Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
S-adenosyl methionine (SAM)
Nicotinamide adenine...
Acetyl-CoA
Tetrahydrofolate (THF)
Flavin adenine dinucleotide (FAD)
Concluding Remarks
Abbreviation
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1187-1199. doi:10.7150/ijbs.33496 This issue Cite
Review
Metabolic Intermediates in Tumorigenesis and Progression
Yuchen He1,2,3*, Menghui Gao1,2,3*, Haosheng Tang1,2,3, Yiqu Cao1,2,3, Shuang Liu4 ![]() , Yongguang Tao1,2,3
, Yongguang Tao1,2,3 ![]()
1. Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008 China
2. Cancer Research Institute, Key Laboratory of Carcinogenesis, Ministry of Health, School of Basic Medicine, Central South University, 110 Xiangya Road, Changsha, Hunan, 410078 China
3. Department of Thoracic Surgery, Second Xiangya Hospital, Central South University, Changsha, China
4. Institute of Medical Sciences, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008 China
*Equal contribution
Received 2019-1-25; Accepted 2019-3-18; Published 2019-5-7
Abstract

Traditional antitumor drugs inhibit the proliferation and metastasis of tumour cells by restraining the replication and expression of DNA. These drugs are usually highly cytotoxic. They kill tumour cells while also cause damage to normal cells at the same time, especially the hematopoietic cells that divide vigorously. Patients are exposed to other serious situations such as a severe infection caused by a decrease in the number of white blood cells. Energy metabolism is an essential process for the survival of all cells, but differs greatly between normal cells and tumour cells in metabolic pathways and metabolic intermediates. Whether this difference could be used as new therapeutic target while reducing damage to normal tissues is the topic of this paper. In this paper, we introduce five major metabolic intermediates in detail, including acetyl-CoA, SAM, FAD, NAD+ and THF. Their contents and functions in tumour cells and normal cells are significantly different. And the possible regulatory mechanisms that lead to these differences are proposed carefully. It is hoped that the key enzymes in these regulatory pathways could be used as new targets for tumour therapy.
Keywords: acetyl-CoA, SAM, FAD, NAD+, THF, cancer
Introduction
Currently, the mechanisms of tumour chemotherapy drugs mainly focus on inhibiting the replication and transcription of tumour cell nucleic acids or the synthesis of proteins. Although these anti-tumour drugs have been achieved success in some degree, we cannot ignore the severe limitations. As we know, most chemotherapy drugs have different degrees of side effects on the normal cells of human body. Patients with important organ dysfunction have difficulty in tolerating these drugs. Moreover, primary or secondary tumour drug resistance, poor effects on G0 stage cells, and difficulty in passing through the blood-brain barrier into the intracranial space are common issues that lead to the overall poor prognosis of cancer patients.
Tumour cells can proliferate in an uncontrolled manner, invade the surrounding tissues and move to distant organs. To achieve this, tumour cells have to significantly change their mechanism of energy metabolism to meet the need of huge amount of energy [1]. This process is known as metabolic reprogramming which makes tumour cells very different from normal ones [2]. We hope that by studying the regulatory mechanisms of metabolic intermediates, we can determine a more effective way to inhibit tumour cell proliferation without affecting normal cells. Tumour cells may not need to be killed; rather, the tumour cell energy supply could be inhibited by changes on metabolic intermediates. This process could put tumour cells in a dormant state, so that they could live in peace with normal cells, thus increasing the survival rate of tumour patients.
Metabolic reprogramming is one of the emerging hallmarks of cancer. In this process, cancer cells alter their metabolic pathway to coordinate with the environmental changes and cell functions [3, 4]. Metabolic reprogramming is one of the non-genetic factors that can alter the epigenetic landscape in cancer by using different metabolites as co-substrates or regulatory factors. Cancer cells utilize metabolic intermediates to alter the post-transcriptional modifications (PTMs) of chromatin [5] to produce more energy and macromolecules, so that they can meet the increasing needs of the rapid cell division and propagation [6]. Emerging data suggests that there are at least three different mechanisms, by which cancer metabolism affects epigenetics to regulate the activity of epigenetic enzymes: (1) alteration of metabolite levels by reprogramming metabolic pathways, (2) nuclear production of metabolites by translocating metabolic enzymes to the nucleus, and (3) generation of onco-metabolites, whose accumulation drives cancer progression [7].
In this review, we summarize the role of several major nuclear metabolic intermediates in tumour progression and metastasis. To provide new ideas for tumour therapy, we summarize the existing pathways and propose possible regulatory pathways that are essential for metabolic reprogramming.
S-adenosyl methionine (SAM)
SAM, also known as AdoMet or SAMe, is generally regarded as a principal methyl donor. It is synthesized from methionine and ATP, which is catalysed by methionine adenosyltransferase (MAT) [8]. SAM is involved in a wide variety of cellular reactions, in which it donates a methyl group to specific amino acid side chains of proteins, nucleotide bases in DNA and RNA, sugars, and many small molecules [9]. SAM mainly involves in three regulatory pathways [10]: (1) in transmethylation reactions, SAM donates its methyl group to acceptor molecules catalysed by methyltransferases (MTs) yielding SAH as a by-product [11]; (2) during the trans-sulfuration process, the sulfur atom of SAM is converted to cysteine (a precursor of taurine and glutathione that are major cellular anti-oxidants) via a series of enzymatic steps [12]; (3) in aminopropylation, SAM is utilized for decarboxylation and then aminopropylation, which leads to polyamine synthesis [10] (Figure 1). SAM can be freely transferred from the cytoplasm to the nucleus through nuclear pore complexes and plays a regulatory role in the nucleus [13].
Synthesis and function of S-adenosyl-L-methionine. In the cytoplasm, MAT catalyses the synthesis of SAM from methionine, with ATP as an energy donor. SAM then participates in transsulfuration, transmethylation, decarboxylation and aminopropylation to produce cysteine, SAH and polyamine, respectively. In the nucleus, SAM participates in the methylation reaction of histone proteins and DNA, which is catalysed by KMTs/PRMTs and DNMTs, respectively, and generates SAH as a by-product. SAM downregulates the ERK1/2 and Stat3 pathways to profoundly slow cell cycle progression. SAM upregulates cell-cycle inhibitors, including p53 and p21, and decreases cyclin A and cyclin E protein levels to arrest the cell cycle. SAM increases the ratio of pro-apoptotic Bax/Bcl-2 and activates caspase-3 and PARP cleavage to induce apoptosis.
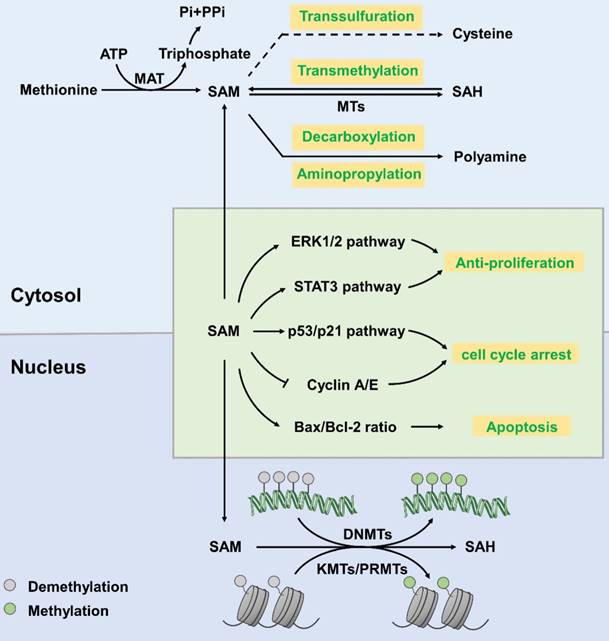
SAM mainly works as a methyl donor in the nucleus. It participates in histone methylation reactions that are mediated by lysine methyltransferases (KMTs; also called histone methyltransferases (HMTs)), protein arginine methyltransferases (PRMTs) and DNA methyltransferases (DNMTs) [13]. As shown in figure 1, after donating its methyl group, SAM is converted into SAH, a product that inhibits methyltransferases. This is a reversible reaction. The Km values of methyltransferases acting upon SAM are close to the physiological concentration of SAM (10-100 µM), which varies in different tissue types [14]. The cellular ratio of SAM/SAH is a potent determinant in regulation of chromatin methylation. Threonine restriction decreases the SAM/SAH ratio, which commonly leads to decreased H3K4me2/3 methylation [15-18].
Hypomethylation of the genome is an important cellular biology feature of tumour cells [19]. The methylation of cytosine at CpG islands in promoter regions strongly implicates the transcriptional silencing. It has been described that aberrant DNA methylation is associated with pathological gene expression in various cancer tissues [20]. In many cases, cancer cells display distinct shifts in DNA methylation patterns towards hypermethylation at CpG islands and hypomethylation within the gene bodies [21]. Specifically, the DNA methylation- mediated silencing of tumour suppressor genes, such as CDKN2A (cyclin-dependent kinase inhibitor 2A) and SFRPs (secreted frizzled-related proteins), have been identified as drivers of lung carcinoma and colorectal cancer progression [22, 23]. What is more, a review shows that RNA polymerase II (Pol II) stalling is influenced by DNA methylation around the proximal sites of promoter [24]. As a major methyl donor, SAM has been shown to exert anti- proliferative effects in various cancer cells by promoting methylation and downregulating proto- oncogene expression [25]. Moreover, SAM inhibits proliferation via downregulating p53, ERK1/2 and Stat3 pathway, slowing down cell cycle progression and inducing apoptosis [26, 27]. Studies have shown that treatment with SAM suppressed the proliferation, migration, and invasion of prostate cancer (PC-3) cells [25] via improving the methylation level of the genome. At present, there is little research on the role of SAM in other types of tumours. Besides, we wonder whether it is possible to increase SAM content in tumour cells by oral administration or local injection of SAM or its synthetic precursor into tumour tissue. Or maybe we can improve the activity of MAT to increase the amount of SAM. Thus, inhibiting the proliferation of tumour cells.
Nicotinamide adenine dinucleotide (NAD+)
NAD+ is a vital coenzyme found in all living cells which serves as a critical coenzyme that carries electrons from one reaction to another in reduction- oxidation reactions. In addition, NAD+ is a co-substrate for sirtuins and polymerases in the modification of proteins and DNA [28]. Adequate compounds, related to NAD+ metabolism, are considered as drug targets for cancer treating [29-31]. The subcellular localization of mammalian nicotinamide mononucleotide adenylyltransferase (NMNAT) isoforms determines and maintains the content and distinctive functions of NAD+ in different organelles. NMNAT has three isoforms and their subcellular localizations are tightly controlled: nuclear NMNAT1, cytosolic NMNAT2 and mitochondrial NMNAT3. Several other enzymes are known to regulate the synthesis of NAD+. For example, 2-amino-3- carboxymuconate-6-semialdehyde decarboxylase (ACMSD) converts 2-amino-3-carboxymuconate-6-semialdehyde into 2-aminomuconate-6-semialdehyde, which negatively regulates NAD+ synthesis from tryptophan [32]. Nicotinamide phosphoribosyltransferase (NAMPT) serves as the first and limiting step in the NAD+ salvage pathway, promoting the conversion of nicotinamide (NAM) into nicotinamide mononucleotide (NMN) (Figure 2).
Synthesis and function of NAD+ in the cytoplasm and nucleus. In the de novo synthesis pathways, NAD+ is synthesized from dietary L-tryptophan (Trp). Trp is converted to quinolinic acid through several reactions. Quinolinic acid is then transformed into NAMN and NAAD; these reactions are catalysed by QPRT and NMNAT, respectively. In the salvage pathway, nicotinamide riboside (NR) or nicotinamide (NAM) is used to produce NAMN and NMN, which are then converted to NAD+. Cellular NAD+ levels are an energy level sensor, and low levels of NAD+ subsequently induce cell death. NAD+ functions as a cofactor of SIRT and PARP. As the cofactor of SIRT, NAD+ participates in protein deacetylation to regulate the gene expression and production of second messengers. As the cofactor of PARP, NAD+ participates in the transfer of ADP-ribose subunits from NAD+ to target proteins to regulate processes such as DNA repair, gene expression, cell survival and cell cycle progression, chromatin remodelling, genomic stability and RNA expression.
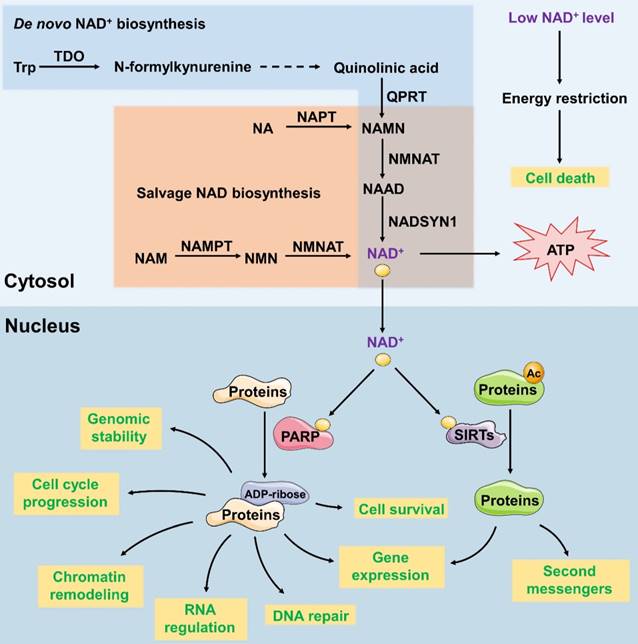
NAD+ functions as both an important redox carrier and the sole substrate of NAD+-dependent enzymes. Thus, it participates in various cell cellular processes, such as metabolism and stress resistance. For example, as a redox carrier, NAD+ drives oxidative phosphorylation and produces ATP. Decreased NAD+ level and the consequent PARP hyperactivation show the depletion of intracellular ATP store, leading to the activation of PARP1 and the release of apoptosis-inducing factor (AIF), passive necrotic cell death inducer ADP- ribosyltransferase enzyme 1 (ARTD1) and consequent cell death due to energy restriction [33]. Therefore, we could reduce the production of NAD+ by downregulating the activity of key enzymes so as to inhibit tumour growth. Besides, as the only substrate of NAD+-dependent enzymes, poly (ADP-Ribose) polymerase (PARP) uses NAD+ exclusively for poly-ADP-ribosylation, which transfers ADP-ribose subunits from NAD+ to target proteins. This process regulates the expression of DNA-repair gene [34], cell survival and cell cycle progression [29], chromatin remodelling [35], genomic stability and RNA expression [36]. PARP's activity is dependent on the amount of NAD+ available. As a nuclear energy sensor, PARP's activity would be impaired due to NAD+ deficiency which leads to functional deficits and subsequently triggers apoptosis. Whether NAD+ is acting as a redox carrier or an enzyme cofactor, reducing its levels in tumour cells can eventually induce cell death.
Nicotinamide adenine dinucleotide phosphate (NADP+), also known as triphosphopyridine nucleotide (TPN), is a final electron acceptor used in anabolic reactions such as the Calvin cycle, nucleic acid and cholesterol syntheses and fatty acid chain elongation, producing NADPH as a reducing agent. NADP+ and NAD+ differ in the presence of an additional phosphate group on the 2' position of the ribose ring that carries the adenine moiety.
NADPH participates in protecting against the toxicity of reactive oxygen species (ROS) and allowing the regeneration of glutathione (GSH) [37]. Studies showed that systemic administration of exogenous NADPH significantly protected neurons against ischemia/reperfusion-induced injury by blocking the elevation of intracellular ROS levels [38]. In addition, The amount of NADPH is an effective biomarker for predicting ferroptosis sensitivity [39], labeled as lipid hydroperoxides accumulated in an iron-dependent way, which is a form of cell death. NADPH is also responsible for generating free radicals in immune cells used to destroy pathogens in a process termed the respiratory burst [40]. Adrenodoxin reductase use NADPH as a coenzyme which transfers two electrons from NADPH to FAD in the chain of mitochondrial P450 systems that synthesize steroid hormones [41].
As a key precursor of DNA synthesis and repair, NADPH has a close relationship with tumor progression. However, current studies on this area are quite limited. Researchers have found that decreasing NADPH levels inhibits viability, induces apoptosis and enhances cisplatin-induced DNA damage in prostate cancer cells [42]. Inhibiting the levels of NADPH increases survival rate and decreases apoptosis, DNA damage and cisplatin-induced cytotoxicity in the nasopharyngeal carcinoma cells [43].
Given the role NADPH plays in lipid metabolism, oxidative stress reaction and DNA synthesis and repair, we hypothesized that it could be applied in a wider field in fighting tumors. In addition to the effects on genetic material, we can also explore its role from the perspective of metabolism and oxidative stress. High levels of fatty acid metabolism in tumor cells requires large amounts of NADPH as substrates. Inhibition of NADPH synthesis may inhibit this process in tumor cells. Meanwhile, low levels of NADPH can reduce the cell's tolerance to oxidative stress, leading to cell death.
Acetyl-CoA
Acetyl-CoA is composed of an acetyl moiety (CH3CO) and coenzyme A (CoA) linked by a thioester bond [44]. As shown in figure 3, acetyl-CoA is a central intermediate in the oxidation of glucose to produce ATP, whose abundance reflects the general energy state of the cells [45]. Moreover, acetyl-CoA is a precursor for the biosynthesis of numerous metabolites. It not only participates in many metabolic pathways, but also influences the activity or specificity of multiple proteins through acetylation or epigenetic regulation of gene expression [46] (Figure 3).
Synthesis, metabolic pathways and function of acetyl-CoA. Acetyl-CoA is predominantly generated in the mitochondrial matrix through glycolysis, fatty acid oxidation and catabolism of branched-chain amino acids (BCAAs), such as valine, leucine and isoleucine. Acetyl-CoA can then be transported from the mitochondria using the “citrate-malate-pyruvate shuttle” to create oxaloacetate and cytoplasm acetyl-CoA. Acetyl-CoA can be generated from the reductive carboxylation of glutamine. In addition, acetate can be used to produce cytoplasmic acetyl-CoA catalysed by acyl-CoA synthetase short-chain family member 2 (ACSS2) in an ATP-dependent manner. Synthesized acetyl-CoA is widely used as a precursor for the biosynthesis of numerous metabolites, such as fatty acids, steroids and some specific amino acids, which include glutamate, proline, and arginine. Reducing the conversion of mitochondrial-derived citrate to acetyl-CoA downregulates lipogenesis. This process also decreases cell proliferation, which is stimulated by cytokines and regulated by the PI3K/Akt signalling pathway. Citrate-derived acetyl-CoA functions as an acetyl donor for protein acetylation. In the cytoplasm, acetylation can activate the function of proteins, resulting in a series of biological activities. In the nucleus, the acetylation of the histone tail neutralizes the charge of the histone, weakens the interaction between histones and DNA, and relaxes the chromatin structure, thus facilitating transcription.
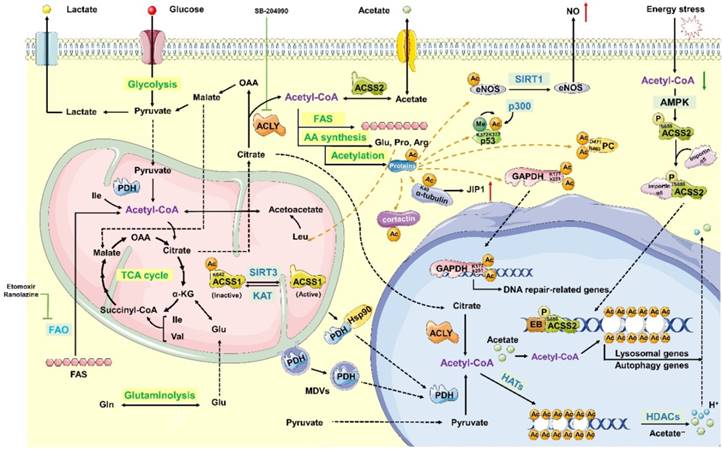
The role of histone acetylation in transcriptional regulation can be described mainly by the following 3 mechanisms: (1) histone acetylation neutralizes its charge, weakens the interaction between histones and DNA, and relaxes the chromatin structure to facilitate transcription; (2) histone acetylation provides specific anchor points for the recruitment of transcription factors; (3) histone acetylation combines with other histone modifications (methylation, phosphorylation, ubiquitination) to form histone codes, affecting the transcription of genes. In contrast, histone acetylation can be reversed by histone deacetylases (HDACs, also called lysine deacetylases [KDACs]), such as Sir2, and by HATs, such as SAS2, which constructs the fine regulating networks of acetylation in living eukaryotic organisms. In cancer cells, the pathological activation of tumorigenic enhancers has been associated with the aberrant accumulation of H3K27 acetylation in particular regions. Some drugs, such as JQ1, that downregulate histone acetylation, have been approved by the FDA, underscoring the value of targeting histone acetylation [47]. In multiple cell types from yeast to human, histone acetylation is highly sensitive to the availability of acetyl-CoA [48-51]. Acetyl-CoA is indeed the obligate cofactor for histone acetyltransferases (HATs, also called lysine acetyltransferases [KATs]) [52], indicating that the abundance of the acetyl-CoA in nucleo-cytosolic pool may have a direct impact on the enzymatic activity of HATs, especially those with a relatively high Kd for acetyl-CoA.
In addition to controlling the balance between cellular anabolism and catabolism, acetyl-CoA also acts as a signal transducer that translocate from the mitochondrial matrix to the cytoplasm or nuclear matrix. In this circumstance, pyruvate dehydrogenase (PDH) is transiently translocated into the nucleus with the help of mitochondrial-derived vesicles (MDVs) [53] or chaperones, such as heat shock protein 90 (Hsp90) [54]. Then, PDH produces nuclear acetyl-CoA in a pyruvate-dependent manner. Besides, ACLY [55] and ACSS2 [56], enzymes participating in generating cytosolic acetyl-CoA, have been found in the nucleus, which is thought to be associated with cell growth and proliferation [57]. Citrate-derived acetyl-CoA acetylates proteins to regulate their activity, interactions, and location. And then it affects signalling pathways, DNA transcription, and chromatin modification to promote cell growth, mitosis, suppressing autophagy and regulating cell death (Figure 3).
Histone acetylation and the concentration of ACLY dependent acetyl CoA are inconstantly sensitive to glucose in cancer cells. Oncogenic signalling pathways are mediated in an ACLY- dependent manner [48, 50]. Knocking down ACLY reduces the conversion of mitochondrial-derived citrate to acetyl-CoA, thus downregulating lipogenesis and decreasing cytokine stimulation and proliferation regulated by PI3K/Akt signalling pathway [58]. Furthermore, the ratio of acetyl-CoA to coenzyme A in the nucleus regulates global histone acetylation levels, which is stimulated by the oncogenic protein Kras or Akt during tumour progression [59]. In addition to acetylation-induced protein-nucleic acid interactions that occur primarily pre-transcription, acetylation also regulates protein- protein interactions in many ways. Acetylation, driven by glucose-derived acetyl-CoA, eliminates the positive charge of the ε-amino group of lysine and causes steric hindrance, thereby modifying proteins post-translation and affecting protein interactions, activity and localization [60]. In addition, the reaction occurs non-enzymatically through direct interactions between lysine and acetyl moieties [61], especially when pH level and acetyl-CoA concentration is high [62-64]. Furthermore, acetylated lysine functions as docking sites for proteins that contain the acetyl- lysine recognition domain of bromodomains [65] and plant homeodomain 1 (PHD1) [66], which is exclusively found in nuclear proteins that participate in DNA transcription and chromatin remodelling.
Lysine acetylation plays a significant role in the regulation of nuclear transcription and other modifications, such as methylation, ubiquitination and phosphorylation. Acetyl-CoA also functions as a donor in the cross-communication regulation pathway. Moreover, a great number of acetylated non-histone proteins also participate in nuclear transcription, including metabolic enzymes such as ACCS, pyruvate carboxylase (PC), glyceraldehyde-3- phosphate dehydrogenase (GAPDH), glutamate dehydrogenase (GDH) and endothelial nitric oxide synthase (eNOS), as well as non-enzymatic proteins, such as p53, α-tubulin, Akt and cortactin [59, 67-69]. For example, the mitochondrial protein sirtuin 3 (SIRT3) deacetylates K642 of ACSS1 to activate the enzyme, which is acetylated at the same site and replaced by glutamine to form the inactive enzyme. This mechanism regulates the activity of ACSS1 by altering the constitutively acetylated lysine residues. Acetyl CoA can regulate both the structure and activities of PC through special residues [68], and the R469 and D471 sites in the allosteric domain of PC directly interact with acetyl CoA [67]. SIRT3-mediated deacetylation of mitochondrial enzymes, such as GDH, in human granulosa and cumulus cells may convert the metabolic milieu surrounding oocytes; therefore, this process may be responsible for the reduction of ovarian reserve at advanced maternal age [69]. Furthermore, in human gliomas and prostate tumours, the Akt phosphorylation level at Ser473 is closely associated with histone acetylation [55, 59]. In fact, acetylation is actually a mechanism that regulates protein localization, activation, and stability, thus participating in energy metabolism, cytoskeleton dynamics, endocytosis, autophagy, and other signalling pathways, such as transforming growth factor-β (TGF-β)-mediated signalling and interferon-α (IFN-α) signalling.
Tetrahydrofolate (THF)
Tetrahydrofolate (THF), the biologically active form of folic acid, exists in the cytoplasm, mitochondria and nucleus and participates in one-carbon metabolism as a carrier for the one-carbon units in N5 or N10 or both N5 and N10. As the core intermediate of one-carbon metabolism, THF is involved in purine and thymidylate (dTMP) biosynthesis and amino acid metabolism, thus playing an important role in DNA replication [70]. The role of anti-folate compounds in cancer has been detected over the past 50 years. However, the role of folate in cancer development and progression has not been found till now.
In mammalian cells, THF is synthesized from dihydrofolate (DHF) or THF derivatives. THF is reduced from DHF by dihydrofolate reductase (DHFR), which requires two molecules of nicotinamide adenine dinucleotide phosphate (NADPH). Serine hydroxymethyltransferase (SHMT), using glycine as a substrate, converts 5,10-methylene-THF into THF and simultaneously generates serine. This reaction is reversible. Additionally, thymidylate synthase (TYMS) catalyses the generation of DHF and dTMP from 5,10-methylene-THF and dUMP. These three reactions constitute a cycle. Methylenetetrahydrofolate dehydrogenase (MTHFD) reversibly catalyses the generation of 5,10-methylene-THF from 10-formyl-THF. MTHFD1 catalyses this reaction in the cytoplasm, and MTHFD2/L catalyses this reaction in the mitochondria (Figure 4).
Synthesis and function of THF. THF is reduced from DHF by dihydrofolate reductase (DHFR) and requires NADPH as a co-factor. SHMT converts 5,10-methylene-THF into THF and simultaneously generates serine; glycine is used as the substrate. In the nucleus, 5,10-methylene-THF is converted into DHF by TYMS. In addition, methylenetetrahydrofolate dehydrogenase (MTHFD) reversibly catalyses the generation of 5,10-methylene-THF from 10-formyl-THF. Enzymes such as TYMS, DHFR and SHMT1/2α and synthesized THF translocate into the nucleus and function as a multienzyme complex for nuclear de novo dTMP synthesis, which is essential for DNA replication and genome stability.
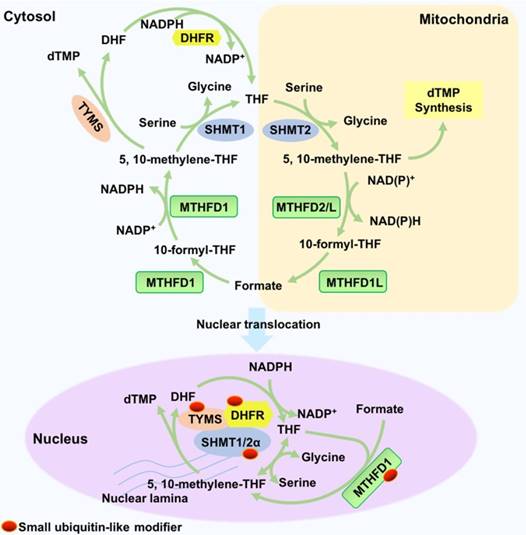
Nuclear THF metabolism is catalysed by enzymes in its special nuclear forms. The enzymes (TYMS, DHFR, and SHMT1/2α) translocate into the nuclei in a small ubiquitin-like modifier (SUMOylation)-dependent manner. In the nucleus, the three enzymes function as a multienzyme complex for nuclear de novo dTMP synthesis, and their metabolic mechanisms are similar to their mechanisms in the cytosol and mitochondria [71]. DHFR reduces DHF to THF in an NADPH-dependent manner. SHMT1 or SHMT2α reversibly synthesizes 5-methyl-THF from THF and serine, which is the rate-limiting step. Furthermore, TYMS catalyses the reaction of dUMP and 5,10-methylene-THF to dTMP and DHF. Nuclear MTHFD1 is also SUMOylated in a cell cycle-dependent manner to produce 5,10- methylene-THF from THF. In addition, the mouse liver MTHFD1 is enriched in the nuclei during folate deficiency [72].
THF is responsible for the nuclear de novo biosynthesis of dTMP, which is essential for DNA replication and genome stability because it prevents uracil misincorporation of DNA [73]. On the other hand, vitamin B12 and folate deficiencies cause 5-methyl-THF trapped in the nucleus; this action inhibits nuclear de novo dTMP biosynthesis and causes DNA damage, which might results in megaloblastic anaemia [74]. Another study demonstrated that arsenic trioxide As2O3 suppresses nuclear de novo dTMP biosynthesis through the degradation of MTHFD1 and SHMT1, leading to increased uracil levels in nuclear DNA and increased genome instability [75].
THF is required for DNA synthesis and cell proliferation and is upregulated in cancer [70]. TYMS is overexpressed in cancer and might be associated with adverse clinical behaviour and poor prognosis [76-79]. TYMS overexpression might predict lymph node metastasis and poor prognosis in colorectal cancer patients [80]. TYMS upregulation is associated with an aggressive prostate cancer phenotype and chromosomal deletions [76]. DHFR, which catalyses the generation of THF, is overexpressed in osteosarcoma stem-like cells; this overexpression enhances their chemoresistance to methotrexate (MTX) [81]. In addition, other THF metabolism enzymes, such as SHMT1, SHMT2, and MTHFD2, are upregulated in cancer to promote DNA synthesis [82-84].
THF metabolic pathway has served as a target for anti-tumour treatment for a long time. 5- fluorouracil (5-FU), MTX and pemetrexed serve as the first-line treatments for several cancers [70, 85-87]. However, they also cause considerable damage to normal cells while treating tumours, which limit effective use in cancer therapy. Tumour cells and normal cells obtain THF in different ways. Whether it is possible for us to specifically inhibit specific enzymes to reduce THF synthesis in tumour cells while reducing damage to normal cells remains to be explored.
Flavin adenine dinucleotide (FAD)
FAD is the most widely utilized 'nucleotide' form of the vitamin riboflavin (Rf, vitamin B2). FAD is capable of carrying out one- and two-electron redox, nucleophilic and electrophilic reactions [88]. As a redox cofactor, FAD participates in reactions in the mitochondria and cytoplasm as well as in nuclear redox activities [89]. The nuclear, mitochondrial, and cytosolic FAD synthase pools constitute a flavin network involved in regulation of cellular metabolism and epigenetic events [90].
De novo synthesis of FAD requires Rf as a precursor [91], which is a type of water-soluble vitamin that must be obtained from food or intestinal microflora in mammals [90]. Catalysed by riboflavin kinase and FAD synthase, the absorbed Rf is converted into FAD via two consequent reactions: one reaction is catalysed by riboflavin kinase, which transfers a phosphoryl group from ATP to Rf to form FMN, and the other reaction adenylates FMN to FAD via the canalization of FAD synthase. Then, FAD is used to produce FAD+ in redox reactions catalysed by FAD synthetase (FLAD1), where FAD acts as the electron donor. FAD also is used to generate FADH2 catalysed by reducing enzymes, such as lysine- specific demethylase 1/2(LSD1/2) [92]. FAD is a cofactor of oxidoreductases, also called as flavoproteins, most of which are metabolic enzymes [93]. For example, under early oxidative stress or proapoptotic stimuli, LSD1 and apoptosis-inducing factor (AIF) translocate to the nucleus from the mitochondrial inner membrane caused by high FAD level, which inhibits the degradation of HIF-1α. And then, HIF-1α promotes the expression of HIF-1α target genes and induces hypoxia responses [94, 95](Figure 5).
The synthesis and function of FAD. The de novo synthesis of FAD requires riboflavin (Rf) as a precursor. Rf is converted into FAD via two consequent reactions: one reaction transfers a phosphoryl group from ATP to Rf to form FMN and is catalysed by riboflavin kinase; the other reaction adenylates FMN to FAD and is catalysed by FAD synthase. The synthesis of FAD can be achieved in the cytoplasm, mitochondrial matrix and nucleus. Mitochondria-derived FAD acts as a hapten and covalently attaches to specific apoproteins, the mitochondrial succinate dehydrogenase complex of the citric acid cycle, to induce a strong immune response. However, nuclear-derived FAD regulates hypoxia responses as the cofactor of lysine-specific demethylase 1 (LSD1); it can decrease the DNA expression of IL-1α, IL-1β, IL-6, and tumour necrosis factor-α via increasing H3K4me1 and H3K4me2 in exon 1. In the hypoxia response, FAD acts as the cofactor of lysine-specific demethylase 1 (LSD1). In early hypoxia, high levels of FAD activate LSD1, which inhibits the degradation of HIF-1α to promote the expression of HIF-1α target genes and induce hypoxia responses. During prolonged hypoxia, decreased cellular FAD levels attenuate the activity of LSD1, leading to the combination of RACK1 and HIF-1α and causing the degradation of HIF-1α.
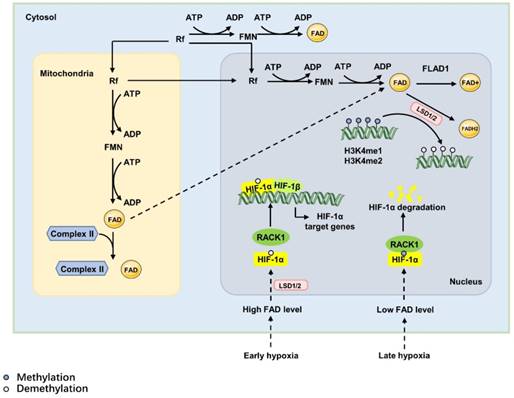
The ratio of FAD+/FADH2, influenced by the oxygen and nutrition supply, modulates the levels of nuclear FAD. In this way, it directly affects epigenetic regulation by regulating the activity of histone demethylases [96]. The nuclear role of FAD is as the cofactor of LSD1/2. In this way, FAD regulates gene expression via demethylation in accordance with the cell state, nutrients or oxygen availability and the intra-/extra cellular environmental conditions to maintain homeostasis.
FAD also acts as an electron acceptor in the nucleus to participate in the demethylation reaction catalysed by LSD1/2 [97, 98]. LSD1/2 catalyses the demethylation reaction by specifically removing methyl groups from mono- or dimethylated H3K4 or H3K9 in histone proteins and DNA in the nucleus. This reaction is coupled with FAD reduction to FADH2, and formaldehyde is released as a by-product [99, 100]. FAD synthase is found in the cytosol and mitochondria, but a recent study demonstrates the existence of a nuclear FAD synthase and FAD- hydrolysing activity, indicating the participation of FAD in nuclear redox activities [101]. This modification directly reprograms the chromatin structure and leads to the context-dependent activation or repression of transcription [100]. FAD acts as a biosensor of the metabolic state, indicating the potential link between epigenetics and metabolism, especially in cancer cells.
Concluding Remarks
Compared with normal cells, tumour cells seem omnipotent and ubiquitous, but this is not the case. The growth of tumour cells requires a huge energy supply and a specific microenvironment, known as a cancer nest. If we cut off the energy supply or destroy the environment, we can simply stop cancer cells from growing. In this process, metabolic reprogramming will play a crucial role. Therefore, we propose that we can take advantage of the significant differences in the metabolism of tumour cells and normal cells to inhibit the energy supply and signal transduction of tumour cells for therapeutic purposes. In addition, the idea of treating tumours by inhibiting the metabolic intermediates of tumour cells is supported by successful models. Methotrexate and fluorouracil are folic acid metabolism inhibitors. They are listed as first-line chemotherapeutic drugs that have been widely used to treat tumours for more than 50 years. They act on cells in S phase and are both cell cycle-specific drugs. However, due to the lack of significant differences in the DNA synthesis processes of tumour cells and normal cells, these anti-metabolites are not selective. Therefore, these drugs are very toxic to normal tissues with rapid proliferation, such as the bone marrow, gastrointestinal mucosa, reproductive system and hair. To solve these problems, we need to know more about the generation process and regulatory function of metabolites to find more effective treatments. New drugs targeting metabolism pathways have been proposed by some researchers, such as the heterogeneous activator of PK-M2 which affects the anaerobic glycolysis pathway and phenformin which can inhibit mitochondrial anaerobic glycolysis. But these are just a few parts of the many metabolic processes. The fat metabolism we mentioned in the paper has not been studied.
We here summarize the synthesis of several major metabolites and their important roles in tumour cells. We hope to find appropriate candidates of therapeutic intervention in their synthesis processes by understanding their underlying mechanisms. We may reduce the production of metabolites by inhibiting the enzyme rates or changing the direction of reversible reactions. We notice that upregulating the production of SAM, and FAD and downregulating acetyl-CoA, NAD+ and THF could be reasonable targets. However, many unsolved problems remain. For example, fatty acid metabolism is obviously enhanced in tumour cells, and we can cut off the energy supply to tumour cells by inhibiting fatty acid metabolism. However, the regulation of fatty acid metabolism in tumour cells is not clear. Another problem is avoiding damage to normal immune cells if anaerobic glycolysis is inhibited. Metabolic pathways are intricately woven, and effective targets must be chosen carefully. The resolution of these problems will provide new, safe and effective treatments for tumours.
Abbreviation
3'-UTR: 3'-untranslated region; 5-FU: 5-fluorouracil; ACC2: acetyl-CoA carboxylase 2; acetyl-CoA: acetyl coenzyme A; Ach: acetylcholine; ACLY: ATP-citrate lyase; ACMSD: 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase; ACSS1: acetyl-CoA synthetase short-chain family member 1; ACSS2: acyl-CoA synthetase short-chain family member 2; ADH1B: alcohol dehydrogenase 1B; ADP: adenosine diphosphate; AIF: apoptosis-inducing factor; α-KG: α-ketoglutarate; ALDH2: aldehyde dehydrogenase 2; AMPK: AMP-activated protein kinase; ARTD1: ADP-ribosyltransferase enzyme 1; As2O3: arsenic trioxide; ATP: adenosine triphosphate; Bax: Bcl-2 assaciated X protein; BCAAs: branched-chain amino acids; Bcl2: B-cell lymphoma 2; cADPR: cyclic adenosine diphosphoribose ribose; CBP: cap binding protein; CH3CO: acetyl moiety; CoA: coenzyme A; Complex II: succinate dehydrogenase; DHF: dihydrofolate; DHFR: dihydrofolate reductase; DNMTs: DNA methyltransferases; dTMP: thymidylate; dUMP: deoxyuridine monophosphate; eNOS: endothelial nitric oxide synthase; ERK1/2: extracellular regulated protein kinases 1/2; ESC: embryonic stem cell; FAD: Flavin adenine dinucleotide; FADH2: flavin adenine dinucleotide, reduced; FAO: fatty acid oxidation; FLAD1: FAD synthetase 1; FMN: flavin mononucleotide; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; GAS: Group A streptococci; GCLC: glutamate-cysteine ligase catalytic; GCLM: glutamate-cysteine ligase modifier; GDH: glutamate dehydrogenase; GS: GSH synthase; GSH: glutathione; H3K4me1/2: mono- or dimethylation of lysine (K)-4 in histone H3; HATs: histone acetyltransferases; HDAC: histone diacetylase; HIF-1α: hypoxia-inducible factor 1α; HIF-1β: hypoxia-inducible factor 1β; HIV: human immunodeficiency virus; Hsp90: heat shock protein 90; IDO: indoleamine 2, 3-dioxygenase; IFN-α: interferon-α; IL: interleukin; iNOS: inducible nitric oxide synthase; IRF9: interferon-regulatory factor 9; ISGF3: IFN- stimulated gene factor 3; JIP1: JNK-interacting protein 1; JNK: c-Jun N-terminal kinase; KMTs: lysine methyltransferases; Kyn: kynurenine; LPS: lipopolysaccharide; LSD1/2: lysine-specific demethylase 1/2; MAT: methionine adenosyl transferase; MDM: murine double minute2; MDVs: mitochondrial-derived vesicles; miRs: microRNAs; MTHFD: methylenetetrahydrofolate dehydrogenase; MTs: methyl transferases; MTX: methotrexate; NAAD: nicotinic acid adenine dinucleotide; NAADP: nicotinic acid adenine dinucleotide phosphate; NAD+: nicotinamide adenine dinucleotide; NADH: reduced form of nicotinamide-adenine dinucleotide; NADP: nicotinamide adenine dinucleotide phosphate; NADPH: reduced nicotinamide adenine dinucleotide phosphate; NADSYN: NAD+ synthetase; NAM: nicotinamide; NAMPT: nicotinamide phosphoribosyltransferase; NF-κB: nuclear factor-κ-gene binding; NLRP: NACHT, LRR and PYD domains-containing protein; NLRs: Nod-like receptors; NMN: nicotinamide mononucleotide; NMNAT: nicotinamide mononucleotide adenylyltransferase; NO: nitric oxide; NR: nicotinamide riboside; NSCLC: non-small-cell lung cancer; O-GlcNAcylation: O-linked N-acetylglucosamine modification; PARP: poly (ADP-Ribose) polymerase; PBMC: peripheral blood mononuclear cell; PC: pyruvate carboxylase; PC-3: prostate cancer cells; PDH: pyruvate dehydrogenase; PGC-1: peroxisome proliferators activated receptor gamma co-activator 1; PHD1: plant homeodomains1; Pi: phosphate; PPi: pyrophosphate; PRMTs: protein arginine methyltransferases; PRRs: pattern recognition receptors; PSCs: Pluripotent stem cells; PTMs: post-translational modifications; RACK1: activated protein-C kinase-1; Rf: riboflavin; RNAi: RNA interference; SAH: S-adenosyl homocysteine; SAM: S-adenosyl methionine; SHMT: serine hydroxymethyltransferase; shRNA: short hairpin RNA; Sir2: silent information regulator 2; SIRT: Sirtuin; STAT: signal transducer and activator of transcription; succinyl-CoA: succinyl coenzyme A; SUMOylation: small ubiquitin-like modifier; TCA: tricarboxylic acid cycle; TDO: tryptophan 2, 3-dioxygenase; TGF-β: transforming growth factor-β; THF: tetrahydrofolate; TLRs: Toll-like receptors; TNF-α: tumor necrosis factor-α; Trp: L-tryptophan; Trp: tryptophan; TYMS: thymidylate synthase; UV: ultraviolet radiation; ZGA: zygotic genome activation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [81672787 (Y. Tao), 81672991 (S. Liu)] and the National Basic Research Program of China [2015CB553903 (Y. Tao)]. We apologize to those colleagues whose work we could not cite due to space constraints.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635-49
2. Zhao L, Mao Y, Zhao Y, Cao Y, Chen X. Role of multifaceted regulators in cancer glucose metabolism and their clinical significance. Oncotarget. 2016;7:31572-85
3. Campbell SL, Wellen KE. Metabolic Signaling to the Nucleus in Cancer. Molecular cell. 2018;71:398-408
4. Sebastian C. Tracking down the origin of cancer: metabolic reprogramming as a driver of stemness and tumorigenesis. Critical reviews in oncogenesis. 2014;19:363-82
5. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nature reviews Clinical oncology. 2017;14:11-31
6. He Y, Gao M, Cao Y, Tang H, Liu S, Tao Y. Nuclear localization of metabolic enzymes in immunity and metastasis. Biochim Biophys Acta Rev Cancer. 2017;1868:359-71
7. Kim JA, Yeom YI. Metabolic Signaling to Epigenetic Alterations in Cancer. Biomolecules & therapeutics. 2018;26:69-80
8. Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Annals of hepatology. 2013;12:183-9
9. Landgraf BJ, McCarthy EL, Booker SJ. Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annual review of biochemistry. 2016;85:485-514
10. Giulidori P, Galli-Kienle M, Catto E, Stramentinoli G. Transmethylation, transsulfuration, and aminopropylation reactions of S-adenosyl-L-methionine in vivo. The Journal of biological chemistry. 1984;259:4205-11
11. Lieber CS, Packer L. S-Adenosylmethionine: molecular, biological, and clinical aspects-an introduction. The American journal of clinical nutrition. 2002;76:1148s-50s
12. Lu SC. S-Adenosylmethionine. The international journal of biochemistry & cell biology. 2000;32:391-5
13. van der Knaap JA, Verrijzer CP. Undercover: gene control by metabolites and metabolic enzymes. Genes & development. 2016;30:2345-69
14. Fan J, Krautkramer KA, Feldman JL, Denu JM. Metabolic regulation of histone post-translational modifications. ACS chemical biology. 2015;10:95-108
15. Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S. et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science (New York, NY). 2013;339:222-6
16. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D. et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell metabolism. 2015;22:861-73
17. Mlika M, Helal I, Laabidi S, Braham E, El Mezni F. Is CD10 antibody useful in the diagnosis of angioimmunoblastic T-cell lymphoma? Journal of immunoassay & immunochemistry. 2015;36:510-6
18. Ding W, Smulan LJ, Hou NS, Taubert S, Watts JL, Walker AK. s-Adenosylmethionine Levels Govern Innate Immunity through Distinct Methylation-Dependent Pathways. Cell metabolism. 2015;22:633-45
19. Ehrlich M, Lacey M. DNA hypomethylation and hemimethylation in cancer. Advances in experimental medicine and biology. 2013;754:31-56
20. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Molecular cell. 2014;54:716-27
21. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239-59
22. Suzuki K, Sakaguchi M, Tanaka S, Yoshimoto T, Takaoka M. Prolyl oligopeptidase inhibition-induced growth arrest of human gastric cancer cells. Biochemical and biophysical research communications. 2014;443:91-6
23. Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E. et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11891-6
24. Liu S, Tao Y. Interplay between chromatin modifications and paused RNA polymerase II in dynamic transition between stalled and activated genes. Biological reviews of the Cambridge Philosophical Society. 2013;88:40-8
25. Schmidt T, Leha A, Salinas-Riester G. Treatment of prostate cancer cells with S-adenosylmethionine leads to genome-wide alterations in transcription profiles. Gene. 2016;595:161-7
26. Ilisso CP, Sapio L, Delle Cave D, Illiano M, Spina A, Cacciapuoti G. et al. S-Adenosylmethionine Affects ERK1/2 and Stat3 Pathways and Induces Apotosis in Osteosarcoma Cells. Journal of cellular physiology. 2016;231:428-35
27. Stiuso P, Bagarolo ML, Ilisso CP, Vanacore D, Martino E, Caraglia M. et al. Protective Effect of Tyrosol and S-Adenosylmethionine against Ethanol-Induced Oxidative Stress of Hepg2 Cells Involves Sirtuin 1, P53 and Erk1/2 Signaling. International journal of molecular sciences. 2016;17:189-96
28. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends in cell biology. 2014;24:464-71
29. Jiménez-García M, Verdugo-Sivianes E, Lucena-Cacace A. Nicotinamide adenine dinucleotide + metabolism biomarkers in malignant gliomas. 2016; 2: 189-93.
30. Kennedy BE, Sharif T, Martell E, Dai C, Kim Y, Lee PW. et al. NAD+ salvage pathway in cancer metabolism and therapy. Pharmacological Research. 2016;114:274-83
31. Garrido A, Djouder N. NAD(+) Deficits in Age-Related Diseases and Cancer. Trends Cancer. 2017;3:593-610
32. Moreira D, Rodrigues V, Abengozar M, Rivas L, Rial E, Laforge M. et al. Leishmania infantum modulates host macrophage mitochondrial metabolism by hijacking the SIRT1-AMPK axis. PLoS pathogens. 2015;11:e1004684-707
33. Abeti R, Duchen MR. Activation of PARP by Oxidative Stress Induced by β-Amyloid: Implications for Alzheimer's Disease. Neurochemical Research. 2012;37:2589-96
34. Bian C, Zhang C, Luo T, Vyas A. NADP(+) is an endogenous PARP inhibitor in DNA damage response and tumor suppression. 2019; 10: 693-706.
35. Wacker DA, Frizzell KM, Zhang T, Kraus WL. Regulation of chromatin structure and chromatin-dependent transcription by poly(ADP-ribose) polymerase-1: possible targets for drug-based therapies. Sub-cellular biochemistry. 2007;41:45-69
36. Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Molecular Aspects of Medicine. 2013;34:1124-37
37. Rush GF, Gorski JR, Ripple MG, Sowinski J, Bugelski P, Hewitt WR. Organic hydroperoxide-induced lipid peroxidation and cell death in isolated hepatocytes. Toxicology & Applied Pharmacology. 1985;78:473-83
38. Li M, Zhou ZP, Sun M, Cao L, Chen J, Qin YY. et al. Reduced Nicotinamide Adenine Dinucleotide Phosphate, a Pentose Phosphate Pathway Product, Might Be a Novel Drug Candidate for Ischemic Stroke. Stroke. 2016;47:187-95
39. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell chemical biology. 2016;23:225-35
40. El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, Dang PM. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunological reviews. 2016;273:180-93
41. Hanukoglu I. Conservation of the Enzyme-Coenzyme Interfaces in FAD and NADP Binding Adrenodoxin Reductase—A Ubiquitous Enzyme. Journal of Molecular Evolution. 2017;85:205-18
42. Huang S, Yang Z, Ma Y, Yang Y, Wang S. miR-101 Enhances Cisplatin-Induced DNA Damage Through Decreasing Nicotinamide Adenine Dinucleotide Phosphate Levels by Directly Repressing Tp53-Induced Glycolysis and Apoptosis Regulator Expression in Prostate Cancer Cells. Dna & Cell Biology. 2017;36:303-10
43. Dong Y, Wang M. Knockdown of TKTL1 additively complements cisplatin-induced cytotoxicity in nasopharyngeal carcinoma cells by regulating the levels of NADPH and ribose-5-phosphate. Biomedicine & Pharmacotherapy. 2017;85:672-8
44. Abo Alrob O, Lopaschuk GD. Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem Soc Trans. 2014;42:1043-51
45. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57-71
46. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805-21
47. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nature reviews Genetics. 2016;17:630-41
48. Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426-37
49. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612-26
50. Friis RM, Wu BP, Reinke SN, Hockman DJ, Sykes BD, Schultz MC. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res. 2009;37:3969-80
51. Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13:270-6
52. Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. 2012;198:155-64
53. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. Embo j. 2014;33:2142-56
54. Hardiville S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014;20:208-13
55. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076-80
56. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK. et al. Acetate dependence of tumors. Cell. 2014;159:1591-602
57. Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207-17
58. Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM. et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157:595-610
59. Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S. et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306-19
60. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536-50
61. Guan KL, Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011;36:108-16
62. Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288:29036-45
63. Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 2014;54:5-16
64. Ghanta S, Grossmann RE, Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit Rev Biochem Mol Biol. 2013;48:561-74
65. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337-56
66. Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258-62
67. Choosangtong K, Sirithanakorn C, Adina-Zada A, Wallace JC, Jitrapakdee S, Attwood PV. Residues in the acetyl CoA binding site of pyruvate carboxylase involved in allosteric regulation. FEBS Lett. 2015;589:2073-9
68. Adina-Zada A, Zeczycki TN, Attwood PV. Regulation of the structure and activity of pyruvate carboxylase by acetyl CoA. Arch Biochem Biophys. 2012;519:118-30
69. Pacella-Ince L, Zander-Fox DL, Lan M. Mitochondrial SIRT3 and its target glutamate dehydrogenase are altered in follicular cells of women with reduced ovarian reserve or advanced maternal age. Hum Reprod. 2014;29:1490-9
70. Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017;25:27-42
71. Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J Biol Chem. 2012;287:7051-62
72. Field MS, Kamynina E, Agunloye OC, Liebenthal RP, Lamarre SG, Brosnan ME. et al. Nuclear enrichment of folate cofactors and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) protect de novo thymidylate biosynthesis during folate deficiency. J Biol Chem. 2014;289:29642-50
73. MacFarlane AJ, Anderson DD, Flodby P, Perry CA, Allen RH, Stabler SP. et al. Nuclear localization of de novo thymidylate biosynthesis pathway is required to prevent uracil accumulation in DNA. J Biol Chem. 2011;286:44015-22
74. Palmer AM, Kamynina E, Field MS, Stover PJ. Folate rescues vitamin B12 depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. Proc Natl Acad Sci U S A. 2017;114:E4095-e102
75. Kamynina E, Lachenauer ER, DiRisio AC, Liebenthal RP, Field MS, Stover PJ. Arsenic trioxide targets MTHFD1 and SUMO-dependent nuclear de novo thymidylate biosynthesis. Proc Natl Acad Sci U S A. 2017;114:E2319-e26
76. Burdelski C, Strauss C, Tsourlakis MC, Kluth M, Hube-Magg C, Melling N. et al. Overexpression of thymidylate synthase (TYMS) is associated with aggressive tumor features and early PSA recurrence in prostate cancer. Oncotarget. 2015;6:8377-87
77. Kotoula V, Krikelis D, Karavasilis V, Koletsa T, Eleftheraki AG, Televantou D. et al. Expression of DNA repair and replication genes in non-small cell lung cancer (NSCLC): a role for thymidylate synthetase (TYMS). BMC cancer. 2012;12:342-56
78. Koumarianou A, Tzeveleki I, Mekras D, Eleftheraki AG, Bobos M, Wirtz R. et al. Prognostic markers in early-stage colorectal cancer: significance of TYMS mRNA expression. Anticancer research. 2014;34:4949-62
79. Yu Y, Ding S, Liang Y, Zheng Y, Li W, Yang L. et al. Expression of ERCC1, TYMS, TUBB3, RRM1 and TOP2A in patients with esophageal squamous cell carcinoma: A hierarchical clustering analysis. Experimental and therapeutic medicine. 2014;7:1578-82
80. Lu Y, Zhuo C, Cui B, Liu Z, Zhou P, Lu Y. et al. TYMS serves as a prognostic indicator to predict the lymph node metastasis in Chinese patients with colorectal cancer. Clinical biochemistry. 2013;46:1478-83
81. Lee YH, Yang HW, Yang LC, Lu MY, Tsai LL, Yang SF. et al. DHFR and MDR1 upregulation is associated with chemoresistance in osteosarcoma stem-like cells. Oncology letters. 2017;14:171-9
82. Wu S, Zhang G, Li P, Chen S, Zhang F, Li J. et al. miR-198 targets SHMT1 to inhibit cell proliferation and enhance cell apoptosis in lung adenocarcinoma. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:5193-202
83. Woo CC, Chen WC, Teo XQ, Radda GK, Lee PT. Downregulating serine hydroxymethyltransferase 2 (SHMT2) suppresses tumorigenesis in human hepatocellular carcinoma. Oncotarget. 2016;7:53005-17
84. Gustafsson Sheppard N, Jarl L, Mahadessian D, Strittmatter L, Schmidt A, Madhusudan N. et al. The folate-coupled enzyme MTHFD2 is a nuclear protein and promotes cell proliferation. Scientific reports. 2015;5:15029-39
85. Chen Q, Meng F, Wang L, Mao Y, Zhou H, Hua D. et al. A polymorphism in ABCC4 is related to efficacy of 5-FU/capecitabine-based chemotherapy in colorectal cancer patients. Scientific reports. 2017;7:7059-65
86. Neradil J, Pavlasova G, Veselska R. New mechanisms for an old drug; DHFR- and non-DHFR-mediated effects of methotrexate in cancer cells. Klinicka onkologie: casopis Ceske a Slovenske onkologicke spolecnosti. 2012;25(Suppl 2):2s87-92
87. Wang T, Chuan Pan C, Rui Yu J, Long Y, Hong Cai X, De Yin X. et al. Association between TYMS expression and efficacy of pemetrexed-based chemotherapy in advanced non-small cell lung cancer: a meta-analysis. PloS one. 2013;8:e74284-98
88. Mishanina TV, Kohen A. Synthesis and application of isotopically labeled flavin nucleotides. Journal of Labelled Compounds & Radiopharmaceuticals. 2015;58:370-5
89. Molano-Arevalo JC, Hernandez DR, Gonzalez WG, Miksovska J, Ridgeway ME, Park MA. et al. Flavin adenine dinucleotide structural motifs: from solution to gas phase. Analytical chemistry. 2014;86:10223-30
90. Giancaspero TA, Busco G, Panebianco C, Carmone C, Miccolis A, Liuzzi GM. et al. FAD synthesis and degradation in the nucleus create a local flavin cofactor pool. Journal of Biological Chemistry. 2013;288:29069-80
91. Hou Y, Hossain GS, Li J, Shin HD, Du G, Chen J. et al. Metabolic engineering of cofactor flavin adenine dinucleotide (FAD) synthesis and regeneration in Escherichia coli for production of alpha-keto acids. 2017; 114: 1928-36.
92. Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S. et al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nature Communications. 2012;3:758-69
93. Yun J, Johnson JL, Hanigan CL, Locasale JW. Interactions between epigenetics and metabolism in cancers. Frontiers in oncology. 2012;2:163-72
94. Yang S, Huang J, Liu P, Li J, Zhao S. Apoptosis-inducing factor (AIF) nuclear translocation mediated caspase-independent mechanism involves in X-ray-induced MCF-7 cell death. International journal of radiation biology. 2017;93:270-8
95. Zheng YC, Ma J, Wang Z, Li J, Jiang B, Zhou W. et al. A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Medicinal research reviews. 2015;35:1032-71
96. Etchegaray JP, Mostoslavsky R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Molecular cell. 2016;62:695-711
97. Niwa H, Umehara T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics. 2017;12:340-52
98. Hou H, Yu H. Structural insights into histone lysine demethylation. Current Opinion in Structural Biology. 2010;20:739-48
99. Zhao ZK, Yu HF, Wang DR, Dong P, Chen L, Wu WG. et al. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World Journal of Gastroenterology. 2012;18:6651-6
100. Kaochar S, Tu BP. Gatekeepers of chromatin: Small metabolites elicit big changes in gene expression. Trends in biochemical sciences. 2012;37:477-83
101. Giancaspero TA, Busco G, Panebianco C, Carmone C, Miccolis A, Liuzzi GM. et al. FAD synthesis and degradation in the nucleus create a local flavin cofactor pool. The Journal of biological chemistry. 2013;288:29069-80
Author contact
![]() Corresponding authors: Liu, S. Email: shuangliu2016edu.cn; Tao, YG. Email: taoyongedu.cn; Tel. +(86) 731-84805448; Fax. +(86) 731-84470589.
Corresponding authors: Liu, S. Email: shuangliu2016edu.cn; Tao, YG. Email: taoyongedu.cn; Tel. +(86) 731-84805448; Fax. +(86) 731-84470589.