its biological interactions. It has a variety of functions in the ECM, cell function, and wound healing [438]. Unlike heparin and heparan sulfate, DS has distinct expression patterns and enzymatic modifications [439].
- Heparin is the oldest and most widely used anticoagulant, discovered in 1926, clinically approved since 1935 [440]. It is derived from animal tissues, primarily pig intestinal and bovine lung, and consists of a highly sulfated GAG backbone [441]. HP exerts its anticoagulant effect by binding to antithrombin III (AT), enhancing its inhibition of thrombin and other coagulation proteases [442]. Beyond anticoagulation, HP interacts with various proteins, leading to additional pharmacological effects, including anti-viral, anti-tumor, anti-inflammatory, and anti-angiogenic properties [443]. Recent research has explored its non-anticoagulant applications in infectious diseases [444], cancer [445], inflammatory conditions [446], Alzheimer´s disease [447], and diabetic nephropathy [448].
- Heparan sulfate (HS) is a widely distributed GAG found in the ECM and on cell surfaces, where it regulates tissue organization, cell signaling, and lipid metabolism [449]. Structurally, HS is less sulfated than heparin and undergoes enzymatic modifications such as epimerization and sulfation in the Golgi, affecting its binding interactions with growth factors, cytokines, and ECM molecules [450]. Its synthesis depends on UDP-sugar availability and is regulated by nucleotide-sugar transporters [451]. HS degradation, mainly by heparanases, modifies ligand recognition and signaling, with dysregulation linked to cancer progression [452]. HS proteoglycans (HSPGs), such as Syndecans and glypicans, have a role in cellular communication and structural integrity [453].
- Hyaluronan/ Hyaluronic Acid is a non-sulfated GAG distributed in connective and epithelial tissues [454]. It is a key component of the ECM, contributing to cell proliferation, migration, and tissue hydration [455]. HA consists of repeating disaccharide units of D-glucuronic acid and N-acetylglucosamine linked by β-1,4 and β-1,3 glycosidic bonds [456]. Its molecular weight ranges from 10 to 1000 kDa, influencing its biological functions. HA interacts with various receptors, including CD44, RHAMM, LYVE-1, and aggrecan, in cellular signaling, wound healing, and inflammation [457]. High molecular weight HA supports tissue integrity and hydration, while lower molecular weight HA is associated with inflammatory responses and scar formation [458]. It is used for skin health, acting as a lubricant in connective tissues and aiding in tissue repair. HA degradation is influenced by UV exposure and inflammatory signals via toll-like receptors (TLR2-TLR4) [458].
- Keratan sulfate (KS) is a mucoid material containing galactose, glucose, acetyl, and sulfate groups [459]. Later studies refined its classification, establishing its structural properties and protein interactions [460]. KS exists in three forms: KS-I in the cornea, KS-II in cartilage, and KS-III in the brain, each differing in linkage types and sulfation patterns [461-463]. It has been found to interact with regulatory proteins, including growth factors and nerve-related molecules [464]
All molecules are summarized and represented in Figures 2 and 3.
The molecules that interact in the extracellular matrix (ECM). A. Molecules in the intersection of the membrane and the ECM. B. Molecules that conform to the ECM. C. Molecules that are in the space between two fibroblasts.
Schematic representation of extracellular matrix (ECM) components and their classification. The diagram illustrates the hierarchical organization of ECM molecules, including cell surface proteoglycans (syndecan, betaglycan, glypican), and pericellular small leucine-rich proteoglycans (SLRPs, Classes I-V). Core proteins and glycosaminoglycan (GAG) chains are indicated, with color-coded domains highlighting structural and functional motifs such as protease-sensitive regions, disulfide bonds, and glycosylation sites. Glycosaminoglycans, including chondroitin sulfate, dermatan sulfate, heparan sulfate, keratan sulfate, and heparin, are shown separately, with corresponding symbols for sugar residues. Multi-adhesive glycoproteins (fibronectin, fibrinogen, fibulin, laminin, osteopontin, nidogen) and collagens (Type I and Type IV) are depicted with their structural organization. This comprehensive schematic provides a reference framework for ECM composition, molecule interactions, and functional domains.
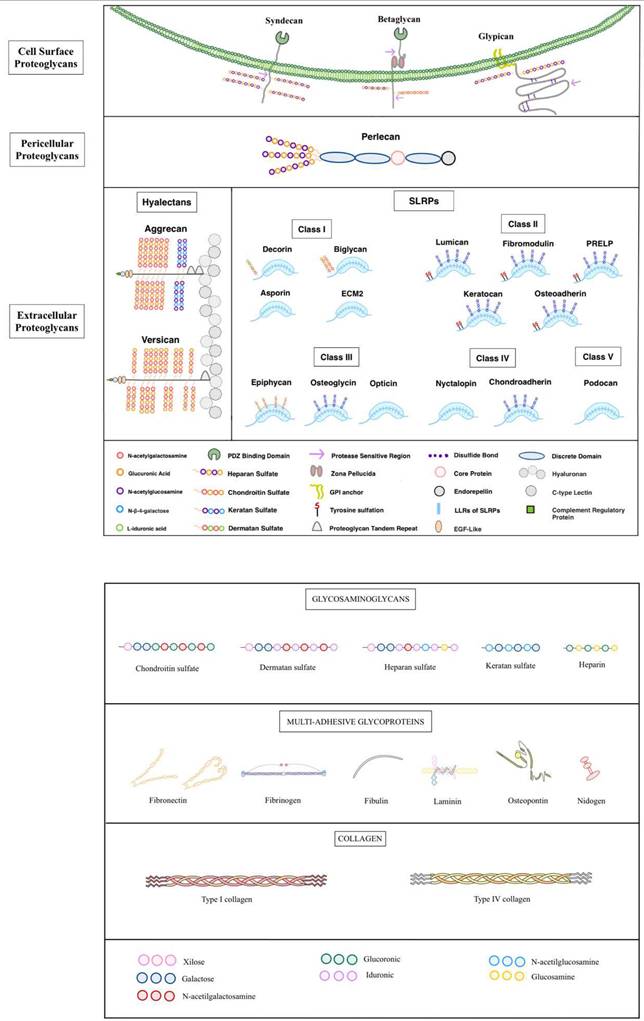
Biological functions of the extracellular matrix
Far from being a passive scaffold, the extracellular matrix (ECM) constitutes a dynamic and interactive interface between cells and their microenvironment. It continuously integrates and responds to mechanical forces and biochemical signals, thus regulating essential cellular processes, maintaining tissue organization, and influencing pathological phenomena such as fibrosis, chronic inflammation, and tumor progression (Table 5). Thus, the ECM is essential not only for structural integrity but also for signal transduction, homeostasis, and disease modulation.
Overview of the biological functions of ECM
| Function | Key ECM components | Mechanism | Biological impact |
|---|---|---|---|
| Structural support | Collagens I, III, elastin | Tensile strength, elasticity | Tissue architecture |
| Cell anchorage | Fibronectin, laminins | Integrin-mediated adhesion | Cell polarity, survival |
| Barrier function | Basement membrane, proteoglycans | Restriction of molecular diffusion | Compartmentalization |
| Reservoir of growth factors | Perlecan, decorin | Binding and controlled release | Morphogenesis, repair |
| Signal transduction | All major ECM ligands | Integrin and receptor activation | Proliferation, survival |
| Migration scaffold | Fibronectin, collagen I | Topography-guided movement | Development, immunity |
| Pathological modulation | Stiff collagen matrix, matrikines | Remodeling via MMPs, ADAMs | Fibrosis, cancer |
Structural support and mechanical integrity
The ECM provides essential mechanical support to tissues by conferring tensile strength, elasticity, and structural resilience. This is especially important in load-bearing tissues such as skin, tendons, cartilage, and blood vessels, where fibrillar collagens (especially types I and III) and elastin are primarily responsible for biomechanical properties [465,466].
Beyond mechanical reinforcement, the ECM functions as a physical scaffold that supports a wide range of cell types, including epithelial, muscle, and bone cells, anchoring them within their native tissue compartments [467]. This anchoring occurs primarily through cell surface integrins, which bind to ECM ligands such as fibronectin, laminins, and collagens [468]. Once bound, integrins form focal adhesions, multiprotein complexes that link the ECM to the actin cytoskeleton and serve as a hub for mechanotransduction and signal transduction pathways [469,470].
In addition, the ECM plays a critical role in maintaining epithelial polarity, orienting tissue patterning, and preserving the structural integrity of specialized compartments such as basement membranes [471]. By defining the spatial organization of cells within tissues, the ECM contributes to maintaining organ function and integrity.
Biochemical barrier and compartmentalization
The ECM also functions as a selective biochemical barrier, regulating the diffusion and local concentration of signaling molecules, nutrients and metabolites [472]. This control is essential for the establishment and maintenance of tissue compartments, as well as for the orchestration of biochemical gradients that guide morphogenesis, immune responses and tissue repair.
ECM components such as proteoglycans, with their high-affinity binding sites for soluble factors, sequester growth factors and cytokines-including TGF-β, FGF, VEGF, and EGF-and release them in a controlled, spatiotemporal manner following matrix remodeling or mechanical stress [473]. This regulation adjusts the local availability of bioactive molecules and modulates processes such as cell migration, proliferation, and angiogenesis.
In pathological contexts such as chronic inflammation or cancer, changes in ECM porosity and composition may facilitate or hinder immune cell infiltration and molecular diffusion, thus modulating local immune response and disease progression [474].
Regulation of cell metabolism, differentiation, and proliferation
The ECM exerts a profound influence on cell behavior through biochemical signals and biomechanical properties (Table 6). The coupling of integrins and other ECM receptors activates intracellular signaling cascades - most notably the PI3K/AKT, MAPK and FAK pathways - that govern cell proliferation, metabolism, differentiation, and survival [475,476].
ECM-regulated signaling pathway and cellular outcomes
| ECM interaction | Receptor/Pathway | Downstream effect | Context |
|---|---|---|---|
| Integrin-collagen I | FAK/PI3K/MAPK | Cell proliferation | Tumor growth |
| Lamini-integrin α6β4 | YAP/TAZ | Lineage commitment | Strem cell fate |
| Fibronectin-integrin α5β1 | MAPK, Rac1 | Cell migration | Wound healing |
| Detachment from ECM | Loss of integrin signaling | Anoikis | Epithelial turnover |
| Proteoglycan-TGF-β | SMADs | Fibroblast activation | Fibrosis |
| Heparan sulfate- FGF/VEGF | RTKs | Angiogenesis | Development, cancer |
A critical phenomenon linked to ECM interactions is anoikis, a form of apoptosis triggered by the loss of cell-matrix adhesion [477]. This mechanism serves as a safeguard against inadequate cell survival and anchorage-independent growth, especially relevant in the context of epithelial homeostasis and tumor suppression [478,479].
In addition, ECM stiffness and topography can instruct lineage specification and modulate stem cell fate through mechanotransduction [480]. Cells sense changes in the mechanical properties of the ECM and transform them into nuclear signals through pathways such as YAP/TAZ, which ultimately influence gene expression programs [481].
The ECM also acts as a dynamic reservoir of soluble growth factors and morphogens, including members of the WNT, EGF, and TGF-β families, which are stored in latent forms and activated locally upon proteolytic cleavage or mechanical deformation of the matrix [482,483]. This spatial and temporal control of signal availability is essential during development, wound healing, and tissue regeneration.
During embryogenesis, ECM composition provides positional information and guidance signals that regulate morphogenetic movements, cell fate decisions, and differentiation trajectories [484,485]. These effects are mediated by coordinated interactions between ECM molecules and specific cell surface receptors, reinforcing the role of the ECM as a critical modulator of developmental biology [486].
The barrier function of the skin depends on the structural integrity of its cellular components and their rapid restoration after injury. Skin wound healing in adult mammalian skin is a highly coordinated multiphasic process involving hemostasis, inflammation, re-epithelialization, granulation tissue formation, neovascularization, and tissue remodeling [487,488]. Under optimal conditions, re-epithelialization begins within hours and continues for several days as the basement membrane is restructured, and the epidermal surface is restored [489]. This regenerative process is regulated by a complex network of signaling pathways involving growth factors, cytokines, matrix metalloproteinases, cell receptors, and ECM components [490].
The ECM as a platform for cell migration
Cell migration is highly dependent on the ECM, which serves as both a structural substrate and a source of directional signals. During processes such as embryogenesis, tissue repair, immune cell trafficking, and cancer dissemination, cells traverse ECM networks through coordinated cycles of adhesion, traction, and detachment.
Cell-ECM interactions during migration are mediated by integrins, proteoglycans (e.g., syndecans, perlecan), and glycoproteins (e.g., fibronectin, laminin), which together regulate adhesion dynamics and cytoskeleton organization [491]. Cells respond to ECM properties, such as stiffness, density, and orientation (collectively termed ECM topography), by modulating their migration speed, directionality, and mode of locomotion (e.g., mesenchymal versus amoeboid migration) [492-494].
ECM remodeling is an integral part of cell migration. MMPs, ADAM family proteases, and other matrix-degrading enzymes cleave ECM components to create migration pathways and release bioactive fragments [495-499]. These degradation products, termed matrikines, can further modulate cell behavior by acting as chemotactic or angiogenic signals.
ECM remodeling in fibrosis, chronic inflammation, and tumor progression
ECM remodeling is a tightly regulated process essential for maintaining tissue homeostasis, especially in response to physiological demands such as growth, wound healing, and regeneration. However, when dysregulated, it contributes to the pathogenesis of various chronic diseases (Table 7).
ECM changes in physiological vs. Pathological remodeling
| Process | ECM composition | Enzymatic activity | Functional outcome |
|---|---|---|---|
| Homeostasis | Balanced ECM synthesis/degradation | Controlled MMP | Tissue integrity |
| Wound healing | Transient increase in fibronectin, collagen III | MMPs upregulated | Regeneration |
| Fibrosis | Excess collagen I, reduced elastin | Persistent fibroblast activation | Stiffness, organ failure |
| Chronic inflammation | Fragmented ECM, increased hyaluronan | MMP overexpression | Tissue disorganization |
| Cancer | Dense, aligned ECM, rich in fibronectin | MMPs, ADAMs active | Invasion, immune evasion |
In fibrotic disorders, there is an excessive and often irreversible accumulation of ECM components, particularly fibrillar collagens, resulting in increased matrix stiffness, loss of tissue elasticity, and distortion of architecture [500-502]. These changes impair organ function and are usually driven by activated fibroblasts and myofibroblasts, which may originate from resident mesenchymal cells, epithelial-mesenchymal transition (EMT) cells, or bone marrow-derived precursors [503,504]. These cells produce large amounts of ECM proteins and secrete profibrotic factors, especially TGF-β, establishing a feed-forward loop that perpetuates matrix deposition.
Chronic inflammation further amplifies ECM dysregulation. Persistently recruited immune cells release cytokines and matrix-modifying enzymes, such as MMPs, that degrade ECM components and alter the biochemical and mechanical landscape of tissues [505-508]. The resulting disorganization of the ECM can promote aberrant healing responses, immune evasion, and, over time, increase susceptibility to malignant transformation.
In cancer, the ECM undergoes extensive biochemical and biomechanical remodeling. Tumor-associated ECM is often denser, stiffer, and biochemically altered, favoring integrin clustering and activation of downstream oncogenic pathways such as FAK and YAP/TAZ [509-511]. These pathways enhance cell proliferation, survival, invasion, and resistance to apoptosis. In addition, physical properties of the tumor ECM, such as altered porosity and fiber alignment, may hinder immune cell infiltration and contribute to an immunosuppressive microenvironment.
The spatial heterogeneity and molecular composition of tumor ECM define distinct niches that may modulate therapeutic responses and metastatic potential [512]. Continuous remodeling through synthesis, degradation, chemical modification, and reassembly of ECM components is a hallmark of cancer progression and therapeutic resistance [513-515]. Importantly, ECM protein fragments generated during remodeling (e.g., endostatin, tumstatin) can act as bioactive mediators that influence angiogenesis, inflammation, and tumor dormancy, further highlighting that the ECM is both a target and effector in pathological remodeling [516,517].
Hidden connections: how ecm interacts with aging, calcification, and cancer
While individual roles of aging, calcification, and cancer have been extensively described, their convergence through ECM dysregulation remains less clearly represented. Strengthening these correlations is essential to understanding how common alterations in the matrix can simultaneously drive tissue degeneration, mineral deposition, and tumor progression.
To address this, it is summarized in the following table the principal ECM changes and shared mechanisms across these conditions (Table 8). In parallel, a schematic model is provided to visualize ECM dysregulation as the central hub linking these dysfunctions, offering a clearer perspective on their interconnected nature (Figure 4).
The principal ECM changes and shared mechanisms across aging, calcification, and cancer
| Process | Key ECM alterations | Shared mechanisms | Pathological outcomes |
|---|---|---|---|
| Aging | Collagen fragmentation, oxydation, glycation, AGEs accumulation Increased ECM stiffness via LOX/LOXL crosslinking Decreased elastin and proteoglycans Accumulation of SASP | Chronic inflammation (inflammaging) ECM stiffening: altered mechanotransduction (YAP/TAZ) SASP-driven MMP activation and ECM degradation | Fibrosis Vascular stiffening and vascular calcification Increased susceptibility to cancer initiation |
| Calcification | Deposition pf calcium-phosphate crystals in ECM Altered collagen/elastin scaffolds favoring mineralization ECM remodeling via MMPs, ADMTs, and EVs | Senescence and inflammation as triggers of mineralization SASP/EVs promoting osteogenic transformation (RUNX2, Sox9, Klf10) ECM stiffness reinforcing mineralization | Vascular calcification: CVD Osteoarthritis Calcific aortic valve disease Tumor microcalcifications |
| Cancer | Excessive ECM deposition (collagen I, fibronectin) Crosslinking by LOX/LOXL enzymes: stiffness MMP/ADAMTS-driven ECM degradation releasing bioactive fragments EV accumulation in ECM | Aging-associated SASP fueling tumor-promoting inflammation Calcification (microcalcifications in tumor) as diagnostic/biological marker Shared role of LOX enzymes in fibrosis, calcification, and tumor progression | Tuor growth and metastasis Therapy resistance Chronic inflammation and immune invasion |
Schematic representation of the interconnections between aging, calcification, and cancer through ECM dysregulation. The diagram illustrates how ECM alterations act as a central hub linking these three processes. Although aging, pathological calcification, and cancer present distinct pathological features, they share common mechanisms such as ECM stiffening driven by LOX/LOXL crosslinking, chronic inflammation and SASP activity, as well as EVs and protease-mediated remodelling. By placing ECM dysregulation at the cancer, the figure highlights the complex and often difficult-to-visualize relationships that underlie tissue dysfunction and disease progression.
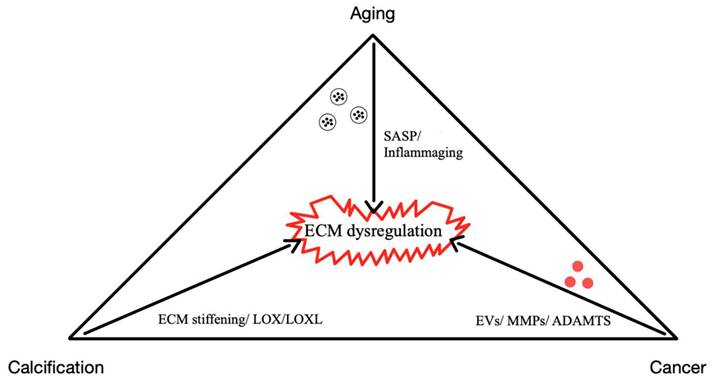
Interaction of ECM and aging
The extracellular matrix (ECM) is a dynamic and versatile network that serves as a key regulator of fundamental cellular functions [518]. It guides cells on how to organize their structure, including adhesion and polarity; when to divide (proliferation) migrate, or undergo programmed cell death (apoptosis); where to release specific molecules (secretion); how to differentiate into specialized cell types; and how to interpret and respond to external signals [519]. Dysfunction of the ECM is linked to various age-related diseases and disorders, making it a valuable target for therapeutic approaches aimed at treating conditions such as fibrosis and cancer [520].
Aging is a widespread process marked by the progressive accumulation of biological changes that gradually impair an organism´s functionality over time [521]. In humans, aging is associated with a steady decline in both cognitive and physical abilities, alongside an increased susceptibility to different diseases, including cancer, diabetes, cardiovascular disorders, musculoskeletal conditions, and neurodegenerative illnesses [522].
These age-related impairments have a bad impact on quality of life, leading to disability, increased morbidity, and a heightened risk of mortality. Aging places a significant burden on individuals, their families, and society [523]. The interaction between the genetic development of an individual, environmental influences, and the stochastic nature of damage accumulation determines susceptibility to age-related diseases [524]. Several biological processes have been identified as hallmarks of aging, including genomic instability, dysregulated nutrient sensing, mitochondrial dysfunction, stem cell depletion, disrupted cellular communication, and excessive cellular senescence [525].
During aging, the structural integrity of the extracellular matrix deteriorates due to the accumulation of fragmented collagens, oxidation, glycation, and aggregated proteins [526]. These changes impair ECM dynamics, leading to tissue fibrosis. The stiffness of the ECM also increases with age because of the gradual formation of enzymatic and nonenzymatic intramolecular and intermolecular covalent bonds between slowly turning over molecules, such as fibrillar collagens and elastin [527]. Interestingly, inhibiting the cross-linking of LOXL2 and LOXL3 has been shown to restore normal collagen fibrillogenesis, reducing tissue stiffness [528]. This suggests that targeting collagen cross-linking can help maintain tissue mechanohomeostasis, limiting the self-perpetuating effects of ECM stiffness on fibrosis and aging.
Also, protein modifications often involve the cleavage of peptide bonds, a process predominantly driven by enzymatic activity [529]. While peptide bond cleavage and protein degradation are components of normal protein homeostasis, this balance can be disrupted in aging tissues, where degradation may outpace synthesis [530]. Furthermore, partially cleaved proteins can persist within the ECM due to physiological or age-related crosslinking, leading to the accumulation of dysfunctional proteins.
Some studies have hypothesized that aging results from the accumulation of cellular and molecular damage caused by the failure of the repair mechanism. This damage is thought to occur randomly, which could explain the variability in aging phenotypes observed, even among genetically identical individuals [531].
Besides these factors that accelerate aging in ECM, the accumulation of senescent fibroblasts with age (a more detailed explanation is provided in the following sections) is ongoing, about whether SASP-driven effects on tumor development are directly attributable to aging [532]. Interestingly, long-lived species such as lobsters and rainbow trout retain telomerase activity, allowing continuous cellular proliferation, which may contribute to slower senescent cell accumulation [533,534]. However, direct evidence supporting this hypothesis remains limited.
Senescence is not a uniform process, and different triggers, including oncogenic activation, replication stress, or environmental factors, can alter the composition of SASP factors [535]. Thus, not all forms of senescence necessarily reflect aging-related decline. In aged tissues, a marked reduction in fibroblast density and proliferative capacity has been observed, yet this loss is highly localized rather than uniformly distributed [536]. Studies in aging mouse skin suggest that fibroblasts do not compensate for this loss by proliferating but instead extend their membrane protrusions to maintain tissue architecture [537].
Beyond numerical decline, aged fibroblast undergoes significant phenotypic change. Recent findings indicate that fibroblasts in aged skin exhibit genomic reprogramming, shifting from ECM-producing cells toward a more adipogenic-like phenotype [538]. Notably, this transformation is metabolically regulated, caloric restriction suppresses this age-related shift, whereas a high-fat diet accelerates it.
Aging is a significant factor that leads to a decline in human health, contributing to the development of various illnesses, including tissue calcification and tumors. This section explores the different pathways through which aging impacts the activity of the ECM. All these concepts are observed in Figure 5.
A. Representation of an extracellular matrix (ECM) before undergoing degradation due to factors associated with aging and senescence. The ECM contains normal levels of collagen and elastin and factors characteristic of the senescence-associated secretory phenotype (SASP). B. Representation of an aged and senescent ECM, showing a reduction and fragmentation of elastin fibers, a lower amount of collagen fibers, the presence of senescence-associated fibroblasts, and the dispersion of molecules synthesized and released into the extracellular matrix by the senescence-associated secretory phenotype (SASP). These molecules promote the senescence of nearby cells near the primary senescent cells.
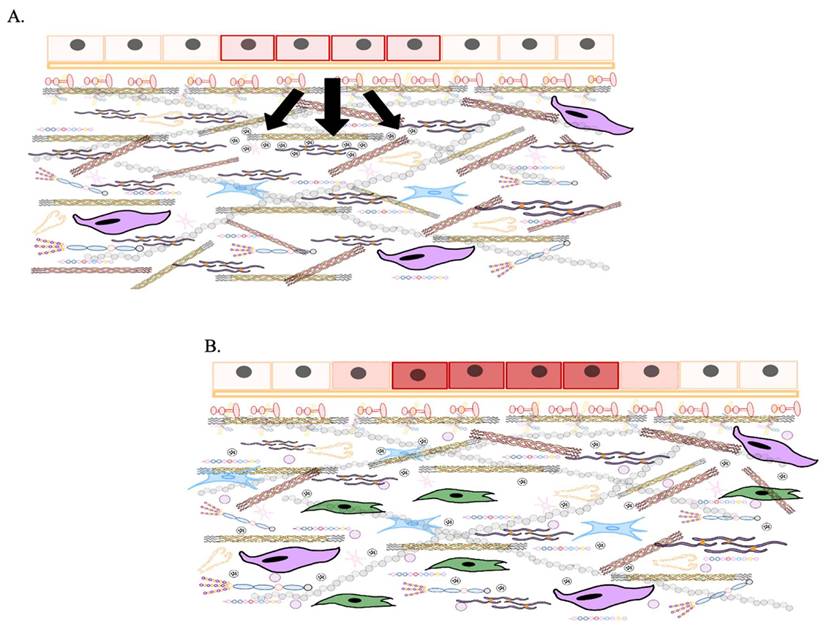
Vascular aging
The fragmentation of elastic fibers is a hallmark of aging in tissues like skin and blood vessels. Vascular aging is characterized by both structural and functional changes in the arteries, often accompanied by increased thickening of the intimal and medial layers of the arterial wall [539]. Alterations in the composition and organization of ECM components primarily drive these changes. Among the various molecular factors that accumulate with age, advanced lipid peroxidation end products (ALEs) are particularly notable [540]. These byproducts are closely linked to oxidative stress, with a role in diseases such as atherosclerosis. Aldehydes, produced through the peroxidation of polyunsaturated fatty acids (PUFAs), including 4-hydroxyphenyl (4-HNE), malondialdehyde (MDA) [541], and acrolein [542], can form adducts with cellular proteins. This leads to progressive protein dysfunction, contributing to the complex pathophysiology of vascular aging [543]. Despite the established role of aldehydes in protein modification, their specific impact on ECM components remains unclear [544]. In one study using immunohistology and confocal immunofluorescence techniques, the research showed that 4-HNE-histidine adducts accumulate in an age-dependent manner across all layers of the human aorta, including the intima, media, and adventitia [545]. These adducts are predominantly localized in vascular smooth muscle cells (VSMC). Interestingly, while elastin fibers in aged vessels exhibit significant structural alterations, the study demonstrated that elastin itself is minimally affected or only weakly modified by 4-HNE. For instance, young human aortas exhibit thick, organized elastic fibers arranged in parallel [545]. In contrast, older aortas show thinner, fragmented, and disorganized fibers interspersed with an increased presence of other ECM components. These findings establish a complex union between lipid peroxidation and ECM components, suggesting that while aldehyde adducts have a role in vascular wall remodeling and the progression of atherosclerosis, their direct impact on elastin integrity appears limited [545].
While Sox9, a transcription factor, is known to influence VSMC differentiation into osteo/chondrogenic lineages, its connection to aging and calcification remains unexplored [546]. Recent research examined human aortic samples and senescent VSCMs to assess Sox9 expression and its impact on ECM properties. Sox9 was not directly linked to vascular calcification but was strongly correlated with cellular senescence markers like p16 [547]. Mechanosensitive responses revealed increased Sox9 expression and nuclear translocation in aged cells and stiff ECM environments. Notably, Sox9 altered ECM stiffness and organization by modulating collagen expression and reducing VSMC contractility, leading to an ECM profile characteristic of senescent cells [547]. An important discovery was the role of procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3 (LH3), as a Sox9 target, mediating ECM stiffness through its secretion in extracellular vesicles. Experimental manipulation demonstrated that Sox9-induced ECM stiffening promotes VSMC senescence, while ECM synthesized from Sox9-depleted cells rescued senescence and restored proliferation [547].
Similarly, in the dermis, long elastic fibers characteristic of younger tissue becomes shorter and more fragmented with age [548]. While mechanical fatigue has been proposed as a direct cause of elastic fiber damage [549], studies on porcine aortic elastic fibers subjected to repetitive cyclic loading in vitro have demonstrated fatigue-related damage [550]. However, mechanical fatigue alone does not account for all observed changes. Notably, increased activity of MMPs with elastase activity in the aortic walls of both rodents and humans suggests a significant role for enzymatic degradation in elastin fragmentation [551].
Alterations of collagen by the effects of aging
In tendons, the ECM provides biochemical signals for tissue growth, repair, and healing, and a physical scaffold to support the biochemical properties of the tendons [552]. However, aging induces significant changes in tendon ECM composition and organization, impacting its function and structure [553]. Collagen accounts for 60-85% of the dry weight in tendons and is central to their tensile strength and viscoelasticity [554]. Collagen type I dominates, with smaller contributions from type III and V, which regulate fibrillogenesis and alignment [555]. With age, collagen biosynthesis declines due to reduced activity of collagen-secreting tenocytes and tendon stem/progenitor cells (TSPCs) [556]. At the same time, remodeling enzymes, oxidative stress, and advanced glycation end products (AGEs) contribute to collagen fragmentation, disorganization, and cross-linking [557]. These processes lead to diminished fibril size, altered fibril alignment, and decreased ECM functionality, ultimately compromising the mechanical performance of the tendons [558]. Enzymes such as MMPs and their inhibitors (TIMPs) have a pivotal role in ECM remodeling. Aging tends to disrupt the balance between MMP and TIMP activity, favoring excessive collagen degradation [559]. For example, increased activity of collagenases (e.g., MMP-1, MMP-13) and gelatinases (MMP-2, MMP-9), combined with reduced TIMP levels, accelerates ECM turnover but also results in disorganized collagen and impaired tissue repair [560,561]. Chronic upregulation of MMP activity has also been linked to tendon degeneration and pathologies [562]. Studies on equine flexor tendons have revealed elevated levels of neo-epitopes (markers produced by the cleavage of collagen´s triple helix by MMP-1 and MMP-13 in older horses) [563]. Also, the expression of MMP-10 at the RNA level and MMP-3 at the protein level increased in aging tendon tissue [564].
AGE formation, which occurs through the non-enzymatic binding of sugars to collagen, further exacerbates the decline in ECM integrity during aging [565]. AGEs increase collagen stiffness, disrupt collagen-proteoglycan interactions, and impair cell adhesion and migration [566]. Hyperglycemia and oxidative stress from senescent cells accelerate AGE accumulation, leading to a more rigid and less functional ECM. Additionally, the long half-life of tendon collagen allows AGE-related cross-linking to accumulate over time, further hindering ECM homeostasis and tendon regeneration [567]. Overall, aging leads to profound alterations in tendon ECM composition and structure, reducing its biochemical properties and repair capacity.
The relationship between aging and tendon biomechanics has been extensively studied in human and animal models. Still, findings are often inconsistent due to differences in study design, subject age, tendon type, and conditions [568].
One of the defining features of tendon aging is the gradual and often irreversible decline in its mechanical properties [569]. This deterioration is largely driven by changes in tendon cellularity, collagen turnover, fibril diameter and alignment, and collagen cross-linking, particularly glycation-induced cross-linking. Key biochemical parameters used to evaluate tendons include tensile strength, modulus, stiffness, and viscoelasticity [570]. These metrics, while interrelated, reflect distinct properties: stiffness measures resistance to deformation under force, while strength quantifies the maximum stress a tendon can endure before permanent damage or failure [571], determining whether a tendon is both string and elastic or strong yet rigid. Proteomic analyses have uncovered age-related neo-peptides derived from collagens and glycoproteins in older equine tendons, absent in younger tissue, suggesting shifts in ECM breakdown patterns with age [572]. A key focus moving forward is identifying and driving the production of these neo-peptides, as they represent a therapeutic target for minimizing ECM fragmentation in aging tissues. Generally, various studies associate aging with unchanged or reduced tensile modulus, stiffness, and strength. For instance, some research has shown minimal differences in maximum strength in aged human tendons or mouse models [573]. In contrast, others report reduced stiffness and modulus in the Achilles tendon of older adults [573]. However, contradictory evidence exists; one study noted stiffer Achilles tendons in elderly participants compared to younger individuals.
Alterations in the production of cartilage caused by aging
The ECM of cartilage has been studied extensively due to its connection to osteoarthritis, a degenerative condition associated with aging [574]. Proteoglycans, such as aggrecan, gradually decline in cartilage and are subject to enzymatic cleavage over time [575]. Aggrecan is broken down between its G1 and G2 globular domains by enzymes like MMP-13 and ADAMTS5, with these fragments accumulating in older tissue [576]. The G3 domain of aggrecan, located at its C-terminal end, also diminishes with age [577]. Similarly, type II collagen, another key structural component of cartilage, undergoes denaturation and fragmentation in aging cartilage, undergoes denaturation and fragmentation in aging cartilage [578].
These observations illustrate the complex processes of ECM degradation across various tissues as they age, emphasizing the importance of further research to uncover the enzymes and pathways responsible. Such insights could enable the development of targeted treatments to maintain ECM integrity and reduce age-related tissue degradation [579].
The role of senescent cells in ECM production
Cellular senescence is a hallmark of aging, characterized by growth arrest and profound changes in gene expression, particularly through the senescence-associated secretory phenotype (SASP) [580]. While it remains difficult to determine whether senescence is driven primarily by aging or external factors, a defining characteristic is the progressive accumulation of senescent cells in aged tissues and stromal microenvironments [581].
It serves as a double-edged sword in physiological processes, providing both protective and detrimental effects depending on its duration and regulation [582]. In its beneficial role, senescence contributes to tissue repair, tumor suppression, and the resolution of fibrosis, ensuring organismal integrity and even playing a role in embryonic development [583]. Under normal conditions, the immune system efficiently clears senescent cells, preventing their excessive accumulation and maintaining tissue homeostasis [584].
However, as aging progresses, the immense system becomes less effective at removing senescent cells, leading to their prolonged persistence and accumulation [585]. This contributes to chronic inflammation, impaired wound healing, and increased susceptibility to tumor progression, turning what was once a protective mechanism into a driver of aging-related diseases [586]. This phenomenon, known as antagonistic pleiotropy, illustrates how biological mechanisms that are beneficial in early life can become detrimental with age [587].
The mechanisms behind this accumulation remain debated. One hypothesis suggests that age-related immune cell decline reduces the clearance of senescent cells, leading to their persistence [588]. However, some studies challenge this notion, arguing that the evidence supporting impaired immune surveillance as the primary cause remains inconclusive [589]. Regardless of the mechanics, senescent cells actively modify their environment by secreting proinflammatory cytokines, chemokines, growth factors, and matrix-degrading enzymes [590]. This secretory phenotype has dual effects: it can signal immune cells to clear damaged cells but also promote chronic inflammation, tumor progression, and ECM remodeling [591,592].
Traditionally, DNA damage has been considered a major trigger for age-related senescence. However, emerging research suggests that epigenetic alterations and chromatin remodeling play equally significant roles [593]. For instance, histone deacetylase (HDAC) modulation in tumor-associated fibroblasts can induce SASP without direct DNA damage [593].
These findings enhance the complex regulatory network governing senescence, emphasizing that genomic instability, epigenetic modifications, and immune dysfunction contribute to senescent cells' persistence [594].
Notably, while increased ECM stiffness may contribute to the development of cellular senescence during aging and chronic fibrotic diseases, ECM derived from young human fibroblasts has been shown to restore a more youthful state in aged, senescent cells [595]. As ECM stiffness increases, the fibrotic process is further driven by excessive secretion of TGFβ, YAP-1, and its paralog WW domain-containing TAZ [596]. YAP and TAZ function as mechanotransducers, activating the expression of pro-fibrotic genes, such as transglutaminase-2 and lysyl oxidases [597].
However, the relationship between aging and YAP/TAZ signaling is complex and tissue-specific [598]. For instance, genetic inactivation of YAP/TAZ in stromal cells accelerates aging, while maintaining YAP activity has been shown to rejuvenate aged cells and prevent age-related changes by mitigating inflammaging [598].
Elastin, an important ECM protein (the most abundant in the lung), has an important role in the formation and structural integrity of alveoli in mice. As a key component of the alveolar architecture, elastin becomes severely dysregulated in chronic obstructive pulmonary disease (COPD), an age-related lung disorder characterized by progressive airflow limitation [599]. This pathology is closely linked to the accumulation of senescent cells and a diminished proliferative capacity of mesenchymal precursor cells in the alveolar parenchyma. Together, these factors significantly impair ECM production, including elastin synthesis [599]. A prominent feature of COPD is emphysema, marked by the abnormal expansion of alveolar spaces and extensive remodeling of lung parenchyma. Evidence suggests that emphysema arises from reduced production and inadequate regulation of ECM components necessary for tissue repair and maintenance [600]. This decline in ECM remodeling disrupts the homeostasis of connective and epithelial tissues, leading to structural insufficiency [601]. Cellular senescence appears to be a central driver of these changes, primarily through its impact on mesenchymal cell function [602]. The resulting decline in ECM protein production, including elastin, exacerbates tissue dysfunction and the role of elastin in preserving normal lung mechanics [601]. These findings emphasize the destructive consequences of senescence-associated elastin dysregulation, providing valuable insights into the progression of COPD and its underlying mechanisms [603].
The accumulation of senescent fibroblasts significantly contributes to skin aging [604]. These fibroblasts lose their cellular identity, exhibit an increased release of the senescence-associated secretory phenotype (SASP) (Figure 3), and disrupt the balance of ECM homeostasis [605,606]. This process triggers a cascade effect as senescence spreads between cells, perpetuating dermal aging [607]. The aged dermis displays prominent clinical characteristics, including decreased thickness, reduced resilience, and diminished mechanical strength, which results in a wrinkled and flabby appearance [608]. These changes are associated with the loss of ECM components during aging, driven by reduced synthesis and increased degradation of ECM in aged fibroblasts. SASP components, such as MMPs, which directly cleave collagen fibrils, significantly contribute to ECM degradation during aging [609]. Research has demonstrated that levels of MMPs, including MMP1, MMP2, and MMP9, are elevated in the aged dermis and fibroblasts derived from older individuals [610]. Overexpression of hMMP1 induces aging phenotypes in ex vivo 3D human skin organ culture and in vivo mouse models. Also, increased MMP levels are accompanied by reduced TIMPs in aged skin, leading to an imbalance in the MMPs/TIMPs ratio and progressive collagen fragmentation [611]. Overexpression of TMP-1 has been shown to protect ECM integrity and maintain elasticity under chronic UVB exposure, whereas neutralizing TIMP-1 produces the opposite effect [612].
Furthermore, fibroblasts in aged skin exhibit reduced production of ECM, particularly the key collagen network components such as collagen types I and III [613]. This decline is attributed to reduced TGF-β signaling. Multiple studies enhance the importance of TGF-β in dermal aging. Physiologically, the aged dermis shows reduced ECM content [614]. TGF-β signaling promotes ECM gene expression while suppressing ECM degradation by downregulating MMPs and upregulating TIMPs [615]. Oxidative stress or UV irradiation can disrupt the TGF-β signaling pathway in fibroblasts, leading to decreased expression of downstream targets such as connective fibers growth factor (CTGF/CCN2) and type I collagen [616,617]. Knockdown of TβRII or Smad3 reduces collagen synthesis, while overexpression of TβRII restores UV-induced collagen loss by activating TGF-β signaling [618]. Disrupted TGF-β signaling also alters CCN1 expression in dermal fibroblast mice, mimicking aged skin characterized by a wrinkled appearance and disrupted collagen network [618]. Reduced fibroblast size (a hallmark of dermal fibroblasts in aged skin) is associated with decreased TβRII expression and diminished ECM production in aged human skin [619]. Thus, a compromised TGF-β signaling pathway is closely linked to dermal aging [620].
Interaction of ECM and calcification
Calcification is the process by which calcium accumulates in tissue, leading to the deposition of calcium salts through the crystallization of phosphate ion (PO43-) and ionized calcium (Ca+2) [621]. While this mechanism is vital for the development and mutation of bones and teeth, pathological calcification can occur in nearly all soft tissues, often associated with aging and diseases [622]. In bone physiology, calcification is fundamental for skeletal growth and mechanical strength. This process involves precisely depositing calcium phosphate crystals within a protein matrix, gradually forming a rigid and durable skeletal framework [623]. The regulation of this process is orchestrated by a complex network of osteoblasts, osteoclasts, stem cells, and signaling molecules, ensuring the production of robust bone tissue. The composite structure of mammalian bones and teeth reflects a sophisticated combination of inorganic and organic components [624]. These tissues have approximately 70% inorganic minerals, 20% proteins, and 10% water by weight [625]. The inorganic crystals confer resistance to compressive forces and enhance tissue toughness by interacting with the protein matrix. Type I collagen, the primary in this matrix, provides tensile strength and is a scaffold for hydroxyapatite deposition, the main inorganic component [625].
Aging is the leading driver of pathological calcification, though this process is also frequently observed in tumors, blood vessels, and joints [626]. Pathological calcification can occur through various pathways, with differing levels of cellular regulation, particularly in non-skeletal tissues such as the vasculature and neoplasms [627]. Calcified deposits, composed of calcium phosphate crystals embedded within the extracellular matrix, can have detrimental effects on tissue function. These deposits contribute to mechanical stress and stiffness, impairing the flexibility and resilience of affected tissues [628]. Additionally, they are associated with cellular damage and inflammation, further exacerbating tissue dysfunction. Despite their adverse effects, calcified deposits are commonly utilized as markers for diagnosing and tracking disease progression [629].
The formation of calcium phosphate crystals can occur through different pathological mechanisms, leading to distinct types of calcifications [630]. Dystrophic dysfunction arises in areas affected by trauma or necrosis despite normal plasma levels of calcium and phosphate [631]. This process can result from various factors, including mechanical injury, inflammation, injections, or parasitic infections, leading to localized calcium accumulation [632]. In contrast, metastatic calcification develops due to systemic disturbances in calcium and phosphate homeostasis [633]. Conditions such as hypercalcemia drive calcium deposition in previously healthy tissues, leading to widespread calcification in multiple organ systems [634].
Also, iatrogenic calcification can occur as an unintended consequence of medical interventions [635]. Surgical procedures, radiation therapy, or the administration of calcium or phosphate-containing agents can trigger pathological calcium deposition [636]. Although abnormal calcification can affect almost any soft tissue in the body, certain areas are particularly susceptible, including blood vessels, heart valves, the brain, breasts, kidneys, gastric mucosa, lungs, and tendons [637-640]. Among these, vascular calcification has been a primary focus of research due to its significant clinical implications.
Vascular calcification refers to the pathological deposition of calcium salts within blood vessels, primarily affecting the intimal and medial layers as well as the aortic valves. Initial calcification is strongly associated with atherosclerosis, where calcium phosphate deposits form within the ECM rather than inside cells [641]. This process is commonly linked to atherosclerosis plaques, leading to vascular stiffening and increased cardiovascular risks. The mineral composition of vascular calcifications varies, with depositions consisting of apatite, whitlockite, or octacalcium phosphate, ranging in size from submicron particles to formations exceeding 0.5 mm [642]. As vascular calcification progresses, it disrupts ECM homeostasis, contributing to arterial stiffening and vessel rupture, both of which significantly impact cardiovascular tissue [643]. Figure 6 shows the main intervention of calcification and its interaction with ECM.
A. Generic image of the muscle cell calcification process. B. Enlarged view of the general image. The left image shows the physiological conditions of the extracellular matrix (ECM) and its interaction with the cell. In contrast, the right image illustrates changes in ECM conditions, where calcification occurs due to ECM alterations. This process begins with the deposition of hydroxyapatite crystals mediated by extracellular vesicles. The accumulation of reactive oxygen species (ROS) promotes this mineralization. These pathological changes are exacerbated by ECM remodeling due to an increase in the presence of matrix metalloproteinases (MMPs), which degrade the ECM, along with other products associated with the senescence-associated secretory phenotype (SASP). C. Calcification process of a muscle cell induced by extracellular matrix (ECM) alterations. The increase in matrix metalloproteinases, the degradation of elastin fibers, and the accumulation of reactive oxygen species create a pro-inflammatory environment that promotes cellular dysfunction. In response, muscle cells release extracellular vesicles rich in calcium and inorganic phosphate, promoting the nucleation of hydroxyapatite crystals through proteins such as osteopontin. Additionally, these vesicles activate macrophages, which amplify inflammation and contribute to the destruction of healthy muscle tissue, further exacerbating the calcification process.
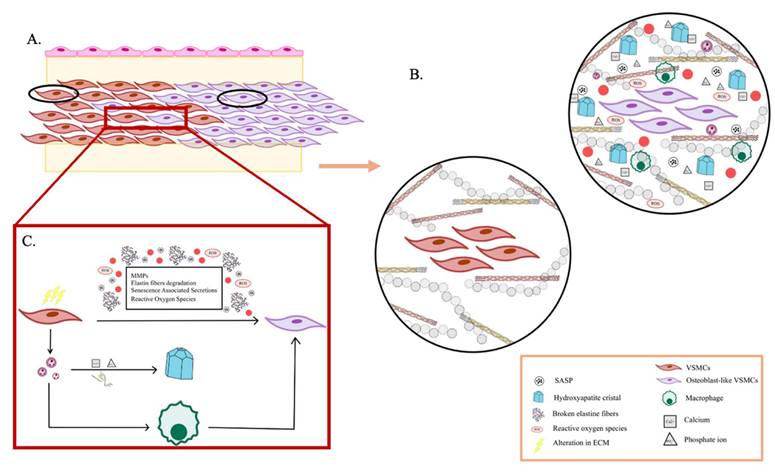
Calcific aortic valve disease
Calcific aortic valve disease (CAVD) is a progressive disorder that ranges from aortic valve (AoV) sclerosis to aortic stenosis (AS), characterized by extensive calcification and impaired leaflet function [644]. It is the most prevalent valvular heart disease in developed countries, particularly affecting aging populations, with a prevalence of up to 13% in individuals over 65 years old [645]. The development of CAVD is influenced by multiple risk factors, including hypertension, smoking, diabetes mellitus, hypercholesterolemia, and male sex. In addition to these well-established contributors, genetic and developmental factors have a significant role [646]. For instance, bicuspid aortic valve (BAV), a common congenital heart defect, has been linked to CAVD through dysregulation of RUNX2 expression, a key regulator of osteogenic differentiation [647].
The progression of the disease follows a continuum, beginning with valve sclerosis, followed by chronic inflammation, and culminating in pathological calcification, which results in aortic stenosis [648]. This pathological process is orchestrated by a dynamic interplay of cellular components, including valve interstitial cells (VICs), vascular endothelial cells (VECs), and inflammatory cells, which collectively mediate ECM remodeling [648]. An abnormal ECM remodeling is a key hallmark of CAVD. In a healthy air valve, ECM integrity is maintained through complex signaling interactions between VICs and VECs, with additional contributions from circulating mesenchymal cells, innate immune cells, and extracellular vesicle release [649]. The ECM composition within the aortic valve varies across its internal layers, each serving distinct mechanical functions. The outflow layer, known as the fibrosa, is rich in type I and III collagen bundles, which provide the tensile strength necessary to withstand mechanical stress [650]. In contrast, the inflow layer to the ventricles contains an advanced concentration of radially oriented elastic fibers, which have an important role in ensuring proper valve closure at the end of the diastolic phase and in absorbing mechanical pressure fluctuations throughout the cardiac cycle [651]. Disruptions in this finely tuned ECM organization contribute to the pathological progression of CAVD, leading to compromised valve function and calcification [652].
This pathological calcification occurs through two primary mechanisms: dystrophic calcification, driven by passive hydroxyapatite deposition, and biomineralization (ossification), an active, cell-mediated process [653]. Microscopic, calcified nodules frequently emerge near sites of endothelial damage, where aggregated cell debris and degraded ECM fibers serve as substrates for calcium and phosphate deposition [654]. Within these nodules, apoptotic cells further contribute to mineral accumulation [655,656]. Myofibroblasts also have a role due to their contractile properties increasing in response to stiffer ECM environments, promoting apoptosis. Meanwhile, oxidative stress has been linked to immune cell apoptosis within the inflamed aortic valve, although the molecular pathways driving ECM mineralization remain poorly understood [657].
Emerging evidence suggests that extracellular vesicles (EVs) are involved in valve mineralization, with two key subtypes identified: mineralized spheroid microparticles and matrix vesicles (MVs) [658]. Mineralized spheroid microparticles (1-3 µm in size), first described in 2013, have been associated with microcalcifications characteristic of dystrophic calcification [659]. VIC cultures undergoing osteogenesis differentiation exhibit these microparticles on their surface, similar to observations in human calcified aortic valves. Although the precise mechanism of their secretion remains unclear, their presence is closely linked to VIC apoptosis and osteogenic transformation [658]. In parallel, MVs, which are enriched in MMPs, likely mediate ECM remodeling and mineralization [660]. Notably, these vesicles overexpress ectonucleotidases such as alkaline phosphatase (ALP), ectonucleotide pyrophosphatase/phosphodiesterase 1 (E-NPP1), and 5´-nucleotidase (CD73), all of which form part of calcium-phosphate nucleation. VICs, macrophages, and VSMC have been shown to release MVs, further promoting biomineralization [661-664]. Studies suggest that mineralization begins within MVs, with Ca+2 and PO43- influx triggering hydroxyapatite formation upon their attachment to the ECM. However, alternative mechanisms propose that mineralization originates intracellularly before propagating to MVs and ECM fibers [665].
As the disease progresses, valve thickening and fibrosis result from excessive ECM deposition, particularly collagen accumulation, leading to tissue hardening and scarring [666]. TGF-β has been identified as a major regulator of this fibrotic phase, promoting ECM synthesis and deposition through canonical Smad signaling. Elevated TGF-β levels have been detected in fibrotic organs, including stenotic aortic valves, where it drives collagen overproduction [667]. However, this fibrotic phase is followed by increased ECM turnover, leading to a decline in collagen I and III content and further ECM remodeling. These events are tightly linked to VIC osteogenic differentiation, reinforcing the pathological transformation of the valve [668]. Nevertheless, hypoxia is involved in ECM remodeling and calcification. Neovascularization in aortic valves restores oxygen supply but also enhances VIC activation and ECM remodeling, contributing to the formation of calcific nodules [669]. A high expression of hypoxia-inducible factor-1 alpha (HIF-1α) has been detected in calcified nodules, driving ECM degradation through pathways involving NF-κB, MMP-2, MMP-9, and neutrophil gelatinase-associated lipocalin (NGAL) [670]. NGAL is known for its ability to enhance MMP-9 activity, accelerate ECM breakdown, and further destabilize the valve microenvironment [671]. In hypoxic aortic valves, the MMP-9-NGAL complex is upregulated, facilitating progressive tissue degradation. Moreover, the presence of ectopic elastic fibers in the fibrosa suggests disruptions in elastin homeostasis, indicating that hypoxia-driven ECM remodeling affects both collagen and elastin metabolism, further contributing to CAVD pathogenesis [672].
Osteoarthritis
Osteoarthritis (OA) is a chronic, degenerative joint disease and a leading cause of disability worldwide [673]. It is characterized by progressive articular cartilage degeneration, subchondral bone remodeling, osteophyte formation, synovial inflammation, and pathological calcification of joint structures, all of which contribute to joint dysfunction. Despite its significant impact, there are currently no effective treatments to halt or reverse OA progression due to its complex biology [674]. While calcification is a normal physiological process in bone formation, its abnormal deposition in cartilage and soft tissues is a key pathological feature of OA [675]. The accumulation of calcium in the ECM exacerbates joint degradation, with calcium deposition in the cartilage matrix being closely linked to disease severity. Inhibiting ECM calcification has been shown to slow OA progression, enhancing the importance of understanding the molecular mechanisms behind cartilage calcification as a potential therapeutic target [676].
Krüppel-like factor 10 (Klf10) is a transcription factor regulated by the TGF-β/Smad signaling pathway and has an important role in bone biology and cartilage homeostasis [677]. It contains a C2H2 zinc finger DNA-binding domain, enabling it to interact with promoter regions such as the CACCC element and GC box [678]. Overexpression of Klf10 in chondrocytes inhibits cell proliferation and migration, while its deletion has been shown to prevent chondrocyte hypertrophy, a key process in cartilage calcification and longitudinal bone growth. However, the role of Klf10 in pathological cartilage calcification remains poorly understood [679].
Recent findings suggest that Frizzled9 (Fzd9), a G-protein-coupled receptor and a member of the Wnt signaling pathway, may be a downstream target of Klf10 [680]. Fzd9 is expressed in various tissues, including the brain, testes, skeletal muscle, and kidneys, and is known to regulate bone formation during fracture healing [681]. Unlike canonical Wnt signaling, Fzd9 functions through a β-catenin-independent mechanism to positively regulate bone remodeling [682]. Studies have shown that Klf10 binds to the Fzd9 promoter, modulating its expression and influencing calcium ion entry into chondrocytes, which is critical for ECM mineralization [683]. In OA, calcium phosphate (BCP) and calcium pyrophosphate dihydrate (CPPD) crystals are the primary components of pathological calcification. These crystals are not only involved in ECM mineralization but also strongly associated with chondrocyte senescence, with CPPD having a more pronounced effect [684]. Experimental data indicate that Klf10-induced chondrocyte senescence is mediated through abnormal calcium deposition and that knockdown of Klf10 reduces ECM calcification in mouse primary chondrocytes [685]. Furthermore, restoring ECM calcification using BCP or CCPD crystals accelerates chondrocyte aging, reinforcing the link between calcification and cellular senescence [686]. In vivo, studies using a destabilization of the medial meniscus (DMM) model of OA In mice demonstrated that Klf10 knockdown mitigated cartilage ECM calcification and slowed cartilage degeneration. These findings suggest that targeting Klf10 and its downstream pathways may provide a novel therapeutic approach for preventing cartilage mineralization and chondrocyte species, thereby attenuating OA progression [686].
Calcific aortic valve
Calcific aortic valve (AoV) disease is a major clinical challenge with poorly understood regulatory mechanisms [687]. Although enhanced cell-cell adhesion is known to contribute to cellular aggregation, its role in calcific lesion formation remains unclear [688]. Cadherin-11 (Cad-11) has been implicated in lesion formation in vitro, but its function in adult valve homeostasis and disease progression has not been established [689]. Using a novel double-transgenic Nfatc1Cre; R26-Cad11Tg/Tg mouse model, researchers induced conditional overexpression of Cad-11 in heart valves [690]. This led to hemodynamically significant aortic stenosis and the development of calcific lesions within the AoV leaflets. Cad-11 upregulation activated RhoA and Sox9 in VICs, promoting pathogenic ECM remodeling and calcification [691]. In vitro, mimicking in vivo lesions, molecular analyses confirmed the upregulation of osteoblastic and myofibroblastic markers, reinforcing the role of Cad-11 in osteogenic differentiation [692]. Further experiments demonstrated that inhibition of Rho-associated protein kinase (ROCK) significantly reduced calcific nodule formation, confirming that Cad-11-driven calcification is mediated through the RhoA/ROCK signaling pathway [693]. These findings identify Cad-11 as a role regulator of AoV calcification and suggest that targeting the Cad-11/RhoA/ROCK axis could serve as a therapeutic strategy to prevent or mitigate aortic valve calcification [692].
Inflammation in the cardiovascular system induces significant alterations in the ECM, similar to its effects in other organ systems [694]. During this process, certain matrix proteins experience increased expression and secretion, while the pre-existing ECM undergoes degradation and remodeling through the action of MMPs [695]. In vascular tissues, inflammation is initiated when lipoprotein particles accumulate within the subendothelial matrix, where they undergo oxidative modifications, triggering an immune response and fueling a chronic inflammatory state [696]. Pro-inflammatory stimuli, including cytokines and lipid oxidation products, have been shown to promote the osteoblastic differentiation of vascular cells, contributing to atherosclerosis and valvular cell calcification in CAVD [697]. Molecular imaging studies further support this link, revealing that inflammation precedes matrix calcification in the vasculature and that inflammatory mediators are closely associated with intimal arterial calcification [698]. Recent research has enhanced the role of smooth muscle cell (SMC)-derived exosomes in vascular calcification [699]. Some studies demonstrated that exposure to TNF-α, a key inflammatory cytokine, enhances the production of exosomes by SMCs, which may contribute to ECM mineralization [700]. Additionally, cell apoptosis often accompanies an inflammatory response, and accumulating evidence suggests that apoptotic cell death further accelerates vascular calcification, reinforcing the connection between chronic inflammation and pathological ECM remodeling [701].
Placenta calcification
Also, in the placenta, the ECM has a pivotal role in its development and function, regulating processes such as angiogenesis, trophoblast invasion, and tissue remodeling [702]. In pathological pregnancies, such as those complicated by chronic venous disease (CVD) or preeclampsia (PE), ECM homeostasis is disrupted, leading to structural and functional alterations in placental tissue [703]. One of the most notable changes is placental villous calcification, which has been associated with abnormal gene expression and signaling pathways involved in osteogenesis, inflammation, and vascular remodeling [704].
Among the key transcription factors involved in calcification, Runt-related transcription factor 2 (RUNX2) has a crucial role in bone and vascular calcification by regulating the expression of osteogenic genes, including type I collagen, osteopontin (OSP), osteocalcin (OSC), and bone sialoprotein [704]. Interestingly, RUNX2, OSP, and OSC have been detected in vascular and placental calcifications, suggesting that pathological mineralization processes in the placenta may share molecular mechanisms with bone tissue [704]. Also, the Wnt/β-catenin signaling pathway, which is essential for embryonic development and tissue homeostasis, is activated in placental calcification and may contribute to dysregulated mineral deposition in CVD and PE [705].
Another key regulator of placental pathology is pigment epithelium-derived factor (PDEF), which has been found at elevated levels in the placenta with venous insufficiency and is implicated in vascular dysfunction and calcification [706]. Transcription factors such as MSX2/HOX8 and SOX9, which are involved in osteogenesis and chondrogenesis, have also been identified as potential contributors to placental ECM remodeling [707]. These molecular pathways, however, remain poorly understood in the context of placental vascular disease, necessitating further investigation into their role in placental function and development.
Beyond calcification, CVD during pregnancy has been linked to increased placental apoptosis, extracellular matrix remodeling, and cellular hypoxia, similar to the mechanism observed in PE and intrauterine growth restriction (IUGR) [708]. Placental angiogenesis is particularly affected, with alterations in VEGF signaling having a pivotal role. Increased expression of VEGF and its receptor (VEGFR-1/Flt-1 and VEGFR-2/KDR) has been reported in CVD placentas, enhancing their involvement in disease progression [709]. Notably, VEGF regulates the expression of soluble Flt-1 (sFlt-1), a decoy receptor for VEGF and placental growth factor (PIGF), which is highly upregulated in PE and other vascular pregnancy complications [710]. Dysregulation of the sFlt-1/PIGF ratio has been proposed as a biomarker for PE and IUGR, as lower PIGF levels correlate with adverse pregnancy outcomes and increased risk of urgent delivery [711].
Another important factor in ECM remodeling and placental pathology is the insulin-like growth factor (IGF) system [712]. IGF-1 and its binding proteins (IGFBPs) are involved in fetal growth and nutrient transport across the placenta, and their dysregulation has been linked to IUGR [713]. In animal models, IGF-1 supplementation has been proposed as a potential therapy to restore placental function in growth-restricted pregnancies [714]. Furthermore, PAPP-A (pregnancy-associated plasma protein-A), a regulator of IGF bioavailability, is overexpressed in the placenta affected by CVD, with ECM alterations contributing to its dysregulated expression. Given that PAPP-A interacts with calcium ions and is involved in proteolysis, its upregulation may further exacerbate placental calcification, reinforcing the pathogenic role of ECM remodeling in vascular pregnancy disorders [715].
The importance of ECM integrity in placental function extends beyond vascular regulation, as it also has a role in perinatal neurodevelopment [716]. Elevated levels of reelin and ECM glycoprotein have been associated with cerebral blood redistribution, suggesting a link between placental ECM changes and fetal neurological outcomes [717]. Additionally, dysregulated ECM composition, including collagen fiber alterations and increased villous calcification, has been consistently observed in the CVD-affected placenta [718]. These findings underscore the systemic impact of vascular pregnancy diseases, not only on the mother but also on fetal development.
Impact of MMPs in calcification
The ECM forms a loose, hydrated network rich in glycoproteins in healthy tissue, maintaining flexibility and structural integrity. However, as the valvular disease progresses, the ECM becomes increasingly dense and fibrotic due to excessive collagen deposition and remodeling [719]. In vitro studies have shown that calcifying vascular cells (CVCs) produce an ECM with elevated levels of collagen I and fibronectin but reduced collagen IV, compared to normal VSMC [720]. This shift in matrix composition creates a stiffer, more adhesive microenvironment, which may further promote mineralization and calcification. In advanced atherosclerosis, the intimal layer thickens and exhibits increased expression of matrix proteins, including thrombospondin, tenascin, osteopontin, osteocalcin, and dentin matrix acidic phosphoprotein 1 (DMP-1) [721]. In the aortic valve, VICs respond to injury by upregulating fibronectin production, further contributing to pathological ECM remodeling. As valvular disease advances, expression of key osteogenic markers, such as osteopontin and osteocalcin, continues to rise, alongside an increase in MMPs (MMP-1, MMP-3, and MMP-9) and their inhibitors, which regulate ECM turnover and degradation. In addition to structural alterations, inflammation-driven matrix changes impact the vascular lipid microenvironment [722]. The elevated fibronectin content in diseased tissue enhances lipoprotein retention, likely through its heparin-binding domain, while proteoglycans also facilitate lipoprotein binding. These interactions contribute to lipid accumulation, further exacerbating vascular dysfunction and disease progression [723].
On the other hand, the MMP family is involved in early bone and dentin formation to fracture healing, and pathological calcification [724]. In osteoblasts and osteocytes, MMP-13 expression has been associated with osteogenic differentiation, and studies in MMP-2-deficient mice have demonstrated progressive loss of bone mineral density and impaired calcification, reinforcing the importance of ECM degradation in matrix mineralization [725]. Similarly, matrix vesicles, which initiate calcification, contain MMP-2 and MMP-9, further improving the contribution of proteolytic enzymes in remodeling the ECM and promoting calcium phosphate deposition. Beyond MMPs, another family of extracellular proteases, the ADAMTS family, has been implicated in ECM turnover during bone development. Enzymes such as ADAMTS-1, ADAMTS-4, and ADAMTS-5 degrade proteoglycans like versican, which accumulate in the ECM and may regulate mineralization [726]. Evidence suggests that ADAMTSs and MMPs function cooperatively, accelerating ECM breakdown and facilitating matrix calcification during bone formation [727]. These findings underscore the complex interplay between ECM degradation, mineralization, and proteolytic enzyme activity [728]. While ECM proteins serve as essential regulators of bone and dentin calcification, further studies are needed to fully elucidate the balance between mineralization inhibitors and the enzymes that degrade them [729]. Understanding these regulatory mechanisms could provide novel insights into bone regeneration, dentin repair, and pathological calcification processes [730].
Interaction of ECM and cancer
Traditional perspectives on cancer have evolved to recognize the ECM as a fundamental regulator of cell proliferation, migration, and apoptosis [731]. The organization and composition of ECM components form a tissue-specific microenvironment that has a primary action in tumor progression [732]. It is established that ECM is not merely a passive scaffold but undergoes continuous remodeling, actively influencing cell adhesion, migration, and signaling pathways [733]. Even minor distribution in ECM homeostasis can significantly affect cancer cell behavior, making ECM remodeling a key determinant in tumor growth and metastasis [734]. Its remodeling is a dynamic process governed by four primary mechanisms that collectively regulate tissue structure, biochemical signaling, and mechanical properties [735]. These processes include ECM deposition, which alters the abundance and composition of ECM components, thereby influencing both biochemical and mechanical properties [736]; post-translational chemical modifications, which affect the structural characteristics and biochemical functionality of the ECM [737]; proteolytic degradation, which releases bioactive ECM fragments and ECM-bound factors, often facilitating processes such as cell migration by eliminating physical constraints [738]; and force-mediated physical remodeling, which reorganizes ECM fibers, aligning them to create pathways for cellular migration [739]. Tissue homeostasis relies on the precise regulation of ECM deposition, modification, degradation, and organization, as even small disturbances in these processes can disrupt the balance and lead to pathological consequences [740]. ECM components serve as key ligands for various cell surface receptors, including integrins, Syndecans, and receptor tyrosine kinases, integrating ECM remodeling into broader cellular signaling networks [741]. Given this intricate interplay, it is not surprising that cancer cells and tumor-associated stromal cells actively manipulate all four remodeling mechanics [735].
Collagen, the most abundant ECM protein, is central to maintaining tissue integrity and function [742]. Changes in collagen deposition, degradation, and crosslinking can lead to a loss of ECM homeostasis, contributing to tumor progression [743]. Tumor-associated ECM remodeling involves increased secretion of fibronectin and collagens I, III, and IV, enhancing the dynamic interplay between tumor cell and their microenvironment [744]. Excessive ECM protein deposition disrupts cell-cell adhesion and polarity, amplifying growth factor signaling and fomenting tumor progression [745]. However, the precise role of collagen is complex (increased collagen crosslinking promotes integrin signaling and tumor progression) [746]. Yet, the depletion of fibrillar collagens can also drive malignancy by altering biomechanical forces within the tumor niche [746]. Collage also exerts its influence through DDR1 and DDR2, which are implicated in cancer progression. DDR1 is essential for collective cell migration, while DDR2 [747]. has been identified in invasive breast tumors, where it stabilizes SNAIL1, promoting epithelial-mesenchymal transition (EMT) and metastasis [748]. The extracellular collagen network, through DDR signaling, thus modifying cell-intrinsic properties, reinforces tumor invasiveness [748].
The ECM serves as both a barrier and a promoter of tumor progression, depending on its composition and structural modifications [735]. ECM gene signatures have also been used to stratify breast cancer subtypes, with tumors exhibiting high protease inhibitor expression correlating with better prognosis [749]. In contrast, those with high MMP expression are linked to poor survival and increased recurrence risk. MMPs have been linked to cancer progression, with initial research suggesting that MMP-mediated ECM degradation facilitates tumor invasion and metastasis [750]. Early studies demonstrated that MMP inhibition reduced tumor invasiveness in animal models, leading to clinical trials to block MMP activity [751]. Beyond degrading physical barriers, MMPs influence multiple signaling pathways, modulating both normal physiological processes and disease progression [752]. Their role extends beyond ECM remodeling to impact cellular communication, immune response, and tumor microenvironment regulation [753]. Also, ADAM and ADAMTS protease families, which share structural similarities with MMPs, have been implicated in tumor progression and are targeted by broad-spectrum inhibitors designed to suppress metzincin protease activity [754]. While MMPs are predominantly associated with cancer promotion, emerging evidence suggests that some MMPs and other extracellular proteases may exhibit tumor-suppressing effects under certain conditions [755]. Their diverse biological functions extend beyond cancer, influencing tissue homeostasis and remodeling in both healthy and diseased states [756]. These findings underscore the dual nature of MMP activity, enhancing the need for a more nuanced therapeutic approach when targeting these enzymes in cancer treatment.
The lethality of most cancers is due to the dissemination of metastatic tumor cells and the growth of secondary tumors in distant organs [757]. Metastatic initiation requires tumor cells to invade peripheral tissues, enter the blood or lymphatic system (intravasation), and establish secondary tumors in receptive environments known as premetastatic niches [758]. MMPs facilitate this process by degrading ECM barriers, such as the endothelial basement membrane, creating pathological metastasis-prone vasculature [759]. MMP-1 expression has been particularly implicated in classifying atypical ductal hyperplasia into being versus pre-malignant lesions, reinforcing the ECM role in cancer progression and risk assessment [760]. Also, MMP-1 activates proteinase-activated receptor-1 (PAR-1), a G-protein-coupled receptor involved in thrombosis and inflammation [761]. This activation promotes cancer cell migration and invasion, particularly in breast, colon, and lung cancers, underscoring the significance of stroma-derived proteinases in tumor progression [762]. Bone metastases, a frequent complication in cancers such as breast and prostate cancer, are also mediated by MMPs [763]. MMP-7, expressed at the tumor-bone interface, facilitates osteolysis by cleaving receptor activator of nuclear factor κB ligand (RANKL), leading to osteoclast activation and enhanced bone degradation [764,765]. Similarly, MMP-1 and ADAMTS-1 activate EGF-like ligands, further stimulating the RANKL pathway and promoting bone metastasis. Beyond ECM degradation, MMPs contribute to tumor progression by reshaping the tumor microenvironment and influencing immune cell interactions [766]. Some studies using high-resolution multimodal microscopy confirm that MMP-14-driven pericellular proteolysis enables single-cell and collective-cell migration, reinforcing the importance of ECM remodeling in tumor invasion [767]. Interestingly, metastatic cells can switch from protease-dependent to protease-independent amoeboid migration, allowing them to navigate the ECM without proteolytic degradation [768]. Additionally, tumor-associated macrophages (TAMs) are key in tumor motility and intravasation [769]. Macrophage-derived MMP-2 and MMP-9 facilitate immune cell migration and may contribute to tumor cells in circulation by degrading the endothelial basement membrane [769].
ECM remodeling extends beyond primary tumor sites, influencing metastatic colonization (Figure 7). Changes in ECM stiffness and elasticity, governed by collagen organization, crosslinking, and chemical modifications, allow tumor cells to interact with and adapt to their environment [770]. Increased ECM stiffness, driven by excessive collagen deposition and crosslinking, disrupts normal tissue architecture and promotes tumor invasion [771]. This process is largely mediated by LOX and LOX-like (LOXL) enzymes, which are frequently overexpressed in primary and metastatic tumors [771]. High LOX expression correlates with poor survival outcomes, particularly in breast cancer, where LOX-induced collagen crosslinking activates β1 integrin clustering, PI3K signaling, and focal adhesion formation, driving tumor progression [772]. Inhibiting LOX has been shown to reduce fibrosis and delay tumor development, demonstrating its potential as a therapeutic target [773]. MicroRNAs (miRNAs) are emerging as potent regulators of ECM remodeling. The miR-29 family, for example, modulates genes involved in ECM dynamics, including collagen chains, LOX, and MMPS, such as MMP2 and MMP9 [774]. In breast cancer, miR-29b overexpression alters the tumor microenvironment and suppresses metastasis [775]. These findings suggest that non-coding RNAs, including long non-coding RNAs, may regulate the chromatin state and transcription of ECM genes, presenting new avenues for research into tumor-associated ECM regulation [776].
The extracellular matrix (ECM) undergoes a series of processes that favor the creation of a tissue-specific microenvironment that promotes tumor progression. These modifications include changes in post-translational chemical modifications (panel a), which lead to changes in biochemical and mechanical properties, promoting epithelial-mesenchymal transition (EMT) (panel b). Excessive ECM protein deposition also contributes to these changes. Furthermore, it leads to alterations in cell adhesion and amplification of growth factor signaling (panel c). The ECM is also subject to proteolytic degradation (panel d). Together with force-mediated physical remodeling (panel e), both lead to ECM fiber remodeling, which, along with collagen deposition and crosslinking, contributes to changes in ECM stiffness and elasticity (panel f). The active modification of the extracellular matrix by stromal and tumor cells generates an inflammatory microenvironment that facilitates tumor invasion. Matrix proteases release bioactive fragments, such as the proteoglycan biglycan, which stimulate Toll-like receptors in macrophages by promoting the release of TNF-α and MIP-2/CXCL2. In addition, antigen-presenting cells (APCs) stimulate the activation of T lymphocytes by presenting tumor antigens (panel g). This sustained inflammation creates a favorable environment for metastasis. All these processes together promote cancer progression and increase its invasiveness, contributing to the transformation of the primary tumor into a distal secondary tumor.
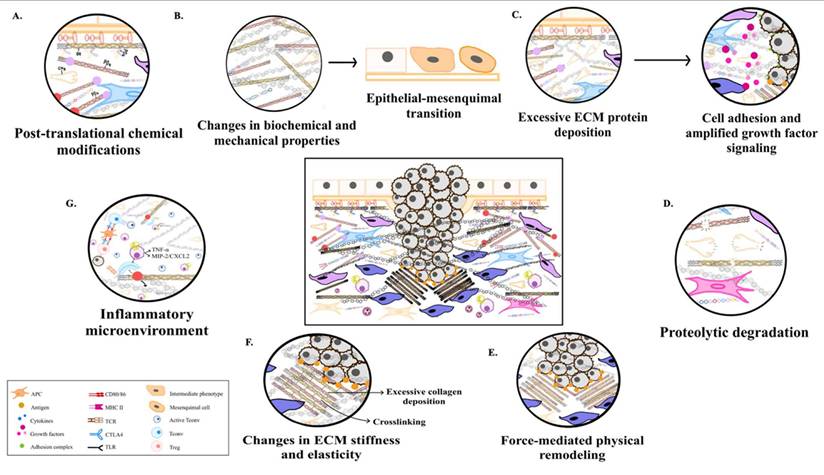
On the other hand, the tumor microenvironment (TME) (Figure 8) has a dynamic action, undergoing continuous ECM remodeling that influences immune evasion, tumor progression, and therapy resistance [777]. Tumor and stromal cells actively modify the ECM, generating an inflammatory microenvironment that facilitates cancer invasion and metastasis [778].
A. Coordination of Tissue Health and Stability: Initial tumor lesion showing the basement membrane, composed of laminin, collagen IV, and nidogen, which separates epithelial cells from the extracellular matrix. The extracellular matrix contains fibroblasts, collagen I/III, fibronectin, hyaluronan, and other proteoglycans. B. Extracellular Matrix Modification in Tumor Progression: Neoplastic cells proliferate rapidly, releasing extracellular vesicles into the extracellular matrix and inducing mechanical strain on the basement membrane, causing it to bulge. Matrix metalloproteinases (MMPs) contribute to tumor progression by reshaping the tumor microenvironment, degrading ECM components locally, and influencing immune cell interactions. Immune cells like tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs) also contribute to ECM remodeling by producing cytokines and growth factors that activate cancer-associated fibroblasts (CAFs). CAFs deposit collagen I, which is aligned by stromal-derived lysyl oxidase (LOX), while hyaluronan, a glycosaminoglycan involved in tissue hydration, absorbs water, increasing interstitial fluid pressure and promoting ECM swelling. C. Cell migration is guided by Collagen Arrangement: Neoplastic cells breach the basement membrane and migrate along aligned collagen fibers. Tumor-derived LOX further stiffens the ECM through collagen crosslinking. The production of cytokines and growth factors by immune cells is increased, and, therefore, the activation of CAFs is also enhanced, intensifying ECM remodeling and facilitating tumor invasion into surrounding tissues.
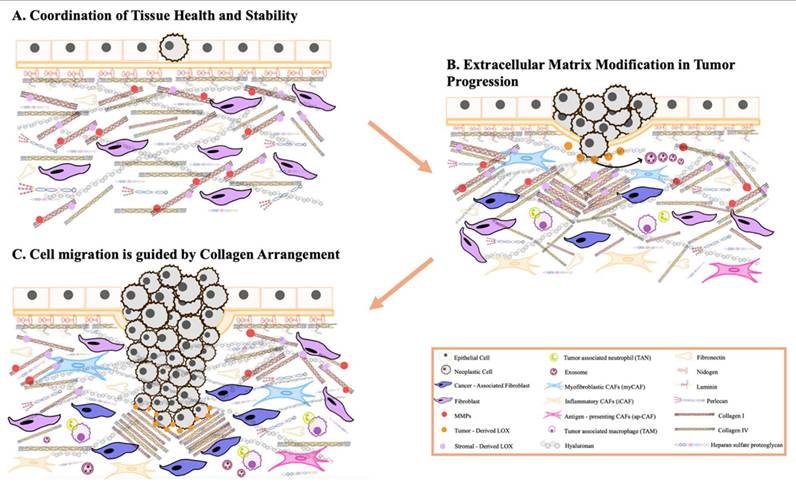
ECM components can act as danger-associated molecular patterns (DAMPs), activating pattern recognition receptors (PPRs) on immune cells and triggering pro-inflammatory responses [779]. ECM-degrading proteases liberate bioactive ECM fragments, including low-molecular-mass hyaluronan (LMM-HA) and biglycan, alongside matrix-bound cytokines and growth factors, fueling chronic inflammation [780]. For instance, biglycan activates Toll-like receptors (TLR4 and TLR2) on macrophages, promoting TNF-α and MIP-2/CXCL2 expression, which sustains tumor-associated inflammation [781].
Beyond their role in modulating immune responses, ECM components also regulate immune cell infiltration and tumor immunity [782]. The ECM proteoglycan versican, when cleaved by ADAMTS proteases, generates versikine, a bioactive fragment that enhances CD8+ T-cell infiltration in colorectal cancer and multiple myeloma [783]. Versikine increases Interferon Regulatory Factor 8 (IRF8) expression in macrophages, promoting the generation of CD103+ CD11c+ MHCIIhi conventional dendritic cells (cDCs), which play a key role in anti-tumor immunity [784]. However, the TAMs that infiltrate tumors contribute to ECM remodeling in ways that both support and suppress tumor progression [785]. M2-like TAMs facilitate proteolytic clearance of interstitial collagen through upregulated MMP expression, including pro-angiogenic TIMP-1-free MMP-9, which, along with neutrophils, increases ECM degradation and tumor invasion [786].
Additionally, TAMs contribute to ECM deposition, upregulating the synthesis and assembly of collagens I, VI, and XIV, leading to enhanced matrix cross-linking and stiffening, which favors tumor cell invasion [787]. Mechanical stress is another factor influencing immune evasion and therapy resistance in the TME, which arises from solid stress, fluid shear stress, and increased intratumoral pressure [788]. These physical forces drive cancer mechanobiology, altering tumor architecture and triggering immune defense mechanisms, including EMT and autophagy, suggesting that targeting these mechanical forces could enhance immunotherapy efficacy.
The adaptive immune system, particularly cytotoxic T cells, is important in immune surveillance, identifying and eliminating tumor cells by recognizing mutated or foreign antigens [789]. However, the ECM exerts dual effects on the tumor immune response, acting as both a facilitator and a barrier to T-cell infiltration and activation [790].
On the one hand, the ECM supports T-cell migration by providing structural pathways (migratory highways) that guide immune cells toward tumors. Also, ECM degradation products, generated through MMP-mediated cleavage, can act as chemoattractants, enhancing immune cell infiltration [791]. This has been demonstrated in inflamed lung tissue, where MMP12 and elastase-driven elastin digestion facilitates monocyte migration [792].
Conversely, ECM stiffening can suppress T-cell function, impairing antigen representation by antigen-present cells (APCs) and disrupting T-cell activation [793]. The ligation of type I collagen to LAIR receptors can directly inhibit T-cell proliferation, while increasing ECM stiffness may interfere with CD3/CD28 signaling, reducing IL-2 production (a cytokine essential for T-cell expression and Th1 differentiation) [794]. As a result, a tumor-associated stiffened ECM may compromise anti-tumor immunity, contributing to immune evasion and cancer progression [795].
Chronic inflammation is a defining feature of tumors and significantly increases the risk of malignant transformation [796]. Notably, persistently inflamed tissue often exhibits fibrotic remodeling, characterized by excessive collagen and fibronectin deposition, which not only alters tissue architecture but also influences immune cell recruitment and activation [797].
The ECM has a pivotal role in regulating immune infiltration. For instance, neutrophil recruitment is severely impaired in the absence of α6β1 integrin, a receptor for laminin [798]. At the same time, macrophage infiltration into atherosclerotic plaques requires DDR1 protein, which inhibits macrophage infiltration, enhancing the regulation of ECM complexity in immune cell migration [799].
Beyond recruitment, ECM composition influences macrophage activation and polarization. A collagen-rich ECM supports macrophage proliferation and promotes M2 polarization, which is associated with pro-tumorigenic activity [800]. In contrast, a fibronectin-rich ECM enhances M1 macrophage polarization, which supports anti-tumor immune responses [801,802]. In tumor microenvironments, ECM stiffening, particularly through type I collagen accumulation, favors an immunosuppressive M2 phenotype, potentially by diminishing TNF-α expression in response to inflammatory stimuli like LPS [803].
For instance, the immune system can prevent tumor formation, yet age-related chronic inflammation (inflammaging) creates an environment that supports tumor initiation and progression [804]. This persistent inflammatory state disrupts acute immune responses, promotes tissue degradation, and is linked to multiple age-related malignancies [805]. Cellular senescence contributes significantly to inflammaging through the SASP, which maintains a low-grade inflammatory response [806]. Other factors, such as gut microbiota alterations, obesity, and ECM remodeling, further drive this process by increasing systemic levels of pro-inflammatory cytokines (IL-1, IL-6, TNF) [807].
A key link between inflammaging and cancer is the recruitment of myeloid-derived suppressor cells (MDSCs), which impair T-cell function through the secretion of ARG1, TGF-β, and ROS [808]. In a melanoma mouse model, upregulated inflaming mediators (IL-1β, GM-CSF, and IFNγ) promoted MDSCS infiltration, accelerating tumor progression and metastasis. Pharmacological inhibition of MDSCS immunosuppressive activity reversed these effects, restoring T-cell function and reducing tumor growth [809].
Similarly, regulatory T cells (Tregs), which suppress effector T-cell responses, are highly enriched in chronic inflammatory environments [810]. In an allergic contact dermatitis (ACD) model, the cytokine IL-33 forms part of the shifting of acute inflammation into a chronic, tumor-promoting state [811]. IL-33-deficient mice were protected against carcinogen-induced skin cancer, suggesting that the IL-33-Treg axis contributes to chronic inflammation-driven tumorigenesis, including colitis-associated colorectal cancer [812].
In breast cancer, IL-6, a major inflammaging cytokine, has been identified as a driver of tumor progression, with high serum levels correlating with poor prognosis [813]. Also, several inflammaging-associated microRNAs (inframammary), such as miR-19, miR-21, miR-126, and miR-146a, have been linked to cancer progression, partially through their regulation of inflammatory cytokines and immune signaling pathways [814-816].
Finally, it is observed that EVs are a crucial component of the SASP, facilitating intercellular communication by transporting proteins, mRNAs, and DNA between cells [817]. Notably, EVs can also accumulate within the ECM, interacting with matrix components and influencing ECM remodeling. The cargo within senescence-associated EVs is influenced by age, sex, and environmental exposures, underscoring their role in age-related diseases [818]. EVs have been implicated in idiopathic pulmonary fibrosis (IPF), a progressive fibrotic lung disease. Elevated exosomal miR-21, a key driver of inflammaging, has been detected in circulating EVs from IPF [819]. Interesting that senescent alveolar epithelial cells (AECs) play a pivotal role in IPF progression, with miR-21 upregulation observed in both fibrotic lung tissues and experimental models Furthermore, EVs from IPF lung fibroblasts contain miR-23 b-3p and miR-494-3p, which inhibit SIRT3, a regulator of mitochondrial homeostasis, and their expression correlated with disease severity [820].
Senescent fibroblasts also increase EV secretion, amplifying paracrine signaling that accelerates mitochondrial damage and epithelial cell senescence, thereby perpetuating lung fibrosis [821]. However, EV effects are highly context-dependent [822]. For instance, EVs derived from inflammatory and myofibroblast cell death suggest a potential antifibrotic role in certain conditions [823].
In essence, senescent cells not only lose their proliferative capacity but actively reshape the ECM in ways that create a fertile ground for cancer development and metastasis, transforming the surrounding tissue into a landscape that favors tumor cells' invasion and survival (Figure 9).
Representation of how a senescent extracellular matrix (ECM) promotes tumor invasion and the metastatic process in cancer. The presence of senescence-associated fibroblasts around tumor cells is observed, which, together with the molecules released by the senescence-associated secretory phenotype (SASP), facilitates the dissemination of these tumor cells.
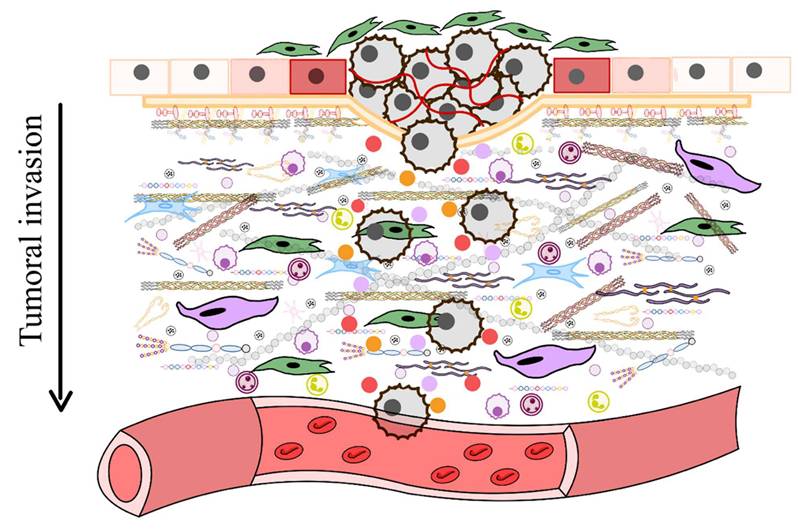
Applications of ECM in different pathologies
The ECM is a highly dynamic, three-dimensional network that plays a crucial role in cell morphology, function, and tissue integrity. Under normal physiological conditions, ECM remodeling is tightly regulated, ensuring proper cellular behavior [824]. However, in pathological states such as cancer, ECM homeostasis is disrupted, leading to uncontrolled remodeling that drives disease initiation and progression [825].
The ECM comprises numerous interacting molecules that communicate with neighboring cells via cell surface receptors, forming complex interaction networks [826]. Understanding these interactions is necessary to identify novel disease biomarkers and develop personalized therapeutic interventions. Recent advancements in big data analytics have facilitated the creation of online databases that enable stochastic evaluations of ECM interactions, helping researchers filter relevant molecular pathways for their specific studies [827].
Some studies have a data-driven approach that addresses existing limitations in ECM research, broadens their understanding of ECM-mediated cellular functions, and has the potential to enhance targeted therapy developments [828].
Ongoing studies continue to highlight the importance of ECM regulation in disease pathology, emphasizing the need for pharmacological strategies to modulate ECM interactions and restore tissue homeostasis.
The extracellular matrix is significantly altered in various diseases, including atherosclerosis, chronic inflammation, and cancer [829]. In recent years, attention has shifted towards targeting the tumor microenvironment, particularly ECM components, as they play a crucial role in tumor progression, drug resistance, and immune evasion [829]. Unlike healthy tissues, cancer-associated ECM undergoes abnormal remodeling, creating physical and biochemical barriers that hinder drug delivery and therapeutic efficacy [830].
Targeting ECM components has emerged as a promising therapeutic strategy. Several clinical trials are investigating approaches such as chimeric antigen receptor (CAR) T-cell therapies against glypican-3 in liver cancer or heparanase inhibitors for multiple myeloma. Also, PGs, MMPs, and CD44/HA signaling pathways have been explored as potential drug targets due to their role in tumor growth and ECM interactions [831]. One major challenge in cancer therapy is inefficient drug transport caused by abnormal vasculature, increased interstitial fluid pressure (IFP), and excessive ECM deposition [832]. Tumor blood vessels are often disorganized and leaky, creating hypoxic conditions that promote tumor survival [833]. Strategies like vascular normalization through VEGF and PDGF inhibitors can improve blood flow and drug distribution [834]. Additionally, collagen-degrading enzymes (MMPs and collagenase) and LOX inhibitors have been shown to reduce ECM stiffness, enhancing drug penetration [835].
Biomaterials are also being investigated as drug carriers to overcome ECM-related resistance. Natural and synthetic matrices, including collagen, gelatin, fibrin, and alginate, have been engineered to improve drug delivery and tissue regeneration [836]. The incorporation of ECM-derived peptides into biomaterials has demonstrated synergistic effects, promoting vascularization, wound healing, and bone regeneration [837]. By interacting ECM-modulating therapies with existing cancer treatments, researchers aim to enhance drug efficacy, reduce metastasis, and improve patient outcomes [838].
Decellularized ECM materials have become versatile in tissue engineering, taking forms such as patches, powders, and injectable hydrogels [839]. With additional processing, these ECM hydrogels offer minimally invasive delivery options for regenerative therapies [840-843]. Derived from various tissue sources, ECM hydrogels have been applied in models of ischemic injury, organ regeneration, and replacement [844]. They promote cellular infiltration, particularly of progenitor cells and macrophages, stimulate neovascularization, and support favorable functional remodeling [845,846].
Although their mechanism of repair remains incompletely understood, ongoing research seeks to optimize the selection, processing, and modification of ECM hydrogels for specific regenerative applications [847]. New technologies like 3D printing and advanced modification protocols are enhancing the complexity of these hydrogels, enabling them to better mimic the target tissue environment [848]. While acellular ECM platforms are closest to clinical application, there is potential for combination therapies incorporating stem cells or growth factors, albeit with increased complexity and cost [849]. Commercial products and standardized manufacturing practices are emerging, setting the stage for broader clinical translation of ECM hydrogel therapies in regenerative medicine [850].
It should be noted, however, that while these emerging interventions are promising, a deeper critique regarding their clinical translatability, limitations, and the available preclinical and clinical evidence would be valuable. More studies are needed to fully understand the ECM functions, remodeling, and impact on human health. Table 9 summarizes all the therapeutic strategies discussed in this section.
Summarized table of the multiple therapist targets against ECM
| Therapeutic strategy | Target/ Mechanism | Application/ Benefit | Status/ Evidence |
|---|---|---|---|
| CAR T-cell therapy | Glypican-3 | Liver cancer targeting tumor ECM | Ongoing clinical trials |
| Heparanase inhibitors | ECM degradation enzyme | Multiple myeloma | Preclinical/ clinical studies |
| PGs, MMPs, CD44/HA signaling modulation | ECM structural proteins and signaling pathways | Tumor growth inhibition, ECM remodeling | Preclinical evidence |
| VEGF/PDGF inhibitors | Vascular normalization | Improve blood flow and drug distribution | Preclinical/ clinical studies |
| Collagen-degrading enzymes (MMPs, collagenases) | ECM stiffness reduction | Enhance drug penetration | Preclinical studies |
| LOX inhibitors | ECM crosslinking and stiffness | Reduce ECM rigidity, facilitate therapy | Preclinical evidence |
| Biomaterials (collagen, gelatin, fibrin, alginate) | Drug carriers, ECM-mimetic scaffolds | Drug delivery tissue regeneration | Preclinical studies |
| ECM-derived peptides in biomaterial | Promote vascularization and tissue regeneration | Synergistic regenerative effects | Preclinical studies |
| Decellularized ECM hydrogels | Structural ECM replacement, progenitor cell support | Tissue regeneration, neovascularization | Preclinical; approaching clinical translation |
| ECM hydrogels + stem cell/ growth factors | Combination therapy | Enhanced regeneration and repair | Preclinical; increased complexity/ cost |
The therapeutic use of ECM in vascularization illness
Cardiac regeneration through stem cell therapy is a promising yet complex approach due to the heart´s limited intrinsic repair mechanism and irreversible remodeling after injury [851]. Despite the early enthusiasm for resident cardiac stem cells (CSCs), their regenerative potential remains insufficient after myocardial infarction [852].
In myocardial infarction, for example, the use of stem cell therapy modifies the stiffness of the infarcted heart tissue by altering ECM composition, making the scar more compliant and cellular [853]. It aims to restore heart function by repopulating cardiomyocytes, with various preclinical and clinical studies underway [854]. Preconditioning strategies, such as heat shock, hypoxia, and chemical treatments, enhance cell survival and engraftment by inducing cytoprotective pathways and promoting angiogenesis. Hypoxia upregulates HIF-1 and CXCR4, improving graft integration and cardiac repair [855]. However, clinical studies primarily focus on medical outcomes, with limited data on the long-term fate of transplanted cells and their effects on ECM remodeling [856].
After this, the strategies of stem cell therapy include cell transplantation (autologous/allogenic stem cells) and stimulation of endogenous progenitor cells via pharmacological, biological, or ECM-based methods [857]. Different cell types, delivery methods, dosages, and administration timing contribute to variability in clinical outcomes, making direct comparisons difficult [858]. While stem cell therapy is known to alter ECM composition, its role in modulating biomechanical properties and facilitating ECM remodeling remains unexplored [859]. Given the unique composition of cardiac ECM, the ability of noncardiac-derived stem cells to restore heart-specific ECM proteins remains unclear, enhancing the need for further research [860].
For instance, cardiac tissue engineering faces key challenges, primarily cell source limitations and scaffold design [861]. The myocardium´s stiffness, ranging from 20 kPa to higher values, is important in cell differentiation and functional maturation [862]. Biodegradable scaffolds must endure the mechanical forces of the cardiac cycle while providing biochemical and structural cues for cell adhesion, migration, and integration [863]. Decellularized ECM is a promising scaffold due to its MMP-sensitive peptides, spatial organization, and growth factors, facilitating vascularization and host cell proliferation [864]. However, construction thickness is a major limitation, as oxygen diffusion constraints restrict the maximum viable thickness to 400 µm.
Several drugs, including omecamtiv mecarbil, spironolactone, and agrin, have shown potential in targeting the cardiac ECM and cytoskeleton for cardiovascular disease (CVD) treatment [865]. Omecamtiv mecarbil, a myosin activator, enhances cardiac contractility and slightly reduces heart failure (HF) risk in patients with low ventricular ejection fraction (LVEF) [866]. Agrin, an ECM proteoglycan, aids in neuromuscular junction formation and has demonstrated protective effects against myocardial infarction and dilated cardiomyopathy (DCM) [867]. Spironolactone, a mineralocorticoid receptor antagonist, limits ECM turnover, reducing cardiac fibrosis and improving congestive HG outcomes. These examples enhance the reduction of cardiac fibrosis of ECM-targeting drugs in CVD management [868]. However, concerns remain regarding unintended effects due to the fundamental roles of ECM and cytoskeletal proteins in cellular function [869]. Despite this, ongoing research, supported by computational analyses, underscores the promise of ECM and cytoskeletal targeting as a novel approach to drug development [870]. Vascularization strategies are being explored to overcome diffusion limits, improving tissue survival and function in engineered cardiac grafts. Table 10 summarizes all the therapeutic agents used.
Main ECM-targeted therapeutic agents and strategies in cardiovascular disease: preclinical and clinical evidence
| Therapeutic agent/ Strategy | Target/ Mechanism | Preclinical evidence | Clinical evidence | Notes |
|---|---|---|---|---|
| Stem cell therapy (autologous/ allogenic CSCs) | Repopulate cardiomyocytes, modify ECM stiffness, promote angiogenesis | Multiple animal models of myocardial infarction; enhanced survival with preconditioning (hypoxia, heat shock, chemical) | Early-phase trials focus on cardiac function; limited long-term ECM data | Variability due to cell type, dose, timing, delivery method |
| Endogenous progenitor cell stimulation | ECM-based or pharmacological activation of cardiac repair pathways | Preclinical studies show improved vascularization and cell integration | Limited clinical translation | Still under investigation |
| Decellularized ECM scaffolds | Provide structural and biochemical cues; MMP-sensitive peptides facilitate remodeling | Preclinical tissues engineering models; improve vascularization and hots cell proliferation | Not widely applied clinically yet | Oxygen diffusion limits max scaffold thicknees (near 400 µm) |
| Omecamtiv mecarbil | Myosin activator; enhances contractility | Preclinical cardiac models | Shown to slightly reduce HF risk in patients with low LVEF | Primarily targets cytoskeleton and contractility |
| Agrin | ECM proteoglycan: supports neuromuscular junction formation, reduced myocardial injury | Animals models of MI and DCM | Not yet in advanced clinical trials | Demonstrates cardioprotective and anti-fibrotic effects |
| Spironolactone | Mineralocorticoid receptor antagonist; limits ECM turnover and fibrosis | Preclinical CDV models | Used in patients with congestive heart failure to improve outcomes | Widely clinically applied; acts on ECM remodeling indirectly |
The strategies of ECM therapy in cancer development
The increasing global burden of cancer enhances the urgent need for precise treatment strategies and the discovery of novel therapeutic targets [871]. Traditional therapeutic approaches, including biomarker-based treatments, immunotherapy, and chemotherapy, have significantly improved patient prognosis [872]. Besides, aberrant gene expression and dysregulated signaling pathways in tumor cells drive pro-tumorigenic ECM remodeling, creating a hostile environment that impedes the infiltration of anti-tumor immune cells and therapeutic agents while simultaneously supporting cancer cell survival under stress induced by anti-cancer treatment [873,874]. The ECM, as shaped by cancer cells, is closely linked to multiple forms of drug resistance.
In this sense, each tumor type is characterized by a unique gene signature, which is closely linked to specific ECM composition [875]. Understanding these ECM-related gene signatures may provide valuable insights into a tumor´s sensitivity to therapy [876]. Several studies have explored this connection; for instance, stromal-related gene signatures have been associated with prognosis in patients with large B-cell lymphoma undergoing chemotherapy [877].
Beyond genetic signatures, proteomic analyses of ECM proteins have also proven useful in distinguishing cancerous tissues from normal ones [878]. More recently, a study revealed that matrix components released into the bloodstream following anti-PD-1 treatment in melanoma patients were predictive of treatment outcomes [879]. Furthermore, TGF frequently controls ECM signature genes-β1 signaling, with COL11A1 showing upregulated expression during ovarian cancer progression [880]. These findings underscore the critical role of ECM remodeling in cancer progression and resistance to therapy [881]. Identifying pivotal genes involved in ECM regulation could be instrumental in designing more effective and personalized therapeutic strategies.
In cancer, immunotherapies harness the body´s immune system to combat cancer, offering promising treatment strategies. These approaches include immune checkpoint inhibitors, such as PD-1, PD-L1, and CTLA-4 blockers, which enhance anti-tumor activity within the TME [882]. Another significant advancement is adoptive cell therapy (ACT), particularly chimeric antigen receptor (CAR) T-cell therapy [883]. This method involves extracting T cells from patients, genetically engineering them to recognize tumor-specific antigens, and reintroducing them to target cancer cells more effectively [884]. While some of these therapies have led to complete tumor remission, their effectiveness varies among patients and tumor types. Understanding the factors influencing treatment response is important for advancing immunotherapy strategies [885]. The ECM plays a pivotal role in regulating immunotherapy efficacy. Studies have shown that ECM-related gene expression can prevent resistance to PD-1 blockade therapy [886]. For example, in metastatic urothelial cancer, TGF-β-induced peritumoral collagen creates a barrier that prevents CD8+ T cells from reaching tumor cells, leading to poor treatment outcomes [886]. In breast cancer, CAFs expressing ECM proteins and TGF-β signaling contribute to immune evasion by upregulating PD-1 and CTLA-4 in regulating T cells [887]. Similarly, collagen-induced CD8+ T cell exhaustion in lung tumors has been linked to immune checkpoint therapy resistance [888].
Interestingly, ECM stiffness in tumors can also influence PD-L1 expression, with studies showing that depleting β3-integrin reduces PD-L1 levels and enhances PD-1 blockade efficacy [889]. These findings suggest that combating ECM-targeting strategies with immunotherapy could improve treatment response [890]. Indeed, targeting key ECM components such as TGF-β and PD-L1 has demonstrated increased ECM remodeling, enhanced CD8+ T cell infiltration, and a shift in macrophage activity towards an anti-tumor phenotype in colon and breast cancer models [891].
ECM remodeling strategies also facilitate the penetration of immunotherapeutic agents into tumors. In preclinical models of breast cancer and melanoma, the use of hyaluronidase to degrade HA enhances the efficacy of PD-L1 inhibitors and cancer vaccines by improving drug delivery [892]. Similarly, oncolytic viruses engineered to express hyaluronidase generate low-molecular-weight HA, which activates NF-κB signaling in macrophages and promotes CD8+ T cell infiltration, ultimately enhancing PD-1 blockade efficacy in glioblastoma models [893].
CAR-T cell therapy has shown limited success in solid tumors, partly due to the EC acting as a physical barrier that restricts T cell infiltration [894]. Expanding CAR-T cells ex vivo reduces their ECM remodeling capabilities, a limitation linked to low heparanase expression [895]. Engineering CAR-T cells to express heparanase has been shown to improve their tumor-penetrating ability and enhance anti-tumor effects [896]. Furthermore, training CAR-T cells enhances their cytotoxicity through AP-1 pathway activation in preclinical studies [897].
Another promising strategy exploits the high collagen content of tumors to direct immunotherapeutic agents, more precisely [898]. By conjugating PD-L1 and CTLA-4 inhibitors, as well as cytokines like IL-2, with a collagen-binding motif derived from the von Willebrand factor A3 domain, these agents can be selectively delivered to tumor sites, improving treatment specificity and efficacy [899,900].
On the other hand, chemotherapy remains one of the most used strategies for cancer treatment, primarily targeting malignant cells by disrupting their proliferation mechanisms [901]. These mechanisms include inducing DNA damage, inhibiting microtubule, and protein function, blocking DNA synthesis, or interfering with the oncogenic signaling pathway [902]. However, chemotherapy is often associated with systemic side effects, such as myelosuppression, due to its impact on blood cell production [903]. Beyond its systemic effects on the immune system, it also alters the local tumor immune microenvironment (TIME), with evidence suggesting it can convert immunologically cold tumors into hot ones [904]. However, this effect is not universal and may vary between tumor types [905]. For instance, in pancreatic cancer, chemotherapy has been observed to promote tumor-supportive immunity by driving the differentiation of monocytes into myeloid-derived suppressive cells (MDSCs) through granulocyte-macrophage colony-stimulating factor (GM-CSF) secretion by cancer cells [906].
Immunogenic cell death (ICD), a key effect of certain cancer therapies such as chemotherapy and radiotherapy, triggers immune activation by releasing damage-associated molecular patterns (DAMPs) and tumor antigens, fostering an anti-tumor immune response [907]. However, chemotherapy can also induce changes in the ECM, including fibrosis, which is closely linked to inflammation [908]. In some cases, chemotherapy has been shown to promote pro-tumor effects within the TIME [909]. For example, paclitaxel treatment can lead CD8+ T cells to secrete LOX, facilitating collagen and elastin crosslinking in the lungs and ultimately promoting metastasis [910]. Despite these findings, the specific impact of chemotherapeutic and targeted therapies on ECM remodeling and TIME modulation remains largely unexplored, and their effects are likely dependent on the tumor´s pre-existing microenvironment.
A clear example of chemotherapy-induced TIME alterations is seen in the effects of neoadjuvant chemotherapy (NACT) [911]. Concurrently, it promotes ECM remodeling, such as the upregulation of collagen VI, which has been associated with chemotherapy resistance and poor prognosis [912]. Notably, these ECM alterations are site-dependent, differing between metastatic sites and primary tumors, suggesting that the local TIME is a crucial determinant of ECM remodeling following chemotherapy [913].
Targeting ECM remodeling has shown the potential to enhance chemotherapy responses. For instance, in a breast cancer mouse model. The absence of MMP-9 improved the response to doxorubicin by increasing vascular permeability [914]. However, doxorubicin treatment has also been linked to the recruitment of MMP-9-expressing macrophages, which promote chemoresistance [915]. These findings highlight the need for further investigation into the relationship between ECM remodeling, immune modulation, and chemoresistance.
As research progresses, integrating ECM-targeting strategies with immunotherapy could open new avenues for more effective and personalized cancer treatments. Altogether, these findings emphasize the complex interplay between chemotherapy, immunotherapy, the immune microenvironment, and ECM remodeling. A deeper understanding of these interactions could lead to novel therapeutic strategies that enhance treatment efficacy, minimize resistance, and improve patient outcomes by co-targeting the ECM and immune components in conjunction with the therapy against cancer. Table 11 summarizes the main therapeutic agents targeting the ECM in cancer.
ECM-targeted and ECM-modulating therapeutic strategies in cancer: preclinical and clinical evidence
| Therapeutic agent/ Strategy | Target/ Mechanism | Preclinical evidence | Clinical evidence | Notes |
|---|---|---|---|---|
| Immune checkpoint inhibitors (PD-1, PD-L1, CTL4-A) | Blockade of inhibitory immune pathways; ECM stiffness and TGF-β can impair efficacy | ECM-related genes linked to resistance; ECM stiffness increases PD-L1 | Approved for multiple cancers; heterogeneous outcomes | ECM modulation may improve infiltration and response |
| CAR-T cell therapy | Genetically engineered T cells to recognize tumor antigens | Limited efficacy in solid tumors due to ECM barrier; engineered CAR-T expressing heparanase improve infiltration | Approved in hematologic cancers; limited success in solid tumors | ECM remodeling is key to enhance efficacy |
| ECM remodeling enzymes (Hyaluronidase) | Degrades hyaluronic acid, reduces stiffness, improves drug penetration | Enhances efficacy of PD-L1 inhibitors, vaccines, and oncolytic viruses in melanoma, breast cancer, glioblastoma | Ongoing preclinical and early translational research | Improves immune infiltration and drug delivery |
| Oncolytic viruses expressing hyaluronidase | ECM degradation; HA breakdown; activation pf NF-κB in macrophages | Improved CD8+ T cell infiltration and PD-1 blockade efficacy in glioblastoma | Preclinical models | Synergistic with checkpoint inhibitors |
| TGF-β inhibitors/ modulators | ECM remodeling and immune invasion | Increased CD8+ infiltration improved anti-tumor immunity in colon and breast cancer models | Early clinical trials in solid tumors | Targeting peritumoral collagen improves immunotherapy |
| Collagen-binding conjugates (PD-L1, CTLA-4 inhibitors, IL-2) | Targeted delivery to collagen-rich tumor ECM | Improved specificity and efficacy in preclinical models | Translational; no large clinical trials yet | Increase immune activation in TME |
| Chemotherapy (doxorubicin, paclitaxel, NACT) | Disrupt cell proliferation; indirect effects on ECM and TIME | ECM remodeling: collagen VI upregulation, MMP-9 effect on resistance | Standard of care; evidence of ECM-driven resistance | May promote fibrosis or metastasis; context-dependent |
| MMP-9 modulation | ECM degradation and vascular remodeling | MMP-9 deletion improves doxorubicin response; macrophage-derived MMP-9 linked to resistance | Preclinical studies | Dual role: sensitization vs chemoresistance |
The use of extracellular vesicles in aging, calcification, and cancer
Extracellular vesicles (EVs) are membrane-enclosed structures released by cells carrying a diverse range of molecular cargo [916]. These vesicles play crucial roles in cell-to-cell communication, influencing the fate of recipient cells in both local and distant tissues and serving as pathophysiological biomarkers [917]. Traditionally, EVs have been classified into three main types: exosomes, microvesicles, and apoptotic bodies [918].
However, recent research has identified additional subtypes, including autophagic EVs, stress-induced EVs, and matrix vesicles [919].
Autophagy has an important role in EV biogenesis and release. During this process, autophagosomes can fuse with endosomes to form amphiboles, which are subsequently secreted as autophagic EVs [920]. Also, processes such as EMT and cancer stemness contribute to EV production. Matrix vesicles and the EMC actively interact with the TME, influencing tumor progression [921].
Extracellular vesicles from healthy cells are being explored as next-generation therapeutics due to their immunogenicity, biocompatibility, and ability to transfer bioactive agents [922]. While no ECM-related EV therapeutics are in clinical trials yet, studies suggest their role in modulating the matrix for tissue regeneration [923]. EVs are found in bodily fluids like blood, saliva, cerebrospinal fluid, and urine, making them valuable for biomarker discovery in diseases, including cancer [924,925]. These biomarkers help in diagnosis, predicting treatment response, and detecting pre-symptomatic conditions. EVs contain RNAs, including miRNA, mRNA, and long noncoding RNAs, which influence recipient cells [926,927]. Other RNA types, like circular RNA (circRNA) and PIWI-interacting RNA (piRNA), have also been identified in EVs [928].
Aging and EVs
EVs serve as fundamental mediators of intracellular communication, playing both beneficial and detrimental roles throughout the aging process [929]. EVs derived from young organisms, healthy tissues, and stem cells often function as positive signaling molecules, promoting cellular repair and regeneration [930]. In contrast, EVs from aged or damaged tissues, like tissues with SASP, can propagate cellular senescence and contribute to chronic inflammation [931].
Over time, the balance between healthy and unhealthy vesicles may shift toward an increased presence of aging-promoting EVs, which reinforce tissue damage and accelerate the onset of age-related diseases [932]. This phenomenon underscores the need for targeted geroptherapeutic interventions.
Two primary strategies have emerged to counteract the detrimental effects of aging-associated EVs [933]. The first approach focuses on eliminating or inhibiting cells that secrete unhealthy EVs [934]. The second strategy, which is focused on EV-based regenerative medicine, aims to replace this harmful vesicle with healthy EVs capable of restoring tissue homeostasis and mitigating age-related degeneration [935].
Aging-related diseases currently account for more than 20% of the global disease burden, with cardiovascular disease leading to 30%, followed by cancer with 15%, and pulmonary, musculoskeletal, and neurodegenerative disorders [936]. Given the promising therapeutic potential of EV-based treatments in addressing key hallmarks of physiological aging, it is not surprising that these vesicles are being explored as next-generation therapies for age-related conditions [937] (Table 12).
Therapeutic strategies involving EVs in aging and age-related diseases
| EV source/ strategy | Role/ mechanism | Impact on aging | Therapeutic potential |
|---|---|---|---|
| EVs from young/ healthy tissues or stem cells | Positive signaling molecules; promote repair and regeneration | Counteract tissue damage, support homeostasis | Regenerative medicine; anti-aging interventions |
| EVs from aged or SASP-associated tissues | Propagate senescence and chronic inflammation | Accelerate tissue dysfunction and age-related diseases | Target for elimination or inhibition |
| Strategy 1: Inhibition/ elimination of harmful EV-secreting cells | Prevent release of aging-promoting EVs | Reduces propagation of senescence signaling | Gerotherapeutic approach under study |
| Strategy 2: EV-based regenerative therapy | Administration of healthy EVs | Restore tissue homeostasis, mitigate degeneration | Next-generation therapies for cardiovascular, cancer, pulmonary, musculoskeletal, neurodegenerative diseases |
Calcification and EVs
Apart from being involved in cancer, EVs interact with vascular calcification (Table 13). This process refers to the pathological mineralization of the ECM within blood vessels, a process commonly associated with the diseases explained above [938]. This phenomenon begins with the transdifferentiation of VSMCs into an osteoblast-like phenotype [939]. Under normal physiological conditions, VSMCs release EVs containing calcification inhibitors, which help maintain vascular homeostasis [940]. However, under pathological conditions driven by chronic inflammation or abnormal mineral metabolism, osteoblast-like VSMCs produce EVs that resemble matrix vesicles, similar to those involved in bone formation [941].
Role of EVs in vascular calcification and ECM remodeling
| EV source/ strategy | Cargo/ mechanism | Impact on ECM and vessels | Clinical relevance |
|---|---|---|---|
| Normal VSMCs | EVs contain calcification inhibitors | Maintain vascular homeostasis, prevent pathological mineralization | Protective mechanism against vascular disease |
| Osteoblast-like VSMCs (pathological state) | EVs enriched with annexin 1; resemble bone matrix vesicles | Aggregate in collagen fibrils, act as nucleation sites for mineralization | Promote vascular calcification, thrombosis, vessel rupture |
| EV aggregation in ECM | Surface phospholipids + annexins enable Ca2+/ PO43- deposition | Destabilizes vessel wall structure, drives pathological mineralization | Increases risk of severe cardiovascular complications |
These pathological EVs have an important role in destabilizing the ECM of blood vessels by promoting calcification in specific regions of the vessel walls [941]. This process increases the risk of thrombosis and vessel rupture, leading to severe cardiovascular complications. Recent studies have demonstrated that annexin AI is highly enriched in EVs released by osteoblast-like VSMCs during vascular calcification [941]. This protein plays a key role in facilitating the aggregation of EVs within collagen fibrils of the ECM [942]. These aggregated EVs serve as nucleation sites for mineralization, which continues in the presence of elevated extracellular Ca+2 phosphate (PO43-) levels [943]. Unlike the mineralization process in bone and cartilage, which occurs within MVs, vascular calcification is driven by the surface phospholipids and annexins of EV aggregates that accumulate with the ECM [944].
Cancer and EVs
In TME, EVs facilitate communication between cancer and stroma cells, carrying bioactive molecules like proteins, mRNA, and miRNA [945]. Some EVs modulate the ECM, contributing to chemotherapy resistance and metastasis, making them potential therapeutic targets and biomarkers (Table 14) [946]. Tumor-derived EVs can be detected in the bloodstream, enhancing their clinical relevance [947]. A subset of ECM-embedded nanovesicles, matrix-bound vesicles (MBVs), influences cell behavior and may have tumor-specific characteristics [948]. MBVs could serve as novel biomarkers, but further research is needed [948]. Cell culture models, particularly 3D cultures, help study EVs, as their molecular content closely resembles EVs found in patient tumors [949].
EVs in the tumor microenvironment: roles in ECM remodeling, resistance, and therapeutic applications
| EV source/ type | Cargo/ mechanism | Impact on TME and ECM | Clinical/ Therapeutic relevance |
|---|---|---|---|
| Tumor-derived EVs | Proteins, mRNA, miRNA | Modulate ECM; promote chemotherapy resistance and metastasis; shape immunologically cold tumors | Circulating biomarkers; potential therapeutic targets |
| Stromal cell-derived EVs (CAFS, TACs) | Deliver bioactive molecules | Induce chemoresistance, immunotherapy resistance, EMT, stemness, and tumor dormancy | Foster immunosuppressive, therapy-resistant microenvironment |
| Matrix bond vesicles (MBVs) | ECM-embedded nanovesicles | Influence cell behavior; niche formation; tumor-specific features | Potential novel biomarkers (need further research) |
| Exosomes (drug delivery systems) | Small molecules, proteins, nucleic acids, gene therapies | Stable and biocompatible carriers; enable precise molecular transport | Enhances drug solubility, targeting and reduces toxicity, promising for personalized therapy |
| ECM-derived vesicles (matrix vesicles) | Collagen, fibronectin, laminin interactions; MMPs on EV surface | Promote tumor stiffness, mechanotransduction, immune modulation, vascular regulation | Involved in poor prognosis tumors (pancreatic ductal adenocarcinoma); biomarkers (glypican-1) |
Through different mechanisms, the cancer cell-derived EVs play a central role in shaping the TME, thanks to tumor-derived EVs that contribute to the formation of immunologically cold tumors [950]. Furthermore, stromal cells in the TME release EVs that deliver bioactive molecules to cancer cells, promoting chemoresistance, immunotherapy resistance, dormancy stemness, and EMT [951]. This reciprocal communication between tumor-associated cells (TACs) and cancer cells via EVs fosters an immunosuppressive and therapy-resistant microenvironment [952].
First, exosomes originate from endosomes through a three-step process: biogenesis, transport, and release [953]. They offer a promising drug delivery system due to their biocompatibility, stability, and ability to transport molecules efficiently [954]. Unlike conventional drugs, which often have low bioavailability and high toxicity, exosome-based delivery systems can enhance therapeutic efficacy while minimizing side effects [955]. They can carry small molecules, proteins, nucleic acids, and gene therapies, improving drug solubility and specificity [956]. Exosomes can be loaded with drugs via direct methods, such as incubation, electroporation, liposome transfection, or indirect methods, like engineering donor cells [957,958]. Various administration routes exist, but modifying exosomes to extend their circulation time enhances their effectiveness [959]. Surface modifications, such as peptide fusions or magnetic therapies, further improve targeting, increasing their potential in personalized cancer therapy [960].
Furthermore, ECM in the TME consists of proteins like collagen, fibronectin, and laminin, supporting cancer progression and immune interactions [961]. ECM-rich tumors, such as pancreatic ductal adenocarcinoma, often show poor prognosis due to their dense stroma, which enhances resistance to therapy [962]. ECM acts as a barrier against drugs and immune responses while increasing tumor stiffness and promoting mechanotransduction signaling [963,964]. Matrix vesicles, including extracellular matrix-bound and matrix-coated vesicles, play key roles in ECM interactions, affecting immune cell function and vascular regulation [965]. CAFs and cancer cells produce matrix vesicles, adding niche formation and metastasis [966]. ECM-bound molecules like glypican-1 serve as biomarkers, helping in early cancer detection. MMPs on EV surface contribute to cancer progression and tumor niche development [967].
Regenerative medicine
The molecular crosstalk between cells and the ECM is inherently dynamic and reciprocal. This complex interaction governs essential physiological processes, which are mechanisms vital for maintaining tissue homeostasis and enabling effective wound healing (Table 15) [968]. In tissue engineering, one of the greatest challenges lies in replicating the native ECM´s biochemical and physical properties [969]. These include surface topology, pore size, mechanical strength, biocompatibility, and degradation profile, factors that collectively influence tissue regeneration.
Decellularized extracellular matrix (dECM) biomaterials in tissue engineering and regeneration
| Strategy/ material | Key properties | Applications/ Impact | Clinical/ Translational relevance |
|---|---|---|---|
| Native ECM (baseline) | Dynamic crosstalk; surface topology, pore size, mechanical strength, degradation profile | Governs homeostasis, wound healing, and tissue regeneration | Basis for biomimetic scaffold desing |
| Decellularized ECM (dECM) | Removal of immunogenic components; preservation of structural proteins and bioactive macromolecules | Provides biologically relevant microenvironment; activated intrinsic regenerative pathways | FDA-approved scaffolds (UBM, SIS) for skin muscle, GI tissue |
| Whole organs/ tissue sheets (early strategies) | Retain native architecture | Limited by poor mechanical stability and incomplete mimicry | Less practical for clinical use |
| Micronized dECM (next-gen approach) | Reconstituted into hydrogels, electrospun scaffolds, 3D-bioprinted constructs | Versatile formats: customizable to tissue-specific needs | Expands application across multiple tissue types |
| Tissue-specific dECM scaffolds | Tailored biochemical & biochemical properties | Cardiac, neural, cartilage muscle, liver, lung (well-developed); pancreas, kidney, vocal folds (emerging) | Significant progress in scaffold design and regenerative outcomes |
As it is said before, decellularized ECM (dECM) materials have emerged as a promising solution, offering a biologically relevant microenvironment that supports specialized cell function and activates the body´s intrinsic regenerative pathways [970]. Through decellularization, immunogenic cellular components are removed, while structural proteins and bioactive macromolecules are largely preserved [971]. FDA-approved dECM scaffolds derived from sources like the urinary bladder matrix (UBM) and small intestinal submucosa (SIS) have already demonstrated clinical efficacy in regenerating skin, muscle, and gastrointestinal tissues [972,973].
While early strategies focused on using entire decellularized organs or tissue sheets, issues related to mechanical stability and native tissue mimicry prompted the development of more adaptable forms [974]. Researchers have shifted toward micronized dECM particles, which are reconstituted into diverse biomaterial formats, including injectable hydrogels, electrospun scaffolds, and bioprinted constructs [975-977]. These newer forms offer improved versatility, enabling customization to match the shape, mechanical properties, and biological demands of specific tissue types.
Overall, dECM-based biomaterials represent a powerful platform for engineering tissue-specific scaffolds that can guide repair and regeneration [978]. The most developed applications so far span nine tissue types, including cardiac, neural, cartilage, muscle, liver, and lung, each showing significant progress in scaffold design and regenerative outcomes [979-984]. Additional tissues, like the pancreas, kidney, and vocal folds, are beginning to show promise as research expands [985-987].
By tailoring scaffold composition and structure to each tissue´s unique physiological demands and leveraging advanced fabrication technologies, the importance of these strategies is to enable functional tissue regeneration through biomaterials that recapitulate the native ECM [988,989].
Discussion
The body of work reviewed underscores the intricate interplay between the extracellular matrix (ECM), cellular communication, and the pathogenesis of a wide array of diseases, including cancer, cardiovascular calcification, and age-related degenerative disorders. Collectively, the data reveal that the ECM is far more than a mere structural scaffold; it is a dynamic, bioactive network that influences cellular behavior, modulates tissue homeostasis, and plays a decisive role in disease progression.
In a healthy state, the ECM provides essential biochemical and mechanical cues to maintain tissue integrity and regulate cell proliferation, migration, differentiation, and apoptosis. Its tightly controlled remodeling processes are fundamental for normal development, repair, and regeneration. However, in pathological conditions, these same remodeling processes become dysregulated. Aberrant ECM remodeling (driven by factors such as chronic inflammation, altered intercellular communication, and cellular senescence) can lead to the formation of fibrotic, stiff, and chemically modified matrices. This shift not only supports tumor progression and metastasis. For instance, in cancer, the ECM is remodeled by cancer cells and stromal components like cancer-associated fibroblasts (CAFs), which deposit excess collagen and other matrix proteins. This remodeling increases tissue stiffness, impedes the infiltration of anti-tumor immune cells, and establishes physical and biochemical barriers that reduce the efficacy of therapeutic agents.
Furthermore, the role of cellular senescence in ECM remodeling is multifaced. While a sentence is a natural, protective mechanism (limiting the proliferation of damaged cells and facilitating tissue repair through the senescence-associated secretory phenotype (SASP)), its chronic accumulation can have deleterious effects. Senescent cells secrete a host of pro-inflammatory cytokines, chemokines, growth factors, and proteases, which contribute to a persistent inflammatory state termed inflammaging. The inflammatory milieu not only accelerates the ECM degradation but also fosters an environment conducive to tumor progression. The balance between beneficial and detrimental outcomes of senescence appears to shift unhealthy with age, leading to an accumulation of unhealthy EVs and other secreted factors that reinforce tissue damage and support disease progression.
Also, cellular senescence, a defining feature of aging, establishes a critical nexus between calcification and cancer. As cells enter senescence, they secrete a complex cocktail of pro-inflammatory cytokines, proteases, and growth factors, which disrupt normal ECM remodeling. This altered ECM not only becomes prone to pathological calcification, as seen in VSMC transforming into osteoblasts-like cells but also fosters a tumor-friendly microenvironment. In the context of cancer, senescence-driven ECM changes contribute to increased tissue stiffness and immune evasion, thereby facilitating tumor initiation, progression, and metastasis. Together, these interrelated processes underscore how age-related cellular senescence can promote both calcification and malignancy, highlighting potential targets for therapeutic intervention.
Extracellular vesicles (EVs) further highlight their complex communication networks within tissues. EVs serve as critical mediators of intercellular signaling, transferring bioactive molecules (such as proteins, mRNA, miRNA, and long noncoding RNAs) across cells. In the tumor microenvironment (TME), EVs from cancer cells can modulate the ECM, promote angiogenesis, drive the polarization of immune cells into protumorigenic phenotypes, and even contribute to chemoresistance. Conversely, EVs derived from healthy cells or stem cells have emerged as promising therapeutic agents capable of promoting tissue repair and beneficially modulating the ECM. Their low immunogenicity, high biocompatibility, and intrinsic cargo-carrying capabilities make them attractive for use as drug delivery vehicles and biomarkers for disease progression.
The potential of ECM-targeting and EV-based therapies is especially compelling in the context of regenerative medicine. Stem cell therapies, for instance, not only aim to replace lost or damaged cardiac cells but also seek to modulate the ECM of injured tissues.
Evidence suggests that the engraftment of stem cells, such as mesenchymal stem cells (MSCs), can alter the stiffness and composition of the infarct zone, transforming a rigid car into a more compliant, heterogeneous matrix. This change in ECM properties is thought to facilitate the proliferation and integration of host cells, thereby improving cardiac function. However, challenges remain regarding the optimization of cell delivery, retention, and functional integration, with factors like scaffold design, oxygen diffusion, and biochemical matching playing pivotal roles.
In oncology, the interplay between ECM remodeling and immune surveillance is becoming increasingly apparent. Traditional chemotherapy, while effective in targeting rapidly dividing tumor cells, can also alter the ECM, sometimes exacerbating chemoresistance and fostering an immunosuppressive environment. The use of immune checkpoint inhibitors and adoptive cell therapies has revolutionized cancer treatment, yet their efficacy is often limited by the physical and biochemical barriers imposed by an aberrantly remodeled ECM. Recent studies suggest that a combination of ECM-targeting strategies with immunotherapies could overcome these hurdles. For instance, agents that degrade excessive ECM components, such as hyaluronidase or collagenase, may improve drug penetration and enhance the infiltration and activation of immune cells. Moreover, strategies aimed at normalizing ECM composition (by targeting key enzymes like lysyl oxidase) could help restore a microenvironment more amenable to therapeutic intervention.
Another promising avenue is leveraging the unique gene and protein signatures of the ECM to predict therapeutic responses and guide personalized treatments. The identification of ECM-specific biomarkers, such as particular collagen subtypes or glycoproteins like glypican-A1, provides valuable prognosis information and may inform the selection of targeted therapies. Additionally, computational and big data approaches have enabled the development of ECM gene expression signatures that correlate with treatment sensitivity or resistance, offering a powerful tool for patient stratification.
In summary, the multifaced roles of ECM in regulating cellular behavior, modulating the immune response, and influencing the efficacy of therapeutic interventions are increasingly recognized as critical determinants of disease progression and treatment outcomes. Whether through direct targeting of ECM components, modulation of intercellular communication via EVs, or the integration of advanced regenerative medicine strategies, the emerging evidence underscores the potential for novel therapeutic approaches that address the underlying ECM dysfunctions in a range of diseases. Future research must continue to elucidate the precise mechanism of ECM remodeling and its interplay with cellular senescence, inflammation, and immune therapy. By doing so, it can pave the way for more effective, personalized interventions that not only treat the symptoms of disease but also restore tissue homeostasis and function.
Conclusion
The body of evidence reviewed establishes the ECM as a central regulator of tissue homeostasis, disease initiation, and therapeutic response. Far from being a passive scaffold, the ECM emerges as a dynamic and bioactive network that integrates mechanical, biochemical, and immunological signals, thereby shaping cellular behavior and influencing the trajectory of health and disease. Its dysregulation, whether through chronic inflammation, senescence, or aberrant remodeling, contributes to fibrosis, calcification, cancer progression, and resistance to therapies, underscoring its dual role as both a mediator of physiological repair and a driver of pathology.
Importantly, the growing underscoring of ECM-cell interactions has opened new therapeutic avenues. Approaches that combine ECM-targeting strategies with immunotherapies or regenerative medicine hold particular promise, as they address not only the tumor or damaged tissue but also the supportive microenvironment that sustains disease. The emerging role of extracellular vesicles as mediators of ECM remodeling and as vehicles for drug delivery adds another layer of therapeutic potential, bridging molecular communication with translational applications.
Looking ahead, the challenge lies in translating these mechanistic insights into clinically effective interventions. This will require refining ECM-targeted therapies, integrating predictive biomarkers, and harnessing big data approaches to better stratify patients and personalize treatments. At the same time, advancing scaffold design, stem cell delivery, and exosome engineering will be essential for regenerative medicine to realize its full potential.
In conclusion, positioning the ECM at the center of therapeutic innovation offers a unique opportunity, not only to combat disease more effectively but also to restore tissue balance and resilience. By continuing to explore its complexities with rigor and creativity, future research can transform the ECM from a barrier to therapy into a gateway for more precise, durable, and patient-tailored interventions.
Acknowledgements
Funding
The study was supported by the Comunidad de Madrid (P2022/BMD-7321), ProACapital, and HALE KULANI, S. L. and MJR.
Author contributions
All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dacks JB, Peden AA, Field MC. Evolution of specificity in the eukaryotic endomembrane system. Int J Biochem Cell Biol. 2009;41:330-40 Available at: https://pubmed.ncbi.nlm.nih.gov/18835459/
2. Fidler AL, Boudko SP, Rokas A, Hudson BG. The triple helix of collagens - an ancient protein structure that enabled animal multicellularity and tissue evolution. J Cell Sci. 2018;131:jcs203950 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5963836/
3. Hynes RO. The evolution of metazoan extracellular matrix. J Cell Biol. 2012;196:671 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3308698/
4. Adams JC, Josephine Adams CC. Thrombospondins: Conserved mediators and modulators of metazoan extracellular matrix. Int J Exp Pathol. 2024;105:136-69
5. Rozario T, DeSimone DW. The Extracellular Matrix in Development and Morphogenesis: A Dynamic View. Dev Biol. 2009;341:126 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2854274/
6. Rousselle P, Montmasson M, Garnier C. Extracellular matrix contribution to skin wound re-epithelialization. Matrix Biol. 2019;75-76:12-26 Available at: https://pubmed.ncbi.nlm.nih.gov/29330022/
7. Wu D, Yamada KM, Wang S. Tissue Morphogenesis Through Dynamic Cell and Matrix Interactions. Annu Rev Cell Dev Biol. 2023;39:123-44 Available at: https://pubmed.ncbi.nlm.nih.gov/37315160/
8. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195
9. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020 11
10. Guo J, Huang X, Dou L. et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduction and Targeted Therapy 2022 7:1. 2022;7:1-40 Available at: https://www.nature.com/articles/s41392-022-01251-0
11. Faleeva M, Ahmad S, Theofilatos K. et al. Sox9 Accelerates Vascular Aging by Regulating Extracellular Matrix Composition and Stiffness. Circ Res. 2024;134:307-24 Available at: https://pubmed.ncbi.nlm.nih.gov/38179698/
12. Alakhdar AA, Sivakumar S, Kopchak RM, Hunter AN, Ambrosio F, Washburn NR. Age-Related ECM Stiffness Mediates TRAIL Activation in Muscle Stem Cell Differentiation. Adv Biol. 2024 8. Available at: https://pubmed.ncbi.nlm.nih.gov/39601528/
13. Selman M, Pardo A. Fibroageing: An ageing pathological feature driven by dysregulated extracellular matrix-cell mechanobiology. Ageing Res Rev. 2021;70:101393
14. Ortega MA, De Leon-Oliva D, Gimeno-Longas MJ. et al. Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities. Biomolecules. 2024 14. Available at: https://pubmed.ncbi.nlm.nih.gov/38540696/
15. Leight JL, Drain AP, Weaver VM. Extracellular matrix remodeling and stiffening modulate tumor phenotype and treatment response. Annu Rev Cancer Biol. 2017;1:313-34
16. Lewin S, Hunt S, Lambert DW. Extracellular vesicles and the extracellular matrix: a new paradigm or old news? Biochem Soc Trans. 2020;48:2335-45 Available at: https://pubmed.ncbi.nlm.nih.gov/33125481/
17. Ortega MA, Fraile-Martinez O, Garcia-Montero C. et al. An Updated View of the Importance of Vesicular Trafficking and Transport and Their Role in Immune-Mediated Diseases: Potential Therapeutic Interventions. Membranes (Basel). 2022 12. Available at: https://pubmed.ncbi.nlm.nih.gov/35736259/
18. Kou M, Huang L, Yang J. et al. Mesenchymal stem cell-derived extracellular vesicles for immunomodulation and regeneration: a next generation therapeutic tool? Cell Death & Disease 2022 13:7. 2022;13:1-16 Available at: https://www.nature.com/articles/s41419-022-05034-x
19. Elsharkasy OM, Nordin JZ, Hagey DW. et al. Extracellular vesicles as drug delivery systems: Why and how? Adv Drug Deliv Rev. 2020;159:332-43
20. Muncie JM, Weaver VM. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr Top Dev Biol. 2018;130:1-37 Available at: https://pubmed.ncbi.nlm.nih.gov/29853174/
21. Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216-9 Available at: https://pubmed.ncbi.nlm.nih.gov/19965464/
22. McKee TJ, Perlman G, Morris M, Komarova S V. Extracellular matrix composition of connective tissues: a systematic review and meta-analysis. Sci Rep. 2019 9. Available at: https://pubmed.ncbi.nlm.nih.gov/31332239/
23. Chun SY, Lim JO, Lee EH. et al. Preparation and Characterization of Human Adipose Tissue-Derived Extracellular Matrix, Growth Factors, and Stem Cells: A Concise Review. Tissue Eng Regen Med. 2019;16:385-93 Available at: https://pubmed.ncbi.nlm.nih.gov/31413942/
24. Gentili C, Cancedda R. Cartilage and bone extracellular matrix. Curr Pharm Des. 2009;15:1334-48 Available at: https://pubmed.ncbi.nlm.nih.gov/19355972/
25. Shaw TJ, Rognoni E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr Rheumatol Rep. 2020 22. Available at: https://pubmed.ncbi.nlm.nih.gov/32562113/
26. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4-27 Available at: https://pubmed.ncbi.nlm.nih.gov/26562801/
27. Theocharis AD, Manou D, Karamanos NK. The extracellular matrix as a multitasking player in disease. FEBS J. 2019;286:2830-69 Available at: https://pubmed.ncbi.nlm.nih.gov/30908868/
28. Chen Z, Du C, Liu S. et al. Progress in biomaterials inspired by the extracellular matrix. Giant. 2024;19:100323
29. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786-801
30. Pozzi A, Yurchenco PD, Iozzo R V. The nature and biology of basement membranes. Matrix Biol. 2017;57-58:1-11
31. Halfter W, Oertle P, Monnier CA. et al. New concepts in basement membrane biology. FEBS J. 2015;282:4466-79
32. Behrens DT, Villone D, Koch M. et al. The Epidermal Basement Membrane Is a Composite of Separate Laminin- or Collagen IV-containing Networks Connected by Aggregated Perlecan, but Not by Nidogens. J Biol Chem. 2012;287:18700
33. Rosset EM, Bradshaw AD. SPARC/osteonectin in mineralized tissue. Matrix Biol. 2016;52-54:78-87
34. Ham SM, Song MJ, Yoon HS, Lee DH, Chung JH, Lee ST. SPARC Is Highly Expressed in Young Skin and Promotes Extracellular Matrix Integrity in Fibroblasts via the TGF-β Signaling Pathway. Int J Mol Sci. 2023 24. Available at: https://pubmed.ncbi.nlm.nih.gov/37569556/
35. Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929-34 Available at: https://pubmed.ncbi.nlm.nih.gov/11306593/
36. Murphy-Ullrich JE. Thrombospondin 1 and Its Diverse Roles as a Regulator of Extracellular Matrix in Fibrotic Disease. J Histochem Cytochem. 2019;67:683-99 Available at: https://pubmed.ncbi.nlm.nih.gov/31116066/
37. Midwood KS, Chiquet M, Tucker RP, Orend G. Tenascin-C at a glance. J Cell Sci. 2016;129:4321-7 Available at: https://pubmed.ncbi.nlm.nih.gov/27875272/
38. Li Z, Chen S, Cui H. et al. Tenascin-C-mediated suppression of extracellular matrix adhesion force promotes entheseal new bone formation through activation of Hippo signalling in ankylosing spondylitis. Ann Rheum Dis. 2021;80:891-902 Available at: https://pubmed.ncbi.nlm.nih.gov/33858850/
39. Wilson SE, Torricelli AAM, Marino GK. Corneal epithelial basement membrane: Structure, function and regeneration. Exp Eye Res. 2020 194. Available at: https://pubmed.ncbi.nlm.nih.gov/32179076/
40. Wright K, Ly T, Kriet M, Czirok A, Thomas SM. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers (Basel). 2023;15:1899 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10047485/
41. Schüler SC, Liu Y, Dumontier S. et al. Extracellular matrix: Brick and mortar in the skeletal muscle stem cell niche. Front Cell Dev Biol. 2022 10. Available at: https://pubmed.ncbi.nlm.nih.gov/36523505/
42. Järveläinen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61:198-223 Available at: https://pubmed.ncbi.nlm.nih.gov/19549927/
43. Kirkpatrick CA, Selleck SB. Heparan sulfate proteoglycans at a glance. J Cell Sci. 2007;120:1829-32 Available at: https://pubmed.ncbi.nlm.nih.gov/17515480/
44. Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol. 2010;341:126-40 Available at: https://pubmed.ncbi.nlm.nih.gov/19854168/
45. Cornfine S, Himmel M, Kopp P. et al. The kinesin KIF9 and reggie/flotillin proteins regulate matrix degradation by macrophage podosomes. Mol Biol Cell. 2011;22:202-15 Available at: https://pubmed.ncbi.nlm.nih.gov/21119006/
46. Lee C, Kim MJ, Kumar A, Lee HW, Yang Y, Kim Y. Vascular endothelial growth factor signaling in health and disease: from molecular mechanisms to therapeutic perspectives. Signal Transduct Target Ther. 2025;10:170 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC12086256/
47. Vlodavsky I, Iozzo R V, Sanderson RD. Heparanase: multiple functions in inflammation, diabetes and atherosclerosis. Matrix Biol. 2013;32:220-2 Available at: https://pubmed.ncbi.nlm.nih.gov/23499526/
48. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395-406 Available at: https://pubmed.ncbi.nlm.nih.gov/22351925/
49. Krymchenko R, Coşar Kutluoğlu G, van Hout N. et al. Elastogenesis in Focus: Navigating Elastic Fibers Synthesis for Advanced Dermal Biomaterial Formulation. Adv Healthc Mater. 2024;13:2400484 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC12344638/
50. Kular JK, Basu S, Sharma RI. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J Tissue Eng. 2014;5:2041731414557112 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4883592/
51. Burk J, Sassmann A, Kasper C, Nimptsch A, Schubert S. Extracellular Matrix Synthesis and Remodeling by Mesenchymal Stromal Cells Is Context-Sensitive. International Journal of Molecular Sciences 2022, Vol 23, Page 1758. 2022;23:1758 Available at: https://www.mdpi.com/1422-0067/23/3/1758/htm
52. Hay ED. Biogenesis and organization of extracellular matrix. FASEB J. 1999 13 Suppl 2. Available at: https://pubmed.ncbi.nlm.nih.gov/10619144/
53. Revel JP, Hay ED. AN AUTORADIOGRAPHIC AND ELECTRON MICROSCOPIC STUDY OF COLLAGEN SYNTHESIS IN DIFFERENTIATING CARTILAGE. Z Zellforsch Mikrosk Anat. 1963;61:110-44 Available at: https://pubmed.ncbi.nlm.nih.gov/14095095/
54. Sipilä L, Ruotsalainen H, Sormunen R. et al. Secretion and Assembly of Type IV and VI Collagens Depend on Glycosylation of Hydroxylysines. Journal of Biological Chemistry. 2007;282:33381-8
55. Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2577133/
56. PORTER KR, PAPPAS GD. Collagen formation by fibroblasts of the chick embryo dermis. J Biophys Biochem Cytol. 1959;5:153-66 Available at: https://pubmed.ncbi.nlm.nih.gov/13630947/
57. Colige A, Li SW, Sieron AL, Nusgens B V, Prockop DJ, Lapière CM. cDNA cloning and expression of bovine procollagen I N-proteinase: A new member of the superfamily of zinc-metalloproteinases with binding sites for cells and other matrix components. Proc Natl Acad Sci U S A. 1997;94:2374-9 Available at: https://www.pnas.org/doi/abs/10.1073/pnas.94.6.2374
58. Li SW, Sieron AL, Fertala A, Hojima Y, Arnold W V, Prockop DJ. The C-proteinase that processes procollagens to fibrillar collagens is identical to the protein previously identified as bone morphogenic protein-1. Proceedings of the National Academy of Sciences. 1996;93:5127-30 Available at: https://www.pnas.org/doi/abs/10.1073/pnas.93.10.5127
59. Chalikias GK, Tziakas DN. Biomarkers of the extracellular matrix and of collagen fragments. Clinica Chimica Acta. 2015;443:39-47
60. Kajava A V. Molecular packing in type I collagen fibrils. A model with neighbouring collagen molecules aligned in axial register. J Mol Biol. 1991;218:815-23 Available at: https://pubmed.ncbi.nlm.nih.gov/2023251/
61. Revell CK, Jensen OE, Shearer T, Lu Y, Holmes DF, Kadler KE. Collagen fibril assembly: New approaches to unanswered questions. Matrix Biol Plus. 2021;12:100079
62. Cai L, Xiong X, Kong X, Xie J. The Role of the Lysyl Oxidases in Tissue Repair and Remodeling: A Concise Review. Tissue Eng Regen Med. 2017;14:15 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6171574/
63. Banushi B, Forneris F, Straatman-Iwanowska A. et al. Regulation of post-Golgi LH3 trafficking is essential for collagen homeostasis. Nature Communications 2016 7:1. 2016;7:1-14 Available at: https://www.nature.com/articles/ncomms12111
64. Wenstrup RJ, Smith SM, Florer JB. et al. Regulation of collagen fibril nucleation and initial fibril assembly involves coordinate interactions with collagens V and XI in developing tendon. J Biol Chem. 2011;286:20455-65 Available at: https://pubmed.ncbi.nlm.nih.gov/21467034/
65. Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nature Reviews Molecular Cell Biology 2014 15:12. 2014;15:771-85 Available at: https://www.nature.com/articles/nrm3902
66. Xiong Y, Li X, Sun B, Zhang J, Wu X, Guo F. Abnormal collagen deposition mediated by cartilage oligomeric matrix protein in the pathogenesis of oral submucous fibrosis. International Journal of Oral Science 2025 17:1. 2025;17:1-12 Available at: https://www.nature.com/articles/s41368-025-00355-x
67. Green H, Hamerman D. Production of Hyaluronate and Collagen by Fibroblast Clones in Culture. Nature 1964 201:4920. 1964;201:710-710 Available at: https://www.nature.com/articles/201710a0
68. Lin X, Patil S, Gao YG, Qian A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front Pharmacol. 2020;11:757 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7264100/
69. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617-27 Available at: https://pubmed.ncbi.nlm.nih.gov/19008372/
70. Nakamura T, Miller D, Ruoslahti E, Border WA. Production of extracellular matrix by glomerular epithelial cells is regulated by transforming growth factor-beta 1. Kidney Int. 1992;41:1213-21 Available at: https://pubmed.ncbi.nlm.nih.gov/1614036/
71. Xu J, Clark RAF. Extracellular matrix alters PDGF regulation of fibroblast integrins. J Cell Biol. 1996;132:239-49 Available at: https://pubmed.ncbi.nlm.nih.gov/8567727/
72. Choi D, Kang W, Park S, Son B, Park T. Identification of Glucocorticoid Receptor Target Genes That Potentially Inhibit Collagen Synthesis in Human Dermal Fibroblasts. Biomolecules. 2023;13:978 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10296022/
73. Revell CK, Jensen OE, Shearer T, Lu Y, Holmes DF, Kadler KE. Collagen fibril assembly: New approaches to unanswered questions. Matrix Biol Plus. 2021;12:100079 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8334717/
74. Nadol JB, Gibbins JR, Porter KR. A reinterpretation of the structure and development of the basement lamella: An ordered array of collagen in fish skin. Dev Biol. 1969;20:304-31
75. Sophia Fox AJ, Bedi A, Rodeo SA. The Basic Science of Articular Cartilage: Structure, Composition, and Function. Sports Health. 2009;1:461 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3445147/
76. Selvaraj V, Sekaran S, Dhanasekaran A, Warrier S. Type 1 collagen: Synthesis, structure and key functions in bone mineralization. Differentiation. 2024;136:100757
77. Eriksen EF. Cellular mechanisms of bone remodeling. Rev Endocr Metab Disord. 2010;11:219 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3028072/
78. Verzijl N, DeGroot J, Thorpe SR. et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027-31 Available at: https://pubmed.ncbi.nlm.nih.gov/10976109/
79. Kanchanawong P, Calderwood DA. Organization, dynamics and mechanoregulation of integrin-mediated cell-ECM adhesions. Nature Reviews Molecular Cell Biology 2022 24:2. 2022;24:142-61 Available at: https://www.nature.com/articles/s41580-022-00531-5
80. Zhu J, Yang G. H2S signaling and extracellular matrix remodeling in cardiovascular diseases: A tale of tense relationship. Nitric Oxide. 2021;116:14-26 Available at: https://pubmed.ncbi.nlm.nih.gov/34428564/
81. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562-73 Available at: https://pubmed.ncbi.nlm.nih.gov/16405877/
82. Van Doren SR. Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol. 2015;0:224 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4466143/
83. Lewis MP, Tippett HL, Sinanan ACM, Morgan MJ, Hunt NP. Gelatinase-B (matrix metalloproteinase-9; MMP-9) secretion is involved in the migratory phase of human and murine muscle cell cultures. J Muscle Res Cell Motil. 2000;21:223-33 Available at: https://link.springer.com/article/10.1023/A:1005670507906
84. Wilson CL, Matrisian LM. Matrilysin: an epithelial matrix metalloproteinase with potentially novel functions. Int J Biochem Cell Biol. 1996;28:123-36 Available at: https://pubmed.ncbi.nlm.nih.gov/8729000/
85. Pang L, Li Q, Li S. et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Scientific Reports 2016 6:1. 2016;6:1-12 Available at: https://www.nature.com/articles/srep22179
86. Curci JA, Liao S, Huffman MD, Shapiro SD, Thompson RW. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J Clin Invest. 1998;102:1900-10 Available at: https://pubmed.ncbi.nlm.nih.gov/9835614/
87. Hite LA, Shannon JD, Fox JW, Bjarnason JB. Sequence of a cDNA clone encoding the zinc metalloproteinase hemorrhagic toxin e from Crotalus atrox: evidence for signal, zymogen, and disintegrin-like structures. Biochemistry. 1992;31:6203-11 Available at: https://pubmed.ncbi.nlm.nih.gov/1378300/
88. Kuno K, Kanada N, Nakashirma E, Fujiki F, Ichimura F, Matsushima K. Molecular Cloning of a Gene Encoding a New Type of Metalloproteinase-disintegrin Family Protein with Thrombospondin Motifs as an Inflammation Associated Gene. Journal of Biological Chemistry. 1997;272:556-62
89. White JM. ADAMs: Modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol. 2003;15:598-606 Available at: https://pubmed.ncbi.nlm.nih.gov/14519395/
90. Bekhouche M, Colige A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biology. 2015;44-46:46-53
91. Crawley JTB, De Groot R, Xiang Y, Luken BM, Lane DA. Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood. 2011;118:3212-21 Available at: https://dx.doi.org/10.1182/blood-2011-02-306597
92. Bond JS, Rojas K, Overhauser J, Zoghbi HY, Jiang W. The structural genes, MEP1A and MEP1B, for the α and β subunits of the metalloendopeptidase meprin map to human chromosomes 6p and 18q, respectively. Genomics. 1995;25:300-3
93. Herzog C, Haun RS, Ludwig A, Shah S V, Kaushal GP. ADAM10 Is the Major Sheddase Responsible for the Release of Membrane-associated Meprin A. J Biol Chem. 2014;289:13308 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4036340/
94. Geurts N, Becker-Pauly C, Martens E. et al. Meprins process matrix metalloproteinase-9 (MMP-9)/gelatinase B and enhance the activation kinetics by MMP-3. FEBS Lett. 2012;586:4264-9 Available at: https://pubmed.ncbi.nlm.nih.gov/23123160/
95. Goldfarb RH, Murano G, Brundage R. et al. Degradation of glycoprotein and collagenous components of the basement membrane: studies with urokinase-type plasminogen activator, alpha-thrombin, and plasmin. Semin Thromb Hemost. 1986;12:335-6 Available at: https://pubmed.ncbi.nlm.nih.gov/2947330/
96. Maiorani O, Pivetta E, Capuano A. et al. Neutrophil elastase cleavage of the gC1q domain impairs the EMILIN1-α4β1 integrin interaction, cell adhesion and anti-proliferative activity. Scientific Reports 2017 7:1. 2017;7:1-13 Available at: https://www.nature.com/articles/srep39974
97. Zoratti GL, Tanabe LM, Varela FA. et al. Targeting matriptase in breast cancer abrogates tumour progression via impairment of stromal-epithelial growth factor signalling. Nature Communications 2015 6:1. 2015;6:1-13 Available at: https://www.nature.com/articles/ncomms7776
98. Vizovišek M, Fonović M, Turk B. Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Biology. 2019;75-76:141-59
99. Fonović M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta. 2014;1840:2560-70 Available at: https://pubmed.ncbi.nlm.nih.gov/24680817/
100. Roy M, Marchetti D. Cell surface heparan sulfate released by heparanase promotes melanoma cell migration and angiogenesis. J Cell Biochem. 2009;106:200-9 Available at: https://onlinelibrary.wiley.com/doi/full/10.1002/jcb.22005
101. Uchimura K, Morimoto-Tomita M, Bistrup A. et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006 7. Available at: https://pubmed.ncbi.nlm.nih.gov/16417632/
102. Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nature Reviews Immunology 2013 13:9. 2013;13:649-65 Available at: https://www.nature.com/articles/nri3499
103. Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 Is a Potent Inhibitor of Aggrecanase 1 (ADAM-TS4) and Aggrecanase 2 (ADAM-TS5). Journal of Biological Chemistry. 2001;276:12501-4
104. Hedrich J, Lottaz D, Meyer K. et al. Fetuin-A and cystatin C are endogenous inhibitors of human meprin metalloproteases. Biochemistry. 2010;49:8599-607 Available at: https://pubmed.ncbi.nlm.nih.gov/20806899/
105. Muller M, Trocme C, Lardy B, Morel F, Halimi S, Benhamou PY. Matrix metalloproteinases and diabetic foot ulcers: the ratio of MMP-1 to TIMP-1 is a predictor of wound healing. Diabet Med. 2008;25:419-26 Available at: https://pubmed.ncbi.nlm.nih.gov/18387077/
106. Perumal S, Antipova O, Orgel JPRO. Collagen fibril architecture, domain organization, and triple-helical conformation govern its proteolysis. Proc Natl Acad Sci U S A. 2008;105:2824 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2268544/
107. Zhao P, Sun T, Lyu C, Liang K, Du Y. Cell mediated ECM-degradation as an emerging tool for anti-fibrotic strategy. Cell Regeneration 2023 12:1. 2023;12:1-16
108. Lee H, Sodek KL, Hwang Q, Brown TJ, Ringuette M, Sodek J. Phagocytosis of collagen by fibroblasts and invasive cancer cells is mediated by MT1-MMP. Biochem Soc Trans. 2007;35:704-6
109. Ohkawara H, Ikeda K, Ogawa K, Takeishi Y. MEMBRANE TYPE 1-MATRIX METALLOPROTEINASE (MT1-MMP) IDENTIFIED AS A MULTIFUNCTIONAL REGULATOR OF VASCULAR RESPONSES. Fukushima J Med Sci. 2015;61:91 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5131593/
110. Richards DM, Endres RG. The Mechanism of Phagocytosis: Two Stages of Engulfment. Biophys J. 2014;107:1542 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4190621/
111. Sorushanova A, Delgado LM, Wu Z. et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv Mater. 2019 31. Available at: https://pubmed.ncbi.nlm.nih.gov/30126066/
112. Myers JC, Amenta PS, Dion AS. et al. The molecular structure of human tissue type XV presents a unique conformation among the collagens. Biochem J. 2007;404:535-44 Available at: https://pubmed.ncbi.nlm.nih.gov/17355226/
113. Peñuela L, Negro C, Massa M. et al. Atomic force microscopy for biomechanical and structural analysis of human dermis: A complementary tool for medical diagnosis and therapy monitoring. Exp Dermatol. 2018;27:150-5 Available at: https://pubmed.ncbi.nlm.nih.gov/29152798/
114. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195-200 Available at: https://pubmed.ncbi.nlm.nih.gov/21123617/
115. Kadler KE, Baldock C, Bella J, Boot-Handford RP. Collagens at a glance. J Cell Sci. 2007;120:1955-8 Available at: https://pubmed.ncbi.nlm.nih.gov/17550969/
116. Hynes RO, Naba A. Overview of the matrisome-an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012 4. Available at: https://pubmed.ncbi.nlm.nih.gov/21937732/
117. Barnes M. Update on Collagens: What You Need to Know and Consider. Plast Surg Nurs. 2019;39:112-5 Available at: https://pubmed.ncbi.nlm.nih.gov/31790038/
118. Malfait F, Francomano C, Byers P. et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8-26 Available at: https://pubmed.ncbi.nlm.nih.gov/28306229/
119. Malfait F, Symoens S, Goemans N. et al. Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome. Orphanet J Rare Dis. 2013 8. Available at: https://pubmed.ncbi.nlm.nih.gov/23692737/
120. Ouyang Z, Dong L, Yao F. et al. Cartilage-Related Collagens in Osteoarthritis and Rheumatoid Arthritis: From Pathogenesis to Therapeutics. Int J Mol Sci. 2023 24. Available at: https://pubmed.ncbi.nlm.nih.gov/37372989/
121. Seegmiller RE, Foster C, Burnham JL. Understanding chondrodysplasia (cho): A comprehensive review of cho as an animal model of birth defects, disorders, and molecular mechanisms. Birth Defects Res. 2019;111:237-47 Available at: https://pubmed.ncbi.nlm.nih.gov/30719872/
122. Kannu P, Bateman J, Savarirayan R. Clinical phenotypes associated with type II collagen mutations. J Paediatr Child Health. 2012 48. Available at: https://pubmed.ncbi.nlm.nih.gov/21332586/
123. Shahri JJ, Saberianpour S, Bayegi SN. Comparison of tissue biomarkers in arterial and vein (arteriovenous fistula) aneurysms. Phlebology. 2022;37:289-95 Available at: https://pubmed.ncbi.nlm.nih.gov/35255762/
124. Merlini L, Bernardi P. Therapy of collagen VI-related myopathies (Bethlem and Ullrich). Neurotherapeutics. 2008;5:613-8 Available at: https://pubmed.ncbi.nlm.nih.gov/19019314/
125. Watanabe M, Natsuga K, Shinkuma S, Shimizu H. Epidermal aspects of type VII collagen: Implications for dystrophic epidermolysis bullosa and epidermolysis bullosa acquisita. J Dermatol. 2018;45:515-21 Available at: https://pubmed.ncbi.nlm.nih.gov/29352483/
126. Darling TN, Yee C, Koh B. et al. Cycloheximide facilitates the identification of aberrant transcripts resulting from a novel splice-site mutation in COL17A1 in a patient with generalized atrophic benign epidermolysis bullosa. J Invest Dermatol. 1998;110:165-9 Available at: https://pubmed.ncbi.nlm.nih.gov/9457913/
127. Nielsen S, Nielsen K, Ivarsen A, Greenhill NS, Davis PF, Hjortdal J. Fuchs Endothelial Corneal Dystrophy: A Systematic Immunofluorescence Study of Collagen Type VIII Suggests Heterogeneous Pathophysiology. Cornea. 2016;35:872-7 Available at: https://pubmed.ncbi.nlm.nih.gov/27078008/
128. Seppinen L, Pihlajaniemi T. The multiple functions of collagen XVIII in development and disease. Matrix Biol. 2011;30:83-92 Available at: https://pubmed.ncbi.nlm.nih.gov/21163348/
129. Kashtan CE. Alport Syndrome: Achieving Early Diagnosis and Treatment. Am J Kidney Dis. 2021;77:272-9 Available at: https://pubmed.ncbi.nlm.nih.gov/32712016/
130. Al Kaissi A, Ghachem MB, Nabil NM. et al. Schmid's Type of Metaphyseal Chondrodysplasia: Diagnosis and Management. Orthop Surg. 2018;10:241-6 Available at: https://pubmed.ncbi.nlm.nih.gov/30027601/
131. Bella J, Hulmes DJS. Fibrillar Collagens. Subcell Biochem. 2017;82:457-90 Available at: https://pubmed.ncbi.nlm.nih.gov/28101870/
132. Richards AJ, Snead MP. Molecular Basis of Pathogenic Variants in the Fibrillar Collagens. Genes (Basel). 2022 13. Available at: https://pubmed.ncbi.nlm.nih.gov/35885981/
133. Chen W, Yang A, Jia J, Popov Y V, Schuppan D, You H. Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology. 2020;72:729-41 Available at: https://pubmed.ncbi.nlm.nih.gov/32176358/
134. Shahrajabian MH, Sun W. Mechanism of Action of Collagen and Epidermal Growth Factor: A Review on Theory and Research Methods. Mini Rev Med Chem. 2024;24:453-77 Available at: https://pubmed.ncbi.nlm.nih.gov/37587815/
135. Holmes DF, Lu Y, Starborg T, Kadler KE. Collagen Fibril Assembly and Function. Curr Top Dev Biol. 2018;130:107-42 Available at: https://pubmed.ncbi.nlm.nih.gov/29853175/
136. Quinlan C, Rheault MN. Genetic Basis of Type IV Collagen Disorders of the Kidney. Clin J Am Soc Nephrol. 2021;16:1101 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8425620/
137. BURGESON RE, LUNSTRUM GP, ROKOSOVA B, RIMBERG CS, ROSENBAUM LM, KEENE DR. The structure and function of type VII collagen. Ann N Y Acad Sci. 1990;580:32-43 Available at: https://pubmed.ncbi.nlm.nih.gov/2186694/
138. Barber RE, Kwan APL. Partial characterization of the C-terminal non-collagenous domain (NC1) of collagen type X. Biochem J. 1996;320(Pt 2):479-85 Available at: https://pubmed.ncbi.nlm.nih.gov/8973556/
139. Knupp C, Squire JM. Molecular packing in network-forming collagens. Adv Protein Chem. 2005;70:375-403 Available at: https://pubmed.ncbi.nlm.nih.gov/15837521/
140. Wu Y, Ge G. Complexity of type IV collagens: from network assembly to function. Biol Chem. 2019;400:565-74 Available at: https://pubmed.ncbi.nlm.nih.gov/30864416/
141. Al-Shaer A, Lyons A, Ishikawa Y, Hudson BG, Boudko SP, Forde NR. Sequence-dependent mechanics of collagen reflect its structural and functional organization. Biophys J. 2021;120:4013-28 Available at: https://pubmed.ncbi.nlm.nih.gov/34390685/
142. Calvo A, Moreno L, Moreno L. et al. Type XIX collagen: a promising biomarker from the basement membranes. Neural Regen Res. 2020;15:988-95 Available at: https://pubmed.ncbi.nlm.nih.gov/31823868/
143. Kassner A, Tiedemann K, Notbohm H. et al. Molecular structure and interaction of recombinant human type XVI collagen. J Mol Biol. 2004;339:835-53 Available at: https://pubmed.ncbi.nlm.nih.gov/15165854/
144. Franzke CW, Tasanen K, Schäcke H. et al. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. 2002;21:5026-35 Available at: https://pubmed.ncbi.nlm.nih.gov/12356719/
145. Khan A, Riaz R, Ashraf S, Akilimali A. Revolutionary breakthrough: FDA approves Vyjuvek, the first topical gene therapy for dystrophic epidermolysis bullosa. Ann Med Surg (Lond). 2023;85:6298-301 Available at: https://pubmed.ncbi.nlm.nih.gov/38098548/
146. Seibel MJ. Biochemical Markers of Bone Turnover Part I: Biochemistry and Variability. The Clinical biochemist Reviews / Australian Association of Clinical Biochemists. 2005;26:97 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC1320175/
147. Theocharis AD, Skandalis SS, Tzanakakis GN, Karamanos NK. Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 2010;277:3904-23 Available at: https://pubmed.ncbi.nlm.nih.gov/20840587/
148. Iozzo R V, Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11-55 Available at: https://pubmed.ncbi.nlm.nih.gov/25701227/
149. Heinz A. Elastic fibers during aging and disease. Ageing Res Rev. 2021;66:101255
150. Nivison-Smith L, Weiss A, Nivison-Smith L, Weiss A. Elastin Based Constructs. Regenerative Medicine and Tissue Engineering - Cells and Biomaterials. 2011 Available at: https://www.intechopen.com/chapters/19028
151. Cohen BE, Geronemus RG, McDaniel DH, Brauer JA. The Role of Elastic Fibers in Scar Formation and Treatment. Dermatol Surg. 2017;43(Suppl 1):S19-24 Available at: https://pubmed.ncbi.nlm.nih.gov/27399940/
152. Ozsvar J, Yang C, Cain SA, Baldock C, Tarakanova A, Weiss AS. Tropoelastin and Elastin Assembly. Front Bioeng Biotechnol. 2021 9. Available at: https://pubmed.ncbi.nlm.nih.gov/33718344/
153. Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239-52 Available at: https://pubmed.ncbi.nlm.nih.gov/20089974/
154. Schräder CU, Heinz A, Majovsky P. et al. Elastin is heterogeneously cross-linked. J Biol Chem. 2018;293:15107 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6166741/
155. Weisel JW. Structure of fibrin: impact on clot stability. Journal of Thrombosis and Haemostasis. 2007;5:116-24
156. Mariencheck MC, Davis EC, Zhang H. et al. Fibrillin-1 and fibrillin-2 show temporal and tissue-specific regulation of expression in developing elastic tissues. Connect Tissue Res. 1995;31:87-97 Available at: https://pubmed.ncbi.nlm.nih.gov/15612324/
157. Lemaire R, Bayle J, Mecham RP, Lafyatis R. Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J Biol Chem. 2007;282:800-8 Available at: https://pubmed.ncbi.nlm.nih.gov/17099216/
158. Boraldi F, Lofaro FD, Cossarizza A, Quaglino D. The 'Elastic Perspective' of SARS-CoV-2 Infection and the Role of Intrinsic and Extrinsic Factors. Int J Mol Sci. 2022 23. Available at: https://pubmed.ncbi.nlm.nih.gov/35163482/
159. Thomson J, Singh M, Eckersley A, Cain SA, Sherratt MJ, Baldock C. Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors. Semin Cell Dev Biol. 2019;89:109-17 Available at: https://pubmed.ncbi.nlm.nih.gov/30016650/
160. Poluzzi C, Casulli J, Goyal A, Mercer TJ, Neill T, Iozzo R V. Endorepellin Evokes Autophagy in Endothelial Cells. J Biol Chem. 2014;289:16114 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4047384/
161. Vindin HJ, Oliver BG, Weiss AS. Elastin in healthy and diseased lung. Curr Opin Biotechnol. 2022;74:15-20 Available at: https://pubmed.ncbi.nlm.nih.gov/34781101/
162. Krasniqi E, Ercolani C, Di Benedetto A. et al. DNA Damage Response in Early Breast Cancer: A Phase III Cohort in the Phobos Study. Cancers (Basel). 2024 16. Available at: https://pubmed.ncbi.nlm.nih.gov/39123356/
163. Calleja-Agius J, Brincat M, Borg M. Skin connective tissue and ageing. Best Pract Res Clin Obstet Gynaecol. 2013;27:727-40 Available at: https://pubmed.ncbi.nlm.nih.gov/23850161/
164. Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397-419 Available at: https://pubmed.ncbi.nlm.nih.gov/20690820/
165. Dalton CJ, Lemmon CA. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells. 2021;10:2443 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8471655/
166. Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861-3 Available at: https://pubmed.ncbi.nlm.nih.gov/12244123/
167. Sabatier L, Chen D, Fagotto-Kaufmann C. et al. Fibrillin assembly requires fibronectin. Mol Biol Cell. 2009;20:846-58 Available at: https://pubmed.ncbi.nlm.nih.gov/19037100/
168. Takahashi S, Leiss M, Moser M. et al. The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J Cell Biol. 2007;178:167-78 Available at: https://pubmed.ncbi.nlm.nih.gov/17591922/
169. Singh P, Schwarzbauer JE. Fibronectin and stem cell differentiation - lessons from chondrogenesis. J Cell Sci. 2012;125:3703-12 Available at: https://pubmed.ncbi.nlm.nih.gov/22976308/
170. Kumra H, Sabatier L, Hassan A. et al. Roles of fibronectin isoforms in neonatal vascular development and matrix integrity. PLoS Biol. 2018 16. Available at: https://pubmed.ncbi.nlm.nih.gov/30036393/
171. Wirth F, Lubosch A, Hamelmann S, Nakchbandi IA. Fibronectin and Its Receptors in Hematopoiesis. Cells. 2020 9. Available at: https://pubmed.ncbi.nlm.nih.gov/33353083/
172. Fish RJ, Neerman-Arbez M. Fibrinogen gene regulation. Thromb Haemost. 2012;108:419-26 Available at: https://pubmed.ncbi.nlm.nih.gov/22836683/
173. Weisel JW, Litvinov RI. Fibrin Formation, Structure and Properties. Subcell Biochem. 2017;82:405-56 Available at: https://pubmed.ncbi.nlm.nih.gov/28101869/
174. Tamura T, Arai S, Nagaya H, Mizuguchi J, Wada I. Stepwise assembly of fibrinogen is assisted by the endoplasmic reticulum lectin-chaperone system in HepG2 cells. PLoS One. 2013 8. Available at: https://pubmed.ncbi.nlm.nih.gov/24040290/
175. Low SWY, Connor TB, Kassem IS, Costakos DM, Chaurasia SS. Small Leucine-Rich Proteoglycans (SLRPs) in the Retina. Int J Mol Sci. 2021;22:7293 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8305803/
176. Hoppe B. Fibrinogen and factor XIII at the intersection of coagulation, fibrinolysis and inflammation. Thromb Haemost. 2014;112:649-58 Available at: https://pubmed.ncbi.nlm.nih.gov/25182841/
177. Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121:1712-9 Available at: https://pubmed.ncbi.nlm.nih.gov/23305734/
178. Cunningham MT, Brandt JT, Laposata M, Olson JD. Laboratory diagnosis of dysfibrinogenemia. Arch Pathol Lab Med. 2002;126:499-505 Available at: https://pubmed.ncbi.nlm.nih.gov/11900586/
179. Coller BS. Historical perspective and future directions in platelet research. J Thromb Haemost. 2011;9(Suppl 1):374-95 Available at: https://pubmed.ncbi.nlm.nih.gov/21781274/
180. Yanagisawa H, Davis EC. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int J Biochem Cell Biol. 2010;42:1084-93 Available at: https://pubmed.ncbi.nlm.nih.gov/20236620/
181. Argraves WS, Tran H, Burgess WH, Dickerson K. Fibulin is an extracellular matrix and plasma glycoprotein with repeated domain structure. J Cell Biol. 1990;111:3155-64 Available at: https://pubmed.ncbi.nlm.nih.gov/2269669/
182. Kobayashi N, Kostka G, Garbe JHO. et al. A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J Biol Chem. 2007;282:11805-16 Available at: https://pubmed.ncbi.nlm.nih.gov/17324935/
183. Zhang H, Hui D, Fu X. Roles of Fibulin-2 in Carcinogenesis. Med Sci Monit. 2020;26:e918099-1 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6977632/
184. PAN T -C, KLUGE M, ZHANG R -Z, MAYER U, TIMPL R, CHU M -L. Sequence of extracellular mouse protein BM-90/fibulin and its calcium-dependent binding to other basement-membrane ligands. Eur J Biochem. 1993;215:733-40 Available at: https://pubmed.ncbi.nlm.nih.gov/8354280/
185. Livingstone I, Uversky VN, Furniss D, Wiberg A. The Pathophysiological Significance of Fibulin-3. Biomolecules. 2020;10:1-24 Available at: https://pubmed.ncbi.nlm.nih.gov/32911658/
186. Chen Q, Zhang T, Roshetsky JF. et al. Fibulin-4 regulates expression of the tropoelastin gene and consequent elastic-fibre formation by human fibroblasts. Biochem J. 2009;423:79-89 Available at: https://pubmed.ncbi.nlm.nih.gov/19627254/
187. Gauster M, Berghold VM, Moser G, Orendi K, Siwetz M, Huppertz B. Fibulin-5 expression in the human placenta. Histochem Cell Biol. 2011;135:203-13 Available at: https://pubmed.ncbi.nlm.nih.gov/21290250/
188. Raja E, Changarathil G, Oinam L. et al. The extracellular matrix fibulin 7 maintains epidermal stem cell heterogeneity during skin aging. EMBO Rep. 2022 23. Available at: https://pubmed.ncbi.nlm.nih.gov/36278510/
189. Zheng Q, Davis EC, Richardson JA. et al. Molecular analysis of fibulin-5 function during de novo synthesis of elastic fibers. Mol Cell Biol. 2007;27:1083-95 Available at: https://pubmed.ncbi.nlm.nih.gov/17130242/
190. De Vega S, Iwamoto T, Yamada Y. Fibulins: multiple roles in matrix structures and tissue functions. Cell Mol Life Sci. 2009;66:1890-902 Available at: https://pubmed.ncbi.nlm.nih.gov/19189051/
191. Sakai LY, Keene DR. Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biol. 2019;80:6-13 Available at: https://pubmed.ncbi.nlm.nih.gov/30219651/
192. van Loon K, Yemelyanenko-Lyalenko J, Margadant C, Griffioen AW, Huijbers EJM. Role of fibrillin-2 in the control of TGF-β activation in tumor angiogenesis and connective tissue disorders. Biochim Biophys Acta Rev Cancer. 2020 1873. Available at: https://pubmed.ncbi.nlm.nih.gov/32119940/
193. Kielty CM, Sherratt MJ, Marson A, Baldock C. Fibrillin microfibrils. Adv Protein Chem. 2005;70:405-36 Available at: https://pubmed.ncbi.nlm.nih.gov/15837522/
194. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene. 2016;591:279-91 Available at: https://pubmed.ncbi.nlm.nih.gov/27437668/
195. Alonso F, Dong Y, Li L. et al. Fibrillin-1 regulates endothelial sprouting during angiogenesis. Proc Natl Acad Sci U S A. 2023 120. Available at: https://pubmed.ncbi.nlm.nih.gov/37252964/
196. Hubmacher D, Sabatier L, Annis DS, Mosher DF, Reinhardt DP. Homocysteine modifies structural and functional properties of fibronectin and interferes with the fibronectin-fibrillin-1 interaction. Biochemistry. 2011;50:5322-32 Available at: https://pubmed.ncbi.nlm.nih.gov/21561146/
197. Summers KM, Bush SJ, Davis MR, Hume DA, Keshvari S, West JA. Fibrillin-1 and asprosin, novel players in metabolic syndrome. Mol Genet Metab. 2023 138. Available at: https://pubmed.ncbi.nlm.nih.gov/36630758/
198. Hohenester E. Structural biology of laminins. Essays Biochem. 2019;63:285-95 Available at: https://pubmed.ncbi.nlm.nih.gov/31092689/
199. Durbeej M. Laminins. Cell Tissue Res. 2010;339:259-68 Available at: https://pubmed.ncbi.nlm.nih.gov/19693542/
200. Yurchenco PD, Kulczyk AW. Polymerizing laminins in development, health, and disease. J Biol Chem. 2024 300. Available at: https://pubmed.ncbi.nlm.nih.gov/38825010/
201. Goddi A, Schroedl L, Brey EM, Cohen RN. Laminins in metabolic tissues. Metabolism. 2021 120
202. Di Russo J, Hannocks MJ, Luik AL. et al. Vascular laminins in physiology and pathology. Matrix Biol. 2017;57-58:140-8
203. Miner JH. Laminins and their roles in mammals. Microsc Res Tech. 2008;71:349-56 Available at: https://pubmed.ncbi.nlm.nih.gov/18219670/
204. Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell Adh Migr. 2013;7:56-63
205. Nonnast E, Mira E, Mañes S. Biomechanical properties of laminins and their impact on cancer progression. Biochim Biophys Acta Rev Cancer. 2024;1879:189181 Available at: https://pubmed.ncbi.nlm.nih.gov/39299492/
206. Dempsey CE, Bigotti MG, Adams JC, Brancaccio A. Analysis of α-Dystroglycan/LG Domain Binding Modes: Investigating Protein Motifs That Regulate the Affinity of Isolated LG Domains. Front Mol Biosci. 2019 6. Available at: https://pubmed.ncbi.nlm.nih.gov/30984766/
207. Domogatskaya A, Rodin S, Tryggvason K. Functional diversity of laminins. Annu Rev Cell Dev Biol. 2012;28:523-53 Available at: https://pubmed.ncbi.nlm.nih.gov/23057746/
208. Goldoni M, Torres B, Pettinato M. et al. A Missense Variant Affecting the N-Terminal Domain of the Laminin-332 β3 Chain Results in a Distinct Form of Junctional Epidermolysis Bullosa with Altered Granulation Tissue Response and No New Blistering: A Second Family Report. Pediatr Dermatol. 2025 42. Available at: https://pubmed.ncbi.nlm.nih.gov/39443834/
209. Niessen CM, Hogervorst F, Jaspars LH. et al. The α6β4 integrin is a receptor for both laminin and kalinin. Exp Cell Res. 1994;211:360-7
210. Silverman LD, Saadia M, Ishal JS. et al. Hydroxyapatite growth inhibition by osteopontin hexapeptide sequences. Langmuir. 2010;26:9899-904 Available at: https://pubmed.ncbi.nlm.nih.gov/20491496/
211. Kurzbach D, Platzer G, Schwarz TC, Henen MA, Konrat R, Hinderberger D. Cooperative unfolding of compact conformations of the intrinsically disordered protein osteopontin. Biochemistry. 2013;52:5167-75 Available at: https://pubmed.ncbi.nlm.nih.gov/23848319/
212. Boskey AL, Villarreal-Ramirez E. Intrinsically disordered proteins and biomineralization. Matrix Biol. 2016;52-54:43-59 Available at: https://pubmed.ncbi.nlm.nih.gov/26807759/
213. Nikel O, Laurencin D, McCallum SA, Gundberg CM, Vashishth D. NMR investigation of the role of osteocalcin and osteopontin at the organic-inorganic interface in bone. Langmuir. 2013;29:13873-82 Available at: https://pubmed.ncbi.nlm.nih.gov/24128197/
214. Whyte MP, Amalnath SD, McAlister WH. et al. Hypophosphatemic osteosclerosis, hyperostosis, and enthesopathy associated with novel homozygous mutations of DMP1 encoding dentin matrix protein 1 and SPP1 encoding osteopontin: The first digenic SIBLING protein osteopathy? Bone. 2020 132. Available at: https://pubmed.ncbi.nlm.nih.gov/31843680/
215. Boukpessi T, Hoac B, Coyac BR. et al. Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone. 2017;95:151-61 Available at: https://pubmed.ncbi.nlm.nih.gov/27884786/
216. Yuan Q, Jiang Y, Zhao X. et al. Increased Osteopontin Contributes to Inhibition of Bone Mineralization in FGF23-Deficient Mice. Journal of Bone and Mineral Research. 2014;29:693-704 Available at: https://dx.doi.org/10.1002/jbmr.2079
217. Yadav MC, Huesa C, Narisawa S. et al. Ablation of osteopontin improves the skeletal phenotype of phospho1(-/-) mice. J Bone Miner Res. 2014;29:2369-81 Available at: https://pubmed.ncbi.nlm.nih.gov/24825455/
218. Sroga GE, Vashishth D. Phosphorylation of Extracellular Bone Matrix Proteins and Its Contribution to Bone Fragility. J Bone Miner Res. 2018;33:2214-29 Available at: https://pubmed.ncbi.nlm.nih.gov/30001467/
219. Durkin ME, Chakravarti S, Bartos BB, Liu SH, Friedman RL, Chung AE. Amino acid sequence and domain structure of entactin. Homology with epidermal growth factor precursor and low density lipoprotein receptor. J Cell Biol. 1988;107:2749-56 Available at: https://pubmed.ncbi.nlm.nih.gov/3264556/
220. Fidler AL, Darris CE, Chetyrkin S V. et al. Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues. Elife. 2017 6. Available at: https://pubmed.ncbi.nlm.nih.gov/28418331/
221. Bechtel M, Keller M V, Bloch W. et al. Different domains in nidogen-1 and nidogen-2 drive basement membrane formation in skin organotypic cocultures. FASEB J. 2012;26:3637-48 Available at: https://pubmed.ncbi.nlm.nih.gov/22623588/
222. Wolfstetter G, Dahlitz I, Pfeifer K. et al. Characterization of Drosophila Nidogen/ entactin reveals roles in basement membrane stability, barrier function and nervous system patterning. Development. 2019 146. Available at: https://pubmed.ncbi.nlm.nih.gov/30567930/
223. Ijezie EC, O'Dowd JM, Kuan MI, Faeth AR, Fortunato EA. HCMV Infection Reduces Nidogen-1 Expression, Contributing to Impaired Neural Rosette Development in Brain Organoids. J Virol. 2023 97. Available at: https://pubmed.ncbi.nlm.nih.gov/37125912/
224. Dong L, Chen Y, Lewis M. et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Invest. 2002;82:1617-30 Available at: https://pubmed.ncbi.nlm.nih.gov/12480912/
225. McNiven V, Ito YA, Hartley T, Kernohan K, Miller E, Armour CM. NID1 variant associated with occipital cephaloceles in a family expressing a spectrum of phenotypes. Am J Med Genet A. 2019;179:837-41 Available at: https://pubmed.ncbi.nlm.nih.gov/30773799/
226. Darbro BW, Mahajan VB, Gakhar L. et al. Mutations in extracellular matrix genes NID1 and LAMC1 cause autosomal dominant Dandy-Walker malformation and occipital cephaloceles. Hum Mutat. 2013;34:1075-9 Available at: https://pubmed.ncbi.nlm.nih.gov/23674478/
227. Dietvorst S, Devriendt K, Lambert J. et al. NID1-related autosomal dominant Dandy-Walker malformation with occipital cephalocele in three generations. Eur J Med Genet. 2023 66. Available at: https://pubmed.ncbi.nlm.nih.gov/36702440/
228. Nguyen M, Panitch A. Proteoglycans and proteoglycan mimetics for tissue engineering. Am J Physiol Cell Physiol. 2022;322:C754 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8993519/
229. Schaefer L. Proteoglycans, key regulators of cell-matrix dynamics. Matrix Biol. 2014;35:1-2 Available at: https://pubmed.ncbi.nlm.nih.gov/24871042/
230. Listik E, Azevedo Marques Gaschler J, Matias M, Neuppmann Feres MF, Toma L, Raphaelli Nahás-Scocate AC. Proteoglycans and dental biology: the first review. Carbohydr Polym. 2019 225. Available at: https://pubmed.ncbi.nlm.nih.gov/31521317/
231. Chen F, Lei L, Chen S. et al. Serglycin secreted by late-stage nucleus pulposus cells is a biomarker of intervertebral disc degeneration. Nat Commun. 2024 15. Available at: https://pubmed.ncbi.nlm.nih.gov/38167807/
232. Song L, Oseid DE, Wells EA, Coaston T, Robinson AS. Heparan Sulfate Proteoglycans (HSPGs) Serve as the Mediator Between Monomeric Tau and Its Subsequent Intracellular ERK1/2 Pathway Activation. J Mol Neurosci. 2022;72:772-91 Available at: https://pubmed.ncbi.nlm.nih.gov/35040015/
233. Fawcett JW, Fyhn M, Jendelova P, Kwok JCF, Ruzicka J, Sorg BA. The extracellular matrix and perineuronal nets in memory. Mol Psychiatry. 2022;27:3192-203 Available at: https://pubmed.ncbi.nlm.nih.gov/35760878/
234. Persson A, Nikpour M, Vorontsov E, Nilsson J, Larson G. Domain Mapping of Chondroitin/Dermatan Sulfate Glycosaminoglycans Enables Structural Characterization of Proteoglycans. Mol Cell Proteomics. 2021 20. Available at: https://pubmed.ncbi.nlm.nih.gov/33757834/
235. Gandley RE, McLaughlin MK, Koob TJ, Little SA, McGuffee LJ. Contribution of chondroitin-dermatan sulfate-containing proteoglycans to the function of rat mesenteric arteries. Am J Physiol. 1997 273. Available at: https://pubmed.ncbi.nlm.nih.gov/9277515/
236. Xu X, Ha P, Yen E, Li C, Zheng Z. Small Leucine-Rich Proteoglycans in Tendon Wound Healing. Adv Wound Care (New Rochelle). 2022;11:202-14 Available at: https://pubmed.ncbi.nlm.nih.gov/34978952/
237. Iozzo R V, Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J. 2010;277:3864-75 Available at: https://pubmed.ncbi.nlm.nih.gov/20840584/
238. Zhang X, Zhao Y, Liu L, He Y. Syndecan-1: A Novel Diagnostic and Therapeutic Target in Liver Diseases. Curr Drug Targets. 2023;24:1155-65 Available at: https://pubmed.ncbi.nlm.nih.gov/37957867/
239. Reszegi A, Tátrai P, Regs E, Kovalszky I, Baghy K. Syndecan-1 in liver pathophysiology. Am J Physiol Cell Physiol. 2022;323:C289-94 Available at: https://pubmed.ncbi.nlm.nih.gov/35704700/
240. Handra-Luca A. Syndecan-1 in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1272:39-53 Available at: https://pubmed.ncbi.nlm.nih.gov/32845501/
241. Yang Z, Chen S, Ying H, Yao W. Targeting syndecan-1: new opportunities in cancer therapy. Am J Physiol Cell Physiol. 2022;323:C29-45 Available at: https://pubmed.ncbi.nlm.nih.gov/35584326/
242. Mytilinaiou M, Nikitovic D, Berdiaki A. et al. Emerging roles of syndecan 2 in epithelial and mesenchymal cancer progression. IUBMB Life. 2017;69:824-33 Available at: https://pubmed.ncbi.nlm.nih.gov/28940845/
243. Szatmári T, Mundt F, Kumar-Singh A. et al. Molecular targets and signaling pathways regulated by nuclear translocation of syndecan-1. BMC Cell Biol. 2017 18. Available at: https://pubmed.ncbi.nlm.nih.gov/29216821/
244. Kamimura K, Maeda N. Glypicans and Heparan Sulfate in Synaptic Development, Neural Plasticity, and Neurological Disorders. Front Neural Circuits. 2021 15. Available at: https://pubmed.ncbi.nlm.nih.gov/33679334/
245. Filmus J, Capurro M. The role of glypicans in Hedgehog signaling. Matrix Biol. 2014;35:248-52 Available at: https://pubmed.ncbi.nlm.nih.gov/24412155/
246. Waghmare I, Page-McCaw A. Regulation of Wnt distribution and function by Drosophila glypicans. J Cell Sci. 2022 135. Available at: https://pubmed.ncbi.nlm.nih.gov/35112708/
247. Capurro M, Shi W, Izumikawa T, Kitagawa H, Filmus J. Processing by convertases is required for glypican-3-induced inhibition of Hedgehog signaling. J Biol Chem. 2015;290:7576-85 Available at: https://pubmed.ncbi.nlm.nih.gov/25653284/
248. Traister A, Shi W, Filmus J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem J. 2008;410:503-11 Available at: https://pubmed.ncbi.nlm.nih.gov/17967162/
249. Vaisfeld A, Neri G. Simpson-Golabi-Behmel syndrome. Am J Med Genet C Semin Med Genet. 2024 196. Available at: https://pubmed.ncbi.nlm.nih.gov/38766979/
250. Vaisfeld A, Pomponi MG, Pietrobono R, Tabolacci E, Neri G. Simpson-Golabi-Behmel syndrome in a female: A case report and an unsolved issue. Am J Med Genet A. 2017;173:285-8 Available at: https://pubmed.ncbi.nlm.nih.gov/27739211/
251. Li N, Gao W, Zhang YF, Ho M. Glypicans as Cancer Therapeutic Targets. Trends Cancer. 2018;4:741-54 Available at: https://pubmed.ncbi.nlm.nih.gov/30352677/
252. Bilandzic M, Stenvers KL. Betaglycan: a multifunctional accessory. Mol Cell Endocrinol. 2011;339:180-9 Available at: https://pubmed.ncbi.nlm.nih.gov/21550381/
253. Ponce-Castañeda MV, Esparza-López J, Vilchis-Landeros MM, Mendoza R. V, López-Casillas F. Murine betaglycan primary structure, expression and glycosaminoglycan attachment sites. Biochim Biophys Acta. 1998;1384:189-96 Available at: https://pubmed.ncbi.nlm.nih.gov/9659379/
254. Kim SK, Henen MA, Hinck AP. Structural biology of betaglycan and endoglin, membrane-bound co-receptors of the TGF-beta family. Exp Biol Med (Maywood). 2019;244:1547-58 Available at: https://pubmed.ncbi.nlm.nih.gov/31601110/
255. Cook LM, Frieling JS, Nerlakanti N. et al. Betaglycan drives the mesenchymal stromal cell osteogenic program and prostate cancer-induced osteogenesis. Oncogene. 2019;38:6959-69 Available at: https://pubmed.ncbi.nlm.nih.gov/31409900/
256. Trout AL, Rutkai I, Biose IJ, Bix GJ. Review of Alterations in Perlecan-Associated Vascular Risk Factors in Dementia. Int J Mol Sci. 2020 21. Available at: https://pubmed.ncbi.nlm.nih.gov/31968632/
257. Lavorgna TR, Gressett TE, Chastain WH, Bix GJ. Perlecan: a review of its role in neurologic and musculoskeletal disease. Front Physiol. 2023 14. Available at: https://pubmed.ncbi.nlm.nih.gov/37324385/
258. Melrose J. Perlecan, a modular instructive proteoglycan with diverse functional properties. Int J Biochem Cell Biol. 2020 128. Available at: https://pubmed.ncbi.nlm.nih.gov/32947020/
259. Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D Complex: A Structural Classification of Protein Complexes. PLoS Comput Biol. 2006;2:e155 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC1636673/
260. Nia HT, Ortiz C, Grodzinsky A. Aggrecan: Approaches to Study Biophysical and Biomechanical Properties. Methods Mol Biol. 2022;2303:209-26 Available at: https://pubmed.ncbi.nlm.nih.gov/34626381/
261. Koch CD, Lee CM, Apte SS. Aggrecan in Cardiovascular Development and Disease. J Histochem Cytochem. 2020;68:777-95 Available at: https://pubmed.ncbi.nlm.nih.gov/32870742/
262. Watanabe H. Aggrecan and versican: two brothers close or apart. Am J Physiol Cell Physiol. 2022;322:C967-76 Available at: https://pubmed.ncbi.nlm.nih.gov/35385326/
263. Liu Y, Huang C, Bai M, Pi C, Zhang D, Xie J. The roles of Runx1 in skeletal development and osteoarthritis: A concise review. Heliyon. 2022 8. Available at: https://pubmed.ncbi.nlm.nih.gov/36636224/
264. Morawski M, Brückner G, Arendt T, Matthews RT. Aggrecan: Beyond cartilage and into the brain. Int J Biochem Cell Biol. 2012;44:690-3 Available at: https://pubmed.ncbi.nlm.nih.gov/22297263/
265. Islam S, Watanabe H. Versican: A Dynamic Regulator of the Extracellular Matrix. J Histochem Cytochem. 2020;68:763-75 Available at: https://pubmed.ncbi.nlm.nih.gov/33131383/
266. Zhang Z wei, Zhang J peng, Zhou T ting, Feng W hua, Jiao B hua. Does the expression of versican isoforms contribute to the pathogenesis of neurodegenerative diseases? Arch Med Res. 2011;42:258-60 Available at: https://pubmed.ncbi.nlm.nih.gov/21722824/
267. Kischel P, Waltregny D, Dumont B. et al. Versican overexpression in human breast cancer lesions: known and new isoforms for stromal tumor targeting. Int J Cancer. 2010;126:640-50 Available at: https://pubmed.ncbi.nlm.nih.gov/19662655/
268. Zhou M, Liu YWY, He YH. et al. FOXO1 reshapes neutrophils to aggravate acute brain damage and promote late depression after traumatic brain injury. Mil Med Res. 2024 11. Available at: https://pubmed.ncbi.nlm.nih.gov/38556884/
269. Feng J, Li Y, Li Y. et al. Versican Promotes Cardiomyocyte Proliferation and Cardiac Repair. Circulation. 2024;149:1004-15 Available at: https://pubmed.ncbi.nlm.nih.gov/37886839/
270. Ween MP, Oehler MK, Ricciardelli C. Role of versican, hyaluronan and CD44 in ovarian cancer metastasis. Int J Mol Sci. 2011;12:1009-29 Available at: https://pubmed.ncbi.nlm.nih.gov/21541039/
271. Wight TN, Kang I, Evanko SP. et al. Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front Immunol. 2020 11. Available at: https://pubmed.ncbi.nlm.nih.gov/32265939/
272. Nikitovic D, Berdiaki K, Chalkiadaki G, Karamanos N, Tzanakakis G. The role of SLRP-proteoglycans in osteosarcoma pathogenesis. Connect Tissue Res. 2008;49:235-8 Available at: https://pubmed.ncbi.nlm.nih.gov/18661350/
273. Zappia J, Joiret M, Sanchez C. et al. From Translation to Protein Degradation as Mechanisms for Regulating Biological Functions: A Review on the SLRP Family in Skeletal Tissues. Biomolecules. 2020 10. Available at: https://pubmed.ncbi.nlm.nih.gov/31947880/
274. Xie C, Mondal DK, Ulas M, Neill T, Iozzo R V. Oncosuppressive roles of decorin through regulation of multiple receptors and diverse signaling pathways. Am J Physiol Cell Physiol. 2022;322:C554-66 Available at: https://pubmed.ncbi.nlm.nih.gov/35171698/
275. Neill T, Schaefer L, Iozzo R V. Decorin: a guardian from the matrix. Am J Pathol. 2012;181:380-7 Available at: https://pubmed.ncbi.nlm.nih.gov/22735579/
276. Nitahara-Kasahara Y, Posadas-Herrera G, Mizumoto S. et al. Myopathy Associated With Dermatan Sulfate-Deficient Decorin and Myostatin in Musculocontractural Ehlers-Danlos Syndrome: A Mouse Model Investigation. Front Cell Dev Biol. 2021 9. Available at: https://pubmed.ncbi.nlm.nih.gov/34708033/
277. Baghy K, Reszegi A, Tátrai P, Kovalszky I. Decorin in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1272:17-38 Available at: https://pubmed.ncbi.nlm.nih.gov/32845500/
278. Dong Y, Zhong J, Dong L. The Role of Decorin in Autoimmune and Inflammatory Diseases. J Immunol Res. 2022. 2022 Available at: https://pubmed.ncbi.nlm.nih.gov/36033387/
279. Miguez PA. Evidence of biglycan structure-function in bone homeostasis and aging. Connect Tissue Res. 2020;61:19-33 Available at: https://pubmed.ncbi.nlm.nih.gov/31597498/
280. Lall SP, Alsafwani ZW, Batra SK, Seshacharyulu P. ASPORIN: A root of the matter in tumors and their host environment. Biochim Biophys Acta Rev Cancer. 2024 1879. Available at: https://pubmed.ncbi.nlm.nih.gov/38008263/
281. Cheng X, Liu Z, Liang W. et al. ECM2, a prognostic biomarker for lower grade glioma, serves as a potential novel target for immunotherapy. Int J Biochem Cell Biol. 2023 158. Available at: https://pubmed.ncbi.nlm.nih.gov/36997057/
282. Heinegård D. Fell-Muir Lecture: Proteoglycans and more-from molecules to biology. Int J Exp Pathol. 2009;90:575-86 Available at: https://pubmed.ncbi.nlm.nih.gov/19958398/
283. Weyers A, Yang B, Solakyildirim K. et al. Isolation of bovine corneal keratan sulfate and its growth factor and morphogen binding. FEBS J. 2013;280:2285-93 Available at: https://pubmed.ncbi.nlm.nih.gov/23402351/
284. Ho LTY, Harris AM, Tanioka H. et al. A comparison of glycosaminoglycan distributions, keratan sulphate sulphation patterns and collagen fibril architecture from central to peripheral regions of the bovine cornea. Matrix Biology. 2014;38:59 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4199143/
285. Nakano T, Scott PG. Proteoglycans of the Articular Disc of the Bovine Temporomandibular Joint: I. High Molecular Weight Chondroitin Sulphate Proteoglycan. Matrix. 1989;9:277-83
286. Tillgren V, Önnerfjord P, Haglund L, Heinegård D. The tyrosine sulfate-rich domains of the LRR proteins fibromodulin and osteoadherin bind motifs of basic clusters in a variety of heparin-binding proteins, including bioactive factors. J Biol Chem. 2009;284:28543-53 Available at: https://pubmed.ncbi.nlm.nih.gov/19700767/
287. Kalamajski S, Oldberg Å. Homologous sequence in lumican and fibromodulin leucine-rich repeat 5-7 competes for collagen binding. J Biol Chem. 2009;284:534-9 Available at: https://pubmed.ncbi.nlm.nih.gov/19008226/
288. Sjöberg A, Önnerfjord P, Mörgelin M, Heinegård D, Blom AM. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem. 2005;280:32301-8 Available at: https://pubmed.ncbi.nlm.nih.gov/16046396/
289. Li W, Vergnes JP, Cornuet PK, Hassell JR. cDNA clone to chick corneal chondroitin/dermatan sulfate proteoglycan reveals identity to decorin. Arch Biochem Biophys. 1992;296:190-7
290. Nikitovic D, Papoutsidakis A, Karamanos NK, Tzanakakis GN. Lumican affects tumor cell functions, tumor-ECM interactions, angiogenesis and inflammatory response. Matrix Biol. 2014;35:206-14 Available at: https://pubmed.ncbi.nlm.nih.gov/24060754/
291. Hayashi Y, Call MK, Chikama TI. et al. Lumican is required for neutrophil extravasation following corneal injury and wound healing. J Cell Sci. 2010;123:2987-95 Available at: https://pubmed.ncbi.nlm.nih.gov/20699360/
292. Bengtsson E, Neame PJ, Heinegård D, Sommarin Y. The primary structure of a basic leucine-rich repeat protein, PRELP, found in connective tissues. J Biol Chem. 1995;270:25639-44 Available at: https://pubmed.ncbi.nlm.nih.gov/7592739/
293. Rucci N, Capulli M, Ventura L. et al. Proline/arginine-rich end leucine-rich repeat protein N-terminus is a novel osteoclast antagonist that counteracts bone loss. J Bone Miner Res. 2013;28:1912-24 Available at: https://pubmed.ncbi.nlm.nih.gov/23559035/
294. Happonen KE, Fürst CM, Saxne T, Heinegård D, Blom AM. PRELP protein inhibits the formation of the complement membrane attack complex. J Biol Chem. 2012;287:8092-100 Available at: https://pubmed.ncbi.nlm.nih.gov/22267731/
295. Birke MT, Lipo E, Adhi M, Birke K, Kumar-Singh R. AAV-mediated expression of human PRELP inhibits complement activation, choroidal neovascularization and deposition of membrane attack complex in mice. Gene Ther. 2014;21:507-13 Available at: https://pubmed.ncbi.nlm.nih.gov/24670995/
296. Corpuz LM, Funderburgh JL, Funderburgh ML, Bottomley GS, Prakash S, Conrad GW. Molecular cloning and tissue distribution of keratocan. Bovine corneal keratan sulfate proteoglycan 37A. J Biol Chem. 1996;271:9759-63 Available at: https://pubmed.ncbi.nlm.nih.gov/8621655/
297. Liu CY, Birk DE, Hassell JR, Kane B, Kao WWY. Keratocan-deficient mice display alterations in corneal structure. J Biol Chem. 2003;278:21672-7 Available at: https://pubmed.ncbi.nlm.nih.gov/12665512/
298. Carlson EC, Sun Y, Auletta J. et al. Regulation of corneal inflammation by neutrophil-dependent cleavage of keratan sulfate proteoglycans as a model for breakdown of the chemokine gradient. J Leukoc Biol. 2010;88:517-22 Available at: https://pubmed.ncbi.nlm.nih.gov/20495072/
299. Wendel M, Sommarin Y, Heinegård D. Bone matrix proteins: isolation and characterization of a novel cell-binding keratan sulfate proteoglycan (osteoadherin) from bovine bone. J Cell Biol. 1998;141:839-47 Available at: https://pubmed.ncbi.nlm.nih.gov/9566981/
300. Sugars R V, Olsson ML, Marchner S, Hultenby K, Wendel M. The glycosylation profile of osteoadherin alters during endochondral bone formation. Bone. 2013;53:459-67 Available at: https://pubmed.ncbi.nlm.nih.gov/23337037/
301. Nikdin H, Olsson ML, Hultenby K, Sugars R V. Osteoadherin Accumulates in the Predentin towards the Mineralization Front in the Developing Tooth. PLoS One. 2012;7:e31525 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3280325/
302. Mierke CT. Extracellular Matrix Cues Regulate Mechanosensing and Mechanotransduction of Cancer Cells. Cells. 2024;13:96 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10777970/
303. Johnson HJ, Rosenberg L, Choi HU, Garza S, Höök M, Neame PJ. Characterization of epiphycan, a small proteoglycan with a leucine-rich repeat core protein. J Biol Chem. 1997;272:18709-17 Available at: https://pubmed.ncbi.nlm.nih.gov/9228042/
304. Kurita K, Shinomura T, Ujita M. et al. Occurrence of PG-Lb, a leucine-rich small chondroitin/dermatan sulphate proteoglycan in mammalian epiphyseal cartilage: Molecular cloning and sequence analysis of the mouse cDNA. Biochemical Journal. 1996;318:909-14
305. Nuka S, Zhou W, Henry SP. et al. Phenotypic characterization of epiphycan-deficient and epiphycan/biglycan double-deficient mice. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2009;18:88 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3013283/
306. Brachvogel B, Zaucke F, Dave K. et al. Comparative proteomic analysis of normal and collagen IX null mouse cartilage reveals altered extracellular matrix composition and novel components of the collagen IX interactome. J Biol Chem. 2013;288:13481-92 Available at: https://pubmed.ncbi.nlm.nih.gov/23530037/
307. Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res. 2014;114:872-88 Available at: https://pubmed.ncbi.nlm.nih.gov/24577967/
308. Petretto E, Sarwar R, Grieve I. et al. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nat Genet. 2008;40:546-52 Available at: https://pubmed.ncbi.nlm.nih.gov/18443592/
309. Tanaka KI, Matsumoto E, Higashimaki Y. et al. Role of osteoglycin in the linkage between muscle and bone. J Biol Chem. 2012;287:11616-28 Available at: https://pubmed.ncbi.nlm.nih.gov/22351757/
310. Barascuk N, Vassiliadis E, Zheng Q. et al. Levels of Circulating MMCN-151, a Degradation Product of Mimecan, Reflect Pathological Extracellular Matrix Remodeling in Apolipoprotein E Knockout Mice. Biomark Insights. 2011;6:97-106 Available at: https://pubmed.ncbi.nlm.nih.gov/22084568/
311. Cheng JM, Akkerhuis KM, Meilhac O. et al. Circulating osteoglycin and NGAL/MMP9 complex concentrations predict 1-year major adverse cardiovascular events after coronary angiography. Arterioscler Thromb Vasc Biol. 2014;34:1078-84 Available at: https://pubmed.ncbi.nlm.nih.gov/24651681/
312. Bengtsson E, Neame PJ, Heinegård D, Sommarin Y. The primary structure of a basic leucine-rich repeat protein, PRELP, found in connective tissues. Journal of Biological Chemistry. 1995;270:25639-44
313. Bech-Hansen NT, Naylor MJ, Maybaum TA. et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000;26:319-23 Available at: https://pubmed.ncbi.nlm.nih.gov/11062471/
314. Haglund L, Tillgren V, Addis L, Wenglén C, Recklies A, Heinegård D. Identification and characterization of the integrin alpha2beta1 binding motif in chondroadherin mediating cell attachment. J Biol Chem. 2011;286:3925-34 Available at: https://pubmed.ncbi.nlm.nih.gov/21127050/
315. Haglund L, Tillgren V, Önnerfjord P, Heinegård D. The C-terminal peptide of chondroadherin modulates cellular activity by selectively binding to heparan sulfate chains. J Biol Chem. 2013;288:995-1008 Available at: https://pubmed.ncbi.nlm.nih.gov/23172228/
316. Hessle L, Stordalen GA, Wenglén C. et al. The skeletal phenotype of chondroadherin deficient mice. PLoS One. 2013 8. Available at: https://pubmed.ncbi.nlm.nih.gov/23755099/
317. Pearring JN, Bojang P, Shen Y. et al. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J Neurosci. 2011;31:10060-6 Available at: https://pubmed.ncbi.nlm.nih.gov/21734298/
318. Audo I, Kohl S, Leroy BP. et al. TRPM1 Is Mutated in Patients with Autosomal-Recessive Complete Congenital Stationary Night Blindness. Am J Hum Genet. 2009;85:720-9
319. Ross MD, Bruggeman LA, Hanss B. et al. Podocan, a novel small leucine-rich repeat protein expressed in the sclerotic glomerular lesion of experimental HIV-associated nephropathy. J Biol Chem. 2003;278:33248-55 Available at: https://pubmed.ncbi.nlm.nih.gov/12796502/
320. Mochida Y, Kaku M, Yoshida K, Katafuchi M, Atsawasuwan P, Yamauchi M. Podocan-like Protein: A novel small leucine-rich repeat matrix protein in bone. Biochem Biophys Res Commun. 2011;410:333 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3159388/
321. Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048-62 Available at: https://pubmed.ncbi.nlm.nih.gov/20551380/
322. Hutter R, Huang L, Speidl WS. et al. Novel small leucine-rich repeat protein podocan is a negative regulator of migration and proliferation of smooth muscle cells, modulates neointima formation, and is expressed in human atheroma. Circulation. 2013;128:2351-63 Available at: https://pubmed.ncbi.nlm.nih.gov/24043300/
323. Kapoor C, Vaidya S, Wadhwan V, Hitesh, Kaur G, Pathak A. Seesaw of matrix metalloproteinases (MMPs). J Cancer Res Ther. 2016;12:28-35 Available at: https://pubmed.ncbi.nlm.nih.gov/27072206/
324. Fallata AM, Wyatt RA, Levesque JM, Dufour A, Overall CM, Crawford BD. Intracellular Localization in Zebrafish Muscle and Conserved Sequence Features Suggest Roles for Gelatinase A Moonlighting in Sarcomere Maintenance. Biomedicines. 2019 7. Available at: https://pubmed.ncbi.nlm.nih.gov/31795436/
325. Ali MAM, Chow AK, Kandasamy AD. et al. Mechanisms of cytosolic targeting of matrix metalloproteinase-2. J Cell Physiol. 2012;227:3397-404 Available at: https://pubmed.ncbi.nlm.nih.gov/22212960/
326. Fischer T, Senn N, Riedl R. Design and Structural Evolution of Matrix Metalloproteinase Inhibitors. Chemistry. 2019;25:7960-80 Available at: https://pubmed.ncbi.nlm.nih.gov/30720221/
327. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161-74 Available at: https://pubmed.ncbi.nlm.nih.gov/11990853/
328. Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463-516 Available at: https://pubmed.ncbi.nlm.nih.gov/11687497/
329. Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44-46:247-54 Available at: https://pubmed.ncbi.nlm.nih.gov/25805621/
330. Chung L, Dinakarpandian D, Yoshida N. et al. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 2004;23:3020-30 Available at: https://pubmed.ncbi.nlm.nih.gov/15257288/
331. Turner NA, Ho S, Warburton P, O'Regan DJ, Porter KE. Smooth muscle cells cultured from human saphenous vein exhibit increased proliferation, invasion, and mitogen-activated protein kinase activation in vitro compared with paired internal mammary artery cells. J Vasc Surg. 2007;45:1022-8 Available at: https://pubmed.ncbi.nlm.nih.gov/17466797/
332. Ayuk SM, Abrahamse H, Houreld NN. The Role of Matrix Metalloproteinases in Diabetic Wound Healing in relation to Photobiomodulation. J Diabetes Res. 2016. 2016 Available at: https://pubmed.ncbi.nlm.nih.gov/27314046/
333. Nam S Il, Yu GI, Kim HJ. et al. A polymorphism at -1607 2G in the matrix metalloproteinase-1 (MMP-1) increased risk of sudden deafness in Korean population but not at -519A/G in MMP-1. Laryngoscope. 2011;121:171-5 Available at: https://pubmed.ncbi.nlm.nih.gov/21154774/
334. Shimizu Y, Kondo S, Shirai A, Furukawa M, Yoshizaki T. A single nucleotide polymorphism in the matrix metalloproteinase-1 and interleukin-8 gene promoter predicts poor prognosis in tongue cancer. Auris Nasus Larynx. 2008;35:381-9 Available at: https://pubmed.ncbi.nlm.nih.gov/18276095/
335. Hoikkala S, Pääkkö P, Soini Y, Mäkitaro R, Kinnula V, Turpeenniemi-Hujanen T. Tissue MMP-2/TIMP-2-complex are better prognostic factors than serum MMP-2, MMP-9 or TIMP-1 in Stage I-III lung carcinoma. Cancer Lett. 2006;236:125-32
336. Mizrachi A, Koren R, Hadar T, Yaniv E, Morgenstern S, Shvero J. Expression of MMP-1 in invasive well-differentiated thyroid carcinoma. Eur Arch Otorhinolaryngol. 2011;268:131-5 Available at: https://pubmed.ncbi.nlm.nih.gov/20652290/
337. Noya M, Qian Y, Jerald Ainsworth A. Molecular and functional characterization of channel catfish (Ictalurus punctatus) neutrophil collagenase. Vet Immunol Immunopathol. 1999;67:303-16
338. Nwomeh BC, Liang HX, Diegelmann RF, Cohen IK, Yager DR. Dynamics of the matrix metalloproteinases MMP-1 and MMP-8 in acute open human dermal wounds. Wound Repair Regen. 1998;6:127-34 Available at: https://pubmed.ncbi.nlm.nih.gov/9776855/
339. Lee W, Aitken S, Sodek J, McCulloch CAG. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo: role of active enzyme in human periodontitis. J Periodontal Res. 1995;30:23-33 Available at: https://pubmed.ncbi.nlm.nih.gov/7722844/
340. Sorsa T, Hernández M, Leppilahti J, Munjal S, Netuschil L, Mäntylä P. Detection of gingival crevicular fluid MMP-8 levels with different laboratory and chair-side methods. Oral Dis. 2010;16:39-45 Available at: https://pubmed.ncbi.nlm.nih.gov/19627514/
341. Dalvie D, Cosker T, Boyden T, Zhou S, Schroeder C, Potchoiba MJ. Metabolism distribution and excretion of a matrix metalloproteinase-13 inhibitor, 4-[4-(4-fluorophenoxy)-benzenesulfonylamino]tetrahydropyran-4-carboxylic acid hydroxyamide (CP-544439), in rats and dogs: assessment of the metabolic profile of CP-544439 in plasma and urine of humans. Drug Metab Dispos. 2008;36:1869-83 Available at: https://pubmed.ncbi.nlm.nih.gov/18566038/
342. Park SJ, Cheon EJ, Lee MH, Kim HA. MicroRNA-127-5p regulates matrix metalloproteinase 13 expression and interleukin-1β-induced catabolic effects in human chondrocytes. Arthritis Rheum. 2013;65:3141-52
343. Mashimo Y, Sakurai-Yageta M, Watanabe M. et al. Induction of the Matrix Metalloproteinase 13 Gene in Bronchial Epithelial Cells by Interferon and Identification of its Novel Functional Polymorphism. Inflammation. 2016;39:949-62 Available at: https://pubmed.ncbi.nlm.nih.gov/26635116/
344. You Y, Shan Y, Chen J. et al. Matrix metalloproteinase 13-containing exosomes promote nasopharyngeal carcinoma metastasis. Cancer Sci. 2015;106:1669-77 Available at: https://pubmed.ncbi.nlm.nih.gov/26362844/
345. Cossins J, Dudgeon TJ, Catlin G, Gearing AJH, Clements JM. Identification of MMP-18, a putative novel human matrix metalloproteinase. Biochem Biophys Res Commun. 1996;228:494-8 Available at: https://pubmed.ncbi.nlm.nih.gov/8920941/
346. Tomlinson ML, Garcia-Morales C, Abu-Elmagd M, Wheeler GN. Three matrix metalloproteinases are required in vivo for macrophage migration during embryonic development. Mech Dev. 2008;125:1059-70 Available at: https://pubmed.ncbi.nlm.nih.gov/18684398/
347. Tonge D, Zhu N, Lynham S. et al. Axonal growth towards Xenopus skin in vitro is mediated by matrix metalloproteinase activity. Eur J Neurosci. 2013;37:519-31 Available at: https://pubmed.ncbi.nlm.nih.gov/23216618/
348. Steffensen B, Wallon UM, Overall CM. Extracellular matrix binding properties of recombinant fibronectin type II-like modules of human 72-kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen. J Biol Chem. 1995;270:11555-66 Available at: https://pubmed.ncbi.nlm.nih.gov/7744795/
349. Shipley JM, Doyle GAR, Fliszar CJ. et al. The structural basis for the elastolytic activity of the 92-kDa and 72-kDa gelatinases. Role of the fibronectin type II-like repeats. J Biol Chem. 1996;271:4335-41 Available at: https://pubmed.ncbi.nlm.nih.gov/8626782/
350. Aimes RT, Quigley JP. Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J Biol Chem. 1995;270:5872-6 Available at: https://pubmed.ncbi.nlm.nih.gov/7890717/
351. Augoff K, Grabowski K, Rabczynskl J, Kolondra A, Tabola R, Sikorski AF. Expression of decorin in esophageal cancer in relation to the expression of three isoforms of transforming growth factor-beta (TGF-beta1, -beta2, and -beta3) and matrix metalloproteinase-2 activity. Cancer Invest. 2009;27:443-52 Available at: https://pubmed.ncbi.nlm.nih.gov/19212830/
352. Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with α5β1 integrin in glioma. Oncogene. 2013;32:327-40 Available at: https://pubmed.ncbi.nlm.nih.gov/22349830/
353. Olszewska E, Matulka M, Mroczko B. et al. Diagnostic value of matrix metalloproteinase 9 and tissue inhibitor of matrix metalloproteinases 1 in cholesteatoma. Histol Histopathol. 2016;31:307-15 Available at: https://pubmed.ncbi.nlm.nih.gov/26490574/
354. Nam S Il, Kwon TK. Dexamethasone inhibits interleukin-1β-induced matrix metalloproteinase-9 expression in cochlear cells. Clin Exp Otorhinolaryngol. 2014;7:175-80 Available at: https://pubmed.ncbi.nlm.nih.gov/25177432/
355. Chen L, Lu Y, Chu Y, Xie J, Ding W, Wang F. Tissue factor expression in rheumatoid synovium: A potential role in pannus invasion of rheumatoid arthritis. Acta Histochem. 2013;115:692-7
356. Qin W, Lu W, Li H. et al. Melatonin inhibits IL1β-induced MMP9 expression and activity in human umbilical vein endothelial cells by suppressing NF-κB activation. Journal of Endocrinology. 2012;214:145-53
357. Donnell SM, Matrisian LM. Stromelysin in tumor progression and metastasis. CANCER AND METASTASIS REVIEW. 1990;9:305-19
358. Becker JW, Marcy AI, Rokosz LL. et al. Stromelysin-1: Three-dimensional structure of the inhibited catalytic domain and of the C-truncated proenzyme. Protein Science. 1995;4:1966-76
359. Pittayapruek P, Meephansan J, Prapapan O, Komine M, Ohtsuki M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int J Mol Sci. 2016 17. Available at: https://pubmed.ncbi.nlm.nih.gov/27271600/
360. Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem. 2007;15:2223-68 Available at: https://pubmed.ncbi.nlm.nih.gov/17275314/
361. Eguchi T, Kubota S, Kawata K. et al. Novel transcription-factor-like function of human matrix metalloproteinase 3 regulating the CTGF/CCN2 gene. Mol Cell Biol. 2008;28:2391-413 Available at: https://pubmed.ncbi.nlm.nih.gov/18172013/
362. Ding L, Guo D, Homandberg GA, Buckwalter JA, Martin JA. A single blunt impact on cartilage promotes fibronectin fragmentation and upregulates cartilage degrading stromelysin-1/matrix metalloproteinase-3 in a bovine ex vivo model. J Orthop Res. 2014;32:811-8 Available at: https://pubmed.ncbi.nlm.nih.gov/24610678/
363. Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giachè V, Mannelli M. Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol. 2012;2012:872713 Available at: http://www.ncbi.nlm.nih.gov/pubmed/22851969
364. Choi DH, Kim JH, Seo JH, Lee J, Choi WS, Kim YS. Matrix metalloproteinase-3 causes dopaminergic neuronal death through Nox1-regenerated oxidative stress. PLoS One. 2014 9. Available at: https://pubmed.ncbi.nlm.nih.gov/25536219/
365. Sokai A, Handa T, Tanizawa K. et al. Matrix metalloproteinase-10: a novel biomarker for idiopathic pulmonary fibrosis. Respir Res. 2015 16. Available at: https://pubmed.ncbi.nlm.nih.gov/26415518/
366. Hirakawa S, Kojima T, Obata K. et al. Marked induction of matrix metalloproteinase-10 by respiratory syncytial virus infection in human nasal epithelial cells. J Med Virol. 2013;85:2141-50 Available at: https://pubmed.ncbi.nlm.nih.gov/24009192/
367. García-Irigoyen O, Latasa MU, Carotti S. et al. Matrix metalloproteinase 10 contributes to hepatocarcinogenesis in a novel crosstalk with the stromal derived factor 1/C-X-C chemokine receptor 4 axis. Hepatology. 2015;62:166-78 Available at: https://pubmed.ncbi.nlm.nih.gov/25808184/
368. Basset P, Bellocq JP, Wolf C. et al. A novel metalloproteinase gene specifically expressed in stromal cells of breast carcinomas. Nature. 1990;348:699-704 Available at: https://pubmed.ncbi.nlm.nih.gov/1701851/
369. Fiorentino M, Fu L, Shi YB. Mutational analysis of the cleavage of the cancer-associated laminin receptor by stromelysin-3 reveals the contribution of flanking sequences to site recognition and cleavage efficiency. Int J Mol Med. 2009;23:389-97 Available at: https://pubmed.ncbi.nlm.nih.gov/19212658/
370. Sharma R, Chattopadhyay TK, Mathur M, Ralhan R. Prognostic significance of stromelysin-3 and tissue inhibitor of matrix metalloproteinase-2 in esophageal cancer. Oncology. 2004;67:300-9 Available at: https://pubmed.ncbi.nlm.nih.gov/15557792/
371. Zhang X, Huang S, Guo J. et al. Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics (Review). Int J Oncol. 2016;48:1783-93 Available at: https://pubmed.ncbi.nlm.nih.gov/26892540/
372. Xian J, Lin Y, Liu Y, Gong P, Liu S. Combined p14ARF and antisense EGFR potentiate the efficacy of adenovirus-mediated gene therapy in laryngeal squamous cell carcinoma (LSCC). DNA Cell Biol. 2007;26:71-9
373. Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617-29 Available at: https://pubmed.ncbi.nlm.nih.gov/15286728/
374. Acioglu E, Yigit Ö, Alkan Z, Server EA, Uzun H, Gelisgen R. The role of matrix metalloproteinases in recurrent tonsillitis. Int J Pediatr Otorhinolaryngol. 2010;74:535-9 Available at: https://pubmed.ncbi.nlm.nih.gov/20233630/
375. Jiang T, Xie P, Liu H. Circulating Anti-Matrix Metalloproteinase-7 Antibodies May Be a Potential Biomarker for Oral Squamous Cell Carcinoma. J Oral Maxillofac Surg. 2016;74:650-7 Available at: https://pubmed.ncbi.nlm.nih.gov/26454036/
376. De Coignac AB, Elson G, Delneste Y. et al. Cloning of MMP-26. A novel matrilysin-like proteinase. Eur J Biochem. 2000;267:3323-9 Available at: https://pubmed.ncbi.nlm.nih.gov/10824119/
377. Vandooren J, Van Den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit Rev Biochem Mol Biol. 2013;48:222-72
378. Marchenko GN, Ratnikov BI, Rozanov D V, Godzik A, Deryugina EI, Strongin AY. Characterization of matrix metalloproteinase-26, a novel metalloproteinase widely expressed in cancer cells of epithelial origin. Biochem J. 2001;356:705-18 Available at: https://pubmed.ncbi.nlm.nih.gov/11389678/
379. Gutschalk CM, Yanamandra AK, Linde N, Meides A, Depner S, Mueller MM. GM-CSF enhances tumor invasion by elevated MMP-2, -9, and -26 expression. Cancer Med. 2013;2:117-29 Available at: https://pubmed.ncbi.nlm.nih.gov/23634280/
380. Zhao YG, Xiao AZ, Park HI. et al. Endometase/matrilysin-2 in human breast ductal carcinoma in situ and its inhibition by tissue inhibitors of metalloproteinases-2 and -4: a putative role in the initiation of breast cancer invasion. Cancer Res. 2004;64:590-8 Available at: https://pubmed.ncbi.nlm.nih.gov/14744773/
381. Itoh Y, Kajita M, Kinoh H, Mori H, Okada A, Seiki M. Membrane type 4 matrix metalloproteinase (MT4-MMP, MMP-17) is a glycosylphosphatidylinositol-anchored proteinase. J Biol Chem. 1999;274:34260-6 Available at: https://pubmed.ncbi.nlm.nih.gov/10567400/
382. Kayano K, Shimada T, Shinomiya T. et al. Activation of pro-MMP-2 mediated by MT1-MMP in human salivary gland carcinomas: possible regulation of pro-MMP-2 activation by TIMP-2. J Pathol. 2004;202:403-11 Available at: https://pubmed.ncbi.nlm.nih.gov/15095267/
383. Imanishi Y, Fujii M, Tokumaru Y. et al. Clinical significance of expression of membrane type 1 matrix metalloproteinase and matrix metalloproteinase-2 in human head and neck squamous cell carcinoma. Hum Pathol. 2000;31:895-904 Available at: https://pubmed.ncbi.nlm.nih.gov/10987249/
384. Sato H, Seiki M. Membrane-type matrix metalloproteinases (MT-MMPs) in tumor metastasis. J Biochem. 1996;119:209-15 Available at: https://pubmed.ncbi.nlm.nih.gov/8882706/
385. Abraham R, Schäfer J, Rothe M, Bange J, Knyazev P, Ullrich A. Identification of MMP-15 as an anti-apoptotic factor in cancer cells. J Biol Chem. 2005;280:34123-32 Available at: https://pubmed.ncbi.nlm.nih.gov/16093241/
386. Szabova L, Son MY, Shi J. et al. Membrane-type MMPs are indispensable for placental labyrinth formation and development. Blood. 2010;116:5752-61 Available at: https://pubmed.ncbi.nlm.nih.gov/20858856/
387. Zhang H, Liu M, Sun Y, Lu J. MMP-14 can serve as a prognostic marker in patients with supraglottic cancer. Eur Arch Otorhinolaryngol. 2009;266:1427-34 Available at: https://pubmed.ncbi.nlm.nih.gov/19283401/
388. Nakada M, Nakamura H, Ikeda E. et al. Expression and tissue localization of membrane-type 1, 2, and 3 matrix metalloproteinases in human astrocytic tumors. American Journal of Pathology. 1999;154:417-28
389. Liu J, van Mil A, Aguor ENE. et al. MiR-155 inhibits cell migration of human cardiomyocyte progenitor cells (hCMPCs) via targeting of MMP-16. J Cell Mol Med. 2012;16:2379-86 Available at: https://pubmed.ncbi.nlm.nih.gov/22348515/
390. Chen MF, Zeng F, Qi L. et al. Transforming growth factor β1 induces epithelial mesenchymal transition and increased expression of matrix metalloproteinase 16 via miR 200b downregulation in bladder cancer cells. Mol Med Rep. 2014;10:1549-54 Available at: https://pubmed.ncbi.nlm.nih.gov/25017509/
391. Liu C, Wu F, Liu Y, Meng C. Catalpol suppresses proliferation and facilitates apoptosis of MCF-7 breast cancer cells through upregulating microRNA-146a and downregulating matrix metalloproteinase-16 expression. Mol Med Rep. 2015;12:7609-14 Available at: https://pubmed.ncbi.nlm.nih.gov/26458573/
392. Hadchouel A, Decobert F, Franco-Montoya ML. et al. Matrix metalloproteinase gene polymorphisms and bronchopulmonary dysplasia: identification of MMP16 as a new player in lung development. PLoS One. 2008 3. Available at: https://pubmed.ncbi.nlm.nih.gov/18784838/
393. Grant GM, Giambernardi TA, Grant AM, Klebe RJ. Overview of expression of matrix metalloproteinases (MMP-17, MMP-18, and MMP-20) in cultured human cells. Matrix Biol. 1999;18:145-8 Available at: https://pubmed.ncbi.nlm.nih.gov/10372554/
394. Kowalska M, Tylicka M, Koper-Lenkiewicz OM, Kamińska J, Dorf J, Matuszczak E. Plasma concentration of MMP-17 is elevated in boys with cryptorchidism and correlates with HSP-70. Sci Rep. 2025 15. Available at: https://pubmed.ncbi.nlm.nih.gov/39747260/
395. Muñoz-Sáez E, Moracho N, Learte AIR. et al. Molecular Mechanisms Driven by MT4-MMP in Cancer Progression. Int J Mol Sci. 2023 24. Available at: https://pubmed.ncbi.nlm.nih.gov/37373092/
396. Fillmore HL, VanMeter TE, Broaddus WC. Membrane-type matrix metalloproteinases (MT-MMPs): Expression and function during glioma invasion. J Neurooncol. 2001;53:187-202
397. Komori K, Nonaka T, Okada A. et al. Absence of mechanical allodynia and Aβ-fiber sprouting after sciatic nerve injury in mice lacking membrane-type 5 matrix metalloproteinase. FEBS Lett. 2004;557:125-8 Available at: https://pubmed.ncbi.nlm.nih.gov/14741353/
398. Benson CS, Babu SD, Radhakrishna S, Selvamurugan N, Sankar BR. Expression of matrix metalloproteinases in human breast cancer tissues. Dis Markers. 2013;34:395-405 Available at: https://pubmed.ncbi.nlm.nih.gov/23568046/
399. Fortin CF, Sohail A, Sun Q, McDonald PP, Fridman R, Fülöp T. MT6-MMP is present in lipid rafts and faces inward in living human PMNs but translocates to the cell surface during neutrophil apoptosis. Int Immunol. 2010;22:637-49 Available at: https://pubmed.ncbi.nlm.nih.gov/20501611/
400. Nagase H. Cell surface activation of progelatinase A (proMMP-2) and cell migration. Cell Res. 1998;8:179-86
401. Sun Q, Weber CR, Sohail A. et al. MMP25 (MT6-MMP) is highly expressed in human colon cancer, promotes tumor growth, and exhibits unique biochemical properties. J Biol Chem. 2007;282:21998-2010 Available at: https://pubmed.ncbi.nlm.nih.gov/17513868/
402. Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A. 1996;93:3942-6 Available at: https://pubmed.ncbi.nlm.nih.gov/8632994/
403. Hou P, Troen T, Ovejero MC. et al. Matrix metalloproteinase-12 (MMP-12) in osteoclasts: New lesson on the involvement of MMPs in bone resorption. Bone. 2004;34:37-47 Available at: https://pubmed.ncbi.nlm.nih.gov/14751561/
404. Li W, Li J, Wu Y. et al. Identification of an orally efficacious matrix metalloprotease 12 inhibitor for potential treatment of asthma. J Med Chem. 2009;52:5408-19 Available at: https://pubmed.ncbi.nlm.nih.gov/19725580/
405. Chelluboina B, Klopfenstein JD, Pinson DM, Wang DZ, Vemuganti R, Veeravalli KK. Matrix Metalloproteinase-12 Induces Blood-Brain Barrier Damage After Focal Cerebral Ischemia. Stroke. 2015;46:3523-31 Available at: https://pubmed.ncbi.nlm.nih.gov/26534974/
406. Kim JM, Kim HJ, Koo BS, Rha KS, Yoon YH. Expression of matrix metalloproteinase-12 is correlated with extracapsular spread of tumor from nodes with metastasis in head and neck squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2013;270:1137-42 Available at: https://pubmed.ncbi.nlm.nih.gov/22907031/
407. Stracke JO, Hutton M, Stewart M. et al. Biochemical characterization of the catalytic domain of human matrix metalloproteinase 19. Evidence for a role as a potent basement membrane degrading enzyme. J Biol Chem. 2000;275:14809-16 Available at: https://pubmed.ncbi.nlm.nih.gov/10809722/
408. Yu G, Herazo-Maya JD, Nukui T. et al. Matrix metalloproteinase-19 promotes metastatic behavior in vitro and is associated with increased mortality in non-small cell lung cancer. Am J Respir Crit Care Med. 2014;190:780-90 Available at: https://pubmed.ncbi.nlm.nih.gov/25250855/
409. Sena P, Mariani F, Marzona L. et al. Matrix metalloproteinases 15 and 19 are stromal regulators of colorectal cancer development from the early stages. Int J Oncol. 2012;41:260-6 Available at: https://pubmed.ncbi.nlm.nih.gov/22576687/
410. Lee DG, Lee SH, Kim JS. et al. Loss of NDRG2 promotes epithelial-mesenchymal transition of gallbladder carcinoma cells through MMP-19-mediated Slug expression. J Hepatol. 2015;63:1429-39 Available at: https://pubmed.ncbi.nlm.nih.gov/26292259/
411. Chan KC, Ko JMY, Lung HL. et al. Catalytic activity of Matrix metalloproteinase-19 is essential for tumor suppressor and anti-angiogenic activities in nasopharyngeal carcinoma. Int J Cancer. 2011;129:1826-37 Available at: https://pubmed.ncbi.nlm.nih.gov/21165953/
412. Ryu OH, Fincham AG, Hu CC. et al. Characterization of recombinant pig enamelysin activity and cleavage of recombinant pig and mouse amelogenins. J Dent Res. 1999;78:743-50 Available at: https://pubmed.ncbi.nlm.nih.gov/10096449/
413. Llano E, Pendás AM, Knäuper V. et al. Identification and structural and functional characterization of human enamelysin (MMP-20). Biochemistry. 1997;36:15101-8 Available at: https://pubmed.ncbi.nlm.nih.gov/9398237/
414. Caterina JJ, Skobe Z, Shi J. et al. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2002;277:49598-604 Available at: https://pubmed.ncbi.nlm.nih.gov/12393861/
415. Barron LA, Giardina JB, Granger JP, Khalil RA. High-salt diet enhances vascular reactivity in pregnant rats with normal and reduced uterine perfusion pressure. Hypertension. 2001;38:730-5 Available at: https://pubmed.ncbi.nlm.nih.gov/11566966/
416. Stracke JO, Fosang AJ, Last K. et al. Matrix metalloproteinases 19 and 20 cleave aggrecan and cartilage oligomeric matrix protein (COMP). FEBS Lett. 2000;478:52-6 Available at: https://pubmed.ncbi.nlm.nih.gov/10922468/
417. Wu T, Li Y, Liu X. et al. Identification of high-risk stage II and stage III colorectal cancer by analysis of MMP-21 expression. J Surg Oncol. 2011;104:787-91 Available at: https://pubmed.ncbi.nlm.nih.gov/21656525/
418. Zhao Z, Yan L, Li S, Sun H, Zhou Y, Li X. Increased MMP-21 expression in esophageal squamous cell carcinoma is associated with progression and prognosis. Med Oncol. 2014 31. Available at: https://pubmed.ncbi.nlm.nih.gov/25015395/
419. Pu Y, Wang L, Wu H, Feng Z, Wang Y, Guo C. High MMP-21 expression in metastatic lymph nodes predicts unfavorable overall survival for oral squamous cell carcinoma patients with lymphatic metastasis. Oncol Rep. 2014;31:2646-50 Available at: https://pubmed.ncbi.nlm.nih.gov/24700287/
420. Suomela S, Koljonen V, Skoog T, Kukko H, Böhling T, Saarialho-Kere U. Expression of MMP-10, MMP-21, MMP-26, and MMP-28 in Merkel cell carcinoma. Virchows Arch. 2009;455:495-503 Available at: https://pubmed.ncbi.nlm.nih.gov/19921252/
421. Bister V, Skoog T, Virolainen S, Kiviluoto T, Puolakkainen P, Saarialho-Kere U. Increased expression of matrix metalloproteinases-21 and -26 and TIMP-4 in pancreatic adenocarcinoma. Mod Pathol. 2007;20:1128-40 Available at: https://pubmed.ncbi.nlm.nih.gov/17873896/
422. Gururajan R, Grenet J, Lahti JM, Kidd VJ. Isolation and characterization of two novel metalloproteinase genes linked to the Cdc2L locus on human chromosome 1p36.3. Genomics. 1998;52:101-6 Available at: https://pubmed.ncbi.nlm.nih.gov/9740677/
423. Liu ZC, Su CM, Xie YL. et al. Intracellular lipid dysregulation interferes with leukocyte function in the ovaries of meat-type hens under unrestricted feed intake. Anim Reprod Sci. 2016;167:40-50 Available at: https://pubmed.ncbi.nlm.nih.gov/26874430/
424. Velasco G, Pendás AM, Fueyo A, Knäuper V, Murphy G, López-Otín C. Cloning and characterization of human MMP-23, a new matrix metalloproteinase predominantly expressed in reproductive tissues and lacking conserved domains in other family members. J Biol Chem. 1999;274:4570-6 Available at: https://pubmed.ncbi.nlm.nih.gov/9988691/
425. Hegedüs L, Cho H, Xie X, Eliceiri GL. Additional MDA-MB-231 breast cancer cell matrix metalloproteinases promote invasiveness. J Cell Physiol. 2008;216:480-5 Available at: https://pubmed.ncbi.nlm.nih.gov/18286480/
426. Cominelli A, Halbout M, N'Kuli F. et al. A unique C-terminal domain allows retention of matrix metalloproteinase-27 in the endoplasmic reticulum. Traffic. 2014;15:401-17 Available at: https://pubmed.ncbi.nlm.nih.gov/24548619/
427. Köhrmann A, Kammerer U, Kapp M, Dietl J, Anacker J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer. 2009 9. Available at: https://pubmed.ncbi.nlm.nih.gov/19531263/
428. Lohi J, Wilson CL, Roby JD, Parks WC. Epilysin, a novel human matrix metalloproteinase (MMP-28) expressed in testis and keratinocytes and in response to injury. J Biol Chem. 2001;276:10134-44 Available at: https://pubmed.ncbi.nlm.nih.gov/11121398/
429. Saarialho-Kere U, Kerkelä E, Jahkola T, Suomela S, Keski-Oja J, Lohi J. Epilysin (MMP-28) expression is associated with cell proliferation during epithelial repair. J Invest Dermatol. 2002;119:14-21 Available at: https://pubmed.ncbi.nlm.nih.gov/12164918/
430. Kevorkian L, Young DA, Darrah C. et al. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004;50:131-41 Available at: https://pubmed.ncbi.nlm.nih.gov/14730609/
431. Momohara S, Okamoto H, Komiya K. et al. Matrix metalloproteinase 28/epilysin expression in cartilage from patients with rheumatoid arthritis and osteoarthritis: comment on the article by Kevorkian et al. Arthritis Rheum. 2004;50:4074-5 Available at: https://pubmed.ncbi.nlm.nih.gov/15593191/
432. Lumi X, Hawlina M, Glavač D. et al. Ageing of the vitreous: From acute onset floaters and flashes to retinal detachment. Ageing Res Rev. 2015;21:71-7
433. Schwartz NB, Domowicz MS, Schwartz NB, Domowicz MS. Chemistry and Function of Glycosaminoglycans in the Nervous System. 2014; 89-115. Available at: https://link.springer.com/chapter/10.1007/978-1-4939-1154-7_5.
434. Sugahara K, Mikami T. Chondroitin/dermatan sulfate in the central nervous system. Curr Opin Struct Biol. 2007;17:536-45
435. Brown JM, Xia J, Zhuang BQ. et al. A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc Natl Acad Sci U S A. 2012;109:4768-73 Available at: https://www.pnas.org/doi/abs/10.1073/pnas.1121318109
436. Trownbridge JM, Gallo RL. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology. 2002 12. Available at: https://pubmed.ncbi.nlm.nih.gov/12213784/
437. Mizumoto S, Yamada S, Sugahara K. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr Opin Struct Biol. 2015;34:35-42 Available at: https://pubmed.ncbi.nlm.nih.gov/26164146/
438. Habicher J, Varshney GK, Waldmann L. et al. Chondroitin/dermatan sulfate glycosyltransferase genes are essential for craniofacial development. PLoS Genet. 2022 18. Available at: https://pubmed.ncbi.nlm.nih.gov/35192612/
439. Attanasio E, Russo P, Carunchio G, Caprino L. Dermatan sulfate versus unfractionated heparin for the prevention of venous thromboembolism in patients undergoing surgery for cancer. A cost-effectiveness analysis. Pharmacoeconomics. 2001;19:57-68 Available at: https://pubmed.ncbi.nlm.nih.gov/11252546/
440. Hao C, Sun M, Wang H, Zhang L, Wang W. Low molecular weight heparins and their clinical applications. Prog Mol Biol Transl Sci. 2019;163:21-39 Available at: https://pubmed.ncbi.nlm.nih.gov/31030749/
441. Aláez-Versón CR, Lantero E, Fernàndez-Busquets X. Heparin: new life for an old drug. Nanomedicine (Lond). 2017;12:1727-44 Available at: https://pubmed.ncbi.nlm.nih.gov/28635544/
442. Shi D, Sheng A, Chi L. Glycosaminoglycan-Protein Interactions and Their Roles in Human Disease. Front Mol Biosci. 2021 8. Available at: https://pubmed.ncbi.nlm.nih.gov/33768117/
443. Torri G, Naggi A. Heparin centenary - an ever-young life-saving drug. Int J Cardiol. 2016;212(Suppl 1):S1-4 Available at: https://pubmed.ncbi.nlm.nih.gov/27264864/
444. Chen CL, Hasan SS, Klose T. et al. Cryo-EM structure of eastern equine encephalitis virus in complex with heparan sulfate analogues. Proc Natl Acad Sci U S A. 2020;117:8890-9 Available at: https://pubmed.ncbi.nlm.nih.gov/32245806/
445. Chhabra M, Wilson JC, Wu L, Davies GJ, Gandhi NS, Ferro V. Structural Insights into Pixatimod (PG545) Inhibition of Heparanase, a Key Enzyme in Cancer and Viral Infections. Chemistry. 2022 28. Available at: https://pubmed.ncbi.nlm.nih.gov/34981584/
446. Li Y, Wan D, Luo X. et al. Circulating Histones in Sepsis: Potential Outcome Predictors and Therapeutic Targets. Front Immunol. 2021 12. Available at: https://pubmed.ncbi.nlm.nih.gov/33868288/
447. Zhao J, Zhu Y, Song X. et al. 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake. Angew Chem Int Ed Engl. 2020;59:1818-27 Available at: https://pubmed.ncbi.nlm.nih.gov/31692167/
448. Stopschinski BE, Thomas TL, Nadji S. et al. A synthetic heparinoid blocks Tau aggregate cell uptake and amplification. J Biol Chem. 2020;295:2974-83 Available at: https://pubmed.ncbi.nlm.nih.gov/31974166/
449. Li JP, Kusche-Gullberg M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int Rev Cell Mol Biol. 2016;325:215-73 Available at: https://pubmed.ncbi.nlm.nih.gov/27241222/
450. Ledin J, Staatz W, Li JP. et al. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem. 2004;279:42732-41 Available at: https://pubmed.ncbi.nlm.nih.gov/15292174/
451. Bishop JR, Stanford KI, Esko JD. Heparan sulfate proteoglycans and triglyceride-rich lipoprotein metabolism. Curr Opin Lipidol. 2008;19:307-13 Available at: https://pubmed.ncbi.nlm.nih.gov/18460924/
452. Caravà E, Moretto P, Caon I. et al. Ha and hs changes in endothelial inflammatory activation. Biomolecules. 2021;11:809 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8229641/
453. Vlodavsky I, Friedmann Y, Elkin M. et al. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5:793-802 Available at: https://pubmed.ncbi.nlm.nih.gov/10395325/
454. CHAFFEY N. Alberts, B, Johnson, A, Lewis, J, Raff, M, Roberts, K. and Walter, P. Molecular biology of the cell. 4th edn. Ann Bot. 2003;91:401 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4244961/
455. Tian X, Azpurua J, Hine C. et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature. 2013;499:346-9 Available at: https://pubmed.ncbi.nlm.nih.gov/23783513/
456. Stecco C, Stern R, Porzionato A. et al. Hyaluronan within fascia in the etiology of myofascial pain. Surg Radiol Anat. 2011;33:891-6 Available at: https://pubmed.ncbi.nlm.nih.gov/21964857/
457. Chang EJ, Kim HJ, Ha J. et al. Hyaluronan inhibits osteoclast differentiation via Toll-like receptor 4. J Cell Sci. 2007;120:166-76 Available at: https://pubmed.ncbi.nlm.nih.gov/17164294/
458. S M-N, J G, S K, M S. Oligosaccharides of hyaluronan induce angiogenesis through distinct CD44 and RHAMM-mediated signalling pathways involving Cdc2 and gamma-adducin. Int J Oncol. 2009 35. Available at: https://pubmed.ncbi.nlm.nih.gov/19724912/
459. Caterson B, Melrose J. Keratan sulfate, a complex glycosaminoglycan with unique functional capability. Glycobiology. 2018;28:182-206 Available at: https://pubmed.ncbi.nlm.nih.gov/29340594/
460. Vitureira N, Andrés R, Pérez-Martínez E. et al. Podocalyxin is a novel polysialylated neural adhesion protein with multiple roles in neural development and synapse formation. PLoS One. 2010 5. Available at: https://pubmed.ncbi.nlm.nih.gov/20706633/
461. Muramatsu T. Embryoglycan: a highly branched poly-N-acetyllactosamine in pluripotent stem cells and early embryonic cells. Glycoconj J. 2017;34:701-12 Available at: https://pubmed.ncbi.nlm.nih.gov/27188587/
462. Melrose J. Glycosaminoglycans, Instructive Biomolecules That Regulate Cellular Activity and Synaptic Neuronal Control of Specific Tissue Functional Properties. International Journal of Molecular Sciences 2025, Vol 26, Page 2554. 2025;26:2554 Available at: https://www.mdpi.com/1422-0067/26/6/2554/htm
463. Krusius T, Reinhold VN, Margolis RK, Margolis RU. Structural studies on sialylated and sulphated O-glycosidic mannose-linked oligosaccharides in the chondroitin sulphate proteoglycan of brain. Biochem J. 1987;245:229-34
464. Graves ML, Cipollone JA, Austin P. et al. The cell surface mucin podocalyxin regulates collective breast tumor budding. Breast Cancer Res. 2016 18. Available at: https://pubmed.ncbi.nlm.nih.gov/26796961/
465. Muiznieks LD, Keeley FW. Molecular assembly and mechanical properties of the extracellular matrix: A fibrous protein perspective. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2013;1832:866-75
466. Trębacz H, Barzycka A. Mechanical Properties and Functions of Elastin: An Overview. Biomolecules. 2023;13:574 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10046833/
467. Pang X, He X, Qiu Z. et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduction and Targeted Therapy 2022 8:1. 2023;8:1-42 Available at: https://www.nature.com/articles/s41392-022-01259-6
468. Chen K, Xu M, Lu F, He Y. Development of Matrix Metalloproteinases-Mediated Extracellular Matrix Remodeling in Regenerative Medicine: A Mini Review. Tissue Eng Regen Med. 2023;20:661 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10352474/
469. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nature Reviews Molecular Cell Biology 2014 15:12. 2014;15:802-12 Available at: https://www.nature.com/articles/nrm3896
470. Saraswathibhatla A, Indana D, Chaudhuri O. Cell-extracellular matrix mechanotransduction in 3D. Nature Reviews Molecular Cell Biology 2023 24:7. 2023;24:495-516 Available at: https://www.nature.com/articles/s41580-023-00583-1
471. Lee JL, Streuli CH. Integrins and epithelial cell polarity. J Cell Sci. 2014;127:3217 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4117227/
472. Muncie JM, Weaver VM. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr Top Dev Biol. 2018;130:1 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6586474/
473. Hynes RO. Extracellular matrix: not just pretty fibrils. Science. 2009;326:1216 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3536535/
474. Puttock EH, Tyler EJ, Manni M. et al. Extracellular matrix educates an immunoregulatory tumor macrophage phenotype found in ovarian cancer metastasis. Nature Communications 2023 14:1. 2023;14:1-16 Available at: https://www.nature.com/articles/s41467-023-38093-5
475. Panneerpandian P, Ganesan K. PI3K/AKT/mTOR inhibitors as potential extracellular matrix modulators for targeting EMT subtype gastric tumors. Med Oncol. 2023 40. Available at: https://pubmed.ncbi.nlm.nih.gov/36934368/
476. Rais A, Husain A, Hasan GM, Hassan MI. A review on regulation of cell cycle by extracellular matrix. Int J Biol Macromol. 2023;232:123426
477. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833:3481-98 Available at: https://pubmed.ncbi.nlm.nih.gov/23830918/
478. Ding W, Zhang M, Zhang P, Zhang X, Sun J, Lin B. Identification of anoikis-related subtypes and immune landscape in kidney renal clear cell carcinoma. Scientific Reports 2023 13:1. 2023;13:1-14 Available at: https://www.nature.com/articles/s41598-023-45069-4
479. Haun F, Neumann S, Peintner L. et al. Identification of a novel anoikis signalling pathway using the fungal virulence factor gliotoxin. Nature Communications 2018 9:1. 2018;9:1-14 Available at: https://www.nature.com/articles/s41467-018-05850-w
480. Piroli ME, Jabbarzadeh E. Matrix Stiffness Modulates Mesenchymal Stem Cell Sensitivity to Geometric Asymmetry Signals. Ann Biomed Eng. 2018;46:888 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5935526/
481. Cai X, Wang KC, Meng Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front Cell Dev Biol. 2021;9:673599 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8182050/
482. Lu P, Takai K, Weaver VM, Werb Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb Perspect Biol. 2011;3:a005058 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3225943/
483. Du J, Zu Y, Li J. et al. Extracellular matrix stiffness dictates Wnt expression through integrin pathway. Scientific Reports 2016 6:1. 2016;6:1-12 Available at: https://www.nature.com/articles/srep20395
484. Yamada KM, Collins JW, Cruz Walma DA. et al. Extracellular matrix dynamics in cell migration, invasion and tissue morphogenesis. Int J Exp Pathol. 2019;100:144-52 Available at: https://pubmed.ncbi.nlm.nih.gov/31179622/
485. Wu D, Yamada KM, Wang S. Tissue Morphogenesis Through Dynamic Cell and Matrix Interactions. Annu Rev Cell Dev Biol. 2023;39:123-44 Available at: https://www.annualreviews.org/content/journals/10.1146/annurev-cellbio-020223-031019
486. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nature Reviews Molecular Cell Biology 2014 15:12. 2014;15:786-801 Available at: https://www.nature.com/articles/nrm3904
487. Diller RB, Tabor AJ. The Role of the Extracellular Matrix (ECM) in Wound Healing: A Review. Biomimetics. 2022;7:87 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9326521/
488. Potekaev NN, Borzykh OB, Medvedev G V. et al. The Role of Extracellular Matrix in Skin Wound Healing. J Clin Med. 2021;10:5947 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8706213/
489. Konttinen YT, Kaivosoja E, Stegaev V. et al. Extracellular Matrix and Tissue Regeneration. Regenerative Medicine: From Protocol to Patient. 2011:21-80 Available at: https://link.springer.com/chapter/10.1007/978-90-481-9075-1_2
490. Yue B. Biology of the Extracellular Matrix: An Overview. J Glaucoma. 2014;23:S20 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4185430/
491. Amar S, Smith L, Fields GB. Matrix metalloproteinase collagenolysis in health and disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2017;1864:1940-51
492. Paluch EK, Aspalter IM, Sixt M. Focal Adhesion-Independent Cell Migration. Annu Rev Cell Dev Biol. 2016;32:469-90 Available at: https://pubmed.ncbi.nlm.nih.gov/27501447/
493. Ruprecht V, Wieser S, Callan-Jones A. et al. Cortical Contractility Triggers a Stochastic Switch to Fast Amoeboid Cell Motility. Cell. 2015;160:673 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4328143/
494. Te Boekhorst V, Preziosi L, Friedl P. Plasticity of Cell Migration In vivo and In Silico. Annu Rev Cell Dev Biol. 2016;32:491-526 Available at: https://pubmed.ncbi.nlm.nih.gov/27576118/
495. McMahon M, Ye S, Pedrina J, Dlugolenski D, Stambas J. Extracellular Matrix Enzymes and Immune Cell Biology. Front Mol Biosci. 2021 8. Available at: https://pubmed.ncbi.nlm.nih.gov/34527702/
496. Mongiat M, Sweeney SM, San Antonio JD, Fu J, Iozzo R V. Endorepellin, a Novel Inhibitor of Angiogenesis Derived from the C Terminus of Perlecan. Journal of Biological Chemistry. 2003;278:4238-49
497. Holmbeck K, Bianco P, Caterina J. et al. MT1-MMP-Deficient Mice Develop Dwarfism, Osteopenia, Arthritis, and Connective Tissue Disease due to Inadequate Collagen Turnover. Cell. 1999;99:81-92
498. Karamanos NK, Theocharis AD, Neill T, Iozzo R V. Matrix modeling and remodeling: a biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2018;75-76:1 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6377817/
499. Zhang M, Zhang B. Extracellular matrix stiffness: mechanisms in tumor progression and therapeutic potential in cancer. Exp Hematol Oncol. 2025;14:54 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11984264/
500. Zhao X, Chen J, Sun H, Zhang Y, Zou D. New insights into fibrosis from the ECM degradation perspective: the macrophage-MMP-ECM interaction. Cell Biosci. 2022;12:117 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9327238/
501. Wight TN, Potter-Perigo S. The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol. 2011;301:G950 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3233785/
502. Wang K, Wen D, Xu X. et al. Extracellular matrix stiffness—The central cue for skin fibrosis. Front Mol Biosci. 2023;10:1132353
503. Allison SJ. ECM remodelling by ADAMTS12 in fibrosis. Nature Reviews Nephrology 2024 20:12. 2024;20:770-770 Available at: https://www.nature.com/articles/s41581-024-00905-2
504. Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013;229:298 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4005341/
505. Vilardi A, Przyborski S, Mobbs C, Rufini A, Tufarelli C. Current understanding of the interplay between extracellular matrix remodelling and gut permeability in health and disease. Cell Death Discovery 2024 10:1. 2024;10:1-11 Available at: https://www.nature.com/articles/s41420-024-02015-1
506. Cantero-Recasens G, Burballa C, Ohkawa Y. et al. The ulcerative colitis-associated gene FUT8 regulates the quantity and quality of secreted mucins. Proc Natl Acad Sci U S A. 2022;119:e2205277119 Available at: https://www.pnas.org/doi/abs/10.1073/pnas.2205277119
507. Júnior C, Ulldemolins A, Narciso M. et al. Multi-Step Extracellular Matrix Remodelling and Stiffening in the Development of Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences 2023, Vol 24, Page 1708. 2023;24:1708 Available at: https://www.mdpi.com/1422-0067/24/2/1708/htm
508. Johansson MEV, Gustafsson JK, Holmen-Larsson J. et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63:281-91 Available at: https://gut.bmj.com/content/63/2/281
509. Urciuoli E, Peruzzi B. Involvement of the FAK Network in Pathologies Related to Altered Mechanotransduction. Int J Mol Sci. 2020;21:9426 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7764271/
510. Novoseletskaya ES, Evdokimov PV, Efimenko AY. Extracellular matrix-induced signaling pathways in mesenchymal stem/stromal cells. Cell Communication and Signaling. 2023;21:1-20 Available at: https://biosignaling.biomedcentral.com/articles/10.1186/s12964-023-01252-8
511. Prakash J, Shaked Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024;14:1375 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11294818/
512. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nature Communications 2020 11:1. 2020;11:1-19 Available at: https://www.nature.com/articles/s41467-020-18794-x
513. Sleeboom JJF, van Tienderen GS, Schenke-Layland K, van der Laan LJW, Khalil AA, Verstegen MMA. The extracellular matrix as hallmark of cancer and metastasis: From biomechanics to therapeutic targets. Sci Transl Med. 2024 16. Available at: https://www.science.org/doi/10.1126/scitranslmed.adg3840
514. Cox TR. The matrix in cancer. Nature Reviews Cancer 2021 21:4. 2021;21:217-38 Available at: https://www.nature.com/articles/s41568-020-00329-7
515. Vijg J. From DNA damage to mutations: all roads lead to aging. Ageing Res Rev. 2021;68:101316 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10018438/
516. Poluzzi C, Iozzo R V, Schaefer L. Endostatin and endorepellin: a common route of action for similar angiostatic cancer avengers. Adv Drug Deliv Rev. 2015;97:156 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4753091/
517. Nielsen SH, Willumsen N, Brix S. et al. Tumstatin, a Matrikine Derived from Collagen Type IVα3, is Elevated in Serum from Patients with Non-Small Cell Lung Cancer. Transl Oncol. 2018;11:528 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5884192/
518. Huang J, Zhang L, Wan D. et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021 6. Available at: https://pubmed.ncbi.nlm.nih.gov/33888679/
519. Walton KL, Johnson KE, Harrison CA. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front Pharmacol. 2017 8. Available at: https://pubmed.ncbi.nlm.nih.gov/28769795/
520. Venning FA, Wullkopf L, Erler JT. Targeting ECM Disrupts Cancer Progression. Front Oncol. 2015 5. Available at: https://pubmed.ncbi.nlm.nih.gov/26539408/
521. Dziechciaż M, Filip R. Biological psychological and social determinants of old age: bio-psycho-social aspects of human aging. Ann Agric Environ Med. 2014;21:835-8 Available at: https://pubmed.ncbi.nlm.nih.gov/25528930/
522. Johnson IP. Age-related neurodegenerative disease research needs aging models. Front Aging Neurosci. 2015 7. Available at: https://pubmed.ncbi.nlm.nih.gov/26388766/
523. Mammoto A, Matus K, Mammoto T. Extracellular Matrix in Aging Aorta. Front Cell Dev Biol. 2022;10:822561
524. Kirkwood TBL, Feder M, Finch CE. et al. What accounts for the wide variation in life span of genetically identical organisms reared in a constant environment? Mech Ageing Dev. 2005;126:439-43 Available at: https://pubmed.ncbi.nlm.nih.gov/15664632/
525. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194 Available at: https://pubmed.ncbi.nlm.nih.gov/23746838/
526. Panwar P, Butler GS, Jamroz A, Azizi P, Overall CM, Brömme D. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. Matrix Biol. 2018;65:30-44 Available at: https://pubmed.ncbi.nlm.nih.gov/28634008/
527. Coelho NM, McCulloch CA. Contribution of collagen adhesion receptors to tissue fibrosis. Cell Tissue Res. 2016;365:521-38 Available at: https://pubmed.ncbi.nlm.nih.gov/27351420/
528. Jones MG, Andriotis OG, Roberts JJW. et al. Nanoscale dysregulation of collagen structure-function disrupts mechano-homeostasis and mediates pulmonary fibrosis. Elife. 2018 7. Available at: https://pubmed.ncbi.nlm.nih.gov/29966587/
529. Ni J, Kanai M. Site-Selective Peptide/Protein Cleavage. Top Curr Chem. 2016;372:103-24 Available at: https://pubmed.ncbi.nlm.nih.gov/26251014/
530. Pulawski W, Ghoshdastider U, Andrisano V, Filipek S. Ubiquitous amyloids. Appl Biochem Biotechnol. 2012;166:1626-43 Available at: https://pubmed.ncbi.nlm.nih.gov/22350870/
531. Shin JW, Kwon SH, Choi JY. et al. Molecular Mechanisms of Dermal Aging and Antiaging Approaches. Int J Mol Sci. 2019;20:2126 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6540032/
532. Faragher RGA, McArdle A, Willows A, Ostler EL. Senescence in the aging process. F1000Res. 2017 6. Available at: https://pubmed.ncbi.nlm.nih.gov/28781767/
533. Klapper W, Kühne K, Singh KK, Heidorn K, Parwaresch R, Krupp G. Longevity of lobsters is linked to ubiquitous telomerase expression. FEBS Lett. 1998;439:143-6 Available at: https://pubmed.ncbi.nlm.nih.gov/9849895/
534. Klapper W, Heidorn K, Kühne K, Parwaresch R, Krupp G. Telomerase activity in 'immortal' fish. FEBS Lett. 1998;434:409-12 Available at: https://pubmed.ncbi.nlm.nih.gov/9742964/
535. Mavrogonatou E, Pratsinis H, Papadopoulou A, Karamanos NK, Kletsas D. Extracellular matrix alterations in senescent cells and their significance in tissue homeostasis. Matrix Biol. 2019;75-76:27-42 Available at: https://pubmed.ncbi.nlm.nih.gov/29066153/
536. MARTIN H, DEAN M. An N-terminal peptide from link protein is rapidly degraded by chondrocytes, monocytes and B cells. Eur J Biochem. 1993;212:87-94 Available at: https://pubmed.ncbi.nlm.nih.gov/8444167/
537. Marsh E, Gonzalez DG, Lathrop EA, Boucher J, Greco V. Positional Stability and Membrane Occupancy Define Skin Fibroblast Homeostasis In vivo. Cell. 2018;175:1620-1633.e13 Available at: https://pubmed.ncbi.nlm.nih.gov/30415836/
538. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. 2019;20:89 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7377404/
539. Ungvari Z, Tarantini S, Sorond F, Merkely B, Csiszar A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J Am Coll Cardiol. 2020;75:931-41 Available at: https://pubmed.ncbi.nlm.nih.gov/32130929/
540. Lacolley P, Regnault V, Segers P, Laurent S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol Rev. 2017;97:1555-617 Available at: https://pubmed.ncbi.nlm.nih.gov/28954852/
541. Haro Girón S, Monserrat Sanz J, Ortega MA. et al. Prognostic Value of Malondialdehyde (MDA) in the Temporal Progression of Chronic Spinal Cord Injury. J Pers Med. 2023 13. Available at: https://pubmed.ncbi.nlm.nih.gov/37109013/
542. Alfarhan M, Jafari E, Priya Narayanan S. Acrolein: A Potential Mediator of Oxidative Damage in Diabetic Retinopathy. Biomolecules. 2020;10:1-17 Available at: https://pubmed.ncbi.nlm.nih.gov/33233661/
543. Jin J, Liu Y, Huang L, Tan H. Advances in epigenetic regulation of vascular aging. Rev Cardiovasc Med. 2019;20:19-25 Available at: https://pubmed.ncbi.nlm.nih.gov/31184092/
544. Bhatia R, Thompson CM, Clement EJ. et al. Malondialdehyde-Acetaldehyde Extracellular Matrix Protein Adducts Attenuate Unfolded Protein Response During Alcohol and Smoking-Induced Pancreatitis. Gastroenterology. 2022;163:1064-1078.e10 Available at: https://pubmed.ncbi.nlm.nih.gov/35788346/
545. Zarkovic K, Larroque-Cardoso P, Pucelle M. et al. Elastin aging and lipid oxidation products in human aorta. Redox Biol. 2015;4:109-17
546. Behzadi P, St. Hilaire C. Aging Two-Step: SOX9's Influence on Vascular Stiffness and Senescence. Circ Res. 2024;134:325-7 Available at: https://pubmed.ncbi.nlm.nih.gov/38300983/
547. Ortega MA, De Leon-Oliva D, Gimeno-Longas MJ. et al. Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities. Biomolecules. 2024;14:275 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10968665/
548. Vitellaro-Zuccarello L, Cappelletti S, Rossi VDP, Sari-Gorla M. Stereological analysis of collagen and elastic fibers in the normal human dermis: Variability with age, sex, and body region. Anat Rec. 1994;238:153-62
549. Van Varik BJ, Rennenberg RJMW, Reutelingsperger CP, Kroon AA, De Leeuw PW, Schurgers LJ. Mechanisms of arterial remodeling: lessons from genetic diseases. Front Genet. 2012 3. Available at: https://pubmed.ncbi.nlm.nih.gov/23248645/
550. Cheng JK, Stoilov I, Mecham RP, Wagenseil JE. A fiber-based constitutive model predicts changes in amount and organization of matrix proteins with development and disease in the mouse aorta. Biomech Model Mechanobiol. 2013;12:497-510 Available at: https://pubmed.ncbi.nlm.nih.gov/22790326/
551. Wang M, Kim SH, Monticone RE, Lakatta EG. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. 2015;65:698-703 Available at: https://pubmed.ncbi.nlm.nih.gov/25667214/
552. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195-200 Available at: https://dx.doi.org/10.1242/jcs.023820
553. Rees SG, Dent CM, Caterson B. Metabolism of proteoglycans in tendon. Scand J Med Sci Sports. 2009;19:470-8
554. Screen HRC, Berk DE, Kadler KE, Ramirez F, Young MF. Tendon functional extracellular matrix. J Orthop Res. 2015;33:793-9 Available at: https://pubmed.ncbi.nlm.nih.gov/25640030/
555. Hudson DM, Archer M, Rai J, Weis MA, Fernandes RJ, Eyre DR. Age-related type I collagen modifications reveal tissue-defining differences between ligament and tendon. Matrix Biol Plus. 2021;12:100070
556. Han W, Wang B, Liu J, Chen L. The p16/miR-217/EGR1 pathway modulates age-related tenogenic differentiation in tendon stem/progenitor cells. Acta Biochim Biophys Sin (Shanghai). 2017;49:1015-21 Available at: https://pubmed.ncbi.nlm.nih.gov/29036495/
557. Svensson RB, Heinemeier KM, Couppé C, Kjaer M, Magnusson SP. Effect of aging and exercise on the tendon. J Appl Physiol. 2016;121:1353-62 Available at: https://journals.physiology.org/doi/10.1152/japplphysiol.00328.2016
558. Minkwitz S, Schmock A, Kurtoglu A. et al. Time-Dependent Alterations of MMPs, TIMPs and Tendon Structure in Human Achilles Tendons after Acute Rupture. International Journal of Molecular Sciences 2017, Vol 18, Page 2199. 2017;18:2199 Available at: https://www.mdpi.com/1422-0067/18/10/2199/htm
559. Lui PPY, Wong CM. Biology of Tendon Stem Cells and Tendon in Aging. Front Genet. 2020 10. Available at: https://pubmed.ncbi.nlm.nih.gov/32010194/
560. Turlo AJ, Ashraf Kharaz Y, Clegg PD, Anderson J, Peffers MJ. Donor age affects proteome composition of tenocyte-derived engineered tendon. BMC Biotechnol. 2018;18:1-11 Available at: https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-018-0414-5
561. Thorpe CT, Mcdermott BT, Goodship AE, Clegg PD, Birch HL. Ageing does not result in a decline in cell synthetic activity in an injury prone tendon. Scand J Med Sci Sports. 2016;26:684-93 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/sms.12500
562. Cabral-Pacheco GA, Garza-Veloz I, Rosa CCD La. et al. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int J Mol Sci. 2020;21:1-53 Available at: https://pubmed.ncbi.nlm.nih.gov/33419373/
563. Thorpe CT, Streeter I, Pinchbeck GL, Goodship AE, Clegg PD, Birch HL. Aspartic acid racemization and collagen degradation markers reveal an accumulation of damage in tendon collagen that is enhanced with aging. J Biol Chem. 2010;285:15674-81 Available at: https://pubmed.ncbi.nlm.nih.gov/20308077/
564. Thorpe CT, Mcdermott BT, Goodship AE, Clegg PD, Birch HL. Ageing does not result in a decline in cell synthetic activity in an injury prone tendon. Scand J Med Sci Sports. 2016;26:684-93 Available at: https://pubmed.ncbi.nlm.nih.gov/26058332/
565. Birch HL, Peffers MJ, Clegg PD. Influence of ageing on tendon homeostasis. Adv Exp Med Biol. 2016;920:247-60 Available at: https://link.springer.com/chapter/10.1007/978-3-319-33943-6_24
566. Wang T, Wagner A, Gehwolf R. et al. Load-induced regulation of tendon homeostasis by SPARC, a genetic predisposition factor for tendon and ligament injuries. Sci Transl Med. 2021 13. Available at: https://www.science.org/doi/10.1126/scitranslmed.abe5738
567. Ghanemi A, Melouane A, Yoshioka M, St-Amand J. Secreted Protein Acidic and Rich in Cysteine (Sparc) KO Leads to an Accelerated Ageing Phenotype Which Is Improved by Exercise Whereas SPARC Overexpression Mimics Exercise Effects in Mice. Metabolites. 2022;12:125 Available at: https://www.mdpi.com/2218-1989/12/2/125/htm
568. Ackerman JE, Bah I, Jonason JH, Buckley MR, Loiselle AE. Aging does not alter tendon mechanical properties during homeostasis, but does impair flexor tendon healing. Journal of Orthopaedic Research. 2017;35:2716-24 Available at: https://onlinelibrary.wiley.com/doi/full/10.1002/jor.23580
569. Delabastita T, Bogaerts S, Vanwanseele B. Age-Related Changes in Achilles Tendon Stiffness and Impact on Functional Activities: A Systematic Review and Meta-Analysis. J Aging Phys Act. 2019;27:116-27 Available at: https://journals.humankinetics.com/view/journals/japa/27/1/article-p116.xml
570. Magnusson SP, Kjaer M. The impact of loading, unloading, ageing and injury on the human tendon. J Physiol. 2019;597:1283-98 Available at: https://onlinelibrary.wiley.com/doi/full/10.1113/JP275450
571. Lacroix AS, Duenwald-Kuehl SE, Brickson S. et al. Effect of age and exercise on the viscoelastic properties of rat tail Tendon. Ann Biomed Eng. 2013;41:1120-8 Available at: https://link.springer.com/article/10.1007/s10439-013-0796-4
572. Peffers MJ, Thorpe CT, Collins JA. et al. Proteomic analysis reveals age-related changes in tendon matrix composition, with age- and injury-specific matrix fragmentation. J Biol Chem. 2014;289:25867-78 Available at: https://pubmed.ncbi.nlm.nih.gov/25077967/
573. Stenroth L, Peltonen J, Cronin NJ, Sipilä S, Finni T. Age-related differences in Achilles tendon properties and triceps surae muscle architecture in vivo. J Appl Physiol. 2012;113:1537-44 Available at: https://journals.physiology.org/doi/10.1152/japplphysiol.00782.2012
574. Diekman BO, Loeser RF. Aging and the emerging role of cellular senescence in osteoarthritis. Osteoarthritis Cartilage. 2024;32:365-71
575. Dudhia J. Aggrecan, aging and assembly in articular cartilage. Cell Mol Life Sci. 2005;62:2241-56 Available at: https://pubmed.ncbi.nlm.nih.gov/16143826/
576. Olszewski J, McDonnell J, Stevens K, Visco D, Moore V. A matrix metalloproteinase-generated aggrecan neoepitope as a marker of skeletal maturation and aging in cartilage. Arthritis Rheum. 1996;39:1234-7 Available at: https://pubmed.ncbi.nlm.nih.gov/8670336/
577. Fan M, Wang C, Kwok B. et al. Impacts of aging on murine cartilage biomechanics and chondrocyte in situ calcium signaling. J Biomech. 2022 144. Available at: https://pubmed.ncbi.nlm.nih.gov/36240656/
578. Wu W, Billinghurst RC, Pidoux I. et al. Sites of collagenase cleavage and denaturation of type II collagen in aging and osteoarthritic articular cartilage and their relationship to the distribution of matrix metalloproteinase 1 and matrix metalloproteinase 13. Arthritis Rheum. 2002;46:2087-94 Available at: https://pubmed.ncbi.nlm.nih.gov/12209513/
579. Birch HL, Birch HL. Extracellular Matrix and Ageing Extracellular Matrix Distribution. Biochemistry and Cell Biology of Ageing: Part I Biomedical Science.; Available at: https://doi.org/10.1007/978-981-13-2835-0_7.
580. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118 Available at: https://pubmed.ncbi.nlm.nih.gov/20078217/
581. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99-114 Available at: https://pubmed.ncbi.nlm.nih.gov/24449267/
582. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685-705 Available at: https://pubmed.ncbi.nlm.nih.gov/23140366/
583. Bartkova J, Hořejší Z, Koed K. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864-70 Available at: https://pubmed.ncbi.nlm.nih.gov/15829956/
584. Favetta LA, Madan P, Mastromonaco GF, St John EJ, King WA, Betts DH. The oxidative stress adaptor p66Shc is required for permanent embryo arrest in vitro. BMC Dev Biol. 2007 7. Available at: https://pubmed.ncbi.nlm.nih.gov/18047664/
585. Morty RE, Prakash YS. Senescence in the lung: is this getting old? Am J Physiol Lung Cell Mol Physiol. 2019;316:L822-5 Available at: https://pubmed.ncbi.nlm.nih.gov/30892079/
586. Aghali A, Khalfaoui L, Lagnado AB. et al. Cellular senescence is increased in airway smooth muscle cells of elderly persons with asthma. Am J Physiol Lung Cell Mol Physiol. 2022;323:L558-68 Available at: https://pubmed.ncbi.nlm.nih.gov/36166734/
587. Mebratu YA, Soni S, Rosas L, Rojas M, Horowitz JC, Nho R. The aged extracellular matrix and the profibrotic role of senescence-associated secretory phenotype. Am J Physiol Cell Physiol. 2023;325:C565 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10511170/
588. De-Carvalho DP, Jacinto A, Saúde L. The right time for senescence. Elife. 2021 10. Available at: https://pubmed.ncbi.nlm.nih.gov/34756162/
589. Burton DGA, Stolzing A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res Rev. 2018;43:17-25 Available at: https://pubmed.ncbi.nlm.nih.gov/29427795/
590. Waaijer MEC, Goldeck D, Gunn DA. et al. Are skin senescence and immunosenescence linked within individuals? Aging Cell. 2019 18. Available at: https://pubmed.ncbi.nlm.nih.gov/31062498/
591. Choi HR, Cho KA, Kang HT. et al. Restoration of senescent human diploid fibroblasts by modulation of the extracellular matrix. Aging Cell. 2011;10:148-57 Available at: https://pubmed.ncbi.nlm.nih.gov/21108727/
592. Horowitz JC, Thannickal VJ. Mechanisms for the Resolution of Organ Fibrosis. Physiology (Bethesda). 2019;34:43-55 Available at: https://pubmed.ncbi.nlm.nih.gov/30540232/
593. Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012;72:2251-61 Available at: https://pubmed.ncbi.nlm.nih.gov/22422937/
594. Karin O, Agrawal A, Porat Z, Krizhanovsky V, Alon U. Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nat Commun. 2019 10. Available at: https://pubmed.ncbi.nlm.nih.gov/31792199/
595. Qin Z, He T, Guo C, Quan T. Age-Related Downregulation of CCN2 Is Regulated by Cell Size in a YAP/TAZ-Dependent Manner in Human Dermal Fibroblasts: Impact on Dermal Aging. JID innovations: skin science from molecules to population health. 2022 2. Available at: https://pubmed.ncbi.nlm.nih.gov/35480397/
596. Qin Z, Xia W, Fisher GJ, Voorhees JJ, Quan T. YAP/TAZ regulates TGF-β/Smad3 signaling by induction of Smad7 via AP-1 in human skin dermal fibroblasts. Cell Commun Signal. 2018;16:18 Available at: https://pubmed.ncbi.nlm.nih.gov/29695252/
597. Ichijo R, Maki K, Kabata M. et al. Vasculature atrophy causes a stiffened microenvironment that augments epidermal stem cell differentiation in aged skin. Nat Aging. 2022;2:592-600 Available at: https://pubmed.ncbi.nlm.nih.gov/37117774/
598. Sladitschek-Martens HL, Guarnieri A, Brumana G. et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature. 2022;607:790-8 Available at: https://pubmed.ncbi.nlm.nih.gov/35768505/
599. Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013;162:156-73 Available at: https://pubmed.ncbi.nlm.nih.gov/23831269/
600. Prado-Audelo ML Del, Mendoza-Muñoz N, Escutia-Guadarrama L. et al. Recent Advances in Elastin-Based Biomaterial. J Pharm Pharm Sci. 2020;23:314-32 Available at: https://pubmed.ncbi.nlm.nih.gov/33751927/
601. Levi N, Papismadov N, Solomonov I, Sagi I, Krizhanovsky V, Krizhanovsky V. The ECM path of senescence in aging: components and modifiers.; Available at: https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.15282
602. Chin T, Lee XE, Ng PY, Lee Y, Dreesen O. The role of cellular senescence in skin aging and age-related skin pathologies. Front Physiol. 2023 14. Available at: https://pubmed.ncbi.nlm.nih.gov/38074322/
603. Blokland KEC, Habibie H, Borghuis T. et al. Regulation of Cellular Senescence Is Independent from Profibrotic Fibroblast-Deposited ECM. Cells. 2021 10. Available at: https://pubmed.ncbi.nlm.nih.gov/34209854/
604. Solé-Boldo L, Raddatz G, Schütz S. et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Communications Biology 2020 3:1. 2020;3:1-12 Available at: https://www.nature.com/articles/s42003-020-0922-4
605. Zou Z, Long X, Zhao Q. et al. A Single-Cell Transcriptomic Atlas of Human Skin Aging. Dev Cell. 2021;56:383-397.e8 Available at: https://www.cell.com/action/showFullText?pii=S1534580720308777
606. Wlaschek M, Maity P, Makrantonaki E, Scharffetter-Kochanek K. Connective Tissue and Fibroblast Senescence in Skin Aging. Journal of Investigative Dermatology. 2021;141:985-92 Available at: https://www.jidonline.org/action/showFullText?pii=S0022202X20323514
607. da Silva PFL, Ogrodnik M, Kucheryavenko O. et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell. 2019;18:e12848 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/acel.12848
608. Farage MA, Miller KW, Elsner P, Maibach HI. Intrinsic and extrinsic factors in skin ageing: a review. Int J Cosmet Sci. 2008;30:87-95 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1468-2494.2007.00415.x
609. Qin Z, Balimunkwe RM, Quan T. Age-related reduction of dermal fibroblast size upregulates multiple matrix metalloproteinases as observed in aged human skin in vivo. British Journal of Dermatology. 2017;177:1337-48 Available at: https://dx.doi.org/10.1111/bjd.15379
610. Quan T, Little E, Quan H, Qin Z, Voorhees JJ, Fisher GJ. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: Impact of altered extracellular matrix microenvironment on dermal fibroblast function. Journal of Investigative Dermatology. 2013;133:1362-6 Available at: https://www.jidonline.org/action/showFullText?pii=S0022202X15362370
611. Xia W, Hammerberg C, Li Y. et al. Expression of catalytically active matrix metalloproteinase-1 in dermal fibroblasts induces collagen fragmentation and functional alterations that resemble aged human skin. Aging Cell. 2013;12:661-71 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/acel.12089
612. Quan T, Xia W, He T. et al. Matrix Metalloproteinase-1 Expression in Fibroblasts Accelerates Dermal Aging and Promotes Papilloma Development in Mouse Skin. Journal of Investigative Dermatology. 2023;143:1700-1707.e1 Available at: https://www.jidonline.org/action/showFullText?pii=S0022202X23018274
613. Yokose U, Hachiya A, Sriwiriyanont P. et al. The endogenous protease inhibitor TIMP-1 mediates protection and recovery from Cutaneous Photodamage. Journal of Investigative Dermatology. 2012;132:2800-9 Available at: https://www.jidonline.org/action/showFullText?pii=S0022202X15355561
614. de Bengy AF, Lamartine J, Sigaudo-Roussel D, Fromy B. Newborn and elderly skin: two fragile skins at higher risk of pressure injury. Biological Reviews. 2022;97:874-95 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/brv.12827
615. Karsdal M, Cox TR, Parker AL. et al. Advances in Extracellular Matrix-Associated Diagnostics and Therapeutics. Journal of Clinical Medicine 2025, Vol 14, Page 1856. 2025;14:1856 Available at: https://www.mdpi.com/2077-0383/14/6/1856/htm
616. He T, Quan T, Shao Y, Voorhees JJ, Fisher GJ. Oxidative exposure impairs TGF-β pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age (Omaha). 2014;36:1079-94 Available at: https://link.springer.com/article/10.1007/s11357-014-9623-6
617. Quan T, He T, Kang S, Voorhees JJ, Fisher GJ. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-β type II receptor/Smad signaling. American Journal of Pathology. 2004;165:741-51 Available at: https://ajp.amjpathol.org/action/showFullText?pii=S0002944010633378
618. Purohit T, He T, Qin Z. et al. Smad3-dependent regulation of type I collagen in human dermal fibroblasts: Impact on human skin connective tissue aging. J Dermatol Sci. 2016;83:80-3 Available at: https://www.jdsjournal.com/action/showFullText?pii=S0923181116300597
619. Aging in the dermis: Fibroblast senescence and its significance | Enhanced Reader
620. Fisher GJ, Shao Y, He T. et al. Reduction of fibroblast size/mechanical force down-regulates TGF-β type II receptor: implications for human skin aging. Aging Cell. 2016;15:67-76 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/acel.12410
621. Proudfoot D. Calcium Signaling and Tissue Calcification. Cold Spring Harb Perspect Biol. 2019 11. Available at: https://pubmed.ncbi.nlm.nih.gov/31138543/
622. De Leon-Oliva D, Barrena-Blázquez S, Jiménez-Álvarez L. et al. The RANK-RANKL-OPG System: A Multifaceted Regulator of Homeostasis, Immunity, and Cancer. Medicina (Kaunas). 2023 59. Available at: https://pubmed.ncbi.nlm.nih.gov/37893470/
623. Murshed M. Mechanism of Bone Mineralization. Cold Spring Harb Perspect Med. 2018 8. Available at: https://pubmed.ncbi.nlm.nih.gov/29610149/
624. De Leon-Oliva D, Boaru DL, Perez-Exposito RE. et al. Advanced Hydrogel-Based Strategies for Enhanced Bone and Cartilage Regeneration: A Comprehensive Review. Gels. 2023 9. Available at: https://pubmed.ncbi.nlm.nih.gov/37998975/
625. Fraile-martínez O, García-montero C, Coca A. et al. Applications of Polymeric Composites in Bone Tissue Engineering and Jawbone Regeneration. Polymers (Basel). 2021 13. Available at: https://pubmed.ncbi.nlm.nih.gov/34641243/
626. Chakrabarti A, Goldstein DR, Sutton NR. Age-associated arterial calcification: the current pursuit of aggravating and mitigating factors. Curr Opin Lipidol. 2020;31:265-72 Available at: https://pubmed.ncbi.nlm.nih.gov/32773466/
627. Vidavsky N, Kunitake JAMR, Estroff LA. Multiple Pathways for Pathological Calcification in the Human Body. Adv Healthc Mater. 2021 10. Available at: https://pubmed.ncbi.nlm.nih.gov/33274854/
628. Veis A, Dorvee JR. Biomineralization mechanisms: a new paradigm for crystal nucleation in organic matrices. Calcif Tissue Int. 2013;93:307-15 Available at: https://pubmed.ncbi.nlm.nih.gov/23241924/
629. Nakahara T, Dweck MR, Narula N, Pisapia D, Narula J, Strauss HW. Coronary Artery Calcification: From Mechanism to Molecular Imaging. JACC Cardiovasc Imaging. 2017;10:582-93 Available at: https://pubmed.ncbi.nlm.nih.gov/28473100/
630. Kwak SY, Green S, Wiedemann-Bidlack FB. et al. Regulation of calcium phosphate formation by amelogenins under physiological conditions. Eur J Oral Sci. 2011;119(Suppl 1):103-11 Available at: https://pubmed.ncbi.nlm.nih.gov/22243235/
631. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34:715-23 Available at: https://pubmed.ncbi.nlm.nih.gov/24665125/
632. Bonetti A, Mora A Della, Contin M, Tubaro F, Marchini M, Ortolani F. Ultrastructural and Spectrophotometric Study on the Effects of Putative Triggers on Aortic Valve Interstitial Cells in In vitro Models Simulating Metastatic Calcification. Anatomical Record. 2012;295:1117-27
633. Cheng K, Li J, Liu X. Metastatic calcification from secondary hyperparathyroidism. Am J Med Sci. 2024;368:e15-6 Available at: https://pubmed.ncbi.nlm.nih.gov/38460923/
634. Turner JJO. Hypercalcaemia - presentation and management. Clin Med (Lond). 2017;17:270-3 Available at: https://pubmed.ncbi.nlm.nih.gov/28572230/
635. Thoma A. Unusual Cause of Iatrogenic Calcifications. J Clin Rheumatol. 2023;29:E6-9 Available at: https://pubmed.ncbi.nlm.nih.gov/36251473/
636. Valenzuela A, Chung L. Calcinosis: pathophysiology and management. Curr Opin Rheumatol. 2015;27:542-8 Available at: https://pubmed.ncbi.nlm.nih.gov/26352733/
637. Ibragimova AG, Stanishevskiy YM, Plakkhin AM. et al. Comparative analysis of calcified soft tissues revealed shared deregulated pathways. Front Aging Neurosci. 2023 15. Available at: https://pubmed.ncbi.nlm.nih.gov/37441678/
638. Ortega MA, Saez MÁ, Asúnsolo Á. et al. Upregulation of VEGF and PEDF in Placentas of Women with Lower Extremity Venous Insufficiency during Pregnancy and Its Implication in Villous Calcification. Biomed Res Int. 2019. 2019 Available at: https://pubmed.ncbi.nlm.nih.gov/31886225/
639. Kosuru V, Mohammed A, Kapoor R. et al. Metastatic Calcinosis of Gastric Mucosa. J Investig Med High Impact Case Rep. 2020 8. Available at: https://pubmed.ncbi.nlm.nih.gov/32677845/
640. Jarjou'i A, Bogot N, Kalak G. et al. Diffuse pulmonary calcifications: A case series and review of literature. Respirol Case Rep. 2021 9. Available at: https://pubmed.ncbi.nlm.nih.gov/34484796/
641. Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008;117:2938-48 Available at: https://pubmed.ncbi.nlm.nih.gov/18519861/
642. Bonetti A, Marchini M, Ortolani F. Ectopic mineralization in heart valves: new insights from in vivo and in vitro procalcific models and promising perspectives on noncalcifiable bioengineered valves. J Thorac Dis. 2019;11:2126-43 Available at: https://pubmed.ncbi.nlm.nih.gov/31285908/
643. Wu M, Rementer C, Giachelli CM. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int. 2013;93:365-73 Available at: https://pubmed.ncbi.nlm.nih.gov/23456027/
644. Atkins SK, Aikawa E. Valve under the microscope: shining a light on emerging technologies elucidating disease mechanisms. Heart. 2019;105:1610-1 Available at: https://pubmed.ncbi.nlm.nih.gov/31296591/
645. Fishbein GA, Fishbein MC. Pathology of the Aortic Valve: Aortic Valve Stenosis/Aortic Regurgitation. Curr Cardiol Rep. 2019;21:1-9 Available at: https://link.springer.com/article/10.1007/s11886-019-1162-4
646. Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental Mechanisms of Aortic Valve Malformation and Disease. Annu Rev Physiol. 2017;79:21-41 Available at: https://www.annualreviews.org/content/journals/10.1146/annurev-physiol-022516-034001
647. Liu T, Xie M, Lv Q. et al. Bicuspid aortic valve: An update in morphology, genetics, biomarker, complications, imaging diagnosis and treatment. Front Physiol. 2019;10:433531 Available at: www.frontiersin.org
648. Latif N, Sarathchandra P, Chester AH, Yacoub MH. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. 2015;36:1335-45 Available at: https://dx.doi.org/10.1093/eurheartj/eht547
649. Perrotta I, Sciangula A, Aquila S, Mazzulla S. Matrix Metalloproteinase-9 Expression in Calcified Human Aortic Valves: A Histopathologic, Immunohistochemical, and Ultrastructural Study. Appl Immunohistochem Mol Morphol. 2016;24:128-37 Available at: https://pubmed.ncbi.nlm.nih.gov/25390353/
650. Clift CL, Blaser MC, Gerrits W. et al. Intracellular proteomics and extracellular vesiculomics as a metric of disease recapitulation in 3D-bioprinted aortic valve arrays. Sci Adv. 2024 10. Available at: https://pubmed.ncbi.nlm.nih.gov/38416823/
651. Büttner P, Feistner L, Lurz P, Thiele H, Hutcheson JD, Schlotter F. Dissecting Calcific Aortic Valve Disease-The Role, Etiology, and Drivers of Valvular Fibrosis. Front Cardiovasc Med. 2021 8. Available at: https://pubmed.ncbi.nlm.nih.gov/34041283/
652. Schoen FJ. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008;118:1864-80 Available at: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.108.805911
653. Hutcheson JD, Goettsch C, Rogers MA, Aikawa E. Revisiting cardiovascular calcification: A multifaceted disease requiring a multidisciplinary approach. Semin Cell Dev Biol. 2015;46:68-77
654. Nagy E, Eriksson P, Yousry M. et al. Valvular osteoclasts in calcification and aortic valve stenosis severity. Int J Cardiol. 2013;168:2264-71 Available at: https://www.internationaljournalofcardiology.com/action/showFullText?pii=S0167527313002672
655. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: Methods, models, and mechanisms. Circ Res. 2011;108:1392-412 Available at: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.110.234138
656. Yip CYY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936-42 Available at: https://pubmed.ncbi.nlm.nih.gov/19304575/
657. Perrotta I, Carito V, Russo E, Tripepi S, Aquila S, Donato G. Macrophage Autophagy and Oxidative Stress: An Ultrastructural and Immunoelectron Microscopical Study. Oxid Med Cell Longev. 2011;2011:282739 Available at: https://onlinelibrary.wiley.com/doi/full/10.1155/2011/282739
658. Bouchareb R, Boulanger MC, Fournier D, Pibarot P, Messaddeq Y, Mathieu P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J Mol Cell Cardiol. 2014;67:49-59 Available at: https://pubmed.ncbi.nlm.nih.gov/24368096/
659. McGough IJ, Vincent JP. Exosomes in developmental signalling. Development. 2016;143:2482-93 Available at: https://dx.doi.org/10.1242/dev.126516
660. Liu Q, Piao H, Wang Y, Zheng D, Wang W. Circulating exosomes in cardiovascular disease: Novel carriers of biological information. Biomedicine & Pharmacotherapy. 2021;135:111148
661. Blaser MC, Aikawa E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front Cardiovasc Med. 2018;5:428908 Available at: www.frontiersin.org
662. Mahmut A, Boulanger MC, Bouchareb R, Hadji F, Mathieu P. Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc Res. 2015;106:109-20 Available at: https://dx.doi.org/10.1093/cvr/cvv027
663. Cardoso L, Weinbaum S. Microcalcifications, their genesis, growth, and biomechanical stability in fibrous cap rupture. Adv Exp Med Biol. 2018;1097:129-55 Available at: https://link.springer.com/chapter/10.1007/978-3-319-96445-4_7
664. Zazzeroni L, Faggioli G, Pasquinelli G. Mechanisms of Arterial Calcification: The Role of Matrix Vesicles. Eur J Vasc Endovasc Surg. 2018;55:425-32 Available at: https://pubmed.ncbi.nlm.nih.gov/29371036/
665. Kapustin AN, Chatrou MLL, Drozdov I. et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312-23 Available at: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.116.305012
666. Latif N, Sarathchandra P, Taylor PM, Antoniw J, Yacoub MH. Molecules mediating cell-ECM and cell-cell communication in human heart valves. Cell Biochem Biophys. 2005;43:275-87
667. Hutson HN, Marohl T, Anderson M, Eliceiri K, Campagnola P, Masters KS. Calcific Aortic Valve Disease Is Associated with Layer-Specific Alterations in Collagen Architecture. PLoS One. 2016 11. Available at: https://pubmed.ncbi.nlm.nih.gov/27685946/
668. Rodriguez KJ, Piechura LM, Porras AM, Masters KS. Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc Disord. 2014;14:1-10 Available at: https://bmccardiovascdisord.biomedcentral.com/articles/10.1186/1471-2261-14-29
669. Salhiyyah K, Sarathchandra P, Latif N, Yacoub MH, Chester AH. Hypoxia-mediated regulation of the secretory properties of mitral valve interstitial cells. Am J Physiol Heart Circ Physiol. 2017;313:H14-23 Available at: https://pubmed.ncbi.nlm.nih.gov/28314761/
670. D'Ignazio L, Bandarra D, Rocha S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016;283:413-24 Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/febs.13578
671. Malara NM, Leotta A, Sidoti A. et al. Ageing, hormonal behaviour and cyclin D1 in ductal breast carcinomas. The Breast. 2006;15:81-9
672. Swaminathan G, Krishnamurthy VK, Sridhar S, Robson DC, Ning Y, Grande-Allen KJ. Hypoxia Stimulates Synthesis of Neutrophil Gelatinase-Associated Lipocalin in Aortic Valve Disease. Front Cardiovasc Med. 2019;6:473567 Available at: www.frontiersin.org
673. Johnson VL, Hunter DJ. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. 2014;28:5-15 Available at: https://pubmed.ncbi.nlm.nih.gov/24792942/
674. Østerås N, Blaker IB, Hjortland T. et al. Improving osteoarthritis management in primary healthcare: results from a quasi-experimental study. BMC Musculoskelet Disord. 2021 22. Available at: https://pubmed.ncbi.nlm.nih.gov/33446167/
675. Hawellek T, Hubert J, Hischke S. et al. Microcalcification of lumbar spine intervertebral discs and facet joints is associated with cartilage degeneration, but differs in prevalence and its relation to age. J Orthop Res. 2017;35:2692-9 Available at: https://pubmed.ncbi.nlm.nih.gov/28467655/
676. Sun Y, Franklin AM, Mauerhan DR, Hanley EN. Biological Effects of Phosphocitrate on Osteoarthritic Articular Chondrocytes. Open Rheumatol J. 2017;11:62-74 Available at: https://pubmed.ncbi.nlm.nih.gov/28659999/
677. Spittau B, Krieglstein K. Klf10 and Klf11 as mediators of TGF-beta superfamily signaling. Cell Tissue Res. 2012;347:65-72 Available at: https://pubmed.ncbi.nlm.nih.gov/21574058/
678. Subramaniam M, Harris SA, Oursler MJ, Rasmussen K, Riggs BL, Spelsberg TC. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995;23:4907-12 Available at: https://pubmed.ncbi.nlm.nih.gov/8532536/
679. Zheng L, Lu H, Li H, Xu X, Wang D. KLF10 is upregulated in osteoarthritis and inhibits chondrocyte proliferation and migration by upregulating Acvr1 and suppressing inhbb expression. Acta Histochem. 2020 122. Available at: https://pubmed.ncbi.nlm.nih.gov/32156482/
680. Sun MMG, Beier F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res C Embryo Today. 2014;102:74-82 Available at: https://pubmed.ncbi.nlm.nih.gov/24677724/
681. Sompel K, Dwyer-Nield LD, Smith AJ. et al. Loss of Frizzled 9 in Lung Cells Alters Epithelial Phenotype and Promotes Premalignant Lesion Development. Front Oncol. 2022 12. Available at: https://pubmed.ncbi.nlm.nih.gov/35924166/
682. Heilmann A, Schinke T, Bindl R. et al. The Wnt serpentine receptor Frizzled-9 regulates new bone formation in fracture healing. PLoS One. 2013 8. Available at: https://pubmed.ncbi.nlm.nih.gov/24391920/
683. Albers J, Schulze J, Beil FT. et al. Control of bone formation by the serpentine receptor Frizzled-9. J Cell Biol. 2011;192:1057-72 Available at: https://pubmed.ncbi.nlm.nih.gov/21402791/
684. Stücker S, Bollmann M, Garbers C, Bertrand J. The role of calcium crystals and their effect on osteoarthritis pathogenesis. Best Pract Res Clin Rheumatol. 2021 35. Available at: https://pubmed.ncbi.nlm.nih.gov/34732285/
685. Shang J, Lin N, Peng R. et al. Inhibition of Klf10 Attenuates Oxidative Stress-Induced Senescence of Chondrocytes via Modulating Mitophagy. Molecules. 2023 28. Available at: https://pubmed.ncbi.nlm.nih.gov/36770589/
686. Peng R, Shang J, Jiang N. et al. Klf10 is involved in extracellular matrix calcification of chondrocytes alleviating chondrocyte senescence. J Transl Med. 2024 22. Available at: https://pubmed.ncbi.nlm.nih.gov/38217021/
687. Towler DA. Oxidation, inflammation, and aortic valve calcification peroxide paves an osteogenic path. J Am Coll Cardiol. 2008;52:851-4 Available at: https://pubmed.ncbi.nlm.nih.gov/18755349/
688. Messika-Zeitoun D, Bielak LF, Peyser PA. et al. Aortic valve calcification: determinants and progression in the population. Arterioscler Thromb Vasc Biol. 2007;27:642-8 Available at: https://pubmed.ncbi.nlm.nih.gov/17185617/
689. Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510-24 Available at: https://pubmed.ncbi.nlm.nih.gov/21659654/
690. Thromb A, Biol V. Results Cad-11 OX Induces ECM Remodeling but Is Not Embryonic Lethal. 2016; Available at: http://ahajournals.org.
691. Kostyunin AE, Yuzhalin AE, Ovcharenko EA, Kutikhin AG. Development of calcific aortic valve disease: Do we know enough for new clinical trials? J Mol Cell Cardiol. 2019;132:189-209 Available at: https://pubmed.ncbi.nlm.nih.gov/31136747/
692. Sung DC, Bowen CJ, Vaidya KA. et al. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arterioscler Thromb Vasc Biol. 2016;36:1627-37 Available at: https://pubmed.ncbi.nlm.nih.gov/27312222/
693. Farrar EJ, Pramil V, Richards JM, Mosher CZ, Butcher JT. Valve interstitial cell tensional homeostasis directs calcification and extracellular matrix remodeling processes via RhoA signaling. Biomaterials. 2016;105:25-37 Available at: https://pubmed.ncbi.nlm.nih.gov/27497058/
694. Li X, Lim J, Lu J, Pedego TM, Demer L, Tintut Y. Protective Role of Smad6 in Inflammation-Induced Valvular Cell Calcification. J Cell Biochem. 2015;116:2354-64 Available at: https://pubmed.ncbi.nlm.nih.gov/25864564/
695. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010;10:712-23 Available at: https://pubmed.ncbi.nlm.nih.gov/20865019/
696. Yu Z, Seya K, Daitoku K, Motomura S, Fukuda I, Furukawa KI. Tumor necrosis factor-α accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther. 2011;337:16-23 Available at: https://pubmed.ncbi.nlm.nih.gov/21205918/
697. Cottignoli V, Relucenti M, Agrosì G. et al. Biological Niches within Human Calcified Aortic Valves: Towards Understanding of the Pathological Biomineralization Process. Biomed Res Int. 2015. 2015 Available at: https://pubmed.ncbi.nlm.nih.gov/26509159/
698. Parhami F, Morrow AD, Balucan J. et al. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680-7 Available at: https://pubmed.ncbi.nlm.nih.gov/9108780/
699. Al-Aly Z, Shao JS, Lai CF. et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler Thromb Vasc Biol. 2007;27:2589-96 Available at: https://pubmed.ncbi.nlm.nih.gov/17932314/
700. Aikawa E, Nahrendorf M, Figueiredo JL. et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841-50 Available at: https://pubmed.ncbi.nlm.nih.gov/18040026/
701. Shroff RC, McNair R, Skepper JN. et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol. 2010;21:103-12 Available at: https://pubmed.ncbi.nlm.nih.gov/19959717/
702. Ortega MA, Fraile-Martínez O, García-Montero C. et al. The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease. Cells. 2022 11. Available at: https://pubmed.ncbi.nlm.nih.gov/35159377/
703. García-Montero C, Fraile-Martinez O, De Leon-Oliva D. et al. Exploring the Role of Mediterranean and Westernized Diets and Their Main Nutrients in the Modulation of Oxidative Stress in the Placenta: A Narrative Review. Antioxidants (Basel). 2023 12. Available at: https://pubmed.ncbi.nlm.nih.gov/38001771/
704. Ortega MA, Asúnsolo Á, Fraile-Martínez O. et al. An increase in elastogenic components in the placental villi of women with chronic venous disease during pregnancy is associated with decreased EGFL7 expression level. Mol Med Rep. 2021 24. Available at: https://pubmed.ncbi.nlm.nih.gov/34080027/
705. Ortega MA, Fraile-Martínez O, Saez MA. et al. Abnormal proinflammatory and stressor environmental with increased the regulatory cellular IGF-1/PAPP-A/STC and Wnt-1/β-Catenin canonical pathway in placenta of women with Chronic venous Disease during Pregnancy. Int J Med Sci. 2021;18:2814-27 Available at: https://pubmed.ncbi.nlm.nih.gov/34220309/
706. Ortega MA, Fraile-Martinez O, García-Montero C. et al. The Placentas of Women Who Suffer an Episode of Psychosis during Pregnancy Have Increased Lipid Peroxidation with Evidence of Ferroptosis. Biomolecules. 2023 13. Available at: https://pubmed.ncbi.nlm.nih.gov/36671505/
707. Ortega MA, Saez MÁ, Asúnsolo Á. et al. Upregulation of VEGF and PEDF in Placentas of Women with Lower Extremity Venous Insufficiency during Pregnancy and Its Implication in Villous Calcification. Biomed Res Int. 2019. 2019 Available at: https://pubmed.ncbi.nlm.nih.gov/31886225/
708. Sharp AN, Heazell AEP, Crocker IP, Mor G. Placental Apoptosis in Health and Disease. Am J Reprod Immunol. 2010;64:159 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3025811/
709. Pietro L, Daher S, Rudge MVC. et al. Vascular endothelial growth factor (VEGF) and VEGF-receptor expression in placenta of hyperglycemic pregnant women. Placenta. 2010;31:770-80 Available at: https://pubmed.ncbi.nlm.nih.gov/20674013/
710. Mlambo ZP, Sebitloane M, Naicker T. Association of angiogenic factors (placental growth factor and soluble FMS-like tyrosine kinase-1) in preeclamptic women of African ancestry comorbid with HIV infection. Arch Gynecol Obstet. 2024;311:259-74 Available at: https://link.springer.com/article/10.1007/s00404-024-07590-3
711. Stepan H, Herraiz I, Schlembach D. et al. Implementation of the sFlt-1/PlGF ratio for prediction and diagnosis of pre-eclampsia in singleton pregnancy: implications for clinical practice. Ultrasound in Obstetrics & Gynecology. 2015;45:241 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4369131/
712. LeRoith D, Holly JMP, Forbes BE. Insulin-like growth factors: Ligands, binding proteins, and receptors. Mol Metab. 2021;52:101245 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8513159/
713. Chassen S, Jansson T. Complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity in IUGR. Biochim Biophys Acta Mol Basis Dis. 2019;1866:165373 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6650384/
714. Davenport BN, Wilson RL, Williams AA, Jones HN. Placental nanoparticle-mediated IGF1 gene therapy corrects fetal growth restriction in a guinea pig model. Gene Ther. 2024 Available at: https://pubmed.ncbi.nlm.nih.gov/39627510/
715. Boldt HB, Conover CA. Pregnancy-associated plasma protein-A (PAPP-A): a local regulator of IGF bioavailability through cleavage of IGFBPs. Growth Horm IGF Res. 2007;17:10-8 Available at: https://pubmed.ncbi.nlm.nih.gov/17218136/
716. Gumina DL, Su EJ. Mechanistic insights into the development of severe fetal growth restriction. Clin Sci (Lond). 2023;137:679 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10241202/
717. Pascual-Mancho J, Pintado-Recarte P, Romero-Román C. et al. Influence of Cerebral Vasodilation on Blood Reelin Levels in Growth Restricted Fetuses. Diagnostics (Basel). 2021 11. Available at: https://pubmed.ncbi.nlm.nih.gov/34199942/
718. Ortega MA, Asúnsolo Á, Álvarez-Rocha MJ. et al. Remodelling of collagen fibres in the placentas of women with venous insufficiency during pregnancy. Histol Histopathol. 2018;33:567-76 Available at: https://www.researchgate.net/publication/321299752_Remodelling_of_collagen_fibres_in_the_placentas_of_women_with_venous_insufficiency_during_pregnancy
719. Lim J, Ehsanipour A, Hsu JJ. et al. Inflammation Drives Retraction, Stiffening, and Nodule Formation via Cytoskeletal Machinery in a Three-Dimensional Culture Model of Aortic Stenosis. Am J Pathol. 2016;186:2378-89 Available at: https://pubmed.ncbi.nlm.nih.gov/27392969/
720. Fayet C, Bendeck MP, Gotlieb AI. Cardiac valve interstitial cells secrete fibronectin and form fibrillar adhesions in response to injury. Cardiovasc Pathol. 2007;16:203-11 Available at: https://pubmed.ncbi.nlm.nih.gov/17637428/
721. Fondard O, Detaint D, Iung B. et al. Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur Heart J. 2005;26:1333-41 Available at: https://pubmed.ncbi.nlm.nih.gov/15827062/
722. Kaden JJ, Dempfle CE, Grobholz R. et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol. 2005;14:80-7 Available at: https://pubmed.ncbi.nlm.nih.gov/15780799/
723. Hsu JJ, Lim J, Tintut Y, Demer LL. Cell-Matrix Mechanics and Pattern Formation in Inflammatory Cardiovascular Calcification. Heart. 2016;102:1710 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5069068/
724. Inoue K, Mikuni-Takagaki Y, Oikawa K. et al. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 2006;281:33814-24 Available at: https://pubmed.ncbi.nlm.nih.gov/16959767/
725. Mosig RA, Dowling O, DiFeo A. et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16:1113-23 Available at: https://pubmed.ncbi.nlm.nih.gov/17400654/
726. Nandadasa S, Foulcer S, Apte SS. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014;35:34-41 Available at: https://pubmed.ncbi.nlm.nih.gov/24444773/
727. Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15-27 Available at: https://pubmed.ncbi.nlm.nih.gov/15554875/
728. Flannery CR. MMPs and ADAMTSs: functional studies. Front Biosci. 2006;11:544-69 Available at: https://pubmed.ncbi.nlm.nih.gov/16146752/
729. Okahata S, Yamamoto R, Yamakoshi Y, Fukae M. A Large Chondroitin Sulfate Proteoglycan, Versican, in Porcine Predentin. J Oral Biosci. 2011;53:72-81 Available at: https://pubmed.ncbi.nlm.nih.gov/22200993/
730. Goldberg M, Kulkarni AB, Young M, Boskey A. Dentin: structure, composition and mineralization. Front Biosci (Elite Ed). 2011;3:711-35 Available at: https://pubmed.ncbi.nlm.nih.gov/21196346/
731. Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol. 2012;226:185-99 Available at: https://pubmed.ncbi.nlm.nih.gov/22006671/
732. Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008;68:7247-9 Available at: https://pubmed.ncbi.nlm.nih.gov/18794108/
733. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165-78 Available at: https://pubmed.ncbi.nlm.nih.gov/21324931/
734. Jayadev R, Sherwood DR. Basement membranes. Curr Biol. 2017;27:R207-11 Available at: https://pubmed.ncbi.nlm.nih.gov/28324731/
735. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nature Communications 2020 11:1. 2020;11:1-19 Available at: https://www.nature.com/articles/s41467-020-18794-x
736. Kai FB, Drain AP, Weaver VM. The Extracellular Matrix Modulates the Metastatic Journey. Dev Cell. 2019;49:332-46 Available at: https://pubmed.ncbi.nlm.nih.gov/31063753/
737. Tikhonov D, Kulikova L, Rudnev V. et al. Changes in protein structural motifs upon post-translational modification in kidney cancer. Diagnostics. 2021;11:1836 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8534394/
738. Pfisterer K, Shaw LE, Symmank D, Weninger W. The Extracellular Matrix in Skin Inflammation and Infection. Front Cell Dev Biol. 2021;9:682414 Available at: www.frontiersin.org
739. Piotrowski-Daspit AS, Nerger BA, Wolf AE, Sundaresan S, Nelson CM. Dynamics of Tissue-Induced Alignment of Fibrous Extracellular Matrix. Biophys J. 2017;113:702-13
740. Lo Buglio G, Lo Cicero A, Campora S, Ghersi G. The Multifaced Role of Collagen in Cancer Development and Progression. International Journal of Molecular Sciences 2024, Vol 25, Page 13523. 2024;25:13523 Available at: https://www.mdpi.com/1422-0067/25/24/13523/htm
741. Pozzi A, Yurchenco PD, Iozzo R V. The nature and biology of basement membranes. Matrix Biol. 2017;57-58:1-11 Available at: https://pubmed.ncbi.nlm.nih.gov/28040522/
742. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015;33:230-6 Available at: https://pubmed.ncbi.nlm.nih.gov/25708906/
743. Levental KR, Yu H, Kass L. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906 Available at: https://pubmed.ncbi.nlm.nih.gov/19931152/
744. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res. 2012;10:1403-18 Available at: https://pubmed.ncbi.nlm.nih.gov/23024188/
745. Du W, Xia X, Hu F, Yu J. Extracellular matrix remodeling in the tumor immunity. Front Immunol. 2023;14:1340634
746. Sun R, Zhao Y, Liu Y. et al. Extracellular matrix stiffness in endometrial cancer: driving progression and modulating treatment sensitivity via the ROCK1/YAP1 axis. Cell Death & Disease 2025 16:1. 2025;16:1-16 Available at: https://www.nature.com/articles/s41419-025-07697-8
747. Hidalgo-Carcedo C, Hooper S, Chaudhry SI. et al. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol. 2011;13:49-58 Available at: https://pubmed.ncbi.nlm.nih.gov/21170030/
748. Zhang K, Corsa CA, Ponik SM. et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15:677-87 Available at: https://pubmed.ncbi.nlm.nih.gov/23644467/
749. Sangaletti S, Tripodo C, Santangelo A. et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell Rep. 2016;17:233-48
750. Niland S, Riscanevo AX, Eble JA. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int J Mol Sci. 2021;23:146 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8745566/
751. Jimenez RE, Hartwig W, Antoniu BA, Compton CC, Warshaw AL, Fernández-Del Castillo C. Effect of Matrix Metalloproteinase Inhibition on Pancreatic Cancer Invasion and Metastasis: An Additive Strategy for Cancer Control. Ann Surg. 2000;231:644 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC1421051/
752. Cabral-Pacheco GA, Garza-Veloz I, Rosa CCD La. et al. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int J Mol Sci. 2020;21:9739 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7767220/
753. Flies DB, Langermann S, Jensen C, Karsdal MA, Willumsen N. Regulation of tumor immunity and immunotherapy by the tumor collagen extracellular matrix. Front Immunol. 2023;14:1199513 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10470046/
754. Pluda S, Mazzocato Y, Angelini A. Peptide-Based Inhibitors of ADAM and ADAMTS Metalloproteinases. Front Mol Biosci. 2021;8:703715 Available at: www.frontiersin.org
755. Quintero-Fabián S, Arreola R, Becerril-Villanueva E. et al. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front Oncol. 2019;9:1370 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6915110/
756. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3954707/
757. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduction and Targeted Therapy 2020 5:1. 2020;5:1-17 Available at: https://www.nature.com/articles/s41392-020-0134-x
758. Houg DS, Bijlsma MF. The hepatic pre-metastatic niche in pancreatic ductal adenocarcinoma. Molecular Cancer 2018 17:1. 2018;17:1-18 Available at: https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-018-0842-9
759. Wang X, Khalil RA. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv Pharmacol. 2017;81:241 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5765875/
760. Roy R, Yang J, Moses MA. Matrix Metalloproteinases As Novel Biomarkers and Potential Therapeutic Targets in Human Cancer. Journal of Clinical Oncology. 2009;27:5287 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2773480/
761. Austin KM, Covic L, Kuliopulos A. Matrix metalloproteases and PAR1 activation. Blood. 2012;121:431 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3548166/
762. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374-403
763. Tauro M, Shay G, Sansil SS. et al. Bone seeking matrix metalloproteinase-2 inhibitors prevent bone metastatic breast cancer growth. Mol Cancer Ther. 2017;16:494 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5337166/
764. Lynch CC, Hikosaka A, Acuff HB. et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485-96 Available at: https://pubmed.ncbi.nlm.nih.gov/15894268/
765. De Leon-Oliva D, Barrena-Blázquez S, Jiménez-Álvarez L. et al. The RANK-RANKL-OPG System: A Multifaceted Regulator of Homeostasis, Immunity, and Cancer. Medicina (Kaunas). 2023 59. Available at: https://pubmed.ncbi.nlm.nih.gov/37893470/
766. Sabit H, Arneth B, Abdel-Ghany S. et al. Beyond Cancer Cells: How the Tumor Microenvironment Drives Cancer Progression. Cells. 2024;13:1666 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11475877/
767. Fisher KE, Pop A, Koh W, Anthis NJ, Saunders WB, Davis GE. Tumor cell invasion of collagen matrices requires coordinate lipid agonist-induced G-protein and membrane-type matrix metalloproteinase-1-dependent signaling. Mol Cancer. 2006;5:1-23 Available at: https://molecular-cancer.biomedcentral.com/articles/10.1186/1476-4598-5-69
768. He L, Wirtz D. Switching from Protease-Independent to Protease-Dependent Cancer Cell Invasion. Biophys J. 2014;107:2484 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC4255217/
769. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. Journal of Hematology & Oncology 2019 12:1. 2019;12:1-16 Available at: https://link.springer.com/articles/10.1186/s13045-019-0760-3
770. Paszek MJ, Zahir N, Johnson KR. et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241-54 Available at: https://pubmed.ncbi.nlm.nih.gov/16169468/
771. Zhang C, Qin M. Extracellular vesicles targeting tumor microenvironment in ovarian cancer. Int J Biol Macromol. 2023;252:126300
772. Erler JT, Bennewith KL, Cox TR. et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35-44 Available at: https://pubmed.ncbi.nlm.nih.gov/19111879/
773. Mouw JK, Yui Y, Damiano L. et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20:360-7 Available at: https://pubmed.ncbi.nlm.nih.gov/24633304/
774. Sengupta S, Den Boon JA, Chen IH. et al. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008;105:5874-8 Available at: https://pubmed.ncbi.nlm.nih.gov/18390668/
775. Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15:201-13 Available at: https://pubmed.ncbi.nlm.nih.gov/23354167/
776. Enerly E, Steinfeld I, Kleivi K. et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One. 2011 6. Available at: https://pubmed.ncbi.nlm.nih.gov/21364938/
777. Mushtaq MU, Papadas A, Pagenkopf A. et al. Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. J Immunother Cancer. 2018 6. Available at: https://pubmed.ncbi.nlm.nih.gov/29970158/
778. Frevert CW, Felgenhauer J, Wygrecka M, Nastase M V, Schaefer L. Danger-Associated Molecular Patterns Derived from the Extracellular Matrix Provide Temporal Control of Innate Immunity. J Histochem Cytochem. 2018;66:213-27 Available at: https://pubmed.ncbi.nlm.nih.gov/29290139/
779. Ouyang X, Ghani A, Mehal WZ. Inflammasome biology in fibrogenesis. Biochim Biophys Acta. 2013;1832:979-88 Available at: https://pubmed.ncbi.nlm.nih.gov/23562491/
780. Babelova A, Moreth K, Tsalastra-Greul W. et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284:24035-48 Available at: https://pubmed.ncbi.nlm.nih.gov/19605353/
781. Schaefer L, Babelova A, Kiss E. et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223-33 Available at: https://pubmed.ncbi.nlm.nih.gov/16025156/
782. Onwudiwe K, Najera J, Siri S, Datta M. Do Tumor Mechanical Stresses Promote Cancer Immune Escape? Cells. 2022 11. Available at: https://pubmed.ncbi.nlm.nih.gov/36497097/
783. Dhakal B, Pagenkopf A, Mushtaq MU. et al. Versican proteolysis predicts immune effector infiltration and post-transplant survival in myeloma. Leuk Lymphoma. 2019;60:2558-62 Available at: https://pubmed.ncbi.nlm.nih.gov/30845856/
784. Hope C, Emmerich PB, Papadas A. et al. Versican-Derived Matrikines Regulate Batf3-Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J Immunol. 2017;199:1933-41 Available at: https://pubmed.ncbi.nlm.nih.gov/28754680/
785. Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia. 2014;16:771-88 Available at: https://pubmed.ncbi.nlm.nih.gov/25379015/
786. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104:20262-7 Available at: https://pubmed.ncbi.nlm.nih.gov/18077379/
787. Afik R, Zigmond E, Vugman M. et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. 2016;213:2315-31 Available at: https://pubmed.ncbi.nlm.nih.gov/27697834/
788. Pankova D, Chen Y, Terajima M. et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol Cancer Res. 2016;14:287-95 Available at: https://pubmed.ncbi.nlm.nih.gov/26631572/
789. Zanotelli MR, Zhang J, Reinhart-King CA. Mechanoresponsive metabolism in cancer cell migration and metastasis. Cell Metab. 2021;33:1307-21 Available at: https://pubmed.ncbi.nlm.nih.gov/33915111/
790. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. 2019;20:89 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7377404/
791. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395-406 Available at: https://pubmed.ncbi.nlm.nih.gov/22351925/
792. Houghton AMG, Quintero PA, Perkins DL. et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753-9 Available at: https://pubmed.ncbi.nlm.nih.gov/16470245/
793. Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol. 2008;83:799-803 Available at: https://pubmed.ncbi.nlm.nih.gov/18063695/
794. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235-71 Available at: https://pubmed.ncbi.nlm.nih.gov/21219185/
795. O'Connor RS, Hao X, Shen K. et al. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189:1330-9 Available at: https://pubmed.ncbi.nlm.nih.gov/22732590/
796. Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol. 2002;83:111-9 Available at: https://pubmed.ncbi.nlm.nih.gov/12383190/
797. Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009-17 Available at: https://pubmed.ncbi.nlm.nih.gov/17435786/
798. Dangerfield J, Larbi KY, Huang MT, Dewar A, Nourshargh S. PECAM-1 (CD31) homophilic interaction up-regulates alpha6beta1 on transmigrated neutrophils in vivo and plays a functional role in the ability of alpha6 integrins to mediate leukocyte migration through the perivascular basement membrane. J Exp Med. 2002;196:1201-11 Available at: https://pubmed.ncbi.nlm.nih.gov/12417630/
799. Franco C, Britto K, Wong E. et al. Discoidin domain receptor 1 on bone marrow-derived cells promotes macrophage accumulation during atherogenesis. Circ Res. 2009;105:1141-8 Available at: https://pubmed.ncbi.nlm.nih.gov/19834008/
800. Wesley RB, Meng X, Godin D, Galis ZS. Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler Thromb Vasc Biol. 1998;18:432-40 Available at: https://pubmed.ncbi.nlm.nih.gov/9514412/
801. Thomassen MJ, Barna BP, Wiedemann HP, Bukowski RM, Farmer M, Ahmad M. Immunologic studies of alveolar macrophages from patients with metastatic renal cell carcinoma. American Review of Respiratory Disease. 1990;141:1256-8
802. Stahl M, Schupp J, Jag̈er B. et al. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One. 2013 8. Available at: https://pubmed.ncbi.nlm.nih.gov/24278429/
803. Patel NR, Bole M, Chen C. et al. Cell elasticity determines macrophage function. PLoS One. 2012 7. Available at: https://pubmed.ncbi.nlm.nih.gov/23028423/
804. Leonardi GC, Accardi G, Monastero R, Nicoletti F, Libra M. Ageing: from inflammation to cancer. Immun Ageing. 2018 15. Available at: https://pubmed.ncbi.nlm.nih.gov/29387133/
805. Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediators Inflamm. 2018. 2018 Available at: https://pubmed.ncbi.nlm.nih.gov/29849499/
806. Zinger A, Cho WC, Ben-Yehuda A. Cancer and Aging - the Inflammatory Connection. Aging Dis. 2017;8:611-27 Available at: https://pubmed.ncbi.nlm.nih.gov/28966805/
807. Greene MA, Loeser RF. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23:1966-71 Available at: https://pubmed.ncbi.nlm.nih.gov/26521742/
808. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108-19 Available at: https://pubmed.ncbi.nlm.nih.gov/29348500/
809. Meyera C, Sevko A, Ramacher M. et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A. 2011;108:17111-6 Available at: https://pubmed.ncbi.nlm.nih.gov/21969559/
810. Ameri AH, Tuchayi SM, Zaalberg A. et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc Natl Acad Sci U S A. 2019;116:2646-51 Available at: https://pubmed.ncbi.nlm.nih.gov/30696763/
811. Garbern JC, Williams J, Kristl AC. et al. Dysregulation of IL-33/ST2 signaling and myocardial periarteriolar fibrosis. J Mol Cell Cardiol. 2019;128:179-86 Available at: https://pubmed.ncbi.nlm.nih.gov/30763587/
812. Kim JW, Won T Bin, Woo H. et al. Age related non-type 2 inflammation and its association with treatment outcome in patients with chronic rhinosinusitis with nasal polyp in Korea. Sci Rep. 2022;12:1671 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8803874/
813. Maggio M, Guralnik JM, Longo DL, Ferrucci L. Interleukin-6 in aging and chronic disease: a magnificent pathway. J Gerontol A Biol Sci Med Sci. 2006;61:575-84 Available at: https://pubmed.ncbi.nlm.nih.gov/16799139/
814. Merline R, Moreth K, Beckmann J. et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal. 2011 4. Available at: https://pubmed.ncbi.nlm.nih.gov/22087031/
815. Olivieri F, Spazzafumo L, Santini G. et al. Age-related differences in the expression of circulating microRNAs: miR-21 as a new circulating marker of inflammaging. Mech Ageing Dev. 2012;133:675-85 Available at: https://pubmed.ncbi.nlm.nih.gov/23041385/
816. Fabbri M, Paone A, Calore F. et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012 109. Available at: https://pubmed.ncbi.nlm.nih.gov/22753494/
817. Wallis R, Mizen H, Bishop CL. The bright and dark side of extracellular vesicles in the senescence-associated secretory phenotype. Mech Ageing Dev. 2020;189:111263 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7347005/
818. Lundy DJ, Liao C. Extracellular Vesicles in Aging and Age-Related Diseases. How Important Are They? Adv Biol. 2025 Available at: https://pubmed.ncbi.nlm.nih.gov/40202045/
819. Xie X, Qu P, Wu H. et al. Circulating exosomal miR-21 mediates HUVEC proliferation and migration through PTEN/PI3K/AKT in Crohn's disease. Ann Transl Med. 2022;10:258-258 Available at: https://pubmed.ncbi.nlm.nih.gov/35402577/
820. Cheresh P, Kim SJ, Jablonski R. et al. SIRT3 overexpression ameliorates asbestos-induced pulmonary fibrosis, mt-DNA damage and lung fibrogenic monocyte recruitment. Int J Mol Sci. 2021;22:6856 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8268084/
821. Kadota T, Yoshioka Y, Fujita Y. et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2020;63:623-36 Available at: https://pubmed.ncbi.nlm.nih.gov/32730709/
822. Kumar MA, Baba SK, Sadida HQ. et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduction and Targeted Therapy 2024 9:1. 2024;9:1-41 Available at: https://www.nature.com/articles/s41392-024-01735-1
823. Martín-Taboada M, Corrales P, Medina-Gómez G, Vila-Bedmar R. Tackling the effects of extracellular vesicles in fibrosis. Eur J Cell Biol. 2022;101:151221
824. Sekiguchi R, Yamada KM. Basement Membranes in Development and Disease. Curr Top Dev Biol. 2018;130:143-91 Available at: https://pubmed.ncbi.nlm.nih.gov/29853176/
825. Hu M, Ling Z, Ren X. Extracellular matrix dynamics: tracking in biological systems and their implications. Journal of Biological Engineering 2022 16:1. 2022;16:1-13 Available at: https://jbioleng.biomedcentral.com/articles/10.1186/s13036-022-00292-x
826. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195-200 Available at: https://dx.doi.org/10.1242/jcs.023820
827. Filipe EC, Chitty JL, Cox TR. Charting the unexplored extracellular matrix in cancer. Int J Exp Pathol. 2018;99:58-76 Available at: https://pubmed.ncbi.nlm.nih.gov/29671911/
828. Kyriakopoulou K, Piperigkou Z, Tzaferi K, Karamanos NK. Trends in extracellular matrix biology. Mol Biol Rep. 2023;50:853-63 Available at: https://pubmed.ncbi.nlm.nih.gov/36342580/
829. Seo BR, DelNero P, Fischbach C. In vitro models of tumor vessels and matrix: engineering approaches to investigate transport limitations and drug delivery in cancer. Adv Drug Deliv Rev. 2014;69-70:205-16 Available at: https://pubmed.ncbi.nlm.nih.gov/24309015/
830. Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release. 2015;201:78-89 Available at: https://pubmed.ncbi.nlm.nih.gov/25526702/
831. Misra S, Heldin P, Hascall VC. et al. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011;278:1429-43 Available at: https://pubmed.ncbi.nlm.nih.gov/21362138/
832. Ma J, Chen CS, Blute T, Waxman DJ. Antiangiogenesis enhances intratumoral drug retention. Cancer Res. 2011;71:2675-85 Available at: https://pubmed.ncbi.nlm.nih.gov/21447737/
833. Kłosowska-Wardȩga A, Hasumi Y, Burmakin M. et al. Combined Anti-Angiogenic Therapy Targeting PDGF and VEGF Receptors Lowers the Interstitial Fluid Pressure in a Murine Experimental Carcinoma. PLoS One. 2009;4:e8149 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2781164/
834. V. Rosca E, E. Koskimaki J, G. Rivera C, B. Pandey N, P. Tamiz A, S. Popel A. Anti-angiogenic peptides for cancer therapeutics. Curr Pharm Biotechnol. 2011;12:1101-16 Available at: https://pubmed.ncbi.nlm.nih.gov/21470139/
835. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205-18 Available at: https://pubmed.ncbi.nlm.nih.gov/23669226/
836. Martino MM, Briquez PS, Maruyama K, Hubbell JA. Extracellular matrix-inspired growth factor delivery systems for bone regeneration. Adv Drug Deliv Rev. 2015;94:41-52 Available at: https://pubmed.ncbi.nlm.nih.gov/25895621/
837. Blanquer SBG, Grijpma DW, Poot AA. Delivery systems for the treatment of degenerated intervertebral discs. Adv Drug Deliv Rev. 2015;84:172-87 Available at: https://pubmed.ncbi.nlm.nih.gov/25451138/
838. Martino MM, Tortelli F, Mochizuki M. et al. Engineering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Sci Transl Med. 2011 3. Available at: https://pubmed.ncbi.nlm.nih.gov/21918106/
839. Brown BN, Badylak SF. Extracellular matrix as an inductive scaffold for functional tissue reconstruction. Transl Res. 2014;163:268-85 Available at: https://pubmed.ncbi.nlm.nih.gov/24291155/
840. D'Amore A, Yoshizumi T, Luketich SK. et al. Bi-layered polyurethane - Extracellular matrix cardiac patch improves ischemic ventricular wall remodeling in a rat model. Biomaterials. 2016;107:1-14 Available at: https://pubmed.ncbi.nlm.nih.gov/27579776/
841. Ohashi K, Yokoyama T, Yamato M. et al. Engineering functional two- and three-dimensional liver systems in vivo using hepatic tissue sheets. Nat Med. 2007;13:880-5 Available at: https://pubmed.ncbi.nlm.nih.gov/17572687/
842. Remlinger NT, Czajka CA, Juhas ME. et al. Hydrated xenogeneic decellularized tracheal matrix as a scaffold for tracheal reconstruction. Biomaterials. 2010;31:3520-6 Available at: https://pubmed.ncbi.nlm.nih.gov/20144481/
843. Nieponice A, Ciotola FF, Nachman F. et al. Patch esophagoplasty: esophageal reconstruction using biologic scaffolds. Ann Thorac Surg. 2014;97:283-8 Available at: https://pubmed.ncbi.nlm.nih.gov/24266951/
844. Singelyn JM, Sundaramurthy P, Johnson TD. et al. Catheter-deliverable hydrogel derived from decellularized ventricular extracellular matrix increases endogenous cardiomyocytes and preserves cardiac function post-myocardial infarction. J Am Coll Cardiol. 2012;59:751-63 Available at: https://pubmed.ncbi.nlm.nih.gov/22340268/
845. Ghuman H, Massensini AR, Donnelly J. et al. ECM hydrogel for the treatment of stroke: Characterization of the host cell infiltrate. Biomaterials. 2016;91:166-81 Available at: https://pubmed.ncbi.nlm.nih.gov/27031811/
846. Wang RM, Johnson TD, He J. et al. Humanized mouse model for assessing the human immune response to xenogeneic and allogeneic decellularized biomaterials. Biomaterials. 2017;129:98-110 Available at: https://pubmed.ncbi.nlm.nih.gov/28334641/
847. Sonnenberg SB, Rane AA, Liu CJ. et al. Delivery of an engineered HGF fragment in an extracellular matrix-derived hydrogel prevents negative LV remodeling post-myocardial infarction. Biomaterials. 2015;45:56-63 Available at: https://pubmed.ncbi.nlm.nih.gov/25662495/
848. Crowe CS, Chattopadhyay A, McGoldrick R, Chiou G, Pham H, Chang J. Characteristics of Reconstituted Lyophilized Tendon Hydrogel: An Injectable Scaffold for Tendon Regeneration. Plast Reconstr Surg. 2016;137:843-51 Available at: https://pubmed.ncbi.nlm.nih.gov/26910664/
849. Sood D, Chwalek K, Stuntz E. et al. Fetal brain extracellular matrix boosts neuronal network formation in 3D bioengineered model of cortical brain tissue. ACS Biomater Sci Eng. 2016;2:131-40 Available at: https://pubmed.ncbi.nlm.nih.gov/29034320/
850. Johnson TD, Dequach JA, Gaetani R. et al. Human versus porcine tissue sourcing for an injectable myocardial matrix hydrogel. Biomater Sci. 2014;2014:735-44 Available at: https://pubmed.ncbi.nlm.nih.gov/24634775/
851. Biteau B, Hochmuth CE, Jasper H. Maintaining tissue homeostasis: dynamic control of somatic stem cell activity. Cell Stem Cell. 2011;9:402-11 Available at: https://pubmed.ncbi.nlm.nih.gov/22056138/
852. Bardelli S, Moccetti M. Remodeling the Human Adult Stem Cell Niche for Regenerative Medicine Applications. Stem Cells Int. 2017. 2017 Available at: https://pubmed.ncbi.nlm.nih.gov/29090011/
853. Don CW, Murry CE. Improving survival and efficacy of pluripotent stem cell-derived cardiac grafts. J Cell Mol Med. 2013;17:1355-62 Available at: https://pubmed.ncbi.nlm.nih.gov/24118766/
854. Zhang Y, Mignone J, Robb Maclellan W. Cardiac Regeneration and Stem Cells. Physiol Rev. 2015;95:1189-204 Available at: https://pubmed.ncbi.nlm.nih.gov/26269526/
855. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int J Mol Sci. 2021 22. Available at: https://pubmed.ncbi.nlm.nih.gov/34071836/
856. Chong JJH, Yang X, Don CW. et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273-7 Available at: https://pubmed.ncbi.nlm.nih.gov/24776797/
857. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840:2506-19 Available at: https://pubmed.ncbi.nlm.nih.gov/24418517/
858. Watt FM, Huck WTS. Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol. 2013;14:467-73 Available at: https://pubmed.ncbi.nlm.nih.gov/23839578/
859. Brizzi MF, Tarone G, Defilippi P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr Opin Cell Biol. 2012;24:645-51 Available at: https://pubmed.ncbi.nlm.nih.gov/22898530/
860. Metra M, Teerlink JR. Heart failure. Lancet. 2017;390:1981-95 Available at: https://pubmed.ncbi.nlm.nih.gov/28460827/
861. Martins AM, Eng G, Caridade SG, Mano JF, Reis RL, Vunjak-Novakovic G. Electrically conductive chitosan/carbon scaffolds for cardiac tissue engineering. Biomacromolecules. 2014;15:635-43 Available at: https://pubmed.ncbi.nlm.nih.gov/24417502/
862. Reis LA, Chiu LLY, Feric N, Fu L, Radisic M. Biomaterials in myocardial tissue engineering. J Tissue Eng Regen Med. 2016;10:11-28 Available at: https://pubmed.ncbi.nlm.nih.gov/25066525/
863. Wainwright JM, Czajka CA, Patel UB. et al. Preparation of cardiac extracellular matrix from an intact porcine heart. Tissue Eng Part C Methods. 2010;16:525-32 Available at: https://pubmed.ncbi.nlm.nih.gov/19702513/
864. Parmaksiz M, Elcin AE, Elcin YM. Decellularization of bovine small intestinal submucosa and its use for the healing of a critical-sized full-thickness skin defect, alone and in combination with stem cells, in a small rodent model. J Tissue Eng Regen Med. 2017;11:1754-65 Available at: https://pubmed.ncbi.nlm.nih.gov/26227678/
865. Olivotto I, Oreziak A, Barriales-Villa R. et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759-69 Available at: https://pubmed.ncbi.nlm.nih.gov/32871100/
866. Kampourakis T, Zhang X, Sun YB, Irving M. Omecamtiv mercabil and blebbistatin modulate cardiac contractility by perturbing the regulatory state of the myosin filament. J Physiol. 2018;596:31-46 Available at: https://pubmed.ncbi.nlm.nih.gov/29052230/
867. Bassat E, Mutlak YE, Genzelinakh A. et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179-84 Available at: https://pubmed.ncbi.nlm.nih.gov/28581497/
868. Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation. 2000;102:2700-6 Available at: https://pubmed.ncbi.nlm.nih.gov/11094035/
869. Villalobos Lizardi JC, Baranger J, Nguyen MB. et al. A guide for assessment of myocardial stiffness in health and disease. Nature cardiovascular research. 2022;1:8-22 Available at: https://pubmed.ncbi.nlm.nih.gov/39196108/
870. Li H, Bao M, Nie Y. Extracellular matrix-based biomaterials for cardiac regeneration and repair. Heart Fail Rev. 2021;26:1231-48 Available at: https://pubmed.ncbi.nlm.nih.gov/32306220/
871. Sung H, Ferlay J, Siegel RL. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49 Available at: https://pubmed.ncbi.nlm.nih.gov/33538338/
872. Li Y, Khuu N, Prince E. et al. Matrix Stiffness-Regulated Growth of Breast Tumor Spheroids and Their Response to Chemotherapy. Biomacromolecules. 2021;22:419-29 Available at: https://pubmed.ncbi.nlm.nih.gov/33136364/
873. Byrne CE, Decombe JB, Bingham GC. et al. Evaluation of Extracellular Matrix Composition to Improve Breast Cancer Modeling. Tissue Eng Part A. 2021;27:500-11 Available at: https://pubmed.ncbi.nlm.nih.gov/33797977/
874. Yang X, Chen L, Mao Y, Hu Z, He M. Progressive and Prognostic Performance of an Extracellular Matrix-Receptor Interaction Signature in Gastric Cancer. Dis Markers. 2020. 2020 Available at: https://pubmed.ncbi.nlm.nih.gov/33178362/
875. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012 11. Available at: https://pubmed.ncbi.nlm.nih.gov/22159717/
876. Vasiukov G, Novitskaya T, Zijlstra A. et al. Myeloid Cell-Derived TGFβ Signaling Regulates ECM Deposition in Mammary Carcinoma via Adenosine-Dependent Mechanisms. Cancer Res. 2020;80:2628-38 Available at: https://pubmed.ncbi.nlm.nih.gov/32312837/
877. Lenz G, Wright G, Dave SS. et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313-23 Available at: https://pubmed.ncbi.nlm.nih.gov/19038878/
878. Zhu P, Lu H, Wang M, Chen K, Chen Z, Yang L. Targeted mechanical forces enhance the effects of tumor immunotherapy by regulating immune cells in the tumor microenvironment. Cancer Biol Med. 2023;20:44 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9843446/
879. Hurkmans DP, Jensen C, Koolen SLW. et al. Blood-based extracellular matrix biomarkers are correlated with clinical outcome after PD-1 inhibition in patients with metastatic melanoma. J Immunother Cancer. 2020 8. Available at: https://pubmed.ncbi.nlm.nih.gov/33093157/
880. Cheon DJ, Tong Y, Sim MS. et al. A collagen-remodeling gene signature regulated by TGF-β signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin Cancer Res. 2014;20:711-23 Available at: https://pubmed.ncbi.nlm.nih.gov/24218511/
881. Yuan Z, Li Y, Zhang S. et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol Cancer. 2023 22. Available at: https://pubmed.ncbi.nlm.nih.gov/36906534/
882. Ortega MA, Boaru DL, De Leon-Oliva D. et al. PD-1/PD-L1 axis: implications in immune regulation, cancer progression, and translational applications. J Mol Med (Berl). 2024;102:987-1000 Available at: https://pubmed.ncbi.nlm.nih.gov/38935130/
883. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807-21 Available at: https://pubmed.ncbi.nlm.nih.gov/32612154/
884. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021 11. Available at: https://pubmed.ncbi.nlm.nih.gov/33824268/
885. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018 9. Available at: https://pubmed.ncbi.nlm.nih.gov/30410077/
886. Azadi S, Aboulkheyr Es H, Razavi Bazaz S, Thiery JP, Asadnia M, Ebrahimi Warkiani M. Upregulation of PD-L1 expression in breast cancer cells through the formation of 3D multicellular cancer aggregates under different chemical and mechanical conditions. Biochim Biophys Acta Mol Cell Res. 2019 1866. Available at: https://pubmed.ncbi.nlm.nih.gov/31398408/
887. Kieffer Y, Hocine HR, Gentric G. et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020;10:1330-51 Available at: https://pubmed.ncbi.nlm.nih.gov/32434947/
888. Horn LA, Chariou PL, Gameiro SR. et al. Remodeling the tumor microenvironment via blockade of LAIR-1 and TGF-β signaling enables PD-L1-mediated tumor eradication. J Clin Invest. 2022 132. Available at: https://pubmed.ncbi.nlm.nih.gov/35230974/
889. Vannini A, Leoni V, Barboni C. et al. αvβ3-integrin regulates PD-L1 expression and is involved in cancer immune evasion. Proc Natl Acad Sci U S A. 2019;116:20141-50 Available at: https://pubmed.ncbi.nlm.nih.gov/31527243/
890. Guan X, Chen J, Hu Y. et al. Highly enhanced cancer immunotherapy by combining nanovaccine with hyaluronidase. Biomaterials. 2018;171:198-206 Available at: https://pubmed.ncbi.nlm.nih.gov/29698869/
891. Miyazawa A, Ito S, Asano S. et al. Regulation of PD-L1 expression by matrix stiffness in lung cancer cells. Biochem Biophys Res Commun. 2018;495:2344-9 Available at: https://pubmed.ncbi.nlm.nih.gov/29274784/
892. Kiyokawa J, Kawamura Y, Ghouse SM. et al. Modification of Extracellular Matrix Enhances Oncolytic Adenovirus Immunotherapy in Glioblastoma. Clin Cancer Res. 2021;27:889-902 Available at: https://pubmed.ncbi.nlm.nih.gov/33257429/
893. Najibi AJ, Shih TY, Zhang DKY. et al. Targeting tumor extracellular matrix activates the tumor-draining lymph nodes. Cancer Immunol Immunother. 2022;71:2957-68 Available at: https://pubmed.ncbi.nlm.nih.gov/35524791/
894. Caruana I, Savoldo B, Hoyos V. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524-9 Available at: https://pubmed.ncbi.nlm.nih.gov/25849134/
895. Peng L, Sferruzza G, Yang L, Zhou L, Chen S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cellular & Molecular Immunology 2024 21:10. 2024;21:1089-108 Available at: https://www.nature.com/articles/s41423-024-01207-0
896. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell. 2020;38:473-88
897. Adu-Berchie K, Liu Y, Zhang DKY. et al. Generation of functionally distinct T-cell populations by altering the viscoelasticity of their extracellular matrix. Nat Biomed Eng. 2023;7:1374-91 Available at: https://pubmed.ncbi.nlm.nih.gov/37365267/
898. Baldari S, Di Modugno F, Nisticò P, Toietta G. Strategies for Efficient Targeting of Tumor Collagen for Cancer Therapy. Cancers (Basel). 2022;14:4706 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9563908/
899. Ishihara J, Ishihara A, Sasaki K. et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Transl Med. 2019 11. Available at: https://pubmed.ncbi.nlm.nih.gov/30971453/
900. Delaunay M, Cadranel J, Lusque A. et al. Immune-checkpoint inhibitors associated with interstitial lung disease in cancer patients. Eur Respir J. 2017 50. Available at: https://pubmed.ncbi.nlm.nih.gov/28798088/
901. Liu B, Zhou H, Tan L, Siu KTH, Guan XY. Exploring treatment options in cancer: tumor treatment strategies. Signal Transduction and Targeted Therapy 2024 9:1. 2024;9:1-44 Available at: https://www.nature.com/articles/s41392-024-01856-7
902. Alhmoud JF, Woolley JF, Al Moustafa AE, Malki MI. DNA Damage/Repair Management in Cancers. Cancers (Basel). 2020;12:1050 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7226105/
903. Crawford J, Herndon D, Gmitter K, Weiss J. The impact of myelosuppression on quality of life of patients treated with chemotherapy. Future Oncol. 2024 20. Available at: https://pubmed.ncbi.nlm.nih.gov/38587388/
904. Deng C, Xu Y, Fu J. et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020;111:1899-909 Available at: https://pubmed.ncbi.nlm.nih.gov/32232912/
905. Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer. 2015;1:66-75 Available at: https://pubmed.ncbi.nlm.nih.gov/26457331/
906. Balachandran VP, Beatty GL, Dougan SK. Broadening the impact of immunotherapy to pancreatic cancer: Challenges and opportunities. Gastroenterology. 2019;156:2056 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6486864/
907. Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. 2020;14:2994-3006 Available at: https://pubmed.ncbi.nlm.nih.gov/33179413/
908. Guarin JR, Fatherree JP, Oudin MJ. Chemotherapy treatment induces pro-invasive changes in liver ECM composition. Matrix Biol. 2022;112:20 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10690958/
909. Karagiannis GS, Condeelis JS, Oktay MH. Chemotherapy-induced metastasis: Mechanisms and translational opportunities. Clin Exp Metastasis. 2018;35:269 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6035114/
910. Haj-Shomaly J, Vorontsova A, Barenholz-Cohen T. et al. T Cells Promote Metastasis by Regulating Extracellular Matrix Remodeling following Chemotherapy. Cancer Res. 2021;82:278 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7612244/
911. Sanford RA, Lei X, Barcenas CH. et al. Impact of Time from Completion of Neoadjuvant Chemotherapy to Surgery on Survival Outcomes in Breast Cancer Patients. Ann Surg Oncol. 2016;23:1515-21 Available at: https://pubmed.ncbi.nlm.nih.gov/26678405/
912. Popova N V, Jücker M. The Functional Role of Extracellular Matrix Proteins in Cancer. Cancers (Basel). 2022;14:238 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8750014/
913. Elgundi Z, Papanicolaou M, Major G. et al. Cancer Metastasis: The Role of the Extracellular Matrix and the Heparan Sulfate Proteoglycan Perlecan. Front Oncol. 2020;9:1482 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6978720/
914. DeNardo DG, Brennan DJ, Rexhepaj E. et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54-67 Available at: https://pubmed.ncbi.nlm.nih.gov/22039576/
915. Abyaneh HS, Regenold M, McKee TD, Allen C, Gauthier MA. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics. 2020;10:1960 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6993244/
916. Théry C, Witwer KW, Aikawa E. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018 7. Available at: https://pubmed.ncbi.nlm.nih.gov/30637094/
917. Eguchi T, Sogawa C, Ono K. et al. Cell Stress Induced Stressome Release Including Damaged Membrane Vesicles and Extracellular HSP90 by Prostate Cancer Cells. Cells. 2020 9. Available at: https://pubmed.ncbi.nlm.nih.gov/32204513/
918. Jeppesen DK, Fenix AM, Franklin JL. et al. Reassessment of Exosome Composition. Cell. 2019;177:428-445.e18 Available at: https://pubmed.ncbi.nlm.nih.gov/30951670/
919. Okusha Y, Eguchi T, Tran MT. et al. Extracellular Vesicles Enriched with Moonlighting Metalloproteinase Are Highly Transmissive, Pro-Tumorigenic, and Trans-Activates Cellular Communication Network Factor (CCN2/CTGF): CRISPR against Cancer. Cancers (Basel). 2020 12. Available at: https://pubmed.ncbi.nlm.nih.gov/32260433/
920. Taha EA, Sogawa C, Okusha Y. et al. Knockout of MMP3 Weakens Solid Tumor Organoids and Cancer Extracellular Vesicles. Cancers (Basel). 2020 12. Available at: https://pubmed.ncbi.nlm.nih.gov/32429403/
921. Minciacchi VR, Spinelli C, Reis-Sobreiro M. et al. MYC Mediates Large Oncosome-Induced Fibroblast Reprogramming in Prostate Cancer. Cancer Res. 2017;77:2306-17 Available at: https://pubmed.ncbi.nlm.nih.gov/28202510/
922. Klyachko NL, Arzt CJ, Li SM, Gololobova OA, Batrakova E V. Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations. Pharmaceutics. 2020;12:1-26 Available at: https://pubmed.ncbi.nlm.nih.gov/33271883/
923. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654-9 Available at: https://pubmed.ncbi.nlm.nih.gov/17486113/
924. Kalluri R, Lebleu VS. Discovery of Double-Stranded Genomic DNA in Circulating Exosomes. Cold Spring Harb Symp Quant Biol. 2016;81:275-80 Available at: https://pubmed.ncbi.nlm.nih.gov/28424339/
925. Park JE, Dutta B, Tse SW. et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. 2019;38:5158-73 Available at: https://pubmed.ncbi.nlm.nih.gov/30872795/
926. Boon RA, Vickers KC. Intercellular transport of microRNAs. Arterioscler Thromb Vasc Biol. 2013;33:186-92 Available at: https://pubmed.ncbi.nlm.nih.gov/23325475/
927. Yang M, Chen J, Su F. et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer. 2011 10. Available at: https://pubmed.ncbi.nlm.nih.gov/21939504/
928. Sheta M, Taha EA, Lu Y, Eguchi T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology (Basel). 2023;12:110 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9856004/
929. Lananna B V, Imai S ichiro. Friends and foes: Extracellular vesicles in aging and rejuvenation. FASEB Bioadv. 2021;3:787-801 Available at: https://onlinelibrary.wiley.com/doi/full/10.1096/fba.2021-00077
930. Childs BG, Gluscevic M, Baker DJ. et al. Senescent cells: an emerging target for diseases of ageing. Nature Reviews Drug Discovery 2017 16:10. 2017;16:718-35 Available at: https://www.nature.com/articles/nrd.2017.116
931. Yin S, Zhou Z, Fu P. et al. Roles of extracellular vesicles in ageing-related chronic kidney disease: Demon or angel. Pharmacol Res. 2023;193:106795
932. Ganesh BH, Padinjarathil H, Rajendran RL, Ramani P, Gangadaran P, Ahn BC. The Role of Extracellular Vesicles in Aging and Age-Related Disorders. Antioxidants. 2025;14:177 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11851802/
933. Nedachi T, Bonod C, Rorteau J. et al. Chronological aging impacts abundance, function and microRNA content of extracellular vesicles produced by human epidermal keratinocytes. Aging (Albany NY). 2023;15:12702 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10713413/
934. Yang P, Peng Y, Feng Y. et al. Immune Cell-Derived Extracellular Vesicles - New Strategies in Cancer Immunotherapy. Front Immunol. 2021;12:771551 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8694098/
935. Li M, Fang F, Sun M, Zhang Y, Hu M, Zhang J. Extracellular vesicles as bioactive nanotherapeutics: An emerging paradigm for regenerative medicine. Theranostics. 2022;12:4879 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9274746/
936. Prince MJ, Wu F, Guo Y. et al. The burden of disease in older people and implications for health policy and practice. The Lancet. 2015;385:549-62 Available at: https://www.thelancet.com/action/showFullText?pii=S0140673614613477
937. Zhang A, Li Q, Chen Z. Therapeutic Efficacy and Promise of Human Umbilical Cord Mesenchymal Stem Cell-Derived Extracellular Vesicles in Aging and Age-Related Disorders. International Journal of Molecular Sciences 2025, Vol 26, Page 225. 2024;26:225 Available at: https://www.mdpi.com/1422-0067/26/1/225/htm
938. Krohn JB, Hutcheson JD, Martínez-Martínez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. 2016;594:2895-903 Available at: https://pubmed.ncbi.nlm.nih.gov/26824781/
939. Ruiz JL, Weinbaum S, Aikawa E, Hutcheson JD. Zooming in on the genesis of atherosclerotic plaque microcalcifications. J Physiol. 2016;594:2915-27 Available at: https://pubmed.ncbi.nlm.nih.gov/27040360/
940. Kapustin AN, Davies JD, Reynolds JL. et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011 109. Available at: https://pubmed.ncbi.nlm.nih.gov/21566214/
941. Rogers MA, Buffolo F, Schlotter F. et al. Annexin A1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci Adv. 2020 6. Available at: https://pubmed.ncbi.nlm.nih.gov/32938681/
942. Huleihel L, Hussey GS, Naranjo JD. et al. Matrix-bound nanovesicles within ECM bioscaffolds. Sci Adv. 2016 2. Available at: https://pubmed.ncbi.nlm.nih.gov/27386584/
943. Weber EMM, Kress T, Abergel D, Sewsurn S, Azaïs T, Kurzbach D. Assessing the Onset of Calcium Phosphate Nucleation by Hyperpolarized Real-Time NMR. Anal Chem. 2020;92:7666-73 Available at: https://pubs.acs.org/doi/full/10.1021/acs.analchem.0c00516
944. Bakhshian Nik A, Hutcheson JD, Aikawa E. Extracellular Vesicles As Mediators of Cardiovascular Calcification. Front Cardiovasc Med. 2017;4:78 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC5732140/
945. Kuhlmann JD, Chebouti I, Kimmig R. et al. Extracellular vesicle-associated miRNAs in ovarian cancer - design of an integrated NGS-based workflow for the identification of blood-based biomarkers for platinum-resistance. Clin Chem Lab Med. 2019;57:1053-62 Available at: https://pubmed.ncbi.nlm.nih.gov/30422797/
946. Huleihel L, Bartolacci JG, Dziki JL. et al. Matrix-Bound Nanovesicles Recapitulate Extracellular Matrix Effects on Macrophage Phenotype. Tissue Eng Part A. 2017;23:1283-94 Available at: https://pubmed.ncbi.nlm.nih.gov/28580875/
947. Manoochehri H, La'ah AS, Babaeizad A. et al. Extracellular vesicles in cancer immunotherapy: Therapeutic, challenges and clinical progress. Asian J Pharm Sci. 2025;20:101065
948. Turner NJ, Quijano LM, Hussey GS, Jiang P, Badylak SF. Matrix Bound Nanovesicles Have Tissue-Specific Characteristics That Suggest a Regulatory Role. Tissue Eng Part A. 2022;28:879-92 Available at: https://pubmed.ncbi.nlm.nih.gov/35946072/
949. Palviainen M, Saari H, Kärkkäinen O. et al. Metabolic signature of extracellular vesicles depends on the cell culture conditions. J Extracell Vesicles. 2019 8. Available at: https://pubmed.ncbi.nlm.nih.gov/31007875/
950. Huang Y, Zucker B, Zhang S. et al. Migrasome formation is mediated by assembly of micron-scale tetraspanin macrodomains. Nat Cell Biol. 2019;21:991-1002 Available at: https://pubmed.ncbi.nlm.nih.gov/31371828/
951. Kalluri R, McAndrews KM. The role of extracellular vesicles in cancer. Cell. 2023;186:1610-26
952. Forder A, Hsing CY, Vazquez JT, Garnis C. Emerging Role of Extracellular Vesicles and Cellular Communication in Metastasis. Cells. 2021;10:3429 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8700460/
953. Guo Q, Jiang C. Delivery strategies for macromolecular drugs in cancer therapy. Acta Pharm Sin B. 2020;10:979-86 Available at: https://pubmed.ncbi.nlm.nih.gov/32642406/
954. Srivastava A, Babu A, Filant J. et al. Exploitation of Exosomes as Nanocarriers for Gene-, Chemo-, and Immune-Therapy of Cancer. J Biomed Nanotechnol. 2016;12:1159-73 Available at: https://pubmed.ncbi.nlm.nih.gov/27319211/
955. Pascucci L, Coccè V, Bonomi A. et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014;192:262-70 Available at: https://pubmed.ncbi.nlm.nih.gov/25084218/
956. Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil Y V, Filatov M V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal. 2013 11. Available at: https://pubmed.ncbi.nlm.nih.gov/24245560/
957. Wiklander OPB, Nordin JZ, O'Loughlin A. et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:1-13 Available at: https://pubmed.ncbi.nlm.nih.gov/25899407/
958. Qi H, Liu C, Long L. et al. Blood Exosomes Endowed with Magnetic and Targeting Properties for Cancer Therapy. ACS Nano. 2016;10:3323-33 Available at: https://pubmed.ncbi.nlm.nih.gov/26938862/
959. Butreddy A, Kommineni N, Dudhipala N, Guerrero-Martínez A, Valpuesta JM. Exosomes as Naturally Occurring Vehicles for Delivery of Biopharmaceuticals: Insights from Drug Delivery to Clinical Perspectives. Nanomaterials. 2021;11:1481 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC8229362/
960. Chehelgerdi M, Chehelgerdi M, Allela OQB. et al. Progressing nanotechnology to improve targeted cancer treatment: overcoming hurdles in its clinical implementation. Mol Cancer. 2023;22:169 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10561438/
961. Karamanos NK, Piperigkou Z, Passi A, Götte M, Rousselle P, Vlodavsky I. Extracellular matrix-based cancer targeting. Trends Mol Med. 2021;27:1000-13 Available at: https://pubmed.ncbi.nlm.nih.gov/34389240/
962. Clayton A, Turkes A, Dewitt S, Steadman R, Mason MD, Hallett MB. Adhesion and signaling by B cell-derived exosomes: the role of integrins. FASEB J. 2004;18:977-9 Available at: https://pubmed.ncbi.nlm.nih.gov/15059973/
963. Al-Akkad W, Acedo P, Vilia MG. et al. Tissue-Specific Human Extracellular Matrix Scaffolds Promote Pancreatic Tumour Progression and Chemotherapy Resistance. Cells. 2022 11. Available at: https://pubmed.ncbi.nlm.nih.gov/36429078/
964. Deng B, Zhao Z, Kong W, Han C, Shen X, Zhou C. Biological role of matrix stiffness in tumor growth and treatment. J Transl Med. 2022 20. Available at: https://pubmed.ncbi.nlm.nih.gov/36419159/
965. Jong AY, Wu CH, Li J. et al. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracell Vesicles. 2017 6. Available at: https://pubmed.ncbi.nlm.nih.gov/28326171/
966. Adnani L, Spinelli C, Tawil N, Rak J. Role of extracellular vesicles in cancer-specific interactions between tumour cells and the vasculature. Semin Cancer Biol. 2022;87:196-213 Available at: https://pubmed.ncbi.nlm.nih.gov/36371024/
967. Melo SA, Luecke LB, Kahlert C. et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177-82 Available at: https://pubmed.ncbi.nlm.nih.gov/26106858/
968. Dhandayuthapani B, Yoshida Y, Maekawa T, Kumar DS. Polymeric Scaffolds in Tissue Engineering Application: A Review. Int J Polym Sci. 2011;2011:290602 Available at: https://onlinelibrary.wiley.com/doi/full/10.1155/2011/290602
969. Aazmi A, Zhang D, Mazzaglia C. et al. Biofabrication methods for reconstructing extracellular matrix mimetics. Bioact Mater. 2024;31:475-96
970. Keane TJ, Swinehart IT, Badylak SF. Methods of tissue decellularization used for preparation of biologic scaffolds and in vivo relevance. Methods. 2015;84:25-34 Available at: https://pubmed.ncbi.nlm.nih.gov/25791470/
971. Liu J, Song Q, Yin W. et al. Bioactive scaffolds for tissue engineering: A review of decellularized extracellular matrix applications and innovations. Exploration. 2024;5:20230078 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11875452/
972. Meng FW, Slivka PF, Dearth CL, Badylak SF. Solubilized extracellular matrix from brain and urinary bladder elicits distinct functional and phenotypic responses in macrophages. Biomaterials. 2015;46:131-40
973. Zhao Y, Peng H, Sun L. et al. The application of small intestinal submucosa in tissue regeneration. Mater Today Bio. 2024;26:101032 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10963656/
974. Chan BP, Leong KW. Scaffolding in tissue engineering: general approaches and tissue-specific considerations. European Spine Journal. 2008;17:467 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2587658/
975. Guo WY, Wang WH, Xu PY, Kankala RK, Chen AZ. Decellularised extracellular matrix-based injectable hydrogels for tissue engineering applications. Biomaterials Translational. 2024;5:114 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11438603/
976. Smoak MM, Hogan KJ, Grande-Allen KJ, Mikos AG. Bioinspired electrospun dECM scaffolds guide cell growth and control the formation of myotubes. Sci Adv. 2021 7. Available at: https://pubmed.ncbi.nlm.nih.gov/33990336/
977. Yeleswarapu S, Dash A, Chameettachal S, Pati F. 3D bioprinting of tissue constructs employing dual crosslinking of decellularized extracellular matrix hydrogel. Biomaterials advances. 2023 152. Available at: https://pubmed.ncbi.nlm.nih.gov/37307772/
978. Ramos-Rodriguez DH, Leach JK. Decellularized Cell-Secreted Extracellular Matrices as Biomaterials for Tissue Engineering. Small Science. 2025;5:2400335 Available at: https://onlinelibrary.wiley.com/doi/full/10.1002/smsc.202400335
979. Bejleri D, Davis ME. Decellularized Extracellular Matrix Materials for Cardiac Repair and Regeneration. Adv Healthc Mater. 2019;8:e1801217 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC7654553/
980. Ortega JA, Soares de Aguiar GP, Chandravanshi P, Levy N, Engel E, Álvarez Z. Exploring the properties and potential of the neural extracellular matrix for next-generation regenerative therapies. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2024;16:e1962 Available at: https://onlinelibrary.wiley.com/doi/full/10.1002/wnan.1962
981. Chen Y, Chen LF, Wang Y. et al. Modeling dECM-based inflammatory cartilage microtissues in vitro for drug screening. Compos B Eng. 2023;250:110437
982. Liang W, Han M, Li G. et al. Perfusable adipose decellularized extracellular matrix biological scaffold co-recellularized with adipose-derived stem cells and L6 promotes functional skeletal muscle regeneration following volumetric muscle loss. Biomaterials. 2024 307. Available at: https://pubmed.ncbi.nlm.nih.gov/38489911/
983. Milton LA, Davern JW, Hipwood L. et al. Liver click dECM hydrogels for engineering hepatic microenvironments. Acta Biomater. 2024;185:144-60 Available at: https://pubmed.ncbi.nlm.nih.gov/38960110/
984. Li J, Zhang J, Ye H. et al. Pulmonary decellularized extracellular matrix (dECM) modified polyethylene terephthalate three-dimensional cell carriers regulate the proliferation and paracrine activity of mesenchymal stem cells. Front Bioeng Biotechnol. 2023;11:1324424
985. Coburn PT, Li X, Li J, Kishimoto Y, Li-Jessen NYK. Progress in Vocal Fold Regenerative Biomaterials: An Immunological Perspective. Adv Nanobiomed Res. 2022 2. Available at: https://pubmed.ncbi.nlm.nih.gov/35434718/
986. Klak M, Łojszczyk I, Berman A. et al. Impact of Porcine Pancreas Decellularization Conditions on the Quality of Obtained dECM. Int J Mol Sci. 2021 22. Available at: https://pubmed.ncbi.nlm.nih.gov/34209772/
987. Su J, Satchell SC, Shah RN, Wertheim JA. Kidney Decellularized Extracellular Matrix Hydrogels: Rheological Characterization and Human Glomerular Endothelial Cell Response to Encapsulation. J Biomed Mater Res A. 2018;106:2448 Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC6376869/
988. Pasini C, Pandini S, Ramorino G, Sartore L. Tailoring the properties of composite scaffolds with a 3D-Printed lattice core and a bioactive hydrogel shell for tissue engineering. J Mech Behav Biomed Mater. 2024;150:106305
989. Brown M, Li J, Moraes C, Tabrizian M, Li-Jessen NYK. Decellularized extracellular matrix: New promising and challenging biomaterials for regenerative medicine. Biomaterials. 2022 289. Available at: https://pubmed.ncbi.nlm.nih.gov/36116171/
Author contact
![]() Corresponding author: Miguel A Ortega; miguelangel.ortegaes.
Corresponding author: Miguel A Ortega; miguelangel.ortegaes.