Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(4):682-693. doi:10.7150/ijbs.37592 This issue Cite
Research Paper
NVP-BEZ235 inhibits thyroid cancer growth by p53- dependent/independent p21 upregulation
Banjun Ruan1,#, Wei Liu1,#, Pu Chen1, Rongrong Cui1, Yu Li1, Meiju Ji2, Peng Hou1,3, ![]() , Qi Yang1,3,
, Qi Yang1,3, ![]()
1. Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China
2. Center for Translational Medicine, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China
3. Key Laboratory for Tumor Precision Medicine of Shaanxi Province, the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China
#Contributed equally
Received 2019-6-14; Accepted 2019-11-30; Published 2020-1-14
Abstract

NVP-BEZ235 is a novel dual PI3K/mTOR inhibitor, currently in phase 1/2 clinical trials, exhibiting clinical efficiency in treatment of numerous malignancies including thyroid cancer. Cancer cells harboring mutant p53 was widely reported to be blunt to pharmaceutical therapies. However, whether this genotype dependent effect also presents in thyroid cancer when treated with NVP-BEZ235 remains unknown. Therefore, in this study, the tumor suppressing effects of NVP-BEZ235 in thyroid cancer cell lines and in-vivo xenograft mouse model harboring different p53 status were examined. The antitumor effects were confirmed in p53 mutant thyroid cancer cells, though less prominent than p53 wild type cells. And for the p53 mutant cells, p53-independent upregulation of p21 plays a critical role in their response to NVP-BEZ235. Moreover, GSK3β/β-catenin signaling inhibition was implicated in the p21-mediated G0/G1 cell cycle arrest in both p53 wild type and mutant thyroid cancer cells treated with NVP-BEZ235.
Keywords: NVP-BEZ235, Thyroid cancer, PI3K/Akt/mTOR, p53, Cell cycle arrest, p21
Introduction
Thyroid cancer is the most frequent endocrine cancer, and its incidence has rapidly increased over the past few decades all over the world, comprising a broad spectrum of diseases with variable prognoses [1-3]. Although the death rate of thyroid cancer is relatively low, the rate of disease recurrence or persistence is high, which is associated with increased incurability and patient morbidity and mortality [4]. Thyroid cancers derive from endocrine thyroid cells, including follicular thyroid cells and parafollicular C cells, and is classified into several histological kinds according to the cellular origins, characteristics and prognoses. Papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC) pertain to well-differentiated carcinomas (WDTCs), which not only could be effectively cured by surgical and radio iodinated therapy, but also acquire a favorable outcome. On the contrary, poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC), belong to the minority malignancies, however with extremely high mortality and lack of efficient therapeutics [5]. Thyroid cancer initiation and progression occurs through gradual accumulation of various genetic and epigenetic alterations, such as activating mutations of the BRAFV600E, RET/PTC rearrangement, PTEN deficiency, and TP53 mutations, that involve the activation of MAPK and PI3K/AKT signaling pathways [5, 6]. Novel compounds and therapeutic strategies that selectively target these pathways have been identified, some of which have been evaluated in preclinical and clinical studies [5-8].
NVP-BEZ235 is an orally bioavailable imidazoquinoline derivative that inhibits the activity of PI3K/mTOR, currently in phase 1/2 clinical trials [9]. Therapeutic potency of NVP-BEZ235 has achieved efficacy as a monotherapeutic drug or in combination with kinds of anticancer drugs. Particularly, NVP-BEZ235 treatments enhance sensitivity to other drugs and improve drug resistance in numerous malignancies, including acute myeloid leukemia [10], prostate cancer [11], ovarian cancer [12], non-small cell lung cancer [13], gastric cancer [14], colorectal cancer [15], and thyroid cancer [16].
Nuclear transcription factor p53 have been identified as a tumor suppressor gene attributed to its role in inducing cell cycle arrest and/or apoptosis [17]. However, loss of function mutations in TP53 gene always occur in over 50 % of human cancers and TP53 then acts as an oncogenic gene in these malignant cancer cells [18]. Tumors carrying TP53 mutations display a chemotherapy-resistant phenotype [19]. In some cases, TP53 mutations are observed in thyroid cancer and exacerbates the malignant phenotype of the tumors [20]. Though NVP-BEZ235 has been verified efficacy in treating thyroid cancer [16], its antitumor effects and mechanisms in thyroid cancer harboring TP53 mutations remain largely unknown. In this study, we used a panel of authenticated thyroid cancer cell lines carrying wild-type TP53 or mutant-type TP53 to test the therapeutic potential of NVP-BEZ235 and attempted to understand its anticarcinogenic mechanisms in thyroid cancer.
Materials and Methods
Cell lines and cell culture
Human thyroid cancer cell lines BCPAP, IHH4, K1 were obtained from Dr. Haixia Guan (The First Affiliated Hospital of China Medical University, Shenyang, China). C643 was provided by Dr. Lei Ye (Ruijin Hospital, Shanghai, China). Among them, IHH4 and K1 cells harbor wild-type TP53, whereas BCPAP and C643 cells carry mutant-type TP53. Genetic alterations including TP53 genomic status for each cell line were summarized in Table 1. All cell lines were cultured in RPMI 1640 medium supplemented with 10 % fetal bovine serum (FBS) at 37 °C in an incubator containing 5 % CO2.
The origins and genetic alterations of thyroid cancer cell lines
| Cell lines | Origins | Genetic alternations | NVP-BEZ235 IC50 (nM ) | ||
|---|---|---|---|---|---|
| MAPK pathway | PI3K/Akt pathway | TP53 status | |||
| IHH4 | PTC | BRAF (V600E) | Akt (E17K) | TP53 (Wild-type) | 38.9 |
| K1 | PTC | BRAF (V600E) | PI3KCA (E542K) | TP53 (Wild-type) | 70.3 |
| BCPAP | PTC | BRAF (V600E) | Akt1 copy gain | TP53 (D259Y/K286E) | 123.5 |
| C643 | ATC | HRAS (G13R) | -- | TP53 (R248Q/K286E) | 221.0 |
NVP-BEZ235 or GSK3β inhibitor treatment
NVP-BEZ235 was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA), and dissolved in dimethylsulfoxide (DMSO). Cells were treated with indicated concentration of NVP-BEZ235 (0 nM, 50 nM, 100 nM, 200 nM, 500 nM and 1000 nM, respectively). The medium and agent were replenished every 24 h at the incubation period. DMSO was added to the medium as the vehicle control and the concentration was always kept below 0.1 %. In addition, SB216763, a GSK3β inhibitor, were purchased from TargetMol (Boston, MA, USA) and used at a concentration of 10 μM in this study.
p21 or p53 knockdown
A double-stranded oligonucleotide of siRNA targeting p21 (5′-AGACCATGTGGACCTGTCA-3′) or p53 (5′-AGACCUAUGGAAACUACUU-3′), and negative control (NC) siRNA were obtained from Ribobio (Guangzhou, China). Cells were transfected at 50 % confluence using X-tremeGENE siRNA transfection reagent (Roche, Germany) with a final siRNA concentration of 50 nM.
RNA extraction and quantitative real-time PCR (RT-qPCR) analysis
Total RNA was extracted from cells using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA samples (1 μg) were reverse transcribed to cDNA by PrimeScriptTM RT reagent Kit (Takara, Dalian, China) according to the instructions of the manufacturer. Real-time quantitative PCR was performed on a CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA) using SYBR Premix Ex TaqTM (Takara, Dalian, China) and results were normalized to β-actin. Each sample was run in triplicate. Primer pairs of CDKN1A and β-actin used for amplification in this study are: 5'-GGCGGCAGACCAGCATGACAGATT-3' (CDKN1A forward) and 5'-GCAGGGGGCGGCCAGGGTAT-3' (CDKN1A reverse); 5'-GCACAGAGCCTCGCCTT-3' (β-actin forward) and 5'-GTTGTCGACGACGAGCG-3' (β-actin reverse).
Western blot analysis
Treated cells were lysed in prechilled RIPA buffer containing protease inhibitors.
Equal amount of proteins was electrophoresed using 10 % SDS-PAGE, and transferred onto polyvinylidene fluoride (PVDF) membranes (Roche Diagnostics, Mannheim, Germany). The membranes were blocked in 5 % BSA dissolved in TBST (1 × TBS buffer and 0.05 % Tween 20) for 2 h, and then incubated at 4 ℃ overnight with the indicated primary antibodies. Anti-phospho-Akt (Ser473 or Thr308; p-Akt) and anti-total Akt (t-Akt) antibodies were purchased from Bioworld Technology (St. Louis, MO, USA). Anti-total mTOR (t-mTOR), anti-p53, anti-β-catenin, anti-phospho-β-catenin (Ser33; p-β-catenin) and anti-Cdk2 antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-phospho-mTOR (Ser2448; p-mTOR), anti-FOXO3a, anti-phospho-FOXO3a (Thr32; p-FOXO3a), anti-p21, anti-GSK3β and anti-phospho-GSK3β (Ser9; p-GSK3β) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-cyclin E1 antibodies was purchased from Sino Biological (Beijing, China). Anti-GAPDH was purchased from Abmart (Shanghai, China). Then species-specific HRP-conjugated secondary antibodies were incubated (ZSGB Biotechnology, Beijing, China) and immunoblotting signals were visualized using the Western Bright ECL detection system (Advansta, CA, USA).
MTT assay
3-(4,5-Dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to evaluate cell proliferation. Cells (3~5 × 103/well) were seeded in 96-well plates and cultured with various concentrations of NVP-BEZ235. IC50 values were calculated using the Reed-Muench method. After treatment, 20 μL of MTT (5 mg/ml; Sigma, Saint Louis, MO, USA) was added and incubated for additional 4 h. Then the MTT containing medium was discarded, followed by addition of 150 μL of DMSO. The plates were then read on a microplate reader using a test wavelength of 570 nm and a reference wavelength of 670 nm. All the experimental sets were performed in triplicate.
Colony formation assays
Cells were seeded onto 6-well plates at a density of 800 cells per well. After 24 h of culture, the cells were treated with NVP-BEZ235 (200 nM) or DMSO vehicle. The medium and drug was refreshed every 3 days. After 14 days of culture, surviving colonies (≥ 50 cells per colony) were stained with 0.5 % crystal violet in 20 % methanol for 20 minutes and counted. Each experiment was performed in triplicate.
Flow cytometry analysis
For cell cycle assay, cells in the exponential growth phase were serum starved for 12 h. After co-culture with 200 nM NVP-BEZ235 for 48 h, cells were harvested, washed twice in phosphate buffered saline (PBS), and fixed in ice-cold 70 % ethanol for at least 30 min. Cells were then stained with propidium iodide (PI) solution (50 μg/ml PI, 50 μg/ml RNase A, 0.1 % Triton X-100, 0.1 mM EDTA). Cell cycle distributions were assessed based on DNA contents using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Immunofluorescence
The human thyroid cancer cells were seeded onto glass coverslips in 6-well plates and cultured until 50-60 % confluence, followed by treatment with 200 nM NVP-BEZ235 for 48 h. Cells were then fixed with 4.0 % paraformaldehyde for 20 min at room temperature and blocked in 5.0 % goat serum for 30 min. Next, the coverslips were incubated at 4 °C with primary FOXO3a antibody (#2497, Cell Signaling Technology,) overnight. Subsequently, the cells were washed in PBS. The coverslips were incubated with goat anti-rabbit IgG H&L (FITC, ab6717, Abcam, San Francisco, CA, USA) for 1.5 h and dyed with Hoechst33342 (Beyotime biotechnology, Shanghai, China). The images were obtained with an Olympus IX71 microscope (Olympus) and color mergence was performed using ImageJ software (ImageJ version 1.44p, NIH, MD, USA).
Immunohistochemical (IHC) staining
IHC staining was performed to determine the protein expression of p53, p21 and Ki67 in the xenograft tumor tissues and done as previously described [21].
Animal studies
Four-week-old female BALB/c athymic mice were purchased from SLAC laboratory Animal Co., Ltd. (Shanghai, China) and randomly divided into four groups (6 mice per group). 6 × 106 K1 cells or 3 × 106 C643 cells were subcutaneously injected into the right groin of the mice to establish tumor xenografts. 7 days after injection, mice were treated with NVP-BEZ235 (25 mg/kg/day) and tumor size was measured every second day. Tumor volume was calculated using the equation: volume (mm3) = (length × width2)/2. The mice were then euthanized on day 15 post-injection and tumors were isolated. Tumor masses were weighed and then subjected to IHC analysis. All animal care and handling procedures were approved by the Animal Ethics Committee of Xi'an Jiaotong University.
Statistical analysis
All results in the present study were presented as the mean ± standard deviation (SD). Statistical significance of difference between the results was assessed using a standard 2-tailed t test, conducted using SPSS statistical package (16.0, Chicago, IL, USA). A value of p < 0.05 was considered statistically significant.
Results
NVP-BEZ235 inhibits PI3K/Akt/mTOR signaling in human thyroid cancer cells
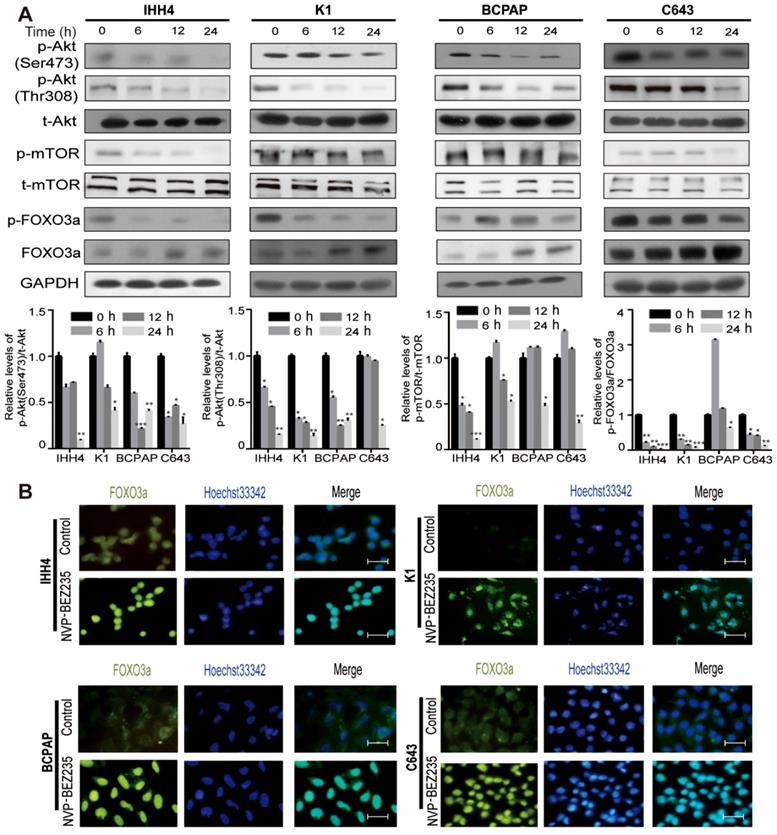
Excessive activation of the PI3K/Akt/mTOR signaling is frequently observed in thyroid tumorigenesis [22]. Using western blot analysis, we first examined thyroid cancer cells response to NVP-BEZ235 by checking the PI3K/Akt/mTOR pathway activity. In both p53 wild type (IHH4 and K1) and mutant (BCPAP and C643) cell lines, 200 nM NVP-BEZ235 treatment decreased the phosphorylation of Akt (p-Akt; Ser473 and Thr308), mammalian target of rapamycin (p-mTOR) and Forkhead box O3 (p-FOXO3a), and increased the FOXO3a expression in a time-dependent manner (Figure 1A). Moreover, the results of immunofluorescence demonstrated that the nuclear translocation of transcription factor FOXO3a, a tumor suppressor and negative downstream effector of PI3K/Akt signaling [23, 24], was significantly increased after NVP-BEZ235 application (Figure 1B). These data indicated PI3K/Akt/mTOR signaling pathway was inhibited by NVP-BEZ235 treatment in both p53 wild type and mutant thyroid cancer cell lines.
The PI3K/Akt/mTOR signaling pathway is inhibited by NVP-BEZ235 in thyroid cancer cells. (A) Western blot showed that NVP-BEZ235 treatment (200 nM) reduced phosphorylation of Akt (p-Akt; Ser473 and Thr308), mammalian target of rapamycin (p-mTOR) and Forkhead box O3 (p-FOXO3a) in IHH4, K1, BCPAP and C643 cells at 0 h, 6 h, 12 h and 24 h (upper). Relative protein levels were quantified by ImageJ software (lower). GAPDH was used as loading control. (B) Immunofluorescence showing that the increased nuclear translocation of transcription factor FOXO3a in IHH4, K1, BCPAP and C643 cells with NVP-BEZ235 treatment (200 nM) for 12 h. Scale bar = 20 μm. *p < 0.05, **p < 0.01, ***p < 0.001, respectively, NVP-BEZ235 versus control.
NVP-BEZ235 suppresses thyroid cancer cell proliferation
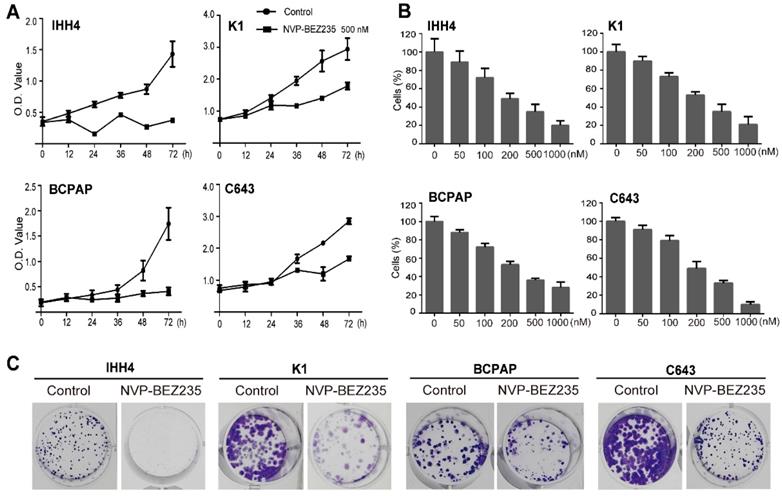
To evaluate the effect of NVP-BEZ235 on cell proliferation in thyroid cancer cells with different TP53 status, we cultured the cells with 500 nM NVP-BEZ235 or vehicle control, and using MTT assays to determine the cell viability on different time points (0, 12, 24, 36, 48 and 72 h). As shown in Figure 2A, NVP-BEZ235 inhibited cell proliferation in all the four thyroid cancer cell lines in a time-dependent manner. And gradient NVP-BEZ235 (0, 50, 100, 200, 500 and 1000 nM) application significantly slowed the growth of both p53 wild type and mutant thyroid cancer cells cultured for 48 h, showing a dose-dependent inhibitory effect (Figure 2B). The data in Table 1 exhibited the corresponding IC50 of the four cell lines, indicating the half maximal inhibitory concentration of NVP-BEZ235 in IHH4, K1, BCPAP, and C643 were 38.9 nM, 70.3 nM, 123.5 nM, 221.0 nM, respectively. We got the phenomenon that the p53 wild type thyroid cancer cells were more sensitive to NVP-BEZ235 than p53 mutant thyroid cancer cells. In addition, as shown in Figure 2C, the long-term anti-proliferative activity on thyroid cancer cell proliferation was further confirmed by colony formation assay.
NVP-BEZ 235 inhibits proliferation in a concentration- and time-dependent manner in thyroid cancer cells. (A) MTT assays suggested that NVP-BEZ235 inhibited cell growth of IHH4, K1, BCPAP and C643 cells at 500 nM in a time-dependent manner. (B) Dose-response curves were drawn in IHH4, K1, BCPAP and C643 cells treated with NVP-BEZ235 from 50 nM to 1000 nM for 48 h. (C) Colony formation assay performed to assess proliferation of IHH4, K1, BCPAP and C643 cells. **p < 0.01, ***p < 0.001, NVP-BEZ235 versus control.
NVP-BEZ235 treatment induces cell cycle arrest in human thyroid cancer cells
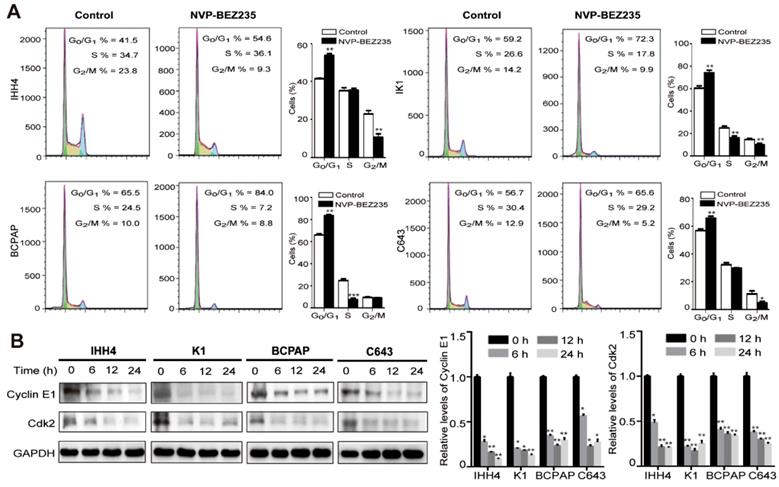
Cell cycle -dependent mitotic rate is a key determinant of cell proliferation. Given the data above, we next tend to detect the cell cycle arrest after NVP-BEZ235 treatment. Compared with vehicle treated control cells, NVP-BEZ235 treatment significantly increased cell fractions at G0/G1 phase in all cell lines (Figure 3A). In specific, the G0/G1 phase proportion of IHH4 lifted from 41.5 % to 54.6 % (p < 0.01), from 59.2 % to 72.3 % (p < 0.01) for K1, from 65.5 % to 84.0 % (p < 0.001) for BCPAP, and from 56.7 % to 65.6 % (p < 0.01) for C643. Briefly, NVP-BEZ235 caused cell cycle arrest in the four thyroid cell lines regardless of TP53 gene status, even more significant in cells carrying mutant TP53. In addition, cyclin E are known to interact with Cdk2 and are involved in triggering cell cycle activity in carcinogenesis [25]. NVP-BEZ235 treatment significantly decreased the protein expression of cyclin E1 and Cdk2 in the human thyroid cancer cell lines in a time-dependent manner (Figure 3B).
NVP-BEZ235 induces cell cycle arrest in thyroid cancer cells. (A) IHH4, K1, BCPAP and C643 cells were treated with 200 nM NVP-BEZ235 for 48 hours, respectively. DNA content was measured by flow cytometry to determine cell cycle fractions. The fraction of cells in each cell cycle phase was indicated in the figures. (B) The protein expressions of cyclin E1 and cyclin-dependent kinase 2 (Cdk2) were examined by western blot in IHH4, K1, BCPAP and C643 cells with 200 nM NVP-BEZ235 treatment (left). Relative protein levels were quantified by ImageJ software (right). *p < 0.05, **p < 0.01, ***p < 0.001, respectively, NVP-BEZ235 versus control.
NVP-BEZ235 inhibits thyroid cancer cell growth through upregulation of p21
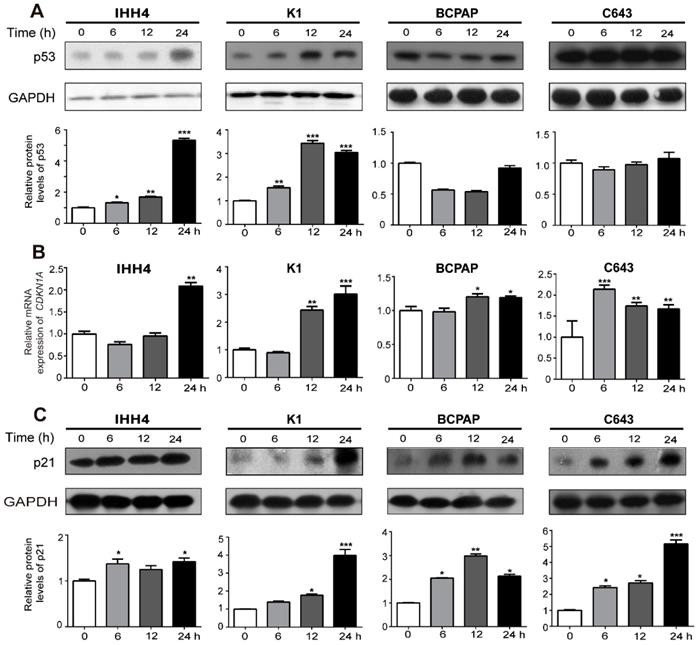
Cell cycle progression is a complex system involving cyclins, cyclin-dependent kinases, cyclin-dependent kinase inhibitors and orther associated proteins [26]. P21, and p27 are both inhibitors of G1 cyclin-dependent kinases [27]. And p21 is a transcription target of p53. Thus we tried to explore wether the inhibitory effect of cell growth induced by NVP-BEZ235 is up to p53, p21 or p27. As shown in western blot assays, NVP-BEZ235 treatment gradually increased the wild type p53 expression in IHH4 and K1 cells in a time dependent manner, while the mutant p53 expression was insusceptible, even slightly decreased in BCPAP and C643 (Figure 4A, representative results in the upper panel, and statistical analysis in the bottom). However, both CDKN1A (p21 coding gene) mRNA level and p21 protein expression levels were remarkably up-regulated by NVP-BEZ235 in all the four cell lines regardless of TP53 status and independent of p53 expression levels (Figure 4B and 4C, for 4C, representative results in the upper panel, and statistical analysis in the bottom), suggesting the inhibitory effect of NVP-BEZ235 on thyroid cancer cell growth is related to transcriptional increase of p21. However, cells treated in the same condition showed no significant response of p27 to NVP-BEZ235 (Supplementary Figure 1).
NVP-BEZ235 up-regulates p21 in both p53 wild type and mutant thyroid cancer cells. (A) p53 protein expression was determined by Western blot in IHH4, K1, BCPAP and C643 cells with 200 nM NVP-BEZ235 treatment. (B) CDKN1A mRNA expression was detected by qRT-PCR in IHH4, K1, BCPAP and C643 cells with 200 nM NVP-BEZ235 treatment. (C) p21 protein expression was tested by Western blot in IHH4, K1, BCPAP and C643 cells with 200 nM NVP-BEZ235 treatment. β-actin was used as a normalized control for qRT-PCR assay. GAPDH was used as loading control in western blot assays. *p < 0.05, **p < 0.01 and ***p < 0.001, respectively, NVP-BEZ235 versus control.
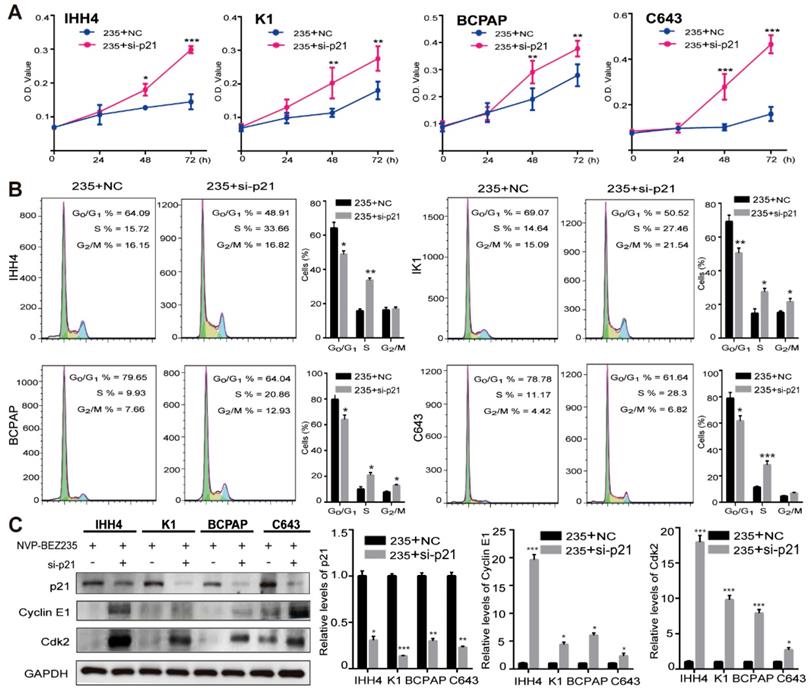
To further clarify the role of p21 in the cell growth inhibition induced by NVP-BEZ235, p21 knockdown was performed by siRNAs in the IHH4, K1, BCPAP and C643 cell lines (Supplementary Figure 2). Then the effect of p21 knockdown on cell proliferation after NVP-BEZ235 treatments was examined by MTT assays. As shown in Figure 5A, p21 knockdown significantly rescued the growth inhibitory effect of NVP-BEZ235 in all the four cell lines. Moreover, the results of flow cytometry suggested that p21 knockdown significantly attenuated cell cycle arrest induced by NVP-BEZ235 in all the four cell lines (Figure 5B). In specific, the G0/G1 phase proportion of IHH4 decreased from 64.09 % to 48.91 % (p < 0.01), from 69.07 % to 50.52 % (p < 0.01) for K1, from 79.65 % to 64.04 % (p < 0.01) for BCPAP, and from 78.78 % to 61.64 % (p < 0.01) for C643. Furthermore, p21 have been identified as a regulator capable of inhibiting cyclin E/Cdk2 complex [28]. Accordingly, the suppression of cyclin E and Cdk2 was either reversed by p21 interfering, additionally demonstrated the cell growth inhibition induced by NVP-BEZ235 was through p21 function (Figure 5C). Therefore, p21 knockdown was capable to abolish the principal effect of NVP-BEZ235 on the thyroid cancer cells regardless of TP53 status.
p21 knockdown abolished the effect of NVP-BEZ235 in both p53 wild type and mutant thyroid cancer cells. (A) MTT assays showed that the inhibitory effect of cell growth on IHH4, K1, BCPAP and C643 cells after NVP-BEZ235 treatment (200 nM) was abolished by p21 knockdown (si-p21). (B) Cell cycle arrest induced by NVP-BEZ235 (200 nM) was reversed by p21 knockdown (si-p21) in IHH4, K1, BCPAP and C643 cell lines. (C) p21 knockdown (si-p21) was performed in IHH4, K1, BCPAP and C643 cells and then cells were treated with NVP-BEZ235. The protein expressions of p21, cyclin E1 and Cdk2 were determined by Western blot (left). Relative protein levels were quantified by ImageJ software (right). GAPDH was used as loading control. 235, NVP-BEZ235; NC, Negative control. *p < 0.05, **p < 0.01, ***p < 0.001, respectively, 235 + si-p21 versus 235 + NC.
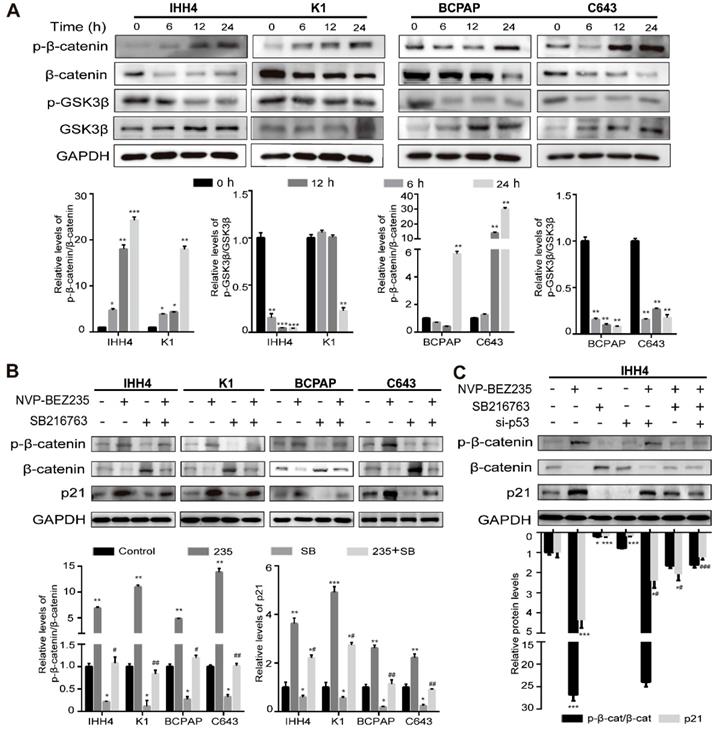
GSK3β/β-catenin signaling inhibition contributed to p21 upregulation induced by NVP-BEZ235 in thyroid cancer cells
It is well known that p21 plays an essential role in anti-tumor progression in p53-dependent and -independent pathways [29, 30] and previous studies have shown that β-catenin was a negative regulator of p21 transcription [31, 32]. As we found a p53-independent expression of p21 in NVP-BEZ235 treated TP53 mutant cells, we supposed the glycogen synthase kinase β (GSK3β)/β-catenin signaling might be affected by NVP-BEZ235. The results of western blot analysis showed the phosphorylation of GSK3β was suppressed by NVP-BEZ235, accompanied by the kinase of GSK3β increased in all the four human thyroid cancer cell lines. And the target of GSK3 kinases, β-catenin was increasingly phosphorylated, suggesting its destabilization, and in accordance with the decrease of β-catenin amount (Figure 6A). These results clearly indicated that GSK3β/β-catenin pathway was inhibited by NVP-BEZ235 in both p53 wild type and mutant cells, and suggesting a potent mechanism of p21 upregulation. To identify the dependency of p21 expression on GSK3β/β-catenin pathway, IHH4, K1, BCPAP and C643 cells were treated with or without GSK3β inhibitor (SB216763) under the application of NVP-BEZ235. As shown in Figure 6B, GSK3β inhibitor abolished the principal effect of NVP-BEZ235 on p21 in p53 mutant cells, however partly prevented the increase of p21 induced by NVP-BEZ235 in p53 wild type cells. Furthermore, to determine the effect of p53 on p21 upregulation in p53 wild type cells, p53 knockdown by siRNAs was performed in IHH4 cells (Supplementary Figure 3). As shown in Figure 6C, western blot assays suggested that p53 knockdown or GSK3β inhibitor both partly prevented the p21 upregulation induced by NVP-BEZ235. However, the combination of p53 knockdown and GSK3β inhibitor completely reversed the increase of p21 induced by NVP-BEZ235 in IHH4 cells. These results indicated the inhibition of GSK3β/β-catenin played a key role in p53 mutant cells whereas p53 additionally contributes to the upregulation of p21 in p53 wild type cells.
NVP-BEZ235 inhibits GSK3β/β-catenin signaling and cell cycle proteins in human thyroid cancer cells. (A) The protein expressions of phosphorylated β-catenin (p-β-catenin), β-catenin, phosphorylated glycogen synthase kinase 3 beta (p-GSK3β) and GSK3β were examined by Western blot in IHH4, K1, BCPAP and C643 cells with 200 nM NVP-BEZ235 treatment (upper). Relative protein levels were quantified by ImageJ software (lower). (B) The protein expressions of p-β-catenin, β-catenin and p21 were determined by Western blot in IHH4, K1, BCPAP and C643 cells treated with GSK3β inhibitor (10 μM; SB216763, SB) and NVP-BEZ235 (235) (upper). Relative protein levels were quantified by ImageJ software (lower). (C) The protein expressions of p-β-catenin, β-catenin and p21 were tested by Western blot in IHH4 cells with various treatments (upper). Relative protein levels were quantified by ImageJ software (lower). GAPDH was used as loading control. p-β-cat, p-β-catenin; β-cat, β-catenin. *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.05 and ##p < 0.01, respectively. * refers to a group versus control, # refers to a group versus 235.
NVP-BEZ235 inhibits xenograft thyroid tumor growth in nude mice
Finally, tumor xenografts were established to verify whether NVP-BEZ235 treatment inhibited tumor growth in vivo. Immuno-deficient mice were subcutaneously injected with p53 wild type K1 cells or p53 mutant C643 cells, treated with NVP-BEZ235 or vehicle, and the growth of the xenograft tumors were followed. Our data demonstrated both K1 and C643 tumors in mice given NVP-BEZ235 grew much slower than the control groups. Weight of the peeled tumors were also lower in the drug treated groups than the control groups (K1, 0.0334 ± 0.007222 g vs. 0.0140 ± 0.003674 g, N = 5, p < 0.05; C643, 0.1900 ± 0.01924 g vs. 0.0940 ± 0.01364 g, N = 5, p < 0.01) (Figure 7A and 7B).
NVP-BEZ235 inhibits tumor growth in nude mice injected with K1 cells or C643 cells. (A) Tumor weight of mice with NVP-BEZ235 (25 mg/kg/day) treatment and the control group. (B) Tumor volume of mice with NVP-BEZ235 treatment and the control group. (C) IHC images of p53, p21 and Ki67 in xenograft tumors, scale bar = 20 μm. N = 5, *p < 0.05, **p < 0.01 and ***p < 0.001, respectively, NVP-BEZ235 versus control. (D) Schematic model of molecular mechanisms underlying tumor suppressive activity of NVP-BEZ235 in thyroid cancer. WT p53, wild type p53.
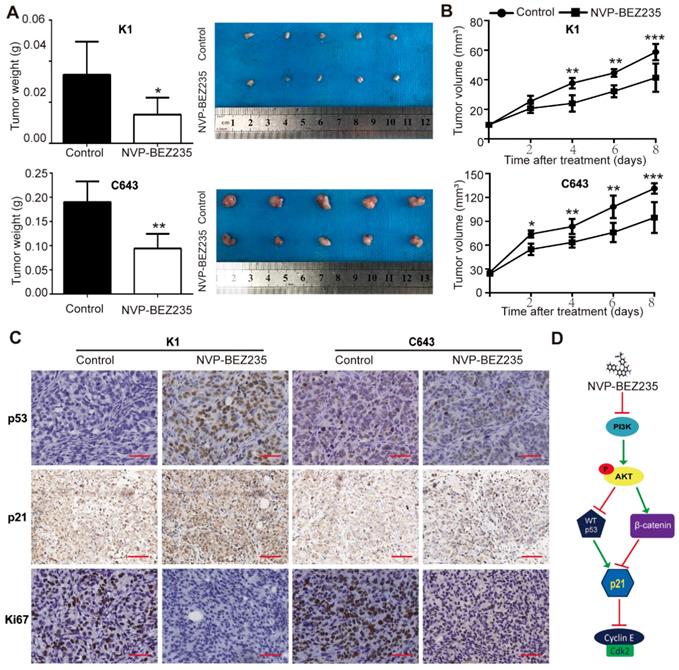
Subsequently, the peeled tumors were subjected to paraffin embedded sections. The protein expressions of p53 and p21 in tumor sections were tested by IHC assays. In both K1 and C643 tumors, p21 expression was higher in NVP-BEZ235 treated mice compared with the controls, since an increased percent and stronger staining of p21 positive cells were observed. While only the mice bearing K1 tumors displayed greater portion and stronger staining of p53 positive cells, rather than C643 tumors. These results suggested a p53-independent mechanism of p21 up-regulation in NVP-BEZ235 treated C643 xenograft tumors, which was consistent with the results of in-vitro studies. Moreover, the mitotic cell marker, Ki67 was shown much weaker in the NVP-BEZ235 treated tumors than the control tumors, further suggested NVP-BEZ235 suppresses thyroid cancer cell proliferation (Figure 7C). Taken together, these results indicated that NVP-BEZ235 treatment inhibited tumor growth in mice injected with K1 cells or C643 cells and substantiate our findings that NVP-BEZ235 inhibits thyroid cancer growth independent of TP53 status.
Altogether, the dual PI3K/mTOR inhibitor NVP-BEZ235 is efficacious against thyroid cancer, among which thyroid tumors with mutant p53 are critically dependent on the inhibition of GSK3β/β-catenin signaling, up-regulating p21 and then inhibiting cell growth in human thyroid cancer. However, p53 additionally contributes to the upregulation of p21 in p53 wild type cells (Figure 7D).
Discussion
Tumorigenesis is a multistep process involving complicate molecular pathogenesis and mechanisms, including the genetic and epigenetic abnormities, involving tumor suppressor genes inactivation as well as oncogenes activation. Thus, great attention was given to molecular targeted cancer therapy in recent decades [8]. The PI3K/mTOR/Akt signaling pathway has a fundamental role in numerous cancers of various histologic origin including thyroid cancer [5, 33, 34]. This pathway appears abnormal to increase the expression and activity of Akt and its downstream effectors, disturbing many biological processes. NVP-BEZ235 as a novel treatment strategy, is a dual PI3K/mTOR inhibitor that has shown great efficacy in suppressing tumor growth in various cancers and currently in phase1/2 clinical trials [9]. It is used alone or in combination to sensitize other drugs, as to achieve synthetic therapeutic effects with reduced side-effects. Yet, the molecular mechanisms that drive antineoplastic function are incompletely understood in thyroid cancer.
p53 have been identified as a tumor suppressor gene which integrates numerous signals controlling cell cycle arrest and apoptosis. The ability of p53 to eliminate excess, damaged or infected cells by apoptosis or induce cell cycle arrest, is critical for maintaining homeostasis of multicellular organisms [35-37]. Nevertheless, loss or mutation of p53 occurs in approximately 50 % of all tumors [35], including thyroid cancer [6, 38]. In our study, NVP-BEZ235 administration in thyroid cancer cell lines successfully inhibited Akt phosphorylation, hence, promoted downstream FOXO3a accumulation and nuclear translocation [24], and demonstrated remarkable anti-proliferation ability via inducing cell cycle arrest. However, we investigated the accumulation of p53 in wild type cell lines, but unaffected even slightly declined p53 was found in mutant cell lines. It is reasonable since Akt enhances MDM2 mediated proteasome degradation always fails for mutant p53 [39]. Meanwhile, p21, the primary effector of p53-mediated G1/S checkpoint control, was up-regulated, although differs in degree, in all the cell lines regardless of the volume change of p53 [30]. Accordantly, p21 expression was enhanced in the p53 wild type and mutant xenograft tumors in the NVP-BEZ235 treated mice. Additionally, previous research have disclosed that susceptibility to NVP-BEZ235 correlates with baseline expression of p27 [16]. However, we found that NVP-BEZ235 treatment failed to give rise to p27 response in IHH4, K1, BCPAP and C643 cells and this might be due to different cell lines used in our study. Taken together, we speculate that the antitumor effect in thyroid cancer cell lines harboring mutant TP53 acts in a p53-independent manner.
p21 is known as a cyclin-dependent kinase (Cdk) inhibitor. It induces cell cycle arrest and represses the G1-to-S phase transition mainly by inhibiting the activity of the cyclin E/Cdk2 complex [27]. The growth arrest mediated by p21 was reported to be negatively regulated by β-catenin via T-cell factor (TCF) or c-Myc transcriptional repression [31, 32, 40]. In the present study, p21 was transcriptionally activated along with GSK3β/β-catenin pathway suppression when cells were treated with NVP-BEZ235. And this phenotype was observed in both p53 wild type and mutant thyroid cancer cells. Therefore, it's very potent to suppose a reasonable mechanism that NVP-BEZ235 induced G0/G1 cell cycle arrest depends on GSK3β/β-catenin/p21 cascade, though the contribution of wild type p53 to p21 in K1 and IHH4 cells is not eliminable. Indeed, the GSK3β inhibitor radically abolished the increase of p21 in mutant cells, and less effective in wild type cells in view of the activity of normal p53. Thus, we supposed the upregulation of p21 roots in GSK3β/β-catenin response and with/out the normal function of p53. However, the specific mechanisms how β-catenin regulated p21 needed to be further explored in our future studies. And p53 dependency analysis based on varying TP53 status in the same cell line must help comprehend the NVP-BEZ235 efficacy.
In summary, the dual PI3K/mTOR inhibitor NVP-BEZ235 has a therapeutic effect on thyroid cancer, among which thyroid tumors with mutant p53 are critically dependent on the response of GSK3β/β-catenin signaling, up-regulating p21 and then inducing G0/G1 cell cycle arrest in human thyroid cancer.
Abbreviations
PTC: papillary thyroid cancer; FTC: follicular thyroid cancer; WDTCs: well-differentiated carcinomas; PDTC: poorly differentiated thyroid cancer; ATC: anaplastic thyroid cancer; IHC: Immunohistochemical; mTOR: mammalian target of rapamycin; FOXO3a: Forkhead box O3; Cdk: cyclin-dependent kinase; TCF: T-cell factor.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (grant nos. 81672645) and the Institutional Foundation of First Affiliated Hospital of Xi'an Jiaotong University (2018MS-01). We sincerely appreciate that Dr. Haixia Guan and Dr. Lei Ye provided us human thyroid cancer cell lines used in this study.
Ethics approval and consent to participate
All animal care and handling procedures were approved by the Animal Ethics Committee of Xi'an Jiaotong University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: A Cancer Journal for Clinicians. 2015;65:5-29
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
3. Sipos JA, Mazzaferri EL. Thyroid cancer epidemiology and prognostic variables. Clinical oncology (Royal College of Radiologists (Great Britain)). 2010;22:395-404
4. Tuttle RM, Ball DW, Byrd D, Dilawari RA, Doherty GM, Duh QY. et al. Thyroid carcinoma. Journal of the National Comprehensive Cancer Network: JNCCN. 2010;8:1228-74
5. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nature reviews Cancer. 2013;13:184-99
6. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nature reviews Endocrinology. 2011;7:569-80
7. Nikiforova MN, Nikiforov YE. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert review of molecular diagnostics. 2008;8:83-95
8. Walsh S, Prichard R, Hill AD. Emerging therapies for thyroid carcinoma. The surgeon: journal of the Royal Colleges of Surgeons of Edinburgh and Ireland. 2012;10:53-8
9. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C. et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular cancer therapeutics. 2008;7:1851-63
10. Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A. et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:5424-35
11. Yasumizu Y, Miyajima A, Kosaka T, Miyazaki Y, Kikuchi E, Oya M. Dual PI3K/mTOR inhibitor NVP-BEZ235 sensitizes docetaxel in castration resistant prostate cancer. The Journal of urology. 2014;191:227-34
12. Santiskulvong C, Konecny GE, Fekete M, Chen KY, Karam A, Mulholland D. et al. Dual targeting of phosphoinositide 3-kinase and mammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic approach in human ovarian carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:2373-84
13. Xu CX, Li Y, Yue P, Owonikoko TK, Ramalingam SS, Khuri FR. et al. The combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PloS one. 2011;6:e20899
14. Zhang C, Awasthi N, Schwarz MA, Schwarz RE. The dual PI3K/mTOR inhibitor NVP-BEZ235 enhances nab-paclitaxel antitumor response in experimental gastric cancer. Int J Oncol. 2013;43:1627-35
15. Roper J, Richardson MP, Wang WV, Richard LG, Chen W, Coffee EM. et al. The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PloS one. 2011;6:e25132
16. Lin SF, Huang YY, Lin JD, Chou TC, Hsueh C, Wong RJ. Utility of a PI3K/mTOR inhibitor (NVP-BEZ235) for thyroid cancer therapy. PloS one. 2012;7:e46726
17. Perri F, Pisconti S, Della Vittoria Scarpati G. P53 mutations and cancer: a tight linkage. Annals of translational medicine. 2016;4:522
18. Yue X, Zhao Y, Xu Y, Zheng M, Feng Z, Hu W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. Journal of molecular biology. 2017;429:1595-606
19. Blandino G, Di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cancer Res. 2018;37:30
20. Manzella L, Stella S, Pennisi MS, Tirro E, Massimino M, Romano C. et al. New Insights in Thyroid Cancer and p53 Family Proteins. International journal of molecular sciences. 2017:18
21. Shi J, Qu Y, Li X, Sui F, Yao D, Yang Q. et al. Increased expression of EHF via gene amplification contributes to the activation of HER family signaling and associates with poor survival in gastric cancer. Cell death & disease. 2016;7:e2442
22. Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Molecular and cellular endocrinology. 2010;321:20-8
23. Santo EE, Stroeken P, Sluis PV, Koster J, Versteeg R, Westerhout EM. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer research. 2013;73:2189-98
24. Qiang W, Zhao Y, Yang Q, Liu W, Guan H, Lv S. et al. ZIC1 is a putative tumor suppressor in thyroid cancer by modulating major signaling pathways and transcription factor FOXO3a. The Journal of clinical endocrinology and metabolism. 2014;99:E1163-72
25. Sonntag R, Giebeler N, Nevzorova YA, Bangen JM, Fahrenkamp D, Lambertz D. et al. Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2018;115:9282-7
26. Santo L, Siu KT, Raje N. Targeting Cyclin-Dependent Kinases and Cell Cycle Progression in Human Cancers. Seminars in oncology. 2015;42:788-800
27. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA repair. 2016;42:63-71
28. Stewart ZA, Leach SD, Pietenpol JA. p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Molecular and cellular biology. 1999;19:205-15
29. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nature reviews Cancer. 2009;9:400-14
30. Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K. et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935-44
31. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A. et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241-50
32. Kamei J, Toyofuku T, Hori M. Negative regulation of p21 by beta-catenin/TCF signaling: a novel mechanism by which cell adhesion molecules regulate cell proliferation. Biochemical and biophysical research communications. 2003;312:380-7
33. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
34. Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid: official journal of the American Thyroid Association. 2010;20:697-706
35. Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nature reviews Clinical oncology. 2011;8:25-37
36. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. Journal of cell science. 2003;116:4077-85
37. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
38. Fagin JA. Tumor suppressor genes in human thyroid neoplasms p53 mutations are associated undifferentiated thyroid cancers. J Endocrinol Invest. 1995;18:140-2
39. Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K. et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. The Journal of biological chemistry. 2002;277:21843-50
40. Zhu Y, Wang W, Wang X. Roles of transcriptional factor 7 in production of inflammatory factors for lung diseases. Journal of translational medicine. 2015;13:273
Author contact
![]() Corresponding authors: Qi Yang, Ph.D, Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China, Tel/Fax: 86-298-532-4039, E-mail: yangqi2015edu.cn; Peng Hou, Ph.D, Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China, Tel/Fax: 86-298-532-4039, E-mail: phouedu.cn
Corresponding authors: Qi Yang, Ph.D, Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China, Tel/Fax: 86-298-532-4039, E-mail: yangqi2015edu.cn; Peng Hou, Ph.D, Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China, Tel/Fax: 86-298-532-4039, E-mail: phouedu.cn