Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(11):5953-5968. doi:10.7150/ijbs.135180 This issue Cite
Research Paper
H3K18la-PSMG1 Axis in Bladder Cancer Progression: Curcumin as a Therapeutic Candidate
Zhe Yu1#, Jinge Zhang2#, Zihuan Wang1#, Shu Wei1, Chen Chen1, Yuan Huang1, Qin Fan3, Fan Deng4, Haiyong Chen5, Zhangfeng Zhong6, Lina Hou7 ![]() , Wanlong Tan1
, Wanlong Tan1 ![]() , Fei Li1
, Fei Li1 ![]()
1. Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China.
2. Department of Urology, Baoshan People's Hospital, Baoshan, Yunnan 678000, P. R. China.
3. School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China.
4. Department of Cell Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China.
5. School of Chinese Medicine, LKS Faculty of Medicine, The University of Hong Kong R619, 3 Sassoon Road, Pokfulam, Hong Kong, SAR, 999077, P. R. China.
6. Macao Centre for Research and Development in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao, SAR, 999078, P. R. China.
7. Huiqiao Medical Center, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China.
#These authors contributed equally to this work.
Received 2026-3-27; Accepted 2026-5-13; Published 2026-5-29
Abstract

Although multiple therapeutic modalities, including surgery, chemotherapy, radiotherapy, immunotherapy, and targeted therapy, have improved the management of bladder cancer, the clinical outcome of muscle-invasive bladder cancer (MIBC) remains unsatisfactory. To address this challenge, we identified MIBC-related genes (MIBC.RGs) through transcriptomic and proteomic analyses and developed a prognostic model to predict patient outcomes. Among the candidate genes, PSMG1 was prioritized through an integrated framework combining machine learning-based screening and single-cell transcriptomic analysis. Experimental analyses revealed that PSMG1 was markedly upregulated in bladder cancer (BCa), progressively upregulated from normal tissue to MIBC, and PSMG1 silencing reduced cell proliferation, invasion, and clonogenic capacity in vitro, while attenuating tumor growth in vivo. Mechanistically, our data suggest that PSMG1 may promote BCa aggressiveness, at least in part, by affecting E-cadherin stability and EMT-related signaling. Epigenetic profiling revealed significant H3K18la enrichment at the PSMG1 promoter, supporting a potential H3K18la-PSMG1 regulatory axis. Finally, molecular docking, proteomic profiling, and Drug Affinity Responsive Target Stability (DARTS) assays prioritized Curcumin as a candidate compound potentially associated with PSMG1 targeting. Overall, our findings indicate that the H3K18la-PSMG1 axis may participate in BCa progression and support further evaluation of Curcumin in PSMG1-associated therapeutic strategies.
Keywords: bladder cancer, curcumin, H3K18la, PSMG1, single cell
Introduction
Bladder cancer (BCa) represents a major cause of cancer-associated mortality and ranked fourth in incidence among men in 2023[1, 2]. According to tumor invasion depth, BCa is broadly classified as either non-muscle-invasive (NMIBC) or muscle-invasive (MIBC)—two subtypes that differ markedly in clinical course and progression risk[3, 4]. Although treatment approaches for MIBC have expanded, including surgery, systemic therapy, radiotherapy, immunotherapy, and targeted therapy, effective disease control remains difficult. For patients with MIBC undergoing radical cystectomy, the 5-year survival rate is approximately 50-60%; however, it declines to 10-15% in the presence of lymph node involvement or metastatic disease[5-7].
Tumor suppressor gene alterations (e.g., TP53, RB1, PTEN) commonly occur in MIBC and drive malignant progression by disrupting cell-cycle control and signaling networks[8]. In addition, the frequent development of drug resistance, particularly to cisplatin-based chemotherapy, further limits treatment efficacy. Resistance mechanisms include enhanced DNA damage repair, evasion of apoptosis, and metabolic reprogramming[9, 10]. MIBC is also defined by strong invasive and metastatic potential, closely tied to epithelial-mesenchymal transition (EMT) activation and the tumor immune microenvironment's remodeling. These unresolved clinical problems emphasize the need to discover additional molecular drivers and actionable therapeutic targets for MIBC.
Advances in multi-omics and gene editing technologies have revolutionized cancer research[11, 12]. Bulk RNA sequencing (RNA-seq) is widely used to characterize overall transcriptomic landscapes[13], while single-cell RNA sequencing (scRNA-seq) resolves transcriptional differences across individual cell populations[14]. Proteomics provides insight into protein function and regulation[15], and CRISPR-Cas9 technology now provides a powerful platform for interrogating gene function in functional genomics studies[16]. The integration of multi-omics data provides a critical link between molecular mechanisms and clinical translation, offering a robust foundation for precision oncology.
Machine learning and artificial intelligence have significantly enhanced biomarker discovery, prognostic modeling, and therapeutic target identification in cancer research[17, 18]. Machine learning offers advantages over conventional statistical approaches by enabling the detection of complex nonlinear associations within high-dimensional transcriptomic and proteomic data, which can improve feature selection and the development of predictive models. By integrating multiple algorithms[19], we improved gene screening and prognostic model development for BCa.
Here, we used an integrated multi-omics strategy to define muscle-invasive bladder cancer-related genes (MIBC.RGs) and identified PSMG1 as a critical factor associated with disease progression from normal bladder tissue through NMIBC to MIBC. We further investigated a potential relationship between H3K18la and PSMG1 expression in association with EMT-related phenotypes during BCa progression. In addition, we explored candidate compounds associated with PSMG1-related therapeutic targeting, including Curcumin. Together, these analyses provide a basis for further investigation of the H3K18la-PSMG1 axis and related therapeutic strategies in BCa.
Methods
Data collection and acquisition
To define MIBC-related genes (MIBC.RGs), bulk RNA-seq data were collected from two GEO datasets, GSE32548 and GSE32894. The proteomic profiles came from the study conducted by Ning Xu et al.[20]. The TCGA-BLCA dataset from The Cancer Genome Atlas (TCGA) served as the training cohort, whereas five GEO datasets, GSE13507, GSE32548, GSE32894, GSE39281, and GSE48075, were used for external validation. Single-cell transcriptomic analyses focusing on PSMG1 were conducted using the GSE129845, GSE130001, GSM4006644, GSE192575, and GSE211388 datasets. Additional data for PSMG1-related analyses were collected from publicly available resources, including BEST[21], Sangerbox[22], SolvingLab, and DepMap[23]. Sangerbox[22] was used for downstream evaluations, including AUC estimation, Kaplan-Meier survival analysis, clinicopathological association analysis, immune infiltration characterization, and mutation profiling. Immune-cell abundance was further estimated with CIBERSORT, followed by Wilcoxon rank-sum testing to assess differences between the high- and low-risk groups.
Normalization of bulk RNA-seq profiles was conducted with the DESeq2 package[24], with size factor estimation applied to adjust for differences in sequencing depth. ScRNA-seq data underwent quality control using Seurat[25], low-quality cells were removed if they contained fewer than 200 detected genes or exhibited mitochondrial gene proportions above 10%. Batch effects across multiple datasets were corrected using the Harmony[26] package.
MIBC.RGs were selected through transcriptomic and proteomic analyses
We separately applied WGCNA[27] to the GSE32548 and GSE32894 datasets to identify MIBC-associated gene modules. The genes within these two modules were combined to form Geneset1 (Table S1). Proteomic profiles were analyzed with the R package DESeq2 to identify genes significantly upregulated in MIBC relative to NMIBC samples, applying a cutoff of log2FC > 1.2 and P < 0.05. The resulting gene set was defined as Geneset2 (Table S2). The intersection of Geneset1 and Geneset2 generated the final set of MIBC.RGs (Table S3).
Integration of multiple machine learning algorithms for prognostic model construction
The MIBC.RGs underwent univariate Cox regression analysis to generate the candidate gene set for model construction (Table S4). Model construction and feature selection were subsequently carried out using an integrated machine-learning framework comprising 101 algorithmic combinations generated from ten survival modeling strategies, including RSF and GBM as tree/boosting-based methods; Enet, LASSO, ridge, and stepwise Cox as Cox regression-based approaches; CoxBoost as a boosting-based Cox model; plsRcox and SPCA as dimension-reduction-based survival models; and survival-SVM as a kernel-based survival learning method[19]. To improve model robustness, each algorithm generated an independent feature ranking, and genes consistently retained across multiple algorithms were prioritized for downstream modeling. Concordance index (C-index) performance across the training and validation cohorts primarily guided the selection of the final model. Among all 101 algorithmic combinations, Ridge regression achieved the highest C-index (0.673) and showed stable performance in external validation datasets; therefore, we selected it as the final prognostic model (Figure 2A). Given the high dimensionality and potential collinearity of transcriptomic features, the regularization property of Ridge regression further supported its suitability for prognostic modeling in this setting. The final Ridge-based model included 34 genes (Table S5). A detailed description of the model selection strategy, algorithm integration framework, and validation procedure is provided in Supplementary Material 1.
Multiple machine learning-integrated analysis. (A) Heatmap of the C-index for 101 algorithm combinations. The Ridge-based model, having the highest C-index, was chosen as the final model. (B) UniCox regression analysis of model genes. (C) ROC curve based on the model. (D) Survival analysis between the high- and low-risk groups. (E-J) Violin plots showing the correlation of risk score with gender, grade, stage, T stage, N stage, and M stage. (K) Box plots were used to compare immune-cell infiltration levels between the high- and low-risk groups, with infiltration scores estimated by CIBERSORT. Statistical significance was assessed using the Wilcoxon rank-sum test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (L) Waterfall plot illustrating the somatic mutation profiles of the high- and low-risk groups.
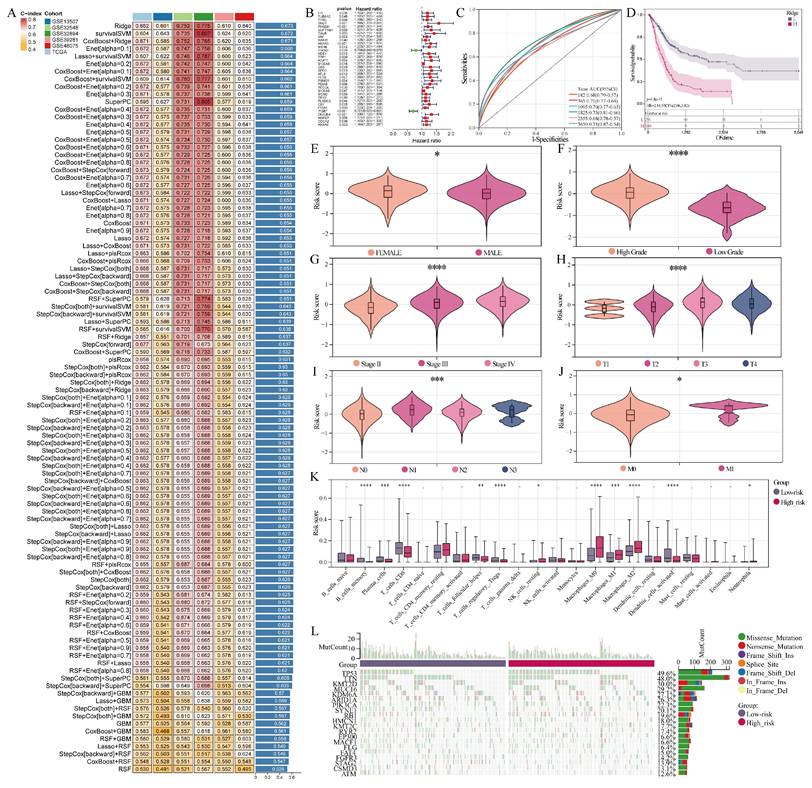
Ten-fold cross-validation, which randomly split the dataset into training (90%) and testing (10%) subsets in each iteration, was used to assess the robustness of our prognostic model. In addition, external validation was conducted using five independent GEO datasets (GSE13507, GSE32548, GSE32894, GSE39281, and GSE48075). Model performance was assessed with the C-index, Kaplan-Meier survival analysis and time-dependent ROC curves (AUC). Calibration plots examined the concordance between predicted and observed survival probabilities.
PSMG1 was selected as a key driver in BCa progression based on pseudo-time analysis
The R packages Seurat, SingleR[28], and Harmony performed dimensionality reduction, clustering, annotation, and subdivision of epithelial subpopulations in scRNA-seq data. For pseudo-time analysis, we employed the R package Monocle3[29]. Initially, uniCox analysis was applied to model genes (Table S6), resulting in the selection of 32 genes with hazard ratios (HR) greater than 1.0. Their temporal trajectories were visualized in the scRNA-seq data. Subsequently, 10 genes demonstrating robust trends from normal tissue to MIBC were chosen. Finally, PSMG1, which exhibited the highest HR of 1.35, was selected for further analysis.
Spatial transcriptomic data were obtained from the study by Kenneth H. Gouin III et al.[30], which were analyzed using STOmicsDB[31]. Gene Set Cancer Analysis (GSCA)[32, 33] was applied to assess whether PSMG1 expression varied according to bladder cancer stage.
A comprehensive analysis of PSMG1 was performed to explore its role in BCa progression
Pan-cancer analysis of PSMG1 was conducted using Sangerbox. Survival analysis was performed through SolvingLab. Correlation analyses with various clinical features were completed using BEST. Representative immunohistochemical staining data for PSMG1 were accessed through The Human Protein Atlas (HPA)[34]. Multi-omics enrichment analyses were conducted employing BEST and ClusterProfiler[35]. Predictive analysis of PSMG1's impact on BCa cell functions was performed using DepMap.
The role of PSMG1 in promoting BCa progression was validated
Lentiviral shRNA constructs were obtained, and UMUC3 cells were transduced with lentiviral particles to generate stable cell lines as per the manufacturer's protocols. Quantitative PCR (qPCR), CCK-8, colony formation, and subcutaneous tumorigenesis experiments were performed as reported in a prior study[36]. The Transwell assay was conducted following the methodology outlined by Li Yi et al.[37]. Changes in the protein expression levels of EMT marker genes, including N-cadherin, Vimentin, SNAIL1, and E-cadherin, following PSMG1 knockdown were validated by Western blotting. The sequences of the shPSMG1 constructs were GTCGACATGTTACCGATTATA and GCAATTCTGTACTTGTGTTAT.
A cycloheximide (CHX) chase assay was used to evaluate E-cadherin protein stability. Following exposure to cycloheximide (50 µg mL⁻¹), UMUC3 cells were harvested at 0, 3, 6, 9, and 12 h. E-cadherin levels were assessed via Western blotting with Actin as the loading control, and representative blots are displayed.
The regulation of PSMG1 by H3K18la was validated
The potential interaction between H3K18la and PSMG1 was investigated using ATAC-seq and ChIP-seq[36]. Additionally, potential interacting proteins of PSMG1 were predicted using the AlphaFold3 artificial intelligence algorithm[38]. Changes in PSMG1 expression following treatment with histone lactylation inhibitors 2-DG and c646, as well as the histone lactylation promoter Nala, were validated through qPCR and Western blotting. Detailed experimental procedures are provided in a previous study[36].
Potential drugs targeting PSMG1 were explored
Candidate drugs targeting PSMG1 were identified using the Beyondcell algorithm[39], with inclusion criteria defined as drug sensitivity showing a significant positive correlation with PSMG1 expression levels (cor > 0.2, P < 0.001). Six compounds meeting these criteria—Curcumin, Emodin, Apigenin, Naringenin, Taxifolin, and Vitexin—were shortlisted based on favorable molecular docking scores obtained using SwissDock[40], which evaluated their binding poses and interaction energies with PSMG1. To further evaluate potential compound-protein interactions, Drug Affinity Responsive Target Stability (DARTS) assays were conducted with proteinase K digestion, followed by Coomassie blue staining and Western blotting. Comparative analyses incorporated both docking results and pharmacological data. Although Emodin exhibited the lowest binding energy, Curcumin demonstrated superior binding stability in DARTS assays, along with extensive evidence of anticancer efficacy and a well-established safety profile. Other candidates, while promising in silico, lacked comparable validation in terms of stability or safety and were therefore not advanced to further experimental evaluation. Therefore, Curcumin was prioritized as the primary candidate for subsequent experimental validation. Sequencing data from Curcumin-treated cell lines are provided in Supplementary Material 2.
Data statistics and analysis
Statistical analyses were conducted with R version 4.2.1 and GraphPad Prism 10, and all in vitro experiments (unless otherwise noted) were independently repeated three or more times. Quantitative data are presented as mean ± SD. Comparisons between two groups were performed using a two-tailed Student's t test, whereas comparisons among multiple groups were performed using one-way ANOVA followed by Tukey's multiple-comparison test. Kaplan-Meier survival curves were compared using the log-rank test. A P value < 0.05 was considered statistically significant. For Western blot-based assays, including CHX chase and DARTS, representative results are shown.
Results
The acquisition of MIBC.RGs was performed through integration of multiple data sources
To comprehensively identify factors driving MIBC in the central pathway, bulk RNA-seq and proteomics data were integrated. Initially, two bulk RNA-seq datasets with comprehensive clinical data were included. In dataset GSE32548, WGCNA identified the module most strongly connected with MIBC. After plotting the distribution of MIBC and NMIBC samples (Figure 1A), scale independence analysis determined a Soft Threshold power of 9 (Figure 1B). Modules were color-coded (Figure 1C). The brown module showed the strongest correlation with MIBC (Figure 1D). The genes corresponding to this module were then extracted. This module also displayed greater gene significance in both overall and MIBC samples (Figure 1E, F), confirming the validity of the analysis. The same methodology was applied to GSE32894 (Figure 1G-L). The union of MIBC-related genes from both bulk RNA-seq datasets formed Geneset1 (Table S1).
The acquisition of MIBC.RGs. (A) The sample dendrogram and trait heatmap of the GSE32548 dataset. (B) Analysis of the scale-free index for various soft-threshold powers. (C) The dynamic tree depicting the gene dendrogram and module colors. (D) Module-trait relationships. (E) Gene significance across modules. (F) The relationship between brown module membership and gene significance. (G) The sample dendrogram and trait heatmap of the GSE32894 dataset. (H) Scale-free topology fit index across different soft-thresholding powers. (I) Gene dendrogram and module colors shown in the dynamic tree. (J) Module-trait relationships. (K) Gene significance across modules. (L) Correlation of black module membership with gene significance. (M) Differential gene analysis between MIBC and NMIBC based on proteomic data. (N) Intersection analysis of MIBC-related genes between transcriptomics and proteomics datasets.
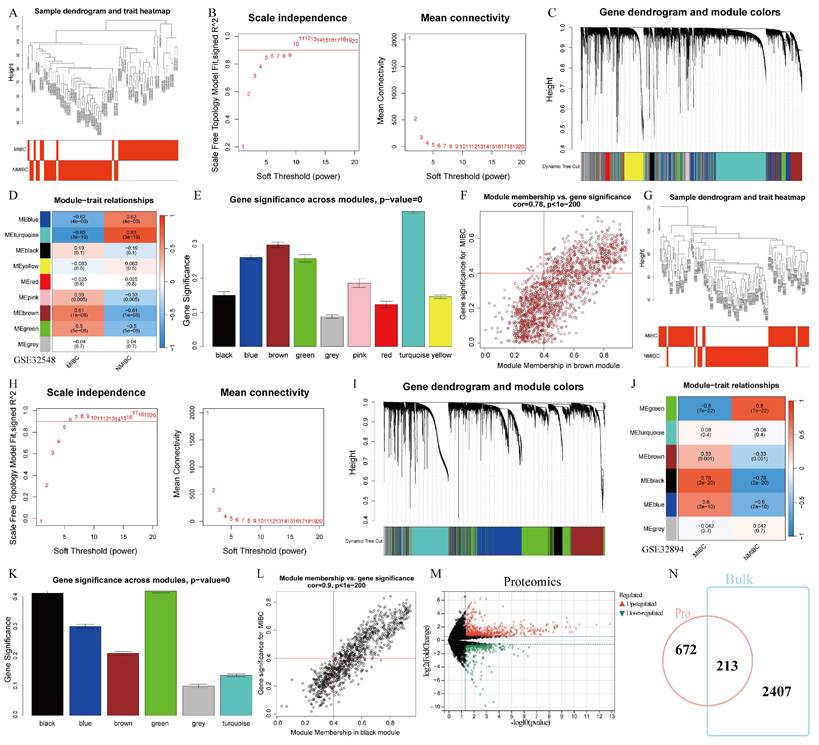
Next, differential genes between MIBC and NMIBC samples were identified from proteomics data, retaining only upregulated genes in MIBC, resulting in Geneset2 (Table S2) (Figure 1M). The intersection of Geneset1 and Geneset2 from both omics analyses yielded the final set of MIBC.RGs (Table S3) (Figure 1N).
A prognostic model was constructed and gene screening was performed using multiple machine learning integration algorithms
Advancements in machine learning algorithms have greatly enhanced the ability to screen gene sets and construct prognostic models based on specific gene expression profiles[19]. From MIBC.RGs, a prognostic model was built by combining multiple machine learning algorithms (Figure 2A, B). We selected the Ridge algorithm from 101 combinations of 10 algorithms, as it achieved the highest concordance index (C-index) of 0.673. The model effectively predicted survival outcomes for BCa patients across six intervals (Figure 2C). We stratified patients into high- and low-risk groups using risk scores, where higher scores correlated with worse prognosis (Figure 2D).
Further analyses incorporated various clinical characteristics. In the female population, a higher risk score was observed (Figure 2E). Notably, elevated risk scores were associated with advanced BCa progression and metastasis (Figure 2F-J). In addition, comparative immune infiltration profiling showed that the high- and low-risk groups were associated with distinct immune-cell composition patterns (Figure 2K). Specifically, the low-risk group exhibited increased CD8+ T-cell infiltration, while the high-risk group showed greater M2 macrophage enrichment. CD8+ T cells are crucial for host defense and tumor cell cytotoxicity[41], while M2 macrophages are associated with pro-tumorigenic effects[42]. These findings suggest that the MIBC.RG-based risk pattern may be associated with differences in the tumor immune microenvironment. Furthermore, the high-risk group exhibited a more active mutation landscape, including a higher frequency of TP53 mutations (Figure 2L).
In conclusion, a prognostic model based on MIBC.RGs was developed by integrating multiple machine learning algorithms. The model effectively predicted BCa patient outcomes and provided insights into patient progression and metastasis through risk score stratification.
PSMG1 was selected as a key driver in BCa progression
ScRNA-seq is a powerful tool for uncovering the heterogeneity of transcriptional profiles at the single-cell level, enabling the identification of distinct cell types and functions within specific tissues[43]. One analysis method used in this technique is pseudo-time analysis, which reconstructs the trajectory of cellular changes over time. This approach maps the dynamic progression of cellular states by considering the relative timing of gene expression changes between individual cells, allowing researchers to track how cells transition from one state to another[44]. Leveraging this approach, comprehensive scRNA-seq data for BCa were collected to investigate mechanisms driving BCa progression.
After data processing steps including dimensionality reduction, clustering, and annotation, we generated a single-cell atlas of BCa (Figure 3A-C). Special focus was placed on epithelial clusters, which were further subdivided into distinct subpopulations (Figure 3D). Among these, epithelial clusters 1, 5, and 7 exhibited a higher proportion of MIBC cells (Figure 3E, F). Pseudo-time analysis reconstructed cellular trajectories, illustrating transitions from normal tissue to NMIBC and eventually to MIBC (Figure 3G, H). Model genes identified as risk factors were mapped onto pseudo-time trajectories (Figure 3I), and those displaying robust trends from normal tissue to MIBC were further analyzed (Figure 3J).
Identification of PSMG1. (A) UMAP plot of single-cell clustering of BCa samples. (B) UMAP plot annotated with cell clusters. (C) Projection of three sample types across various cell clusters. (D) Subdivision of epithelial cell cluster into subgroups. (E) Projection of three sample types across various epithelial subgroups. (F) Proportions of three sample types across various epithelial subgroups. (G) UMAP plot showing pseudo-temporal trajectory changes of epithelial subgroups. (H) Pseudo-temporal trajectory UMAP plot incorporating sample type information. (I) Pseudo-temporal trajectory plots of 32 model genes with HR > 1.0 in uniCox analysis. (J) Ten model genes exhibiting a trend from normal to MIBC in expression. (K) Pseudo-temporal trajectory plot of PSMG1. (L) Bubble plot of PSMG1 expression across different sample types. (M) Heatmap of PSMG1 expression levels across different stages. (N) Trend plot of PSMG1 expression levels across different stages. (O) Box plot of PSMG1 scores across different stages. (P) Left: spatial transcriptomic clustering of MIBC data. Right: projection of PSMG1 expression in MIBC spatial transcriptomic data.
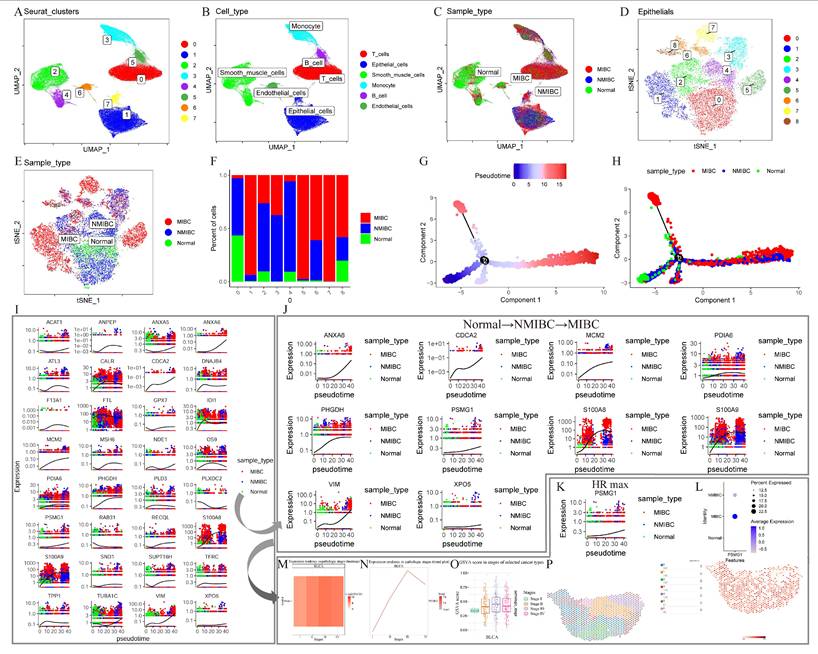
From this analysis, PSMG1, which had the highest HR of 1.35, was identified as a key focus for subsequent investigations. Elevated expression of PSMG1 in MIBC was validated using scRNA-seq, bulk RNA-seq, and spatial transcriptomic analyses (Figure 3K-P).
In conclusion, through an integrative multi-omics analysis of MIBC.RGs, a prognostic model was constructed and gene selection was performed using advanced machine learning algorithms. By mapping cellular trajectories within the BCa single-cell atlas, PSMG1 was identified as a potential key regulator influencing BCa progression.
BCa progression was promoted by PSMG1
Currently, research on PSMG1 is limited, with only a few reports in inflammatory bowel disease (IBD), and studies in tumors are scarce. To explore the role of PSMG1 in cancer, particularly in BCa, we conducted further comprehensive analysis. Intriguingly, PSMG1 was identified as an independent risk factor across various cancers (Figure 4A). Large-scale pan-cancer survival analysis demonstrated that high PSMG1 expression was significantly associated with poor prognosis (Figure 4B). High PSMG1 expression was associated with worse overall survival (OS), disease-free survival (DFS), and progression-free survival (PFS) in BCa patients compared to those with low expression (Figure 4C). Beyond BCa, PSMG1 was found to be upregulated in a range of cancer types, reinforcing its association with malignancy (Figure 4D).
The comprehensive analysis of PSMG1. (A) Pan-cancer uniCox regression analysis of PSMG1. (B) Large-scale survival analysis integrating tumor cohorts where PSMG1 is considered an independent risk factor. (C) Analysis of PSMG1 in BCa for OS (left one), DFS (left two), and PFS (left three). (D) Violin plot of pan-cancer expression levels of PSMG1. (E) Box plot of PSMG1 expression levels in BCa. (F-I) Box plots showing the correlation between PSMG1 expression levels and grade. (J-K) Box plots illustrating the correlation between PSMG1 expression levels and stage. (L) KEGG pathway enrichment analysis of proteins positively correlated with PSMG1 in the proteomic dataset. (M) Hallmark pathway enrichment analysis of PSMG1-associated genes in the bulk RNA-seq dataset. (N) KEGG pathway enrichment analysis of PSMG1-correlated genes identified via bulk RNA-seq. (O) KEGG pathway enrichment analysis of genes positively correlated with PSMG1 at the single-cell RNA level. Colored boxes indicate recurrently enriched pathways shared across multiple datasets, highlighting the cross-platform consistency of PSMG1-related biological programs. (P) Prediction of PSMG1 gene effects based on DepMap.
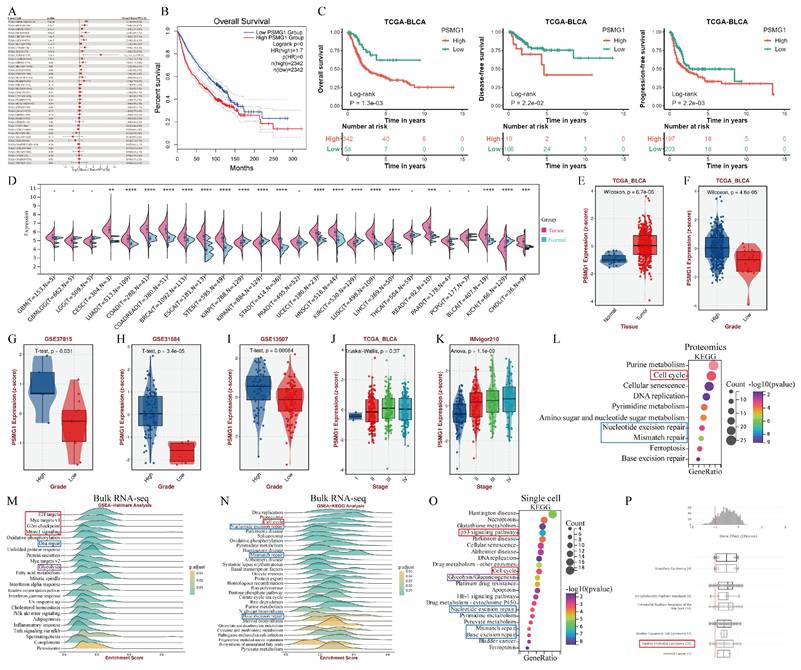
BCa tumor tissues showed significantly higher PSMG1 expression than normal tissues (Figure 4E; Figure S1). Moreover, elevated PSMG1 expression correlated with higher malignancy grades in BCa (Figures 4F-K). Functional enrichment analyses of PSMG1 were performed across proteomic, bulk RNA-seq, and single-cell RNA-seq datasets to evaluate the robustness and consistency of its associated biological programs from multiple molecular layers. As shown in Figure 4L-O, these independent analyses convergently highlighted pathways related to tumor proliferation and progression, including cell cycle, E2F/MYC-related programs, G2/M checkpoint, mTORC1 signaling, and p53 signaling[45-50]. In addition, pathways associated with DNA damage repair and glycolytic metabolism were also recurrently enriched across datasets. Collectively, these findings support the cross-platform consistency of PSMG1-associated biological programs. Analysis using DepMap predicted a gene effect score < 0, indicating that PSMG1 knockdown or inhibition could suppress BCa cell proliferation (Figure 4P).
In summary, these findings underscored the critical role of PSMG1 in promoting BCa progression and highlighted its potential as a therapeutic target.
The role of PSMG1 in promoting BCa progression was validated
To validate PSMG1's role in promoting BCa progression, we knocked down PSMG1 using shRNA and confirmed knockdown efficiency by qPCR (Figure 5A). CCK-8 and Transwell assays showed that PSMG1 knockdown significantly inhibited tumor proliferation, invasion, and metastasis (Figure 5B-D). Additionally, the proliferation of BCa cells was reduced following PSMG1 knockdown (Figure 5E), and tumor volume was significantly decreased (Figure 5F), suggesting that elevated PSMG1 expression promotes BCa progression.
Validation of BCa progression promoted by PSMG1. (A) PSMG1 was knocked down using shRNA technology, and the effectiveness of knockdown was validated by qPCR. *: P < 0.05, **: P < 0.01, ***: P < 0.001. (B) Assessment of proliferative function by CCK-8 assay. (C-D) Evaluation of migratory and invasive abilities using Transwell assays. (E) Colony formation assay reflecting proliferative function changes. (F) Subcutaneous tumorigenesis assay in mice. (G) Western blotting assessed EMT marker expression (N-cadherin, Vimentin, SNAIL, E-cadherin) after PSMG1 knockdown. (H) CHX-chase Western blotting was performed in the control group (shNC) and the PSMG1 knockdown group (shPSMG1). Cells were treated with 50 µg mL-¹ CHX and harvested at 0, 3, 6, 9, and 12 h. E-cadherin, PSMG1, and Actin were detected, with Actin used as the loading control. Representative results are shown.
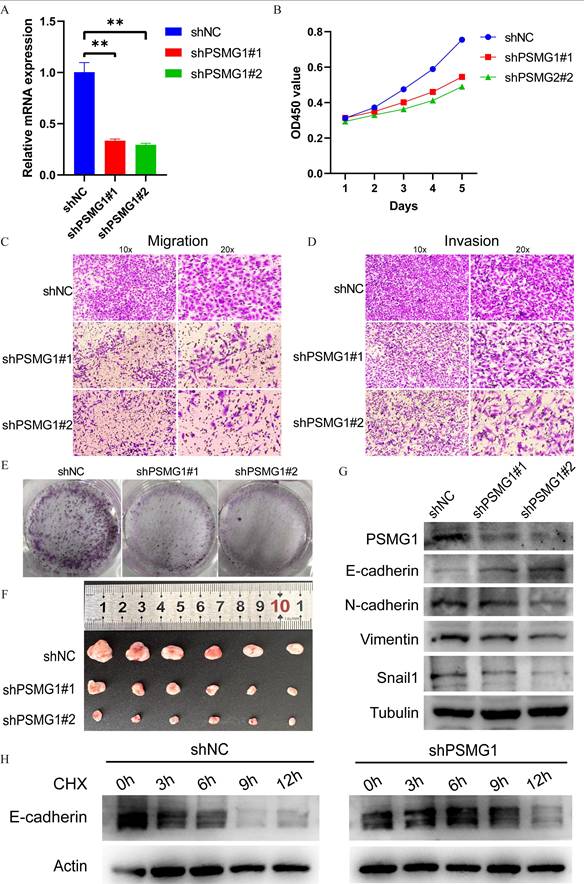
Given the close association between EMT and MIBC aggressiveness, we examined the effect of PSMG1 on EMT-associated proteins. Western blot results showed that knockdown of PSMG1 led to reduced expression of N-cadherin, Vimentin, and SNAIL, whereas E-cadherin levels were increased (Figure 5G). Among these EMT-related proteins, E-cadherin showed the clearest and most reproducible change after PSMG1 silencing. In light of this observation, together with the known function of the proteasome in protein turnover[51], we investigated whether PSMG1 could influence E-cadherin stability. CHX chase analysis demonstrated that E-cadherin degradation was delayed in PSMG1-knockdown cells, resulting in a longer half-life than that observed in control cells (Figure 5H). These findings suggest that PSMG1 may promote EMT-related phenotypes by reducing E-cadherin stability.
The H3K18la-PSMG1 axis was associated with BCa progression
Histone lactylation is a recently recognized post-translational modification that plays a key role in gene expression regulation and tumor progression[52-54]. Consistent with enrichment analysis, single-cell metabolism profiling revealed enhanced glycolysis in tumor cells with high PSMG1 expression, along with elevated co-expression of lactate-associated factors such as EP300, KAT8, and AARS2 (Figure 6A). To further examine the potential link between lactylation and PSMG1 expression, we assessed publicly available histone H3K18la modification data. This analysis revealed overlapping chromatin open regions between H3K18la and PSMG1 (Figure 6B), suggesting a possible association between H3K18la enrichment and PSMG1 transcriptional regulation.
Evidence supporting an association between H3K18la and PSMG1 in BCa. (A) Bubble plot of glycolysis-related genes and lactate-associated factors between groups with high and low PSMG1 expression in tumor epithelial cells. (B) Visualization of chromatin open regions of H3K18la and PSMG1. (C) The enrichment of H3K18la sites was observed in the promoter region of PSMG1. (D-E) The changes in PSMG1 expression levels after the addition of histone lactylation inhibitors 2-DG and c646, and the histone lactylation promoter Nala, were validated using qPCR and Western blotting.
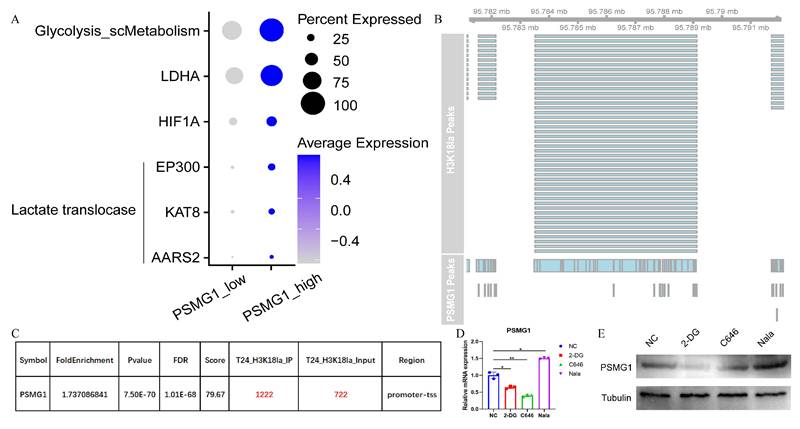
ChIP-seq analysis identified H3K18la-enriched regions in gene promoter areas, including the promoter region of PSMG1 (Figure 6C). These findings support a potential regulatory link between H3K18la and PSMG1 in BCa.
To further assess this association, we treated cells with histone lactylation inhibitors 2-DG and c646, which resulted in decreased PSMG1 mRNA expression, whereas treatment with the histone lactylation promoter Nala increased PSMG1 expression (Figure 6D). Western blotting showed similar trends at the protein level (Figure 6E). Collectively, these findings indicate that histone lactylation status correlates with PSMG1 expression, pointing to an H3K18la-PSMG1 regulatory axis in BCa. However, the direct causal mechanism requires further experimental validation.
Curcumin was prioritized as a candidate compound associated with PSMG1 targeting in BCa
Natural products derived from traditional Chinese herbs have attracted increasing attention because of their potential anti-tumor effects[55]. These compounds have the potential to improve the effectiveness of chemotherapy, radiotherapy, and immunotherapy while mitigating treatment-related toxicities. They also lay a foundation for future therapeutic studies[56-59].
Using the Beyondcell algorithm, compounds associated with PSMG1-related drug sensitivity were identified. To estimate the binding potential of these candidate compounds, molecular docking analysis was performed using SwissDock. This analysis identified six candidate compounds with favorable binding poses and interaction energies with PSMG1 (Curcumin: -7.3 kCal/mol, Apigenin: -7.0 kCal/mol, Naringenin: -7.1 kCal/mol, Emodin: -8.2 kCal/mol, Taxifolin: -7.5 kCal/mol, and Vitexin: -7.8 kCal/mol), and the predicted interaction patterns were visualized (Figure 7A-F).
Screening and preliminary evaluation of candidate compounds associated with PSMG1. Molecular docking and DARTS assays were used as preliminary approaches to assess potential compound-PSMG1 interactions. For the docking results, interaction diagrams were generated using SwissDock and visualized with PyMOL and LigPlot+. Binding interfaces between drugs and PSMG1 were annotated to indicate hydrogen bonds, hydrophobic contacts, and relevant residues involved in the interaction. (A-F) Six drugs with their respective binding poses and interactions with PSMG1. From A to F, they were respectively: Curcumin, Apigenin, Naringenin, Emodin, Taxifolin, and Vitexin. (G-H) Coomassie blue staining of the DARTS assay. The regions corresponding to Emodin and Curcumin are shown at approximately 35 kDa. (I-J) Western blot analysis of the DARTS assay. Representative results are shown. (K) Protein expression of PSMG1 under different doses of Curcumin. (L) Effect of Curcumin on BCa cell proliferation assessed by CCK-8 assay. (M) Transwell assays showing the effects of Curcumin on BCa cell migration and invasion.
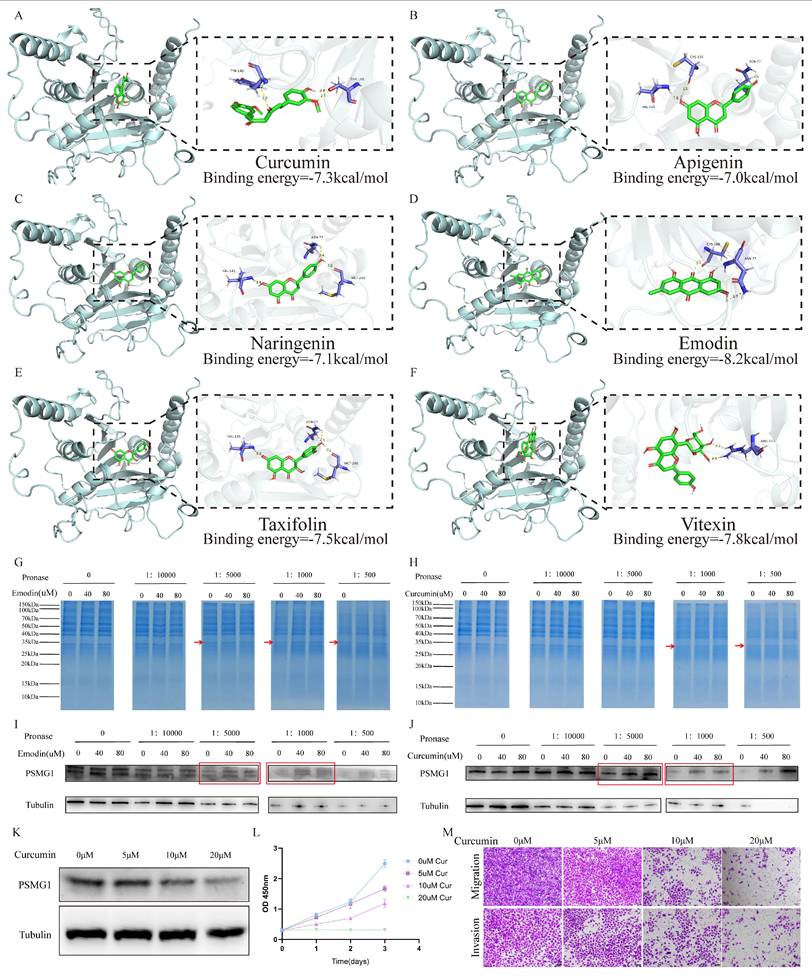
Among the shortlisted compounds, Emodin, which showed the lowest predicted binding energy, and Curcumin, a molecule with a favorable safety profile and documented anti-tumor activity, were selected for further investigation[58, 60, 61]. DARTS assays were subsequently performed to examine whether these compounds were associated with altered protease susceptibility of PSMG1. In the Emodin-treated group, degradation of the PSMG1-corresponding region was reduced relative to the control group (Figure 7G, I). A similar pattern was observed in Curcumin-treated cells (Figure 7H, J), supporting the possibility of an interaction between Curcumin and PSMG1. To further assess the biological relevance of Curcumin, we conducted proteomic sequencing (Supplementary Material 2). The sequencing data indicated decreased PSMG1 expression in Curcumin-treated cell lines, which was further supported by Western blot analysis (Figure 7K). Moreover, functional assays demonstrated that Curcumin inhibited BCa cell invasion and migration in a dose-dependent manner (Figure 7L-M). However, the present study did not directly establish that the observed decrease in PSMG1 expression was mechanistically caused by Curcumin-PSMG1 interaction. Overall, these findings support Curcumin as a prioritized candidate compound within the PSMG1-related therapeutic screening framework, while the mechanistic relationship between the putative Curcumin-PSMG1 interaction and reduced PSMG1 expression remains to be clarified.
Discussion
PSMG1 (Proteasome Assembly Chaperone 1), also known as PAC1, is a chaperone protein involved in 20S proteasome assembly[51]. Our study identified PSMG1 as a key oncogene in BCa, showing progressive upregulation from normal tissue to NMIBC and MIBC, where it was associated with tumor aggressiveness and poor prognosis. Functional assays confirmed that PSMG1 promotes proliferation, invasion, and EMT, reinforcing its oncogenic role. Additionally, a prognostic model was developed from MIBC.RGs via multi-omics integration and machine learning, which effectively categorized BCa patients as high- or low-risk. Immune infiltration analysis further showed that the MIBC.RG-related prognostic risk pattern was associated with lower CD8⁺ T-cell infiltration and higher M2 macrophage infiltration, indicating a potential immunosuppressive state in advanced BCa. Nevertheless, our data do not directly demonstrate that PSMG1 regulates M2 macrophage polarization or CD8⁺ T-cell infiltration. Thus, these immune-related findings represent preliminary associations and do not constitute definitive proof that PSMG1 directly modulates the tumor immune microenvironment.
Our findings suggest that PSMG1 may contribute to BCa aggressiveness, at least partly, by affecting E-cadherin turnover and thereby facilitating EMT-related changes. In this study, E-cadherin was not identified through an unbiased substrate-screening approach. Instead, it was selected as a candidate downstream effector because analysis of EMT-associated markers showed that E-cadherin increased consistently after PSMG1 knockdown, whereas mesenchymal markers were reduced. CHX chase experiments further supported the notion that PSMG1 influences E-cadherin stability. In parallel, our data support a possible association between histone lactylation, particularly H3K18la, and PSMG1 expression. ChIP-seq analysis revealed H3K18la enrichment in the promoter region of PSMG1, while single-cell metabolic profiling showed that elevated PSMG1 expression was accompanied by increased glycolytic activity and higher expression of lactate-associated factors, including EP300, KAT8, and AARS2. Moreover, pharmacological modulation of histone lactylation altered PSMG1 expression at both the transcript and protein levels. Taken together, these findings suggest that glycolysis-related lactylation may participate in the transcriptional regulation of PSMG1 in BCa. However, because these observations are derived mainly from public epigenomic datasets and pharmacological perturbation, they should be regarded as supportive rather than definitive evidence of direct causality. Although H3K18la has previously been implicated in other malignancies[62], our results extend these observations by suggesting a potential H3K18la-PSMG1 regulatory relationship in BCa.
Previous studies have indicated that Curcumin can modulate epigenetic and metabolic pathways, including histone acetyltransferase-related regulation and glycolytic activity[63-67]. In light of our results, these reports suggest that Curcumin may influence BCa progression through multiple interconnected mechanisms. Nevertheless, the relative importance of direct PSMG1 regulation compared with broader upstream effects remains unclear. Although molecular docking and DARTS assays in our study supported a possible interaction between Curcumin and PSMG1, we did not directly determine whether the reduction in PSMG1 expression observed after Curcumin treatment was caused by compound-protein interaction itself or by indirect regulatory events. We also did not directly assess E-cadherin expression following Curcumin treatment. Since Curcumin treatment was associated with reduced PSMG1 expression, and PSMG1 knockdown prolonged E-cadherin stability, it is conceivable that Curcumin may affect E-cadherin-related EMT signaling; however, this inference remains speculative and requires direct experimental confirmation. Future studies, including rescue experiments using PSMG1 overexpression after Curcumin exposure, as well as direct assessment of Curcumin's effects on epigenetic enzymes such as EP300 and KAT8 in BCa, will be necessary to clarify the underlying mechanism.
The clinical implications of these findings remain preliminary. Targeting histone lactylation, particularly H3K18la, may represent a potential strategy to suppress PSMG1-associated BCa progression. Curcumin, given its favorable safety profile and reported anticancer activity[58, 60, 61], was prioritized in our screening framework as a candidate compound associated with reduced PSMG1 expression and attenuation of aggressive BCa phenotypes. In addition, combination strategies involving Curcumin and other therapeutic modalities, such as immune checkpoint inhibitors, epigenetic modulators, or cisplatin-based chemotherapy, may warrant further investigation in future preclinical studies[58, 68, 69]. However, the efficacy and mechanistic basis of such combinations were not evaluated in the present study.
We also acknowledge the limitations of this study. Although IHC validation from the Human Protein Atlas supports our findings, validation in larger independent cohorts (IHC or TMA) is lacking. Moreover, our functional evidence is derived from in vitro assays; xenograft and patient-derived organoid models will be essential for confirming Curcumin's therapeutic efficacy. Another key challenge is Curcumin's poor bioavailability due to low solubility and rapid metabolism[70]. Nanocarrier-based delivery systems[70-72] (liposomes, nanoparticles, exosome encapsulation) and Curcumin derivatives (e.g., EF-24, morpholine-modified analogs)[73] have shown improved pharmacokinetics and anticancer potency in preclinical models, and represent promising strategies for clinical translation.
In summary, our study identified PSMG1 as a key oncogene in BCa, supported a potential association between H3K18la and PSMG1 in BCa progression, and prioritized Curcumin as a candidate compound for further investigation in PSMG1-related therapeutic strategies.
Conclusion
In this study, we found PSMG1 to be a key oncogenic driver in BCa progression and uncovered a potential link between the H3K18la-PSMG1 axis and BCa aggressiveness. Our findings suggest that histone lactylation, particularly H3K18la, may contribute to BCa aggressiveness in association with increased PSMG1 expression and subsequent activation of the EMT program. Furthermore, our data prioritized Curcumin as a candidate therapeutic compound associated with reduced PSMG1 expression and suppression of aggressive BCa phenotypes. These findings provide preliminary support for further investigation of Curcumin in the context of PSMG1-related therapeutic strategies.
Abbreviations
BCa: Bladder cancer; NMIBC: Non-muscle-invasive bladder cancer; MIBC: Muscle-invasive bladder cancer; MIBC.RGs: Muscle-invasive bladder cancer-related genes; PSMG1: Proteasome assembly chaperone 1; H3K18la: Histone H3 lysine 18 lactylation; EMT: Epithelial-mesenchymal transition; RNA-seq: RNA sequencing; scRNA-seq: Single-cell RNA sequencing; WGCNA: Weighted gene co-expression network analysis; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; CIBERSORT: Cell-type identification by estimating relative subsets of RNA transcripts; AUC: Area under the curve; ROC: Receiver operating characteristic; C-index: Concordance index; HR: Hazard ratio; OS: Overall survival; DFS: Disease-free survival; PFS: Progression-free survival; qPCR: Quantitative polymerase chain reaction; CHX: Cycloheximide; DARTS: Drug affinity responsive target stability; ChIP-seq: Chromatin immunoprecipitation sequencing; ATAC-seq: Assay for transposase-accessible chromatin using sequencing; KEGG: Kyoto Encyclopedia of Genes and Genomes; IHC: Immunohistochemistry; HPA: Human Protein Atlas; CCK-8: Cell Counting Kit-8.
Supplementary Material
Supplementary figure.
Supplementary material 1 and 2.
Supplementary tables.
Acknowledgements
This study was funded by the Natural Science Foundation of Guangdong Province of China (2026A1515010715) (F.L.), the National Natural Science Foundation of China (82573186) (F.L.) (82372867) (WL.T.), the President Foundation of Nanfang Hospital, Southern Medical University (2025A013) (LN.H.) (2023B027) (Z.Y.), and the Hangzhou Oriental Clinical Oncology Research Center (ECCO-KY-24003) (F.L.). The authors thank the various institutions and organizations that had provided funding and public data.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material. All software applications used are included in this article. Relevant sequencing data and analysis data results can be downloaded from the attachment or obtained by contacting corresponding authors.
Author contributions
Zhe Yu: Conceptualization, Methodology, Software, Validation, Formal analysis, Writing - Original Draft, Visualization, Funding acquisition. Jinge Zhang: Methodology, Software, Validation, Formal analysis, Investigation, Data Curation, Writing - Original Draft, Visualization. Zihuan Wang: Writing - Original Draft, Methodology, Validation, Resources, Visualization, Investigation. Shu Wei: Validation, Visualization. Chen Chen: Resources, Validation. Yuan Huang: Investigation, Software, Visualization. Qin Fan: Investigation. Fan Deng: Investigation. Haiyong Chen: Software, Visualization. Zhangfeng Zhong: Software, Visualization. Lina Hou: Supervision, Project administration, Funding acquisition. Wanlong Tan: Writing - Review & Editing, Supervision, Project administration, Funding acquisition. Fei Li: Conceptualization, Methodology, Writing - Review & Editing, Supervision, Project administration, Funding acquisition.
Ethics statement
The data used in this study were sourced from public databases. The patients included in these databases had obtained ethical approval. Users can freely download relevant data for research purposes. Our research was grounded in open-access data and complied with ethical requirements. The experimental study received support from the Ethics Committee of Southern Medical University.
Declaration of generative AI and AI-assisted technologies in the writing process
Purpose and scope of AI use
Generative AI (ChatGPT-5.2, OpenAI) was employed solely for improving the clarity, grammar, and readability of the manuscript text during the drafting and revision stages. It was not used to generate scientific hypotheses, design or conduct experiments, perform statistical analyses, or interpret study findings. The AI assistance was limited to language refinement and structuring of text for better academic readability. No figures, tables, or original scientific content were generated by AI. The authors confirm that all scientific content, data interpretation, and conclusions are the result of the authors' independent work.
AI tool(s) and configuration
The tool used was ChatGPT-5.2 (OpenAI), accessed via the official cloud-based API during manuscript drafting and revision in 2026. The tool operated under standard default settings (temperature = 0.2; maximum token length = 4096). No plug-ins, fine-tuning, or third-party integrations were employed.
Data inputs and safeguards
The AI system was provided only with de-identified manuscript drafts, literature abstracts, and general scientific text. No patient-level data, identifiable images, or sensitive clinical information were shared with the AI tool. All inputs were fully anonymized and handled in compliance with GDPR and HIPAA regulations. No institutional approvals were required, as no protected health information was used.
Human oversight and verification
All AI-assisted outputs were reviewed, verified, and edited by the corresponding author and the full author team. Each suggested revision was cross-checked against source literature and experimental data to ensure factual accuracy and clinical validity. Where AI-generated content did not meet scientific accuracy or clarity standards, it was either revised extensively or discarded. The authors take full responsibility for the integrity, accuracy, and originality of the final manuscript.
Bias, ethics, and regulatory compliance
The authors recognize the potential for algorithmic bias in generative AI tools. To mitigate this risk, all AI-assisted outputs were cross-referenced with peer-reviewed scientific literature and verified for consistency with established clinical guidelines. The use of AI complies with the ethical frameworks outlined by the TITAN Guidelines 2025[74]. The authors declare no financial or institutional conflicts of interest related to the use of AI tools.
Consent
Written informed consent for publication was obtained from all participants.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74:12-49
2. Huang Y, Chen C, Su M, Ye W, Wei S, Long K. et al. TMEM11 promotes cisplatin resistance by inhibiting BNIP3-mediated mitophagy in bladder cancer. Cancer Lett. 2026;641:218271
3. McConkey DJ, Choi W. Molecular subtypes of bladder cancer. Curr Oncol Rep. 2018;20:77
4. Lin J, Jiang S, Chen B, Du Y, Qin C, Song Y. et al. Tertiary lymphoid structures are linked to enhanced antitumor immunity and better prognosis in muscle-invasive bladder cancer. Adv Sci (Weinh). 2025;12:e2410998
5. Li F, Zheng Z, Chen W, Li D, Zhang H, Zhu Y. et al. Regulation of cisplatin resistance in bladder cancer by epigenetic mechanisms. Drug Resist Updat. 2023;68:100938
6. Xu W, Liang T, Fang H, Fu L, Deng D, Tan X. et al. Single-cell RNA sequencing identifies MMP11+ cancer-associated fibroblasts as drivers of angiogenesis and bladder cancer progression. Adv Sci (Weinh). 2025;12:e02774
7. Hammouda K, Tokuyama N, Corredor G, Pathak T, Dakarapu R, Genega E. et al. AI-informed computational pathology classifier predicts outcomes across treatment modalities in muscle-invasive urothelial carcinoma. Cancer Lett. 2025;634:218059
8. Lopez-Beltran A, Cookson MS, Guercio BJ, Cheng L. Advances in diagnosis and treatment of bladder cancer. BMJ. 2024;384:e076743
9. Lin H, Fu L, Zhou X, Yu A, Chen Y, Liao W. et al. LRP1 induces anti-PD-1 resistance by modulating the DLL4-NOTCH2-CCL2 axis and redirecting M2-like macrophage polarisation in bladder cancer. Cancer Lett. 2024;593:216807
10. Wu Y, Ying Y, Zhang F, Shu X, Qi Z, Wang J. et al. NSUN2-mediated R-loop stabilization as a key driver of bladder cancer progression and cisplatin sensitivity. Cancer Lett. 2024;611:217416
11. Che G, Yin J, Wang W, Luo Y, Chen Y, Yu X. et al. Circumventing drug resistance in gastric cancer: A spatial multi-omics exploration of chemo and immuno-therapeutic response dynamics. Drug Resist Updat. 2024;74:101080
12. Huang Y, Chen C, Su M, Li D, Zhang J, Long K. et al. MCUB Inhibits PRKN-Dependent Mitophagic Degradation of PD-L1 to Promote Immune Evasion in Bladder Cancer. Adv Sci (Weinh). 2026;13:e14764
13. Li X, Wang C-Y. From bulk, single-cell to spatial RNA sequencing. Int J Oral Sci. 2021;13:36
14. Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52:1428-1442
15. Creighton CJ. Clinical proteomics towards multiomics in cancer. Mass Spectrom Rev. 2024;43:1255-1269
16. Lu Y, Chan Y-T, Wu J, Feng Z, Yuan H, Li Q. et al. CRISPR/Cas9 screens unravel miR-3689a-3p regulating sorafenib resistance in hepatocellular carcinoma via suppressing CCS/SOD1-dependent mitochondrial oxidative stress. Drug Resist Updat. 2023;71:101015
17. Akhoundova D, Rubin MA. Clinical application of advanced multi-omics tumor profiling: Shaping precision oncology of the future. Cancer Cell. 2022;40:920-938
18. Wang S-W, Gao C, Zheng Y-M, Yi L, Lu J-C, Huang X-Y. et al. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer. 2022;21:57
19. Wang M, Chen X, Tan P, Wang Y, Pan X, Lin T. et al. Acquired semi-squamatization during chemotherapy suggests differentiation as a therapeutic strategy for bladder cancer. Cancer Cell. 2022;40:1044-1059.e8
20. Xu N, Yao Z, Shang G, Ye D, Wang H, Zhang H. et al. Integrated proteogenomic characterization of urothelial carcinoma of the bladder. J Hematol Oncol. 2022;15:76
21. Liu Z, Liu L, Weng S, Xu H, Xing Z, Ren Y. et al. BEST: a web application for comprehensive biomarker exploration on large-scale data in solid tumors. J Big Data. 2023;10:165
22. Shen W, Song Z, Zhong X, Huang M, Shen D, Gao P. et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta. 2022;1:e36
23. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS. et al. Defining a Cancer Dependency Map. Cell. 2017;170:564-576.e16
24. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
25. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495-502
26. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289-1296
27. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
28. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163-172
29. Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496-502
30. Gouin KH 3rd, Ing N, Plummer JT, Rosser CJ, Ben Cheikh B, Oh C. et al. An N-cadherin 2-expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer. Nat Commun. 2021;12:4906
31. Xu Z, Wang W, Yang T, Li L, Ma X, Chen J. et al. STOmicsDB: a comprehensive database for spatial transcriptomics data sharing, analysis and visualization. Nucleic Acids Res. 2024;52:D1053-D1061
32. Liu C-J, Hu F-F, Xie G-Y, Miao Y-R, Li X-W, Zeng Y. et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 2023;24:bbac558
33. Liu C-J, Hu F-F, Xia M-X, Han L, Zhang Q, Guo A-Y. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34:3771-3772
34. Uhlen M, Fagerberg L, Hallstroem BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Tissue-based map of the human proteome. Science. 2015;347:1260419
35. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS. 2012;16:284-287
36. Li F, Zhang H, Huang Y, Li D, Zheng Z, Xie K. et al. Single-cell transcriptome analysis reveals the association between histone lactylation and cisplatin resistance in bladder cancer. Drug Resist Updat. 2024;73:101059
37. Yi L, Zhou X, Li T, Liu P, Hai L, Tong L. et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J Exp Clin Cancer Res. 2019;38:339
38. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493-500
39. Fustero-Torre C, Jimenez-Santos MJ, Garcia-Martin S, Carretero-Puche C, Garcia-Jimeno L, Ivanchuk V. et al. Beyondcell: targeting cancer therapeutic heterogeneity in single-cell RNA-seq data. Genome Med. 2021;13:187
40. Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270-W277
41. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374-403
42. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:78
43. Lei Y, Tang R, Xu J, Wang W, Zhang B, Liu J. et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J Hematol Oncol. 2021;14:91
44. Macnair W, Gupta R, Claassen M. psupertime: supervised pseudotime analysis for time-series single-cell RNA-seq data. Bioinformatics. 2022;38:i290-i298
45. Icard P, Fournel L, Wu Z, Alifano M, Lincet H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem Sci. 2019;44:490-501
46. Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer. 2019;19:326-338
47. Schulze A, Oshi M, Endo I, Takabe K. MYC Targets Scores Are Associated with Cancer Aggressiveness and Poor Survival in ER-Positive Primary and Metastatic Breast Cancer. Int J Mol Sci. 2020;21:8127
48. Oshi M, Takahashi H, Tokumaru Y, Yan L, Rashid OM, Matsuyama R. et al. G2M Cell Cycle Pathway Score as a Prognostic Biomarker of Metastasis in Estrogen Receptor (ER)-Positive Breast Cancer. Int J Mol Sci. 2020;21:2921
49. Jeong M-H, Urquhart G, Lewis C, Chi Z, Jewell JL. Inhibition of phosphodiesterase 4D suppresses mTORC1 signaling and pancreatic cancer growth. JCI Insight. 2023;8:e158098
50. Borrero LJH, El-Deiry WS. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta Rev Cancer. 2021;1876:188556
51. Hirano Y, Hendil KB, Yashiroda H, Iemura S, Nagane R, Hioki Y. et al. A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature. 2005;437:1381-1385
52. Wu H, Huang H, Zhao Y. Interplay between metabolic reprogramming and post-translational modifications: from glycolysis to lactylation. Front Immunol. 2023;14:1211221
53. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y. et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-580
54. Li J, Chen Z-S, Pan Y, Zeng L. The important role of lactylation in regulating DNA damage repair and tumor chemotherapy resistance. Drug Resist Updat. 2025;78:101148
55. Luo H, Vong CT, Chen H, Gao Y, Lyu P, Qiu L. et al. Naturally occurring anti-cancer compounds: shining from Chinese herbal medicine. Chin Med. 2019;14:48
56. Zhang X, Qiu H, Li C, Cai P, Qi F. The positive role of traditional Chinese medicine as an adjunctive therapy for cancer. Biosci Trends. 2021;15:283-298
57. Wang S, Long S, Deng Z, Wu W. Positive Role of Chinese Herbal Medicine in Cancer Immune Regulation. Am J Chin Med. 2020;48:1577-1592
58. Ming T, Tao Q, Tang S, Zhao H, Yang H, Liu M. et al. Curcumin: An epigenetic regulator and its application in cancer. Biomed Pharmacother. 2022;156:113956
59. Jiang H, Li M, Du K, Ma C, Cheng Y, Wang S. et al. Traditional Chinese Medicine for adjuvant treatment of breast cancer: Taohong Siwu Decoction. Chin Med. 2021;16:129
60. Gupta SC, Patchva S, Aggarwal BB. Therapeutic Roles of Curcumin: Lessons Learned from Clinical Trials. AAPS J. 2013;15:195-218
61. Tomeh MA, Hadianamrei R, Zhao X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int J Mol Sci. 2019;20:1033
62. Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X. et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22:85
63. Banerjee S, Ji C, Mayfield JE, Goel A, Xiao J, Dixon JE. et al. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc Natl Acad Sci U S A. 2018;115:8155-8160
64. Choi H, Chun Y-S, Kim S-W, Kim M-S, Park J-W. Curcumin inhibits hypoxia-inducible factor-1 by degrading aryl hydrocarbon receptor nuclear translocator: A mechanism of tumor growth inhibition. Mol Pharmacol. 2006;70:1664-1671
65. Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U. et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163-51171
66. Han JH, Lee E-J, Park W, Ha K-T, Chung H-S. Natural compounds as lactate dehydrogenase inhibitors: potential therapeutics for lactate dehydrogenase inhibitors-related diseases. Front Pharmacol. 2023;14:1275000
67. Das L, Vinayak M. Long Term Effect of Curcumin in Regulation of Glycolytic Pathway and Angiogenesis via Modulation of Stress Activated Genes in Prevention of Cancer. PLoS One. 2014;9:e99583
68. Wang X, Yu J, Liu X, Luo D, Li Y, Song L. et al. PSMG2-controlled proteasome-autophagy balance mediates the tolerance for MEK-targeted therapy in triple-negative breast cancer. Cell Rep Med. 2022;3:100741
69. Zhou L, Huang X, Zhang Y, Wang J, Li H, Huang H. PSMG3-AS1 enhances glioma resistance to temozolomide via stabilizing c-Myc in the nucleus. Brain Behav. 2022;12:e2531
70. Gera M, Sharma N, Ghosh M, Huynh DL, Lee SJ, Min T. et al. Nanoformulations of curcumin: an emerging paradigm for improved remedial application. Oncotarget. 2017;8:66680-66698
71. Yakubu J, Pandey AV. Innovative Delivery Systems for Curcumin: Exploring Nanosized and Conventional Formulations. Pharmaceutics. 2024;16:637
72. Moballegh Nasery M, Abadi B, Poormoghadam D, Zarrabi A, Keyhanvar P, Khanbabaei H. et al. Curcumin Delivery Mediated by Bio-Based Nanoparticles: A Review. Molecules. 2020;25:689
73. Kobylka P, Bakun P, Kuzminska J, Goslinski T, Murias M, Kucinska M. Insights into the Mode of Action of Novel Morpholinated Curcumin Derivatives Exhibiting Potent Antitumor Activity in Bladder Cancer Cells In Vitro. Molecules. 2025;30:295
74. Agha RA, Mathew G, Rashid R, Kerwan A, Al-Jabir A, Sohrabi C. et al. Transparency in the reporting of artificial intelligence - the TITAN guideline. Premier J Sci. 2025;10:100082
Author contact
![]() Corresponding authors: Dr. Fei Li, Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China. Email: feili20700338com. Dr. Wanlong Tan, Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China. E-mail: tanwanlongcom. Dr. Lina Hou, Department of Healthy Management, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China. E-mail: lnhou2010com.
Corresponding authors: Dr. Fei Li, Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China. Email: feili20700338com. Dr. Wanlong Tan, Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China. E-mail: tanwanlongcom. Dr. Lina Hou, Department of Healthy Management, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P. R. China. E-mail: lnhou2010com.