Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(7):845-859. doi:10.7150/ijbs.11921 This issue Cite
Research Paper
bFGF Promotes the Migration of Human Dermal Fibroblasts under Diabetic Conditions through Reactive Oxygen Species Production via the PI3K/Akt-Rac1- JNK Pathways
Hongxue Shi1, Yi Cheng1, Jingjing Ye1, Pingtao Cai1, Jinjing Zhang1, Rui Li 1, Ying Yang1, Zhouguang Wang1, Hongyu Zhang1, Cai Lin2, Xianghong Lu3, Liping Jiang1, Aiping Hu1, Xinbo Zhu1, Qiqiang Zeng2, Xiaobing Fu4, Xiaokun Li1 ![]() , Jian Xiao1
, Jian Xiao1 ![]()
1. School of Pharmaceutical Sciences, Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, 325035 China
2. The First Affiliate Hospital, Wenzhou Medical University, Wenzhou, 325035, China
3. Translation Medicine Research Center, Lishui People's Hospital, Wenzhou Medical University, Lishui, 323000, China
4. Wound Healing and Cell Biology Laboratory, Institute of Basic Medical Science, Chinese PLA General Hospital, Beijing, PR China
Received 2015-2-17; Accepted 2015-4-13; Published 2015-6-1
Abstract

Fibroblasts play a pivotal role in the process of cutaneous wound repair, whereas their migratory ability under diabetic conditions is markedly reduced. In this study, we investigated the effect of basic fibroblast growth factor (bFGF) on human dermal fibroblast migration in a high-glucose environment. bFGF significantly increased dermal fibroblast migration by increasing the percentage of fibroblasts with a high polarity index and reorganizing F-actin. A significant increase in intracellular reactive oxygen species (ROS) was observed in dermal fibroblasts under diabetic conditions following bFGF treatment. The blockage of bFGF-induced ROS production by either the ROS scavenger N-acetyl-L-cysteine (NAC) or the NADPH oxidase inhibitor diphenylene iodonium chloride (DPI) almost completely neutralized the increased migration rate of dermal fibroblasts promoted by bFGF. Akt, Rac1 and JNK were rapidly activated by bFGF in dermal fibroblasts, and bFGF-induced ROS production and promoted dermal fibroblast migration were significantly attenuated when suppressed respectively. In addition, bFGF-induced increase in ROS production was indispensable for the activation of focal adhesion kinase (FAK) and paxillin. Therefore, our data suggested that bFGF promotes the migration of human dermal fibroblasts under diabetic conditions through increased ROS production via the PI3K/Akt-Rac1-JNK pathways.
Keywords: bFGF, human dermal fibroblast
Introduction
Impaired wound healing is an emerging global public concern with considerable social, health, and economic consequences. One of the most common diseases associated with impaired tissue repair is diabetes mellitus, and foot ulcerations are the most frequent cause of hospitalization in patients with diabetes [1]. The healing of cutaneous wounds is a complex and highly orchestrated process that includes four main integrated and overlapping phases: hemostasis, inflammation, proliferation and tissue remodeling [2]. However, this orderly progression of the healing process is impaired in diabetic patients [3].
Cutaneous wounds require a well-orchestrated interaction of cell migration and proliferation from numerous different tissues and cell lineages. Among these cell types, fibroblasts play a pivotal role in all four phases of wound healing. After wounding, fibroblasts are attracted from the edge of the wound or from the bone marrow [4]. At the inflammation stage, fibroblasts produce a variety of chemokines [5]. At the stage of new tissue formation, fibroblasts are stimulated by macrophages, and some differentiate into myofibroblasts. Fibroblasts interact with myofibroblasts to produce extracellular matrix, mainly in the form of collagen [5]. At the tissue remodeling stage, most of the myofibroblasts, macrophages and endothelial cells undergo apoptosis or exit from the wound, leaving a mass that contains few cells and consists mostly of collagen and other extracellular matrix proteins [6]. However, diabetes has an adverse effect on the proliferation of fibroblasts [7, 8]. Accumulating evidence also suggests that the fibroblasts from diabetic mice and rats exhibit a marked reduction in migratory ability compared with those from normal control mice [9, 10].
Growth factors have been shown to play multiple and critical roles in the process of wound healing, and their decreased expression in diabetes mellitus may disrupt the normal healing process [11]. Among growth factors, basic fibroblast growth factor (bFGF) is a potent mitogen that stimulates the migration, proliferation, and differentiation of cells of mesenchymal and neuroectodermal origin, such as keratinocytes, fibroblasts, melanocytes and endothelial cells [12]. Animal studies have shown that recombinant human bFGF can promote wound healing in genetically diabetic mice and streptozotocin-induced diabetic rats [13, 14]. It has been suggested that recombinant bFGF promotes granulation and epithelialization and shortens the time required for healing in patients with diabetic ulcers as well as in patients with other types of skin ulcers [15, 16]. However, the mechanisms underlying the therapeutic effect of bFGF on skin wounds under diabetic conditions are not completely understood.
The migration of various cells mediated by bFGF and the underlying signal transduction pathways have been studied extensively [17]. However, the effect of bFGF on human dermal fibroblast migration under diabetic conditions remains unknown. In this study, we investigated the effect of bFGF on the migration of human dermal fibroblasts under diabetic conditions and further examined its molecular mechanism.
Materials and Methods
Cell culture
Human foreskins were obtained from the First Affiliated Hospital of Wenzhou Medical University with approval by the Ethic Committee of First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China. All of the experiments were performed in accordance with the relevant approved guidelines and regulations. The isolation and primary culture of human dermal fibroblasts were performed as described previously [16]. Briefly, the foreskins were washed three times in phosphate-buffered saline (PBS) solution containing 1% penicillin and streptomycin sulfate. Subsequently, the excised tissues were digested with 0.5% dispase II at 4°C overnight. The epidermis and subcutaneous tissue were then removed from the excised tissues, cut into pieces with a size of approximately 1×1×0.5 cm and placed into T25 tissue culture flasks as explants. Dulbecco's modified eagle medium (DMEM) containing 5.5 mM D-glucose, 20% fetal bovine serum (FBS), 1% penicillin, streptomycin sulfate and 2 mM L-glutamine was used as the growth medium. The medium was replaced every three days. The primary fibroblasts were grown at 37°C in an atmosphere of 5% CO2 and were passaged every other day by trypsinization. Cells that had undergone three to six passages were used for all of our experiments.
Cell proliferation assay
Human dermal fibroblasts were plated in 35-mm tissue culture dishes at a density of 2.4×104 cells/ml and cultured overnight. The culture medium was modified according to the experimental groups to include the following constituents: 5.5 mM D-glucose for the normal-glucose group, 30 mM D-glucose for the high glucose group and 5.5 mM D-glucose and 24.5 mM mannitol for the osmotic control group. Twenty-four hours later, the cells were treated with 100 ng/ml bFGF for 24 h in the presence or absence of 5 μg/ml mitomycin-C. The cells were then washed three times with PBS and harvested using 0.1% trypsin. The number of cells was counted in a microscopic counting chamber.
Wound-healing assay
Human dermal fibroblasts were plated overnight in six-well tissue culture dishes under standard culture conditions. The culture medium was modified as described previously for a period of 24 h. After another 24 h of serum starvation, the cells were wounded with a linear scratch using a sterile pipette tip and treated with 100 ng/ml bFGF in the presence of 5 μg/ml mitomycin-C. Twelve and 24 hours after wounding, the cells were washed with PBS to remove any floating cells and cellular debris. Images of the wound closure or cell migration were immediately photographed using an inverted microscope equipped with a digital camera. To observe the role of PI3K/Akt, JNK, ROS, and NADPH oxidases in the migration of human dermal fibroblasts, the cells were pretreated with 10 μM LY294002, 10 μM SP600125, 10 mM NAC or 10 μM diphenyleneiodonium chloride (DPI), respectively, for 1 h before wounding. The distances between the front edges of the selected cells and the wound edge 12 or 24 h after wounding were measured using the ImageJ software. The migration rate was expressed as the migration distance/time (μm/h). All of the wound-healing assays were performed in the presence of 5 μg/ml mitomycin-C to inhibit cell proliferation.
Analysis of cell polarity index
To migrate, cells must possess a defined front and rear to move in one direction. Without this front-rear polarity, the cells will be unable to coordinate a directed migration. To assess the effect of bFGF on cell polarity, the polarity index was calculated as the length of the major migration axis (parallel to the direction of movement) divided by the length of the perpendicular axis that intersects the center of the cell nucleus after treatment with 100 ng/ml bFGF for 1 h in the presence of 5 μg/ml mitomycin-C [10, 18].
Rac 1 pull-down assay
The Rac1 pull-down assay was performed using the manufacturer's protocol. In brief, the cells were scraped into ice-cold lysis buffer and centrifuged for 10 min at 12,000 rpm. The cleared lysates (containing at least 500 μg) were incubated with 20 μg of Pak1-PBD agarose (Thermo scientific, Hangzhou, China) for 1 h at 4°C with gentle rocking. The beads were washed three times with wash buffer, heated for 5 min at 100°C in reducing SDS-PAGE sample buffer, and then centrifuged for 2 min at 6,000 rpm. Rac1-GTP expression was analyzed by western blot using anti-Rac1 antibody.
siRNA knockdown of Rac1
Scrambled control siRNA (sc-37007) and Rac1 siRNA (sc-36351) were purchased from Santa Cruz. Primary human fibroblasts were transfected with Rac1 siRNA (10 nM) using Lipofectamine 2000 reagent (Invitrogen, Shanghai, China) with the manufacturer's protocol. Twenty-four hours after transfection, the cells were subjected to bFGF treatment. Specific silencing was confirmed by western blot.
Measurement of intracellular ROS production
The membrane-permeable indicator H2DCF-DA was used to detect intracellular ROS production. The cells were cultured in DMEM containing 5.5 mM glucose overnight. The cells were then cultured in DMEM containing 30 mM glucose for 24 h. After another 24 h of serum starvation, the cells were treated with 100 ng/ml bFGF for 24 h in the presence or absence of DPI or NAC. The cells were loaded with 10 μmol/l H2DCF-DA in serum-free DMEM at 37°C for 30 min and washed three times with PBS. Fluorescent images were obtained using an upright fluorescence microscope.
Western blot analysis
The cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% Na deoxycholate, 1% NP-40, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, and complete protease inhibitor cocktail), incubated for 10 min on ice and centrifuged at 12,000 rpm for 10 min at 4°C. The protein samples were denatured by incubating at 100°C for 10 min in 5× loading buffer, separated on 10.6% polyacrylamide gels, and transferred to a polyvinylidene difluoride membrane. The membranes were incubated in TBS containing 5% nonfat milk and 0.05% Tween-20 for 1 h and blotted with primary antibodies at 4°C overnight (p-Akt/Akt, Santa cruz, Shanghai, China. 1:500 dilution; p-JNK/JNK, p-FAK/FAK and p-Paxillin/Paxillin, Cell signaling technology, Shanghai, China. 1:1000 dilution; NOX4, Abcam, Shanghai, China. 1:1000 dilution). The membranes were washed with TBST for 15 min (three times for 5 min each) the next day. Subsequently, the membranes were incubated with second antibody (Santa cruz, Shanghai, China. 1:3000 dilution) for 1 h at room temperature and washed with TBST for 21 min (three times for 7 min each). The membranes were then detected using ECL ECL (Bio-Rad, Hangzhou, China). The western blot results were further analyzed using the Quantity One software 4.1.1 (Bio-Rad, Hangzhou, China).
Immunofluorescence
The cells were plated in six-well tissue culture dishes. The cells were maintained in a high-glucose medium containing 10% FBS for 24 h and cultured in serum-free DMEM for 24 h. The cells were then treated with or without 100 ng/ml bFGF for 6 and 12 h after wounding in the presence of 5 μg/ml mitomycin-C. The cells were fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.3% Triton X-100 in PBS for 15 min and incubated with 5% bovine serum albumin (BSA) in PBS for 45 min to block nonspecific antibody binding at room temperature. The cells were incubated with fluorescein isothiocyanate-labeled phalloidin staining solution in PBS containing 1% BSA for 35 min and washed with PBS for 15 min. The nuclei were stained with DAPI. Fluorescence images were obtained using an upright fluorescence microscope. All of the illustrations were assembled and processed digitally.
Statistical analysis
Data are expressed as the mean ± SEM. Statistical significance was determined using Student's t-test for two experimental groups. For more than two groups, the statistical evaluation of data was performed using one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test. For all tests, P < 0.05 was considered significant.
Results
bFGF promoted the migration of human dermal fibroblasts under diabetic conditions
High glucose mediated oxidative stress and impaired cell migration. To investigate the effect of high glucose on human dermal fibroblast migration, a wound-healing assay was performed. The migratory ability of dermal fibroblasts was markedly impaired in the presence of a high concentration of glucose for 12 and 24 h, and the migration rates of dermal fibroblasts incubated with mannitol showed no significant decrease (Supplementary Material: Figs. S1A and S1B). These results suggest that the high-glucose-impaired migration of dermal fibroblasts does not result from changes in osmotic pressure.
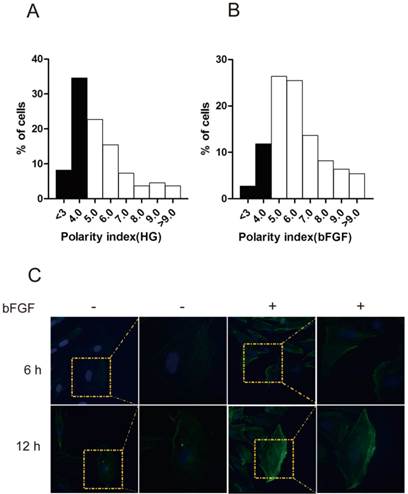
bFGF is a well-known potent mitogen for most cell types. In this study, we focused on investigating the effect of bFGF on the migration of human dermal fibroblasts under high-glucose conditions. Therefore, to eliminate the impact of cell proliferation on the process of evaluating bFGF-induced dermal fibroblast migration, mitomycin C at a final concentration of 5 μg/ml was selected and used in all of our subsequent experimental procedures. Under such concentrations, mitomycin C completely blocked the proliferation of dermal fibroblasts while exhibiting no visible damage on cell viability (Supplementary Material: Fig. S1C). To migrate, a cell must normally establish morphological polarity and continuously protrude a single lamellipodium polarized in the direction of migration. To explore the impact of bFGF on cell polarity, the polarity index of dermal fibroblasts with or without bFGF treatment was determined. The results indicate that bFGF treatment significantly increased the cell number with elongated morphology. The fraction of those dermal fibroblasts under high-glucose conditions with a polarity index greater than 5 was 57.28%. When treated with bFGF for only 1 h, the percentage of cells with a polarity index greater than 5 sharply increased to 85.44% (Figs. 1A and 1B). To migrate, cells also require reorganization of the cytoskeleton, such as F-actin. bFGF promotes the remodeling of F-actin in dermal fibroblasts under high-glucose conditions and thus further boosts the formation of lamellipodia (Fig. 1C). These results suggest that the migratory ability of dermal fibroblasts is significantly impaired under high-glucose conditions and that bFGF promotes the migration of dermal fibroblasts impaired by high glucose by increasing the number of cells with a high polarity index and boosts cytoskeletal rearrangement, including F-actin.
bFGF altered human dermal fibroblast polarity impaired by high glucose. (A, B) The front-rear polarity index of dermal fibroblasts treated without or with 100 ng/ml bFGF under high-glucose conditions was calculated in single cell in scratch wound edge. (C) F-actin was labeled by FITC-conjugated phalloidin 6 h or 12 h after wounding with or without bFGF treatment.
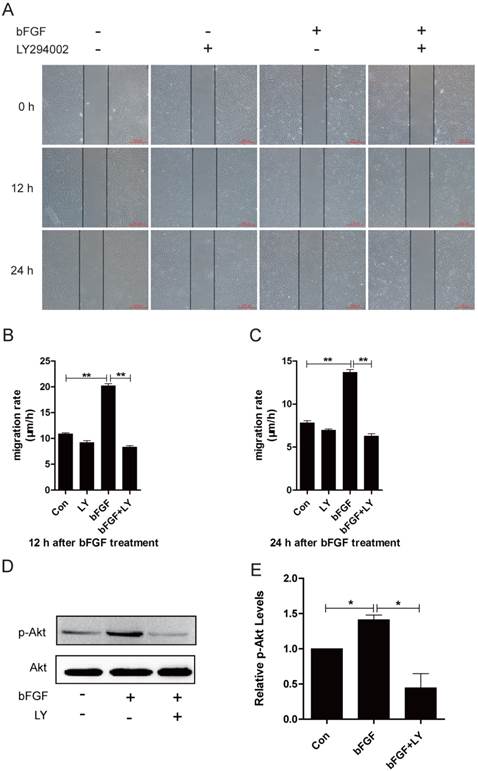
PI3K/Akt is involved in bFGF-promoted dermal fibroblast migration under high-glucose conditions
It has been widely reported that phosphatidylinositol 3-kinase (PI3K) is associated with cell polarization and migration in certain cell types [19]. However, it remains unclear whether PI3K is involved in bFGF-induced human dermal fibroblast migration under high-glucose conditions. The addition of bFGF at a final concentration of 100 ng/ml sharply increased the activity of Akt after 15 min of treatment. The level of phosphorylated Akt enhanced by bFGF in cultured human dermal fibroblasts was completely inhibited by LY294002, which is an Akt-specific inhibitor (Figs. 2D and 2E). Through a wound-healing assay, we found that bFGF treatment notably promoted the migration of dermal fibroblasts under high-glucose conditions with migratory rates of 20.200 ± 0.428 and 13.680 ± 0.333 µm/h 12 h or 24 h after wounding compared with 10.901 ± 0.200 and 7.812 ± 0.253 µm/h for the untreated control, respectively. However, when pretreated with LY294002, bFGF-induced fibroblast migration was completely blocked. The migratory rates were determined to be 8.343 ± 0.282 and 6.294 ± 0.252 µm/h at 12 h or 24 h after wounding, respectively (Figs. 2A-2C). These results demonstrate that PI3K is involved in bFGF-promoted dermal fibroblast migration under high-glucose conditions.
PI3K/Akt was involved in bFGF-promoted dermal fibroblast migration under high-glucose conditions. (A) PI3K inhibitor LY294002 (10 µM) blocked bFGF-induced dermal fibroblast migration at 12 or 24 h after wounding as detected using a wound-healing assay. (B, C) The migration rate of dermal fibroblasts in the presence of LY294002 was expressed as the migrating distance per hour and analyzed. The data are presented as the means ± S.E.M. from six independent experiments. (D, E) bFGF enhanced the activity of Akt but was markedly inhibited by LY294002, as detected by western blot. *P < 0.05 and **P < 0.01 compared with the indicated control group.
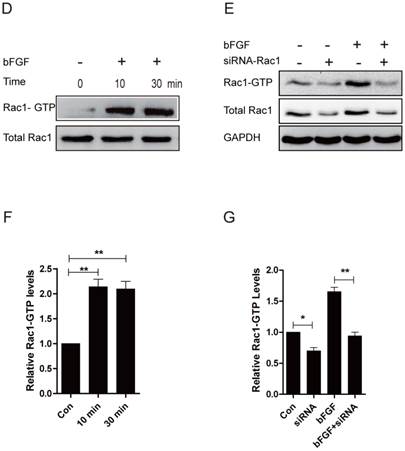
Rac1 is involved in bFGF-promoted dermal fibroblast migration under high-glucose conditions
Rac1 is a member of the small GTPases of the Rho family and regulates cell cytoskeletal dynamics and cell migration [20]. To investigate the involvement of Rac1 in bFGF-induced fibroblast migration, we measured the activities of Rac1 in fibroblasts after treatment with bFGF. Rac1 activity was increased two-fold 10 min after bFGF stimulation and remained high for 30 min (Figs. 3D and 3F). The activity of Rac1 enhanced by bFGF in cultured human dermal fibroblasts was partially inhibited by Rac1 interference (Figs. 3E and 3G). The transfection of Rac1 siRNA led to a significant reduction in the Rac1 protein levels and Rac1-GTP levels in response to bFGF stimulation. The inhibition of Rac1 activity also significantly affected bFGF-induced dermal fibroblast migration under high-glucose conditions in a wound-healing assay, with the migratory rate decreasing to 8.995 ± 0.742 and 6.733 ± 0.485 µm/h compared with 16.741 ± 1.259 and 14.660 ± 0.513 µm/h for the non-treated control at 12 h or 24 h after wounding (Figs. 3A-3C).
Rac1 was involved in bFGF-promoted dermal fibroblast migration under high glucose conditions. (A) Rac1-siRNA (10 nM) blocked bFGF-induced dermal fibroblast migration at 12 or 24 h after wounding, as detected through a wound-healing assay. (B, C) The migration rate of dermal fibroblast in the presence of Rac1 siRNA is expressed as the migrating distance per hour and analyzed. The data are presented as the means ± S.E.M. from six independent experiments. (D, F) bFGF enhanced the activity of Rac1, as detected by western blot. (E, G) Rac1-siRNA reduced the total Rac1 protein and the activity of Rac1 after bFGF treatment. *P < 0.05 and **P < 0.01 compared with the indicated group.

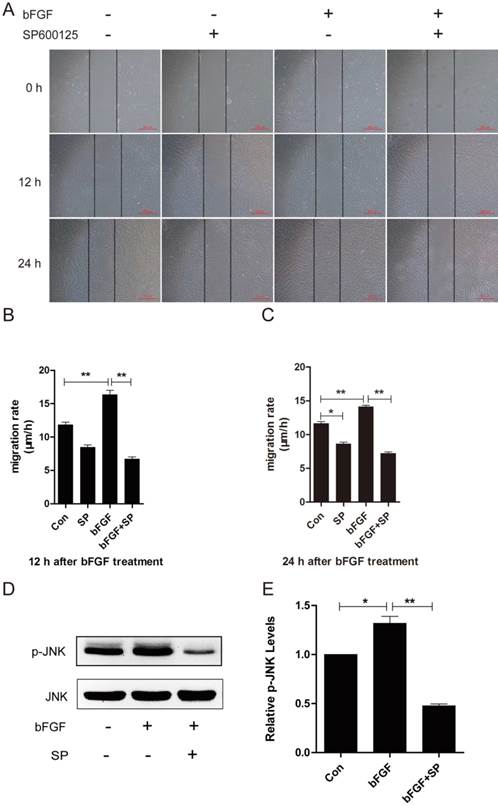
JNK is involved in bFGF-promoted dermal fibroblast migration under high-glucose conditions
JNK was initially identified and purified as a p54 microtubule-associated protein kinase and is activated by a wide range of stresses. JNK has also been reported to be involved in a wide variety of cellular processes, such as cell proliferation, migration and cytoskeleton reorganization [21]. To explore whether JNK also participates in the process of dermal fibroblast migration promoted by bFGF under high-glucose conditions, SP600125, a specific JNK inhibitor, was added into the culture medium for the subsequent detection of phosphorylated JNK levels and the migratory rate. bFGF treatment increased the activity of JNK, and its activation was completely blocked by SP600125 (Figs. 4D and 4E). The inhibition of phosphorylated JNK activity also significantly affected bFGF-induced dermal fibroblast migration under high-glucose conditions in a wound-healing assay, with the migratory rate decreasing to 6.679 ± 0.319 and 7.197 ± 0.214 µm/h compared with 16.310 ± 0.669 and 14.100 ± 0.228 µm/h for the non-treated control at 12 h or 24 h after wounding, respectively (Figs. 4A-4C).
JNK mediated bFGF-induced dermal fibroblast migration under diabetic conditions. (A) JNK inhibitor SP600125 (10 µM) suppressed bFGF-stimulated dermal fibroblast migration at 12 or 24 h after wounding, as detected through a wound-healing assay. (B, C) The migration rate of dermal fibroblast in the presence of SP600125 is expressed as the migrating distance per hour and analyzed. The data are represented as the means ± S.E.M. from six independent experiments. (D, E) bFGF increased the activity of JNK but was markedly inhibited by SP600125, as detected by western blot. *P < 0.05 and **P < 0.01 compared with the indicated control group.
PI3k/Akt contributed to Rac1 activation, and JNK was downstream of Rac1 in bFGF signaling
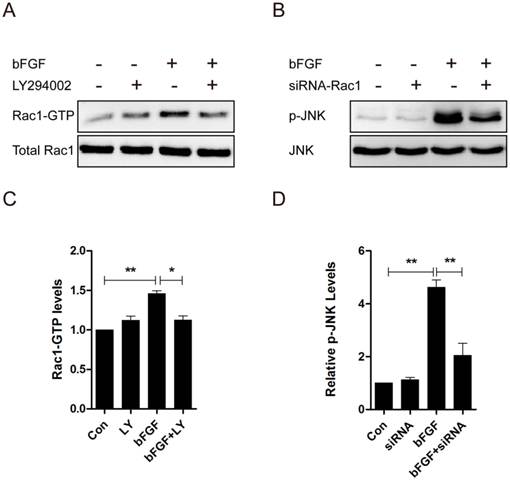
Rac1 acts as a downstream effector of PI3-kinase in several growth factor-stimulated pathways, and the activity of JNK is regulated by the small GTPases [22]. Consequently, we measured the activation of Rac1 treated with LY294002 and the activation of JNK through treatment with Rac1 siRNA in dermal fibroblasts after bFGF stimulation to examine whether PI3-kinase is upstream of Rac1, and JNK was found to be downstream of Rac1 in bFGF-induced dermal fibroblast migration. The level of Rac1-GTP showed a 30% reduction in the presence of LY294002 (Figs. 5A and 5C), and the activity level of JNK showed a 50% reduction in the presence of Rac1 siRNA after bFGF stimulation (Figs. 5B and 5D). These data indicate that PI3K/Akt contributes to bFGF-induced Rac1 activation and that JNK is downstream of Rac1 in bFGF signaling.
PI3k/Akt contributed to Rac1 activation, and JNK was downstream of Rac1 in bFGF signaling. (A, C) The activities of Rac1 after treatment with or without 10 µM LY294002 were analyzed 15 min after stimulation with 100 ng/ml bFGF. (B, D) The activities of JNK after treatment with or without 10 nM Rac1-siRNA were analyzed 15 min after stimulation with 100 ng/ml bFGF.
Increased ROS production was required for bFGF-promoted dermal fibroblast migration under diabetic conditions
ROS is linked to cell migration in certain cell types. To identify whether ROS production was also involved in the bFGF-induced migration of dermal fibroblasts under high-glucose conditions, the intracellular ROS generation following bFGF treatment was detected using the fluorescent probe H2DCFDA. bFGF can significantly induce the generation of intracellular ROS after 24 h of incubation. Treatment with 10 μM N-acetyl-l-cysteine (NAC), a potent ROS scavenger, strongly suppressed the increased ROS levels induced by bFGF in dermal fibroblasts (Fig. 6A). In the wound-healing assay, the inhibition of bFGF-induced ROS generation by NAC apparently attenuated the migration of dermal fibroblasts promoted by bFGF with their migratory rate decreasing to 6.952 ± 0.6419 µm/h compared with a migratory rate of 11.27 ± 0.6327 µm/h in the control group at 24 h after wounding (Figs. 6B and 6D).
Increased reactive oxygen species (ROS) production was required for bFGF-promoted dermal fibroblast migration under high-glucose conditions. (A) bFGF induced the generation of intracellular ROS in dermal fibroblasts but was suppressed by the ROS scavenger N-acetyl-l-cysteine (NAC) or the NADPH oxidase inhibitor DPI, as detected using the fluorescent probe H2DCFDA. (B, C, D) The suppression of bFGF-induced intracellular ROS production by NAC or DPI blocked bFGF-promoted dermal fibroblast migration. *P < 0.05 and **P < 0.01 compared with the indicated control group. (E, F) The NOX4 mRNA and protein levels in dermal fibroblasts after treatment with bFGF were determined by RT-PCR and western blot. *P < 0.05 compared with the indicated control group.
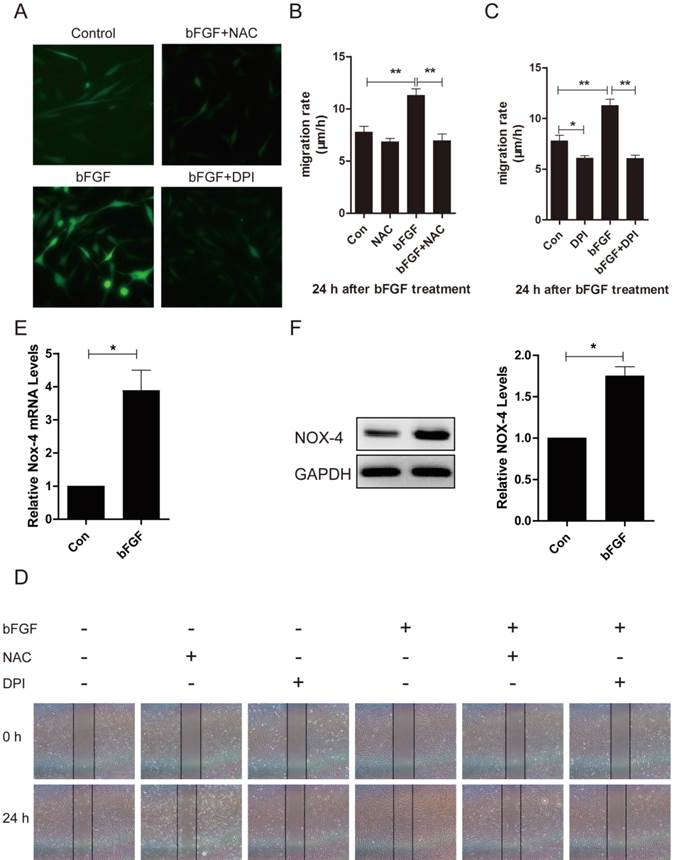
NADPH oxidase (NOX) is considered a major source of ROS in several physiological and pathological processes. Thus, we determined whether bFGF-upregulated ROS production was dependent on NADPH oxidase [23]. Pretreatment with DPI, a potent inhibitor of NADPH oxidase, completely blocked bFGF-induced ROS production (Fig. 6A). DPI also significantly suppressed bFGF-boosted dermal fibroblast migration, with the migratory rate decreasing to 6.069 ± 0.3205 µm/h at 24 h after wounding in the wound-healing assay (Figs. 6C and 6D). In addition, the NOX4 mRNA and protein levels were significantly up-regulated after bFGF treatment compared with the control group (Figs. 6E and 6F). These results suggest that bFGF induces ROS production in a NADPH oxidase-depending manner to promote the migration of dermal fibroblast under high-glucose conditions.
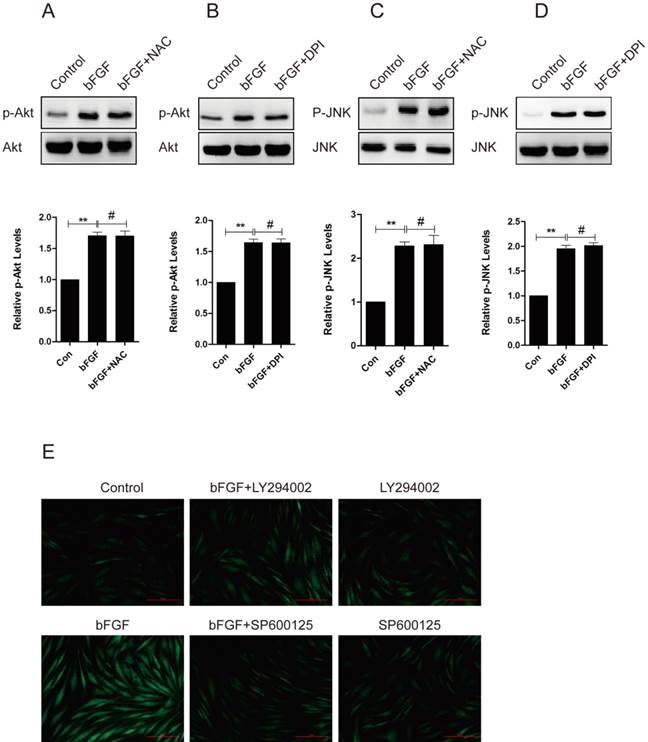
bFGF induced intracellular ROS production via the PI3K/Akt and JNK pathways
To evaluate whether ROS is involved in the bFGF-induced migration of human dermal fibroblasts, we investigated the potential interaction between ROS production and the phosphorylation of PI3K/Akt and JNK. The pretreatment of dermal fibroblasts with NAC or DPI did not block the phosphorylation of either Akt or JNK induced by bFGF (Figs. 7A-7D), suggesting that NADPH oxidase-derived ROS are not necessary for the bFGF-induced activation of both Akt and JNK. However, the addition of LY294002 or SP600125 sharply suppressed bFGF-induced ROS production (Fig. 7E). These results indicate that bFGF induces the generation of ROS via the Akt and JNK pathways to promote the migration of human dermal fibroblasts under high-glucose conditions.
bFGF induced intracellular ROS production via the PI3K/Akt and JNK pathways. (A-D) The ROS scavenger N-acetyl-l-cysteine (NAC) or NADPH oxidase inhibitor DPI did not block the phosphorylation of either Akt or JNK induced by bFGF, as detected by western blot. The optical density of p-Akt or p-JNK was normalized to Akt or JNK using the Quantity One software. The results are presented as fold changes compared with the control. (E) The PI3K inhibitor LY294002 or JNK inhibitor SP600125 suppressed bFGF-increased reactive oxygen species production, as detected using the fluorescent probe H2DCFDA. *P < 0.05 and **P < 0.01 compared with the indicated control group.
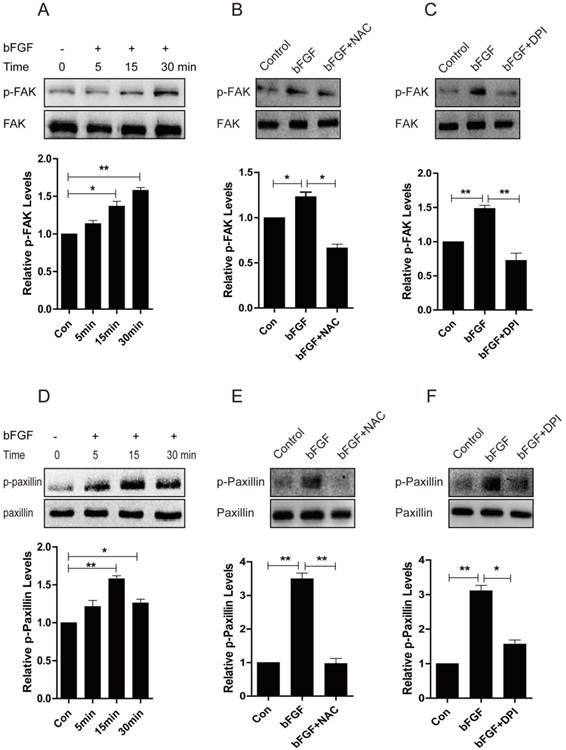
Increased ROS production is indispensible for the activation of FAK and paxillin induced by bFGF
Focal adhesion kinase (FAK) has been widely reported to be involved in the control of several biological processes, including cell proliferation and migration. Paxillin is phosphorylated by FAK and thus provides a docking site for the recruitment of other signaling molecules to focal adhesions to promote cell migration [23, 24]. Therefore, we further investigated whether these molecules mediate the bFGF-induced migration of dermal fibroblasts in the presence of a high concentration of glucose. The level of phosphorylated FAK was significantly up-regulated after 15 min of treatment with bFGF compared with the level of total FAK (Fig. 8A). Nevertheless, both the ROS scavenger NAC and the NADPH oxidase inhibitor DPI nearly completely blocked the phosphorylation of FAK induced by bFGF, but these had no effect on the level of total FAK (Figs. 8B and 8C). Paxillin began to be phosphorylated after incubation with bFGF for 5 min and reached a maximum level 15 min after bFGF addition (Fig. 8D). Similarly, the phosphorylation of paxillin following treatment with bFGF was also significantly inhibited by NAC and DPI, suggesting that increased ROS production by bFGF was indispensable for the activation of FAK and paxillin (Figs. 8E and 8F). These data suggested that the activation of FAK and paxillin is involved in the process of bFGF-induced fibroblast migration under high-glucose conditions. Furthermore, bFGF-induced ROS production is required for the activation of FAK and paxillin to promote the migration of dermal fibroblasts under high-glucose conditions.
Increased ROS production was necessary for the activation of FAK and paxillin induced by bFGF. (A, D) The activity of focal adhesion kinase (FAK) or paxillin was significantly up-regulated following bFGF treatment at the indicated time point, as detected by western blot. The optical density of p-FAK or p-paxillin was measured using the Quantity One software and normalized to that of FAK or paxillin. The results are presented as fold changes compared with the non-treated control. (B, E) The ROS scavenger N-acetyl-l-cysteine (NAC) blocked the activity of FAK or paxillin induced by bFGF, as detected by western blot. (C, F) The NADPH oxidase inhibitor DPI suppressed the phosphorylation of FAK or paxillin stimulated by bFGF. *P < 0.05 and **P < 0.01 compared with the indicated control group.
Discussion
The proliferation and migration of dermal fibroblasts are essential for cutaneous wound repair because dermal fibroblasts migrate to damaged sites, repopulate the wound, and remodel fibrin and collagen deposits [25]. Cutaneous wound healing is severely impaired in diabetic patients, and high glucose is thought to be main reason for delaying wound healing in diabetes, we investigated the effect of a high concentration of glucose on the migration of human dermal fibroblasts. In this study, we showed that the migratory ability of dermal fibroblasts from human dermal skin was markedly impaired in a high-glucose environment and that bFGF activated the PI3K/Akt-Rac1-JNK signal pathway and significantly promoted the migration of human dermal fibroblasts by increasing the number of cells with a high polarity index and boosting cytoskeletal rearrangement under diabetic conditions. In addition, we demonstrated that bFGF induces dermal fibroblast migration by upregulating the NOX4 level to mediate ROS production in the presence of a high concentration of glucose.
Akt is a major transducer of the phosphoinositide 3-kinase pathway and plays a crucial role in the regulation of cellular processes, including growth, metabolism, survival, proliferation and migration [19]. The activation of the PI3K/Akt pathway plays a central role in establishing cell polarity and migration speed and is therefore required for the migration of various cell types, including fibroblasts [26]. Rac1 is required at the front of the cell to regulate actin polymerization and membrane protrusion [20], and c-Jun N-terminal kinase (JNK) is involved in the regulation of inflammation, differentiation and apoptosis [21]. Accumulating evidence also suggests that the JNK pathway is important for regulating cell migration [27]. bFGF has been shown to stimulate the directed migration of periodontal ligament cells via PI3K/Akt [28]. Epidermal growth factor stimulates Rac1 activation through Src and PI3K to promote colonic epithelial cell migration [29] and induces the ERK/Rac1 signaling pathway to promote cell migration in human hepatoma HepG2 cells [30]. In our study, the cultured human dermal fibroblasts showed significant increases in Akt, Rac1 and JNK activity following bFGF treatment. The inhibition of their activities by a chemical inhibitor or siRNA interference significantly inhibited the migration of dermal fibroblasts induced by bFGF under diabetic conditions, implying that PI3K/Akt, Rac1 and JNK are involved in the regulation of bFGF-promoted dermal fibroblast migration. Several studies showed that Rac1 activation was dependent on PI3K activity and that inhibitors of PI3K/Akt blocked Rac1 activation [22, 31]. Moreover, inhibition of the activity of Rac1 reduced JNK activity [32, 33]. In our study, the activity of Rac1 was abolished by the PI3-kinase/Akt inhibitor LY294002, suggesting that PI3-kinase/Akt contributes to bFGF-induced Rac1 activation. Furthermore, the inhibitory activity of Rac1 achieved with Rac1 siRNA down-regulated JNK activation after treatment with bFGF. Thus, we concluded that bFGF promotes dermal fibroblast migration through the PI3K/Akt-Rac1-JNK pathway under diabetic conditions. Shigeyuki Kanazawa et al. also showed that bFGF regulates the PI3K/Akt-Rac1-JNK pathway and promotes dermal fibroblast migration in rodents [34]. However, the difference between our studies is the culture medium conditions. Primary rat dermal fibroblasts were cultured in normal DMEM medium in their experiments, whereas human dermal fibroblasts were cultured in high-glucose medium in our study to mimic the diabetic conditions in diabetes patients. The data demonstrated that the PI3K/Akt-Rac1-JNK signal pathway is a cell-dependent pathway in bFGF that promotes dermal fibroblast migration.
ROS are a family of molecules that have historically been viewed as purely harmful metabolites resulting in oxidative stress and damage in aging and diseases, such as diabetes mellitus [35, 36]. Diabetes mellitus is a group of metabolic disorders that cause chronic hyperglycemia, and hyperglycemia has been linked to impaired wound healing, particularly altered angiogenesis and extracellular matrix remodeling [10, 37]. It has been shown that alterations in cell function associated with diabetic conditions and oxidative stress are the result of diabetes that damage the cell [38, 39]. High glucose modulates ROS formation and induces inflammation and cell apoptosis [40, 41], and a high-glucose environment enhances oxidative stress, increased interleukin-8 secretion and impaired keratinocyte migration. Fibroblasts from diabetic mice migrate 75% less than those from normoglycemic mice, and Lamers et al. showed that high glucose mediates oxidative stress, causes cell polarity loss and impairs cell migration [10]. In addition, advanced glycation end-products (AGEs) mediate the activation of ROS and blocks wound healing through impairing dermal fibroblast proliferation and migration [42]. Anti-AGE agents appear to facilitate wound healing by reducing AGE-associated inflammation and promoting the recovery process [43, 44]. However, there is no difference in ROS production between normal glucose and high glucose group in our study. Unexpectedly, we found that bFGF increased ROS generation in high glucose, the reasons for this maybe the different kinds of cell, the glucose concentration and cell culture time in high glucose condition. Wealth of evidence shows that ROS produced in certain situations may also function as important physiological regulators of intracellular signaling pathways [45]. Furthermore, studies have shown that ROS play a pivotal role in mediating the migration of certain cell types. It has been reported that ROS are involved in vascular endothelial growth factor (VEGF)-mediated endothelial migration [46], platelet-derived growth factor-induced smooth muscle cell migration [47], vascular cell adhesion molecule-1 (VCAM-1)-regulated leukocyte migration [48], and insulin-like growth factor-I (IGF-I)-stimulated vascular smooth muscle cell migration [49]. In accordance with the literature, our data suggest that increased ROS production is required for bFGF-induced human dermal fibroblast migration and that NAC significantly inhibits bFGF-promoted human dermal fibroblast migration under diabetic conditions. There are numerous potential sources of ROS within the cell, and NADPH oxidases are one of the major enzymatic sources of ROS in different tissues [50]. Our data show that DPI significantly inhibits bFGF-promoted dermal fibroblast migration, suggesting that bFGF-induced ROS production in human dermal fibroblasts under high-glucose conditions is dependent on NDAPH oxidases. Classic NADPH oxidase consist of the two membrane bound subunits Nox2 and p22phox and the cytosolic components p47phox, p67phox, p40phox, and Rac-1, and several isoforms of the NADPH oxidase, such as NOX1, NOX3, and NOX4, have been reported [51]. Sampson et al. showed that NADPH oxidase 4 (NOX4)-derived ROS mediate fibroblast-to-myofibroblast transdifferentiation [52]and that NOX4 expression is increased in pulmonary fibroblasts and mediates TGF-beta1-induced fibroblast differentiation into myofibroblasts [53]. Thus, we tested whether NOX4 is up-regulated and mediates ROS production after bFGF treatment in dermal fibroblasts under diabetic conditions. Our data showed that the NOX4 mRNA and protein levels were upregulated in the presence of bFGF, indicating that bFGF induces ROS production and mediates dermal fibroblast migration by up-regulating the NOX4 mRNA and protein levels. In adherent cells, ROS production participates in actin cytoskeletal reorganization by increasing FAK activity during cell spreading and participates in further control of gene expression and cell proliferation and migration [54]. In our study, bFGF was found to promote the migration of dermal fibroblasts through the activation of FAK and paxillin, and NAC and DPI inhibited the bFGF-induced activation of FAK and paxillin, suggesting that intracellular ROS are necessary for the activation of FAK and paxillin and participate in actin cytoskeletal reorganization in the presence of bFGF. The data demonstrated that the NOX-ROS signal pathway is another cell-dependent pathway in bFGF-induced dermal fibroblast migration under high-glucose conditions.
Endogenously generated ROS following treatment with peptide growth factors leads to the activation of the PI3K/Akt/JNK pathway [55, 56]. For example, the bFGF-induced migration of rodent vascular smooth muscle cells depends on the ROS-mediated activation of JNK [57]. The data indicated that ROS is upstream of PI3K/Akt and JNK activation and regulates cell proliferation, migration and apoptosis. The ROS-mediated activation of Akt induces apoptosis in prostate cancer cells [58], and cathepsin S plays an important role in the regulation of autophagy and apoptosis via ROS and serves upstream of the PI3K/AKT/mTOR/p70S6K and JNK signaling pathways in human glioblastoma cells[21]. However, in our study, we found that the blockade of intracellular ROS production with NAC or DPI did not affect the bFGF-induced activation of Akt or JNK. On the contrary, inhibition of the PI3K/Akt or JNK pathway significantly suppressed bFGF-induced ROS formation, suggesting that bFGF induces the generation of ROS in human dermal fibroblasts through the PI3K/Akt-Rac1-JNk pathway. All of the above findings indicated that ROS formation and the activation of PI3K/Akt and JNK are involved in cell proliferation, survival and migration. In addition, most of these results showed the ROS is upstream of the activation of PI3K/Akt and JNK and modulates cell physiological activity. However, our data are contrary to the specificity of the cells, and the cell culture microenvironment differences may be taken into consideration, but the PI3K/Akt and JNK mechanisms that regulate ROS formation in dermal fibroblasts under diabetic environment remain to be elucidated.
A duality in the roles of ROS with respect to wound healing. Injury to the skin initiates a series of events, such as inflammation, tissue regeneration, and matrix remodeling. During the early inflammatory phase, leukocytes and macrophages infiltrate the wounded tissue and produce large amounts of reactive oxygen species (ROS) as part of their defense mechanism once activated [59]. ROS are essential during various stages of the healing process, ranging from the initial signal that instigates the immune response, to the triggering of intracellular redox-dependent signalling pathways and the defence against invading bacteria [60]. For example, galectin-1 induced myofibroblast activation, migration, and proliferation by upregulating NOX4, triggered intracellular ROS production and accelerated the healing of general and pathological wounds and decreased the mortality of diabetic mice with skin wounds [61]. Further, study showed that ROS were essential mediators in epidermal growth factor (EGF)-stimulated corneal epithelial cell proliferation, adhesion, migration, and wound healing [55]. Although this process is beneficial, increased levels of ROS can inhibit cell migration and proliferation and even cause severe tissue damage, such as diabetes and diabetic complications [62, 63]. Based on above, increased appropriate ROS are helpful in promoting wound healing, excessive ROS cause cell dysfunction and impair wound healing. In our study, bFGF increased ROS formation which were essential for promoting dermal fibroblasts migration, indicating that the beneficiary of increased ROS in the wound healing may be wound stage or context dependent. Therefore, we are going to observe the synergistic effect during cutaneous wound repair by combining use low level of hydrogen peroxide (H2O2) and bFGF in future study, which could be helpful for clinical use of bFGF.
It should be noted that the detailed interaction of bFGF with ROS and PI3K/Akt-Rac1-JNK has not been fully established in the present study. In addition, why AKT and JNK activation lead to ROS production via NADPH oxidase activation are also unclear. A further clarification of these issues utilizing knock down of Akt, JNK and NOX4 will provide novel insights into these questions and provide more evidence to support our conclusions. In addition, whether NOX4 is the source of ROS responsible for bFGF-induced dermal fibroblast migration and whether the Rac1, p47phox, and p67phox cytosolic subunits have to assemble with the membrane-bound subunits to induce ROS formation after bFGF treatment in dermal fibroblasts under high glucose. The knock down of NOX4 protein should be performed to confirm whether NOX4 is the specific NADPH oxidase that induces ROS formation. Meanwhile, co-immunoprecipitation assays should also be performed to verify whether Rac1, p47phox, and p67phox transfer from the cytoplasm to the membrane, assemble with the membrane-bound subunits, allow the transfer of electrons from NADPH to molecular oxygen and induce ROS formation in the presence of bFGF.
In conclusion, our results indicate that bFGF induces ROS formation and promotes the migration of dermal fibroblasts, leading to the activation of FAK and paxillin in human dermal fibroblasts under high-glucose conditions (Fig. 9). In addition, we have shown that inhibition of the activation of PI3K/Akt, Rac1, JNK and ROS using special inhibitors or siRNA interference can block the bFGF-induced migratory properties of human dermal fibroblasts under diabetic conditions. Taken together, our results suggest that bFGF promotes the migration of dermal fibroblasts by NOX4 and mediates ROS production via the PI3K/Akt-Rac1-JNK pathways in a diabetic environment.
The proposed mechanism of bFGF promotes the migration of human dermal fibroblasts under diabetic conditions. Schematic representation of the mechanism through which bFGF promotes human dermal fibroblast migration under diabetic conditions.
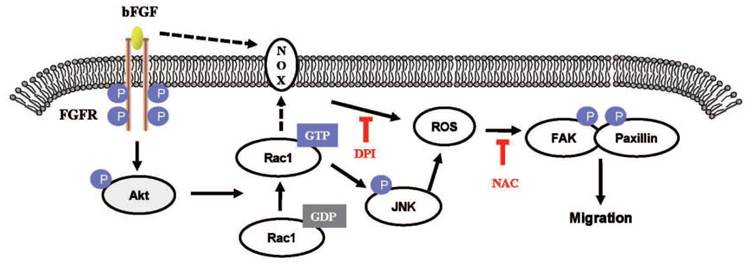
Acknowledgements
This work was supported by Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents (to J. X), This work was supported by National Natural Science Funding of China (81372112, 81372064, 81302775, 81472165), Zhejiang Provincial Natural Science Foundation of China (Y14H150023, Y14H090062, LY13H030008, LY14H150008), State Key Basic Research Development Program (2012CB518105), Zhejiang Provincial Project of Key Group (2010R50042). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
J X, H-X S and X-K L conceived and designed the experiments. H-X S, Y C, J-J Y, R L, J-J Z, Y Y, P-T C, C L, X-H L, L-P J, A-P H and X-B Z performed the experiments. H-X S, X-B F, Z-G W, H-Y Z, Q-Q Z and J X analyzed the data. J X and H-X S wrote the paper.
Supplementary Material
Figure S1.
Competing interests
The authors have declared that no competing interests exist.
References
1. Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden of diabetic foot disease. Lancet. 2005;366:1719-24
2. Sun DP, Yeh CH, So E, Wang LY, Wei TS, Chang MS. et al. Interleukin (IL)-19 promoted skin wound healing by increasing fibroblast keratinocyte growth factor expression. Cytokine. 2013;62:360-8
3. Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736-43
4. Opalenik SR, Davidson JM. Fibroblast differentiation of bone marrow-derived cells during wound repair. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1561-3
5. Werner S, Krieg T, Smola H. Keratinocyte-fibroblast interactions in wound healing. The Journal of investigative dermatology. 2007;127:998-1008
6. Wang T, Feng Y, Sun H, Zhang L, Hao L, Shi C. et al. miR-21 regulates skin wound healing by targeting multiple aspects of the healing process. The American journal of pathology. 2012;181:1911-20
7. Hehenberger K, Hansson A. High glucose-induced growth factor resistance in human fibroblasts can be reversed by antioxidants and protein kinase C-inhibitors. Cell Biochem Funct. 1997;15:197-201
8. Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 1998;6:135-41
9. Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. The American journal of pathology. 2003;162:303-12
10. Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF, Santos MF. High glucose-mediated oxidative stress impairs cell migration. PloS one. 2011;6:e22865
11. Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabetic medicine: a journal of the British Diabetic Association. 2006;23:594-608
12. Bennett SP, Griffiths GD, Schor AM, Leese GP, Schor SL. Growth factors in the treatment of diabetic foot ulcers. The British journal of surgery. 2003;90:133-46
13. Liu Y, Cai S, Shu XZ, Shelby J, Prestwich GD. Release of basic fibroblast growth factor from a crosslinked glycosaminoglycan hydrogel promotes wound healing. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2007;15:245-51
14. Mizuno K, Yamamura K, Yano K, Osada T, Saeki S, Takimoto N. et al. Effect of chitosan film containing basic fibroblast growth factor on wound healing in genetically diabetic mice. Journal of biomedical materials research Part A. 2003;64:177-81
15. Tan Y, Xiao J, Huang ZF, Xiao YC, Lin SQ, Jin LT. et al. Comparison of the therapeutic effects recombinant human acidic and basic fibroblast growth factors in wound healing in diabetic patients. Journal of Health Science. 2008;54:432-40
16. Shi HX, Lin C, Lin BB, Wang ZG, Zhang HY, Wu FZ. et al. The Anti-Scar Effects of Basic Fibroblast Growth Factor on the Wound Repair In Vitro and In Vivo. PloS one. 2013:8
17. Boosani CS, Nalabothula N, Sheibani N, Sudhakar A. Inhibitory effects of arresten on bFGF-induced proliferation, migration, and matrix metalloproteinase-2 activation in mouse retinal endothelial cells. Current eye research. 2010;35:45-55
18. Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. Segregation and activation of myosin IIB creates a rear in migrating cells. The Journal of cell biology. 2008;183:543-54
19. Lu JW, Liao CY, Yang WY, Lin YM, Jin SL, Wang HD. et al. Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PloS one. 2014;9:e85318
20. Magi S, Takemoto Y, Kobayashi H, Kasamatsu M, Akita T, Tanaka A. et al. 5-Lipoxygenase and cysteinyl leukotriene receptor 1 regulate epidermal growth factor-induced cell migration through Tiam1 upregulation and Rac1 activation. Cancer science. 2014;105:290-6
21. Zhang L, Wang H, Xu J, Zhu J, Ding K. Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicology letters. 2014;228:248-59
22. Murga C, Zohar M, Teramoto H, Gutkind JS. Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-kappa B. Oncogene. 2002;21:207-16
23. Wang H, Yang Z, Jiang Y, Hartnett ME. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Molecular vision. 2014;20:231-41
24. Ben Mahdi MH, Andrieu V, Pasquier C. Focal adhesion kinase regulation by oxidative stress in different cell types. IUBMB life. 2000;50:291-9
25. Hata S, Okamura K, Hatta M, Ishikawa H, Yamazaki J. Proteolytic and non-proteolytic activation of keratinocyte-derived latent TGF-beta1 induces fibroblast differentiation in a wound-healing model using rat skin. Journal of pharmacological sciences. 2014;124:230-43
26. Yu J, Wang Q, Wang H, Lu W, Li W, Qin Z. et al. Activation of liver X receptor enhances the proliferation and migration of endothelial progenitor cells and promotes vascular repair through PI3K/Akt/eNOS signaling pathway activation. Vascular pharmacology. 2014;62:150-61
27. Mendes KN, Wang GK, Fuller GN, Zhang W. JNK mediates insulin-like growth factor binding protein 2/integrin alpha5-dependent glioma cell migration. International journal of oncology. 2010;37:143-53
28. Shimabukuro Y, Terashima H, Takedachi M, Maeda K, Nakamura T, Sawada K. et al. Fibroblast growth factor-2 stimulates directed migration of periodontal ligament cells via PI3K/AKT signaling and CD44/hyaluronan interaction. Journal of cellular physiology. 2011;226:809-21
29. Dise RS, Polk DB. Epidermal growth factor stimulates Rac1 activation in intestinal epithelial cells through SRC and PI 3-kinase to promote cell migration. Gastroenterology. 2006;130:A124-A
30. Hu ZZ, Du J, Yang L, Zhu YC, Yang Y, Zheng DT. et al. GEP100/Arf6 Is Required for Epidermal Growth Factor-Induced ERK/Rac1 Signaling and Cell Migration in Human Hepatoma HepG2 Cells. PloS one. 2012:7
31. Huang JS, Cho CY, Hong CC, Yan MD, Hsieh MC, Lay JD. et al. Oxidative stress enhances Axl-mediated cell migration through an Akt1/Rac1-dependent mechanism. Free Radical Bio Med. 2013;65:1246-56
32. Yamauchi J, Miyamoto Y, Kokubu H, Nishii H, Okamoto M, Sugawara Y. et al. Endothelin suppresses cell migration via the JNK signaling pathway in a manner dependent upon Src kinase, Rac1, and Cdc42. FEBS letters. 2002;527:284-8
33. Chen JC, Lin BB, Hu HW, Lin C, Jin WY, Zhang FB. et al. NGF accelerates cutaneous wound healing by promoting the migration of dermal fibroblasts via the PI3K/Akt-Rac1-JNK and ERK pathways. BioMed research international. 2014;2014:547187
34. Kanazawa S, Fujiwara T, Matsuzaki S, Shingaki K, Taniguchi M, Miyata S. et al. bFGF regulates PI3-kinase-Rac1-JNK pathway and promotes fibroblast migration in wound healing. PloS one. 2010;5:e12228
35. Rojas A, Mercadal E, Figueroa H, Morales MA. Advanced glycation and ROS: A link between diabetes and heart failure. Curr Vasc Pharmacol. 2008;6:44-51
36. Rovira-Llopis S, Rocha M, Falcon R, de Pablo C, Alvarez A, Jover A. et al. Is Myeloperoxidase a Key Component in the ROS-Induced Vascular Damage Related to Nephropathy in Type 2 Diabetes? Antioxid Redox Sign. 2013;19:1452-8
37. Browning AC, Alibhai A, McIntosh RS, Rotchford AP, Bhan A, Amoaku WM. Effect of diabetes mellitus and hyperglycemia on the proliferation of human Tenon's capsule fibroblasts: implications for wound healing after glaucoma drainage surgery. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2005;13:295-302
38. Takahashi A, Aoshiba K, Nagai A. Apoptosis of wound fibroblasts induced by oxidative stress. Experimental lung research. 2002;28:275-84
39. Lan CC, Wu CS, Huang SM, Wu IH, Chen GS. High-glucose environment enhanced oxidative stress and increased interleukin-8 secretion from keratinocytes: new insights into impaired diabetic wound healing. Diabetes. 2013;62:2530-8
40. Liu D, Zhang H, Gu W, Zhang M. Effects of exposure to high glucose on primary cultured hippocampal neurons: involvement of intracellular ROS accumulation. Neurological sciences: official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2014;35:831-7
41. Jayakumar T, Chang CC, Lin SL, Huang YK, Hu CM, Elizebeth AR. et al. Brazilin ameliorates high glucose-induced vascular inflammation via inhibiting ROS and CAMs production in human umbilical vein endothelial cells. BioMed research international. 2014;2014:403703
42. Loughlin DT, Artlett CM. Precursor of advanced glycation end products mediates ER-stress-induced caspase-3 activation of human dermal fibroblasts through NAD(P)H oxidase 4. PloS one. 2010;5:e11093
43. Chang PC, Tsai SC, Jheng YH, Lin YF, Chen CC. Soft-tissue wound healing by anti-advanced glycation end-products agents. Journal of dental research. 2014;93:388-93
44. Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG. et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. The American journal of pathology. 2001;159:513-25
45. Finkel T. Signal transduction by reactive oxygen species. The Journal of cell biology. 2011;194:7-15
46. Yamaoka-Tojo M, Ushio-Fukai M, Hilenski L, Dikalov SI, Chen YE, Tojo T. et al. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species--dependent endothelial migration and proliferation. Circulation research. 2004;95:276-83
47. Weber DS, Taniyama Y, Rocic P, Seshiah PN, Dechert MA, Gerthoffer WT. et al. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circulation research. 2004;94:1219-26
48. Morise Z, Ueda M, Kitajima M, Epstein CJ, Granger DN, Grisham MB. Reactive oxygen species and vascular cell adhesion molecule-1 in distant organ failure following bile duct obstruction in mice. Digestive diseases and sciences. 2002;47:607-13
49. Meng D, Lv DD, Fang J. Insulin-like growth factor-I induces reactive oxygen species production and cell migration through Nox4 and Rac1 in vascular smooth muscle cells. Cardiovascular research. 2008;80:299-308
50. Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature chemical biology. 2011;7:504-11
51. Nam HJ, Park YY, Yoon G, Cho H, Lee JH. Co-treatment with hepatocyte growth factor and TGF-beta1 enhances migration of HaCaT cells through NADPH oxidase-dependent ROS generation. Experimental & molecular medicine. 2010;42:270-9
52. Sampson N, Plas E, Berger P. NADPH oxidase 4 (NOX4) derived ROS mediate fibroblast to myofibroblast transdifferentiation in the diseased prostatic stroma. Faseb Journal. 2009:23
53. Rocic P, Lucchesi PA. NAD(P)H oxidases and TGF-beta-induced cardiac fibroblast differentiation: Nox-4 gets Smad. Circulation research. 2005;97:850-2
54. Gregg D, de Carvalho DD, Kovacic H. Integrins and coagulation: a role for ROS/redox signaling? Antioxid Redox Signal. 2004;6:757-64
55. Huo Y, Qiu WY, Pan Q, Yao YF, Xing K, Lou MF. Reactive oxygen species (ROS) are essential mediators in epidermal growth factor (EGF)-stimulated corneal epithelial cell proliferation, adhesion, migration, and wound healing. Experimental eye research. 2009;89:876-86
56. Yang Y, Du J, Hu Z, Liu J, Tian Y, Zhu Y. et al. Activation of Rac1-PI3K/Akt is required for epidermal growth factor-induced PAK1 activation and cell migration in MDA-MB-231 breast cancer cells. Journal of biomedical research. 2011;25:237-45
57. Schroder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1736-43
58. Chetram MA, Bethea DA, Odero-Marah VA, Don-Salu-Hewage AS, Jones KJ, Hinton CV. ROS-mediated activation of AKT induces apoptosis via pVHL in prostate cancer cells. Molecular and cellular biochemistry. 2013;376:63-71
59. Steiling H, Munz B, Werner S, Brauchle M. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Experimental cell research. 1999;247:484-94
60. Vermeij WP, Backendorf C. Skin Cornification Proteins Provide Global Link between ROS Detoxification and Cell Migration during Wound Healing. PloS one. 2010:5
61. Lin YT, Chen JS, Wu MH, Hsieh IS, Liang CH, Hsu CL. et al. Galectin-1 accelerates wound healing by regulating the neuropilin-1/Smad3/NOX4 pathway and ROS production in myofibroblasts. The Journal of investigative dermatology. 2015;135:258-68
62. Li JM, Shah AM. ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy. Journal of the American Society of Nephrology: JASN. 2003;14:S221-6
63. Bitar MS, Al-Mulla F. ROS constitute a convergence nexus in the development of IGF1 resistance and impaired wound healing in a rat model of type 2 diabetes. Disease models & mechanisms. 2012;5:375-88
Author contact
![]() Corresponding authors: School of Pharmaceutical Sciences, Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, 325035 China. Tel.: +86-577-85773087; Fax.: +86-577-85773087; E-mail address: xfxj2000com (J. Xiao), xiaokunlinet (X. Li)
Corresponding authors: School of Pharmaceutical Sciences, Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, 325035 China. Tel.: +86-577-85773087; Fax.: +86-577-85773087; E-mail address: xfxj2000com (J. Xiao), xiaokunlinet (X. Li)