Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(12):6417-6433. doi:10.7150/ijbs.136823 This issue Cite
Research Paper
Targeting Kupffer Cell CD44-Mediated Ammonia Death via the ELAVL1-GLS Metabolic Circuit in Radiation-Induced Liver Disease
Yanyan Lin1,2*, Shuxuan Wang1,2*, Bufu Tang3*, Pan Zhou1,2, Xiaojun Zhang1,2, Huanliang Chen1,2, Jiaoyang Yang1,2, Shisuo Du1,2, Qian-Qian Zhao1,2 ![]() , Zhao-Chong Zeng1,2
, Zhao-Chong Zeng1,2 ![]()
1. Department of Radiation Oncology, Zhongshan Hospital, Fudan University, Shanghai, 200032, China.
2. Cancer Center, Zhongshan Hospital, Fudan University, Shanghai, 200032, China.
3. Department of Interventional Radiology, Zhongshan Hospital, Fudan University, Shanghai, 200032, China.
* These authors contributed equally to this work.
Received 2026-4-26; Accepted 2026-6-3; Published 2026-6-17
Abstract

Radiation-induced liver disease (RILD) remains a major dose-limiting toxicity in liver cancer radiotherapy. However, the underlying mechanisms, particularly the immunometabolic reprogramming that sustains inflammatory amplification following irradiation, are still poorly understood. Here, we report that Kupffer cell (KC) depletion significantly attenuated radiation-induced liver inflammation and injury. Irradiation markedly upregulated CD44 expression in KCs, and single-cell RNA sequencing revealed a CD44⁺ macrophage subset associated with injury, inflammatory activation, and metabolic stress signatures. Consistently, genetic deletion or pharmacological inhibition of CD44 protected against RILD by suppressing KC M1 polarization, oxidative stress accumulation, and inflammatory cytokine output. Furthermore, radiation exposure profoundly disrupted nitrogen homeostasis and increased intracellular ammonia levels, effects that were abrogated by CD44 deficiency. Mechanistically, CD44 stabilized the RNA-binding protein ELAVL1 by inhibiting its proteasomal degradation, which subsequently increased the abundance and activity of glutaminase (GLS), elevated the intracellular ammonia burden, and triggered macrophage death. Pharmacological inhibition of GLS with CB-839 rescued the injurious phenotype, while exogenous NH4Cl supplementation confirmed the ammonia dependence of the downstream phenotypes. Our findings reveal a CD44-ELAVL1-GLS immunometabolic circuit that links radiation stress to ammonia dysregulation in KCs and subsequent liver injury, suggesting a potential therapeutic target for RILD.
Keywords: radiation-induced liver disease, Kupffer cells, CD44, glutaminase, ammonia-induced cell death
Introduction
Hepatocellular carcinoma imposes a substantial clinical burden, and many patients present at stages when curative resection or ablation is not feasible[1-3]. Liver-directed radiotherapy is increasingly used for liver cancer, particularly for unresectable lesions, to balance local control with functional hepatic reserve[4-6]. However, exceeding established thresholds of irradiated liver volume and dose can lead to radiation-induced liver disease (RILD), a pathological process driven by severe oxidative stress and inflammation, which presents as hepatic dysfunction and can progress to liver failure[7-9]. The biological programs driving RILD remain incompletely understood, and current clinical management mainly relies on supportive therapy[10]. This diagnostic and therapeutic limitation highlights the urgent need to elucidate core injury mechanisms for developing targeted preventative strategies.
Emerging evidence indicates that RILD is a multicellular process driven not only by parenchymal stress, but also by nonparenchymal cell (NPC) remodeling within the hepatic microenvironment[11-13]. Kupffer cells (KCs), the resident macrophages of the liver, constitute a prominent fraction of NPCs and serve as sentinels of tissue homeostasis[14]. Beyond direct target-cell radiotoxicity, the radiation-induced bystander effect propagates sustained sterile inflammation across neighboring tissue zones[15]. Specifically, radiation-induced parenchymal damage triggers massive release of damage-associated molecular patterns (DAMPs) into the extracellular space. Acting as danger signals, these DAMPs are rapidly intercepted by KCs, thereby translating the initial physical injury into an active inflammatory cascade[16]. Consequently, activated KCs rapidly reprogram the local microenvironment through cytokine release and reactive oxygen species (ROS) production[17, 18]. However, during RILD, the mechanistic coupling between inflammatory programs and cellular metabolic stress in KCs remains insufficiently defined.
Radiation effects on immune cells can extend beyond DNA damage responses to include metabolic remodeling that reinforces inflammatory activation. In the liver, nitrogen handling is tightly coupled to both tissue homeostasis and immune-cell fitness. Crucially, excessive ammonia accumulation is a potent trigger of mitochondrial dysfunction and oxidative stress in immune cells[19, 20]. Recent studies have highlighted the disruption of nitrogen balance as a critical determinant of immune cell survival[21, 22], suggesting that impaired ammonia buffering is an underappreciated contributor to oxidative injury in RILD. Macrophages are particularly vulnerable to metabolic stress because of the absence of a functional urea cycle for enzymatic detoxification. Unlike parenchymal hepatocytes, which possess an immense capacity to clear free ammonia, KCs are intrinsically vulnerable to this nitrogenous stress[23]. Unbuffered free ammonia acts as a lipophilic weak base that readily permeates and alkalinizes highly acidic endolysosomal compartments[24, 25]. This rapid neutralization disrupts autophagic flux, triggers mitochondrial permeability transition, and drives an unresolvable collapse of organelle homeostasis, defined as ammonia-induced cell death[26, 27]. Paradoxically, this lethal stress is a direct byproduct of immune activation. During M1 polarization, KCs undergo profound metabolic reprogramming to support inflammatory cascades, becoming highly dependent on glutaminolysis as an alternative cellular fuel[28-30]. Driven by this metabolic shift, glutaminase (GLS) converts glutamine to glutamate with concomitant release of NH₃, functioning as a major intracellular source of ammonia and a potential regulator of redox balance[31].
CD44, a hyaluronan receptor best known for its role in adhesion and tumor biology, is widely expressed on myeloid cells and has emerged as a regulator of macrophage activation[32]. Rather than serving solely as a structural anchor, CD44 functions as an advanced microenvironmental sensor, converting extracellular physical stress into profound intracellular remodeling[33]. Recent studies have shown that CD44 can directly bridge these pathological cues to dynamic alterations in mitochondrial metabolism and downstream programs, thereby sustaining inflammatory macrophage phenotypes[34, 35]. However, whether CD44 couples radiation stress to nitrogen metabolic imbalance in the liver and whether such an immunometabolic axis contributes to RILD remain unclear. Given these immunometabolic perspectives, we hypothesized that radiation triggers detrimental metabolic reprogramming in KCs driven by CD44 activation, which in turn increases the stability of the ELAVL1-GLS circuit and promotes ammonia accumulation and mitochondrial oxidative injury, ultimately leading to KC death. By identifying the specific CD44-ELAVL1-GLS cascade, our study reveals how highly activated macrophages drive inflammatory amplification during RILD and establishes this pathway as a precise immunometabolic target for mitigating RILD.
Materials and Methods
Animals and mouse model construction
Male C57BL/6N mice (6-8 weeks old) were used to establish the RILD model. The mice were anesthetized and subjected to targeted liver irradiation using an Elekta Axesse™ linear accelerator. A single fraction of 25 Gy was delivered to the whole liver (6 MV, 3 Gy/min) to induce acute liver injury, as previously described[36-38], with lead shielding for nonhepatic organs. For KC depletion, clodronate liposomes (Cld-Lipo, 5 mg/ml; Yeasen, China) were administered intraperitoneally (200 μl per mouse) 24 h before treatment. Cd44 knockout (Cd44⁻/⁻) mice on a C57BL/6N background were generated by CRISPR/Cas9 and obtained from Cyagen Biosciences (Suzhou, China). For pharmacological inhibition, a CD44 inhibitor (7 mg/kg, MCE, USA) was administered intraperitoneally starting 24 h before irradiation and continued daily until 48 h after irradiation. All mice were maintained under specific pathogen-free (SPF) conditions, and all animal procedures were approved by the Animal Ethics Committee of Zhongshan Hospital, Fudan University (Approval No. 2023-263).
Hematoxylin and eosin (HE), immunofluorescence (IF) and immunohistochemistry (IHC) staining
The mice were fasted overnight before the experiment. We first perfused the mouse livers with PBS to remove blood and then fixed them in 4% paraformaldehyde. Clinical specimens were obtained from Zhongshan Hospital with approval from its Institutional Review Board (Approval No. B2025-547). Following paraffin embedding, the tissues were sectioned and subjected to corresponding staining. We then captured images using light or fluorescence microscopy. Antibody information is provided in Supplementary Table S2.
Flow cytometry
Single-cell suspensions from mouse liver tissues were prepared by perfusion followed by collagenase IV digestion, red blood cell lysis, and filtration. Cultured cells were gently detached, filtered through a 40 μm cell strainer, and collected as single-cell suspensions. Cells were stained with the indicated antibodies or fluorescent probes protected from light and analyzed on a BD FACSAria III flow cytometer. Antibody information is provided in Supplementary Table S3.
Single-cell RNA sequencing
Following vascular perfusion, mouse liver tissues were immediately collected and dissociated into single-cell suspensions. After filtration and viability assessment, single-cell libraries were generated using the 10× Genomics platform and sequenced by a commercial service provider (OE Biotech, Shanghai, China).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9 and R software. Comparisons between two groups were conducted using unpaired two-tailed Student's t tests where appropriate. Correlation analyses were performed using Pearson's correlation coefficient, and correlation strength and significance are reported as R and P values. The data are presented as the mean ± SD. Statistical significance was defined as follows: ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Radiation induces M1 KC polarization, shaping the inflammatory microenvironment in RILD
Transcriptomic profiling of liver tissues revealed that irradiation broadly upregulated immune and inflammatory genes (Fig. 1A). Functional analyses indicated strong enrichment of immune activation, chemotaxis, NF-κB signaling, and inflammatory response pathways (Fig. 1B-D). Macrophage activation, including increased expression of M1-associated markers (e.g., Cd86 and Nos2) and inflammatory mediators (Ifng, Tnf, Il6, and Il1b), was prominent (Fig. 1E and F, S1A and B). Consistent with the transcriptomic changes, KCs adopted an M1-like phenotype after irradiation, as evidenced by increased iNOS levels without a marked change in CD163 levels (Fig. 1G and H). Flow cytometry confirmed a significant increase in M1-like KC polarization after irradiation (Fig. 1I). Concurrently, irradiated KCs exhibited markedly elevated expression of the key recruitment chemokines Ccl2 and Cxcl1, indicating their active role in reshaping the hepatic immune microenvironment (Fig. S1C). These findings indicate that KC M1 polarization is closely associated with the dysregulated inflammatory microenvironment of the irradiated liver.
Irradiation activates KCs toward an M1 phenotype and exacerbates the severity of RILD. (A) Heatmap showing distinct transcriptional profiles between non-irradiated (NIR) and irradiated (IR) mouse livers. (B) GO enrichment analysis of upregulated differentially expressed genes (DEGs) in IR livers (top 30 terms). (C) KEGG enrichment of upregulated DEGs in IR group (top 20 pathways). (D) GSEA of inflammatory related gene sets. (E) WikiPathways enrichment of the top 20 upregulated pathways including macrophage markers. (F) Heatmap of representative inflammation-related genes elevated in IR livers. (G) IF images of F4/80 (yellow) with iNOS (purple) and CD163 (green) co-staining in the indicated groups. DAPI, blue. (H) IHC staining for iNOS and CD163 signals in IR and NIR livers. (I) Flow cytometric analysis of CD86⁺ KCs. (J) WikiPathways enrichment identifying pathways activated by irradiation and reduced after KCs depletion. (K) GO biological process enrichment showing radiation-induced pathways reversed by Cld-Lipo treatment. (L) Liver histology changes as shown by HE staining. (M) Serum ALT and AST levels (n=5). (N) Hepatic TNF-α and IL-1β concentrations (n=5). (O) γH2AX staining (green) indicating changes of DNA damage signals. Statistical significance: ns, not significant; **P < 0.01, ****P < 0.0001; two-tailed Student's t-test.
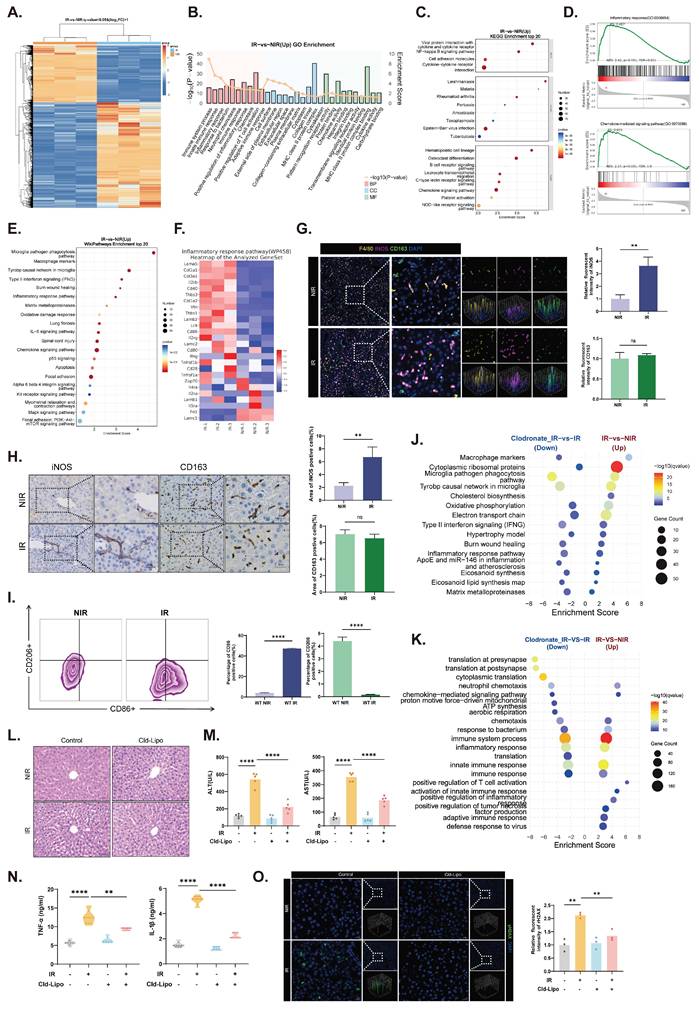
To determine the functional contribution of KCs, we depleted liver macrophages using Cld-Lipo (Fig. S1D)[39]. Depletion of KCs substantially disrupted the radiation-induced transcriptional program, downregulated the expression of macrophage-related genes, and suppressed the activity of inflammatory pathways (Fig. 1J and K). The pathways attenuated by KC depletion overlapped with those activated by radiation exposure, demonstrating that KCs are essential mediators of the hepatic inflammatory cascade in response to irradiation. Depletion of KCs alleviated radiation-induced liver histologic damage (Fig. 1L). Similarly, the elevations in serum ALT/AST and hepatic TNF-α/IL-1β levels were significantly reduced after KC depletion (Fig. 1M and N). γH2AX staining also revealed markedly reduced DNA damage in irradiated livers after Cld-Lipo treatment, indicating a reduction in radiation-associated genomic injury (Fig. 1O).
Collectively, these data illustrate that radiation-induced inflammation, liver injury, and DNA damage are strongly associated with KCs, indicating that KCs are central drivers of the pathological microenvironment in RILD.
CD44 is a KC-enriched regulator of RILD pathogenesis
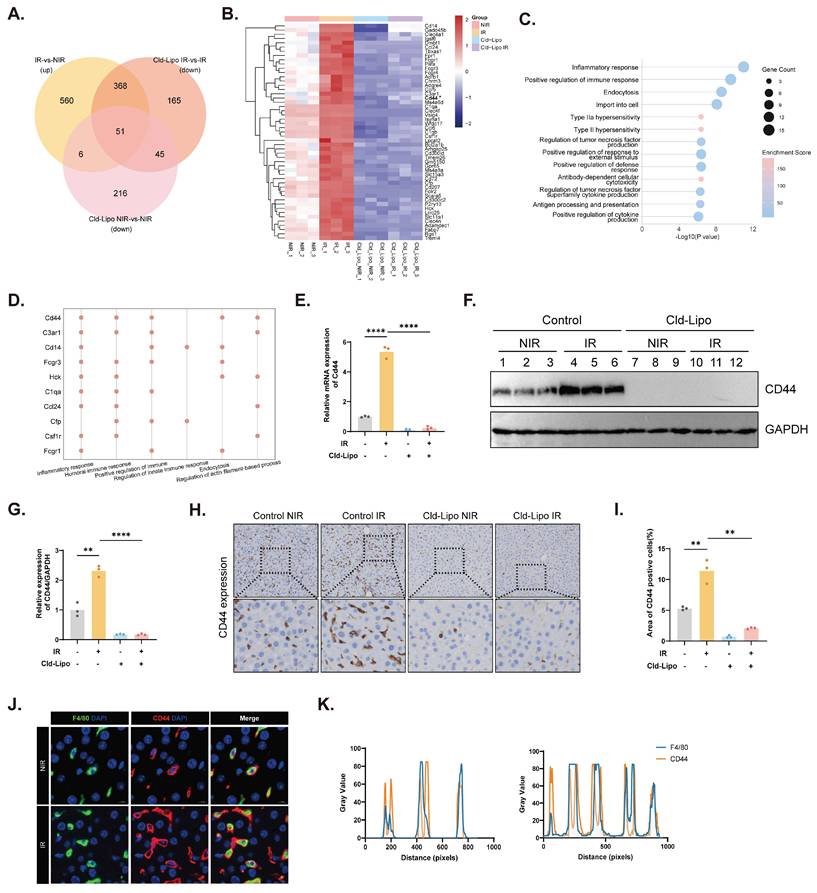
An intersection analysis strategy yielded 51 KC-dependent candidate genes whose expression responded to irradiation but was lost when KCs were removed (Fig. 2A and B). Metascape enrichment analysis revealed that these genes were predominantly involved in immune and inflammatory pathways (Fig. 2C). Among the top-ranked candidates, Cd44 emerged together with C3ar1, Cd14, Fcgr3, Hck, C1qa, Ccl24, Cfp, Csf1r, and Fcgr1 (Fig. 2D). We confirmed marked CD44 upregulation at both the RNA and protein levels after irradiation, whereas this was largely abolished by KC depletion (Fig. 2E-G, S2A). IHC staining further confirmed these results (Fig. 2H and I), and co-immunofluorescence localized CD44 to F4/80⁺ macrophages (Fig. 2J and K). These findings establish CD44 as a KC-enriched mediator activated during RILD.
CD44 defines a potential regulatory node in KCs during RILD. (A)Venn diagram overlapping 51 candidate genes associated with KC-dependent regulatory node of RILD. (B) Heatmap of the 51 candidate genes expression in NIR, IR, Cld-Lipo NIR, and Cld-Lipo IR groups. (C) Metascape enrichment analysis of the 51 candidate genes. (D) Top 10 immune-associated genes out of the 51 candidates. (E) mRNA expression of Cd44 under the indicated experimental conditions (n=3). (F and G) Protein expression of CD44 in liver tissues (F) with corresponding quantification (G). (H and I) CD44 expression in different conditions as shown by IHC staining (H) with corresponding quantification (I) (n=3). (J and K) IF staining results (J) for CD44 (red) localizing in KCs (F4/80, green) within liver tissues with line scan quantification (K). Statistical significance: **P < 0.01, ****P < 0.0001; two-tailed Student's t-test.
Analysis of normal liver samples from the TCGA cohort revealed a significant positive correlation between CD44 expression and an aggregate M1 macrophage signature (Fig. 3A). Consistently, correlation heatmaps further demonstrated that CD44 expression was positively associated with the expression of individual M1 markers and inflammatory mediators, including CD86, IFNG, CCL2, IL1B, IL6, and TNF (Fig. 3B). The clinical relevance of CD44 was supported by paired human liver specimens collected before and after radiotherapy. Postirradiation samples displayed characteristic pathological features of RILD together with pronounced CD44 upregulation in irradiated regions (Fig. 3C). Radiologic assessment revealed MRI features characteristic of RILD, including a T1-hypointense hepatic lesion with peripheral enhancement, heterogeneous internal signals, and accompanying perfusion abnormalities (Fig. 3C).
CD44 drives M1 macrophage polarization after radiation. (A) Analysis of normal liver tissue from TCGA between CD44 expression and the M1 macrophage gene signature. (B) Correlation heatmap of positive relationships between CD44 and representative M1 markers and inflammatory cytokines including CD86, IFNG, CCL2, IL1B, IL6, TNF from TCGA normal liver tissues. (C) Representative samples from RILD patients' livers before (POR-) and after (POR+) radiotherapy, showing HE staining (top panel), IHC staining of CD44 (bottom panel), and MRI images (right panel). (D) UMAP visualization of single-cell RNA sequencing data of macrophage subpopulations in mouse livers based on CD44 expression. (E) DEG analysis between CD44⁺ and CD44⁻ macrophages. (F) GO analysis revealed CD44⁺ macrophages were specifically associated with immune-related pathways. (G) Pathway enrichment analysis in CD44⁺ macrophages. (H, I and K) Expression of CD44 and pro-inflammatory cytokines in KCs under indicated treatment. (J) Protein expression of iNOS in KCs with quantification (n=3). (L) Levels of cytokines in KCs conditioned media (n=5). (M and N) Expression of CD44 in BMDMs in response to radiation dose (M) and over time (N). (O and P) ROS levels in BMDMs (O) and liver tissue (P) detected by DCFH-DA with corresponding quantification. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-tailed Student's t-test.
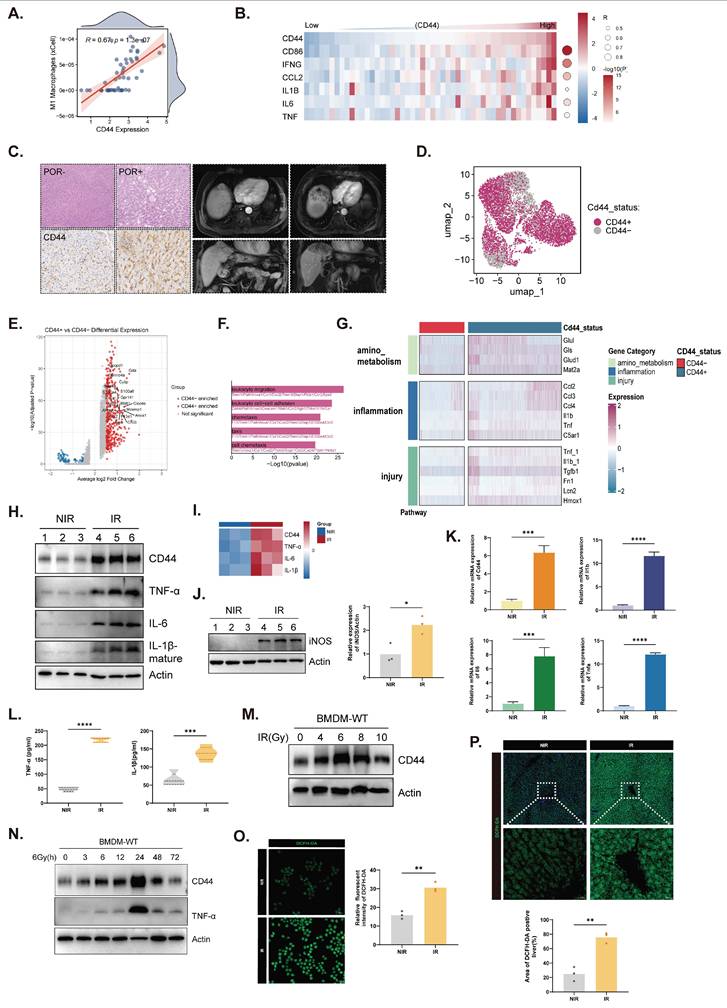
Single-cell RNA sequencing of mouse livers further distinguished CD44⁺ from CD44⁻ macrophage subpopulations, revealing distinct transcriptional signatures (Fig. 3D and E). CD44⁺ macrophages were enriched in inflammatory, injury, and amino acid metabolic pathways (Fig. 3F and G), indicating the emergence of a specialized proinflammatory subset during RILD. In KCs, irradiation increased the expression of CD44 and the proinflammatory cytokines TNF-α, IL-6, and IL-1β at the mRNA and protein levels (Fig. 3H, I, and K), as well as the M1 marker iNOS (Fig. 3J). These mediators were also elevated in KC-conditioned media (Fig. 3L). In bone marrow-derived macrophages (BMDMs), irradiation also induced CD44 expression, which peaked at 6 Gy and 24 h (Fig. 3M and N). Increased ROS production in irradiated BMDMs and livers reflected heightened oxidative stress, consistent with these activated states (Fig. 3O and P). Cell-cell communication analysis further mapped the extensive ligand-receptor interactions between this CD44⁺ subset and NPCs, highlighting its core role in orchestrating immune remodeling (Fig. S3A-C). Together, these findings support CD44 as a radiation-response node associated with proinflammatory macrophage polarization in RILD.
CD44 deficiency protects against RILD by suppressing KC M1 polarization
Cd44⁻/⁻ mice were constructed to evaluate the functional contribution of CD44 (Fig. S4A). Under physiological conditions, CD44 deficiency did not cause obvious histological abnormalities in major organs (Fig. S4B). Compared with those in control mice, M1 polarization was suppressed in both KCs and BMDMs derived from Cd44⁻/⁻ mice (Fig. 4A-C), indicating that CD44-dependent macrophage activation was attenuated. Furthermore, conditioned media from irradiated Cd44⁻/⁻ KCs contained lower concentrations of secreted inflammatory mediators (Fig. 4D). Cd44⁻/⁻ KCs also presented reduced TNF-α, IL-6, and IL-1β expression at both the protein and mRNA levels (Fig. 4E, S4C and D). A diminished inflammatory response was similarly observed in Cd44⁻/⁻ BMDMs, as confirmed by immunoblot analyses and IF (Fig. 4F and G). Compared with control cells, ROS formation in irradiated Cd44⁻/⁻ BMDMs was reduced (Fig. 4H). Moreover, the suppression of M1 polarization in Cd44⁻/⁻ KCs was accompanied by reduced expression of Ccl2 and Cxcl1 (Fig. S4E), further supporting the role of CD44 in KC-driven immune remodeling.
Effects of CD44 deficiency on liver injury and macrophage activation following irradiation. (A and B) CD86⁺ cells in KCs (A) and BMDMs (B) under different conditions with representative images (left) and quantification (right) (n=3). (C) Immunoblot analysis of iNOS in KCs isolated from WT and Cd44⁻/⁻ mice under irradiation (left) and corresponding quantification (right). (D) Cytokine concentrations in conditioned media from KCs (n=5). (E and F) Expression of CD44 and cytokines in KCs and BMDMs. (G) IF images (left) of inflammatory mediator expression in BMDMs across the indicated groups with corresponding quantification (right). (H) ROS levels in BMDMs. (I) Representative liver histology from WT and Cd44⁻/⁻ mice under NIR and IR conditions. (J) Serum ALT and AST levels in mice with or without irradiation (n=5). (K) IF staining of iNOS, CD163, and F4/80 in mice liver sections under IR conditions. (L) γH2AX staining in liver sections in the four groups. (M) ROS levels in liver tissues under NIR and IR conditions (left) with corresponding quantification (right). (N) Body-weight changes of mice before and after irradiation (n=5). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-tailed Student's t-test.
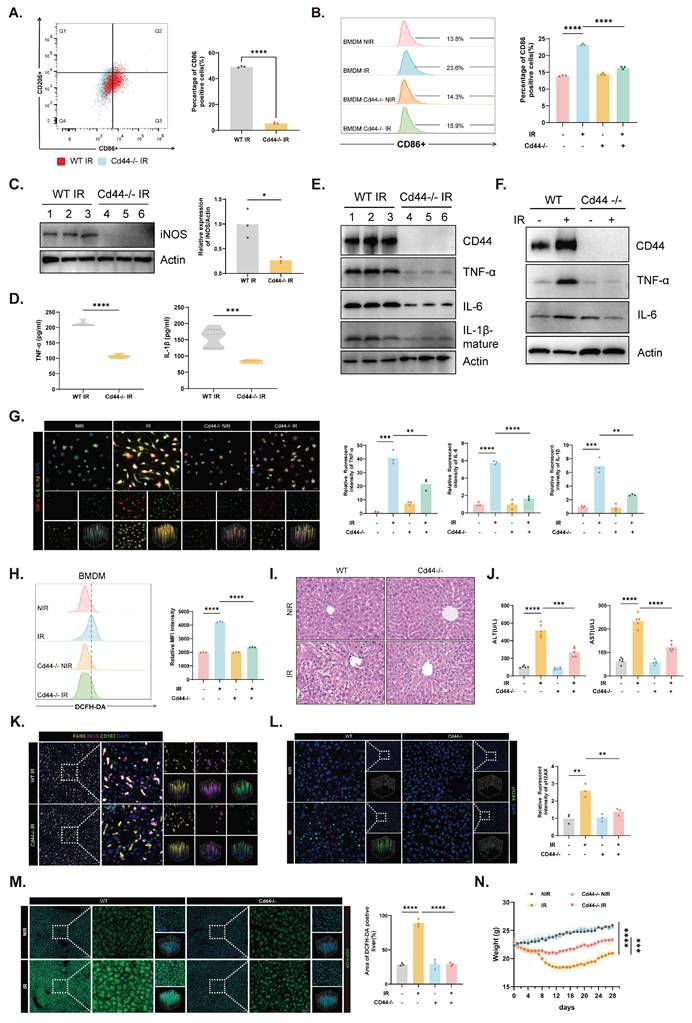
In vivo, compared with control mice, irradiated Cd44⁻/⁻ mice developed less liver injury, with improved histology and serum ALT/AST levels (Fig. 4I and J). Lower iNOS expression was observed in liver sections from irradiated Cd44⁻/⁻ mice than in those from control mice (Fig. 4K and S4F). The percentage of γH2AX-positive cells was also reduced in irradiated Cd44⁻/⁻ livers, indicating less DNA damage (Fig. 4L). Furthermore, less ROS formation in irradiated Cd44⁻/⁻ liver tissues was consistent with the cellular phenomenon (Fig. 4M). Concurrently, CD44 deficiency alleviated overall body weight loss and promoted faster recovery during the postirradiation period (Fig. 4N and S4G).
These data collectively reveal that CD44 is required for the irradiation-induced M1 polarization of KCs and that CD44 deficiency broadly protects against RILD by restraining macrophage-driven inflammatory signaling and oxidative stress.
CD44 regulates ammonia dysregulation and macrophage death following irradiation
To define the downstream mechanisms of CD44 in RILD, we performed transcriptomic profiling of KCs isolated from WT and Cd44⁻/⁻ mice. Functional enrichment revealed attenuated inflammatory, immune, and oxidative stress pathways in irradiated Cd44⁻/⁻ KCs compared with controls (Fig. 5A and B). GSEA further revealed broad metabolic perturbations in irradiated KCs, including changes in nitrogen metabolism, amino acid metabolism, the urea cycle, and arginine metabolism (Fig. 5C, S5A). In particular, primary KCs exhibited minimal expression of core urea cycle enzymes (Cps1, Otc, Ass1, and Asl) compared with hepatocytes (Fig. S5B-E), indicating a limited intrinsic capacity for ammonia clearance. Further analysis at the single-cell level revealed that a glutamine/ammonia-stress module was predominantly enriched within the CD44⁺ KC subpopulation (Fig. S5F). Notably, these metabolic disturbances were largely reversed by CD44 deficiency (Fig. 5C, S5A), suggesting that irradiation reprograms KC metabolism toward a CD44-dependent nitrogen-stress state, setting the stage for impaired ammonia homeostasis.
CD44 links irradiation to ammonia metabolic dysregulation and macrophage death. (A and B) GO biological process and KEGG pathway enrichment analysis of genes downregulated in Cd44⁻/⁻ KCs compared to WT KCs under irradiation. (C) GSEA results showing suppression of arginine and nitrogen metabolism in IR KCs compared with NIR KCs, with restoration upon Cd44⁻/⁻ under IR conditions. (D) TEM images of mitochondrial structural damage in BMDMs following IR treatment. (E and F) Time- and dose-dependent changes in intracellular ammonia levels in BMDMs following irradiation. (G) Intracellular ammonia levels in BMDMs. (H and I) Lysosomal (H) and mitochondrial (I) signals in BMDMs. (J) Flow cytometric analysis of mitochondrial membrane potential (top), followed by quantification (bottom). (K) Mitochondrial ROS levels of BMDMs, with or without NH4Cl (12 mM, 24 h). Representative histograms, top; quantification, bottom. (L) Macrophage death under NIR or IR conditions in BMDMs, with or without NH4Cl. Histograms, left; quantification, right. Statistical significance: **P < 0.01, ***P < 0.001, ****P < 0.0001; n=3; two-tailed Student's t-test.
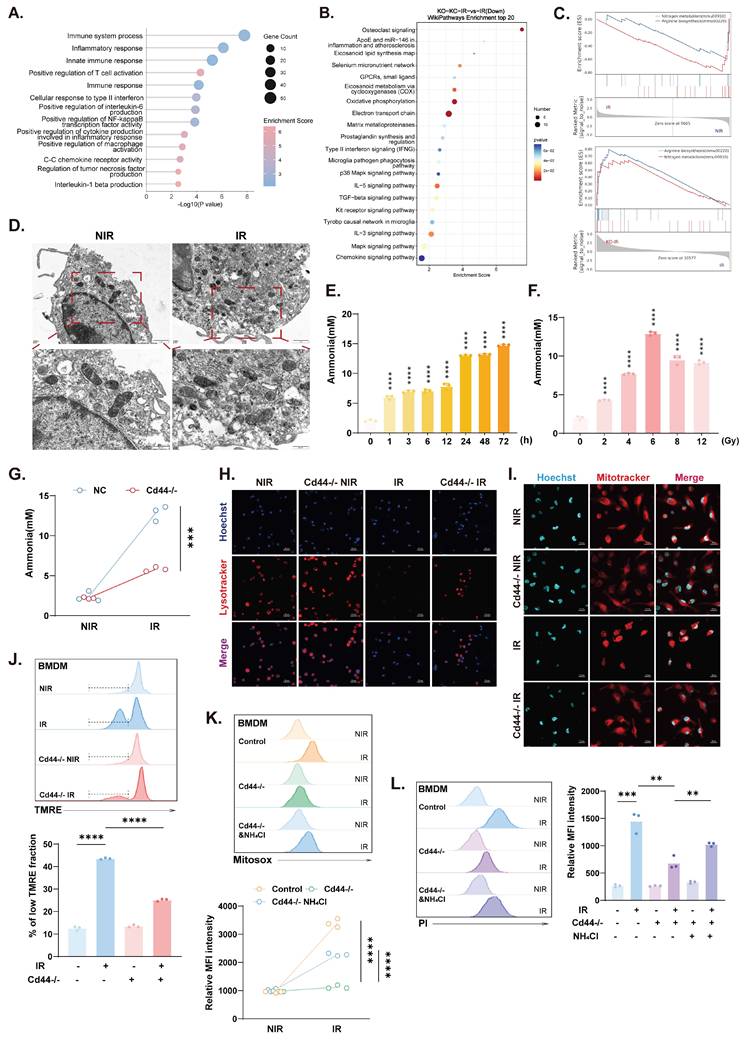
Transmission electron microscopy (TEM) revealed mitochondrial swelling and cristae disruption in irradiated BMDMs (Fig. 5D), which is consistent with the ultrastructural features of ammonia-induced cell death[21]. Intracellular ammonia levels increased in irradiated BMDMs in a time- and dose-dependent manner (Fig. 5E and F), and this effect was markedly reversed in Cd44⁻/⁻ cells (Fig. 5G), indicating that CD44-dependent disruption of ammonia metabolism occurred. Crucially, direct measurement of hepatic ammonia confirmed that the irradiation-induced ammonia burden was significantly attenuated in Cd44⁻/⁻ mice compared with controls (Fig. S5G), consistent with the protection observed in vitro. Furthermore, ammonia dysregulation in irradiated macrophages was accompanied by lysosomal impairment and mitochondrial defects, including structural disruption, depolarization, and oxidative stress (Fig. 5H-K). Depletion of CD44 significantly alleviated this multi-organelle stress and protected macrophages from ammonia-induced cell death (Fig. 5L). To assess the causal role of ammonia toxicity, exogenous NH4Cl was applied at 12 mM for 24 h to recapitulate the pathological ammonia accumulation and stress observed in our irradiated BMDMs. The addition of NH4Cl alone had a minimal effect, while it abolished the protection conferred by CD44 deficiency, restoring mitochondrial ROS levels and cell death to those observed in the irradiated controls (Fig. 5K and L).
As a result, irradiation disrupts ammonia homeostasis in macrophages in a CD44-dependent manner. CD44 depletion partially restored ammonia metabolic balance, attenuated organelle damage, and protected macrophages from radiation-induced death, indicating that the a CD44-ammonia axis is involved in this process.
CD44 engages an ELAVL1-GLS circuit to drive ammonia-dependent macrophage dysfunction
We next examined whether CD44 regulates GLS, the major ammonia-producing enzyme. In BMDMs, irradiation clearly increased GLS expression and enzymatic activity, which were markedly attenuated in Cd44⁻/⁻ cells (Fig. 6A and B). Pharmacological inhibition of GLS with CB-839[40] reduced macrophage death (Fig. 6C), decreased ammonia accumulation (Fig. 6D), alleviated mitochondrial superoxide stress (Fig. 6E), and suppressed inflammatory cytokine release under irradiation (Fig. 6F), indicating that GLS-dependent ammonia production occurs downstream of CD44.
CD44 stabilizes ELAVL1 to enhance GLS-dependent metabolic dysfunction in macrophages. (A) Immunoblot analysis of GLS and CD44 with or without irradiation. (B) GLS enzymatic activity measured in BMDMs under the indicated conditions. (C) Propidium iodide staining showing macrophage death in response to irradiation and GLS inhibitor CB-839; representative histograms (left) and quantification (right). (D) Measurement of intracellular ammonia levels in BMDMs under irradiation with or without CB-839 (n=3). (E) Mitochondrial ROS levels in BMDMs following irradiation and GLS inhibition (CB-839, 5 μM). (F) Levels of TNF-α and IL-1β concentration of BMDM-conditioned media following irradiation and GLS inhibition (n=3). (G) Silver staining gel images of immunoprecipitated complexes from CD44-overexpressing 293T cells. (H) PPI network constructed from the intersection of CD44 pull-down candidates and STRING-annotated CD44 interactors. (I) Three major functional modules identified by cluster analysis of the PPI network. (J) Hub analysis highlighting ELAVL1 as a top-ranked candidate within the network. (K) Protein expression of GLS and MYC-ELAVL1 overexpression in 293T cells. (L) Molecular docking model of the CD44-ELAVL1 interaction. Docking score: -268.83. (M and N) IF images (M) showing co-localization of CD44 (red) and ELAVL1 (green) in macrophages with corresponding line scan quantification (N). (O) Coimmunoprecipitation in 293T cells with or without Flag-CD44 overexpression. (P) Protein expression of CD44 and ELAVL1 under NIR or IR conditions. (Q) Immunoblot analysis showing dose-dependent induction of ELAVL1 and CD44 in 293T cells. (R) Increased CD44-ELAVL1 association under IR condition in 293T cells endogenous coimmunoprecipitation. (S) ELAVL1 protein stability measured by cycloheximide (CHX, 100 µg/mL) chase assay in THP-1 macrophages. (T) Protein expression of ELAVL1 in THP-1 with or without treatment of MG132 (10 μM). (U) Reduced polyubiquitination of ELAVL1 with Flag-CD44 overexpression. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
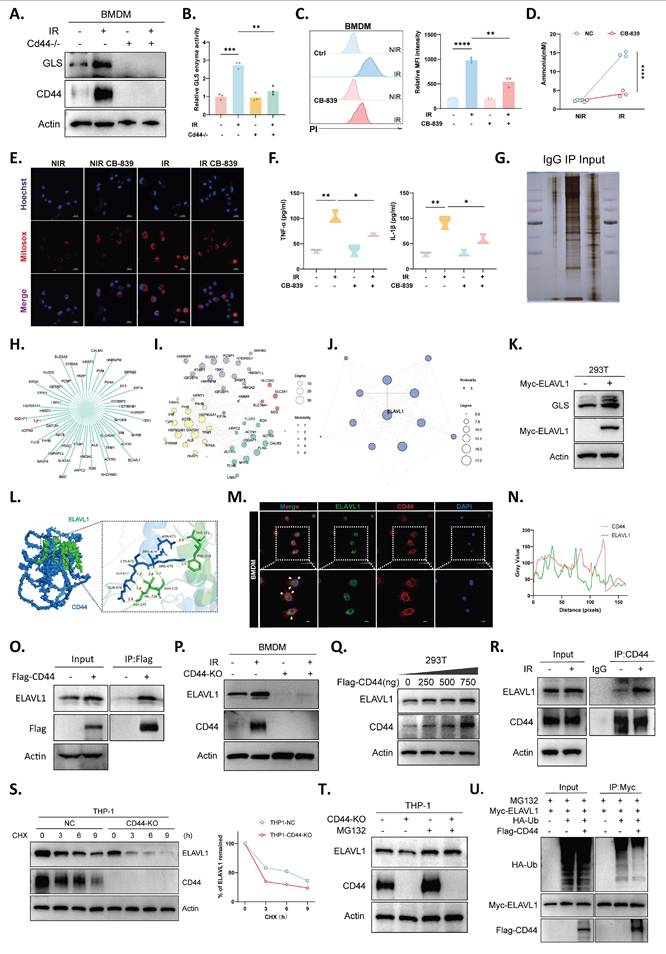
An LC-MS interactome screen in CD44-overexpressing cells identified a set of candidates linking CD44 to GLS regulation (Fig. 6G). A protein-protein interaction (PPI) network integrating these candidates with STRING-predicted interactors was constructed (Fig. 6H) and organized into several distinct functional modules (Fig. 6I), with hub analysis highlighting ELAV-like RNA binding protein 1 (ELAVL1/HuR) as a central node (Fig. 6J). Since ELAVL1 is known to stabilize GLS mRNA and increase GLS expression[41], we considered the CD44-ELAVL1-GLS axis as a potential mechanism linking CD44 to ammonia production. Indeed, ELAVL1 overexpression increased GLS protein levels (Fig. 6K). Structural modeling revealed a binding interface between CD44 and ELAVL1 (Fig. 6L). Consistent with this prediction, confocal imaging revealed their intracellular colocalization (Fig. 6M and N), and their direct interaction was confirmed by coimmunoprecipitation (Fig. 6O). In macrophages, irradiation drove parallel upregulation of both proteins, whereas CD44 deficiency blunted the induction of ELAVL1 expression (Fig. 6P), supporting the role of ELAVL1 as a downstream component regulated by CD44.
Mechanistically, increasing CD44 expression increased ELAVL1 protein level in a dose-dependent manner without altering ELAVL1 mRNA level (Fig. 6Q, S6A). Radiation also strengthened the endogenous interaction between CD44 and ELAVL1 (Fig. 6R). Protein stability assays revealed accelerated ELAVL1 degradation upon CD44 loss (Fig. 6S), whereas proteasome inhibition restored ELAVL1 levels in CD44⁻/⁻ cells (Fig. 6T). Moreover, ubiquitination assays revealed reduced ELAVL1 polyubiquitination in the presence of CD44 overexpression (Fig. 6U), indicating that CD44-dependent regulation of ELAVL1 stability occurs via the ubiquitin-proteasome pathway. Together, these data support a mechanistic cascade in which CD44 stabilizes ELAVL1, thereby enhancing GLS-driven ammonia production and promoting macrophage dysfunction under irradiation. Notably, this coordinate upregulation of ELAVL1 and GLS and its dependence on CD44 were also observed in vivo, as irradiated whole-liver lysates from Cd44⁻/⁻ mice showed markedly reduced ELAVL1 and GLS protein levels compared with controls (Fig. S6B). Analysis of public liver transcriptomic cohorts further revealed a positive correlation between CD44 and GLS expression (Fig. S6C), underscoring the translational value of this immunometabolic axis.
Targeting CD44 alleviates RILD by restoring inflammatory and metabolic homeostasis
CD44 was targeted with a hyaluronic acid-based inhibitor both in vitro and in vivo[42]. Pharmacological inhibition of CD44 significantly suppressed intracellular ammonia accumulation, along with radiation-induced GLS expression and enzymatic activity in macrophages (Fig. 7A-C). This metabolic restoration coincided with improved mitochondrial membrane integrity (Fig. 7D), a marked reduction in both mitochondrial and cellular ROS generation (Fig. 7E and F), and a subsequent decrease in macrophage death (Fig. 7G). Furthermore, CD44 blockade effectively restrained the proinflammatory response, as evidenced by downregulated iNOS expression in both macrophages and liver tissues (Fig. 7H and I) and reduced hepatic TNF-α and IL-1β levels (Fig. 7J). Consequently, this intervention significantly decreased serum ALT and AST levels (Fig. 7K), which is consistent with diminished inflammatory infiltration and preserved tissue architecture upon HE staining (Fig. 7L). At the molecular level, CD44 inhibitor-treated livers exhibited mitigated DNA damage, as indicated by a substantially diminished γH2AX-positive area (Fig. 7M).
Inhibition of CD44 protects against RILD through the inflammatory-metabolic axis. (A) Intracellular ammonia levels in BMDMs treated with or without CD44 inhibitor (CD44i) following irradiation (n=3). (B) Protein expression of GLS in BMDMs under indicated conditions. (C) Enzymatic activity of GLS in BMDM lysates following various treatment (n=3). (D) Mitochondrial membrane potential assessed by TMRE staining. (E) Mitochondrial superoxide levels presented as histograms (left) and quantification (right) (n=3). (F) Measurement of total cellular ROS levels in BMDMs (left), with quantification (right) (n=3). (G) Propidium iodide staining showing cell death rate, shown as representative histograms (left) and quantification (right) (n=3). (H) Protein expression levels of iNOS in BMDMs under CD44i- and CD44i+. (I) iNOS (red) expression in liver tissues (left) with relative quantification (right) (n=3). (J) Hepatic concentrations of TNF-α and IL-1β (n=5). (K) Serum ALT and AST levels across groups (n=5). (L) Histology changes of liver sections with or without CD44i treatment. (M) DNA damage in liver sections assessed by γH2AX (green) IF staining (left) and corresponding quantification (right) (n=3). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-tailed Student's t-test.
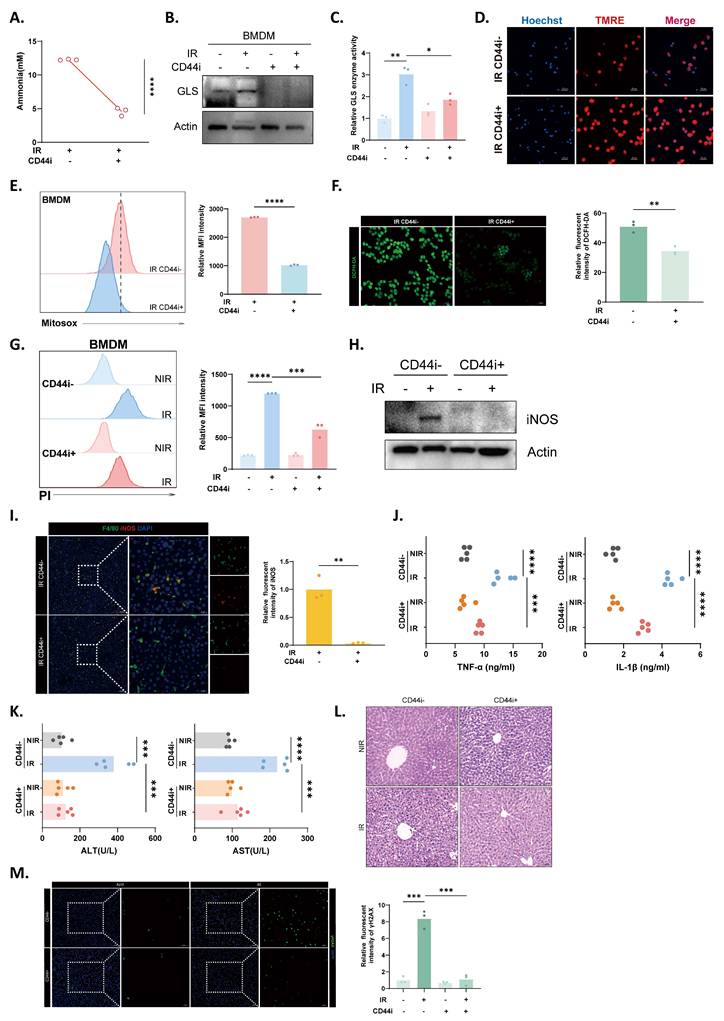
In summary, our findings suggest that targeting CD44 exerts a protective effect against RILD through the suppression of inflammatory activation in macrophages and the restoration of metabolic homeostasis. These results provide preclinical evidence that CD44 is a potential therapeutic target for RILD.
Discussion
RILD represents a complex pathology driven by sustained oxidative stress and inflammatory propagation, which limits the efficacy of liver-directed radiotherapy[7, 43]. While physical dose constraints are currently the primary preventive measure, they fail to address the underlying molecular and metabolic drivers of injury[44]. Although inflammatory signaling and microenvironmental reprogramming are increasingly recognized as integral to RILD pathobiology, the dominant cellular sources of ROS and metabolic checkpoints that enhance this oxidative burst remain incompletely defined[45-47]. Evaluating the molecular programs that connect radiation exposure to immunometabolic stress and inflammatory amplification therefore remains a critical need.
A central contribution of this study is the establishment of a conceptual framework linking radiation-induced oxidative stress to immunometabolic reprogramming in RILD. Within this framework, we identify KCs not only as immune responders but also as the primary drivers of redox imbalance and inflammatory amplification. We defined CD44 as a key driver linking radiation exposure to metabolic dysfunction in KCs. As guardians of hepatic innate immunity, KCs swiftly recognize injury signals and initiate inflammatory cascades that influence hepatic homeostasis and damage pathways[48, 49]. This study connects KC-driven inflammatory amplification to a defined and pharmacologically tractable molecular axis, providing a mechanistic basis for therapeutic targeting in RILD.
CD44 is best known as an adhesion and migration receptor involved in tumor stemness and therapeutic resistance[50], but accumulating evidence supports its active regulatory functions in macrophages rather than serving as a passive marker. Emerging studies support a pathogenic contribution of myeloid CD44 to hepatic inflammation and injury through the modulation of local stress environments[51, 52]. However, whether CD44 plays a KC-centered role in RILD and how it relates radiation stress to metabolic stress and inflammatory amplification have not been defined. Our study addresses this gap by placing CD44 upstream of a mechanistically actionable KC immunometabolic cascade.
In accordance with this hypothesis, CD44 expression was strongly induced in irradiated livers and isolated KCs, which is consistent with the upregulation of oxidative stress markers and M1 polarization. Mechanistically, CD44 functions upstream of an immunometabolic switch by stabilizing ELAVL1/HuR, thereby increasing GLS activity and driving excessive ammonia accumulation. The resulting toxic ammonia burden induces mitochondrial impairment, amplifies inflammation, and leads to ammonia-induced macrophage death, thereby worsening RILD (Fig. 8). While our ubiquitination data clearly reveal that CD44 protects ELAVL1 from proteasomal degradation, the precise molecular dynamics governing this process remain unclear. We speculate that the intracellular domain of CD44 may physically interact with ELAVL1 to induce steric hindrance, thereby shielding critical lysine residues from specific E3 ubiquitin ligases known to target ELAVL1, such as β-TrCP1[53]. Alternatively, CD44 might act through competitive inhibition by sequestering the responsible E3 ligase or indirectly by recruiting specific deubiquitinases such as USP20[54] to dynamically reverse ELAVL1 ubiquitination. Collectively, these findings suggest that CD44 is not merely a passive biomarker but a decisive node that couples radiation stress to metabolic and inflammatory dysfunction in KCs, linking macrophage failure to tissue-level hepatic injury. Beyond post-transcriptional regulation, the upstream transcriptional circuitry directing the inflammatory CD44⁺ KC state remains to be identified. Preliminary regulon analysis of our single-cell data suggests that transcription factors linked to myeloid activation and stress responses, including Nfil3, Klf4, Cebpb, and Stat2, are selectively active in CD44⁺ KCs. Elucidating how these factors intersect with the CD44-ELAVL1-GLS axis will further clarify the initiation of immunometabolic amplification during RILD.
A working model depicting CD44 as a central node driving KCs cell dysfunction, linking metabolic stress to inflammatory signaling in RILD.
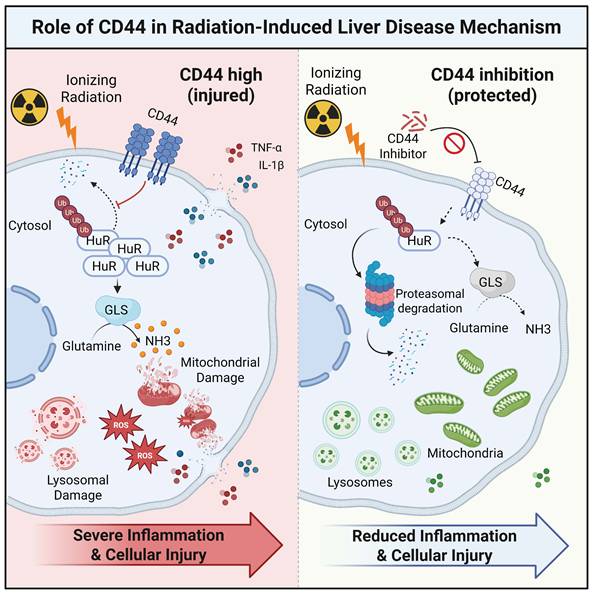
Ammonia-induced cell death is a recently described immune cell fate linked to ammonia imbalance and has rarely been considered in the context of RILD[21, 27, 55]. Our data underscore that radiation injury in macrophages extends beyond canonical inflammatory signaling or DNA damage. Radiation also perturbs metabolic homeostasis, promotes toxic ammonia accumulation, and induces organelle failure. This provides a mechanistic route through which metabolic and oxidative stress intensifies inflammatory escalation, sustaining a feedforward loop within the irradiated liver microenvironment. Both genetic and pharmacological inhibition of CD44 led to improved ammonia homeostasis, decreased oxidative inflammatory burden, and reduced liver tissue injury. Moreover, NH4Cl add-back and CB-839 experiments revealed ammonia handling downstream of CD44 and upstream of mitochondrial dysfunction, supporting a model in which irradiation activates CD44 to disrupt ammonia homeostasis in KCs and thereby create a self-reinforcing inflammatory state during RILD. Pharmacologically targeting this glutaminolysis checkpoint is a compelling dual therapeutic opportunity, particularly considering the established clinical profile of CB-839 in advanced oncology trials. Nevertheless, the ubiquitous expression of CD44 across primary tissues raises valid concerns regarding systemic off-target toxicity. Future therapeutic avenues must therefore prioritize localized delivery strategies. Employing vehicles targeted specifically to the liver, such as peptide-functionalized nanocarriers or engineered liposomes tailored for selective uptake by KCs[56-58], represents an optimal path to harness this protective mechanism without compromising systemic safety. By establishing dysregulated ammonia metabolism and GLS activity as core components of KC pathology, we expand the mechanistic landscape of RILD toward the immunometabolic dimension.
In conclusion, we identified a CD44-ELAVL1-GLS axis that links radiation stress to abnormal ammonia accumulation, mitochondrial collapse, and oxidative stress in KCs. This metabolic program leads to ammonia-induced cell death, thereby amplifying inflammatory liver injury. These findings expand the immunometabolic landscape of RILD and provide a tractable mechanistic foundation for KC-focused preventive strategies. Importantly, CD44 provides a molecular entry point for preventive intervention, complementing current physical dose constraints. It remains to be determined whether ammonia dysregulation extends to other hepatic immune subsets and how KCs coordinate with hepatocytes and stellate cells within a spatially organized injury niche. Apart from normal tissue injury, this conserved immunometabolic axis likely operates within tumor-associated macrophages in irradiated hepatocellular carcinoma. Unlike resident KCs, which experience fatal ammonia toxicity, tumor-associated macrophages might divert glutaminolysis products for survival while utilizing secreted ammonia to suppress infiltrating cytotoxic T lymphocytes. Although the specific metabolic adaptations in these complex microenvironments warrant further investigation, strategically combining targeted CD44 or GLS inhibitors with localized radiotherapy is poised to offer a dual benefit, simultaneously protecting normal liver tissue and creating a more favorable immunological context for tumor control. Additionally, comprehensive preclinical evaluation and prospective clinical studies are needed to define safety, efficacy, and patient selection.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This study was supported by grants as follows: National Natural Science Foundation of China (Grant No. 82373519); National Key Research and Development Program of China (Grant No. 2022YFC2503704); Scientific Research Development Fund of Zhongshan Hospital, Fudan University (2024ZRFZ25; 2025XKPT50-1); National Natural Science Foundation of China (Grant No. 82303886). Thanks for Mingjun Ji for his appreciation and understanding.
Availability of data and materials
All data are available in the main text, the supplementary materials, and from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. European Association for the Study of the Liver. EASL clinical practice guidelines on the management of hepatocellular carcinoma. J Hepatol. 2025;82:315-74
2. Chan SL, Sun HC, Xu Y, Zeng H, El-Serag HB, Lee JM. et al. The Lancet commission on addressing the global hepatocellular carcinoma burden: Comprehensive strategies from prevention to treatment. Lancet. 2025;406:731-78
3. Vitale A, Cabibbo G, Iavarone M, Vigano L, Pinato DJ, Ponziani FR. et al. Personalised management of patients with hepatocellular carcinoma: A multiparametric therapeutic hierarchy concept. Lancet Oncol. 2023;24:e312-e22
4. Dudzinski SO, Newman NB, McIntyre J, Engineer R, Sanford NN, Wo JY. et al. Emerging evidence-based role for external-beam radiation therapy in hepatocellular carcinoma. Lancet Gastroenterol Hepatol. 2025;10:387-98
5. Vogel A, Chan SL, Dawson LA, Kelley RK, Llovet JM, Meyer T. et al. Hepatocellular carcinoma: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2025;36:491-506
6. Singal AG, Llovet JM, Yarchoan M, Mehta N, Heimbach JK, Dawson LA. et al. AASLD practice guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology. 2023;78:1922-65
7. Miften M, Vinogradskiy Y, Moiseenko V, Grimm J, Yorke E, Jackson A. et al. Radiation dose-volume effects for liver SBRT. Int J Radiat Oncol Biol Phys. 2021;110:196-205
8. Apisarnthanarax S, Barry A, Cao M, Czito B, DeMatteo R, Drinane M. et al. External beam radiation therapy for primary liver cancers: An ASTRO clinical practice guideline. Pract Radiat Oncol. 2022;12:28-51
9. Li JX, Zhang RJ, Qiu MQ, Yan LY, He ML, Long MY. et al. Non-classic radiation-induced liver disease after intensity-modulated radiotherapy for Child-Pugh grade B patients with locally advanced hepatocellular carcinoma. Radiat Oncol. 2023;18:48
10. Koay EJ, Owen D, Das P. Radiation-induced liver disease and modern radiotherapy. Semin Radiat Oncol. 2018;28:321-31
11. Zhou L, Zhu J, Liu Y, Zhou PK, Gu Y. Mechanisms of radiation-induced tissue damage and response. MedComm. 2024;5:e725
12. Gan C, Yuan Y, Shen H, Gao J, Kong X, Che Z. et al. Liver diseases: Epidemiology, causes, trends and predictions. Signal Transduct Target Ther. 2025;10:33
13. Chen G, Zhao Q, Yuan B, Wang B, Zhang Y, Li Z. et al. ALKBH5-modified HMGB1-STING activation contributes to radiation induced liver disease via innate immune response. Int J Radiat Oncol Biol Phys. 2021;111:491-501
14. Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwalder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77:1136-60
15. Azzam EI, de Toledo SM, Little JB. Oxidative metabolism, gap junctions and the ionizing radiation-induced bystander effect. Oncogene. 2003;22:7050-7
16. Zhao J, Yi Z, Deng G, Li Y, Li J, Qin M. et al. STING modulates iron metabolism to promote liver injury and inflammation in acute immune hepatitis. Free Radic Biol Med. 2024;210:367-77
17. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. 2022;55:1515-29
18. Taru V, Szabo G, Mehal W, Reiberger T. Inflammasomes in chronic liver disease: Hepatic injury, fibrosis progression and systemic inflammation. J Hepatol. 2024;81:895-910
19. Gao B, Zheng D, Liu H, Guo Y, Ye Y, Chen Z. et al. Asparagine synthetase modulates glutaminase inhibitor sensitivity through metabolic reprogramming and serves as a prognostic biomarker in hepatocellular carcinoma. Redox Biol. 2025;86:103813
20. Kerbert AJC, Engelmann C, Habtesion A, Kumar P, Hassan M, Qi T. et al. Hyperammonemia induces programmed liver cell death. Sci Adv. 2025;11:eado1648
21. Zhang H, Liu J, Yuan W, Zhang Q, Luo X, Li Y. et al. Ammonia-induced lysosomal and mitochondrial damage causes cell death of effector CD8(+) T cells. Nat Cell Biol. 2024;26:1892-902
22. Xu Y, Wang J, Wu A, Wang M. Ammonia metabolism and ammonia-induced cell death: Role in cancer therapy. Cell Commun Signal. 2025;23:468
23. Schreiber R, Zhang F, Haussinger D. Regulation of vesicular pH in liver macrophages and parenchymal cells by ammonia and anisotonicity as assessed by fluorescein isothiocyanate-dextran fluorescence. Biochem J. 1996;315:385-92
24. Moriwaki T, Terawaki S, Otomo T. Impaired lysosomal acidity maintenance in acid lipase-deficient cells leads to defective autophagy. J Biol Chem. 2024;300:105743
25. Zhang YX, Wan H, Shan GY, Gao ZC, Li HJ. Hyperuricemia increases susceptibility to chronic kidney injury exacerbation via autophagic flux blockade-mediated ammonia death pathway. J Adv Res. 2025 [Epub ahead of print]
26. Albrecht J, Norenberg MD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology. 2006;44:788-94
27. Rehman U, Gupta G, Sahebkar A, Kesharwani P. Ammonia-induced cell death: A novel frontier to enhance cancer immunotherapy. Immunology. 2025;175:16-20
28. Jiang Q, Qiu Y, Kurland IJ, Drlica K, Subbian S, Tyagi S. et al. Glutamine is required for M1-like polarization of macrophages in response to Mycobacterium tuberculosis infection. mBio. 2022;13:e0127422
29. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: Metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10:1564
30. Chen X, Li L, Deng Y, Liao J, Meng H, Liang L. et al. Inhibition of glutaminase 1 reduces M1 macrophage polarization to protect against monocrotaline-induced pulmonary arterial hypertension. Immunol Lett. 2025;272:106974
31. Du B, Dong J, Gao Q, Yin Z, Yang J, Ma S. et al. Comprehensive analysis of ammonia-induced cell death and GLS1 in gastric adenocarcinoma: Implications for prognosis and therapeutic strategies. Cancer Cell Int. 2025;25:342
32. Chen Z, Zhang W, Wang D, Luo R, Yao Y, Tao X. et al. CD44 is a macrophage receptor for TcdB from Clostridioides difficile that via its lysine-158 succinylation contributes to inflammation. Gut Microbes. 2025;17:2506192
33. Frascogna C, Mottareale R, La Verde G, Arrichiello C, Muto P, Netti PA. et al. Role of the mechanical microenvironment on CD-44 expression of breast adenocarcinoma in response to radiotherapy. Sci Rep. 2024;14:391
34. Solier S, Muller S, Caneque T, Versini A, Mansart A, Sindikubwabo F. et al. A druggable copper-signalling pathway that drives inflammation. Nature. 2023;617:386-94
35. Huang J, Meng P, Liang Y, Li X, Zhou S, Li J. et al. Tubular CD44 plays a key role in aggravating AKI through NF-kappaB p65-mediated mitochondrial dysfunction. Cell Death Dis. 2025;16:119
36. Huang XW, Yang J, Dragovic AF, Zhang H, Lawrence TS, Zhang M. Antisense oligonucleotide inhibition of tumor necrosis factor receptor 1 protects the liver from radiation-induced apoptosis. Clin Cancer Res. 2006;12:2849-55
37. Malik G, Wilting J, Hess CF, Ramadori G, Malik IA. PECAM-1 modulates liver damage induced by synergistic effects of TNF-alpha and irradiation. J Cell Mol Med. 2019;23:3336-44
38. Wu Q, Li L, Wei Y, Fu Q, Zhou T, Chen K. et al. Dynamic metabolomics atlas of radiation-induced liver injury and hepatocyte proliferation process in rat. Radiat Res. 2026;205:100-12
39. Wang H, Li L, Li Y, Li Y, Sha Y, Wen S. et al. Intravital imaging of interactions between iNKT and kupffer cells to clear free lipids during steatohepatitis. Theranostics. 2021;11:2149-69
40. Yang WH, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends Cancer. 2021;7:790-804
41. Adamoski D, L MDR, Mafra ACP, Correa-da-Silva F, Moraes-Vieira PMM, Berindan-Neagoe I. et al. HuR controls glutaminase RNA metabolism. Nat Commun. 2024;15:5620
42. Dai Y, Zhao F, Chen Q, Zeng B, Gu W, Zhang Y. et al. Microfluidic chip-integrated vascularized endometrial complexes: Mitochondrial function and paracrine crosstalk enhance regenerative potential. Bioact Mater. 2025;54:551-69
43. Bae SH, Chun SJ, Chung JH, Kim E, Kang JK, Jang WI. et al. Stereotactic body radiation therapy for hepatocellular carcinoma: Meta-analysis and international stereotactic radiosurgery society practice guidelines. Int J Radiat Oncol Biol Phys. 2024;118:337-51
44. Dawson LA, Winter KA, Knox JJ, Zhu AX, Krishnan S, Guha C. et al. Stereotactic body radiotherapy vs sorafenib alone in hepatocellular carcinoma: The NRG oncology/RTOG 1112 phase 3 randomized clinical trial. JAMA Oncol. 2025;11:136-44
45. Dong A, Wei G, Liang Z, Di Y, Tang Y, Ling Y. et al. Gasdermin D aggravates a mouse model of radiation-induced liver disease by promoting chemokine secretion and neutrophil recruitment. Nat Commun. 2025;16:6064
46. Wang B, Zhang Y, Niu H, Zhao X, Chen G, Zhao Q. et al. METTL3-mediated STING upregulation and activation in Kupffer cells contribute to radiation-induced liver disease via pyroptosis. Int J Radiat Oncol Biol Phys. 2024;119:219-33
47. Zhu W, Zhang X, Yu M, Lin B, Yu C. Radiation-induced liver injury and hepatocyte senescence. Cell Death Discov. 2021;7:244
48. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18:45-56
49. Kostallari E, Schwabe RF, Guillot A. Inflammation and immunity in liver homeostasis and disease: A nexus of hepatocytes, nonparenchymal cells and immune cells. Cell Mol Immunol. 2025;22:1205-25
50. Zhou HM, Zhang JG, Zhang X, Li Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct Target Ther. 2021;6:62
51. Sutherland TE, Dyer DP, Allen JE. The extracellular matrix and the immune system: A mutually dependent relationship. Science. 2023;379:eabp8964
52. Rousseau D, Bonnafous S, Soysouvanh F, Sarrail D, Bourinet M, Strazzulla A. et al. CD44 in myeloid cells is a major driver of liver inflammation and injury in alcohol-associated liver disease. Hepatology. 2025;82:1211-28
53. Lan Y, Xiao X, He Z, Luo Y, Wu C, Li L. et al. Long noncoding RNA OCC-1 suppresses cell growth through destabilizing HuR protein in colorectal cancer. Nucleic Acids Res. 2018;46:5809-21
54. Chen Y, Wu S, Deng L, Zhou Y, Zhang Y, Mei J. et al. Ubiquitin-specific protease 20 (USP20) mitigates doxorubicin-induced cardiotoxicity by deubiquitinating and stabilizing HuR. Int J Biol Macromol. 2026;358:151642
55. Li Z, Lin J, Yin P. Ammonia death: A novel potential strategy to augment immunotherapy in cancer. Cancer Gene Ther. 2024;31:1751-3
56. Guo Y, Huang J, Lin M, Yin Q, Zhang T, Guo Z. et al. Nano particle loaded EZH2 inhibitors: Increased efficiency and reduced toxicity for malignant solid tumors. J Transl Int Med. 2025;13:156-69
57. Tong T, Gao W, Jian H, Yang R, Zhang J, Li K. et al. The role and potential mechanisms of exosomes in the progression of hepatocellular carcinoma. Holistic Integrative Oncology. 2025;4:36
58. Ma C, Zhou Y, Yang Z, Wang J, Zhu L, Chen H. et al. Kupffer cells biomimetic nanoparticles alleviate sepsis related liver injury. J Nanobiotechnology. 2025;23:756
Author contact
![]() Corresponding authors: Zhao-Chong Zeng, E-mail: zeng.zhaochongsh.cn; Qian-Qian Zhao, E-mail: zhao.qianqian1sh.cn. Department of Radiation Oncology, Zhongshan Hospital, Fudan University, Shanghai, 200032, China; Postal address: No.180, Fenglin Road, Xuhui District, Shanghai, China. Phone: 86-21-64041990; Fax: 86-21-64048472.
Corresponding authors: Zhao-Chong Zeng, E-mail: zeng.zhaochongsh.cn; Qian-Qian Zhao, E-mail: zhao.qianqian1sh.cn. Department of Radiation Oncology, Zhongshan Hospital, Fudan University, Shanghai, 200032, China; Postal address: No.180, Fenglin Road, Xuhui District, Shanghai, China. Phone: 86-21-64041990; Fax: 86-21-64048472.