Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Result
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(12):6521-6538. doi:10.7150/ijbs.135145 This issue Cite
Research Paper
High density-lipoprotein regulates liquid-liquid phase separation of heat shock protein β-1 by lncRNA HDRACA to affect vascular inflammation and atherosclerosis
Zhi-Wei Mo, MD, PhD1,2,3#, Yi-Fang Liu, BS1,3#, Yi-Xin Zhang, MD, PhD3,4#, Yan Li, PhD1,3#, Yang Cao, MD1,3#, Chu-Yu Liu, MD, PhD6#, Le Li, BS1,3, Yue-Ting Kang, MD3,4, Hong-Yu Cao, MD1,3, Zhen-Sheng Ma, MD1,3, Yue-Ming Peng, MD, PhD1,3, Zhi-Jun Ou, MD, PhD3,4 ![]() , Guang-Qi Chang, MD, PhD2,3
, Guang-Qi Chang, MD, PhD2,3 ![]() , Jing-Song Ou, MD, PhD1,3,5
, Jing-Song Ou, MD, PhD1,3,5 ![]()
1. Division of Cardiac Surgery, Cardiovascular Diseases Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510080, P.R. China.
2. Division of Vascular Surgery, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510080, P.R. China.
3. National-Guangdong Joint Engineering Laboratory for Diagnosis and Treatment of Vascular Diseases, NHC key Laboratory of Assisted Circulation and Vascular Diseases (Sun Yat-sen University), Key Laboratory of Assisted Circulation and Vascular Diseases, Chinese Academy of Medical Sciences, Guangdong Provincial Engineering and Technology Center for Diagnosis and Treatment of Vascular Diseases, Guangzhou, 510080, P.R. China.
4. Division of Hypertension and Vascular Diseases, Department of Cardiology, Cardiovascular Diseases Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510080, P.R. China.
5. Guangdong Provincial Key Laboratory of Brain Function and Disease, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, 510080, P.R. China.
6. Department of Biomedical Sciences, Faculty of Health Sciences, University of Macau, Taipa, Macau SAR, P.R. China.
# These authors contributed equally to this study.
Received 2026-3-26; Accepted 2026-6-3; Published 2026-7-1
Abstract

High-density lipoprotein (HDL) from healthy subjects (HDLhealthy) has anti-inflammatory effects, whereas HDL from patients with coronary artery disease (HDLCAD) is functionally impaired, which may affect atherosclerosis formation differently. We previously demonstrated that HDLhealthy inhibits the expression of the long noncoding RNA high-density lipoprotein-regulated angiogenesis in coronary artery disease (HDRACA), whereas HDLCAD is much less effective. HDRACA is highly expressed in endothelial cells (ECs) of arteriosclerosis obliterans and is associated with the expression of adhesion molecules and chemokines. However, whether HDLhealthy and HDLCAD affect vascular inflammation and atherosclerosis by regulating HDRACA expression remains unclear. Here, we found that HDLhealthy suppressed HDRACA expression in ECs, thereby inhibiting endothelial chemotactic and adhesive effects, whereas HDLCAD had less of an effect. Mechanistically, HDRACA bound to heat shock protein β-1 (HSPB1), an anti-inflammatory protein that underwent liquid-liquid phase separation (LLPS) mediated by its N-terminal domain (NTD) and C-terminal domain (CTD). Overexpression of HDRACA promoted LLPS of HSPB1, inhibited HSPB1-IKKβ interaction, and activated the NF-ĸB pathway, leading to increased expression of adhesion molecules and chemokines. HDLhealthy enhanced the phosphorylation of serine residues 15, 78, and 82 in the NTD of HSPB1 to inhibit its LLPS by suppressing HDRACA expression, whereas HDLCAD was less effective. Delivering HDRACA into mouse aortic ECs also promoted LLPS of HSPB1, leading to enhanced vascular inflammation and accelerated atherosclerosis formation in low-density lipoprotein receptor null mice. Our findings identified HDRACA-induced LLPS of HSPB1 as a novel mechanism linking HDL dysfunction and atherosclerosis development. HDLhealthy suppressed HDRACA-induced LLPS of HSPB1 to inhibit vascular inflammation and exerted anti-atherosclerotic effects, whereas HDLCAD lost its protective role because of its inefficacy in HDRACA inhibition. These findings suggest that improving HDL function, inhibiting HDRACA, and blocking LLPS of HSPB1 may represent promising therapeutic strategies for atherosclerosis.
Keywords: high-density lipoprotein, inflammation, long noncoding RNA, liquid-liquid phase separation, atherosclerosis
Introduction
Atherosclerosis is a core vascular pathological change associated with atherosclerotic cardiovascular disease (ASCVD). Traditional risk factors, including hypercholesterolemia, diabetes mellitus, smoking, and obesity, can cause persistent production of oxidative stress, inflammatory factors, and oxidized lipids, all of which contribute to the development of atherosclerosis [1-4]. Recent studies have confirmed that risk factors, including mental stress, dysbiosis, and viral infections, also induce the production of inflammatory factors, thereby increasing the risk of atherosclerosis [5-7]. These proinflammatory mediators can cause dysfunction of endothelial cells (ECs) that sustainably express adhesion molecules and chemokines, which induce immune cells to continuously infiltrate the vascular wall, further causing atherosclerosis progression [2, 4, 8]. In healthy subjects, high density-lipoprotein (HDL) exerts anti-atherogenic effects by inhibiting the adhesive and chemotactic activities of ECs induced by proinflammatory mediators [9-11]. However, we and other researchers found that HDL undergoes multiple modifications, losing its protective ability and becoming proinflammatory or dysfunctional in ASCVD [9]. While previous studies have primarily focused on the compositional alterations of HDL in ASCVD, the mechanistic changes underlying HDL-mediated regulation of endothelial adhesion and chemotaxis in ASCVD remain poorly understood.
Long noncoding RNA (lncRNA) is arbitrarily defined as a class of transcripts comprising more than 200 nucleotides with limited protein-coding potential [12]. Recent studies have demonstrated that lncRNAs participate in the development of atherosclerosis [13-16]. We previously demonstrated that HDL from healthy subjects (HDLhealthy) inhibits the expression of lncRNA high-density lipoprotein-regulated angiogenesis in coronary artery disease (HDRACA) in ECs, whereas HDL from patients with coronary artery disease (HDLCAD) is much less effective because of sphingosine 1-phosphate (S1P) deficiency [17]. Specifically, HDRACA is upregulated in the arterial ECs of patients with arteriosclerosis obliterans, and its silencing alters the expression of genes associated with the TNF signaling, cell adhesion, and chemokine signaling pathways [17]. Additionally, we found putative interactions between HDRACA and multiple proteins, most notably the highly enriched heat shock protein β-1 (HSPB1) [17]. HSPB1, a canonical small heat shock protein (sHSP), has been recognized for its anti-inflammatory effects and role in stabilizing atherosclerotic plaques [18-20]. A previous study demonstrated that HSPB1 overexpression effectively decreases de novo atherosclerotic lesion formation and increases plaque stability [21]. HSPB1 can interact with the inhibitor of κB kinase (IKK) complex, thereby inhibiting inhibitor of κB α (IκBα) degradation and nuclear factor-ĸB (NF-κB) nuclear translocation, ultimately suppressing the expression of adhesion molecules and chemotaxis-related genes [22]. Thus, the distinct regulatory effects of HDLhealthy and HDLCAD on the HDRACA/HSPB1/IKKβ/IκBα/NF-κB pathway may account for their different anti-inflammatory properties.
The concept of liquid-liquid phase separation (LLPS) is relatively novel in the field of biomedicine. It refers to the process that biological polymers including DNA, RNA and proteins form liquid-like condensates through weak multivalent interactions, which dynamically coexists with a dilute phase, enabling spatial organization of biomolecules [23]. This either concentrates functionally related biomolecules to activate signal transduction or sequesters them to inhibit interactions [23]. Recent studies found that LLPS of proteins mediates vascular inflammation and atherosclerosis formation [24, 25]. Increasing evidence highlights that lncRNAs are important drivers of LLPS-mediated regulation of protein activity [26, 27]. However, it is unclear whether HDLhealthy and HDLCAD differentially affect atherosclerosis by regulating LLPS-mediated protein activity via lncRNAs.
In the present study, we found that HDLhealthy downregulated HDRACA to inhibit LLPS of HSPB1 in ECs, whereas HDLCAD was much less effective. Non-aggregated HSPB1 interacted with IKKβ to inhibit NF-κB signaling activation and suppressed the expression of adhesion molecules and chemokines, thus attenuating the adhesive and chemotactic effects of ECs and ultimately inhibiting atherosclerosis formation. Our findings reveal that HDLhealthy may inhibit atherosclerosis by attenuating HDRACA-induced LLPS of HSPB1, whereas HDLCAD cannot effectively inhibit atherosclerosis because it is less effective in suppressing HDRACA expression, which provides a novel entry point for developing effective anti-inflammatory-based therapeutics for atherosclerosis.
Materials and Methods
More detailed materials and methods are available in the Supplementary Materials.
Study populations and HDL isolation
Age- and sex-matched patients with CAD and healthy subjects without cardiovascular risk factors were enrolled to isolate HDL. To avoid the confounding effects of medications, the enrolled patients were all newly diagnosed with CAD and had not received any drug treatment in the three months prior to HDL isolation. Detailed information on the patients and healthy volunteers is provided in the Supplementary Table S1. The levels of S1P in HDL were quantified using a previously described method [17]. Written informed consent was obtained from all the participants. Plasma was isolated from individual venous blood samples and then subjected to HDL isolation through sequential ultracentrifugation, as described previously [17, 28-30]. For functional and molecular biological assays involving HDL, HDL preparations were randomly selected from the HDL sample pool. The study protocol was approved by the Ethics Review Board of the First Affiliated Hospital, Sun Yat-sen University (approval number: [2023]374) and was conducted in accordance with the principles outlined in the Declaration of Helsinki.
Assessment of nHDLox
HDL lipid peroxide content (HDLox) was measured, and normalized HDLox (nHDLox) was calculated to assess HDL function, as described previously [31].
Cell culture
Human umbilical vein endothelial cells (HUVECs), mouse aortic endothelial cells (MAECs), and THP-1 monocytes were cultured at 37 °C in a humidified incubator with 5% CO2. HUVECs were serum-starved overnight prior to treatment with 100 μg protein/mL HDLhealthy or HDLCAD, followed by incubation with 100 ng/mL tumor necrosis factor-α (TNF-α). The concentration used in this study was based on the HDL concentrations used in our previous studies, which approximate physiological levels in human plasma [17, 29, 32].
Adhesion assay and chemotaxis assay
Adhesion and chemotaxis assays were performed to evaluate adhesive and chemotactic effects of ECs.
RNAi and antisense silencing studies
Cells were transfected with lncRNA Smart Silencers or small interfering RNAs (siRNAs) following the manufacturer's instructions for the Lipofectamine RNAiMAX Transfection Reagent. The target sequences of lncRNA Smart Silencers and siRNAs are listed in the Supplementary Table S2.
Lentiviral transduction
Cells at 30-50% confluence were exposed to lentiviral particles and cultured for 72 h prior to subsequent experiments unless otherwise indicated. Enhanced green fluorescent protein (EGFP) linked HSPB1 wild-type or mutants were expressed in ECs via lentiviral transduction. To reduce potential interference between EGFP and wild-type or mutant HSPB1 domains, a linker was inserted between them to maintain their independent functions [33].
RNA extraction, reverse transcription, and RT-qPCR
Total RNA was extracted and reverse-transcribed into complementary DNA. Real-time quantitative polymerase chain reaction (RT-qPCR) was performed as described previously [17]. Primers used for RT-qPCR are listed in the Supplementary Table S3.
Western blot
The western blot assays were performed as described previously [34-37].
Enzyme-linked immunosorbent assay
The levels of monocyte chemoattractant protein-1 (MCP-1) in the culture medium of HUVECs and MAECs were measured using enzyme-linked immunosorbent assays (ELISA).
Immunoprecipitation
The immunoprecipitation assays were performed as described previously [38, 39].
Nuclear/cytoplasmic fractionation
Nuclear and cytoplasmic fractions were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents following the manufacturer's instructions.
RNA pull-down assay
RNA pull-down assays were performed according to the manufacturer's instructions for the Pierce™ Magnetic RNA-Protein Pull-Down Kit, as described previously [17].
RNA immunoprecipitation
RNA immunoprecipitation (RIP) assays were performed according to the manufacturer's instructions for the Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit, as described previously [40, 41].
Fluorescence in situ hybridization and immunofluorescence assays
Fluorescence in situ hybridization (FISH) assays were performed to analyze the expression of HDRACA in HUVECs and aortic roots, according to the manufacturer's instructions for the RiboTM Fluorescent In Situ Hybridization Kit. Immunofluorescence staining was performed to detect HSPB1 expression in HUVECs, MAECs, and VE-cadherin-positive cells in whole-mount en face mouse aortic preparations. Additionally, vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), and MCP-1 were detected in the VE-cadherin-positive cells of aortic roots.
Recombinant protein purification
Purification of EGFP-linked HSPB1 recombinant protein was performed by constructing a pET-SUMO vector through fusing EGFP and His-SUMO tags to the N-terminus of HSPB1, transforming Rosetta (DE3) cells, inducing expression with isopropyl β-D-1-thiogalactopyranoside, and purifying the target protein via Ni-NTA affinity chromatography.
In vitro phase separation assay
In vitro phase separation assays were performed to evaluate the LLPS of EGFP-linked HSPB1 recombinant protein, as previously described, with modifications [42].
Fluorescence recovery after photobleaching assay
Fluorescence recovery after photobleaching (FRAP) assays were performed to evaluate the dynamics of EGFP-linked HSPB1 in vitro and in HUVECs.
Molecular docking analysis
HDRACA and HSPB1 structures were docked using the HDOCK module on the WeMol platform (https://wemol.wecomput.com/).
Animal experiments
Low-density lipoprotein receptor null (Ldlr-/-) mice were fed a high-fat diet for 12 weeks after transduction with adeno-associated virus 9 (AAV9) carrying human HDRACA full-length (AAV-HDRACA) or negative control AAV9 (AAV-NC). Six-week-old male Ldlr-/- mice were randomly (simple randomization) assigned to two groups. The investigators were unaware of the allocation. AAV9 vectors were modified by incorporating an EC-specific TIE1 promoter sequence to enhance the specificity of target gene expression in ECs. AAV-NC and AAV-HDRACA were intravenously injected into Ldlr-/- mice. After anesthesia induction via intraperitoneal pentobarbital sodium (60 mg/kg), the mice were euthanized and carefully dissected to collect the intact aorta for further analysis, including FISH assays, immunofluorescence assays, immunohistochemistry assays, and aortic Oil Red O staining, as previously described, with modifications [17, 43-45]. Following anesthesia, mice were euthanized by cervical dislocation, except for whole-mount aortic preparations, in which mice were euthanized by intracardiac perfusion with KCl and formaldehyde after anesthesia. All experiments were approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University (approval number: SYSU-IACUC-2023-000830) and were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Statistical analyses were performed using SPSS version 13.0 or GraphPad Prism 9.0. Data were first tested for normality. For normally distributed data, differences among groups were assessed using unpaired two-tailed Student's t-test for comparisons between two groups and one-way or two-way ANOVA followed by Tukey's multiple comparisons test for comparisons among multiple groups. Non-normally distributed data were analyzed using nonparametric tests. The Mann-Whitney U test was used for comparisons between two groups, and the Kruskal-Wallis test followed by Dunn's multiple comparisons test was used for comparisons among multiple groups. The chi-square test was used to compare age and smoking status between CAD patients and healthy controls. Statistical significance was defined as p < 0.05.
Result
HDLhealthy and HDLCAD differentially affect adhesive and chemotactic effects of ECs by differential regulation of HDRACA expression
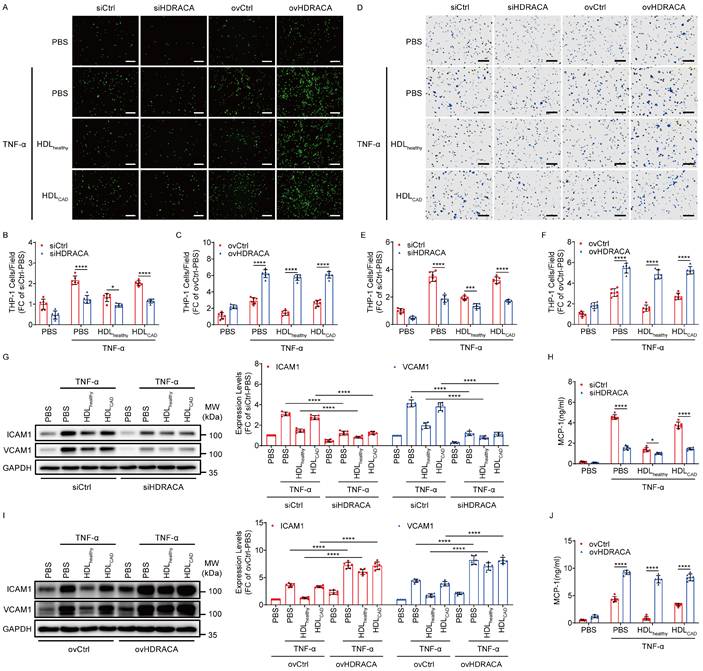
HDL was extracted from healthy subjects and patients with CAD. HDL-bound S1P was lower in patients with CAD than in healthy individuals (Supplementary Table S1), which is consistent with our previous findings [17]. The nHDLox level was significantly higher in patients with CAD than in healthy individuals, demonstrating that lipid peroxide levels in HDLCAD are elevated and HDL function is impaired in CAD (Supplementary Figure S1). In our previous study, silencing of HDRACA affected the levels of genes related to the inflammatory signaling pathway for the adhesive and chemotactic effects of ECs [17]. Therefore, the role of HDRACA in HDLhealthy and HDLCAD regulating the adhesive and chemotactic effects of ECs was further investigated. Supplementary Figure S2 shows that tumor necrosis factor-α (TNF-α) did not affect HDRACA levels in HUVECs. However, HDLhealthy significantly downregulated HDRACA levels in TNF-α-treated HUVECs, whereas HDLCAD was less effective. We silenced HDRACA by transfecting HUVECs with a lncRNA smart silencer and overexpressed HDRACA by transducing HUVECs with lentivirus carrying the full-length sequence of HDRACA, as previously described [17]. HDLhealthy and HDRACA silencing reduced THP-1 monocyte adhesion to and migration towards HUVECs treated with TNF-α, whereas HDLCAD was less effective than HDLhealthy (Figures 1A, B, D, and E). Moreover, overexpression of HDRACA promoted THP-1 monocyte adhesion to and migration towards HUVECs treated with TNF-α and attenuated the inhibitory effect of HDLhealthy (Figures 1A, C, D, and F). In addition, the expression of ICAM1, VCAM1, and MCP-1 in ECs was consistent with the effect of THP-1 monocyte adhesion to and migration towards ECs (Figures 1G-J). Taken together, these findings indicate that HDRACA is a positive regulator of the adhesive and chemotactic effects of ECs. HDLhealthy inhibits the adhesive and chemotactic effects of ECs by inhibiting HDRACA expression, whereas HDLCAD has less of an effect in reducing HDRACA, resulting in a reduced ability to attenuate the adhesive and chemotactic effects of ECs.
HDLhealthy and HDLCAD differentially affect adhesive and chemotactic effects of ECs by differential regulation of HDRACA expression. A. Representative images of THP-1 monocytes (green) adhering to HUVECs. HUVECs were subjected to HDRACA silencing or overexpression and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. Scale bars, 200 μm. B, C. Quantification of the adhesion assays shown in (A) for HDRACA silencing (B) or overexpression (C). D. Representative images of THP-1 monocytes (violet) migrating to the HUVEC culture medium. HUVECs were treated as described in (A). Scale bars, 100 μm. E, F. Quantification of the chemotaxis assay shown in (D) for HDRACA silencing (E) or overexpression (F). G. Representative blots (left) and quantification (right) of the western blot analysis of ICAM1 and VCAM1 in HUVECs. HUVECs were subjected to HDRACA silencing and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. H. Quantification of ELISA analysis of MCP-1 in HUVEC culture medium. HUVECs were treated as described in (G). I. Representative blots (left) and quantification (right) of the western blot analysis of ICAM1 and VCAM1 in HUVECs. HUVECs were subjected to HDRACA overexpression and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. J. Quantification of ELISA analysis of MCP-1 in HUVEC culture medium. HUVECs were treated as in (I). Data are presented as mean ± SD. For all experiments, n=6. Statistical significance was determined using two-way ANOVA with Tukey's multiple comparisons test (B, C, E-J). *p<0.05; ***p<0.001; ****p<0.0001.
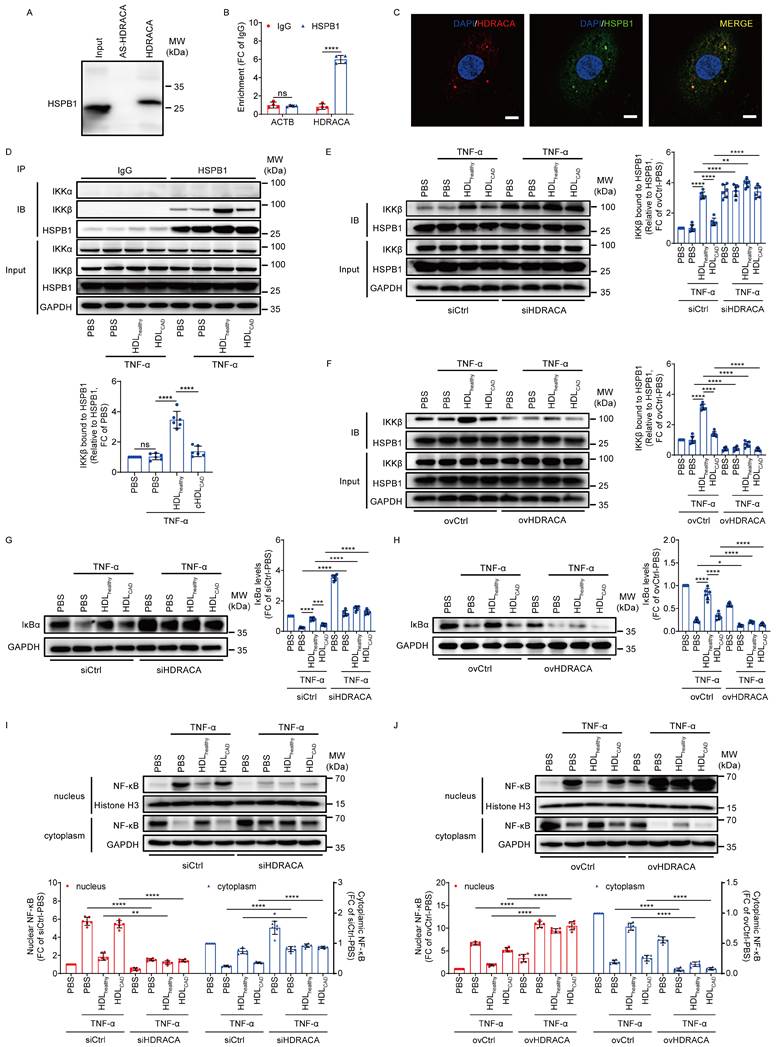
HDLhealthy and HDLCAD differentially affect the interaction between HSPB1 and IKKβ to mediate NF-κB pathway activation by differential regulation of HDRACA expression
The interaction between HDRACA and HSPB1 was confirmed by RNA pull-down-western blot and RIP-RT-qPCR assays (Figures 2A and B). FISH and immunofluorescence assays showed that HDRACA and HSPB1 co-localized in the cytoplasm of HUVEC (Figure 2C). Moreover, HSPB1 knockdown promoted the expression of VCAM1, ICAM1, and MCP-1, whereas it attenuated the inhibitory effect of HDLhealthy (Supplementary Figure S3), suggesting that HSPB1 is an important anti-inflammatory protein involved in the anti-inflammatory activity of HDLhealthy. However, neither HDLhealthy and HDLCAD treatment nor silencing and overexpression of HDRACA affected the levels of HSPB1 in cultured HUVECs (Supplementary Figure S4). IKKα and IKKβ are the two catalytic subunits of IKK complex [22]. We performed co-immunoprecipitation assays and found that HSPB1 bound to IKKβ in HUVECs without binding to IKKα (Figure 2D). Additionally, the interaction between HSPB1 and IKKβ in HUVECs did not change after 6 h of TNF-α treatment (Figure 2D). However, HDLhealthy promoted the interaction between HSPB1 and IKKβ, whereas HDLCAD had less of an effect (Figure 2D). Silencing of HDRACA promoted the interaction between HSPB1 and IKKβ, whereas overexpression of HDRACA not only inhibited the interaction between HSPB1 and IKKβ but also attenuated the promoting effect of HDLhealthy (Figures 2E and F). Furthermore, HDLhealthy inhibited TNF-α-induced IκBα reduction and NF-κB nuclear translocation, whereas HDLCAD had much less of an effect (Figures 2G-J). Silencing of HDRACA inhibited TNF-α-induced IκBα reduction and NF-κB nuclear translocation, whereas overexpression of HDRACA not only enhanced TNF-α-induced IκBα reduction and NF-κB nuclear translocation but also attenuated the inhibitory effect of HDLhealthy (Figures 2G-J). Collectively, these data suggest that HDLhealthy inhibits HDRACA expression to promote the interaction between HSPB1 and IKKβ, leading to IκBα upregulation and reduced NF-κB translocation into the nuclei, which reduces the transcription of NF-κB target genes. HDLCAD has much less of an effect in inhibiting HDRACA expression, which cannot inhibit the transcription of NF-κB genes via the HSPB1-IKKβ-IκBα-NF-κB pathway.
HDLhealthy and HDLCAD differentially affect the interaction between HSPB1 and IKKβ to mediate NF-κB pathway activation by differential regulation of HDRACA expression. A. HSPB1 detection by western blot in protein lysates from HUVECs after RNA pull-down with biotin-labeled HDRACA or antisense control (AS-HDRACA). B. RIP assays demonstrated the interaction between HDRACA and HSPB1 in HUVECs. C. Confocal images showing the colocalization of HDRACA (red) and HSPB1 (green) in HUVECs. Nuclei were stained with DAPI (blue). Scale bars, 10 μm. D. Immunoprecipitation analysis of the interaction between HSPB1 and IKKα or IKKβ in HUVECs. HUVECs were treated with HDLhealthy or HDLCAD for 24 h, followed by TNF-α incubation for 6 h. Representative blots (up) and quantification of IKKβ bound to HSPB1 (down) are shown. E, F. Immunoprecipitation analysis of the HSPB1/IKKβ interaction in HUVECs. HUVECs were subjected to HDRACA silencing (E) or overexpression (F), and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. Representative blots (left) and quantification (right) are shown. G, H. Western blot analysis of IκBα in HUVECs. HUVECs were subjected to HDRACA silencing (G) or overexpression (H), and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. Representative blots (left) and quantification (right) are shown. I, J. Western blot analysis of nuclear and cytoplasmic NF-κB in HUVECs. HUVECs were subjected to HDRACA silencing (I) or overexpression (J), and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. Histone H3 was used as internal reference in nucleus. GAPDH was used as internal reference in cytoplasm. Representative blots (up) and quantification (down) are shown. Data are presented as mean ± SD. For A-C, n=4; For D-J, n=6. Statistical analysis was performed using unpaired Student's t-test (B), one-way ANOVA with Tukey's multiple comparisons test (D) and two-way ANOVA with Tukey's multiple comparisons test (E-J). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns, not significant.
NTD and CTD of HSPB1 regulate its LLPS
As shown in Figure 2C, HSPB1 aggregated to form puncta in HUVECs. Furthermore, silencing of HDRACA reduced the formation of HSPB1 puncta, whereas overexpression of HDRACA increased the formation of HSPB1 puncta (Figure 3A). The structure of HSPB1 is characterized by a central conserved α-crystallin domain (ACD) with variable N-terminal (NTD) and C-terminal domains (CTD) at its flanks [46, 47]. Bioinformatics analyses of the human HSPB1 protein sequence showed that both the NTD and CTD contain several regions with multiple degrees of disorder, which are responsible for driving the LLPS of proteins (Figure 3B). LLPS is typically characterized by three features: the formation of spherical condensates, the ability of these condensates to fuse with others, and the recovery of fluorescence after photobleaching [23]. We exogenously expressed EGFP-linked human HSPB1 wild-type (EGFP-HSPB1-WT) in HUVECs and found that EGFP-HSPB1-WT formed puncta and underwent fusion (Figure 3C). FRAP assays demonstrated that EGFP-HSPB1-WT puncta formation was dynamic (Figure 3D). In vitro, purified EGFP-HSPB1-WT protein formed droplets and fused into larger droplets over time (Figures 3E and G). The FRAP assays confirmed the dynamic properties of the droplets (Figure 3F). These results demonstrate that HSPB1 undergoes LLPS in ECs.
NTD and CTD of HSPB1 regulate its LLPS. A. Representative images (left) of HSPB1 (green) in HUVECs after silencing (up) or overexpressing (down) HDRACA. The nuclei were stained with DAPI (blue). Scale bar, 10 μm. Quantification of HSPB1 puncta area per cell is shown (right). B. Schematic illustration of human HSPB1 domains and IDR prediction using D2P2 algorithms (http://d2p2.pro/). C. Time-series images showing the fusion of EGFP-HSPB1-WT puncta in HUVECs. Scale bar, 20 μm. D. FRAP analysis of EGFP-HSPB1-WT puncta in HUVECs. Scale bar, 10 μm. Representative images (left) and quantification (right) of the fluorescence intensity recovery in the bleached region are shown. E. Representative images of in vitro phase separation assays for EGFP-HSPB1-WT at 10 μM and 50 μM captured using differential interference contrast (DIC) and fluorescence microscopy (λ488 nm). Scale bar, 10 μm. F. FRAP analysis of EGFP-HSPB1-WT puncta in vitro. Scale bar, 5 μm. Representative images (left) and quantification (right) of the fluorescence intensity recovery in the bleached region are shown. G. Time-series images showing the fusion of EGFP-HSPB1-WT puncta in vitro. Scale bar, 20 μm. H. Schematic illustration of EGFP-HSPB1-WT, EGFP-HSPB1-ΔNTD, and EGFP-HSPB1-ΔCTD (up). Representative western blots for EGFP-HSPB1-WT, EGFP-HSPB1-ΔNTD, and EGFP-HSPB1-ΔCTD in HUVECs 72 h post-lentiviral transduction are shown (down). I, J. Representative images of HUVECs transduced with EGFP-HSPB1-WT, EGFP-HSPB1-ΔNTD, and EGFP-HSPB1-ΔCTD for 48 and 72 h (I). Quantification of EGFP-linked HSPB1 puncta area per cell is shown (J). Scale bar, 10 μm. K. FRAP analysis of EGFP-HSPB1-ΔCTD puncta after 72 h transduction. Scale bar, 10 μm. Representative images (left) and quantification (right) of the fluorescence intensity recovery in the bleached region are shown. Data are presented as mean ± SD. For all experiments, n=4. Statistical significance was determined using unpaired Student's t-test (A) and two-way ANOVA with Tukey's multiple comparisons test (J). **p<0.01; ****p<0.0001; ns, not significant.
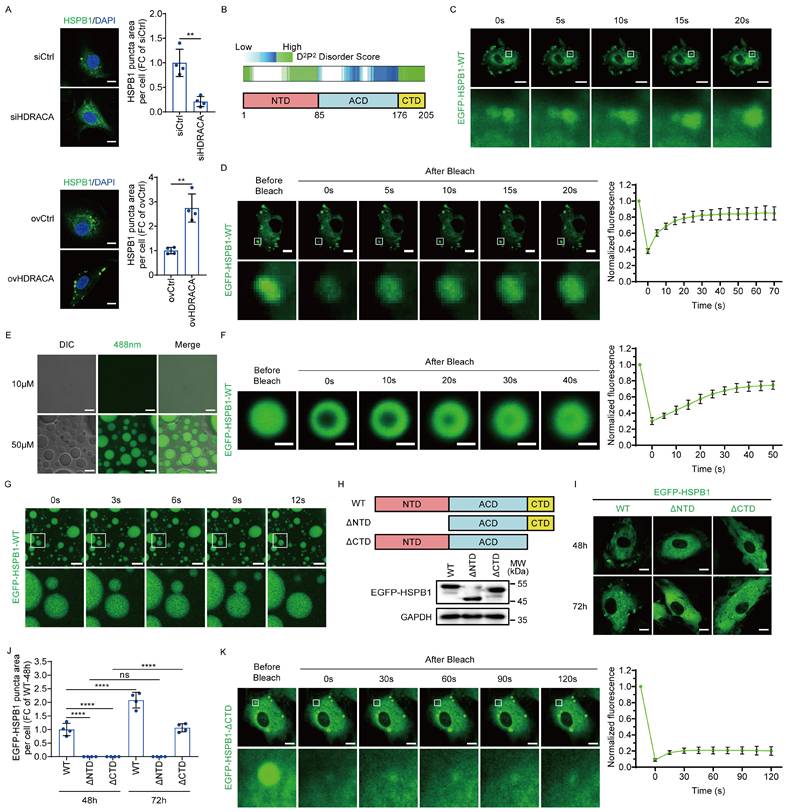
We further investigated the roles of the NTD and CTD in the LLPS of HSPB1. EGFP-HSPB1-WT and EGFP-linked HSPB1 mutants lacking NTD (EGFP-HSPB1-ΔNTD) or CTD (EGFP-HSPB1-ΔCTD) were expressed in HUVECs (Figure 3H). We found that EGFP-HSPB1-WT containing both the NTD and CTD formed puncta in HUVECs, with a significant increase in puncta number over time (Figures 3I and J). EGFP-HSPB1-ΔNTD containing CTD did not form puncta, whereas EGFP-HSPB1-ΔCTD containing NTD showed a delay in puncta formation (Figures 3I and J). Intriguingly, EGFP-HSPB1-ΔCTD puncta were not dynamic (Figure 3K). These data indicate that both the NTD and CTD are important domains for HSPB1 to undergo LLPS. The NTD is essential for HSPB1 aggregation initiation, whereas the CTD not only enhances aggregation but also imparts dynamic characteristics to the aggregates.
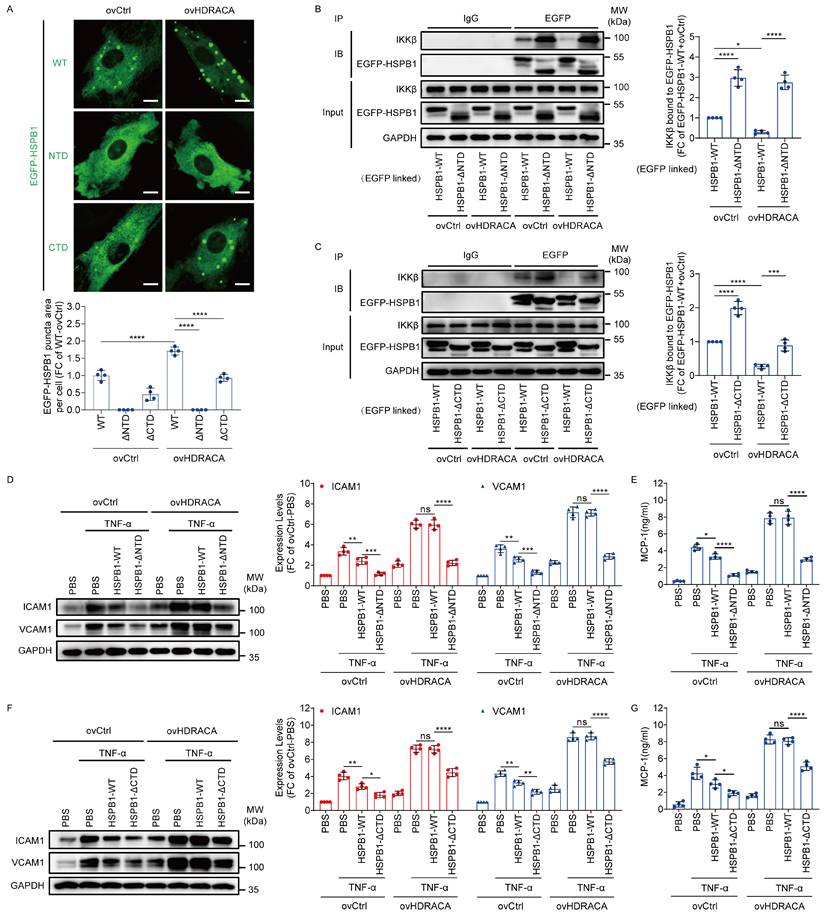
HDRACA induces the LLPS of HSPB1 to promote the adhesive and chemotactic effects of ECs
Because the NTD and CTD of HSPB1 play critical roles in LLPS, we systematically investigated the effects of HSPB1 LLPS on HDRACA-mediated HSPB1/IKKβ interaction and expression of adhesion molecules and chemokines via NTD or CTD deletion. Overexpression of HDRACA increased the formation of EGFP-HSPB1-WT puncta as endogenous HSPB1 (Figure 4A). However, deletion of the NTD significantly inhibited HDRACA overexpression-induced EGFP-HSPB1 puncta formation, whereas deletion of the CTD partially attenuated this effect (Figure 4A). Moreover, deletion of the NTD significantly inhibited HDRACA overexpression-induced suppression of HSPB1/IKKβ interaction, whereas deletion of the CTD partially attenuated this effect (Figures 4B and C). Furthermore, deletion of the NTD significantly inhibited HDRACA overexpression-induced promotion of ICAM1, VCAM1, and MCP-1 expression, whereas deletion of the CTD partially attenuated these effects (Figures 4D-G). Collectively, these data suggest that HDRACA induces LLPS of HSPB1 to inhibit the interaction between HSPB1 and IKKβ, consequently promoting the expression of adhesion molecules and chemokines in ECs.
HDRACA induces the LLPS of HSPB1 to promote the adhesive and chemotactic effects of ECs. A. Representative images of HUVECs transduced with EGFP-linked HSPB1-WT, HSPB1-ΔNTD and HSPB1-ΔCTD for 72 h with or without HDRACA overexpression (up). Quantification of EGFP-linked HSPB1 puncta area per cell is shown (down). Scale bar, 10 μm. B. Representative blots of immunoprecipitation analysis for the interaction between EGFP-HSPB1-WT or EGFP-HSPB1-ΔNTD and IKKβ in HUVECs after overexpressing HDRACA (left). Quantification of IKKβ bound to EGFP-HSPB1-WT or EGFP-HSPB1-ΔNTD after overexpressing HDRACA (right). C. Representative blots of immunoprecipitation analysis for the interaction between EGFP-HSPB1-WT or EGFP-HSPB1-ΔCTD and IKKβ in HUVECs after overexpressing HDRACA (left). Quantification of IKKβ bound to EGFP-HSPB1-WT or EGFP-HSPB1-ΔCTD after overexpressing HDRACA (right). D. Representative blots (left) and quantification (right) of western blot analysis of ICAM1 and VCAM1 in HUVECs transduced with HSPB1-WT or HSPB1-ΔNTD after HDRACA overexpression. E. The quantification of ELISA analysis of MCP-1 from HUVECs transducing with HSPB1-WT or HSPB1-ΔNTD after HDRACA overexpression. F. Representative blots (left) and quantification (right) of western blot analysis of ICAM1 and VCAM1 in HUVECs transduced with HSPB1-WT or HSPB1-ΔCTD after HDRACA overexpression. G. The quantification of ELISA analysis of MCP-1 from HUVECs transducing with HSPB1-WT or HSPB1-ΔCTD after HDRACA overexpression. Data are presented as the mean ± SD. For all experiments, n=4. Statistical significance was determined using two-way ANOVA with Tukey's multiple comparisons test (A-G). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns, not significant.
HDLhealthy and HDLCAD differentially affect the phosphorylation of HSPB1 within NTD to mediate its LLPS by differential regulation of HDRACA expression
Interestingly, HDLhealthy inhibited the formation of HSPB1 puncta, whereas HDLCAD had less of an effect (Figure 5A). Additionally, the overexpression of HDRACA prompted HSPB1 puncta formation and attenuated the inhibitory effect of HDLhealthy (Figure 5A). Next, we investigated the effect of HDRACA in regulating the LLPS of HSPB1.We performed RIP assays after transducing EGFP-HSPB1-WT and EGFP-HSPB1 mutants into HUVECs (Figure 5B and Supplementary Figure S5). The data showed that EGFP-HSPB1-ΔNTD failed to bind to HDRACA, whereas EGFP-HSPB1-WT, EGFP-HSPB1-ΔACD, and EGFP-HSPB1-ΔCTD did (Figure 5B). This finding indicates that the NTD is necessary for the binding of HSPB1 and HDRACA in ECs. Molecular docking results showed that the NTD of HSPB1 binds to multiple sites on HDRACA via various modes (Figure 5C and Supplementary Figure S6). A previous study indicated that phosphorylation of specific N-terminal serine residues 15 (S15), 78 (S78), and 82 (S82) can inhibit HSPB1 aggregation (Figure 5D) [48]. Therefore, we evaluated whether HDLhealthy and HDLCAD regulated the phosphorylation of NTD to affect the LLPS of HSPB1 by mediating HDRACA expression. As shown in Figures 5E-H, HDLhealthy significantly increased the phosphorylation of S15, S78, and S82 in the NTD of HSPB1, whereas HDLCAD had less of an effect. Overexpression of HDRACA decreased the phosphorylation of S15, S78, and S82 and attenuated the promoting effect of HDLhealthy (Figures 5E-H). A previous study indicated that mutating S15, S78, and S82 to alanine effectively inhibits phosphorylation at these sites [49]. Therefore, we expressed EGFP-HSPB1-WT and EGFP-HSPB1 mutant with S15, S78, and S82 mutated to alanine (S15A/S78A/S82A) in HUVECs by lentiviral transfection (Figure 5I). HDLhealthy inhibited the formation of EGFP-HSPB1-WT puncta, whereas HDLCAD was less effective (Figure 5J). Replacing S15, S78, and S82 with alanine significantly promoted the formation of EGFP-HSPB1 puncta and abolished the inhibitory effect of HDLhealthy (Figure 5J). Collectively, these data suggest that HDLhealthy promotes phosphorylation at N-terminal S15, S78, and S82 of HSPB1 to inhibit its LLPS by downregulating HDRACA, whereas HDLCAD has less of an effect because of its inability to decrease HDRACA.
HDLhealthy and HDLCAD differentially affect the phosphorylation of HSPB1 within NTD to mediate its LLPS by differential regulation of HDRACA expression. A. Representative images of HSPB1 (green) in HUVECs. HUVECs were subjected to HDRACA overexpression and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. The nuclei were stained with DAPI (blue). Scale bar, 10 μm. Quantification of HSPB1 puncta area per cell is shown (right). B. RIP assays verified the interaction between EGFP-linked HSPB1 truncated mutants and HDRACA. C. Schematic illustration showing the molecular docking results with the highest interaction score between HDRACA and NTD of HSPB1. D. Schematic illustration showing S15, S78, and S82 in the NTD of HSPB1. E-H. Western blot analysis of p15-HSPB1, p78-HSPB1, p82-HSPB1, and HSPB1 in HUVECs. HUVECs were treated as described in (A). Representative blots (E) and quantification (F-H) are shown. I. Schematic illustration showing that mutation of serine residues at positions 15, 78, and 82 of HSPB1 to alanine can effectively block phosphorylation of these three sites. J. Representative images (left) of wild type or S15A/S78A/S82A EGFP-linked HSPB1 puncta in HUVECs. HUVECs were transduced with EGFP-linked HSPB1-WT or HSPB1- S15A/S78A/S82A, and subsequently treated with either HDLhealthy or HDLCAD for 24 h, followed by a 6 h incubation with TNF-α. The nuclei were stained with DAPI (blue). Scale bar, 10 μm. Quantification of EGFP-linked HSPB1 puncta area per cell is shown (right). Data are presented as mean ± SD. For A, E-H, J, n=6; For B, n=4. Statistical significance was determined using two-way ANOVA with Tukey's multiple comparisons test (A, F-H, J) and unpaired Student's t-test (B). *p<0.05; **p<0.01; ****p<0.0001; ns, not significant.
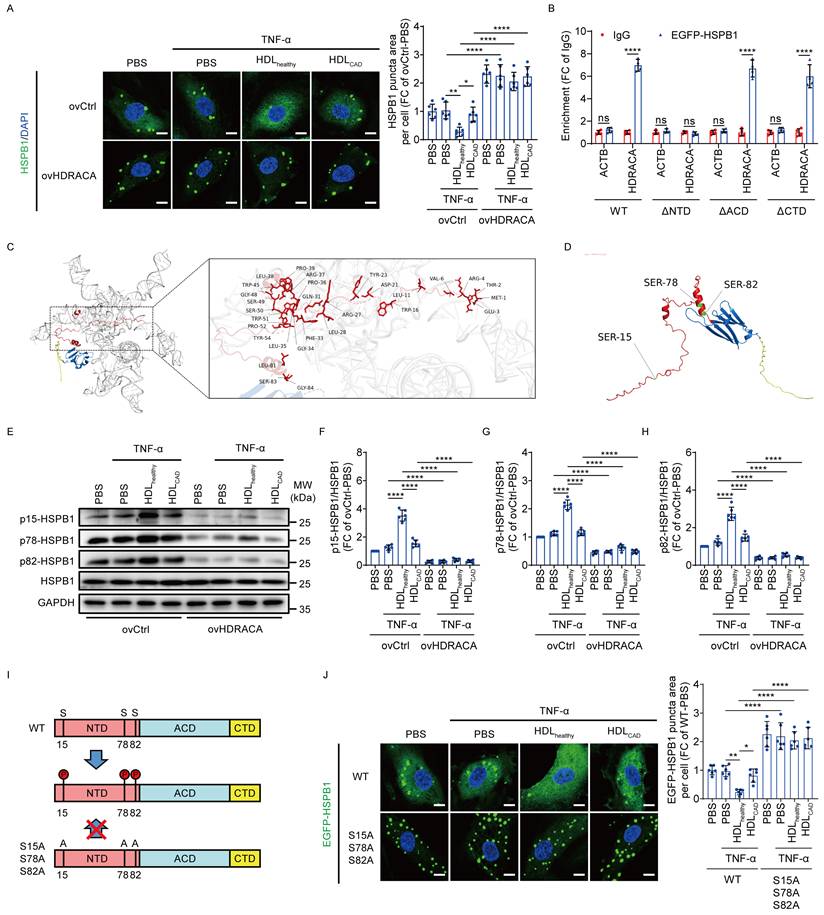
HDRACA induces the formation of HSPB1 puncta and activates NF-κB pathway in mouse ECs
HDRACA is not conserved across humans and other species. Therefore, we could not directly inhibit HDRACA expression in the murine models. However, our previous study demonstrated that HDRACA interacts with evolutionarily conserved protein partners and exerts consistent functional effects on ECs across species [17]. Mouse HSPB1 also contains a central ACD flanked by the NTD and CTD with multiple degrees of disorder (Figure 6A). In the present study, we ectopically expressed HDRACA in MAECs via lentiviral transduction (Figure 6B). RNA pull-down assays indicated that HDRACA bound to HSPB1 in MAECs (Figure 6C). Ectopic expression of HDRACA in MAECs promoted the formation of HSPB1 puncta (Figure 6D). Indeed, ectopic expression of HDRACA inhibited the interaction between HSPB1 and IKKβ, decreased IκBα levels, and promoted the nuclear translocation of NF-κB in MAECs (Figures 6E-G). In addition, ectopic expression of HDRACA increased ICAM1, VCAM1, and MCP-1 expression (Figures 6H and I). These data suggest that HDRACA can also induce LLPS of HSPB1 to activate the NF-κB pathway in mouse ECs, thus promoting the expression of adhesion molecules and chemokines.
HDRACA induces the formation of HSPB1 puncta and activates NF-κB pathway in mouse ECs. A. Schematic illustration of mouse HSPB1 domains and IDR prediction using D2P2 algorithms (http://d2p2.pro/). B. Schematic illustration showing the ectopic expression of HDRACA in MAECs via lentiviral transduction. C. HSPB1 detection by western blot analysis in protein lysates from MAECs with HDRACA transduction subjected to RNA pull-down with biotin-labeled HDRACA or AS-HDRACA. D. Representative images of HSPB1 (green) in MAECs after transduction with HDRACA. The nuclei were stained with DAPI (blue). Scale bar, 10 μm. Quantification of HSPB1 puncta area per cell is shown (right). E. Representative blots of immunoprecipitation analysis for the interaction between HSPB1 and IKKβ in MAECs after transduction with HDRACA (left). Quantification of IKKβ bound to HSPB1 is shown (right). F. Western blot analysis of IκBα expression in MAECs after transduction with HDRACA. Representative blots (left) and quantification (right) are shown. G. Western blot analysis of NF-κB in the nuclei and cytoplasm of MAECs after transduction with HDRACA. Histone H3 was used as an internal reference for the nuclei. GAPDH was used as an internal cytoplasmic reference. Representative blots (left) and quantification (right) are shown. H. Representative blots (left) and quantification (right) of western blot analysis of ICAM1 and VCAM1 in MAECs after transduction with HDRACA. I. Quantification of ELISA analysis of MCP-1 in MAECs after transduction with HDRACA. Data are presented as mean ± SD. For C-I, n=4. Statistical significance was determined using an unpaired Student's t-test (D-I). **p<0.01; ***p<0.001; ****p<0.0001.
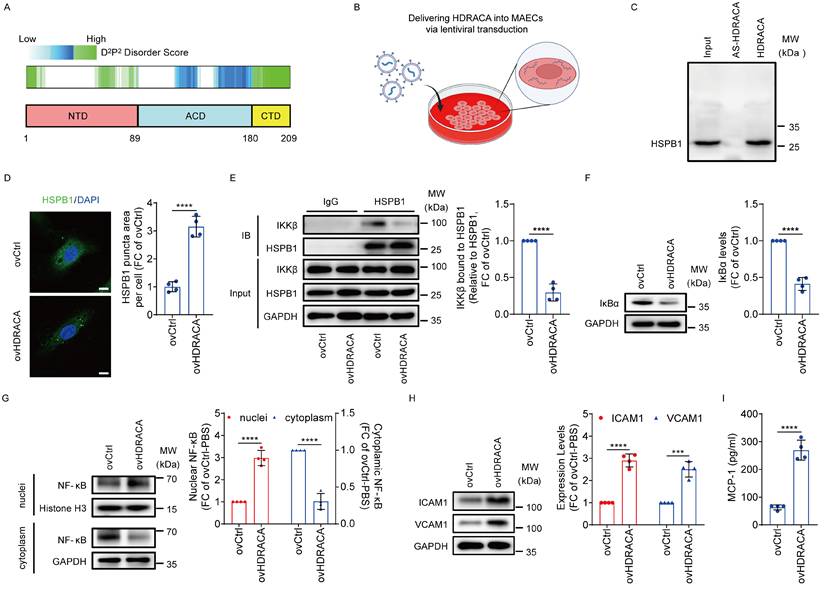
Ectopically expressing HDRACA promotes atherosclerosis
Furthermore, in Ldlr-/- mice fed a high-fat diet, we investigated the effects of HDRACA delivered via AAV9 transduction on vascular inflammation and atherosclerosis formation (Figure 7A). HDRACA was successfully expressed in mouse aortic ECs (Supplementary Figure S7). Transduction of HDRACA promoted the formation of HSPB1 puncta in aortic ECs (Figure 7B). ICAM1, VCAM1, and MCP-1 levels were upregulated in ECs of the AAV-HDRACA group (Figure 7C). The number of F4/80 positive cells (predominantly macrophages and macrophage-derived foam cells) was higher in the plaques of AAV-HDRACA mice than that in AAV-NC controls (Figure 7D). Staining of aortic valve sections with Oil Red O revealed that ectopic expression of HDRACA significantly increased the Oil Red O-positive lesion area (Figure 7E). Lesions stained with Oil Red O in aortic en face preparations showed that ectopic expression of HDRACA caused a significant elevation in the lesion area compared to the AAV-NC group (Figure 7F). Collectively, these findings suggest that HDRACA promotes HSPB1 puncta formation, vascular inflammation, and atherosclerosis in vivo.
Ectopically expressing HDRACA promotes atherosclerosis. A. Schematic illustration of atherosclerotic model establishment in Ldlr-/- mice transduced with AAV-NC or AAV-HDRACA. B. Representative immunofluorescence staining images (left) of HSPB1 (red) in ECs of whole mount en face mouse aortic preparations from Ldlr-/- mice transduced with AAV-NC or AAV-HDRACA. The ECs were stained with VE-cadherin (green) and the nuclei were stained with DAPI (blue). Scale bars, 20 μm. Quantification of HSPB1 puncta area in ECs is shown (right). C. Representative immunofluorescence staining images of VCAM1 (left, red), ICAM1 (middle, red) and MCP-1 (right, red) in ECs of whole mount en face mouse aortic preparations from Ldlr-/- mice transduced with AAV-NC or AAV-HDRACA. The ECs were stained with VE-cadherin (green) and the nuclei were stained with DAPI (blue). Scale bars, 20 μm. Quantitative analysis of the fluorescence intensities of VCAM1 (left), ICAM1 (middle) and MCP-1 (right) in the ECs is shown. D. Representative images (left) of immunohistochemical staining for F4/80 in aortic roots from Ldlr-/- mice transduced with AAV-NC or AAV-HDRACA. Scale bars, 200 μm. Quantitative analysis of the F4/80-positive areas in the plaque regions is shown (right). E. Representative images (left) of Oil Red O staining in aortic roots from Ldlr-/- mice transduced with AAV-NC or AAV-HDRACA. Scale bars, 200 μm. Quantitative analysis of the Oil Red O-positive areas in the aortic root regions is shown (right). F. Representative en face images (left) of Oil Red O staining in aorta from Ldlr-/- mice transduced with AAV-NC or AAV-HDRACA. Quantitative analysis of aortic lesion areas is shown (right). G. Graphical illustration of HDL-HDRACA-HSPB1 regulatory mechanism. (1) HDLhealthy inhibits HDRACA expression to enhance the phosphorylation of S15, S78 and S82 in the NTD of HSPB1 in the ECs, thus promoting the interaction between HSPB1 and IKKβ, reducing the interaction between IKKβ and IκBα and increasing the interaction between IκBα and NF-κB. Consequently, reduced nuclear translocation of NF-κB attenuates the expression of downstream target genes encoding key chemokines and adhesion molecules, ultimately inhibiting immune cells infiltration and atherosclerosis formation. (2) HDLCAD does not inhibit HDRACA expression in the ECs. HDRACA binds to the NTD of HSPB1 to inhibit its phosphorylation at S15, S78 and S82 sites, promoting the aggregation of HSPB1 to undergo LLPS and thus inhibit the interaction between HSPB1 and IKKβ. IKKβ binds to IκBα to induce its degradation, reducing the interaction between IκBα and NF-κB. NF-κB translocates into the nucleus to induce the expression of downstream target genes encoding key chemokines and adhesion molecules. As a result, HDLCAD loses ability to inhibit immune cells infiltration and atherosclerosis formation. Data are presented as the mean ± SD. For B-F, n=6. Statistical significance was determined using unpaired Student's t-test (B-F). **p<0.01; ***p<0.001; ****p<0.0001.
Discussion
Our present study is the first to report that HDLhealthy downregulates the expression of HDRACA to inhibit LLPS of HSPB1 in ECs, leading to reduced NF-κB-induced expression of adhesion molecules and chemokines, which attenuates monocytes adhesion to and migration towards ECs, thereby inhibiting atherosclerosis. However, HDLCAD cannot decrease the expression of HDRACA to inhibit LLPS of HSPB1 in ECs, resulting in the NF-κB-induced expression of adhesion molecules and chemokines, which stimulates the migration and adhesion of monocytes to ECs and promotes atherosclerosis.
HDL exerts multiple cardiovascular protective effects, primarily via reverse cholesterol transport, alongside anti-inflammatory, antioxidant, endothelial-protective, and pro-angiogenic activities [9, 17, 29, 50]. However, several clinical studies have demonstrated that increasing HDL cholesterol levels does not reduce cardiovascular events [51-53]. We and other researchers previously found that HDL undergoes compositional alterations, loses its protective ability, and becomes proinflammatory or dysfunctional in ASCVD [9, 17, 29, 54, 55] Indeed, a recent study found that HDL raised by niacin contains more atherogenic proteins [56]. In addition, previous studies demonstrated that S1P levels in HDL were decreased in patients with CAD [17, 57, 58]. We recently found that HDLhealthy-bound S1P inhibits HDRACA expression to promote angiogenesis, whereas HDLCAD is much less effective because of S1P deficiency [17]. S1P is a bioactive lipid that can activate receptors on ECs to reduce adhesion molecules expression and attenuate inflammation [10, 59]. Silencing KLF5 in EC has been shown to inhibit inflammation [60]. We demonstrated that KLF5 is a transcription factor for HDRACA, and HDL-bound S1P induces ubiquitination and degradation of KLF5 through the S1P1 receptor, thereby suppressing HDRACA transcription [17]. Therefore, it is possible that HDLhealthy may inhibit inflammation to suppress atherosclerosis through S1P-inhibited KLF5 expression to reduce the expression of HDRACA in ECs, whereas HDLCAD cannot inhibit inflammation due to loss of S1P, which cannot inhibit KLF5 expression and reduce the expression of HDRACA, leading to atherosclerosis formation. Moreover, our previous study demonstrated that exogenous supplementation of S1P with HDLCAD can significantly restore its inhibitory effect on HDRACA expression [17]. Therefore, the level of HDL-bound S1P is a key factor in determining its inhibitory effect on HDRACA expression. Previous studies have demonstrated that the decreased S1P content in HDLCAD may be associated with the oxidation of HDL [58]. Our recent and present study showed increased nHDLox levels in CAD patients compared to healthy subjects [61]. nHDLox represents the HDL lipid peroxide content, which can result from HDL oxidation [31]. Therefore, increased nHDLox levels in CAD indicate elevated HDL oxidation, which can lead to a reduction in the S1P content in HDLCAD. Moreover, patients with CAD usually have disorders of lipid metabolism. Notably, fatty acids, including myristic acid, compete with S1P for binding to apolipoprotein M (ApoM) in HDL [62]. These lipids may promote S1P release from HDL by competing with S1P for binding to ApoM in patients with CAD. Thus, increased nHDLox and changes in lipid composition in CAD may be critical factors for the reduced S1P levels in HDLCAD, which attenuates the inhibitory effect of HDLCAD on HDRACA expression. Collectively, this is the first novel finding that HDLhealthy inhibits inflammation and atherosclerosis by suppressing HDRACA expression, whereas HDLCAD loses its atheroprotective ability owing to its reduced ability to inhibit HDRACA.
Recent studies reported that LLPS can mediate vascular inflammation and atherosclerosis formation [24, 25]. Additionally, LLPS plays a crucial role in regulating various endothelial functions [24, 63, 64]. Importantly, lncRNAs may be important drivers of LLPS [26, 27]. lncRNAs can initiate condensate formation by binding to RNA-binding proteins to form ribonucleoprotein particles, which subsequently recruit additional non-condensed proteins through intrinsically disordered regions (IDRs) to drive LLPS [26, 27]. Previous studies demonstrated that lncRNAs can efficiently regulate protein activities by recruiting far outnumbered protein molecules to undergo LLPS [26, 27]. LLPS is recognized as a regulatory mechanism that can enrich specific factors while excluding others, thereby coordinating the specificity and efficiency of signal transduction [23]. The present study demonstrates that HDRACA can induce LLPS of HSPB1, which sequesters the interaction between HSPB1 and IKKβ and activates the NF-κB pathway. Additionally, other studies have reported that HSPB1 can co-phase separate with TDP-43 and FUS to regulate amyloid fibril aggregation of these proteins [46, 47]. Therefore, HSPB1 may form compartmentalized reaction hubs through LLPS to bind to specific molecules and exert distinct regulatory functions.
Small heat shock proteins, including HSPB1, act as molecular chaperones that regulate protein folding and aggregation. Previous studies have suggested that the ACD of HSPB1 is the major active domain for its chaperone activity, whereas the NTD and CTD modulate this activity [65-67]. Additionally, HSPB1 can aggregate to form oligomers, which reduce its affinity for substrates [68-70]. Previous studies have shown that phosphorylation within the NTD of HSPB1 at serine residues 15, 78, and 82 decreases oligomerization and promotes interaction with IKKβ, thereby limiting NF-κB activation [49, 68-70]. The present study further demonstrates that inhibiting the phosphorylation of serine residues 15, 78, and 82 in the NTD of HSPB1 promotes its LLPS. Moreover, a recent study reported that CCF0054500 can inhibit the phosphorylation of HSPB1 at serine residues 15, 78, and 82 in platelets, thereby disrupting the actin cytoskeleton and reducing platelet aggregation [71]. Thus, LLPS-mediated compartmentalization of HSPB1 may be critical for cytoskeletal maintenance. This requires further investigation into the functions of HSPB1 LLPS-formed reaction hubs in the future.
Our present study found that HDRACA inhibits the phosphorylation of HSPB1 at S15, S78, and S82 sites of the NTD, likely through multiple binding interactions with the NTD. The direct binding of HDRACA and HSPB1 may create steric hindrance, physically blocking upstream kinases, including MAPKAPK2/3, from accessing serine residues [72]. Additionally, HDRACA promotes LLPS of HSPB1 into biomolecular condensates, which may alter the conformational accessibility of its NTD, further limiting kinase accessibility. Thus, HDRACA may suppress the phosphorylation of HSPB1 through a combination of binding-induced blockade and promotion of a phase-separated state. HDRACA may serve as a scaffold that promotes the interaction and aggregation of multiple HSPB1 monomers, thereby forming an HDRACA-HSPB1 complex that serves as a core for driving LLPS. Moreover, the CTD of HSPB1 within the complex can enhance the formation of LLPS condensates via weak multivalent interactions with free HSPB1 or HSPB1 from the other complexes. Interestingly, the absence of the CTD causes HSPB1 condensates to lose their dynamic liquid-like state, potentially leading to amyloid aggregation, insoluble aggregation, or liquid-solid phase separation [73-75]. This is the second novel finding that HDRACA induces vascular inflammation to promote atherosclerosis by increasing LLPS of HSPB1 to activate the NF-κB pathway.
An important finding is that inhibition of HSPB1 aggregation may be a critical therapeutic step to reduce vascular inflammation and atherosclerosis. Therefore, the direct targeting of HSPB1 is a potential strategy for inhibiting atherosclerosis. Furthermore, we not only found that deletion of the NTD more effectively reduced the formation of HSPB1 condensates than deletion of the CTD but also demonstrated that HDRACA primarily targets the NTD of HSPB1. Therefore, the development of NTD-targeted therapeutic agents to inhibit HSPB1 aggregation may offer a promising strategy for inhibiting atherosclerosis. Currently, technologies, including virtual screening and surface plasmon resonance, facilitate high-throughput screening of large chemical libraries, providing a strong foundation for the development of NTD-targeted drugs. Alternatively, improving HDL function or inhibiting HDRACA represent two other promising strategies. These strategies can not only attenuate atherosclerosis but also promote angiogenesis, facilitating tissue revascularization following atherosclerotic stenosis.
This study had several limitations. First, although HUVECs are widely used in atherosclerosis research because of their accessibility and high proliferative capacity, our findings require further validation in human artery endothelial cells, which are more directly relevant to atherosclerosis. Second, although our study supports a functional role for HDRACA in promoting atherosclerosis, we acknowledge that direct inhibition of HDRACA is essential to conclusively establish its role as a key therapeutic target. We recently made significant findings regarding this matter. The human HDRACA-encoding gene (LOC105373383) is located on the sense strand of the X chromosome between the ATP2B3 and DUSP9 genes. We identified an expressed sequence tag, CF424538, located on the antisense strand of the mouse X chromosome between the Atp2b3 and Dusp9 genes. Although CF424538 shares only 45.8% homology with human HDRACA, Mfold analysis showed that their secondary structures were highly similar (data not shown). Since HDRACA primarily regulates substrate protein function via conformational binding, we hypothesized that CF424538 may be a potential structural homolog candidate for HDRACA [76]. Next, we will investigate whether CF424538 binds to murine HSPB1 and then evaluate atherosclerosis development following CF424538 silencing in high-fat diet-fed Ldlr-/- mice. Moreover, advanced structural validation, including comparative analysis of RNA three-dimensional structures should be performed to confirm the structural consistency between HDRACA and CF424538. Additionally, three-dimensional organoid or artery-on-a-chip models derived from human cells provide a promising platform for investigating the effect of HDRACA silencing on atherosclerosis formation [77-79].
Overall, this study highlights that HDRACA plays a critical regulatory role in HDL-mediated vascular inflammation and atherosclerosis. The differential regulation of HDRACA by HDLhealthy and HDLCAD may explain their distinct anti-inflammatory effects. Furthermore, our findings directly demonstrate that HDRACA promotes atherosclerosis formation, positioning it as a key link between HDL dysfunction and atherosclerosis development. Additionally, this study uncovered an important role of lncRNA-regulated LLPS in vascular inflammation, offering a novel perspective for exploring the mechanisms of atherosclerosis. We proposed three potential anti-atherosclerosis strategies: improving HDL function, inhibiting HDRACA, and blocking the LLPS of HSPB1. The clinical application of these strategies holds great promise for advancing ASCVD therapy.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
The authors are sincerely grateful to the patients and staff of the First Affiliated Hospital, Sun Yat-sen University for their invaluable contributions to this study. This research was funded by the National Natural Science Foundation of China (Grant Nos. 92268202, 82400526, 82495171, 82270485, 82570485), National Key R&D Program of China (2021YFA0805100), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2023-PT320-03), Guangdong Basic and Applied Basic Research Foundation, China (2024A1515030041, 2023A1515110400), The Science and Technology Planning Project of Guangdong Province (2023B1212060018), the Sun Yat-sen University Basic Research Funds (24qnpy354) and Clinical Research 5010 Program (2014002) and the Program of National Key Clinical Specialties.
BioRender.com was used to generate Figures 6B, 7A and 7G.
Data availability
The authors will provide the data supporting the findings of this study upon reasonable request to corresponding authors.
Author contributions
J.S.O., G.Q.C, Z.J.O. and Z.W.M. conceived and designed the study. Z.W.M., Y.F.L., Y.X.Z., Y.L., Y.C., C.Y.L., L.L., Y.T.K., Z.S.M. and Y.M.P. performed the experiments. Z.W.M., Y.F.L., Y.X.Z., Y.L., Y.C., C.Y.L., L.L., Y.T.K., Z.S.M., Y.M.P., Z.J.O., G.Q.C. and J.S.O. generated or analyzed the data. Z.W.M. and J.S.O. wrote the manuscript. Z.J.O., G.Q.C and J.S.O. supervised the project and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS. et al. Atherosclerosis. Nature reviews Disease primers. 2019;5:56
2. Björkegren JLM, Lusis AJ. Atherosclerosis: Recent developments. Cell. 2022;185:1630-45
3. Csordas A, Bernhard D. The biology behind the atherothrombotic effects of cigarette smoke. Nature reviews Cardiology. 2013;10:219-30
4. Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524-33
5. Li X, Ma G, Chen H, Zhang L, He J, Liu S. et al. From Heart to Mind: Research Advances in the Link between Coronary Artery Disease and Mental Stress - A Narrative Review. Heart and Mind. 2025;9:328-43
6. Sanchis-Gomar F, Pareja-Galeano H, Moya-Morales JM, Vázquez-González S, Perez-Quilis C. Dietary Trends, Microbiome, and Cardiovascular Health: A Narrative Review. Heart and Mind. 2025;9:237-54
7. Gregório C, Caldeira D, Brito J, Plácido R, Pinto FJ. How COVID-19 Can Damage the Heart? - Association of Cardiac Injury with COVID-19: A Narrative Review. Heart and Mind. 2025;9:255-63
8. Gianopoulos I, Daskalopoulou SS. Macrophage profiling in atherosclerosis: understanding the unstable plaque. Basic research in cardiology. 2024;119:35-56
9. Rosenson RS, Brewer HB Jr, Ansell BJ, Barter P, Chapman MJ, Heinecke JW. et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nature reviews Cardiology. 2016;13:48-60
10. Ruiz M, Frej C, Holmér A, Guo LJ, Tran S, Dahlbäck B. High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:118-29
11. Gharavi NM, Gargalovic PS, Chang I, Araujo JA, Clark MJ, Szeto WL. et al. High-density lipoprotein modulates oxidized phospholipid signaling in human endothelial cells from proinflammatory to anti-inflammatory. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1346-53
12. Liu L, Wang Q, Qiu Z, Kang Y, Liu J, Ning S. et al. Noncoding RNAs: the shot callers in tumor immune escape. Signal transduction and targeted therapy. 2020;5:102
13. Li S, He RC, Wu SG, Song Y, Zhang KL, Tang ML. et al. LncRNA PSMB8-AS1 Instigates Vascular Inflammation to Aggravate Atherosclerosis. Circulation research. 2024;134:60-80
14. Ni H, Ge Y, Zhuge Y, Liu X, Chen H, Liu J. et al. LncRNA MIR181A1HG Deficiency Attenuates Vascular Inflammation and Atherosclerosis. Circulation research. 2025;136:862-83
15. Fasolo F, Jin H, Winski G, Chernogubova E, Pauli J, Winter H. et al. Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization. Circulation. 2021;144:1567-83
16. Vacante F, Rodor J, Lalwani MK, Mahmoud AD, Bennett M, De Pace AL. et al. CARMN Loss Regulates Smooth Muscle Cells and Accelerates Atherosclerosis in Mice. Circulation research. 2021;128:1258-75
17. Mo ZW, Peng YM, Zhang YX, Li Y, Kang BA, Chen YT. et al. High-density lipoprotein regulates angiogenesis by long non-coding RNA HDRACA. Signal transduction and targeted therapy. 2023;8:299
18. Martin-Ventura JL, Nicolas V, Houard X, Blanco-Colio LM, Leclercq A, Egido J. et al. Biological significance of decreased HSP27 in human atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1337-43
19. He W, Tu S, Han J, Cui H, Lai L, Ye Y. et al. Mild phototherapy mediated by IR780-Gd-OPN nanomicelles suppresses atherosclerotic plaque progression through the activation of the HSP27-regulated NF-κB pathway. Acta biomaterialia. 2024;182:199-212
20. Inia JA, O'Brien ER. Role of Heat Shock Protein 27 in Modulating Atherosclerotic Inflammation. Journal of cardiovascular translational research. 2021;14:3-12
21. Seibert TA, Hibbert B, Chen YX, Rayner K, Simard T, Hu T. et al. Serum heat shock protein 27 levels represent a potential therapeutic target for atherosclerosis: observations from a human cohort and treatment of female mice. Journal of the American College of Cardiology. 2013;62:1446-54
22. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344-62
23. Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 2019;176:419-34
24. Liu J, Zhao C, Xiao X, Li A, Liu Y, Zhao J. et al. Endothelial discoidin domain receptor 1 senses flow to modulate YAP activation. Nature communications. 2023;14:6457
25. Li X, Zhu R, Jiang H, Yin Q, Gu J, Chen J. et al. Autophagy enhanced by curcumin ameliorates inflammation in atherogenesis via the TFEB-P300-BRD4 axis. Acta pharmaceutica Sinica B. 2022;12:2280-99
26. Lin Y, Protter DS, Rosen MK, Parker R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Molecular cell. 2015;60:208-19
27. Elguindy MM, Mendell JT. NORAD-induced Pumilio phase separation is required for genome stability. Nature. 2021;595:303-8
28. Liu X, Wang TT, Li Y, Shi MM, Li HM, Yuan HX. et al. High density lipoprotein from coronary artery disease patients caused abnormal expression of long non-coding RNAs in vascular endothelial cells. Biochemical and biophysical research communications. 2017;487:552-9
29. Li HM, Mo ZW, Peng YM, Li Y, Dai WP, Yuan HY. et al. Angiogenic and Antiangiogenic mechanisms of high density lipoprotein from healthy subjects and coronary artery diseases patients. Redox biology. 2020;36:101642
30. Kang BA, Li HM, Chen YT, Deng MJ, Li Y, Peng YM. et al. High-density lipoprotein regulates angiogenesis by affecting autophagy via miRNA-181a-5p. Science China Life sciences. 2024;67:286-300
31. Sasko B, Scharow L, Mueller R, Jaensch M, Dammermann W, Seibert FS. et al. Reduced high-density lipoprotein antioxidant function in patients with coronary artery disease and acute coronary syndrome. JCI insight. 2025 10
32. Atamanalp SS, Keles MS, Atamanalp RS, Acemoglu H, Laloglu E. The effects of serum cholesterol, LDL, and HDL levels on gallstone cholesterol concentration. Pakistan journal of medical sciences. 2013;29:187-90
33. Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Advanced drug delivery reviews. 2013;65:1357-69
34. Yuan HX, Chen YT, Li YQ, Wang YS, Ou ZJ, Li Y. et al. Endothelial extracellular vesicles induce acute lung injury via follistatin-like protein 1. Science China Life sciences. 2024;67:475-87
35. Liu ZL, Li Y, Lin YJ, Shi MM, Fu MX, Li ZQ. et al. Aging aggravates aortic aneurysm and dissection via miR-1204-MYLK signaling axis in mice. Nature communications. 2024;15:5985
36. Chen S, Gao JJ, Liu YJ, Mo ZW, Wu FY, Hu ZJ. et al. The oxidized phospholipid PGPC impairs endothelial function by promoting endothelial cell ferroptosis via FABP3. Journal of lipid research. 2024;65:100499
37. Gong K, Song K, Wang H, Li L, Sun X, Sun L. et al. Triptonide-mediated PTGS2 Inhibition Induces Autophagic Cell Death to Suppress the Progression of Triple-negative Breast Cancer and Epithelial Ovarian Cancer. International journal of biological sciences. 2026;22:2215-32
38. Li Y, Chen YT, Liu JS, Liang KF, Song YK, Cao Y. et al. Oncoprotein-induced transcript 3 protein-enriched extracellular vesicles promotes NLRP3 ubiquitination to alleviate acute lung injury after cardiac surgery. Journal of molecular and cellular cardiology. 2024;195:55-67
39. Gao JJ, Wu FY, Liu YJ, Li L, Lin YJ, Kang YT. et al. Increase of PCSK9 expression in diabetes promotes VEGFR2 ubiquitination to inhibit endothelial function and skin wound healing. Science China Life sciences. 2024;67:2635-49
40. Xiao R, Lu X, Huang F, Zhao Y, Jin H, Jia X. et al. METTL3-mediated m6A methylation on lncRNA H19 inhibits intrahepatic cholangiocarcinoma progression through PPARγ downregulation. International journal of biological sciences. 2025;21:6062-80
41. Lu Q, Lv X, Wang J, Xia B, Yan H, Wang Z. et al. Methylation-mediated LncRNA CRAT40 promotes colorectal cancer progression by recruiting YBX1 to initiate RelA transcription. International journal of biological sciences. 2025;21:4834-50
42. Li RH, Tian T, Ge QW, He XY, Shi CY, Li JH. et al. A phosphatidic acid-binding lncRNA SNHG9 facilitates LATS1 liquid-liquid phase separation to promote oncogenic YAP signaling. Cell research. 2021;31:1088-105
43. Ning DS, Ma J, Peng YM, Li Y, Chen YT, Li SX. et al. Apolipoprotein A-I mimetic peptide inhibits atherosclerosis by increasing tetrahydrobiopterin via regulation of GTP-cyclohydrolase 1 and reducing uncoupled endothelial nitric oxide synthase activity. Atherosclerosis. 2021;328:83-91
44. Liu Z, Cao Y, Liao XL, Ou ZJ, Mo ZW, Liu YF. et al. Apolipoprotein A-I Mimetic Peptide Restores VEGF-induced Angiogenesis in Hypercholesterolemic Ischemic Heart by Reducing HDL Proinflammatory Properties. Journal of cardiovascular translational research. 2025;18:58-69
45. Hamczyk MR, Nevado RM, Gonzalo P, Andrés-Manzano MJ, Nogales P, Quesada V. et al. Endothelial-to-Mesenchymal Transition Contributes to Accelerated Atherosclerosis in Hutchinson-Gilford Progeria Syndrome. Circulation. 2024;150:1612-30
46. Liu Z, Zhang S, Gu J, Tong Y, Li Y, Gui X. et al. Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nature structural & molecular biology. 2020;27:363-72
47. Lu S, Hu J, Arogundade OA, Goginashvili A, Vazquez-Sanchez S, Diedrich JK. et al. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nature cell biology. 2022;24:1378-93
48. Hayes D, Napoli V, Mazurkie A, Stafford WF, Graceffa P. Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. The Journal of biological chemistry. 2009;284:18801-7
49. Park KJ, Gaynor RB, Kwak YT. Heat shock protein 27 association with the I kappa B kinase complex regulates tumor necrosis factor alpha-induced NF-kappa B activation. The Journal of biological chemistry. 2003;278:35272-8
50. Pownall HJ, Rosales C, Gillard BK, Gotto AM Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nature reviews Cardiology. 2021;18:712-23
51. Kastelein JJ, van Leuven SI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ. et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. The New England journal of medicine. 2007;356:1620-30
52. Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J. et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. The New England journal of medicine. 2012;367:2089-99
53. Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J. et al. Effects of extended-release niacin with laropiprant in high-risk patients. The New England journal of medicine. 2014;371:203-12
54. Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M. et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127:891-904
55. Rohatgi A, Westerterp M, von Eckardstein A, Remaley A, Rye KA. HDL in the 21st Century: A Multifunctional Roadmap for Future HDL Research. Circulation. 2021;143:2293-309
56. Ronsein GE, Vaisar T, Davidson WS, Bornfeldt KE, Probstfield JL, O'Brien KD. et al. Niacin Increases Atherogenic Proteins in High-Density Lipoprotein of Statin-Treated Subjects. Arteriosclerosis, thrombosis, and vascular biology. 2021;41:2330-41
57. Sattler KJ, Elbasan S, Keul P, Elter-Schulz M, Bode C, Gräler MH. et al. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic research in cardiology. 2010;105:821-32
58. Sattler K, Gräler M, Keul P, Weske S, Reimann CM, Jindrová H. et al. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content: Correction by Sphingosine-1-Phosphate-Loading. Journal of the American College of Cardiology. 2015;66:1470-85
59. Kimura T, Tomura H, Mogi C, Kuwabara A, Damirin A, Ishizuka T. et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. The Journal of biological chemistry. 2006;281:37457-67
60. McCoy MG, Jamaiyar A, Sausen G, Cheng HS, Pérez-Cremades D, Zhuang R. et al. MicroRNA-375 repression of Kruppel-like factor 5 improves angiogenesis in diabetic critical limb ischemia. Angiogenesis. 2023;26:107-27
61. Cao HY, Mo ZW, Ma ZS, Liu YF, Wang C, Peng YM. et al. High-density lipoprotein antioxidant function is associated with lipid profiles and lipid-lowering therapy. Science China Life sciences. 2026 [Epub ahead of print]
62. Sevvana M, Ahnström J, Egerer-Sieber C, Lange HA, Dahlbäck B, Muller YA. Serendipitous fatty acid binding reveals the structural determinants for ligand recognition in apolipoprotein M. Journal of molecular biology. 2009;393:920-36
63. Ma N, Wu F, Liu J, Wu Z, Wang L, Li B. et al. Kindlin-2 Phase Separation in Response to Flow Controls Vascular Stability. Circulation research. 2024;135:1141-60
64. Zhang H, Chen Q, Zhang Q, Gan H, Li H, Chen S. et al. DDX24 Mutation Alters NPM1 Phase Behavior and Disrupts Nucleolar Homeostasis in Vascular Malformations. International journal of biological sciences. 2023;19:4123-38
65. Alderson TR, Roche J, Gastall HY, Dias DM, Pritišanac I, Ying J. et al. Local unfolding of the HSP27 monomer regulates chaperone activity. Nature communications. 2019;10:1068
66. Freilich R, Betegon M, Tse E, Mok SA, Julien O, Agard DA. et al. Competing protein-protein interactions regulate binding of Hsp27 to its client protein tau. Nature communications. 2018;9:4563
67. Carver JA, Grosas AB, Ecroyd H, Quinlan RA. The functional roles of the unstructured N- and C-terminal regions in αB-crystallin and other mammalian small heat-shock proteins. Cell stress & chaperones. 2017;22:627-38
68. Jovcevski B, Kelly MA, Rote AP, Berg T, Gastall HY, Benesch JL. et al. Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chemistry & biology. 2015;22:186-95
69. Shashidharamurthy R, Koteiche HA, Dong J, McHaourab HS. Mechanism of chaperone function in small heat shock proteins: dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. The Journal of biological chemistry. 2005;280:5281-9
70. Garrido C. Size matters: of the small HSP27 and its large oligomers. Cell death and differentiation. 2002;9:483-5
71. Aggarwal A, Josyula V, Wang N, Yang M, Shim YJ, Kennedy QP. et al. Olfactory Receptor Activation Reduces Platelet Reactivity and Arterial Thrombosis Through Actin Cytoskeleton Remodeling. Circulation. 2026;153:1827-44
72. Rada CC, Mejia-Pena H, Grimsey NJ, Canto Cordova I, Olson J, Wozniak JM. et al. Heat shock protein 27 activity is linked to endothelial barrier recovery after proinflammatory GPCR-induced disruption. Science signaling. 2021;14:eabc1044
73. Jiang Y, Gu J, Niu X, Hu J, Zhang Y, Li D. et al. Phosphorylation-Regulated Dynamic Phase Separation of HIP-55 Protects Against Heart Failure. Circulation. 2024;150:938-51
74. Li X, Yu L, Liu X, Shi T, Zhang Y, Xiao Y. et al. β-synuclein regulates the phase transitions and amyloid conversion of α-synuclein. Nature communications. 2024;15:8748
75. Shao F, Li Z, Xiao H, Chen Y, Zhang Y, Li L. et al. Suppressing protein damage response to overcome multidrug resistance in cancer therapy. Cell discovery. 2025;11:80
76. Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen LL. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nature reviews Molecular cell biology. 2023;24:430-47
77. Kong D, Ryu JC, Shin N, Lee SE, Kim NG, Kim HY. et al. In Vitro Modeling of Atherosclerosis Using iPSC-Derived Blood Vessel Organoids. Advanced healthcare materials. 2025;14:e2400919
78. Chen P, Zhou JB, Chu XP, Feng YY, Zeng QB, Lei JH. et al. Establishing a cryopreserved biobank of living tumor tissues for drug sensitivity testing. Bioactive materials. 2025;46:582-96
79. Paloschi V, Pauli J, Winski G, Wu Z, Li Z, Botti L. et al. Utilization of an Artery-on-a-Chip to Unravel Novel Regulators and Therapeutic Targets in Vascular Diseases. Advanced healthcare materials. 2024;13:e2302907
Author contact
![]() Corresponding authors: Jing-Song Ou, MD, Ph.D, Division of Cardiac Surgery, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhong Shan Er Road, Guangzhou, 510080, P.R. China; Tel: 86-20-87755766-8238; Fax: 86-20-87333122; Email: oujssysu.edu.cn, oujs2000com; and Guang-Qi Chang, MD, Ph.D, Division of Vascular Surgery, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhong Shan Er Road, Guangzhou, 510080, P.R. China; Tel: 86-20-87755766-6250; Fax: 86-20-87333122; Email: changgqsysu.edu.cn; and Zhi-Jun Ou, MD, Ph.D, Division of Vascular Surgery, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhong Shan Er Road, Guangzhou, 510080, P.R. China; Tel: 86-20-87755766-6250; Fax: 86-20-87333122; Email: Zhijunoucom.
Corresponding authors: Jing-Song Ou, MD, Ph.D, Division of Cardiac Surgery, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhong Shan Er Road, Guangzhou, 510080, P.R. China; Tel: 86-20-87755766-8238; Fax: 86-20-87333122; Email: oujssysu.edu.cn, oujs2000com; and Guang-Qi Chang, MD, Ph.D, Division of Vascular Surgery, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhong Shan Er Road, Guangzhou, 510080, P.R. China; Tel: 86-20-87755766-6250; Fax: 86-20-87333122; Email: changgqsysu.edu.cn; and Zhi-Jun Ou, MD, Ph.D, Division of Vascular Surgery, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhong Shan Er Road, Guangzhou, 510080, P.R. China; Tel: 86-20-87755766-6250; Fax: 86-20-87333122; Email: Zhijunoucom.