Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. TAMs as architects of the...
3. Reciprocal regulation of TAMs...
4. Therapeutic strategies
5. Challenges and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(10):5083-5118. doi:10.7150/ijbs.132476 This issue Cite
Review
Targeting Tumor-Associated Macrophages to Reshape the Immuno-Mechanical Landscape: Molecular Mechanisms and Therapeutic Strategies
Guanghui Liu1,*, Yang Yu2,*, Zichen Guo1, Lijuan Liu3, ![]() , Wujin Chen4,
, Wujin Chen4, ![]() , Changgang Sun3,5,
, Changgang Sun3,5, ![]()
1. College of First Clinical Medicine, Shandong University of Traditional Chinese Medicine, Jinan, Shandong, 250000, China.
2. Faculty of Chinese Medicine and State Key Laboratory of Mechanism and Quality of Chinese Medicine, Macau University of Science and Technology, Macau, 999078, China.
3. Department of Oncology, Weifang Traditional Chinese Hospital, Weifang, Shandong, 261000, China.
4. The Affiliated People's Hospital of Fujian University of Traditional Chinese Medicine, Fuzhou, Fujian, 350122, China.
5. College of Traditional Chinese Medicine, Shandong Second Medical University, Weifang, Shandong, 261000, China.
* Guanghui Liu, and Yang Yu contributed equally to this work.
Received 2026-2-1; Accepted 2026-4-15; Published 2026-5-11
Abstract

The progression and therapeutic resistance of solid tumors are profoundly influenced by the mechanical microenvironment, in which extracellular matrix stiffening, elevated interstitial pressure, and aberrant mechanotransductive signaling constitute critical physical barriers. Tumor-associated macrophages (TAMs) occupy a central position in this process. They not only act as active architects that remodel the matrix and exacerbate fibrosis, but their phenotypes and functions are also reciprocally regulated by the mechanical microenvironment, thereby forming a self-reinforcing malignant loop. Accordingly, targeting TAMs to mechanically soften tumors has emerged as an important therapeutic strategy, encompassing TAMs depletion, reprogramming, inhibition of TAM-mediated the extracellular matrix (ECM) modification, and disruption of mechanosensing pathways. In addition, mechanical immunoengineering and combination therapeutic strategies provide new tools for modulating the tumor mechanical-immune microenvironment. This review systematically examines the bidirectional regulatory mechanisms of TAMs within the mechanical microenvironment and the corresponding therapeutic strategies, and highlights that overcoming spatiotemporal heterogeneity and developing precision intervention paradigms are key to achieving future clinical translation.
Keywords: tumor-associated macrophages (TAMs), mechanical microenvironment, extracellular matrix remodeling, tumor softening, therapeutic strategies
1. Introduction
The initiation and progression of tumors are not determined solely by intrinsic genetic alterations within tumor cells, but are instead rooted in a highly abnormal tumor microenvironment (TME). This microenvironment comprises multiple cellular components, the extracellular matrix (ECM), vascular and lymphatic networks, as well as a range of physical and mechanical signals[1]. In recent years, research has gradually expanded its focus from biochemical signals to mechanical signals, leading to the proposal of the concept of the tumor mechanical microenvironment[2]. This concept emphasizes that ECM composition and crosslinking, overall tissue stiffness, solid stress, interstitial fluid pressure (IFP), and the permeability and perfusion status of blood and lymphatic vessels together constitute the mechanical basis of tumor growth and evolution[3, 4]. Across multiple solid tumors, high collagen deposition and fibrosis, increased tissue stiffness and interstitial pressure, and structurally abnormal and leaky vascular networks have all been demonstrated to be closely associated with enhanced tumor aggressiveness, increased metastatic risk, and poor prognosis. These findings suggest that the mechanical microenvironment itself has become a key dimension for assessing tumor malignancy[5, 6].
An abnormal mechanical microenvironment is not merely a passive consequence of tumor progression, but can actively promote tumor progression and attenuate therapeutic responses through multiple levels of mechanisms[7]. On the one hand, increased matrix stiffness and altered mechanical tension can, via mechanotransduction pathways involving integrins, focal adhesion kinase (FAK), and yes-associated protein (YAP)/ (transcriptional coactivator with PDZ-binding motif) TAZ, accelerate epithelial-mesenchymal transition, maintenance of stemness, and enhancement of invasive capacity in tumor cells, thereby driving local invasion and distant metastasis[8]. On the other hand, dense fibrosis and elevated IFP together form dual physical and functional barriers that compress blood vessels, reduce perfusion, and impede the delivery of chemotherapeutic agents, targeted therapies, and immune cells (particularly CD8+ T cells) to the tumor core[9]. In addition, hypoxia and metabolic dysregulation caused by abnormal vasculature and ECM remodeling can further activate immunosuppressive pathways[10], leading a substantial proportion of patients to develop primary or acquired resistance to immunotherapies such as immune checkpoint inhibitors (ICIs).
Within this complex network, tumor-associated macrophages (TAMs) occupy a critical hub position linking the immune microenvironment and the mechanical microenvironment[11, 12]. TAMs are among the most abundant populations of tumor-infiltrating immune cells, and their high-density infiltration is strongly associated with poor patient prognosis, metastasis, and resistance to chemotherapy and radiotherapy[13, 14]. Single-cell omics and spatial transcriptomic studies have shown that typical M2-like or immunosuppressive TAMs highly express vascular endothelial growth factor (VEGF), and transforming growth factor-β (TGF-β), interleukin-10 (IL-10), arginase 1 (ARG1), as well as various matrix metalloproteinases (MMPs) and cathepsins, thereby simultaneously driving angiogenesis, suppressing effector T-cell function, and remodeling the ECM[15-17]. Recent studies further indicate that fibrotic and hypoxic regions in multiple solid tumors are often enriched with M2-like or lineage-specific TAMs; these cells closely co-localize with cancer-associated fibroblasts (CAFs) and, through the secretion of factors such as MMPs, TGF-β, and C-C motif chemokine ligand 2 (CCL2)/ C-C motif chemokine ligand 5 (CCL5), induce collagen deposition and crosslinking, increase matrix stiffness, and maintain a malignant mechanobiological ecosystem characterized by immune exclusion and high fibrosis[18, 19]. Collectively, this evidence indicates that TAMs not only support tumors through immune pathways but also directly participate in the establishment and reinforcement of an abnormal mechanical microenvironment.
On the other hand, TAMs themselves act as integrative responders to mechanical and metabolic signals, and their recruitment, polarization, and functions are profoundly regulated by the mechanical properties of the microenvironment[20, 21]. Hypoxia, lactate accumulation, cell-matrix adhesion, and fluid shear stress within the TME collectively shape a highly heterogeneous TAM population[22, 23]. Among them, M2-like immunosuppressive TAMs tend to accumulate in regions with high stiffness and high fibrosis that are distant from functional blood vessels[24]. Within these specific mechanobiological niches, TAMs exhibit enhanced transcriptional programs associated with tissue repair and pro-fibrotic activity, thereby further exacerbating ECM remodeling and immunosuppression[15]. Recent reviews have pointed out a close causal relationship between TAM polarization states and multiple physical attributes of the TME, including tissue stiffness, matrix density, and perfusion status[25]. The reciprocal shaping of TAMs by the microenvironment and the remodeling of the microenvironment by TAMs are intricately intertwined, forming a highly dynamic, plastic, yet difficult-to-spontaneously-reverse malignant positive feedback loop[13].
Despite some progress having been made, there remain several gaps in the current understanding of the interplay between TAMs and the tumor mechanical microenvironment. First, existing studies tend to investigate TAM-mediated immunoregulation and tumor mechanical microenvironment remodeling as relatively independent processes, lacking an integrated theoretical framework that systematically connects immune function with biomechanical evolution. Second, the dynamic and stage-dependent interactions between TAMs and the tumor mechanical microenvironment during tumor progression have not yet been sufficiently elucidated, with most studies focusing on static features and thus failing to capture the temporal characteristics of this bidirectional regulatory system. Third, although both TAM-targeted therapeutic strategies and stroma-modulating approaches have demonstrated certain potential, they have not yet been effectively integrated into a systematic therapeutic framework with clear mechanistic guidance, particularly in overcoming mechano-immunological resistance. These limitations suggest that it is necessary to adopt a more unified and dynamic perspective to re-examine and integrate the TAM-tumor mechanical microenvironment axis, in order to promote the coordinated advancement of mechanistic understanding and therapeutic strategy design.
Therefore, viewing TAMs as a key hub connecting immune networks and the mechanical microenvironment, and reinterpreting their roles in tumor progression and therapeutic responses from a mechanical perspective, may provide new theoretical foundations and potential intervention windows for the development of combined mechanics-immunity intervention strategies. This article systematically elucidates the dual roles of TAMs in both actively shaping the tumor mechanical microenvironment and sensitively responding to mechanical signals within it, and further explores potential approaches and associated challenges in modulating the tumor mechanical microenvironment by targeting TAMs.
2. TAMs as architects of the tumor mechanical microenvironment
2.1 ECM remodeling and modification
Within the tumor mechanical microenvironment, the ECM is not only a physical scaffold supporting tumor cell proliferation and migration, but also a key determinant that integrates biochemical and biomechanical signals and governs tissue stiffness and topological architecture[26, 27]. In this process, TAMs play a central processing role. On the one hand, they drive collagen deposition and crosslinking, leading to the formation of a high-stiffness fibrotic matrix[20, 25]. On the other hand, through proteolysis and physical traction, they enable localized ECM degradation and fiber rearrangement[28, 29]. This continuous dynamic process of deposition, degradation, and reorganization is often carried out cooperatively by TAMs and stromal cells such as CAFs[19, 30], together shaping an ECM microenvironment with highly heterogeneous composition and mechanical properties, thereby influencing tumor invasion patterns, immune cell infiltration, and the efficiency of drug delivery[5, 18] (Fig. 1).
TAM-driven ECM remodeling shapes a stiff, immunosuppressive mechanical niche in solid tumors. Schematic illustration of TAMs-mediated ECM remodeling within the TME. TAMs promote collagen deposition and crosslinking through profibrotic enzymes and cytokines, leading to matrix stiffening and increased interstitial pressure. In parallel, TAMs cooperate with CAFs to orchestrate localized ECM degradation and fiber reorganization, releasing bioactive matrix fragments and growth factors. These dynamic deposit-degrade-reorganize processes generate a mechanically heterogeneous ECM that enhances tumor cell invasion, restricts immune cell infiltration and drug penetration, and favors the accumulation of immunosuppressive myeloid and lymphoid populations. The stiffened ECM further reinforces profibrotic and immunosuppressive TAM states, forming a self-amplifying mechanical-immune feedback loop that drives tumor progression.
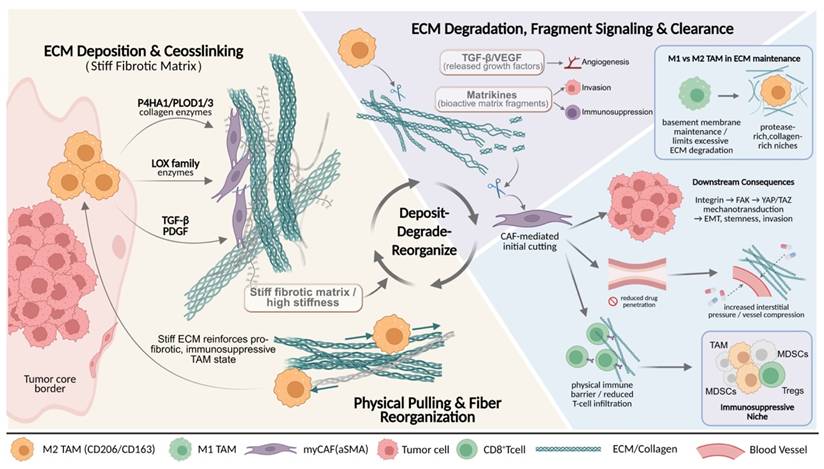
ECM remodeling, as a core event in tumor progression, mainly comprises four categories of processes: re-deposition of ECM components and quantity, chemical modifications such as crosslinking, proteolytic degradation, and physical rearrangement of fibers. These changes collectively determine the three-dimensional architecture, stiffness, and degradability of the ECM[31, 32]. With respect to ECM synthesis and modification, studies have confirmed that TAMs are an important cellular source of collagen synthesis and maturation[33]. In tumor tissues with high TAM infiltration and in transcriptomic analyses, upregulated expression of multiple ECM components or their regulatory factors (such as type I and type III collagen, fibronectin, and proteoglycans), as well as enzymes that promote collagen crosslinking, can be detected[5, 11, 34]. In breast cancer and colorectal cancer models, TAMs highly express key enzymes involved in collagen maturation, including Prolyl 4-Hydroxylase Subunit Alpha 1 and PLOD1/3, and initiate collagen biosynthesis through TGF-β-dependent programs, thereby further enhancing stromal fibrosis and tissue stiffness[35, 36]. Meanwhile, factors such as TGF-β and platelet-derived growth factor (PDGF) secreted by M2-like TAMs can activate CAFs and induce their differentiation into myofibroblast-like CAFs (myCAFs) with a high ECM-synthetic phenotype[18, 37, 38]. myCAFs abundantly secrete type I, VI, and XIV collagen, fibronectin, and proteoglycans[6]. In addition, this CAF subset highly expresses ECM signature genes such as COL1A2, POSTN, and SPP1, and spatially co-localizes with TAMs, forming ECM synthesis-centered processing units that continuously reinforce matrix crosslinking and densification[38, 39]. Notably, such a stiffened matrix, on the one hand, promotes epithelial-mesenchymal transition and maintenance of stemness through mechanotransduction pathways involving integrins, FAK, and YAP/TAZ[35, 40](6,35,40). On the other hand, by increasing interstitial tissue pressure and compressing blood vessels, it restricts immune cell infiltration and drug delivery[24]. Therefore, ECM stiffening is regarded as an important physical basis driving malignant tumor progression and immune exclusion[36]. More importantly, the stiffened ECM can in turn regulate TAMs, promoting their polarization toward pro-fibrotic and immunosuppressive phenotypes, thereby forming a self-reinforcing malignant cycle.
With respect to ECM degradation, TAM-mediated proteolysis is not a simple destructive process, but rather a finely regulated, stage-specific and stepwise process[28, 41]. Multiple studies have shown that M2-like TAMs are an important source of various MMPs, such as MMP 2, MMP 9, and MMP 14, as well as other proteolytic enzymes[15, 19]. These enzymes not only directly cleave structural proteins including collagen, laminin, and fibronectin, but also release growth factors stored within the ECM, such as TGFβ and VEGF, and generate bioactive matrix fragments[42]. This further amplifies pro-angiogenic, pro-invasive, and immunosuppressive signals, thereby endowing ECM degradation with dual functions of structural remodeling and signal regulation[18, 34]. The contribution of TAMs to ECM degradation is not unidirectional, but functionally complementary with stromal components such as CAFs[18]. Quantitative collagen degradation models have shown that, in multiple solid tumors, the initial cleavage of collagen fibers is primarily mediated by MMPs secreted by CAFs, whereas TAMs cooperate with CAFs to handle the subsequent uptake and lysosomal degradation of collagen fragments[43]. During this process, TAMs highly express multiple ECM receptors and endocytic receptors, among which the mannose receptor (CD206) is particularly critical for the uptake of collagen fragments[44, 45]. In tumor models using CD206-deficient mice, collagen uptake by TAMs is significantly impaired, indicating that this receptor is a core molecule mediating TAM-driven ECM degradation[28, 46]. Notably, the function of TAMs in ECM degradation exhibits a clear phenotypic bias. M2-like TAMs, marked by CD206 and CD163, are enriched in regions with high collagen content and high proteolytic enzyme activity, whereas M1-like TAMs with antigen-presenting and cytotoxic potential tend to maintain basement membrane integrity and limit excessive degradation[14, 29, 47]. This phenotype-function association suggests that reprogramming or selectively depleting M2-like TAMs may represent a potential strategy to attenuate the formation of pathological ECM networks[48].
The ECM collagen network remodeled by TAMs not only constitutes a functional physical barrier, hindering effector T cells and drugs from penetrating into the tumor core, but also shapes an immunosuppressive niche by altering ECM contextual signals and mechanical properties[49, 50]. Single-cell and spatial multi-omics analyses show that SPP1+ or CD206+ M2-like TAMs are highly co-localized with αSMA+ CAFs at the tumor boundary and in collagen-enriched regions[51, 52]. The two synergistically drive ECM fibrosis and the formation of immune barrier structures, limiting effector T-cell infiltration into the interior of tumors[18, 25]. Among them, CAFs mainly dominate matrix densification by secreting large amounts of ECM proteins and regulating the MMP/TIMP balance[53, 54]. TAMs are more inclined to synthesize specific ECM components, secrete degradative enzymes, and regulate mechanical signaling, thereby finely controlling ECM plasticity and spatial organizational patterns[18, 25]. Together, they constitute a continuous and dynamic ECM processing system. On this basis, abnormal ECM can also remodel the activation threshold and migratory behavior of T cells through receptor signaling such as integrins, and selectively provide anchoring sites and survival signals for immunosuppressive cells such as TAMs, myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs), ultimately jointly promoting an immune cold tumor phenotype and leading to limited efficacy of ICIs[5, 55, 56].
2.2 Collagen architecture and biomechanical signal guidance
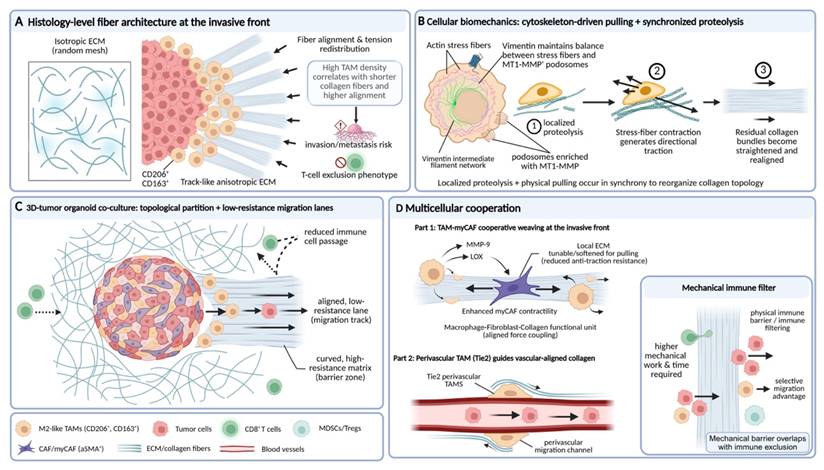
In the construction of the tumor mechanical microenvironment, the function of TAMs goes beyond simply synthesizing or clearing ECM components, and the more central role lies in serving as an organizer of fibrous structures. Through cytoskeleton-mediated active pulling and rearrangement, TAMs can reshape the orientation, spatial connectivity, and tension distribution of collagen fibers[57, 58]. This pulling transforms an originally relatively isotropic matrix network into a physical barrier that is mechanically favorable for tumor cell invasion, but at the same time hinders immune cell infiltration[59, 60].
Histological and advanced imaging studies provide intuitive evidence for the close association between TAMs and specific fiber configurations. In multiple solid tumors such as breast cancer and ovarian cancer, regions with high-density TAM infiltration, especially the tumor invasive front, often show characteristic collagen structural changes[51, 58]. Specifically, collagen fibers become shorter and thinner, and their arrangement shows a trend toward high parallelism and alignment[59, 61]. For example, a study comparing canine and human breast cancer showed that the degree of TAM infiltration is negatively correlated with the average length of collagen fibers, and positively correlated with the alignment of fiber arrangement[58]. This suggests that TAMs not only participate in matrix degradation, but also dominate the systematic rearrangement of fiber architecture, transforming an originally randomly interwoven reticular ECM into an orderly arranged linear structure[34, 62]. Such collagen bundles arranged radially along the tumor edge or perpendicular to the boundary have been confirmed to be significantly associated with stronger tumor invasiveness, higher metastatic risk, and a T-cell exclusion phenotype, and thus have become an important morphological indicator for evaluating patient prognosis[63].
The molecular mechanism by which TAMs achieve ECM fiber rearrangement is rooted in their unique cytoskeletal architecture and mechanosensing capacity. Studies show that when TAMs synergistically carry out ECM degradation and reorganization, they rely on the precise coordination of actin stress fibers, invadopodia, and the intermediate filament network[64, 65]. Among these, the intermediate filament network formed by vimentin is crucial for maintaining the functional balance between stress fibers and MT1-MMP-rich invadopodia[66]. As a mechanical scaffold, it can ensure the spatiotemporal coordination of ECM endocytosis, lysosomal degradation, and local export of degradative enzymes[67, 68]. Under this mechanism, TAMs first anchor and locally soften collagen fibers through invadopodia, and then apply directional mechanical pulling force via contractile stress fibers, thereby straightening the remaining collagen fiber bundles and realigning them along the direction of cell migration[19, 69]. The latest three-dimensional tumor organoid-macrophage co-culture model has intuitively reproduced this process. M2-like TAMs can pull and comb collagen fibers along the migration path of tumor cells, leading to local fiber straightening and alignment. Meanwhile, surrounding fibers that are not directly acted upon maintain their curved conformation, forming a microenvironment with obvious topological partitioning, thereby opening up a directionally migratory channel with low mechanical resistance for tumor cells[62].
This complex fibrotic process is not an isolated action of TAMs, but rather the result of their precise coordination with other cellular components within the TME. Spatial transcriptomic analyses have revealed that, in triple-negative breast cancer, regions enriched with myCAFs, characterized by high expression of contractile proteins and strong synthetic capacity, are typically associated with denser and more highly aligned collagen bundles[70-72]. These regions also correspond to the invasive front, where tumor cells and M2-like TAMs co-accumulate[73, 74]. In contrast, areas enriched with immunomodulatory CAFs (iCAFs) exhibit a relatively looser matrix architecture, which is more permissive for the recruitment of various immunosuppressive cell populations[75]. Within this specific ecological niche, TAMs are frequently positioned within the interstices or termini of collagen bundles. By secreting factors such as MMP-9 and lysyl oxidase (LOX), these TAMs not only locally modify the ECM to reduce its resistance to tensile forces, but also enhance the contractile activity of adjacent myCAFs[34, 53]. Acting in concert along the same directional axis, TAMs and myCAFs cooperatively exert mechanical forces that efficiently weave large-scale collagenous microstructures with high structural uniformity[38, 76]. Emerging evidence suggests that, beyond the aforementioned biochemical paracrine circuits, direct mechanical crosstalk between TAMs and myCAFs may exist in the form of a “cell-matrix-cell” signaling axis. However, the in situ quantitative dissection of such intercellular mechanical interactions remains technically challenging and continues to be an active area of investigation[18, 38].
Given that both cell types anchor to the same ECM network via integrin-mediated adhesions, contractile forces generated by one cell population can be transmitted over long distances along collagen fibers to the other, thereby establishing a mechanically coupled unit. For instance, myCAFs are capable of extensively depositing and crosslinking nascent collagen, thereby constructing a stiff and anisotropic fibrillar scaffold[77, 78]; TAMs, in contrast, exhibit pronounced myosin-dependent contractility and possess refined mechanosensory structures, such as invasive protrusions and stress fibers, enabling them to exert directional traction forces on the same ECM fibers via integrins including αvβ3 and α5β1[79].
This process not only promotes collagen fiber straightening and realignment, but also generates tensile stress along the fibers[80]. Such forces can be directly sensed by adjacent myCAFs through their focal adhesions and integrin-dependent mechanotransduction machinery, thereby further reinforcing their contractile activity and matrix-producing programs[81]. Similarly, perivascular TAMs, such as Tie2⁺ TAMs, through their interactions with endothelial cells, can guide the reorganization of the perivascular basement membrane and collagen fibers along the vascular axis, thereby establishing a structural pattern that facilitates tumor cell migration along vessel walls[82, 83]. This ECM scaffold, cooperatively constructed by multiple cell types, provides a structural basis for the directional distribution of IFP and the local amplification of mechanical signaling[84, 85].
Importantly, the fibrous networks orchestrated predominantly by TAMs ultimately generate a highly selective physical microenvironment. This selectivity is reflected in the differential regulation of migratory capacities among distinct cell types. For instance, the reduction and heterogeneity of interfiber pore size markedly increase the mechanical work and time required for effector immune cells, such as CD8⁺ T cells, to traverse the matrix[59]. Their relatively rigid and larger cellular morphology results in inefficient migration within such dense and anisotropic structures. In contrast, tumor cells, TAMs themselves, and MDSCs, which possess greater deformability and matrix adaptability, are able to exploit—or even actively remodel—these channels more effectively[86, 87]. Consequently, the fiber rearrangement driven by TAMs effectively constructs a selective physical barrier that spatially integrates mechanical obstruction with immune exclusion, thereby coupling biomechanical constraints to immunosuppressive organization (Fig. 2).
TAM-orchestrated collagen fiber alignment guides biomechanical signaling and establishes selective migration tracks at the invasive front. Schematic illustration of how TAMs reorganize collagen architecture to shape biomechanical signal guidance and immune exclusion. (A) At the tumor invasive front, high densities of CD206⁺/CD163⁺ TAMs associate with shortened, highly aligned collagen fibers, forming anisotropic, track-like ECM structures linked to increased invasion and T-cell exclusion. (B) At the cellular level, TAMs synchronize localized proteolysis with cytoskeleton-driven pulling, generating directional traction that straightens and realigns residual collagen bundles. (C) In 3D tumor organoid co-culture models, TAM-mediated fiber reorganization creates topologically partitioned matrices, opening low-resistance migration lanes for tumor cells while restricting immune cell passage. (D) Cooperative interactions between TAMs and myofibroblast-like CAFs, as well as perivascular TAMs, further reinforce aligned force coupling and vascular-guided collagen remodeling, collectively forming a mechanical immune filter that selectively favors tumor and myeloid cell migration over cytotoxic T-cell infiltration.
2.3 Regulation of interstitial pressure
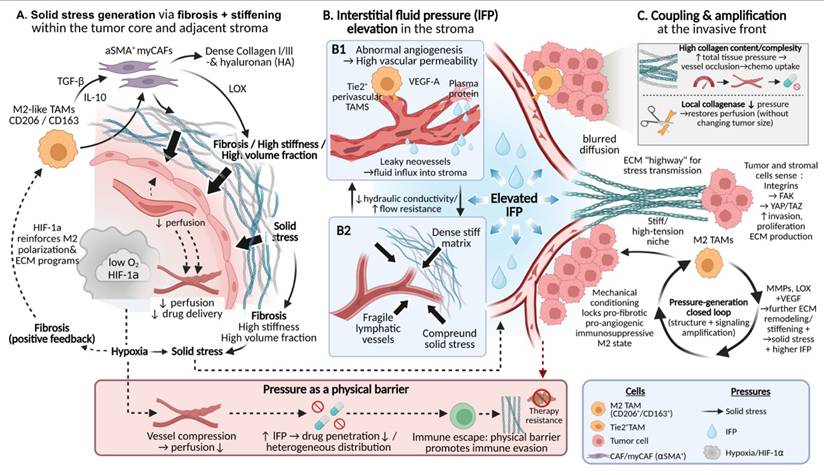
Abnormally elevated mechanical pressures within the TME constitute a critical physical barrier that restricts drug delivery and promotes immune evasion[2]. These pressures primarily comprise three interrelated forms: solid stress generated by tumor volumetric expansion and matrix contraction[88], IFP arising from impaired vascular and lymphatic function[89], and aberrant stress transmission and relaxation resulting from altered material properties of the ECM[90]. Accumulating evidence indicates that TAMs play indispensable and active roles in the generation, maintenance, and amplification of these distinct pressure components, rather than serving merely as passive responders to the mechanical milieu.
TAMs are central regulatory cells driving matrix fibrosis and stiffening, thereby generating and amplifying solid stress. M2-like TAMs secrete key mediators such as TGF-β and IL-10[11], which not only directly stimulate collagen synthesis and the expression of cross-linking enzymes within TAMs themselves, but, more importantly, robustly activate CAFs, inducing their differentiation toward an α-smooth muscle actin (α-SMA)-expressing myofibroblastic phenotype[51, 91]. Activated CAFs produce large amounts of type I and type III collagen, hyaluronic acid, and other ECM components, while concomitantly upregulating cross-linking enzymes such as LOX, collectively resulting in marked increases in matrix stiffness and physical volume[6, 92]. Notably, ECM thickening and enhanced cross-linking substantially limit the compressibility of tumor tissue during growth. This structural rigidification leads to the accumulation of solid stress within the tumor core and perivascular regions, thereby compressing microvessels, reducing blood perfusion, and promoting the formation of hypoperfused and hypoxic areas[93]. These hypoxic regions, in turn, further reinforce M2-like TAM polarization and the expression of ECM biosynthetic programs through hypoxia-inducible factor-1α (HIF-1α)-dependent signaling, establishing a self-reinforcing positive feedback loop linking tissue stiffness, interstitial pressure, hypoxia, and TAM activation[12, 94].
At the level of fluid dynamics, TAMs are also major drivers of elevated IFP within tumors, primarily through disruption of normal vascular and lymphatic function. On the one hand, TAMs—particularly perivascularly enriched Tie2⁺ subsets—are a major source of VEGF-A. The structurally disorganized and highly permeable neovasculature promoted by TAM-derived VEGF-A leads to continuous extravasation of protein-rich plasma components into the interstitial space[95, 96]. On the other hand, TAM-driven fibrosis and the associated solid stress directly compress and damage the already fragile intratumoral lymphatic vessels, severely impairing interstitial fluid drainage and clearance[11, 25]. In parallel, matrix densification and pore size reduction mediated cooperatively by TAMs and CAFs markedly decrease the mobility of water and solutes within the interstitium, increasing hydraulic resistance and fluid retention time, thereby sustaining elevated IFP[35, 97, 98]. Studies in animal models of pancreatic cancer and other solid tumors have demonstrated that increased local collagen content and structural complexity are positively correlated with total tissue pressure and are associated with vascular collapse and a pronounced reduction in chemotherapeutic drug uptake[99]. Conversely, localized administration of collagenase can acutely reduce tissue pressure and restore drug perfusion without altering overall tumor volume, directly demonstrating that collagen accumulation and cross-linking per se are key drivers of interstitial hypertension and heterogeneous drug distribution[100-102].
More importantly, the biological activities of TAMs tightly couple solid stress with fluid pressure and endow this system with potent signal amplification capacity. The ECM remodeled cooperatively by TAMs and CAFs serves not only as a source of mechanical pressure but also as an efficient conduit for the propagation of aberrant mechanical signals throughout tumor tissue. Dense and highly aligned collagen fiber networks enable more effective transmission of localized solid stress to distant cells, while elevated IFP influences the spatial distribution of chemical gradients by modulating convection and diffusion processes[97, 103]. Tumor cells and stromal cells sense these mechanical cues through mechanotransduction pathways involving integrins, FAK, and downstream effectors such as YAP/TAZ, which in turn induce enhanced invasive, proliferative, and ECM-synthetic programs[8, 104]. Notably, TAMs themselves constitute a core component of this mechanical feedback circuitry. High-stiffness and high-tension microenvironments actively promote the stabilization of TAMs toward pro-fibrotic, pro-angiogenic, and immunosuppressive M2-like phenotypes through the same mechanotransduction pathways[105, 106]. These TAMs subsequently secrete increased levels of matrix-modifying factors (e.g., MMPs and LOX) and pro-angiogenic mediators (e.g., VEGF), further driving matrix stiffening, contraction, and vascular leakage[107, 108]. Together, these processes culminate in a pressure-generating loop that is self-sustaining and self-amplifying at both structural and signaling levels (Fig. 3).
TAM-driven interstitial pressure as a physical barrier in tumors. Schematic illustration of how TAMs generate and amplify interstitial pressure within the TME. (A) M2-like TAMs promote fibrosis and matrix stiffening by activating αSMA⁺ myofibroblastic CAFs via TGF-β and IL-10, leading to collagen accumulation, solid stress generation, vascular compression, and hypoxia-driven positive feedback. (B) TAMs elevate IFP by inducing leaky angiogenesis through VEGF-A secretion and impairing lymphatic drainage, while dense ECM increases hydraulic resistance and fluid retention. (C) At the invasive front, stiff, collagen-rich matrices couple solid stress with fluid pressure and transmit mechanical signals sensed via integrin-FAK-YAP/TAZ pathways, reinforcing pro-fibrotic and immunosuppressive TAM states and establishing a self-amplifying pressure loop that restricts drug penetration and immune cell infiltration.
2.4 Mechanotransduction and signal amplification
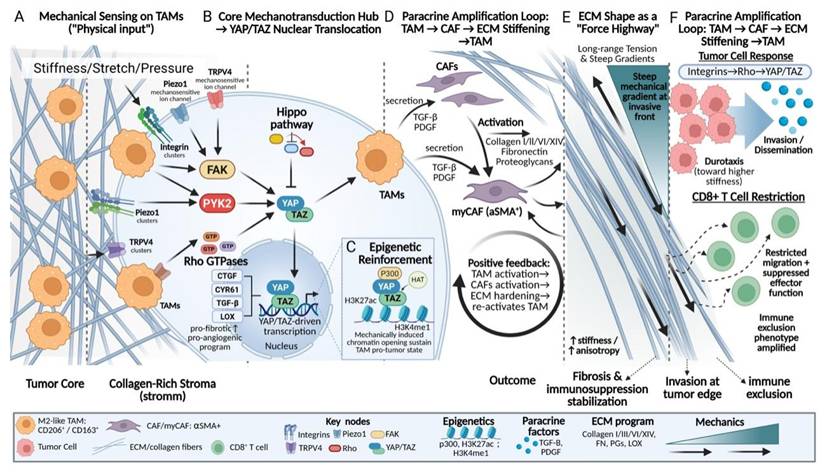
Within the mechanically aberrant TME, TAMs do not merely passively respond to ECM stiffness and pressure. Instead, they translate local physical perturbations into systemic and sustained transcriptional and immunological reprogramming signals. This process of “amplification” enables TAMs to function as a central hub linking the physical microenvironment to biochemical responses and represents one of the core mechanisms underlying the maintenance of a highly fibrotic and immunosuppressive malignant steady state.
The initiation of this amplification process relies on diverse mechanosensors expressed on the surface of TAMs, including integrin clusters and mechanosensitive ion channels such as Piezo1 and TRPV4. Acting as highly sensitive antennas, these sensors continuously detect the physical properties of the microenvironment[109]. Upon signal input, mechanotransduction rapidly converges on key intracellular nodes, including FAK, proline-rich tyrosine kinase 2 (PYK2), and Rho GTPases[110, 111], thereby driving the activation of downstream mechanotransduction pathways such as Hippo-YAP/TAZ signaling[104]. Notably, recent studies suggest that PYK2 not only occupies a central position in the transduction of integrin- and mechanics-derived signals, but also couples inflammatory and differentiation-related pathways, positioning it as a critical coordinator linking mechanical signal input to immune phenotypic polarization[112, 113]. Concurrently, increased stiffness and tensile forces suppress the Hippo kinase cascade, thereby promoting the nuclear translocation of YAP/TAZ[114]. Once translocated into the nucleus, YAP/TAZ drive the transcription of a series of target genes, including connective tissue growth factor (CTGF), cysteine-rich angiogenic inducer 61 (CYR61), TGF-β, and LOX[115, 116], effectively converting the physical stiffness of the matrix into explicit biochemical instructions that promote fibrosis and angiogenesis[117].
More importantly, mechanically induced phenotypic changes in TAMs are not transient events, but are accompanied by profound epigenetic remodeling. Accumulating evidence indicates that YAP/TAZ and their co-activators can function as scaffolds to recruit histone acetyltransferases and methyltransferase complexes, such as p300, to the promoters and enhancers of mechanosensitive genes, catalyzing active chromatin modifications including H3K27ac and H3K4me1, thereby maintaining an open chromatin state at these loci[118-120]. This process, initiated by mechanical cues and ultimately consolidated into a defined epigenetic landscape, provides a stable molecular basis for the sustained pro-tumorigenic functions of TAMs in a dynamically changing microenvironment and partially explains the persistence and self-maintaining nature of fibrotic tumor niches.
Following the completion of their own mechanical signal transduction and phenotypic reprogramming, TAMs propagate the amplified mechanical signals throughout the microenvironment in a paracrine manner. The secretion of key cytokines, such as TGF-β and PDGF, constitutes a central signal for the activation and maintenance of CAFs[121]. Activated CAFs, in turn, synthesize and deposit large amounts of ECM components, including type I and type III collagen, thereby markedly increasing ECM stiffness and mechanical anisotropy[37]. The resulting increase in matrix stiffness subsequently feeds back through mechanosensors such as integrins to further reinforce the pro-fibrotic phenotype of TAMs, forming a self-sustaining malignant feedback loop[38]. Notably, TAM-mediated shaping of tumor mechanical properties is not solely dependent on cytokine secretion, but is also achieved through direct physical remodeling of ECM spatial architecture. The linearly aligned and densely packed collagen fiber bundles formed with TAM participation exhibit higher elastic moduli and are more efficient at transmitting contractility-generated mechanical tension[122]. Such specialized ECM structures enable long-range transmission and focalization of local stress along fiber axes, thereby generating steep mechanical gradients at critical regions such as the tumor-stroma interface and the invasive front[62]. Tumor cells can sense these mechanical gradients through their integrin-Rho-YAP/TAZ axis and subsequently undergo directed durotactic migration, invading surrounding tissues along high-stiffness fiber tracks[35, 123, 124]. In contrast, effector immune cells, such as CD8⁺ T cells, exhibit severely restricted migration within these high-tension and structurally heterogeneous fibrous networks, accompanied by impaired activation and effector functions[11, 59]. Through these mechanisms, TAMs synergistically exacerbate tumor immune exclusion at both physical and immunological levels (Fig. 4).
Mechanotransduction and signal amplification in TAMs within stiff tumor matrices. Schematic overview of how TAMs convert mechanical cues into sustained pro-fibrotic and immunosuppressive programs. (A-B) Increased matrix stiffness, stretch, and pressure are sensed by integrins and mechanosensitive ion channels (Piezo1, TRPV4), converging on FAK-PYK2-Rho signaling to activate YAP/TAZ nuclear translocation. (C) YAP/TAZ-driven transcription is stabilized by epigenetic reinforcement, including p300-mediated H3K27ac and H3K4me1, sustaining TAM pro-tumor states. (D-F) Paracrine activation of CAFs by TAM-derived TGF-β and PDGF promotes ECM deposition, alignment, and stiffening, forming a positive feedback loop. Aligned collagen fibers act as “force highways,” amplifying mechanical gradients that drive tumor cell durotaxis while restricting CD8⁺ T-cell migration, thereby reinforcing invasion and immune exclusion.
3. Reciprocal regulation of TAMs by the biomechanical microenvironment
3.1 Mechanical sensing and initial signal transduction in TAMs
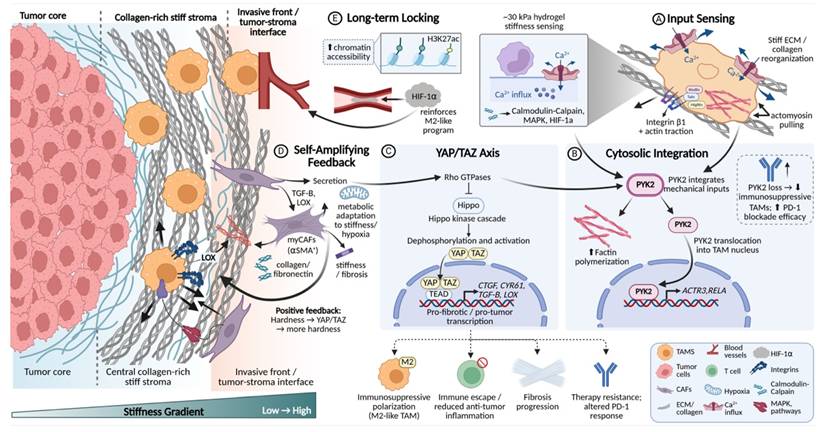
During tumor progression, ECM stiffening is not merely a passive background alteration but functions as a potent physical cue that is actively sensed by TAMs and their precursor cells. The decoding of mechanical signals is initiated at the plasma membrane, where collagen network reorganization induced by matrix stiffening increases the tensile load borne by integrin clusters and mechanosensitive ion channels such as Piezo1.
Studies have demonstrated that, in hydrogels mimicking the high stiffness of pancreatic cancer and other solid tumors, monocytes rapidly respond to matrix stiffness of approximately 30 kPa through Piezo1-mediated Ca2+ influx[125, 126]. This influx activates downstream signaling pathways, including calmodulin-calpain, mitogen-activated protein kinase (MAPK), and HIF-1α[127, 128]. These early events fundamentally reshape the inflammatory cytokine secretion profile and metabolic gene expression programs of macrophages, thereby influencing their differentiation trajectory[129]. In parallel, integrin engagement with ECM ligands induces focal adhesion assembly, recruiting proteins such as talin, vinculin, and FAK to form a mechanically coupled signal transduction platform linked to the cytoskeleton[130].
These membrane-proximal events are further integrated within the cytoplasm. Piezo1-Ca²⁺ signaling cooperates with integrin β1-mediated actin traction to activate the immunomechanical checkpoint PYK2[131]. Concurrently, integrin cluster-activated FAK further engages Rho GTPase signaling, which suppresses the Hippo pathway and promotes dephosphorylation and nuclear translocation of its downstream effectors YAP/TAZ[132, 133].
3.2 Mechanotransductive phenotypic remodeling and stabilization of TAM programs
Following the initial sensing and transduction of mechanical cues, these signals are progressively integrated into gene regulatory programs that drive stable phenotypic remodeling of TAMs.
In pancreatic cancer models, PYK2 not only integrates upstream mechanical inputs derived from Piezo1 and integrins, but also drives F-actin polymerization and undergoes nuclear translocation[134]. This directly regulates the promoters of mechanosensitive and differentiation-associated genes, such as ACTR3 and RELA, thereby determining both the efficiency of monocyte-to-TAM differentiation and the direction of macrophage polarization. Genetic or functional ablation of PYK2 markedly impairs the generation of monocyte-derived TAMs and their immunosuppressive activity, while enhancing the therapeutic efficacy of programmed cell death protein 1 (PD-1) blockade, underscoring its central role in mechanically driven macrophage polarization[113].
Nuclear translocation of YAP/TAZ represents a pivotal step in converting mechanical signals into sustained transcriptional programs. Across multiple tumor models, including breast and colorectal cancer, macrophages cultured on rigid substrates exhibit significantly increased nuclear localization of YAP/TAZ[135, 136], which correlates with enrichment of M2-like TAMs and poor patient prognosis[137]. Once in the nucleus, YAP/TAZ function as transcriptional co-activators in cooperation with TEAD family proteins[119], robustly inducing the expression of genes such as CTGF, CYR61, TGF-β, and LOX[138]. The resulting production of these factors further exacerbates ECM cross-linking and fibrosis[139], thereby establishing a self-reinforcing loop in which matrix stiffness initiates YAP/TAZ signaling, which is subsequently amplified to drive further matrix stiffening and progressive magnification of the original mechanical input[140, 141]. More profoundly, the effects of this pathway are inscribed into long-term TAM functional memory through epigenetic remodeling and metabolic reprogramming. Specifically, YAP/TAZ regulate chromatin accessibility, enabling sustained high-level expression of mechanosensitive genes even after mechanical stimulation diminishes[142, 143]. In parallel, metabolic reprogramming jointly mediated by Piezo1-dependent Ca2+ signaling and YAP/TAZ endows TAMs with metabolic traits adapted to high-stiffness and hypoxic microenvironments[126, 144]. Thus, through the integrin/Piezo1-PYK2-YAP/TAZ axis, the tumor matrix systematically decodes physical stiffness into biological instructions that drive and stabilize specific TAM polarization states (Fig. 5).
Mechanotransductive programming of TAM phenotypes along tumor stiffness gradients. Schematic illustration of how ECM stiffening instructs TAMs phenotypic programming. (A) At collagen-rich stiff stroma and the invasive front, reorganized ECM increases tensile load on integrins and activates mechanosensitive ion channels, including Piezo1, inducing Ca²⁺ influx. (B) Mechanical inputs are integrated through integrin β1-actomyosin traction and the mechanosensitive checkpoint PYK2, promoting cytoskeletal remodeling and nuclear signaling. (C) PYK2- and FAK-dependent activation of Rho GTPases suppresses Hippo signaling and drives YAP/TAZ nuclear translocation, initiating pro-fibrotic and immunosuppressive transcriptional programs. (D) TAM-derived TGF-β and LOX activate myCAFs, enhancing ECM deposition and stiffening. (E) YAP/TAZ-associated epigenetic remodeling locks TAMs into stable M2-like states, establishing a self-amplifying stiffness-mechanotransduction feedback loop.
3.3 Physical barrier-driven metabolic and functional output
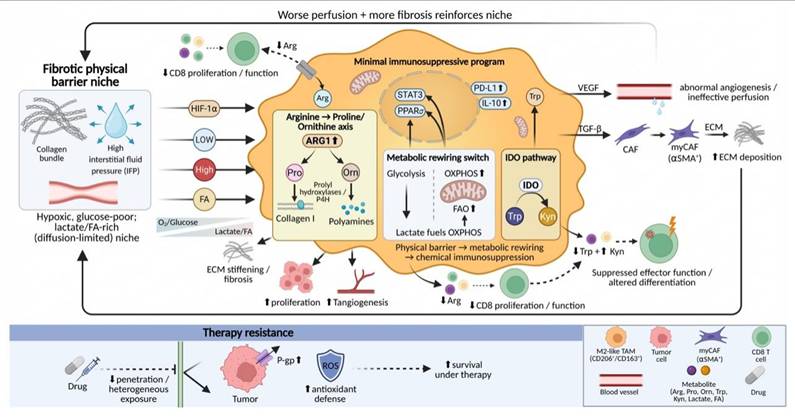
Following mechanically driven phenotypic remodeling, TAMs translate these internal transcriptional changes into profound metabolic and functional outputs. In densely fibrotic tumors, physical barriers formed by aberrantly deposited collagen and elevated IFP exert effects that extend beyond simply altering cellular mechanosensing. More importantly, these barriers reshape the spatiotemporal distribution of nutrients, metabolic substrates, and signaling molecules within the TME, thereby driving characteristic metabolic reprogramming in TAMs.
TAMs themselves are active architects of the fibrotic barrier, a process that is tightly coupled to their own metabolic remodeling. Recent studies have shown that, under conditions of matrix stiffening and TGF-β signaling, TAMs can initiate intrinsic collagen biosynthetic programs, upregulating the expression of type I collagen and key enzymes such as prolyl hydroxylases[145, 146]. Collagen biosynthesis is highly dependent on arginine metabolism[147, 148], leading TAMs to consume large amounts of arginine from the microenvironment and convert it into proline and ornithine. As a direct consequence, extracellular arginine concentrations are markedly reduced in TAM-enriched regions, whereas levels of proline and ornithine are increased[148, 149]. Arginine depletion directly suppresses the proliferation and function of CD8⁺ T cells, as T-cell activation and expansion are strictly dependent on this amino acid[149, 150]. Meanwhile, accumulated ornithine can be further converted into polyamines, which not only promote tumor cell proliferation and angiogenesis but also serve as important facilitators of collagen cross-linking, thereby synergistically exacerbating matrix stiffening at both metabolic and structural levels[147, 151].
Diffusion restriction and impaired perfusion imposed by physical barriers place TAMs under a distinctive and sustained metabolic stress environment, characterized by chronic hypoxia, limited glucose availability, and concomitant enrichment of metabolic by-products such as lactate and fatty acids[152, 153]. To adapt to this environment, TAMs undergo a fundamental metabolic shift, transitioning from the glycolysis-dependent pro-inflammatory phenotype toward a greater reliance on oxidative phosphorylation and fatty acid oxidation[154-156]. For example, large amounts of lactate produced by tumor cells and stromal cells can be taken up by TAMs and utilized to fuel oxidative phosphorylation[157]. At the molecular level, this metabolic transition is associated with activation of signaling pathways such as STAT3 and peroxisome proliferator-activated receptor δ (PPARδ), which together stabilize the immunosuppressive gene expression program of TAMs, including upregulation of programmed death-ligand 1 (PD-L1) and high expression of ARG1 and IL-10[157, 158]. Metabolically reprogrammed TAMs further promote aberrant angiogenesis and matrix deposition through the secretion of factors such as VEGF and TGF-β, leading to poorer tissue perfusion and more restricted molecular diffusion, thereby reinforcing the adverse microenvironment that initially drove their metabolic adaptation[159, 160].
Beyond passive adaptation, TAMs actively convert physical obstruction into functional immune exclusion through their metabolic outputs. Within dense matrix regions, ARG1-high TAMs exacerbate local arginine depletion[149]. In addition, TAMs can deplete tryptophan through the indoleamine 2,3-dioxygenase (IDO) pathway, generating immunosuppressive metabolites such as kynurenine[161, 162]. These metabolic activities superimpose a chemical barrier onto the physical barrier, systemically suppressing effector immune cell function and influencing their differentiation fate[163]. This metabolic remodeling process also represents one of the central mechanisms underlying therapeutic resistance. Dense stroma impedes the uniform diffusion of therapeutic agents, while M2-like TAMs localized within these regions promote tumor cell survival under drug exposure by upregulating drug efflux pumps, such as P-glycoprotein, and enhancing antioxidant metabolic capacity[161, 164] (Fig. 6).
Fibrotic physical barriers mechanotransductively program TAM metabolism to enforce immune exclusion and therapy resistance. Dense collagen bundles and elevated IFP restrict diffusion and impair perfusion, creating a hypoxic, glucose-poor but lactate/FA-rich niche. TAMs adapt by shifting from glycolysis toward OXPHOS/FAO (lactate-fueled) and activating STAT3/PPARδ, reinforcing an immunosuppressive program (PD-L1, IL-10, ARG1). In stiff, TGF-β-rich regions, TAMs also engage an arginine→proline/ornithine axis to support collagen maturation (prolyl hydroxylases) and generate ornithine-derived polyamines that promote tumor growth, angiogenesis, and collagen cross-linking, further stiffening ECM. Metabolic outputs superimpose a chemical barrier: ARG1-driven arginine depletion and IDO-mediated Trp to Kyn suppress CD8⁺ T-cell function. TAM-derived VEGF/TGF-β sustains abnormal angiogenesis, myCAF activation, ECM deposition, limited drug penetration, and TAM-assisted survival under therapy.
3.4 Mechanically guided spatial distribution and accumulation
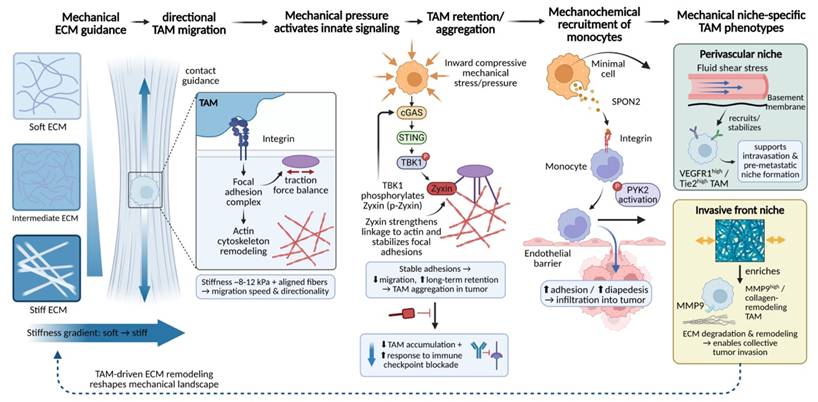
Ultimately, the cascade of mechanical sensing and functional remodeling culminates in the precise spatial distribution of TAMs. Within tumors, heterogeneity in ECM stiffness, the preferential alignment of collagen fibers, and local differences in tissue pressure collectively generate a complex mechanical microenvironment. By regulating TAM adhesive properties, migratory behavior, and ultimate positioning, this mechanical landscape drives TAM enrichment in specific functional niches—such as the invasive front, perivascular regions, or highly fibrotic tumor cores—thereby establishing a suppressive microenvironmental architecture in which spatial distribution is tightly coupled to function.
Highly aligned collagen fiber bundles within the tumor stroma provide structural physical tracks that support directed TAM migration[165, 166]. This process depends on the active sensing and response of cells to the physical properties of the matrix. Integrin-mediated cell-matrix adhesion transduces information regarding fiber orientation and tension into intracellular signals that govern actin cytoskeletal reorganization through focal adhesion complexes[167]. When a precise balance is achieved between local adhesion strength and actomyosin contractility-generated traction forces, TAMs preferentially undergo persistent, directional migration along the longitudinal axis of collagen fibers rather than random movement[168, 169]. Experimental evidence demonstrates that, in regions with matrix stiffness of approximately 8-12 kPa and highly ordered fiber alignment, both the migration speed and directionality of macrophages are significantly enhanced[170-172]. This enables TAMs to migrate systematically along fibrotic tracks pre-remodeled by tumor cells or CAFs, ultimately accumulating at mechanically active tumor-stroma interfaces.
Notably, the molecular mechanisms governing TAM migration and retention frequently integrate classical immune signaling pathways with mechanotransduction circuits, thereby employing distinct modalities to coordinate the sequential processes of directional navigation and site-specific anchoring. Recent studies have revealed a direct role for innate immune signaling in regulating macrophage mechanical behavior. Specifically, mechanical stress within the TME can activate the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING)-TANK-binding kinase 1 (TBK1) signaling axis in TAMs[173]. Activated TBK1 directly phosphorylates the focal adhesion protein zyxin, thereby strengthening its association with the actin cytoskeleton and stabilizing focal adhesion structures. This signaling cascade ultimately suppresses macrophage motility and promotes their long-term retention within tumor tissue. Genetic or pharmacological disruption of this pathway effectively reduces intratumoral TAM accumulation and enhances the efficacy of immune checkpoint blockade therapy[174]. These findings illustrate that immune signaling pathways can directly determine TAM spatial distribution by modulating the physical properties of the cytoskeleton. Similarly, the initial recruitment of monocytes is regulated by coupled mechanical and chemical cues[113]. For example, the matricellular protein spondin-2 (SPON2), secreted by tumor cells, can bind integrin β1 on the surface of monocytes, activating downstream PYK2 signaling and markedly enhancing cellular adhesion and transendothelial migration[175-177], thereby driving monocyte infiltration into tumor tissue[178].
Interestingly, the integrin-PYK2 axis and the cGAS-STING-TBK1 axis converge on Zyxin, a key regulator of actin dynamics and focal adhesion turnover, yet impose markedly divergent functional outputs. Activation of the integrin-PYK2 pathway promotes the recruitment of Zyxin to nascent focal adhesions at the leading edge, thereby accelerating adhesion turnover and supporting efficient, directional migration along ECM fibers[179, 180]. In contrast, TBK1-mediated phosphorylation of Zyxin at Ser143 enhances its localization to mature focal adhesions, stabilizing these structures and attenuating actin cytoskeletal dynamics, ultimately constraining TAMs in a sessile, tissue-resident state[181]. Thus, rather than operating in a purely competitive or cooperative manner, these pathways are functionally integrated: PYK2-driven signaling directs TAMs toward specific mechanical niches, whereas TBK1-Zyxin signaling stabilizes their retention at these sites, together ensuring precise spatial positioning of TAMs within the heterogeneous mechanical landscape of the tumor microenvironment.
The ultimate outcome of such mechanically guided processes is the establishment of finely tuned functional spatial organization of TAMs within tumors. TAM subpopulations residing in microenvironments with distinct mechanical characteristics exhibit markedly different phenotypes and functions. In perivascular regions, unique combinations of fluid shear stress and basement membrane components preferentially recruit and stabilize TAMs with high expression of receptors such as VEGFR1 and Tie2[25, 182, 183]. These cells play critical roles in facilitating tumor cell intravasation and the formation of pre-metastatic niches[183, 184]. In contrast, at the tumor invasive front, high tensile forces and densely cross-linked ECM selectively enrich TAM subsets characterized by high expression of MMP9 and collagen remodeling-associated genes[34, 185], with functions centered on ECM degradation and restructuring to enable collective tumor cell migration[186-188]. Thus, local mechanical properties act as a selective pressure that shapes both the spatial and functional heterogeneity of the TAM population. Importantly, TAMs do not merely respond passively to pre-existing mechanical environments; as described above, they actively participate in the dynamic construction of the tumor mechanical landscape through continuous ECM remodeling activities (Fig. 7).
Mechanically driven TAM migration and spatial accumulation establish niche-specific TAM programs. ECM stiffness gradients and collagen fiber alignment provide mechanical guidance cues that promote persistent, directional TAM migration via integrin-focal adhesion signaling and actin remodeling, with maximal speed/directionality in ~8-12 kPa, highly ordered matrices. Local compressive stress activates the innate cGAS-STING-TBK1 axis; TBK1 phosphorylates zyxin to strengthen actin coupling and stabilize adhesions, suppressing motility and promoting long-term TAM retention/aggregation—an effect whose disruption reduces TAM accumulation and improves checkpoint blockade responses. Mechanical cues also cooperate with chemotactic signals to recruit monocytes: tumor-derived SPON2 engages integrin β1, activates PYK2, and enhances adhesion, diapedesis, and infiltration. Distinct mechanics then select functional TAM states, including perivascular VEGFR1-high/Tie2-high TAMs (shear/basement membrane; intravasation, pre-metastatic niche) and invasive-front MMP9-high collagen-remodeling TAMs enabling collective invasion.
3.5 Self-Reinforcing Malignant Loop of Bidirectional Regulation
The bidirectional interplay between TAMs and the tumor mechanical microenvironment, as delineated in Sections 2 and 3, is not merely a sequential coupling of independent processes, but rather constitutes a self-reinforcing malignant system. In this system, TAM-driven remodeling of the ECM and mechanically induced TAM reprogramming are intrinsically interdependent, forming a closed-loop circuit that progressively stabilizes both stromal fibrosis and immunosuppression.
At the structural level, TAMs, in concert with CAFs, continuously reshape the ECM through coordinated deposition, crosslinking, degradation, and fiber reorganization, leading to increased matrix stiffness, elevated interstitial pressure, and the establishment of highly anisotropic collagen architectures. These alterations, in turn, redefine the mechanical landscape of the tumor microenvironment. At the signaling level, such persistent mechanical cues are sensed and transduced by TAMs through integrin-dependent adhesions and mechanosensitive pathways, thereby reinforcing pro-fibrotic and immunosuppressive transcriptional programs. At the functional level, the resulting TAM populations further amplify matrix remodeling, metabolic reprogramming, and immune exclusion, collectively consolidating a tumor-permissive niche.
Importantly, this feedback loop is not only molecularly self-sustaining but also spatially and temporally propagative. Mechanically defined niches characterized by high stiffness and dense fibrosis preferentially recruit and retain TAMs, while simultaneously imposing metabolic and physical constraints that suppress effector immune cell infiltration. As a consequence, newly recruited monocytes entering these regions are rapidly conditioned by the pre-established mechanical context, thereby perpetuating the expansion of pro-tumorigenic TAM populations.
Taken together, this self-reinforcing loop provides a unifying framework to understand how mechanical and immune dysregulation co-evolve during tumor progression. It also implies that disrupting this bidirectional coupling—rather than targeting individual components in isolation—may represent a critical prerequisite for effectively overcoming mechano-immunological resistance.
4. Therapeutic strategies
4.1 Targeting TAMs to mechanically soften tumors
Directly interfering with the pro-fibrotic activities of TAMs through strategies such as depletion, reprogramming, functional suppression, or signaling blockade can modulate tumor matrix stiffening at its cellular source. By alleviating aberrant stromal hardening, these approaches create a more permissive physical microenvironment for improved drug delivery and immune cell infiltration.
4.1.1 TAMs depletion and inhibition of monocyte recruitment
Given that aberrant physical cues and chemotactic axes (e.g., CCL2-CCR2) continuously drive monocyte recruitment to fuel early stromal stiffening, depleting established TAM populations and blocking their influx represent foundational therapeutic strategies for alleviating tumor immunosuppression and matrix densification. The core of this approach lies in blocking key signaling axes, such as CCL2-CCR2 and colony-stimulating factor 1 (CSF-1)/CSF-1 receptor (CSF-1R), thereby directly reducing the abundance and supply of immunosuppressive TAMs and weakening their support for tumor growth, fibrosis, and tissue mechanical stiffening at the source.
With respect to suppressing monocyte recruitment to tumor sites, targeting the CCL2-CCR2 axis is among the most extensively investigated strategies[15, 189]. In stroma-rich tumors such as pancreatic ductal adenocarcinoma (PDAC), CCL2 is highly expressed by both tumor cells and stromal components. Through engagement of its receptor CCR2, CCL2 continuously recruits CCR2+ monocytes to the tumor, where they subsequently differentiate into TAMs[190, 191]. Preclinical studies have demonstrated that treatment with the CCR2 small-molecule inhibitor PF-04136309[192] or the anti-CCL2 monoclonal antibody Carlumab[193] significantly reduces intratumoral TAMs and MDSCs, while enhancing the efficacy of chemotherapy or immunotherapy. Notably, the redundancy of the chemokine network suggests that simultaneous targeting of multiple signaling axes may yield greater therapeutic benefit[194]. For example, in hepatocellular carcinoma models, blockade of the CCL5/CCR5 axis not only suppresses the recruitment and function of polymorphonuclear MDSCs, but also indirectly modulates the immunosuppressive TME[195]. Although early clinical trials of CCL2-CCR2 inhibitors failed to meet expectations due to limited efficacy or toxicity, these outcomes underscore the presence of complex compensatory mechanisms within the TME and have prompted the development of next-generation agents and combination strategies.
In contrast to inhibiting monocyte recruitment, direct elimination of established TAM populations is primarily achieved by targeting the CSF-1/CSF-1R signaling pathway, which is central to macrophage survival, proliferation, and differentiation[15, 196] Studies have shown that small-molecule CSF-1R inhibitors, such as PLX3397 and BLZ945, markedly reduce the density of CD11b⁺F4/80⁺ TAMs across multiple murine solid tumor models, accompanied by enhanced CD8⁺ T-cell infiltration and reduced tumor burden[197-199]. Similarly, in head and neck squamous cell carcinoma models, BLZ945 and PLX3397 not only induce TAM apoptosis but also downregulate CD206 and IL-10 expression, thereby alleviating immunosuppression and synergizing with cisplatin to suppress tumor growth[200, 201]. In sonic hedgehog (SHH)-subtype medulloblastoma and sarcoma models, treatment with PLX5622 or PLX3397 likewise reduces TAM abundance and prolongs survival, indicating broad therapeutic potential of CSF-1R blockade in tumors highly dependent on TAMs[197, 202]. However, reduction in TAM numbers alone does not always translate into durable antitumor efficacy, in part because residual TAMs may undergo compensatory adaptation or promote the recruitment of alternative immunosuppressive cell populations[203, 204]. Accordingly, next-generation strategies aim to achieve more sustained and comprehensive remodeling of the TAM ecosystem. For instance, a recently reported covalent CSF-1R inhibitor, FF-10101, achieves prolonged and profound suppression of CSF-1R signaling through irreversible target engagement. In animal models, FF-10101 not only markedly reduces immunosuppressive TAMs but also expands TAM subsets with antitumor potential, while significantly enhancing antigen-specific CD8⁺ T-cell responses. Importantly, combination treatment with anti-PD-1 antibodies demonstrate pronounced synergistic antitumor effects[205]. Similarly, the next-generation CSF-1R inhibitor PXB17 exhibits superior pharmacokinetic properties and efficacy compared with PLX3397 in colorectal cancer models, significantly reducing M2-like TAMs and promoting CD8+ T-cell infiltration, thereby enhancing and stabilizing the therapeutic effects of PD-1 blockade[197]. It should be noted, however, that CSF-1R inhibitors are not universally effective as monotherapies across all tumor types and may exert adverse effects on dendritic cells and macrophages in normal tissues[197, 206, 207]. These findings indicate that TAM depletion or recruitment-inhibition strategies are more suitably applied in combination with ICIs or chemotherapy to effectively remodel the tumor immune microenvironment.
Beyond these canonical pathways, certain chemotactic signaling circuits with greater spatial selectivity offer additional opportunities for precision intervention. For example, in triple-negative breast cancer, the interleukin-17A (IL-17A)-osteopontin (OPN)-LYVE-1 axis has been shown to coordinately regulate the accumulation of TAMs from distinct origins. Specifically, IL-17A-induced OPN recruits peripheral monocytes through integrins such as αvβ3, promoting their differentiation into pro-fibrotic TAMs, while simultaneously stimulating the proliferation of tissue-resident macrophages[208]. Targeting such pathways may enable selective suppression of fibrosis-driving TAM subsets without complete macrophage depletion (Table 1).
Summary of therapeutic strategies targeting TAMs for tumor mechanical softening
| Drug | Target | Core Mechanism | Ref. |
|---|---|---|---|
| i. TAMs Depletion and Inhibition of Monocyte Recruitment | |||
| FF-10101 | Covalent small-molecule inhibitor (FLT3 / CSF-1R) | Sustained inhibition of CSF-1R to deplete immunosuppressive TAMs. | [205] |
| BLZ945 | Small-molecule inhibitor (CSF-1R) | Inhibits CSF-1R to reduce TAMs/microglia and attenuate stromal support in brain metastases. | [281] |
| PLX5622 | Small-molecule inhibitor (CSF-1R) | Depletes a subset of CSF-1R-dependent TAMs to delay tumor progression. | [202] |
| GW2580 | Small-molecule inhibitor (CSF-1R) | Reduces infiltration of profibrotic (Ly6C⁺ M2-like) macrophages to alleviate fibrosis. | [282] |
| Unnamed c-FMS inhibitor | Small-molecule inhibitor (CSF-1R) | Blocks CSF-1R to reduce TAM infiltration and CAF-rich dense stroma, partially restoring tissue structure. | [283] |
| PXB17 | CSF-1R | Next-gen inhibition; depletes M2-TAMs and stabilizes T-cells. | [197] |
| PF-04136309 | CCR2 | Blocks monocyte recruitment; weakens fibrotic stromal support. | [192] |
| Anti-OPN/αvβ3 | OPN-αvβ3 | Spatial targeting; selectively suppresses pro-fibrotic TAM subsets. | [208] |
| Anti-GM-CSF mAb | Neutralizing antibody (GM-CSF) | Blocks the GM-CSF-bone marrow axis to inhibit monopoiesis and TAM/MDSC replenishment. | [284] |
| ii. TAMs Reprogramming | |||
| Anti-CD40 agonist (PDA, mouse) | Agonistic antibody (CD40) | Activates myeloid/DC cells to convert "cold" tumors into T-cell-sensitive state. | [285] |
| Anti-CD40 agonist (PDA + T-cell therapy) | Agonistic antibody (CD40) | Reprograms TAMs towards an inflammatory phenotype to enhance engineered T-cell longevity (superior to depletion). | [286] |
| Anti-CD40 + Anti-PD-1 | Antibody combination (CD40 + PD-1) | CD40 activation reprograms TAMs/DCs; PD-1 blockade synergizes to enhance cytotoxic T-cell response. | [287-289] |
| FLT3L + Anti-CD40 | Growth factor + Agonistic antibody (Flt3L + CD40) | Restores and activates cDCs to trigger a type 1 immune response in fibrotic tumors. | [290] |
| poly(I:C) + R848 | TLR agonist combination (TLR3 + TLR7/8) | Synergistically reprograms M2-like macrophages to M1-like, cytotoxic effectors. | [291] |
| R848 + poly(I:C) (lung cancer) | TLR agonist combination (TLR7/8 + TLR3) | Drives TAM repolarization towards M1 phenotype and promotes T-cell recruitment. | [292] |
| 1V270 | Small-molecule TLR7 agonist | Intratumoral injection increases M1/M2 ratio in TAMs and enhances antigen presentation. | [293] |
| Lipo-MP-LPS | Liposome-encapsulated LPS analog (TLR4 agonist) | Induces M2→M1 conversion systemically, promoting a pro-inflammatory TME. | [294] |
| AZD2796 (anti-LILRB2) | Antibody (LILRB2) | Reprograms TAMs towards an immunostimulatory phenotype by blocking the myeloid checkpoint LILRB2. | [295] |
| iii. Inhibition of TAM-Mediated ECM Modification | |||
| BAPN | Small-molecule LOX inhibitor | Inhibits collagen cross-linking to reduce tumor stiffness and improve T-cell migration. | [59] |
| Anti-LOXL2 neutralizing antibody | Antibody (targets extracellular LOXL2) | Inhibits collagen stabilization and promotes macrophage-mediated "on-fiber" collagenolysis. | [296] |
| PXS-5153A | LOX/LOXL2 | Dual inhibition; blocks collagen crosslinking and reduces stiffness. | [234, 241] |
| DX-2400 | MT1-MMP | Selective MMP block; inhibits pathological matrix remodeling. | [246] |
| CAR-147 macrophages | Cellular therapy (CAR-M) | HER2-targeted CAR-M cells are engineered to upregulate MMPs for localized, precise ECM degradation. | [49] |
| iv. Disruption of TAM Mechanosensing Pathways | |||
| VS-4718 | Small-molecule FAK inhibitor | Inhibits integrin-FAK signaling driven by matrix stiffness, reducing YAP/TAZ activity. | [262] |
| Defactinib, GSK2256098 | Small-molecule FAK inhibitors | Block FAK-mediated adhesion, migration, and stiffness-sensing pathways. | [297] |
| iTEAD | Small-molecule TEAD inhibitor | Blocks the terminal transcriptional output (YAP/TAZ-TEAD) of ECM stiffness signaling. | [298] |
| Verteporfin | YAP/TAZ | Interrupts transcriptional decoding of extracellular mechanical cues. | [270, 271] |
| Fasudil | ROCK | Relaxes cytoskeletal tension; suppresses YAP-mediated activation. | [274] |
| TB511 | Peptide-drug conjugate (targets activated CD18) | Targets and deletes M2-TAMs with high conformational activation of integrin CD18. | [299] |
4.1.2 TAMs reprogramming
Because elevated matrix stiffness actively engages mechanotransduction networks to lock macrophages into a pro-fibrotic M2-like state, therapeutic reprogramming aims to counteract this mechanically driven polarization by redirecting TAMs toward an M1-like state with antitumor functions. Rather than eliminating TAMs, this strategy fundamentally alters their functional identity, thereby dismantling their support for the tumor mechanical microenvironment at its core.
Agonist-based approaches initiate reprogramming by activating innate immune signaling pathways. Among these, agonistic CD40 antibodies represent one of the most advanced strategies. CD40, a member of the tumor necrosis factor receptor superfamily, effectively drives macrophage polarization toward an M1-like phenotype upon activation and upregulates molecules involved in antigen presentation[209-211]. In fibrotic tumor models such as PDAC, the CD40 agonist APX005M not only induces phenotypic conversion of TAMs but also triggers pronounced stromal remodeling[212]. This includes reduced collagen density, improved vascular perfusion, and concomitant deep infiltration of CD8+ T cells[213]. Clinical studies further demonstrate that CD40 agonists combined with chemotherapy induce tumor stromal alterations and elicit T-cell responses in patients with PDAC[214]. Toll-like receptor (TLR) agonists constitute another important class of reprogramming agents. Encapsulation of the TLR7/8 agonist resiquimod (R848) into CD206-targeting nanoparticles enables selective activation of TLR signaling within TAMs. In breast cancer models, this targeted delivery system successfully reprograms TAMs toward an M1 phenotype and significantly suppresses pulmonary metastasis. Such targeted delivery strategies enhance therapeutic efficacy while minimizing systemic toxicity associated with widespread immune activation[215]. Notably, TAM reprogramming exhibits pronounced temporal dependence. M1-like states induced by CD40 or TLR agonists tend to revert to mixed or suppressive phenotypes in the absence of sustained signaling[216, 217]. This limitation indicates that molecular agonists alone are insufficient to maintain long-term functional conversion of TAMs and provides a rationale for integrating reprogramming strategies with delivery systems or biomaterial-based platforms.
Adoptive cell therapies, particularly chimeric antigen receptor macrophage (CAR-M) technologies, offer an engineered solution for TAM reprogramming. Through genetic modification, macrophages are endowed with CAR constructs that enable specific recognition of tumor antigens. Beyond conferring targeted cytotoxicity, CAR signaling itself drives macrophages toward a pro-inflammatory, antigen-presenting state[218]. Preclinical studies of human anti-HER2 CAR-M cells (CT-0508) demonstrate that infused CAR-Ms efficiently infiltrate tumors and remodel the immunosuppressive TME into a pro-inflammatory state, as evidenced by increased expression of M1 markers and pro-inflammatory cytokines[219]. Notably, CAR-Ms can also secrete MMPs, directly degrading dense ECM and thereby physically reducing tumor stiffness[49, 220]. Based on these findings, CT-0508 has advanced into a phase I clinical trial (NCT04660929), with preliminary data indicating favorable safety profiles and early signs of TME remodeling[221]. In addition, more advanced cellular therapies are under exploration, including macrophages engineered to release encapsulated reprogramming factors only in response to specific tumor microenvironmental cues, such as low pH or elevated reactive oxygen species, enabling safer and more precise in situ phenotypic conversion[222].
Metabolic and epigenetic interventions are critical for stabilizing reprogrammed phenotypes and preventing reversion to M2-like states. Metabolites within the TME are key drivers maintaining the M2 phenotype of TAMs[223], and targeting these metabolic pathways can effectively facilitate reprogramming. For example, phosphoinositide 3-kinase γ (PI3Kγ) is highly active in M2-like TAMs, and its inhibitor IPI-549 (eganelisib) has been shown in preclinical models to reprogram TAMs and synergize with ICIs[224, 225]. Epigenetic agents further lock in reprogrammed gene expression profiles by modifying chromatin architecture. Histone deacetylase (HDAC) inhibitors, for instance, increase histone acetylation at M1-associated gene loci, thereby promoting sustained transcriptional activation[226]. Specifically, the selective HDAC6 inhibitor ACY-1215, when combined with PD-1 blockade, synergistically enhances antitumor efficacy in ovarian cancer models, accompanied by a shift of TAM phenotypes toward a pro-inflammatory state[227] (Table 1).
4.1.3 Inhibition of TAM-mediated ECM modification
Building on the finding that TAMs function as key stromal "architects" by secreting crosslinking enzymes to weave highly aligned collagen bundles, inhibiting these ECM-modifying functions represents the most direct strategy to halt tumor mechanical stiffening. Rather than altering TAM abundance or globally reprogramming their phenotype, this approach focuses on targeting key enzymes secreted by TAMs that are responsible for ECM crosslinking, degradation, and remodeling. By disrupting aberrant collagen deposition and structural reorganization, this strategy aims to physically reduce tumor stiffness and improve tissue perfusion.
Targeting the LOX family constitutes a central strategy for suppressing ECM crosslinking. LOX and its homologs, such as LOXL2, are highly expressed in multiple tumor types and promote collagen fiber thickening, alignment, and overall matrix stiffening[228]. TAMs represent a major cellular source of LOX/LOXL2 within the TME[229]. Small-molecule LOXL2 inhibitors, including simtuzumab and PAT-1251, have demonstrated efficacy in preclinical fibrotic and selected tumor models by reducing tissue stiffness, attenuating collagen crosslinking, and suppressing metastasis[230, 231]. Furthermore, inhibition of LOX activity can downregulate integrin-FAK/Src signaling in tumor and stromal cells, thereby weakening the mechanically driven positive feedback loop that sustains fibrosis[232, 233]. However, phase II/III clinical trials combining simtuzumab with chemotherapy in pancreatic and colorectal cancers failed to improve overall survival[234, 235]. These disappointing outcomes highlight the functional complexity and redundancy of the LOX family, as well as the limitations of single-target interventions in advanced, highly fibrotic tumors[236]. In fact, the failure of selective LOXL2 inhibitors is closely associated with the compensatory upregulation of other family members, including LOXL1, LOXL3, and LOXL4. Studies have shown that even when a single isoform is inhibited, they may still collectively maintain collagen crosslinking activity[237, 238]. In addition, in established tumors in which the ECM has already undergone extensive crosslinking and stiffening, simply inhibiting further enzymatic activity may be insufficient to reverse the existing physical barrier, because the accumulated matrix remains structurally intact[6, 239]. Therefore, current research efforts are shifting toward the development of broader-spectrum LOX family inhibitors, such as dual LOX/LOXL2 inhibitors (e.g., PXS-5153A), or strategies that combine ECM-modulating agents with conventional chemotherapy or immunotherapy, aiming to take advantage of the transient window of matrix remodeling to enhance drug penetration and immune cell infiltration[240, 241].
Modulating the balance between MMPs and their endogenous inhibitors represents another strategy to control pathological ECM degradation and aberrant remodeling[242, 243]. TAMs secrete multiple MMPs, including MMP-2, MMP-9, and MMP-12, whose excessive activation and dysregulation contribute to ECM disorganization, enhanced tumor invasion, and the release of pro-tumorigenic factors[244]. Recent studies indicate that MMP-13, through coordinated interactions among CAFs and TAMs in breast cancer, can amplify fibrotic deposition and crosslinking, while under specific spatial and temporal contexts it may also exert antifibrotic or antimetastatic effects[245, 246]. However, first-generation broad-spectrum MMP inhibitors, such as marimastat, failed in clinical trials due to limited efficacy and severe musculoskeletal toxicity, largely attributable to their non-selective inhibition of MMPs and consequent disruption of normal tissue homeostasis[247]. Accordingly, next-generation approaches emphasize selective targeting. For instance, the monoclonal antibody DX-2400, which targets membrane-type 1 MMP (MT1-MMP/MMP-14), has demonstrated specific inhibition of tumor growth and metastasis with manageable toxicity in breast cancer models[42, 246]. An alternative strategy involves restoring physiological MMP/TIMP balance rather than abolishing enzymatic activity, either by upregulating tissue inhibitor of metalloproteinases-3 (TIMP-3) or by designing TIMP mimetics[248]. In addition, nanoparticle-based systems delivering MMP-9-specific small interfering RNA (siRNA) selectively to TAMs have been developed. In hepatocellular carcinoma models, such approaches achieved localized silencing of MMP-9, effectively suppressing tumor invasion and angiogenesis while avoiding systemic toxicity[249] (Table. 1).
4.1.4 Disruption of TAM mechanosensing pathways
Because extreme mechanical forces in advanced tumors must ultimately be transduced through cellular receptors to drive malignant progression, directly disrupting fundamental mechanosensing pathways (e.g., integrins, FAK, and YAP/TAZ) offers an ultimate strategy to overcome established mechanoresistance, even when TAM abundance is constrained[20]. This constitutes a therapeutic strategy that does not rely on directly altering ECM structure.
Integrins are a major class of mechanosensors that physically connect the ECM to the intracellular cytoskeleton. In TAMs, integrins—particularly αvβ3 and α5β1—undergo clustering upon binding to fibrotic ECM components such as fibronectin and vitronectin, thereby initiating downstream signaling cascades[250]. Small-molecule integrin antagonists and monoclonal antibodies have shown potential in preclinical studies to suppress macrophage-mediated pro-angiogenic and pro-metastatic functions[251, 252]. However, cilengitide failed to demonstrate a survival benefit in a phase III clinical trial in glioblastoma, partly due to pathway redundancy and compensatory mechanisms[253, 254]. Consequently, current research has shifted toward disrupting the assembly and function of integrin-mediated mechanosignaling complexes rather than simply blocking adhesive interactions. For example, peptides that interfere with the interactions between integrins and cytoskeletal adaptor proteins such as talin and kindlin may more specifically inhibit mechanotransduction while preserving other integrin-dependent biological functions[255, 256].
FAK is a central downstream signaling hub of integrins, responsible for converting mechanical cues into biochemical signals that promote cell survival, proliferation, and migration. In TAMs, FAK activation is closely associated with an immunosuppressive phenotype[257, 258]. FAK inhibitors (e.g., defactinib, VS-6063) not only suppress tumor cells in preclinical models of pancreatic cancer and mesothelioma, but also exert significant effects on TAMs[259, 260]. Specifically, they reduce the expression of M2 polarization markers in TAMs, decrease the secretion of the pro-fibrotic factor TGF-β, and improve CD8⁺ T-cell infiltration into tumors[261, 262]. A phase II clinical trial (NCT02758587) evaluated defactinib in combination with the anti-PD-1 antibody pembrolizumab and chemotherapy in advanced pancreatic cancer, with preliminary results indicating modulation of the immune microenvironment[263, 264]. However, because FAK is broadly expressed throughout the body, systemic inhibition may cause off-target toxicity[265]. Therefore, developing strategies that selectively target FAK signaling nodes within macrophages represents an important future direction.
The Hippo pathway effectors YAP/TAZ are key transcriptional co-activators that translate mechanical signals into gene expression programs[266, 267]. When TAMs sense a high-stiffness matrix, cytoskeletal tension suppresses the Hippo pathway, leading to YAP/TAZ dephosphorylation and nuclear translocation[267, 268], which in turn drives the expression of a series of pro-fibrotic and pro-proliferative genes, including CTGF, CYR61, and LOX[269]. Accordingly, inhibition of YAP/TAZ represents a critical step in blocking mechanical signal amplification. Verteporfin, an inhibitor of the interaction between YAP/TAZ and TEAD transcription factors[270, 271], has been shown in breast cancer and hepatocellular carcinoma models to suppress TAM-associated tumor progression and fibrosis[272, 273]. However, direct pharmacological targeting of transcription factors remains challenging. Alternative approaches include targeting upstream regulatory pathways, such as using the Rho-associated kinase (ROCK) inhibitor fasudil to relax cytoskeletal tension and thereby indirectly suppress YAP/TAZ activation[274].
Mechanosensitive ion channels provide an intervention point closer to the cell membrane. Channels such as Piezo1 and TRPV4 mediate the sensing of shear stress, stretch, and stiffness changes in multiple innate immune cell types, and activate downstream pathways including MAPK, NF-κB, HIF-1α, and YAP/TAZ through Ca²⁺ influx[275, 276]. In the tumor context, selectively blocking Piezo/TRPV channels expressed by TAMs using small-molecule inhibitors or nanoparticle-based delivery systems may attenuate their aberrant responses to elevated interstitial pressure and fluid shear stress, thereby suppressing HIF-1α-driven expression of pro-fibrotic factors such as SPP1 and LOX[277]. It must be noted, however, that mechanosensing pathways are highly conserved across cell types and play essential roles in tissue homeostasis[278]; thus, their systemic inhibition may result in widespread toxicity. Future research should therefore focus on developing interventions with greater cell-type specificity or enabling precise local delivery (Table 1).
Taken together, the reciprocal interaction between TAMs and the mechanical microenvironment is not static but evolves dynamically during tumor progression, forming a progressively reinforced biomechanical-immunological feedback system. In this context, presenting TAM-targeted therapeutic strategies as independent or parallel options may obscure their potential stage-specific relevance in clinical settings. To enhance translational applicability, it is therefore valuable to conceptualize these strategies within a longitudinal intervention framework aligned with the mechanical evolution of the tumor stroma.
During the early phase of tumor development, when inflammation-driven matrix remodeling and active monocyte recruitment predominate and the stromal barrier remains relatively permissive[279, 280], interventions that limit TAM accumulation or reprogram their highly plastic phenotypes may effectively prevent the initial establishment of pro-fibrotic niches. As tumors progress toward an intermediate stage characterized by active ECM crosslinking and progressive tissue stiffening, TAMs cooperate with stromal cells to promote dense collagen deposition and matrix remodeling[47]. At this stage, targeting TAM-derived ECM-modifying enzymes, such as LOX and MMPs, may help restrain the rapid escalation of stromal rigidity. In more advanced tumors, where highly crosslinked matrices generate sustained mechanical stress that stabilizes immunosuppressive TAM programs[11, 20], therapeutic strategies may need to shift toward disrupting core mechanotransduction pathways—such as integrin-FAK-YAP signaling—often in combination with matrix-modulating approaches to overcome established mechano-immunological resistance.
Despite the diverse strategies targeting TAMs at the cellular and molecular levels, these approaches are often limited by insufficient spatial precision, transient effects, and the inability to directly reconstitute the physical architecture of the tumor microenvironment. As tumor progression is governed not only by cellular behaviors but also by dynamically evolving biomechanical contexts, there is an increasing need for strategies that can actively and precisely modulate the physical and immunological properties of the TME in situ. Indeed, given the inherent limitations of single-agent TAM-targeted strategies-such as off-target effects, incomplete mechanical remodeling, and compensatory resistance-biomaterial-based mechano-immunological engineering offers a novel paradigm for the spatiotemporally precise modulation of the tumor microenvironment.
4.2 Mechanical immunoengineering and biomaterial-based strategies
Mechanical immunoengineering aims to exploit rationally designable biomaterials to actively create or mimic specific physicochemical microenvironments, thereby enabling precise, in situ regulation of TAMs and other immune cells. This approach represents a transformative strategy for overcoming intrinsic tumor mechanical barriers and implementing “active softening” of the TME.
4.2.1 Mechanically programmable hydrogels and scaffolds
Mechanically programmable hydrogels or scaffolds enable in situ recruitment and controllable phenotypic reprogramming of TAMs by precisely regulating matrix stiffness, degradation kinetics, and three-dimensional architecture, without the need for systemic administration. This strategy is a representative paradigm within the field of mechanical immunoengineering.
One major approach focuses on the development of injectable hydrogels with spatiotemporally controllable immunomodulatory functions. These materials typically exhibit favorable injectability and in situ gelation properties, allowing them to fill irregular tissue cavities and form mechanically tunable network structures through chemical or physical crosslinking, thereby serving as platforms for local sustained drug release and mechanical signal presentation. For example, an injectable hydrogel composite system based on carbon dots and proteins (TTF-L-C) has been used for multistage regulation of TAMs. In this system, the carbon dot component mediates chemotactic effects by upregulating Ctnnd1 expression, promoting selective macrophage accumulation within the gel. Subsequently, lipopolysaccharide encapsulated in the hydrogel locally activates macrophages and drives their polarization toward an M1-like phenotype, while concurrent administration of a PD-L1 blockade prevents potential compensatory upregulation of PD-L1 following reprogramming. As the hydrogel gradually degrades, these “trained” M1-like macrophages are continuously released into the tumor site, thereby promoting dendritic cell maturation and T-cell activation. In both primary and recurrent 4T1 breast cancer models, this strategy significantly enhanced immunotherapeutic efficacy and demonstrated favorable safety profiles[300]. This design elevates hydrogels from passive carriers to in situ activation platforms capable of actively modulating the local immune microenvironment. Another representative study developed a radiation-responsive self-assembling peptide hydrogel that undergoes conformational changes upon radiotherapy, enabling on-demand release of TLR7/8 agonists[301]. This allows precise temporal coordination between radiotherapy-induced immunogenic cell death (ICD) and TAM reprogramming, thereby markedly enhancing antitumor efficacy when combined with PD-1 blockade[302]. Collectively, these studies demonstrate that coupling material degradation kinetics with drug release dynamics enables single-intervention yet long-term regulation of TAMs, achieving temporally precise therapeutic control.
Another class of strategies focuses on the development of implantable scaffold systems that integrate both mechanical support and immunomodulatory functions, particularly suited for clinical scenarios combining postoperative tumor control with tissue regeneration. These scaffolds not only provide macroscopic mechanical support to defect sites, but their porous three-dimensional structures also offer an ideal matrix for regulating cell behavior and local drug delivery. For example, in postoperative bone tumor models, researchers constructed 3D-printed calcium phosphate scaffolds loaded with the CSF-1R inhibitor BLZ945. During the early post-implantation phase, the scaffold functions as a local drug reservoir, continuously releasing the inhibitor to selectively block the CSF-1/CSF-1R axis, thereby effectively depleting TAMs and suppressing pro-tumorigenic M2-like polarization to alleviate local immunosuppression. Meanwhile, the scaffold structure itself serves as a physical barrier. At later stages, as the scaffold surface gradually degrades, its internal architecture guides bone tissue regeneration[303]. Similarly, a self-healing hydrogel based on RADA peptides capable of loading the CaMKII inhibitor KN93 has been shown, in models such as malignant ascites, to induce immunogenic tumor cell death and directly reprogram M2-like TAMs (via inhibition of their highly expressed CaMKII) following a single intraperitoneal injection, thereby synergistically enhancing antitumor immune responses[304]. Such multifunctional integrated systems, combining chemotherapy, TAM reprogramming, and immune activation, fully illustrate the substantial potential of biomaterials as active therapeutic platforms.
4.2.2 TAM-targeted biomaterial delivery systems
Material-based delivery systems targeting TAMs constitute another central pillar of mechanobiological immunoengineering. Unlike hydrogels or scaffolds that provide fixed mechanical frameworks, these systems achieve selective recognition and intervention of TAMs and their monocyte precursors by precisely tuning the physicochemical properties of nanocarriers and their surface ligands. Relying on systemic or intracavitary administration, such platforms enable immune modulation across broader spatial scales of the TME and can act synergistically with immune checkpoint blockade, chemotherapy, or radiotherapy.
In terms of targeting strategies, current designs predominantly exploit receptors or surface molecules that are highly expressed on pro-tumoral TAM subsets, including CD163, CD206/MRC1, Siglecs, and MGL[305, 306]. Selective uptake and intratumoral enrichment are achieved through antibodies, short peptides, or glycan-based modifications[305]. These receptors not only mark immunosuppressive phenotypes but are also functionally linked to matrix-degrading enzymes and pro-angiogenic factors, thereby directly participating in tissue remodeling and alterations of mechanical properties. For example, polymeric prodrug nanoparticles loaded with doxorubicin and functionalized with anti-CD163 monoclonal antibodies undergo disassembly within acidic endosomal compartments, enabling preferential drug release in CD163⁺ M2-like TAMs and enhanced tumor suppression[305, 307]. Similarly, liposomes engineered based on the sialic acid-Siglec axis increase uptake by circulating monocytes and TAMs via surface-grafted sialic acids, thereby attenuating the continuous recruitment of immunosuppressive myeloid cells into tumors and slowing cell influx associated with M2 polarization and tissue remodeling[306, 308]. Mannose- or galactose-modified albumin and high-density lipoprotein-mimetic nanoparticles selectively bind CD206/MGL-positive TAMs, enriching clodronate, Toll-like receptor (TLR) agonists, or small-molecule immunomodulators within this population to enable selective depletion or inflammatory phenotype conversion[307, 309, 310].
In recent years, the delivery of informational biomolecules—such as nucleic acids and proteins—to rewire TAM signaling networks and transcriptional programs has emerged as a major direction. These strategies typically target key nodes involved in metabolism, epigenetic regulation, and cytoskeletal remodeling, thereby synchronously reprogramming immune and mechanical behaviors[311]. Multiple studies have demonstrated that ligand-modified nanoparticles encapsulating mRNAs encoding interferon regulatory factor 5 (IRF5) and IκB kinase β (IKKβ) can stably drive TAM polarization toward an M1-like state in ovarian cancer, melanoma, and glioblastoma models, accompanied by increased CD8⁺ T-cell infiltration and marked tumor regression[312]. Such M1-like TAMs generally secrete fewer pro-fibrotic and pro-angiogenic factors, thereby restraining collagen deposition and aberrant vascular expansion and alleviating stromal stiffening[48, 313]. CRISPR/Cas9-based delivery platforms further enable gene-level intervention of critical regulators such as PI3Kγ, a key modulator of TAM metabolism and migration that is closely associated with cytoskeletal reorganization and chemotactic motility[314, 315]. In murine solid tumor models, co-delivery of Cas9 complexes targeting Pik3cg with CpG-rich bacterial-derived nanovesicles simultaneously blocks PI3Kγ signaling and activates TLR9. These specialized vesicles markedly reduce the proportion of immunosuppressive TAM subsets and, when combined with PD-1 blockade, convert immunologically cold tumors into states highly responsive to immunotherapy[309, 316]. In addition, mannose-decorated hierarchical micelles have been used to deliver TLR7/8 agonists such as R848 to TAMs in gliomas, enabling M2-to-M1 reprogramming across the blood-brain barrier and reversing resistance to temozolomide[317, 318]. Activated M1-like TAMs in gliomas further modulate ECM secretion by stromal and astrocytic cells, improving local tissue compliance and drug diffusion[319].
Representative nanomedicine-based combination strategies that integrate immunosuppressive microenvironment remodeling with ICD induction typically operate through two synergistic mechanisms. First, TAM-targeting nanocarriers are used to deliver polarization cues that directly reprogram pro-tumorigenic M2-TAMs into pro-inflammatory M1-TAMs, thereby suppressing tumor growth and limiting metastatic dissemination[154, 320]. Multiple platforms, including M2pep-modified ferritin nanocages delivering CpG(311), HA-PEI nanoparticles loaded with miR-125b[321], and HA-mannose-modified nanocapsules co-encapsulating poly(I:C) and R848[291], have consistently demonstrated robust M1 polarization and significant inhibition of primary and metastatic tumor progression across diverse solid tumor models. Second, chemotherapeutic agents or photothermal/photosensitizing components are incorporated to exploit ICD-mediated immune amplification. Nanocarrier-delivered anthracyclines such as doxorubicin (DOX), as well as taxanes, efficiently induce hallmark ICD signals, including DAMP exposure and HMGB1 release, leading to enhanced dendritic cell activation, increased CD8⁺ T-cell infiltration, and elevated effector cytokine production[322, 323]. In parallel, these systems frequently reduce the infiltration of Tregs, MDSCs, and TAMs, while suppressing angiogenic programs, resulting in pronounced tumor regression and metastasis inhibition[324]. Importantly, combining TAM-targeted immunostimulatory agents (e.g., R848) with conventional chemotherapy within coordinated nanodelivery systems further amplifies these effects, effectively converting “cold” tumors into immunotherapy-responsive states and enhancing the efficacy of radiochemotherapy or PD-1/PD-L1 blockade[302, 310]. Collectively, these findings underscore that coupling nanodelivery, TAM reprogramming, and ICD induction represents a streamlined and broadly applicable strategy for synergistically potentiating chemo-immunotherapy in solid tumors.
Overall, TAM-targeted material delivery systems achieve indirect yet pivotal regulation of tumor immune and mechanical microenvironments through systematic design of targeting recognition, cargo loading, environmental responsiveness, and effector cell reprogramming. Future integration of mechano-responsive structural units, TAM-targeting modules, and immune checkpoint regulatory elements within a single platform may enable more compact therapeutic strategies capable of coordinately reshaping tumor mechanical and immune landscapes (Table 2).
Advanced Biomaterial Delivery Systems Targeting TAMs for Immune-Mechanical Landscape Reshaping
| Therapeutic Cargo | Delivery Vehicle/Platform | Targeting & Engineering Strategy | Therapeutic Outcomes & Mechanobiological Readouts | Ref. |
|---|---|---|---|---|
| BLZ945 (CSF-1R inhibitor) | Hydroxyl-terminated PAMAM dendrimers (D-BLZ) | Selective Intracellular Delivery: Leveraging nanoscale dimensions and hydrophilic shells for preferential accumulation within glioblastoma TAMs. | Achieved sustained intracellular release; significantly downregulated M2 markers, enhanced CD8⁺ T cell infiltration, and prolonged survival. | [325] |
| PLX3397 (CSF-1R inhibitor) | Nebulized/Localized pulmonary delivery system | Regional Spatial Targeting: Overcoming the pulmonary epithelial barrier via aerosolization to minimize systemic exposure and off-target toxicity. | Potently suppressed tumor burden at ultra-low doses (1 mg/kg); reduced M2-TAM density and enhanced M1-like antigen-presenting capacity. | [326] |
| anti-CSF-1R siRNA | anti-CSF-1R siRNA | Dual-Ligand Recognition: Surface-functionalized with SR-B1 targeting peptides and M2pep for high-fidelity identification of M2-like TAMs. | Induced a ~52% reduction in M2-TAMs and an 87% decrease in tumor volume; downregulated IL-10/TGF-β while promoting effector T cell responses. | [327] |
| Poly(I:C) + R848 (TLR7/8 agonists) | Mannose-hyaluronic acid coated polymer nanocapsules | Receptor-Mediated Endocytosis: Utilizing mannose-functionalized surfaces to target CD206⁺ TAMs for specific cellular internalization. | Triggered a robust M2-to-M1 phenotypic switch (CD86↑/CD206↓); suppressed primary tumor growth and distal metastasis in lung cancer models. | [328] |
| Chloroquine (CQ) + Polysaccharides | Redox-responsive PLGA nanoparticles | Metabolic-Immune Reprogramming: Exploiting the natural affinity of polysaccharides for pro-tumorigenic macrophages to co-deliver metabolic modulators. | Shifted TAM metabolic profiles from OXPHOS toward glycolysis; elevated M1-like polarization and augmented antitumor efficacy in breast cancer. | [329] |
| R848 (TLR7/8 agonist) | Cross-linked carboxymethyl dextran nanoparticles (Macrins) | Intrinsic Phagocytic Preference: Leveraging the inherent avidity of macrophages for dextran-based architectures to ensure targeted delivery. | Markedly upregulated IL-12 expression in TAMs; effectively inhibited tumor progression in MC38 colorectal cancer models. | [217] |
| R848 (TME-responsive NP) | M0 Macrophages as cell carriers (PR-M) | Cell-Mediated "Trojan Horse": Utilizing the innate tumor-homing capacity of macrophages for deep tissue penetration and targeted cargo release. | Achieved a 5.5-fold increase in M1/M2 ratio; robustly activated CD4⁺/CD8⁺ T cells and reduced tumor volume by 87.6%. | [330] |
| Doxorubicin (DOX) | Acid-sensitive PEGylated and mannose-modified PLGA NPs | Environment-Triggered Exposure: PEG shedding in the acidic TME to reveal mannose ligands for CD206-targeted uptake. | Enhanced TAM-specific internalization while minimizing hepatic and splenic sequestration; improved intratumoral drug distribution. | [331] |
| Sialic acid / Drug | Sialic acid-grafted liposomes | Siglec-axis targeting: Exploiting sialic acid affinity for circulating monocytes and TAMs. | Attenuates monocyte recruitment; slows M2-driven tissue remodeling. | [306] |
| IRF5 / IKKβ mRNA | Ligand-modified nanoparticles | Transcriptional rewiring: Encapsulating informational mRNAs to reprogram TAM signaling networks. | Drives stable M1 polarization; restrains collagen deposition and stiffening. | [312] |
| CRISPR/Cas9 (Pik3cg) | CpG-rich bacterial nanovesicles | Gene-level intervention: Co-delivery of Cas9 and TLR9 agonists for metabolic blockade. | Blocks PI3Kγ signaling; converts "cold" tumors to ICI-responsive states. | [316] |
| miR-125b | HA-PEI nanoparticles | CD44-mediated uptake: Utilizing hyaluronic acid (HA) for targeted miRNA delivery. | Induces robust M1-TAM conversion; potently inhibits metastatic progression. | [321] |
| siYTHDF1 (RNA methylation regulator) | RGD/mannose dual-targeted chromium nanoparticles | Multimodal Surface Engineering: Combining dual-receptor targeting with photothermal therapy (PTT)-triggered siRNA release. | Modulated the STAT3/STAT1 signaling axis; reshaped the myeloid landscape by reducing Treg and M2-TAM frequencies while enriching CD8⁺ T cells. | [332] |
While biomaterial-based and mechanical immunoengineering strategies provide powerful platforms for localized and programmable modulation of TAMs and tumor mechanics, their therapeutic efficacy as standalone interventions may still be constrained by the multifactorial nature of tumor progression and resistance. Given that TAM-driven immunosuppression and mechanical remodeling are tightly intertwined with angiogenesis, metabolic reprogramming, and treatment-induced adaptation, integrating TAM-targeted approaches with established therapeutic modalities has become a critical direction for achieving more durable and systemic antitumor effects.
4.3 Combination therapeutic strategies
As mechanistic insights into TAM biology and the efficacy of single-agent interventions have matured, combination therapeutic strategies have emerged as a critical avenue for improving treatment outcomes, particularly for remodeling the immunosuppressive and mechanically aberrant TME. TAMs contribute to tumor progression through dual mechanisms: on the one hand, they promote angiogenesis, ECM remodeling, and interstitial pressure elevation, thereby establishing a rigid, poorly perfused physical barrier; on the other hand, they sustain profound immune suppression. Accordingly, integrating TAM depletion or reprogramming strategies with ICIs, anti-angiogenic or anti-fibrotic therapies, and radiochemotherapy holds promise not only for overcoming immune tolerance but also for reshaping the mechanical microenvironment, ultimately restoring effective antitumor immune surveillance.
4.3.1 Combination with ICIs
Among combination paradigms, the integration of TAM-targeting strategies with ICIs is supported by the most substantial body of clinical and preclinical evidence. Multiple studies have demonstrated that M2-like TAMs are a major driver of both primary resistance and acquired refractoriness to PD-1/PD-L1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade. M2-like TAMs suppress effector T-cell activity through high expression of PD-L1, Arg1, and IDO, as well as through secretion of immunosuppressive cytokines such as IL-10 and TGF-β. In parallel, TAM-derived VEGF, MMPs, and chemokines sustain abnormal vasculature, dense ECM deposition, and restricted immune cell infiltration, collectively reinforcing an immunosuppressive and mechanically hostile tumor niche.
On this basis, combining TAM-targeted interventions with ICIs can generate synergistic effects at both the immune and tissue-structural levels. On the one hand, reducing the abundance and suppressive functions of M2-like TAMs through CSF-1R inhibition, blockade of the CCR2/CCL2 axis, or PI3Kγ inhibitors attenuates their antagonistic effects on ICIs[197, 205, 299]. On the other hand, reprogramming TAMs toward an M1-like phenotype using TLR or CD40 agonists, small-molecule immunomodulators, or nanodelivery platforms enhances antigen-presenting capacity and pro-inflammatory cytokine production, while promoting the recruitment and functional activity of dendritic cells and CD8⁺ T cells within tumors[292, 333].
In the phase II MARIO-3 trial in metastatic triple-negative breast cancer, the PI3Kγ inhibitor eganelisib combined with an anti-PD-L1 antibody and nab-paclitaxel not only induced TAM reprogramming and T-cell activation, but was also accompanied by downregulation of ECM-related transcriptional signatures and reorganization of vascular-immune gene programs, suggesting that TAM-targeted therapy combined with ICIs and chemotherapy may concurrently improve immune and stromal structural features(NCT03961698)[334]. Similarly, in melanoma and other animal models, interventions targeting macrophage-associated receptors such as MARCO in combination with CTLA-4 blockade shifted chemokine profiles from an M2-biased immunosuppressive pattern toward one favoring M1-like inflammatory infiltration, thereby significantly increasing effector T-cell entry into tumors and enhancing tumor burden control[335]. Taken together, current evidence indicates that TAM-targeted strategies combined with ICIs not only potentiate antitumor immune responses, but may also optimize the immune and mechanical properties of tumors by alleviating abnormal vascular leakage and excessive ECM deposition, improving local perfusion, and facilitating immune cell migration within dense stromal regions.
4.3.2 Combination with anti-angiogenic or anti-fibrotic therapies
The combination of TAM-targeted strategies with anti-angiogenic or anti-fibrotic therapies focuses on modulating the mechanical state of the TME at the interface of vasculature, ECM, and immune cells. Classical VEGF/VEGFR inhibitors, when administered within an appropriate dosing window, can partially normalize vascular structure and function, thereby improving perfusion and enhancing the delivery of therapeutics and immune cells. However, prolonged or excessive angiogenesis inhibition readily induces secondary hypoxia, which drives the accumulation of M2-like TAMs and activation of pro-fibrotic programs, accelerates ECM deposition, and ultimately reinforces tissue stiffness and immunosuppression[336].
Recent perspectives emphasize that therapeutic strategies simultaneously targeting angiogenesis and TAMs can more effectively disrupt these pathological feedback loops. On the one hand, inhibiting TAM-derived VEGF, PDGF, MMPs, LOX, or their upstream regulatory pathways (such as HIF-1α and PI3Kγ signaling) may reduce aberrant angiogenesis and collagen crosslinking, thereby alleviating ECM densification and elevated interstitial pressure at their source[314, 337]. On the other hand, combining anti-VEGF/VEGFR agents or multi-target tyrosine kinase inhibitors with TAM reprogramming approaches can, while improving vascular morphology and reducing leakage and tissue edema, attenuate pro-fibrotic interactions between M2-like TAMs and CAFs, leading to decreased collagen deposition and matrix crosslinking[338, 339].
For example, the heme oxygenase-1 (HO-1) inhibitor KCL-HO-1i suppresses LYVE-1⁺ perivascular TAMs and, when combined with cytotoxic chemotherapy, significantly enhances therapeutic responses in chemotherapy-resistant breast cancer and sarcoma models. This is accompanied by a shift of tumors from an immune-excluded state toward an inflamed microenvironment enriched in CD8⁺ T cells, together with vascular structural and functional parameters approaching a more physiological state[340]. Clinical practice in hepatocellular carcinoma, renal cell carcinoma, and endometrial cancer has demonstrated that combining anti-angiogenic therapy with ICIs improves objective response rates and survival outcomes. Building on this foundation, further incorporation of TAM-targeted interventions may reduce residual M2 TAM-mediated mechanical and immune resistance, thereby enabling more durable microenvironmental remodeling.
4.3.3 Combination with radiotherapy and chemotherapy
The combination of TAM modulation with radiotherapy and chemotherapy reflects an evolution of therapeutic strategies that incorporate microenvironmental regulation on the basis of conventional local or systemic cytotoxic treatments. Various chemotherapeutic agents and radiotherapy can induce ICD of tumor cells, leading to the release of tumor-associated antigens and damage-associated molecular patterns (DAMPs), thereby providing an antigenic source for the initiation and amplification of antitumor T-cell responses[341]. However, these treatments can also promote a shift of TAMs toward pro-angiogenic and pro-fibrotic M2-like phenotypes through vascular damage and inflammation-associated cytokine release, resulting in ECM remodeling and aberrant vascular repair[342]. Over certain temporal scales, this process gives rise to tolerance that encompasses both mechanical and immune dimensions.
Integrating TAM depletion or reprogramming interventions into radiotherapy or chemotherapy can re-direct tissue repair pathways while maintaining or enhancing cytotoxic efficacy. On the one hand, delivery platforms such as liposomal or polymeric nanoparticles can co-load ICD inducers, including doxorubicin, together with TAM-depleting or reprogramming agents, enabling a single therapeutic regimen to simultaneously trigger ICD within tumor cells and reduce M2-like TAM abundance and regulatory T-cell recruitment in the microenvironment. This restores local pro-inflammatory cytokine profiles and amplifies ICD-driven T-cell responses and memory formation. In 4T1 breast cancer mouse models, such “dual-targeting” designs significantly reduce M2-like TAMs, increase M1-like TAMs and effector T-cell infiltration, and suppress distant lung metastasis[324, 343].
On the other hand, radiotherapy alters tumor-region hemodynamics and matrix architecture through endothelial injury and parenchymal inflammation[344], during which TAMs participate in post-irradiation vascular repair and fibrosis and critically determine the modes of inflammation resolution and necrotic tissue clearance[345]. Available evidence indicates that adding CD40 or Toll-like receptor (TLR) agonists to local radiotherapy activates TAMs and drives their polarization toward M1-like states, thereby enhancing both local and abscopal antitumor responses and improving long-term tumor control[216]. If further combined with small-molecule or nanomedicine-based inhibitors targeting LOX, MMPs, or cathepsins, this strategy may restrain excessive ECM crosslinking and fibrosis during the early phase of radiation-induced tissue remodeling, preventing sustained loss of stromal compliance and restricted drug diffusion.
4.3.4 Additional combination strategies
Building on these approaches, integrating TAM-targeted nanoplatforms with the above combination strategies provides a feasible route for multilevel interventions spanning cellular, tissue, and organ scales. Recent studies have proposed that ligand-modified nanoparticles or vesicular systems can selectively deliver TLR/CD40 agonists, PI3Kγ inhibitors, or CRISPR/Cas9 components to specific TAM subsets, thereby enhancing antitumor immune responses while simultaneously influencing how these cells regulate ECM-producing cells and vascular endothelial cells. Ultimately, such strategies alter collagen fiber organization, vascular permeability, and local tissue compliance.
For example, in glioblastoma models, TAM-targeted TLR7/8 agonists are able to cross the blood-brain barrier and induce the conversion of M2-like TAMs toward M1-like phenotypes, thereby modifying their regulation of astrocytes and ECM components and improving the mechanical properties of brain tissue and drug diffusion[346]. When combined with ICIs, anti-angiogenic agents, or temozolomide, these platforms can simultaneously optimize immune activity and physical barrier characteristics within intracranial microenvironments characterized by high stiffness and restricted geometric space, leading to improved overall therapeutic responses in refractory tumors.
In summary, TAM-centered combination strategies are evolving from purely immunological synergy toward integrated regulatory paradigms that jointly address immune and mechanical dimensions. Whether combined with ICIs, anti-angiogenic/anti-fibrotic therapies, or radiotherapy, chemotherapy, and nanodelivery platforms, these approaches share a common objective: to selectively deplete or reprogram pro-tumorigenic TAMs, thereby relieving physical barriers to drug delivery and immune cell infiltration while attenuating TAM-driven immunosuppressive networks (Fig. 8).
TAM-centered tumor softening strategies to recondition a stiff, fibrotic, immune-suppressive TME. Left, TAM-myCAF-driven collagen deposition elevates ECM stiffness and IFP, induces HIF-1α-associated hypoxia, and sustains abnormal vessels with poor perfusion, leading to immune exclusion. Center, intervention axes that reduce fibrosis/pressure and restore perfusion/immune access: (i) limit TAM input or deplete TAMs (CCL2-CCR2; CSF-1/CSF-1R), (ii) reprogram TAMs toward anti-tumor states (CD40/TLR; CAR-M; PI3KPPAR/HDAC), (iii) inhibit TAM-mediated ECM remodeling (LOX/LOXL2; MMPs/cathepsins), and (iv) block TAM mechanosensing/mechanotransduction (integrin/FAK, Rho, YAP/TAZ, Piezo/TRPV). Right, reduced stiffness improves porosity, drug penetration, CD8⁺ infiltration, and response to PD-1/PD-L1 blockade.
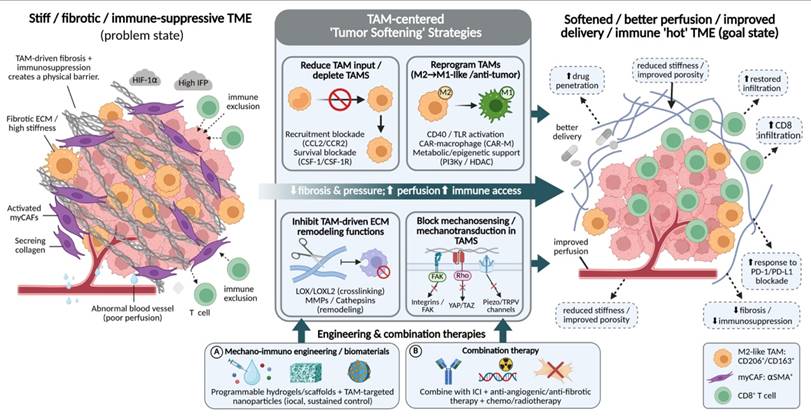
4.4 Comparative analysis and key challenges in clinical translation
Although the aforementioned therapeutic strategies have demonstrated encouraging efficacy in preclinical models, their translation into clinical practice remains limited by the multifactorial and adaptive nature of tumor progression. A critical evaluation indicates that, despite distinct mechanistic targets, these approaches share fundamental constraints arising from biological redundancy, spatial heterogeneity, and systemic complexity.
From a comparative perspective, TAM-targeted strategies differ across several key dimensions, including targeting specificity, spatiotemporal controllability, durability, and translational feasibility. TAM depletion strategies (e.g., CSF-1R or CCL2-CCR2 axis blockade) directly reduce immunosuppressive populations within the tumor microenvironment; however, their clinical efficacy is often limited by immune plasticity and functional compensation, including the recruitment of alternative suppressive myeloid populations such as PMN-MDSCs, as well as potential depletion of tissue-resident macrophages, leading to systemic toxicity[347, 348].
In contrast, TAM reprogramming strategies (e.g., CD40/TLR agonists or CAR-M approaches) preserve macrophage populations while redirecting their functional states toward antitumor activity and matrix remodeling. However, these effects are frequently transient, as the persistent mechanical and metabolic pressures within the tumor microenvironment can drive phenotypic reversion toward pro-tumoral states in the absence of sustained stimulation or engineered retention systems[265, 349]. Mechanobiology-targeted strategies (e.g., LOX, MMP, FAK, or integrin inhibition) directly address the physical basis of tumor progression and stromal stiffening; nevertheless, their clinical application is challenged by pathway redundancy and systemic pleiotropy, given the essential roles of these signaling axes in normal tissue homeostasis, often resulting in dose-limiting toxicities prior to achieving effective intratumoral exposure[350].
Collectively, these approaches exhibit distinct but complementary limitations: depletion strategies are limited by compensatory immune responses and toxicity; reprogramming strategies are constrained by temporal instability; and mechanobiology-targeted interventions face challenges related to redundancy and safety.
Beyond target-specific limitations, three overarching challenges further constrain clinical translation. First, the physical delivery paradox: dense, highly crosslinked collagen networks and elevated interstitial fluid pressure significantly restrict intratumoral penetration, particularly limiting access to poorly vascularized regions enriched in M2-like TAMs and cancer-associated fibroblasts[351]. Second, the lack of mechanobiology-informed biomarkers: current clinical stratification relies primarily on disease stage rather than tumor mechanical properties or TAM functional states, underscoring the need for validated non-invasive indicators such as ECM-derived circulating fragments, mechanotransduction-associated gene signatures, or elastography-based parameters[59]. Third, the context-dependent duality of ECM remodeling: while matrix deposition restricts tumor expansion and immune infiltration in certain contexts, excessive or untimely degradation may paradoxically facilitate tumor dissemination and metastatic progression[342].
Taken together, these limitations highlight the intrinsic constraints of single-modality approaches in the tumor mechano-immune ecosystem. Although emerging biomaterial-based and combination strategies offer improved spatiotemporal control and therapeutic synergy, they also introduce additional translational barriers, including manufacturing complexity, scalability, immunogenicity risks, and challenges in dose optimization and mechanistic deconvolution. Accordingly, the rational integration of complementary strategies, supported by mechanistic biomarkers and multi-omics monitoring, represents a promising yet still technically demanding direction for future clinical translation.
5. Challenges and future directions
TAMs serve as the central hub linking the tumor immune microenvironment and mechanical microenvironment[69, 352]. By shaping ECM stiffness, fiber alignment, and interstitial pressure, TAMs drive tumor progression and therapeutic resistance. Simultaneously, TAMs themselves are reciprocally regulated by mechanical signals, forming a vicious cycle[13]. Therefore, TAMs represent a key therapeutic target, and strategies such as depletion, reprogramming, the blockade of mechanical sensing, and material engineering aim to "soften" the tumor, thereby improving drug delivery and enhancing immune infiltration.
Despite the promising therapeutic strategy of targeting TAMs to remodel the tumor mechanical microenvironment, this field still faces a series of profound and interconnected challenges. Firstly, both the tumor mechanical microenvironment and TAM populations exhibit high spatiotemporal heterogeneity, which is the primary obstacle to precise intervention. Spatially, distinct tumor regions differ significantly in ECM stiffness, architecture, and immune composition[59]. Recent studies have revealed that profibrotic SPP1+ TAM subsets and proangiogenic TIE2+ TAM subsets occupy completely distinct tumoral regions, each colocalizing with specific mechanical characteristics[353]. Temporally, both the mechanical microenvironment and TAM phenotypes undergo dynamic evolution with tumor progression and therapeutic intervention[354]. This heterogeneity implies that generalized TAM-targeting strategies may fail to eliminate key pathogenic subsets and may disrupt macrophages with antitumor potential. Therefore, a core future challenge lies in developing technologies capable of real-time identification and targeting of specific functional TAM subsets. This may require integrating strategies such as activatable antibody-drug conjugates (ADCs), chimeric antigen receptor macrophage (CAR-M) design based on specific activation markers on TAM surfaces, or smart material delivery systems responsive to local mechanical properties to achieve precise intervention.
Current experimental studies on the interactions between immune cells/macrophages and the mechanical microenvironment are still largely based on mouse tumor models or static 2D/3D in vitro systems, whereas these models exhibit systematic deviations from human tumors in key parameters such as matrix stiffness, collagen crosslinking, and interstitial fluid mechanics[59, 355]. For example, elastography measurements show that human tumors exhibit substantially higher stiffness than corresponding mouse models[356]. Conventional 2D plastic culture completely lacks three-dimensional ECM topology, while commonly used hydrogels are limited in stiffness range, reproducibility, and architectural control, making it difficult to recapitulate the complex, mature collagen networks and their dynamic stiffening processes observed in human solid tumor[357]. In recent years, tumor-/organ-on-chip platforms integrating patient-derived organoids with CAFs, endothelial cells, and immune cells, together with perfusable microvasculature and controllable interstitial flow, have enabled the establishment of vascular perfusion, pressure gradients, and drug transport environments that more closely resemble in vivo conditions at the microscale. These systems have already been applied to evaluate drug penetration and efficacy within 3D matrices, providing tools with greater translational potential for dissecting and modulating TAM-associated mechanobiological niches[358].
Addressing these limitations increasingly relies on emerging advanced technologies that enable high-resolution, dynamic, and systems-level interrogation of tumor mechanobiology. Recent advances in spatial transcriptomics allow mapping of TAM subpopulations within intact tumor architectures, linking integrin-associated signaling states with local mechanical niches and immune landscapes[359]. In parallel, in vivo mechanical imaging techniques enable dynamic visualization of mechanical properties in living tissues, bridging static measurements with the spatiotemporal evolution of mechanotransductive signaling[360, 361]. Furthermore, machine learning-based frameworks integrate multi-omics, spatial, and biomechanical data to reconstruct mechanotransductive networks and predict TAM behavior under varying conditions[362, 363]. Collectively, these approaches provide a foundation for translating mechanistic insights into quantitatively testable and clinically actionable models.
In the future, it is necessary to vigorously develop and standardize these advanced models that can accurately simulate the mechanical properties of human tumors, including dynamic stiffness-tunable hydrogels, organoids integrated with biomechanical sensors, and bioreactor systems capable of applying cyclic solid stress or interstitial pressure within physiological ranges. These models will more reliably predict the efficacy of drugs in softening tumor mechanical barriers and reprogramming TAMs in humans.
Translating laboratory findings into clinically applicable therapies remains a long and arduous journey. First, identifying predictive biomarkers is crucial. We need to clarify which patients are most likely to benefit from TAM-targeted "softening" strategies. Peripheral blood monocyte subsets, circulating collagen metabolites, or imaging-derived mechanical parameters may serve as potential biomarkers. Second, treatment timing and sequencing need optimization. Key questions include: At which stage, before, during, or after immune checkpoint inhibitor therapy, is TAM-targeted intervention most effective? These questions require exploration in well-designed clinical trials. Recent clinical trials have provided preliminary clues. For example, early-phase clinical trials of CD40 agonists combined with chemotherapy in pancreatic cancer have shown changes in the immune and stromal microenvironments, but the efficacy awaits verification in larger cohorts[213]. In the future, more clinical trials incorporating mechanical microenvironment parameters as endpoints are needed—for instance, using serial imaging to assess changes in tumor stiffness before and after treatment, and correlating these changes with pathological responses and survival outcomes.
Targeting TAMs to remodel the tumor mechanical microenvironment is a cutting-edge field full of opportunities and challenges. It requires us to go beyond the traditional biochemical perspective and recognize mechanics as a therapeutic dimension equally important to immunity. With the deepening understanding of TAMs as "mechano-immune hubs" and the emergence of new technologies and strategies, we have reason to anticipate that interventions targeting the tumor mechanical microenvironment will evolve from auxiliary approaches to pillar strategies—providing a novel breakthrough for improving drug delivery, overcoming immune resistance, and ultimately combating solid tumors.
Abbreviations
ADCs: Antibody-drug conjugates; α-SMA: Alpha-smooth muscle actin; ARG1: Arginase 1; CAFs: Cancer-associated fibroblasts; CAR-M: Chimeric antigen receptor macrophages; cGAS: Cyclic GMP-AMP synthase; CTGF: Connective tissue growth factor; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; CYR61: Cysteine-rich angiogenic inducer 61; DAMPs: Damage-associated molecular patterns; ECM: Extracellular matrix; FAK: Focal adhesion kinase; FAO: Fatty acid oxidation; HDAC: Histone deacetylase; HIF-1⍺: Hypoxia-inducible factor 1 alpha; ICD: Immunogenic cell death; ICIs: Immune checkpoint inhibitors; IDO: Indoleamine 2,3-dioxygenase; IFP: Interstitial fluid pressure; IL-10: Interleukin 10; LOX: Lysyl oxidase; LOXL2: Lysyl oxidase-like 2; MAPK: Mitogen-activated protein kinase; MDSCs: Myeloid-derived suppressor cells; MMPs: Matrix metalloproteinases; MT1-MMP: Membrane-type 1 matrix metalloproteinase (MMP-14); myCAFs: Myofibroblast-like cancer-associated fibroblasts; OXPHOS: Oxidative phosphorylation; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; PDAC: Pancreatic ductal adenocarcinoma; PDGF: Platelet-derived growth factor; PI3Kγ: Phosphoinositide 3-kinase gamma; PPARδ: Peroxisome proliferator-activated receptor delta; PYK2: Proline-rich tyrosine kinase 2; ROCK: Rho-associated coiled-coil-containing protein kinase; STING: Stimulator of interferon genes; TAMs: Tumor-associated macrophages; TAZ: Transcriptional coactivator with PDZ-binding motif; TBK1: TANK-binding kinase 1; TGF-β: Transforming growth factor beta; TLR: Toll-like receptor; TME: Tumor microenvironment; Tregs: Regulatory T cells; VEGF: Vascular endothelial growth factor; YAP: Yes-associated protein.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (No. 82430123; 82174222), Noncommunicable Chronic Diseases-National Science and Technology Major Project (Grant No. 2024ZD0521400), Taishan Scholars Distinguished Expert Program of Shandong Province (Grant No. tstp20221166), the Collaborative Scientific and Technological Project of the Science and Technology Department of the National Administration of Traditional Chinese Medicine (Grant No. GZY-KJS-SD-2023-023), Shandong University of Traditional Chinese Medicine graduate quality improvement and innovation project (Grant No. YJSTZCX2025180).
Author contributions
Guanghui Liu: Writing-original draft
Yang Yu: Writing-review& editing
Zichen Guo: Visualization
Lijuan Liu: Investigation
Wujin Chen: Supervision
Changgang Sun: Funding acquisition, Project administration
AI usage statement
Figures were created using BioRender (www.biorender.com). During the preparation of this manuscript, the authors used ChatGPT-4o to assist with text polishing. The tool was employed solely for the purpose of checking grammar, spelling, and improving the clarity of language in the completed manuscript text. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the final content of the publication. ChatGPT-4o was not used to generate images, design experimental content, collect data, or perform data analysis. No AI tools were used as authors of this paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Khalaf K, Hana D, Chou JT, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front Immunol. 2021;12:656364
2. Nia HT, Munn LL, Jain RK. Physical traits of cancer. Science. 2020 370
3. Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S. et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6:153
4. Nia HT, Munn LL, Jain RK. Probing the physical hallmarks of cancer. Nat Methods. 2025;22:1800-18
5. Yuan Z, Li Y, Zhang S, Wang X, Dou H, Yu X. et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol Cancer. 2023;22:48
6. Zhang M, Zhang B. Extracellular matrix stiffness: mechanisms in tumor progression and therapeutic potential in cancer. Exp Hematol Oncol. 2025;14:54
7. Kalli M, Poskus MD, Stylianopoulos T, Zervantonakis IK. Beyond matrix stiffness: targeting force-induced cancer drug resistance. Trends Cancer. 2023;9:937-54
8. Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15:34
9. Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321-46
10. Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las Rivas J. et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updat. 2021;59:100787
11. Tharp KM, Kersten K, Maller O, Timblin GA, Stashko C, Canale FP. et al. Tumor-associated macrophages restrict CD8(+) T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat Cancer. 2024;5:1045-62
12. He Z, Zhang S. Tumor-Associated Macrophages and Their Functional Transformation in the Hypoxic Tumor Microenvironment. Front Immunol. 2021;12:741305
13. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:78
14. Huang R, Kang T, Chen S. The role of tumor-associated macrophages in tumor immune evasion. J Cancer Res Clin Oncol. 2024;150:238
15. Xu J, Ding L, Mei J, Hu Y, Kong X, Dai S. et al. Dual roles and therapeutic targeting of tumor-associated macrophages in tumor microenvironments. Signal Transduct Target Ther. 2025;10:268
16. Ghebremedhin A, Athavale D, Zhang Y, Yao X, Balch C, Song S. Tumor-Associated Macrophages as Major Immunosuppressive Cells in the Tumor Microenvironment. Cancers (Basel). 2024 16
17. Li M, He L, Zhu J, Zhang P, Liang S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. 2022;12:85
18. Yu S, Wang S, Wang X, Xu X. The axis of tumor-associated macrophages, extracellular matrix proteins, and cancer-associated fibroblasts in oncogenesis. Cancer Cell Int. 2024;24:335
19. Basak U, Sarkar T, Mukherjee S, Chakraborty S, Dutta A, Dutta S. et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. 2023;14:1295257
20. Xiong J, Xiao R, Zhao J, Zhao Q, Luo M, Li F. et al. Matrix stiffness affects tumor-associated macrophage functional polarization and its potential in tumor therapy. J Transl Med. 2024;22:85
21. Qian Y, Yin Y, Zheng X, Liu Z, Wang X. Metabolic regulation of tumor-associated macrophage heterogeneity: insights into the tumor microenvironment and immunotherapeutic opportunities. Biomark Res. 2024;12:1
22. Wang H, Yung MMH, Ngan HYS, Chan KKL, Chan DW. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int J Mol Sci. 2021 22
23. Laviron M, Petit M, Weber-Delacroix E, Combes AJ, Arkal AR, Barthelemy S. et al. Tumor-associated macrophage heterogeneity is driven by tissue territories in breast cancer. Cell Rep. 2022;39:110865
24. Yu KX, Yuan WJ, Wang HZ, Li YX. Extracellular matrix stiffness and tumor-associated macrophage polarization: new fields affecting immune exclusion. Cancer Immunol Immunother. 2024;73:115
25. Chu X, Tian Y, Lv C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol Cancer. 2024;23:150
26. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120
27. Mohan V, Das A, Sagi I. Emerging roles of ECM remodeling processes in cancer. Semin Cancer Biol. 2020;62:192-200
28. Thorseth ML, Carretta M, Jensen C, Molgaard K, Jurgensen HJ, Engelholm LH. et al. Uncovering mediators of collagen degradation in the tumor microenvironment. Matrix Biol Plus. 2022;13:100101
29. Liguori M, Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (Basel). 2011;3:3740-61
30. Caligiuri G, Tuveson DA. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell. 2023;41:434-49
31. Wu H, Li J, Yao R, Liu J, Su L, You W. Focusing on the interplay between tumor-associated macrophages and tumor microenvironment: from mechanism to intervention. Theranostics. 2025;15:7378-408
32. Saraswathibhatla A, Indana D, Chaudhuri O. Cell-extracellular matrix mechanotransduction in 3D. Nat Rev Mol Cell Biol. 2023;24:495-516
33. Du W, Xia X, Hu F, Yu J. Extracellular matrix remodeling in the tumor immunity. Front Immunol. 2023;14:1340634
34. Garcia APV, Salvi M, Reis LA, Ribeiro BRM, Nunes CB, de Paula AM. et al. Tumor-Associated Macrophages and Collagen Remodeling in Mammary Carcinomas: A Comparative Analysis in Dogs and Humans. Int J Mol Sci. 2025 26
35. Zhang Y, Fu Q, Sun W, Yue Q, He P, Niu D. et al. Mechanical forces in the tumor microenvironment: roles, pathways, and therapeutic approaches. J Transl Med. 2025;23:313
36. Najafi M, Farhood B, Mortezaee K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J Cell Biochem. 2019;120:2782-90
37. Mai Z, Lin Y, Lin P, Zhao X, Cui L. Modulating extracellular matrix stiffness: a strategic approach to boost cancer immunotherapy. Cell Death Dis. 2024;15:307
38. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131
39. Ren Y, Lu D, Wang F, Wang Z, Li J, Huang R. et al. A multi-omics atlas of CAF subtypes reveals apCAF-M2 macrophage interactions driving immune resistance in glioma. PLoS One. 2025;20:e0329801
40. Deng B, Zhao Z, Kong W, Han C, Shen X, Zhou C. Biological role of matrix stiffness in tumor growth and treatment. J Transl Med. 2022;20:540
41. Cui N, Hu M, Khalil RA. Biochemical and Biological Attributes of Matrix Metalloproteinases. Progress in Molecular Biology and Translational Science. 2017;147:1-73
42. Niland S, Riscanevo AX, Eble JA. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. International Journal of Molecular Sciences. 2021;23:146
43. Madsen DH, Jürgensen HJ, Siersbæk MS, Kuczek DE, Grey Cloud L, Liu S. et al. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Reports. 2017;21:3662-71
44. Arlt A, von Bonin F, Rehberg T, Perez-Rubio P, Engelmann JC, Limm K. et al. High CD206 levels in Hodgkin lymphoma-educated macrophages are linked to matrix-remodeling and lymphoma dissemination. Molecular Oncology. 2020;14:571-89
45. Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ, Peters DE. et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. The Journal of Cell Biology. 2013;202:951-66
46. Vujičić M, Broderick I, Salmantabar P, Perian C, Nilsson J, Sihlbom Wallem C. et al. A macrophage-collagen fragment axis mediates subcutaneous adipose tissue remodeling in mice. Proceedings of the National Academy of Sciences of the United States of America. 2024;121:e2313185121
47. Toledo B, Zhu Chen L, Paniagua-Sancho M, Marchal JA, Perán M, Giovannetti E. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. Journal of Hematology & Oncology. 2024;17:44
48. Han S, Wang W, Wang S, Yang T, Zhang G, Wang D. et al. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics. 2021;11:2892-916
49. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H. et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. British Journal of Cancer. 2019;121:837-45
50. Juric V, O'Sullivan C, Stefanutti E, Kovalenko M, Greenstein A, Barry-Hamilton V. et al. MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PloS One. 2018;13:e0207255
51. Tan Y, Yang Y-G, Zhang X, Zhao L, Wang X, Liu W. Tumor cell-derived osteopontin promotes tumor fibrosis indirectly via tumor-associated macrophages. Journal of Translational Medicine. 2025;23:432
52. Blise KE, Sivagnanam S, Banik GL, Coussens LM, Goecks J. Single-cell spatial architectures associated with clinical outcome in head and neck squamous cell carcinoma. NPJ precision oncology. 2022;6:10
53. Wright K, Ly T, Kriet M, Czirok A, Thomas SM. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers. 2023;15:1899
54. Kay EJ, Koulouras G, Zanivan S. Regulation of Extracellular Matrix Production in Activated Fibroblasts: Roles of Amino Acid Metabolism in Collagen Synthesis. Frontiers in Oncology. 2021;11:719922
55. Puttock EH, Tyler EJ, Manni M, Maniati E, Butterworth C, Burger Ramos M. et al. Extracellular matrix educates an immunoregulatory tumor macrophage phenotype found in ovarian cancer metastasis. Nature Communications. 2023;14:2514
56. Mei J, Cai Y, Xu R, Li Q, Chu J, Luo Z. et al. Conserved immuno-collagenic subtypes predict response to immune checkpoint blockade. Cancer Communications (London, England). 2024;44:554-75
57. Alonso-Nocelo M, Ruiz-Cañas L, Sancho P, Görgülü K, Alcalá S, Pedrero C. et al. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut. 2023;72:345-59
58. Burke RM, Madden KS, Perry SW, Zettel ML, Brown EB. Tumor-associated macrophages and stromal TNF-α regulate collagen structure in a breast tumor model as visualized by second harmonic generation. Journal of Biomedical Optics. 2013;18:86003
59. Nicolas-Boluda A, Vaquero J, Vimeux L, Guilbert T, Barrin S, Kantari-Mimoun C. et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. eLife. 2021;10:e58688
60. Liu J, Chiang H-C, Xiong W, Laurent V, Griffiths SC, Dülfer J. et al. A highly selective humanized DDR1 mAb reverses immune exclusion by disrupting collagen fiber alignment in breast cancer. Journal for Immunotherapy of Cancer. 2023;11:e006720
61. Heaton AR, Burkard NJ, Sondel PM, Skala MC. Quantifying in vivo collagen reorganization during immunotherapy in murine melanoma with second harmonic generation imaging. Biophotonics Discovery. 2024;1:015004
62. Szulczewski JM, Inman DR, Proestaki M, Notbohm J, Burkel BM, Ponik SM. Directional cues in the tumor microenvironment due to cell contraction against aligned collagen fibers. Acta Biomaterialia. 2021;129:96-109
63. Fan Y, Chiu A, Zhao F, George JT. Understanding the interplay between extracellular matrix topology and tumor-immune interactions: Challenges and opportunities. Oncotarget. 2024;15:768-81
64. Gudneppanavar R, Di Pietro C, H Öz H, Zhang P-X, Cheng E-C, Huang PH. et al. Ezrin drives adaptation of monocytes to the inflamed lung microenvironment. Cell Death & Disease. 2024;15:864
65. Meirson T, Genna A, Lukic N, Makhnii T, Alter J, Sharma VP. et al. Targeting invadopodia-mediated breast cancer metastasis by using ABL kinase inhibitors. Oncotarget. 2018;9:22158-83
66. Jiu Y, Peränen J, Schaible N, Cheng F, Eriksson JE, Krishnan R. et al. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. Journal of Cell Science. 2017;130:892-902
67. Huang X, Li Z, Huang Y, Zhang Q, Cui Y, Shi X. et al. Vimentin intermediate filaments coordinate actin stress fibers and podosomes to determine the extracellular matrix degradation by macrophages. Developmental Cell. 2025;60:1669-85.e6
68. Strouhalova K, Přechová M, Gandalovičová A, Brábek J, Gregor M, Rosel D. Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers. 2020;12:184
69. Yang Y, Li S, To KKW, Zhu S, Wang F, Fu L. Tumor-associated macrophages remodel the suppressive tumor immune microenvironment and targeted therapy for immunotherapy. Journal of experimental & clinical cancer research: CR. 2025;44:145
70. Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS. et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. The EMBO journal. 2020;39:e104063
71. Sebastian A, Hum NR, Martin KA, Gilmore SF, Peran I, Byers SW. et al. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers. 2020;12:1307
72. Wang X, Venet D, Lifrange F, Larsimont D, Rediti M, Stenbeck L. et al. Spatial transcriptomics reveals substantial heterogeneity in triple-negative breast cancer with potential clinical implications. Nature Communications. 2024;15:10232
73. Wu Y, Shi Y, Luo Z, Zhou X, Chen Y, Song X. et al. Spatial multi-omics analysis of tumor-stroma boundary cell features for predicting breast cancer progression and therapy response. Frontiers in Cell and Developmental Biology. 2025;13:1570696
74. Jamiyan T, Kuroda H, Yamaguchi R, Abe A, Hayashi M. CD68- and CD163-positive tumor-associated macrophages in triple negative cancer of the breast. Virchows Archiv: An International Journal of Pathology. 2020;477:767-75
75. Ma C, Yang C, Peng A, Sun T, Ji X, Mi J. et al. Pan-cancer spatially resolved single-cell analysis reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment. Molecular Cancer. 2023;22:170
76. Shakiba M, Tuveson DA. Macrophages and fibroblasts as regulators of the immune response in pancreatic cancer. Nature Immunology. 2025;26:678-91
77. Zhang J-Y, Zhu W-W, Wang M-Y, Zhai R-D, Wang Q, Shen W-L. et al. Cancer-associated fibroblasts promote oral squamous cell carcinoma progression through LOX-mediated matrix stiffness. Journal of Translational Medicine. 2021;19:513
78. Xiao W, Pahlavanneshan M, Eun C-Y, Zhang X, DeKalb C, Mahgoub B. et al. Matrix stiffness mediates pancreatic cancer chemoresistance through induction of exosome hypersecretion in a cancer associated fibroblasts-tumor organoid biomimetic model. Matrix Biology Plus. 2022;14:100111
79. Dalpati N, Rai SK, Dash SP, Kumar P, Singh D, Sarangi PP. Integrins α5β1 and αvβ3 Differentially Participate in the Recruitment and Reprogramming of Tumor-associated Macrophages in the In Vitro and In Vivo Models of Breast Tumor. Journal of Immunology (Baltimore, Md: 1950). 2024;213:1553-68
80. Pakshir P, Alizadehgiashi M, Wong B, Coelho NM, Chen X, Gong Z. et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nature Communications. 2019;10:1850
81. Ezzo M, Spindler K, Wang JB, Lee D, Pecoraro G, Cowen J. et al. Acute contact with profibrotic macrophages mechanically activates fibroblasts via αvβ3 integrin-mediated engagement of Piezo1. Science Advances. 2024;10:eadp4726
82. Duran CL, Borriello L, Karagiannis GS, Entenberg D, Oktay MH, Condeelis JS. Targeting Tie2 in the Tumor Microenvironment: From Angiogenesis to Dissemination. Cancers. 2021;13:5730
83. Bahri M, Anstee JE, Opzoomer JW, Arnold JN. Perivascular tumor-associated macrophages and their role in cancer progression. Essays in Biochemistry. 2023;67:919-28
84. Mahlbacher G, Curtis LT, Lowengrub J, Frieboes HB. Mathematical modeling of tumor-associated macrophage interactions with the cancer microenvironment. Journal for Immunotherapy of Cancer. 2018;6:10
85. Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A. et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood "resident" monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood. 2009;114:901-14
86. Chen W, Wang Y, Gu H, Zhang Y, Chen C, Yu T. et al. Molecular characteristics, clinical significance, and immune landscape of extracellular matrix remodeling-associated genes in colorectal cancer. Frontiers in Oncology. 2023;13:1109181
87. Nambiar DK, Viswanathan V, Cao H, Zhang W, Guan L, Chamoli M. et al. Galectin-1 Mediates Chronic STING Activation in Tumors to Promote Metastasis through MDSC Recruitment. Cancer Research. 2023;83:3205-19
88. Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Research. 2013;73:3833-41
89. Stylianopoulos T. The Solid Mechanics of Cancer and Strategies for Improved Therapy. Journal of Biomechanical Engineering. 2017 139
90. Kharaishvili G, Simkova D, Bouchalova K, Gachechiladze M, Narsia N, Bouchal J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell International. 2014;14:41
91. Tung NTC, Nogami M, Iwasaki M, Yahara Y, Seki S, Makino H. et al. M2-like macrophages derived from THP-1 cells promote myofibroblast differentiation of synovial fibroblasts in association with the TGF-β1/SMAD2/3 signaling pathway. Scientific Reports. 2025;15:25505
92. Ishihara S, Haga H. Matrix Stiffness Contributes to Cancer Progression by Regulating Transcription Factors. Cancers. 2022;14:1049
93. Pirentis AP, Polydorou C, Papageorgis P, Voutouri C, Mpekris F, Stylianopoulos T. Remodeling of extracellular matrix due to solid stress accumulation during tumor growth. Connective Tissue Research. 2015;56:345-54
94. Bai R, Li Y, Jian L, Yang Y, Zhao L, Wei M. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: mechanisms and clinical treatment strategies. Molecular Cancer. 2022;21:177
95. Kazakova A, Sudarskikh T, Kovalev O, Kzhyshkowska J, Larionova I. Interaction of tumor-associated macrophages with stromal and immune components in solid tumors: Research progress (Review). International Journal of Oncology. 2023;62:32
96. Malekghasemi S, Majidi J, Baghbanzadeh A, Abdolalizadeh J, Baradaran B, Aghebati-Maleki L. Tumor-Associated Macrophages: Protumoral Macrophages in Inflammatory Tumor Microenvironment. Advanced Pharmaceutical Bulletin. 2020;10:556-65
97. Ferruzzi J, Sun M, Gkousioudi A, Pilvar A, Roblyer D, Zhang Y. et al. Compressive Remodeling Alters Fluid Transport Properties of Collagen Networks - Implications for Tumor Growth. Scientific Reports. 2019;9:17151
98. Soko GF, Kosgei BK, Meena SS, Ng YJ, Liang H, Zhang B. et al. Extracellular matrix re-normalization to improve cold tumor penetration by oncolytic viruses. Frontiers in Immunology. 2024;15:1535647
99. Nieskoski MD, Marra K, Gunn JR, Hoopes PJ, Doyley MM, Hasan T. et al. Collagen Complexity Spatially Defines Microregions of Total Tissue Pressure in Pancreatic Cancer. Scientific Reports. 2017;7:10093
100. Voutouri C, Polydorou C, Papageorgis P, Gkretsi V, Stylianopoulos T. Hyaluronan-Derived Swelling of Solid Tumors, the Contribution of Collagen and Cancer Cells, and Implications for Cancer Therapy. Neoplasia (New York, NY). 2016;18:732-41
101. Li X, Shepard HM, Cowell JA, Zhao C, Osgood RJ, Rosengren S. et al. Parallel Accumulation of Tumor Hyaluronan, Collagen, and Other Drivers of Tumor Progression. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2018;24:4798-807
102. Dolor A, Szoka FC. Digesting a Path Forward: The Utility of Collagenase Tumor Treatment for Improved Drug Delivery. Molecular Pharmaceutics. 2018;15:2069-83
103. Zeltz C, Primac I, Erusappan P, Alam J, Noel A, Gullberg D. Cancer-associated fibroblasts in desmoplastic tumors: emerging role of integrins. Seminars in Cancer Biology. 2020;62:166-81
104. Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen D. et al. Periostin Promotes Colorectal Tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Reports. 2020;30:793-806.e6
105. Li M, Zhang X, Wang M, Wang Y, Qian J, Xing X. et al. Activation of Piezo1 contributes to matrix stiffness-induced angiogenesis in hepatocellular carcinoma. Cancer Communications (London, England). 2022;42:1162-84
106. Sun L, Zhang X, Song Q, Liu L, Forbes E, Tian W. et al. IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in pancreatic ductal adenocarcinoma through the STAT3 pathway. Cancer Letters. 2021;500:132-46
107. Zeng X-Y, Xie H, Yuan J, Jiang X-Y, Yong J-H, Zeng D. et al. M2-like tumor-associated macrophages-secreted EGF promotes epithelial ovarian cancer metastasis via activating EGFR-ERK signaling and suppressing lncRNA LIMT expression. Cancer Biology & Therapy. 2019;20:956-66
108. Le F, Yang L, Han Y, Zhong Y, Zhan F, Feng Y. et al. TPL Inhibits the Invasion and Migration of Drug-Resistant Ovarian Cancer by Targeting the PI3K/AKT/NF-κB-Signaling Pathway to Inhibit the Polarization of M2 TAMs. Frontiers in Oncology. 2021;11:704001
109. Wang Y, Lee BH, Yang Z, Ho TJ, Ci H, Jackson B. et al. Chewing-Activated TRPV4/PIEZO1-HIF-1α-Zn Axes in a Rat Periodontal Complex. Journal of Dental Research. 2025;104:398-407
110. Kamen LA, Schlessinger J, Lowell CA. Pyk2 is required for neutrophil degranulation and host defense responses to bacterial infection. Journal of Immunology (Baltimore, Md: 1950). 2011;186:1656-65
111. Kim J, Kang W, Kang SH, Park SH, Kim JY, Yang S. et al. Proline-rich tyrosine kinase 2 mediates transforming growth factor-beta-induced hepatic stellate cell activation and liver fibrosis. Scientific Reports. 2020;10:21018
112. Lee J-W, Mizuno K, Watanabe H. et al. Enhanced phagocytosis associated with multinucleated microglia via Pyk2 inhibition in an acute β-amyloid infusion model. Journal of neuroinflammation. 2024 21
113. Muller AK, Kohler UA, Trzebanski S, Vinik Y, Raj HM, Girault JA. et al. Mouse Modeling Dissecting Macrophage-Breast Cancer Communication Uncovered Roles of PYK2 in Macrophage Recruitment and Breast Tumorigenesis. Adv Sci (Weinh). 2022;9:e2105696
114. Günay KA, Silver JS, Chang T-L, Bednarski OJ, Bannister KL, Rogowski CJ. et al. Myoblast mechanotransduction and myotube morphology is dependent on BAG3 regulation of YAP and TAZ. Biomaterials. 2021;277:121097
115. Wang K-C, Yeh Y-T, Nguyen P, Limqueco E, Lopez J, Thorossian S. et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:11525-30
116. Miranda MZ, Bialik JF, Speight P, Dan Q, Yeung T, Szászi K. et al. TGF-β1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. The Journal of Biological Chemistry. 2017;292:14902-20
117. Azad T, Ghahremani M, Yang X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells. 2019;8:407
118. Merritt N, Garcia K, Rajendran D, Lin Z-Y, Zhang X, Mitchell KA. et al. TAZ-CAMTA1 and YAP-TFE3 alter the TAZ/YAP transcriptome by recruiting the ATAC histone acetyltransferase complex. eLife. 2021;10:e62857
119. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nature Cell Biology. 2015;17:1218-27
120. Kim M, Kim T, Johnson RL, Lim D-S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Reports. 2015;11:270-82
121. Chandra Jena B, Sarkar S, Rout L, Mandal M. The transformation of cancer-associated fibroblasts: Current perspectives on the role of TGF-β in CAF mediated tumor progression and therapeutic resistance. Cancer Letters. 2021;520:222-32
122. Taufalele PV, VanderBurgh JA, Muñoz A, Zanotelli MR, Reinhart-King CA. Fiber alignment drives changes in architectural and mechanical features in collagen matrices. PloS One. 2019;14:e0216537
123. Zhang H, Lin F, Huang J, Xiong C. Anisotropic stiffness gradient-regulated mechanical guidance drives directional migration of cancer cells. Acta Biomaterialia. 2020;106:181-92
124. Li P, Zhou H, Yan R, Yan W, Yang L, Li T. et al. Aligned fibrous scaffolds promote directional migration of breast cancer cells via caveolin-1/YAP-mediated mechanosensing. Materials Today Bio. 2024;28:101245
125. Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W. et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nature Communications. 2021;12:3519
126. Leng S, Zhang X, Wang S, Qin J, Liu Q, Liu A. et al. Ion channel Piezo1 activation promotes aerobic glycolysis in macrophages. Frontiers in Immunology. 2022;13:976482
127. Chen X, Jiang J, He B, Luo S, Tan Q, Yao Y. et al. Piezo1 aggravates ischemia/reperfusion-induced acute kidney injury by Ca2+-dependent calpain/HIF-1α/Notch signaling. Renal Failure. 2025;47:2447801
128. Lan Y, Lu J, Zhang S, Jie C, Chen C, Xiao C. et al. Piezo1-Mediated Mechanotransduction Contributes to Disturbed Flow-Induced Atherosclerotic Endothelial Inflammation. Journal of the American Heart Association. 2024;13:e035558
129. Luo S, Zhao X, Jiang J, Deng B, Liu S, Xu H. et al. Piezo1 specific deletion in macrophage protects the progression of liver fibrosis in mice. Theranostics. 2023;13:5418-34
130. Klapholz B, Brown NH. Talin - the master of integrin adhesions. Journal of Cell Science. 2017;130:2435-46
131. Zhu X, Bao Y, Guo Y, Yang W. Proline-Rich Protein Tyrosine Kinase 2 in Inflammation and Cancer. Cancers. 2018;10:139
132. Holland EN, Fernández-Yagüe MA, Zhou DW, O'Neill EB, Woodfolk AU, Mora-Boza A. et al. FAK, vinculin, and talin control mechanosensitive YAP nuclear localization. Biomaterials. 2024;308:122542
133. Sabra H, Brunner M, Mandati V, Wehrle-Haller B, Lallemand D, Ribba A-S. et al. β1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. The Journal of Biological Chemistry. 2017;292:19179-97
134. Xie W, Yu X, Yang Q, Ke N, Wang P, Kong H. et al. The Immunomechanical Checkpoint PYK2 Governs Monocyte-to-Macrophage Differentiation in Pancreatic Cancer. Cancer Discovery. 2025;15:1740-65
135. Meli VS, Atcha H, Veerasubramanian PK, Nagalla RR, Luu TU, Chen EY. et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Science Advances. 2020;6:eabb8471
136. Huang Y-J, Yang C-K, Wei P-L, Huynh T-T, Whang-Peng J, Meng T-C. et al. Ovatodiolide suppresses colon tumorigenesis and prevents polarization of M2 tumor-associated macrophages through YAP oncogenic pathways. Journal of Hematology & Oncology. 2017;10:60
137. Zhang Y, Fan Y, Jing X, Zhao L, Liu T, Wang L. et al. OTUD5-mediated deubiquitination of YAP in macrophage promotes M2 phenotype polarization and favors triple-negative breast cancer progression. Cancer Letters. 2021;504:104-15
138. Wagner DE, Alsafadi HN, Mitash N, Justet A, Hu Q, Pineda R. et al. Inhibition of epithelial cell YAP-TEAD/LOX signaling attenuates pulmonary fibrosis in preclinical models. Nature Communications. 2025;16:7099
139. Noguchi S, Saito A, Nagase T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. International Journal of Molecular Sciences. 2018;19:3674
140. Chu C-Q, Quan T. Fibroblast Yap/Taz Signaling in Extracellular Matrix Homeostasis and Tissue Fibrosis. Journal of Clinical Medicine. 2024;13:3358
141. Chen G, Xia B, Fu Q, Huang X, Wang F, Chen Z. et al. Matrix Mechanics as Regulatory Factors and Therapeutic Targets in Hepatic Fibrosis. International Journal of Biological Sciences. 2019;15:2509-21
142. Seavey CN, Pobbati AV, Hallett A, Ma S, Reynolds JP, Kanai R. et al. WWTR1(TAZ)-CAMTA1 gene fusion is sufficient to dysregulate YAP/TAZ signaling and drive epithelioid hemangioendothelioma tumorigenesis. Genes & Development. 2021;35:512-27
143. Takahashi H, Yang G, Yoneshiro T, Abe Y, Ito R, Yang C. et al. MYPT1-PP1β phosphatase negatively regulates both chromatin landscape and co-activator recruitment for beige adipogenesis. Nature Communications. 2022;13:5715
144. Bai J, Yan M, Xu Y, Wang Y, Yao Y, Jin P. et al. YAP enhances mitochondrial OXPHOS in tumor-infiltrating Treg through upregulating Lars2 on stiff matrix. Journal for Immunotherapy of Cancer. 2024;12:e010463
145. Shen L, Li H, Shi Y, Wang D, Gong J, Xun J. et al. M2 tumour-associated macrophages contribute to tumour progression via legumain remodelling the extracellular matrix in diffuse large B cell lymphoma. Scientific Reports. 2016;6:30347
146. Zhang Y, Manouchehri Doulabi E, Herre M, Cedervall J, Qiao Q, Miao Z. et al. Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment. Cancers. 2022;14:1947
147. Ren B, Van Kampen E, Van Berkel TJC, Cruickshank SM, Van Eck M. Hematopoietic arginase 1 deficiency results in decreased leukocytosis and increased foam cell formation but does not affect atherosclerosis. Atherosclerosis. 2017;256:35-46
148. Miska J, Rashidi A, Lee-Chang C, Gao P, Lopez-Rosas A, Zhang P. et al. Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Science Advances. 2021;7:eabc8929
149. Menjivar RE, Nwosu ZC, Du W, Donahue KL, Hong HS, Espinoza C. et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. eLife. 2023;12:e80721
150. Aaboe Jørgensen M, Ugel S, Linder Hübbe M, Carretta M, Perez-Penco M, Weis-Banke SE. et al. Arginase 1-Based Immune Modulatory Vaccines Induce Anticancer Immunity and Synergize with Anti-PD-1 Checkpoint Blockade. Cancer Immunology Research. 2021;9:1316-26
151. Bhatta A, Yao L, Xu Z, Toque HA, Chen J, Atawia RT. et al. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovascular Research. 2017;113:1664-76
152. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A. et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017;32:377-91.e9
153. Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61:942-53
154. Ramesh A, Malik V, Brouillard A, Kulkarni A. Supramolecular nanotherapeutics enable metabolic reprogramming of tumor-associated macrophages to inhibit tumor growth. Journal of Biomedical Materials Research Part A. 2022;110:1448-59
155. Sun R, Lei C, Xu Z, Gu X, Huang L, Chen L. et al. Neutral ceramidase regulates breast cancer progression by metabolic programming of TREM2-associated macrophages. Nature Communications. 2024;15:966
156. Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RY. et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. The Journal of Clinical Investigation. 2018;128:3794-805
157. Geeraerts X, Fernández-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y. et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Reports. 2021;37:110171
158. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M. et al. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Research. 2020;80:1438-50
159. Okikawa S, Morine Y, Saito Y, Yamada S, Tokuda K, Teraoku H. et al. Inhibition of the VEGF signaling pathway attenuates tumor-associated macrophage activity in liver cancer. Oncology Reports. 2022;47:71
160. Ludwig N, Yerneni SS, Azambuja JH, Pietrowska M, Widłak P, Hinck CS. et al. TGFβ+ small extracellular vesicles from head and neck squamous cell carcinoma cells reprogram macrophages towards a pro-angiogenic phenotype. Journal of Extracellular Vesicles. 2022;11:e12294
161. Kim MS, Kang H, Baek J-H, Cho M-G, Chung EJ, Kim S-J. et al. Disrupting Notch signaling related HES1 in myeloid cells reinvigorates antitumor T cell responses. Experimental Hematology & Oncology. 2024;13:122
162. Zhang X, Liu X, Zhou W, Du Q, Yang M, Ding Y. et al. Blockade of IDO-Kynurenine-AhR Axis Ameliorated Colitis-Associated Colon Cancer via Inhibiting Immune Tolerance. Cellular and Molecular Gastroenterology and Hepatology. 2021;12:1179-99
163. Wang Y-C, Wang X, Yu J, Ma F, Li Z, Zhou Y. et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nature Communications. 2021;12:3530
164. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T. et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8+ T Cell-Derived Interferon-γ. Immunity. 2019;51:381-97.e6
165. Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M. et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. The Journal of Experimental Medicine. 2016;213:2315-31
166. Callaway MK, Noonan BJ, Schwertfeger KL, Provenzano PP. Extracellular matrix architecture promotes immunosuppressive microenvironments in pancreatic cancer. Matrix Biology: Journal of the International Society for Matrix Biology. 2025;141:114-26
167. Kanchanawong P, Calderwood DA. Organization, dynamics and mechanoregulation of integrin-mediated cell-ECM adhesions. Nature Reviews Molecular Cell Biology. 2023;24:142-61
168. Cervero P, Wiesner C, Bouissou A, Poincloux R, Linder S. Lymphocyte-specific protein 1 regulates mechanosensory oscillation of podosomes and actin isoform-based actomyosin symmetry breaking. Nature Communications. 2018;9:515
169. Hemkemeyer SA, Vollmer V, Schwarz V, Lohmann B, Honnert U, Taha M. et al. Local Myo9b RhoGAP activity regulates cell motility. The Journal of Biological Chemistry. 2021;296:100136
170. Hsieh JY, Keating MT, Smith TD, Meli VS, Botvinick EL, Liu WF. Matrix crosslinking enhances macrophage adhesion, migration, and inflammatory activation. APL bioengineering. 2019;3:016103
171. Wang WY, Davidson CD, Lin D, Baker BM. Actomyosin contractility-dependent matrix stretch and recoil induces rapid cell migration. Nature Communications. 2019;10:1186
172. Ford AJ, Orbach SM, Rajagopalan P. Fibroblasts stimulate macrophage migration in interconnected extracellular matrices through tunnel formation and fiber alignment. Biomaterials. 2019;209:88-102
173. Liu J, Xiang J, Jin C, Ye L, Wang L, Gao Y. et al. Medicinal plant-derived mtDNA via nanovesicles induces the cGAS-STING pathway to remold tumor-associated macrophages for tumor regression. Journal of Nanobiotechnology. 2023;21:78
174. Wang M, Huang Y, Chen M, Wang W, Wu F, Zhong T. et al. Inhibition of tumor intrinsic BANF1 activates antitumor immune responses via cGAS-STING and enhances the efficacy of PD-1 blockade. Journal for Immunotherapy of Cancer. 2023;11:e007035
175. Ellert-Miklaszewska A, Pilanc P, Poleszak K, Roura A-J, Cyranowski S, Ghosh M. et al. 7aaRGD - a novel SPP1/integrin signaling-blocking peptide reverses immunosuppression and improves anti-PD-1 immunotherapy outcomes in experimental gliomas. Journal of experimental & clinical cancer research: CR. 2025;44:132
176. Grimm TM, Dierdorf NI, Herbinger M, Baumgärtner S, Sontowski E, Paone C. et al. The phosphatase PPM1F, a negative regulator of integrin activity, is essential for embryonic development and controls tumor cell invasion. BMC biology. 2025;23:166
177. Zhao X-K, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H. et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Scientific Reports. 2016;6:19276
178. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D. et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Research. 2016;76:5671-82
179. Miao L, Xin X, Xin H, Shen X, Zhu YZ. Hydrogen Sulfide Recruits Macrophage Migration by Integrin beta1-Src-FAK/Pyk2-Rac Pathway in Myocardial Infarction. Sci Rep. 2016;6:22363
180. Huang Y, Zhao H, Zhang Y, Tang Y, Shi X, Jiang S. et al. Enhancement of Zyxin Promotes Skin Fibrosis by Regulating FAK/PI3K/AKT and TGF-beta Signaling Pathways via Integrins. Int J Biol Sci. 2023;19:2394-408
181. Zhou R, Wang M, Li X, Liu Y, Yao Y, Wang A. et al. TBK1-Zyxin signaling controls tumor-associated macrophage recruitment to mitigate antitumor immunity. The EMBO journal. 2024;43:4984-5017
182. Chen XW, Yu TJ, Zhang J, Li Y, Chen HL, Yang GF. et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene. 2017;36:5045-57
183. Ma L, Jin Y, Xu J, Zhang J. Tumor-Associated Macrophage in Breast Tumor Microenvironment. International Journal of Molecular Sciences. 2025;26:5973
184. Bied M, Ho WW, Ginhoux F, Blériot C. Roles of macrophages in tumor development: a spatiotemporal perspective. Cellular & Molecular Immunology. 2023;20:983-92
185. Vinnakota K, Zhang Y, Selvanesan BC, Topi G, Salim T, Sand-Dejmek J. et al. M2-like macrophages induce colon cancer cell invasion via matrix metalloproteinases. Journal of Cellular Physiology. 2017;232:3468-80
186. Guiet R, Van Goethem E, Cougoule C, Balor S, Valette A, Al Saati T. et al. The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumor cells. Journal of Immunology (Baltimore, Md: 1950). 2011;187:3806-14
187. Zhang L, Abdi S, Szafraniec HM, Provenzano PP, Schwertfeger KL, Wood DK. Collagen-Based Tumor Spheroid Model for Investigating Tumor-Macrophage Interactions through Extracellular Matrix Remodeling. bioRxiv: The Preprint Server for Biology. 2025. 2025 10.01.679587
188. Hingorani DV, Lippert CN, Crisp JL, Savariar EN, Hasselmann JPC, Kuo C. et al. Impact of MMP-2 and MMP-9 enzyme activity on wound healing, tumor growth and RACPP cleavage. PloS One. 2018;13:e0198464
189. Fei L, Ren X, Yu H, Zhan Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Frontiers in Immunology. 2021;12:771210
190. Wang J, Saung MT, Li K, Fu J, Fujiwara K, Niu N. et al. CCR2/CCR5 inhibitor permits the radiation-induced effector T cell infiltration in pancreatic adenocarcinoma. J Exp Med. 2022 219
191. Xie M, Lin Z, Ji X, Luo X, Zhang Z, Sun M. et al. FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. Journal of Hepatology. 2023;79:109-25
192. Grossman JG, Nywening TM, Belt BA, Panni RZ, Krasnick BA, DeNardo DG. et al. Recruitment of CCR2+ tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology. 2018;7:e1470729
193. Regan DP, Coy JW, Chahal KK, Chow L, Kurihara JN, Guth AM. et al. The Angiotensin Receptor Blocker Losartan Suppresses Growth of Pulmonary Metastases via AT1R-Independent Inhibition of CCR2 Signaling and Monocyte Recruitment. Journal of Immunology (Baltimore, Md: 1950). 2019;202:3087-102
194. Domingos-Pereira S, Sathiyanadan K, Polak L, Haefliger J-A, Schmittnaegel M, Ries CH. et al. Tumor-Microenvironment Characterization of the MB49 Non-Muscle-Invasive Bladder-Cancer Orthotopic Model towards New Therapeutic Strategies. International Journal of Molecular Sciences. 2022;24:123
195. Chen J, Lin Q, Lan R, Wu J, Wang Z, Chen R. et al. A CCR5 antagonist enhances the radiosensitivity of hepatocarcinoma in a mouse model. Journal of Radiation Research. 2025;66:396-407
196. Tomassetti C, Insinga G, Gimigliano F, Morrione A, Giordano A, Giurisato E. Insights into CSF-1R Expression in the Tumor Microenvironment. Biomedicines. 2024;12:2381
197. Fujiwara T, Yakoub MA, Chandler A, Christ AB, Yang G, Ouerfelli O. et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Molecular Cancer Therapeutics. 2021;20:1388-99
198. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N. et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology. 2013;2:e26968
199. Zhang H, Almuqbil RM, Alhudaithi SS, Sunbul FS, da Rocha SRP. Pulmonary administration of a CSF-1R inhibitor alters the balance of tumor-associated macrophages and supports first-line chemotherapy in a lung cancer model. International Journal of Pharmaceutics. 2021;598:120350
200. Chen K, Li X, Dong S, Guo Y, Luo Z, Zhuang S-M. et al. Modulating tumor-associated macrophages through CSF1R inhibition: a potential therapeutic strategy for HNSCC. Journal of Translational Medicine. 2025;23:27
201. Zhang J, Huang X, Li M, Zhang W, Yang H. CSF1R inhibition agents protect against cisplatin ototoxicity and synergize with immunotherapy for Head and Neck Squamous Cell Carcinoma. International Immunopharmacology. 2025;152:114428
202. Tan I-L, Arifa RDN, Rallapalli H, Kana V, Lao Z, Sanghrajka RM. et al. CSF1R inhibition depletes tumor-associated macrophages and attenuates tumor progression in a mouse sonic Hedgehog-Medulloblastoma model. Oncogene. 2021;40:396-407
203. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nature Reviews Clinical Oncology. 2017;14:399-416
204. Saeed AF. Tumor-Associated Macrophages: Polarization, Immunoregulation, and Immunotherapy. Cells. 2025;14:741
205. Sato T, Sugiyama D, Koseki J, Kojima Y, Hattori S, Sone K. et al. Sustained inhibition of CSF1R signaling augments antitumor immunity through inhibiting tumor-associated macrophages. JCI insight. 2025;10:e178146
206. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Research. 2014;74:5057-69
207. Pfirschke C, Zilionis R, Engblom C, Messemaker M, Zou AE, Rickelt S. et al. Macrophage-Targeted Therapy Unlocks Antitumoral Cross-talk between IFNγ-Secreting Lymphocytes and IL12-Producing Dendritic Cells. Cancer Immunology Research. 2022;10:40-55
208. Zhao L, Wang Z, Tan Y, Ma J, Huang W, Zhang X. et al. IL-17A/CEBPβ/OPN/LYVE-1 axis inhibits anti-tumor immunity by promoting tumor-associated tissue-resident macrophages. Cell Reports. 2024;43:115039
209. Djureinovic D, Wang M, Kluger HM. Agonistic CD40 Antibodies in Cancer Treatment. Cancers. 2021;13:1302
210. Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annual Review of Medicine. 2020;71:47-58
211. Lim CY, Chang JH, Lee WS, Kim J, Park IY. CD40 Agonists Alter the Pancreatic Cancer Microenvironment by Shifting the Macrophage Phenotype toward M1 and Suppress Human Pancreatic Cancer in Organotypic Slice Cultures. Gut and Liver. 2022;16:645-59
212. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W. et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (New York, NY). 2011;331:1612-6
213. Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA. et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2021;27:4574-86
214. O'Hara MH, O'Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH. et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. The Lancet Oncology. 2021;22:118-31
215. Figueiredo P, Lepland A, Scodeller P, Fontana F, Torrieri G, Tiboni M. et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomaterialia. 2021;133:231-43
216. Jardim C, Bica M, Reis-Sobreiro M, Teixeira da Mota A, Lopes R, Pinto MF. et al. Sustained Macrophage Reprogramming Is Required for CD8+ T cell-Dependent Long-Term Tumor Eradication. Cancer Immunology Research. 2025;13:1207-25
217. Sarkar B, Arlauckas SP, Cuccarese MF, Garris CS, Weissleder R, Rodell CB. Host-functionalization of macrin nanoparticles to enable drug loading and control tumor-associated macrophage phenotype. Frontiers in Immunology. 2024;15:1331480
218. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nature Biotechnology. 2020;38:947-53
219. Pierini S, Gabbasov R, Gabitova L, Ohtani Y, Shestova O, Gill S. et al. Abstract 63: Chimeric antigen receptor macrophages (CAR-M) induce anti-tumor immunity and synergize with T cell checkpoint inhibitors in pre-clinical solid tumor models. Cancer Research. 2021;81:63
220. Jiang M, Bao W, He L, Liang Y, Zhou N, Zhang C. et al. Ginsenoside F3 alleviates T cell exhaustion via RIPOR2-mediated immunometabolic reprogramming to potentiate anti-PD-1 therapy. Phytomedicine. 2026;150:157649
221. Pierini S, Gabbasov R, Oliveira-Nunes MC, Qureshi R, Worth A, Huang S. et al. Chimeric antigen receptor macrophages (CAR-M) sensitize HER2+ solid tumors to PD1 blockade in pre-clinical models. Nature Communications. 2025;16:706
222. Gu K, Liang T, Hu L, Zhao Y, Ying W, Zhang M. et al. Intraperitoneal programming of tailored CAR macrophages via mRNA lipid nanoparticle to boost cancer immunotherapy. Nature Communications. 2025
223. Luo Q, Zheng N, Jiang L, Wang T, Zhang P, Liu Y. et al. Lipid accumulation in macrophages confers protumorigenic polarization and immunity in gastric cancer. Cancer Science. 2020;111:4000-11
224. Jiang C, Liu R, Li Z. A Phosphatidylinositol 3-Kinase Gamma Inhibitor Enhances Anti-Programmed Death-1/Programmed Death Ligand-1 Antitumor Effects by Remodeling the Tumor Immune Microenvironment of Ovarian Cancer. MedComm. 2025;6:e70223
225. Xu H, Russell SN, Steiner K, O'Neill E, Jones KI. Targeting PI3K-gamma in myeloid driven tumour immune suppression: a systematic review and meta-analysis of the preclinical literature. Cancer immunology, immunotherapy: CII. 2024;73:204
226. Han Y, Sun J, Yang Y, Liu Y, Lou J, Pan H. et al. TMP195 Exerts Antitumor Effects on Colorectal Cancer by Promoting M1 Macrophages Polarization. International Journal of Biological Sciences. 2022;18:5653-66
227. Noonepalle SKR, Gracia-Hernandez M, Aghdam N, Berrigan M, Coulibaly H, Li X. et al. Cell therapy using ex vivo reprogrammed macrophages enhances antitumor immune responses in melanoma. Journal of experimental & clinical cancer research: CR. 2024;43:263
228. Vallet SD, Ricard-Blum S. Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays in Biochemistry. 2019;63:349-64
229. Ye M, Song Y, Pan S, Chu M, Wang Z-W, Zhu X. Evolving roles of lysyl oxidase family in tumorigenesis and cancer therapy. Pharmacology & Therapeutics. 2020;215:107633
230. Ikenaga N, Peng Z-W, Vaid KA, Liu SB, Yoshida S, Sverdlov DY. et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66:1697-708
231. Chen W, Yang A, Jia J, Popov YV, Schuppan D, You H. Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology (Baltimore, Md). 2020;72:729-41
232. Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE. et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nature Communications. 2020;11:2416
233. Gong L, Zhang Y, Yang Y, Yan Q, Ren J, Luo J. et al. Inhibition of lysyl oxidase-like 2 overcomes adhesion-dependent drug resistance in the collagen-enriched liver cancer microenvironment. Hepatology Communications. 2022;6:3194-211
234. Ferreira S, Saraiva N, Rijo P, Fernandes AS. LOXL2 Inhibitors and Breast Cancer Progression. Antioxidants (Basel, Switzerland). 2021;10:312
235. Wang W, Wang X, Yao F, Huang C. Lysyl Oxidase Family Proteins: Prospective Therapeutic Targets in Cancer. International Journal of Molecular Sciences. 2022;23:12270
236. Liburkin-Dan T, Toledano S, Neufeld G. Lysyl Oxidase Family Enzymes and Their Role in Tumor Progression. International Journal of Molecular Sciences. 2022;23:6249
237. Ma HY, Li Q, Wong WR, N'Diaye EN, Caplazi P, Bender H. et al. LOXL4, but not LOXL2, is the critical determinant of pathological collagen cross-linking and fibrosis in the lung. Sci Adv. 2023;9:eadf0133
238. Tjin G, White ES, Faiz A, Sicard D, Tschumperlin DJ, Mahar A. et al. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis Model Mech. 2017;10:1301-12
239. Prakash J, Shaked Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024;14:1375-88
240. Ferreira S, Saraiva N, Rijo P, Fernandes AS. LOXL2 Inhibitors and Breast Cancer Progression. Antioxidants (Basel). 2021 10
241. Smithen DA, Leung LMH, Challinor M, Lawrence R, Tang H, Niculescu-Duvaz D. et al. 2-Aminomethylene-5-sulfonylthiazole Inhibitors of Lysyl Oxidase (LOX) and LOXL2 Show Significant Efficacy in Delaying Tumor Growth. Journal of Medicinal Chemistry. 2020;63:2308-24
242. Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, Ramirez-Acuña JM, Perez-Romero BA, Guerrero-Rodriguez JF. et al. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. International Journal of Molecular Sciences. 2020;21:9739
243. Fields G B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells. 2019 8
244. Almutairi S, Kalloush HMd, Manoon NA, Bardaweel SK. Matrix Metalloproteinases Inhibitors in Cancer Treatment: An Updated Review (2013-2023). Molecules (Basel, Switzerland). 2023;28:5567
245. Avalle L, Raggi L, Monteleone E, Savino A, Viavattene D, Statello L. et al. STAT3 induces breast cancer growth via ANGPTL4, MMP13 and STC1 secretion by cancer associated fibroblasts. Oncogene. 2022;41:1456-67
246. Kwon MJ. Matrix metalloproteinases as therapeutic targets in breast cancer. Frontiers in Oncology. 2022;12:1108695
247. Winer A, Adams S, Mignatti P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Molecular Cancer Therapeutics. 2018;17:1147-55
248. Radisky ES, Raeeszadeh-Sarmazdeh M, Radisky DC. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. Journal of Cellular Biochemistry. 2017;118:3531-48
249. Wang B, Li Q, Wang J, Zhao S, Nashun B, Qin L. et al. Plasmodium infection inhibits tumor angiogenesis through effects on tumor-associated macrophages in a murine implanted hepatoma model. Cell communication and signaling: CCS. 2020;18:157
250. Lin X, Sun Y, Yang S, Yu M, Pan L, Yang J. et al. Omentin-1 Modulates Macrophage Function via Integrin Receptors αvβ3 and αvβ5 and Reverses Plaque Vulnerability in Animal Models of Atherosclerosis. Frontiers in Cardiovascular Medicine. 2021;8:757926
251. Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. The American Journal of Pathology. 2000;156:1345-62
252. Gvozdenovic A, Boro A, Meier D, Bode-Lesniewska B, Born W, Muff R. et al. Targeting αvβ3 and αvβ5 integrins inhibits pulmonary metastasis in an intratibial xenograft osteosarcoma mouse model. Oncotarget. 2016;7:55141-54
253. Weller M, Nabors LB, Gorlia T, Leske H, Rushing E, Bady P. et al. Cilengitide in newly diagnosed glioblastoma: biomarker expression and outcome. Oncotarget. 2016;7:15018-32
254. Batchelor TT, Reardon DA, de Groot JF, Wick W, Weller M. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2014;20:5612-9
255. Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH. et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nature Immunology. 2015;16:505-16
256. Xu Z, Cai J, Gao J, White GC, Chen F, Ma Y-Q. Interaction of kindlin-3 and β2-integrins differentially regulates neutrophil recruitment and NET release in mice. Blood. 2015;126:373-7
257. Cheng B, Wan W, Huang G, Li Y, Genin GM, Mofrad MRK. et al. Nanoscale integrin cluster dynamics controls cellular mechanosensing via FAKY397 phosphorylation. Science Advances. 2020;6:eaax1909
258. Gregor M, Osmanagic-Myers S, Burgstaller G, Wolfram M, Fischer I, Walko G. et al. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:715-29
259. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA. et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature Medicine. 2016;22:851-60
260. Liu X, Baer JM, Stone ML, Knolhoff BL, Hogg GD, Turner MC. et al. Stromal reprogramming overcomes resistance to RAS-MAPK inhibition to improve pancreas cancer responses to cytotoxic and immune therapy. Science Translational Medicine. 2024;16:eado2402
261. Jiang H, Liu X, Knolhoff BL, Hegde S, Lee KB, Jiang H. et al. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut. 2020;69:122-32
262. Wei Y, Wang Y, Liu N, Qi R, Xu Y, Li K. et al. A FAK Inhibitor Boosts Anti-PD1 Immunotherapy in a Hepatocellular Carcinoma Mouse Model. Frontiers in Pharmacology. 2021;12:820446
263. Wang-Gillam A, Lim K-H, McWilliams R, Suresh R, Lockhart AC, Brown A. et al. Defactinib, Pembrolizumab, and Gemcitabine in Patients with Advanced Treatment Refractory Pancreatic Cancer: A Phase I Dose Escalation and Expansion Study. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2022;28:5254-62
264. Davelaar J, Gai J, Brown Z, Levi A, Linden S, Minasyan A. et al. Preliminary translational immune and stromal correlates in a randomized phase II trial of pembrolizumab with or without defactinib for resectable pancreatic ductal adenocarcinoma (PDAC). Journal of Clinical Oncology. 2023
265. de Jonge MJA, Steeghs N, Lolkema MP, Hotte SJ, Hirte HW, van der Biessen DAJ. et al. Phase I Study of BI 853520, an Inhibitor of Focal Adhesion Kinase, in Patients with Advanced or Metastatic Nonhematologic Malignancies. Targeted Oncology. 2019;14:43-55
266. Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skládal P. et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nature Communications. 2017;8:15321
267. Plouffe SW, Lin KC, Moore JL, Tan FE, Ma S, Ye Z. et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. The Journal of Biological Chemistry. 2018;293:11230-40
268. Chakraborty S, Njah K, Pobbati AV, Lim YB, Raju A, Lakshmanan M. et al. Agrin as a Mechanotransduction Signal Regulating YAP through the Hippo Pathway. Cell Reports. 2017;18:2464-79
269. Wei Y, Hui VLZ, Chen Y, Han R, Han X, Guo Y. YAP/TAZ: Molecular pathway and disease therapy. MedComm. 2023;4:e340
270. Gibault F, Bailly F, Corvaisier M, Coevoet M, Huet G, Melnyk P. et al. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem. 2017;12:954-61
271. Wei C, Li X. Verteporfin inhibits cell proliferation and induces apoptosis in different subtypes of breast cancer cell lines without light activation. BMC cancer. 2020;20:1042
272. Golino JL, Wang X, Bian J, Ruf B, Kelly M, Karim BO. et al. Anti-Cancer Activity of Verteporfin in Cholangiocarcinoma. Cancers. 2023;15:2454
273. Talukdar PD, Roy H, Chatterji U. Targeting breast cancer stem cells in ER-positive breast cancer by repurposing the benzoporphyrin derivative verteporfin as a YAP/TAZ small molecule inhibitor. Molecular Biology Reports. 2025;52:154
274. Wu Y, Jiang Y, Jiang L, Peng Y, Zhou T, Xia X. et al. Phospho-cofilin predicts efficiency of Fasudil for oral squamous cell carcinoma treatment through Yes-associated protein inhibition. Archives of Oral Biology. 2025;172:106185
275. Qu P, Zhang H. The dual role of Piezo1 in tumor cells and immune cells: a new target for cancer therapy. Frontiers in Immunology. 2025;16:1635388
276. Hamza A, Amit J, Elizabeth L E, Medha M P, Michael D C, Wendy F L. Ion channel mediated mechanotransduction in immune cells. Current Opinion in Solid State & Materials Science. 2021;25:100951
277. Mo J, Zhang Y, Cao C, Zheng W, Zheng L, Wang X. et al. Activation of mechanosensitive ion channel Piezo1 linking metabolic reprogramming and pro-inflammatory responses in hepatocellular carcinoma. Cell communication and signaling: CCS. 2025;23:280
278. Liu H, Hu J, Zheng Q, Feng X, Zhan F, Wang X. et al. Piezo1 Channels as Force Sensors in Mechanical Force-Related Chronic Inflammation. Frontiers in Immunology. 2022;13:816149
279. Elahi-Gedwillo KY, Carlson M, Zettervall J, Provenzano PP. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Research. 2019;79:372-86
280. Andreuzzi E, Fejza A, Polano M, Poletto E, Camicia L, Carobolante G. et al. Colorectal cancer development is affected by the ECM molecule EMILIN-2 hinging on macrophage polarization via the TLR-4/MyD88 pathway. Journal of experimental & clinical cancer research: CR. 2022;41:60
281. Zhang W, Rahman S, Wu AML, Isanogle K, Robinson C, Kumar D. et al. A CSF-1R inhibitor both prevents and treats triple-negative breast cancer brain metastases in hematogenous preclinical models. Clinical & Experimental Metastasis. 2025;42:45
282. Deng X, Yang Q, Wang Y, Zhou C, Guo Y, Hu Z. et al. CSF-1R inhibition attenuates ischemia-induced renal injury and fibrosis by reducing Ly6C+ M2-like macrophage infiltration. International Immunopharmacology. 2020;88:106854
283. Ryder M, Gild M, Hohl TM, Pamer E, Knauf J, Ghossein R. et al. Genetic and pharmacological targeting of CSF-1/CSF-1R inhibits tumor-associated macrophages and impairs BRAF-induced thyroid cancer progression. PloS One. 2013;8:e54302
284. Ruffolo LI, Jackson KM, Kuhlers PC, Dale BS, Figueroa Guilliani NM, Ullman NA. et al. GM-CSF drives myelopoiesis, recruitment and polarisation of tumour-associated macrophages in cholangiocarcinoma and systemic blockade facilitates antitumour immunity. Gut. 2022;71:1386-98
285. Byrne KT, Vonderheide RH. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Reports. 2016;15:2719-32
286. Stromnes IM, Burrack AL, Hulbert A, Bonson P, Black C, Brockenbrough JS. et al. Differential Effects of Depleting versus Programming Tumor-Associated Macrophages on Engineered T Cells in Pancreatic Ductal Adenocarcinoma. Cancer Immunology Research. 2019;7:977-89
287. Ma HS, Poudel B, Torres ER, Sidhom J-W, Robinson TM, Christmas B. et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunology Research. 2019;7:428-42
288. Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q. et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. Journal of Hepatology. 2021;74:1145-54
289. Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS. et al. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunology Research. 2017;5:1109-21
290. Hogg GD, Weinstein AG, Kingston NL, Liu X, Dres OM, Kang L-I. et al. Combined Flt3L and CD40 agonism restores dendritic cell-driven T cell immunity in pancreatic cancer. Science Immunology. 2025;10:eadp3978
291. Anfray C, Mainini F, Digifico E, Maeda A, Sironi M, Erreni M. et al. Intratumoral combination therapy with poly(I:C) and resiquimod synergistically triggers tumor-associated macrophages for effective systemic antitumoral immunity. Journal for Immunotherapy of Cancer. 2021;9:e002408
292. Chen S, Li L, Yuan H, Gui H, Wan Q, Wang M. et al. Intratumoral injection of R848 and poly(I:C) synergistically promoted antitumor immune responses by reprogramming macrophage polarization and activating DCs in lung cancer. Clinical and Experimental Immunology. 2025;219:uxae110
293. Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM. et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI insight. 2017;2:e93397
294. Richert I, Berchard P, Abbes L, Novikov A, Chettab K, Vandermoeten A. et al. A TLR4 Agonist Induces Osteosarcoma Regression by Inducing an Antitumor Immune Response and Reprogramming M2 Macrophages to M1 Macrophages. Cancers. 2023;15:4635
295. Jones DC, Irving L, Dudley R, Blümli S, Wolny M, Chatzopoulou EI. et al. LILRB2 blockade facilitates macrophage repolarization and enhances T cell-mediated antitumor immunity. Journal for Immunotherapy of Cancer. 2025;13:e010012
296. Klepfish M, Gross T, Vugman M, Afratis NA, Havusha-Laufer S, Brazowski E. et al. LOXL2 Inhibition Paves the Way for Macrophage-Mediated Collagen Degradation in Liver Fibrosis. Frontiers in Immunology. 2020;11:480
297. Wan Z, Zhu Z, Wang P, Xu X, Ma T, Li H. et al. Targeting Focal Adhesion Kinase in Lung Diseases: Current Progress and Future Directions. Biomolecules. 2025;15:1233
298. Rahhal R, Schmidt MO, Moussa M, Asemi T, Battina R, Kiliti A. et al. TEAD inhibition alters the lung immune microenvironment and attenuates metastasis. bioRxiv: The Preprint Server for Biology. 2025. 2025 02.26.639326
299. Han I-H, Choi I, Choi H, Kim S, Jeong C, Yang J. et al. Conformation-sensitive targeting of CD18 depletes M2-like tumor-associated macrophages resulting in inhibition of solid tumor progression. Journal for Immunotherapy of Cancer. 2025;13:e011422
300. Li L, Wu J, Wu X, Li Z, Zhang X, Yan Z. et al. Carbon Dot-Linked Hydrogel for TAMs Transform: Spatiotemporal Manipulation to Reshape Tumor Microenvironment. Advanced Materials (Deerfield Beach, Fla). 2025;37:e2420068
301. Liu X, Huangfu Y, Wang J, Kong P, Tian W, Liu P. et al. Supramolecular Polymer-Nanomedicine Hydrogel Loaded with Tumor Associated Macrophage-Reprogramming polyTLR7/8a Nanoregulator for Enhanced Anti-Angiogenesis Therapy of Orthotopic Hepatocellular Carcinoma. Advanced Science (Weinheim, Baden-Wurttemberg, Germany). 2023;10:e2300637
302. Zhang Y, Feng Z, Liu J, Li H, Su Q, Zhang J. et al. Polarization of tumor-associated macrophages by TLR7/8 conjugated radiosensitive peptide hydrogel for overcoming tumor radioresistance. Bioactive Materials. 2022;16:359-71
303. Li C, Li C, Ma Z, Chen H, Ruan H, Deng L. et al. Regulated macrophage immune microenvironment in 3D printed scaffolds for bone tumor postoperative treatment. Bioactive Materials. 2023;19:474-85
304. Dai X, Meng J, Deng S, Zhang L, Wan C, Lu L. et al. Targeting CAMKII to reprogram tumor-associated macrophages and inhibit tumor cells for cancer immunotherapy with an injectable hybrid peptide hydrogel. Theranostics. 2020;10:3049-63
305. Scodeller P, Simón-Gracia L, Kopanchuk S, Tobi A, Kilk K, Säälik P. et al. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Scientific Reports. 2017;7:14655
306. Ding J, Sui D, Liu M, Su Y, Wang Y, Liu M. et al. Sialic acid conjugate-modified liposomes enable tumor homing of epirubicin via neutrophil/monocyte infiltration for tumor therapy. Acta Biomaterialia. 2021;134:702-15
307. Li Y, Wu H, Ji B, Qian W, Xia S, Wang L. et al. Targeted Imaging of CD206 Expressing Tumor-Associated M2-like Macrophages Using Mannose-Conjugated Antibiofouling Magnetic Iron Oxide Nanoparticles. ACS applied bio materials. 2020;3:4335-47
308. Tang X, Sui D, Liu M, Zhang H, Liu M, Wang S. et al. Targeted delivery of zoledronic acid through the sialic acid - Siglec axis for killing and reversal of M2 phenotypic tumor-associated macrophages - A promising cancer immunotherapy. International Journal of Pharmaceutics. 2020;590:119929
309. Shan H, Dou W, Zhang Y, Qi M. Targeted ferritin nanoparticle encapsulating CpG oligodeoxynucleotides induces tumor-associated macrophage M2 phenotype polarization into M1 phenotype and inhibits tumor growth. Nanoscale. 2020;12:22268-80
310. Zhang X, Wei Z, Yong T, Li S, Bie N, Li J. et al. Cell microparticles loaded with tumor antigen and resiquimod reprogram tumor-associated macrophages and promote stem-like CD8+ T cells to boost anti-PD-1 therapy. Nature Communications. 2023;14:5653
311. Xiao H, Guo Y, Li B, Li X, Wang Y, Han S. et al. M2-Like Tumor-Associated Macrophage-Targeted Codelivery of STAT6 Inhibitor and IKKβ siRNA Induces M2-to-M1 Repolarization for Cancer Immunotherapy with Low Immune Side Effects. ACS central science. 2020;6:1208-22
312. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME. et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nature Communications. 2019;10:3974
313. Li Z, Ning F, Wang C, Yu H, Ma Q, Sun Y. Normalization of the tumor microvasculature based on targeting and modulation of the tumor microenvironment. Nanoscale. 2021;13:17254-71
314. Russell SN, Demetriou C, Valenzano G, Evans A, Go S, Stanly T. et al. Induction of macrophage efferocytosis in pancreatic cancer via PI3Kγ inhibition and radiotherapy promotes tumour control. Gut. 2025;74:825-39
315. Han MG, Jang B-S, Kang MH, Na D, Kim IA. PI3Kγδ inhibitor plus radiation enhances the antitumour immune effect of PD-1 blockade in syngenic murine breast cancer and humanised patient-derived xenograft model. European Journal of Cancer (Oxford, England: 1990). 2021;157:450-63
316. He S, Song W, Cui S, Li J, Jiang Y, Chen X. et al. Modulation of miR-146b by N6-methyladenosine modification remodels tumor-associated macrophages and enhances anti-PD-1 therapy in colorectal cancer. Cellular Oncology (Dordrecht, Netherlands). 2023;46:1731-46
317. Liu Z, Xie Y, Xiong Y, Liu S, Qiu C, Zhu Z. et al. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Letters. 2020;469:173-85
318. Li T, Xu D, Ruan Z, Zhou J, Sun W, Rao B. et al. Metabolism/Immunity Dual-Regulation Thermogels Potentiating Immunotherapy of Glioblastoma Through Lactate-Excretion Inhibition and PD-1/PD-L1 Blockade. Advanced Science (Weinheim, Baden-Wurttemberg, Germany). 2024;11:e2310163
319. Zhou M, Wu J, Shao Y, Zhang J, Zheng R, Shi Q. et al. Short-chain fatty acids reverses gut microbiota dysbiosis-promoted progression of glioblastoma by up-regulating M1 polarization in the tumor microenvironment. International Immunopharmacology. 2024;141:112881
320. Zhang S, Xie F, Li K, Zhang H, Yin Y, Yu Y. et al. Gold nanoparticle-directed autophagy intervention for antitumor immunotherapy via inhibiting tumor-associated macrophage M2 polarization. Acta Pharmaceutica Sinica B. 2022;12:3124-38
321. Parayath NN, Parikh A, Amiji MM. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Letters. 2018;18:3571-9
322. Song J, Cheng M, Xie Y, Li K, Zang X. Efficient tumor synergistic chemoimmunotherapy by self-augmented ROS-responsive immunomodulatory polymeric nanodrug. Journal of Nanobiotechnology. 2023;21:93
323. Zhang D, Song J, Jing Z, Qin H, Wu Y, Zhou J. et al. Stimulus Responsive Nanocarrier for Enhanced Antitumor Responses Against Hepatocellular Carcinoma. International Journal of Nanomedicine. 2024;19:13339-55
324. Zang X, Song J, Yi X, Piyu J. Polymeric indoximod based prodrug nanoparticles with doxorubicin entrapment for inducing immunogenic cell death and improving the immunotherapy of breast cancer. Journal of Materials Chemistry B. 2022;10:2019-27
325. Liaw K, Reddy R, Sharma A, Li J, Chang M, Sharma R. et al. Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma. Bioengineering & Translational Medicine. 2021;6:e10205
326. Alhudaithi SS, Almuqbil RM, Zhang H, Bielski ER, Du W, Sunbul FS. et al. Local Targeting of Lung-Tumor-Associated Macrophages with Pulmonary Delivery of a CSF-1R Inhibitor for the Treatment of Breast Cancer Lung Metastases. Molecular Pharmaceutics. 2020;17:4691-703
327. Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L. et al. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS nano. 2017;11:9536-49
328. Anfray C, Varela CF, Ummarino A, Maeda A, Sironi M, Gandoy S. et al. Polymeric nanocapsules loaded with poly(I:C) and resiquimod to reprogram tumor-associated macrophages for the treatment of solid tumors. Frontiers in Immunology. 2023;14:1334800
329. Zhan C, Jin Y, Xu X, Shao J, Jin C. Antitumor therapy for breast cancer: Focus on tumor-associated macrophages and nanosized drug delivery systems. Cancer Medicine. 2023;12:11049-72
330. Zhang X, Yue L, Cao L, Liu K, Yang S, Liang S. et al. Tumor microenvironment-responsive macrophage-mediated immunotherapeutic drug delivery. Acta Biomaterialia. 2024;186:369-82
331. Niu M, Naguib YW, Aldayel AM, Shi Y-c, Hursting SD, Hersh MA. et al. Biodistribution and in vivo activities of tumor-associated macrophage-targeting nanoparticles incorporated with doxorubicin. Molecular Pharmaceutics. 2014;11:4425-36
332. Chen S, He Y, Huang X, Shen Y, Zou Q, Yang G. et al. Photosensitive and dual-targeted chromium nanoparticle delivering small interfering RNA YTHDF1 for molecular-targeted immunotherapy in liver cancer. Journal of Nanobiotechnology. 2024;22:348
333. Luo F, Li H, Ma W, Cao J, Chen Q, Lu F. et al. The BCL-2 inhibitor APG-2575 resets tumor-associated macrophages toward the M1 phenotype, promoting a favorable response to anti-PD-1 therapy via NLRP3 activation. Cellular & Molecular Immunology. 2024;21:60-79
334. O'Connell BC, Hubbard C, Zizlsperger N, Fitzgerald D, Kutok JL, Varner J. et al. Eganelisib combined with immune checkpoint inhibitor therapy and chemotherapy in frontline metastatic triple-negative breast cancer triggers macrophage reprogramming, immune activation and extracellular matrix reorganization in the tumor microenvironment. Journal for Immunotherapy of Cancer. 2024;12:e009160
335. Takahashi H, Perez-Villarroel P, Falahat R, Mulé JJ. Targeting MARCO in combination with anti-CTLA-4 leads to enhanced melanoma regression and immune cell infiltration via macrophage reprogramming. Journal for Immunotherapy of Cancer. 2025;13:e011030
336. Kabir AU, Subramanian M, Lee DH, Wang X, Krchma K, Wu J. et al. Dual role of endothelial Myct1 in tumor angiogenesis and tumor immunity. Science Translational Medicine. 2021;13:eabb6731
337. Oršolić N, Kunštić M, Jazvinšćak Jembrek M. Antitumor and Antiangiogenic Effect of Tannic Acid in the Advanced Stage of Ehrlich Ascites Tumor in Mice. International Journal of Molecular Sciences. 2025;26:9070
338. Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA. et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2009;27:2542-52
339. Kato Y, Tabata K, Kimura T, Yachie-Kinoshita A, Ozawa Y, Yamada K. et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PloS One. 2019;14:e0212513
340. Bahri M, Al-Adhami T, Demirel E, Sarkar J, Feehan KT, Anstee JE. et al. An oral heme oxygenase inhibitor targets immunosuppressive perivascular macrophages in preclinical models of cancer. Science Translational Medicine. 2025;17:eads3085
341. Nikolos F, Hayashi K, Hoi XP, Alonzo ME, Mo Q, Kasabyan A. et al. Cell death-induced immunogenicity enhances chemoimmunotherapeutic response by converting immune-excluded into T-cell inflamed bladder tumors. Nature Communications. 2022;13:1487
342. Haj-Shomaly J, Vorontsova A, Barenholz-Cohen T, Levi-Galibov O, Devarasetty M, Timaner M. et al. T Cells Promote Metastasis by Regulating Extracellular Matrix Remodeling following Chemotherapy. Cancer Research. 2022;82:278-91
343. Pawar VK, Singh Y, Sharma K, Shrivastav A, Sharma A, Singh A. et al. Improved chemotherapy against breast cancer through immunotherapeutic activity of fucoidan decorated electrostatically assembled nanoparticles bearing doxorubicin. International Journal of Biological Macromolecules. 2019;122:1100-14
344. Guipaud O, Jaillet C, Clément-Colmou K, François A, Supiot S, Milliat F. The importance of the vascular endothelial barrier in the immune-inflammatory response induced by radiotherapy. The British Journal of Radiology. 2018;91:20170762
345. Brown JM. Radiation Damage to Tumor Vasculature Initiates a Program That Promotes Tumor Recurrences. International Journal of Radiation Oncology, Biology, Physics. 2020;108:734-44
346. Turco V, Pfleiderer K, Hunger J, Horvat NK, Karimian-Jazi K, Schregel K. et al. T cell-independent eradication of experimental glioma by intravenous TLR7/8-agonist-loaded nanoparticles. Nat Commun. 2023;14:771
347. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C. et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130:5380-96
348. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21:799-820
349. Mak G, Soria JC, Blagden SP, Plummer R, Fleming RA, Nebot N. et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br J Cancer. 2019;120:975-81
350. Zhao J, Hu S, Qi Z, Xu X, Long X, Huang A. et al. Mitochondrial metabolic reprogramming of macrophages and T cells enhances CD47 antibody-engineered oncolytic virus antitumor immunity. J Immunother Cancer. 2024 12
351. Wang S, Li Y, Xu C, Dong J, Wei J. An oncolytic vaccinia virus encoding hyaluronidase reshapes the extracellular matrix to enhance cancer chemotherapy and immunotherapy. J Immunother Cancer. 2024 12
352. Yang Q, Guo N, Zhou Y, Chen J, Wei Q, Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharmaceutica Sinica B. 2020;10:2156-70
353. Pan Z, Chen J, Xu T, Cai A, Han B, Li Y. et al. VSIG4+ tumor-associated macrophages mediate neutrophil infiltration and impair antigen-specific immunity in aggressive cancers through epigenetic regulation of SPP1. Journal of experimental & clinical cancer research: CR. 2025;44:45
354. Qiao T, Yang W, He X, Song P, Chen X, Liu R. et al. Dynamic differentiation of F4/80+ tumor-associated macrophage and its role in tumor vascularization in a syngeneic mouse model of colorectal liver metastasis. Cell Death & Disease. 2023;14:117
355. Nabavizadeh A, Payen T, Iuga AC, Sagalovskiy IR, Desrouilleres D, Saharkhiz N. et al. Noninvasive Young's modulus visualization of fibrosis progression and delineation of pancreatic ductal adenocarcinoma (PDAC) tumors using Harmonic Motion Elastography (HME) in vivo. Theranostics. 2020;10:4614-26
356. Li J, Zormpas-Petridis K, Boult JKR, Reeves EL, Heindl A, Vinci M. et al. Investigating the Contribution of Collagen to the Tumor Biomechanical Phenotype with Noninvasive Magnetic Resonance Elastography. Cancer Research. 2019;79:5874-83
357. Wan L, Neumann CA, LeDuc PR. Tumor-on-a-chip for integrating a 3D tumor microenvironment: chemical and mechanical factors. Lab on a Chip. 2020;20:873-88
358. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M. et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell communication and signaling: CCS. 2020;18:59
359. Xun Z, Ding X, Zhang Y, Zhang B, Lai S, Zou D. et al. Reconstruction of the tumor spatial microenvironment along the malignant-boundary-nonmalignant axis. Nat Commun. 2023;14:933
360. Massey A, Stewart J, Smith C, Parvini C, McCormick M, Do K. et al. Mechanical properties of human tumour tissues and their implications for cancer development. Nat Rev Phys. 2024;6:269-82
361. Angeli S, Neophytou C, Kalli M, Stylianopoulos T, Mpekris F. The mechanopathology of the tumor microenvironment: detection techniques, molecular mechanisms and therapeutic opportunities. Front Cell Dev Biol. 2025;13:1564626
362. Zuo C, Xia J, Xu Y, Xu Y, Gao P, Zhang J. et al. stClinic dissects clinically relevant niches by integrating spatial multi-slice multi-omics data in dynamic graphs. Nat Commun. 2025;16:5317
363. Wang Q, Ni Y, Lu S, Zhang B, Ji J, Cai Q. et al. Multi-dimensional omics integrated machine learning framework identifies macrophage-fibroblast-tumor co-infiltration patterns to predict prognosis in gastric cancer. NPJ Digit Med. 2025;9:10
Author contact
![]() Corresponding authors: Lijuan Liu, hxdhxd19852003com; Wujin Chen, chenwj8077com; Changgang Sun, zyxyscgedu.cn.
Corresponding authors: Lijuan Liu, hxdhxd19852003com; Wujin Chen, chenwj8077com; Changgang Sun, zyxyscgedu.cn.