Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):882-894. doi:10.7150/ijbs.30290 This issue Cite
Research Paper
Glycolytic Enzyme PKM2 Mediates Autophagic Activation to Promote Cell Survival in NPM1-Mutated Leukemia
Lu Wang1*, Liyuan Yang1*, Zailin Yang2, Yuting Tang1, Yao Tao1, Qian Zhan3, Li Lei1, Yipei Jing1, Xueke Jiang1, Hongjun Jin1, Qin Zou1, Jingrong Xian1, Ling Zhang1 ![]()
1. Key Laboratory of Laboratory Medical Diagnostics Designated by the Ministry of Education, School of Laboratory Medicine, Chongqing Medical University, Chongqing, China.
2. Center for Hematology, Southwest Hospital, Third Military Medical University, Chongqing, China.
3. The Center for Clinical Molecular Medical detection, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
*Lu Wang and Liyuan Yang contributed equally to this work.
Received 2018-9-29; Accepted 2019-1-12; Published 2019-3-1
Abstract

Acute myeloid leukemia (AML) with mutated nucleophosmin (NPM1) has been defined as a distinct leukemia entity in the 2016 updated WHO classification of myeloid neoplasm. Our previous report showed that autophagic activity was elevated in NPM1-mutated AML, but the underlying molecular mechanisms remain elusive. Mount of study provides evidence that glycometabolic enzymes are implicated in the autophagic process. Pyruvate kinase isoenzyme M2 (PKM2), a key glycolytic enzyme, has been recently reported as a tumor supporter in leukemia. However, little is known about the roles of PKM2 in autophagic activity in NPM1-mutated AML. In this study, PKM2 highly expressed in NPM1-mutated AML, and partially, high levels of PKM2 were upregulated by PTBP1. Further experiments demonstrated that PKM2 mediated autophagic activation and increased the phosphorylation of key autophagy protein Beclin-1. Importantly, functional experiments demonstrated that PKM2 contributed to cell survival via autophagic activation. Ultimately, high PKM2 expression was associated with short overall and event-free survival time in NPM1-mutated AML patients. Our findings indicate for the first time that glycolytic enzyme PKM2 mediates autophagic activation and further contributes to cell survival in NPM1-mutated AML, suggesting that PKM2 may serve as a promising target for treatment of NPM1-mutated AML.
Keywords: PKM2, Autophagy, PTBP1, Cell survival, Nucleophosmin, Acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous disease characterized by genes mutations that affect proliferation, differentiation and apoptosis [1]. Recently, AML with nucleophosmin (NPM1) gene mutations is recognized as a separate entity in the 2016 updated World Health Organization (WHO) classification of myeloid neoplasms [2]. Notably, NPM1 mutation is present in 50-60% of cytogenetically normal AML (CN-AML) cases [3]. Although it has been well established NPM1 mutation as an AML-driving lesion, this mutation alone is not sufficient to cause leukemogenesis and it requires cooperative events that aid leukemogenesis [4]. Our newly published report showed that autophagic activity was elevated in NPM1-mutated AML [5]. However, investigations conducted on the molecular mechanism of autophagic activation in the pathogenesis of NPM1-mutated leukemia are limited.
Autophagy is primarily a degradative pathway that clears malfunctioning cellular components in response to various types of stress [6]. Preclinical investigations have shown that the dysregulation of autophagy is associated with hematological malignancies[7]. Of note, mount of evidence has demonstrated that an essential role of autophagy in the regulation of AML progression [8]. In a study by Torgersen et al. [9], autophagic activation induced by histone deacetylase inhibitors (HDACi) has a prosurvival role in t (8;21) AML cells. It has been well acknowledged that autophagy is highly regulated through the action of various protein kinases, phosphatases, and guanosine triphosphatases (GTPases) [10]. Supporting this idea is a recent study showing that phosphoglycerate kinase 1 (PGK1), one of two ATP-generating enzymes in the glycolytic pathway, mediated the autophagy keeper Beclin-1 phosphorylation to initiate autophagy and was instrumental for brain tumorigenesis [11]. Additionally, pyruvate dehydrogenase kinase-1 (PDK1) was found to interact with autophagy protein ULK1 to induce autophagy in AML [12]. Another report also revealed that lactate dehydrogenase B (LDHB) sustained lysosomal acidification and controlled the fusion between lysosomes and autophagosomes, thus to promote autophagy and cancer cell proliferation [13]. As mentioned above, a number of studies have shown the role of glycometabolic key enzymes during autophagy process. In particular, pyruvate kinase (PK), another ATP- generating enzyme in the glycolytic pathway, is uniquely important to the molecular feature of tumor development [14]. More specifically, the PK isoenzyme M2 (PKM2), governed by polypyrimidine tract binding protein (PTBP1/PTB) mutually exclusive alternative splicing of pyruvate kinase M (PKM) gene, has been reported to play a critical role in protecting tumor against stress in primary hematopoietic cells as its nonglycolytic functions [15]. In this regard, we explore whether PKM2 participates in the regulation of the autophagic process in NPM1-mutated AML.
Autophagy includes several key steps and each of these steps is tightly regulated by autophagy- related proteins (Atgs) [16]. An early step in autophagy initiation is the binding of Beclin-1 (mammalian homolog of yeast Atg6) to the class III-type phosphoinositide 3-kinase (Class III PI3K, also known as Vps34), which promotes the recruitment of other Atg proteins to the phagophore membrane [17]. It has been well documented that the regulation of Beclin-1 may be critical for autophagy induction [18]. Under normal steady-state growth conditions, Beclin-1 binds to Bcl-2 anti-apoptotic family members, which represses Beclin-1-dependent autophagy [19]. Accordingly, the dissociation of Beclin-1 from its inhibitor Bcl-2 is essential for its autophagic activity. Increasing lines of evidence suggest that the interaction between Beclin-1 and Bcl-2 are subject to protein post-translational modifications, including TRAF6-mediated ubiquitination of Beclin-1[20] and JNK-mediated phosphorylation of Bcl-2[21]. Noticeably, Zalckvar et al.[22] demonstrated that death-associated protein kinase (DAPK) phosphorylated Beclin-1 at Thr119, which displaced Bcl-2 from Beclin-1 and thus led to autophagic activation. Given that PKM2 acts as a dual-specificity protein kinase that phosphorylates both the Ser/Thr and Tyr residues of its substrates [23, 24], further efforts are required to reveal whether PKM2 phosphorylates Beclin-1 to activate autophagy in NPM1-mutated AML cells.
Herein, we show that PKM2 highly expresses in NPM1-mutated AML, and high levels of PKM2 are partially upregulated by PTBP1. Further experiments demonstrate that PKM2 mediates autophagic activation and increases the phosphorylation of key autophagy protein Beclin-1. Importantly, PKM2 contributes to cell survival via autophagic activation in vitro. Ultimately, high PKM2 expression is associated with poor clinical outcomes in NPM1-mutated AML patients. This study suggests that the nonglycolytic role of PKM2 in autophagic cell survival and PKM2 may be a promising therapeutic target for NPM1-mutated AML.
Materials and Methods
Oncomine and TCGA database gene expression analysis
Oncomine database (https://www.oncomine. org/resource/login.html), an online tumor microarray database, was utilized to analyze the transcription levels of PKM2 in AML. According to the French- American-British (FAB) classification, a total of 43 AML cases was divided into eight leukemia subtypes and PKM2 expression levels were analyzed. The expression fold change of PKM2 in each AML subtype was obtained as the parameters of p-value < 0.05, fold change ≥ 2, and gene ranking in the top 10%.
Gene expression levels and clinical information of 200 AML patients were retrieved from The Cancer Genome Atlas (TCGA, http://www.cancergenome. nih.gov). A total of 171 samples had IlluminaGA RNA-Seq gene expression data. Clinical data and PKM2 mRNA expression data for AML samples were analyzed using the cBioPortal for Cancer Genomics. The PKM2 mRNA expression was compared between AML cases (years > 55) with the NPM1 mutation (n = 22) and those without the NPM1 mutation (n = 25).
Clinical patient samples
The study was carried out on diagnostic bone marrow samples from 30 AML patients: 16 NPM1- unmutated and 14 NPM1-mutated cases were obtained from Southwest Hospital of the Third Military Medical University and the First Affiliated Hospital of Chongqing Medical University. The mononuclear cells were enriched by Ficoll gradient purification and used for analysis of genes mRNA relative expression. Details of the clinical characteristics of patients are provided in Table 1. The mRNA expression levels were analyzed using the 2- ΔΔCt method and expressed as a fold change.
Clinical characteristics of newly diagnosed AML patients
| Characteristics | Median(range) | No. of cases |
|---|---|---|
| Sex | ||
| Female | 14 | |
| Male | 16 | |
| Total | 30 | |
| Median age, y | 55.5(8-79) | |
| Younger than 40 y | 8 | |
| 40-60 y | 9 | |
| Older than 60 y | 13 | |
| Median WBC, 109/L | 44 (0.3-295) | |
| Median platelets, 109/L | 57.3 (3.0 - 655.0) | |
| AML FAB subtype | ||
| AML without maturation: M1 | 3 | |
| AML with maturation: M2 | 7 | |
| Acute promyelocytic leukemia: M3 | 4 | |
| Acute myelomonocytic leukemia: M4 | 5 | |
| Acute monoblastic or monocytic leukemia: M5 | 10 | |
| Other subtypes | 1 | |
| Karyotype | ||
| Normal | 14 | |
| t(8;21) | 3 | |
| t(15;17) | 4 | |
| inv(16) | 7 | |
| Unknown | 2 | |
| Gene mutations | ||
| NPM1 | 14 | |
| FLT3/ITD | 10 | |
| WT1 | 4 | |
| CBFB-MYH11 | 3 |
Abbreviations: AML, acute myeloid leukemia; y, year old; WBC, white blood cell; FAB classification, French-American-British classification, a classification of acute leukemia produced by three-nation joint collaboration.
Cell lines and culture
The myelogenous leukemia cell lines, including THP-1, NB4, HL60, K562, KG1a were procured from the American Type Culture Collection (ATCC, USA). The OCI-AML3 AML cells naturally harboring NPM1-mA were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Germany). All of the cell lines were routinely cultured in RPMI 1640 medium (Gibco, MD, USA), supplemented with 10% fetal bovine serum (FBS; Gibco, MD, USA) and 1% penicillin and streptomycin (Beyotime, Shanghai, China), and maintained in a humidified incubator at 37 °C with 5% CO2.
Reverse transcription and real-time PCR
RNA was extracted using TRIzol (Takara, Kyoto, Japan) according to the manufacturer's procedures. The concentration and purity of the total RNA were evaluated by ultraviolet spectrophotometry. Total RNA was stored at -80 °C until analysis could be performed. After RNA extraction, 1 μg of total RNA was retrotranscribed using PrimeScript™ RT Reagent Kit (Takara, Kyoto, Japan), used as a template for conventional PCR reactions. Real-time PCR analysis was performed using MJ Mini™ Gradient Thermal Cycler Real-Time PCR machine (Bio-Rad, CA, USA) with the SYBR Green reaction kit (KAPA Biosystems, MA, USA), according to the manufacturer's instructions. Primers used are listed in Table 2.
The PCR primer sequences for each gene used in this study
| Genes | Sequences(5' - 3') |
|---|---|
| PKM2 | F: 5'-GCCTGCTGTGTCGGAGAAG-3' |
| R: 5'-CAGATGCCTTGCGGATGAATG-3' | |
| Beclin-1 | F: 5'-CAGGAGAGGAGCCATTTA-3' |
| R: 5'-CCGATCAGAGTGAAGCTA-3' | |
| LC3 | F: 5'-GACCGCTGTAAGGAGGTGC-3' |
| R: 5'-CTTGACCAACTCGCTCATGTTA-3' | |
| p62 | F: 5'-GGGGACTTGGTTGCCTTTT-3' |
| R: 5'-CAGCCATCGCAGATCACATT-3' | |
| PTBP1 | F: 5'-ATCAGGCCTTCATCGAGATGCACA-3' |
| R: 5'-TGTCTTGAGCTCCTTGTGGTTGGA-3' | |
| PKM1 | F: 5'-CGAGCCTCAAGTCACTCCAC-3' |
| R: 5'-GTGAGCAGACCTGCCAGACT-3' | |
| β-actin | F: 5'-TAGTTGCGTTACACCCTTTCTTG-3' |
| R: 5'-TGCTGTCACCTTCACCGTTC-3' |
Abbreviations: F stands for forward; R stands for reverse
Protein extracts and western blot
The cultured cells were washed and lysed in cell extraction buffer. Cells were resuspended in RIPA buffer (Selleck, TX, USA). After 10 mins on ice, the extracts were centrifuged for 10 mins at 12,000 g, the supernatants were collected and used for western blot as described. Equal amounts of proteins were separated on 12% polyacrylamide gels by SDS PAGE and then transferred to PVDF membranes (Thermo Fisher, MA, USA). The membranes were thereafter blocked with 5% nonfat dry milk and incubated with the desired primary antibody (1:1000) at 4 °C overnight. Primary antibody as following: rabbit monoclonal antibody PKM2 (Cell Signaling, MA, USA ), rabbit monoclonal antibody LC3 І/II (Novus, Co, USA), rabbit monoclonal antibody p62 (Abcam, Cambridge, UK), Beclin-1 (Cell Signaling, MA, USA ), p-Beclin-1Thr119 (Merck KGaA, Darmstadt, Germany), PTBP1 (Abcam, Cambridge, UK), PKM1 (Cell Signaling, MA, USA ), and rabbit polyclonal antibody β-actin (Proteintech, IL, USA) as loading control. After washed with 1× TBST for two times and 1×TBS once, membranes were incubated with HRP-conjugated secondary antibodies (Proteintech, IL, USA) and visualized using ECL detection kit (Millipore, MA, USA). The densitometric analysis of the band gray scales was performed by the Bio-Rad Gel Imaging System on cool image workstation II (Viagene, FL, USA). Protein expression quantification was normalized against the β-actin protein expression using image software.
Lentiviral vectors and cell infection
The lentivirus-based short hairpin RNA (shRNA) vectors targeting PKM2 (5'- CATCTACCACTTGCAATTA -3') and scramble lentiviral vectors were purchased from Genechem (Shanghai, China). The OCI-AML3 cells were infected with lentivirus for 48 hrs in the presence of 5 μg/mL polybrene (Sigma, CA, USA), after which they were subjected to 2 μg/mL puromycin selection for 7 d (Sigma, CA, USA). The puromycin-resistant cells were isolated and propagated for further analysis.
Plasmids and cell transfection
The PKM2 expression vectors (pcDNA3.1-PKM2 and pcDNA 3.1-PKM2 K367M) were gifts from Dr. Lu Z (Department of Neuro-Oncology, Division of Cancer Medicine, the University of Texas MD Anderson Cancer Center, USA). PTBP1 expression vector (Myc-tagged WT PTB) was purchased from Addgene (http://www.addgene.org, # 23024). All transfection experiments were performed using the Neofect™ reagent (Neofect, Beijing, China) according to the manufacturer's instructions. After 48 hrs of transfection, the cells were collected for real-time PCR or western blot analysis.
Small interfering RNA (siRNA) and cell transfection
The siRNA targeting PTBP1 (PTBP1#1: 5′-GCACAGUGUUGAAGAUCAU-3′ and PTBP1#2: 5′- AACUUCCAUCAUUCCAGAGAA-3′) and scramble siRNA (5′-UUCUCCGAACGUGUCACGU-3′) were purchased from Invitrogen (CA, USA). Transfection was performed using the RfectPM siRNA Transfection Reagent (BaiDai, Changzhou, China) according to the manufacturer's instructions. The transfected cells were harvested for mRNA and for protein expression, respectively.
Cell viability assay
Cells were seeded into a 96-well plate (Corning, NY, USA) at a density of 1 × 103 cells per well with RPMI-1640 containing 10% FBS, and subsequently treated with rapamycin (5 μM) or 3-methyladenine (3-MA) (2 mM) and Tat- Beclin-1 (30 μM) reagents purchased from Selleck (TX, USA) for indicated times. Cell viability was evaluated by Cell Counting Kit-8 (CCK8, Dojindo Laboratories, Japan) every 12 hrs following the manufacturer's protocols. The absorbance was measured at 450 nm using the microplate reader (Eon, BioTeck, CA, USA). The cell growth curves were plotted with the cell number values as the ordinate and time as the abscissa. Each experiment was performed in triplicate.
Colony formation assay
The methylcellulose clonogenic assay was carried out to determine cell colony formation ability by plating 1×103 cells per well in triplicate in 24-well plate, and maintained in RPMI 1640 medium containing 20% FBS at 37 °C in an incubator. Colony numbers were scored 10 days later. The colony forming units (CFU), defined as cell clusters consisting of more than 5 cells, were counted using an inverted microscope.
Flow cytometric (FCM) analysis
The Annexin V FITC-PI staining assay was used to detect apoptosis. In brief, after silencing PKM2, the cells were harvested and washed with PBS. Apoptosis staining was performed using an Annexin V FITC-PI apoptosis detection kit (BD Biosciences, Piscataway, NJ, USA) according to the manufacturer's instructions. Stained cells were analyzed using FACSCaliburTM Flow cytometry (BD Biosciences) with Cell- Quest software.
Survival analysis
Total of 45 NPM1-mutated AML cases were obtained from TCGA dataset. The NPM1 mutation occurs frequently in normal karyotype AMLs and cytogenetic abnormalities are responsible for poor clinical outcomes in AML. Therefore, only 36 NPM1-mutated AML cases with normal karyotype were included in our study. Finally, all patients were stratified by PKM2 expression levels into quartiles and median, to categorize patients into either a high cohort or low cohort. The overall survival (OS) and the three-year event-free survival (EFS) curves were plotted according to the Kaplan-Meier methods.
Statistical analysis
All data were derived from three independent experiments. Data were presented as the means ± standard deviation (SD). Significant differences between groups were evaluated using Student's t-test (two-tailed). A p-value < 0.05 was considered statistically significant. The Kaplan-Meier estimation and log-rank test were used to compare the survival difference. The SPSS (Version 22.0) software and GraphPad (Prism 5.0) was used for statistical analyses.
Results
PKM2 highly expresses in leukemia with NPM1 mutation
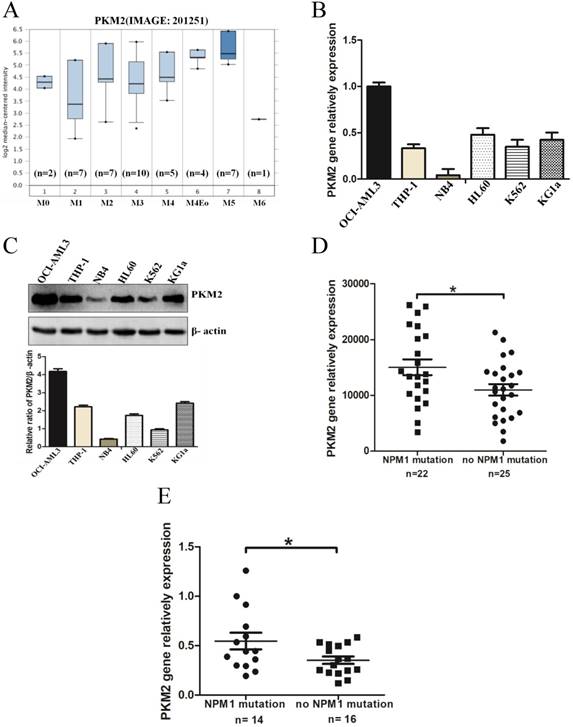
To determine the expression levels of PKM2 in AML with NPM1 mutation, we analyzed an independent gene expression dataset using Oncomine (https://www.oncomine.org/resource/login.html). The data showed higher PKM2 expression in M4/5 consist of monocyte blasts, which classified by FAB in AML samples (Fig. 1A). Next, we detected the higher expression of PKM2 in OCI-AML3 cells with the NPM1 mutation by real-time PCR, compared to those leukemic cell lines without the NPM1 mutation (Fig. 1B). In support, the high protein levels of PKM2 in OCI-AML3 cells were determined by western blot (Fig. 1C). To further validate the above results, we identified the transcript mRNA of PKM2 from the publically available TCGA RNA-seq dataset of AML patients. As a result, PKM2 was preferentially higher in the cases of AML with NPM1 mutation than in those without the NPM1 mutation (p = 0.0235, Fig. 1D). Furthermore, we examined the mRNA levels of PKM2 in 30 AML primary blasts. PKM2 mRNA expression was significantly increased in NPM1-mutated AML cases (n = 14), as compared to NPM1-unmutated AML (n = 16) (p = 0.0367, Fig. 1E). These findings suggest that PKM2 highly expresses in NPM1-mutated leukemia.
PKM2 highly expresses in leukemia with NPM1 mutation. A PKM2 mRNA expression of AML samples was analyzed using Oncomine (https://www.oncomine.org/resource/login.html). B The mRNA expression of PKM2 was detected by real-time PCR in five indicated myeloid leukemia cell lines. C PKM2 protein expression was detected by western blot in leukemia cell lines. D RNA-seq mRNA expression data from the TCGA database were used to compare PKM2 expression between AML patients with (n = 22) and without NPM1 mutation (n = 25). (E) Dot plot showing the relative expression levels of PKM2 in 30 AML patients, consisting of 14 cases with NPM1 mutation and 16 cases with no NPM1 mutation. The data are expressed as the mean ± SD (n = 3). *p < 0.05.
High expression of PKM2 is upregulated by PTBP1 in leukemic cells
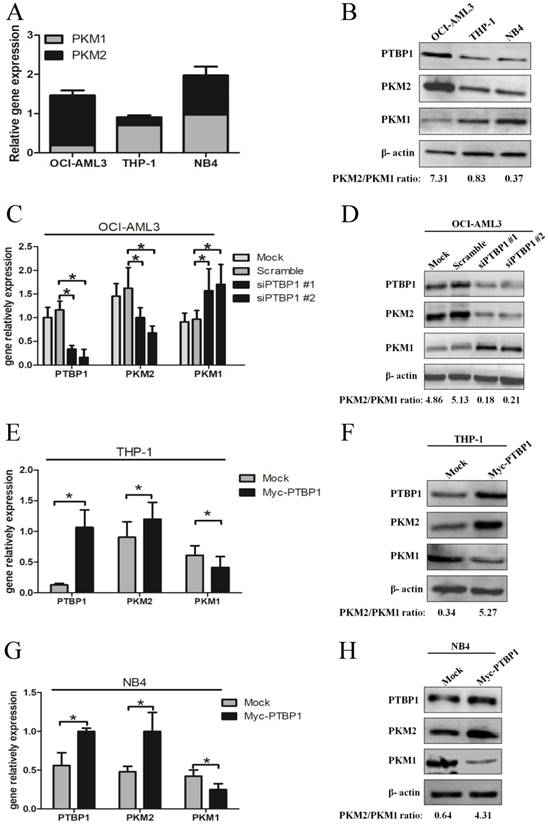
Based on PTBP1 as a critical splicing factor determining the relative expression of PK isoforms, PKM1 and PKM2 [25], we explored whether the PKM2 expression is regulated by PTBP1 in NPM1-mutated AML. Firstly, we measured the basal levels of PTBP1, PKM1 and PKM2. The results showed higher PTBP1 and PKM2 levels in OCI-AML3 cells, as compared to THP-1 and NB4 cells (Fig. 2A-B). Next, knockdown the expression of PTBP1 by siPTBP1 decreased the mRNA levels of PKM2 and increased the levels of PKM1 in OCI-AML3 cell lines (Fig. 2C). In addition, PKM2/PKM1 protein ratio was downregulated by siPTBP1 (Fig. 2D). To confirm this notion even further, we overexpressed PTBP1 by transfecting myc-PTBP1 plasmids and found that enforced PTBP1 expression. Expectedly, enforced PTBP1 expression upregulated the PKM2/PKM1 ratio in THP-1 cells (Fig. 2E-F) and NB4 cells (Fig. 2G-H). Overall, these observations show that high expression of PKM2 might be upregulated by PTBP1 in leukemic cells.
High expression of PKM2 is regulated by PTBP1 in leukemic cells. A-B Basal mRNA and protein levels of PTBP1, PKM2 and PKM1 were detected by real-time PCR and western blot. C-D The mRNA and protein expression of PTBP1 and PKM2/PKM1 after PTBP1 knockdown were detected in OCI-AML3 cells. E-H The mRNA and protein expression of PTBP1, PKM2/PKM1 were detected after PTBP1 overexpression in THP-1 and NB4 cells. The data are expressed as the mean ± SD (n = 3). *p < 0.05.
PKM2 mediates autophagic activation in leukemic cells
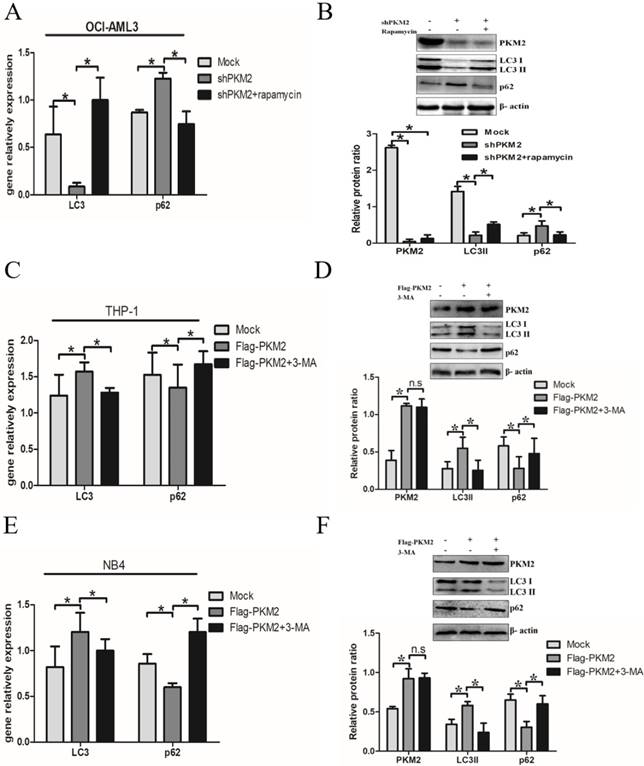
Next, we elucidate whether PKM2 is involved in the autophagic process. Silencing PKM2 by shRNA was performed to observe the effect of PKM2 on autophagic activity. The results showed that depletion of PKM2 significantly decreased LC3-II gene levels and increased p62 levels in OCI-AML3 cells, and similar results were obtained by western blot results. In addition, treatment with autophagy activator rapamycin reversed the changes in autophagic activity caused by PKM2 knockdown (Fig. 3A-B). On the other hand, the plasmids Flag-PKM2 were transiently transfected into THP-1 cells to enforce PKM2 expression. The results showed that PKM2 overexpression increased LC3-II and decreased p62 levels. In addition, exposure to autophagy inhibitor 3-MA abrogated the enhancement of PKM2-induced autophagic activation in THP-1 cells (Fig. 3C-D). Consistent with previous observation in THP-1 cells, similar results were obtained from NB4 cells (Fig. 3E-F). Collectively, these results demonstrate that PKM2 mediates autophagic activation in leukemic cells.
PKM2 mediates autophagic activation in leukemic cells. A Autophagy markers (LC3 II and p62) were detected by real-time PCR assay after silenced PKM2 and then treated with autophagy activator rapamycin (5 μM) for 6 hrs in the different groups. B LC3 II and p62 protein levels were detected by western blot, the bands were quantified and data were shown as mean ± SD of three independent experiments. C LC3 II and p62 mRNA were detected by real-time PCR after enforced PKM2 and treated with autophagy inhibitor 3-MA (2 mM) in THP-1 cells. D LC3 II and p62 protein levels were detected by western blot, the bands were quantified and data were shown as mean ± SD of three independent experiments. E-F LC3 II and p62 mRNA and protein were detected in NB4 cells with different treatments. The data are expressed as the mean ± SD (n = 3). *p < 0.05. n.s represents no significance.
PKM2 increases the phosphorylation of Beclin-1 in leukemic cells
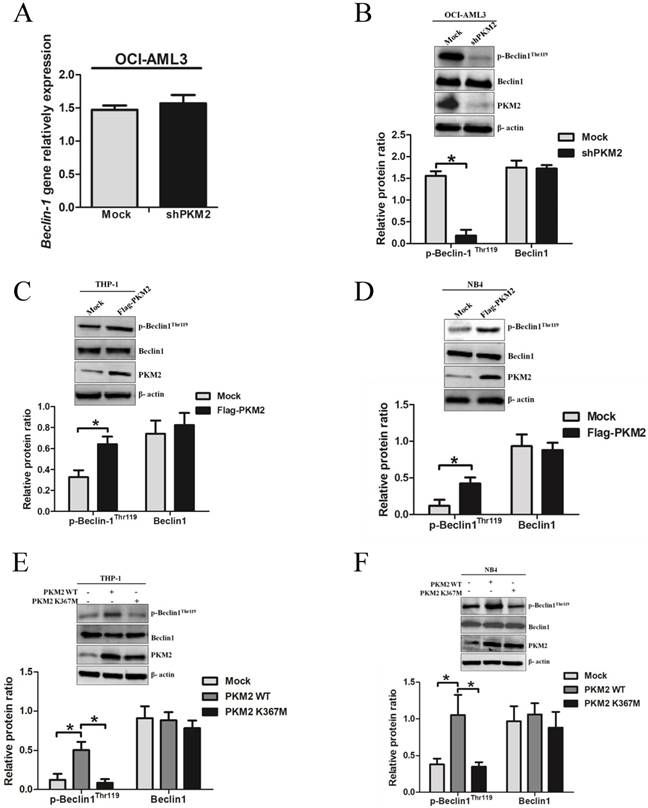
To explore the molecular mechanisms underlying PKM2-mediated autophagic activation, we explored whether PKM2 regulated the phosphorylation of autophagy protein Beclin-1. As showed in Fig. 4A, loss of PKM2 didn't affect the mRNA levels of Beclin-1. Interestingly, PKM2 depletion reduced p-Beclin-1Thr119 levels, but no obvious reduction in total protein Beclin-1 levels (Fig. 4B). And conversely, overexpression of PKM2 increased the p-Beclin-1Thr119 levels in THP-1 cells (Fig. 4C). Similar results were observed in NB4 cells (Fig. 4D). To further validate the effect of PKM2 on Beclin-1 phosphorylation, we transfected the PKM2 wild type (PKM2 WT) or inactive PKM2 mutant (PKM2 K367M) into THP-1 and NB4 cells, respectively. As expected, the results showed that PKM2 WT, but not PKM2 K367M, increased the phosphorylation of Beclin-1 (Fig. 4E-F). Altogether, these data indicate that PKM2 increases the phosphorylation of Beclin-1 in leukemic cells.
PKM2 increases the phosphorylation of Beclin-1 in leukemic cells. A The gene expression of Beclin-1 was determined by real-time PCR in OCI-AML3 cells following PKM2 knockdown. B The phosphorylation levels of Beclin-1 were assessed by western blot in PKM2-silenced OCI-AML3 cells. C-D The expression of p-Beclin-1Thr119, Beclin-1, and PKM2 proteins were determined by western blot in THP-1 and NB4 cells after PKM2 overexpression. E-F The protein expression was assessed from the THP-1 and NB4 cells transfected with the plasmids expressing PKM2 WT or PKM2 K367M. The data are expressed as the mean ± SD (n = 3). *p < 0.05.
PKM2 promotes cell survival via autophagic activation
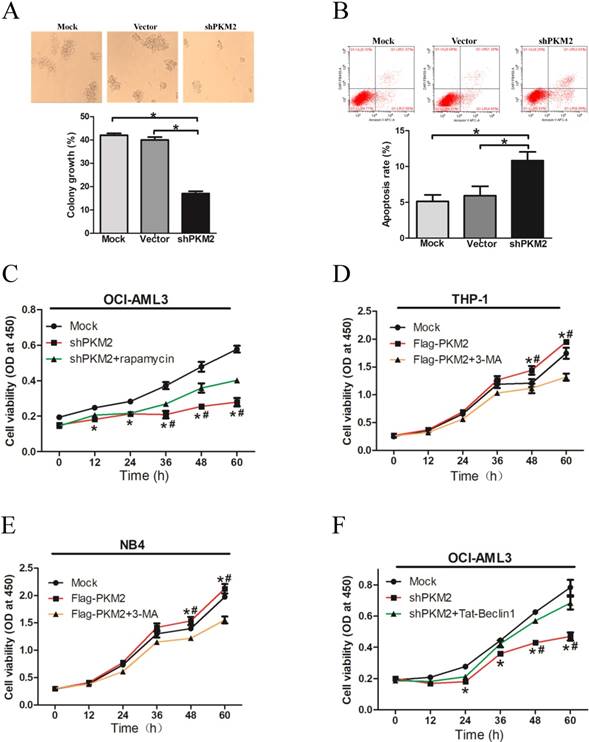
To explore the biological role of PKM2 in NPM1-mutated leukemia, we observed the effect of PKM2 on OCI-AML3 cell survival in vitro. The results showed decreased number of cell clusters morphologically and a lower proportion of colony forming units in the PKM2-silenced OCI-AML3 cells as compared to the controls (Fig. 5A). The FACS results revealed that PKM2 knockdown induced the apoptosis of OCI-AML3 cells (Fig. 5B). In addition, PKM2 knockdown inhibited cell proliferation in vitro, however, rapamycin reversed the changes in OCI-AML3 cells (Fig. 5C). Conversely, PKM2 overexpression promoted cell proliferation whereas 3-MA abrogated the enhancement of PKM2-induced growth advantage in THP-1 and NB4 cells (Fig. 5D-E). Based on the aforementioned results that PKM2 could increase the phosphorylation of Beclin-1, we further performed a rescue assay to illustrate the effect of Beclin-1 on PKM2-mediated cell survival. Tat-Beclin- 1, the Beclin-1 activator derived from a region of protein Beclin-1 [26], was used to treat the PKM2- silenced OCI-AML3 cells. The results showed that Tat-Beclin-1 could reverse the effect on cell survival due to knockdown of PKM2 (Fig. 5F). These results indicate that PKM2 promotes cell survival via autophagic activation.
PKM2 promotes cell survival via autophagic activation. A Cell proliferation was assessed by colony formation in PKM2-silenced OCI-AML3 cells. B Cell apoptosis was evaluated by flow cytometry analysis in PKM2-silenced OCI-AML3 cells. C Cell viability was detected in the PKM2-silenced OCI-AML3 cells and then treated with rapamycin for indicated times by CCK-8 assay. D Cell viability was detected by CCK-8 in THP-1 cells and then treated with 3-MA for indicated times by CCK-8 assay. E Cell viability was detected by CCK-8 in NB4 cells. F Cell proliferation was revealed by CCK-8 assay in PKM2-silenced OCI-AML3 cells and then treated with Tat-Beclin-1. The data are expressed as the mean ± SD (n = 3). *p < 0.05.
High PKM2 expression is associated with poor clinical outcomes in patients with NPM1-mutated leukemia
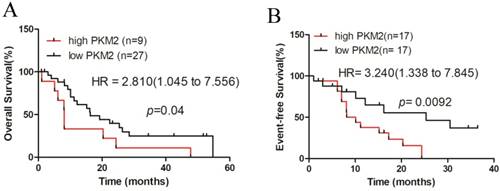
Finally, we investigated the prognostic value of PKM2 in AML patients using the dataset of TCGA, for which clinical data were available. A total of 36 NPM1-mutated AML cases with normal karyotype were selected and stratified by PKM2 expression levels. Kaplan-Meier survival analysis revealed that the NPM1-mutated AML patients with high PKM2 expression had a significantly shorter overall survival (OS), compared to those with low PKM2 expression (quartiles, p = 0.04, Fig. 6A). Additionally, the hazard ratio (HR = 2.810) from TCGA samples confirmed the poor outcome value of high PKM2 in AML. Moreover, analysis of the three-year event-free survival (EFS) showed that high PKM2 expression was evidently associated with a poorer prognosis in NPM1-mutated AML (median, p = 0.0092, HR = 3.240, Fig. 6B). Collectively, these findings suggest that high PKM2 expression is associated with poor clinical outcomes in patients with NPM1-mutated leukemia.
High PKM2 expression is associated with poor clinical outcome in patients with NPM1-mutated leukemia. A-B The survival outcome was showed by OS and EFS according to the levels of PKM2 in NPM1-mutated leukemia patients with the log-rank test applied for comparison.
Discussion
Autophagy plays an important role in developmental processes, human disease, and cellular response to nutrient deprivation [27]. Over the last few years, autophagy is emerging and receiving great research attraction in leukemia [28]. Recent studies point out the glycometabolic enzymes are involved in the autophagy process [29]. In the present study, we found high expression of glycolytic enzyme PKM2 in NPM1-mutated AML. Further experiments demonstrated that PKM2 mediated autophagic activation and increased the phosphorylation levels of key autophagy protein Beclin-1. Importantly, PKM2 contributed to cell survival via autophagic activation in vitro. Ultimately, the high levels of PKM2 were positively associated with poor clinical outcomes in AML patients with NPM1 mutation.
PKM2 is an isoform of PK which regulates the final rate-limiting step of glycolysis [30]. Noticeably, PKM2 is expressed in all proliferating cells including normal proliferating cells and especially in tumor cells [31]. In this work, we analyzed PKM2 mRNA levels in NPM1-mutated AML from the two public datasets (Oncomine and TCGA) and further detected PKM2 expression in leukemic cell lines and AML primary blasts. These results showed that PKM2 was highly expressed in NPM1-mutated AML. Supporting our idea is that Wang et al. [32] reported that PKM2 was abundantly expressed in leukemic cell lines and hematopoietic stem cells. Furthermore, we explored the potential molecular mechanisms underlying the increased expression of PKM2 in NPM1-mutated AML. It is well known that PKM2 (exon 10 inclusion) and PKM1 (exon 9 inclusion) are different products of PKM mRNA transcript spliced by a heterogeneous nuclear ribonucleoprotein PTBP1 [33]. In the present study, we found that knockdown of PTBP1 decreased the PKM2/PKM1 ratio and overexpression of PTBP1 increased the PKM2/PKM1 ratio in leukemic cells, which indicates that PKM2 is partially up-regulated by PTBP1 in NPM1-mutated AML. These results are consistent with those of a previous report by He et al. [34], who observed that knockdown of PTBP1 expression led to an increase in the ratio of PKM1 vs PKM2. Moreover, David et al. [35] reported that a decrease in the expression of PTB, also known as PTBP1, brought about switching from PKM2 to PKM1 expression in the mouse myoblast cell line C2C12. Important previous data also revealed that PTB resulted in the generation of PKM2 by controlling exon 10 inclusion and exon 9 exclusion of PKM gene in HeLa cells [36]. Certainly, further studies are warranted to investigate what binding sites PTBP1 recognizes in the PKM gene leading to the mutually exclusive splicing in NPM1-mutated leukemic cells.
Considering that glycometabolic key enzymes constituted an important regulatory force in autophagy process [29] and the high levels of PKM2 in NPM1-mutated AML, we next assessed the role of PMK2 on autophagy in NPM1-mutated leukemic cells. The results showed that silencing PKM2 resulted in the inhibition of autophagic activity (decreased LC3II and increased p62), and conversely, overexpression PKM2 enforced autophagic activity. More notably, autophagy activator rapamycin or autophagy inhibitor 3-MA could rescue the above effects, respectively. A case in point is that expression of PKM2 enhanced LC3B levels to promote autophagic response in glioma cells [37]. Given that Beclin-1 is the key protein in autophagy initiation [38], we further explored whether PKM2 functioned as a protein kinase to regulate the phosphorylation levels of Beclin-1. Our results showed that there was no significant change in the mRNA levels of Beclin-1 between the group of silenced PKM2 and the control. Of significance, p-Beclin-1Thr119 levels were repressed by PKM2 knockdown while the p-Beclin-1Thr119 levels were increased by PKM2 overexpression. Additionally, the inactive PKM2 mutant (K367M) couldn't affect the phosphorylation of Beclin-1, which further confirmed that PKM2 might increase Beclin-1 phosphorylation. Of note, the phosphorylation of Thr119 is required for the dissociation of Beclin-1 from Bcl-2 [39], we next will use a Beclin-1 mutant, in which Thr119 is substituted by alanine (T119A) [22], to identify PKM2-mediated phosphorylation at this site. In addition, further studies are also needed to test the contribution of PKM2-mediated Beclin-1 phosphorylation to autophagic activity. Collectively, our observations for the first time indicate that PKM2 promotes autophagic activity and Beclin-1 phosphorylation.
Next, we investigated the biological roles of PKM2 via autophagy in NPM1-mutated leukemic cells. Our data firstly showed that PKM2 knockdown inhibited colony forming potential of OCI-AML3 cells and induced cell apoptosis. Previous work demonstrated that knockdown of PKM2 resulted in decreased viability and increased apoptosis in hepatocellular carcinoma and ovarian carcinoma [40]. Furthermore, we explored the effect of PKM2-mediated autophagy on leukemic cell survival. The results revealed that autophagy activator rapamycin reversed the inhibiting effect on cell proliferation caused by PKM2 knockdown, while autophagy inhibitor 3-MA rescued the inducement of cell proliferation caused by PKM2 overexpression. Pioneer studies demonstrated that PKM2 knockdown caused impairment of the autophagic process, and consequently inhibited pancreatic cancer cell growth [41]. Based on the aforementioned results, we further investigate whether Beclin-1 is involved in PKM2-mediated autophagic cell survival. In this study, we used Tat-Beclin-1, a Beclin-1 activator, to treat PKM2-silenced OCI-AML3 cells and found that Tat-Beclin-1 rescued the inhibitory cell proliferation due to PKM2 knockdown. Our data indicate a potential function of PKM2 in promoting cell survival by sustaining autophagy in NPM1- mutated AML. In our experiments, OCI-AML3 cell line is accessible to be selected for research. In a future study, we will use another NPM1 mutant positive IMS-M2 cell line [42] to further verify the role of PKM2 in the autophagic process in NPM1-mutated AML. Certainly, a major challenge in the future will be to elucidate the effects of PKM2 on autophagic activity in mouse knock-in models that mimic human NPM1-mutated AML.
Finally, we explored the relationship between high PKM2 expression and prognosis. The results revealed that NPM1-mutated AML patients expressing higher PKM2 levels had a shorter overall survival rate and event-free survival. Consistently, Mohammad et al. [43] observed that PKM2 had strong expression, and found that overexpression of PKM2 was associated with poor prognosis in pancreatic cancer. In addition, according to Panchabhai et al. [44], patients with higher levels of PKM2 gene expression had a poor prognosis as compared to those with lower levels in multiple myeloma. These findings and our data imply that the glycolytic enzyme PKM2 could function as a potential prognostic value in NPM1- mutated leukemia.
Conclusions
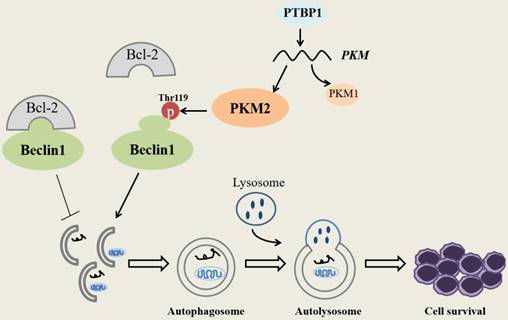
In summary, our research demonstrates that PKM2 highly expresses in NPM1-mutated AML and was partially upregulated by the expression of PTBP1. We identify for the first time that PKM2 mediates autophagic activation and increases the phosphorylation of key autophagy protein Beclin-1. Importantly, PKM2 contributes to cell survival via autophagic activation in vitro (Fig. 7). Ultimately, the upregulation of PKM2 is significantly associated with poor clinical outcomes in patients with NPM1-mutated leukemia. These discoveries suggest that PKM2 might be a potential therapeutic target in NPM1- mutated AML.
Schematic diagram describes the potential biological role of PKM2 in NPM1-mutated leukemic cells.
Abbreviations
AML: acute myeloid leukemia; ATCC: American Type Culture Collection; Atg: autophagy-related proteins; CCK8: Cell Counting Kit-8; CFU: colony forming units; Class III PI3K: class III-type phosphoinositide 3-kinase ( also known as Vps34); CN-AML: cytogenetically normal AML; DAPK: death-associated protein kinase; DSMZ: Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH; EFS: three-year event free survival; F: forward; FAB: French-American-British; FBS: fetal bovine serum; FCM: flow cytometric; GTPases: guanosine triphosphatases; HR: hazard ratio; JNK: c-Jun N-terminal protein kinase 1; LC3: microtubule-associated protein 1 light chain 3; LDHB: lactate dehydrogenase B; mRNA: messenger ribonucleic acid; NPM1: nucleophosmin; n.s: no significance; OS: overall survival; PCR: polymerase chain reaction; PDK1: pyruvate dehydrogenase kinase-1; PGK1: phosphoglycerate kinase 1; PK: pyruvate kinase; PKM: pyruvate kinase M; PKM1: pyruvate kinase isoenzyme M1; PKM2: pyruvate kinase isoenzyme M2; PTBP1/PTB: polypyrimidine tract binding protein; R: reverse; SD: standard deviation; Ser: serine; shRNA: lentiviral short hairpin RNA; TBS: Tris-buffered saline; TCGA: The Cancer Genome Atlas; Thr: threonine; TRAF6: tumor necrosis factor receptor-associated factor 6; Tyr: tyrosine; ULK1: unc-51-like kinase 1; WHO: World Health Organization; 3-MA: 3-methylademine.
Acknowledgements
The authors thank Dr. Lu Z (Department of Neuro-Oncology, Division of Cancer Medicine, the University of Texas MD Anderson Cancer Center, USA) for providing pcDNA3.1-PKM2 and pcDNA 3.1-PKM2 K367M plasmids.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (grant no. 81873973), Natural Science Foundation of Chongqing Yuzhong District (grant no. 20170411), and Graduate Fellowship in research innovation from the Chongqing Municipal Education Commission (grant no. CYS17155).
Authorship
LZ, LW and LYY initiated the work and designed the experiments. LW and LYY performed the experiments and wrote the manuscript. ZLY and QZ prepared the patient samples. YTT, HJJ, and YT contributed to analysis of data from datasets. LL, YPJ, XKJ, QZ and JRX provided assistance with revising the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All samples were collected with informed consent and the experiments were approved by the institutional ethics committee of Chongqing medical university and Third Military medical university.
Availability of data and material
Literature collection was performed using PubMed. Statistical analyses were executed using SPSS 22.0 software (IBM, Chicago, IL, USA) and GraphPad (Prism 5.0). Raw and processed data are stored in corresponding author and are available upon request. The RNAseq datasets and clinical information of NPM1-mutated AMLs in the current study were retrieved from the TCGA database (http://www.cancergenome.nih.gov). The expression level of PKM2 gene in AML was analyzed using Oncomine (https://www.oncomine.org/resource/ login.html), a cancer microarray database and web-based data mining platform from genome-wide expression analyses.
Competing Interests
The authors have declared that no competing interest exists.
References
1. De Kouchkovsky I, Abdul-Hay M. 'Acute myeloid leukemia: a comprehensive review and 2016 update'. Blood Cancer J. 2016;6:e441
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405
3. Cagnetta A, Adamia S, Acharya C, Patrone F, Miglino M, Nencioni A. et al. Role of genotype-based approach in the clinical management of adult acute myeloid leukemia with normal cytogenetics. Leuk Res. 2014;38:649-659
4. Heath EM, Chan SM, Minden MD, Murphy T, Shlush LI, Schimmer AD. Biological and clinical consequences of NPM1 mutations in AML. Leukemia. 2017;31:798-807
5. Zou Q, Tan S, Yang Z, Zhan Q, Jin H, Xian J. et al. NPM1 Mutant Mediated PML Delocalization and Stabilization Enhances Autophagy and Cell Survival in Leukemic Cells. Theranostics. 2017;7:2289-2304
6. Rossin F, D'Eletto M, Falasca L, Sepe S, Cocco S, Fimia GM. et al. Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic glycolysis. Cell Death Differ. 2015;22:408-418
7. Orsini M, Morceau F, Dicato M, Diederich M. Autophagy as a pharmacological target in hematopoiesis and hematological disorders. Biochem Pharmacol. 2018;152:347-361
8. Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle. 2011;10:1719-1725
9. Torgersen ML, Engedal N, Boe SO, Hokland P, Simonsen A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood. 2013;122:2467-2476
10. Daniel J. Klionsky SDE. Autophagy as a Regulated Pathway of Cellular Degradation. Science. 2000;290:1717-1721
11. Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang Y. et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol Cell. 2017;65:917-931 e916
12. Qin L, Tian Y, Yu Z, Shi D, Wang J, Zhang C. et al. Targeting PDK1 with dichloroacetophenone to inhibit acute myeloid leukemia (AML) cell growth. Oncotarget. 2015;7:1395-1407
13. Brisson L, Banski P, Sboarina M, Dethier C, Danhier P, Fontenille MJ. et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell. 2016;30:418-431
14. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med. 2013;45:e45
15. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014-1020
16. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913-1930
17. Suzuki H, Osawa T, Fujioka Y, Noda NN. Structural biology of the core autophagy machinery. Curr Opin Struct Biol. 2017;43:10-17
18. Duffy A, Le J, Sausville E, Emadi A. Autophagy modulation: a target for cancer treatment development. Cancer Chemother Pharmacol. 2015;75:439-447
19. Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268-277
20. Shi C, Kehrl J. TRAF6 and A20 Regulate Lysine 63-Linked Ubiquitination of Beclin-1 to Control TLR4-Induced Autophagy. Sci Signal. 2010;3:ra42
21. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates StarvationInduced Autophagy. Mol Cell. 2008;30:678-688
22. Zalckvar E, Berissi H, Eisenstein M, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285-292
23. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D. et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685-696
24. Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598-609
25. Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107:1894-1899
26. Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L. et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201-206
27. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233-242
28. Evangelisti C, Evangelisti C, Chiarini F, Lonetti A, Buontempo F, Neri LM. et al. Autophagy in acute leukemias: a double-edged sword with important therapeutic implications. Biochim Biophys Acta. 2015;1853:14-26
29. Lu Z, Hunter T. Metabolic Kinases Moonlighting as Protein Kinases. Trends Biochem Sci. 2018;43:301-310
30. Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969-980
31. Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300-308
32. Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB. et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309-1323
33. Chen M, David C, Manley J. Tumor metabolism_ hnRNP proteins get in on the act. Cell Cycle. 2010;9:1863-1864
34. He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, Beck WT. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis. 2014;3:e84
35. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364-368
36. Chen M, David CJ, Manley JL. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat Struct Mol Biol. 2012;19:346-354
37. Ahmad F, Dixit D, Joshi SD, Sen E. G9a inhibition induced PKM2 regulates autophagic responses. Int J Biochem Cell Biol. 2016;78:87-95
38. Maejima Y, Isobe M, Sadoshima J. Regulation of autophagy by Beclin 1 in the heart. J Mol Cell Cardiol. 2016;95:19-25
39. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927-939
40. Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012;209:217-224
41. Xu Y, Chu L, Yuan S, Yang Y, Yang Y, Xu B. et al. RGD-modified oncolytic adenovirus-harboring shPKM2 exhibits a potent cytotoxic effect in pancreatic cancer via autophagy inhibition and apoptosis promotion. Cell Death Dis. 2017;8:e2835
42. Chi HT, Vu HA, Iwasaki R, Nagamura F, Tojo A, Watanabe T. et al. Detection of exon 12 type A mutation of NPM1 gene in IMS-M2 cell line. Leuk Res. 2010;34:261-262
43. Mohammad GH, Olde Damink SW, Malago M, Dhar DK, Pereira SP. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS One. 2016;11:e0151635
44. Panchabhai S, Schlam I, Sebastian S, Fonseca R. PKM2 and other key regulators of Warburg effect positively correlate with CD147 (EMMPRIN) gene expression and predict survival in multiple myeloma. Leukemia. 2017;31:991-994
Author contact
![]() Corresponding author: Dr. Ling Zhang, Email: lingzhangedu.cn, School of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, China. No.1, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 023-68485240, Fax: +86 023-68485239
Corresponding author: Dr. Ling Zhang, Email: lingzhangedu.cn, School of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, China. No.1, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 023-68485240, Fax: +86 023-68485239