Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Definition and classification...
3. Effects of Exercise on Liver...
4. The Specific Effects of...
5. Potential Molecular Therapies
6. Summary and Outlook
Abbreviations
Author Contributions
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(8):4148-4167. doi:10.7150/ijbs.126841 This issue Cite
Review
Exercise Impacts Liver Disease: Balancing Metabolic and Immune Homeostasis
Chen Cheng1,3†, Wentao Xu2,3†, Shanshan Wu3,4†, Jia Gao2,3†, Jianshang Huang3,5, Yunsheng Dong1,3, Jingjing Ji1,3, Li Zuo5,6* ![]() , Hua Wang2*
, Hua Wang2* ![]()
1. School of Pharmacy, Anhui Medical University, Hefei, 230022, China.
2. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, 230022, China.
3. Innovation and Entrepreneurship Laboratory for College Students, Anhui Medical University, Hefei, 230032, China.
4. Department of Gastroenterology, The First Affiliated Hospital of Anhui Medical University, Hefei, 230022, China.
5. Laboratory of Molecular Biology, and Department of Biochemistry, School of Basic Medical Science, Anhui Medical University, Hefei, 230032, China.
6. Center for Experimental Animals, Anhui Medical University, Hefei, 230032, China.
† The authors contributed equally and share first authorship.
* The authors contributed equally and share corresponding authorship.
Received 2025-10-1; Accepted 2026-2-19; Published 2026-4-3
Abstract

Liver diseases pose a significant global health burden. This review systematically elucidates the crucial role of exercise as a non-pharmacological intervention in the prevention and treatment of various liver conditions, including metabolic dysfunction-associated steatotic liver disease (MASLD), alcohol-related liver disease (ALD), viral hepatitis, liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). Exercise effectively delays disease progression and improves patients' quality of life through multi-targeted mechanisms, such as improving glucose and lipid metabolism, enhancing insulin sensitivity, regulating immune-inflammatory responses, inhibiting hepatic stellate cell activation, and remodeling the tumor microenvironment. Future research should focus on developing individualized, precise exercise prescriptions and further exploring its molecular mechanisms by integrating multi-omics technologies, thereby providing innovative strategies for the comprehensive management of liver diseases.
Keywords: liver diseases, exercise intervention, molecular pathways, metabolic regulation, immune regulation
1. Introduction
As a core organ of the human body, the liver plays a key role in energy metabolism, protein synthesis, and toxin excretion. Liver diseases are primarily characterized by pathological features such as hepatocyte injury, inflammatory cell infiltration, and activation of hepatic stellate cells. The persistent progression of these conditions can lead to impaired liver function and structural damage [1]. Globally, nearly 2 million people die from liver diseases each year, accounting for approximately 4% of total global mortality [2]. Among these, metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease (ALD) have a large patient base, with incidence rates continuing to rise [3, 4]. Without timely intervention, these conditions may progress to end-stage liver diseases such as cirrhosis and hepatocellular carcinoma (HCC) [2]. Therefore, early intervention is crucial for controlling the progression of liver diseases and alleviating the burden on healthcare systems. Studies have shown that regular exercise not only effectively prevents and treats various liver diseases [4-9], but also reduces the risk of liver cancer and improves the quality of life in patients with chronic liver diseases through multiple mechanisms, including improving lipid profiles, regulatory myokines, maintaining muscle mass, and increasing insulin sensitivity [10]. In summary, the necessity of studying exercise lies in its multi-target effects, such as regulating metabolism, inhibiting hepatocyte inflammation, and reducing fibrosis, thereby safely, economically, and non-pharmacologically delaying the progression of liver diseases and effectively improving patients' quality of life.
This review summarizes the role and related mechanisms of exercise in the prevention and treatment of liver diseases, discussing the positive effects of exercise on MASLD, cirrhosis, and HCC [Table 1]. It aims to elucidate how exercise, through multiple mechanisms such as improving glucose and lipid metabolism, enhancing insulin sensitivity, and modulating the immune system, not only reduces the risk of liver diseases and improves the quality of life in patients with chronic liver diseases and liver transplant recipients, but also provides a scientific basis for the comprehensive management of liver diseases.
Key metabolites, immune cells, and inflammatory mediators involved in liver diseases.
| Liver Disease | Metabolites | Immune Cells | Inflammatory Mediators | Reference |
|---|---|---|---|---|
| MASLD | TAG | MoMFs | TNF-α | [31,79,80,82-85] |
| ACC | IL-1β | [32,71,82-85] | ||
| SCD-1 | [31, 33] | |||
| Acox-1 | [38, 39] | |||
| CPT-1 | [38, 39] | |||
| ALD | Kupffer cells | [86] | ||
| Viral hepatitis | NK cells | TNF-α | [68,86,87] | |
| IL-6 | [86] | |||
| Liver fibrosis/cirrhosis | Kupffer cells | TGF-β | [182,183,184] | |
| TNF-α | [83] | |||
| HCC | NK cells | [88] | ||
| γδ T cells | [90] |
2. Definition and classification of exercise
Physical activity refers to bodily movement that effectively increases metabolic rate and energy expenditure, with its intensity quantifiable and graded using metabolic equivalents (METs): light (1.6 < 3.0 METs), moderate (3 < 6 METs), and vigorous (6.0-9.0 METs). Assessment can also be based on heart rate reserve or percentage of maximal heart rate (%HRmax), roughly corresponding to < 55%, 55 < 70%, and 70-90% HRmax, respectively. An exercise prescription should comprehensively consider exercise mode, frequency, duration, total energy expenditure, as well as activity structure and characteristics of the energy supply systems [11].
Aerobic exercise primarily relies on aerobic metabolism for energy, characterized by moderate intensity and sustainability, such as running, brisk walking, cycling, and swimming. It improves liver function through multiple pathways: enhancing insulin sensitivity [12, 13], inhibiting hepatic gluconeogenesis [13], reducing the deposition of free fatty acids (FFAs) in the liver [12, 13], and promoting glucose utilization in skeletal muscle [14]. It also regulates the hypothalamic-pituitary-adrenal axis to reduce stress hormones like cortisol [15, 16], thereby decreasing visceral fat breakdown and FFAs influx to the liver [15, 16].
Anaerobic exercise primarily involves high-intensity, short-duration activities, typically exceeding 60%-80% of an individual's maximal oxygen uptake (VO₂max) [17, 18]. Energy is mainly derived from the phosphagen and glycolytic systems [17, 18], exemplified by resistance training and power training [14, 17, 19]. Its potential hepatic effects include maintaining blood glucose stability by promoting gluconeogenesis, increasing hepatic glycogen storage in the long term [20, 21], and indirectly reducing hepatic lipid accumulation by improving insulin sensitivity [19], although its independent effects are generally weaker than those of aerobic exercise [14, 19].
High-intensity interval training (HIIT, intensity 80%-100% VO2max), sprint interval training (SIT, intensity > 100% VO2max), and moderate-intensity continuous training (MICT, intensity 40%-59% VO2max) are three common modalities [22, 23]. They can all reduce the risk of MASLD and lower liver enzyme levels through shared pathways such as improving insulin sensitivity and regulating lipid metabolism [20, 23-25]. [Table 2]
Classification of exercise intensities and their underlying mechanisms for improving liver function.
| Exercise Category | Intensity Classification/Assessment Indicators | Specific Exercise Forms | Effects/Mechanisms on Liver Function | Reference |
|---|---|---|---|---|
| Aerobic Exercise | Moderate intensity, highly sustainable | Running, brisk walking, cycling, swimming | Enhances insulin sensitivity, inhibits hepatic gluconeogenesis, reduces hepatic free fatty acid deposition, promotes skeletal muscle glucose utilization, lowers cortisol levels, reduces visceral fat breakdown | [12-16] |
| Anaerobic Exercise | High intensity, short duration (> 60%-80% VO2max) | Resistance training, plyometric training | Promotes gluconeogenesis to maintain blood glucose, increases hepatic glycogen reserves, improves insulin sensitivity (indirectly reduces hepatic lipid accumulation) | [14, 17-21] |
| HIIT | 80%-100% VO2max | Intermittent high-intensity exercise | Improves insulin sensitivity, regulates lipid metabolism, reduces MAFLD risk, improves liver enzyme levels | [20, 22-25] |
| SIT | > 100% VO2max | Extremely high-intensity sprints | Improves insulin sensitivity, regulates lipid metabolism, reduces MAFLD risk, improves liver enzyme levels | [22-25] |
| MICT | 40%-59% VO2max | Continuous moderate-intensity exercise | Improves insulin sensitivity, regulates lipid metabolism, reduces MAFLD risk, improves liver enzyme levels | [22-25] |
3. Effects of Exercise on Liver Health and Molecular Mechanisms
3.1 Glucose and lipid metabolism
Exercise energy metabolism exhibits intensity dependence. According to the "crossover concept," low intensity (< 40% VO2max) primarily relies on lipid oxidation, which peaks at moderate intensity (45%-65% VO2max), while high intensity (> 65% VO2max) shifts to carbohydrate dominance [26]. During the initial phase of exercise (10-15 minutes), skeletal muscle uptake causes a transient decrease in plasma fatty acids [27]; subsequently, activation of the β-adrenergic system increases lipolysis efficiency threefold [28], and sympathetic nervous system excitation increases adipose tissue blood flow 2-7 times [29, 30], synergistically promoting lipid mobilization.
At the molecular level, AMPK plays a significant regulatory role: exercise training significantly reduces hepatic diacylglycerol levels, particularly when AMPK function is normal [31]. AMPK inhibits fatty acid synthesis by phosphorylating acetyl-CoA carboxylase (ACC) [32] and promotes mitochondrial biogenesis to enhance β-oxidation capacity. AMPK-deficient mice exhibit hepatic triglyceride (TAG) accumulation post-exercise, confirming its necessity [31]. This kinase also restricts glycerolipid synthesis by inhibiting the SREBP-1c/SCD-1 axis [31, 33]. In glucose metabolism, AMPK promotes hepatic glycogen storage via a dual mechanism: upregulating UDP-glucose pyrophosphorylase 2 (UGP2) expression [34] and activating glycogen synthase by increasing glucose-6-phosphatase (G6Pase) levels [31]. It is noteworthy that core evidence for AMPK's role primarily comes from gene knockout animal models. Human AMPK isoforms are diverse and subject to complex regulation by genetics, nutrition, and disease states, posing challenges for directly validating its central role in the human liver.
Within the nuclear receptor pathway, exercise bidirectionally regulates the PPAR family: it inhibits PPARγ-mediated lipogenesis by upregulating HNF1A/IRF6 and balancing PGC1α/RORα [35-38]; while simultaneously activating the PPARα signaling axis to promote CPT-1/Acox-1 expression, enhancing fatty acid oxidation [38, 39]. High-intensity interval training (HIIT) can coordinately regulate PPAR family members, promoting PPARα and its downstream targets CPT1α and ACOX1 expression while suppressing PPARγ-mediated lipogenesis [40]. However, differences exist in expression patterns, ligand specificity, and physiological functions of PPAR isoforms between humans and rodents, necessitating caution when extrapolating animal model results to human clinical contexts [41-45].
In insulin signal transduction, exercise improves glucose metabolism through multiple targets. Exercise significantly enhances the hepatic IRS/PI3K/AKT pathway, specifically by reducing IRS1 Ser307 phosphorylation [46]and increasing AKT Ser473 phosphorylation [38]. This improved signaling cascade effectively enhances insulin sensitivity and glucose tolerance [47]. Additionally, exercise suppresses gluconeogenesis by downregulating phosphoenolpyruvate carboxykinase (PEPCK) and G6Pase expression, while increasing hepatic glycogen storage [38].
The mTORC1 signaling axis also plays a significant role in exercise-mediated metabolic regulation. Overactivation of this pathway promotes lipid accumulation, whereas exercise intervention reduces hepatic triglyceride content by decreasing phosphorylation levels of the mTORC1 pathway marker S6K1 [48]. When exercise inhibits the Akt/mTORC1 - SREBP-1 pathway, it can reduce the expression and activity of SREBP-1, thereby diminishing the transcriptional activation of lipogenic genes like ACC and FAS, leading to decreased hepatic lipid synthesis and accumulation [49]. [Fig 1]
Core regulatory role of exercise in hepatic energy metabolism. In terms of glucose and lipid metabolism, exercise activates AMPK and PPARα, improves insulin signaling (IRS/PI3K/AKT), and suppresses the mTORC1-SREBP-1 axis. These coordinated actions enhance hepatic fatty acid oxidation, inhibit lipid synthesis, and thereby improve insulin sensitivity. Regarding amino acid metabolism, exercise accelerates muscle-liver crosstalk, driving hepatic gluconeogenesis with alanine as a key precursor. Concurrently, it enhances muscle protein synthesis and sensitivity to amino acids—particularly leucine—which synergistically promotes post-exercise anabolism and tissue repair.
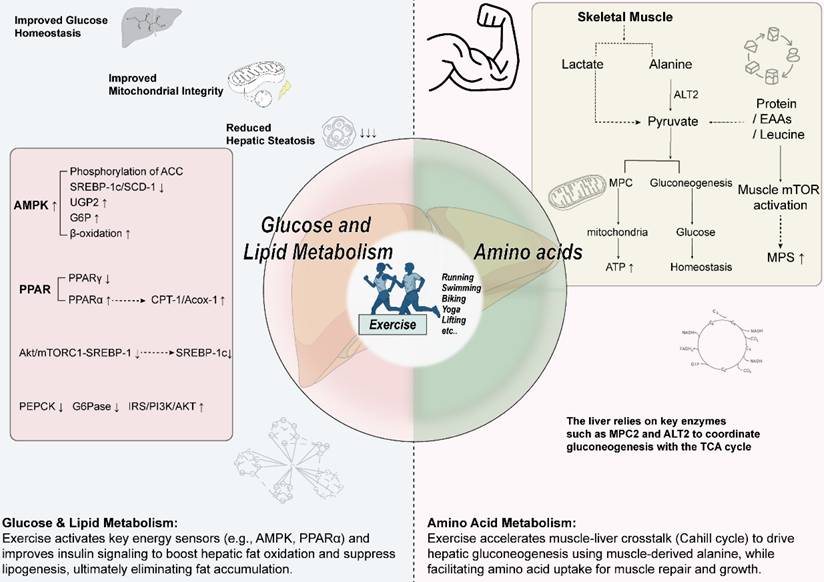
3.2 Amino acids metabolism
Exercise significantly increases the body's demand for glucose to meet the energy requirements of muscle contraction [50]. This process generates more pyruvate/lactate in skeletal muscle, with most lactate released into the bloodstream and taken up by the liver as a gluconeogenic substrate [51]. Concurrently, alanine release from muscle increases and is transported via circulation to the liver [52]. The liver processes alanine via deamination; its carbon skeleton is converted to pyruvate, which is then used for glucose production in the gluconeogenic pathway mediated by the Cahill cycle [52, 53]. Exercise accelerates this cycle, promoting the efficient transport of alanine to the liver for gluconeogenesis to meet the body's glucose demand [53]. The enhancement of hepatic gluconeogenesis relies on metabolic cross-talk between muscle and liver [51].
Research has identified key enzymes in this metabolic coordination: the mitochondrial pyruvate carrier (MPC) mediates pyruvate entry into mitochondria [54, 55], while alanine aminotransferase (ALT) catalyzes the conversion of alanine to pyruvate. Inhibiting hepatic MPC (particularly MPC2) and ALT2 activity impairs amino acid metabolism and gluconeogenic function. Liver-specific double knockout mice exhibit reduced gluconeogenic efficiency, decreased endogenous glucose production, impaired exercise endurance, and abnormal blood glucose levels [51]. This study leveraged the advantages of animal models to clarify the necessity of MPC and ALT in metabolic coordination during exercise. However, complete loss-of-function mutations in such genes are rare in humans, and human glucose homeostasis regulation is more redundant and compensatory (e.g., renal gluconeogenesis). The severe phenotypes in knockout models may overestimate the absolute importance of single targets in human physiology. Research indicates that this inhibition also leads to a decline in tricarboxylic acid (TCA) cycle flux, primarily through the following mechanisms: (1) Insufficient substrate supply: MPC inhibition hinders pyruvate entry into mitochondria for conversion to acetyl-CoA (a key TCA cycle substrate); ALT2 inhibition reduces the conversion of alanine to pyruvate, collectively weakening TCA cycle substrate input [51]. (2) Impaired metabolic flux: Reduced gluconeogenic efficiency disrupts the metabolism of shared intermediates (e.g., pyruvate, oxaloacetate) with the TCA cycle; concurrently, fluxes through anaplerotic reactions (replenishing intermediates) and cataplerotic reactions (removing intermediates) decrease, disrupting TCA cycle intermediate homeostasis [51]. (3) Systemic metabolic dysregulation: Inhibition of MPC and ALT2 disrupts the coordination between hepatic gluconeogenesis, the TCA cycle, and inter-organ metabolic signaling (e.g., Cori cycle and Cahill cycle), ultimately compromising TCA cycle function [51].
Beyond regulating glucose metabolism, acute exercise (particularly resistance training) effectively stimulates muscle protein synthesis (MPS) [56]. Protein supplementation around exercise synergizes with this stimulation to promote MPS [56]. Studies confirm that acute amino acid intake significantly increases MPS rates [57]. In the fasted state, muscle protein balance is often negative after a single bout of resistance exercise [58], but can become positive when combined with protein intake [59-61]. Muscle sensitivity to amino acids is significantly enhanced post-exercise (lasting about 24 hours) [62]. Supplementing protein during this period, compared to non-exercise periods, more efficiently provides a net stimulus for MPS and promotes protein accretion [28]. Data show that immediate post-exercise protein intake can peak MPS rates within 3 hours and maintain elevated levels for 24 to 72 hours [62, 63]. Therefore, timely and regular (every 3-4 hours) protein intake post-exercise maximizes its positive impact on muscle protein metabolism.
Furthermore, essential amino acids (EAAs), particularly leucine among the branched-chain amino acids (BCAAs), are crucial for regulating protein metabolism [56]. Oral BCAAs rapidly increase plasma concentrations, providing key substrates for skeletal muscle MPS [56]. Leucine intake can prolong the duration of protein synthesis [64-66], with its stimulatory effect being dose-dependent: plateauing at about 2g at rest [60, 64], but requiring about 3.5g after 60 minutes of moderate-intensity cycling [66]. However, the regulation of MPS rate duration depends not only on leucine concentration but also on ATP status and the availability of other EAAs. Therefore, balanced intake of all EAAs is most effective for sustaining elevated MPS rates [67]. [Fig 1]
3.3 Injury and repair
Exercise exerts protective effects against metabolic-associated liver injury through synergistic multi-mechanisms, with both common and specific pathways involved in models of diabetes and fatty liver disease.
For diabetes-associated liver injury, mechanisms primarily include metabolic regulation, oxidative stress alleviation, apoptosis inhibition, and inflammation modulation. Twelve weeks of aerobic exercise significantly improved metabolic disorders in db/db mice, manifested as reduced body weight and fasting blood glucose, and enhanced insulin sensitivity. Phenotypic validation confirmed that exercise ameliorated hepatic fat accumulation and functional injury [68]. Regarding oxidative stress regulation, exercise can activate the Nrf2/ARE pathway, upregulating Nrf2 expression to promote SOD activity and reduce oxidative stress markers like MDA, exerting a protective effect [68-70]. db/db mice are a classic model for studying type 2 diabetes, but their pathology progresses rapidly and severely, with liver injury often accompanied by extreme metabolic disturbances. The significant exercise effects observed in these genetically deficient animals may be less prominent in human diabetic patients, who typically have milder and slower-progressing conditions. Additionally, differences may exist in the target gene repertoire and activation thresholds of the Nrf2 pathway between rodents and humans. The anti-apoptotic effect of exercise on hepatocytes is mainly reflected in downregulating the phosphorylation levels of signaling proteins like p-JAK2, p-STAT3, reducing Bax/cleaved caspase-3 expression, and decreasing TUNEL-positive cell numbers, thereby inhibiting apoptosis pathway activation [68]. At the inflammation level, exercise not only inhibits NF-κB pathway activation and reduces p-P65 expression but also modulates the concentrations of inflammatory mediators like IL-6, TNF-α and macrophage infiltration, forming a multi-dimensional anti-inflammatory effect [68].
In MASLD models, exercise optimizes hepatic lipid homeostasis through bidirectional regulatory mechanisms: on one hand, promoting fatty acid β-oxidation by increasing ACC phosphorylation levels and cytochrome c content; on the other hand, inhibiting FAS expression to reduce lipid synthesis [71]. Regarding cytoprotective mechanisms, exercise inhibits the mitochondrial apoptosis pathway by modulating the Bcl-2/Bax balance, reducing caspase-3 activity, thereby decreasing hepatocyte programmed cell death; and induces protective autophagy to promote lipid droplet degradation, which is particularly important for MASLD with impaired autophagy function [72-75]. In oxidative stress regulation, exercise reduces ROS generation, enhances antioxidant enzyme expression, downregulates pro-inflammatory factors like TNF-α, and increases anti-inflammatory mediators like IL-10 via the MAPK/NF-κB pathway, thereby inhibiting macrophage infiltration and alleviating liver injury caused by MASLD [76, 77]. However, most MASLD animal models are induced by high-fat diets or genetic modifications, developing significant steatosis and inflammation within weeks, which differs from the often years-long development of human MASLD. The rapid inhibition of apoptosis and induction of autophagy observed in short-term animal models may reflect an acute adaptive response to stress rather than long-term regulation of chronic human disease. Measuring hepatic autophagy flux in humans is extremely challenging. [Fig 2]
Mechanisms by which exercise improves metabolic liver injury and the immune microenvironment. Exercise exerts protective effects against metabolic liver injury and remodels the hepatic immune microenvironment through multi-pathway synergism. The key mechanisms include: (1) Antioxidant effects: Activation of the Nrf2/ARE pathway upregulates antioxidant enzymes such as SOD, reducing oxidative stress and ROS levels. (2) Anti-apoptotic and reparative effects: Upregulation of the Bcl-2/Bax ratio, inhibition of caspase-3 activity, and downregulation of signaling pathways such as p-JAK2/p-STAT3 collectively suppress hepatocyte apoptosis. (3) Immunomodulation and anti-inflammatory effects: Inhibition of the NF-κB pathway (reducing p-IκBα and p-p65) decreases the production of pro-inflammatory cytokines such as TNF-α and IL-1β. Meanwhile, exercise modulates macrophage polarization, promoting a shift from the pro-inflammatory M1 phenotype (CD86⁺) to the anti-inflammatory M2 phenotype (CD163⁺, CD206⁺), and enhances Kupffer cell function, thereby alleviating hepatic inflammation.
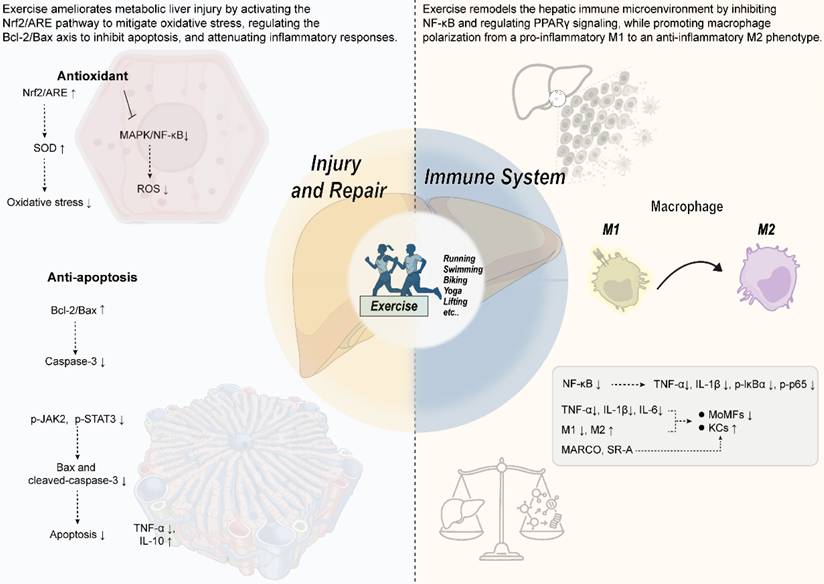
3.4 Immune system
Exercise significantly modulates the hepatic immune microenvironment, effectively attenuating liver inflammation through multi-target regulation of the NF-κB and PPARγ signaling pathways. Studies have found that long-term high-fat diet significantly activates the NF-κB signaling pathway, manifested as increased hepatic p-IκBα and p-p65 protein levels, and markedly elevated expression of pro-inflammatory factors TNF-α and IL-1β in serum and liver tissue [38]. Swimming exercise can significantly suppress NF-κB pathway activation and reduce the levels of these inflammatory markers [38]. In terms of PPARγ signaling regulation, exercise intervention works through multiple mechanisms: directly downregulating PPARγ and its downstream target genes (e.g., CD36, SCD1, PLIN2), reducing hepatic fatty acid uptake and lipid droplet accumulation [38]; upregulating transcriptional repressors like HNF1A and IRF6, which bind to the PPARγ promoter region to inhibit its transcriptional activity [38]; modulating the dynamic balance between the coactivator PGC1α [78]and the negative regulator RORα [35], thereby suppressing PPARγ transcriptional activity. The synergistic action of these molecular mechanisms ultimately leads to reduced lipogenesis and attenuated inflammatory response, improving hepatic metabolic homeostasis.
In the development of MASLD, the accumulation and activation of pro-inflammatory monocytes (MoMFs) play a key role in hepatic inflammation [79, 80]. Exercise intervention significantly reduces the risk of progression to metabolic dysfunction-associated steatohepatitis (MASH) by modulating the migration and activation of MoMFs. Specific mechanisms include: (1) lowering the expression levels of hepatic pro-inflammatory cytokines (e.g., TNF-α, IL-1β); (2) reducing pro-inflammatory monocyte infiltration; (3) alleviating hepatic oxidative stress [81, 82]. These mechanisms collectively inhibit inflammation triggered by immune cells, protecting the liver from MASH damage [82-85].
Additionally, exercise enhances immune function and anti-inflammatory effects through multiple mechanisms: on one hand, upregulating the expression of CD68 and scavenger receptors (such as MARCO, SR-A) on Kupffer cells (KCs), enhancing their phagocytic and endotoxin clearance capacity, while increasing blood dehydroepiandrosterone and inhibiting NFκB-p65 phosphorylation to reduce inflammation; on the other hand, accelerating endotoxin clearance, lowering levels of pro-inflammatory factors like TNF-α and IL-6, further optimizing KCs function and modulating immune responses [86]. Exercise also has significant immunostimulatory effects in cancer patients. Studies show that acute exercise can significantly increase circulating natural killer (NK) cell numbers [87] and may induce lasting functional changes in NK cells, thereby enhancing the body's immune defense against cancer [88]. Simultaneously, γδ T cells exhibit enhanced expansion efficiency and phenotypic remodeling upon bisphosphonate antigen activation following exercise-induced stress stimulation, consequently amplifying their cytotoxic effects against various hematologic malignancies [89]. Notably, as pivotal components of the CD4/CD8 double-negative T cell subpopulation, the antitumor activity of γδ T cells is closely associated with IL-15 signaling pathway: IL-15 can activate surface effector molecules including NKG2D, DNAM-1, and NKp30, while promoting intracellular expression of perforin and granzyme B, ultimately augmenting this subpopulation's tumor-targeted cytolytic capacity [90].
Research indicates that HIIT has significant anti-inflammatory effects, downregulating the gene expression of pro-inflammatory cytokines such as TNF-α, MCP-1, IL-6, and IL-1β, thereby alleviating liver inflammation [40]. Moreover, HIIT modulates macrophage polarization by reducing the expression of total macrophage markers (F4/80, CD68) and M1 markers (CD86), while increasing M2 markers (CD163, CD206), thereby improving inflammation. Mechanistic studies suggest that HIIT may promote hepatic macrophage polarization towards the M2 phenotype via an RORα-dependent KLF4 pathway, exerting anti-inflammatory effects [91]. It is important to note that research on macrophage polarization heavily relies on flow cytometry and specific markers (e.g., CD86, CD206), whose expression and function are not entirely consistent between rodent and human macrophages. The clear M1-to-M2 shift observed in animal models may appear more blurred and dynamic in the complex human liver immune microenvironment. Additionally, the comparability of HIIT protocols (e.g., intensity, interval duration) between animal and human studies is problematic, as animals often endure higher absolute exercise loads, potentially leading to stronger immune stress responses. [Fig 2]
Additionally, the mechanism by which exercise suppresses hepatic immune cell activation through the regulation of lipotoxic metabolites has garnered increasing attention. Research has demonstrated that exercise effectively reduces hepatic levels of lipotoxic metabolites, particularly FFAs and ceramides, thereby elevating the activation threshold of immune cells and curbing excessive inflammatory responses. The underlying mechanisms involve exercise-induced upregulation of PGC-1α, which enhances mitochondrial fatty acid β-oxidation and reduces FFAs accumulation in hepatocytes [92]. Moreover, chronic exercise lowers levels of long-chain ceramides, such as C16:0 and C18:0, in both serum and liver tissue; these molecules are critical mediators of apoptosis and inflammation [93]. Furthermore, exercise activates the AMPK/mTOR pathway to induce autophagy, facilitating the clearance of damaged mitochondria and surplus lipids, thereby alleviating lipotoxic burden [92].
The reduction in these lipotoxic metabolites profoundly influences the hepatic immune microenvironment. On one hand, ceramides act as activating factors for the NLRP3 inflammasome; by downregulating ceramide levels, exercise suppresses NLRP3 activation, reduces the release of IL-1β and IL-18, and elevates the threshold for immune cell response to damage signals [93]. On the other hand, exercise remodels the composition of hepatic immune cells, diminishing the infiltration of pro-inflammatory CD4+, T cells, B cells, and macrophages, thereby reducing susceptibility to acute injury [94]. At the signaling pathway level, exercise enhances the interaction between macrophage migration inhibitory factor and CD74, inhibiting the activation of the lipotoxic stress-associated kinase JNK [95]. Concurrently, it upregulates anti-apoptotic proteins, counteracting TNF-α-mediated inflammatory damage and reinforcing immune homeostasis [94]. In summary, by systematically reducing levels of lipotoxic metabolites, exercise synergistically suppresses excessive hepatic immune activation at the levels of metabolic substrates, immune cell composition, and signaling pathways, thereby exerting a protective effect.
3.5 Myokines, sarcopenia and the muscle-liver axis
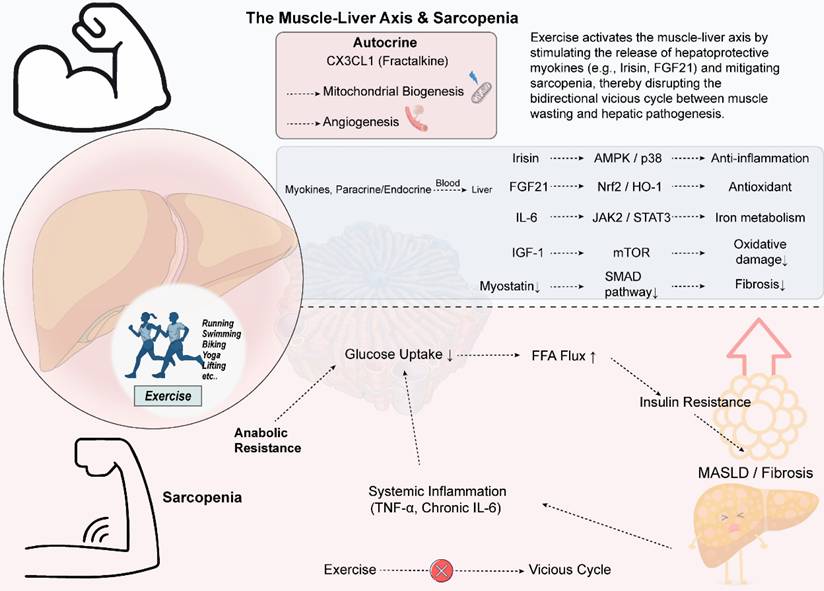
Exercise-derived myokines exert hepatoprotective effects through synergistic multi-pathways [96-98]. Long-term regular exercise reduces myostatin levels, promoting intrahepatic fatty acid oxidation and alleviating fibrosis via the SMAD-AMPK/G6PD signaling axis [96-98]. Acute exercise induces an increase in interleukin-6 (IL-6), improving iron metabolism via the IL-6/JAK2/STAT3 pathway and potentially attenuating ferroptosis by inhibiting tumor necrosis factor-α (TNF-α) activity [99-101]. Endurance training promotes irisin secretion, activating the AMPK/p38 MAPK pathway and inhibiting the NLRP3 inflammasome, thereby reducing lipid peroxidation and inflammation [102-105]. Exercise also upregulates FGF21, enhancing antioxidant capacity via the Nrf2/HO-1/GPX4 pathway [106-108]. In the elderly, insulin-like growth factor 1 (IGF-1) elevated by resistance exercise alleviates oxidative damage via the IGF-1/mTOR pathway [109, 110]. In summary, various myokines synergistically improve hepatic lipid metabolism and fibrosis by enhancing antioxidant defense (e.g., activating Nrf2/GPX4 [101, 106]), promoting fatty acid breakdown, inhibiting inflammation, and cell death. Different forms of exercise differentially affect myokine release, with resistance training potentially being more effective than aerobic exercise [4, 111], while combined endurance training may further optimize mitochondrial function and lipid metabolism, comprehensively enhancing liver protection [112].
The chemokine CX3CL1 plays a key role in regulating skeletal muscle metabolic adaptation and repair. In skeletal muscle, CX3CL1 primarily functions in an autocrine manner, driving the transition of fast-twitch fibers from the fast-twitch oxidative-glycolytic to purely glycolytic (type IIB), thereby enhancing anaerobic energy supply capacity; this transition is mainly mediated by modulating mitochondrial activity and myosin ATPase, with minimal impact on slow-twitch fibers [113, 114]. Exercise training upregulates skeletal muscle CX3CL1 expression, a mechanism potentially related to Ca²⁺/CaMKII signal activation, which subsequently promotes glucose uptake and mitochondrial biogenesis, regulating muscle metabolism [114-116]. Furthermore, the CX3CL1/CX3CR1 signaling axis is crucial in muscle injury repair, including promoting angiogenesis, mediating macrophage clearance of necrotic tissue, and coordinating muscle satellite cells with immune cells to accelerate regeneration [117, 118]. It is worth noting that CX3CL1 not only exerts autocrine effects locally in skeletal muscle, but recent studies have revealed that it can also act on the liver and participate in regulating immune cell functions, suggesting that CX3CL1 may play an important role in muscle-liver crosstalk [119].
Sarcopenia, an age-related progressive loss of skeletal muscle mass and function, is closely associated with MASLD. The prevalence of sarcopenia in MASLD patients increases with the progression of liver fibrosis [120-122]. The two conditions form a bidirectional vicious cycle via the "muscle-liver axis," with core pathological mechanisms including: skeletal muscle anabolic resistance, i.e., reduced responsiveness to nutritional and hormonal signals, which can be exacerbated by MASLD-related inflammation and metabolic disturbances [123-125]; decreased muscle glucose uptake leading to insulin resistance, causing ectopic deposition of free fatty acids in the liver and promoting steatosis, while muscle loss further worsens systemic insulin resistance [123, 126]; impaired metabolic flexibility in MASLD/MASH patients, manifesting as difficulty switching between fatty acid oxidation and glucose utilization, thereby worsening intrahepatic lipid accumulation and insulin resistance [127]; systemic inflammation and signaling dysregulation, where factors like TNF-α and IL-6 promote both muscle catabolism and liver fibrosis, coupled with imbalances in myokine and hepatokine secretion and factors like vitamin D deficiency, collectively constituting this complex pathological network [128-130]. Therefore, comprehensive interventions targeting the "muscle-liver axis" (e.g., nutritional and exercise therapies) can provide multi-target strategies for MASLD/MASH by modulating these mechanisms [123]. [Fig 3]
Exercise modulates the muscle-liver axis to interrupt the vicious cycle between sarcopenia and liver disease. Exercise stimulates the release of various myokines, such as Irisin, FGF21, and IGF-1, from skeletal muscle. These myokines act on the liver through the systemic circulation and exert protective effects via distinct molecular pathways: Irisin activates the AMPK/p38 pathway to reduce inflammation, FGF21 enhances the Nrf2-mediated antioxidant defense system, and IGF-1 alleviates oxidative damage through the mTOR pathway. Collectively, these mechanisms improve hepatic lipid metabolism and suppress fibrosis. Moreover, exercise upregulates the chemokine CX3CL1, which promotes mitochondrial biogenesis and angiogenesis within muscle tissue. Conversely, sarcopenia is characterized by anabolic resistance, reduced muscle glucose uptake, and systemic inflammation—marked by elevated levels of TNF-α and chronic IL-6. These alterations exacerbate insulin resistance and increase the influx of free fatty acids into the liver, thereby promoting hepatic steatosis and injury, and perpetuating a vicious cycle.Various forms of exercise can effectively target this axis, offering a therapeutic strategy to break the reciprocal worsening of sarcopenia and liver disease.
4. The Specific Effects of Exercise on Different Liver Diseases
4.1 Exercise and MASLD
MASLD is a liver disease caused by multi-system metabolic imbalances. Its diagnosis requires hepatic steatosis ≥ 5% accompanied by conditions such as overweight/obesity, type 2 diabetes, or metabolic dysregulation, while excluding other liver diseases and alcohol abuse [131]. The disease is categorized into simple steatosis and MASH, with the latter carrying a higher risk of progression to cirrhosis and HCC due to accompanying inflammation and fibrosis [132]. Epidemiological data show a global MASLD prevalence exceeding 30%, with significant regional variations: highest in South America (> 40%), lowest in Africa (14%), and fastest growth in the Middle East and North Africa [133, 134]. Incidence is particularly high among males and obese populations, while weight loss of 5%-10% can effectively reverse hepatic steatosis and fibrosis, highlighting the necessity of preventive interventions for high-risk groups [135].
Lifestyle interventions centered on weight loss and regular exercise are foundational treatment strategies [136]. Aerobic exercise improves liver fat content through dual mechanisms: on one hand, enhancing cardiorespiratory function, promoting hepatocyte and skeletal muscle mitochondrial proliferation and functional optimization, thereby increasing fatty acid oxidation efficiency [137]; on the other hand, modulating hormone balance, such as stimulating growth hormone secretion to accelerate visceral fat breakdown and reduce fatty acid transport to the liver via the portal system [138]. Exercise also enhances insulin sensitivity, inhibiting lipolysis and FFAs release [139, 140]. Common aerobic exercise forms include walking, treadmill, and stationary cycling, which offer controllable intensity and strong site adaptability. Compound exercises like rowing machines and ellipticals are increasingly popular due to their ability to activate multiple muscle groups. Although high-intensity aerobic training is less commonly applied, studies confirm that performing it three times weekly (for 8 weeks) or five times weekly (for 6 months) can reduce liver fat by 2.4% and 5.0% respectively, suggesting its potential value [141, 142].
Research on exercise dosage shows that 150-240 minutes of moderate-intensity aerobic exercise per week can reduce hepatic fat by 2%-4%, and even 135 minutes still yields significant effects [4, 142]. This approach improves central obesity and cardiopulmonary function even without weight loss, demonstrating clinical feasibility. Resistance training, as an important complement to aerobic exercise, although less stimulating to the cardiopulmonary system, effectively enhances muscle mass and metabolic regulation capacity [143, 144]. Population studies have confirmed a negative correlation between resistance training and hepatic fat levels, with high-intensity resistance training showing superior effects [145, 146]. An 8-week intervention can reduce hepatic steatosis by 13% independent of weight change [147] but due to study heterogeneity, its exact efficacy still needs to be verified [146]. Currently, there is a lack of direct mechanistic research on the effects of resistance training on intrahepatic lipids and metabolism [143, 148]. HIIT, which alternates high-intensity exercise (reaching 80%-100%HRmax) with rest, shows lipid-lowering effects comparable to MICT [137, 149]. However, due to limitations in sample size and study dimensions, more evidence is needed to compare their efficacy [24, 149]. MICT, as a traditional model, requires maintaining 45%-80%HRmax for 20-60 minutes [24, 150]. Studies have confirmed that its regular implementation (≥ 5 times per week) can reduce MASLD incidence by 16% and increase remission rates by 40%, with exercise volume accumulation positively correlated with disease improvement [151].
In summary, the comprehensive application of aerobic exercise, resistance training, and high-intensity interval training holds significant value for the prevention and treatment of MASLD. Regular moderate-to-high-intensity physical exercise not only reduces the risk of disease onset but also promotes disease remission, providing evidence-based support for clinical practice. [Fig 4]
Specific Effects of Exercise on Different Types of Liver Diseases. In MASLD, exercise activates the AMPK pathway to promote ACC phosphorylation and enhance beta-oxidation. It also facilitates mitochondrial biogenesis via the IRS/PI3K/AKT pathway and PGC-1α. Furthermore, exercise modulates macrophage polarization through mTOR and reduces levels of NF-κB-related inflammatory factors, thereby alleviating hepatic inflammation. In ALD, exercise improves mitochondrial function, increases the tricarboxylic acid (TCA) cycle rate and cytochrome oxidase activity, and protects the liver from ethanol-induced oxidative damage. For viral hepatitis, exercise regulates inflammatory factors such as TNF-α and IL-6, mitigating chronic inflammation. It also enhances muscle metabolism, which may help alleviate sarcopenia. Regarding fibrosis/cirrhosis, exercise upregulates CCN1, which binds to TGF-β and induces endoplasmic reticulum stress, thereby reducing HSCs. It also decreases TGF-β1 mRNA levels and suppresses the expression of macrophage markers and chemokines. In HCC, exercise inhibits Akt and S6-RP phosphorylation via the AMPK/mTORC1 pathway and blocks cell proliferation signals through the P53/P21 pathway. It also suppresses the JNK/AP-1 pathway via phosphorylated c-Jun (p-c-Jun), thereby impeding HCC progression. Additionally, exercise negatively regulates Akt signaling through PTEN, reducing invasive tumor growth, and inhibits the STAT3 pathway, thereby diminishing angiogenesis and metastasis.
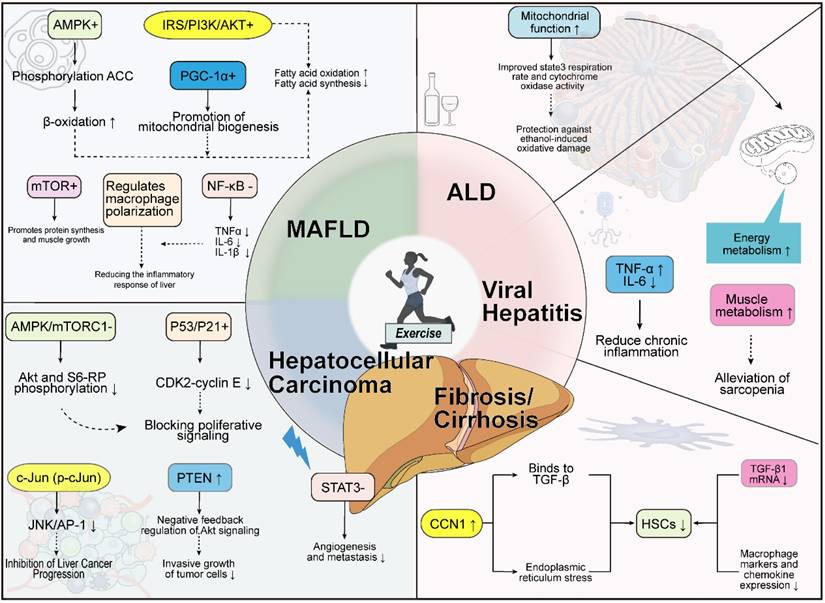
4.2 Exercise and ALD
ALD is a group of liver disorders caused by chronic excessive alcohol consumption. Its clinical manifestations and histopathological features include alcohol-associated steatohepatitis (ASH), cirrhosis, and HCC [152]. Among individuals with long-term heavy alcohol consumption, over 90% develop alcohol-associated fatty liver, characterized by abnormal fat accumulation in hepatocytes [152]. The mechanisms underlying steatosis are complex, involving impaired mitochondrial fatty acid β-oxidation, abnormal lipid transport from peripheral tissues to the liver, and dysregulation of lipid metabolism-related transcription factors [152].
An 8-week animal study explored the interactive effects of exercise and ethanol on hepatic mitochondrial function in female Sprague-Dawley rats. Results showed that ethanol exposure specifically impaired mitochondrial function: state 3 respiratory rate (with glutamate as substrate) and cytochrome oxidase activity significantly decreased, with the most pronounced decline in the sedentary ethanol (S/E) group compared to others (p < 0.05). Notably, although ethanol intervention reduced maximal oxygen consumption rate, indicators reflecting oxidative phosphorylation coupling efficiency, such as respiratory control ratio (RCR) and phosphorus-to-oxygen ratio (P:O), showed no significant change, suggesting the damage primarily affects the electron transport chain rather than ATP synthesis coupling [153].
Regarding the effect of exercise intervention, daily 2-hour swimming training itself did not improve baseline mitochondrial function parameters (state 3 respiration, RCR, and P:O) but demonstrated significant protective effects under ethanol exposure. The swim-ethanol combined group maintained better mitochondrial oxidative function compared to the S/E group, with a 21.3%±3.2% increase in state 3 respiratory rate (p < 0.01) and cytochrome oxidase activity restored to 89.7%±5.4% of control levels. This confirms that endurance exercise can effectively counteract ethanol-induced oxidative damage by enhancing mitochondrial functional adaptability [153]. These findings reveal a unique protective mechanism of exercise intervention in alcohol-associated liver injury: regular endurance training, while not directly enhancing baseline mitochondrial function, improves mitochondrial stress tolerance, maintaining functional homeostasis under pathological conditions. It is noteworthy that the study used female rats, while significant gender differences exist in human ALD (higher prevalence in males, but faster progression in females) [154]. Sex hormones have important effects on liver metabolism and exercise responses [155, 156], thus gender factors must be considered when inferring results [157, 158]. Furthermore, in animal experiments, ethanol administration is a controlled, forced intake, differing from the complex and variable drinking patterns in humans. The role of exercise in preventing (rather than treating) established alcohol-associated hepatitis or cirrhosis still requires more human prospective studies for confirmation. [Fig 4]
Furthermore, when discussing the protective effects of exercise, it is essential to acknowledge the dual nature of oxidative stress. While regular moderate exercise can induce adaptive protective effects by upregulating the endogenous antioxidant system [159], acute, high-intensity, or unaccustomed exercise may trigger a transient burst of ROS, thereby exacerbating tissue damage [159]. Studies have shown that even a single bout of acute exercise in heavy drinkers can further deplete their already low GSH levels, accompanied by a significant increase in oxidative damage markers such as thiobarbituric acid reactive substances, indicating a severe deficiency in antioxidant reserves under acute stress [160].
For patients with severe alcoholic hepatitis, whose livers are already in a state of extreme oxidative stress and intense inflammation, the timing and intensity of exercise interventions require careful consideration. During the acute phase of the disease, treatment should primarily focus on bed rest and nutritional support, avoiding any high-intensity physical activity that could lead to a surge in ROS and potentially exacerbate hepatocyte apoptosis and necrosis [161]. Once the patient's condition stabilizes and they enter the rehabilitation phase, exercise prescriptions should adhere to the principle of "low-intensity initiation and gradual progression" [162]. Current clinical consensus suggests that patients with advanced liver disease can engage in low-to-moderate intensity aerobic training, supplemented by light resistance exercises, with close monitoring of liver function throughout the process [163]. This approach aims to harness the long-term antioxidant benefits of exercise while effectively avoiding the potential risks associated with acute increases in oxidative load.
4.3 Exercise and viral hepatitis
Acute hepatitis is a common liver disease with diverse but regular clinical manifestations. Its early symptoms mainly include gastrointestinal symptoms such as anorexia, malaise, nausea, and myalgia, typically occurring 1-2 weeks before the onset of jaundice [164].
Regarding rest and exercise for acute hepatitis patients, traditional views advocate strict bed rest to avoid physical exertion. Clinical observations have found that vigorous exercise during the incubation period may lead to rapid deterioration of the condition, even endangering life [165, 166]. To explore the impact of exercise on the course of acute viral hepatitis, researchers conducted a controlled study. Twenty-five hospitalized patients were randomly assigned to an exercise group and a control group. The exercise group performed moderate-intensity exercise twice daily (reaching 70% of maximal exercise intensity), while the control group adhered to strict bed rest. Results showed no significant difference between the two groups in the recovery time of biochemical markers or histopathological changes [167]. This finding provides new insights into rehabilitation strategies for acute hepatitis patients.
In the field of chronic liver disease (CLD), the diagnosis is made when liver inflammation and/or necrosis persist for more than 6 months [164]. CLD is a significant cause of premature death in the working-age population [168, 169]. This disorder induces a plurality of pathophysiological alterations in skeletal muscle, encompassing metabolic, cellular, vascular, and inflammatory changes, which culminate in sarcopenia [170]. These changes, combined with malnutrition and physical inactivity [171], contribute to a high prevalence of frailty (40%-70%) secrete fibrogenic factors and inflammatory mediators [172]. Although theoretically exercise might have positive effects on chronic hepatitis, studies show only a neutral impact on chronic persistent hepatitis [173]. To assess the effect of exercise intervention on chronic hepatitis, researchers implemented a 12-week standardized interval training program for 17 patients with chronic active hepatitis, monitoring liver function tests, maximal oxygen uptake, and exercise tolerance. Results showed stable serum transaminase levels during training, while maximal oxygen consumption and exercise tolerance significantly improved [174]. Therefore, patients with chronic active hepatitis can not only tolerate regular exercise but may also benefit from its effects on liver health. However, this study had a relatively small sample size, and the historical classification "chronic active hepatitis" has now been replaced by more specific etiological and pathological staging systems. Physiological and pathological responses to exercise may differ among chronic liver diseases of various etiologies. [Fig 4]
4.4 Exercise and liver fibrosis, cirrhosis
Liver fibrosis is a common dynamic pathological process in various chronic liver injuries, characterized by excessive extracellular matrix accumulation and reduced degradation by metalloproteinases [175]. Activated hepatic stellate cells (HSCs) play a critical role in this process, serving as the primary source of extracellular matrix [176] and transforming into myofibroblasts [177]. Although multiple etiologies of chronic liver disease can directly activate HSCs, the core driver of sustained fibrosis progression is the inflammatory microenvironment.
Studies indicate that intracellular lipid accumulation in hepatocytes promotes the transition from steatosis to MASH [178, 179]. Lipotoxicity causes hepatocyte damage, triggering cellular stress, apoptosis, and death [180, 181]. HSCs and Kupffer cells, upon uptake of apoptotic bodies released by hepatocytes, secrete fibrogenic factors and inflammatory mediators [182]. Substances secreted by Kupffer cells attract immune cell aggregation, exacerbating inflammation, activating HSCs, and inducing metabolic-associated liver fibrosis [183].
Exercise offers multiple benefits for liver fibrosis. Studies show that exercise increases the expression of CCN1, a senescence marker in HSCs [85]. CCN1 can bind to TGF-β, inhibiting SMAD signaling and reducing collagen deposition [184]. It is also associated with endoplasmic reticulum stress-induced HSC apoptosis [185], thereby reducing HSC activation and inhibiting fibrogenesis. Another study showed that 16-week of aerobic exercise intervention decreased hepatic TNF-α mRNA levels, alleviated inflammation, suppressed TGF-β1 mRNA expression, and reduced collagen deposition and macrophage markers and chemokine expression in the liver [83]. It is noteworthy that animal models used to induce liver fibrosis are based on acute or subacute injury, whose pathophysiology fundamentally differs from the slow, multi-year progression of fibrosis in human chronic liver disease. The anti-fibrotic effects demonstrated by exercise in acute injury models are not equivalent to its ability to reverse established fibrosis or even cirrhosis in human livers. Human studies primarily rely on non-invasive indicators like liver elastography to indirectly assess fibrosis changes, which differs from the molecular analysis of HSC phenotype and extracellular matrix components possible in animal experiments.
Cirrhosis is a global disease with high morbidity and mortality, characterized by the replacement of healthy liver parenchyma with fibrous tissue and regenerative nodules, leading to portal hypertension and liver failure [183, 186]. Treatment focuses on controlling the etiology and managing complications, with liver transplantation as a last resort [187-189]. In recent years, the role of exercise intervention in the treatment and rehabilitation of cirrhosis has gained increasing attention. Research confirms that regular aerobic exercise significantly improves physical function in cirrhotic patients. If patients engage in supervised aerobic exercise at least twice weekly, such as slow jogging on a treadmill or whole-body aerobic training using a bicycle ergometer, for 2-3 months, their peak oxygen uptake can increase by 1.7 to 5.3 mL/kg/min [190, 191]. Clinically, doctors often recommend that patients with compensated cirrhosis develop and implement a progressive exercise plan. Various aerobic exercises are effective and safe for this population. Walking is favored for its simplicity; swimming reduces body burden due to water buoyancy and minimizes joint stress; and spinning effectively enhances cardiorespiratory function and is also popular [173, 192]. To ensure safety and effectiveness, patients are advised to start with low-load training, initially choosing short-duration, low-intensity activities, and gradually increasing duration, intensity, and difficulty as the body adapts. This exercise intervention strategy helps improve physical function and quality of life. [Fig 4]
4.5 Exercise and liver cancer
Early clinical symptoms of HCC are often insidious, with approximately 70% of patients losing the opportunity for curative treatment at diagnosis. The high five-year postoperative recurrence rate severely impacts patients' physiological function and mental health [193].
Exercise intervention inhibits the occurrence and progression of HCC through a multidimensional molecular regulatory network, involving cell cycle regulation, signaling pathway inhibition, and tumor microenvironment remodeling. In the molecular regulatory network, the AMPK/mTORC1 signaling axis plays a central role: exercise-induced AMPK phosphorylation disrupts mTOR complex stability, inhibits mTORC1 activity, and blocks cell proliferation signals [194]. This process is accompanied by reduced phosphorylation levels of key downstream effectors Akt and S6-RP, with Akt inhibition forming a cascade amplification effect as an upstream activator of mTORC1. Simultaneously, STAT3 signaling inhibition significantly weakens tumor angiogenesis and metastatic potential [195].
In cell cycle regulation, exercise induces cell cycle arrest by activating the p53/p27 pathway. Experiments confirm that exercise significantly increases hepatic p53 Ser15 phosphorylation; with activated p53 upregulating the cell cycle inhibitor p27, specifically inhibiting CDK2-cyclin E complex function, thereby curbing abnormal hepatocyte proliferation. Additionally, exercise exerts anti-cancer effects by inhibiting the JNK1 signaling pathway: reducing phosphorylated c-Jun (p-cJun) accumulation and blocking JNK/AP-1 signaling axis activation, which is closely associated with MASH and HCC progression [196].
The tumor suppressor PTEN plays a key regulatory role in exercise intervention. Exercise upregulates PTEN expression, forming a negative feedback loop on Akt signaling pathway, effectively inhibiting invasive tumor growth. Phenotypically, exercise significantly reduces the proportion of active tumor areas, decreases CD31-positive vascular density, and suppresses tumor angiogenesis. Notably, exercise-induced metabolic improvements (e.g., enhanced insulin sensitivity, reduced hepatic lipid deposition) may indirectly inhibit HCC progression by reshaping the tumor microenvironment. However, the clear pathway inhibition observed in animal experiments may be invalidated or altered in human HCC due to factors like frequent TP53 mutations, PTEN loss, or Wnt/β-catenin pathway activation. Therefore, when developing exercise as an adjunctive therapy for human HCC, tumor molecular subtypes must be considered [194].
It is noteworthy that the relationship between exercise intensity and anti-tumor effects is nonlinear. Experimental data show that moderate-intensity training can significantly activate tumor-suppressive pathways, inducing tumor cell apoptosis more efficiently than low-intensity training (< 50% VO2max), while high-intensity training (> 80% VO2max) may exert pro-carcinogenic effects through mechanisms like oxidative stress [197, 198]. This dose-dependent characteristic highlights the need for individualized exercise prescriptions in clinical practice. [Fig 4]
5. Potential Molecular Therapies
Exercise brings multiple clinical benefits to liver disease patients, including improved aerobic capacity, increased muscle mass, reduced fatigue, and enhanced quality of life [199]. The systemic metabolic remodeling induced by exercise suggests that key molecular changes during this process could serve as potential drug targets for treating liver diseases. However, when translating these targets into clinical therapies, their respective limitations and safety issues must be fully considered.
AMPK, as a core energy-sensing molecule, is an important intervention target. Its direct activator, AICAR, has shown potential in preclinical studies to mimic exercise benefits [200]. However, as an "exercise mimetic," its long-term safety, potential to interfere with exercise adaptation, and drug specificity (avoiding off-target effects) remain major challenges. Although the clinically common drug metformin can indirectly activate AMPK, its inhibition of mitochondrial complex I may attenuate exercise-induced metabolic adaptations. Furthermore, metformin is not yet approved for treating MASLD, and its liver-specific effects and long-term benefits require more evidence.
Thiazolidinediones (TZDs) improve insulin resistance and hepatic steatosis by activating PPARγ [201-203]. However, their clinical application is limited by significant safety concerns, including weight gain, edema, increased risk of congestive heart failure, and fracture risk. Rosiglitazone has also been controversial due to potential cardiovascular risks. Although its hepatic benefits may stem from preferential effects on peripheral adipose tissue [204, 205], side effects associated with systemic PPARγ activation limit its widespread use in MASLD/MASH. Developing liver-specific PPARγ modulators or partial agonists may be a direction, but their efficacy-safety balance still needs validation.
Inhibitors targeting stearoyl-CoA desaturase 1 (SCD1), such as CAY10566, show potential in preclinical models for reducing hepatic steatosis and enhancing lipophagy [206]. However, SCD1 plays important roles in lipid synthesis in various tissues (e.g., skin, pancreas). Systemic inhibition may lead to adverse effects like skin barrier dysfunction and pancreatitis. Developing liver-targeted SCD1 inhibitors is a key challenge in translational research.
Overactivation of the mTORC1 signaling pathway is closely associated with metabolic diseases [48]. However, its inhibitors (e.g., rapamycin) face significant limitations in clinical application. mTORC1 is widely involved in systemic cell growth, proliferation, and immune regulation; inhibiting it can cause metabolic side effects like hyperglycemia, hyperlipidemia, and exacerbated insulin resistance, as well as issues like oral ulcers and immunosuppression [207, 208]. Therefore, developing tissue-specific (especially liver-specific) mTOR modulation strategies or identifying more selective downstream effectors is key to improving the therapeutic window.
Evidence indicates that bioactive molecules capable of mimicking the effects of exercise may contain components beneficial to hepatic metabolism. For instance, SERCA activators can markedly reduce hepatic lipid accumulation and inflammatory responses by improving mitochondrial function and alleviating endoplasmic reticulum stress, thereby playing an important protective role in maintaining liver metabolic health [209]. On the other hand, the mitochondrial DNA-derived peptide MOTS-c has been identified as a key signaling molecule mediating communication between mitochondria and the nucleus as well as other organs. As a potent exercise mimetic, MOTS-c can significantly enhance insulin sensitivity and modulate lipid metabolism. In models of liver disease, this peptide exerts anti-diabetic hepatic fibrotic effects by regulating the Keap1-Nrf2-Smad2/3 signaling pathway, offering a potential therapeutic strategy for liver injury induced by metabolic disorders [210].
In summary, although several highly promising molecular targets have been identified based on exercise biology mechanisms, their translation into clinical therapies still faces challenges related to specificity, safety, and uncertainty of long-term benefits [211]. Future research needs to focus on developing tissue-specific delivery systems, exploring more precise downstream effector molecules, and advancing clinical translation based on solid preclinical safety assessments [212]. Simultaneously, exercise itself, as a safe and multi-beneficial foundational intervention, warrants in-depth exploration of its synergistic effects with these potential targeted therapies [213].
6. Summary and Outlook
The liver, as a core organ of the human body, plays key roles in energy metabolism, protein synthesis, and toxin excretion. Liver injury can lead to issues like hypoglycemia and lipid metabolism disorders, severely impacting health. Common liver diseases include MASLD, ALD, various viral hepatitis, liver fibrosis, cirrhosis, and liver cancer. These diseases not only have high incidence rates and serious consequences but also possess unique pathogenesis, transmission routes, symptoms, and prognoses.
Compared to pharmacotherapy alone, exercise intervention demonstrates unique comprehensive advantages in liver disease treatment, offering multi-target. Exercise activates signaling pathways like AMPK/mTORC1 and p53/p27, improving insulin resistance, regulating lipid metabolism, and directly inhibiting abnormal hepatocyte proliferation and tumor angiogenesis. This multi-dimensional regulatory effect is difficult to achieve with a single drug. Furthermore, exercise intervention can synchronously improve systemic metabolic status, including weight reduction, amelioration of glucose and lipid metabolism disorders, and reduction of oxidative stress and inflammation, thereby creating a more favorable microenvironment for the liver. Additionally, exercise possesses a unique "exercise adaptation" effect, enhancing mitochondrial functional reserve and improving the liver's tolerance to various injurious factors—a form of protection that drugs cannot replicate. Moreover, exercise intervention avoids potential side effects and drug resistance issues associated with long-term medication and is more cost-effective. Most importantly, exercise improves patients' quality of life, achieving holistic enhancement of physical and mental health through overall modulation of the neuro-endocrine-immune network. These advantages make exercise intervention an indispensable component of liver disease management.
Future research can utilize multi-omics technologies to systematically analyze the mechanisms underlying exercise-induced hepatic protective adaptation, particularly the regulatory effects of exercise on the hepatic stellate cell-immune cell interaction network. Concurrently, developing artificial intelligence-based models to predict exercise efficacy, integrating biomarkers such as gut microbiota and circulating exosomes, will provide new paradigms for precision exercise medicine. It is noteworthy that while exercise mimetic drugs can partially replicate exercise effects, their potential interference with adaptive exercise responses requires careful evaluation. At the translational application level, there is an urgent need for large-scale clinical studies to validate the long-term benefits of different exercise modalities for patients with end-stage liver disease and to establish remote exercise monitoring systems based on wearable devices to improve treatment adherence.
Looking ahead, exercise intervention will transcend its traditional positioning as an adjunctive therapy. Through multi-dimensional integration with molecular targeted therapies, nutritional modulation, and psychological interventions, it will become a crucial pillar of comprehensive liver disease management. The development of this field requires not only deepening fundamental mechanistic research but also collaboration across multiple disciplines including clinical medicine, exercise science, and bioinformatics, ultimately achieving a shift from "population recommendations" to "individualized prescriptions" and providing innovative solutions for global liver health management.
Abbreviations
MASLD: metabolic dysfunction-associated steatotic liver disease; ALD: alcohol-related liver disease; ASH: alcohol-associated steatohepatitis; HCC: hepatocellular carcinoma; METs: metabolic equivalents; %HRmax: percentage of maximal heart rate; FFAs: free fatty acids; VO2max: maximal oxygen uptake; HIIT: high-intensity interval training; SIT: sprint interval training; MICT: moderate-intensity continuous training; MPC: mitochondrial pyruvate carrier; ALT: alanine aminotransferase; MPS: muscle protein synthesis; EAAs: essential amino acids; BCAAs: branched-chain amino acids; ATP: adenosine triphosphate; Nrf2: nuclear factor erythroid 2-related factor 2; ARE: antioxidant response element; SOD: superoxide dismutase; MDA: malondialdehyde; p-JAK2: phosphorylated Janus kinase 2; p-STAT3: phosphorylated signal transducer and activator of transcription 3; Bax: BCL2-associated X protein; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; IL-6: interleukin-6; TNF-α: tumor necrosis factor alpha; ACC: acetyl-CoA carboxylase; FAS: fatty acid synthase; Bcl-2: B-cell lymphoma 2; ROS: reactive oxygen species; IL-10: interleukin-10; MAPK: mitogen-activated protein kinase; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; IL-1β: interleukin-1 beta; CD36: cluster of differentiation 36; SCD1: stearoyl-CoA desaturase 1; PLIN2: perilipin 2; HNF1A: hepatocyte nuclear factor 1 alpha; IRF6: interferon regulatory factor 6; PGC1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; RORα: RAR-related orphan receptor alpha; MoMFs: pro-inflammatory monocytes; MASH: metabolic dysfunction-associated steatohepatitis; CD68: cluster of differentiation 68; MARCO: macrophage receptor with collagenous structure; SR-A: scavenger receptor class A; KCs: Kupffer cells; NK: natural killer cells; γδ T cells: gamma delta T cells; IL-15: interleukin-15; NKG2D: natural killer group 2D; DNAM-1: DNAX accessory molecule-1; NKp30: natural cytotoxicity triggering receptor 3; MCP-1: monocyte chemoattractant protein-1; F4/80: EGF-like module-containing mucin-like hormone receptor-like 1; CD80: cluster of differentiation 80; CD163: cluster of differentiation 163; CD206: cluster of differentiation 206; PGC-1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; KLF4: Krüppel-like factor 4; CD86: cluster of differentiation 86; SMAD: mothers against decapentaplegic homolog; AMPK: AMP-activated protein kinase; G6PD: glucose-6-phosphate dehydrogenase; JAK2: Janus kinase 2; STAT3: signal transducer and activator of transcription 3; NLRP3: NACHT, LRR and PYD domains-containing protein 3; FGF21: fibroblast growth factor 21; HO-1: heme oxygenase-1; GPX4: glutathione peroxidase 4; IGF-1: insulin-like growth factor 1; mTOR: mechanistic target of rapamycin; CX3CL1: C-X3-C motif chemokine ligand 1; CaMKII: Ca²⁺/calmodulin-dependent protein kinase II; CX3CR1: C-X3-C motif chemokine receptor 1; S/E: sedentary ethanol; RCR: respiratory control ratio; P:O: phosphorous-to-oxygen ratio; GSH: Glutathione; CLD: chronic liver disease; HSCs: hepatic stellate cells; CCN1: cellular communication network factor 1; TGF-β: transforming growth factor beta; mTORC1: mechanistic target of rapamycin complex 1; Akt: protein kinase B; S6-RP: ribosomal protein S6; p53: tumor protein p53; p27: cyclin-dependent kinase inhibitor 1B; CDK2: cyclin-dependent kinase 2; JNK1: c-Jun N-terminal kinase 1; AP-1: activator protein 1; PTEN: phosphatase and tensin homolog; CD31: cluster of differentiation 31; TP53: tumor protein p53; β-catenin: beta-catenin; AICAR: 5-aminoimidazole-4-carboxamide ribonucleotide; TZDs: thiazolidinediones; MASLD/MASH: metabolic dysfunction-associated steatotic liver disease/metabolic dysfunction-associated steatohepatitis; MOTS-c: mitochondrial open reading frame of the 12S rRNA-c; Smad2/3: intracellular signaling proteins.
Author Contributions
Chen Cheng, Wentao Xu made substantial contributions to the conception and design of the work and drafted the manuscript; Shanshan Wu and Jia Gao critically revised the manuscript for important intellectual content; Li Zuo and Hua Wang finally approved the version to be published; Jianshang Huang and Yunsheng Dong and Jingjing Ji agreed to be accountable for all aspects of the work, ensuring that any issues related to the accuracy or integrity of any part of the work are appropriately investigated and resolved
Competing Interests
The authors have declared that no competing interest exists.
References
1. Han H, Desert R, Das S, Song Z, Athavale D, Ge X. et al. Danger signals in liver injury and restoration of homeostasis. J Hepatol. 2020;73:933-51
2. Devarbhavi H, Asrani SK, Arab JP, Nartey YA, Pose E, Kamath PS. Global burden of liver disease: 2023 update. J Hepatol. 2023;79:516-37
3. Wong MCS, Huang JLW, George J, Huang J, Leung C, Eslam M. et al. The changing epidemiology of liver diseases in the Asia-Pacific region. Nat Rev Gastroenterol Hepatol. 2019;16:57-73
4. Hashida R, Kawaguchi T, Bekki M, Omoto M, Matsuse H, Nago T. et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J Hepatol. 2017;66:142-52
5. Kawaguchi T, Kawaguchi A, Hashida R, Nakano D, Tsutsumi T, Kawaguchi M. et al. Resistance exercise in combination with aerobic exercise reduces the incidence of serious events in patients with liver cirrhosis: a meta-analysis of randomized controlled trials. J Gastroenterol. 2024;59:216-28
6. Lee YT, Wang JJ, Luu M, Tseng HR, Rich NE, Lu SC. et al. State-Level HCC Incidence and Association with Obesity and Physical Activity in the United States. Hepatology. 2021;74:1384-94
7. Diao X, Ling Y, Zeng Y, Wu Y, Guo C, Jin Y. et al. Physical activity and cancer risk: a dose-response analysis for the Global Burden of Disease Study 2019. Cancer Commun (Lond). 2023;43:1229-43
8. Sung KC, Ryu S, Lee JY, Kim JY, Wild SH, Byrne CD. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J Hepatol. 2016;65:791-7
9. Choo YJ, Cho CW, Chang MC. Effects of supervised exercise on aerobic capacity and quality of life in patients with chronic liver disease and patients who underwent liver transplantation: a systematic review and meta-analysis. Int J Rehabil Res. 2022;45:1-11
10. Garber CE, Blissmer B, Deschenes MR, Franklin BA, Lamonte MJ, Lee IM. et al. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43:1334-59
11. Norton K, Norton L, Sadgrove D. Position statement on physical activity and exercise intensity terminology. J Sci Med Sport. 2010;13:496-502
12. Guo R, Liong EC, So KF, Fung ML, Tipoe GL. Beneficial mechanisms of aerobic exercise on hepatic lipid metabolism in non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int. 2015;14:139-44
13. van der Windt DJ, Sud V, Zhang H, Tsung A, Huang H. The Effects of Physical Exercise on Fatty Liver Disease. Gene Expr. 2018;18:89-101
14. Mambrini SP, Grillo A, Colosimo S, Zarpellon F, Pozzi G, Furlan D. et al. Diet and physical exercise as key players to tackle MASLD through improvement of insulin resistance and metabolic flexibility. Front Nutr. 2024;11:1426551
15. Bergasa NV, Mehlman J, Bir K. Aerobic exercise: a potential therapeutic intervention for patients with liver disease. Med Hypotheses. 2004;62:935-41
16. Bagnato CB, Bianco A, Bonfiglio C, Franco I, Verrelli N, Carella N. et al. Healthy Lifestyle Changes Improve Cortisol Levels and Liver Steatosis in MASLD Patients: Results from a Randomized Clinical Trial. Nutrients. 2024 16
17. Sahlin K. Muscle energetics during explosive activities and potential effects of nutrition and training. Sports Med. 2014;44(Suppl 2):S167-73
18. Duffield R, Dawson B, Goodman C. Energy system contribution to 100-m and 200-m track running events. J Sci Med Sport. 2004;7:302-13
19. Liu C, Liu CJ. Effects of Exercise Intervention in Subjects with Metabolic Dysfunction-Associated Steatotic Liver Disease. J Obes Metab Syndr. 2025;34:239-52
20. Trefts E, Williams AS, Wasserman DH. Exercise and the Regulation of Hepatic Metabolism. Prog Mol Biol Transl Sci. 2015;135:203-25
21. Hargreaves M, Spriet LL. Skeletal muscle energy metabolism during exercise. Nat Metab. 2020;2:817-28
22. MacInnis MJ, Gibala MJ. Physiological adaptations to interval training and the role of exercise intensity. J Physiol. 2017;595:2915-30
23. Houttu V, Bouts J, Vali Y, Daams J, Grefhorst A, Nieuwdorp M. et al. Does aerobic exercise reduce NASH and liver fibrosis in patients with non-alcoholic fatty liver disease? A systematic literature review and meta-analysis. Front Endocrinol (Lausanne). 2022;13:1032164
24. Sabag A, Barr L, Armour M, Armstrong A, Baker CJ, Twigg SM. et al. The Effect of High-intensity Interval Training vs Moderate-intensity Continuous Training on Liver Fat: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. 2022;107:862-81
25. Fu J, Liu C, Yang L, Zhang B, Zhou R, Deng C. et al. Effect of high-intensity interval training on clinical parameters in patients with metabolic dysfunction-associated steatotic liver disease: a systematic review and meta-analysis of randomized controlled trials. Eur J Gastroenterol Hepatol. 2025;37:789-98
26. Kiens B, Alsted TJ, Jeppesen J. Factors regulating fat oxidation in human skeletal muscle. Obes Rev. 2011;12:852-8
27. Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E. et al. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol. 1993;265:E380-91
28. Wolfe RR, Klein S, Carraro F, Weber JM. Role of triglyceride-fatty acid cycle in controlling fat metabolism in humans during and after exercise. Am J Physiol. 1990;258:E382-9
29. Bülow J, Madsen J. Adipose tissue blood flow during prolonged, heavy exercise. Pflugers Arch. 1976;363:231-4
30. Mulla NA, Simonsen L, Bülow J. Post-exercise adipose tissue and skeletal muscle lipid metabolism in humans: the effects of exercise intensity. J Physiol. 2000 524 Pt 3: 919-28
31. Hughey CC, Bracy DP, Rome FI, Goelzer M, Donahue EP, Viollet B. et al. Exercise training adaptations in liver glycogen and glycerolipids require hepatic AMP-activated protein kinase in mice. Am J Physiol Endocrinol Metab. 2024;326:E14-e28
32. Fullerton MD. AMP-activated protein kinase and its multifaceted regulation of hepatic metabolism. Curr Opin Lipidol. 2016;27:172-80
33. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B. et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376-88
34. Hughey CC, James FD, Bracy DP, Donahue EP, Young JD, Viollet B. et al. Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J Biol Chem. 2017;292:20125-40
35. Kim K, Boo K, Yu YS, Oh SK, Kim H, Jeon Y. et al. RORα controls hepatic lipid homeostasis via negative regulation of PPARγ transcriptional network. Nat Commun. 2017;8:162
36. Tong J, Han CJ, Zhang JZ, He WZ, Zhao GJ, Cheng X. et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology. 2019;69:2471-88
37. Patitucci C, Couchy G, Bagattin A, Cañeque T, de Reyniès A, Scoazec JY. et al. Hepatocyte nuclear factor 1α suppresses steatosis-associated liver cancer by inhibiting PPARγ transcription. J Clin Invest. 2017;127:1873-88
38. Zhang Y, Xu J, Zhou D, Ye T, Zhou P, Liu Z. et al. Swimming exercise ameliorates insulin resistance and nonalcoholic fatty liver by negatively regulating PPARγ transcriptional network in mice fed high fat diet. Mol Med. 2023;29:150
39. Kim KH, Kim SH, Min YK, Yang HM, Lee JB, Lee MS. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS One. 2013;8:e63517
40. Wang Y, Guo Y, Xu Y, Wang W, Zhuang S, Wang R. et al. HIIT Ameliorates Inflammation and Lipid Metabolism by Regulating Macrophage Polarization and Mitochondrial Dynamics in the Liver of Type 2 Diabetes Mellitus Mice. Metabolites. 2022 13
41. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289-312
42. Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2-8
43. Jun M, Foote C, Lv J, Neal B, Patel A, Nicholls SJ. et al. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010;375:1875-84
44. Foreman JE, Koga T, Kosyk O, Kang BH, Zhu X, Cohen SM. et al. Species Differences between Mouse and Human PPARα in Modulating the Hepatocarcinogenic Effects of Perinatal Exposure to a High-Affinity Human PPARα Agonist in Mice. Toxicol Sci. 2021;183:81-92
45. Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, Staels B. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta. 2007;1771:1065-81
46. Zhang Y, Wan J, Xu Z, Hua T, Sun Q. Exercise ameliorates insulin resistance via regulating TGFβ-activated kinase 1 (TAK1)-mediated insulin signaling in liver of high-fat diet-induced obese rats. J Cell Physiol. 2019;234:7467-74
47. Garin-Shkolnik T, Rudich A, Hotamisligil GS, Rubinstein M. FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues. Diabetes. 2014;63:900-11
48. Gosis BS, Wada S, Thorsheim C, Li K, Jung S, Rhoades JH. et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science. 2022;376:eabf8271
49. Onaka GM, Carvalho MR, Onaka PK, Barbosa CM, Martinez PF, Oliveira-Junior SA. Exercise, mTOR Activation, and Potential Impacts on the Liver in Rodents. Biology (Basel). 2024 13
50. Radziuk J, Pye S. Hepatic glucose uptake, gluconeogenesis and the regulation of glycogen synthesis. Diabetes Metab Res Rev. 2001;17:250-72
51. Martino MR, Habibi M, Ferguson D, Brookheart RT, Thyfault JP, Meyer GA. et al. Disruption of hepatic mitochondrial pyruvate and amino acid metabolism impairs gluconeogenesis and endurance exercise capacity in mice. Am J Physiol Endocrinol Metab. 2024;326:E515-e27
52. Wasserman DH, Williams PE, Lacy DB, Green DR, Cherrington AD. Importance of intrahepatic mechanisms to gluconeogenesis from alanine during exercise and recovery. Am J Physiol. 1988;254:E518-25
53. Felig P, Owen OE, Wahren J, Cahill GF Jr. Amino acid metabolism during prolonged starvation. J Clin Invest. 1969;48:584-94
54. Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC. et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96-100
55. Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B. et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93-6
56. Jäger R, Kerksick CM, Campbell BI, Cribb PJ, Wells SD, Skwiat TM. et al. International Society of Sports Nutrition Position Stand: protein and exercise. J Int Soc Sports Nutr. 2017;14:20
57. Tipton KD, Ferrando AA, Phillips SM, Doyle D Jr, Wolfe RR. Postexercise net protein synthesis in human muscle from orally administered amino acids. Am J Physiol. 1999;276:E628-34
58. Biolo G, Maggi SP, Williams BD, Tipton KD, Wolfe RR. Increased rates of muscle protein turnover and amino acid transport after resistance exercise in humans. Am J Physiol. 1995;268:E514-20
59. Churchward-Venne TA, Murphy CH, Longland TM, Phillips SM. Role of protein and amino acids in promoting lean mass accretion with resistance exercise and attenuating lean mass loss during energy deficit in humans. Amino Acids. 2013;45:231-40
60. Moore DR, Robinson MJ, Fry JL, Tang JE, Glover EI, Wilkinson SB. et al. Ingested protein dose response of muscle and albumin protein synthesis after resistance exercise in young men. Am J Clin Nutr. 2009;89:161-8
61. Tipton KD, Borsheim E, Wolf SE, Sanford AP, Wolfe RR. Acute response of net muscle protein balance reflects 24-h balance after exercise and amino acid ingestion. Am J Physiol Endocrinol Metab. 2003;284:E76-89
62. Burd NA, West DW, Moore DR, Atherton PJ, Staples AW, Prior T. et al. Enhanced amino acid sensitivity of myofibrillar protein synthesis persists for up to 24 h after resistance exercise in young men. J Nutr. 2011;141:568-73
63. Miller BF, Olesen JL, Hansen M, Døssing S, Crameri RM, Welling RJ. et al. Coordinated collagen and muscle protein synthesis in human patella tendon and quadriceps muscle after exercise. J Physiol. 2005;567:1021-33
64. Glynn EL, Fry CS, Drummond MJ, Timmerman KL, Dhanani S, Volpi E. et al. Excess leucine intake enhances muscle anabolic signaling but not net protein anabolism in young men and women. J Nutr. 2010;140:1970-6
65. Norton LE, Layman DK, Bunpo P, Anthony TG, Brana DV, Garlick PJ. The leucine content of a complete meal directs peak activation but not duration of skeletal muscle protein synthesis and mammalian target of rapamycin signaling in rats. J Nutr. 2009;139:1103-9
66. Pasiakos SM, McClung HL, McClung JP, Margolis LM, Andersen NE, Cloutier GJ. et al. Leucine-enriched essential amino acid supplementation during moderate steady state exercise enhances postexercise muscle protein synthesis. Am J Clin Nutr. 2011;94:809-18
67. Churchward-Venne TA, Burd NA, Mitchell CJ, West DW, Philp A, Marcotte GR. et al. Supplementation of a suboptimal protein dose with leucine or essential amino acids: effects on myofibrillar protein synthesis at rest and following resistance exercise in men. J Physiol. 2012;590:2751-65
68. Sun M, Zhao X, Li X, Wang C, Lin L, Wang K. et al. Aerobic Exercise Ameliorates Liver Injury in Db/Db Mice by Attenuating Oxidative Stress, Apoptosis and Inflammation Through the Nrf2 and JAK2/STAT3 Signalling Pathways. J Inflamm Res. 2023;16:4805-19
69. Qi X, Lu XT, Sun XH, Lin CQ, Cui CB. The regulatory effect of total flavonoids of Sedum aizoon L. on oxidative stress in type 1 diabetic mice. Curr Res Food Sci. 2022;5:1140-7
70. Liu J, Wu KC, Lu YF, Ekuase E, Klaassen CD. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid Med Cell Longev. 2013;2013:305861
71. Rector RS, Thyfault JP, Morris RT, Laye MJ, Borengasser SJ, Booth FW. et al. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. 2008;294:G619-26
72. Almeida OF, Condé GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH. et al. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. Faseb j. 2000;14:779-90
73. Kim YA, Kim YS, Oh SL, Kim HJ, Song W. Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem. 2013;69:697-705
74. Czaja MJ. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig Dis Sci. 2016;61:1304-13
75. Chun SK, Lee S, Yang MJ, Leeuwenburgh C, Kim JS. Exercise-Induced Autophagy in Fatty Liver Disease. Exerc Sport Sci Rev. 2017;45:181-6
76. Gomez-Cabrera MC, Domenech E, Viña J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med. 2008;44:126-31
77. Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev. 2010;16:105-18
78. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78-90
79. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N. et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59:1060-72
80. Tamura Y, Sugimoto M, Murayama T, Minami M, Nishikaze Y, Ariyasu H. et al. C-C chemokine receptor 2 inhibitor improves diet-induced development of insulin resistance and hepatic steatosis in mice. J Atheroscler Thromb. 2010;17:219-28
81. Farzanegi P, Dana A, Ebrahimpoor Z, Asadi M, Azarbayjani MA. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur J Sport Sci. 2019;19:994-1003
82. Guarino M, Kumar P, Felser A, Terracciano LM, Guixé-Muntet S, Humar B. et al. Exercise Attenuates the Transition from Fatty Liver to Steatohepatitis and Reduces Tumor Formation in Mice. Cancers (Basel). 2020 12
83. Kawanishi N, Yano H, Mizokami T, Takahashi M, Oyanagi E, Suzuki K. Exercise training attenuates hepatic inflammation, fibrosis and macrophage infiltration during diet induced-obesity in mice. Brain Behav Immun. 2012;26:931-41
84. Jeong JH, Lee YR, Park HG, Lee WL. The effects of either resveratrol or exercise on macrophage infiltration and switching from M1 to M2 in high fat diet mice. J Exerc Nutrition Biochem. 2015;19:65-72
85. Linden MA, Sheldon RD, Meers GM, Ortinau LC, Morris EM, Booth FW. et al. Aerobic exercise training in the treatment of non-alcoholic fatty liver disease related fibrosis. J Physiol. 2016;594:5271-84
86. Komine S, Akiyama K, Warabi E, Oh S, Kuga K, Ishige K. et al. Exercise training enhances in vivo clearance of endotoxin and attenuates inflammatory responses by potentiating Kupffer cell phagocytosis. Sci Rep. 2017;7:11977
87. Radom-Aizik S, Zaldivar F, Haddad F, Cooper DM. Impact of brief exercise on peripheral blood NK cell gene and microRNA expression in young adults. J Appl Physiol (1985). 2013;114:628-36
88. Saran U, Humar B, Kolly P, Dufour JF. Hepatocellular carcinoma and lifestyles. J Hepatol. 2016;64:203-14
89. Baker FL, Bigley AB, Agha NH, Pedlar CR, O'Connor DP, Bond RA. et al. Systemic β-Adrenergic Receptor Activation Augments the ex vivo Expansion and Anti-Tumor Activity of Vγ9Vδ2 T-Cells. Front Immunol. 2019;10:3082
90. Yao J, Ly D, Dervovic D, Fang L, Lee JB, Kang H. et al. Human double negative T cells target lung cancer via ligand-dependent mechanisms that can be enhanced by IL-15. J Immunother Cancer. 2019;7:17
91. Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS. et al. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep. 2017;20:124-35
92. McCaffrey JM, Ibdah JA. Effects of Diet and Exercise on Mitochondrial Health in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): Role of Ceramides. Nutrients. 2025 17
93. Mardare C, Krüger K, Liebisch G, Seimetz M, Couturier A, Ringseis R. et al. Endurance and Resistance Training Affect High Fat Diet-Induced Increase of Ceramides, Inflammasome Expression, and Systemic Inflammation in Mice. J Diabetes Res. 2016;2016:4536470
94. Huber Y, Gehrke N, Biedenbach J, Helmig S, Simon P, Straub BK. et al. Voluntary distance running prevents TNF-mediated liver injury in mice through alterations of the intrahepatic immune milieu. Cell Death Dis. 2017;8:e2893
95. Cui N, Li H, Dun Y, Ripley-Gonzalez JW, You B, Li D. et al. Exercise inhibits JNK pathway activation and lipotoxicity via macrophage migration inhibitory factor in nonalcoholic fatty liver disease. Front Endocrinol (Lausanne). 2022;13:961231
96. Delogu W, Caligiuri A, Provenzano A, Rosso C, Bugianesi E, Coratti A. et al. Myostatin regulates the fibrogenic phenotype of hepatic stellate cells via c-jun N-terminal kinase activation. Dig Liver Dis. 2019;51:1400-8
97. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int J Obes (Lond). 2016;40:434-42
98. Zhu L, Wang X, Wei Z, Yang M, Zhou X, Lei J. et al. Myostatin Deficiency Enhances Antioxidant Capacity of Bovine Muscle via the SMAD-AMPK-G6PD Pathway. Oxid Med Cell Longev. 2022;2022:3497644
99. Pedersen BK, Fischer CP. Beneficial health effects of exercise-the role of IL-6 as a myokine. Trends Pharmacol Sci. 2007;28:152-6
100. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204-9
101. Li Z, Wang B, Li X. Irisin prevents liver injury during exhausting physical activity by suppressing ferroptosis via Nrf2/GPX4 signaling. BMC Gastroenterol. 2025;25:516
102. Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K. et al. Irisin Mediates Effects on Bone and Fat via αV Integrin Receptors. Cell. 2018;175:1756-68.e17
103. Bi J, Zhang J, Ren Y, Du Z, Li Q, Wang Y. et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019;20:296-306
104. Li Q, Tan Y, Chen S, Xiao X, Zhang M, Wu Q. et al. Irisin alleviates LPS-induced liver injury and inflammation through inhibition of NLRP3 inflammasome and NF-κB signaling. J Recept Signal Transduct Res. 2021;41:294-303
105. Zhu W, Sahar NE, Javaid HMA, Pak ES, Liang G, Wang Y. et al. Exercise-Induced Irisin Decreases Inflammation and Improves NAFLD by Competitive Binding with MD2. Cells. 2021 10
106. Wu A, Feng B, Yu J, Yan L, Che L, Zhuo Y. et al. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131
107. Tanimura Y, Aoi W, Takanami Y, Kawai Y, Mizushima K, Naito Y. et al. Acute exercise increases fibroblast growth factor 21 in metabolic organs and circulation. Physiol Rep. 2016 4
108. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F. et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271-81
109. Barclay RD, Burd NA, Tyler C, Tillin NA, Mackenzie RW. The Role of the IGF-1 Signaling Cascade in Muscle Protein Synthesis and Anabolic Resistance in Aging Skeletal Muscle. Front Nutr. 2019;6:146
110. Pyka G, Wiswell RA, Marcus R. Age-dependent effect of resistance exercise on growth hormone secretion in people. J Clin Endocrinol Metab. 1992;75:404-7
111. Bucarey JL, Trujillo-González I, Paules EM, Espinosa A. Myokines and Their Potential Protective Role Against Oxidative Stress in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). Antioxidants (Basel). 2024 13
112. Bórquez JC, Díaz-Castro F, La Fuente FP, Espinoza K, Figueroa AM, Martínez-Ruíz I. et al. Mitofusin-2 induced by exercise modifies lipid droplet-mitochondria communication, promoting fatty acid oxidation in male mice with NAFLD. Metabolism. 2024;152:155765
113. Swalsingh G, Pani P, Senapati U, Sahu B, Pani S, Pati B. et al. Intramuscular administration of fractalkine modulates mitochondrial properties and promotes fast glycolytic phenotype. Biofactors. 2025;51:e2092
114. Swalsingh G, Pani P, Sadayappan S, Bal NC. Fractalkine is a key player in skeletal muscle metabolism and pathophysiology. Febs j. 2025
115. Strömberg A, Olsson K, Dijksterhuis JP, Rullman E, Schulte G, Gustafsson T. CX3CL1-a macrophage chemoattractant induced by a single bout of exercise in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2016;310:R297-304
116. Chin ER. Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. J Appl Physiol (1985). 2005;99:414-23
117. Arnold L, Perrin H, de Chanville CB, Saclier M, Hermand P, Poupel L. et al. CX3CR1 deficiency promotes muscle repair and regeneration by enhancing macrophage ApoE production. Nat Commun. 2015;6:8972
118. Zhao W, Lu H, Wang X, Ransohoff RM, Zhou L. CX3CR1 deficiency delays acute skeletal muscle injury repair by impairing macrophage functions. Faseb j. 2016;30:380-93
119. Ni Y, Zhuge F, Ni L, Nagata N, Yamashita T, Mukaida N. et al. CX3CL1/CX3CR1 interaction protects against lipotoxicity-induced nonalcoholic steatohepatitis by regulating macrophage migration and M1/M2 status. Metabolism. 2022;136:155272
120. Zambon Azevedo V, Silaghi CA, Maurel T, Silaghi H, Ratziu V, Pais R. Impact of Sarcopenia on the Severity of the Liver Damage in Patients with Non-alcoholic Fatty Liver Disease. Front Nutr. 2021;8:774030
121. Koo BK, Kim D, Joo SK, Kim JH, Chang MS, Kim BG. et al. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J Hepatol. 2017;66:123-31
122. Lee YH, Kim SU, Song K, Park JY, Kim DY, Ahn SH. et al. Sarcopenia is associated with significant liver fibrosis independently of obesity and insulin resistance in nonalcoholic fatty liver disease: Nationwide surveys (KNHANES 2008-2011). Hepatology. 2016;63:776-86
123. Chakravarthy MV, Siddiqui MS, Forsgren MF, Sanyal AJ. Harnessing Muscle-Liver Crosstalk to Treat Nonalcoholic Steatohepatitis. Front Endocrinol (Lausanne). 2020;11:592373
124. Wall BT, Dirks ML, van Loon LJ. Skeletal muscle atrophy during short-term disuse: implications for age-related sarcopenia. Ageing Res Rev. 2013;12:898-906
125. Bhanji RA, Narayanan P, Allen AM, Malhi H, Watt KD. Sarcopenia in hiding: The risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis. Hepatology. 2017;66:2055-65
126. Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98:2133-223
127. Gastaldelli A. Insulin resistance and reduced metabolic flexibility: cause or consequence of NAFLD? Clin Sci (Lond). 2017;131:2701-4
128. Katsarou A, Moustakas, II, Pyrina I, Lembessis P, Koutsilieris M, Chatzigeorgiou A. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J Gastroenterol. 2020;26:1993-2011
129. Arias-Loste MT, Ranchal I, Romero-Gómez M, Crespo J. Irisin, a link among fatty liver disease, physical inactivity and insulin resistance. Int J Mol Sci. 2014;15:23163-78
130. Barchetta I, Cimini FA, Cavallo MG. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): An Update. Nutrients. 2020 12
131. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M. et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol. 2020;73:202-9
132. Keating SE, Sabag A, Hallsworth K, Hickman IJ, Macdonald GA, Stine JG. et al. Exercise in the Management of Metabolic-Associated Fatty Liver Disease (MAFLD) in Adults: A Position Statement from Exercise and Sport Science Australia. Sports Med. 2023;53:2347-71
133. Yip TC, Lee HW, Lin H, Tsochatzis E, Petta S, Bugianesi E. et al. Prognostic performance of the two-step clinical care pathway in metabolic dysfunction-associated steatotic liver disease. J Hepatol. 2025;83:304-14
134. Golabi P, Paik JM, AlQahtani S, Younossi Y, Tuncer G, Younossi ZM. Burden of non-alcoholic fatty liver disease in Asia, the Middle East and North Africa: Data from Global Burden of Disease 2009-2019. J Hepatol. 2021;75:795-809
135. Le MH, Le DM, Baez TC, Wu Y, Ito T, Lee EY. et al. Global incidence of non-alcoholic fatty liver disease: A systematic review and meta-analysis of 63 studies and 1,201,807 persons. J Hepatol. 2023;79:287-95
136. Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:8713-42
137. Sabag A, Little JP, Johnson NA. Low-volume high-intensity interval training for cardiometabolic health. J Physiol. 2022;600:1013-26
138. Sabag A, Chang D, Johnson NA. Growth Hormone as a Potential Mediator of Aerobic Exercise-Induced Reductions in Visceral Adipose Tissue. Front Physiol. 2021;12:623570
139. Kurioka S, Murakami Y, Nishiki M, Sohmiya M, Koshimura K, Kato Y. Relationship between visceral fat accumulation and anti-lipolytic action of insulin in patients with type 2 diabetes mellitus. Endocr J. 2002;49:459-64
140. Thomas SH, Wisher MH, Brandenburg D, Sönksen PH. Insulin action on adipocytes. Evidence that the anti-lipolytic and lipogenic effects of insulin are mediated by the same receptor. Biochem J. 1979;184:355-60
141. Keating SE, Hackett DA, Parker HM, O'Connor HT, Gerofi JA, Sainsbury A. et al. Effect of aerobic exercise training dose on liver fat and visceral adiposity. J Hepatol. 2015;63:174-82
142. Zhang HJ, He J, Pan LL, Ma ZM, Han CK, Chen CS. et al. Effects of Moderate and Vigorous Exercise on Nonalcoholic Fatty Liver Disease: A Randomized Clinical Trial. JAMA Intern Med. 2016;176:1074-82
143. Larose J, Sigal RJ, Boulé NG, Wells GA, Prud'homme D, Fortier MS. et al. Effect of exercise training on physical fitness in type II diabetes mellitus. Med Sci Sports Exerc. 2010;42:1439-47
144. Williams MA, Haskell WL, Ades PA, Amsterdam EA, Bittner V, Franklin BA. et al. Resistance exercise in individuals with and without cardiovascular disease: 2007 update: a scientific statement from the American Heart Association Council on Clinical Cardiology and Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2007;116:572-84
145. Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Zvibel I, Goldiner I. et al. Role of leisure-time physical activity in nonalcoholic fatty liver disease: a population-based study. Hepatology. 2008;48:1791-8
146. Couret A, King JA, Pereira B, Courteix D, Obert P, Vinet A. et al. Effect of different modalities of exercise on Fatty Liver Index in patients with metabolic syndrome: The RESOLVE randomized trial. Clin Res Hepatol Gastroenterol. 2024;48:102461
147. Hallsworth K, Fattakhova G, Hollingsworth KG, Thoma C, Moore S, Taylor R. et al. Resistance exercise reduces liver fat and its mediators in non-alcoholic fatty liver disease independent of weight loss. Gut. 2011;60:1278-83
148. Gordon BA, Benson AC, Bird SR, Fraser SF. Resistance training improves metabolic health in type 2 diabetes: a systematic review. Diabetes Res Clin Pract. 2009;83:157-75
149. Khalafi M, Symonds ME. The impact of high intensity interval training on liver fat content in overweight or obese adults: A meta-analysis. Physiol Behav. 2021;236:113416
150. Sultana RN, Sabag A, Keating SE, Johnson NA. The Effect of Low-Volume High-Intensity Interval Training on Body Composition and Cardiorespiratory Fitness: A Systematic Review and Meta-Analysis. Sports Med. 2019;49:1687-721
151. Keating SE, Adams LA. Exercise in NAFLD: Just do it. J Hepatol. 2016;65:671-3
152. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572-85
153. Ardies CM, Morris GS, Erickson CK, Farrar RP. Effects of exercise and ethanol on liver mitochondrial function. Life Sci. 1987;40:1053-61
154. Kezer CA, Simonetto DA, Shah VH. Sex Differences in Alcohol Consumption and Alcohol-Associated Liver Disease. Mayo Clin Proc. 2021;96:1006-16
155. Kasarinaite A, Sinton M, Saunders PTK, Hay DC. The Influence of Sex Hormones in Liver Function and Disease. Cells. 2023 12
156. Fu MH, Maher AC, Hamadeh MJ, Ye C, Tarnopolsky MA. Exercise, sex, menstrual cycle phase, and 17beta-estradiol influence metabolism-related genes in human skeletal muscle. Physiol Genomics. 2009;40:34-47
157. Smiriglia A, Lorito N, Serra M, Perra A, Morandi A, Kowalik MA. Sex difference in liver diseases: How preclinical models help to dissect the sex-related mechanisms sustaining NAFLD and hepatocellular carcinoma. iScience. 2023;26:108363
158. Shen M, Shi H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int J Endocrinol. 2015;2015:294278
159. Radak Z, Chung HY, Koltai E, Taylor AW, Goto S. Exercise, oxidative stress and hormesis. Ageing Res Rev. 2008;7:34-42
160. Georgakouli K, Manthou E, Fatouros IG, Deli CK, Spandidos DA, Tsatsakis AM. et al. Effects of acute exercise on liver function and blood redox status in heavy drinkers. Exp Ther Med. 2015;10:2015-22
161. Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG Clinical Guideline: Alcoholic Liver Disease. Am J Gastroenterol. 2018;113:175-94
162. Tandon P, Ismond KP, Riess K, Duarte-Rojo A, Al-Judaibi B, Dunn MA. et al. Exercise in cirrhosis: Translating evidence and experience to practice. J Hepatol. 2018;69:1164-77
163. EASL Clinical Practice Guidelines on nutrition in chronic liver disease. J Hepatol. 2019; 70: 172-93.
164. Harrington DW. Viral hepatitis and exercise. Med Sci Sports Exerc. 2000;32:S422-30
165. de Celis G, Casal J, Latorre X, Angel J. Hepatitis A and vigorous physical activity. Lancet. 1998;352:325
166. Krikler DM, Zilberg B. Activity and hepatitis. Lancet. 1966;2:1046-7
167. Graubaum HJ, Metzner C, Ziesenhenn K. [Physical exertion and the course of hepatitis]. Dtsch Med Wochenschr. 1987;112:47-9
168. Williams R, Aspinall R, Bellis M, Camps-Walsh G, Cramp M, Dhawan A. et al. Addressing liver disease in the UK: a blueprint for attaining excellence in health care and reducing premature mortality from lifestyle issues of excess consumption of alcohol, obesity, and viral hepatitis. Lancet. 2014;384:1953-97
169. Waki K, Tamura S, Sugawara Y, Yamashiki N, Kadowaki T, Kokudo N. An analysis of the OPTN/UNOS Liver Transplant Registry. Clin Transpl. 2009:55-64
170. Phu S, Boersma D, Duque G. Exercise and Sarcopenia. J Clin Densitom. 2015;18:488-92
171. Dunn MA, Josbeno DA, Schmotzer AR, Tevar AD, DiMartini AF, Landsittel DP. et al. The gap between clinically assessed physical performance and objective physical activity in liver transplant candidates. Liver Transpl. 2016;22:1324-32
172. Sinclair M, Gow PJ, Grossmann M, Angus PW. Review article: sarcopenia in cirrhosis-aetiology, implications and potential therapeutic interventions. Aliment Pharmacol Ther. 2016;43:765-77
173. Ritland S, Foss NE, Skrede S. The effect of a standardized work load on 'liver tests' in patients with chronic active hepatitis. Scand J Gastroenterol. 1982;17:1013-6
174. Ritland S, Foss NE, Gjone E. Physical activity in liver disease and liver function in sportsmen. Scand J Soc Med Suppl. 1982;29:221-6
175. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151-66
176. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823
177. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411
178. Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016;28:53-61
179. Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in NASH. J Hepatol. 2018;68:238-50
180. Yamada K, Mizukoshi E, Sunagozaka H, Arai K, Yamashita T, Takeshita Y. et al. Characteristics of hepatic fatty acid compositions in patients with nonalcoholic steatohepatitis. Liver Int. 2015;35:582-90
181. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147:765-83.e4
182. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF. et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188-98
183. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181-94
184. Borkham-Kamphorst E, Schaffrath C, Van de Leur E, Haas U, Tihaa L, Meurer SK. et al. The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim Biophys Acta. 2014;1843:902-14
185. Borkham-Kamphorst E, Steffen BT, Van de Leur E, Haas U, Tihaa L, Friedman SL. et al. CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress-related apoptosis. Cell Signal. 2016;28:34-42
186. The global, regional, national burden of cirrhosis by cause in 195 countries, territories, 1990-2017. a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5:245-66
187. D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217-31
188. Allen AM, Kim WR, Moriarty JP, Shah ND, Larson JJ, Kamath PS. Time trends in the health care burden and mortality of acute on chronic liver failure in the United States. Hepatology. 2016;64:2165-72
189. Arroyo V, Moreau R, Jalan R. Acute-on-Chronic Liver Failure. N Engl J Med. 2020;382:2137-45
190. Gracia-Sancho J, Marrone G, Fernández-Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol. 2019;16:221-34
191. Zenith L, Meena N, Ramadi A, Yavari M, Harvey A, Carbonneau M. et al. Eight weeks of exercise training increases aerobic capacity and muscle mass and reduces fatigue in patients with cirrhosis. Clin Gastroenterol Hepatol. 2014;12:1920-6.e2
192. Kelbaek H, Eriksen J, Brynjolf I, Raboel A, Lund JO, Munck O. et al. Cardiac performance in patients with asymptomatic alcoholic cirrhosis of the liver. Am J Cardiol. 1984;54:852-5
193. Lu SD, Li L, Liang XM, Chen W, Chen FL, Fan LL. et al. Updates and advancements in the management of hepatocellular carcinoma patients after hepatectomy. Expert Rev Gastroenterol Hepatol. 2019;13:1077-88
194. Saran U, Guarino M, Rodríguez S, Simillion C, Montani M, Foti M. et al. Anti-tumoral effects of exercise on hepatocellular carcinoma growth. Hepatol Commun. 2018;2:607-20
195. Subramaniam A, Shanmugam MK, Perumal E, Li F, Nachiyappan A, Dai X. et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim Biophys Acta. 2013;1835:46-60
196. Arfianti A, Pok S, Barn V, Haigh WG, Yeh MM, Ioannou GN. et al. Exercise retards hepatocarcinogenesis in obese mice independently of weight control. J Hepatol. 2020;73:140-8
197. Zhang QB, Zhang BH, Zhang KZ, Meng XT, Jia QA, Zhang QB. et al. Moderate swimming suppressed the growth and metastasis of the transplanted liver cancer in mice model: with reference to nervous system. Oncogene. 2016;35:4122-31
198. Siewierska K, Malicka I, Kobierzycki C, Paslawska U, Cegielski M, Grzegrzolka J. et al. The Impact of Exercise Training on Breast Cancer. In Vivo. 2018;32:249-54
199. Sirisunhirun P, Bandidniyamanon W, Jrerattakon Y, Muangsomboon K, Pramyothin P, Nimanong S. et al. Effect of a 12-week home-based exercise training program on aerobic capacity, muscle mass, liver and spleen stiffness, and quality of life in cirrhotic patients: a randomized controlled clinical trial. BMC Gastroenterol. 2022;22:66
200. Linecker M, Frick L, Kron P, Limani P, Kambakamba P, Tschuor C. et al. Exercise Improves Outcomes of Surgery on Fatty Liver in Mice: A Novel Effect Mediated by the AMPK Pathway. Ann Surg. 2020;271:347-55
201. Stefanovic-Racic M, Perdomo G, Mantell BS, Sipula IJ, Brown NF, O'Doherty RM. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am J Physiol Endocrinol Metab. 2008;294:E969-77
202. Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L. et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100-10
203. Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G. et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445-53
204. Bian F, Ma X, Villivalam SD, You D, Choy LR, Paladugu A. et al. TET2 facilitates PPARγ agonist-mediated gene regulation and insulin sensitization in adipocytes. Metabolism. 2018;89:39-47
205. Saraf N, Sharma PK, Mondal SC, Garg VK, Singh AK. Role of PPARg2 transcription factor in thiazolidinedione-induced insulin sensitization. J Pharm Pharmacol. 2012;64:161-71
206. Zhou Y, Zhong L, Yu S, Shen W, Cai C, Yu H. Inhibition of stearoyl-coenzyme A desaturase 1 ameliorates hepatic steatosis by inducing AMPK-mediated lipophagy. Aging (Albany NY). 2020;12:7350-62
207. Shah H, Stankov M, Panayotova-Dimitrova D, Yazdi A, Budida R, Klusmann JH. et al. Autolysosomal activation combined with lysosomal destabilization efficiently targets myeloid leukemia cells for cell death. Front Oncol. 2023;13:999738
208. Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C. et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018;9:3312
209. Pati B, Sendh S, Sahu B, Pani S, Jena N, Bal NC. Recent advancements in pharmacological strategies to modulate energy balance for combating obesity. RSC Med Chem. 2023;14:1429-45
210. Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J. et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21:443-54
211. Hawley JA, Joyner MJ, Green DJ. Mimicking exercise: what matters most and where to next? J Physiol. 2021;599:791-802
212. Zhu Y, Song G. Molecular origin and biological effects of exercise mimetics. J Exerc Sci Fit. 2024;22:73-85
213. Fan W, Evans RM. Exercise Mimetics: Impact on Health and Performance. Cell Metab. 2017;25:242-7
Author contact
![]() Corresponding authors: Li Zuo, Email: zuoliedu.cn; Hua Wang, Email: wanghuaedu.cn.
Corresponding authors: Li Zuo, Email: zuoliedu.cn; Hua Wang, Email: wanghuaedu.cn.