Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1923-1934. doi:10.7150/ijbs.27022 This issue Cite
Research Paper
Saikosaponin A Protects From Pressure Overload-Induced Cardiac Fibrosis via Inhibiting Fibroblast Activation or Endothelial Cell EndMT
Yuan Liu1, Lu Gao1, Xiaoyan Zhao1, Sen Guo1, Yuzhou Liu1, Ran Li1, Cui Liang1, Ling Li1, Jianzeng Dong1, Lina Li2 ![]() , Haibo Yang1
, Haibo Yang1 ![]()
1. Department of Cardiology, The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Zhengzhou, China
2. Department of Anesthesiology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Received 2018-5-2; Accepted 2018-7-16; Published 2018-10-31
Abstract

Saikosaponin A (SSA) is a triterpenoid saponin with many pharmacological activities, including anti-inflammatory and antioxidant effects. The effect of SSA on cardiac remodeling and fibrosis, however, remains unclear. Aortic banding surgery was used to establish a mouse cardiac remodeling and fibrosis model. Mice were subjected to an intraperitoneal (i.p.) injection of SSA (5 mg/kg/d or 40 mg/kg/d) 2 weeks after surgery for 28 days. As a result, SSA had limited effect on cardiac hypertrophy but decreased cardiac fibrosis remarkably. Neonatal rat cardiomyocytes were isolated and cultured with SSA (1 and 30 μM). Both 1 and 30 μM SSA reduced atrial natriuretic peptide transcription induced by angiotensin II. Adult mouse cardiac fibroblasts were isolated and cultured with SSA (1, 3, 10 and 30 μM). Only 10 and 30 μM SSA ameliorated transforming growth factor β (TGFβ)-induced fibroblast activation and function. Mouse heart endothelial cells were isolated and stimulated with TGFβ and cocultured with SSA (1, 3, 10 and 30 μM). Only 1 and 3 μM SSA ameliorated TGFβ-induced endothelium-mesenchymal transition (EndMT). Consistently, only the 5 mg/kg/d treatment relieved pressure overload-induced EndMT in vivo. Furthermore, we found that high dosages of SSA (10 and 30 μM) inhibited the TGFβ/smad pathway in fibroblasts, while low dosages of SSA (1 and 3 μM) inhibited the Wnt/β-catenin pathway in endothelial cells. The Smad pathway activator SRI-011381 eliminated SSA (30 μM)-induced protective effects on fibroblasts. The Wnt pathway activator WAY-262611 eliminated SSA (1 μM)-induced protective effects on endothelial cells. In summary, this study indicates the potential application of SSA in the treatment of myocardial fibrosis in cardiac fibrosis, with different target effects associated with different dosages.
Keywords: Saikosaponin A, pressure overload, cardiac fibrosis, fibroblast, endothelium-mesenchymal transition
Introduction
Heart failure is a clinical manifestation of various cardiovascular diseases. It is a devastating disease characterized by interstitial fibrosis, ventricular remodeling and decreased ventricular compliance. During the pathology of cardiac remodeling, activation of cardiac fibroblasts (CFs) results in excessive deposition of extracellular matrix (ECM) protein, decreased tissue compliance and accelerated heart failure [1, 2]. After myocardial injury, the secretion of various proinflammatory cytokines and fibrotic factors increase, leading to fibroblast proliferation and activation into myofibroblasts[3, 4]. In myofibroblasts, alpha-smooth muscle actin (α-SMA) is assembled into stress fibers that can remodel the surrounding ECM and then contribute to pathologic cardiac remodeling [5]. Therefore, the development of drugs targeting fibroblasts is essential for reducing cardiac remodeling and delaying the development of heart failure.
During the pathological process of various cardiovascular diseases, the sources of activated fibroblasts include the presence of fibroblasts, vascular origin cells, hematopoietic cells and pericytes[6]. Of these cells, cardiac endothelial cells undergoing endothelial-mesenchymal transition (EndMT) have been shown to contribute 27~33% of all cardiac fibroblasts following pressure-overload[7]. During the EndMT process, endothelial cells lose polarity and expression of intercellular adhesion complexes, such as CD31 and E-Cadherin, but activate the expression of additional mesenchymal genes and proteins, such as α-SMA, collagen I and III, and vimentin[8]. Inhibition of EndMT/EMT is a promising target for clinical therapeutic applications in cardiac fibrosis[9, 10]; thus, drugs targeting EndMT may become a new strategy for the treatment of cardiac fibrosis.
Saikosaponin A (SSA) is a triterpenoid saponin isolated from Radix Bupleuri (Rb). At present, SSA has been reported to have many pharmacological activities, such as anti-inflammatory and antioxidant effects. During activation of hepatic stellate cells, SSA was reported to regulate the expression of bone morphogenetic protein 4 (BMP-4)[11]. In the chemical-induced liver inflammation and fibrosis rat model, SSA was also reported to exert protective effects[12]. Moreover, in human umbilical vein endothelial cells, SSA inhibited oxidative stress and inflammation by suppressing TLR4 translocation into lipid rafts[13]. These data suggest a protective effect of SSA on tissue fibrosis as well as on endothelial cells. Thus, we suppose SSA would exert some extension of these effects on cardiac fibrosis. In this study, aortic banding surgery was used to establish the mouse cardiac fibrosis model. The effects of SSA on cardiac fibrosis as well as the effect of SSA on fibroblast and endothelial cells were investigated.
Materials and Methods
Materials
National Institutes for Food and Drug Control (Beijing, China) supplied the Saikosaponin A. Antibodies specific for GAPDH, TGFβ, smad2, smad3, smad4, Wnt, β-catenin, GSK3β and laminin B came from Cell Signaling Technology (Beverly, MA). Antibodies including VE-cadherin, CD31, α-SMA and vimentin came from Abcam (Cambridge, CB4 0FL, UK). SRI-011381 and WAY-262611 were purchased from MechemExpress (Monmouth Junction, NJ 08852, USA).
Animal and Animal models
We followed the Guide for the Care and Use of Laboratory Animals, which was published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The Animal Care and Use Committee of The First Affiliated Hospital of Zhengzhou University approved our experiment. The Institute of Laboratory Animal Science, CAMS& PUMC (Beijing, China) supplied us with male C57BL/6 mice that were 8-10 weeks old. The animals were randomly assigned to four groups: a. sham surgery group; b. vehicle-aortic banding (AB) group; c. SSA-low dosage (LD)-AB group (intraperitoneal injection, 5 mg/kg/d) after 2 weeks of AB for 28 consecutive days. d. SSA-high dosage (HD)-AB group (intraperitoneal injection, 40 mg/kg/d) after 2 weeks of AB for 28 consecutive days.
Echocardiography and Hemodynamics
Echocardiography was performed as described in a previous study with MyLab 30CV ultrasound (Biosound Esaote, Genoa, Italy) and a 10-MHz linear array ultrasound transducer[10]. Hemodynamics were measured with a microtip catheter transducer (SPR-839; Millar Instruments, Houston, TX) as described in a previous study[10]. The PVAN data analysis software was used to process data.
Histology and Immunofluorescence
HE and PSR staining were performed according to the protocol in our previous study[10]. For immunofluorescence, the antigen was heated by pressure cooker, incubated with anti-CD31 and alpha-SMA, and then incubated with goat anti-mouse / rabbit secondary antibody. DAPI was used to stain nuclear material and prevent fading.
RT-PCR and Western blot
RNA and RT-PCR were performed according to the protocol in our previous study [10]. A LightCycler 480 SYBR Green 1 Master Mix (Roche, 04707516001) was used for amplification prior to PCR. GAPDH gene expression was used as a reference.
Western blotting was performed according to the protocol in our previous study [10]. Protein samples were separated with SDS electrophoresis and transferred to immobilon-FL transfer membranes (IPFL00010, Millipore, Billerica, MA). After blocking with 5% milk, membranes were incubated with primary antibodies. Goat anti-rabbit (LI-COR) IgG or anti-mouse IgG (LI-COR) were used. A two-color infrared imaging system (Odyssey, LI-COR, Lincoln, NE) was used to scan the blots.
Cell culture
Neonatal rat cardiomyocytes (NRCMs)
NRCMs were isolated according to the protocol in our previous study[14]. Briefly, 1- to 3-day-old Sprague-Dawley rat hearts were harvested. Ventricles were digested four times for fifteen min each in 0.125% trypsin-EDTA in PBS. Following centrifugation, cells were resuspended and incubated for 90 min in a 100-mm dish to allow noncardiac myocytes (mainly cardiac fibroblasts) to adhere to the plastic. After culture with 1% bromodeoxyuridine for 48 h, NRCMs were treated with AngII (1 μM) for 24 h and then with SSA (1, 3, 10, 30, 50 μM) for 12 h.
Mouse adult CF culture
Mouse adult CFs were isolated according to the protocol in our previous study [15]. Briefly, 0.125% trypsin and collagenase were used to digest the left ventricles from 8-week-old mice. DMEM/F12 medium containing 10% FBS was used to suspend and culture adult CFs. Ninety minutes after attachment, the attached CFs were cultured and passaged. Cells were cultured in serum-free DMEM/F12 for 8 h and then stimulated with TGFβ1 (10 ng/ml) for 24 h. SSA (1, 3, 10, 30 and 50 μM) was applied for another 12 h. To activate TGFβ/smad signaling, after culturing with TGFβ1 for 24 h, the cells were treated with SSA (30 μM) and SRI-011381 (10 μM) for another 12 h.
Primary mouse heart EC (MHEC) culture
MHECs were isolated as described in the previous study [16]. Briefly, mouse hearts were cut in Hanks' balanced salt solution buffer. Collagenase A was used to digest the heart tissue, and 10% FBS-DMEM was used to stop digestion. After filtering with a nylon mesh (70-mm pores), cells were resuspended in Hanks' solution. CD31 beads were used to bind ECs. ECs were then washed and cultured in dishes precoated with 2% gelatin (Sigma, Oakville, ON, Canada) in endothelial basal medium with 10% FBS. MHECs were starved for 12 h and then cultured with TGFβ1 (10 ng/ml) for 24 h. SSA (1, 3, 10, 30 and 50 μM) was applied for another 12 h. To activate Wnt/β-catenin signaling, after culture with TGFβ1 for 24 h, cells were treated with SSA (1 μM) and WAY-262611(1 μM) for another 12 h.
Cell viability detection
The MTT assay was used to detect cell viability. Briefly, after treatment, cells were incubated with MTT (5 mg/ml) at 37°C for 4 h. Following additional incubation with dimethyl sulfoxide, an ELISA reader was used to measure absorbance at 495 nm.
Immunofluorescence
After being washed with PBS and fixed in 4% polyoxymethylene, cells were permeabilized with 0.1% Triton X-100 (Amresco). The following primary antibodies were used to stain the cells: anti-actin (in NRCMs), anti-α-SMA (in fibroblasts), or anti-CD31 and vimentin (in MHECs). After incubation with Goat anti-Mouse/Rabbit IRdye 800CW secondary antibody for 60 min, cells were cultured with DAPI to stain nuclear material and prevent fading.
Statistical analysis
All data are expressed as the mean ± standard deviation (SD). The differences between groups were evaluated by one-way ANOVA and post-tests. Student's unpaired t test was used to compare differences between the two groups. P < 0.05 had statistical significance.
Results
The effects of SSA on cardiac hypertrophy in vivo
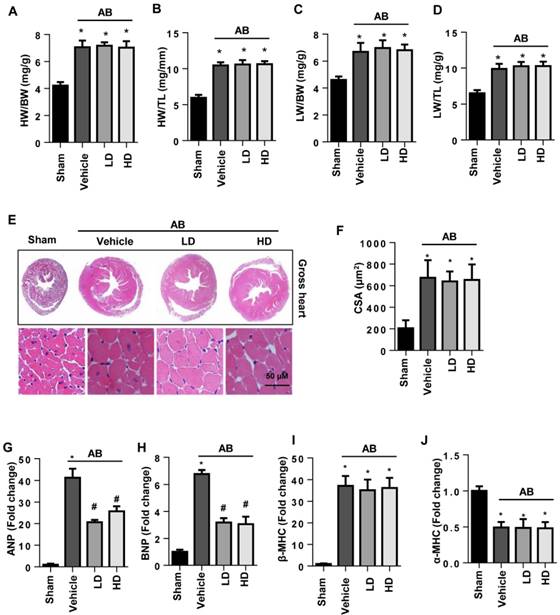
Six weeks after surgery, severe cardiac hypertrophy was observed in vehicle-treated mice with increased HW/BW, HW/TL, LW/BW, and LW/TL ratios, increased cell surface area (CSA), and increased transcription of hypertrophy markers. The extent of cardiac hypertrophy in both the LD and HD SSA-treated mice showed no significant difference from that in the vehicle-treated mice since the extent of HW/BW, HW/TL, LW/BW, LW/TL, and CSA was the same for all three groups (Fig. 1A-F). The transcription levels of ANP and BNP, but not β type-MHC and α type-MHC, were reduced by both LD and HD SSA treatment (Fig. 1G-J). These data indicate that SSA exerted a limited effect on cardiac hypertrophy.
The effects of SSA on cardiac hypertrophy in vivo. A-D. HW/BW, HW/TL, LW/BW, and LW/TL ratios in groups 6 weeks after surgery (n=8, LD: low dosage, 5 mg/kg/d; HD: high dosage, 40 mg/ kg/d). E and F. HE staining (n=6) and quantitative results (n=100+ cells per group). G-J. Transcription level of hypertrophic markers (n=6). *P<0.05 vs sham; #P<0.05 vs vehicle-AB.
The effects of SSA on cardiac fibrosis in vivo
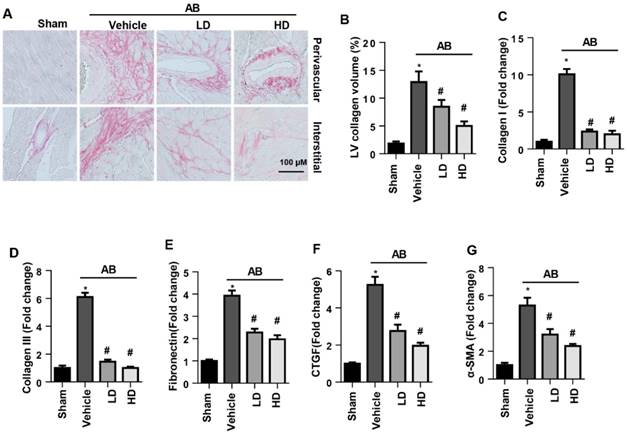
The perivascular and interstitial fibrosis levels were increased in vehicle-treated AB mice compared with the sham group. Both LD and HD SSA decreased perivascular and interstitial fibrosis levels as well as LV collagen volume compared with those of the vehicle-treated mice (Fig. 2A, B). Moreover, the increased expression level of fibrosis markers was reduced by both LD and HD SSA treatment (Fig. 2C-G). These data suggest that SSA affected cardiac fibrosis.
The effects of SSA on cardiac fibrosis in vivo. A and B. PSR staining (n=6) and quantification of the total collagen volume in mouse hearts (n=25+ fields per experimental group). C-G. Transcription level of fibrotic markers in mouse hearts (n=6). *P<0.05 vs sham; #P<0.05 vs vehicle-AB.
The effects of SSA on cardiac function
Cardiac dysfunction is the basic characteristic of heart failure. Thus, echocardiography and hemodynamics measurement were used to evaluate cardiac function. Increased ventricular wall thickness (increased IVSd, LVPWd), dilatation (increased LVEDd, LVESd) and systolic (decreased EF, FS, dp/dtmax, dp/dtmin) and diastolic dysfunction (increased Tau value) were observed in vehicle-treated mice after 6 weeks of AB. Both LD and HD SSA treatment did not change the increased ventricular wall thickness but did ameliorate the ventricular dilatation (decreased LVEDd, LVESd) and improved systolic (increased EF, FS, dp/dtmax, dp/dtmin) and diastolic function (decreased Tau value)(Table 1). Collectively, our results suggest that SSA protects against cardiac fibrosis and improves cardiac function.
Echocardiography and hemodynamics measurement data for different groups after 6 weeks of AB.
| Sham (n=15) | Vehicle-AB (n=15) | LD-AB (n=15) | HD-AB (n=15) | |
|---|---|---|---|---|
| HR (bpm) | 465±6.4 | 457±11 | 463±10 | 470±11 |
| IVSd (mm) | 0.64±0.01 | 0.84±0.01* | 0.85±0.01* | 0.86±0.01* |
| LVPWd (mm) | 0.67±0.03 | 0.87±0.01* | 0.86±0.01* | 0.86±0.01* |
| EF (%) | 65.2±0.77 | 36.7±0.53* | 52.3±0.88# | 53.8±0.69# |
| LVEDd (mm) | 3.51±0.04 | 4.31±0.03* | 3.95±0.02# | 3.93±0.03# |
| LVEDs (mm) | 2.18±0.03 | 2.84±0.03* | 2.55±0.03# | 2.49±0.02# |
| FS (%) | 41.0±0.60 | 28.1±0.62* | 31.6±0.41# | 33.6±0.82# |
| dp/dtmax (mmHg/s) | 10241±230 | 6426±153* | 8040±149# | 7804±2443# |
| dp/dtmin (mmHg/s) | -9316±213 | -6704±135* | -7455±214# | 7576±120# |
| Tau (Weiss; ms) | 8.77±0.34 | 17.1±1.56* | 10.6±0.34# | 11.6±0.94# |
HR, heart rate; IVSd, interventricular septal thickness at diastole; LVPWd, left ventricular posterior wall thickness at end-diastole; LVEDd, left ventricular end-diastolic diameter; LVESd, left ventricular end-systolic diameter; EF, left ventricular ejection fraction; FS, fractional shortening; dp/dtmax, maximal rate of pressure development; dp/dtmin, maximal rate of pressure decay; Tau, time constant of LV pressure decay *P<0.05 for difference from sham group. #P<0.05 vs vehicle-AB group.
The effects of SSA on AngII-induced cardiomyocyte hypertrophy in NRCMs
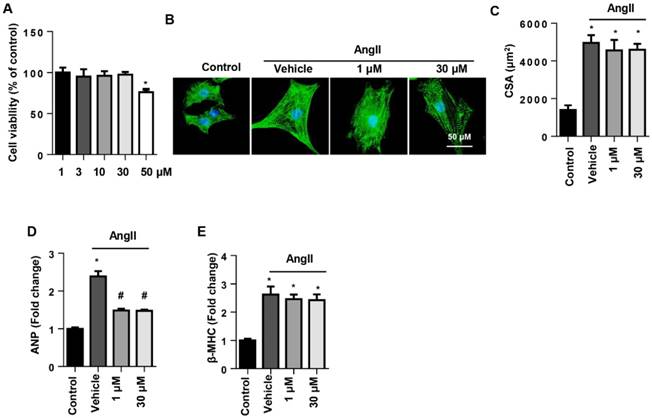
To determine the effect of SSA on cell types, NRCMs were stimulated with AngII to induce cardiomyocyte hypertrophy. Cell viability in the 1~30 μM SSA groups revealed no significant difference from the control group, though the 50 µM group did differ (Fig. 3A). Thus, we chose 1 and 30 μM SSA for further study. AngII induced deteriorated hypertrophy as assessed by the increase in CSA and increased transcription levels of hypertrophic markers. Nevertheless, none of the concentrations of SSA could attenuate AngII-induced increased CSA and transcription of β-MHC (Fig. 3B, C, E). However, both 1 and 30 μM SSA attenuated the transcription of ANP (Fig. 3D).
The effects of SSA on AngII induced cardiomyocyte hypertrophy in NRCMs. NRCMs were stimulated with AngII for 24 h and then treated with SSA for 12 h. A. Cell viability in different groups (n=5). B and C. Immunofluorescence staining of α-actin (n=6). B, representative images; C, quantitative results (n=50+ cells per group). D and E. Transcription level of hypertrophic markers in each group (n=6). *P<0.05 vs control group; #P<0.05 vs vehicle-AngII group. All experiments were repeated 3 independent times.
SSA relieves TGFβ1-induced fibroblast activation and function in adult mouse CFs
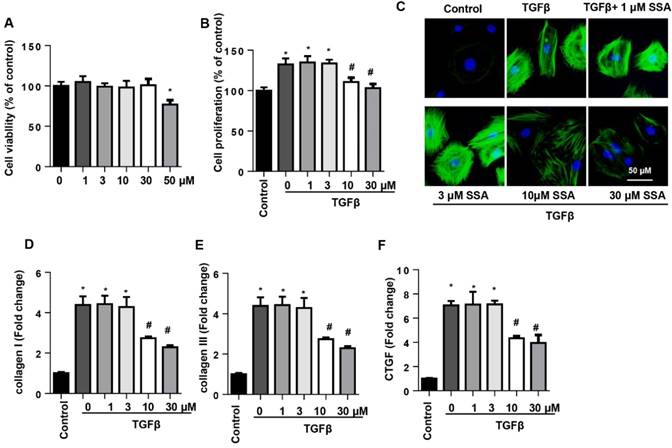
To determine whether SSA directly affected CFs to protect against cardiac fibrosis, CFs were isolated and cultured with TGFβ1 and then treated with SSA (1, 3, 10, 30 and 50 μM). Different concentrations of SSA did not affect cell viability except for 50 μM SSA (Fig. 4A). Thus, 1, 3, 10 and 30 μM SSA were used. TGFβ1 induced remarkable proliferation of CFs, but only 10 and 30 μM SSA decreased TGFβ1-induced cell proliferation (Fig. 4B). Increased expression of α-SMA and transcription of collagen I, collagen III, and CTGF were observed in CFs after TGFβ1 stimulation, suggesting a myofibroblast transition and function after TGFβ1 stimulation. Treatment with 10 and 30 μM SSA, but not 1 and 3 μM SSA, inhibited CF transition and function (Fig. 4C-F).
SSA relieves TGFβ1-induced fibroblast activation and function in adult mouse CFs. CFs were stimulated with TGFβ for 24 h and then treated with SSA for 12 h. A. Cell viability in different groups (n=5). B. Cell proliferation in different groups (n=5). C. Immunofluorescence staining of α-SMA (n=6). D-F. Transcription level of fibrotic markers in each group (n=6). *P<0.05 vs control group; #P<0.05 vs vehicle-TGFβ group. All experiments were repeated 3 independent times.
SSA suppresses TGFβ1-induced EndMT in MHECs
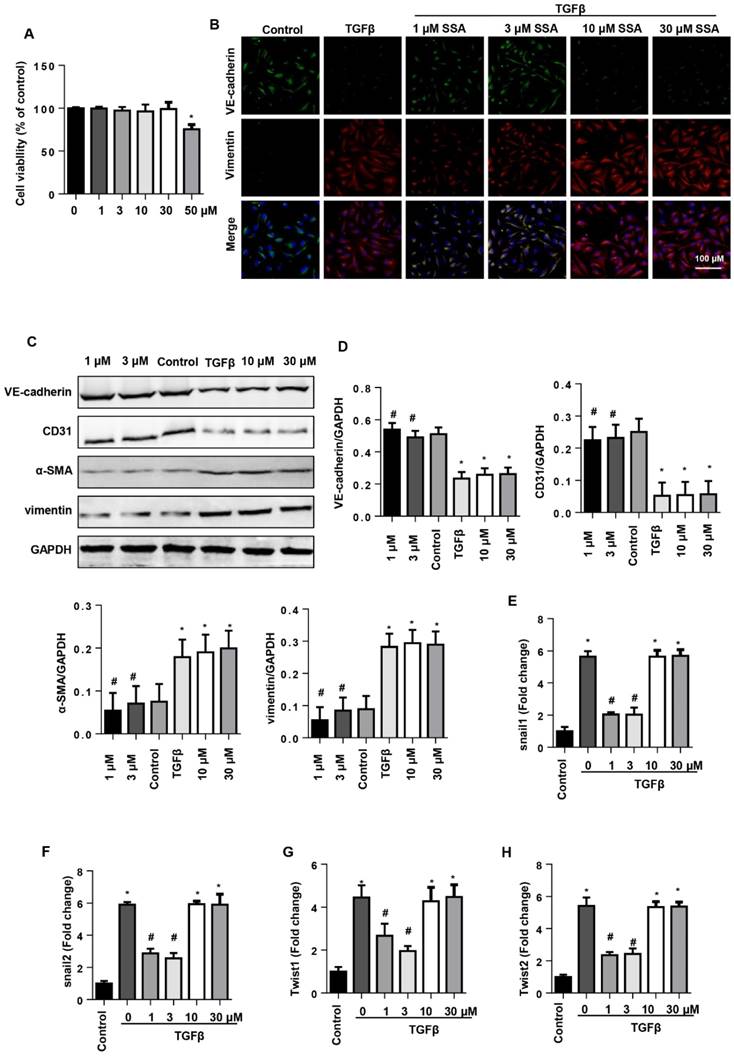
EndMT is an important source of CFs in the pressure overload induced cardiac fibrosis model. Thus, we investigated whether SSA affected EndMT in endothelial cells. MHECs were isolated and cultured with TGFβ1 and then treated with SSA (1, 3, 10, 30 and 50 μM). Different concentrations of SSA did not affect cell viability except 50 μM SSA (Fig. 5A). Thus, 1, 3, 10, 30 μM SSA were used. TGFβ1 induced remarkable EndMT in MHECs as assessed by increased CF markers (α-SMA and vimentin) and decreased endothelial cell markers (VE-cadherin and CD31), as well as increased transcription levels of EndMT markers (snail1, snail2, twist1, and twist2). Interestingly, only low concentrations (1 and 3 μM), but not higher concentrations (10 and 30 μM), of SSA ameliorated the increased EndMT induced by TGFβ1 as evidenced by decreased CF markers and increased endothelial cell markers and decreased transcription levels of EndMT markers (Fig. 5B-H).
SSA suppresses TGFβ1-induced EndMT in MHECs. MHECs were stimulated with TGFβ for 24 h and then treated with SSA for 12 h. A. Cell viability in each group (n=5). B. Immunofluorescence staining of VE-cadherin and vimentin (n=6). C and D. Protein level of VE-cadherin, CD31, α-SMA, and vimentin (n=5). E-H. RT-PCR analysis of snail1, snail2, twist1, and twist2 mRNA expression levels was performed for each group (n=6). *P<0.05 vs control group; #P<0.05 vs vehicle-TGFβ group. All experiments were repeated 3 independent times.
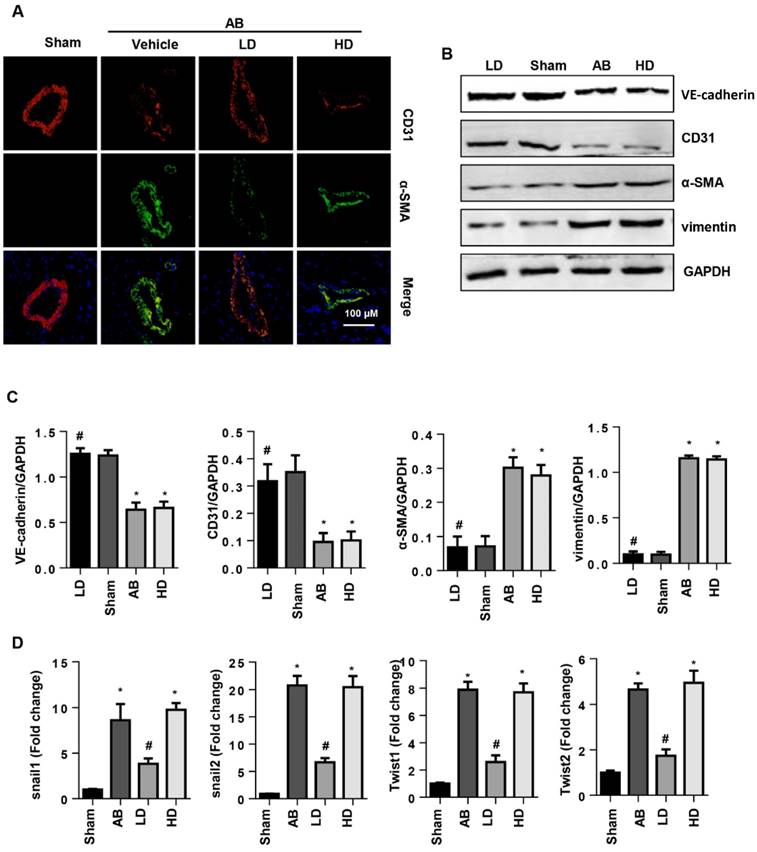
SSA attenuates EndMT in vivo
To confirm the influence of SSA on EndMT in vivo, EndMT markers were evaluated in mouse hearts after 6 weeks of AB. Increased EndMT was observed in vehicle-treated mouse hearts with increased CF markers (α-SMA and vimentin) and decreased endothelial cell markers (VE-cadherin and CD31) (Fig. 6A, B), as well as increased transcription levels of EndMT markers (snail1, snail2, twist1, and twist2) (Fig. 6D). Consistent with the in vitro study, only LD SSA treatment attenuated these EndMT transitions in mouse hearts (Fig. 6A-D). These data indicate that different concentrations of SSA may target different cell types to protect against cardiac fibrosis.
SSA attenuates EndMT in vivo. A. CD31 and α-SMA immunofluorescence staining in hearts (n=6). B and C. Protein level of VE-cadherin, CD31, α-SMA and vimentin in hearts (n=5). D. Transcription level of snail1, snail2, twist1, and twist2 in mouse hearts (n=6). *P<0.05 vs sham; #P<0.05 vs vehicle-AB.
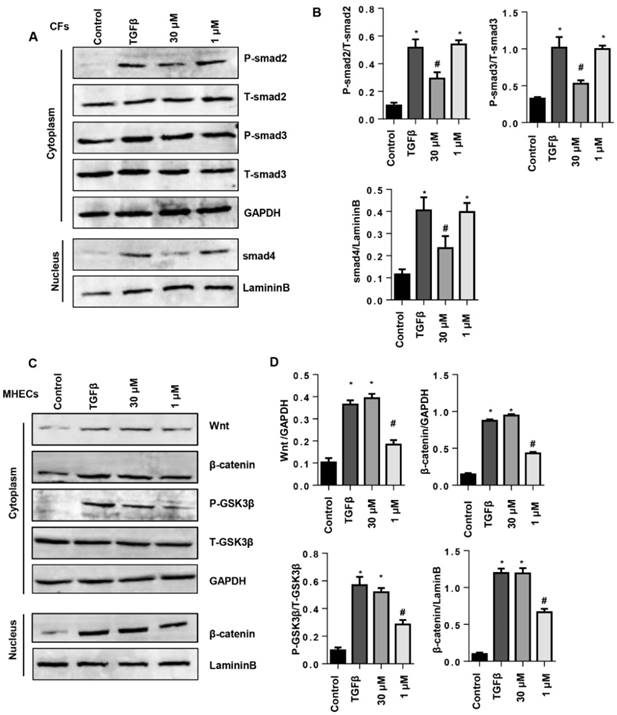
The effects of SSA on TGFβ/smad and Wnt/β-catenin pathway
To evaluate the protective effects of SSA on CFs and MHECs, the associated signaling pathways were screened by western blot. As a result, 30 μM SSA treatments decreased the phosphorylation level of smad2, smad3 and nuclear expression levels of smad4, while 1 μM SSA treatment did not affect this pathway (Fig. 7A, B). In MHECs, 1 μM SSA treatment decreased the expression of Wnt, β-catenin, the phosphorylation level of GSK3β as well as the nuclear expression of β-catenin; 30 μM SSA treatments did not affect this pathway (Fig. 7C, D).
The effects of SSA on TGFβ/smad and the Wnt/β-catenin pathway. A and B. CFs were stimulated with TGFβ for 24 h and then treated with SSA (1 and 30 μM) for 12 h. Protein levels of smad2, smad3 and smad4 in CFs (n=5). C and D. MHECs were stimulated with TGFβ for 24 h and then treated with SSA (1 and 30 μM) for 12 h. Protein levels of Wnt, β-catenin and GSK3β in MHECs (n=5). *P<0.05 vs control group; #P<0.05 vs vehicle-TGFβ group. All experiments were repeated 3 independent times.
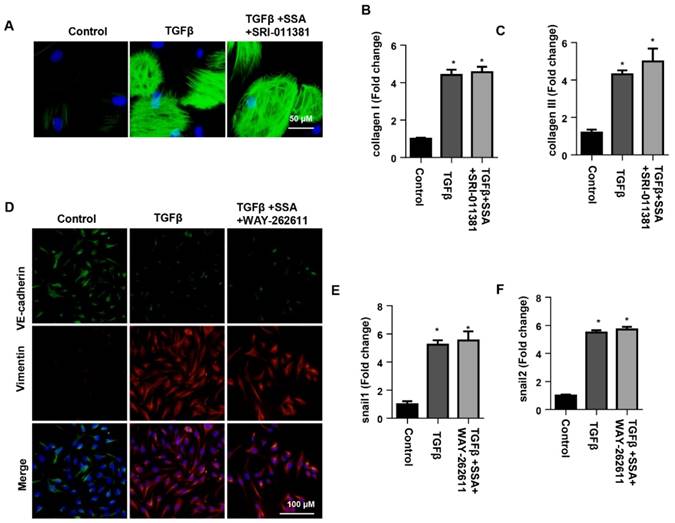
The effects of smad activator on fibroblast activation and function in CFs or Wnt activator on EndMT in MHECs
To confirm the effect of SSA on smad signaling in CFs, CFs were stimulated with TGFβ1 and then treated with 30 μM SSA and SRI-011381. The Smad activator SRI-011381 eliminated the protective effects of high concentrations of SSA on CFs as shown by the same expression levels of α-SMA, collagen I and collagen III in the TGFβ group and the TGFβ +SSA+SRI-011381 group (Fig. 8A-C). To confirm the effect of SSA on Wnt/β-catenin signaling in MHECs, MHECs were stimulated with TGFβ1 and then treated with 1 μM SSA and WAY-262611. The Wnt activator WAY-262611 eliminated the protective effects of low concentrations of SSA on EndMT in MHECs as shown by the same expression levels of increased vimentin, snail1 and snail2 and decreased expression levels of VE-cadherin in the TGFβ group and the TGFβ +SSA+ WAY-262611 group (Fig. 8D-F).
The effects of smad activator on fibroblast activation and function in CFs or Wnt activator on EndMT in MHECs. A-C. CFs were stimulated with TGFβ for 24 h and then treated with SSA (30 μM) and SRI-011381 for 12 h. A. Immunofluorescence staining of α-SMA (n=6 per experiment). B and C. Transcription levels of collagen I and collagen III in CFs (n=6). #P<0.05 vs vehicle-TGFβ group. D-F. MHECs were stimulated with TGFβ for 24 h and then treated with SSA (1 μM) and WAY-262611 for 12 h. D. Immunofluorescence staining of VE-cadherin and vimentin (n=6 per experiment). E and F. Transcription levels of snail1 and snail2 in MHECs (n=6). *P<0.05 vs control group; #P<0.05 vs vehicle-TGFβ group. All experiments were repeated 3 independent times.
Discussion
In this study, we found that SSA alleviated long-term stress overload in cardiac dysfunction and cardiac fibrosis. Supplementation with high-dose SSA can block the transformation of CFs to MFs, inhibit the proliferation and activation of fibroblasts induced by TGF-beta, and inhibit collagen secretion. Supplementation with low dosages of SSA blocked the EndMT process in TGF-β-treated MHECs and pressure overload-induced heart remodeling. High dosages of SSA inhibited phosphorylation and activation of Smad signaling in CFs. Smad activators can make these protective effects vanish. We also found that low doses of SSA inhibited the activation of Wnt/ β-catenin signaling in MHECs, while the protective effect of LD SSA was eliminated by a Wnt/β-catenin activator. Therefore, our current research finds that SSA is a new therapeutic agent for cardiac fibrosis.
More and more evidence indicates that SSA may play a role in cardiac remodeling due to its pleiotropic biological activity, including powerful antioxidant and anti-inflammatory effects[17, 18]. SSA protects lung tissue against inflammation induced by cigarette smoke by inhibiting NF-kappa B and upregulating Nrf2 and HO-1 expression[17]. By activating LXRα, SSA protects human osteoarthritis chondrocytes from injury induced by IL-1β[19]. However, our study shows that the protective effect of SSA on cardiac fibrosis is not related to these factors. The antifibrosis effect of SSA does not depend on cardiomyocytes because we show negative results in the NRCM experiments.
ECM derived from CFs provides the structural scaffold for cardiomyocytes in heart tissue[6]. However, during various pathologies, increased ECM enhanced ventricular stiffness and can lead to cardiac dysfunction[5]. In addition, excessive ECM and fibroblasts damage the electromechanical coupling of CMS, thereby reducing the risk of cardiac contractions and increasing the occurrence and mortality of arrhythmias[20]. Herein, we found that a higher dosage of SSA prevented against cardiac fibrosis via directly inhibiting CFs activation and function. The TGF-beta growth factor family may be the most extensively activated mediator of fibroblasts, and TGF-beta 1 may play the most important role in pathological fibrosis[21]. The signal transduction pathway of TGF-beta 1 includes the phosphorylation of Smad 2/3, and the binding of Smad2 and Smad4 leads to the migration of Smad4 to the nucleus[21]. The complex acts as a transcription factor to induce the activation of many fibroblast genes[22]. In this setting, we found that the effect of the higher dosage of SSA on CF activation and function was dependent on the inhibition of TGFβ/smad signaling since a smad activator eliminated the protective effects of SSA on CFs.
Evidence implicating EndMT in cardiac fibrosis has been mounting for several years[8]. In a landmark publication in 2007, Kalluri and co-workers demonstrated that EndMT makes a significant contribution to myocardial fibrosis in the adult heart[7]. Many findings suggest that the inhibition of EndMT/EMT may be a promising target for clinical therapeutic applications in cases of cardiac fibrosis[9, 10]. In this study, we found that lower dosages of SSA, but not higher ones, could inhibit TGFβ1-induced EndMT both in vitro and in vivo. Wnt/β-catenin plays a major causal role in EndMT in endothelial cells[7]. Under inactivation, cytoplasmic β-catenin is bound to APC, Axin and GSK3β, leading to the phosphorylation, ubiquitination and decomposition of β-catenin. The binding of Wnt to its receptor Frizzled and LRP inhibited the degradation of the complex and induced the signal transduction of β-catenin[23]. Binding of nuclear β-catenin to members of the transcription factor TCF/LEF family promotes EndMT[24]. Studies have found that by downregulating AXIN2, TGFβ activation primes canonical Wnt signaling in fibroblasts[25]. In our study, we found that in endothelial cells, TGFβ1 induced activation of the Wnt/β-catenin pathway. Lower dosages of SSA did not affect smad signaling (data not given) but inhibited the Wnt/β-catenin pathway. These anti-EndMT effects of lower dosages of SSA were dependent on inhibition of the Wnt/β-catenin pathway since a β-catenin activator eliminated the protective effects of SSA on MHECs.
The target of SSA in cardiac fibrosis was unclear. Studies have reported that SSA stimulates bone marrow stromal cells to differentiate into osteoblasts by activating the Wnt/β-catenin pathway[18]. However, in our study, lower dosages of SSA inhibited EndMT in heart endothelial cells by blocking the Wnt/β-catenin pathway. These results suggested diverse effects of SSA on different cell types at different dosages. Further studies are needed to determine why different dosages of SSA exert different effects on diverse cell types.
In conclusion, this study shows that supplementation with SSA reduces cardiac dysfunction and inhibits cardiac fibrosis after prolonged stress overload in vivo. The beneficial effects of SSA on cardiac fibrosis may be attributed to its inhibition of TGFβ/Smad signaling in CFs when using higher dosage treatment and inhibition of the Wnt/β-catenin signaling in endothelial cells when using lower dosage treatment.
Abbreviations
AngII: angiotensin II; ANP: atrial natriuretic peptide; α-SMA: alpha-smooth muscle actin; BMP-4: bone morphogenetic protein 4; BNP: brain natriuretic peptide; BW: body weight; CFs: cardiac fibroblasts; CTGF: connective tissue growth factor; dp/dtmax: maximum descending rate of left ventricular pressure; dp/dtmin: minimum descending rate of left ventricular pressure; ECM: extracellular matrix; ECs: endothelial cells; EF: ejection fraction; EndMT: endothelium-mesenchymal transition; FS: fractional shortening; HE: hematoxylin and eosin; HW: heart weight; IVSd: interventricular septal end diastolic dimension; LV: left ventricular; LVEDd: left ventricular end-diastolic dimension; LVESd: left ventricular end-systolic dimension; LVPWd: left ventricular end diastolic posterior wall dimension; LW: lung weight; MHC: myosin heavy chain; MHECs: mouse heart endothelial cells; PSR: PicroSirius red; SSA: Saikosaponin A; TGFβ: transforming growth factor β; TL: tibia length.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No: 81600189, 81600191) and the Scientific and Technological Project of Henan province (NO: 172102310531 and 182102310495).
Authors' contributions
Yuan Liu, Lina Li, and Haibo Yang contributed to the conception and design of the experiments; Yuan Liu, Lu Gao, Xiaoyan Zhao and Sen Guo carried out the experiments; Yuzhou Liu, Ran Li and Cui Liang analyzed the experimental results. Ling Li, Jianzeng Dong and Lina Li revised the manuscript; Lina Li and Yuan Liu wrote and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. A. L. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 2015;116:1269-76
2. Al Hattab D CM. A primer on current progress in cardiac fibrosis. Can J Physiol Pharmacol. 2017;95:1091-9
3. Hara H TN, Komuro I. Pathophysiology and therapeutic potential of cardiac fibrosis. Inflamm Regen. 2017;37:13
4. therapy. Mocmdifcfa. Mechanoregulation of cardiac myofibroblast differentiation: implications for cardiac fibrosis and therapy. Am J Physiol Heart Circ Physiol. 2015;309:H532-42
5. Kurose H MS. Myofibroblasts and inflammatory cells as players of cardiac fibrosis. Arch Pharm Res. 2016;39:1100-13
6. Travers JG KF, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021-40
7. Zeisberg EM TO, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952-61
8. Sánchez-Duffhues G GdVA, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev Dyn. 2018;247:492-508
9. Lai YJ CI, Li HH, Huang CC. EP4 Agonist L-902,688 Suppresses EndMT and Attenuates Right Ventricular Cardiac Fibrosis in Experimental Pulmonary Arterial Hypertension. Int J Mol Sci. 2018;19:E727
10. Liu Y HZ, Liao HH, Liu W, Liu J, Ma ZG, Wu QQ, Xu M, Zhang N, Zhang Y, Bian ZY, Tang QZ. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim Biophys Acta. 2015;1852:2456-66
11. Wang X WQ, Burczynski FJ, Kong W, Gong Y. Saikosaponin A of Bupleurum chinense (Chaihu) elevates bone morphogenetic protein 4 (BMP-4) during hepatic stellate cell activation. Phytomedicine. 2013;20:1330-5
12. Wu SJ TK, Tsai YH, Chang CC, Chao JC. Curcumin and saikosaponin a inhibit chemical-induced liver inflammation and fibrosis in rats. Am J Chin Med. 2010;38:99-111
13. Fu Y, Hu X, Cao Y, Zhang Z, Zhang N. Saikosaponin a inhibits lipopolysaccharide-oxidative stress and inflammation in Human umbilical vein endothelial cells via preventing TLR4 translocation into lipid rafts. Free radical biology & medicine. 2015;89:777-85
14. Wu QQ, Xiao Y, Duan MX, Yuan Y, Jiang XH, Yang Z. et al. Aucubin protects against pressure overload-induced cardiac remodelling via the beta3 -adrenoceptor-neuronal NOS cascades. British journal of pharmacology. 2018;175:1548-66
15. Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS, Tang QZ. Piperine Attenuates Pathological Cardiac Fibrosis Via PPAR-gamma/AKT Pathways. EBioMedicine. 2017;18:179-87
16. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b Mediates Endothelial-to-Mesenchymal Transition in Diabetic Cardiomyopathy. Diabetes. 2016;65:768-79
17. Chen RJ GX, Cheng BH, Gong YQ, Ying BY, Lin MX. Saikosaponin A inhibits compound 48/80-induced pseudo-allergy via the Mrgprx2 pathway in vitro and in vivo. Inflammation. 2018 [Epub ahead of print]
18. Huang W ZX, Yang X, Fan S. Stimulation of Osteogenic Differentiation by Saikosaponin-A in Bone Marrow Stromal Cells Via WNT/β-Catenin Pathway. Calcif Tissue Int. 2017;100:392-401
19. Gao H SY, Li D, Feng W, Liu J. Saikosaponin A inhibits IL-1β-induced inflammatory mediators in human osteoarthritis chondrocytes by activating LXRα. Oncotarget. 2017;8:88941-50
20. Chaturvedi RR HT, Simmons R, Shore D, Kumar P, Sethia B, Chua F, Vassiliadis E, Kentish JC. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation. 2010;121:979-88
21. Leask A AD. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816-27
22. Wipff PJ RD, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311-23
23. Burgy O, Konigshoff M. The WNT signaling pathways in wound healing and fibrosis. Matrix biology: journal of the International Society for Matrix Biology. 2018
24. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Science signaling. 2014;7:re8
25. Gillespie J RR, Corinaldesi C, Esteves F, Derrett-Smith E, McDermott MF, Doody GM, Denton CP, Emery P, Del Galdo F. TGFβ activation primes canonical Wnt signaling through the downregulation of AXIN2. Arthritis Rheumatol. 2018 [Epub ahead of print]
Author contact
![]() Corresponding authors: Professor Lina Li and Haibo Yang. Lina Li: Department of Anesthesiology, The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Zhengzhou (China). Email: fccliuyedu.cn and Haibo Yang: Department of Cardiology, The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Zhengzhou (China). Email:yhb160320com
Corresponding authors: Professor Lina Li and Haibo Yang. Lina Li: Department of Anesthesiology, The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Zhengzhou (China). Email: fccliuyedu.cn and Haibo Yang: Department of Cardiology, The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Zhengzhou (China). Email:yhb160320com