Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
Methods
Results
Discussion
Conclusion
Acknowledgements
Supplementary Material
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(6):782-793. doi:10.7150/ijbs.18732 This issue Cite
Research Paper
Dioscin Induces Gallbladder Cancer Apoptosis by Inhibiting ROS-Mediated PI3K/AKT Signalling
Xiaoling Song1, 2*, Zheng Wang1, 2*, Haibin Liang1, 2*, Wenjie Zhang1*, Yuanyuan Ye1, 2, HuaiFeng Li1, 2, Yunping Hu1, 2, Yijian Zhang1, 2, Hao Weng1, Jianhua Lu1, Xuefeng Wang1, Maolan Li1, 2, Yingbin Liu1, 2 ![]() , Jun Gu1
, Jun Gu1 ![]()
1. Department of General Surgery and Laboratory of General Surgery, Xinhua Hospital affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, People's Republic of China;
2. Institute of Biliary Tract Disease, Shanghai Jiao Tong University School of Medicine, Shanghai, People's Republic of China.
* Xiaoling Song, Zheng Wang, Haibin Liang and Wenjie Zhang contributed equally to this work.
Received 2016-12-13; Accepted 2017-3-25; Published 2017-6-1
Abstract

Gallbladder cancer (GBC), highly aggressive form of cancer with an extremely poor prognosis, is the most common malignancy of the biliary tract. In this study, we investigated the effects of dioscin (DSN) on human GBC and the potential mechanisms underlying these effects. The results showed that DSN significantly inhibited GBC cell proliferation and migration. Moreover, DSN induced GBC cell apoptosis via mitochondrial dependent apoptotic signalling. Reactive oxygen species (ROS) and glutathione (GSH) levels were measured, and ROS scavengers completely inhibited DSN-induced apoptosis and migration, indicating that ROS play an essential role in GBC progression. Western blot analysis showed that AKT activity was significantly downregulated after DSN treatment, and that inhibition/ectopic expression of AKT enhanced/abolished DSN-induced apoptosis but not migration. Furthermore, we confirmed the relationship between ROS and the PI3K/AKT pathway and found that DSN induced apoptosis by regulating ROS-mediated PI3K/AKT signaling. Taken together, these findings indicate that DSN induces GBC apoptosis through inhibiting ROS-mediated PI3K/AKT signalling.
Keywords: gallbladder cancer, apoptosis, dioscin, reactive oxygen species.
Background
GBC is the most common and aggressive biliary tract malignancy and the fifth gastrointestinal cancer. The only available curative treatment is complete surgical resection; however, only 10% of patients are eligible for surgery because of its asymptomatic characteristics and chemoresistance. As a result, the 5-year survival rate for GBC remains between 0% and 10% in most reported series [4]. Additionally, among those patients who undergo surgical resection, recurrence rates remain high [4]. Therefore, novel and effective therapies are urgently needed.
Natural compounds, especially plant-derived compounds, have been widely used as therapeutic agents against cancer. Dioscin (DSN), a plant glucoside saponin, extracted from Dioscorea nipponica Makino and Dioscorea zingiberensis Wright, has been shown to exert many biological and pharmacological effects. Previous studies have shown that DSN has anti-fungal, anti-virus and hepatoprotective properties. DSN has recently attracted increasing amount of attention because of its anti-cancer effects on lung cancer, colon cancer, breast cancer and gastric cancer [9]. However, the effects of DSN on GBC has not been determined.
Reactive oxygen species (ROS) are a group of reactive, short-lived, oxygen-containing species, such as superoxide, singlet oxygen atoms, hydrogen peroxide, hydroxyl radicals and peroxyl radicals. ROS can activate intracellular signal transduction pathways in cancer, such as inflammation, cell cycle progression, apoptosis, migration and invasion. A previous investigation showed that DSN induced generation of ROS through mitochondria dysfunction [10]. Whether ROS generation exerts anti-cancer effects on GBC has not yet been illustrated. In this study, we investigated the effects of DSN on GBC cells and their potential mechanisms underlying the induction of GBC cell apoptosis and migration. Our results showed that DSN induced GBC cell apoptosis by regulating ROS-mediated PI3K/AKT signalling, thus we deduced DSN may represent a novel therapeutic intervention for GBC.
Methods
Drugs and reagents
DSN was purchased from Sigma-Aldrich (St. Louis, MO, USA); the purity was at least 98%, as determined by high performance liquid chromatography. DSN was dissolved in dimethyl sulfoxide (DMSO) in a 100 mM stock solution and stored at -20°C. The final DMSO concentration was less than 0.1%. Cell Counting Kit-8 (CCK-8), Hoechst 33342 and Rhodamine 123 kits were purchased from Sigma-Aldrich. Annexin V/Propidium Iodide (PI) Apoptosis Kit was purchased from Invitrogen (Carlsbad, CA, USA). A pan-caspase inhibitor (Z-VAD-FMK) and PI3K inhibitor (LY294002) as well as GSH, NAC and GSH/GSSH and ROS detection kits were purchased from Beyotime Institute of Biotechnology Company (Suzhou, Jiangsu, China). All antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All cell culture supplies were obtained from Invitrogen (Carlsbad, CA, USA).
Cell culture
The human GBC cell lines NOZ and SGC996 were purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, Shanghai, China). NOZ cells were cultured in William's medium, and SGC996 cells were cultured in 1640 medium. All media were supplemented with 100 μg/ml streptomycin and 100 U/ml penicillin and 10% foetal bovine serum. The cells were cultured at 37°C in a humidified incubator with 5% CO2.
Cell viability assay
A CCK-8 assay was used to evaluate NOZ and SGC996 cell viability. Human kidney epithelial cells (293T) was used to determine the change of normal cells. Cells were seeded into 96-well plates at a density of 4000 cells/well and were cultured for approximately 24h. Various concentrations of DSN (0, 1, 2, 4, 6, 8 μM) were subsequently added, and the cells were incubated for 24h, 48h or 72h. After treatment, CCK-8 (10μl) was added and incubated for 3h in the dark. Absorbance was measured at 450 nm using a microplate reader (Norcross, GA, USA). IC50 was measured by Graphpad Prism 5.
Colony formation assay
SGC996 and NOZ cells were seeded into 6-well plates with DSN (0, 2, 4, 6μM) for 15 days. Then, the cells were fixed with 10% formalin and stained with 0.1% crystal violet (Sigma-Aldrich, St. Louis, MO, USA). After washing, the plates were dried and the colonies (with more than 50 cells) were observed under a microscope.
Annexin V/PI staining assay for apoptosis
SGC996, NOZ and 293T cells were treated with DSN for 48h. After centrifugation (1500rpm, 5min), the cells were combined with 1x Annexin V binding buffer and then incubated with 5ul Annexin V and PI at 37°C for 30min in the dark. Cell apoptosis was measured using flow cytometry.
Hoechst 33342 staining
SGC996 and NOZ cells were treated with DSN (0, 2, 4, 6μM) for 48h. After the cells were fixed with 1 ml methanol/acetic acid (3:1) and stained with 5μg/ml Hoechst 33342, a fluorescence microscope was used to analyse the morphological changes.
Mitochondrial membrane potential (ΔΨm) assay
Cells were treated with DSN (0, 2, 4, 6μM) for 48 h, collected and washed with cold PBS. Then, the cells were incubated with Rhodamine 123 in a 5% CO2 incubator at 37°C for 20 min in the dark. Finally, the cells were analysed by flow cytometry.
Transwell migration analysis
To prevent effects of proliferation and apoptosis, we chose the indicated concentrations (0, 0.5, 1, 2μM) for subsequent assays. After the cells were resuspended, 200μl of medium containing 2 × 105 cells with different treatment was added into the upper well and incubated for 24-72h. Images of the stained cells were captured by microscopy.
ROS and GSH detection
Intracellular ROS generation was assessed using a ROS assay kit, and GSH generation was assessed using a GSH/GSSH assay kit according to the manufacturer's instructions.
Plasmid transfection
Two eukaryotic expression vectors containing cDNA encoding wild-type (WT) and constitutively active (CA) mouse AKT and an empty vector (pcDNA 3.1) were purchased from Era Biotech (Shanghai, Shanghai, China). Cells were seeded on 24-well plates and transfected for 24h using Viafect transfection reagent, according to the manufacturer's protocol.
Western blot analysis
Proteins were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked for 1h at 37°C and incubated with primary antibodies against Bcl-2, Bax, cleaved caspase9, cleaved caspase3, cleaved poly(ADP ribose) polymerase (PARP), PI3K, pAKT, p-AKT and GAPDH overnight at 4°C. Next, the membranes were incubated with secondary antibodies for 1h at 37 °C. Proteins were observed using a Gel Doc 2000 (Berkeley, California, USA).
In vivo tumour xenograft study
All animal treatments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee of Shanghai Jiaotong University. Male nude mice (aged 4-6 weeks, weighting 18-22 g) were purchased from Shanghai SLAC Laboratory Animal Co Ltd (Shanghai, Shanghai, China). The animals were housed at 25°C±2°C at a relative humidity of 70%±5% under natural light/dark conditions for 1 week and were allowed free access to food and water. NOZ cells (at a density of 1x106 cells in 0.2 ml) were injected into the right axilla of each mouse. Twenty-four hours later, the mice were randomly divided into 3 groups (control, 5mg/kg, 10mg/kg). Mice received DSN at the appropriate dose (0, 5 or 10mg/kg) every 3 days for up to 25 days. On day 26, the animals were sacrificed, and their tumours were dissected and weighed.
HE staining and Immunohistochemistry
After the mice were sacrificed, their tumours, livers and spleens were resected and immediately fixed in 10% formalin, embedded in paraffin, cut into 5mm sections and mounted on slides. The expression patterns of PI3K and p-AKT were analysed using immunohistochemical (IHC) streptavidin-peroxidase staining.
Statistical analysis
All experiments were performed at least 3 times, and the results are expressed as the means ± standard deviations unless otherwise stated. Student's t-test was used to compare differences between the treated groups and the corresponding control groups; p<0.05 was considered statistically significant.
Results
DSN inhibits GBC cell proliferation and migration
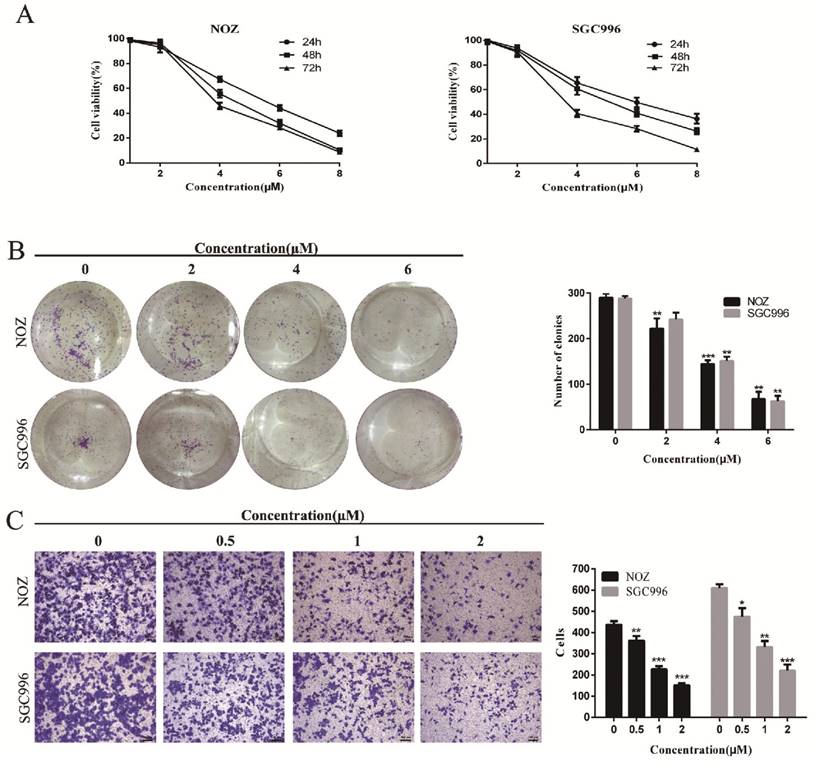
CCK-8 assay and colony formation assays were performed to determine the effects of DSN on GBC cell proliferation. As shown in Figure 1A-1B, NOZ and SGC996 cell proliferation was significantly inhibited by DSN in a dose-dependent manner, with an IC50 value of 4.465μM (NOZ) and 5.049 μM (SGC996), and the numbers and sizes of the colonies treated with DSN were substantially smaller than those of the control group. Migration was evaluated by transwell migration analysis. We chose concentrations (0, 0.5, 1, 2μM) that would not affect cell viability. As shown in Figure 1C, DSN repressed NOZ and SGC996 cell migration in a dose-dependent manner. Therefore, DSN inhibits GBC cell proliferation and migration.
DSN inhibits GBC cell proliferation and migration. A) NOZ and SGC996 cells were treated with DSN (0, 1, 2, 4, 6, 8μM) for 24h, 48h and 72h. CCK-8 assays were carried out to assess proliferation. B) DSN suppressed NOZ and SGC996 cell colony formation. Cells were exposed to DSN (0, 2, 4, 6μM) and were allowed to form colonies for 15 days. C) The effects of DSN on GBC cell migration were assessed by transwell migration analysis. All data are presented as the means ± standard deviations, and each experiment was repeated 3 times. Significant differences compared with the control are indicated by *p<0.05, **p<0.01, and ***p<0.001.
DSN induces GBC cell apoptosis
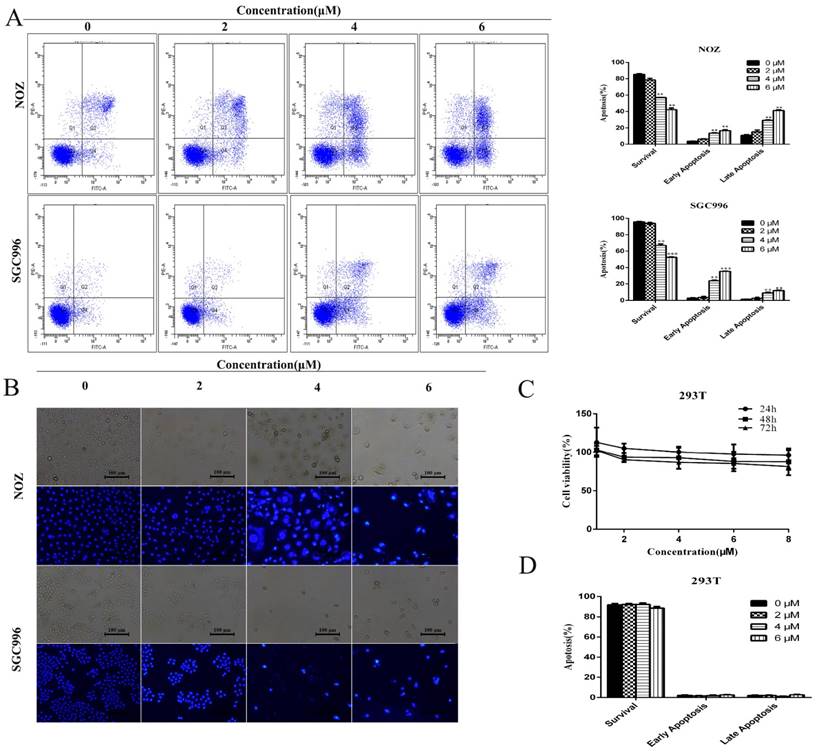
We investigated the effects of DSN on GBC cell apoptosis by flow cytometry and Hoechst 33342 staining. Compared with control group, DSN-treated cells exhibited significant chromatin condensation and fragmentation, and the percentage of early and late apoptosis cells were strikingly elevated in a dose-dependent manner (Figure 2A-2B). As no normal gallbladder cells were available, we used human kidney epithelial cells (293T) to determine whether normal cells undergo apoptosis following DSN treatment [1]; CCK-8 and flow cytometry assays revealed that DSN-treated 293T cells did not undergo apoptosis( Figure 2C -2D).
DSN induces GBC cell apoptosis. A) NOZ and SGC996 cells were treated with DSN for 48 h. Apoptosis was analysed by flow cytometry. B) NOZ and SGC996 cells were exposed to DSN for 48 h, and the nuclear morphological changes associated with apoptosis were evaluated by Hoechst33342 staining. C-D) 293T cell viability and apoptosis were evaluated by CCK-8 analysis and flow cytometry. All data are presented as the means ± standard deviations, and each experiment was repeated 3 times. Significant differences compared with the control are indicated by *p<0.05, **p<0.01, and ***p<0.001.
DSN induces mitochondrial dependent apoptosis in NOZ and SGC996 cell
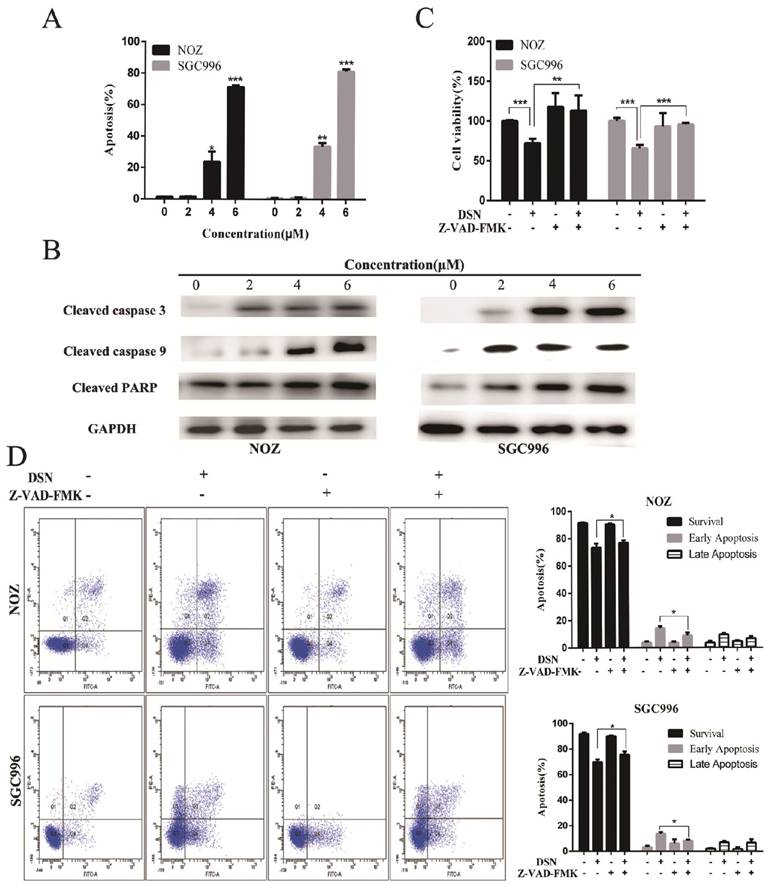
Mitochondria regulates the mitochondrial permeability transition and the release of toxic components, such as caspases to regulate apoptosis. The loss of ΔΨm was indicated by a decrease in Rhodamine 123 fluorescent staining intensity. Compared with control group, DSN-treatment increased numbers of Rhodamine 123-negative cells (Figure 3A). Moreover, DSN treatment upregulated the levels of cleaved caspase PARP, 3 and 9, which may have facilitated mitochondrial dependent apoptosis (Figure 3B). To confirm these results, we evaluated cell viability after DSN treatment in the presence or absence of Z-VAD-FMK, a caspase inhibitor. As shown in Figure 3D, Z-VAD-FMK abolished DSN-induced GBC cell cytotoxity. Together, these results indicate that DSN induces mitochondrial dependent apoptosis in NOZ and SGC996 cell.
DSN induces mitochondrial dependent apoptosis in NOZ and SGC996 cell. A) NOZ and SGC996 cells were treated with DSN and stained with Rhodamine 123. Cells with high ΔΨm are marked “survival”, and those with low ΔΨm are marked “apoptosis”. B) Apoptosis-related protein expression in NOZ and SGC996 cells was analysed by Western blot analysis. GAPDH was used as a loading control. C-D) After treatment with DSN in the presence or absence of Z-VAD-FMK, GBC cell viability and apoptosis ware evaluated by CCK-8 analysis and flow cytometry.
ROS accumulation is associated with DSN-induced apoptosis and migration inhibition
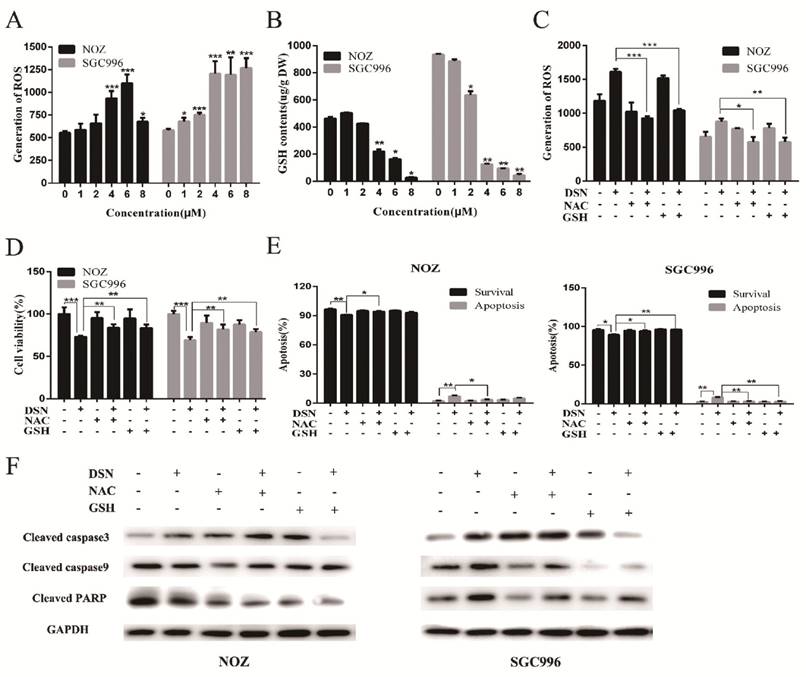
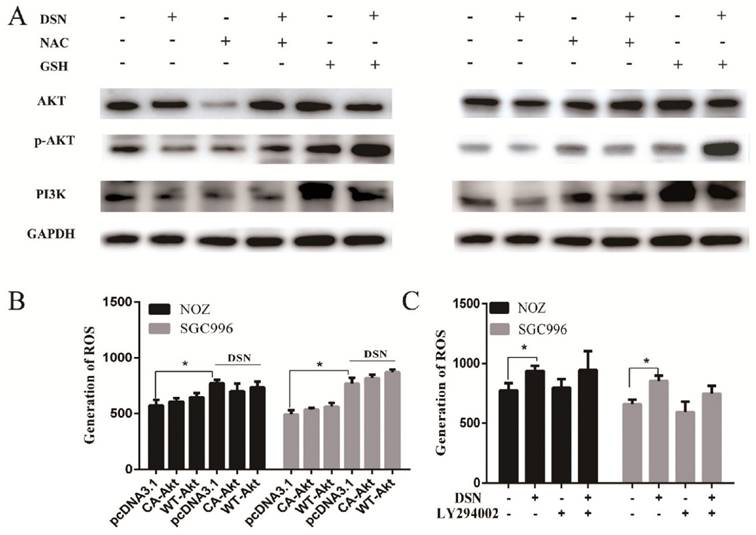
Many studies have revealed that ROS accumulation can induce cell death in many types of cancers after treatment with anti-cancer drugs. Therefore, we investigated whether ROS are associated with DSN-induced apoptosis. We detected intracellular ROS generation following different DSN treatment intervals, and we found that ROS generation was most significant at 4h of treatment (Figure S1A). As shown in Figure 4A, increasing amounts of ROS were generated in a dose-dependent manner at lower concentrations than at higher concentrations, as higher doses were ineffective at inducing ROS because of their strong cytotoxic effect. Consistent with these results, fluorescence analysis by microscopy also demonstrated that ROS generation was dose-dependent (Figure S1B). GSH is the major cellular ROS-scavenger, treatment strategies inducing deceases in the level of reduced GSH may have a profound effect on cell survival and drug sensitivity by altering the ability of cells to detoxify ROS [15]. Thus, we investigated whether DSN-induced apoptosis is related to GSH levels. After treatment with DSN for 48h, dose-dependent decreases in intracellular GSH levels were observed, as shown in Figure 4B. Moreover, we treated GBC cell in the presence or absence of the ROS scavengers NAC and GSH, and then assessed intracellular ROS production and cytotoxicity. As shown in Figure 4C -4E, NAC and GSH abolished DSN-induced ROS accumulation and cytotoxicity. Furthermore, NAC and GSH also abolished DSN-induced mitochondrial dependent apoptosis (Figure 4E -4F, Figure S1C). Consistent with the findings of previous studies, we found that ROS accumulation was related to DSN-induced migration inhibition (Figure S2A-S2B).
ROS accumulation is associated with DSN-induced apoptosis and migration inhibition. A) NOZ and SGC996 cells were treated with various concentrations of DSN for 4h, followed by incubation with the fluorescent probe DCFH-DA (10μM) for 30min, the ROS generation was detected using a microplate reader. B) GSH generation was determined in the absence or presence of 5mM NAC or 5mM GSH for 1h. C-D) ROS generation and cell viability were determined in the absence or presence of 5mM NAC or 5mM GSH for 1h. E) NOZ and SGC996 cells were treated in the absence or presence of NAC and GSH for 24h. Apoptosis was analysed by flow cytometry. F) Apoptosis-related protein expression in NOZ and SGC996 cells was analysed by western blot. GAPDH was used as a loading control.
Collectively, our results showed that DSN-induced apoptosis and migration are related to mitochondrial ROS accumulation.
DSN induces GBC cell apoptosis by inhibiting the PI3K/AKT signalling pathway
The PI3K/AKT pathway is one of the major signalling pathways associated with the growth of various tumours, and it is also associated with cancer progression and invasion. To determine whether the PI3K/AKT signalling pathway is associated with DSN-induced apoptosis, we estimated the effects of DSN on PI3K/AKT-related protein expression by western blot analysis. As shown in Figure 5A, DSN significantly inhibited PI3K/AKT signalling. To further confirm whether DSN-induced GBC apoptosis is mediated by Akt activity inhibition, we next transfected wild-type AKT (WT-AKT) and constitutively active AKT (CA-AKT) into GBC cells. As shown in Figure 5C, S3A-3C, ectopic expression of AKT (Figure 5B) abolished DSN-induced apoptosis and cytotoxicity. After treatment with DSN in the presence or absence of LY294002, a PI3K/AKT inhibitor, we found that LY294002 significantly enhanced DSN-induced cell death (Figure 5D-5E, S4A-S4B). Furthermore, we found that the PI3K/AKT signalling pathway is not associated with DSN-induced migration inhibition (Figure S5A-S5D). Together, these results indicate that the PI3K/AKT signalling pathway is associated with DSN-induced apoptosis but not migration.
DSN induces GBC cell apoptosis by inhibiting the PI3K/AKT signalling pathway. A) Protein lysates from cells treated with various concentrations of DSN for 48h were subjected to western blot analysisto measure AKT phosphorylation. B) AKT and p-AKT expression was analysed by western blotting after transfection with WT-AKT and CA-AKT. C) Cell viability was determined after transfection with WT-AKT and CA-AKT in the absence or presence of DSN for 24h. D) Cell viability was determined in the absence or presence of 50μM LY294002. E) AKT and p-AKT were analysed by western blotting in the absence or presence of 50μM LY294002.
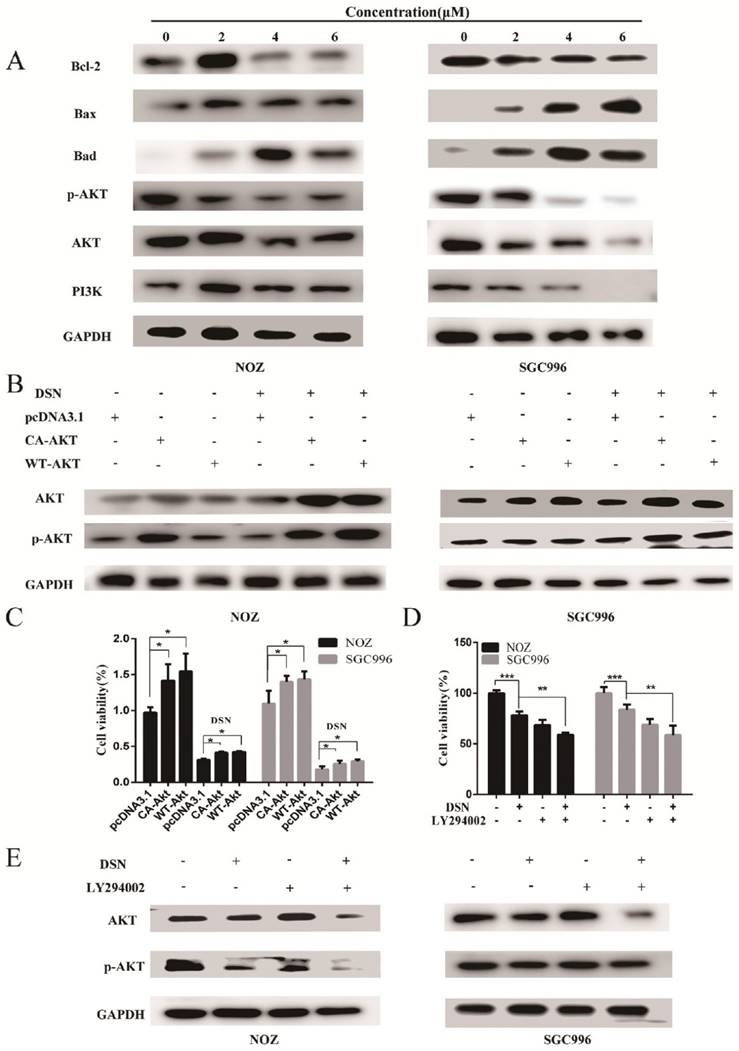
DSN induces GBC cell apoptosis by regulating ROS-mediated PI3K/AKT signalling
Our previous results showed that DSN-induced GBC apoptosis is related to ROS accumulation and PI3K/AKT signalling pathway inhibition. To confirm the relationship between ROS and the PI3K/AKT signalling pathway, we examined GBC cells in the presence or absence of NAC or GSH. As showed in Figure 6A, NAC and GSH enhanced PI3K, p-AKT and AKT expression. However, ectopic AKT and LY294002 had no effect on ROS generation (Figure 6C-6D). Thus, we believe that DSN induces GBC cell apoptosis by regulating ROS-mediated PI3K/AKT signalling not AKT-mediated ROS accumulation.
DSN induces GBC cell apoptosis by regulating ROS-mediated PI3K/AKT signalling. A) NOZ and SGC996 cells were treated with 4μM DSN in the absence or presence of NAC and GSH, PI3K/AKT signaling pathway-related protein expression was analysed by western blotting. B) NOZ and SGC996 cell ROS generation after transfection with CA-AKT and WT-AKT. C) NOZ and SGC996 cell ROS generation in the absence or presence of LY294002. All data are presented as the means ± standard deviations, and each experiment was repeated 3 times. Significant differences compared with the control are indicated by *p<0.05, **p<0.01, and ***p<0.001.
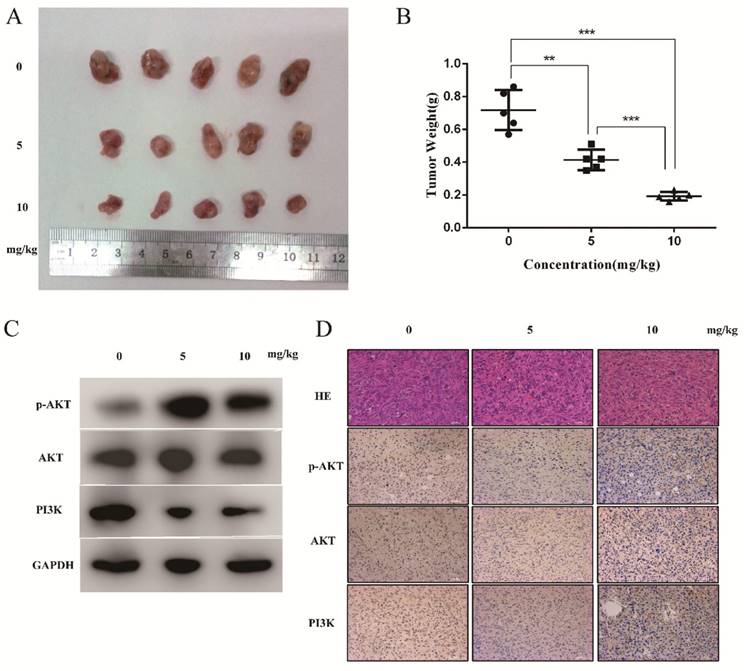
DSN inhibits tumour growth in vivo
To evaluate the anti-cancer effects of DSN in vivo, we injected mice with DSN at a concentration of 0, 5mg/kg or 10mg/kg every 2 days after inoculation with NOZ cells. We found that DSN inhibited tumour growth in a dose-dependent manner without other obvious appearance changes (Figure 7A- 7B, S6). Based on this observation, we performed western blot analysis, HE staining and IHC analysis. As shown in Figure 7C-7D, PI3K, p-AKT and AKT expression levels were significantly reduced compared with those of the control group. Moreover, from HE staining of the mice's liver and spleen, dioscin showed no significant effect on organ structures compared to the control group (Figure S6). These results are consistent with the in vitro effects of DSN.
DSN inhibits tumour growth in vivo. A) Different concentrations (0, 5 mg/kg and 10 mg/kg) of DSN were injected into nude mice after NOZ cell inoculation every 2 days. Images of 5 representative mice from each group are presented to show the sizes of the resultant tumours. B) Tumours were excised from the animals and weighed. C, D) AKT, p-AKT and PI3K expression levels were analysed using western blot analysis(C) and HE ,IHC staining(D).
Discussion
Despite significant improvements in the technical aspects of GBC detection and management, there are no known effective adjuvant therapies for GBC, although various combinations have been used clinically. The GBC metastatic cascade is complex and regulated at multiple levels. However, studies have shown that natural Chinese medicines play an important role in GBC therapy. The traditional Chinese medicine DSN has been shown to present anti-fungal, anti-virus and hepatoprotective effects. Therefore, we aimed to determine whether DSN exerts anti-cancer effects on GBC.
In this study, we investigated NOZ and SGC996 cell proliferation and viability using CCK-8 analysis and colony formation assays. As no normal gallbladder cells were available, human kidney epithelial cells (293T) was used as a control. We found that DSN significantly inhibited GBC cell proliferation, but it was not highly toxic to 293T even at higher doses (8 μM). Moreover, DSN inhibited GBC cell migration at concentrations without effects of proliferation and apoptosis. Therefore, we propose that DSN represents a new and promising therapeutic agent for GBC. In the following, we evaluated the effects of DSN treatment in mice with xenografted tumours. Based on the weights and volumes, HE staining of livers and spleen of three groups, we concluded that DSN exerted anti-cancer effects on GBC cells in vivo.
ROS are an important mediator of many anti-cancer agents, which are derived from at least two sources: the oxidative protein folding machinery in the endoplasmic reticulum (ER) and mitochondria. Mitochondria possess a total of nine potential ROS generation sites that can leak single electrons to oxygen and convert it into a superoxide anion, a progenitor ROS during the depletion of mitochondrial transmembrane potential (∆Ψm). Attenuation of ∆Ψm can induce caspase9 activation, which subsequently induces the caspase3 cleavage. These caspases are believed to cause cell death through PARP cleavage, chromatin condensation, and DNA fragmentation. In our study, after treatment with different concentration of DSN for 48h, DSN-treated cells were obviously chromatin condensation and fragmentation compared with control group (Figure 2B). Moreover, we found that ROS accumulated in conjunction with ∆Ψm decrease, and the level of cleaved PARP, caspase3, 9 increased in a dose-dependent manner. Furthermore, the inhibition of apoptosis with pan-caspase inhibitor, Z-VAD-FMK, effectively blocked DSN-induced cell death, indicating that DSN induced mitochondrial dependent apoptosis.
In the following, we treated GBC cells with DSN and measured ROS generation. We found that DSN induced ROS accumulation at lower concentrations at 4h after DSN treatment. However, ROS scavengers NAC and GSH, can repress ROS generation and cytotoxicity, indicating that ROS was involved in DSN-induced apoptosis. GSH acts as an anti-oxidant by quenching reactive oxygen species and is involved in the ascorbate-glutathione cycle, which eliminates damaging peroxides. We measured GSH generation and noted decreased GSH depletion. Thus, we concluded that DSN inhibits ROS elimination and then induces significant ROS accumulation in GBC cells, leading to oxidative damage and cell death. As DSN inhibits GBC cells migration, we treated GBC cells with DSN and NAC/GSH, and found that DSN-induced migration inhibition via ROS generation. Furthermore, it is noteworthy that blocking ROS generation prevented the DSN-induced phosphorylation of PARP, caspase3 and caspase9, demonstrating that DSN stimulated the production of ROS, which subsequently actived DSN induced mitochondrial dependent apoptosis.
PI3K/AKT signalling is frequently deregulated in many human cancers, and AKT is a key downstream effector of PI3K that regulates a variety of biological processes, including survival, proliferation, apoptosis, and differentiation. Western blot analysis indicated that DSN treatment strikingly reduced PI3K/AKT pathway activation in GBC cells. Moreover, AKT and p-AKT overexpression/inhibition abolished/enhanced DSN-induced apoptosis. However, DSN-induced migration inhibition is not related to the PI3K/AKT signalling pathway. All of these findings demonstrate that DSN inhibits GBC cell proliferation and apoptosis by regulating the PI3K-AKT signalling pathway.
Evidence indicates that transient or moderate ROS production serves as a second messenger that regulates AKT activation in various types of cells, such as haematopoietic stem cells and cardiac cells. Thus we performed experiments to confirm the relationship between ROS and the PI3K/AKT pathway. Our results showed that NAC and GSH enhanced PI3K, p-AKT and AKT expression, whereas ectopic AKT and LY294002 expression had no effect on ROS generation. Therefore, we concluded that DSN induces GBC cell apoptosis via regulating ROS-mediated PI3K/AKT signalling.
Conclusion
Taken together, these findings indicate that DSN induces GBC cell apoptosis by inhibiting of PI3K/AKT signaling through a ROS-dependent mechanism. Furthermore, DSN inhibits GBC cell migration via ROS generation. Therefore, we believe that DSN may be a novel and effective therapy for GBC.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81572819,81172026, 81272402, 81301816, 81172029, 81402403, 81502433 and 31501127), China Postdoctoral Science Foundation (No. 2015M571577), the Program for Changjiang Scholars, the Natural Science Research Foundation of Shanghai Jiao Tong University School of Medicine (No. 13XJ10037), the Leading Talent program of Shanghai and Specialized Research Foundation for the PhD Program of Higher Education-Priority Development Field (No. 20130073130014), the Interdisciplinary Program of Shanghai Jiao Tong University (No.14JCRY05), and the Shanghai Rising-Star Program (No. 15QA1403100).
Supplementary Material
Supplementary figures.
Abbreviations
ROS: reactive oxygen species
GSH: glutathione
PARP: poly ADP-ribose polymerase
AKT: protein kinase B
p-AKT: phosphorylated protein kinase B
DMSO: dimethyl sulfoxide
CCK-8: Cell Counting Kit-8
Z-VAD-FMK: Pan-caspase inhibitor
FITC: fluorescein isothiocyanate
PI: propidium iodide
IHC: immunohistochemical streptavidin-peroxidase staining
HE: hematoxylin and eosin
SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
PVDF: polyvinylidene difluoride
LY294002: 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one
PBS: phosphate buffered saline
PI3K: phosphatidylinositol 3-kinase
GAPDH: glyceraldehyde 3-phosphate dehydrogenase
Competing Interests
The authors have declared that no competing interest exists.
References
1. Li M, Zhang Z, Li X. et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nature Genetics. 2014;46(8):872-6
2. Kanthan R, Senger J L, Ahmed S. et al. Gallbladder Cancer in the 21st Century. Journal of Oncology. 2015;2015:967472
3. Lai CHE, Lau W Y. Gallbladder cancer — A comprehensive review. Surgeon. 2008;6(2):101-10
4. Zhu A X. et al. Current management of gallbladder carcinoma. Oncologist. 2010;15(2):168-81
5. Pandey M. et al. Carcinoma of the Gallbladder. Digestive Diseases & Sciences. 2001;46(6):1145-51
6. Li M. et al. Yes-associated protein 1 (YAP1) promotes human gallbladder tumor growth via activation of the AXL/MAPK pathway. Cancer Lett. 2014;355(2):201-9
7. Shu Y J. et al. SPOCK1 as a potential cancer prognostic marker promotes the proliferation and metastasis of gallbladder cancer cells by activating the PI3K/AKT pathway. Mol Cancer. 2015;14:12
8. Subramani R. et al. Nimbolide inhibits pancreatic cancer growth and metastasis through ROS-mediated apoptosis and inhibition of epithelial-to-mesenchymal transition. Sci Rep. 2016;6:19819
9. Hsieh M J. et al. Dioscin-induced autophagy mitigates cell apoptosis through modulation of PI3K/Akt and ERK and JNK signaling pathways in human lung cancer cell lines. Arch Toxicol. 2013;87(11):1927-37
10. Wang Y. et al. Dioscin (Saponin)-Induced Generation of Reactive Oxygen Species through Mitochondria Dysfunction: A Proteomic-Based Study. J Proteome Res. 2007;6(12):4703-10
11. Tong Q. et al. Dioscin inhibits colon tumor growth and tumor angiogenesis through regulating VEGFR2 and AKT/MAPK signaling pathways. Toxicol Appl Pharm. 2014;281(2):166-73
12. Kim EA. et al. Dioscin induces caspase-independent apoptosis through activation of apoptosis-inducing factor in breast cancer cells. Apoptosis. 2014;19(7):1165-75
13. Zhao X. et al. Potent effects of dioscin against gastric cancer in vitro and in vivo. Phytomedicine. 2016;23(3):274-82
14. Titus E A. Degradation of Misfolded Proteins Prevents ER-Derived Oxidative Stress and Cell Death. Mol Cell. 2004;15(5):767-76
15. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579-91
16. Gong K, Li W. Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: A potential new treatment for hepatocellular carcinoma. Free Radical Bio Med. 2011;51(12):2259-71
17. Sabharwal S S, Schumacker P T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer. 2014;14(11):709-21
18. Hengartner M O. The biochemistry of apoptosis. Nature. 2000;407(6805):770-6
19. Jacobson M D. Reactive oxygen species and programmed cell death. Trends Biochem Sci. 1996;21(3):83-6
20. Meng Q. et al. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18(12):2262-71
21. Chan E, Berlin J. Biliary Tract Cancers: Understudied and Poorly Understood. J Clin Oncol. 2015;33(16):1845-8
22. Jiang L. et al. Bufalin induces cell cycle arrest and apoptosis in gallbladder carcinoma cells. Tumour Biol. 2014;35(11):10931-41
23. Maolan Li. et al. Magnolol inhibits growth of gallbladder cancer cells through the p53 pathway. Cancer Sci. 2015;106(10):1341-50
24. Sena L, Chandel N. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol Cell. 2012;48(2):158-67
25. Qiyan Cai, Teng Ma, Chengren Li. et al. Catalpol Protects Pre-Myelinating Oligodendrocytes against Ischemia-induced Oxidative Injury through ERK1/2 Signaling Pathway. Int. J. Biol.Sci. 2016;12(12):1415-1426
26. Guzy RD. et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401-8
27. D'Autréaux B, Toledano M B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Bio. 2007;8(10):813-24
28. Fulda S. Targeting apoptosis for anticancer therapy. Semin Cancer Biol. 2015;31:84-8
29. Deepak M. Kasote, Surendra S. Katyare, Mahabaleshwar V. Hegde, et al. Significance of Antioxidant Potential of Plants and its Relevance to Therapeutic Applications. Int J Biol Sci. 2015;11(8):982-991
30. Fresno Vara. et al. PI3K/Akt signalling pathway and cancer. Cancer Treat. 2004;30(2):193-204
31. Yuzhen Yao. et al. α-Lipoic acid increases tolerance of cardiomyoblasts to glucose/glucose oxidase-induced injury via ROS-dependent ERK1/2 activation. Biochimica Et Biophysica Acta. 2012;1823(4):920-9
32. Surong Jiang. et al. α-Lipoic acid protected cardiomyoblasts from the injury induced by sodium nitroprusside through ROS-mediated Akt/Gsk-3β activation. Toxicology in Vitro. 2014;28(8):1461-73
Author contact
![]() Corresponding authors: Yingbin Liu email: liuybphdcom; Jun Gu email: gujun6666com
Corresponding authors: Yingbin Liu email: liuybphdcom; Jun Gu email: gujun6666com