Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results and Discussions
4. Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(6):365-374. doi:10.7150/ijbs.3.365 This issue Cite
Research Paper
An mRNA-protein Fusion at N-terminus for Evolutionary Protein Engineering
Shingo Ueno, Hidenao Arai, Miho Suzuki, Yuzuru Husimi ![]()
Department of Functional Materials Science, Saitama University, 255 Shimo-okubo, Sakura-ku, Saitama 338-8570, Japan
Received 2007-7-8; Accepted 2007-8-27; Published 2007-8-29
Abstract
A novel method to link a nascent protein (phenotype) to its mRNA (genotype) covalently through the N-terminus was developed. The mRNA harboring amber stop codon at just downstream of initiation site was hybridized with hydrazide-modified ssDNA at upstream of coding region and was ligated to the DNA. This construct was then modified with 4-acetyl-phenylalanyl amber suppressor tRNA. This modified construct was fused with the nascent protein via the phenylalanine derivative when the mRNA uses the amber suppressor tRNA to decode the amber stop codon. The obtained fusion molecule was used successfully in selective enrichment experiments. It will be applicable for high-through-put screening in evolutionary protein engineering. In contrast to fusion molecules generated by other methods in which the protein is linked to genotype molecule through the C- terminus, our fusion molecule will serve to select a protein for which the C-terminus is essential to be active.
Keywords: In vitro selection, mRNA-display, in vitro virus, C-terminus, Non-natural amino acid, T4 RNA Ligase
1. Introduction
In evolutionary protein engineering, in vitro selection using a cell-free translation system carries advantages of large library size and applicability to cytotoxic proteins. Although various in vitro protein selection techniques such as in vitro virus [1], mRNA display [2] and ribosome display [3] have been developed, most of them are the techniques in which 3'-terminus of the genotype molecule is linked to the decoded peptide via its C-terminus. Thus, they are not adequate to the evolution of the protein, of which C-terminus is essential for its function (such as various peptide ligands, protein-protein interaction domain, membrane proteins, peptide hormones, etc). Therefore, it is worthwhile to devise the method in which the genotype molecule is linked to the decoded protein via its N-terminus, i.e. C-terminus of the protein is displayed freely.
Also in phage display method, the peptide is fused usually at the N-terminus of coat protein. However, the display method in which the peptide library was fused at the C-terminus of M13 phage major coat protein [4,5] and T7 phage D-coat protein [6] has been also reported. The carboxyl terminal end of PDZ domain-ligands, which is essential to be recognized by PDZ domain, was screened [7,8] and mapping of the C-terminal epitope of the Alzheimer's disease specific antibody [9] was carried out using such carboxyl-terminal phage display.
Several methods that link the protein to its mRNA (or DNA) via N-terminus have been reported. Sawata and Taira reported the binding procedure between MS2 coat protein dimer fused at N-terminus of the protein and the specific recognition sequence in its mRNA [10]. Yanagawa and coworkers developed the method named “STABLE” in which the streptavidin fused at N-terminus of a peptide was bound to biotin labeled gene DNA in emulsion [11,12]. These methods are simple and effective; however, screening condition is limited because its linkage between the peptide and its genotype molecule is non-covalent. Baskerville and Bartel made the ribozyme that catalyzes a phosphoamide bond formation between the 5'-terminus of the encoding RNA and the N-terminus of the decoded polypeptide [13]. Nester and coworkers developed the method that employed the property of VirD2 protein, which covalently binds to the specific DNA sequence [14]. These methods employ a covalent bond to linkage the peptide and its genotype molecule; however, these have not been applied yet to in vitro selection.
To date, many studies on the non-natural amino acid incorporation into a protein using nonsense [15,16] or frame-shift [17] suppression have been reported. And recently, these have been applied to in vitro protein display methods to introduce the mRNA-peptide fusion molecule library containing non-natural amino acid [18,19].
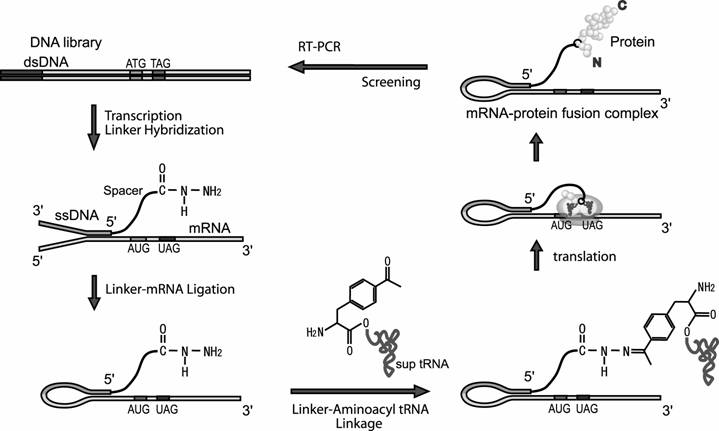
In this article we report the novel method to link the N-terminus of the peptide to its mRNA. In our method, the mRNA is linked via ssDNA and spacer to phenylalanine derivative, which is acylated to amber sup tRNA. When this modified mRNA is subjected to a cell-free translation system, this phenylalanine derivative would be incorporated into the N-terminus region of the nascent peptide at the amber stop codon inserted at the beginning of the encoded region. Consequently, C-terminus of the peptide is displayed on its mRNA (Fig.1).
Screening cycle of the mRNA-protein fusion in this study. The dsDNA library is transcribed to mRNA. The mRNA is hybridized to the DNA moiety of the linker having hydrazide group and ligated with T4 RNA ligase. Hydrazide group of the ligated product and acetyl group of the phenylalanine derivative that is acylated to sup tRNA are ligated chemically and the modified mRNA is translated. The modified aminoacyl sup tRNA tends to occupy the A-site of ribosome at UAG codon inserted near downstream of initiation codon and the phenylalanine is incorporated into the growing peptide. Thus, linkage between N-terminus of the nascent peptide and 5'-terminus of its mRNA is achieved. Screening of mRNA-peptide fusion library according to property of the displayed peptide and amplify the genotype molecules of the screened fusions by RT-PCR.
2. Materials and Methods
Synthesis of the DNA-linker having a hydrazide group
The DNA-linker having a hydrazide group at 5'-terminus (hydrazide-linker) was synthesized as follows. 5'-NH2-(CH2)6-(CH2CH2O)6-(dA)30-(CH2CH2O)6-GGG-(dT-FITC)-CGGGGGGCAAAA-3', where (dA)30 is 30 nucleotides of deoxy adenosine, (dT-FITC) is fluorescein-modified deoxy thymidine, was purchased from Tsukuba Oligo Service Co., Ltd (Ibaraki, Japan). Single riboadenosine was added to 3'-terminus of the oligomer (1.0 nmol) using terminal deoxynucleotidyl transferase (24 U, Takara Bio) with 20 µmol ATP at 37 °C for overnight in 50 µl of supplied TdT buffer. The product was purified on P6 column (Bio Rad) to remove excess ATP, followed by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation. The precipitate was dissolved in 65 µl of 0.5 M phosphate buffer (pH 9.0) and then mixed with 10 µl of 0.025 M succinic anhydride in DMSO for overnight at room temperature and purified on P6 column (Bio Rad) pre-equilibrated with H2O. Purified sample was mixed with 2 mg adipodihydrazide and 5 mg EDC (1-ethyl-3- (3-dimethylaminopropyl) carbodiimide) in 70 µl of 0.1 M phosphate buffer (pH 7.2) for 4 hour at room temperature and purified on P6 column pre-equilibrated with H2O.
The length of the DNA-linker used in this study was designed as follow. The local maximum concentration of two sites on a linear ssRNA chain (polyU) is described as:
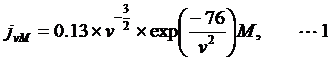
where ν is the distance-through-bond in nucleotides [20].
Probability distribution of local concentration jν (r) is described as :
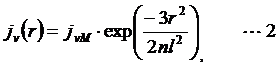
where r is the distance-through-space between the two sites, n is the distance-through-bond in the number of Kuhn segment and l is statistical segment length or Kuhn length. For ssRNA, the number of Kuhn segment and Kuhn length were reported for polyU as ν/6 and 4 nm, respectively.
The value of r for our case is 14 nm that is the distance-through-space between the exit position of mRNA on ribosome and the entrance of A-site of ribosome [21]. On the assumption that mRNA and the linker were composed of ssRNA, the probability distribution of local concentration of linked aminoacyl sup tRNA on the entrance of A-site of ribosome is given as:
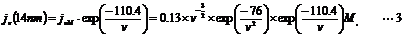
According to Equation 3, the maximum local concentration 45.5 µM is obtained when the distance-through-bond ν is 75 nt. Thus, the linker length was designed so that the chain length from the mRNA exit site to the 5'-terminus of the linker become equivalent to 75 nt.
Construction of the mRNA-templates
The His-tag encoding DNA (178 bp, Fig.2A) was constructed by three steps of PCR amplification. The first step PCR was performed using three oligomers; NL-temp (5'-ATGGGCTAGGGTGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGCAGCGCATCAC-3'), prNL+ (5'-CGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATATCCATGGGCTAGGGTGTGAGCA-3') and prNL- (5'-ACTCCTTAATGATGGTGATGGTGATGCGCTGCCCCGGT-3') and the resulting dsDNA was further amplified using two primers; prNLφ2.5GC+ (5'- CAGTAATACGACTCACTATTAGGGCCCCCCGACCCCGGTTTCCCTCTAGAAATAATTTTGT–3') and prNLHis- (5'-GCATCGACTCCTTAATGATGGTGATGGTGATGCGCTGCCCCGGTG-3'). The final step PCR was performed using the second step product dsDNA and two primers; prRT+ (5'-CAGTAATACGACTCACTATTAGGGCCCCCCGAC-3') and prRT- (5'-GAATTCGCCCTTGCATCGACTC-3'). The FLAG encoding DNA (190 bp, Fig.2A) was constructed by two steps of PCR amplification. The first step PCR was performed using the His-tag encoding DNA and two primers; prRT+ and prNLFLAG- (5'-GCATCGACTCCTTATTACTTGTCATCGTCATCCTTGTAATCCGCCGCTGCCCCGGTGAACAG-3') and the resulting dsDNA was further amplified using two primers; prRT+ and prRT-. These PCR products were purified using QIAquick PCR purification kit (Qiagen) and transcribed in vitro using RiboMAX TM Large Scale RNA Production System - T7 (Promega) in the presence of 36 mM AMP and 3.6 mM each NTPs. The RNA transcript was purified on P6 column to remove excess AMP, followed by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation.
Constructs of DNA and mRNA used in this study. (A) Construct of the model dsDNA. (B) Constructs of mRNA and DNA-linker having hydrazide group and the complex in the ligation reaction. T7φ2.5, T7φ2.5 promoter; clump, GC-rich sequence for hybridization to the DNA-linker; ε, T7 gene 10 translation enhancer; SD, Shine-Dalgarno sequence; ATG or AUG, initiation codon; TAG or UAG, amber stop codon; GFPN, N-terminus 10 amino acids of Green Fluorescent Protein gene; 6x His, Histidine hexamer tag; FLAG, FLAG epitope tag, TAA or UAA, ocher stop codon; RTP, hybridization region for RT-primer prRT-; FITC, fluorescein isothiocyanate labeled to deoxythymidine.
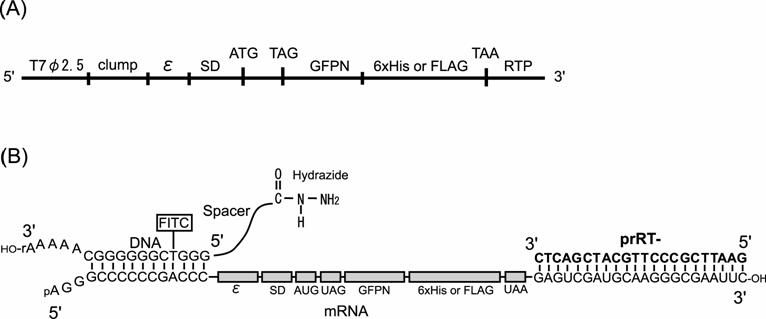
Ligation between the mRNA and the hydrazide-linker
The mRNA (2 µM) was hybridized to the DNA moiety of the hydrazide-linker (4 µM) and prRT- (4 µM) by heating at 95 °C and cooling to 25 °C in 50 µl of T4 RNA ligase buffer (Takara Bio) and ligation reaction was started by adding T4 RNA ligase (40 U, Takara Bio) and ribonuclease inhibitor (40 U, Takara Bio). Ligation reaction was performed at 25 °C for overnight and the ligated product was analyzed by 8 M urea 8 % PAGE using TBE running buffer at 65 °C and were visualized with fluorescence of FITC and then visualized again after staining by SYBR Green II (Cambrex) using a fluorescence imager (Pharos FX; Bio-Rad). Ligated product was purified using RNeasy Mini Kit (Qiagen).
Preparation of amino acid substrate
4-acetyl-L-phenylalanine cyanomethyl ester was synthesized by as follows; 10 mg of Boc-4-acetyl-L-phenylalanine (Chem Impex) was dissolved in 1 ml of acetonitrile and mixed with 350 µl of triethylamine and 100 µl of chloroacetonitrile. This mixture was stirred at room temperature for over night and the reaction mixture was diluted with 10 ml of ethyl acetate and extracted with 20 ml of 0.5 N HCl. The organic layer was then washed with 20 ml of 4 % NaHCO3 and 10 ml of saturated aqueous sodium chloride and dried using a rotary evaporator. The resulting residue was mixed with 250 µl of TFA/anisole mixture (9:1) and stirred at room temperature for 30 min. The solvent was removed using a rotary evaporator and 1 ml of 4 M HCl in dioxane was added to form the hydrochloride salt. The solution was concentrated using a rotary evaporator and 2 ml of ether was added to precipitate the hydrochloride salt. These reactions were traced by Thin Layer Chromatography.
Preparation of aminoacyl-sup tRNA
Aminoacyl-sup tRNA was prepared by the same procedure previously reported [22] with minor modification using 4-acetyl-L-phenylalanine cyanomethyl ester as the amino acid substrate.
The template DNA encoding the aminoacyl ribozyme was constructed by two steps of PCR amplification. The first PCR was performed using three oligomers; Fx (5'-ACCTAACGCCAATACCCTTTCGGGCCTGCGGAAATCTTTCGATCC-3'), P5-1 (5'-ACGCATATGTAATACGACTCACTATAGGATCGAAAGATTTCCGC-3') and P5-2 (5'-GGTAACACGCATATGTAATACGACTC-3'). The resulting dsDNA was further amplified using two primers; p5-2 and p3-2 (5'-T20-ACCTAACGCC-3'). The template DNA encoding the engineered tRNAAsnCUA was constructed by two steps of PCR amplification. The first PCR was performed using three oligomers; tR (5'-TGGTGCCTCTGACTGGACTCGAACCAGTGACATACGGATTTAGAGTCCGCCGTTCTACCGACTGAACTACAGAGGC-3'), p5-3 (5'-ACGCATATGTAATACGACTCACTATAGCCTCTGTAGTTCAGTCGGT-3') and p3-3 (5'-TGGTGCCTCTGACTGGACTC-3'). The resulting dsDNA was further amplified using two primers; p5-2 and p3-3. Amplified these two dsDNAs, encoding aminoacyl ribozyme or tRNAAsnCUA, were transcribed in vitro using RiboMAX TM Large Scale RNA transcription System – T7 in the presence of 7.5 mM GMP and 3.75 mM each NTPs. The RNA transcript was purified on P6 column to remove excess GMP, followed by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation.
400 µl of 0.1 M NaIO4 was added to 1 ml of 30 µM ribozyme, and the mixture was incubated on ice for 20 min. The oxidized RNA was then precipitated with 14 ml of 2 % LiClO4 in acetone and washed with 1 ml acetone. The pellet was dissolved in 1.4 ml of 0.1 M sodium acetate (pH 5.0) and added to 0.7 ml of adipic acid dihydrazide-agarose (Sigma) that was prewashed with RNase-free water. The mixture was stirred at room temperature for three hour and then 300 µl of 1 M NaBH3CN was added and the mixture was incubated for 30 min to stabilize the imine bond. The resin-immobilized ribozyme was washed with 1.4 ml of W1 (0.1 M sodium acetate, 300 mM NaCl, 7.5 M urea, 0.1% SDS, pH 5.0) and suspended in 2.1 ml of W1.
30 µl of 50 µM tRNAAsnCUA was folded by heating at 95 °C and cooling to room temperature in a buffer (50 mM EPPS, 12.5 mM KCl, 12 mM MgCl2, pH 7.0) and then diluted in 15 µl of a buffer (50 mM EPPS, 12.5 mM KCl, 3.6 M MgCl2, pH 7.0) and 20 µl of a buffer (50 mM EPPS, 12.5 mM KCl, 1.2 M MgCl2, pH 7.0). The folded tRNA solution was mixed with 5 nmol of resin-immobilized ribozyme that was prewashed with RNase-free water, and then 12.5 µl of 100 mM 4-acetyl-L-phenylalanine cyanomethyl ester in DMSO was added to this mixture followed by adding 1.6 µl of 0.25 M KOH (to adjust pH to 7.0-7.2). After 2 hour incubation under rotation at 4 °C, EK buffer (50 mM EPPS, 12.5 mM KCl, 10 mM EDTA, pH 7.0) was added and the supernatant, containing aminoacyl-tRNA product, was recovered and ethanol precipitated. Confirmation of aminoacylation was performed as follows. 50 pmol of aminoacylated tRNA was dissolved in 5 µl of EPPS (0.1 M, pH 5.9) containing 20 mM 6-(Biotinylamino)hexanoic acid N-hydroxy-sulfosuccinimide ester (Dojindo), and 1.0 µl of EPPS-KOH (0.3 M, pH 9.1) was added to adjust pH to 8.0. The mixture was incubated for one hour on ice and ethanol precipitated. The pellet of biotinylated aminoacyl-tRNA was dissolved in 10 µl of the loading buffer (167 pmol streptavidin, 0.1 M sodium acetate, 8 M urea, 0.05 % bromophenol blue, 0.05 % xylene cyanol, pH 5.0) and analyzed by 8 M urea 15 % PAGE using 50 mM sodium acetate (pH 5.0) as a running buffer at 30 °C and visualized after staining by SYBR Green II.
Selective enrichment analysis
10 pmol of the mRNA-linker ligation product encoding His-tag sequence was mixed with 100 pmol of 4-acetyl-phenylalanyl tRNAAsnCUA in 10 µl of 1 mM sodium acetate (pH 5.2) for overnight at room temperature and translated in 16 µl volume of E. coli S30 Extract System for Linear Templates (Promega) for 5 min at 37 °C with SUPERase-In RNase inhibitor (20 U, Ambion). The translated sample was incubated in 100 µl of Ni-NTA agarose (Qiagen) suspended in wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) for 1 hour at 4 °C using a rotary mixer. The Ni-NTA agarose was washed two times with 200 µl of wash buffer and eluted with 50 µl of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). The eluted sample was desalted with MicroSpin G-25 Column (GE Healthcare) and amplified by RT-PCR using C.therm. Polymerase One-Step RT-PCR System (Roche) using prRT+ and prRT- as primers. As negative controls, the translation product without methionine and the translation product without modification of 4-acethyl-phenylalanine tRNAAsnCUA to the linker before translation were screened with Ni-NTA agarose and amplified by RT-PCR. The amplified products were analyzed by 8 M urea 8 % PAGE using TBE running buffer at 65 °C and visualized after staining by SYBR Green I (Cambrex).
In selective enrichment analysis of the DNA encoding the His-tag sequence, Mixture of the mRNA-linker ligation products (1 pmol of the His-tag encoding molecule and 10 pmol of the FLAG-tag encoding molecule) was reacted with 100 pmol of 4-acetyl-phenylalanyl tRNAAsnCUA in 10 µl of 1 mM sodium acetate (pH 5.2) for overnight at room temperature and translated in 16 µl volume of E. coli S30 Extract System for Linear Templates for 5 min at 37 °C with SUPERase-In RNase inhibitor (20U). The translated sample was incubated in 100 µl of Ni-NTA agarose suspended in wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) for 1 hour at 4 °C using a rotary mixer. The Ni-NTA agarose was washed two times with 200 µl of wash buffer and eluted with 50 µl of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). The eluted sample was desalted with MicroSpin G-25 Column and amplified by RT-PCR using C.therm. Polymerase One-Step RT-PCR System using prRT+ and prRT- as primers. The amplified product was transcribed and ligated with hydrazide-linker for the next round of screening. The amplified product of each screening round was analyzed by 8 M urea 8 % PAGE using TBE running buffer at 65 °C and visualized after staining by SYBR Green I.
In selective enrichment analysis of the DNA encoding the FLAG-tag sequence, Mixture of the mRNA-linker ligation products (1 pmol of FLAG-tag encoding molecule and 10 pmol of His-tag encoding molecule) was reacted with 100 pmol of 4-acetyl-phenylalanyl tRNAAsnCUA in 10 µl of 1 mM sodium acetate (pH 5.2) for overnight at room temperature and translated in 16 µl reaction volume of E. coli S30 Extract System for Linear Templates (Promega) for 5 min at 37 °C with SUPERase-In RNase inhibitor (20U, Ambion). The translated sample was diluted in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 % Triton X-100, pH 8.0) and added to 40 µl of anti-FLAG M2 agarose (Sigma), followed by incubation for 1 hour at 4 °C using a rotary mixer. The anti-FLAG agarose was washed three times with 500 µl lysis buffer and incubated with 100 µl of TBS containing 0.1 mg / ml of FLAG peptide for 1 hour at 4 °C. The supernatant was desalted with MicroSpin G-25 Column and amplified by RT-PCR using C.therm. Polymerase One-Step RT-PCR System using prRT+ and prRT- as primers. The amplified product was transcribed and ligated with hydrazide-linker for the next round of screening. The amplified product of each screening round was analyzed by 8 M urea 8 % PAGE using TBE running buffer at 65 °C and visualized after staining by SYBR Green I.
Selective enrichment of the DNA encoding the His-tag sequence using PURESYSTEM [23] was performed as follows. The mixture of the mRNA-linker ligation products (0.5 pmol of His-tag encoding molecule and 5 pmol of FLAG-tag encoding molecule) was reacted with 50 pmol of 4-acetyl-phenylalanyl tRNAAsnCUA in 5 µl of 1 mM sodium acetate (pH 5.2) for overnight at room temperature and the reaction product was ethanol precipitated. The precipitated sample was translated in 3.13 µl reaction volume of PURESYSTEM classic II (Post Genome Institute Co., Ltd.) for 15 min at 37 °C with SUPERase-In RNase inhibitor. The translated sample was incubated in 50 µl of Ni-NTA agarose suspended in wash buffer for 4 hour at 4 °C using a rotary mixer. The Ni-NTA agarose was washed five times with 400 µl of wash buffer and eluted with 100 µl of elution buffer. The eluted sample was desalted with MicroSpin G-25 Column and amplified by RT-PCR using C.therm. Polymerase Two-Step RT-PCR System (Roche) and KOD dash DNA polymerase (Toyobo) using prRT+ and prRT-FITC (fluorescein was modified on the 5'-terminus of prRT-) as primers. The amplified product was analyzed by 8 M urea 8 % PAGE using TBE running buffer at 65 °C and visualized with fluorescence of FITC using a fluorescence imager.
Selective enrichment of the DNA encoding the FLAG-tag sequence using PURESYSTEM was performed as follows. The mixture of the mRNA-linker ligation products (0.5 pmol of FLAG-tag encoding molecule and 5 pmol of His-tag encoding molecule) was reacted with 50 pmol of 4-acetyl-phenylalanyl tRNAAsnCUA in 5 µl of 1 mM sodium acetate (pH 5.2) for overnight at room temperature and the reaction product was ethanol precipitated. The precipitated sample was translated in 3.13 µl reaction volume of PURESYSTEM classic II for 15 min at 37 °C with SUPERase-In RNase inhibitor. The translated sample was diluted in TBS buffer and added to 40 µl of anti-FLAG M2 agarose, followed by incubation for 4 hour at 4 °C using a rotary mixer. The anti-FLAG M2 agarose was washed five times with 400 µl of TBS buffer and incubated with 100 µl of Elution buffer (0.1 M glycine-HCl, pH3.5) for 10 min at room temperature. The supernatant was desalted with MicroSpin G-25 Column and amplified by RT-PCR using C.therm. Polymerase Two-Step RT-PCR System and KOD dash DNA polymerase using prRT+ and prRT-FITC as primers. The amplified product was analyzed by 8 M urea 8 % PAGE using TBE running buffer at 65 °C and visualized with fluorescence of FITC using a fluorescence imager.
Detection of the mRNA-peptide fusion
5 pmol of mRNA-linker ligation product was reacted with 50 pmol of 4-acetyl-phenylalanyl tRNAAsnCUA in 0.1 M sodium acetate (pH 5.2) for overnight at room temperature and the reaction product was ethanol precipitated. The precipitated pellet was translated in 6.25 µl reaction volume of PURESYSTEM classic II for 30 min at 37 °C with SUPERase-In RNase inhibitor. The translation product was blotted to PVDF-membrane after 15 % SDS-PAGE and labeled with anti-FLAG M2 monoclonal antibody (Sigma) as primary antibody and Cy3-linked anti-mouse IgG (GE Healthcare) as secondary antibody. Fluorescence of Cy3 was detected using a fluorescence imager.
3. Results and Discussions
Ligation between the mRNA and the hydrazide-linker
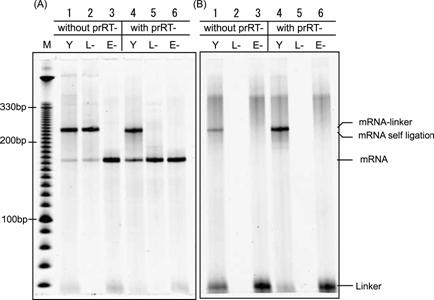
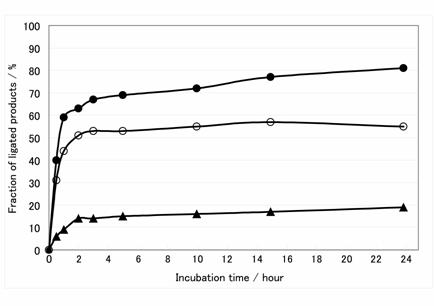
The connecting efficiency between the mRNA and the linker-molecule affects the diversity of the library. Thus, various sophisticated procedures have been devised [24-27]. In this report, we applied the Y-ligation method [25], which is the method of two single-strands ligation in the presence of a double-stranded stem to make a stem-loop structure in high efficiency. Because the transcripts were used as a phosphate donor substrate and the DNA moiety of hydrazide-linker were used as an acceptor substrate for T4 RNA ligase (Fig.2B), several modifications were required. First, transcription must be performed with excessive AMP (for T7φ2.5 promoter, GMP for normal T7 promoter and SP6 promoter) to generate the monophosphate at the 5'-terminus of the transcript and a riboadenosine must be attached to the 3'-terminus of the DNA moiety of hydrazide-linker by terminal deoxyribonucleotidyl transferase to be recognized by T4 RNA ligase (Fig.2B) [28]. Unless these modifications were performed, the ligated product was hardly observed (data not shown). Second, since 3'-OH of the transcript can be ligated with 5'-monophosphate of itself, self-ligation of the transcript was occurred (Fig.3, lane 1-2). For blocking of the 3'-OH of the transcript, the DNA oligomer prRT- was hybridized to 3'-terminus region of the transcript before ligation reaction (Fig.2B). Self-ligation was effectively prevented (Fig.3, lane 4-5) and yield of desired product was increased from 10 % to 90 % (Fig.3B, lane 1 and 4). This blocking method is simple and effective compared with other methods (e.g., making 2', 3'-cyclic phosphate group by self-cleavage of ribozyme placed at 3'-terminus [29,30] or cleavage with deoxyribozyme [31,32].). Third, it is reported that T4 RNA ligase does not prefer guanine residue as a phosphate donor [33,34], but common promoters for in vitro transcription start the transcription with guanine residue, which is located at 5'-terminus of transcripts and act as phosphate donor in ligation reaction. Therefore, it is suspected that the transcripts generated by guanine-initiation type promoter are not suitable for donor substrate of T4 RNA ligase. Hence, We applied T7φ2.5 promoter, which initiates the transcription with adenine residue [35], because adenine is better than guanine as a donor substrate of T4 RNA ligase [33,34]. We compared the ligation efficiency of transcripts generated by three kind of promoters; T7, SP6 and T7φ2.5. These promoters generate the fixed triplet sequence at 5'-terminus of the transcripts; GGG, GAA and AGG respectively. The transcript by T7φ2.5 promoter gave the best efficiency of over 80 %. For SP6 and T7, the efficiency saturated at about 55 % and 20 %, respectively (Fig.4).
Gel electrophoresis pattern of mRNA-linker ligation. The ligation products reacted with or without prRT- DNA oligomer used as a blocker of the 3'-end of mRNA were electrophoresis on 8 M urea 8 % PAGE at 65 °C and were visualized with fluorescence of (A) SYBR Green II and (B) FITC. Lane M: DNA ladder, Lane Y: ligation product, Lane L-: negative control, reaction product without DNA-linker, Lane E-: negative control, reaction product without T4 RNA ligase. Mobility of the mRNA-linker and the self-ligation product of mRNA are shown to be equivalent.
Time course of ligation reactions using three mRNAs transcribed by different promoters. Filled circle, open circle and triangle are ligation product of mRNA transcribed by T7φ2.5 promoter, normal T7 promoter and SP6 promoter, respectively.
Aminoacylation of tRNA and conjugation between hydrazide-linker and aminoacyl-tRNA
tRNAAsnCUA was acylated with 4-acetyl-L-phenylalanine using resin-immobilized ribozyme and aminoacylation efficiency was analyzed using a streptavidin-dependent gel-shift assay after biotinylation of the α-amino group of 4-acetyl-L-phenylalanine. The efficiency was approximately 15 % in our procedure (Fig.5A).
Aminoacylation of tRNAAsnCUA and conjugation between aminoacyleted tRNAAsnCUA and hydrazide-linker. (A) Amino acylation of tRNAAsnCUA analyzed by streptavidin-dependent gel-shift assay. Approximately 15 % of tRNA was aminoacylated (Lane 3). Lane 1, tRNAAsnCUA in the presence of SAv; Lane 2 and 5, tRNAAsnCUA in the absence of SAv; Lane 3, aminoacylated tRNAAsnCUA in the presence of SAv; Lane 4, aminoacylated tRNAAsnCUA in the absence of SAv. (B) Conjugation between aminoacylated tRNAAsnCUA and hydrazide-linker analyzed by acidic denatured PAGE. Approximately 10 % of hydrazide-linker was conjugated with aminoacylated tRNAAsnCUA (Lane 2). Lane 1, aminoacylated tRNAAsnCUA; Lane 2, conjugation product between aminoacylated tRNAAsnCUA and hydrazide-linker; Lane 3, hydrazide-linker.

The yield of conjugation between mRNA-linker-hydrazide and 4-acetyl-L-phenylalanyl tRNAAsnCUA was estimated as follows. 7.5 pmol of hydrazide-linker and 75 pmol of 4-acetyl-L-phenylalanyl tRNAAsnCUA were incubated in 5 µl of 0.1 M sodium acetate (pH 5.2) for overnight at room temperature. The mixture was mixed with equal volume of loading buffer (0.1 M sodium acetate, 8 M urea, 0.05 % bromophenol blue, 0.05 % xylene cyanol, pH 5.0) and analyzed by 8 M urea 15 % PAGE using 50 mM sodium acetate (pH 5.0) as a running buffer at 30 °C and visualized with fluorescence of FITC and then visualized again after staining by SYBR Green II. The yield of the conjugation was approximately 10 % because of 90 % of hydrazide-linker was left without conjugation (Fig. 5B). This yield may be including the event of hydrolysis of aminoacyl-tRNA that has been conjugated to hydrazide-linker.
Selective enrichment analysis
We demonstrated a model selection experiment using a histidine-tag having high affinity to Ni2+. The translation product displaying the His-tag peptide was screened using a Ni-NTA agarose column. The bound sample was eluted and used as a template for RT-PCR. As negative controls, the translation product without methionine that is necessary to initiate translation and the translation product without modification of 4-acethyl-phenylalanine sup tRNA to the linker before translation were screened with Ni-NTA agarose. The RT-PCR products were analyzed by electrophoresis and the desired product with higher amount was confirmed than one of negative controls generated by non-specific binding (Fig.6A).
Results of the selective enrichment experiment. (A) Result of RT-PCR after single-round Ni-NTA screening of the mRNA-peptide fusion displaying 6x His-tag sequence. Left panel, RT-PCR of the screened samples translated with or without methionine. Right panel, RT-PCR of the screened samples translated using mRNA with or without modification of 4-acethyl-phenylalanyl tRNAAsnCUA before translation. Lane M: DNA ladder, Lane PC: positive control using mRNA before translation as template of RT-PCR. (B) Results of selective enrichment experiment for binary mixture: the FLAG-tag displaying fusion molecule and the 6x His-tag displaying fusion molecule. Initial molar ratio of the former to the latter is 87:13 for left panel and 17:83 for right panel. RT-PCR product at each screening round was analyzed by electrophoresis. Left panel; screening with Ni-NTA column. Right panel; screening with Anti-FLAG antibody column. (C) Results of selective enrichment experiment using PURESYSTEM for binary mixture: the FLAG-tag displaying fusion molecule and the 6x His-tag displaying fusion molecule. Initial molar ratio of the former to the latter is 76:24 for left panel and 6:94 for right panel. RT-PCR product at each screening round was analyzed by electrophoresis. Left panel; screening with Ni-NTA column. Right panel; screening with Anti-FLAG antibody column.
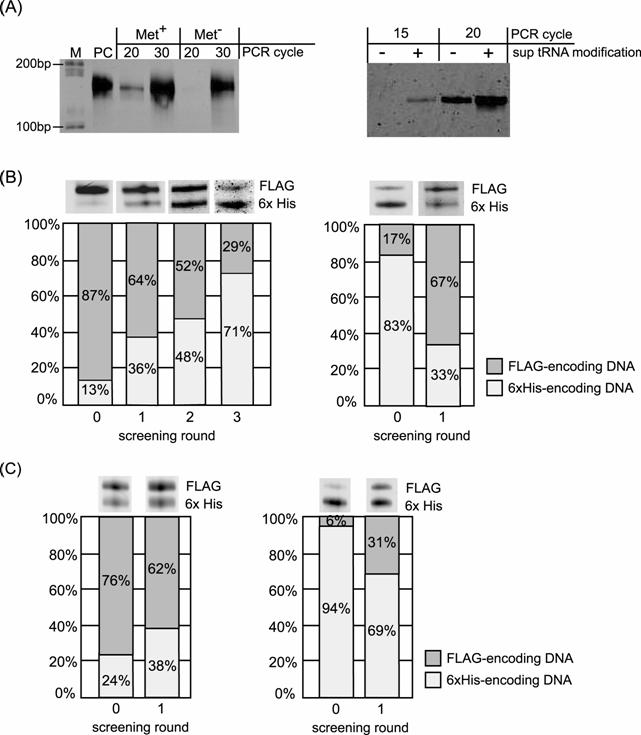
Next, we performed enrichment experiments for the DNA mixture. It was achieved to enrich the DNA encoding the His-tag sequence from a mixture containing an order of magnitude excess of the other DNA encoding FLAG-tag sequence (Fig.2A) in three rounds screening with Ni-NTA (Fig. 6B). Furthermore, enrichment of the DNA encoding the FLAG-tag sequence from a mixture containing an order of magnitude excess of the other DNA encoding His-tag sequence was also accomplished in single round screening with anti-FLAG M2 antibody (Fig. 6B). Additionally, we also performed enrichment experiments using another translation system (PURESYSTEM) and achieved the enrichment of desired sequence (Fig. 6C). These results indicated that the enrichment was not an artefact by nonspecific binding of displayed peptide but a fact by specific binding of each peptide sequence.
Detection of the mRNA-peptide fusion
The mRNA-linker ligation product encoding the FLAG peptide was translated and the mRNA-peptide fusion product was analyzed by SDS-PAGE and western blotting. Although the migration of the band caused by fusion formation was not detected on SDS-PAGE (Fig. 7A), the fusion molecule was detected by the western blotting (Fig. 7B). This low yield of the fusion formation was corresponding to relatively low efficiency of the selective enrichment (Fig. 6B, C). The low yield (~10 %) of the linker-aminoacyl-tRNA conjugate (Fig. 5B) affected directly the yield of the final product. In general, non-natural amino acid possessing large side group was incorporated into protein with relatively low efficiency. However, it is also reported that incorporation efficiency was not dependent on size of side group but on its shape, and the linear shape was appropriate [36]. In addition, non-natural amino acids possessing relatively long side chain (BODIPY FL-X-aminophenylalanine [37] or ε-N-biotinyl-L-lysine [18,19,38]) were also successfully incorporated into the polypeptide. The side chain of the amino acid used in this study was linear and did not contain any large aromatic ring. However, since the side chain of the non-natural amino acid used in this study was extraordinarily long, this amino acid may be difficult to be incorporated into polypeptide or premature termination may be occur during peptide chain elongation if this amino acid was incorporated. The linker length also must affect the formation efficiency of the mRNA-peptide fusion. Therefore, experimental optimization of the linker length will give better efficiency although the length of the linker used in this study was optimized theoretically.
Detection of mRNA-peptide fusion. The mRNA-linker ligation product that encoded FLAG peptide was translated and analyzed by the western blotting after SDS-PAGE separation. The fusion molecule was detected by the fluorescence of the Cy3 labeled on the secondary anti-body. (A) SDS-PAGE separation visualized by the fluorescence of FITC labeled on the hydrazide-linker. (B) Western blotting analysis visualized by the fluorescence of the Cy3 labeled on the secondary anti-body. The arrowhead and asterisk indicate the fusion molecule and nonspecific band, respectively. Lane 1: mRNA-linker-hydrazide. Lane 2: translated sample. Lane 3: proteinase K treated sample after translation. Lane 4: translated sample incubated with unaminoacylated sup tRNA in place of aminoacyl-sup tRNA before translation. Lane 5: translated sample without mRNA template.

4. Conclusion
We have developed a new method to prepare an mRNA-peptide fusion molecule covalently linked via N-terminus during in vitro translation. Because this fusion molecule displays the C-terminus of the peptide, it makes it possible to selection of the peptides for which function the C-terminus is essential. And also because this fused molecule has the free 3'-terminus of mRNA, it would make it possible to select an RNA replicase of de novo initiation-type.
And because a stop codon is inserted near downstream of the initiation codon, the translation reaction terminates just after initiation unless the sup tRNA is incorporated. Therefore unfused peptides that must compete with the fused peptides are never generated. Furthermore, this system is able to work even if the mRNA contains a termination codon. This method may be extended to the fusion formation linked at any position of protein by changing the position of amber codon that corresponds to the incorporation site of the linked aminoacyl sup tRNA. Thus, it may be possible that displaying the protein which having free terminus at both end and that arbitrary selection of the linking point on the protein according to its structure.
Abbreviations
FITC: fluorescein isothiocyanate; NTA: nitrilotriacetic acid; sup tRNA: suppressor tRNA; SAv: streptavidin.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (C) and by a Grant-in-Aid for Scientific Research on Priority Area No. 14085203 from MEXT, Japan.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Nemoto N, Miyamoto-Sato E, Husimi Y, Yanagawa H. In vitro virus: Bonding of mRNA bearing puromycin at the 3'-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997;414:405-408
2. Roberts R.W, Szostak J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci USA. 1997;94:12297-12302
3. Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci USA. 1997;94:4937-4942
4. Fuh G, Sidhu S.S. Efficient phage display of polypeptides fused to the carboxy-terminus of the M13 gene-3 minor coat protein. FEBS Lett. 2000;480:231-234
5. Weiss G.A, Sidhu S.S. Design and evolution of artificial M13 coat proteins. J Mol Biol. 2000;300:213-219
6. Sokoloff A.V, Bock I, Zhang G, Sebestyen M.G, Wolff J.A. The interactions of peptides with the innate immune system studied with use of T7 phage peptide display. Mol Ther. 2000;2:131-139
7. Fuh G, Pisabarro M.T, Li Y, Quan C, Lasky L.A, Sidhu S.S. Analysis of PDZ domain-ligand interactions using carboxyl-teminal phage display. J Biol Chem. 2000;275:21486-21491
8. Laura R.P, Witt A.S, Held H.A, Gerstner R, Deshayes K, Koehler M.F, Kosik K.S, Sidhu S.S, Lasky L.A. The Erbin PDZ domain binds with high affinity and specificity to carboxyl termini of -catenin and ARVCF. J Biol Chem. 2002;277:12906-12914
9. Khuebachova M, Verzillo V, Skrabana R, Ovecka M, Vaccaro P, Panni S, Bradbury A, Novak M. Mapping the C terminal epitope of the Alzheimer's disease specific antibody MN423. J Immunol Methods. 2002;262:205-215
10. Sawata S.Y, Taira K. Modified peptide selection in vitro by introduction of a protein-RNA interaction. Protein Eng. 2003;16:1115-1124
11. Doi N, Yanagawa H. STABLE: protein-DNA fusion system for screening of combinatorial protein libraries in vitro. FEBS Lett. 1999;457:227-230
12. Yonezawa M, Doi N, Kawahashi Y, Higashinakagawa T, Yanagawa H. DNA display for in vitro selection of diverse peptide libraries. Nucleic Acids Res. 2003;31:e118
13. Baskerville S, Bartel D.P. A ribozyme that ligates RNA to protein. Proc Natl Acad Sci USA. 2002;99:9154-9159
14. de Figueiredo P, Roberts R.L, Nester E.W. DARTs: A DNA-based in vitro polypeptide display technology. Proteomics. 2004;4:3128-3140
15. Bain J.D, Diala E.S, Glabe C.G, Dix T.A, Chamberlin A.R. Biosynthetic site-specific incorporation of a non-natural amino acid into a polypeptide. J Am Chem Soc. 1989;111:8013-8014
16. Noren C.J, Anthony-Cahill S.J, Griffith M.C, Schultz P.G. A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 1989;244:182-188
17. Hohsaka T, Ashizuka Y, Murakami H, Sisido M. Incorporation of nonnatural amino acids into streptavidin through in vitro frame-shift suppression. J Am Chem Soc. 1996;118:9778-9779
18. Li S, Millward S, Roberts R. In vitro selection of mRNA display libraries containing an unnatural amino acid. J Am Chem Soc. 2002;124:9972-9973
19. Muranaka N, Hohsaka T, Sisido M. Four-base codon mediated mRNA display to construct peptide libraries that contain multiple nonnatural amino acids. Nucleic Acids Res. 2006;34:e4
20. Rippe K. Making contacts on a nucleic acid polymer. Trends Biochem Sci. 2001;26:733-740
21. Yusupov M.M, Yusupova G.Z, Baucom A, Lieberman K, Earnest T.N, Cate J.H, Noller H.F. Crystal structure of the ribosome at 5.5 Å resolution. Science. 2001;292:883-896
22. Murakami H, Kourouklis D, Suga H. Using a solid-phase ribozyme aminoacylation system to reprogram the genetic code. Chem Biol. 2003;10:1077-1084
23. Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat Biotechnol. 2001;19:751-755
24. Kurz M, Gu K, Lohse P.A. Psoralen photo-crosslinked mRNA-puromycin conjugates: a novel template for the rapid and facile preparation of mRNA-protein fusions. Nucleic Acids Res. 2000;28:e83
25. Tabuchi I, Soramoto S, Suzuki M, Nishigaki K, Nemoto N, Husimi Y. An efficient ligation method in the making of an in vitro virus for in vitro protein evolution. Biol Proced Online. 2002;4:49-54
26. Tabuchi I, Soramoto S, Nemoto N, Husimi Y. An in vitro DNA virus for in vitro protein evolution. FEBS Lett. 2001;508:309-312
27. Miyamoto-Sato E, Takashima H, Fuse S, Sue K, Ishizaka M, Tateyama S, Horisawa K, Sawasaki T, Endo Y, Yanagawa H. Highly stable and efficient mRNA templates for mRNA-protein fusions and C-terminally labeled proteins. Nucleic Acids Res. 2003;31:e78
28. Kinoshita Y, Nishigaki K, Husimi Y. Enzymatic synthesis of sequencing primers based on a library of tetramers. Chem Express. 1992;7:149-152
29. Kim I, Lukavsky P.J, Puglisi J.D. NMR study of 100kDa HCV IRES RNA using segmental isotope labeling. J Am Chem Soc. 2002;124:9338-9339
30. Schurer H, Lang K, Schuster J, Morl M. A universal method to produce in vitro transcripts with homogeneous 3' ends. Nucleic Acids Res. 2002;30:e56
31. Santoro S.W, Joyce G.F. A general purpose RNA-cleaving DNA enzyme. Proc Natl Acad Sci USA. 1997;94:4262-4266
32. Pyle A.M, Chu V.T, Jankowsky E, Boudvillain M. Using DNAzyme to cut, process, and map RNA molecules for structural studies or modification. Methods Enzymol. 2000;317:140-146
33. Romaniuk E, McLaughlin L.W, Neilson T, Romaniuk P.J. The effect of acceptor oligoribonucleotide sequence on the T4 RNA ligase reaction. Eur J Biochem. 1982;125:639-643
34. England T.E, Uhlenbeck O.C. Enzymatic oligoribonucleotide synthesis with T4 RNA ligase. Biochem. 1978;17:2069-2076
35. Huang F, Bugg C.W, Yarus M. RNA-catalyzed CoA NAD, and FAD synthesis from phosphopantetheine NMN, and FMN. Biochem. 2000;39:15548-15555
36. Hohsaka T, Kajihara D, Ashizuka Y, Murakami H. and Sisido M. Efficient incorporation of nonnatural amino acids with large aromatic group into streptavidin in in vitro protein synthesizing systems. J Am Chem Soc. 1999;121:34-40
37. Hohsaka T, Abe R, Shiraga K. and Sisido M. Incorporation of fluorescently labeled nonnatural amino acids into proteins in an E.coli in vitro translation system. Nucleic Acids Research Supplement. 2003;3:271-272
38. Murakami H, Ohta A, Ashigai H. and Suga H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nature Methods. 2006;3:357-359
Author contact
![]() Correspondence to: Yuzuru Husimi, Department of Functional Materials Science, Saitama University, 255 Shimo-okubo, Sakura-ku, Saitama 338-8570, Japan. Phone/Fax: +81-48-858-3531, E-mail: husimisaitama-u.ac.jp
Correspondence to: Yuzuru Husimi, Department of Functional Materials Science, Saitama University, 255 Shimo-okubo, Sakura-ku, Saitama 338-8570, Japan. Phone/Fax: +81-48-858-3531, E-mail: husimisaitama-u.ac.jp