Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(2):156-167. doi:10.7150/ijbs.10283 This issue Cite
Research Paper
Effects of Nitric Oxide on Notexin-Induced Muscle Inflammatory Responses
XingHui Liu 1*, Gang Wu2*, DanDan Shi 1*, Rong Zhu1, HuiJun Zeng1, Biao Cao1, MeiXian Huang1, Hua Liao1 ![]()
1. Department of Anatomy, Guangdong Provincial Key Laboratory of Construction and Detection in Tissue Engineering, Southern Medical University, Guangzhou, 510515 China
2. Department of Emergency, NanFang Hospital, Southern Medical University, Guangzhou, 510515 China
* The first three authors contributed equally to this work
Received 2014-8-6; Accepted 2014-11-25; Published 2015-1-5
Abstract

Excessive inflammatory response may delay the regeneration and damage the normal muscle fibers upon myoinjury. It would be important to be able to attenuate the inflammatory response and decrease inflammatory cells infiltration in order to improve muscle regeneration formation, resulting in better muscle functional recovery after myoinjury. This study was undertaken to explore the role of Nitric oxide (NO) during skeletal muscle inflammatory process, using a mouse model of Notexin induced myoinjury. Intramuscular injection (tibialis anterior, TA) of Notexin was performed for preparing mice myoinjury. NO synthase inhibitor (L-NAME) or NO donor (SNP) was intraperitoneally injected into model mice. On day 4 and 7 post-injury, expression of muscle-autoantigens and toll-like receptors (TLRs) was evaluated from muscle tissue by qRT-PCR and Western Blot; the intramuscular infiltration of monocytes/macrophage (CD11b+ or F4/80+ cells), CD8+ T cell (CD3ε+CD8α+), apoptotic cell (CD11b+caspase3+), and MHC-I molecule H-2Kb-expressing myofibers in damaged muscle were assessed by imunoflourecence analysis; the mRNAs expression of cytokines and chemokines associated with the preferential biological role during the muscle damage-induced inflammation response, were assessed by qRT-PCR. We detected the reduced monocytes/macrophages infiltration, and increased apoptotic cells in the damaged muscle treated with SNP comparing to untreatment. As well, SNP treatment down-regulated mRNA and protein levels of muscle autoantigens, TLR3, and mRNA levels of TNF-α, IL-6, MCP-1, MCP-3, and MIP-1α in damaged muscle. On the contrary, L-NAME induced more severe intramuscular infiltration of inflammatory cells, and mRNA level elevation of the above inflammatory mediators. Notably, we observed an increased number of MHC-I (H2-Kb) positive new myofibers, and of the infiltrated CD8+ T cells in damaged muscle at the day 7 after L-NAME treatment. The result herein shows that, NO can act as an endogenous anti-inflammatory molecule during the ongoing muscle inflammation. Our finding may provide new insight to optimize NO-based therapies for improving muscle regeneration after myoinjury.
Keywords: Nitric Oxide, inflammation, skeletal muscle
Introduction
The acute muscle injuries, including crush, contusion, laceration or freezing are common. They are classified on the basis of inflammatory process in the early stage. On the one hand, infiltration of inflammatory cells following muscle injury promotes regeneration by clearing debris, engulfment and digestion of necrotic cell bodies. The other hand, inflammatory cells can produce growth factors and cytokines involved in the muscle regeneration process [1]. Inflammation of injured muscle has been interpreted as indicating an early stage of muscle regeneration because of the ability of neutrophils and macrophages accumulated at the site of damage to release soluble molecules, which are capable of affecting the transcriptional activity and the viability of regenerating muscle cells. However, excessive and prolonged inflammatory response can delay the regeneration, damage the normal muscle fibers, and even lead to muscle fibrosis, or chronic myoinjury [2]. It would be important to be able to attenuate the inflammatory response and decrease inflammatory cells infiltration reasonably in order to coordinate between inflammatory and regeneration process, resulting in better muscle functional recovery after myoinjury.
Nitric oxide (NO), the smallest signaling molecule known, synthesized from L-arginine by NO synthase (NOS) enzymes and plays an important role in skeletal muscle regeneration after injury [3]. In addition, NO is the characteristic key molecule that have been linked to the mechanical-stretch signal pathway of skeletal muscle [4]. Neuronal NOS (nNOS) and the endothelial NOS (eNOS) isoforms are constitutively expressed under non-pathological skeletal muscle. Inducialbe NOS (iNOS) is expressed in skeletal muscle primarily under severe inflammatory conditions, such as in the course of autoimmune inflammatory myopathies and acute myoinjury (e.g. crush injury and cardiotoxin injection) [5, 6, 7]. NO has been shown to enhance repair of the damaged muscle via actions on survival, activation and differentiation of myogenic precursor cell, via different signalling pathways [8]. NO action in muscle inflammation has been investigated and found to be multifaceted. For example, Hickey et al. showed that, in iNOS-deficient mice inflammatory cell infiltration of the skeletal muscle appears to be reduced [9]. However, McCafferty et al. [10] and Rigamonti et al. [7] supported iNOS-derived NO contribute to the protective response to injury by reducing the extent of leukocyte infiltration and by establishing an effective homeostatic inflammatory response of the skeletal muscle upon damage. The report from the team of Rovere-Querini P suggested that, expression levels of chemokines in iNOS-deficient muscles significantly higher than their wild type counterparts and display more infiltrating neutrophils after injury and a persistence of macrophages at later time points [11]. Recently we induced the proliferation of C2C12 myoblasts through the in vitro mechanical-stretch and detected the increased expression of NOS proteins. Our further work indicates that NO donor SNP reduced the levels of TLR3 and of proteins known to represent potential autoantigens [12]. Collectively, these findings show that the beneficial role of NO in muscle inflammation and repair processes after injury. However, the more detailed and definite data are necessary to clarify the effects of NO, which greatly increased its level in local muscle upon damage, on inflammatory process after myoinjury.
In this study, we explored the role of NO during inflammation process in Notexin model of skeletal muscle injury using an NO synthase inhibitor (nitro-L-arginine methylester: L-NAME) and an NO donor (sodium nitroprusside: SNP). Multiple inflammatory parameters, including mononuclear cell infiltration and apoptosis, the expression of muscle-autoantigens and TLRs, cytokines and chemokines associated with the preferential muscle tissue infiltration by inflammatory cells, were explored on day 4 and 7 post-injury. We observed the reduced monocytes/macrophages infiltration, and the increase number of apoptotic cells in the damaged muscle treated with SNP comparing to untreatment. As well, SNP treatment down-regulated mRNA and protein levels of autoantigens and TLR3, and mRNA levels of TNF-α, IL-6, MCP-1, MCP-3, and MIP-1α in damaged muscle. On the contrary, L-NAME induced more severe intramuscular infiltration of inflammatory cells, and mRNA level elevation of the above inflammatory mediators. Notably, we observed an increased number of MHC-I (H2-Kb) positive new myofibers, and of the infiltrated CD8+ T cells in damaged muscle at the day 7 after L-NAME treatment. Taken together, these results support a beneficial role of NO in skeletal muscle repair through the down-regulation of muscle inflammatory responses.
Materials and Methods
Animals and experimental groups
In all experiments, 6-to 8-wk-old female wild mice (C57BL/6) were used. The animals were randomly divided into four groups: (a) Control, (b) Notexin, (c) L-NAME (Notexin+L-NAME) and (d) SNP (Notexin+SNP). Animals were anesthetized and subsequently injected with 10ul of Notexin (25μg/ml in 0.9% NaCl; Latoxan, Rosans, France) into tibialis anterior (TA) muscles except for the first group. Mice were sacrificed at day 4 and 7 after Notexin injection. Animals of the L-NAME and SNP groups respectively received a 100 mg/kg dose of L-NAME (Santa Cruz, USA) or a 0.5 mg/kg dose of SNP (Sigma, USA) i.p. one day after Notexin injection. For animals sacrificed at day 7, additional one dose of injection i.p. of L-NAME or SNP was performed on day 4 post-injury. All experiments were performed in accordance with the guidelines of the Animal Experimentation Ethics Committee of the Southern Medical University. Injured TA muscles were collected and snap frozen for NO levels, RNA and protein analyses. For histology, muscles were collected and directly frozen in liquid nitrogen-cool isopentane.
Measurement of NO levels of muscle tissue
The muscle samples were collected as described above and tissue homogenates were made. After centrifugation, the supernatant of muscle tissue homogenate were used for next treatment. Protein concentrations were evaluated using a BCA assay kit (KeyGEN, China). The total concentration of nitrate and nitrite in the muscle supernatant were determined by Nitric Oxide Metabolite Detection Kit (PanEra, China) and the absorbance of sample was measured at 550nm wavelength using Multiscan Go microplate reader (ThermoFisher, Finland). Finally, the NO concentration was calculated according to the manufacturer's instructions.
RNA extraction and quantitative real-time PCR analysis
Total RNA was extracted from muscle tissue using RNA extraction kit (DongSheng, China), following manufacturer's recommendations. Total RNA (1ug) was then used for reverse transcription (RT) with commercially available kit (Revert Aid First Strand cDNA Synthesis Kit, Fermentas). Real-time polymerase chain reaction (PCR) was performed in triplicate with an ABI Step One Plus system (Applied Biosystems, USA) and a fluorescence-labeled SYBR Green/ROX qPCR Master Mix kit (Fermentas) using specific primers. Muscle autoantigens (Mi-2, HARS, Ku-70, DNA-pkcs), eNOS, iNOS, nNOS,TLR3, TLR7, TNF-α, IL-1, IL-10, IL-6, TGF-β, MCP-1, MCP-3, MIP-1α and with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) taken as an endogenous control (primer sequences and sizes of product are listed in table 1) were detected. The results were analyzed with SOS2.1 software (Applied Biosystems). Expression of the genes was calculated from the accurate threshold cycle (Ct), which is the PCR cycle at which an increase in fluorescence from SYBR Green probes above the baseline signal can first be detected. The Ct values for GAPDH were compared with those from Mi-2, HARS, Ku-70, DNA-pkcs, eNOS, iNOS, nNOS, TLR3, TLR7, TNF-α, IL-10, IL-1, IL-6, TGF-β, MCP-1, MCP-3 and MIP-1α and in each well to calculate ΔCt. Data of the treated conditions were expressed relative to the signal obtained for the average of the untreated controls by the ΔΔCt calculation. The triplicate ΔΔCt values for each sample were averaged.
Primer sequences used for qRT-PCR
| Gene name | Forward(5'→3') | Reverse(5'→3') | product size |
|---|---|---|---|
| eNOS | GTTTGTCTGCGGCGATGT | GTGCGTATGCGGCTTGTC | 192bp |
| iNOS | GAGCGAGTTGTGGATTGTC | CCAGGAAGTAGGTGAGGG | 133bp |
| nNOS | ACAACCTCGCTACTATTCCA | CCTTCTCCATCTCGGGTA | 102bp |
| Mi-2 | CCCGAGGAGTGTGGA ACTA | CCCTACCACCCTAGCCAAG | 62bp |
| HARS | GATGGGATGTTTGCTGTCTG | TCCCACCATCTCATTCTTCA | 114bp |
| Ku-70 | GACAGCAGGAAGTGGAGACA | GCCACGAACAGAGTCTTGAA | 91bp |
| DNA-pkcs | ATATCCTTGGCAGGACTTGG | AGGTCCTCTCGGAGACAGAA | 92bp |
| TLR3 | TCTGTTTGCGAAGAGGAATG | AATTCCGAGATCCAAGTGCT | 114bp |
| TLR7 | TTGCAACTGTGATGCTGTGT | TTTGTGTGCTCCTGGACCTA | 106bp |
| TNF-α | GGCGGTGCCTATGTCTCA | CCTCCACTTGGTGGTTTGT | 117bp |
| IL-6 | GGCAATTCTGATTGTATG | CTCTGGCTTTGTCTTTCT | 208bp |
| IL-1 | GCCCATCCTCTGTGACTC | TGTGCCGTCTTTCATTAC | 283 bp |
| IL-10 | TTTCAAACAAAGGACCAG | GGATCATTTCCGATAAGG | 100bp |
| TGF-β | GGCGGTGCTCGCTTTGTA | TCCCGAATGTCTGACGTATTGA | 201bp |
| MCP-1 | GGGTCCAGACATACATTAA | ACGGGTCAACTTCACATT | 119bp |
| MCP-3 | GCTTCTGTGCCTGCTGCTC | CTTCCATGCCCTTCTTTG | 197bp |
| MIP-1α | CTGCCCTTGCTGTTCTTC | CAAAGGCTGCTGGTTTCA | 154bp |
| GAPDH | CAATGTGTCCGTCGTGGATCT | GTCCTCAGTGTAGCCCAAGATG | 124bp |
Western blot analysis
Skeletal muscle protein extraction was performed according to the manufacturer's protocol (KeyGEN, China). Protein concentrations were evaluated using a BCA assay kit (KeyGEN, China). Equal amounts of proteins were electrophoresed on 6-12% SDS-polyacrylamide gel and transferred to Immobilon P membrane (Millipore, USA). Membranes were blocked in 5% non-fat dried milk in Tris-buffered saline/Tween-20 (TBS-T: 20mM Tris, pH7.5, 150 mM NaCl, 0.05% Tween-20) for 1h at RT. The following rabbit polyclonal or mouse monoclonal antibody were used for detection: Rabbit polyclonal anti-Mi-2 (1:2000, abcam, USA); Rabbit polyclonal anti-HARS (1:500, NOVUS, USA); Rabbit polyclonal anti-Ku-70 (1:800, NOVUS, USA); Rabbit polyclonal anti-DNA-PKcs (1:100, Santa Cruz, USA) ;Rabbit polyclonal anti-TLR3 (1:800, abcam, USA); Rabbit polyclonal anti-TLR7 (1:200, Santa Cruz, USA); mouse monoclonal anti-GAPDH (1:3000, KANGCHEN, China). Primary antibodies were incubated for 20h at 4℃ in 5% non-fat dried milk in TBS-T. The membrane was then washed three times in TBS-T and incubated for 1h at RT with horseradish peroxidase conjugated goat anti-rabbit IgG (1:4000, Fudebio, China) or goat anti-mouse IgG (1:2000, CST, USA), in 5% non-fat dried milk in TBS-T. After three washes in TBS-T, the protein bands were visualized by enhanced chemiluminescence (ECL) detection reagents (Applygen Technologic Inc., China). Immunoreactive bands was detected by the ECL detection system (Protein Simple, USA), and densitometric values were analyzed with Quantity One (Bio-Rad, USA). Relative expression of each immunoreactive band was calculated by comparison with GAPDH.
Histological and immunofluorescence detection
Snap-frozen whole TA muscle was transversely cryosectioned (8μm), and either stained with hematoxylin and eosin or prepared for immunofluorescence. For immunofluorescence, muscle was fixed with cold acetone and incubated with rat anti-mouse F4/80 (1:200, eBioscience, USA); rat anti-mouse CD11b (1:200, eBioscience, USA); rat anti-mouse CD8α (1:200, eBioscience, USA); rat anti-mouse H-2Kb (1:100, BD, USA); rabbit polyclonal anti-caspase3 (1:500, abcam, USA);rabbit polyclonal anti-dystrophin (1:200, Santa Cruz, USA); or rabbit polyclonal anti-CD3ε (1:200, Abcam, USA). Rhodamine-conjugated goat anti-rat IgG (1:200, Santa Cruz, USA), FITC-conjugated goat anti-rabbit IgG (1:200, Santa Cruz, USA) or Alexa 568-conjugated rat anti-mouse IgG (1:1000, Invitrogen, USA) were used as secondary antibodies. Nuclei were counterstained with DAPI. Slides were viewed with an Olympus BX51 fluorescence microscope (Olympus, Japan). For results analysis, the Image J software was performed to quantify the intensity of staining. The integrated optical density (IOD) and area of interest (AOI) of all the positive staining (intense red staining) were measured, respectively. The mean density (IOD/AOI) was then calculated. Cell apoptosis was determined by counting the number of CD11b+caspase-3+ cells and expressed as a percentage of total CD11b+cells counted.
Statistical Analysis
All data are expressed as mean ± standard deviation (SD). One-way ANOVA was used for multiple comparisons (Duncan's multiple range test) using SPSS ver.13.0 software. P values<0.05 were considered as statistically significant.
Results
SNP and L-NAME Treatment Regulates NO and NOS levels in Notexin-Damaged TA Muscle
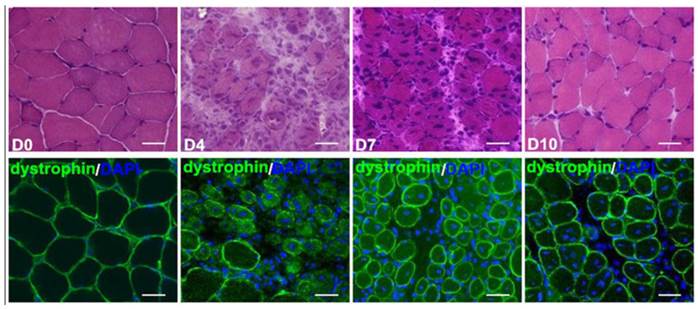
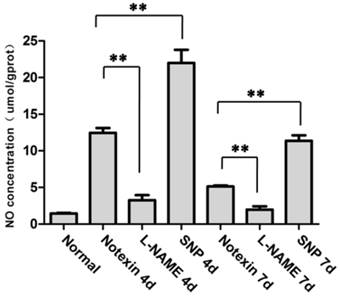
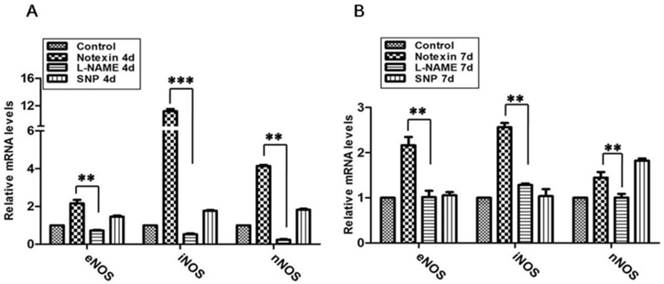
Using H&E and dystrophine fluorescence staining, we observed on day 4 post-injury, Notexin injection in TA muscle in WT mice induced myofiber necrosis and degeneration. Damage was gradually replaced by smaller regenerating myofibers, and centrally nucleated myofibers became prominent on day 7. Myorepair was almost completed on day 10 (Fig.1). We therefore took day 4 and 7 as the detection time points of necrosis and regeneration, respectively. As expected, mean NO concentration (umol/g muscle protein) in damaged muscle tissue significantly increased to 21.98 and 11.36 after NO donor SNP injection, but decreased to 3.26 and 1.97 after L-NAME injection, compared to animals with Notexin-treatment alone (12.46 and 5.13 ) on day 4 and 7 post-injury, respectively (Fig.2). Our qRT-PCR analysis further proved that, the mRNA expression of nNOS, eNOS and iNOS greatly increased in Notexin-damaged muscle at day 4 and 7, with the highest level of iNOS. L-NAME treatment caused the significant down-regulation of three NOS isoforms in damaged muscle, compared to untreated but damaged ones (Fig.3).
Histopathologic features of Notexin-induced acute myoinjury of TA. The top images: standard H&E staining for morphologic assessment. The bottom images: Dystrophin immunofluorescence staining (green). On day 0 (d0) (prior to myoinjury), the muscle appears normal. On day 4, full-blown necrosis and myophagocytosis are evident. On day 7, small regenerating myofibers with central nuclei neighbored by mononuclear inflammatory cells have appeared. On day 10, decreased inflammatory changes are seen in healing muscle. Bar = 50μm.
SNP and L-NAME treatment influence on the total Nitric Oxide level in TA muscle. Nitrate/Nitrite Assay was performed to evaluate the total NO concentration in TA muscle on day 4 and 7 post-injury. One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (**p<0.01)
L-NAME treatment interfere with mRNA levels of NOS isoforms in Notexin-damaged TA muscle. mRNA levels of NOS isoforms were evaluated in 4d (A) and 7d (B) damaged muscle by qRT-PCR. One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (**p<0.01, ***p<0.001).
Nitric Oxide Interfere with Muscle-Autoantigens and TLR3 Expression in Notexin-Damaged Skeletal Muscle tissue
Using an in vitro culturing system of C2C12 myoblasts, we recently found the expression of TLR3 and of proteins known to represent autoantigens in inflammatory autoimmune myopathies were down-regulated by SNP and, conversely, up-regulated by L-NAME [12]. This data suggested a role of NO on help to control skeletal muscle inflammation and immune response. For further investigating the in vivo effect of NO on myoinjury induced inflammation response, in this study, Notexin-induced mice myoinjuy model were prepared and treated with SNP, or L-NAME respectively. The expression levels of muscle autoantigens (Mi-2, HRS, DNA-PKcs and Ku-70) and the proinflammatory TLR3 and TLR7 were analyzed in damaged muscle tissue by qRT-PCR and immunoblotting.
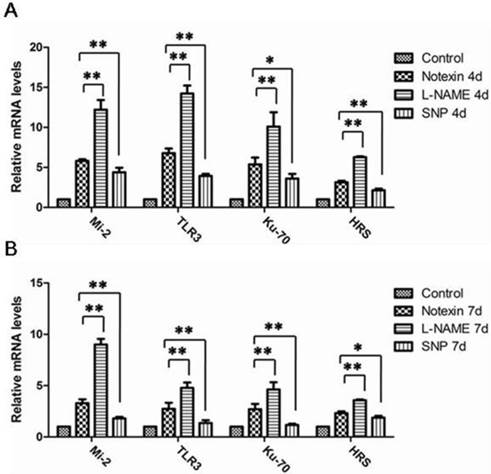
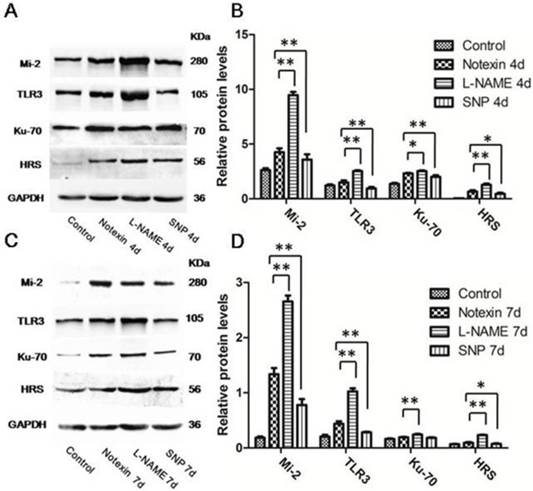
Although DNA-PKcs and TLR7 expression has no change (data not shown), we found most of autoantigens and TLR3 up-regulated in damaged muscle compared to normal muscle. As shown in Figure 4 and Figure 5, mRNA and protein levels of Mi-2, HRS, Ku-70 and TLR3 peaked around day 4 following Notexin injection, and gradually down-regulated as myofibers had begun to reform (day 7), but still higher than normal muscle. It's worth noting that the expression levels of Mi-2, HRS, Ku-70 and TLR3 in damaged muscle decreased significantly after SNP treating, but increased after L-NAME treating, compared to muscle with Notexin-damage alone, especially at the muscle degenerating period (day 4). Because autoantigens and TLR3 represent the important immune/inflammatory mediators in damaged muscle [13, 14], this data suggested the inhibition effects of NO on muscle inflammation.
SNP and L-NAME treatment interfere with mRNA levels of muscle-autoantigens and TLR3 in Notexin-damaged TA muscle. mRNA levels corresponding to proteins that can serve as muscle autoantigens and TLR3 were evaluated in 4d (A) and 7d (B) damaged muscle by qRT-PCR. One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (*p<0.05, **p<0.01).
SNP and L-NAME treatment interfere with protein expression of muscle-autoantigens and TLR3 in Notexin-damaged TA muscle. Western blot analysis showing autoantigens and TLR3 expression in Notexin-damaged muscle on day 4 (A) and 7 (C). The relative band intensities from western blots experiments were normalized to the level of GAPDH and analyzed with Quantity One software (B and D). One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (*p<0.05, **p<0.01).
Nitric Oxide Suppresses Notexin-Induced Intramuscular Infiltration of Monocytes/Macrophages
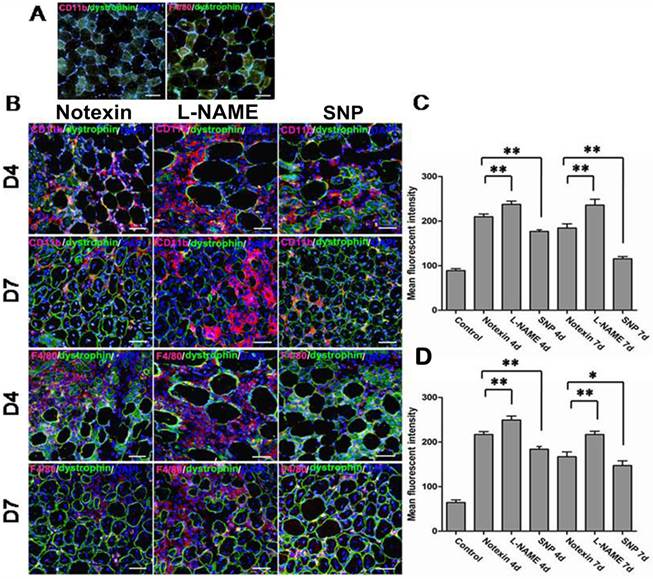
Because muscle injury and healing encompass the recruitment of inflammatory cells, and the inflammatory response to acute muscle damage is an innate immune response [15], we next investigated whether SNP and L-NAME treatment interferes with the intramuscular infiltration of monocytes/macrophages after Notexin injection. We conducted immunostaining to assess the infiltration of CD11b+ and F4/80+ cells on damaged and intact muscles of B6 mice on day 4 and 7 post-injury. The extent of infiltration was evaluated by fluorescent staining intensity analysis. As shown in figure 6, SNP treatment caused a significant decrease of the number of CD11b+ and F4/80+ cells in damaged muscles. On day 4 and 7 post-injury, the mean value of fluorescence intensity (Rhodamine) of infiltrated monocytes/macrophages reduced to 180 and 131 for CD11b, 184 and 147 for F4/80 respectively, comparing to that of Notexin damage alone at the same detecting time point (213 and 176 for CD11b, 217 and 167 for F4/80, respectively). Nevertheless, a powerful increase in the number of CD11b+ and F4/80+ cells was detected in damaged muscle tissue of B6 mice with L-NAME treatment (the mean value of fluorescence intensity of infiltration in muscle elevated to 243 and 266 for CD11b, and 249 and 217 for F4/80 at day 4 and 7, respectively), comparing to muscle damaged alone. Indeed, at the later regenerating time point (10d, myorepair was almost complete at this stage), persistent necrotic fiber segments contained massive inflammatory cell infiltration was still observed in muscle tissue of mice treated with L-NAME (data not shown).
SNP and L-NAME treatment interfere with Notexin-induced intramuscular infiltration of monocytes/macrophages. Representative immunofluorescence-stained muscle sections obtained at day 0 (A), 4 (B) and 7 (B). Quantification of the mean fluorescent intensity values of CD11b (C) and F4/80 (D) in muscle cross sections was shown. One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (*p<0.05, **p<0.01). Bar = 50μm.
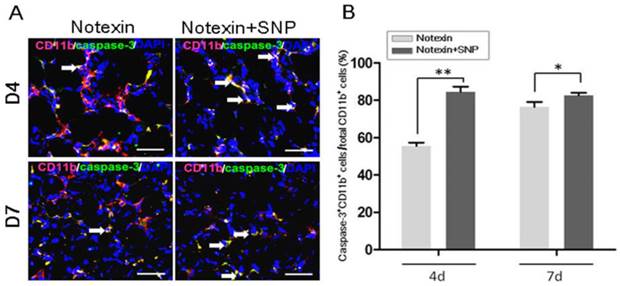
Because NO is one of modulators of apoptosis of inflammatory cells [16], we next examined whether SNP treatment induce apoptosis of myeloid cells infiltrated in damaged muscle. Using immunofluorecence staining and cell counting, we found SNP injection induced more severe inflammatory cells apoptosis in damaged muscle: CD11b+caspase-3+ cells were about 55.6%, and 76.4% of the total of CD11b+ cells, on the day 4 and 7 post-injury, respectively. However, double staining cells increased to 84.5%, and 82.5% respectively after SNP treating (Fig.7). These data suggest that NO acts as an anti-inflammatory role by blocking the intramuscular infiltration, and by inducing the apoptosis of innate leukocytes.
SNP treatment induce apoptosis of infiltrated cells in the Notexin-damaged TA muscle. Representative caspase 3 and CD11b immunofluorescence-stained muscle sections obtained on day 4 and 7 (A). Caspase-3+CD11b+ cells increased after SNP treating, compared to that of myoinjury alone. Arrows indicate caspase-3+CD11b+ cells. Cell apoptosis was determined by counting the number of CD11b+caspase-3+ cells and expressed as a percentage of total CD11b+cells counted (B). One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (*p<0.05, **p<0.01). Bar = 100μm.
Nitric Oxide Inhibits the Production of Pro-inflammatory Cytokines and Chemokines in the Notexin-Injuried Muscle
Monocytes and macrophages dominate a basic inflammatory response to muscle damage, which are a rich source of diverse cytokines and chemokines at the site of inflammation [17]. To evaluate more directly how the muscle inflammation response is affected by NO molecule, we next analyzed the expression of selected pro-inflammatory cytokines involving TNF-α, IL-1 and IL-6; anti-inflammatory cytokines involving TGF-β and IL-10, and the macrophage-sourced chemokines involving MIP-1α, MCP-1 and MCP-3, which have been associated with the preferential biological role during the muscle damage-induced inflammation response. In agreement with previous reports [15], our qPCR analysis demonstrated the up-regulation of mRNA level of TGF-β (on day 4 and 7) and IL-10 (on day 4) in TA muscle after myoinjury. However, we did not find SNP and L-NAME treatment effected on mRNA levels of TGF-β and IL-10 in Notexin-injected TA muscle (data not shown).
Noteworthy, SNP treatment resulted in a significant down-regulation of mRNA levels of pro-inflammatory TNF-α and IL-6 in damaged muscle, compared to that of muscle damaged alone, and this inhibition tendency maintained during the muscle necrosis (day 4) and repair processes (day 7) (Fig. 8A). In contrast, L-NAME stimulated a conspicuous production of TNF-α and IL-6, as mRNA levels of them in damaged TA muscle of mice treating with L-NAME were nearly the double of that damaged alone (Fig. 8A).
SNP and L-NAME treatment interfere with the production of pro-inflammatory cytokines and chemokines in the Notexin-damaged TA muscle. mRNA levels of TNF-α and IL-6 (A), MCP-1, MCP-3 and MIP-1α (B) were quantified by qRT-PCR. One-way ANOVA was used for multiple comparisons. All data are presented as mean ± SD (n=3). (*p<0.05, **p<0.01)
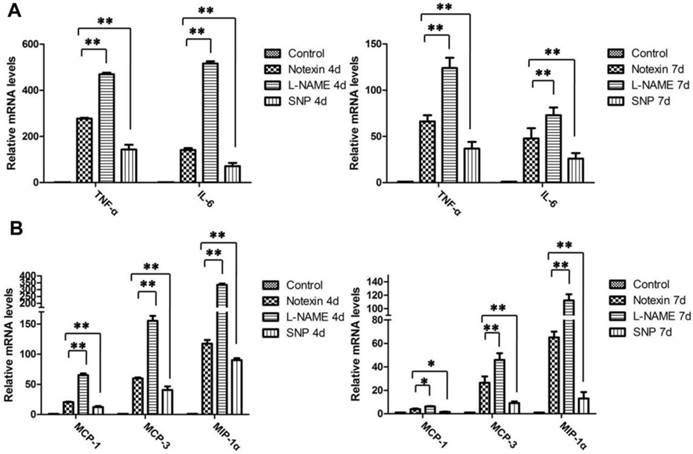
Similarly to what was observed above, we noticed that, after changing NO level in vivo artificially, the expression of macrophage-sourced chemokines in TA muscle was changed on day 4 and 7 post-injury: SNP treatment effectively diminished mRNA levels of MIP-1α, MCP-1, and MCP-3 (Fig. 8B). Conversely, L-NAME treatment resulted in a striking increase in the production of these genes (Fig. 8B). Taken together, these results collectively confirm that NO is crucial for the negative regulation of pro-inflammatory cytokines and chemokines production in damaged muscle tissue.
Nitric Oxide Interfere with Intramuscular Infiltration of CD8+ T cell and MHC-I Expression on New Myofibers after Myoinjury
Muscle antigens have been shown to induce migration of T lymphocytes to the endomysium or perimysium after myoinjury [13, 14, 18-20], and MHC-I gene expression on regenerated myofibers is a possible reason of intramuscular CD8+ T cells infiltration [21, 22]. We have found NO concentration change by SNP or L-NAME injection i.p. influences on mRNA and protein expression of muscle autoantigens and TLR3 in Notexin-damaged TA muscle. We next wish to find out whether NO is involved in the myofiber MHC-I expression and intramuscular CD8+ T cell infiltration.
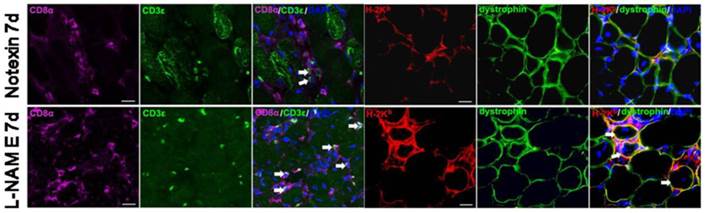
We conducted double immunostaining to assess the presence of CD3ε+CD8α+ T cells, and MHC-I molecule H-2Kb-expressing myofibers on Notexin-damaged muscle of B6 mice with or without SNP and L-NAME treatment on day 4 and 7. Sarcolemmal H-2Kb expression was assessed by comparison to basement membrane staining with dystrophine. As shown in figure 9, the number of CD3ε+CD8α+ T cells and H-2Kb+ myofiber were rather less in Notexin-injuried TA muscle (0-2/per slide), and only observed on day 7 post-injury. Interestingly, L-NAME treatment resulted in an increased number of H-2Kb positive myofibers (3-7/per slide), and of the infiltrated CD3ε+CD8α+ T cells in damaged muscle on day 7 post-injury. However, we did not found the effects of SNP treatment on the number of CD8+ T cell and H-2Kb+ myofibers in damaged TA muscle (data not shown). All together, these results suggest the inhibition effects of NO on CD8+ T cell intramuscular infiltration.
L-NAME treatment interferes with intramuscular infiltration of CD8+ T cell and MHC-I expression on regenerated myofibers after myoinjury. The number of CD3ε+CD8α+ T cells and of H-2Kb+ myofiber increased after L-NAME treating, compared to that of myoinjury alone. Arrows indicate CD3ε+CD8α+ T cells, or H-2Kb positive myofibers. Bar = 50μm.
Discussion
It has long been recognized that inflammatory processes are essential in skeletal muscle injury and repair. New findings confirmed that this processes not wholly beneficial for muscle regeneration. For example, the invading mononuclear cells can product proinflammatory cytokines, and release cytolytic and cytotoxic molecules, such as superoxide or its conversion hydrogen peroxide, which can lyse the cell membranes and damage the muscle even further. Persistence of inflammatory cells leads to damage of the surrounding normal tissue and exacerbation of inflammation, even lead to secondary injury [23]. It would be helpful to coordinate between inflammatory and regeneration process, resulting in better muscle functional recovery after myoinjury. Nitric Oxide-dependent satellite cell activation and myofiber regeneration during acute and chronic myoinjuries have been extensively studied [8, 24]. However, there are still few published studies investigating the role of NO in post-injury muscle inflammation response. Until now, the role of NO in muscle inflammation remains controversial [25]. Some scholars believed that NO is an anti-inflammatory molecule [7, 10, 11] and our previous in vitro experiments also support the view [12]. In this study, we examined whether and how NO participate in the mediation of muscle inflammatory response using the mice model of Notexin induced myoinjury.
Since the concomitant up-regulation of TLRs or of potential muscle autoantigens could potentially aggravate muscle inflammation and damage in immune-mediated muscle disease by stimulating cytokines and chemokines production, as well as the activation of autoimmune T cells [13, 14], in this study, we firstly explored whether the blocking of NO production by L-NAME, or the increasing of NO output by SNP during the myoinjury phases may influence on the expression of muscle autoantigens and TLRs. In our work, autoantigens and TLR-3 were detected to up-regulate persistently from day 4 to day 7 post-injury, which supports the opinion that Notexin induced a rapid muscle necrosis and sequentially reformation of new myofibers, and those new immature muscle cells preferentially express autoantigens and TLR3 during the conditions of muscle inflammation [19]. Previous studies have shown that NO promotes myogenic precursor cell activation, regeneration and fusion in acutely and chronically damaged muscles [8, 24]. It seems that we can reasonably presume that the increase of NO concentration in damaged muscle should contribute to up-regulate autoantigens and TLR3 expression, by promoting new myofiber formation. Unexpectedly, we found mRNAs and protein levels of Mi-2, HRS, Ku-70 and TLR3 in Notexin-damaged skeletal muscle were significantly down-regulated by SNP treatment, but up-regulated by L-NAME treatment at the muscle necrosis and regeneration phages. This observation suggests NO can negatively regulate immune response by down-regulating autoantigens and pro-inflammatory TLR3 after myoinjury.
Several studies have confirmed that, during the muscle inflammatory phase there is a great increase in local NO production [26, 27]. Muscle-derived NO appears to be a particularly important regulator of muscle inflammation and damage by invading inflammatory cells [28]. McCafferty et al. demonstrated iNOS-derived NO regulate the inflammation response to injury by reducing the extent of leukocyte infiltration [10]. Our results support this opinion, because we observed SNP treatment decreased the number of infiltrated CD11b+ and F4/80+ cells in Notexin-damaged muscle tissue, but L-NAME resulted in a significant increase in cell infiltration. In vitro and in vivo studies have shown that muscle-derived NO reduces of inflammatory damage by increasing the apoptosis of infiltrated cells and inhibiting the expression of adhesion molecules, reduces neutrophil-mediated lysis of muscle cells and decreases superoxide concentration, forming less reactive intermediates [29]. Our results clearly show the capacity of NO donor SNP to induce apoptosis in intramuscular infiltrated mononuclear cells. By exploring the expression of selected pro- and anti-inflammatory cytokines, and the macrophage-sourced chemokines in Notexin-damaged muscle tissue with or without SNP or L-NAME treatment, we confirmed that SNP treatment resulted in a significant production inhibition of TNF-α, IL-6, MIP-1α, MCP-1, and MCP-3 in damaged muscle. In contrast, L-NAME stimulated a conspicuous production of those molecules. Therefore, it is conceivable that endogenous NO functions as a negative modulator of muscle inflammatory response.
One peculiar immunologic feature of myofibers is that they do not express detectable levels of major histocompatibility complex class I (MHC-I) molecules. In contrast, cultured myogenic precursors [30] and regenerating myofibers from myopathic muscle show surface reexpression of MHC-I [31-34]. Hence, surface MHC-I likely participates in the pathogenesis of myoinjury by its relationship with autoreactive cytotoxic CD8+ T cells. However, in acutely damaged skeletal muscle, intramuscular CD8+ T cell infiltration and MHC-I expression on regenerated myofibers is rather occasional, despite of heavy macrophage/monocyte infiltration. This reflects that normal muscle tissue is resistant to autoimmunity. Interesting, in patients and animal models of autoimmune myositis, nNOS is absent from the sarcolemma, and relocated to the cytosol, leading to a reduction of total muscle NOS activity [35, 36]. The restoration of NO generation by transgenic expression of nNOS ameliorates the dystrophic phenotype [37]. Whether muscle-derived NO participates in governing immune priming/tolerance toward muscle antigens are still poorly defined. From our data that NO interfere with muscle autoantigens and TLR3 expression, it is possible NO has a role on regulating muscle auto-tolerance. Using fluorescent staining to detect H-2Kb expression in damaged muscle of B6 mice treating with L-NAME, we observed an obviously increased number of H-2Kb positive myofibers, compared to that of Notexin-damaged muscle alone. In accord with this result, we found the increased number of CD3ε+CD8α+ T cells in damaged muscle on day 7 post-injury. We therefore assume that muscle-derived NO reduces of muscle damage through inhibiting the MHC-I expression on regenerated myofibers, and therefore blocking the infiltration of CD8+ T cell. However, further experimental studies are needed to verify this assumption.
Collectively the data presented here show that, NO, a multi-functional molecule, is an intrinsic negative regulator of muscle inflammatory response. Muscle-derived NO helps to reduce inflammatory damage of skeletal muscle through suppressing muscle-autoantigens and TLR3 expression, decreasing the infiltration of leukocytes, inducing the apoptosis of infiltrated cells, and down-regulating the production of pro-inflammatory cytokines and chemokines. Our founding also imply a possible inhibition effects of NO on MHC-I expression on new myofibers and intramuscular infiltration of CD8+ T cell after myoinjury.
Abbreviations
NO, nitric oxide; NOS, nitric oxide synthase; iNOS, inducible nitric oxide synthase; nNOS, neuronal nitric oxide synthase; TLRs, toll-like receptors; HRS, histidyl tRNA synthetase; DNA-PKcs, the catalytic subunit of DNA-dependent protein kinase; SNP, sodium nitroprusside dehydrate; L-NAME, L-NG-Nitroarginine Methyl Ester; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Acknowledgements
This study has been supported by grants from the National Natural Science Foundation of China (81171724) and (81371924), GuangDong Provincial Science and Technology Plan Projects (2012B031800146) and (2011B031800085).
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Hua Liao had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design: Hua Liao. Acquisition of data: XingHui Liu, Gang Wu, DanDan Shi. Analysis and interpretation of data: XingHui Liu, Gang Wu, DanDan Shi, Rong Zhu, HuiJun Zeng, Biao Cao, MeiXian Huang, Hua Liao.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Charge S.B, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209-38
2. Porter JD, Khanna S, Kaminski HJ. et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263-72
3. Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859-74
4. Tatsumi R, Hattori A, Allen RE. et al. Mechanical stretch-induced activation of skeletal muscle satellite cells is dependent on nitric oxide production in vitro. Anim Sci J. 2002;73:235-239
5. De Paepe B, Racz GZ, Schröder JM. et al. Expression and distribution of the nitric oxide synthases in idiopathic inflammatory myopathies. Acta Neuropathol. 2004;108:37-42
6. Filippin L. et al. The role of nitric oxide during healing of trauma to the skeletal muscle. Inflamm. Res. 2011;60:347-356
7. Rigamonti E, Touvier T, Clementi E. et al. Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J. Immunol. 2013;190:1767-1777
8. Buono R, Vantaggiato C, Pisa V. et al. Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells. 2012;30:197-209
9. Hickey M.J. Role of inducible nitric oxide synthase in the regulation of leucocyte recruitment. Clin. Sci. 2001;100:1-12
10. McCafferty D.M, Mudgett J.S, Swain M.G. et al. Inducible nitric oxide synthase plays a critical role in resolving intestinal inflammation. Gastroenterology. 1997;112:1022-1027
11. Rovere-Querini P, Clementi E, Brunelli S. Nitric oxide and muscle repair: multiple actions converging on therapeutic efficacy. Eur J Pharmacol. 2014;730:181-5
12. Chen R, Feng L, Ruan M. et al. Mechanical-Stretch of C2C12 Myoblasts Inhibits Expression of Toll-Like Receptor 3 (TLR3) and of Autoantigens Associated with Inflammatory Myopathies. PLoS One. 2013;8:e79930
13. Tournadre A, Lenief V, Miossec P. Expression of Toll-like receptor 3 and Toll-like receptor 7 in muscle is characteristic of inflammatory myopathy and is differentially regulated by Th1 and Th17 cytokines. Arthritis Rheum. 2010;62:2144-2151
14. Betteridge ZE, Gunawardena H, McHugh NJ. Pathogenic mechanisms of disease in myositis: autoantigens as clues. Curr Opin Rheumatol. 2009;21:604-609
15. Arnold L, Henry A, Poron F. et al. Inflammatory monocytes recruited after skeletal muscle injury switch into anti-inflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057-1069
16. Ward C, Wong TH, Murray J, Rahman I, Haslett C, Chilvers ER, Rossi AG. Induction of human neutrophil apoptosis by nitric oxide donors: evidence for a caspase-dependent, cyclic-GMP-independent, mechanism. Biochem Pharmacol. 2000;59(3):305-14
17. Lu H. et al. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. 2011;25:3344-3355
18. Mammen AL, Casciola-Rosen LA, Hall JC. et al. Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum. 2009;60:3784-3793
19. Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum. 1996;39:292-296
20. Casciola-Rosen L, Nagaraju K, Plotz P. et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201:591-601
21. Emslie-Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol. 1989;20:224-31
22. Confalonieri P, Oliva L, Andreetta F. et al. Muscle inflammation and MHC class I up-regulation in muscular dystrophy with lack of dysferlin: an immunopathological study. J Neuroimmunol. 2003;142:130-36
23. Tiidus PM. Radical species in inflammation and overtraining. Can J Physiol Pharmacol. 1998;76:533-538
24. Filippin L. et al. Nitric oxide and repair of skeletal muscle injury. Nitric Oxide. 2009;21:157-163
25. Tripathi P, Kashyap L, Singh V. The role of nitric oxide in inflammatory reactions. FEMS Immunol Med Microbiol. 2007;51:443-452
26. Kaminski H.J, Andrade F.H. Nitric oxide: biologic effects on muscle and role in muscle diseases. Neuromuscul Disord. 2001;11:517-524
27. Rubinstein I. et al. Involvement of nitric oxide systemin experimental muscle crush injury. J. Clin.Invest. 1998;101:1325-1333
28. Tidball J.G. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345-R353
29. Stamler J.S, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev. 2001;81:209-237
30. Hohlfeld R, Engel AG. HLA expression in myoblasts. Neurology. 1991;41:2015
31. Appleyard ST, Dunn MJ, Dubowitz V. et al. Increased expres-sion of HLA ABC class I antigens by muscle fibres in Duchenne muscular dystrophy, inflammatory myopathy, and other neuromus-cular disorders. Lancet. 1985;1:361-363
32. McDouall RM, Dunn MJ, Dubowitz V. Expression of class I and class II MHC antigens in neuromuscular diseases. J Neurol Sci. 1989;89:213-226
33. Emslie-Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol. 1989;20:224-231
34. Confalonieri P, Oliva L, Andreetta F. et al. Muscle inflammation and MHC class I up-regulation in muscular dystrophy with lack of dysferlin: an immunopathological study. J Neuro-immunol. 2003;142:130-136
35. Brenman J.E, Chao D.S, Xia H. et al. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743-752
36. Chang W.J, Iannaccone S.T, Lau K.S. et al. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci U S A. 1996;93:9142-7
37. Wehling-Henricks M, Tidball J.G. Neuronal nitric oxide synthase-rescue of dystrophin/utrophin double knockout mice does not require nNOS localization to the cell membrane. PloS One. 2011;6:e25071
Author contact
![]() Corresponding author: Hua Liao, MD, Ph.D. Department of Anatomy, Guangdong Provincial Key Laboratory of Construction and Detection in Tissue Engineering, School of Basic Medical Science, Southern Medical University, No.1838, GuangZhou Avenue North, GuangZhou, 510515, China. Tel: +862062789092; Fax: +862061648202; E-mail: hua-liaocom
Corresponding author: Hua Liao, MD, Ph.D. Department of Anatomy, Guangdong Provincial Key Laboratory of Construction and Detection in Tissue Engineering, School of Basic Medical Science, Southern Medical University, No.1838, GuangZhou Avenue North, GuangZhou, 510515, China. Tel: +862062789092; Fax: +862061648202; E-mail: hua-liaocom