Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(4):543-552. doi:10.7150/ijbs.35256 This issue Cite
Research Paper
Inhibition of PERK Signaling Prevents Against Glucocorticoid-induced Endotheliocyte Apoptosis and Osteonecrosis of the Femoral Head
Yanchun Gao, Hongyi Zhu, Qiyang Wang, Yong Feng ![]() , Changqing Zhang
, Changqing Zhang ![]()
Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China
Received 2019-3-27; Accepted 2019-6-23; Published 2020-1-1
Abstract

Vascular injury is considered an important pathological process during glucocorticoid (GC)-induced osteonecrosis of the femoral head (ONFH). In this study, we tried to investigate whether the endoplasmic reticulum (ER) stress is triggered in the GC-induced endotheliocyte (EC) apoptosis and ONFH. The results showed that a GC upregulated the expression of ER stress-related proteins, and PERK-CHOP signaling played an important role and induced EC apoptosis. The inhibition of PERK by GSK2656157 significantly decreased the GC-induced EC apoptosis in vitro and in vivo, thus protecting a rat model from vascular injury and significantly preventing GC-induced ONFH.
Keywords: glucocorticoid, osteonecrosis of femoral head, endoplasmic-reticulum stress, PERK signaling, endotheliocyte, apoptosis
Introduction
Glucocorticoids (GCs) are widely used against a variety of diseases and are some of the most common causes of nontraumatic osteonecrosis of the femoral head (ONFH) (6, 15, 22, 26). It has been reported that osteonecrosis develops in 9-40% of patients receiving long-term GC therapy (35). Due to the lack of effective drugs for ONFH, patients often need surgical treatment. The long duration of this condition imposes persistent pain and an economic burden on the patients.
The adverse effects of GCs on bone are primarily due to direct actions, in particular, apoptosis of osteoblasts, osteoclasts, and endotheliocytes (ECs) (20, 42, 43). Many studies have suggested that the proapoptotic effect of GCs is cell type dependent (1, 13, 16, 34). Growing evidence shows that ECs are injured by a GC via induction of apoptosis and dysfunction during the pathological process of GC-induced ONFH (10, 20, 28, 46, 47). Thus, a better understanding of the mechanisms of action of GCs on ECs may lead to a better treatment option for ONFH.
The occurrence of endoplasmic reticulum (ER) stress-induced apoptosis has been proven in many diseases (7, 33). ER stress activates stress sensors, including protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring kinase 1 (IRE1), involved in the regulation of cell homeostasis. On the other hand, prolonged and unmitigated ER stress may cause apoptosis (2, 14, 33). Although ER stress correlates with various diseases, the role of ER stress in the pathogenesis of GC-induced ONFH remains unclear. A few studies have indicated that treatment with GCs leads to ER stress and results in various changes, including dysfunction and apoptosis of osteoblasts, osteocytes, and trabecular-meshwork cells (34, 44, 45). Nevertheless, few studies have focused on the GC-induced EC apoptosis mediated by the ER stress signaling pathway.
We were thus prompted to investigate whether ER stress is triggered during GC-induced EC apoptosis and ONFH, and, if so, whether GC-induced EC apoptosis and ONFH could be prevented by inhibition of the GC-induced ER stress signaling pathway.
Materials and Methods
In Vitro Experiments
Cell culture, treatment, and small interfering RNA (siRNA) transfection
The human EC line EAhy926 and human alveolar-bone-derived osteoblasts (OBs) were obtained from KeyGENE BIOTECH (Nanjing, China). The cell lines were cultured (37°C, 5% CO2) in DMEM supplemented with 10% of fetal bovine serum (Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). Human bone marrow stromal cells (BMSCs) were obtained from patients after amputation according to the method described by Kodama (21). Informed consent was obtained from all the patients. The BMSCs and OBs were also cultured (37°C, 5% CO2) in DMEM supplemented with 10% of FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco).
The siRNA experiment was conducted via delivery of plasmids. To construct siRNA expression vectors, the sequences were purchased from GenePharma (Shanghai, China). The siRNA sequences are listed in Appendix Table s1. ECs were transfected with siRNA against PERK, IRE1α, or ATF6 or scrambled siRNA (GenePharma, Shanghai, China). At 8 h after transfection, the transfection medium was replaced with a fresh culture medium, and the cells were cultured for 40 h before treatment with dexamethasone (DEX; Selleck, Houston, TX, USA). The transfection efficiency was >80%.
Annexin V/Propidium Iodide (PI) Fluorescence-Activated Cell Sorting (FACS) Analysis
Cells were analyzed for phosphatidylserine exposure by the annexin-V fluorescein isothiocyanate (FITC)/PI double-staining method according to the manufacturer's instructions (Dojindo Molecular Technologies, Inc. Gaithersburg, MD). Briefly, ECs were harvested by gentle trypsinization and then washed with PBS twice and resuspended in annexin-coupling buffer at a concentration of ~106 cells/mL. A total of 100 μL of the cell suspension was incubated with 5 μL of the annexin V-FITC conjugate and 5 μL of a PI solution at room temperature for 15 min. A FACS machine was employed to evaluate the rate of apoptosis. Approximately 5,000 events were analyzed for apoptotic, necrotic, and live cells. All the experiments were repeated three times, and the results are expressed as a percentage of all the events in each experiment.
Western Blot Analysis
ECs were treated with DEX under different conditions. The cells were harvested and lysed with cell lysis buffer supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO) on ice for 15 min. Protein samples were diluted 1:5 with protein loading buffer (Transgen Biotech, Beijing, China). A total of 30 µg of protein was subjected to SDS-PAGE after denaturation at 95°C for 5 min. The cell lysates were analyzed on a 10% gel (based on Tris-HCl buffer) under reducing conditions. After electrophoresis, the proteins were electrophoretically transferred to 0.22 μm polyvinylidene difluoride membranes (Merck, Darmstadt, Hesse, Germany) and blocked with 5% nonfat dry milk at 4°C overnight. The membranes were then incubated for 3 h at 37°C with anti-ATF6 (Thermo Fisher Scientific, Waltham, MA), anti-phosphorylated-IRE1α, (p-IRE1α; Abcam, Cambridge, MA), anti-PERK, anti-phosphorylated-PERK (p-PERK), anti-IRE1α, anti-CHOP, anti-BIP, anti-XBP1-s, anti-Caspase-3 (Casp3), anti-cleaved Caspase-3 (cCasp3), anti-β-Tubulin, or anti-GAPDH (Cell Signaling Technology, Danvers, MA) antibodies. The membranes were next immersed in a solution of a secondary antibody: an anti-rabbit or anti-mouse IgG antibody (Cell Signaling Technology) for 1 h at 37°C.
After three washes with Tris-buffered saline containing 0.1% of Tween 20, the membranes were added to an ECL substrate in a dark room for imaging on a FluorChem M Gel Documentation System (ProteinSimple, San Jose, CA, USA). The results were analyzed in densitometric analysis software Quantity One (Bio-Rad Laboratories, Inc., Hercules, CA, USA) β-Tubulin or GAPDH served as an internal reference.
In Vivo Experiments
Establishment of the Osteonecrosis Model and Treatment
A short-term GC treatment model was set up as follows. The Sprague-Dawley (SD) rats were randomly and equally divided into the following three groups: (1) Control group (n = 10); (2) methylprednisolone (MPS) group (rats treated with MPS, n = 10); and (3) treatment group (osteonecrotic rats treated with MPS and PERK inhibitor GSK2656157, n = 10). In parallel, 0.2 mL of normal saline was intramuscularly injected into the rats in the control group. MPS (20 mg/[kg•d], Pfizer, New York NY) was intramuscularly injected once a day for 3 days.
The in vivo animal model of ONFH was constructed according to the description by Guo (11). The rats were randomly and equally subdivided into the control group (n = 10), MPS group (n = 10), and treatment group (n = 10).
According to a manufacturer's protocol, PERK inhibitor GSK2656157 (Selleck) was intragastrically administered (25 mg/kg) 8 h before every MPS injection in the treatment group.
None of the rats died before the scheduled euthanasia under 4% chloral hydrate anesthesia. Next, the femoral heads were collected for micro-computed tomography (CT) scanning, immunostaining, and immunohistochemical staining. All the experimental and animal care procedures were approved by the Animal Research Ethics Committee of Shanghai Sixth People's Hospital and were in compliance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Angiography and Micro-CT Scanning
Before euthanasia, the rats were anesthetized and successively perfused with 4% heparinized saline and Microfil (MV-112, Flow Tech, Carver, MA) according to the manufacturer's protocol. The bodies of the rats were stored at 4°C overnight, and then the bilateral femoral heads were collected for further experiments. The femoral heads were subjected to micro-CT scanning before and after decalcification in a 10% EDTA solution for 4 weeks. The micro-CD scanner was set to a resolution of 9 μm per pixel. The trabecular bone was segmented from the bone marrow and analyzed to determine trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), trabecular bone pattern factor (Tb.pf), bone volume per tissue volume (BV/TV), and trabecular number (Tb.N). Three planes (coronal section, sagittal section, and transverse section) of the representative samples from each group were generated in the DataViewer software (Bruker Micro-CT). The total vessel volume was calculated by CTAn.
Immunofluorescent Staining
The femoral heads were sectioned at 5µm thickness in the coronal plane. The deparaffinized sections were processed by 0.25% trypsin antigen retrieval and were blocked with 10% FBS for 1 h at 37°C. The sections were incubated with anti-CD31 (1:200, Sigma-Aldrich) and anti-cleaved caspase 3 (cCasp3) (1:600, Cell Signaling Technology) antibodies at 4°C overnight (primary antibodies), and then the appropriate secondary antibodies (Servicebio, Wuhan, China) were applied for incubation at room temperature for 1 h. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Servicebio) for 5 min. Immunofluorescence photomicrographs were captured by means of a fluorescence microscope (Leica DMI6000B, Germany).
Immunohistochemical Staining
After 1 h 0.25% trypsin antigen retrieval and 1 h 10% FBS incubation at room temperature, the sections were incubated with the anti-cCasp3 (1:200, Cell Signaling Technology) primary antibody overnight at 4°C. After that, the sections were incubated with a biotinylated secondary antibody (Servicebio, Wuhan, China) according to the manufacturer's instructions. The sections were stained with a 3,3-diaminobenzidine precipitate and counterstained with hematoxylin. Photomicrographs were acquired using a LEICA DM 4000 (Leica Microsystems, Germany).
Statistical Methods
Means of multiple groups were compared by one-way analysis of variance (ANOVA). Fisher's exact test was conducted to compare the incidence of the disease between two groups. The independent-sample t test was performed to compare means between two different groups. Statistical analysis was conducted in SPSS 20.0 software (IBM Corp., Armonk, NY, USA). Data with P values <0.05 were considered statistically significant.
Results
DEX Induces EC Apoptosis
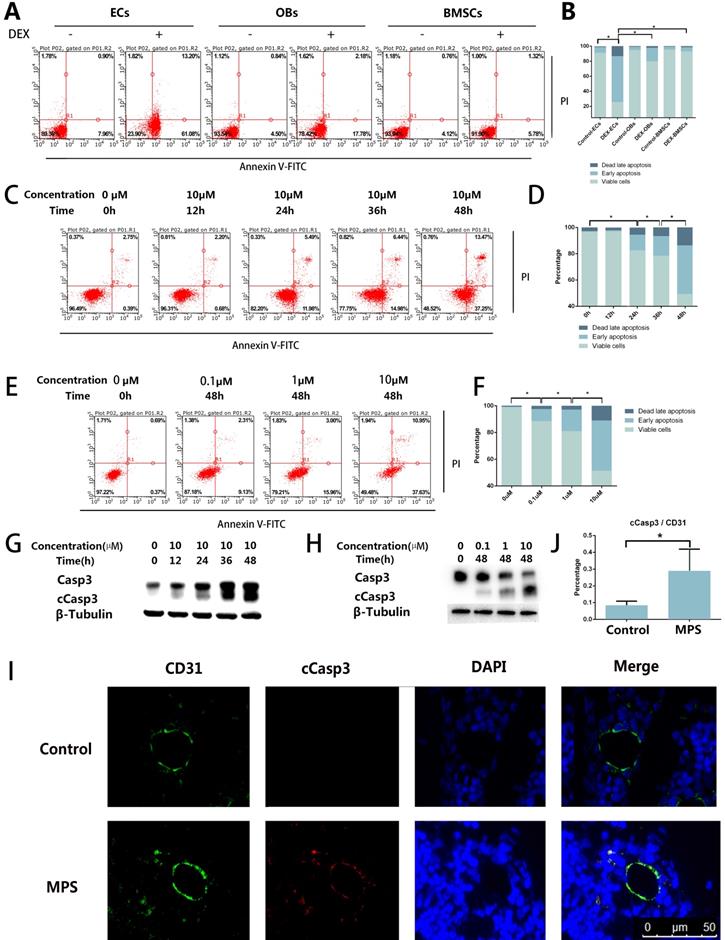
The results of the flow cytometric analysis revealed DEX-induced apoptosis in ECs, BMSCs, and OBs. Compared with OBs and BMSCs, the ECs showed a stronger apoptotic tendency (Figure 1A, B). The flow cytometric analysis also revealed that the apoptosis of ECs increased with the increase of DEX stimulation time and concentration (Figure 1C-F). cCasp3, a representative activated Caspase involved in several types of cell death, was also detected here by western blotting, which supports the conclusions from the flow cytometric analysis (Figure 1G, H). To observe apoptosis in vivo, we performed cCasp3 and CD31 double-label immunofluorescence staining on rat femoral head sections. The results meant that short-term GC treatment induced apoptosis in the inner wall of blood vessels, whereas no apoptosis was found in other tissues of a femoral head (Figure 1I, J). We concluded that apoptosis occurs in the ECs in response to GC treatment, which revealed that the apoptosis was stronger in ECs than in other cells in bone tissue.
Apoptosis in the ECs in response to GC treatment. A. ECs, BMSCs, and MC3T3 were treated with DEX (10 µM) for 48 h. The FACS chart of Annexin V-FITC/PI staining is shown. B. The histogram of FACS results indicates the apoptotic status of different cells. C. ECs were treated with DEX (10 µM) for different periods (0-48 h). The FACS chart of Annexin V-FITC/PI staining is shown. D. The histogram of FACS results indicates the apoptotic status of the cells. E. Additional ECs were treated with different concentrations of DEX (0-10 µM) for 48 h. The results are presented as a FACS chart. F. The histogram of FACS results indicates the apoptotic status of the cells. G, H. ECs under the same treatment conditions presented in A and B were harvested from 6-well plates. The expression levels of Casp3 and β-tubulin are shown in a western blot. I. A representative image of cCasp3 immunohistochemical staining in the femoral head from a 3-day steroid-treated rat shows a pattern consistent with the shape of a microvessel. J. The histogram of immunofluorescence results indicates the percentage of apoptotic ECs in the femoral head. *p < 0.05.
DEX Induces ER Stress in ECs
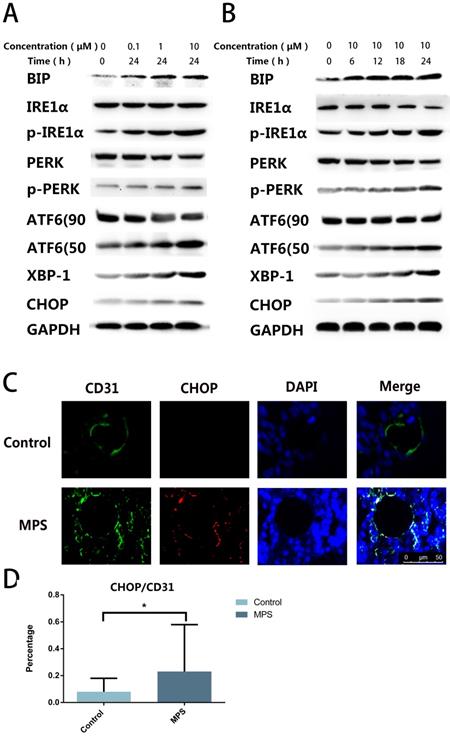
To find out the cause of apoptosis and to confirm the presence of ER stress, we quantified ER stress-related proteins including PERK, p-PERK, IRE1α, p-IRE1α, ATF6, CHOP, XBP-1s, and BIP. The western blot revealed that ER stress-related proteins were upregulated with the increase of stimulation time and concentration, and three classical signaling pathways were found to be activated simultaneously after DEX treatment (Figure 2A, B). The CHOP and CD31 double-label immunofluorescence staining indicated that CHOP expression in ECs increased after short-tern GC treatment (Figure 2C, D). These findings confirmed GC-induced ER stress in ECs.
A. DEX Induces ER stress in ECs. ECs were treated with DEX at different concentrations (0-10 μm) for 24 h. ER stress-related proteins (PERK, p-PERK, IRE1α, p-IRE1α, ATF6, CHOP, XBP-1s, and BIP) were examined by western blotting. B. ER stress-related proteins were also observed among ECs treated with DEX for different periods (0-24 h, 10 μM). C. A representative image of CHOP immunohistochemical analysis in the femoral head from a 3-day steroid-treated rat shows a pattern consistent with the shape of a microvessel. D. The histogram of immunofluorescence results reveals the CHOP-positive percentage of ECs in the femoral head. *p < 0.05.
PERK Inhibition Prevents EC Apoptosis In Vitro
Given that we observed the activation of all three ER stress-related downstream signaling pathways, further experiments were necessary to determine the specific pathway associated with the apoptosis. We blocked the three signaling pathways respectively by transfection of short interfering RNAs against PERK, IRE1α, or ATF6. The siRNA-treated ECs were then stimulated with DEX and assessed by flow cytometry. Compared with the control group, neither the IRE1α-deficient nor ATF6-deficient ECs showed a decrease of apoptosis, whereas the inhibition of PERK significantly decreased the apoptosis after DEX treatment (Figure 3A, B).
PERK inhibition by siRNA or PERK inhibitor GSK2656157 decreased the apoptosis of ECs in vitro. A. ECs were transfected with siRNAs to knockdown PERK, IRE1α, or ATF6, followed by treatment with vehicle or DEX (10 µM) for 48 h. The FACS charts of the Annexin V-FITC/PI staining are presented. B. The histogram indicates the apoptotic status of the cells. C. The ECs were treated under the same above-mentioned conditions, and the expression levels of Casp3 were examined by western blotting. D. The cells were pretreated with the PERK inhibitor GSK2656157 8 h before the DEX treatment (10 µM; 48 h). The FACS charts are shown. E. The histogram of the Annexin V-FITC/PI staining is shown. *p < 0.05.
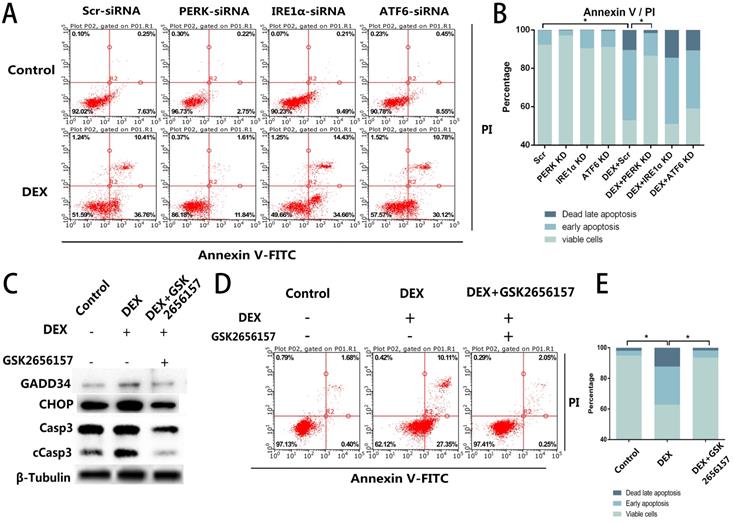
After that, we identified a PERK inhibitor, GSK2656157, and pretreated the cells with the inhibitor 8 h before the DEX treatment. Downstream of PERK signaling pathway. We quantified GADD34 and CHOP, which are downstream of the PERK signaling pathway and participate in apoptosis. The western blot showed that the expression of GADD34 and CHOP diminished after GSK2656157 treatment. Besides, the decreased expression of cCasp3 indicated the antiapoptotic effect of PERK inhibition (Figure 3C). The flow cytometric analysis also showed the decrease of apoptosis in the GSK2656157-pretreated ECs, indicating that the PERK inhibitor exerted a protective action against GC-induced apoptosis (Figure 3D, E).
PERK Inhibition Prevents the GC-induced Vascular Damage In Vivo
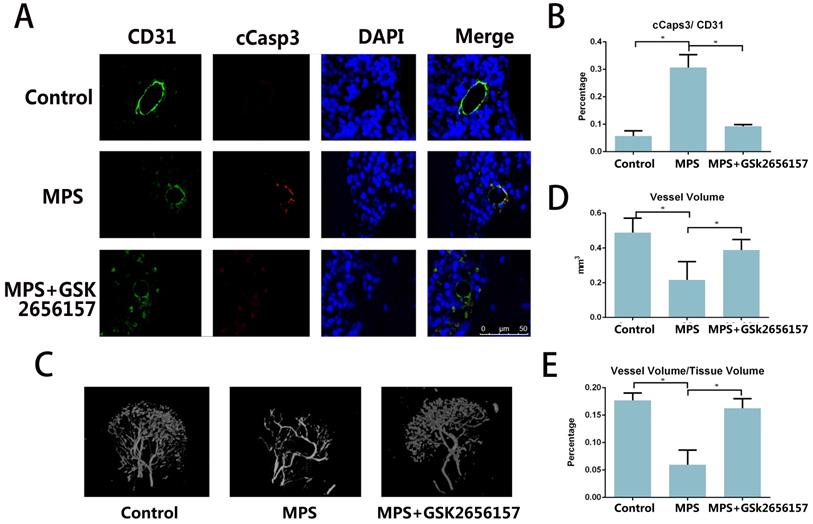
The immunofluorescent staining of CD31 and cCasp3 revealed that PERK inhibitor GSK26556157 reduced the EC apoptosis after short-term MPS treatment. The PERK inhibitor provided protection against GC-induced apoptosis, as verified by our findings in vitro (Figure 4A, B) The micro-CT analysis of the perfused vessels in the femoral heads also suggested that the long-term GC treatment injured the vessels in the femoral head. The PERK inhibitor GSK2656157 had a protective effect against GC-induced vascular injury (Figure 4C-E).
PERK inhibitor GSK2656157 prevented vascular damage in an ONFH model. A. Representative immunostaining of the femoral heads from the rats that received 3-day steroid injections shows the colocalization of CD31 and cCasp3 in the GC group, whereas the immunostaining of the femoral heads from both the control and treatment groups shows low expression levels of cCasp3. B. The histogram based on densitometric analysis of immunostaining from various groups (n = 10; * p < 0.05). C. A representative 3D reconstructed microangiographic image shows the general form of the intraosseous microvessels in the femoral head. D, E. Quantification of the total vessel volume (mm3) and the ratio of total vessel volume to total tissue volume in the femoral heads from the different groups are shown (n = 10). *p < 0.05
PERK inhibitor GSK2656157 Prevents GC-induced ONFH In Vivo
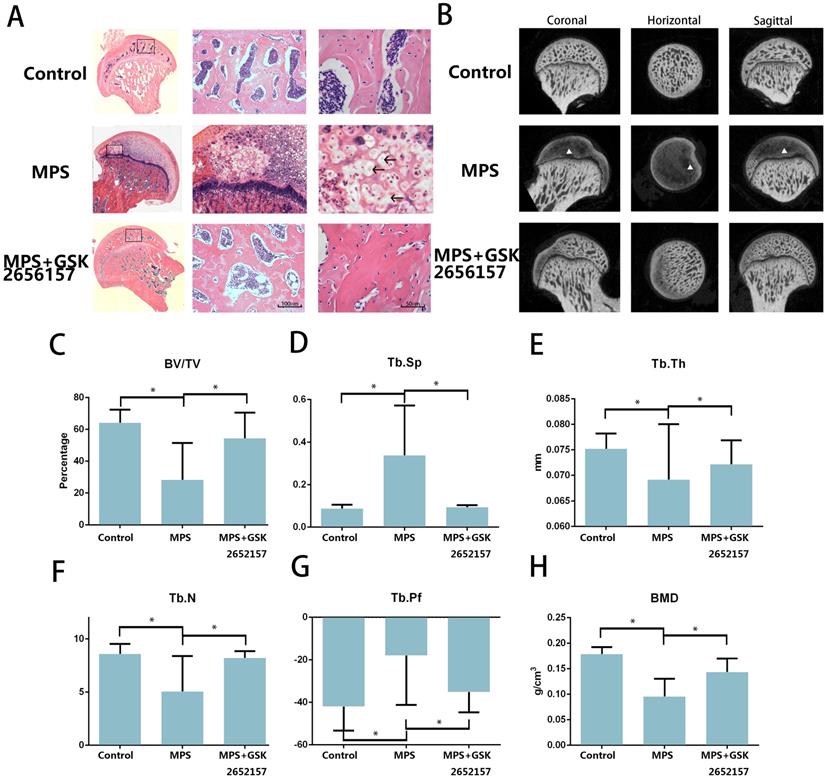
The results of an in vitro experiment revealed that 13 in 20 femoral heads (10 Sprague-Dawley rats) in the MPS group showed visible signs of osteonecrosis, while only one osteonecrotic femoral head was found in the treatment group (p < 0.001). Hematoxylin and eosin (H&E) staining revealed visible subchondral necrosis with fatty-tissue invasion in the subchondral bone trabecular area, whereas PERK inhibitor GSK2656157 successfully prevented ONFH in the treatment group (Figure 5A). The trabecular changes in the subchondral region of the femoral heads were detected by micro-CT (Figure 5B). Besides, the micro-CT analysis yielded results on bone parameters, including Tb.Th, Tb.Sp, Tb.pf, and BV/TV. We noticed a significant preventive effect of GSK2656157 (Figure 5C-G). The bone mineral density (BMD) of the rats in the model group was 0.076 g/cm3, which was significantly lower than that of the control group, whereas the PERK inhibitor significantly prevented the reduction in BMD (Figure 5H). The results above led to the conclusion that PERK signaling blockage (by GSK2656157) may prevent ONFH.
PERK inhibitor GSK2656157 protected the femoral head from osteonecrosis in vivo. A. The H&E staining of coronal sections of the representative femoral heads from each group. The arrows indicate the empty lacunae and hypertrophic fat cells, which indicate osteonecrosis. B. Representative images from the micro-CT analyses in the sagittal, horizontal, and coronal planes. The triangle indicates lower density changes in the subchondral area in the ONFH group. C-H. The histograms of the morphometric analysis show the BMD and bone parameters (BV/TV, Tb.Th, Tb.N, Tb.Pf, and Tb.Sp) of the upper outer subchondral bones of the femoral heads (n = 20). *p < 0.05
Discussion
The most important findings of this study are that ER stress is strongly involved in GC-induced EC apoptosis and ONFH. ECs are more sensitive to apoptosis than other cells in bone tissue when stimulated by GCs. PERK-CHOP signaling plays a critical part in this process. The PERK inhibitor GSK2656157 was demonstrated to be effective in preventing GC-induced vessel injury and ONFH in vivo, thereby further proving the decisive participation of PERK signaling in GC-induced ONFH. This study provides new insight into the inherent relation among EC apoptosis, vascular injury, and GC-induced ONFH. Namely, GCs cause ER stress in ECs and induce apoptosis, which leads to microvascular damage and eventually causes ONFH.
Apoptosis in bone is thought to be the key determinant of GC-induced ONFH (17, 18, 43). Some studies have mainly focused on the direct adverse effects of administered GCs on BMSCs, osteoblasts, and osteoclasts; GCs decrease the formation of both osteoblasts and osteoclasts and increase the apoptosis of osteoblasts while prolonging the lifespan of osteoclasts (12, 34, 36, 37, 43, 48). However, recent studies pointed out that apoptosis of ECs in ONFH are essential but ignored. One of the possible reasons is that the apoptotic effect of a GC is cell-type dependent (9, 46). Compared to other cells, ECs show a stronger and earlier apoptotic tendency in response to GC treatment. Furthermore, there is an even higher GC concentration in blood vessels than in other tissues in vivo (30, 38). It has been reported that during high-dose GC treatment (blood concentration of 15-100 µM, according to a GC treatment guide) (39), the vessels are more affected than any other tissues in the femoral head. This observation is consistent with earlier studies, which suggest that vessel injury is an initiating factor for the pathological processes of osteonecrosis (19, 25, 47), and that osteonecrosis is a series of secondary pathological changes in response to ischemia (4).
ER stress is known to activate three major signaling pathways, such as the PERK-ATF4 axis, ATF6 signal transduction, and IRE1a cascade (24, 33, 41). ER stress may act as a two-edged sword in such diseases as ONFH. The ER responds to stress by activating the unfolded protein response (UPR). If various UPR-induced mechanisms fail to alleviate ER stress, both the intrinsic and extrinsic pathways of apoptosis can get activated. When activated upon sensing ER stress, PERK oligomerizes and phosphorylates itself and the ubiquitous translation initiation factor eIF2α, thereby indirectly inactivating eIF2α and inhibiting mRNA translation. In this way, PERK helps reduce the flux of protein entering the ER to alleviate ER stress (40). It has been demonstrated that a dephosphorylation inhibitor of eIF2α can reduce ER stress (3, 25, 34). Nevertheless, strong or prolonged ER stress may break the homeostatic balance of UPR (32, 40). Subsequently, the PERK-CHOP signaling pathway is activated, inducing apoptosis (5, 8, 24, 31).
PERK is the major protein responsible for attenuation of mRNA translation under ER stress, preventing the influx of newly synthesized protein molecules into the already stressed ER compartment. The activation of eIF2α phosphorylation has been demonstrated to reduce the flux of protein into the ER and thus alleviate ER stress. Nonetheless, different levels of stimulation have been found to cause various ER stress responses. Prolonged or extra strong stimulation is thought to break the PERK-eIF2α regulatory mechanism, mediated by the activation of ATF4-CHOP signaling, which has a proapoptotic activity and is crucial for triggering apoptosis in response to ER stress (29, 31, 33, 40). The experiment on the relation between GC-induced ER stress and stimulus intensity (time/concentration) was under consideration. Because apoptosis in patients with steroid-induced ONFH has been widely reported, it is believed that long-term or large-dose GC treatment may directly cause this apoptosis (27).
More recently, a few studies described the participation of ER stress in ONFH. Sato AY et al. have reported that salubrinal or guanabenz, specific inhibitors of eIF2α dephosphorylation, block apoptosis of osteocytic MLO-Y4 and osteoblastic OB-6 cells, when this apoptosis is induced by either GC or ER stress inducers (34). Liu et al. have reported that ER stress is an important pathological outcome in the surgery-induced ONFH model, and salubrinal alleviates ONFH symptoms by enhancing angiogenesis and bone healing via suppression of ER stress (23). These two studies show that inhibition of eIF2α dephosphorylation reduces ER stress and is proven to be effective at launching the ER stress-related response (3). Furthermore, they focused on the direct effects of ONFH on osteoblasts and osteoclasts. In this study, we proved that a GC activates PERK-CHOP signaling, indicating that GC-induced ER stress breaks ER homeostasis and leads to apoptosis. Furthermore, we confirmed that ER stress is strongly involved in GC-induced EC apoptosis and ONFH. GSK2656157, an inhibitor of PERK phosphorylation, has been proven to be effective against ER stress-induced apoptosis (23). In our study, GSK2656157 blocked the whole downstream signaling of PERK and attenuated GC-induced cell apoptosis. GSK2656157 was demonstrated to be effective in the protection from steroid-induced vessel injury and from ONFH in vivo. These findings confirmed the decisive role of PERK signaling in ONFH.
Our study has several limitations. First, a CHOP knockout transgenic model was not constructed due to the lack of osteonecrotic murine models. Second, although the therapeutic effect is clear-cut, PERK inhibitor GSK2656157 is still not an approved drug and further clinical studies are required.
This work should improve the understanding of the relation between intraosseous microvascular injuries and steroid-induced ONFH. GC-induced ER stress was found to induce EC apoptosis, which triggers other secondary changes and finally causes osteonecrosis. Our experimental results also suggest a promising drug for the prevention of ONFH. PERK signaling, as a brand-new research direction for ONFH prevention, requires much lucubration in the future. Furthermore, a more in-depth study on ONFH may change the traditional paradigm of several bone diseases and could contribute to the discovery of a novel therapeutic method.
Supplementary Material
Supplementary figures and tables.
Abbreviations
ATF6: activating transcription factor 6; BIP: immunoglobulin heavy-chain-binding protein; BMSCs: bone marrow stromal cells; Casp3: caspase 3; CHOP: C/EBP-homologous protein; cCasp3: cleaved caspase 3; DEX: dexamethasone; EC: endotheliocyte; ER: endoplasmic reticulum; FACS: fluorescence-activated cell sorting; GC: glucocorticoid; IRE1: inositol-requiring kinase 1; OBs: osteoblasts; ONFH: osteonecrosis of the femoral head; PERK: protein kinase-like ER kinase; p-IRE1α: phosphorylated IRE1α; p-PERK: phosphorylated-PERK; XBP1s: X-box-binding protein 1 sliced.
Acknowledgements
This work was supported by National Natural Science Foundation of China Grant 81371959.
Authors' contributions
YC Gao and HY Zhu carried out the design and conception of the study and drafted the manuscript. Y Feng and CQ Zhang participated in the drafting of the manuscript and its critical revision. YC Gao and QY Wang conducted all data acquisition, analysis, and interpretation. All the authors read and approved the final manuscript. YC Gao and HY Zhu contributed equally to this work; Y Feng and CQ Zhang are co-senior authors and contributed equally to this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Andric SA, Kojic Z, Bjelic MM, Mihajlovic AI, Baburski AZ, Sokanovic SJ. et al. The opposite roles of glucocorticoid and alpha1-adrenergic receptors in stress triggered apoptosis of rat Leydig cells. American journal of physiology Endocrinology and metabolism. 2013Jan01;304(1):E51-9
2. Binet F, Sapieha P. ER Stress and Angiogenesis. Cell metabolism. 2015Oct6;22(4):560-75
3. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D. et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005Feb11;307(5711):935-9
4. Brandi ML, Collin-Osdoby P. Vascular biology and the skeleton. J Bone Miner Res. 2006Feb;21(2):183-92
5. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. Journal of hepatology. 2013Sep;59(3):583-94
6. Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE. et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis & rheumatology. 2017Aug;69(8):1521-37
7. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & redox signaling. 2014Jul20;21(3):396-413
8. Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnology and bioengineering. 2011Dec;108(12):2777-93
9. El Zaoui I, Behar-Cohen F, Torriglia A. Glucocorticoids exert direct toxicity on microvasculature: analysis of cell death mechanisms. Toxicological sciences: an official journal of the Society of Toxicology. 2015Feb;143(2):441-53
10. Gao Y, Zhu H, Yang F, Wang Q, Feng Y, Zhang C. Glucocorticoid-activated IRE1alpha/XBP-1s Signaling: An Autophagy-Associated Protective Pathway Against Endotheliocyte Damage. American journal of physiology Cell physiology. 2018
11. Guo SC, Tao SC, Yin WJ, Qi X, Sheng JG, Zhang CQ. Exosomes from Human Synovial-Derived Mesenchymal Stem Cells Prevent Glucocorticoid-Induced Osteonecrosis of the Femoral Head in the Rat. International journal of biological sciences. 2016;12(10):1262-72
12. He M, Wang J, Wang G, Tian Y, Jiang L, Ren Z. et al. Effect of glucocorticoids on osteoclast function in a mouse model of bone necrosis. Molecular medicine reports. 2016Aug;14(2):1054-60
13. Heidari N, Miller AV, Hicks MA, Marking CB, Harada H. Glucocorticoid-mediated BIM induction and apoptosis are regulated by Runx2 and c-Jun in leukemia cells. Cell death & disease. 2012Jul19;3:e349
14. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nature reviews Drug discovery. 2013Sep;12(9):703-19
15. Hoes JN, Jacobs JW, Boers M, Boumpas D, Buttgereit F, Caeyers N. et al. EULAR evidence-based recommendations on the management of systemic glucocorticoid therapy in rheumatic diseases. Annals of the rheumatic diseases. 2007Dec;66(12):1560-7
16. Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R. et al. Glucocorticoid dose determines osteocyte cell fate. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011Oct;25(10):3366-76
17. Kabata T, Kubo T, Matsumoto T, Nishino M, Tomita K, Katsuda S. et al. Apoptotic cell death in steroid induced osteonecrosis: an experimental study in rabbits. The Journal of rheumatology. 2000Sep;27(9):2166-71
18. Kerachian MA, Harvey EJ, Cournoyer D, Chow TY, Nahal A, Seguin C. A rat model of early stage osteonecrosis induced by glucocorticoids. Journal of orthopaedic surgery and research. 2011Dec21;6:62
19. Kerachian MA, Harvey EJ, Cournoyer D, Chow TY, Seguin C. Avascular necrosis of the femoral head: vascular hypotheses. Endothelium: journal of endothelial cell research. 2006;13(4):237-44
20. Kerachian MA, Seguin C, Harvey EJ. Glucocorticoids in osteonecrosis of the femoral head: a new understanding of the mechanisms of action. J Steroid Biochem Mol Biol. 2009Apr;114(3-5):121-8
21. Kodama A, Kamei N, Kamei G, Kongcharoensombat W, Ohkawa S, Nakabayashi A. et al. In vivo bioluminescence imaging of transplanted bone marrow mesenchymal stromal cells using a magnetic delivery system in a rat fracture model. The Journal of bone and joint surgery British volume. 2012Jul;94(7):998-1006
22. Larson E, Jones LC, Goodman SB, Koo KH, Cui Q. Early-stage osteonecrosis of the femoral head: where are we and where are we going in year 2018? International orthopaedics. 2018Jul;42(7):1723-8
23. Liu D, Zhang Y, Li X, Li J, Yang S, Xing X. et al. eIF2alpha signaling regulates ischemic osteonecrosis through endoplasmic reticulum stress. Sci Rep. 2017;7(1):5062
24. Liu Z, Lv Y, Zhao N, Guan G, Wang J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell death & disease. 2015Jul30;6:e1822
25. Matsuoka M, Komoike Y. Experimental Evidence Shows Salubrinal, an eIF2alpha Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. International journal of molecular sciences. 2015Jul17;16(7):16275-87
26. McNally EM, Kaltman JR, Benson DW, Canter CE, Cripe LH, Duan D. et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. Circulation. 2015May5;131(18):1590-8
27. Mutijima E, De Maertelaer V, Deprez M, Malaise M, Hauzeur JP. The apoptosis of osteoblasts and osteocytes in femoral head osteonecrosis: its specificity and its distribution. Clinical rheumatology. 2014Dec;33(12):1791-5
28. Okada Y, Tanikawa T, Iida T, Tanaka Y. [Vascular injury by glucocorticoid; involvement of apoptosis of endothelial cells]. Clinical calcium. 2007Jun;17(6):872-7
29. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation. 2004Apr;11(4):381-9
30. Petersen MC, Nation RL, McBride WG, Ashley JJ, Moore RG. Pharmacokinetics of betamethasone in healthy adults after intravenous administration. European journal of clinical pharmacology. 1983;25(5):643-50
31. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Current molecular medicine. 2016;16(6):533-44
32. Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends in biochemical sciences. 2007Oct;32(10):469-76
33. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochimica et biophysica acta. 2013Dec;1833(12):3460-70
34. Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone. 2015Apr;73:60-8
35. Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmunity reviews. 2013Mar;12(5):629-32
36. Shi C, Qi J, Huang P, Jiang M, Zhou Q, Zhou H. et al. MicroRNA-17/20a inhibits glucocorticoid-induced osteoclast differentiation and function through targeting RANKL expression in osteoblast cells. Bone. 2014Nov;68:67-75
37. Shi J, Wang L, Zhang H, Jie Q, Li X, Shi Q. et al. Glucocorticoids: Dose-related effects on osteoclast formation and function via reactive oxygen species and autophagy. Bone. 2015Oct;79:222-32
38. Tsuei SE, Petersen MC, Ashley JJ, McBride WG, Moore RG. Disporition of synthetic glucocorticoids. II. Dexamethasone in parturient women. Clinical pharmacology and therapeutics. 1980Jul;28(1):88-98
39. van der Goes MC, Jacobs JW, Bijlsma JW. The value of glucocorticoid co-therapy in different rheumatic diseases-positive and adverse effects. Arthritis research & therapy. 2014Nov13;16(Suppl 2):S2
40. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011Nov25;334(6059):1081-6
41. Wang C, Wang Y, Meng H, Gou W, Yuan X, Xu X. et al. Microstructure and Nanomechanical Properties of Single Trabecular Bone in Different Regions of Osteonecrosis of the Femoral Head. Journal of nanoscience and nanotechnology. 2016Mar;16(3):2264-9
42. Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. The New England journal of medicine. 2011Jul07;365(1):62-70
43. Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine. 2012Apr;41(2):183-90
44. Whirledge S, Senbanjo LT, Cidlowski JA. Genistein disrupts glucocorticoid receptor signaling in human uterine endometrial Ishikawa cells. Environmental health perspectives. 2015Jan;123(1):80-7
45. Yang J, Wu Q, Lv J, Nie H. 4-Phenyl butyric acid prevents glucocorticoid-induced osteoblast apoptosis by attenuating endoplasmic reticulum stress. Journal of bone and mineral metabolism. 2016
46. Yu QS, Guo WS, Cheng LM, Lu YF, Shen JY, Li P. Glucocorticoids Significantly Influence the Transcriptome of Bone Microvascular Endothelial Cells of Human Femoral Head. Chinese medical journal. 2015Jul20;128(14):1956-63
47. Zhang Y, Yin J, Ding H, Zhang C, Gao YS. Vitamin K2 Ameliorates Damage of Blood Vessels by Glucocorticoid: a Potential Mechanism for Its Protective Effects in Glucocorticoid-induced Osteonecrosis of the Femoral Head in a Rat Model. International journal of biological sciences. 2016;12(7):776-85
48. Zou W, Yang S, Zhang T, Sun H, Wang Y, Xue H. et al. Hypoxia enhances glucocorticoid-induced apoptosis and cell cycle arrest via the PI3K/Akt signaling pathway in osteoblastic cells. Journal of bone and mineral metabolism. 2015Nov;33(6):615-24
Author contact
![]() Corresponding authors: Yong Feng, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China. E-mail: fengyongxshcom or Changqing Zhang, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China. E-mail: zhangcqedu.cn
Corresponding authors: Yong Feng, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China. E-mail: fengyongxshcom or Changqing Zhang, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China. E-mail: zhangcqedu.cn