Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(7):752-761. doi:10.7150/ijbs.11880 This issue Cite
Research Paper
Valproate Attenuates 25-kDa C-Terminal Fragment of TDP-43-Induced Neuronal Toxicity via Suppressing Endoplasmic Reticulum Stress and Activating Autophagy
Xuejing Wang1*, Mingming Ma2*, Junfang Teng1, Xiangqian Che4, Wenwen Zhang1, Shuman Feng2, Shuang Zhou3, Ying Zhang3, Erxi Wu3 ![]() , Xuebing Ding1
, Xuebing Ding1 ![]()
1. Department of neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, China
2. Department of neurology, People's Hospital of Zhengzhou University, Zhengzhou, Henan 450003, China
3. Department of Pharmaceutical Sciences, North Dakota State University, Fargo, ND, 58105, USA
4. Department of Neurology, Ruijin Hospital, Shanghai Jiaotong University, Shanghai 200025, China
*Equal contribution
Received 2015-2-12; Accepted 2015-4-17; Published 2015-5-19
Abstract

Amyotrophic lateral sclerosis (ALS) is a fatal adult-onset neurodegenerative disease. To date, there is no any effective pharmacological treatment for improving patients' symptoms and quality of life. Rapidly emerging evidence suggests that C-terminal fragments (CTFs) of TAR DNA-binding protein of 43 kDa (TDP-43), including TDP-35 and TDP-25, may play an important role in ALS pathogenesis. Valproate (VPA), a widely used antiepileptic drug, has neuroprotective effects on neurodegenerative disorders. As for ALS, preclinical studies also provide encouraging evidence for multiple beneficial effects in ALS mouse models. However, the potential molecular mechanisms have not been explored. Here, we show protective effects of VPA against TDP-43 CTFs-mediated neuronal toxicity and its underlying mechanisms in vitro. Remarkably, TDP-43 CTFs induced neuronal damage via endoplastic reticulum (ER) stress-mediated apoptosis. Furthermore, autophagic self-defense system was activated to reduce TDP-43 CTFs-induced neuronal death. Finally, VPA attenuated TDP-25-induced neuronal toxicity via suppressing ER stress-mediated apoptosis and enhancing autophagy. Taken together, these results demonstrate that VPA exerts neuroprotective effects against TDP-43 CTFs-induced neuronal damage. Thus, we provide new molecular evidence for VPA treatment in patients with ALS and other TDP-43 proteinopathies.
Keywords: Amyotrophic lateral sclerosis, Valproate, 25 kDa C-terminal fragment of TDP-43, Endoplasmic reticulum stress, Autophagy
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset devastating neurodegenerative disease, characterized by selective loss of both upper and lower motor neurons, resulting in generalized muscular atrophy, paralysis, and muscle weakness [1-4]. Underlying mechanisms of motor neuron degeneration in ALS is still unknown. Abnormal autophagy [5], endoplastic reticulum (ER) -induced cellular stress [6], neuroinflammation [7], protein aggregation [8], and oxidative stress [9] are hypothesized to be possible causes. Therapeutic options against ALS are currently very limited. To date, riluzole is the only drug approved for the treatment of ALS with a modest survival extent [10]. However, it does not improve patients' symptoms and quality of life [11]. Therefore, finding new effective therapeutic agents for ALS is imperative.
TAR DNA-binding protein of 43 kDa (TDP-43) has been identified as the major component of ubiquitin-positive cytoplasmic inclusions in central nervous system (CNS) of TDP-43 proteinopathies, including ALS [12]. Pathological TDP-43 polypeptides undergo post-translational modifications, including cleavage, ubiquitination, and phosphorylation, ultimately lead to its misfolding and aggregation [13]. C-terminal fragments (CTFs) of TDP-43, including TDP-35 and TDP-25, are the cleaved fragments of TDP-43 by activated caspases-3 [14]. Immunocytochemical studies demonstrate that cytoplasmic inclusions in CNS of ALS contain phosphorylated and ubiquitinated TDP-43 CTFs [15, 16]. Actually, TDP-43 CTFs, containing a “prion-like domain” at the C-terminal end (residues 277-414), are prone to form cytoplasmic aggregates [17]. A previous study showed that overexpression of TDP-25 induced mitochondrial autophagy and ubiquitin-positive inclusions [18]. Furthermore, TDP-25 transgenic rats suffered selective forelimb impairments to mimic ALS by increasing levels of TDP-25 [19]. However, the definitive pathological role of TDP-43 CTFs involved in ALS remains unclear.
Valproate (VPA), an antiepileptic drug, is also discovered to inhibit histone deacetylases (HDACs) for promoting gene transcription [20]. VPA inhibits death of neuronal cells by its ability to counterbalance oxidative stress, autophagy, ER stress, apoptosis, and glutamate toxicity [21-26]. Recently, VPA has been shown to be beneficial in various models of neurodegenerative diseases [27]. Notably, evidence accumulating from mouse models has indicated that VPA may have therapeutic potential for the treatment of ALS via activating transcription factor, modulating the expression of splicing factors, inhibiting glutamate toxicity and so on [22, 28-31]. However, clinical trials have provided inconclusive results [23, 28, 29, 32]. To solve these issues, investigation of the precise mechanisms involved in the effect of VPA on ALS is the first important step.
In this study, we show that overexpression of TDP-43 CTFs induces inclusion body formation and neuronal toxicity via activating ER stress-mediated apoptosis in SH-SY5Y cells. Moreover, we identify that VPA attenuates TDP-25-induced neuronal toxicity via suppressing ER stress and enhancing autophagy in SH-SY5Y cells. Our findings demonstrate that VPA protects neurons against TDP-43 CTFs- induced damage. Thus, we provide the new evidence for VPA in treatment of TDP-43 proteinopathy, such as ALS and frontotemporal lobar degeneration (FTLD).
Materials and Methods
Cell culture, transfections, and drug treatments
SH-SY5Y cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, USA) containing 15% fetal bovine serum (GIBCO, USA), 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen, USA), maintained at 37°C in a humidified incubator of 5% CO2. Plasmids (pEGFP-TDP-43, pEGFP-TDP-35, and pEGFP-TDP-25) were kindly donated by Professor Guanghui Wang (Soochow University, China). The mammalian expression plasmids HA-TDP-43, HA-TDP-35, and HA-TDP-25 were subclones of PCR products. Transfections were performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. Forty-eight hours after transfection, cells were treated with an autophagy inhibitor 3-methyladenine (3-MA) (10 mM; Sigma-Aldrich, USA) or VPA (0 mM, 0.5 mM, 1.0 mM, and 2.0 mM; Sigma-Aldrich, USA).
Immunoblotting
Cells were collected from the plates and centrifuged at 500 × g for 5 min. The cell pellets were resuspended in TSPI buffer [50 mM Tris-HCl (pH7.5), 150 mM NaCl, 1 mM EDTA, 1 mg/ml aprotinin, 10 mg/ml leupeptin, 0.5 mM Pefabloc SC, 10 mg/ml pepstatin, and 1% NP-40]. The samples were heated in loading buffer, and equal amounts of total protein were loaded and separated by SDS-PAGE. After transferred to nitrocellulose membranes, blots were blocked with 5% nonfat dry milk in TBST (0.25% Triton X-100 in PBS, pH 7.4) for 30 min, then incubated with primary antibodies overnight at 4°C. After washed three times in TBST, the membranes were incubated with anti-rabbit IgG (Cell Signaling Technology, USA) or anti-mouse IgG (Cell Signaling Technology, USA) for 1.5 h. Membranes were washed three times and proteins were visualized after ECL (Pierce Chemical, USA) treatment. The primary antibodies used were rabbit polyclonal anti-caspase-12 antibody (Abcam, UK), rabbit polyclonal anti-phospho-eIF2α antibody (Cell Signaling Technology, USA), mouse monoclonal anti-CHOP antibody (Santa Cruz Biotechnology, USA), rabbit polyclonal anti-LC3 antibody (Cell Signaling Technology, USA), mouse monoclonal anti-Beclin1 antibody (Cell Signaling Technology, USA), rabbit polyclonal anti-Bcl-2 antibody (Santa Cruz Biotechnology, USA), rabbit polyclonal anti-Bcl-xL antibody (Cell Signaling Technology, USA), and mouse monoclonal anti-GAPDH antibody (Cell Signaling Technology, USA).
MTT and LDH assays
According to the procedure previously described [33], we used the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to evaluate cell viability. After incubation with VPA for 48 h, 0.5 mg/ml MTT (Sigma-Aldrich, USA) was added to each well at 37 °C for 2 h. Formazan salt formed was dissolved in DMSO, and colorimetric determination was performed at 540 nm. Cell death and lysis were evaluated on lactate dehydrogenase (LDH) activity released into the supernatant. LDH activity was determined with a commercial LDH Assay kit (Sigma-Aldrich, USA).
Flow cytometry assay
For the apoptosis assay, cells were washed twice with 1 × PBS. The cells were double-stained with Annexin V conjugated to FITC and PI, using Annexin V-FITC apoptosis detection kit (Sigma-Aldrich, USA) according to the manufacturer's instruction and analyzed on a Cytomics FC 500 flow cytometer (Beckman Coulter, USA).
Loading of lysotracker red and MDC
LysoTracker (Molecular Probes, USA) was added to cultured media for 30 min, and each well was washed three times with DMEM, and fixed with 2% paraformaldehyde for 10 min at 4°C. The red fluorescence of LysoTracker was visualized using Nikon Labphoto-2 fluorescence microscope.
Cells were incubated with 0.05 mM monodansylcadaverine (MDC) (Sigma-Aldrich, USA) at 37°C for 1 h, and the changes of fluorescence were observed by Nikon Labphoto-2 fluorescence microscope at excitation wave length 380 nm with emission filter 525 nm.
Statistical analysis
All statistical analyses were conducted using SPSS statistical software package (SPSS version 8.0). Data were shown as mean±SD. A p value of less than 0.05 indicated a significant difference.
Results
Overexpression of TDP-43 CTFs induces formation of inclusions and neuronal toxicity
To demonstrate the involvement of TDP-43 CTFs in inclusion formation, SH-SY5Y cells were made to express 35-kDa or 25-kDa TDP-43 fragments, corresponding to the C-terminal truncated products generated when TDP-43 is cleaved by caspases-3 [34]. As shown in Figure 1A, wild-type GFP-TDP-43 was evenly distributed in the nucleus, and no TDP-43 positive aggregates were detected. GFP-TDP-35-positive immunoreactivity showed both cytoplasmic and nuclear distribution, and GFP-TDP-35-positive aggregates were detected in both cytoplasm and nucleus. Furthermore, GFP-TDP-25 formed compact cytoplasmic inclusions. Moreover, more inclusions observed in cells expressing GFP-TDP-25 (42.8%) compared with GFP-TDP-35 (22.2%) (Figure 1B).
TDP-43 CTFs enhance aggregation and induce neuronal toxicity. (A) Formation of cytoplasmic inclusion bodies by overexpressing of GFP-TDP-35 or GFP-TDP-25. GFP-tagged proteins are green; nuclei are stained with Hoechst33258 (blue). Fluorescent confocal microscopy images demonstrate the predominant nuclear localization of wild-type GFP-TDP-43, as well as cytoplasmic inclusions formed in cells transfected with pEGFP-TDP-35 or pEGFP-TDP-25 in SH-SY5Y cells. (B) Percentage of aggregate-containing neurons increases in cells expressing GFP-TDP-35 and GFP-TDP-25. Values shown are the mean ± S.E.M. from three experiments. (C) The release of LDH into the media was used as an indicator of neuronal toxicity. Values shown are the mean ± S.E.M. from five experiments. (D) SH-SY5Y cells viability measured by MTT assay after cells transfected with the indicated constructs. Values shown are the mean ± S.E.M. from five experiments. (E) Neuronal apoptosis detected by Annexin V-FITC/PI after cells transfected with the indicated constructs. Values shown are the mean ± S.E.M. from five experiments. Level of statistical significance: *p < 0.05, ** p < 0.01.
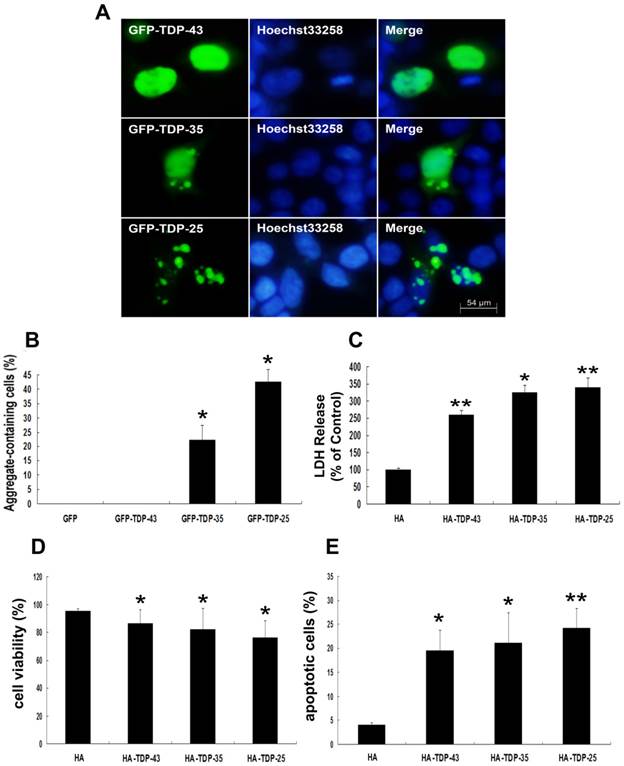
Next, we examined whether TDP-43 CTFs increase neuronal toxicity. Neuronal death was assessed by measuring LDH release and cell viability using LDH assay and MTT, respectively. LDH levels were increased in cells expressing HA-TDP-43, HA-TDP-35, and HA-TDP-25 compared with cells expressing HA (Figure 1C). Notably, LDH release was markedly increased in cells expressing HA-TDP-25 compared with cells transfected with HA-TDP-43 or HA-TDP-35 (Figure 1C). For cell viability using MTT assay, we observed that overexpression of HA-TDP-43, HA-TDP-35 or HA-TDP-25 significantly suppressed the viability of SH-SY5Y cells compared to HA (Figure 1D). Similarly, cell viability was decreased in cells expressing HA-TDP-25 compared with cells transfected with HA-TDP-43 or HA-TDP-35 (Figure 1D). We then used Annexin V/PI staining to examine the extent of apoptosis (Figure 1E). In addition, we examined the expression levels of both antiapoptotic proteins Bcl-2, Bcl-xL and pro-apoptotic protein cleaved caspase-3 by Western blot analysis (Figure 2A, B). The result showed that protein levels of Bcl-2 and Bcl-xL were decreased and cleaved caspase-3 was increased in cells expressing GFP-TDP-35 and GFP-TDP-25, compared with cells expressing GFP-TDP-43 or GFP. These findings confirm that apoptosis was significantly enhanced in cells expressing TDP-43 CTFs, especially TDP-25.
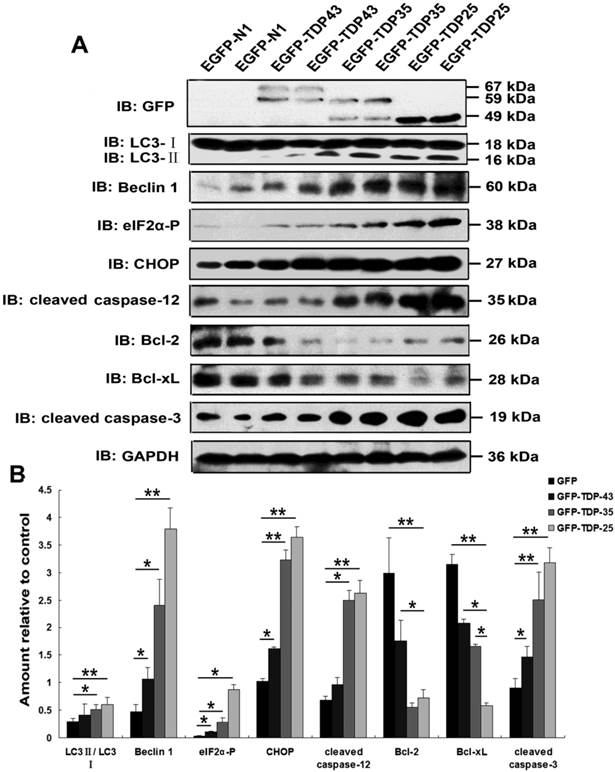
Overexpression of TDP-43 CTFs induces ER stress and autophagy. (A) Western blot analysis performed using lysis of SH-SY5Y cells following transfection of pEGFP, pEGFP-TDP-43, pEGFP-TDP-35, and pEGFP-TDP-25 for 72 h. Cell lysates were examined by immunoblotting with the indicated antibodies. (B) Quantification showing significant increases in LC3II/LC3I, Beclin 1, phosphorylated eIF2α, CHOP, cleaved caspase-12, and cleaved caspase-3 levels in pEGFP-TDP-35- and pEGFP-TDP-25-transfected cells in comparison to pEGFP-transfected cells, whereas expression levels of Bcl-2 and Bcl-xL significantly decreased. Compared pEGFP-transfected cells with pEGFP-TDP-43-transfected cells, expression levels of Beclin-1, phosphorylated eIF2α, CHOP, and cleaved caspase-3 were significantly increased. Values shown are the mean ± S.E.M. from three experiments. Level of statistical significance: *p < 0.05, ** p < 0.01.
Overexpression of TDP-43 CTFs activates ER stress and autophagy in neuronal cells
Previous studies suggested a critical role of ER stress in the pathology of ALS [35, 36]. A recent study demonstrated that overexpression of mutant TDP-43 induced ER stress via activation of XBP-1 and ATF6, thus linking ER stress to neurodegeneration [6]. As shown in Figure 2A, our findings demonstrate that expression of phosphorylated eIF2α, CHOP, and cleaved caspase-12 was markedly increased by overexpression of GFP-TDP-35 or GFP-TDP-25, especially in the cells expressing GFP-TDP-25 (Figure 2A, B). These data indicate that TDP-43 CTFs induce ER stress-associated apoptosis.
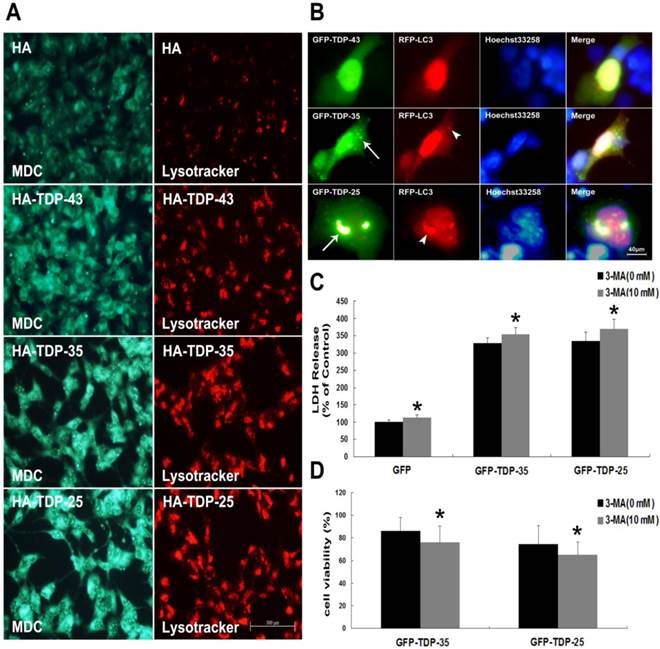
Macroautophagy participates in TDP-43 proteinopathy pathogenesis. Recent reports show that autophagy-related proteins are pathologically altered in the brain of patients with ALS [5, 37]. To investigate the change of autophagic activity, we first performed Western blot analysis to determine the expression levels of macroautophagic markers, LC3 and Beclin-1, in cells transfected with pEGFP, pEGFP-TDP-43, pEGFP-TDP-35 or pEGFP-TDP-25 (Figure 2A, B). Immunoblot analysis showed that the conversion from LC3-I to LC3-II and expression of Beclin-1 were increased by overexpression of GFP-TDP-35 or GFP-TDP-25 (Figure 2A, B). Next, we used MDC and LysoTracker Red assays to detect autophagic vacuoles in the transfected SH-SY5Y cells (Figure 3A). We found that the number of MDC-labeled fluorescent particles and autophagosomes (Lysotracker red fluorescent dye) were markedly increased in the cells transfected with HA-TDP-35 and HA-TDP-25, compared with cells transfected with HA and HA-TDP-43. Double immunofluorescent staining revealed co-localization of TDP-43 positive aggregates and RFP-LC3 in the cells expressing GFP-TDP-35 and GFP-TDP-25 (Figure 3B). Moreover, 3-MA treatment dramatically increased LDH release, and suppressed the viability of SH-SY5Y cells transfected with pEGFP-TDP-35 or pEGFP-TDP-25 (Figure 3C, D). These findings demonstrate that activation of macroautophagy is involved in self-defense mechanism for neuron survival in cells overexpressing TDP-43 CTFs.
Inhibition of autophagy enhances TDP-43 CTFs-induced neuronal damage in SH-SY5Y cells. (A) Following transfection of HA, HA-TDP-43, HA-TDP-35, or HA-TDP-25 for 48 h, changes of cellular autophagy were observed by fluorescence microscopy with MDC and Lysotracker staining. (B) Co-localization of GFP-TDP-35 or GFP-TDP-25 with punctuate RFP-LC3 was observed. (C) The release of LDH was used as an indicator of neuronal toxicity following 3-MA treatment. Values shown are the mean ± S.E.M. from five experiments, asterisks indicate LDH release form 3-MA treated cells statistically increased compared to untreated cells. (D) SH-SY5Y cells viability was measured by MTT assay in cells treated with 3-MA. Values shown are the mean ± S.E.M. from five experiments. Level of statistical significance: *p < 0.05.
VPA suppresses ER stress and enhances autophagy in neuronal cells overexpressing TDP-25
Since Sugai et al., reported the beneficial effect of VPA in a mouse model of ALS [24], VPA has be regarded as a promising candidate drug for the treatment of ALS. However, clinical studies about effect of VPA on survival in patients with ALS revealed inconsistent conclusions [23, 32]. To investigate whether VPA exerts neuroprotective effects in vitro, and to clarify the precise mechanisms involved in the VPA treatment, we first examined the effect of VPA on TDP-25-induced ER stress. Since TDP-25 is believed to be the pathologic species in FTLD-U and ALS [19, 38], and our above findings suggest that GFP-TDP-25 caused the more serious neuronal damage than GFP-TDP-35, we thus focused our following studies on TDP-25. Results show that exposure of cells transfected with pEGFP-TDP-25 to VPA resulted in significant decrease of CHOP and cleaved caspase-12 in a dose-dependent manner. We conclude that VPA suppresses GFP-TDP-25-induced ER stress (Figure 4A, B).
Exposure to VPA inhibits ER stress and enhances autophagy in SH-SY5Y cells overexpressing TDP-25. (A) Western blot analysis performed using lysates of pEGFP-TDP-25-transfected cells following treatment of VPA for 48 h with different doses. Cell lysates were examined by immunoblotting with the indicated antibodies. (B) Quantification showing significant increase in expression levels of LC3II/LC3I and Beclin-1, and decrease of CHOP and cleaved caspase-12 in GFP-TDP-25-transfected cells following VPA treatment in a dose-dependent manner. (C) Following exposure to VPA (2 mM) for 48 h, autophagy was evaluated by fluorescence microscopy with Lysotracker and MDC staining in HA-TDP-25-transfected cells. Values shown are the mean ± S.E.M. from three experiments. Level of statistical significance: *p < 0.05, ** p < 0.01.
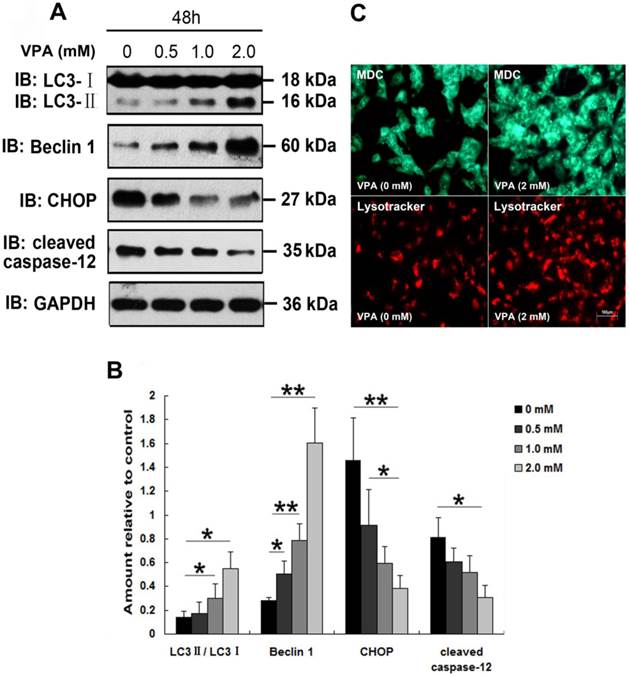
A previous study suggested that autophagy activation was a potentially useful route for the therapy of neurodegenerative diseases with TDP-43 proteinopathies [39]. Therefore, we next investigated the effect of VPA on TDP-25-induced autophagy. Immunoblot analysis revealed that exposure of cells transfected with pEGFP-TDP-25 to VPA resulted in significant increase of conversion from LC3-I to LC3-II and expression of Beclin-1 in a dose-dependent manner (Figure 4A, B). Further LysoTracker Red and MDC assays confirmed that treatment of VPA activated autophagy in SH-SY5Y cells transfected with HA-TDP-25 (Figure 4C).
VPA attenuates TDP-25-induced neuronal toxicity
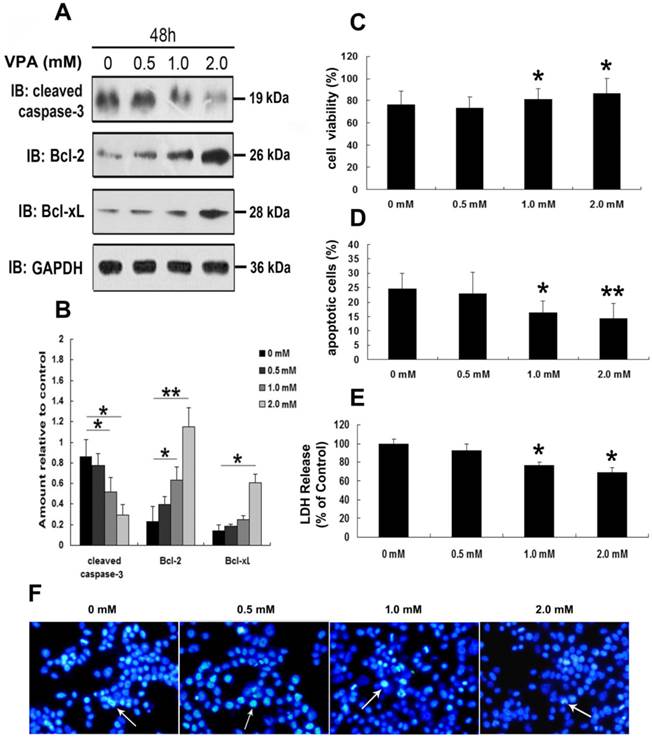
To further address the neurotoxic effects of VPA on TDP-25-overexpressed cells, we first detected the expression of cleaved caspase-3, Bcl-2 and Bcl-xL in cells expressing HA-TDP-25 following treatment of VPA. As shown in Figure 5A, VPA treatment decreased cleaved caspase-3 expression, while increased Bcl-2 and Bcl-xL expression in a dose-dependent manner in cells transfected with HA-TDP-25. Consistent with our Western blot analysis, Hoechst33258 staining and Annexin V/PI flow cytometry assay also revealed that VPA treatment significantly decreased the undergoing apoptosis population in a dose-dependent manner (Figure 5D, F). We next examined cell viability using MTT assay. We observed that exposure to VPA significantly increased the viability of SH-SY5Y cells transfected with HA-TDP-25 in a dose-dependent manner (Figure 5C). Furthermore, we showed that VPA decreased LDH release in a dose-dependent manner in HA-TDP-25-overexpressed cells (Figure 5E). We conclude that VPA protects cells from TDP-25-induced neuronal toxicity via suppressing ER stress and activating autophagy.
VPA attenuates TDP-25-induced neuronal toxicity. (A) Western blot analysis performed using lysates of HA-TDP-25-transfected cells following treatment of VPA for 48 h with different doses. Cell lysates were examined by immunoblotting with the indicated antibodies. (B) Quantification showing significant increase in expression levels of Bcl-2 and Bcl-xL, and decrease of cleaved caspase-3 in a dose-dependent manner. (C) Cell viability measured by MTT assay following treated with VPA. Quantification indicated VPA inhibited TDP-25-induced neuronal death in a dose-dependent manner. Values shown are the mean ± S.E.M. from three experiments. (D) Neuronal apoptosis detected by Annexin V-FITC/PI following cells treated with VPA. Quantification showed that VPA inhibited TDP-25-induced neuronal apoptosis in a dose-dependent manner. Values shown are the mean ± S.E.M. from three experiments. (E) The release of LDH used as an indicator of neuronal toxicity following VPA treatment. Quantification showed that VPA inhibited TDP-25-induced neuronal damage in a dose-dependent manner. Values shown are the mean ± S.E.M. from three experiments. (F) Hochest33258 staining performed to detect apoptosis in HA-TDP-25-transfected cells following VPA treatment. Values shown are the mean ± S.E.M. from three experiments. Level of statistical significance: *p < 0.05, ** p < 0.01.
Discussion
Abnormally cleaved CTFs of TDP-43 accumulate in neurons of affected CNS regions is a common pathological feature of the two neurodegenerative TDP-43 proteinopathies, FTLD-U, and ALS [40]. CTFs contain the prion-related domains, being rich in glutamine and asparagine residues, which results in aggregation of CTFs [40-42]. Our study confirms that TDP-35 and TDP-25 fragments formed cytoplasm aggregates in SH-SY5Y cells. These observations demonstrate that TDP-43 CTFs play an important role in the formation of cytoplasmic inclusions in TDP-43 proteinopathies. However, little is known about whether and how TDP-43 CTFs exert harmful effects on neurons. Our work shows that TDP-43 CTFs induce neuronal death via ER stress-mediated apoptosis, and autophagy is activated for neuronal survival in TDP-35- and TDP-25-overexpressed neuronal cells.
ER stress is predominantly associated with intracellular inclusion bodies in neurodegenerative disease. Accumulating evidence suggests the involvement of ER stress in the pathogenic mechanisms of TDP-43 proteinopathies [6, 36]. Our results demonstrate that TDP-43 CTFs induces ER stress-mediated apoptosis via increasing expression of phosphorylated eIF2α, CHOP and cleavage of caspase-12. Previous studies showed that ER stress could be triggered by overexpression of wild-type TDP-43, and involved in TDP-43 subcellular distribution and TDP-43-positive aggregates formation [6]. The accumulation of misfolded proteins triggers ER stress responses which protect cells against the toxic buildup of such proteins and inhibit general protein translation and promote the degradation of misfolded proteins [43]. However, if the ER stress response is unable to rescue the cells, it will eventually lead to cell death [44]. Autophagy is an intracellular degradation process, and responsible for the clearance of dysfunctional organelles and protein aggregates [45]. Induction of autophagy promotes the clearance of protein aggregates that cause neurodegeneration in cells and animal models [46]. Our results show that overexpression of GFP-TDP-35 or GFP-TDP-25 in SH-SY5Y cells induces macroautophagy. Moreover, inhibition of autophagy by 3-MA enhances TDP-43 CTFs-induced neuronal death. Consistent with previous observations, our findings indicate that activation of autophagy is self-defense mechanism for neuron survival in TDP-35- or TDP-25-overexpressed cells. Thus, autophagy attenuates TDP-43 CTFs-induced neuronal toxicity.
VPA is primarily used to treat epilepsy. However, accumulated evidence indicates that this agent also holds promise for treating neurodegenerative diseases via their diverse mechanisms of action [27]. In ALS mice, treatment with VPA was found to prolong lifespan in a G93A mouse model of ALS [24]. Another study investigated the effect of a chronic VPA treatment on ALS mouse model, it was found that VPA exerted neuroprotective effects in vivo through CREB-Binding protein-dependent mechanisms, but not improved mouse survival [22]. Although results from the preclinical studies raise the possibility that VPA has putative utility for the treatment of ALS, clinical trials have provided inconclusive results. A randomized sequential trial found it to be ineffective of VPA in patients with ALS [32]. However, the latest clinical trial in randomized patients with ALS found that lithium and VPA co-treatment significantly increased survival [29]. These contradictory results may be explained by using different VPA dosing, timing of initiation treatment, and histopathological subtypes. However, studies should first clarify the precise mechanisms involved in the treatment of VPA to ALS. In the present study, we demonstrate that VPA treatment significantly decreases ER stress-associated apoptosis and enhances autophagy in TDP-25-overexpressed cells. As a result, VPA reduces TDP-25-induced neuronal toxicity. In our study, SH-SY5Y cells were exposed to various concentrations form 0 mM to 2.0 mM of VPA for 48 h. Our findings show that VPA at concentrations of 1.0 mM or 2.0 mM acts protective effect on TDP-25-induced neurons damage without cytotoxicity. To our knowledge, this is the first report that delineates the protective role of VPA against TDP-43 CTFs-induced neuronal damage.
Previous studies showed that regulation of ER stress by VPA might prove to be important to the action mechanism of this drug [47]. In addition, VPA was shown to exert its neuroprotective action in a rat model of spinal cord injury via prevention of ER stress signaling mediated by CHOP [48]. A recent study exhibited that VPA protects β-cells from palmitate-induced ER stress via inhibition of ATF4/CHOP pathway [25]. Our present results demonstrate that treatment with VPA results in decrease of CHOP and cleaved caspase-12 in a dose-dependent manner in TDP-25-overexpressed SH-SY5Y cells. Emerging evidence supports the notion that activation of autophagy may be protective in some neurodegenerative diseases by enhancing the removal of toxic protein aggregates [49]. Recent studies revealed that TDP-43 is involved in the abnormal autophagy regulation [5], and stimulation of neuronal autophagy enhances TDP-43 clearance and improves neuronal survival [50]. Accumulated evidence suggests that VPA is involved in regulation of autophagy. In diabetic nephropathy, VPA treatment ameliorates the podocyte and renal injuries via facilitating the autophagy [26]. In malignant gliomas, VPA was also proven to exert cytoprotective functions via potentiating autophagy [51]. Our results demonstrate that autophagy inhibitor 3-MA increases TDP-CTFs-induced neuronal death. Moreover, VPA treatment facilitates TDP-25-induced autophagy. In conclusion, our work provides new preclinical evidence for the therapeutic effect of VPA on ALS. Further research is warranted to verify the neuroprotective effect of VPA on animal models of TDP-43 proteinopathy.
Conclusions
In summary, our results demonstrate that TDP-43 CTFs induced neuronal death via ER stress-mediated apoptosis. Furthermore, VPA attenuates TDP-25-induced neuronal damage via suppressing ER stress-mediated apoptosis and enhancing autophagy. Our work proffers the new molecular evidence for the neuroprotective effects of VPA on TDP-43 proteinopathy in vitro. It further suggests that clinical trials using VPA to treat patients with ALS may be effective according to the pathological characterization of TDP-43 positive neuronal cytoplasmic inclusions. Therefore, these findings indicate that VPA may be a promising therapeutic strategy for treatment of TDP-43 proteinopathies.
Acknowledgements
This work was supported by project grants from the National Natural Science Foundation of China (No. 81471307, 81301086, 81100881, and 81100949) and the Youth Innovation Fund of The First Affiliated Hospital of Zhengzhou University (Xuejing Wang and Xuebing Ding), as well as 5451 Project of Health Department of Henan Province (201201007 and Xuebing Ding). The authors would like to extend their thanks to Dr. Fengfei Wang for her thoughtful reading and inputs.
Authors' contributions
XD, EW, and XW conceived the research design and carried out the experiments, as well as analyzed the data. XW and EW wrote, reviewed and/or revised this manuscript. MM, JT, XC, WZ, SF, SZ, and YZ provided administrative, technical, and material support. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
References
1. Bakkar N, Boehringer A, Bowser R. Use of biomarkers in ALS drug development and clinical trials. Brain Res. 2014
2. Malaspina A, Puentes F, Amor S. Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol. 2014
3. de Carvalho M, Eisen A, Krieger C, Swash M. Motoneuron firing in amyotrophic lateral sclerosis (ALS). Front Hum Neurosci. 2014;8:719
4. Kim C, Lee HC, Sung JJ. Amyotrophic lateral sclerosis - cell based therapy and novel therapeutic development. Exp Neurobiol. 2014;23:207-14
5. Chen S, Zhang X, Song L, Le W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 2012;22:110-6
6. Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA. et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One. 2013;8:e81170
7. Consilvio C, Vincent AM, Feldman EL. Neuroinflammation, COX-2, and ALS--a dual role? Exp Neurobiol. 2004;187:1-10
8. Ruble BK, Richards JL, Cheung-Lau JC, Dmochowski IJ. Mismatch Discrimination and Efficient Photomodulation with Split 10-23 DNAzymes. Inorganica chimica acta. 2012;380:386-91
9. Pollari E, Goldsteins G, Bart G, Koistinaho J, Giniatullin R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:131
10. Sreedharan J, Brown RH Jr. Amyotrophic lateral sclerosis: Problems and prospects. Ann Neurol. 2013;74:309-16
11. Poppe L, Rue L, Robberecht W, Van Den Bosch L. Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Exp Neurobiol. 2014;262(Pt B):138-51
12. Yang C, Tan W, Whittle C, Qiu L, Cao L, Akbarian S. et al. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One. 2010;5:e15878
13. Forman MS, Trojanowski JQ, Lee VM. TDP-43: a novel neurodegenerative proteinopathy. Curr Opin Neurobiol. 2007;17:548-55
14. Suzuki H, Lee K, Matsuoka M. TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J Biol Chem. 2011;286:13171-83
15. Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001-4
16. Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15:772-80
17. Budini M, Buratti E, Stuani C, Guarnaccia C, Romano V, De Conti L. et al. Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J Biol Chem. 2012;287:7512-25
18. Hong K, Li Y, Duan W, Guo Y, Jiang H, Li W. et al. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci Lett. 2012;530:144-9
19. Dayton RD, Gitcho MA, Orchard EA, Wilson JD, Wang DB, Cain CD. et al. Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Mol Ther. 2013;21:1324-34
20. Cincarova L, Zdrahal Z, Fajkus J. New perspectives of valproic acid in clinical practice. Expert Opin Investig Drugs. 2013;22:1535-47
21. Wang W, Liao XL, Chen JH, Li DD, Lin CL, Yan YX. et al. Sodium valproate induces mitochondria-dependent apoptosis in human hepatoblastoma cells. Chin Med J (Engl). 2011;124:2167-72
22. Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L. et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535-45
23. Tremolizzo L, Rodriguez-Menendez V, Sala G, Di Francesco JC, Ferrarese C. Valproate and HDAC inhibition: a new epigenetic strategy to mitigate phenotypic severity in ALS? Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:185-6
24. Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T. et al. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur J Neurosci. 2004;20:3179-83
25. Huang S, Zhu M, Wu W, Rashid A, Liang Y, Hou L. et al. Valproate pretreatment protects pancreatic beta-cells from palmitate-induced ER stress and apoptosis by inhibiting glycogen synthase kinase-3beta. J Biomed Sci. 2014;21:38
26. Khan S, Jena G, Tikoo K, Kumar V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of NF-kappaB/iNOS signaling in diabetic rat. Biochimie. 2015
27. Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65:105-42
28. Tremolizzo L, Sala G, Conti E, Rodriguez-Menendez V, Fogli A, Michelucci A. et al. Valproate Treatment in an ALS Patient Carrying a c.194G>A Spastin Mutation and SMN2 Homozygous Deletion. Case Rep Neurol Med. 2014;2014:216094
29. Boll MC, Bayliss L, Vargas-Canas S, Burgos J, Montes S, Penaloza-Solano G. et al. Clinical and biological changes under treatment with lithium carbonate and valproic acid in sporadic amyotrophic lateral sclerosis. J Neurol Sci. 2014;340:103-8
30. Crochemore C, Virgili M, Bonamassa B, Canistro D, Pena-Altamira E, Paolini M. et al. Long-term dietary administration of valproic acid does not affect, while retinoic acid decreases, the lifespan of G93A mice, a model for amyotrophic lateral sclerosis. Muscle Nerve. 2009;39:548-52
31. Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155:567-72
32. Piepers S, Veldink JH, de Jong SW, van der Tweel I, van der Pol WL, Uijtendaal EV. et al. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann Neurol. 2009;66:227-34
33. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55-63
34. Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469:112-6
35. Lautenschlaeger J, Prell T, Grosskreutz J. Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:166-77
36. Mutihac R, Alegre-Abarrategui J, Gordon D, Farrimond L, Yamasaki-Mann M, Talbot K. et al. TARDBP pathogenic mutations increase cytoplasmic translocation of TDP-43 and cause reduction of endoplasmic reticulum Ca signaling in motor neurons. Neurobiol Dis. 2014;75C:64-77
37. Song CY, Guo JF, Liu Y, Tang BS. Autophagy and Its Comprehensive Impact on ALS. Int J Neurosci. 2012;122:695-703
38. Liu Y, Duan W, Guo Y, Li Z, Han H, Zhang S. et al. A new cellular model of pathological TDP-43: The neurotoxicity of stably expressed CTF25 of TDP-43 depends on the proteasome. Neuroscience. 2014;281C:88-98
39. Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ. et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109:15024-9
40. Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y. et al. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J Biol Chem. 2009;284:8516-24
41. Ugras SE, Shorter J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis and Neurodegeneration. Neurol Res Int. 2012;2012:432780
42. King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61-80
43. Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653-62
44. Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M. et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111-23
45. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885-9
46. Garcia-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010;21:691-8
47. Bown CD, Wang JF, Chen B, Young LT. Regulation of ER stress proteins by valproate: therapeutic implications. Bipolar Disord. 2002;4:145-51
48. Penas C, Verdu E, Asensio-Pinilla E, Guzman-Lenis MS, Herrando-Grabulosa M, Navarro X. et al. Valproate reduces CHOP levels and preserves oligodendrocytes and axons after spinal cord injury. Neuroscience. 2011;178:33-44
49. Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases - recent advances and future perspectives. J Neurochem. 2011;118:317-25
50. Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM. et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677-85
51. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. 2010;12:328-40
Author contact
![]() Corresponding author: Xuebing Ding: E-mail: ballet8114com, Tel: +0017017935935. Erxi Wu: E-mail: Erxi.wuedu, Tel: +0017012317250
Corresponding author: Xuebing Ding: E-mail: ballet8114com, Tel: +0017017935935. Erxi Wu: E-mail: Erxi.wuedu, Tel: +0017012317250