Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):87-99. doi:10.7150/ijbs.13229 This issue Cite
Research Paper
Retinoic Acid Induced-Autophagic Flux Inhibits ER-Stress Dependent Apoptosis and Prevents Disruption of Blood-Spinal Cord Barrier after Spinal Cord Injury
Yulong Zhou1,2, Hongyu Zhang2, Binbin Zheng1,2, Libing Ye2, Sipin Zhu1,2, Noah R Johnson3, Zhouguang Wang2, Xiaojie Wei4, Daqing Chen5, Guodong Cao6, Xiaobing Fu7, Xiaokun Li 2, Hua-Zi Xu1, Jian Xiao2 ![]()
1. Department of Orthopaedics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou, Zhejiang, 325035 China
2. Molecular Pharmacology Research Center, School of Pharmacy, Wenzhou Medical University, Wenzhou, Zhejiang, 325035 China
3. Department of Bioengineering and the McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA 15219, USA
4. Department of Neurosurgery, Cixi People's Hospital, Wenzhou Medical University, Ningbo, 315300, China
5. Department of Emergency, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325035, China
6. Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
7. Institute of Basic Medical Science, Chinese PLA General Hospital, Beijing 100853, China
The first two authors contributed equally to this study.
Received 2015-7-13; Accepted 2015-10-7; Published 2016-1-1
Abstract

Spinal cord injury (SCI) induces the disruption of the blood-spinal cord barrier (BSCB) which leads to infiltration of blood cells, an inflammatory response, and neuronal cell death, resulting spinal cord secondary damage. Retinoic acid (RA) has a neuroprotective effect in both ischemic brain injury and SCI, however the relationship between BSCB disruption and RA in SCI is still unclear. In this study, we demonstrated that autophagy and ER stress are involved in the protective effect of RA on the BSCB. RA attenuated BSCB permeability and decreased the loss of tight junction (TJ) molecules such as P120, β-catenin, Occludin and Claudin5 after injury in vivo as well as in Brain Microvascular Endothelial Cells (BMECs). Moreover, RA administration improved functional recovery in the rat model of SCI. RA inhibited the expression of CHOP and caspase-12 by induction of autophagic flux. However, RA had no significant effect on protein expression of GRP78 and PDI. Furthermore, combining RA with the autophagy inhibitor chloroquine (CQ) partially abolished its protective effect on the BSCB via exacerbated ER stress and subsequent loss of tight junctions. Taken together, the neuroprotective role of RA in recovery from SCI is related to prevention of of BSCB disruption via the activation of autophagic flux and the inhibition of ER stress-induced cell apoptosis. These findings lay the groundwork for future translational studies of RA for CNS diseases, especially those related to BSCB disruption.
Keywords: blood-spinal cord barrier (BSCB), retinoic acid (RA), autophagy, endocytoplasmic reticulum (ER) stress, spinal cord injury (SCI)
Introduction
The blood-spinal cord barrier (BSCB) is composed of highly specialized endothelial cells (ECs) that line the vascular wall of the spinal cord capillaries. ECs form a tight structural barrier due to the presence of well-developed tight junction (TJ) proteins on the luminal side of the endothelium that effectively block the intercellular movement of large molecules, including plasma proteins [1-3]. Thus the BSCB is necessary and particularly important in the precise control of the microenvironment [4-6]. Central nervous system (CNS) trauma, including SCI, is a leading cause of death and long-term disability among young adults worldwide [7, 8]. SCI is a severe trauma of the CNS and is accompanied by the disruption of the BSCB [9, 10]. The initial physical damage to the BSCB allows infiltration of macrophages and other immune cells to the area of injury, resulting in secondary injury involving a complex cascade of molecular events such as ionic imbalance, excitatory amino acid release, and oxidative species formation in the injury area [11]. These secondary events induce the 'programmed death' of neurons and glia, leading to permanent neurological deficits [12]. Thus, drugs that prevent the BSCB disruption may limit nervous system complications and improve the prognosis in SCI cases.
Retinoic acid (RA), an active metabolite of vitamin A, is vital for embryonic development and maintenance of many organ systems in the adult body [13]. It is particularly crucial during development of the central nervous system. Radial glial cell-derived RA is critical in providing the brain with the correct developmental pattern during neurogenesis [14-16]. Systemic administration of retinoic acid reduces early transcript levels of pro-inflammatory cytokines after experimental SCI [17]. RA treatment also led to an increase in the expression of TJ proteins in brain ECs [1, 18]. Thus, we suppose RA prevents disruption of BSCB after SCI via stimulate TJ proteins expression.
Endocytoplasmic reticulum (ER) stress activation is a part of the inherent response within the spinal cord acutely following traumatic SCI [19-21]. At the early phase of ER stress or in the case of mild ER stress, cells exhibit a self-protective signal transcription pathway by upregulating expression of ER-localized molecular chaperones [22, 23]. However, at the late phase of ER stress or during excessive ER stress, an apoptotic pathway may be activated via CHOP and caspase-12 which leads to secondary injury after SCI [24]. ER stress signal may have a direct role in promoting cell death in secondary injury after SCI [19-21]. Autophagy, a lysosome-dependent cellular degradation pathway, is an essential process for the maintenance of cellular homeostasis in the CNS, both under physiological conditions and pathological conditions [25, 26]. Autophagy activation reduced the rate of apoptosis in acute SCI [27, 28], contradictory, inhibiting autophagy abolished the anti-ER stress effects of rapamycin after SCI [28, 29]. Furthermore, autophagy can be induced by ER stress [30, 31], and inhibition of autophagy leads to increased ER stress after SCI [32]. Taken together, these reports suggest that severe ER stress is involved in neuronal injury by inducing ER stress-dependent apoptosis, while autophagy activation may reduce the expression of apoptosis proteins induced by severe ER stress in CNS trauma. Many reports showed that RA exerted a cell type-dependent stimulatory effect on autophagy flux via Beclin-1 upregulation and inhibition of the mTOR pathway, or by enhancing autophagosome maturation [33-35]. In addition to neurons, markers of autophagy and ER stress are increased in other cell types after CNS trauma, including all major glial sub-types (astrocytes, oligodendrocytes, and microglia) [36-39], However, much less is known about autophagy and ER stress in ECs, especially the mechanism and pathway involved in treatment with exogenous RA.
In this study, we investigated the protective effects of exogenous RA on the BSCB both in vivo and in vitro and demonstrated that autophagy and ER stress are involved in these effects after SCI. Our data show that RA attenuated BSCB permeability and also attenuated the loss of TJ molecules after SCI. We also elucidate the signaling pathways and molecular mechanisms involved in ER stress and autophagy following SCI, especially related to the BSCB. These studies support the translation of RA as a therapy for SCI and potentially for other CNS diseases.
Materials and methods
Reagents and antibodies
DMSO (Sigma-Aldrich) was used to dissolve non-water soluble reagents and as a vehicle control. Endothelial Cell Medium (ECM) which contains 500 ml of basal medium, 25 ml of fetal bovine serum (FBS, Cat. No. 0025), 5 ml of endothelial cell growth supplement (ECGS, Cat. No. 1052) and 5 ml of penicillin/streptomycin solution (P/S, Cat. No. 0503) were purchased from ScienCell Research Laboratories. Antibodies against Occludin, Claudin5, CHOP, GRP78, and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p62 and microtubule-associated protein-1 light-chain 3 (LC3) were purchased from Cell Signaling Technologies (Danvers, MA, USA). Anti-β-catenin, P120-catenin, cleaved caspase-12, and PDI were purchased from Abcam. All chemicals including RA were from Sigma Chemical Company.
SCI model and drug treatment
Adult female Sprague-Dawley rats (220 - 250 g) purchased from Animal Center of Chinese Academy of Sciences, Shanghai, China, were used for surgical procedures. All the rats were fed under controlled temperature (21 - 23°C), 12 h light/dark cycles and ad libitum access to food and water. The protocol for animal care and use conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and was approved by the Animal Care and Use Committee of Wenzhou Medical University. All rats were anaesthetized by intraperitoneal injection of 10 % chloral hydrate (3.6 ml/kg) and the model of a T9 half-cut spinal cord injury was made. For each rat, the back was shaved and sterilized and an incision was made posterior to the lower thoracic region and the vertebral column was exposed. A laminectomy was performed at the T9 level. The exposed spinal cord was subjected to crush injury by compressing with a vascular clip (30 g force, Oscar, China) for 2 minutes. Sham group rats received the same surgical procedure but sustained no injury. The incision sites were then closed in layers and a topical antibiotic (cefazolin sodium salt) was applied to the incision site. Drug treatments were administered according to previous reports [17]. RA was diluted to a stock solution of 100 mg/mL in 100% DMSO. To prevent oxidation, air above the aliquots was replaced with nitrogen and stock aliquots were stored at -80 ◦C. All handling of RA was performed in the dark. RA stock solution was further diluted in 100% ethanol to a concentration of 1 mg/mL. RA was administered into the injured rat via intraperitoneal injection (15 mg/kg) immediately after spinal cord injury and then re-administered once a day for 2 weeks for behavioral tests. CQ, where applicable, was administered in similar fashion via intraperitoneal injection (50 mg/kg). Equivalent ethanol and DMSO injections were administered for vehicle control. Animals either received vehicle (n = 25), RA (n = 25), RA together with CQ (n = 15), or CQ (n = 15). Postoperative care involved manual urinary bladder emptying twice daily until the return of bladder function. All animals showed no significant side effects resulting from drug treatment such as mortality or signs of infectious disease during these experiments.
Cell culture and cell viability assay
Primary cultures of Human Brain Microvascular Endothelial Cells (HBMVEC) were purchased from ScienCell Research Laboratories. BMVEC cultures were expanded and maintained in Endothelial Cell Medium (ECM) which contains 500 ml of basal medium, 25 ml of fetal bovine serum (FBS), 5 ml of endothelial cell growth supplement (ECGS,) and 5ml of penicillin/streptomycin solution (P/S). They were then incubated in a humidified atmosphere containing 5 % CO2 at 37 °C. RA was diluted to a stock solution of 10mM in 100% DMSO. Cells were treated with Thapsigargin (TG, 10 μM), TG compound with RA (1 μM), or RA (5 μM). Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after treatment.
Locomotion recovery assessment
Locomotor outcome after spinal cord contusion injury was conducted as previously described [40]. Two trained investigators who were blind to the experimental conditions scored locomotion recovery in an open field, according to the Basso, Beattie and Bresnahan (BBB) scale. BBB scores range from 0, indicative of no observed hindlimb movements, to 21, representative of a normal ambulating rodent. Animals were placed individually in an open field and allowed to move freely for 5 min. The animals were evaluated every two days from day 0 to day 14 following the surgical procedure.
Measurement of BSCB disruption
The integrity of the BSCB was investigated by Evans blue dye extravasation according to previous reports [41, 42] with minor modifications. 1 ml of 2% Evan's Blue dye solution in saline was injected intravenously into the tail vein. Two hours later, animals were anaesthetized and killed by intracardiac perfusion with saline. One centimeter of the spinal cord surrounding the T9 injury site was extracted and homogenized in a 50% trichloroacetic acid solution. Samples (400 mg) were then homogenized in 400 μL of N,N-dimethylformamide (DMF) and incubated at 70℃ for 72 h. Samples were centrifuged at 18,000 rpm for 20 min twice. The supernatant was collected, aliquotted (200 μl) into a 96-well glass plate, and its fluorescence was quantified using a spectrophotometer at an excitation wavelength of 620 nm and an emission wavelength of 680 nm. EB concentration (μg/g of tissue) was determined from a standard curve of EB in DMF.
Western blot analysis
Spinal cord tissue samples were removed 1 day after surgery [40, 43] and the spinal cords from the T7- T10 level around the lesion epicenter were excised, a spinal cord segment (0.5 cm length) at the contusion epicenter was dissected and rapidly stored at -80 °C for western blotting [44]. Briefly, frozen animal spinal cord tissues and cells were homogenized in ice-cold lysis buffer containing 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 10 mM Na2P2O7, 10 mM NaF, 1 m g/ml aprotinin, 10 mg/ml leupeptin, 1 mM sodium vanadate and 1 mM PMSF. Tissue homogenates were incubated for 15 min at 4℃, and centrifuged at 12,000 rpm, for 15 min at 4℃. The equivalent of 60 μg of total protein was loaded onto SDS-PAGE and transferred to PVDF membrane (Bio-Rad). The membrane was blocked with 5 % non-fat milk in TBS with 0.1 % Tween 20 for 90 min, and then incubated overnight at 4℃ with primary antibody solutions according to the manufacturer's recommendations. Then the membranes were washed with TBS for 5 min three times and primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies. Signals were visualized using the ChemiDicTM XRS + Imaging System (Bio-Rad). Experiments were repeated three times.
Immunofluorescence staining
Spinal cord segments obtained from animals 1 day after surgery were cryoprotected in 30% sucrose in phosphate buffer and embedded in OCT compound (ProSciTech). Spinal cord sections were cut transversely at 7 μm on a cryostat. Sections were thawed at room temperature for 30 minutes, then washed twice for 10 minutes in PBS. Then sections were incubated with 5% albumin from bovine serum in PBS containing 0.1 % Triton X-100 in a 37°C oven for 30 min. They were then incubated with the appropriate primary antibodies overnight at 4°C in the same buffer. The following primary antibodies were used, based on differing targets: Occludin (1:200 Bioworld), CD31, CHOP, NeuN (1:100 Santa Cruz Biotechnology), LC3-II (1:400 Cell Signaling Technology). Sections were rinsed three times in PBS after primary antibody (Ab) incubation and then incubated with either fluorescent Alexa 568, 647 donkey anti-mouse/rabbit, or 488 goat anti-mouse/rabbit secondary Ab (1:500; Invitrogen) for 1 h at room temperature. Sections were rinsed three times with PBS and incubated with 4,6-diamidino-2-phenylindole (DAPI) for 10 min and finally washed in PBS and sealed with a coverslip. All images were captured on a Nikon ECLIPSE Ti microscope (Nikon, Japan).
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) from three independent experiments. Statistical significance was examined using Student's t-test when there were two experimental groups. When more than two groups were compared, statistical evaluation of the data was performed using one-way analysis of variance (ANOVA) and Dunnett's post hoc test. P values < 0.05 were considered statistically significant.
Results
RA reduces BSCB permeability and improves functional recovery after SCI
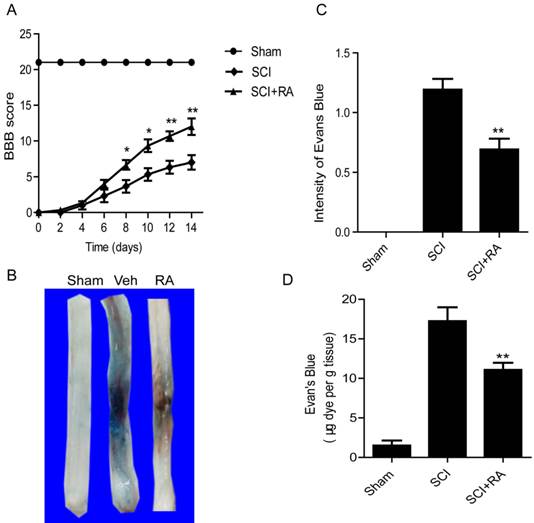
To evaluate the therapeutic effect of RA on SCI, rats were treated with RA by intraperitoneal injection. Functional recovery was then evaluated for 2 weeks after injury using the 21-point BBB rating scale. RA treatment significantly increased the BBB scores within 7-14 days after injury, compared to vehicle-treated controls (Figure 1A). At 14 d after injury, the RA group BBB scores reached at 12.34 ± 0.85, while the vehicle-treated control group was 6.89 ± 0.72. This indicates that RA improves functional recovery after SCI. The BSCB is disrupted very soon after SCI (as early as 5 minutes after injury), it is maximally injured at 24 h post-SCI, and appears to be restored 14 days later [9]. Thus, we examined the effect of RA on BSCB permeability at 1 day after injury by Evan's Blue assay. As shown in Figure 1B and C, BSCB permeability was increased significantly in response to spinal cord compression injury as compared with uninjured sham control, and RA significantly reduced the increase in BSCB permeability. In addition, after treatment with RA the content of EB in spinal cord tissue was significantly decreased (10.81 ± 1.04, μg/g) compared to the SCI group (10.81 ± 1.04 vs. 17.29 ± 1.27, μg/g, P < 0. 01) (Figure 1D). These results imply that RA can effectively prevent BSCB disruption and promote functional improvement of locomotor recovery after SCI.
RA inhibits the increase in BSCB permeability and improves functional recovery after SCI. After SCI, rats were treated with RA and BSCB permeability was measured at 24 h post-SCI by using Evan's Blue dye (n = 5/group). (A) The BBB scores of sham, SCI group and SCI rat treated with RA group. * P < 0.05 versus the SCI group, and ** P < 0.01 versus the SCI group, n = 5. (B) Representative whole spinal cords showing Evan's Blue dye permeabilized into the spinal cord at 24 h post-SCI. (C) Quantification of BSCB permeability data using B by ImageJ software **P < 0.01, versus the SCI group, n=5, F=1.848. (D) Quantification of the EB content of the spinal cord (μ g/g), **P < 0.01, versus the SCI group, n=5, F=1.482. All data represent mean values ± SEM.
RA induces autophagic flux in acute SCI
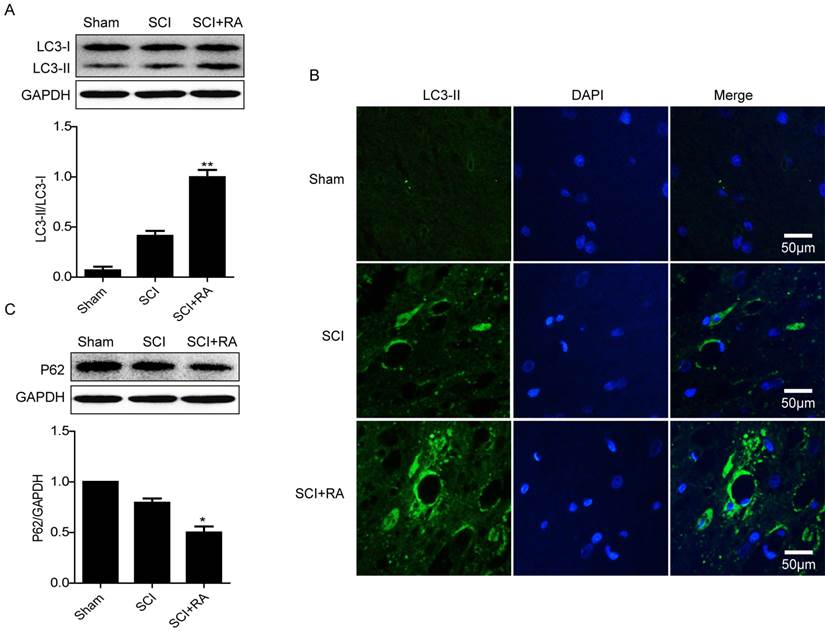
Autophagy is an essential process for the maintenance of cellular homeostasis in the central nervous system, both under physiological conditions and pathological conditions [45]. It has been reported that autophagy activation is involved in SCI [36], however its role has not been clearly defined in the impaired spinal cord or with respect to the BSCB. In our model, we first detected the level of LC3 protein which stably associates with the membranes of autophagosomes. Compared with the SCI-only group, the ratio of LC3-II/LC3-I was lower in the sham group but was significantly higher in the RA group (Figure 2A). Immunofluorescence staining results also showed that RA treatment increased the fluorescence intensity of LC3-II compared with the SCI group (Figure 2B). P62 is a substrate of the autophagic process and its presence is a marker of autophagic flux in vivo [46, 47]. Western blot results showed that the level of P62 was decreased early (1 day) after SCI, and this decrease was significantly exacerbated in the RA-treated group as compared with the vehicle-treated group (Figure 2C). These results indicate that RA effectively activates autophagic flux in acute SCI.
RA induces “Autophagic Flux” in acute SCI. (A) Representative western blots and quantification data of autophagy marker LC3-II in Sham group, SCI rats and treated with RA group. **P < 0.01, versus the SCI group, n=5, F=2.122. (B) Representative micrographs showing immunofluorescence with LC3-II. Nuclei are labeled with DAPI (blue) in each group. (C) Representative western blots and quantification data of autophagy marker P62 in Sham group, SCI rats and treated with RA group. *P < 0.05, versus the SCI group, n=5, F=1.788.
RA prevents the loss of TJ and AJ proteins after SCI
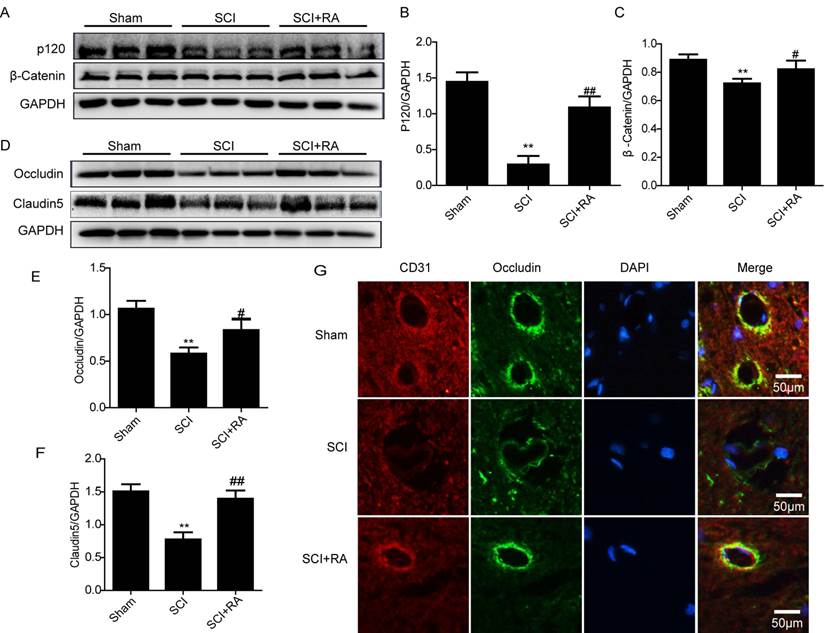
The TJ and AJ in the ECs of blood vessels are involved in BSCB integrity [48]. Below the TJ, in the basal region of lateral plasma membrane there are adherens junctions (AJ) that mediate events such as the adhesion of BMVEC to each other, and the regulation of paracellular permeability [3]. The increased permeability of the BSCB may be explained by the disruption of TJ between BSCB ECs. We investigated whether RA attenuates the increased permeability by preventing the loss of TJ and AJ proteins after SCI. The expression of the TJ (Occludin, Claudin5) and AJ (β-catenin, P120-catenin) proteins were examined in spinal lysates by western blot. Western blot results showed that the levels of AJ (β-catenin, P120-catenin) (Figure 3A, B and C) were decreased 1 day after SCI, as well as the levels of TJ (Occluding, Claudin5) (Figure 3D, E and F). Furthermore, RA-treated groups showed significantly higher levels of P120, β-catenin, Occludin, and Claudin5 at 1 day after injury compared to the vehicle control (Figure 3 A-F), indicating that RA prevents loss of TJ and AJ proteins after injury. Double labeling immunofluorescence also showed that the fluorescence intensity of Occludin and CD31 immunoreactivity was decreased after injury as compared to sham controls, and the fluorescence intensity was greatly increased in RA treatment group compared with the SCI-only group (Figure 3G). These data suggest that RA preserves BSCB integrity by inhibiting degradation of TJ and AJ proteins after SCI.
RA prevents the loss of TJ and AJ proteins after SCI. (A) Representative western blots of AJ proteins β-catenin and P120-catenin in the sham, SCI model and SCI model treated RA groups. (B), (C) Quantification of western blot data from A. (D) Representative western blots of TJ proteins Occludin and Claudin5 in the sham, SCI model and SCI model treated RA groups. (E), (F) Quantification of western blot data from D. All data represent Mean values ± SEM, n =5, **P < 0.01, versus the Sham group, #P < 0.05, ##P < 0.01 versus the SCI group, F < 2.749 in all T- tests. (G) Representative micrographs showing double immunofluorescence with Occludin (green) and CD31 (endothelial cell marker, red), nuclei are labeled with DAPI (blue ) in each group, n=5.
RA inhibits ER stress-associated CHOP and caspase-12 expression after SCI
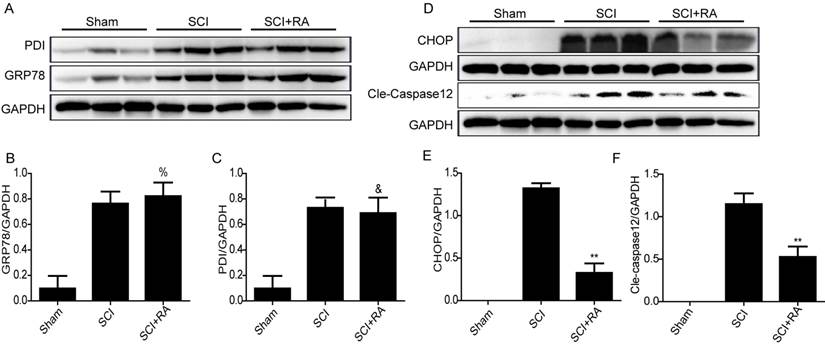
A recent report indicated that RA might attenuate ER stress-induced CHOP expression by modulating the phosphorylation of p38 MAPK in FRTL5 thyroid cells [49]. However, the relationship between RA and ER stress in SCI has not been reported. Next, we examined the alterations of ER stress-associated proteins including GRP78, PDI, and CHOP induced by SCI and the effect of RA on these alterations by western blot. However, the results from in vivo experiments showed that RA had no significant effects on protein expression of GRP78 and PDI compared with the SCI-only group (Figure 4A-C). Although a slight increase in GRP78 protein level in the RA treatment group was observed (Figure 4B), there was no statistical significance. Interestingly, RA significantly reduced the expression of CHOP and caspase-12 compared with the SCI-only group (Figure 4D and E) (P < 0.01). These findings illustrate that ER-stress dependent apoptosis protein CHOP and caspase-12 inhibition by RA is involved in acute SCI protective effects.
RA inhibits ER stress-associated CHOP and caspase-12 expression after SCI. (A) Representative western blots of ER stress markers GRP78, and PDI in the sham, SCI model and SCI model treated RA groups. (B), (C) Quantification of western blot data from A. There were no significant alterations on protein expression of GRP78 and PDI between SCI model and RA treated group %P = 0.45, &P = 0.70. FGRP78 = 1.247, FPDI = 1.026. (D) Representative western blots of CHOP and caspase-12 in the sham, SCI model and SCI model treated RA groups. (E), (F) Quantification of western blot data from D. **P < 0.01, versus the SCI group, FCHOP = 3.569, Fcaspase-12 =1.369. All data represent Mean values ± SEM, n =5.
Inhibition of autophagy by CQ abolishes the BSCB protective effect of RA
Our data showed that RA had no significant effect on protein expression of GRP78 and PDI, the proteins most representative of ER stress, while it significantly reduced the expression of CHOP compared with the SCI group. These results indicate that RA might indirectly affect ER stress through other signaling pathways. It has been reported that the rapamycin, a specific inducer of autophagy, increased the expression of GRP78 while reducing the expression of cleaved caspase-12 and CHOP. In addition, inhibition of autophagy with 3-MA resulted in excessive ER stress, leading to increases in CHOP and caspase-12 [29]. Since RA activates autophagic flux in acute SCI, we tested if RA inhibits the expression of CHOP and caspase-12 after SCI by activating autophagy. Chloroquine (CQ), a lysosome inhibitor, was used to block the autophagic flux. As shown in Figure 5A, BSCB permeability was increased significantly in response to CQ compared with the RA-treated group. Evans Blue density in the spinal cord tissue indicated that, compared with the RA-treated group, Evans Blue density was significantly higher in the RA and CQ co-treated group (Figure 5B, P<0.01). We next detected the alterations of TJ and AJ proteins when RA treatment was combined with CQ at 1 day after SCI by western blot. As shown, the levels of TJ proteins (Occludin, Claudin5, β-catenin) were significantly decreased in the RA and CQ co-treated group as compared with the RA alone group (Figure 5C and D). These data indicate that inhibition of autophagy by CQ abolishes the protective effect of RA on the BSCB by increasing the loss of TJ proteins after SCI.
Inhibition of autophagy by CQ abolishes the BSCB protective effect of RA and it's inhibition of apoptotic proteins expression after SCI. (A) Representative whole spinal cords showing Evan's Blue dye permeabilized into spinal cord at 24 h post-SCI. (B) Quantification of BSCB permeability data from B by ImageJ software, ##P < 0.01 versus SCI group, F =1.653; **P < 0.01 versus treated RA groups, n = 4, F = 1.516. (C), (D) Representative western blots and quantification data of TJ proteins Occludin, Claudin5 and β-catenin in each group rats. !!P< 0.01, @@P< 0.01, $$P< 0.01 versus SCI group. **P< 0.01, ##P< 0.01, &&P< 0.01 versus treated RA groups, F < 2.139 in all T-tests. (E), (F), (G), (H) Representative western blots and quantification data of ER stress markers GRP78, PDI, CHOP, and Caspase-12 in the sham, SCI model and SCI model treated RA groups, RA compound with CQ, and CQ alone groups. FPDI =0.723, P = 0.566; FGRP78 = 2.171, P =0.169. &P< 0.05, %P< 0.05, versus SCI group. **P< 0.01, ##P< 0.01, versus treated RA groups. All data represent Mean values ± SEM, n = 5, F < 3.034 in all T-tests. (I) Representative micrographs showing double immunofluorescence with NeuN (neuron cell marker, green) and CHOP (red), nuclei are labeled with DAPI (blue ) in each group.
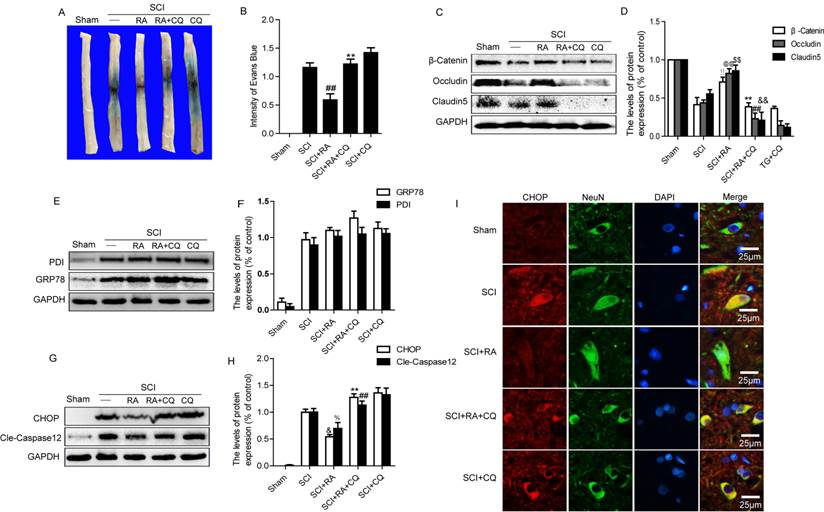
We then evaluated the alterations of ER stress-associated proteins when treatment was combined with CQ in vivo by western blot. As shown in Figure 5E and F, these results showed that there was no significant alteration of protein expression of GRP78 and PDI between the RA treated group and the RA combined with CQ group. Contrary to the effects of RA on these molecular chaperones, apoptosis proteins CHOP and caspase-12 were significantly increased following the inhibition of autophagy by treatment with CQ (Figure 5G and H). To further assess the influence of RA induced autophagy on the expression of CHOP, we performed double labeling immunofluorescence staining of CHOP and NeuN in the Sham, SCI-only, RA, RA combined with CQ and CQ-only groups. The results showed that the fluorescence intensity of CHOP immunoreactivity was increased after injury as compared to sham controls, and RA treatment significantly attenuated the increased fluorescence intensity. However, the fluorescence intensity of CHOP was significantly higher in the RA and CQ co-treated group compared with the RA-only group. These results illustrate that inhibition of autophagy by CQ efficiently abolishes the protective effect of RA with respect to apoptosis proteins CHOP and caspase-12 expression after SCI.
RA protects TG-treated ECs by inducing autophagic flux in vitro
To further confirm the effect of RA, ECs were treated with thapsigargin (TG), a specific inducer of ER stress, alone or combined with RA. In MTT assays, after exposure to TG (10 μM) for 6 h, cell viability was reduced by TG but was partially restored after RA addition (Figure 6A). The effect of RA on autophagy was assessed by western blot. Our data showed that compared with the TG-treated cells, levels of LC3B-II expression were lower in the control cells but were significantly higher in the RA-treated cells (Figure 6B and C). The level of P62 was decreased after exposure to TG (10 μM) for 6 h, and the level of P62 was further lowered in the RA-treated cells as compared with the TG-treated group (Figure 6B and C).
RA protects TG-treated ECs by inducing “Autophagic Flux” in vitro. ECs were treated with TG (10μM) or treated with RA (1 μM) or RA (5 μM), or compound with CQ (100μM) for 6h. (A) MTT results of RA-treated endothelial cells induced by TG. All experiments were repeated four times. All data represent Mean values ± SEM, **P< 0.01, versus TG treated cells, F = 1.225. (B), (C), (D) Representative western blots and quantification data of autophagy marker LC3-II and P62 in each group cells. **P< 0.01, versus TG treated cells, FLC3-II = 6.288; FP62 = 1.067. (E), (F) Representative western blots and quantification data of tight junction proteins Occludin, P120 and β-catenin in each group cells. !!P< 0.01, #P< 0.05, $$P< 0.01 versus TG group. **P< 0.01, ##P< 0.01, &&P< 0.01 versus TG groups, F < 2.834 in all T- tests. (G) immunofluorescence staining of Occludin (green) in ECs treated with TG for 6 h, nuclei are labeled with DAPI (blue). (H), (I) Representative western blots and quantification data of ER stress markers GRP78, PDI, CHOP, and Caspase-12 in each group cells, FPDI =0.678, P = 0.543; FGRP78 = 0.021, P =0.979. %%P< 0.01, &&P< 0.01, versus TG group. **P< 0.01, ##P< 0.01, versus treated RA groups. All data represent Mean values ± SEM, n = 4, F < 3.517 in all T-tests.
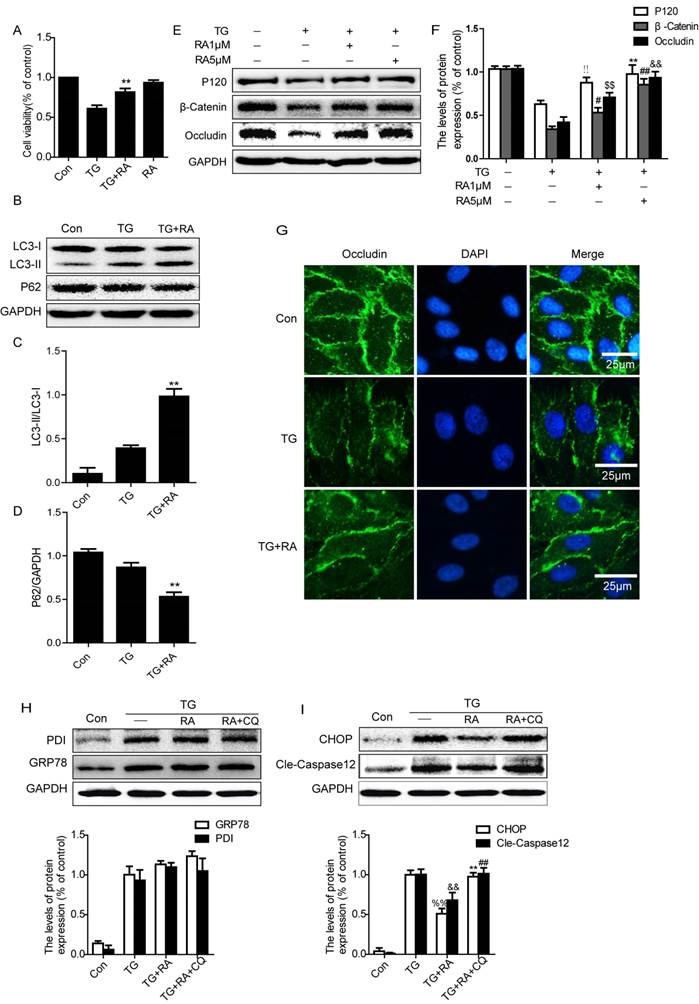
To examine the protective effect of RA in BSCB recovery in vitro, we detected the change of TJ and AJ in ECs. ECs were treated with TG alone (10 μM, 6 h) or combined with RA (1 μM, 5μM). Western blot results showed that the levels of P120, β-catenin and Occludin were decreased after treatment with TG as compared with the control group (Figure 6E and F). On the other hand, RA (1 and 5 μM) significantly attenuated the decrease in P120, β-catenin and Occludin induced by TG. Immunofluorescence staining also revealed that the intensity of Occludin expression decreased by TG compared to control cells, and RA treatment prevented the loss of Occludin expression (Figure 6G).
Furthermore, GRP78 and PDI were significantly higher in the TG-treated cells as compared with the control group, however there was no significant alteration in protein expression of GRP78 and PDI among TG-treated cells, RA-treated cells and RA combined with CQ cells (Figure 6H). On the contrary, the levels of apoptosis proteins CHOP and caspase-12 significantly increased in the TG-treated cells as compared with the control group, and RA treatment significantly decreased the level of CHOP and caspase-12. However, these decreases of CHOP and caspase-12 were reversed after the autophagy inhibitor CQ was added.
Discussion
Growing evidence shows that the BSCB might play a pivotal role in the development or progression of several diseases of the CNS [6, 48]. Disruption of BSCB has been reported to contribute to neuronal cell death both in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) [50], radiation injury to the spinal cord, multiple sclerosis (MS) [51], spinal cord ischemia, neuropathic pain, and traumatic SCI [9, 42]. Following the mechanical disruption of capillaries at the moment of the impact, the damage to the BSCB allows infiltration of macrophages and other immune cells to the area of injury, resulting in secondary injury in SCI [5, 10]. RA is a powerful vitamin A-derived morphogen in early CNS development and radial glia-derived RA is crucial in normal neurogenesis [1]. In addition, radial glial cells supply the brain with RA during the developmental cascade and associate closely with the developing vasculature. RA is also important for Blood-Brain Barrier (BBB) properties in brain ECs [52] and leads to an increase in TJ protein expression yielding a 4-fold increase in Occludin expression [18]. Recent studies have shown that RA synthesis by reactive astrocytes represents an endogenous protective response to neuroinflammation, possibly aimed at protecting the BBB against inflammatory insult [51]. Based on this evidence, we hypothesized that exogenous RA may have protective effect towards BSCB disruption after SCI. In our study, treatment with exogenous RA increased the locomotor function progressively during the experimental period, decreased the BSCB permeability, and increased the level of Occludin, Claudin5, β-catenin, and P120 proteins which are considered the most important membranous components of TJs. Altogether, these results indicate that exogenous RA administration has a protective effect on the BSCB and can improve recovery following SCI. To the best of our knowledge, the current study is the first time to demonstrate the association between BSCB disruption and RA under pathological conditions in acute injury.
It has been reported that autophagy activation is involved in SCI [36, 37, 53]. However, until recently its mechanisms and function remained unknown, especially with respect to the BSCB. Autophagy, a lysosome-dependent cellular degradation pathway, is an essential process for the maintenance of cellular homeostasis in the central nervous system under both physiological and pathological conditions [25, 26, 45]. Disruption of autophagy flux-defined as the progress of cargo through the autophagy system towards its subsequent degradation, has been reported to contribute to neuronal cell death in neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's disease [54-56]. Increased autophagy flux may be protective after mild injury [37], however excessive activation of autophagy is detrimental to neuronal cells [36, 55], indicating that disruption of autophagy is involved in the secondary damage mechanism. Most reports describe concomitant accumulation of both LC3-II and P62 after injury, suggesting uniform inhibition of autophagy flux [32]. However, in contrast to previous studies, in the present study the level of P62 was slightly decreased 1 day after SCI. These differences in autophagy flux may be attributed to the different injury models or injury severity which may differentially affect autophagy activation and the ability of autophagy flux to proceed to completion. Further investigation is needed to elucidate these discrepancies. Induction of autophagy flux by rapamycin can produce neuroprotective effects in acute SCI in rats via inhibition of apoptosis and improve functional recovery after SCI [27]. RA exerts a cell-type dependent stimulatory effect on autophagy flux via Beclin-1 up-regulation and inhibition of the mTOR pathway or by enhancing autophagosome maturation [33-35]. In the present study, the effects of RA induction on autophagy flux after SCI were consistent with previous reports. Therefore, we assume that the neuronal protective effect of RA might be related to its induction of autophagy flux after SCI. The current study showed that RA prevented loss of TJ and AJ proteins by induction of autophagy flux and inhibition of autophagy by CQ abolished the protective effect of RA on the BSCB both in vivo and in vitro. However, to the best of our knowledge, our group is the first to report the effect of exogenous RA on ECs and BSCB integrity by induction of autophagy flux after SCI. Further work is needed to determine how autophagy affect EC survival and TJ and AJ proteins after SCI.
Previous reports show that induction of GRP78 prevents neuronal death induced by severe ER stress both in vitro and in vivo [57, 58]. As a molecular chaperone, GRP78 regulates protein folding and facilitates protein translocation in the ER, which is involved in autophagy activation induced by ischemic preconditioning in neural cells [31]. PDI is an enzyme in the ER in eukaryotes that catalyzes the formation and breakage of disulfide bonds between cysteine residues within proteins as they fold, loss of PDI activity has been associated with the pathogenesis of numerous disease states including neurological protein folding disorders, most commonly related to the unfolded protein response (UPR) [59]. In current study there was no significant difference in protein expression of GRP78 and PDI between RA treated group and SCI-only group. Opposing the effects of these beneficial molecular chaperones, CHOP and caspase-12 are involved in ER stress-dependent apoptosis [60, 61]. The pro-apoptotic transcription factor CHOP plays a critical role in ER stress-induced apoptosis and is believed to play a central role in ER stress-induced cell death in CNS trauma [20, 60]. Caspase-12, a protein associated with the ER membrane, normally exists in an inactive pro-caspase form. Rapamycin is a specific inducer of autophagy and increased the expression of GRP78 while reducing the expression of cleaved caspase-12 and CHOP [28, 62]. In addition, inhibition of autophagy with 3-MA resulted in excessive ER stress leading to increases in CHOP and caspase-12 [29, 63]. Previous studies showed that rapamycin but not 3-MA treatment improved locomotor function after SCI and reduced ER stress-dependent apoptosis [27, 37]. In agreement with previous findings, our results showed that CHOP and caspase-12 expression were significantly inhibited after treatment with RA both in vivo and in vitro, while CQ significantly abolished RA-induced decreases in CHOP and caspase-12. The signaling mechanisms by which autophagy inhibits CHOP and caspase-12 expression remained unknown. We suspect that: (1) In UPR, autophagy performs a protective role by transporting misfolded proteins for degradation to attenuate severe ER stress, thus inhibiting ER stress-dependent apoptosis protein expression. (2) Apoptosis proteins are targeted by P62 and then act as a substrate for autophagy and were degraded during autophagy. (3) In present study, RA may inhibit the expression of CHOP and caspase-12 via activation of autophagy after SCI. However, the precise signaling mechanisms by which autophagy inhibits CHOP and caspase-12 expression remain to be determined.
In the clinic, RA is widely used in the treatment of acute leukemia and various skin diseases. Even so, over the past few decades RA has generated considerable excitement for its potential as a therapy for a wide variety of neurological disorders for which there are currently no treatments. In this study, we first demonstrated that RA plays a role in maintaining BSCB integrity by preventing the loss of TJ and AJ proteins under pathological conditions such as SCI, and improves functional recovery after SCI. Thus, the present study lays the ground work for future translation of RA in CNS diseases, especially those related to BSCB disruption. However, much work remains to be done. Whether RA promotes neural axon regeneration after SCI? How about the effect of RA on reactive astrocytosis and glial scar after SCI? Moreover, further work is needed to determine how RA affect changes in autophagy flux after SCI. Functional studies in transgenic animals with defects in autophagy, such as the Becn1-/- or Atg5 -/- mice, should also be performed.
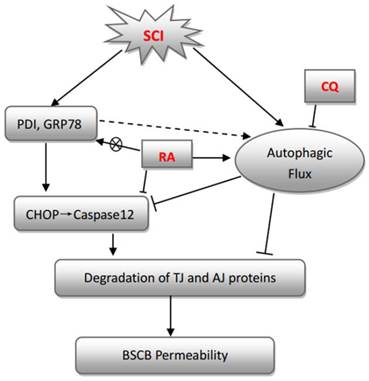
In conclusion, this study examined the effect of RA on autophagic flux, ER stress, and BSCB integrity after SCI. Our study shows that RA significantly attenuated BSCB permeability and degradation of TJ molecules such as P120, β-catenin, Occludin, and Claudin5 1 day after injury, and thus improved functional recovery after SCI. RA inhibited the expression of CHOP and caspase-12 by induction of autophagic flux both in vivo and in vitro, although it had no effect on expression of GRP78 and PDI. Furthermore, combination therapy with the autophagy inhibitor CQ partially abolished the BSCB protective effect of RA by exacerbating ER stress and the loss of TJs both in vivo and in vitro (Figure 7). Our results suggest that RA may provide potential therapeutic interventions for preventing BSCB disruption after SCI.
A model illustrating the BSCB protective effect of RA after SCI. Both autophagy and ER stress activation were involved in SCI, which aim to either repair or further damage the BSCB. Autophagy plays a central role in protecting SCI-induced BSCB disruption, which was induced by RA. ER stress-induced expression of CHOP and caspase-12 activation contributed to ECs death after SCI, which was inhibited by RA.
Acknowledgements
This study was partially supported by a research grant from the National Natural Science Funding of China (81572237, 81372112, 81302775, 81472165,), Zhejiang Provincial Natural Science Foundation of China (LY14H090013, LY14H150010, LY14H170002), Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents (to J.X.) and State Key Basic Research Development Program (2012CB518105).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mizee MR, Vries aHEd. Blood-brain barrier regulation. Tissue Barriers. 2013;1:e26882 1-6
2. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of disease. 2010;37:13-25
3. Cardoso FL, Brites D, Brito MA. Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain research reviews. 2010;64:328-63
4. Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: morphology and clinical implications. Annals of neurology. 2011;70:194-206
5. Palmer AM. The role of the blood-CNS barrier in CNS disorders and their treatment. Neurobiology of disease. 2010;37:3-12
6. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nature medicine. 2013;19:1584-96
7. Varma AK, Das A, Wallace Gt, Barry J, Vertegel AA, Ray SK. et al. Spinal cord injury: a review of current therapy, future treatments, and basic science frontiers. Neurochemical research. 2013;38:895-905
8. Robert AA, Zamzami MM. Traumatic spinal cord injury in Saudi Arabia: a review of the literature. The Pan African medical journal. 2013;16:104
9. Figley SA, Khosravi R, Legasto JM, Tseng YF, Fehlings MG. Characterization of vascular disruption and blood-spinal cord barrier permeability following traumatic spinal cord injury. Journal of neurotrauma. 2014;31:541-52
10. Sharma HS. Early microvascular reactions and blood-spinal cord barrier disruption are instrumental in pathophysiology of spinal cord injury and repair: novel therapeutic strategies including nanowired drug delivery to enhance neuroprotection. Journal of neural transmission. 2011;118:155-76
11. Penas C, Guzman MS, Verdu E, Fores J, Navarro X, Casas C. Spinal cord injury induces endoplasmic reticulum stress with different cell-type dependent response. Journal of neurochemistry. 2007;102:1242-55
12. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. British journal of anaesthesia. 2007;99:4-9
13. Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nature reviews Genetics. 2008;9:541-53
14. Kornyei Z, Gocza E, Ruhl R, Orsolits B, Voros E, Szabo B. et al. Astroglia-derived retinoic acid is a key factor in glia-induced neurogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2007;21:2496-509
15. Shearer KD, Fragoso YD, Clagett-Dame M, McCaffery PJ. Astrocytes as a regulated source of retinoic acid for the brain. Glia. 2012;60:1964-76
16. Paschaki M, Lin SC, Wong RL, Finnell RH, Dolle P, Niederreither K. Retinoic acid-dependent signaling pathways and lineage events in the developing mouse spinal cord. PloS one. 2012;7:e32447
17. van Neerven S, Mey J, Joosten EA, Steinbusch HW, van Kleef M, Marcus MA. et al. Systemic but not local administration of retinoic acid reduces early transcript levels of pro-inflammatory cytokines after experimental spinal cord injury. Neuroscience letters. 2010;485:21-5
18. Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Scientific reports. 2014;4:4160
19. Ohri SS, Maddie MA, Zhao Y, Qiu MS, Hetman M, Whittemore SR. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia. 2011;59:1489-502
20. Matsuyama D, Watanabe M, Suyama K, Kuroiwa M, Mochida J. Endoplasmic reticulum stress response in the rat contusive spinal cord injury model-susceptibility in specific cell types. Spinal cord. 2014;52:9-16
21. Lee JY, Maeng S, Kang SR, Choi HY, Oh TH, Ju BG. et al. Valproic acid protects motor neuron death by inhibiting oxidative stress and endoplasmic reticulum stress-mediated cytochrome C release after spinal cord injury. Journal of neurotrauma. 2014;31:582-94
22. Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z. et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy. 2014;10:1801-13
23. Mosquera L, Colon JM, Santiago JM, Torrado AI, Melendez M, Segarra AC. et al. Tamoxifen and estradiol improved locomotor function and increased spared tissue in rats after spinal cord injury: their antioxidant effect and role of estrogen receptor alpha. Brain research. 2014;1561:11-22
24. Liu L, Liu C, Zhong Y, Apostolou A, Fang S. ER stress response during the differentiation of H9 cells induced by retinoic acid. Biochemical and biophysical research communications. 2012;417:738-43
25. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology. 2010;12:814-22
26. Emr DJKSD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717-21
27. Chen HC, Fong TH, Hsu PW, Chiu WT. Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. The Journal of surgical research. 2013;179:e203-10
28. Yin J, Gu L, Wang Y, Fan N, Ma Y, Peng Y. Rapamycin improves palmitate-induced ER stress/NF kappa B pathways associated with stimulating autophagy in adipocytes. Mediators of inflammation. 2015;2015:272313
29. Zhang R, Wang R, Chen Q, Chang H. Inhibition of autophagy using 3methyladenine increases cisplatininduced apoptosis by increasing endoplasmic reticulum stress in U251 human glioma cells. Molecular medicine reports. 2015
30. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S. et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and cellular biology. 2006;26:9220-31
31. Zhang XY, Zhang TT, Song DD, Zhou J, Han R, Qin ZH. et al. Endoplasmic reticulum chaperone GRP78 is involved in autophagy activation induced by ischemic preconditioning in neural cells. Mol Brain. 2015;8:20
32. Liu S, Sarkar C, Dinizo M, Faden AI, Koh EY, Lipinski MM. et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell death & disease. 2015;6:e1582
33. Zeng M, Zhou JN. Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cellular signalling. 2008;20:659-65
34. Trocoli A, Mathieu J, Priault M, Reiffers J, Souquere S, Pierron G. et al. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy. 2011;7:1108-14
35. Yogendra Rajawat, Bossis ZHaI. Autophagy A target for retinoic acids. Autophagy. 2010;6:1224-6
36. Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine. 2011;36:E1427-34
37. Tang P, Hou H, Zhang L, Lan X, Mao Z, Liu D. et al. Autophagy reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis after spinal cord injury in rats. Molecular neurobiology. 2014;49:276-87
38. Smith CM, Mayer JA, Duncan ID. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:8088-100
39. Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell death and differentiation. 2013;20:515-23
40. Zhang H, Wu F, Kong X, Yang J, Chen H, Deng L. et al. Nerve growth factor improves functional recovery by inhibiting endoplasmic reticulum stress-induced neuronal apoptosis in rats with spinal cord injury. Journal of translational medicine. 2014;12:130
41. Xanthos DN, Pungel I, Wunderbaldinger G, Sandkuhler J. Effects of peripheral inflammation on the blood-spinal cord barrier. Molecular pain. 2012;8:44
42. Lee JY, Kim HS, Choi HY, Oh TH, Yune TY. Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood-spinal cord barrier after spinal cord injury. Brain: a journal of neurology. 2012;135:2375-89
43. Lee JY, Kim HS, Choi HY, Oh TH, Ju BG, Yune TY. Valproic acid attenuates blood-spinal cord barrier disruption by inhibiting matrix metalloprotease-9 activity and improves functional recovery after spinal cord injury. Journal of neurochemistry. 2012;121:818-29
44. Zhang H-Y, Wang Z-G, Wu F-Z, Kong X-X, Yang J, Lin B-B. et al. Regulation of Autophagy and Ubiquitinated Protein Accumulation by bFGF Promotes Functional Recovery and Neural Protection in a Rat Model of Spinal Cord Injury. Molecular neurobiology. 2013;48:452-64
45. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-41
46. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062-75
47. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. The Journal of biological chemistry. 2007;282:24131-45
48. Dobrowolska JBA. Physiology and pharmacological role of the blood-brain barrier. Pharmacological Reports. 2008;60:600-22
49. Lee SJ, Kim SH, Kang JG, Kim CS, Ihm SH, Choi MG. et al. Effects of all-trans retinoic acid on sodium/iodide symporter and CCAAT/enhancer-binding protein-homologous protein under condition of endoplasmic reticulum stress in FRTL5 thyroid cells. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2011;43:331-6
50. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178-201
51. Mizee MR, Nijland PG, van der Pol SM, Drexhage JA, van Het Hof B, Mebius R. et al. Astrocyte-derived retinoic acid: a novel regulator of blood-brain barrier function in multiple sclerosis. Acta neuropathologica. 2014;128:691-703
52. Mizee MR, Wooldrik D, Lakeman KA, van het Hof B, Drexhage JA, Geerts D. et al. Retinoic acid induces blood-brain barrier development. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:1660-71
53. Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y. et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321-33
54. Alirezaei M, Kemball CC, Whitton JL. Autophagy, inflammation and neurodegenerative disease. The European journal of neuroscience. 2011;33:197-204
55. Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends in neurosciences. 2006;29:528-35
56. Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nature reviews Neuroscience. 2011;12:437-52
57. Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Chan PH. Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:949-61
58. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. The Journal of biological chemistry. 2003;278:20915-24
59. Pharmacology Do. The human protein disulfide isomerase gene family. Human Genomics. 2012;6:1479-7364
60. Fassbender JM. et al. Deletion of Endoplasmic Reticulum Stress-Induced CHOP Protects Microvasculature Post-Spinal Cord Injury. Current Neurovascular Research. 2012;9:274-81
61. Zhang HY, Wang ZG, Lu XH, Kong XX, Wu FZ, Lin L. et al. Endoplasmic Reticulum Stress: Relevance and Therapeutics in Central Nervous System Diseases. Molecular neurobiology. 2014
62. Rui She ng, Xiao-Qi an Liu. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2015;8:310-25
63. Song L, Liu H, Ma L, Zhang X, Jiang Z, Jiang C. Inhibition of autophagy by 3-MA enhances endoplasmic reticulum stress-induced apoptosis in human nasopharyngeal carcinoma cells. Oncology letters. 2013;6:1031-8
Author contact
![]() Corresponding author: Jian Xiao, School of Pharmacy, Wenzhou Medical University, Wenzhou 325035, Zhejiang 325035, PR China. Tel.:+86-577-85773087; Fax.: +86-577-85773087; E-mail: xfxj2000com. Hua-Zi Xu, Department of Orthopaedics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, PR China. Tel.: +86-577-88816381; Fax.: +86-577-88816381; E-mail: spinexucom
Corresponding author: Jian Xiao, School of Pharmacy, Wenzhou Medical University, Wenzhou 325035, Zhejiang 325035, PR China. Tel.:+86-577-85773087; Fax.: +86-577-85773087; E-mail: xfxj2000com. Hua-Zi Xu, Department of Orthopaedics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, PR China. Tel.: +86-577-88816381; Fax.: +86-577-88816381; E-mail: spinexucom