Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(8):1637-1653. doi:10.7150/ijbs.33790 This issue Cite
Research Paper
PERK/eIF-2α/CHOP Pathway Dependent ROS Generation Mediates Butein-induced Non-small-cell Lung Cancer Apoptosis and G2/M Phase Arrest
Shouyin Di1*, Chongxi Fan2*, Zhiqiang Ma1*, Mingyang Li3, Kai Guo1, Donghui Han4, Xiaofei Li1 ![]() *, Deguang Mu5
*, Deguang Mu5 ![]() *, Xiaolong Yan1
*, Xiaolong Yan1 ![]() *
*
1. Department of Thoracic Surgery, Tangdu Hospital, The Fourth Military Medical University, 1 Xinsi Road, Xi'an 710038, China
2. Department of Biomedical Engineering, The Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China
3. Department of Pathology, Xijing Hospital, The Fourth Military Medical University, 127 Changle West Road, Xi'an 710032, China
4. Department of Urology, Xijing Hospital, The Fourth Military Medical University, 127 Changle West Road, Xi'an 710032, China
5. Department of Respiratory Medicine, Zhejiang Provincial People's Hospital, Hangzhou Medicine College, 158 Shangtang Road, Hangzhou 310014, China
*These authors contributed equally to this work.
Received 2019-2-2; Accepted 2019-5-4; Published 2019-6-4
Abstract

Butein, a member of the chalcone family, is a potent anticarcinogen against multiple cancers, but its specific anti-NSCLC mechanism remains unknown. The present study examined the effects of butein treatment on NSCLC cell lines and NSCLC xenografts. Butein markedly decreased NSCLC cell viability; inhibited cell adhesion, migration, invasion, and colony forming ability; and induced cell apoptosis and G2/M phase arrest in NSCLC cells. Moreover, butein significantly inhibited PC-9 xenograft growth. Both in vivo and in vitro studies verified that butein exerted anti-NSCLC effect through activating endoplasmic reticulum (ER) stress-dependent reactive oxygen species (ROS) generation. These pro-apoptotic effects were reversed by the use of 4- phenylbutyric acid (4-PBA), CHOP siRNA, N-acetyl-L-cysteine (NAC) and Z-VAD-FMK (z-VAD) in vitro. Moreover, inhibition of ER stress markedly reduced ROS generation. In addition, in vivo studies further confirmed that inhibition of ER stress or oxidative stress partially abolished the butein-induced inhibition of tumor growth. Therefore, butein is a potential therapeutic agent for NSCLC, and its anticarcinogenic action might be mediated by ER stress-dependent ROS generation and the apoptosis pathway.
Keywords: Butein, Non-small-cell lung cancer, Endoplasmic reticulum stress, Oxidative stress, Apoptosis
Introduction
The morbidity and mortality rate of lung cancer both rank the first in all kinds of malignant tumors worldwide. Recently, although tremendous advances have been made in lung cancer therapy, such as the development of targeted therapies, less than 20% patients can survive more than 5 years (1). Lung cancer mainly consists of lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, adenosquamous carcinoma and small cell lung cancer, with the adenocarcinoma being the most diagnosed subtype (2). Considering the high mortality rate and the unsatisfactory treatment outcomes of non-small-cell lung cancer (NSCLC), greater efforts should be made to investigate the underlying mechanisms of NSCLC and to develop new anti-NSCLC agents. Recently, butein has drawn increasing attention for its satisfying anticancer effect and slight side effect.
Butein, or 3,4,2',4'-tetrahydroxychalcone, is a polyphenol that can be isolated from many kinds of Chinese herbal medicine (3). Butein belongs to the chalcone family of flavonoids. Chalcones and their derivatives are known to have numerous biological properties, including anticancer, anti-inflammatory, antioxidant, anti-hyperlipidemia, and other properties (4). Studies have shown that Butein exerts potent anticancer effect in ovarian cancer, hepatocellular carcinoma, and bladder cancer (5-11). In NSCLC, butein has been proved to induce A549 cells apoptosis (12), and enhance the anti-lung cancer efficiency of gefitinib (13). These evidences showed that butein might be a good anti-NSCLC agent. However, exact effects of butein on various NSCLC cell types and xenografts, as well as the underlying mechanisms, remain unknown.
Endoplasmic reticulum (ER) is a type of organelle in eukaryotic cells, which is mainly responsible for the balance of intracellular Ca2+, modification of proteins, lipid metabolism, carbohydrate metabolism and detoxification (14, 15). ER stress can be activated by various physiological and pathological stressors, and induces accumulation of unfolded proteins (16). Accumulation of unfolded proteins activates unfolded protein response (UPR), which is the molecular basis of ER stress. Moderate activation of UPR helps to address the unfolded proteins and restore ER homeostasis (17, 18). However, if ER stress prolongs, UPR will be over activated and further trigger cell apoptosis (14). Previous studies have confirmed that some flavonoids and chalcones exert anticancer effects by activating ER stress (14, 19-21). However, whether ER stress mediates butein-induced NSCLC apoptosis still remains unclear.
Superoxide anionradical (O2-) is a by-product of mitochondria, which can form other reactive oxygen species (ROS) (22). ROS accumulation can activate redox-signaling pathways, some of which are often associated with ER stress (10). Previous studies have shown that, butein induces ROS generation in multiple cancer types, which leads to cell apoptosis and cell cycle arrest (10, 23). However, the roles of ROS in the anti-NSCLC effects induced by butein have not been totally illuminated.
In the present study, we investigated the anti-NSCLC effects of butein, and the vital roles of ER stress and ROS signaling in this process both in vitro and in vivo.
Materials and Methods
Animals
8-week-old male nude mice weighing 19-21 g were purchased from the Laboratory Animal Centre of the Fourth Military Medical University (FMMU, Xi'an, China). The in vivo study design was approved by the Ethics Committee of FMMU. All the in vivo experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (National Institutes of Health Publication No. 85-23, revised 1996). All the mice were raised in a specific-pathogen-free environment at 26 °C with a 12 h light and 12 h dark cycle in the Laboratory Animal Centre of FMMU. All the mice had free access to regular rodent chow and tap water.
Materials
Butein, N-acetyl-L-cysteine (NAC), 4',6-diamidino-2-phenylindole (DAPI), dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell counting kit 8 (CCK8) was purchased from 7-sea Biotechnology (Shanghai, China). Propidium Iodide (PI) was purchased from Merck Millipore (Darmstadt, Germany). RNase A, Triton X-100 and DCFH-DA were purchased from Solarbio life science (Beijing, China). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) kits were purchased from Roche Diagnostics (Mannheim, Germany). JC-1, crystal violet and goat serum were purchased from the Beyotime Institute of Biotechnology (Nanjing, Jiangsu, China). Caspase-3, caspase-8 and caspase-9 activity assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). A glutathione (GSH) assay kit was obtained from Shanghai Enzyme-linked Biotechnology Co., Ltd. (Shanghai, China). Anti-cell division cyclin 25 homolog C (Cdc25C), Cyclin B1, Bax, Bcl-2, ATF4, C/EBP homologous protein (CHOP), X-box binding protein 1 (XBP1) antibodies were purchased from Abcam (Cambridge, UK). Anti-cell division cycle 2 (Cdc2), β-actin, p53 upregulated modulator of apoptosis (PUMA), superoxide dismutase 2 (SOD2), protein kinase RNA-like ER kinase (PERK), phospho-PERK(Thr980), eukaryotic translation initiation factor 2α (eIF2α), phospho-eIF2α (Ser51), inositol-requiring kinase 1α (IRE1α) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). The Cy3 goat anti-rabbit IgG was purchased from Abbkine (California, USA). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). Penicillin/streptomycin was purchased from Thermo Fisher Scientific (Breda, Netherlands).
Cell culture
NSCLC cell lines were purchased from the cell bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in complete medium [90% DMEM, 10% FBS, penicillin and streptomycin (100 units/ml, respectively)] and incubated at 37 °C with 95% air and 5% CO2. Butein, NAC (10 mM), and 4-PBA (10 mM) were diluted in DMSO first and further diluted in FBS-free DMEM before added to cells (the final concentration of DMSO in medium is 0.1%). Cells in the control group were cultured with PBS-free DMEM containing 0.1% DMSO. The dosages of these drugs were determined according to previous studies and our previous data (24, 25).
Cell viability assessment
Spectrophotometry was utilized to detect cell viability in adherence to the CCK-8 manufacturer's instructions. NSCLC cells were taken and seeded in 96-well plates (10,000 cells per well). After 12 h attachment, the medium was replaced with FBS-free DMEM (with 0.1% DMSO) or butein (20 μM, 40 μM or 60 μM) and further incubate for 24 h or 48 h. Then the medium was discarded and 100 μl of DMEM and 10 μl of CCK-8 was added to each well. After further incubation for 2 h, cells were subjected to optical density (OD) values detection at 450 nm. Then the data was collected and analyzed. The OD value of the wells in control group was normalized to 100%. All the experiments were repeated 6 times.
Cell wound-healing, adhesion and matrigel invasion assay
According to our previous data, treatment with lower dose of butein (less than 20 μM) for 24 h exerted little influence on cell viability. To confirm the effects of butein on NSCLC cell adhesion, migration, invasion and proliferation, low butein concentration (5 μM, 10 μM and 20 μM) was used in this part. Cells were cultured in 6-well plates (5×105 cells per well) in the logarithmic growth phase. When the cells had grown to confluence, a 200 μl micropipette tip was used to make a linear wound in the middle of the well. The floating cells were washed with PBS 3 times. After exposed to butein for 24 h, the images of the wounds were captured (×100 magnification), and the distance between the wound edges were calculated and analyzed.
For the adhesion assay, cells were collected, seeded in a 96-well plate and allowed to attach for 30 min. The medium was then discarded and adherent cells were stained with MTT for 4 h. Then images were obtained and 100 μl of DMSO were added to each well to dissolve the formazan. After 15 min of dissolution, the OD values were measured using a spectrophotometer at 490 nm. The OD value reflects the number of adherent cells, which is directly proportional to the cell adhesive capacity.
Matrigel invasion assay was performed to detect the invasion ability of tumor cells. Cells were collected and seeded in a transwell chamber (Corning, NY, USA). The bottom of the transwell chamber was covered with 300 ng/ml Matrigel (BD Biosciences, San Jose, CA, USA). Medium with butein or other drugs was added into the upper chamber, while medium with 20% FBS was added to the lower chamber. After incubating in 37 °C with 5% CO2 for 24 h, the upper chamber was taken for detection. Cells adhered to the underside of the chamber were visualized with 0.5% (w/v) crystal violet for 20 min. After that, images were captured (×200) in 5 random fields to calculate cell numbers.
Colony formation assay
A 14-day long colony formation assay was performed as previously described (26). Briefly, cells were cultured in 6-well plates (2×103 cells per well) with complete medium. 12 h later, the medium was discarded and cells were subjected to Butein or other treatment. The medium was renewed every 3 days and the exposure lasted for 14 days. Then, the cells were fixed and further visualized with 0.5% (w/v) crystal violet.
Analysis of cell apoptosis by TUNEL staining
The apoptosis rate was assessed with TUNEL staining. It was conducted in adherence to the manufacturer's instructions. In brief, cells were fixed and subjected to 0.5% Trixon-X 100 for 20 min. Working solution of TUNEL was prepared and add to each dish for 1 h at 37 °C. After that, cells were stained with DAPI for 10 min. An Olympus FV1000 microscope was used to capture images and Image Pro Plus software was used to count the apoptotic cells.
Cell cycle detection
Cells were cultured in complete medium for attachment. Then, the cells were exposed to relative treatments for 24 h. After exposure, cells were harvested, centrifuged and suspended in 400 μl of PBS with 0.2% Triton X-100, 100 μg/ml RNase A and 50 μg/ml PI for 30 min at 4 °C away from light. All samples were detected on a NovoCyte flow cytometer (ACEA Bioscience, San Diego, CA, USA).
Mitochondrial membrane potential (MMP) detection
The loss of MMP has been proven to indicate early apoptosis. Fluorescent dye JC-1 was used to detect MMP levels as previously described (27). Briefly, cells were seeded in Petri dishes and treatments were given. Cells were then subjected to JC-1 fluorescent dye for 20 min in the dark. An Olympus FV1000 microscope was used to capture images, and the ratio of green fluorescence intensity to red fluorescence intensity was calculated to analyze the early cell apoptosis.
Measurement of ROS, NADPH oxidase activity, and GSH level
For ROS detection, cells were seeded in specific dishes (Corning, NY 14831 USA) designed for laser scanning confocal microscopy. After treatment, cells were subjected to 5 μM DCFH-DA at 37 °C for 30 min. An Olympus FV1000 microscope was then used to capture images (Ex: 488 nm, Em: 522). Fluorescence of the control group was defined as 100% and fluorescence of other groups was normalized to that of control group.
NADPH oxidase activity assay was conducted in adherence to the manufacturer's instructions (28). Briefly, cells were collected, lysed, and the supernatants in Krebs-HEPES buffer were added with 5 μM lucigenin and further incubated away from light for 10 min at 37 °C. After that, 100 μM NADPH was added and allowed to react for 1 min. Then the chemiluminescences were determined immediately. The chemiluminescence of control group was normalized to 100%.
GSH levels were determined in adherence to the manufacturer's instructions (27). The fluorescent intensity was measured using an FLX 800 fluorescence microplate reader (Ex: 335 nm, Em: 541 nm). The GSH level in the control group was normalized to 100%.
Detection of the activities of caspases
The activities of caspases were determined using commercial kits. It was performed in adherence to the instructions. Briefly, cells were collected, lysed and added with a specific buffer. Ac-DEVD-pNA, Ac-IETD-pNA, LEDH-pNA were used for activity detection of caspase 3, caspase 8, caspase 9 respectively. The pNA-conjugated substrate was prepared before detection. After reaction, the tubes were placed at 37 °C for 2 h. Then the OD value of each tube was determined at 405 nm. A calibration curve was made and the caspase activity in each sample was calculated.
RNA-seq
The RNA-seq was entrusted to Chi-Biotech (Shenzhen, Guangdong, China). The heatmap of representative gene was conducted by Multiple experiment viewer 4.9.0 (29-31).
Western blotting
Western blotting was conducted to asses protein expression as previously described. In brief, after treatment, the cells were collected and homogenized in lysis buffer with 1% phosphatase inhibitor and 1% protease inhibitor cocktail for 20 min at 4 °C. After that, the lysates were centrifuged (12,000 ×g, 15 min), and the supernatant was collected. For xenograft protein extraction, the xenografts were taken and cut into pieces before being added to the lysis buffer; the other procedures were the same as the cell protein extraction. The extracted protein solutions were boiled with loading buffer. And then the equal proteins were subjected to SDS-PAGE and further transferred to PVDF membrane. The membranes were incubated with relative primary and secondary antibodies. The anti-Cdc25 (1:1000), Cyclin B1 (1:1000), Cdc2 (1:1000), β-actin (1:2000), Bax (1:1500), Bcl2 (1:1500), PUMA (1:1000), SOD2 (1:1000), p-PERK (1:1000), PERK (1:1000), p-eIF2α(1:1000), ATF4 (1:1000), CHOP (1:1000), IRE1α (1:1000), XBP1 (1:1000) were used as primary antibodies. Expressions of proteins were determined using an enhanced chemiluminescence method with a Bio-Rad imaging system (Bio-Rad, Hercules, CA, USA).
siRNA transfection
A human CHOP siRNA was purchased from GenePharma (Shanghai, China). For the siRNA transfection, cells seeded in six-well plates were cultured overnight before being transfected with a mixture of siRNAs (90 pg), Lipofectamine 2000 (Invitrogen, Life Technologies, Carlsbad, CA) and antibiotics-free medium for 90 min. Then the medium was discarded and the cells were washed with PBS. After that, the cells were cultured with complete medium in 37 °C for 24 h. Finally, the target mRNA levels and protein expressions of targeted genes were detected to confirm the transfection efficiency.
Establishment of the xenograft model
The node mice were divided into 4 groups, namely the control group, the butein treatment group, the butein + 4-PBA group, and the butein + NAC group. PC-9 (1× 107) cells were subcutaneously injected into right rear limbs. The mice were weighted and the xenografts were measured every 3 days. The tumor size was estimated as (length × width2)/2. After cells were injected, mice were given 100 μl of PBS (i.p.), butein (10 mg/kg, i.p.) (32), butein plus 4-PBA (50 mg/kg, i.p.) (33), or butein plus NAC (1 g/L, in drinking water) (34) every day. On the 28th day, mice were sacrificed and the xenografts were harvested for further detection.
Statistical analyses
All data are presented as the means ± SEM. Data were analyzed with SPSS 18.0 software for windows (SPSS Inc., Chicago, USA). Statistical significance for each variable was evaluated by one-way ANOVA. P value <0.05 was considered statistically significant.
Results
Butein treatment reduced the viability of NSCLC cells in a dose-dependent manner
We first assessed the sensitivity of HBE cells and NSCLC cells to butein. HBE cells, A549, PC-9, SPC-A1 and H1299 cells were treated with butein (20 μM, 40 μM, and 60 μM) for 24 or 48 h. The dose of butein was based on our previous data (Figure S1). Cells showed a significant shrinkage after butein treatment (Fig. 1A, A549 and PC-9 cell morphology images are shown). Cell viability was detected and the result showed that butein treatment markedly reduced cell viability of NSCLC cells and HBE cells in a dose-dependent manner (Fig. 1B - 1C). A549, PC-9 and SPC-A1 cells were more sensitive to butein than HBE cells for both 24 h and 48 h (Fig. 1B - 1C). These results indicated that butein significantly inhibited cell growth and reduced cell viability of NSCLC and HBE cells, and butein exerted a greater inhibitory effect on NSCLC cells at specific range of doses, which makes it potentially useful for cancer treatment.
Effects of butein treatment on NSCLC cell viability and adhesion, migration, invasion ability. (A) Morphology of A549 and PC-9 cells after butein treatment for 24 h. (B) Viability of NSCLC cells and HBE cells after exposed to butein for 24 h. (C) Viability of NSCLC cells and HBE cells after exposed to butein for 48 h. The results are expressed as the mean ± SEM, n=6. aP < 0.05 versus control group, bP < 0.05 versus 20 μM butein group, cP < 0.05 versus 40 μM butein group. (D) Representative adhesion images of A549 and PC-9 cells after butein treatment. (E) The adhesion ability of A549 and PC-9 cells after butein treatment. (F) Representative wound healing images of A549 and PC-9 cells after butein treatment. (G) The migration ability of A549 and PC-9 cells after butein treatment. (H) Representative matrigel invasion images of A549 and PC-9 cells after butein treatment. (I) The migration ability of A549 and PC-9 cells after butein treatment. In Figure D-I, the results are expressed as the mean ± SEM, n=6. aP < 0.05 versus control group, bP < 0.05 versus 5 μM butein group, cP < 0.05 versus 10 μM butein group.
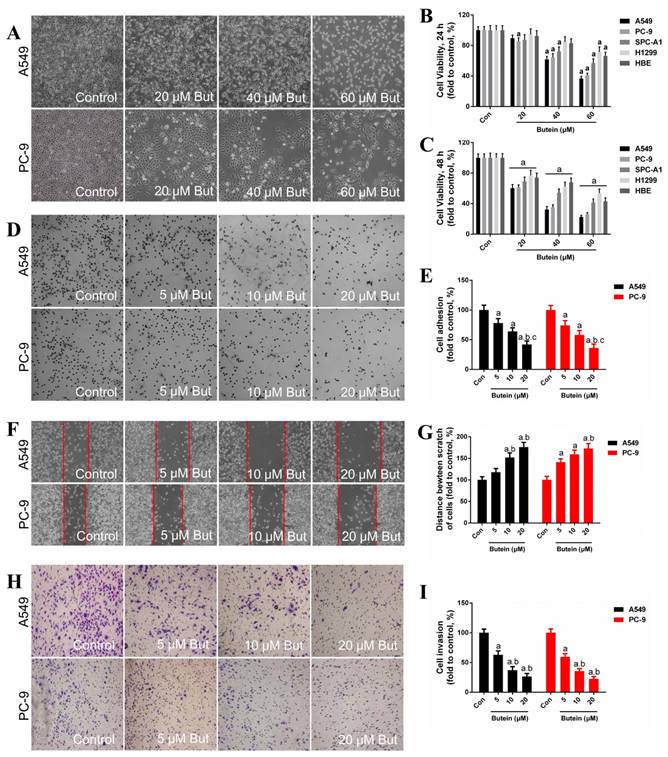
Butein treatment reduced A549 and PC-9 cell adhesion, migration and invasion ability
The adhesion ability, migration ability and invasion ability of these two NSCLC cell lines were determined by adhesion assay, wound-healing assay and transwell invasion assay, respectively. In our previous study, low concentrations of butein (5 μM, 10 μM, and 20 μM) had very little damage on the viability of NSCLC cells after 24 h. Therefore, these doses were used for the cell adhesion, migration and invasion assays. Treatment with butein for 24 h dose-dependently inhibited cell adhesion capacity of both A549 and PC-9 cells (Fig. 1D - 1E). Furthermore, incubation with butein for 24 h significantly reduced the cell migration capacity of A549 and PC-9 cells, as indicated by wider wounds after butein treatment in contrast to that of the control group (Fig. 1F - 1G). Moreover, transwell invasion assay showed that butein treatment reduced cell invasion capacity in a dose-dependent manner (Fig. 1H-1I).
Butein inhibited NSCLC cell proliferation
To examine the anti-proliferative effect of butein, we analyzed the clonogenicity of A549 and PC-9 cells after butein treatment. To avoid cell death induced by high concentrations of butein, low doses of butein (5 μM, 10 μM, and 20 μM) was used. As shown in Fig. 2A-2B, butein reduced the clonogenicity of these two cell lines in a dose-dependent manner.
Effects of butein treatment on NSCLC cell proliferation ability and cell cycle distribution. (A) Representative images of clonogenic survival assay after butein treatment. (B) The clonogenicity of A549 and PC-9 cells after butein treatment. the results are expressed as mean ± SEM, n=6. aP < 0.05 versus control group, bP < 0.05 versus 5 μM butein group, cP < 0.05 versus 10 μM butein group. (C) Representative images of cell cycle distribution after butein treatment. (D) Cell cycle distribution of A549 cells after butein treatment. (E) Cell cycle distribution of PC-9 cells after butein treatment. (F) Expressions of cell cycle markers. In Figure C-F, all of the results are expressed as mean ± SEM, n=6. aP < 0.05 versus control group, bP < 0.05 versus 20 μM butein group, cP < 0.05 versus 40 μM butein group.
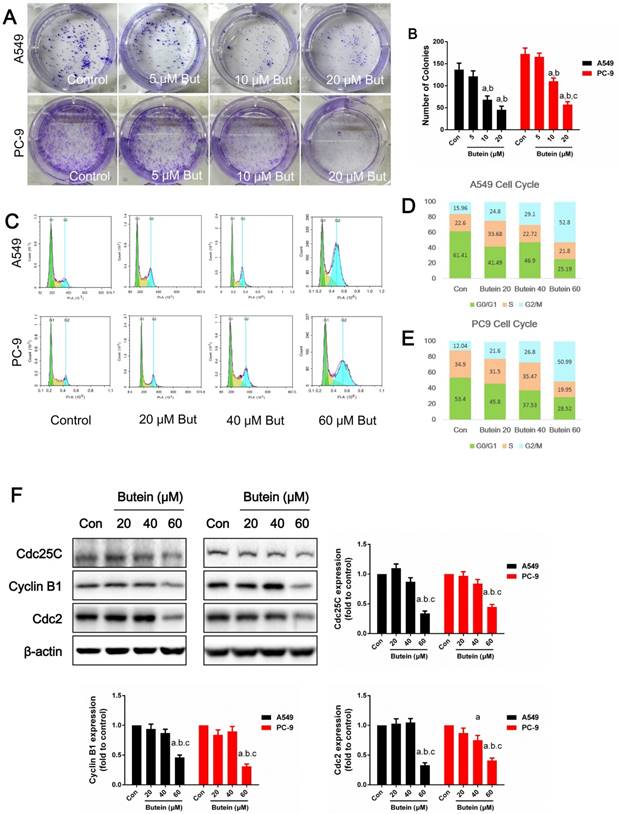
Butein induced A549 and PC-9 cells G2/M phase arrest
To illuminate the mechanisms involved in the cytotoxic effect of butein, we detected the cell cycle distribution after butein treatment for 24 h. Cells in G0/G1 phase were notably reduced and cells in G2/M phase were significantly increased after butein treatment, especially in the 60 μM group (Fig. 2C-2D). Furthermore, protein expression of cell cycle markers confirmed this effect. As shown in Fig. 2E-2G, the expression of cdc25c, Cylin-B1 and cdc2 significantly decreased after butein treatment.
Butein induced A549 and PC-9 cell mitochondrial membrane depolarization and apoptosis
Cell apoptosis was investigated through MMP detection, TUNEL staining, and caspase-3, caspase-8 and caspase-9 activity detection in adherence to manufacturers' instructions. The expression of key apoptotic regulators, such as Bcl2, Bax and PUMA was detected by western blot analysis. Butein induced a notable increase in the apoptosis rate. After treated with 20 μM, 40 μM, and 60 μM butein for 24 h, the apoptosis rate increased to 13.5 ± 1.1 %, 21.3 ± 1.6 %, 32.6 ± 2.2 % in A549 cell line and to 9.7 ± 1.5 %, 18.7 ± 1.9 %, 29.9 ± 2.5 % in PC-9 cell line, respectively (Fig. 3A-3B). This effect was further validated by MMP detection. As shown in Fig. 2C-2D, with the increase of butein concentrations, red fluorescence intensity decreased and green fluorescence intensity grew stronger in both the A549 and PC-9 cell lines, indicating that apoptosis was induced by butein. Consistent with results of TUNEL staining and MMP detection, butein increased Bax and PUMA expressions and decreased Bcl2 expression. Furthermore, detection of caspase-3, caspase-8 and caspase-9 activity was conducted to assess the activation of pro-apoptotic pathways. After exposed to butein (20 μM, 40 μM, and 60 μM) for 24 h, the caspase-3 activities increased to (168.6 ± 16.2) %, (267.4 ± 17.9) %, (431.8 ± 20.1) % in A549 cells and to (142.3 ± 14.6) %, (195.7 ± 15.3) %, (367.8 ± 17.2) % in PC-9 cells, compared to that of the control group, respectively; The caspase-8 activities increased to (133.1 ± 13.1) %, (131.2 ± 13.5) %, (143.2 ± 13.7) % in A549 cells and to (148.1 ± 14.1) %, (162.4 ± 13.2) %, (171.3 ± 14.8) % in PC-9 cells, compared to that of the control group, respectively; The caspase-9 activities increased to (164.8 ± 12.9) %, (192.6 ± 13.6) %, (267.2 ± 15.7) % in A549 cells and to (158.9 ± 13.2) %, (212.3 ± 15.8) %, (248.2 ± 16.4) % in PC-9 cells compared to that of the control group, respectively.
Effects of butein treatment on NSCLC cell apoptosis rate and MMP. (A) Representative images of A549 and PC-9 TUNEL staining. (B) Analysis of A549 and PC-9 apoptosis rate. (C) Representative images of A549 and PC-9 MMP detection. (D) Analysis of A549 and PC-9 MMP lose. All of the results are expressed as mean ± SEM, n=6. aP < 0.05 versus control group, bP < 0.05 versus 20 μM butein group, cP < 0.05 versus 40 μM butein group.
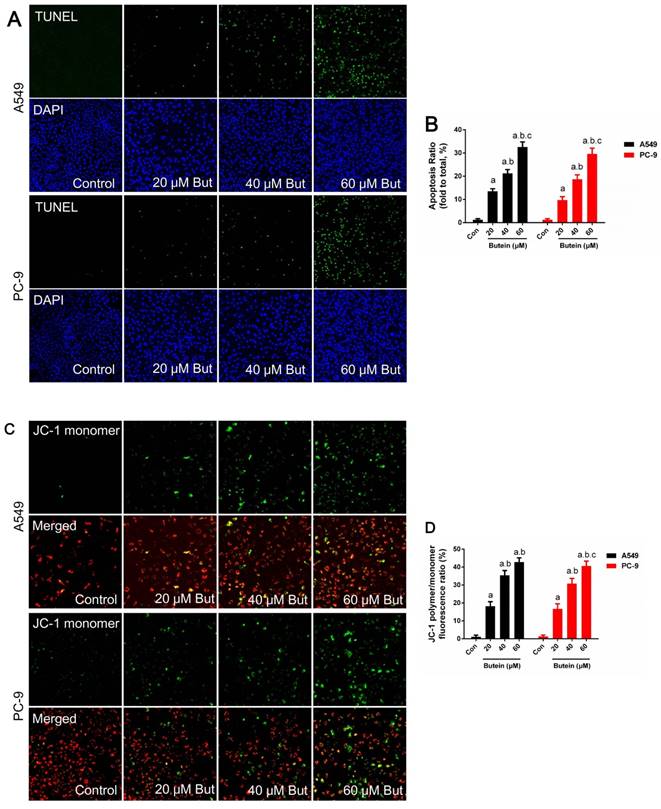
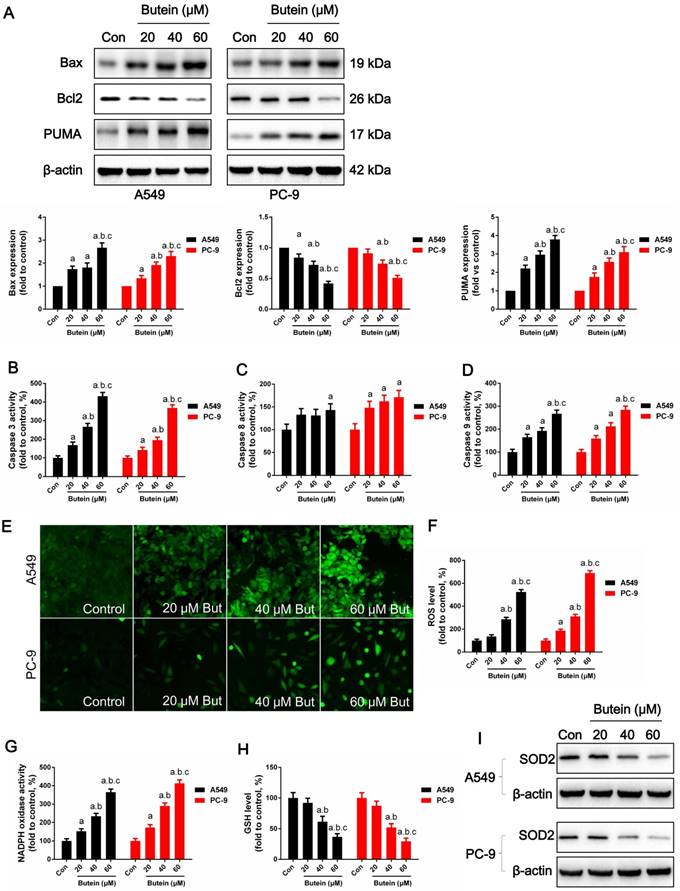
Butein induces A549 and PC-9 cell apoptosis by activating oxidative stress
Cell oxidative stress is an endogenous regulator of cell fate and cell cycle distribution. Previous studies have shown that butein increases ROS generation in hepatoma cancer cells (10). Here we determined whether ROS plays a role in butein induced NSCLC apoptosis. ROS were detected utilizing a DCFH-DA fluorescent dye. As shown in Fig 4E-4F, after butein treatment, intracellular ROS generation was significantly increased. NADPH oxidase activity was also significantly increased after Butein treatment, comparing to the control group (Fig. 4G). SOD2, an anti-redox enzyme that locates in cell mitochondria, was significantly decreased by butein (Fig. 4I). In addition, the cellular GSH level was decreased by butein (Fig. 4H), indicating the occurrence of oxidative stress after butein treatment.
Effects of butein treatment on NSCLC cell apoptosis pathways and oxidative stress. (A) Expressions of apoptosis associated proteins. (B) Analysis of caspase-3 activities. (C) Analysis of caspase-8 activities. (D) Analysis of caspase-9 activities. (E) Representative images of cellular ROS detection in A549 and PC-9 cells. (F) Analysis of ROS level in A549 and PC-9 cells. (G) Analysis of NADPH oxidase activities. (H) Analysis of GSH concentrations. (I) Expressions of SOD2. All of the results are expressed as the mean ± SEM, n=6. aP < 0.05 versus control group, bP < 0.05 versus 20 μM butein group, cP < 0.05 versus 40 μM butein group.
Butein activated cellular endoplasmic reticulum stress
To further explore the underlying molecular basis of the effects of butein on NSCLC cells, RNA-seq based on transcriptome analysis was conducted, and the differential expressions of ER stress markers, apoptosis-related genes and cell cycle-related genes are shown in the heatmap (Fig. 5A). ER stress markers, which are marked with black arrows, were significantly elevated after butein treatment. In addition, some apoptosis-related genes, which are marked with red arrows, were also increased by butein treatment.
Effects of butein treatment on NSCLC cell ER stress pathways. (A) Representative Heatmap of gene expression levels. The black arrows indicate ER stress associated genes; the red arrows indicate apoptosis associated genes. (B-C) Representative bands and quantitative analysis of ER stress markers are shown. All of the results are expressed as the mean ± SEM, n=6. aP < 0.05 versus Control, bP < 0.05 versus 20 μM butein group, cP < 0.05 versus 40 μM butein group.
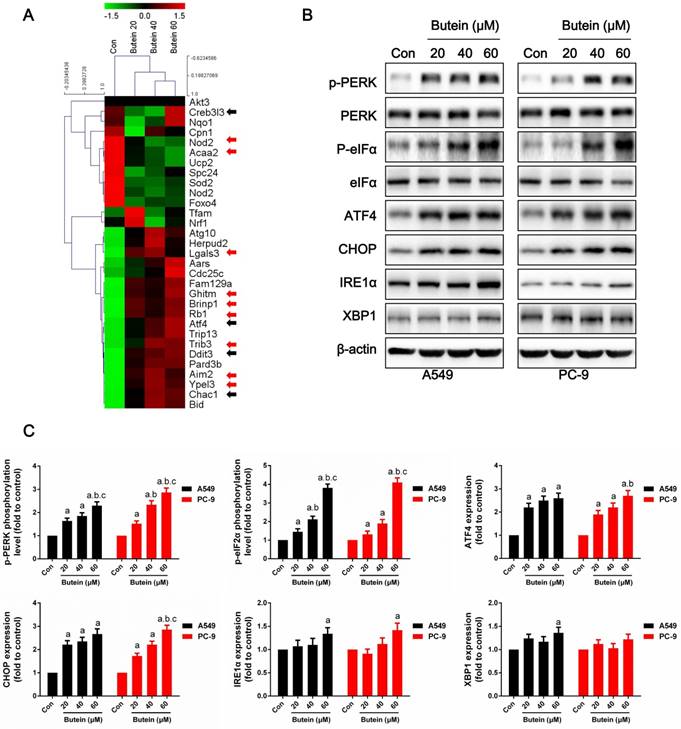
ROS accumulation can be both a stimulator and result of ER stress (35, 36). It is well known that activation of the ER stress pathway by small molecules can trigger cancer cell apoptosis. Under non-stress conditions, PERK is banded with BIP and remains inactivated. Stress activation can separate BIP from PERK, causing PERK to become auto-phosphorylated and further activates eIF2α. Activated eIF2α increases the expression of ATF4 and CHOP, which promote cell apoptosis under ER stress. IRE1α sis banded with BIP under non-stress conditions and is separated from BIP under stress conditions. Activated IRE1α slices the mRNA of XBP1 and promoting XBP1 mRNA translation. In this study, we detected key indicators of the three pathways of ER stress. After treated with butein for 24 h, the phosphorylation levels of PERK and eIF2α were dose-dependently increased (Fig. 5B-5C). Moreover, expressions of ATF4 and CHOP were increased by butein (Fig. 5B-5C). IRE1α and XBP1 showed slight increases in expression after exposure to butein in comparison with the control group (Fig. 5B-5C).
ER stress inhibition attenuated butein-induced ROS generation and apoptosis
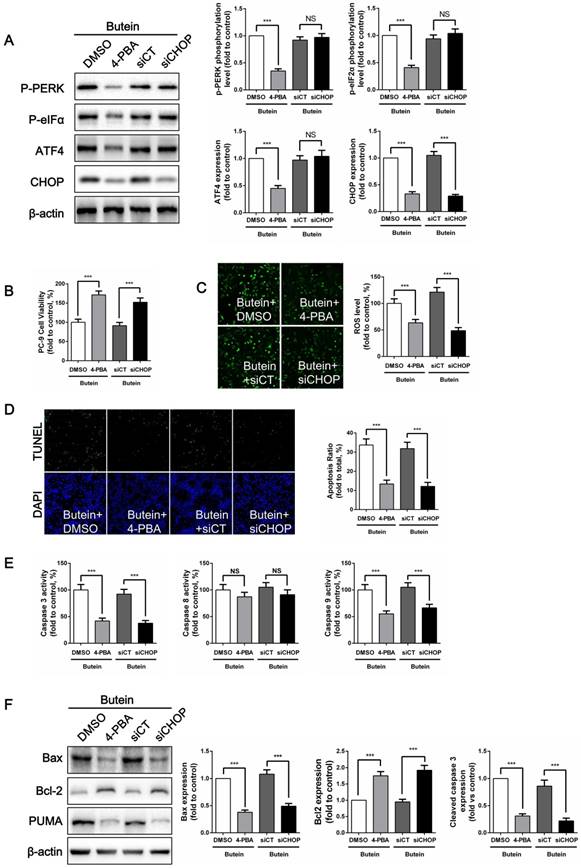
To confirm whether butein induces NSCLC cell apoptosis through the ER stress pathway, we used 4-PBA, an ER stress inhibitor, to suppress ER stress activation and used siRNA to knock down CHOP expression in PC-9 cells. 4-PBA decreased PERK and eIF2α phosphorylation levels, and decreased ATF4 and CHOP expressions. CHOP siRNA significantly reduced CHOP expression without affecting other ER stress markers. (Fig. 6A). Cell viability detection showed that, both ER stress inhibition and CHOP silencing attenuated the butein-induced cell viability loss (Fig. 6B). Previously, we found that ROS activation mediated butein induced cell injury. Since oxidative stress can be cause or consequence of ER stress, ROS generation was detected after ER stress inhibition. We demonstrated that both 4-PBA and CHOP silencing markedly reversed butein induced ROS generation (Fig. 6C). This result indicated that ROS generation induced by butein was ER stress dependent. ER stress might be an upstream regulator of ROS generation. Furthermore, compared with the butein group, inhibition of ER stress and knocking down of CHOP expression both reduced butein induced apoptosis, as indicated by the decreased apoptosis rate (Fig. 6D), decreased caspase activity (Fig. 6E), decreased Bax/Bcl2 ratio and decreased PUMA expression (Fig. 6F).
Effects of ER stress inhibition on butein induced NSCLC cell apoptosis. (A) Representative band and quantitative analysis of ER stress markers. (B) Cell viability of PC-9 cells. (C) ROS detection in PC-9 cells. (D) Apoptosis detection via TUNEL staining in PC-9 cells. (E) Analysis of activities of caspase-3, caspase-8 and caspase-9 in PC-9 cells. (F) Representative bands and quantitative analysis of apoptosis associated proteins. All of the results are expressed as the mean ± SEM, n=6. ***P < 0.01.
NAC treatment impaired the pro-apoptotic effects of butein
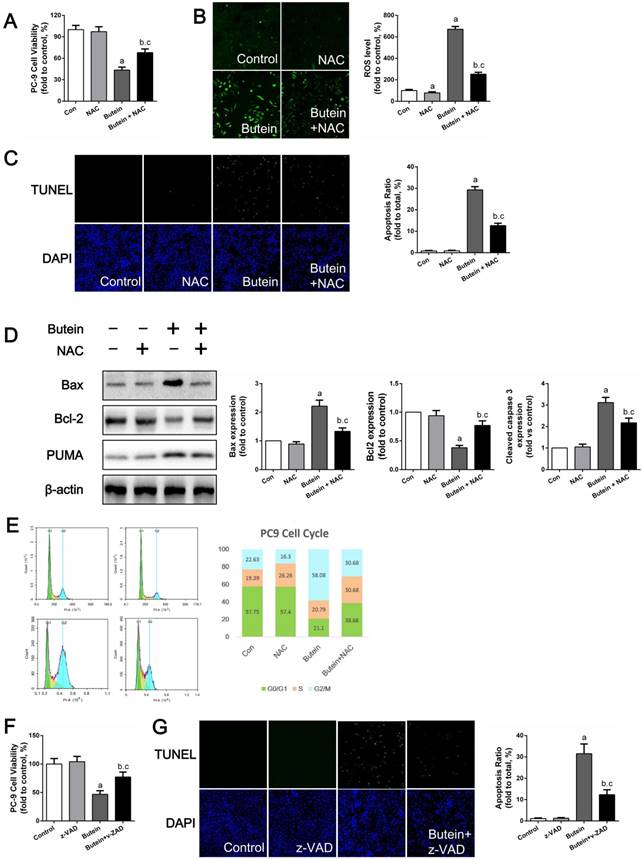
To test whether ROS mediated butein-induced apoptosis, NAC, a specific ROS scavenger, was used to reduce ROS generation. As shown in Fig. 7A, the use of NAC attenuated the butein induced reduction in cell viability. DCFH-DA staining showed that NAC treatment significantly decreased butein-induced ROS generation (Fig. 7B) and TUNEL staining showed that NAC reduced butein-induced cell apoptosis (Fig. 7C). In addition, western blot analysis of apoptosis markers showed that co-treatment with NAC decreased Bax/Bcl2 ratio and PUMA expression (Fig. 7D). Furthermore, cell cycle detection showed that ROS inhibition helped to reverse the G2/M phase arrest inducted by butein (Fig. 7E). These results demonstrated that ROS activation is a key process in butein induced cell inhibition.
Effects of NAC or z-VAD treatment on butein induced anti-NSCLC activity. (A) Cell viability of PC-9 cells after co-treatment of butein and NAC. (B) ROS detection in PC-9 cells. (C) Apoptosis rate detection via TUNEL staining in PC-9 cells. (D) Representative bands and quantitative analysis of apoptosis associated proteins. (E) Representative images and analysis of cell cycle distribution of PC-9 cells. All of the results in (A-E) are expressed as the mean ± SEM, n=6. aP < 0.05 versus Control group, bP < 0.05 versus NAC group, cP < 0.05 versus butein group. (F) Cell viability of PC-9 cells after co-treatment of butein and z-VAD. (G) Apoptosis detection via TUNEL staining in PC-9 cells. All of the results in (F, G) are expressed as the mean ± SEM, n=6. aP < 0.05 versus Control group, bP < 0.05 versus z-VAD group, cP < 0.05 versus butein group.
The pan-caspase inhibitor z-VAD partially blocked butein-induced cell apoptosis
Previously, we confirmed that caspase 3, caspase 8, and caspase 9 activities were activated by butein. Since caspase pathways are the ultimate mediators of cell apoptosis, we further validated their roles in butein induced apoptosis using z-VAD, a pan-caspase inhibitor. After treated with butein and z-VAD, cell viability loss induced by butein was notably decreased but not totally abolished, indicating that caspase related apoptosis pathway played a crucial role in butein induced cell inhibition.
Butein significantly inhibited the growth of PC-9 tumor xenografts in nude mice, and blocking ER stress or ROS impaired the in vivo inhibitory effect of butein
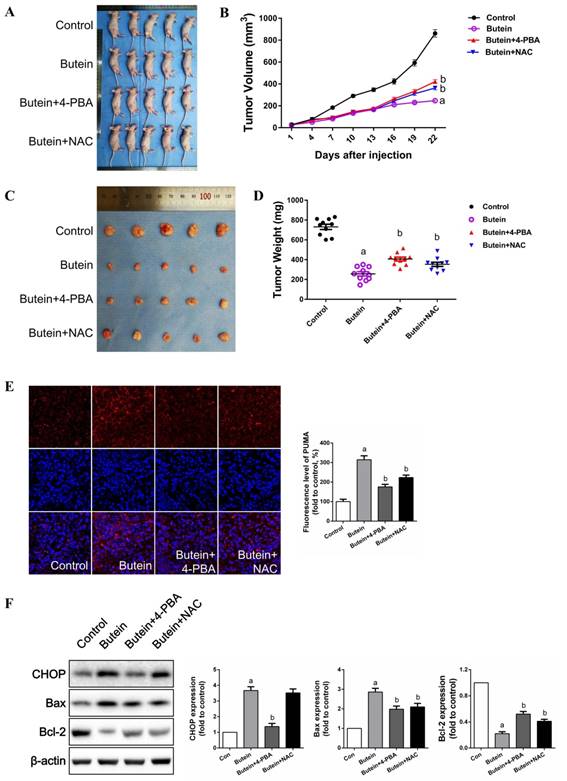
As shown in Fig. 8A-8D, butein treatment significantly inhibited PC-9 tumor xenograft growth. Co-treatment with 4-PBA or NAC markedly impaired the inhibitory effects of butein. Immunofluorescent staining showed that butein treatment induced an abrupt increase in PUMA expression (Fig. 8E). Both 4-PBA and NAC attenuated this effect (Fig. 8E). Western bolt detection showed that 4-PBA and NAC reversed the butein-induced increase in CHOP expression and the Bax/Bcl2 ratio (Fig. 8F). Taken together, these results showed that butein inhibited PC-9 tumor xenograft growth in vivo by triggering ER stress and ROS generation.
Inhibitory effects of butein on in vivo xenografts and the role of ER strass and ROS generation. PC-9 cells were subcutaneously injected into right rear limbs. Over the course of 22 days, butein significantly suppressed the growth of PC-9 tumor xenografts compared to the vehicle control; Co-treatment of 4-PBA or NAC impaired this effect. (A) Photograph of the nude mice on day 22. (B) Tumor volumes of the xenografts. (C) Photographs of the collected xenografts following 22 days of treatment. (D) Tumor weights of the collected xenografts. (E) Immunofluorescent staining of apoptosis associated protein PUMA. (E) Representative band and quantitative analysis of apoptosis associated proteins. All of the results are expressed as the mean ± SEM, n=6. aP < 0.05 versus Control group, bP < 0.05 versus butein group.
Discussion
Butein is a member of the chalcone family of flavonoids and exists in some traditional Chinese herbal medicine plants (37). Such plants have been widely used in Chinese traditional medicine to cure inflammatory diseases, atherosclerosis, cough and even cancers (38). Experimentally, butein has been proved to be an organ protective agent, as well as a tumor suppressor. These very different functions of butein might be attributed to different tissue types and different pathological processes. Zhu et al. found that butein alleviates sepsis induced brain injury through activating SIRT1 (39). Song et al. discovered that butein promotes white adipose tissue browning via increasing Prdm4 expression (40). These studies indicate that butein plays a role in anti-inflammation and lipid metabolism. While, experimentally, butein has also been reported to inhibit a wide range of tumor types. Studies reported that multiple signaling pathways are associated with the anticancer activity of butein. For instance, Yit and colleagues firstly proved that butein exerts cytotoxic effects on human colon adenocarcinoma cell proliferation (41); additionally, butein inhibits telomerase activity by downregulating hTERT gene expression and further inhibits human leukemia cell proliferation and differentiation (42). Butein also increase the apoptosis level of human hepatoma cells treated by TRAIL through upregulating DR5 and inhibiting NF-κB (43). In NSCLC, butein induces A549 cell apoptosis via inhibiting cyclooxygenase 2 expression (12) and overcomes gefitinib-resistance in HCC827 cells through targeting both EGFR and MET (13). However, investigation of the effects of butein in NSCLC has been far from sufficient, and a comprehensive study is needed to elucidate the exact anti-NSCLC effects of butein and the detailed underlying mechanisms. In our present study, butein treatment dose- and time-dependently suppressed cell viability of NSCLC cells (Fig. 1A-1C). Additionally, butein markedly inhibited PC-9 tumor growth in vivo (Fig. 8A-8D). We also observed that butein significantly reduced NSCLC cell migration, adhesion, invasion and proliferation (Fig. 1D-1I, Fig. 2A-2B), which are major activities associated with tumor metastasis. Furthermore, butein arrested NSCLC cells at G2/M phase through regulating Cdc25C, Cyclin B1 and Cdc2 (Fig. 2C-2F). Similar results have been reported in other cancer types (10, 44, 45). In addition, butein treatment induced NSCLC cell apoptosis, which was indicated by TUNEL staining results, MMP lose, increased Bax/Bcl2 ratio and increased caspases activity (Fig. 3, Fig. 4A-4D).
ROS plays a vital role in cell division, cellular senescence and cell signaling. ROS accumulation has been proven to induce cell injury and many other impacts. Many bioactive agents exert anticancer effects via over-activating ROS generation. Studies have illuminated that butein might also exerts anticancer effects via regulating ROS generation. Moon et. al. found butein triggers hepatoma cancer cells G2/M phase arrest and induced cell death by increasing ROS level. Chen et. al. demonstrated that ROS generation mediates butein-induced neuroblastoma cell apoptosis. We observed that butein markedly triggered ROS accumulation in NSCLC cells and further induced cell apoptosis (Fig. 4E-4I). More specifically, butein treatment increased A549 and PC-9 cells ROS generation, activated activity of NADPH oxidase and decreased GSH content. SOD2 was also decreased by butein, indicating that butein might induce ROS and cell injury via alterations in mitochondrial integrity. NAC, a ROS scavenger, was used to confirm the roles of ROS in the anti-NSCLC effects of butein and we observed that treatment with NAC impaired the butein-induced ROS burst and cell cycle arrest (Fig. 7A-7E). These results showed that ROS generation is an important mediator in the anti-NSCLC effect of butein. However, the signaling pathways between butein and ROS still remain unclear.
ER stresses represent a condition, in which unfolded protein accumulates and UPR was activated. UPR activation is the molecular basis of ER stress. During growth of solid cancers, more proteins were synthesized and folded in ER due to the high metabolic level. However, blood supply is often deficient (14). Therefore, hypoxia and nutrient deprivation induce ER stress. Moderate level of ER stress helps to address unfolded proteins accumulated in the ER and restore ER function, while server ER stress will induce cell apoptosis. ER stress mainly contains 3 pathways which can be initiated by PERK, IRE1 and ATF6 (18, 46). Chalcone derivatives other than butein have been demonstrated to exert anticancer effects via regulating the UPR; for example, Lee et al. found that a synthetic chalcone activates UPR and cell apoptosis in breast cancer (20), and Gil et al. proved that another synthetic chalcone derivative triggers UPR-associated apoptosis in A549 cells (47). The present study first confirmed that butein induced ER stress and activated the UPR pathways in NSCLC cells. RNA-seq and western blot showed that butein increased the mRNA and protein levels of some UPR markers and apoptosis related proteins (Fig. 5A-5C). Interestingly, western blot showed that butein mainly affected PERK pathway, indicated by activated eIF2α phosphorylation levels, and increased ATF4, CHOP expressions (Fig. 5B-5C). To confirm the exact role of ER stress in butein-induced cell apoptosis, siRNA targeting CHOP and 4-PBA, a chemical ER stress inhibitor were used. Pre-treatment of CHOP siRNA or 4-PBA could reverse the loss in cell viability and the increase in apoptosis rate induced by butein (Fig. 6B, Fig. 6D-6F). Moreover, ER stress inhibition reduced ROS generation triggered by butein, as indicated by reduced ROS levels (Fig. 6C). These results demonstrated that ER stress and ROS generation mediated the anti-NSCLC effect of butein.
ROS plays a dual role in ER stress signaling. ROS act as signaling intermediates and report ER stress to the UPR, which helps to alleviate the protein burden in ER. Prolonged activation of ER stress induces pro-apoptotic proteins such as CHOP. CHOP then initiate a secondary rise in ROS. This secondary increase in ROS, together with CHOP associated apoptosis pathway, contributes to cell death (48, 49). In the present study, butein treatment triggered ER stress and increased ROS generation in NSCLC cells. More, CHOP siRNA and 4-PBA significantly reduced ROS generation and alleviated cell injury, which indicated that ROS generation in this process is ER stress dependent. Our previous studies also proved that triggering ROS generation is a pro-apoptotic approach of some anticancer flavonoids or their derivatives (14, 19, 27).
Mitochondria-dependent apoptosis and caspase pathways coordinately occur in multiple pathological processes. The change of permeability of mitochondrial membrane can trigger caspase pathway and further induce cell apoptosis (20, 50). Recently, activation of Bcl2 family and caspase family pathway in chalcone-induced apoptosis has been described (20, 21). In the present study, the Bax/Bcl2 ratio was increased by butein (Fig. 4A). In addition, activation of caspase-3, caspase-8 and caspase-9 were also detected (Fig. 4B-4D), indicating that receptor-dependent and mitochondrial apoptotic pathways were activated by butein treatment. We further confirmed the action of caspases in butein-induced NSCLC inhibition through utilizing z-VAD, a pan-caspase inhibitor. We observed that pretreatment with z-VAD significantly preserved cell viability and attenuated butein-induced apoptosis (Fig. 7F-7G). These results were consistent with Khan and collages' study demonstrating that z-VAD reversed butein-induced caspases activation in prostate cancer cells (51). Notably, recent studies by Moon et al. reported that z-VAD did not significantly affect butein-induced apoptosis in human hepatoma cancer cells (10). This difference might due to different tumors, different dosage of butein and the complex mechanisms regulated by butein.
The present study mainly investigated the mechanisms involved in butein-induced apoptosis and cell cycle arrest, while the molecular basis of the anti-migration and anti-invasion effects of butein was not further detected in the present study. To fully display the effects of butein on NSCLC cells, we showed the anti-migration and anti-invasion roles of butein at the beginning of the study. We found that low concentrations of butein (lower than 20 μM) can inhibit cell migration and invasion, without affecting cell viability. The mechanisms that butein inhibits NSCLC cell migration and invasions still need further investigation.
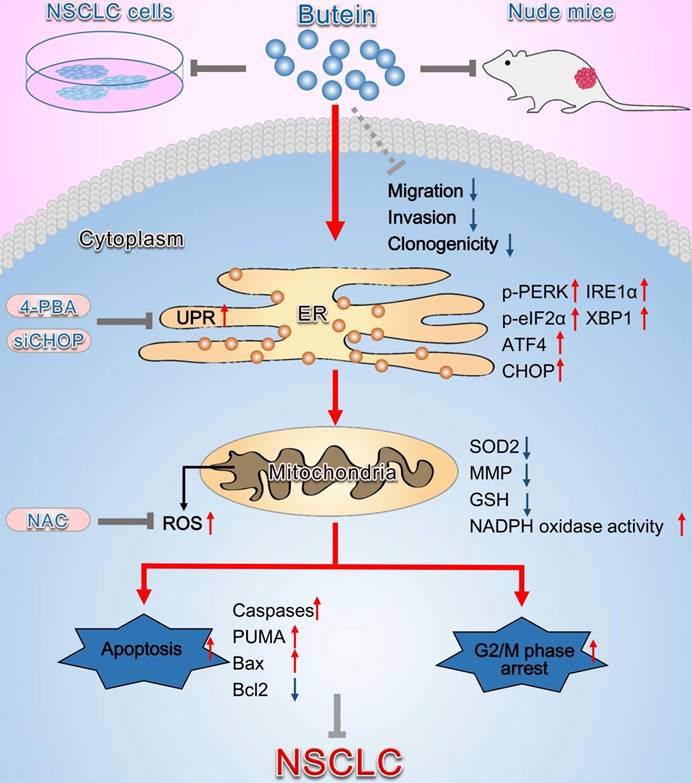
In summary, the present study demonstrates that butein induces NSCLC apoptosis and cell cycle arrest by activating ER stress and ROS pathway (Fig. 9). Inhibition of ER stress, ROS generation, and caspase activity can reverse butein-induced NSCLC cell apoptosis and cell cycle arrest. Consequently, butein might be a promising anti-NSCLC agent, while more comprehensive studies on mechanisms and side effects are needed before applied in clinic.
Schematic diagram summarizing the anti-NSCLC action of butein via activation of ER stress and ROS generation. The in vivo and in vitro experiments showed that butein triggered ER stress and upregulated p-PERK, p-eIF2α levels and ATF4, CHOP, IRE1α, XBP1 expressions. Activation of ER stress further induced oxidative stress, indicated by increased ROS concentration, down-regulated SOD2 expression, decreased the MMP, decreased GSH and increased NADPH oxidase activity, which contributed to cell apoptosis, cell cycle arrest and inhibition of tumor behaviors. However, administration of 4-PBA, CHOP siRNA or NAC partially blocked the anti-NSCLC action of butein. Butein also inhibited cell adhesion, migration and invasion, and the underlying mechanisms still needs further investigation.
Abbreviations
ATF6: activating transcription factor 6; Cdc2: cell division cycle 2; Cdc25C: cell division cyclin 25 homolog C; CHOP: C/EBP homologous protein; eIF2α: eukaryotic translation initiation factor 2α; ER: endoplasmic reticulum; GSH: glutathione; IRE1α: inositol-requiring kinase 1α; MMP: mitochondrial membrane potential; NSCLC: non-small-cell lung cancer; PERK: protein kinase RNA-like ER kinase; PUMA: p53 upregulated modulator of apoptosis; ROS: reactive oxygen species; SOD2: superoxide Dismutase 2; UPR: unfolded protein response; XBP1: X-box binding protein 1; ATF4: activating transcription factor 4; Bcl2: B-cell lymphoma-2; Bax: Bcl2-associated X protein; 4-PBA: 4-phenylbutyric acid; NAC: N-acetyl-L-cysteine.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81871866, 81572252 and 81702731) and the Natural Science Foundation of Shaanxi Province (General Project, 2018SF-159). All data included in this study are available upon request by contact with the corresponding author.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ettinger DS, Akerley W, Borghaei H. et al. Non-small cell lung cancer, version 2.2013. Journal of the National Comprehensive Cancer Network: JNCCN. 2013;11:645-53
2. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. The Lancet Oncology. 2015;16:e342-51
3. Pandey MK, Sandur SK, Sung B. et al. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. The Journal of biological chemistry. 2007;282:17340-50
4. Alshammari GM, Balakrishnan A, Chinnasamy T. Butein protects the nonalcoholic fatty liver through mitochondrial reactive oxygen species attenuation in rats. BioFactors. 2018;44:289-98
5. Bai X, Ma Y, Zhang G. Butein suppresses cervical cancer growth through the PI3K/AKT/mTOR pathway. Oncology reports. 2015;33:3085-92
6. Cho SG, Woo SM, Ko SG. Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. Journal of experimental & clinical cancer research: CR. 2014;33:51
7. Choi HS, Kim MK, Choi YK. et al. Rhus verniciflua Stokes (RVS) and butein induce apoptosis of paclitaxel-resistant SKOV-3/PAX ovarian cancer cells through inhibition of AKT phosphorylation. BMC complementary and alternative medicine. 2016;16:122
8. Huang YT, Lin CI, Chien PH. et al. The depletion of securin enhances butein-induced apoptosis and tumor inhibition in human colorectal cancer. Chemico-biological interactions. 2014;220:41-50
9. Moon DO, Choi YH, Moon SK. et al. Butein suppresses the expression of nuclear factor-kappa B-mediated matrix metalloproteinase-9 and vascular endothelial growth factor in prostate cancer cells. Toxicology in vitro : an international journal published in association with BIBRA. 2010;24:1927-34
10. Moon DO, Kim MO, Choi YH. et al. Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer letters. 2010;288:204-13
11. Zhang L, Chen W, Li X. A novel anticancer effect of butein: inhibition of invasion through the ERK1/2 and NF-kappa B signaling pathways in bladder cancer cells. FEBS letters. 2008;582:1821-8
12. Li Y, Ma C, Qian M. et al. Butein induces cell apoptosis and inhibition of cyclooxygenase2 expression in A549 lung cancer cells. Molecular medicine reports. 2014;9:763-7
13. Jung SK, Lee MH, Lim DY. et al. Butein, a novel dual inhibitor of MET and EGFR, overcomes gefitinib-resistant lung cancer growth. Molecular carcinogenesis. 2015;54:322-31
14. Di S, Fan C, Yang Y. et al. Activation of endoplasmic reticulum stress is involved in the activity of icariin against human lung adenocarcinoma cells. Apoptosis: an international journal on programmed cell death. 2015;20:1229-41
15. Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends in cell biology. 2011;21:709-17
16. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature reviews Cancer. 2014;14:581-97
17. Hu W, Ma Z, Di S. et al. Snapshot: implications for melatonin in endoplasmic reticulum homeostasis. British journal of pharmacology. 2016;173:3431-42
18. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081-6
19. Feng Y, Yang Y, Fan C. et al. Pterostilbene Inhibits the Growth of Human Esophageal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;38:1226-44
20. Lee DH, Jung Jung Y, Koh D. et al. A synthetic chalcone, 2'-hydroxy-2,3,5'-trimethoxychalcone triggers unfolded protein response-mediated apoptosis in breast cancer cells. Cancer letters. 2016;372:1-9
21. Ji T, Lin C, Krill LS. et al. Flavokawain B, a kava chalcone, inhibits growth of human osteosarcoma cells through G2/M cell cycle arrest and apoptosis. Molecular cancer. 2013;12:55
22. Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer research. 1994;54:1890s-4s
23. Yang LH, Ho YJ, Lin JF. et al. Butein inhibits the proliferation of breast cancer cells through generation of reactive oxygen species and modulation of ERK and p38 activities. Molecular medicine reports. 2012;6:1126-32
24. Scheffel MJ, Scurti G, Simms P. et al. Efficacy of Adoptive T-cell Therapy Is Improved by Treatment with the Antioxidant N-Acetyl Cysteine, Which Limits Activation-Induced T-cell Death. Cancer research. 2016;76:6006-16
25. Ma W, Goldberg E, Goldberg J. ER retention is imposed by COPII protein sorting and attenuated by 4-phenylbutyrate. eLife. 2017:6
26. Zumsteg ZS, Morse N, Krigsfeld G. et al. Taselisib (GDC-0032), a Potent beta-Sparing Small Molecule Inhibitor of PI3K, Radiosensitizes Head and Neck Squamous Carcinomas Containing Activating PIK3CA Alterations. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:2009-19
27. Ma Z, Yang Y, Di S. et al. Pterostilbene exerts anticancer activity on non-small-cell lung cancer via activating endoplasmic reticulum stress. Scientific reports. 2017;7:8091
28. Hoshino A, Ariyoshi M, Okawa Y. et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:3116-21
29. Wang T, Cui Y, Jin J. et al. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic acids research. 2013;41:4743-54
30. Zhang G, Fedyunin I, Kirchner S. et al. FANSe: an accurate algorithm for quantitative mapping of large scale sequencing reads. Nucleic acids research. 2012;40:e83
31. Xiao CL, Mai ZB, Lian XL. et al. FANSe2: a robust and cost-efficient alignment tool for quantitative next-generation sequencing applications. PloS one. 2014;9:e94250
32. Zhou Y, Li M, Yu X. et al. Butein suppresses hepatocellular carcinoma growth via modulating Aurora B kinase activity. International journal of biological sciences. 2018;14:1521-34
33. Liu Y, Jiang ZY, Zhou YL. et al. beta-elemene regulates endoplasmic reticulum stress to induce the apoptosis of NSCLC cells through PERK/IRE1alpha/ATF6 pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;93:490-7
34. Jiang Y, Cao Y, Wang Y. et al. Cysteine transporter SLC3A1 promotes breast cancer tumorigenesis. Theranostics. 2017;7:1036-46
35. Tang JY, Farooqi AA, Ou-Yang F. et al. Oxidative stress-modulating drugs have preferential anticancer effects - involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Seminars in cancer biology. 2018
36. Delaunay-Moisan A, Appenzeller-Herzog C. The antioxidant machinery of the endoplasmic reticulum: Protection and signaling. Free radical biology & medicine. 2015;83:341-51
37. Padmavathi G, Roy NK, Bordoloi D. et al. Butein in health and disease: A comprehensive review. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2017;25:118-27
38. Padmavathi G, Rathnakaram SR, Monisha J. et al. Potential of butein, a tetrahydroxychalcone to obliterate cancer. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2015;22:1163-71
39. Zhu Y, Wang K, Ma Z. et al. SIRT1 activation by butein attenuates sepsis-induced brain injury in mice subjected to cecal ligation and puncture via alleviating inflammatory and oxidative stress. Toxicology and applied pharmacology. 2019;363:34-46
40. Song NJ, Choi S, Rajbhandari P. et al. Prdm4 induction by the small molecule butein promotes white adipose tissue browning. Nature chemical biology. 2016;12:479-81
41. Yit CC, Das NP. Cytotoxic effect of butein on human colon adenocarcinoma cell proliferation. Cancer letters. 1994;82:65-72
42. Moon DO, Kim MO, Lee JD. et al. Butein suppresses c-Myc-dependent transcription and Akt-dependent phosphorylation of hTERT in human leukemia cells. Cancer letters. 2009;286:172-9
43. Moon DO, Kim MO, Choi YH. et al. Butein sensitizes human hepatoma cells to TRAIL-induced apoptosis via extracellular signal-regulated kinase/Sp1-dependent DR5 upregulation and NF-kappaB inactivation. Molecular cancer therapeutics. 2010;9:1583-95
44. Ishikawa C, Senba M, Mori N. Butein inhibits NF-kappaB, AP-1 and Akt activation in adult T-cell leukemia/lymphoma. International journal of oncology. 2017;51:633-43
45. Tang YL, Huang LB, Lin WH. et al. Butein inhibits cell proliferation and induces cell cycle arrest in acute lymphoblastic leukemia via FOXO3a/p27kip1 pathway. Oncotarget. 2016;7:18651-64
46. Santos CX, Nabeebaccus AA, Shah AM. et al. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxidants & redox signaling. 2014;20:121-34
47. Gil HN, Koh D, Lim Y. et al. The synthetic chalcone derivative 2-hydroxy-3',5,5'-trimethoxychalcone induces unfolded protein response-mediated apoptosis in A549 lung cancer cells. Bioorganic & medicinal chemistry letters. 2018;28:2969-75
48. Chan CM, Huang DY, Huang YP. et al. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. Journal of cellular and molecular medicine. 2016;20:1749-60
49. Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Molecular aspects of medicine. 2018
50. Fan C, Pan Y, Yang Y. et al. HDAC1 inhibition by melatonin leads to suppression of lung adenocarcinoma cells via induction of oxidative stress and activation of apoptotic pathways. Journal of pineal research. 2015;59:321-33
51. Khan N, Adhami VM, Afaq F. et al. Butein induces apoptosis and inhibits prostate tumor growth in vitro and in vivo. Antioxidants & redox signaling. 2012;16:1195-204
Author contact
![]() Corresponding authors: Xiaolong Yan, Department of Thoracic Surgery, Tangdu Hospital, Fourth Military Medical University, 1 Xinsi Road, Xi'an, 710038, China. Deguang Mu, Department of Respiratory Medicine, Zhejiang Provincial People's Hospital, Hangzhou Medicine College, 158 Shangtang Road, Hangzhou 310014, China. E-mail address: yanxiaolongedu.cn (Xiaolong. Yan), lxfchestedu.cn (Xiaofei Li), mudeguangcom (Deguang Mu)
Corresponding authors: Xiaolong Yan, Department of Thoracic Surgery, Tangdu Hospital, Fourth Military Medical University, 1 Xinsi Road, Xi'an, 710038, China. Deguang Mu, Department of Respiratory Medicine, Zhejiang Provincial People's Hospital, Hangzhou Medicine College, 158 Shangtang Road, Hangzhou 310014, China. E-mail address: yanxiaolongedu.cn (Xiaolong. Yan), lxfchestedu.cn (Xiaofei Li), mudeguangcom (Deguang Mu)